 Yuqing Liu1
Yuqing Liu1 Rongcun Yang
Rongcun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1518779
This article is part of the Research Topic Mechanism Explorations of Enhancing Immunotherapeutic Sensitivity via Mediating Immune Infiltration and Programmed Cell Death in Solid Tumor Microenvironment View all 8 articles
Anti-tumor immunity, including innate and adaptive immunity is critical in inhibiting tumorigenesis and development of tumor. The adaptive immunity needs specific lymph organs such as tertiary lymphoid structures (TLSs), which are highly correlated with improved survival outcomes in many cancers. In recent years, with increasing attention on the TLS in tumor microenvironment, TLSs have emerged as a novel target for anti-tumor therapy. Excitingly, studies have shown the contribution of TLSs to the adaptive immune responses. However, it is unclear how TLSs to form and how to more effectively defense against tumor through TLS formation. Recent studies have shown that the inflammation plays a critical role in TLS formation. Interestingly, studies have also found that gut microbiota can regulate the occurrence and development of inflammation. Therefore, we here summarize the potential effects of gut microbiota- mediated inflammation or immunosuppression on the TLS formation in tumor environments. Meanwhile, this review also explores how to manipulate mature TLS formation through regulating gut microbiota/metabolites or gut microbiota associated signal pathways for anti-tumor immunity, which potentially lead to a next-generation cancer immunotherapy.
Anti-tumor immunity, including innate and adaptive immune responses is critical in inhibiting tumorigenesis and tumor development. Notably, anti-tumor adaptive immunity needs specific lymph organs such as lymph nodes (secondary lymphoid organs) and tertiary lymphoid structures (TLSs), also known as ectopic lymphoid structures (ELSs) or tertiary lymphoid organs (TLOs) (1). When TLSs are located in close vicinity of tumor, they are active sites of inducing adaptive immune responses against tumor (2, 3). Indeed, emerging studies have already revealed the contribution of the TLSs to adaptive immune response against tumor (4) such as the production of antibodies that can mark tumor cells for complement-mediated lysis, antibody-dependent cellular cytotoxicity or opsonization (5). Increased activation markers has also been observed on the T cells in TLSs, as compared with other tumor-resident T cells in melanoma (6). These TLSs were highly correlated with improved survival outcomes in many tumors, such as breast cancer, hepatocellular cancer (HCC), colorectal cancer (CRC), melanoma, gastric cancer, head and neck squamous cell cancer (HNSCC), lung cancer and sarcoma (7, 8). However, TLS formation remains to be further clear.
Although the factor(s) and mechanism(s) influencing TLS formation are incompletely clear, some studies have shown that TLSs are generally derived from hematopoietic LTi (lymphoid tissue inducers), which can interact with stromal cells known as LTo (lymphoid tissue organizers) to form mature TLS through immature TLS (Figure 1) (2, 8, 9). The stromal cells can express adhesion molecules and chemokines to recruit immune cells from adjacent high endothelial venules (HEV) for TLS formation. Notably, LTi is also converted from nature killer (NK) cells under the stimulation of transforming growth factor (TGF)-β and IL-12 (10–13). In addition, chronic inflammation also plays a critical role in TLS formation (14). It not only induces immune cells such as T helper cells secreting IL-17 (TH17) to become LTi but also produces widely cytokines and chemokines, which are necessary to TLS formation (14). The requirement of chronic inflammation for TLS formation not only emerges from cancer mouse models but also from clinical observations (15–17).
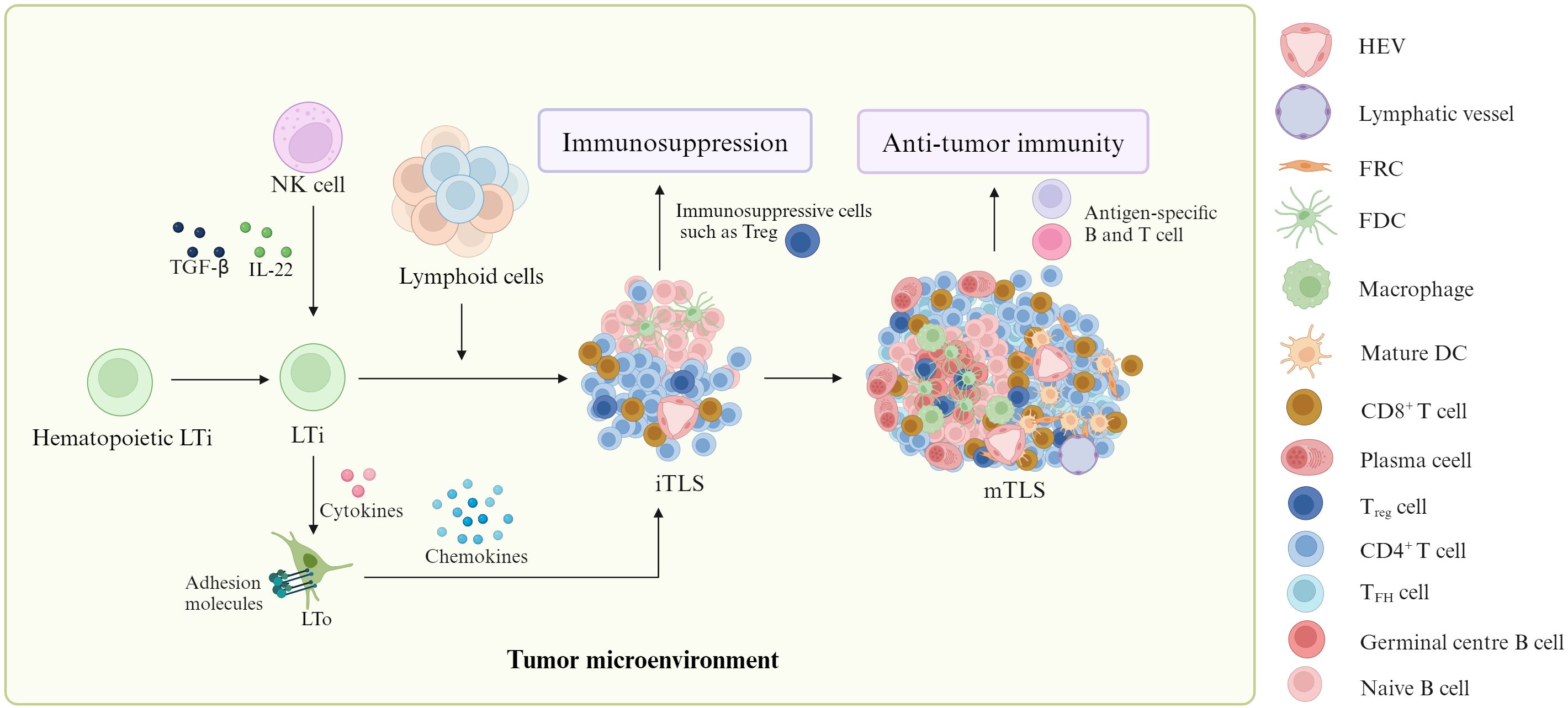
Figure 1. Formation of TLS. In tumor environment, lymphoid tissue inducers (LTi) can produce cytokines to work on lymphoid tissue organizers (LTo). These LTo express adhesion molecules and chemokines to recruit immune cells from adjacent high endothelial venules (HEV) for mature tertiary lymphoid structure (mTLS) formation through immature TLS (iTLS) containing a lot of immunosuppressive cells such as T regulatory cells (Tregs). MTLSs can produce antigen-specific T and B cells to promote anti-tumor immunity. Notably, natural killer (NK) cells could be converted into LTi. ITLS can cause immunosuppression through immunosuppressive cells such as Treg.
Gut microbiota can regulate the occurrence and development of inflammation, which potentially affect TLS formation. Indeed, the gut microbes, which appear in tumor tissue can promote TLS formation. For example, Helicobacter pylori (H. pylori) inside tumors could promotes the formation of TLS to trigger anti-tumor immune responses (18). The gut microbiota Lachnoclostridium was also related to the existence of intratumoral TLSs in hepatocellular carcinoma (19). Besides the microbes from gut microbiota in tumor tissues, the metabolites derived from gut microbiota also potentially affect the TLS formation. These metabolites can enter tumor tissues through bloodstream. Since gut microbiota and its metabolites (microbiota/metabolites) are one of the major environmental factors that affects TLS formation (20), we here summarize the potential contribution of gut microbiota/metabolites mediated inflammation or immunosuppression on the TLS formation in tumor environments, especially on the different immune cells such as macrophages (Macs), dendritic cells (DCs), Tregs (regulatory cells), LTi, Tfh (follicular helper T cell) and B cell in TLSs (21). Since there exists the association of TLS with a favorable response to immunotherapy against tumor, studies are employing different approaches to induce TLS formation in vivo. This review also explores the potential opportunity to promote TLS formation for defensing against tumor through regulating gut microbiota/metabolites or their signal pathway(s) in immune cells. Notably, the effects of gut microbiota/metabolites on TLS formation have only just begun. Of course, besides gut microbiota, others such as tumor-derived factors also influence TLS formation.
TLSs, as ectopic lymphoid organs develop in non-lymphoid tissues at the sites of chronic inflammation such as tumors (22). They share the vasculature, chemokines, cellular compartments, spatial organization, and function with secondary lymphoid organs, especially lymph nodes, where adaptive anti-tumor cellular and humoral responses can be generated (23).
TLSs in cancer tissues are heterogeneous (7). Their cell composition and distribution is also different in different cancer types (24), ranging from disorganized cellular aggregates such as early TLS or immature (iTLS) to well-organized and structured organs which are similar to secondary lymphoid organs (SLOs) such as lymphoid nodes (2, 25). Thus, TLSs include multiple lymphoid structures, from immune cell aggregates to organized structures, which form primary follicle (PFL) or secondary follicle (SFL) with a germinal center (GC) in the tumor microenvironment (TME) (2).
Lymphoid aggregates are composed of a few B cells and T cells without any follicular dendritic cells (FDCs) (23). In immature TLS (iTLS), besides T, B cells and FDCs, there often are aggregates of immune suppressive cells that suppress anti-tumor immunity such as higher programmed death-ligand 1 tumor-associated Macs (26), immature DCs (26), regulatory T cells (26, 27), PD-1high CD8 T cells (26, 28) and PD-1highCD4 T cells (28). Typically, a low CD8/Treg cell ratio in lung squamous cell carcinoma (LUSC) (29), interleukin (IL)-10 expressing monocytes and T cells in pancreatic ductal adenocarcinoma (PDAC) (30) and phosphoprotein 1 (SPP1)+ Macs in the lung cancer activation modules (LCAMs) (31) have also been reported. Notably, one study also exhibited that there were only minor differences in immune cell frequencies in immature versus mature TLS with the exception of B cells (28).
Signatures of mature TLSs have been described in other papers (2, 23). They have typical structures comprised of an internal B-cell zone surrounded by T cell-rich area, which includes CD8 cytotoxic T, CD4 Th-1 and Tfh lymphocytes, as well as LAMP3+ DCs (32). The B cell zone has a network of follicular DCs (FDCs), which express CD21 in PFL as well as CD23 in SFL. These TLSs reveal clear active GCs within the B-cell area with the mature DCs (33), antigen-experienced CD4, CD8 and B cells (34).
Inflammation plays a critical role in TLS formation (Figure 2). TLS formation relies on the cytokine signaling network between heterogeneous cell populations such as lymphocytes, stromal cells and cancer cells (35). TLSs can be generally derived from LTi, which can produce cytokines such as lymphotoxin α1β2 (LTα1β2), TNFα and IL-17 (8, 36), which interact with the receptors on the LTo cells such as stromal cells. These LTo cells express adhesion molecules such as VCAM1, ICAM1, and MADCAM1, and chemokines such as CXCL13, CXCL12, CCL21 and CCL19, which can recruit immune cells from adjacent HEV to participate in TLS formation. Interestingly, HEV can express PNAD (peripheral lymph node addressin), a L-selectin receptor, which is necessary for leukocytes to the TLSs. However, in response to chronic inflammatory, several immune cell populations can potentially replace LTi cells, including TH17 (37, 38), natural cytotoxicity receptor (NCR)+ ILC3 (NCR+ILC3) (39), M1-polarized Macs (40), effector CD8 T cells and NK cells (41), as well as B lymphocytes (42). These cells can interact with stromal cells to induce TLS formation in the absence of LTi cells. Some local stromal cells such as fibroblasts can be acted as LTo (43). In addition, inflammation also promotes immature TLSs into mature TLSs through cytokines and chemokines (23) such as interleukin-17A (IL-17A), which has been shown to be involved in the formation of TLSs (44). Indeed, the requirement of chronic inflammation for TLS formation not only emerges from cancer mouse models but also from clinical observations (45, 46).
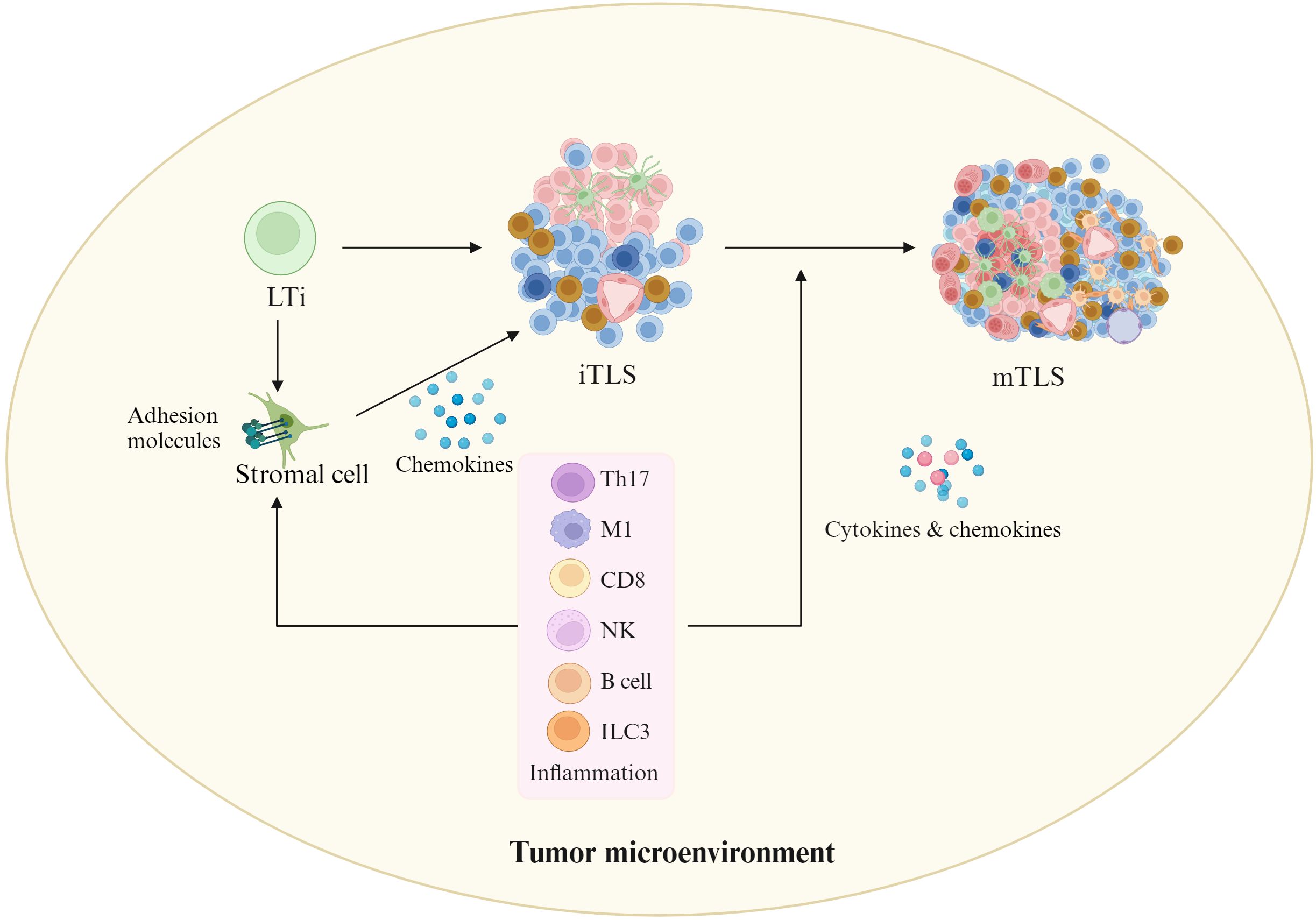
Figure 2. Effects of inflammation on TLS formation. In tumor environments, inflammatory cells such as ILC3 (innate lymphoid cell 3), T helper cells secreting IL-17 (Th17), M1-polarized Macs, effector CD8 T cells and natural killer (NK) cells, as well as B lymphocytes may replace LTi cells. They can interact with stromal cells to initiate TLS formation in the absence of LTi cells. In addition, inflammation also promotes immature TLSs into mature TLSs through cytokines and chemokines. Cells in iTLS and mTLS, see Figure 1.
Gut microbiota/metabolites play a critical role in the development of LTO. Since TLSs are similar to secondary lymphoid organs such as LN (47, 48), it may be useful to predict the effects of gut microbiota/metabolites on the TLS formation. Indeed, the microbiota has been shown to promote formation of TLSs (23). For instance, H. hepaticus promoted enrichment of TLSs in a mouse model of colorectal cancer (CRC) (18). Notably, although the effects of gut microbiota/metabolites on the differentiation and function of immune cells have been widely studies, the role of gut microbiota/metabolites in TLS formation has only just begun.
Gut microbiota/metabolites not only affect inflammatory immune cells but also immunosuppressive cells, which can potentially affect formation of TLSs, typically metabolites derived from gut microbiota such as short chain fatty acids (SCFAs) (49, 50), bile acid (BA) (50–52) and tryptophan (Trp) metabolites (53, 54), and other metabolites, which have been reviewed by us and others. SCFAs such as butyrate, propionate, and acetate are from dietary fiber fermentation by gut microbiota in the cecum and colon (49, 50). The primary BAs cholic acid (CA) and chenodeoxycholic acid (CDCA) generated in the liver can conjugate CDCA, DCA or CA to one or more amino acids by bacteria (55). Four distinct ways including deconjugation, oxidation, dehydroxylation and epimerization in human are used to transform BAs (56). While BAs are deconjugated, BAs can be converted into secondary BAs deoxycholic acid (DCA) and lithocholic acid (LCA) through bacterial bile acid hydrolases (57) and dehydroxylases (58) to remove 7α or 7β-hydroxyl groups from primary BAs in the colon. In addition, a range of oxo-, iso- and epi-derivatives such as 3-oxoLCA, 7-oxoCDCA, 12-oxoCA, 7-oxoCA, 12-oxo-DCA, 3-oxo-LCA, 3-oxo-allo-LCA, iso-LCA, iso-alloLCA, allo-LCA and 3-ketoLCA are also generated by gut bacteria (56, 59, 60). Gut microbiota also metabolizes Trp into tryptamine and indole derivatives, such as indole, indole-3-acid-acetic (IAA), indole acetic acid, indole-3-propionic acid (IPA), indole-acrylic acid (IA), indole-3-aldehyde (IAld), indole-3-pyruvate (IPy), indole-3-acetamide (IAM), indoxyl sulfate and tryptamine (61, 62). In addition, some bacteria also produce kynurenine (Kyn) and downstream metabolites such as 3-hydroxyanthranilic acid (3-HAA) through enzymes homologous to those of the eukaryotic kyn pathway (63).
These gut microbial metabolites can regulate the differentiation and function of immune cells through various receptors. SCFAs are through G-protein coupled receptor (GPR)41 (also known as free fatty acid receptor (FFAR3)), GPR109a (also called hydroxycarboxylic acid receptor 2 (HCA2)) and GPR43; Whereas cellular membrane receptors such as G-protein BA receptor 1 (GPBAR1) known as Takeda G protein-coupled receptor 5 (TGR5), and nuclear receptors such as farnesoid X receptor (FXR), pregnane X receptor (PXR), vitamin D receptor (VDR), liver-X-receptor (LXR), constitutive androstane receptor (CAR), and retinoid related orphan receptor (RORγt) (64), are used by BA derivatives. A variety of Trp-indole metabolites are also through receptors such as aryl hydrocarborn receptor (AhR) and PXR to exert their functions (65).
The gut microbiota is a complex ecosystem of approximately 100 trillion microorganisms inhabiting the human gut, including bacteria, viruses, fungi and protozoa (66). The vast majority is represented by Firmicutes (gram-positive, 60–80%) and Bacteroidetes (gram-negative, 20–30%) along with Proteobacteria and Actinobacteria (67), which can induce either inflammation or immunosuppression in the individuals. The gut microbiota plays a critical role in the development of lymph organs, including primary lymphoid organs such as thymus (68) and bone marrow cells (69), and second lymphoid organs such as lymph node (LN) and Peyer’s patches (70), spleen, tonsils and mucosa-associated lymphoid tissues, which support immune surveillance in the mammalian organisms. Indeed, studies have found that secondary lymphoid organs of germ-free mice are under-developed (71). Antibiotic treatment, which resulted in decreased gut microbiota, also reduced DC and effector CD8 T cell responses, and attenuated responses to immune checkpoint blockade therapy (72).
Since inflammation plays a critical role in TLS formation, inflammation-associated gut microbiota/metabolites can potentially promote TLS formation (Figure 3).
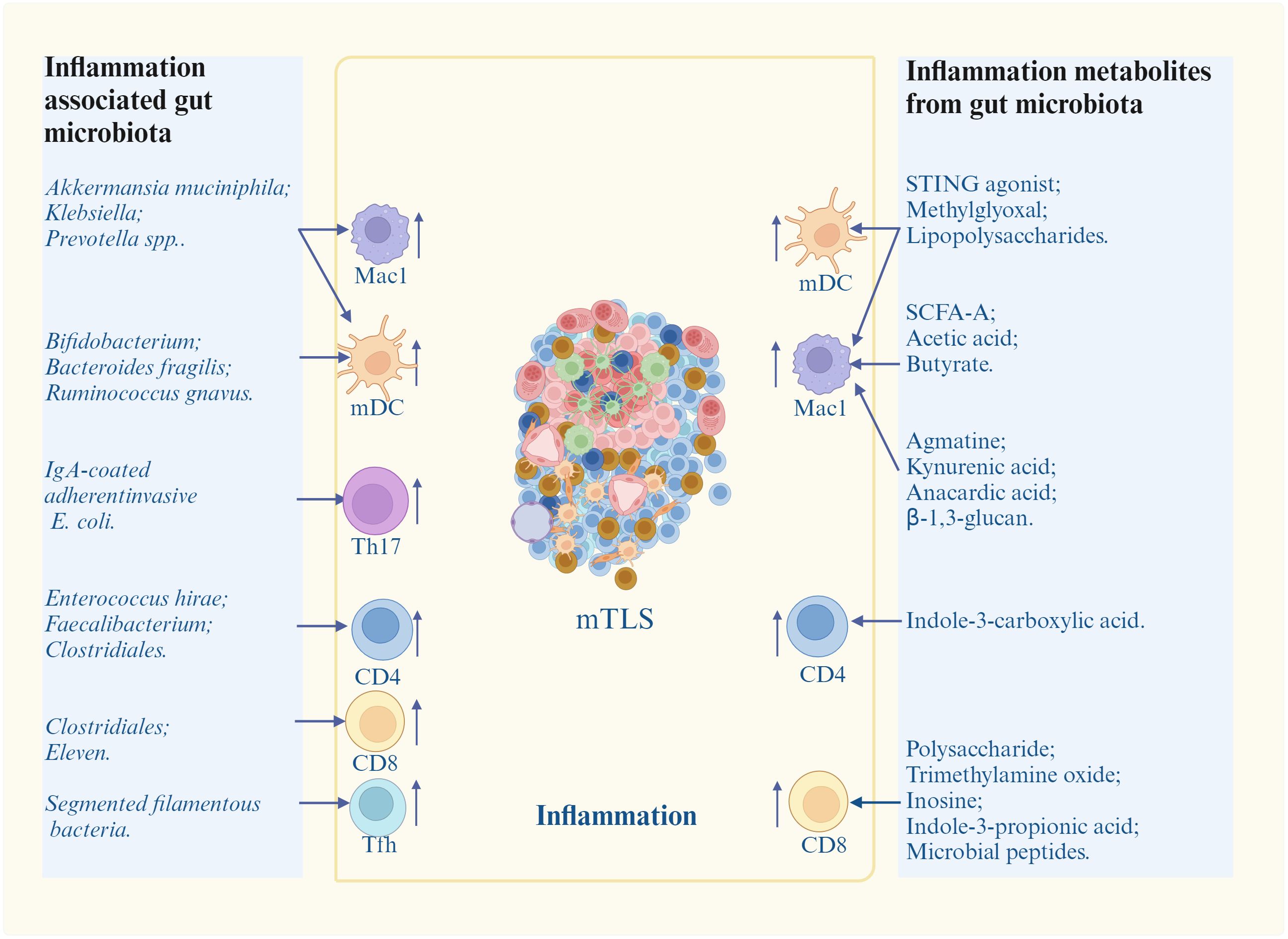
Figure 3. Promotion of inflammation-associated gut microbiota/metabolites on mature TLS (mTLS) formation. Inflammation associated gut microbiota/metabolites induce and/or promote mature TLS formation through different inflammatory immune cells. mDC, nature dendritic cells; Mac1, inflammatory macrophages; Th17, T helper 17 cells; Tfh, T follicle helper cells; STING, stimulator of interferon genes; SCFA-A, short chain fatty acid A. Cells in mTLS, see Figure 1.
Multiple bacteria can cause inflammation such as Akkermansia muciniphila (A. muciniphila) (73, 74), Prevotella spp (75), Klebsiella (76), Bifidobacterium (77, 78), Bacteroides fragilis (B. fragilis) (79), B. fragilis (80), Ruminococcus gnavus (81), Segmented filamentous bacteria (SFB) (82), IgA-coated adherent-invasive E. coli (AIEC) (83), Clostridiales (84), Faecalibacterium (85), Enterococcus hirae (86, 87) and Eleven strains (79). A. muciniphila could produce cdAMP to activate stimulator of interferon genes (STING)-interferon pathways of immune cells such as DCs and Macs to produce cytokines (73, 74). Prevotella spp up-regulated pro-inflammatory responses in Macs (75). Klebsiella activated NF-κB and promoted the secretion of pro-inflammatory IL-1, IL-6 and TNF-α in Macs (76). Bifidobacterium could alter the functional capacity of DCs to induce CD8 T cell proliferation and Th1 differentiation, and also IFNγ production (77, 78); Bacteroides fragilis (B. fragilis) could stimulate DC maturation to induce IL-12-dependent Th1 cell immune responses (79). B. fragilis also induced Mac polarization to M1 (80). Ruminococcus gnavus induced TNF-α secretion of DCs through pro-inflammatory polysaccharide (PSA) (81). Segmented filamentous bacteria (SFB) induced differentiation of Tfh cells in Peyer’s patches thorough DC-mediated inhibition on IL-2-related pathway, and also directed Tfh trafficking to lymphoid tissues responsible for antibody production (82). IgA-coated adherent-invasive E. coli (AIEC) triggered Th17 cells activation to increase systemic immune responses (83); Clostridiales promoted antigen presentation to promote CD4 and CD8 T cell function in melanoma patients (84). Faecalibacterium increased CD4 T cell proportion in peripheral blood in metastatic melanoma patients (85). Enterococcus hirae could induce the polarization of immune cells towards a Th1 phenotype in secondary lymphoid organs in P815 mastocytomas established in syngenic DBA2 mice (86, 87). Eleven strains robustly induced IFN γ+CD8 T cells in syngeneic tumor models (79). Bacteria C. rodentium induced pro-inflammatory Th17 cells (88).
Bacterial structural components also modulate immune cells through different receptors such as Toll-like receptors (TLR) and nod-like receptors (NLR). There have existed considerable literatures on various TLR and NLR ligands from gut microbiota impacting immune cells. For example, lipopolysaccharides (LPSs), a component of the Gram-negative bacterial outer membrane, was identified as a key contributing factor in the initiation and progression of inflammation (89). It could promote production of pro-inflammatory cytokines (TNF, IL-17, IL-22, etc.) in immune cells such as monocytes, Macs, and Kupffer cells, which expressed LPS receptors such as Toll-like receptor 4 (TLR4) (90). Bacterial-derived lipoteichoic acid could exert strong effects on immune cells via host receptors and targeted molecules. Microbial peptides also activated tumor-infiltrating lymphocytes in the tumor such as glioblastoma (91).
In conclusion, since the gut microbiota and its structural components can promote inflammation through stimulating different immune cells, they should be effective in promoting TLS formation against tumors.
There are a lot of gut microbiota derived metabolites, which can cause inflammation such as stimulator of interferon gene (STING) agonists (92), methylglyoxal (93), SCFAs (94), agmatine (95), kynurenic acid (KYNA) (96), anacardic acid (97), tryptophan (Trp) metabolites (98), trimethylamine oxide (TMA) (99), inosine, microbial peptides (91) and β-1, 3 –glucan (100). STING agonists can promote TLS formation (Figure 4). Chelvanambi et al. demonstrated that intratumoral administration of STING agonist ADU-S100 could cause sustained inflammation in the TME of B16 melanomas, and production of cytokines/chemokines (LTα, LTβ, CCL19 and CCL21, as well as LIGHT), which could promote TLS formation (92). CD11c+ DCs activated by STING agonist ADU S-100 also upregulated expression of lymphotoxin-α (LTA), IL-36, chemokines and type I interferons in vitro and in vivo, which could promote TLS formation (92). The cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)-STING pathway also initiated TLS formation through chemokine mediated cross-talk between endothelial cells and T cells (101). Mechanically, cell-intrinsic cGAS could lead to CCL5 production in vascular endothelial cells. Then, peripheral CD8 T cells were recruited to produce CXCL13 and interferon-γ to promote TLS formation (101). Methylglyoxal from gut microbes could boost radioimmunotherapy in rectal cancer by triggering endoplasmic reticulum stress and cGAS-STING activation, which could induce TLS formation (93). There existed a positive correlation between anti-PD-1/PD-L1 responses and bacteria that produce SCFAs like Eubacterium, Lactobacillus, and Streptococcus (102). SCFA-A, a metabolite of gut microbiota was also involved in the activation of T cells and induction of M1 Macs, thereby enhancing the anti-tumor effects of anti- PD-1 antibody therapy (103). B. thetaiotaomicron-derived acetic acid had the potential to modulate the polarization of pro-inflammatory Macs, which promoted CD8 T cell function in hepatocellular carcinoma (104). Gut microbiota-derived butyrate inhibited the immunosuppressive factors PD-L1 and IL-10 in tumor-associated Macs in gastric cancer (94). Butyrate also promoted the production of antitumor cytokines in cytotoxic CD8 T cells by regulating T-cell receptor (TCR) signaling pathway in patients with non-small cell lung cancer (NSCLC) (105). Gut microbiota-derived metabolite agmatine could suppress the Rnf128-mediated ubiquitination of β-catenin to upregulate the genes, which could activate Wnt pathway, including Cyclin D1, Lgr5, CD44 and C-myc. The activated Wnt signaling pathway could upregulate pro-inflammatory cytokines (IL-6 and TNF-α) and downregulate anti-inflammatory cytokine (IL-10) in colorectal tumorigenesis (95). Gut microbiota-induced kynurenic acid induced GPR35-positive Macs to promote inflammation (96). Anacardic acid could also activate Macs thorough mitogen-activated protein kinases (MAPKs) (97). Anacardic acid promoted tumor-infiltrating immune cells such as tumor-infiltrated NK cells and CTLs through inducing production of a neutrophil extracellular trap in breast cancer models (97). L. gallinarum-derived Trp metabolite indole-3-carboxylic acid (ICA) could compete with Kyn for binding site on AhR on CD4 T cells to inhibit Treg differentiation in vitro (98). I3AA, a Trp metabolite made by a gut microbia induced pro-inflammatory T cells, and reduced Treg subset in vivo and iTreg development in vitro via regulating response to TGFβ (106). IPA derived from L. johnsonii, could enhance the differentiation of CD8 T cells through H3K27 acetylation at the super-enhancer (SE) of Tcf7 gene in pan-cancer (107). In mice and humans, PSA generated by L. delbrueckii could induce CCR6+CD8 T cells (108). The choline or carnitine in foods were metabolized to generate trimethylamine (TMA) by the gut microbiota, which enters the liver through portal vein. Then TAM, which was catalyzed to produce trimethylamine oxide (TMAO) in liver, was demonstrated to promote CD8 T cell-mediated anti-tumor immunity in mouse models of triple-negative breast cancer (99). Inosine, a purine metabolite of A. muciniphila and B. pseudolongum, strengthened differentiation and proliferation of T cells (109), induced differentiation of B cells, and antibody production via activating Macs (109), and promoted antitumor immunity through Th1 differentiation and effector function of T cells (77). Some bacterial species, which could produce inosine or its metabolite hypoxanthine, induced T-helper 1 differentiation and effector functions via inosine-A2AR-cAMP-PKA pathway (110). The systemic administration of β-1,3-glucan from a fungal element promoted cytokine secretion (100). Thus, metabolites derived from gut microbiota, which can induce inflammation, also potentially result in mature TLS formation.
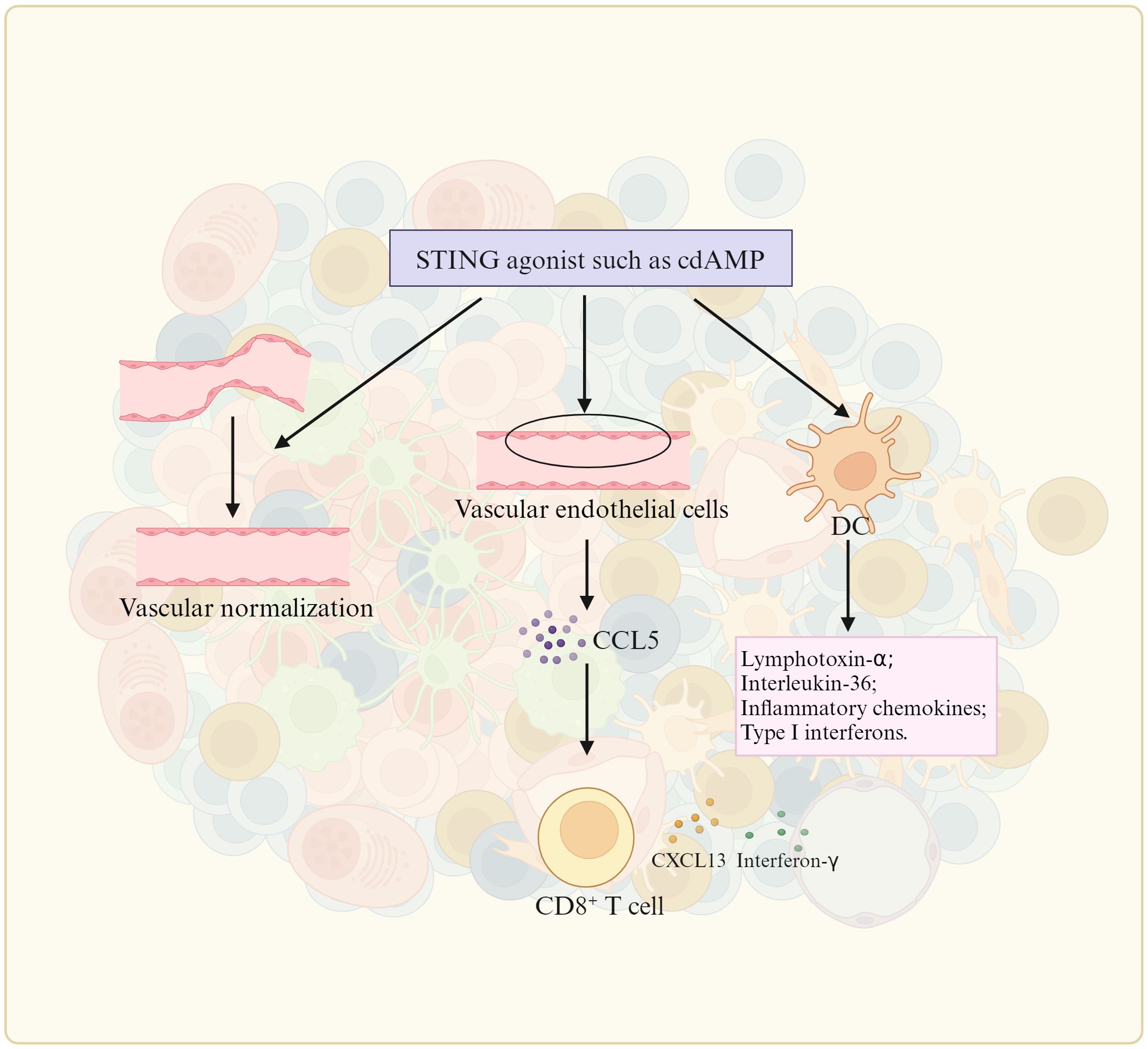
Figure 4. STING agonist promotes TLS formation. The stimulator of interferon genes (STING) promote TLS formation by upregulating the expression of TLS-promoting factors such as lymphotoxin-alpha (LTa), IL-36, type I IFN, inflammatory chemokines by DC (dendritic cells), vascular normalization, and CCL5 (CC chemokine ligand-5) production in vascular endothelial cells.
Besides stromal cell and HEVs, mature TLSs are mainly composed of the different kinds of immune cells, including Macs, DCs, T cells such as (CD8, Tfh, Th1, Th17, and Treg) and B cells; Whereas in iTLS, besides T and B cells, there often are aggregates of immature and immune suppressive cells such as immunosuppressive Macs, immature DCs and Treg (26, 27), which can potentially inhibit mature TLS formation. Indeed, in a mouse LUAD model, Treg cell depletion not only enlarged the lung area covered by TLS but also increased levels of T cell proliferation and co-stimulatory ligand expression by DCs in tumor-associated TLSs (111). Repressing immunosuppressive cells such as anti-inflammatory macrophage or Breg cells also favored formation of TLS. Notably, the immunosuppressive cells can be regulated by gut microbiota/metabolites. Here, we mainly discuss the effects of anti-inflammatory gut microbiota or gut microbiota derived metabolites such as SCFAs, BA and Trp metabolites on these immunosuppressive cells, which can potentially inhibit the TLS formation (Figure 5).
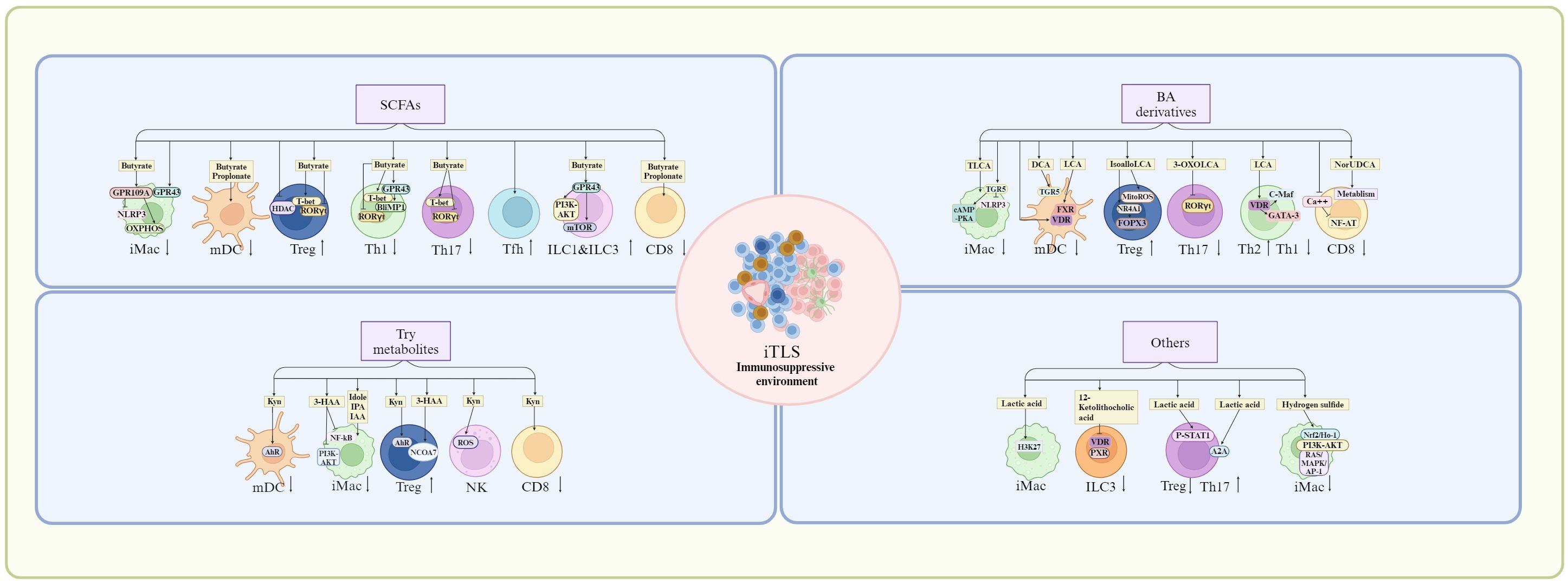
Figure 5. Suppression of anti-inflammation gut microbiota/metabolites on TLS formation. Anti-inflammation gut microbiota/metabolites can potentially inhibit the TLS formation by immunosuppressive cells or immature immune cells through their receptors such as short chain fatty acid (SCFA) receptors such as G-protein coupled receptor (GPR)43 and GPR109, bile acid (BA) derivative receptors such as farnesoid X receptor (FXR), VDR (Vitamin D receptor), retinoid related orphan receptor (RORγt), and tryptophan-indole metabolites receptors such as aryl hydrocarborn receptor (AhR). mDC, mature dendritic cells; iMac, inflammatory macrophages; Th17, T helper 17 cells; Tfh, T follicle helper cells; Treg, T regulatory cells; ILC, innate lymphoid cells; NLRP3, nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain-containing 3; OXPHOS, oxidative phosphorylation; HDAC, histone deacetylase; PI3K, phosphoinositol-3 kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NR4A1, nuclear receptor subfamily 4 group A member 1; cAMP-PKA, cyclic adenosine monophosphate/protein kinase A; NCOA7, nuclear receptor co-activator protein 7; Nrf/Ho-2, nuclear factor E2-related factor/heme-oxygenase-2; A2A, A2A adenosine receptor; ROS, reactive oxygen species; RAS/MAPK, rat Sarcoma mitogen-activated protein kinase; AP-1, activator protein 1; ERK, extracellular-signal regulated kinase; JAK/STAT, Janus Kinase/Signal transducer of activated T-cells; TLCA, taurolithocholic acids; DCA, deoxycholic acid; LCA, lithocholic acid; 3-HAA, 3-hydroxyanthranilic acid; IPA, 3-indolepropionic acid; IAA, indole-3-acetic acid; Kyn, kynurenine. Cells in iTLS, see Figure 1.
Some bacteria from gut microbiota are resistant to inflammation such as Bacteroides (112, 113), Faecalibacterium prausnitzii (112, 113), Parabacteroides distasonis (114), Segmented filamentous bacteria (90) and Bacteroides genus (115). Bacteroides, and Faecalibacterium prausnitzii could inhibit NF-κB to reduce pro-inflammatory cytokines such as IL-8 and TNF-α (112, 113). Parabacteroides distasonis inhibited the expression of pro-inflammatory cytokines (IL-1β, IL-6, IL-17A, and TNF-α), increased anti-inflammatory cytokines (IL-10), reversed Th17/Treg imbalance in the mesenteric LN (MLN) of mice with arthritis, and even induced M2-type polarization of Macs (114). Segmented filamentous bacteria was involved in the promotion of maturation of Treg cells, and also performed a mutualistic interaction with the gut microbiota by anti-inflammatory cytokines, such as IL-10 and TGF-β (90).
Interaction between segmented filamentous bacteria (SFB) and epithelial cells promoted formation of endocytic vesicles containing bacterial cell wall proteins, causing generation of non-inflammatory helper T (Th) 17 cells (116). Bacteroides genus was correlated with increased myeloid derived suppression cells (MDSCs) and the cytokines IL-8 and IL-13, which had roles in MDSC recruitment and proliferation, respectively (115). Lactobacillus acidophilus improved intestinal inflammation by modulating the balance of Th17 and Treg cells (117). The polysaccharide A from Bacteroides fragilis (118) facilitated the expansion and differentiation of intestinal Foxp3+ Tregs in addition to the production of IL-10 and TGF-β that regulate the functions of intestinal myeloid cells (119). Thus, the anti-inflammatory gut microbiota can potentially inhibit the formation of TLS in tumor environment.
Gut microbiota metabolites such as SCFAs, BA and Trp metabolites can inhibit inflammation. After interacting with GPR43 or GPR109A, SCFAs could regulate the activation of the NLRP3 and production of IL-18 to inhibit the activation of the Macs (120, 121). Butyrate also reprogramed Mac metabolism toward oxidative phosphorylation, causing an anti-inflammatory tolerant phenotype (122) through oxidative phosphorylation (OXPHOS) (123). Notably, SCFAs could down-regulate DC-secreted chemokines such as C-C chemokine ligand 5, up-regulate anti-inflammatory IL-10 and suppress DC maturation (124). SCFAs butyrate and propionate also inhibited LPS-mediated maturation of human monocyte-derived DCs in vitro (125, 126). SCFAs could promote differentiation and expansion of Treg cells. SCFA butyrate induced Treg cell differentiation through histone H3 acetylation at the Foxp3 promoter of the Foxp3 locus (127, 128), and fatty acid oxidation (129). Butyrate also supported DC-induced differentiation of Treg cells through butyrate receptor GPR109 and the butyrate transporter SLC5A8 in the DCs (130). Notably, the IL-10 expression in Th1, Th17 and Treg cells was promoted by SCFAs by suppression of histone deacetylases and regulation of the mTOR-S6K pathway (117). SCFAs also enhanced the generation of CXCR5+ T follicular helper cells in vitro and in vivo, which supported B cell differentiation (131). SCFAs, particularly butyrate stimulated ILCs via upregulating expression of AhR and HIF (132), the activation of the PI3K–AKT and mTOR signaling pathways in a GPR43-dependent manner to promote the proliferation of intestinal ILC1s and ILC3s (133). SCFA butyrate and propionate also regulated CD8 T cell activation via inhibiting IL-12 production in DCs. Taken together, SCFAs such as butyrate may inhibit TLS formation through immunosuppressive cells.
Through BA receptors TGR5, BA metabolites were essential to maintain tolerant phenotypes of the Macs (134–136). They modulated anti-inflammatory macrophage phenotype transformation and inhibited pro-inflammatory cytokine production through cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway and regulating NLRP3 (137). The taurine-conjugated DCA (TLCA) also inhibited the expression of cytokines, such as IL-12 and TNFα and the expression of chemokines, CCL2, CCL 3, CCL4, CCL5, CXCL9, and CXCL10 in LPS-activated human Macs (138). DCA also inhibited the production of proinflammatory cytokines, such as IL-1β, IL-6, IL-12, IL-23, and TNF-α from LPS-activated DCs via TGR5 (139), whereas LCA suppressed the expression of proinflammatory cytokines via FXR (139). DC maturation and the inflammatory cytokine production were inhibited by VDR (140). DCA treatment also impeded Th1 and Th17 differentiation although LPS-activated DCs induced the differentiation of naïve CD4 T cells into Th1 or Th17 (141). BA derivatives isoalloLCA could selectively upregulate the expression of FoxP3 (142) through NR4A1, which could bind to 500 bp upstream of the Foxp3 transcriptional start site (143, 144). It also promoted the differentiation of Tregs through mitochondrial reactive oxygen species (mitoROS) (142, 145, 146). 3-oxoLCA inhibited the differentiation of Th17 cells through RORγt (142). Notably, Foxp3 expression was also induced by BA metabolite isoDCA by reducing DC immune-stimulatory properties (145). Through increased transcription factors c-Maf and GATA-3, a shift from the Th1 to the Th2 phenotype was promoted by VDR activation (147). Through a VDR-dependent mechanism, the CD4 Th1 cell activation was suppressed by unconjugated BA metabolite LCA, causing diminished TNFα and IFNγ (148). BA metabolites also disrupted intracellular calcium homeostasis. The intracellular calcium was critical for NFAT (nuclear factor of activated T cells) signaling, which was necessary for T cells activation (149). The immune-metabolism in CD8 T cells could be reshaped by 24-Norursodeoxycholic acid (NorUDCA) to alleviate inflammation (150). Thus, these BA derivatives can also potentially inhibit the formation of TLS.
Trp metabolites from gut microbiota were essential in regulating the function of Macs via receptor AhR. Through suppressing histamine production, Trp metabolites could mediate suppression on inflammatory responses in the Macs (151). Trp metabolite 3-HAA inhibited LPS mediated NF-κB (nuclear factor κ gene binding) and PI3K (phosphatidylinositol 3 kinase)/Akt (protein kinase B)/mTOR (mammalian target of rapamycin) signaling pathways to reduce inflammatory cytokine production in the Macs (152). A recent report has demonstrated that the gut-bacterial-released indole and its metabolites (IPA and IAA) interacted with myeloperoxidase to inhibit its inflammatory function in the polymorphonuclear leukocytes at physiological concentrations (153). The kynurenine (Kyn), as an endogenous ligand of AhR, could induce AhR activation when generated in the tumor microenvironment (154), which promote tolerant phenotype in DCs to mediate the generation and expansion of Tregs.
Through AhR-ligand-Treg axis (155, 156), gut microbiota derived Trp metabolites promoted differentiation and function of Tregs. Kyn and its metabolites also enhanced Treg differentiation through the AhR (157–159), direct transactivation and the induction of epigenetic modifications, which controlled foxp3 transcription (160–162). 3-HAA in Kyn pathway also induced the differentiation and production of Treg cells via a nuclear coactivator 7 (NCOA7)-dependent pathway (163), and caused immune suppression by inducing apoptosis in T-cells through glutathione depletion (164). The expression of PD-1 in CD8 T cells, an immune inhibitory molecule, could be upregulated through Kyn (165). Through ROS pathway, the activity of NK cells was suppressed by Kyn, which could lead to cell death (166). Kyn also suppressed cytokine-mediated receptors, which were responsible for NK cells-mediated killing (167). Thus, Trp-indole derived metabolites also prevent the TLS formation through immunosuppressive cells.
Other some gut microbiota derived metabolites also induce anti-inflammation responses, such as lactic acid (168), 12-ketolithocholic acid (169), linoleic acid (170), 5-hydroxyindoleacetic acid (171), polyamines (putrescine, spermidine, and spermine) (172), inosine (173) and hydrogen sulfide (174). Gut microbiota derived lactic acid induced transcriptional repression of macrophage inflammatory response via histone acetylation (168). It also served as a primary fuel source to promote histone H3K27 acetylation, which allowed the expression of immunosuppressive genes (168). Gut microbiota-derived 12-ketolithocholic acid could suppress the IL-17A secretion from colonic group ILC3 through down-regulated bile acid receptors, including vitamin D receptor (VDR) and pregnane X receptor (PXR) (169). The linoleic acid (LA) pathway in the gut microbiota determined the degree of inflammation and functions by suppressing Th17 differentiation and promoting Treg cell differentiation via the phosphorylation of Stat1 at Ser727 (170). Gut microbiota-derived 5-hydroxyindoleacetic acid alleviated colitis via MAPKs-PPARγ/NF-κB inhibition (171). The spermidine was produced by collective metabolic pathways of gut bacteria in immune cell regulation. It could facilitate polarization of Macs toward an anti-inflammatory phenotype, thus contributing to attenuation of inflammation (172). The inosine was a purine metabolite of Akkermansia muciniphila and Bifidobacetrium pseudolongum (77). The combination of A. muciniphila and inosine could modulate Treg cells and imbalance of Treg/Th17/Th1 cells through partly adenosine A2A receptor (173). The bacteria metabolized taurine or cysteine to form hydrogen sulfide (174). It could exert significant immune-regulatory roles by inhibiting the p38/ERK MAPK, p65 NF-κB, and JAK-STAT3 pathways and activating pathways such as Nrf2/HO-1, PI3K-AKT, and RAS/MAPK/AP-1 (175).
Taken together, besides inflammation associated gut microbiota and its structural components/metabolites, which can potentially promote the TLS formation, gut microbiota derived anti-inflammation metabolites such as SCFAs, BA and Trp metabolites, and other some metabolites also potentially inhibit the mature TLS formation through immunosuppressive cells in tumor environments. Notably, some metabolites derived gut microbiota such as SCFAs not only promote inflammation but also induce immunosuppression through different pathways.
In immature TLS, besides T and B cells, there often exist immature and immune suppressive cells such as immunosuppressive Macs, myeloid-derived suppressive cells (MDSCs), immature DCs and Treg (26, 27). These immunosuppressive cells can potentially inhibit mature TLS formation. Follicular regulatory T cells (TFR cells), characterized by the expression of CD25, FOXP3, glycoprotein A repetitions predominant and CXCR5, could suppress TFH cells in B cell follicles with the involvement of TGF-β (176). DC–lysosome-associated membrane protein (DC-LAMP)+ mature DCs, which were potent antigen-presenting cells could form tight cell contacts with T cells, and were responsible for the activation of naive T cells and reactivation of TCM cells (177). By contrast, Treg cells found in this zone could disrupt the cross-talk, inhibiting the anti-tumor immune responses generated in TLS (111). Interestingly, following Treg cell depletion, increased proliferation could be observed among CD4+ and CD8+ T cells within the TLS. Treg cells present within TLS strongly expressed CTLA4 and CD39, suggesting that two inhibitory pathways were triggered by Treg cells in TLS, through interaction with DCs and the production of adenosine, a potent inhibitor of T cells (111).
Besides gut microbiota metabolites, which can directly enter bloodstream to arrive in different tissues and organs, the microbes from the intestines can be transported via the bloodstream to tumor tissues through damaged blood vessels. Microorganisms include bacteria, viruses, fungus and others, among which viruses and bacteria have been found to be linked to the formation of TLSs (19, 178, 179). In tumor tissues, bacteria can release PAMPs (pathogen-associated molecular patterns) such as flagellins and LPSs to activate signaling pathways (MAPK, JAK–STAT, NF-κB) to release cytokines and chemokines, which promote the maturation of TLS. Typically, H. hepaticus colonization could induce H. hepaticus-specific T follicular helper (Tfh) cells, supporting the maturation of H. hepaticus + tumor-adjacent tertiary lymphoid structures in colorectal cancer (18). In hepatocellular carcinoma, the enrichment of the gut microbiota Lachnoclostridium was also associated with the presence of intratumoral TLS (19). Intra-tumor injection of E. coli MG1655 reprogramed tumor-associated macrophages into M1 phenotype that produce abundant CCL5, together facilitating tumor infiltration of adoptively transferred T cells. This effectively eradicated early-stage melanoma and inhibited the progression of pancreatic tumors (180). In addition, Salmonella-specific resident CX3CR1(hi) macrophages also induced tertiary lymphoid structures in situ (181). E. coli strain Nissle 1917, which could deliver cyclic di-AMP (a STING agonist) was shown to promote antigen presentation and type I interferon production by DCs, which were necessary for TLS formation (182). Innate immune cells activated by S. Typhimurium also released cytokines, such as IL-1β, TNF and IFNγ to induce inflammation, hereby transforming immune ‘cold’ tumors into ‘hot’ ones, which cause TLS (183).
Both antigen-specific antibody and T cell responses can be mounted in mature TLS. Various cancer treatments can also trigger TLS formation such as immunotherapy and cytokines (184). For instance, vaccination with human papillomavirus (HPV) oncoprotein vaccine could induce the formation of TLS (185). In pancreatic ductal adenocarcinoma (PDAC) mice, an anti-fibroblastic protein nanoparticle encoding LIGHT (Tumor necrosis factor superfamily member 14) induced intra-tumor TLS to inhibit abnormal collagen secretion (186). Low-dose radiotherapy could also trigger the development of immature TLS in a mouse model of lung cancer (184). Given the inextricable link between the gut microbiota and TLS, several potential strategies can be used to regulate mature TLS formation (Figure 6).
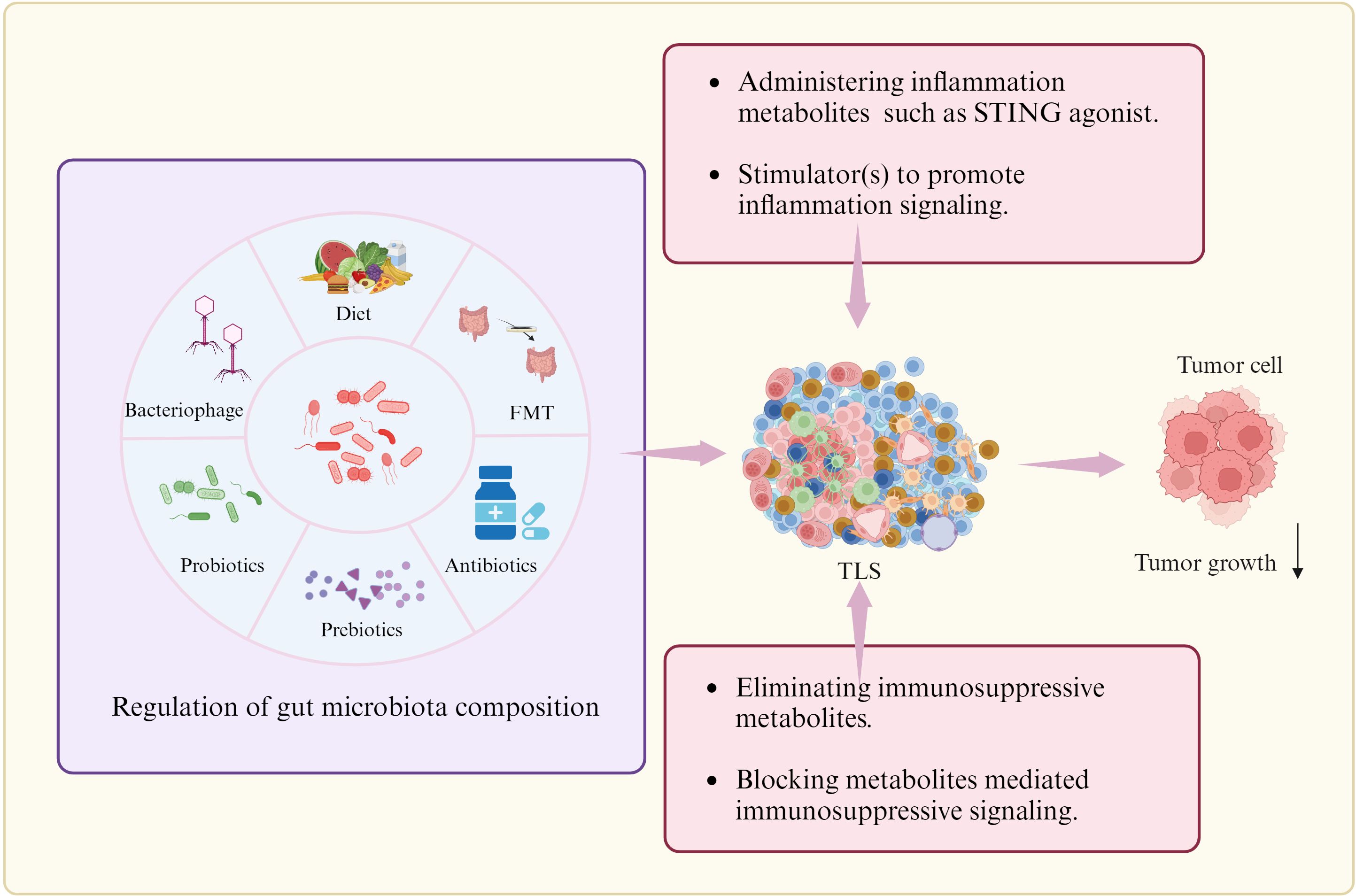
Figure 6. Potential strategies to regulate TLS formation. There are multiple strategies to promote TLS formation such as regulating the composition of gut microbiome through diet, FMT (Faecal microbiota transplantation), probiotics and prebiotics, and antibiotics or bacteriophages, administrating inflammation associated gut microbiota metabolites or stimulator to promote inflammation signaling, and eliminating immunosuppressive metabolites or blocking metabolites-mediated immunosuppressive signaling. Cells in TLS, see Figure 1.
Regulation on gut microbiome composition. Regulation on the gut microbiome though diet, FMT (Faecal microbiota transplantation), probiotics and prebiotics, and antibiotics or bacteriophages can potentially improve the TLS formation. In murine models of CRC and melanoma, oral gavage with commensal Clostridiales strains could potently induce antitumor immunity through infiltration and activation of intratumoral CD8+ T cells. Accumulating studies have observed that the diversity and composition of host gut microbiota are associated with the efficacy of immunotherapy as well as the incidence of immune-related adverse events (irAEs). The commensal microbial community could also positively affect patient’s outcomes through activating CD8+ T cells-dependent antitumor response (187), enhancing antitumor T cell immunity by activating DCs via toll-like receptor 4 (TLR4) signaling in melanoma mice receiving radiation (188). Intact commensal bacteria were also found to support immune surveillance in mice with lung carcinoma partially via enhancing γδT17 cell response (189). Diet with fructooligosaccharides, the structural units of inulin fiber, can activate human Macs to produce pro-inflammatory cytokines (190). In mice, an insulin-based high-fiber diet upregulates microbiota-derived bile acid metabolites, which promote IL-33 production (191). Antibiotic therapy in animal studies has demonstrated its capacity to modify gut microbiota composition and associated metabolites (192). The next-generation probiotics, specifically Eubacterium hallii, Faecalibacterium prausnitzii, Roseburia spp., Akkermansia muciniphila, and Bacteroides fragilis can have an impact on the development of various diseases (193). Bacteriophages possess specific characteristics, including specificity for particular or closely related bacterial species. They can be as control agents in gut microbiota environments (194).
Administration of inflammation metabolite(s). Intra-tumoral administration of STING agonist ADU-S100 could promote TLS formation (92). STING agonist ADU S-100 -activated CD11c+ DCs also showed upregulated expression of TLS promoting factors in vitro and in vivo (92). cGAS-STING pathway initiated TLS formation through chemokine (101). Boosting Tfh cell numbers by initiating their differentiation, has also been proven as an efficient strategy to promote establishment of TLS in tumors.
Elimination of anti-inflammation metabolites. Immunosuppressive metabolites can be eliminated through multiple methods such as neutralization.
Blockage of immunosuppressive signaling(s). The activation or inhibition of immune-related signaling may cause immune cell aggregation and TLS formation, contributing to anti- tumor immunity (184). Elimination of immunosuppressive metabolites or blockage of metabolites-mediated immunosuppressive signaling should potentially improve the TLS formation. Repressing other regulatory cells such as macrophage subsets or Breg cells may also favor formation of TLS.
We here summarize the potential effects of gut microbiota/metabolites on the TLS formation, including inflammation-associated gut microbiota/metabolites, and also immune-suppressive gut microbiota/metabolites. Meanwhile, different strategies to promote the TLS formation also are suggested through regulating gut microbiota or gut microbiota associated pathways for anti-tumor immunity such as diet, FMT, probiotics and prebiotics, and antibiotics or bacteriophages, or administering inflammation metabolites or immunosuppressive metabolite inhibitor, or regulating inflammation or immunosuppressive mediated signaling. Notably, further studying the relationship between TLSs and microorganisms in cancer, especially the molecular network between them, will be helpful to fully understand the etiology and immune environment of cancer, and search for effective biomarkers and new treatments. Spatial transcriptomics can offer us a powerful tool to study the regulation of gut microbiota on TLS formation, which can capture the spatial distribution of RNA transcripts within the TME to phenotype all cell types and study their spatial organization within the tumor niche. Advancements in these technologies have paved the way for a deeper understanding of TLS, and hold great promise for precision medicine initiatives.
RY: Conceptualization, Supervision, Writing – review & editing. YL: Investigation, Writing – original draft. FL: Investigation, Writing – original draft. JW: Investigation, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NSFC grants (grant number 82271779, 91842302, 81970457, 81901677and 91629102); The Tianjin Science and Technology Commission (grant number, 18JCZDJC35300); A Ministry of Science and Technology (grant number, 2016YFC1303604); The State Key Laboratory of Medicinal Chemical Biology and the Fundamental Research Funds for the Central University, Nankai university (63191724).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. (2012) 33:297–305. doi: 10.1016/j.it.2012.04.006
2. Fridman WH, Meylan M, Pupier G, Calvez A, Hernandez I, Sautes-Fridman C. Tertiary lymphoid structures and B cells: An intratumoral immunity cycle. Immunity. (2023) 56:2254–69. doi: 10.1016/j.immuni.2023.08.009
3. Playoust E, Remark R, Vivier E, Milpied P. Germinal center-dependent and -independent immune responses of tumor-infiltrating B cells in human cancers. Cell Mol Immunol. (2023) 20:1040–50. doi: 10.1038/s41423-023-01060-7
4. Ruffin AT, Cillo AR, Tabib T, Liu A, Onkar S, Kunning SR, et al. B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat Commun. (2021) 12:3349. doi: 10.1038/s41467-021-23355-x
5. Bruno TC. New predictors for immunotherapy responses sharpen our view of the tumour microenvironment. Nature. (2020) 577:474–6. doi: 10.1038/d41586-019-03943-0
6. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
7. You X, Koop K, Weigert A. Heterogeneity of tertiary lymphoid structures in cancer. Front Immunol. (2023) 14:1286850. doi: 10.3389/fimmu.2023.1286850
8. Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science. (2022) 375:eabf9419. doi: 10.1126/science.abf9419
9. Yang L, Li A, Wang Y, Zhang Y. Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy. Signal Transduct Target Ther. (2023) 8:35. doi: 10.1038/s41392-022-01304-4
10. Cuff AO, Sillito F, Dertschnig S, Hall A, Luong TV, Chakraverty R, et al. The obese liver environment mediates conversion of NK cells to a less cytotoxic ILC1-like phenotype. Front Immunol. (2019) 10:2180. doi: 10.3389/fimmu.2019.02180
11. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
12. Pelletier A, Stockmann C. The metabolic basis of ILC plasticity. Front Immunol. (2022) 13:858051. doi: 10.3389/fimmu.2022.858051
13. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol. (2017) 18:995–1003. doi: 10.1038/ni.3809
14. Sato Y, Silina K, van den Broek M, Hirahara K, Yanagita M. The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol. (2023) 19:525–37. doi: 10.1038/s41581-023-00706-z
15. Kucukkose E, Heesters BA, Villaudy J, Verheem A, Cercel M, van Hal S, et al. Modeling resistance of colorectal peritoneal metastases to immune checkpoint blockade in humanized mice. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-005345
16. Zhang Y, Liu G, Zeng Q, Wu W, Lei K, Zhang C, et al. CCL19-producing fibroblasts promote tertiary lymphoid structure formation enhancing anti-tumor IgG response in colorectal cancer liver metastasis. Cancer Cell. (2024) 42:1370–85 e9. doi: 10.1016/j.ccell.2024.07.006
17. van Hooren L, Vaccaro A, Ramachandran M, Vazaios K, Libard S, van de Walle T, et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat Commun. (2021) 12:4127. doi: 10.1038/s41467-021-24347-7
18. Overacre-Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A, et al. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity. (2021) 54:2812–24 e4. doi: 10.1016/j.immuni.2021.11.003
19. Zhao R, Li J, Chen B, Zhao J, Hu L, Huang K, et al. The enrichment of the gut microbiota Lachnoclostridium is associated with the presence of intratumoral tertiary lymphoid structures in hepatocellular carcinoma. Front Immunol. (2023) 14:1289753. doi: 10.3389/fimmu.2023.1289753
20. Fernandes MR, Aggarwal P, Costa RGF, Cole AM, Trinchieri G. Targeting the gut microbiota for cancer therapy. Nat Rev Cancer. (2022) 22:703–22. doi: 10.1038/s41568-022-00513-x
21. Winkler F, Venkatesh HS, Amit M, Batchelor T, Demir IE, Deneen B, et al. Cancer neuroscience: State of the field, emerging directions. Cell. (2023) 186:1689–707. doi: 10.1016/j.cell.2023.02.002
22. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
23. Teillaud JL, Houel A, Panouillot M, Riffard C, Dieu-Nosjean MC. Tertiary lymphoid structures in anticancer immunity. Nat Rev Cancer. (2024) 24:629–46. doi: 10.1038/s41568-024-00728-0
24. Dominiak A, Chelstowska B, Olejarz W, Nowicka G. Communication in the cancer microenvironment as a target for therapeutic interventions. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12051232
25. Lin JR, Wang S, Coy S, Chen YA, Yapp C, Tyler M, et al. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell. (2023) 186:363–81 e19. doi: 10.1016/j.cell.2022.12.028
26. Xu W, Lu J, Liu WR, Anwaier A, Wu Y, Tian X, et al. Heterogeneity in tertiary lymphoid structures predicts distinct prognosis and immune microenvironment characterizations of clear cell renal cell carcinoma. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2023-006667
27. Meylan M, Petitprez F, Lacroix L, Di Tommaso L, Roncalli M, Bougouin A, et al. Early hepatic lesions display immature tertiary lymphoid structures and show elevated expression of immune inhibitory and immunosuppressive molecules. Clin Cancer Res. (2020) 26:4381–9. doi: 10.1158/1078-0432.CCR-19-2929
28. Tietscher S, Wagner J, Anzeneder T, Langwieder C, Rees M, Sobottka B, et al. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat Commun. (2023) 14:98. doi: 10.1038/s41467-022-35238-w
29. Zhang H, AbdulJabbar K, Moore DA, Akarca A, Enfield KSS, Jamal-Hanjani M, et al. Spatial positioning of immune hotspots reflects the interplay between B and T cells in lung squamous cell carcinoma. Cancer Res. (2023) 83:1410–25. doi: 10.1158/0008-5472.CAN-22-2589
30. Mi H, Sivagnanam S, Betts CB, Liudahl SM, Jaffee EM, Coussens LM, et al. Quantitative spatial profiling of immune populations in pancreatic ductal adenocarcinoma reveals tumor microenvironment heterogeneity and prognostic biomarkers. Cancer Res. (2022) 82:4359–72. doi: 10.1158/0008-5472.CAN-22-1190
31. Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. (2021) 184:4734–52 e20. doi: 10.1016/j.cell.2021.08.003
32. Schlosser HA, Thelen M, Lechner A, Wennhold K, Garcia-Marquez MA, Rothschild SI, et al. B cells in esophago-gastric adenocarcinoma are highly differentiated, organize in tertiary lymphoid structures and produce tumor-specific antibodies. Oncoimmunology. (2019) 8:e1512458. doi: 10.1080/2162402X.2018.1512458
33. Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res. (2015) 21:3031–40. doi: 10.1158/1078-0432.CCR-14-2926
34. Workel HH, Lubbers JM, Arnold R, Prins TM, van der Vlies P, de Lange K, et al. A transcriptionally distinct CXCL13(+)CD103(+)CD8(+) T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol Res. (2019) 7:784–96. doi: 10.1158/2326-6066.CIR-18-0517
35. Li H, Ding JY, Zhang MJ, Yu HJ, Sun ZJ. Tertiary lymphoid structures and cytokines interconnections: The implication in cancer immunotherapy. Cancer Lett. (2023) 568:216293. doi: 10.1016/j.canlet.2023.216293
36. Ladanyi A, Kiss J, Somlai B, Gilde K, Fejos Z, Mohos A, et al. Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol Immunother. (2007) 56:1459–69. doi: 10.1007/s00262-007-0286-3
37. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest. (2014) 124:953–9. doi: 10.1172/JCI71611
38. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
39. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. (2015) 6:8280. doi: 10.1038/ncomms9280
40. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, et al. M1 macrophages act as LTbetaR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res. (2014) 101:434–43. doi: 10.1093/cvr/cvt263
41. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun. (2015) 6:7114. doi: 10.1038/ncomms8114
42. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med. (2011) 208:125–34. doi: 10.1084/jem.20100052
43. Guedj K, Abitbol Y, Cazals-Hatem D, Morvan M, Maggiori L, Panis Y, et al. Adipocytes orchestrate the formation of tertiary lymphoid organs in the creeping fat of Crohn's disease affected mesentery. J Autoimmun. (2019) 103:102281. doi: 10.1016/j.jaut.2019.05.009
44. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. (2011) 12:639–46. doi: 10.1038/ni.2053
45. Dong Y, Wang T, Wu H. Tertiary lymphoid structures in autoimmune diseases. Front Immunol. (2023) 14:1322035. doi: 10.3389/fimmu.2023.1322035
46. Kinker GS, Vitiello GAF, Diniz AB, Cabral-Piccin MP, Pereira PHB, Carvalho MLR, et al. Mature tertiary lymphoid structures are key niches of tumour-specific immune responses in pancreatic ductal adenocarcinomas. Gut. (2023) 72:1927–41. doi: 10.1136/gutjnl-2022-328697
47. Wang M, Rajkumar S, Lai Y, Liu X, He J, Ishikawa T, et al. Tertiary lymphoid structures as local perpetuators of organ-specific immune injury: implication for lupus nephritis. Front Immunol. (2023) 14:1204777. doi: 10.3389/fimmu.2023.1204777
48. Zhang Q, Wu S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front Immunol. (2022) 13:1063711. doi: 10.3389/fimmu.2022.1063711
49. Mann ER, Lam YK, Uhlig HH. Short-chain fatty acids: linking diet, the microbiome and immunity. Nat Rev Immunol. (2024) 24:577–95. doi: 10.1038/s41577-024-01014-8
50. Wang J, Zhu N, Su X, Yang R. Gut microbiota: A double-edged sword in immune checkpoint blockade immunotherapy against tumors. Cancer Lett. (2024) 582:216582. doi: 10.1016/j.canlet.2023.216582
51. Jia W, Li Y, Cheung KCP, Zheng X. Bile acid signaling in the regulation of whole body metabolic and immunological homeostasis. Sci China Life Sci. (2024) 67:865–78. doi: 10.1007/s11427-023-2353-0
52. Fleishman JS, Kumar S. Bile acid metabolism and signaling in health and disease: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. (2024) 9:97. doi: 10.1038/s41392-024-01811-6
53. Hou Y, Li J, Ying S. Tryptophan metabolism and gut microbiota: A novel regulatory axis integrating the microbiome, immunity, and cancer. Metabolites. (2023) 13. doi: 10.3390/metabo13111166
54. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
55. Lucas LN, Barrett K, Kerby RL, Zhang Q, Cattaneo LE, Stevenson D, et al. Dominant bacterial phyla from the human gut show widespread ability to transform and conjugate bile acids. mSystems. (2021) e0080521. doi: 10.1128/mSystems.00805-21
56. Guzior DV, Quinn RA. Review: microbial transformations of human bile acids. Microbiome. (2021) 9:140. doi: 10.1186/s40168-021-01101-1
57. Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A. (2008) 105:13580–5. doi: 10.1073/pnas.0804437105
58. Ridlon JM, Hylemon PB. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7alpha-dehydroxylating intestinal bacterium. J Lipid Res. (2012) 53:66–76. doi: 10.1194/jlr.M020313
59. Sato Y, Atarashi K, Plichta DR, Arai Y, Sasajima S, Kearney SM, et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature. (2021) 599:458–64. doi: 10.1038/s41586-021-03832-5
60. Paik D, Yao L, Zhang Y, Bae S, D'Agostino GD, Zhang M, et al. Human gut bacteria produce TauEta17-modulating bile acid metabolites. Nature. (2022) 603:907–12. doi: 10.1038/s41586-022-04480-z
61. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. (2012) 336:1262–7. doi: 10.1126/science.1223813
62. Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. (2018) 23:716–24. doi: 10.1016/j.chom.2018.05.003
63. Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. (2013) 5:193ra91. doi: 10.1126/scitranslmed.3006438
64. Wang J, Zhu N, Su X, Gao Y, Yang R. Gut-microbiota-derived metabolites maintain gut and systemic immune homeostasis. Cells. (2023) 12. doi: 10.3390/cells12050793
65. Dvorak Z, Sokol H, Mani S. Drug mimicry: promiscuous receptors PXR and ahR, and microbial metabolite interactions in the intestine. Trends Pharmacol Sci. (2020) 41:900–8. doi: 10.1016/j.tips.2020.09.013
66. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. (1977) 31:107–33. doi: 10.1146/annurev.mi.31.100177.000543
67. Pitocco D, Di Leo M, Tartaglione L, De Leva F, Petruzziello C, Saviano A, et al. The role of gut microbiota in mediating obesity and diabetes mellitus. Eur Rev Med Pharmacol Sci. (2020) 24:1548–62. doi: 10.26355/eurrev_202002_20213
68. Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, Wu WH, Saldana-Morales FB, et al. Thymic development of gut-microbiota-specific T cells. Nature. (2021) 594:413–7. doi: 10.1038/s41586-021-03531-1
69. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. (2010) 16:228–31. doi: 10.1038/nm.2087
70. Fenton TM, Jorgensen PB, Niss K, Rubin SJS, Morbe UM, Riis LB, et al. Immune profiling of human gut-associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region-specific immunity. Immunity. (2020) 52:557–70 e6. doi: 10.1016/j.immuni.2020.02.001
71. Zhang Z, Li J, Zheng W, Zhao G, Zhang H, Wang X, et al. Peripheral lymphoid volume expansion and maintenance are controlled by gut microbiota via RALDH+ Dendritic cells. Immunity. (2016) 44:330–42. doi: 10.1016/j.immuni.2016.01.004
72. Choi Y, Lichterman JN, Coughlin LA, Poulides N, Li W, Del Valle P, et al. Immune checkpoint blockade induces gut microbiota translocation that augments extraintestinal antitumor immunity. Sci Immunol. (2023) 8:eabo2003. doi: 10.1126/sciimmunol.abo2003
73. Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell. (2021) 184:5338–56 e21. doi: 10.1016/j.cell.2021.09.019
74. Si W, Liang H, Bugno J, Xu Q, Ding X, Yang K, et al. Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut. (2022) 71:521–33. doi: 10.1136/gutjnl-2020-323426
75. Jiang L, Shang M, Yu S, Liu Y, Zhang H, Zhou Y, et al. A high-fiber diet synergizes with Prevotella copri and exacerbates rheumatoid arthritis. Cell Mol Immunol. (2022) 19:1414–24. doi: 10.1038/s41423-022-00934-6
76. Lee IA, Kim DH. Klebsiella pneumoniae increases the risk of inflammation and colitis in a murine model of intestinal bowel disease. Scand J Gastroenterol. (2011) 46:684–93. doi: 10.3109/00365521.2011.560678
77. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. (2020) 369:1481–9. doi: 10.1126/science.abc3421
78. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. (2015) 350:1084–9. doi: 10.1126/science.aac4255
79. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
80. Deng H, Li Z, Tan Y, Guo Z, Liu Y, Wang Y, et al. A novel strain of Bacteroides fragilis enhances phagocytosis and polarises M1 macrophages. Sci Rep. (2016) 6:29401. doi: 10.1038/srep29401
81. Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. (2010) 105:2420–8. doi: 10.1038/ajg.2010.281
82. Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL, et al. Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of peyer's patch T follicular helper cells. Immunity. (2016) 44:875–88. doi: 10.1016/j.immuni.2016.03.013
83. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn's disease spondyloarthritis promote T(H)17-dependent inflammation. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aaf9655
84. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
85. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
86. Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity. (2016) 45:931–43. doi: 10.1016/j.immuni.2016.09.009
87. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. (2013) 342:971–6. doi: 10.1126/science.1240537
88. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory th17 cells. Immunity. (2019) 51:77–89 e6. doi: 10.1016/j.immuni.2019.05.004
89. Cao S, Zhang Q, Wang C, Wu H, Jiao L, Hong Q, et al. LPS challenge increased intestinal permeability, disrupted mitochondrial function and triggered mitophagy of piglets. Innate Immun. (2018) 24:221–30. doi: 10.1177/1753425918769372
90. Di Vincenzo F, Del Gaudio A, Petito V, Lopetuso LR, Scaldaferri F. Gut microbiota, intestinal permeability, and systemic inflammation: a narrative review. Intern Emerg Med. (2024) 19:275–93. doi: 10.1007/s11739-023-03374-w
91. Naghavian R, Faigle W, Oldrati P, Wang J, Toussaint NC, Qiu Y, et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature. (2023) 617:807–17. doi: 10.1038/s41586-023-06081-w
92. Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2020-001906
93. Zhou H, Wang L, Lin Z, Jiang C, Chen X, Wang K, et al. Methylglyoxal from gut microbes boosts radiosensitivity and radioimmunotherapy in rectal cancer by triggering endoplasmic reticulum stress and cGAS-STING activation. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2023-007840
94. Lee SY, Jhun J, Woo JS, Lee KH, Hwang SH, Moon J, et al. Gut microbiome-derived butyrate inhibits the immunosuppressive factors PD-L1 and IL-10 in tumor-associated macrophages in gastric cancer. Gut Microbes. (2024) 16:2300846. doi: 10.1080/19490976.2023.2300846
95. Lu Y, Cui A, Zhang X. Commensal microbiota-derived metabolite agmatine triggers inflammation to promote colorectal tumorigenesis. Gut Microbes. (2024) 16:2348441. doi: 10.1080/19490976.2024.2348441
96. Miyamoto K, Sujino T, Harada Y, Ashida H, Yoshimatsu Y, Yonemoto Y, et al. The gut microbiota-induced kynurenic acid recruits GPR35-positive macrophages to promote experimental encephalitis. Cell Rep. (2023) 42:113005. doi: 10.1016/j.celrep.2023.113005
97. Gnanaprakasam JNR, Estrada-Muniz E, Vega L. The anacardic 6-pentadecyl salicylic acid induces macrophage activation via the phosphorylation of ERK1/2, JNK, P38 kinases and NF-kappaB. Int Immunopharmacol. (2015) 29:808–17. doi: 10.1016/j.intimp.2015.08.038
98. Fong W, Li Q, Ji F, Liang W, Lau HCH, Kang X, et al. Lactobacillus gallinarum-derived metabolites boost anti-PD1 efficacy in colorectal cancer by inhibiting regulatory T cells through modulating IDO1/Kyn/AHR axis. Gut. (2023) 72:2272–85. doi: 10.1136/gutjnl-2023-329543
99. Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab. (2022) 34:581–94 e8. doi: 10.1016/j.cmet.2022.02.010
100. Benham H, Rehaume LM, Hasnain SZ, Velasco J, Baillet AC, Ruutu M, et al. Interleukin-23 mediates the intestinal response to microbial beta-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheumatol. (2014) 66:1755–67. doi: 10.1002/art.38638
101. Zhao R, Zhang J, Ma J, Qu Y, Yang Z, Yin Z, et al. cGAS-activated endothelial cell-T cell cross-talk initiates tertiary lymphoid structure formation. Sci Immunol. (2024) 9:eadk2612. doi: 10.1126/sciimmunol.adk2612
102. Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H, et al. The gut microbiome is associated with clinical response to anti-PD-1/PD-L1 immunotherapy in gastrointestinal cancer. Cancer Immunol Res. (2020) 8:1251–61. doi: 10.1158/2326-6066.CIR-19-1014
103. Hosonuma M, Murayama M, Yoshimura K. The gut microbiota metabolite A enhances the anti-tumor effects of anti-PD-1 antibody therapy through immune modulation. Gan To Kagaku Ryoho. (2023) 50:960–4.
104. Ma H, Yang L, Liang Y, Liu F, Hu J, Zhang R, et al. B. thetaiotaomicron-derived acetic acid modulate immune microenvironment and tumor growth in hepatocellular carcinoma. Gut Microbes. (2024) 16:2297846. doi: 10.1080/19490976.2023.2297846
105. Zhu X, Li K, Liu G, Wu R, Zhang Y, Wang S, et al. Microbial metabolite butyrate promotes anti-PD-1 antitumor efficacy by modulating T cell receptor signaling of cytotoxic CD8 T cell. Gut Microbes. (2023) 15:2249143. doi: 10.1080/19490976.2023.2249143
106. Wojciech L, Png CW, Koh EY, Kioh DYQ, Deng L, Wang Z, et al. A tryptophan metabolite made by a gut microbiome eukaryote induces pro-inflammatory T cells. EMBO J. (2023) 42:e112963. doi: 10.15252/embj.2022112963
107. Jia D, Wang Q, Qi Y, Jiang Y, He J, Lin Y, et al. Microbial metabolite enhances immunotherapy efficacy by modulating T cell stemness in pan-cancer. Cell. (2024) 187:1651–65 e21. doi: 10.1016/j.cell.2024.02.022
108. Kawanabe-Matsuda H, Takeda K, Nakamura M, Makino S, Karasaki T, Kakimi K, et al. Dietary lactobacillus-derived exopolysaccharide enhances immune-checkpoint blockade therapy. Cancer Discovery. (2022) 12:1336–55. doi: 10.1158/2159-8290.CD-21-0929
109. Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat Metab. (2020) 2:635–47. doi: 10.1038/s42255-020-0219-4
110. Samami E, Aleebrahim-Dehkordi E, Mohebalizadeh M, Yaribash S, Saghazadeh A, Rezaei N. Inosine, gut microbiota, and cancer immunometabolism. Am J Physiol Endocrinol Metab. (2023) 324:E1–8. doi: 10.1152/ajpendo.00207.2022
111. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity. (2015) 43:579–90. doi: 10.1016/j.immuni.2015.08.006
112. Pan M, Barua N, Ip M. Mucin-degrading gut commensals isolated from healthy faecal donor suppress intestinal epithelial inflammation and regulate tight junction barrier function. Front Immunol. (2022) 13:1021094. doi: 10.3389/fimmu.2022.1021094
113. Dahal RH, Kim S, Kim YK, Kim ES, Kim J. Insight into gut dysbiosis of patients with inflammatory bowel disease and ischemic colitis. Front Microbiol. (2023) 14:1174832. doi: 10.3389/fmicb.2023.1174832
114. Sun H, Guo Y, Wang H, Yin A, Hu J, Yuan T, et al. Gut commensal Parabacteroides distasonis alleviates inflammatory arthritis. Gut. (2023) 72:1664–77. doi: 10.1136/gutjnl-2022-327756
115. Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, Del Chierico F, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. (2019) 69:107–20. doi: 10.1002/hep.30036
116. Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, Soualhi S, et al. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science. (2019) 363. doi: 10.1126/science.aat4042
117. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. (2015) 8:80–93. doi: 10.1038/mi.2014.44
118. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
119. Kayama H, Takeda K. Regulation of intestinal homeostasis by innate and adaptive immunity. Int Immunol. (2012) 24:673–80. doi: 10.1093/intimm/dxs094
120. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. (2015) 6:6734. doi: 10.1038/ncomms7734
121. Shao X, Sun S, Zhou Y, Wang H, Yu Y, Hu T, et al. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the NLRP3 axis. Cancer Lett. (2021) 523:170–81. doi: 10.1016/j.canlet.2021.10.002
122. Scott NA, Andrusaite A, Andersen P, Lawson M, Alcon-Giner C, Leclaire C, et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aao4755
123. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. (2019) 50:432–45 e7. doi: 10.1016/j.immuni.2018.12.018
124. Yang W, Cong Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol Immunol. (2021) 18:866–77. doi: 10.1038/s41423-021-00661-4
125. Nastasi C, Fredholm S, Willerslev-Olsen A, Hansen M, Bonefeld CM, Geisler C, et al. Butyrate and propionate inhibit antigen-specific CD8(+) T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci Rep. (2017) 7:14516. doi: 10.1038/s41598-017-15099-w
126. Wang R, Li B, Huang B, Li Y, Liu Q, Lyu Z, et al. Gut microbiota-derived butyrate induces epigenetic and metabolic reprogramming in myeloid-derived suppressor cells to alleviate primary biliary cholangitis. Gastroenterology. (2024) 167(4):733–49. doi: 10.1053/j.gastro.2024.05.014
127. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
128. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
129. Hao F, Tian M, Zhang X, Jin X, Jiang Y, Sun X, et al. Butyrate enhances CPT1A activity to promote fatty acid oxidation and iTreg differentiation. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2014681118
130. Gurav A, Sivaprakasam S, Bhutia YD, Boettger T, Singh N, Ganapathy V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem J. (2015) 469:267–78. doi: 10.1042/BJ20150242
131. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. (2016) 20:202–14. doi: 10.1016/j.chom.2016.07.001
132. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. (2020) 11:4457. doi: 10.1038/s41467-020-18262-6
133. Sepahi A, Liu Q, Friesen L, Kim CH. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. (2021) 14:317–30. doi: 10.1038/s41385-020-0312-8
134. Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. (2018) 9:1853. doi: 10.3389/fimmu.2018.01853
135. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. (2003) 278:9435–40. doi: 10.1074/jbc.M209706200
136. Haselow K, Bode JG, Wammers M, Ehlting C, Keitel V, Kleinebrecht L, et al. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol. (2013) 94:1253–64. doi: 10.1189/jlb.0812396
137. Sun R, Zhang Z, Bao R, Guo X, Gu Y, Yang W, et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J Hepatol. (2022) 77:453–66. doi: 10.1016/j.jhep.2022.02.030
138. Kiriyama Y, Nochi H. The role of gut microbiota-derived lithocholic acid, deoxycholic acid and their derivatives on the function and differentiation of immune cells. Microorganisms. (2023) 11. doi: 10.3390/microorganisms11112730
139. Hu J, Zhang Y, Yi S, Wang C, Huang X, Pan S, et al. Lithocholic acid inhibits dendritic cell activation by reducing intracellular glutathione via TGR5 signaling. Int J Biol Sci. (2022) 18:4545–59. doi: 10.7150/ijbs.71287
140. Szeles L, Keresztes G, Torocsik D, Balajthy Z, Krenacs L, Poliska S, et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J Immunol. (2009) 182:2074–83. doi: 10.4049/jimmunol.0803345
141. Hu J, Wang C, Huang X, Yi S, Pan S, Zhang Y, et al. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. (2021) 36:109726. doi: 10.1016/j.celrep.2021.109726
142. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature. (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
143. Li W, Hang S, Fang Y, Bae S, Zhang Y, Zhang M, et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe. (2021) 29:1366–77 e9. doi: 10.1016/j.chom.2021.07.013
144. Fassett MS, Jiang W, D'Alise AM, Mathis D, Benoist C. Nuclear receptor Nr4a1 modulates both regulatory T-cell (Treg) differentiation and clonal deletion. Proc Natl Acad Sci U S A. (2012) 109:3891–6. doi: 10.1073/pnas.1200090109
145. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. (2020) 581:475–9. doi: 10.1038/s41586-020-2193-0
146. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature. (2020) 577:410–5. doi: 10.1038/s41586-019-1865-0
147. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. (2001) 167:4974–80. doi: 10.4049/jimmunol.167.9.4974
148. Pols TWH, Puchner T, Korkmaz HI, Vos M, Soeters MR, de Vries CJM. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PloS One. (2017) 12:e0176715. doi: 10.1371/journal.pone.0176715
149. Ding C, Hong Y, Che Y, He T, Wang Y, Zhang S, et al. Bile acid restrained T cell activation explains cholestasis aggravated hepatitis B virus infection. FASEB J. (2022) 36:e22468. doi: 10.1096/fj.202200332R
150. Zhu C, Boucheron N, Muller AC, Majek P, Claudel T, Halilbasic E, et al. 24-Norursodeoxycholic acid reshapes immunometabolism in CD8(+) T cells and alleviates hepatic inflammation. J Hepatol. (2021) 75:1164–76. doi: 10.1016/j.jhep.2021.06.036
151. Masuda K, Kimura A, Hanieh H, Nguyen NT, Nakahama T, Chinen I, et al. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int Immunol. (2011) 23:637–45. doi: 10.1093/intimm/dxr072
152. Lee K, Kwak JH, Pyo S. Inhibition of LPS-induced inflammatory mediators by 3-hydroxyanthranilic acid in macrophages through suppression of PI3K/NF-kappaB signaling pathways. Food Funct. (2016) 7:3073–82. doi: 10.1039/c6fo00187d
153. Alexeev EE, Dowdell AS, Henen MA, Lanis JM, Lee JS, Cartwright IM, et al. Microbial-derived indoles inhibit neutrophil myeloperoxidase to diminish bystander tissue damage. FASEB J. (2021) 35:e21552. doi: 10.1096/fj.202100027R
154. Leclerc D, Staats Pires AC, Guillemin GJ, Gilot D. Detrimental activation of AhR pathway in cancer: an overview of therapeutic strategies. Curr Opin Immunol. (2021) 70:15–26. doi: 10.1016/j.coi.2020.12.003
155. Huang Z, Jiang Y, Yang Y, Shao J, Sun X, Chen J, et al. 3,3'-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol Immunol. (2013) 53:335–44. doi: 10.1016/j.molimm.2012.09.007
156. Ehrlich AK, Pennington JM, Wang X, Rohlman D, Punj S, Lohr CV, et al. Activation of the aryl hydrocarbon receptor by 10-cl-BBQ prevents insulitis and effector T cell development independently of foxp3+ Regulatory T cells in nonobese diabetic mice. J Immunol. (2016) 196:264–73. doi: 10.4049/jimmunol.1501789
157. de Araujo EF, Feriotti C, Galdino NAL, Preite NW, Calich VLG, Loures FV. The IDO-ahR axis controls th17/treg immunity in a pulmonary model of fungal infection. Front Immunol. (2017) 8:880. doi: 10.3389/fimmu.2017.00880
158. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
159. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. (2014) 511:184–90. doi: 10.1038/nature13323
160. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
161. Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. (2016) 17:1318–29. doi: 10.1016/j.celrep.2016.09.082
162. Tashita C, Hoshi M, Hirata A, Nakamoto K, Ando T, Hattori T, et al. Kynurenine plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. World J Gastroenterol. (2020) 26:918–32. doi: 10.3748/wjg.v26.i9.918
163. Gargaro M, Vacca C, Massari S, Scalisi G, Manni G, Mondanelli G, et al. Engagement of nuclear coactivator 7 by 3-hydroxyanthranilic acid enhances activation of aryl hydrocarbon receptor in immunoregulatory dendritic cells. Front Immunol. (2019) 10:1973. doi: 10.3389/fimmu.2019.01973
164. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
165. Campesato LF, Budhu S, Tchaicha J, Weng CH, Gigoux M, Cohen IJ, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun. (2020) 11:4011. doi: 10.1038/s41467-020-17750-z
166. Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. (2011) 11:932–8. doi: 10.1016/j.intimp.2011.02.005
167. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
168. Shi W, Cassmann TJ, Bhagwate AV, Hitosugi T, Ip WKE. Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell Rep. (2024) 43:113746. doi: 10.1016/j.celrep.2024.113746
169. Li N, Ma P, Li Y, Shang X, Nan X, Shi L, et al. Gut microbiota-derived 12-ketolithocholic acid suppresses the IL-17A secretion from colonic group 3 innate lymphoid cells to prevent the acute exacerbation of ulcerative colitis. Gut Microbes. (2023) 15:2290315. doi: 10.1080/19490976.2023.2290315
170. Jia L, Jiang Y, Wu L, Fu J, Du J, Luo Z, et al. Porphyromonas gingivalis aggravates colitis via a gut microbiota-linoleic acid metabolism-Th17/Treg cell balance axis. Nat Commun. (2024) 15:1617. doi: 10.1038/s41467-024-45473-y
171. Wu M, Wang Q, Li X, Yu S, Zhao F, Wu X, et al. Gut microbiota-derived 5-hydroxyindoleacetic acid from pumpkin polysaccharides supplementation alleviates colitis via MAPKs-PPARgamma/NF-kappaB inhibition. Int J Biol Macromol. (2024) 264:130385. doi: 10.1016/j.ijbiomac.2024.130385
172. Chamoto K, Zhang B, Tajima M, Honjo T, Fagarasan S. Spermidine - an old molecule with a new age-defying immune function. Trends Cell Biol. (2024) 34:363–70. doi: 10.1016/j.tcb.2023.08.002
173. Wei L, Pan Y, Guo Y, Zhu Y, Jin H, Gu Y, et al. Symbiotic combination of Akkermansia muciniphila and inosine alleviates alcohol-induced liver injury by modulating gut dysbiosis and immune responses. Front Microbiol. (2024) 15:1355225. doi: 10.3389/fmicb.2024.1355225
174. Laue H, Denger K, Cook AM. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol. (1997) 63:2016–21. doi: 10.1128/aem.63.5.2016-2021.1997
175. Jin T, Lu H, Zhou Q, Chen D, Zeng Y, Shi J, et al. H(2)S-releasing versatile montmorillonite nanoformulation trilogically renovates the gut microenvironment for inflammatory bowel disease modulation. Adv Sci (Weinh). (2024) 11:e2308092. doi: 10.1002/advs.202308092
176. Noel G, Fontsa ML, Garaud S, De Silva P, de Wind A, Van den Eynden GG, et al. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J Clin Invest. (2021) 131. doi: 10.1172/JCI139905
177. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. (2014) 74:705–15. doi: 10.1158/0008-5472.CAN-13-1342
178. He T, Hao Z, Lin M, Xin Z, Chen Y, Ouyang W, et al. Oncolytic adenovirus promotes vascular normalization and nonclassical tertiary lymphoid structure formation through STING-mediated DC activation. Oncoimmunology. (2022) 11:2093054. doi: 10.1080/2162402X.2022.2093054
179. Houel A, Foloppe J, Dieu-Nosjean MC. Harnessing the power of oncolytic virotherapy and tertiary lymphoid structures to amplify antitumor immune responses in cancer patients. Semin Immunol. (2023) 69:101796. doi: 10.1016/j.smim.2023.101796
180. Zhu C, Liu C, Wu Q, Sheng T, Zhou R, Ren E, et al. Remolding the tumor microenvironment by bacteria augments adoptive T cell therapy in advanced-stage solid tumors. Signal Transduct Target Ther. (2024) 9:307. doi: 10.1038/s41392-024-02028-3
181. Koscso B, Kurapati S, Rodrigues RR, Nedjic J, Gowda K, Shin C, et al. Gut-resident CX3CR1(hi) macrophages induce tertiary lymphoid structures and IgA response. situ Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aax0062
182. Leventhal DS, Sokolovska A, Li N, Plescia C, Kolodziej SA, Gallant CW, et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat Commun. (2020) 11:2739. doi: 10.1038/s41467-020-16602-0
183. Guo Y, Chen Y, Liu X, Min JJ, Tan W, Zheng JH. Targeted cancer immunotherapy with genetically engineered oncolytic Salmonella typhimurium. Cancer Lett. (2020) 469:102–10. doi: 10.1016/j.canlet.2019.10.033
184. Chen Y, Wu Y, Yan G, Zhang G. Tertiary lymphoid structures in cancer: maturation and induction. Front Immunol. (2024) 15:1369626. doi: 10.3389/fimmu.2024.1369626
185. Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, et al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med. (2014) 6:221ra13. doi: 10.1126/scitranslmed.3007323
186. Huang Y, Chen Y, Zhou S, Chen L, Wang J, Pei Y, et al. Dual-mechanism based CTLs infiltration enhancement initiated by Nano-sapper potentiates immunotherapy against immune-excluded tumors. Nat Commun. (2020) 11:622. doi: 10.1038/s41467-020-14425-7
187. Zhou CB, Zhou YL, Fang JY. Gut microbiota in cancer immune response and immunotherapy. Trends Cancer. (2021) 7:647–60. doi: 10.1016/j.trecan.2021.01.010
188. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. (2007) 117:2197–204. doi: 10.1172/JCI32205
189. Cheng M, Qian L, Shen G, Bian G, Xu T, Xu W, et al. Microbiota modulate tumoral immune surveillance in lung through a gammadeltaT17 immune cell-dependent mechanism. Cancer Res. (2014) 74:4030–41. doi: 10.1158/0008-5472.CAN-13-2462
190. Armstrong HK, Bording-Jorgensen M, Santer DM, Zhang Z, Valcheva R, Rieger AM, et al. Unfermented beta-fructan fibers fuel inflammation in select inflammatory bowel disease patients. Gastroenterology. (2023) 164:228–40. doi: 10.1053/j.gastro.2022.09.034
191. Arifuzzaman M, Won TH, Li TT, Yano H, Digumarthi S, Heras AF, et al. Inulin fibre promotes microbiota-derived bile acids and type 2 inflammation. Nature. (2022) 611:578–84. doi: 10.1038/s41586-022-05380-y
192. Singh V, Yeoh BS, Abokor AA, Golonka RM, Tian Y, Patterson AD, et al. Vancomycin prevents fermentable fiber-induced liver cancer in mice with dysbiotic gut microbiota. Gut Microbes. (2020) 11:1077–91. doi: 10.1080/19490976.2020.1743492
193. Al-Fakhrany OM, Elekhnawy E. Next-generation probiotics: the upcoming biotherapeutics. Mol Biol Rep. (2024) 51:505. doi: 10.1007/s11033-024-09398-5
Keywords: gut microbiota, tertiary lymphoid structure, immunotherapy, Tregs, tumor
Citation: Liu Y, Li F, Wang J and Yang R (2025) Exploring effects of gut microbiota on tertiary lymphoid structure formation for tumor immunotherapy. Front. Immunol. 15:1518779. doi: 10.3389/fimmu.2024.1518779
Received: 28 October 2024; Accepted: 20 December 2024;
Published: 07 March 2025.
Edited by:
Baochi Ou, First Affiliated Hospital of Anhui Medical University, ChinaReviewed by:
Nan Ding, Chinese Academy of Sciences (CAS), ChinaCopyright © 2025 Liu, Li, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rongcun Yang, cnlhbmdAbmFua2FpLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.