
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 December 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1509195
This article is part of the Research Topic Harnessing Molecular Insights for Enhanced Drug Sensitivity and Immunotherapy in Cancer View all 27 articles
 Taotao Yan1,2,3
Taotao Yan1,2,3 Jiahai Shi2,3*
Jiahai Shi2,3*Lung cancer remains the primary cause of cancer-related mortality, with factors such as postoperative tumor recurrence, metastasis, and therapeutic drug resistance exacerbating patient outcomes. Immunotherapy has emerged as a transformative approach, challenging conventional treatment paradigms for lung cancer. Consequently, advancing research in lung cancer immunotherapy is imperative. Recent studies indicate that numerous regulators within the tumor microenvironment (TME) drive tumor angiogenesis and epithelial-mesenchymal transition (EMT); these processes are interdependent, reciprocal, and collectively contribute to tumor progression. Tumor angiogenesis not only supplies adequate oxygen and nutrients for cellular proliferation but also establishes pathways facilitating tumor metastasis and creating hypoxic regions that foster drug resistance. Concurrently, EMT enhances metastatic potential and reinforces drug-resistance genes within tumor cells, creating a reciprocal relationship with angiogenesis. This interplay ultimately results in tumor invasion, metastasis, and therapeutic resistance. This paper reviews key regulators of angiogenesis and EMT, examining their impact on lung cancer immunotherapy and progression, and investigates whether newly identified regulators could influence lung cancer treatment, thus offering valuable insights for developing future therapeutic strategies.
Lung cancer ranks as the second most prevalent cancer globally, leading in incidence among men and following breast cancer among women (1). Non-small cell lung cancer (NSCLC), the predominant form, is a highly malignant subtype with a poor prognosis, encompassing adenocarcinoma, large cell carcinoma, and squamous cell carcinoma, which together constitute approximately 80%-85% of all lung cancers (2, 3). While early-stage cancers are often managed effectively with surgical resection or radiotherapy, advanced cancers can only be treated by chemotherapy (4), which becomes progressively less effective as resistance develops and the disease worsens (5). Recent advancements in cancer immunotherapy, particularly immune checkpoint inhibitors (ICIs), have introduced promising alternatives for lung cancer management. ICIs, effective in treating unresectable advanced lung cancer as well as in perioperative settings, target PD-1 or PD-L1 pathways (6) to mitigate treatment side effects and improve survival rates. Nevertheless, achieving a definitive cure for lung cancer remains a distant goal (7). Research into molecular mechanisms underlying lung cancer progression is essential for identifying potential therapeutic targets, offering significant insights for future treatment modalities.
The tumor microenvironment (TME) plays a pivotal role in tumor cell proliferation and invasion, constituting a complex system composed of cellular components such as tumor, immune, and stromal cells, and non-cellular elements including tumor-associated fibroblasts, adjacent mesenchymal tissues, vascular networks, and various chemokines (8, 9). Within the TME, immune cells contribute to cancer progression through mechanisms that support tumor proliferation and metastasis, such as immune evasion, epithelial-mesenchymal transition (EMT), angiogenesis, and immunosuppression (10). In recent years, strategies targeting TME regulation have gained significant interest in cancer immunotherapy. Despite the initial efficacy of immunotherapeutic agents, therapeutic success is frequently compromised by emerging drug resistance within the host (11). Continued research on angiogenesis and EMT regulatory mechanisms offers potential pathways to address these challenges, advancing the effectiveness of lung cancer therapies.
The development of the human vascular system is an intricately orchestrated process, necessitating precise temporal and spatial coordination among various cell types to form functional blood vessels. Angiogenesis, the formation of new vasculature from pre-existing vessels, is fundamental to both physiological and pathological processes, such as wound healing, organ development, ischemic conditions, inflammatory diseases, fibrosis, and cancer (12). This multistep process initiates new capillary growth from multifunctional pre-existing vessels, which significantly contributes to tumor recurrence and metastasis (13). Tumor expansion demands substantial nutrients and oxygen, necessitating an adequate blood supply within the TME. This supply is facilitated through angiogenesis, where the recruitment of new vessels from existing ones provides tumors with essential resources, a process driven by a complex signaling network of growth factors (14). Recently, anti-angiogenesis has emerged as a promising immunotherapeutic strategy, aiming to normalize abnormal vasculature, inhibit tumor growth and metastasis, and restrict tumor blood supply through anti-angiogenic agents (15). This approach is now applied in treating various solid tumor types (16).
EMT is a form of cellular reprogramming that allows epithelial cells to acquire a mesenchymal phenotype, essential for embryonic development and adult tissue maintenance. This process triggers cytoskeletal remodeling and mitochondrial division to meet the high energy demands of EMT, fueling further transition. EMT plays a pivotal role in tumor progression, endowing cancer cells with enhanced invasiveness and relative drug resistance (17, 18).
Emerging experimental data increasingly demonstrate that the interaction between angiogenesis and EMT in tumors significantly enhances tumor invasion, metastasis, and drug resistance. For instance, hypoxic conditions stimulate EMT through hypoxia-inducible factors (HIFs), which mediate diverse signaling pathways pivotal to angiogenesis (19). Hypoxia or HIF overexpression alone can induce EMT and promote invasiveness across various cell types. The HIF pathway indirectly drives EMT via multiple cellular signaling pathways, including Notch, TGF-β, integrin-linked kinases, tyrosine kinase receptors, Wnt, and Hedgehog (20). In hypoxic environments, HIF-1α upregulates anti-apoptotic genes and activates PD-L1 in tumor cells, enabling immune evasion and enhancing invasion and migration (21). Angiogenesis is frequently accompanied by an inflammatory response, with pro-inflammatory chemokines like IL-8 prompting EMT in tumor cells (22). This review consolidates current research on key regulatory factors in lung cancer, examining various pathways that drive tumor angiogenesis and EMT, ultimately contributing to tumor growth, metastasis, drug resistance, and advancements in immunotherapy.
Angiogenesis, the formation of new capillaries from preexisting blood vessels, is crucial for the growth and metastasis of many solid tumors. Tumor-derived angiogenic factors drive endothelial cell migration and proliferation, establishing new capillaries that support tumor expansion, invasion, and metastasis. This process initiates when pro-angiogenic molecules outweigh anti-angiogenic counterparts (23, 24). Key angiogenic regulators, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), HIF, and angiopoietin, are vital in hypoxia-induced angiogenesis (Table 1) (25). Under hypoxic conditions, signaling pathways activate relevant receptors, and HIF directly promotes transcription of angiogenesis-related genes (e.g., VEGF), upregulating these regulators to advance angiogenesis and mitigate tissue hypoxia (26). Investigating these regulatory and signaling pathways may provide insights for enhancing therapeutic drug development for lung cancer (Figure 1).
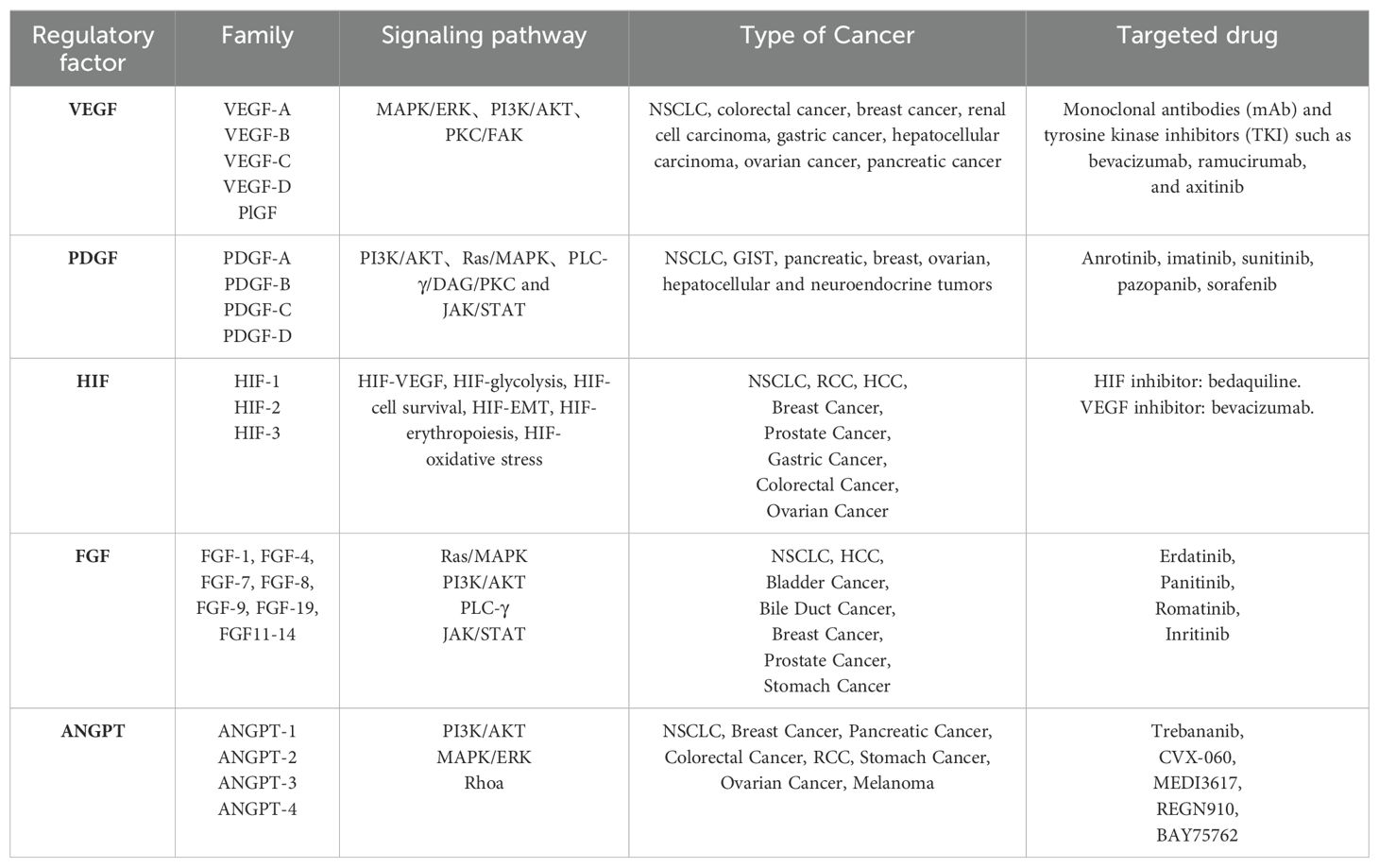
Table 1. Regulators of angiogenesis.
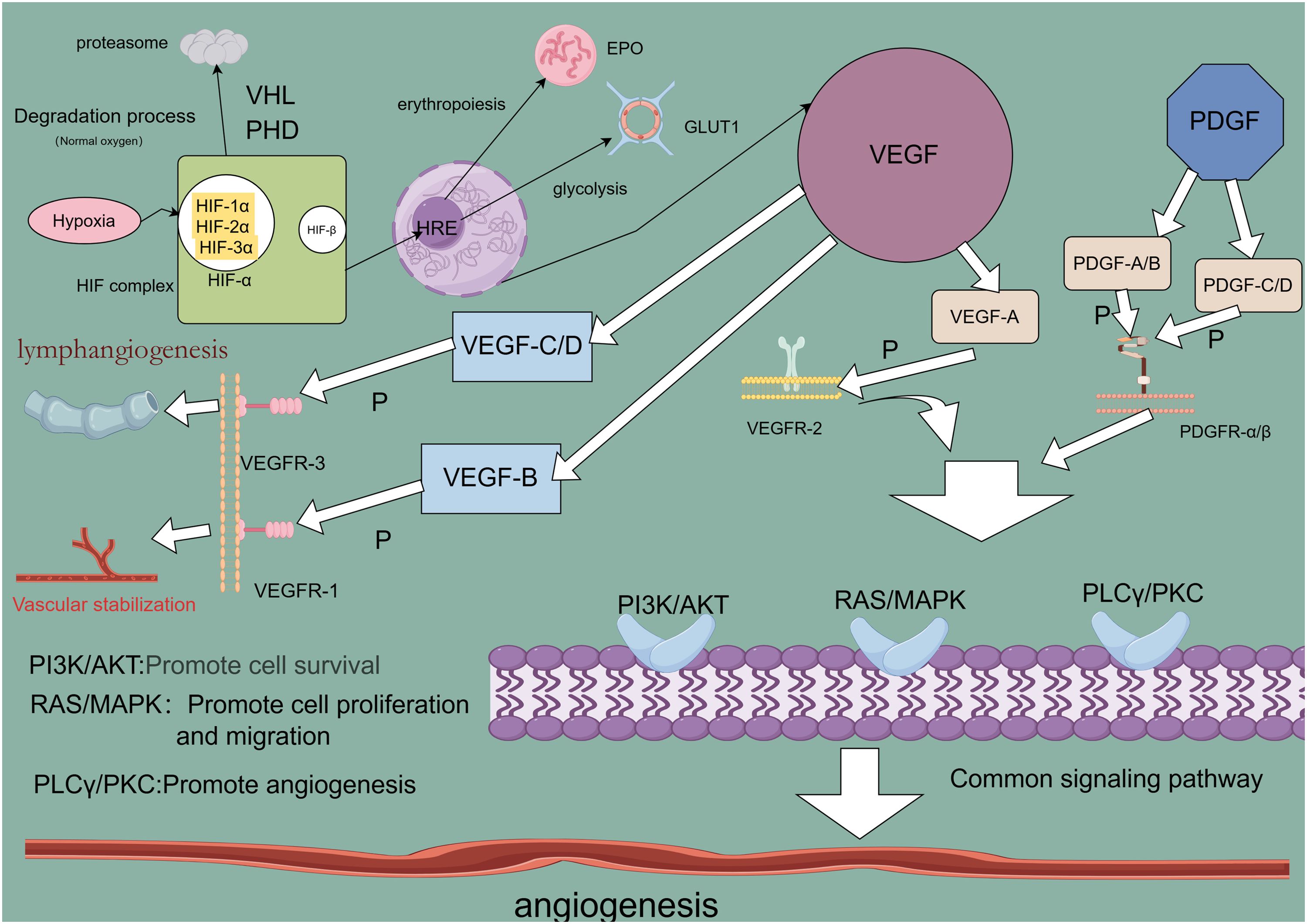
Figure 1. Angiogenesis-related regulatory factor signaling pathway.
Angiogenesis is a critical driver of lung cancer cell growth, invasion, and metastasis, with VEGF serving as a key mediator in this process. In NSCLC, elevated levels of angiogenic markers, including VEGF, correlate with poorer prognoses (27), underscoring VEGF’s central role in tumor neovascularization (28). The VEGF family, comprising VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF), exhibits specific functions within various tissues. VEGF-A, the most potent angiogenic factor, is essential for regulating endothelial cell sprouting, mitogenesis, migration, vasodilation, and vascular permeability (29, 30). In the heart, VEGF-B supports neuronal survival and cardiovascular growth through angiogenesis. It regulates cardiac angiogenesis and sympathetic innervation by inducing tissue-specific angiogenic responses in the myocardium, upregulating nerve ciliary protein (Nrp-1) expression, and mediating VEGFR-1 and Nrp-1 to promote nerve growth and protection. This mechanism contributes to alleviating ischemic heart disease (31), thereby aiding ischemic heart disease recovery (32). VEGF-C and VEGF-D facilitate tumor growth and metastasis through VEGFR-3-mediated lymphangiogenesis and lymphatic metastasis (33). Upon binding to its receptor, VEGF activates downstream signaling pathways such as MAPK/ERK, PI3K/AKT, PKC, and FAK, which collectively support angiogenesis. Although VEGF/VEGFR is not the sole regulatory axis, it plays an indispensable role in angiogenic signaling (34). Consequently, antivascular drugs targeting VEGF or VEGFR have emerged as promising therapeutic options, primarily including monoclonal antibodies (mAb) and tyrosine kinase inhibitors (TKIs), such as bevacizumab, ramucirumab, and acitretinib. Monoclonal antibodies exhibit high specificity by binding directly to VEGF or VEGFR, preventing receptor interaction and thereby exerting anti-angiogenic effects through receptor blockade. Tyrosine kinase inhibitors, conversely, bind to receptor tyrosine kinases, inhibiting their kinase activity and thus impeding downstream signaling (35, 36). Research confirms that VEGF and VEGFR are key regulators in lung cancer angiogenesis, serving as primary therapeutic targets for antivascular drug development to inhibit tumor growth, metastasis, and drug resistance. However, tumors often develop multiple drug resistance (MDR) through mechanisms such as decreased intracellular drug concentrations, altered drug targets, and imbalances in metabolic detoxification and DNA repair. Overexpression of transporter proteins in tumor cells further limits drug efficacy by blocking drug entry and actively expelling intracellular drugs. These transporters expel lipophilic chemotherapeutic agents, reducing their effective intracellular concentrations and establishing resistance (37). To counteract drug resistance, current treatments employ sequential maximal dose-density regimens to maximize cell destruction and minimize resistance onset (38). Dual targeting of the VEGF and ANG2 pathways has proven more effective than single-target approaches, enhancing antiangiogenic therapy outcomes (39). Despite progress, many molecular mechanisms underlying VEGF-targeted antivascular therapies remain to be elucidated, necessitating further investigation (40).
PDGF, a fundamental protein stored in platelet α-granules (41), along with its receptors (PDGFRα and PDGFRβ), is expressed in numerous malignant cells and tissues, including NSCLC, gastrointestinal stromal tumors (GIST), and pancreatic, breast, ovarian, hepatocellular, and neuroendocrine cancers (42). In NSCLC, overexpression of PDGFRα/β and PDGF-A/B correlates with poor prognosis; PDGF-AA, for instance, regulates VEGF expression via autocrine signaling, advancing the transformation of precancerous lesions into aggressive malignancies (43). Additionally, mutations in the PDGFR-α gene enhance PDGFRα expression, triggering ligand-independent PDGF signaling that fosters tumor growth in NSCLC (44). In GIST, PDGF ligand binding to PDGFRα and PDGFRβ activates the STAT pathway, influencing disease progression (45). In breast cancer, high PDGF-C expression correlates with lymph node metastasis, HER2 amplification, and elevated Ki-67 proliferation, driving progression via autocrine and paracrine signaling (46). In cholangiocarcinoma, hypoxia-induced PDGF-D upregulation in cholangiocarcinoma (CAA) cells activates a paracrine loop in the tumor-associated stroma, coordinating lymphangiogenesis and accelerating regional lymph node metastasis (47). PDGF activates signaling through PDGFR-mediated cellular pathways, where receptor binding initiates dimerization and phosphorylation, creating sites for downstream signaling molecule attachment and activating pathways such as PI3K/AKT, Ras/MAPK, PLC-γ, DAG, PKC, and JAK/STAT (48). Studies reveal that inhibiting the PDGF/PDGFR pathway effectively hinders tumor cell proliferation and angiogenesis (49). TKIs, which neutralize PDGFR antibodies and antagonize PDGFR kinase activity (50), have shown promise in targeted therapies, notably improving the outcomes of patients with NSCLC. Anlotinib, for example, is utilized as a third-line treatment for advanced NSCLC, targeting VEGFR to inhibit angiogenesis and lung cancer cell proliferation, demonstrating efficacy in advanced cases (51, 52). However, TKI-related toxicity and resistance present significant challenges in prolonged therapy, necessitating further in-depth research to optimize efficacy and mitigate these issues (53).
Hypoxia is often indicative of solid tumor presence, activating the HIF family to modulate gene expression in both tumor cells and immune cells within the TME, thereby influencing tumor progression and therapeutic response (54). The HIF family consists of isoforms HIF-1, HIF-2, and HIF-3, each with distinct functions and transcriptional activities (55). Among these, HIF-1 has been widely identified across various cancers and plays a pivotal role in cancer development, acting as a key transcription factor (TF) that drives cancer progression and serves as a target for therapeutic intervention. HIF-1 promotes cancer cell growth, survival, angiogenesis, metastasis, and treatment resistance (56). In the hypoxic TME, rapid tumor cell proliferation outpaces the oxygen supply from surrounding blood vessels, creating an imbalance that triggers a cellular adaptive response coordinated by HIF-1 (57). HIF-1 itself is a heterodimer of α and β subunits, with HIF-1α particularly induced under hypoxia to regulate genes related to cancer cell proliferation and angiogenesis (58). HIF-1α transcriptionally activates several pro-angiogenic molecules by binding directly to promoter regions. Specifically, HIF-1α binds to the hypoxia response element (HRE) on VEGF and VEGFR1 gene promoters, inducing VEGFA and VEGFR1 expression, which promotes tumor angiogenesis through VEGF and ANGPTL4 (59). Under hypoxic conditions, HIF-1α stabilizes and initiates the expression of multiple genes through a gene expression cascade involving the MAPK pathway and VEGF signaling (60). In this process, HIF-1α binds to the hypoxia-responsive element (HRE) within the VEGF promoter, forming an HIF-1α/HRE complex that directly upregulates VEGFR-1 expression in tumor cells. This amplification of VEGF signaling promotes both solid tumor angiogenesis and pathological angiogenesis (61). Ropivacaine, a local anesthetic, has been shown to inhibit HIF-1α signaling in lung cancer cells, along with downstream VEGF signaling, thus reducing angiogenesis in malignant lung cancers (62). Additionally, HIF-1α activation of the Hippo-YAP pathway accelerates malignant progression in NSCLC, while silencing HIF-1α induces ferroptosis and inhibits NSCLC invasion (63). These findings underscore the extensive interplay of HIF-1 signaling pathways in lung cancer development, suggesting that targeting HIF-1 could open new avenues for the development of effective HIF inhibitors and therapeutic strategies (64).
Among the various regulators of angiogenesis, basic fibroblast growth factor (FGF2) is considered the first identified pro-angiogenic molecule, promoting angiogenesis by activating FGF receptor 1 (FGFR1) signaling in endothelial cells (65). To counteract the effects of the highly expressed FGF2/FGFR1 pathway, research indicates that VEGF-B can act as a unique angiogenic factor; although it typically has limited angiogenic activity, it can inhibit tumor growth and angiogenesis under specific conditions by suppressing FGF2-induced Erk phosphorylation and thus reducing FGF2-driven angiogenesis (66, 67). This mechanism offers a potential therapeutic strategy for controlling excessive angiogenesis, contributing to targeted therapies aimed at preventing lung cancer cell metastasis and dissemination. In the context of advancing lung cancer immunotherapy research, miRNAs, a subset of non-coding RNAs, have emerged as key regulators of cancer cell growth, metastasis, angiogenesis, and apoptosis. For instance, miR-937-3p, often highly expressed in patients with lung adenocarcinoma (LUAD), activates the PI3K/AKT pathway by targeting the downstream gene SOX11, thereby enhancing NSCLC angiogenesis and facilitating metastasis (68, 69). Additionally, dietary compounds such as flavonoids, retinoids, triterpenoids, omega fatty acids, and carotenoids have demonstrated promising roles in anti-angiogenic therapy within current cancer immunotherapy approaches (70).
EMT is a process in which epithelial cells lose their connectivity and polarity while gaining mesenchymal characteristics and invasive potential. EMT progresses through distinct states—fully epithelial, partial EMT, partial MET, and fully mesenchymal (full EMT)—each with unique functional traits, plasticity, and heterogeneity that contribute to cancer invasion, recurrence, and drug resistance (71, 72). EMT is generally mediated by multiple signaling pathways, including the TGF-β, bone morphogenetic protein (BMP), receptor tyrosine kinase (RTK), STAT3, extracellular matrix (ECM)-mediated, and hypoxia signaling pathways. These pathways regulate TFs, influencing gene expression to increase EMT-related markers (73). Consequently, targeting EMT presents a promising therapeutic strategy, potentially offering improved recovery opportunities for patients with cancer.
TGF-β is widely recognized as a central driver of cancer cell plasticity through EMT. The TGF-β family includes 33 evolutionarily conserved proteins, such as TGF-β1, TGF-β2, TGF-β3, activins, bone morphogenetic proteins (BMPs), inhibins, growth and differentiation factors (GDFs), and mullerian inhibitory substances (MISs) (74). TGF-β, particularly prominent in advanced cancers, is closely linked to metastasis and chemotherapy resistance. During EMT, TGF-β activates the SMAD pathway following ligand-induced receptor activation, where SMAD proteins transmit signals to the nucleus to regulate target gene expression (75). TGF-β signaling begins with the activation of membrane-bound type I (TGFβRI) and type II (TGFβRII) receptors, leading to SMAD2 and SMAD3 activation. These form a complex with SMAD4, translocating to the nucleus to interact with DNA-binding TFs and co-regulators, modulating gene expression (76). Additionally, the TGF-β receptor complex activates non-SMAD pathways, including RAS/MAPK, TAK1/JNK/p38MAPK/IKK, and PI3K/Akt (77) (Figure 2). Given its overexpression and pro-tumorigenic effects across many tumor types, TGF-β is a promising therapeutic target. Combining TGF-β inhibitors with immune checkpoint blockade or chemotherapy can effectively reduce cancer cell plasticity (78). Neferine, a bisbenzylisoquinoline alkaloid, has been found to downregulate TGF-β in NSCLC, modulating MST1 to induce ROS formation, thereby promoting apoptosis and preventing proliferation, metastasis, and EMT (79). Additionally, TGFβ1-induced upregulation of PD-L1 in tumor cells has emerged as a novel mechanism of immunosuppression in NSCLC. Bintrafusp alfa (M7824), targeting both PD-L1 and TGF-β, has shown efficacy in inhibiting tumor mesenchymalization, reducing PD-L1-dependent immunosuppression, and overcoming chemoresistance in NSCLC (80). These studies, along with the development of more potent and specific TGF-β inhibitors, hold potential for treating tumors that thrive in TGF-β-rich environments (81).
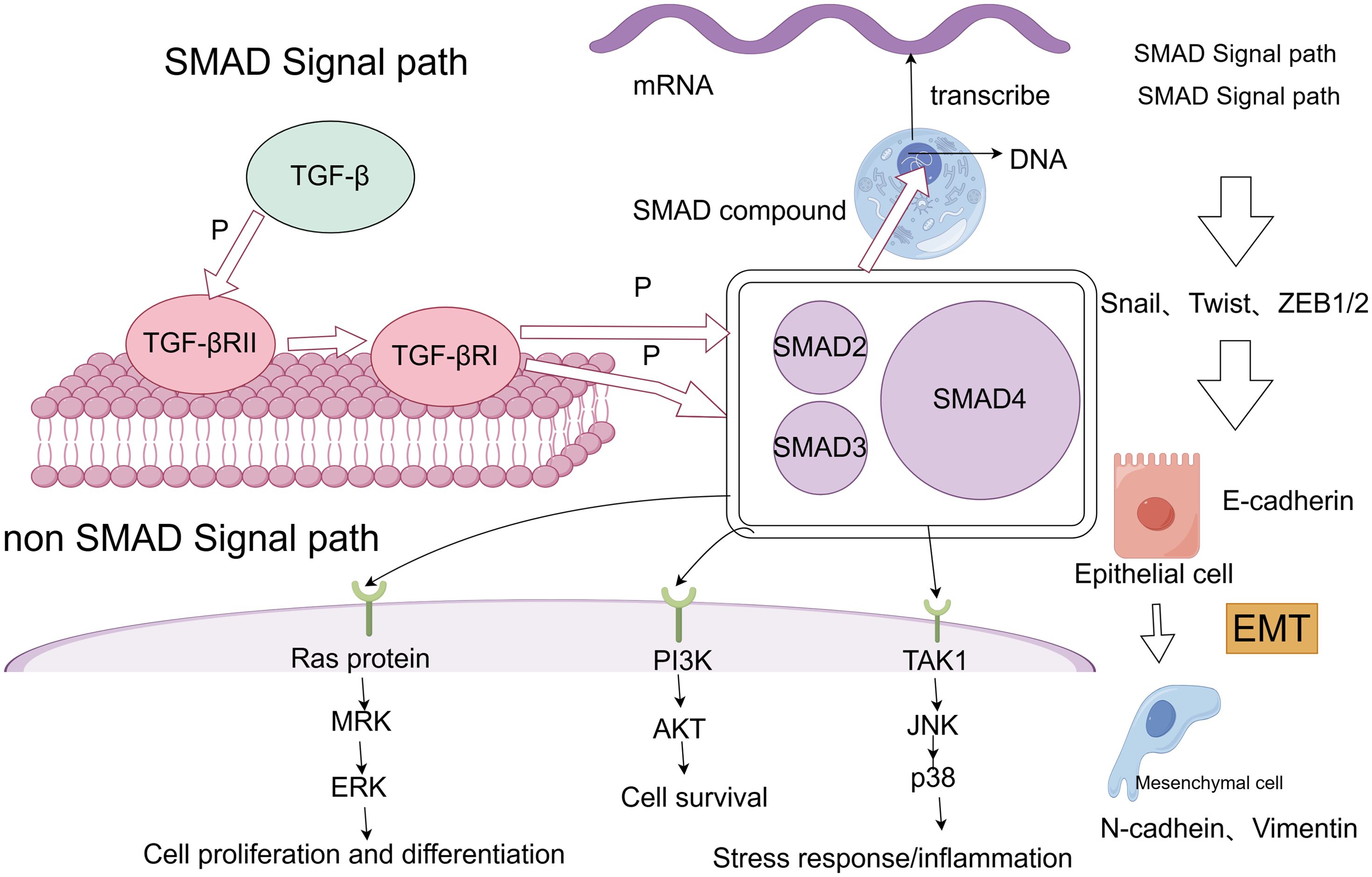
Figure 2. SMAD and non-SMAD pathways of TGF-β lead to EMT process.
EMT-TFs (Table 2) include the zinc finger proteins SNAI1 (Snail1) and SNAI2 (Snail2, or Slug), twist-related proteins 1 and 2 (Twist1/2), and the zinc finger e-box binding homology cassette 1/2 (ZEB1/2). Upregulation of these EMT-TFs drives cancer cells to transition from a differentiated epithelial state to a stem cell-like state, enhancing metastatic potential (82). Among them, Snail1 plays a critical role, with its expression preceding other EMT-TFs. Snail1 activates additional EMT-TFs and suppresses epithelial genes like epithelial cadherin (E-cadherin), allowing tumors to adopt a mesenchymal morphology and invasive capacity (83). Snail1 also recruits chromatin-modifying enzymes to the E-cadherin promoter, promoting DNA methylation and transcriptional repression of E-cadherin, thereby driving EMT and promoting dedifferentiation of cancer cells into cancer stem cell-like (CSC) phenotypes. Snail1 expression is linked to increased invasion and metastasis across various cancer types, including lung cancer. Notably, reducing Snail1 expression has been shown to enhance the efficacy of numerous chemotherapies and immunotherapies. Although direct chemical inhibitors targeting Snail1 are scarce, inhibitors targeting Snail1-induced EMT have demonstrated promising results (84, 85). For instance, Entestat (ENT) can reverse Snail1-induced EMT, leading to increased E-cadherin expression and decreased levels of Twist, Snail, and other EMT-TFs, thereby reducing metastatic potential (86). ZEB1 acts as a key regulator, functioning as both an activator and repressor of target genes, depending on its interaction with the CDH1 promoter, miR-190 promoter, and TGF-β signaling intermediates like Smad, p300, and P/CAF (87). Twist1 is recognized as a key regulator of oncogenesis and metastasis. During EMT, Twist1 promotes the expression of EMT-related genes, such as type I collagen and N-cadherin, by directly binding to their promoters (88), enabling epithelial cells to transition to a mesenchymal phenotype. Twist1 is also crucial in regulating intercellular adhesion by influencing downstream targets like E-cadherin. Twist1 promotes EMT by inhibiting E-cadherin expression through Snail1 activity (89). In smokers, exposure to the nicotine-derived carcinogen nitrosamine ketone (NNK) upregulates Twist mRNA and protein expression, which correlates with increased migration and invasion of lung cancer cells. This underscores Twist’s role in regulating NNK-induced changes in EMT marker expression in lung cancer (90).
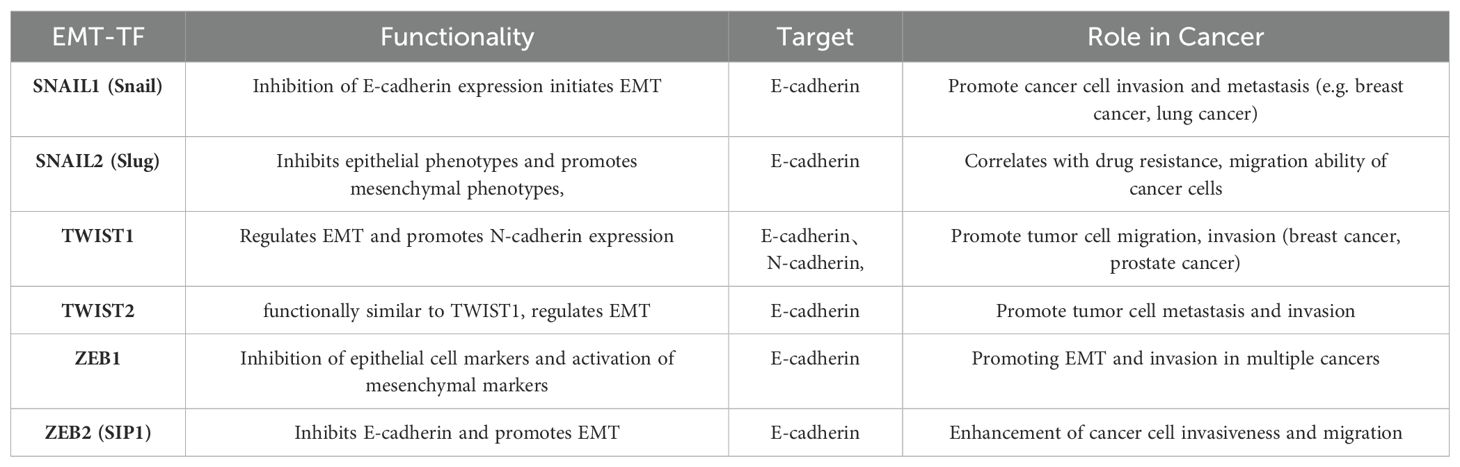
Table 2. EMT-related transcription factors and their functions, targets, and roles played in cancer.
With expanding research on EMT, numerous TFs have emerged as novel EMT regulators, such as specificity protein 1 (SP1) and E2F1. Recent studies highlight E2F1 as a pivotal TF for cell cycle progression in cancer, closely linked to metastasis. In NSCLC tissues and cell lines, E2F1 is notably upregulated and controls ZEB2 expression via an E2F1 binding site on the ZEB2 promoter, ultimately driving EMT and enhancing tumor invasion and metastasis (91, 92). SP1, part of a TF family that includes Sp2, Sp3, and Sp4, is critical for various biological functions such as cell growth, differentiation, apoptosis, and carcinogenesis, activating numerous cellular genes (93). In lung adenocarcinoma (LADC), aberrant SP1 expression induces EMT (94). Specifically, SP1-activated SGPP2 promotes LADC cell proliferation and invasion while inhibiting apoptosis (95). SP1 also functions as a direct target of miR-145-5p in NSCLC, where its overexpression decreases drug sensitivity, promotes EMT, and heightens drug resistance in cancer cells (96). As more EMT-related markers are identified, these insights pave the way for improved therapeutic protocols for cancer treatment.
This study investigated the pivotal roles of angiogenesis and EMT regulators within the tumor microenvironment in lung cancer progression. Key angiogenic regulators, including VEGF, PDGF, and HIF, significantly contribute to promoting angiogenesis, tumor growth, and metastasis in lung cancer. Concurrently, EMT regulators such as TGF-β, Snail, and Twist intensify cancer progression by enhancing the invasive and drug-resistant characteristics of tumor cells. Together, these processes synergize to drive tumor malignancy and facilitate immune evasion.
These findings lay a theoretical foundation for the potential application of combined anti-angiogenic and EMT-targeted therapies, particularly within immunotherapy. Targeting both angiogenesis and EMT modulators may enable future therapeutic strategies to not only suppress tumor growth and metastasis but also improve responsiveness to conventional treatments, offering more effective options for patients with lung cancer.
TY: Data curation, Software, Writing – original draft, Writing – review & editing. JS: Conceptualization, Investigation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Postgraduate Research & Practice Program of Jiangsu Province (KYCX23_3427), National Natural Science Foundation of China (82370253), Jiangsu Provincial Research Hospital (YJXYY202204), Innovation Team Project of Affiliated Hospital of Nantong University (XNBHCX31773).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gao K, Chen Z, Zhang N, Jiang P. High throughput virtual screening and validation of Plant-Based EGFR L858R kinase inhibitors against Non-Small cell lung Cancer: An integrated approach Utilizing GC-MS, network Pharmacology, Docking, and molecular dynamics. Saudi Pharm journal: SPJ. (2024) 32:102139. doi: 10.1016/j.jsps.2024.102139
2. Wang Y, Zhu H, Zhang L, He J, Bo J, Wang J, et al. Common immunological and prognostic features of lung and bladder cancer via smoking-related genes: PRR11 gene as potential immunotherapeutic target. J Cell Mol Med. (2024) 28:e18384. doi: 10.1111/jcmm.18384
3. Zheng Y, Ji H, Yi W, Chen Z, Hu X, Zhou J, et al. PRMT5 facilitates angiogenesis and EMT via HIF-1α/VEGFR/Akt signaling axis in lung cancer. Aging. (2023) 15:6163–78. doi: 10.18632/aging.204826
4. de Nijs K, de Koning HJ, van der Aalst C, Ten Haaf K. Medical costs of lung cancer by stage, histology and first-line treatment modality in the Netherlands (2012-2021). Eur J Cancer (Oxford England: 1990). (2024) 208:114231. doi: 10.1016/j.ejca.2024.114231
5. Jin X, Liu Z, Yang D, Yin K, Cahng X. Recent progress and future perspectives of immunotherapy in advanced gastric cancer. Front Immunol. (2022) 13:948647. doi: 10.3389/fimmu.2022.948647
6. Wang Y, Ma L, He J, Gu H, Zhu H. Identification of cancer stem cell-related genes through single cells and machine learning for predicting prostate cancer prognosis and immunotherapy. Front Immunol. (2024) 15:1464698. doi: 10.3389/fimmu.2024.1464698
7. Mandlik DS, Mandlik SK, Choudhary HB. Immunotherapy for hepatocellular carcinoma: Current status and future perspectives. World J Gastroenterol. (2023) 29:1054–75. doi: 10.3748/wjg.v29.i6.1054
8. Wang Y, Li C, He J, Zhao Q, Zhou Y, Sun H, et al. Multi-omics analysis and experimental validation of the value of monocyte-associated features in prostate cancer prognosis and immunotherapy. Front Immunol. (2024) 15:1426474. doi: 10.3389/fimmu.2024.1426474
9. Wang Y, Ji B, Zhang L, Wang J, He J, Ding B, et al. Identification of metastasis-related genes for predicting prostate cancer diagnosis, metastasis and immunotherapy drug candidates using machine learning approaches. Biol Direct. (2024) 19:50. doi: 10.1186/s13062-024-00494-x
10. Wang Y, He J, Zhao Q, Bo J, Zhou Y, Sun H, et al. Evaluating the predictive value of angiogenesis-related genes for prognosis and immunotherapy response in prostate adenocarcinoma using machine learning and experimental approaches. Front Immunol. (2024) 15:1416914. doi: 10.3389/fimmu.2024.1416914
11. Yang M, Li J, Gu P, Fan X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioactive Materials. (2020) 6:1973–87. doi: 10.1016/j.bioactmat.2020.12.010
12. Francescone R, Vendramini-Costa DB. In vitro models to study angiogenesis and vasculature. In: Marques dos Reis E, Berti F, editors. Vasculogenic Mimicry: Methods and Protocols. Springer US, New York, NY (2022). p. 15–28. doi: 10.1007/978-1-0716-2403-6_2
13. La Mendola D, Trincavelli ML, Martini C. Angiogenesis in disease. Int J Mol Sci. (2022) 23:10962. doi: 10.3390/ijms231810962
14. Shaw P, Dwivedi SKD, Bhattacharya R, Mukherjee P, Rao G. Vegf signaling: Role in angiogenesis and beyond. Biochim Biophys Acta (BBA) - Rev Cancer. (2024) 1879:189079. doi: 10.1016/j.bbcan.2024.189079
15. Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduction Targeted Ther. (2023) 8:198. doi: 10.1038/s41392-023-01460-1
16. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sciences: CMLS. (2019) 77:1745–70. doi: 10.1007/s00018-019-03351-7
17. Lu J, Kornmann M, Traub B. Role of epithelial to mesenchymal transition in colorectal cancer. Int J Mol Sci. (2023) 24:14815. doi: 10.3390/ijms241914815
18. Qiu Y, Ye W, Wang C, Zang A. Prognostic significance and immunoinfiltration analysis of genes associated with epithelial-mesenchymal transition and energy metabolism in bladder urothelial carcinoma. Aging (Albany NY). (2023) 15:13312–28. doi: 10.18632/aging.205242
19. Tirpe AA, Gulei D, Ciortea SM, Crivii C. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci. (2019) 20:6140. doi: 10.3390/ijms20246140
20. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Sci (New York N.Y.). (2016) 352:175. doi: 10.1126/science.aaf4405
21. Lin YT, Wu KJ. Epigenetic regulation of epithelial-mesenchymal transition: focusing on hypoxia and TGF-β signaling. J Biomed Sci. (2020) 27:39. doi: 10.1186/s12929-020-00632-3
22. Fousek K, Horn LA, Palena C. Interleukin-8: a chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol Ther. (2021) 219:107692. doi: 10.1016/j.pharmthera.2020.107692
23. Altorki NK, Markowitz GJ, Gao D, Port JL, Sanena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. (2019) 19:9–31. doi: 10.1038/s41568-018-0081-9
24. Li S, Xu HX, Wu CT, Wang WQ, Jin W, Gao HL, et al. Angiogenesis in pancreatic cancer: current research status and clinical implications. Angiogenesis. (2019) 22:15–36. doi: 10.1007/s10456-018-9645-2
25. Yao C, Wu S, Kong J, Sun Y, Bai Y, Zhu R, et al. Angiogenesis in hepatocellular carcinoma: mechanisms and anti-angiogenic therapies. Cancer Biol Med. (2023) 20:25–43. doi: 10.20892/j.issn.2095-3941.2022.0449
26. Zimna A, Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. BioMed Res Int. (2015) 2015:549412. doi: 10.1155/2015/549412
27. Gadgeel SM. Targeted therapy and immune therapy for small cell lung cancer. Curr Treat Options Oncol. (2018) 19:53. doi: 10.1007/s11864-018-0568-3
28. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 23:1011–27. doi: 10.1200/JCO.2005.06.081
29. Shukla NA, Yan MN, Hanna N. The story of angiogenesis inhibitors in non–small-cell lung cancer: the past, present, and future. Clin Lung Cancer. (2020) 21:308–13. doi: 10.1016/j.cllc.2020.02.024
30. Patel SA, Nilsson MB, Le X, Cascone T, Jain RK, Heymach JV. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin Cancer research. (2023) 29:30–9. doi: 10.1158/1078-0432.CCR-22-1366
31. Lähteenvuo J, Hätinen OP, Kuivanen A, Kuuso J, Paananen J, Lähteenvuo M, et al. Susceptibility to cardiac arrhythmias and sympathetic nerve growth in VEGF-B overexpressing myocardium. Mol Ther. (2020) 28:1731. doi: 10.1016/j.ymthe.2020.03.011
32. Kivelä R, Bry M, Robciuc MR, Räsänen M, Taavitsainen M, Silvola JM, et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. (2014) 6:307. doi: 10.1002/emmm.201303147
33. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. (2011) 473:298–307. doi: 10.1038/nature10144
34. Malekan M, Haass NK, Rokni GR, Gholizadeh N, Ebrahimzadeh M, Kazeminejad A. VEGF/VEGFR axis and its signaling in melanoma: Current knowledge toward therapeutic targeting agents and future perspectives. Life Sci. (2024) 345:122563. doi: 10.1016/j.lfs.2024.122563
35. Li Y, Lin M, Wang S, Cao B, Li C, Li G. Novel angiogenic regulators and anti-angiogenesis drugs targeting angiogenesis signaling pathways: perspectives for targeting angiogenesis in lung cancer. Front Oncol. (2022) 12:842960. doi: 10.3389/fonc.2022.842960
36. Alevizakos M, Kaltsas S, Syrigos KN. The VEGF pathway in lung cancer. Cancer Chemotherapy Pharmacol. (2013) 72:1169–81. doi: 10.1007/s00280-013-2298-3
37. Duan C, Yu M, Xu J, Li BY, Zhao Y, Kankala RK. Overcoming Cancer Multi-drug Resistance (MDR): Reasons, mechanisms, nanotherapeutic solutions, and challenges. Biomedicine Pharmacotherapy. (2023) 162:114643. doi: 10.1016/j.biopha.2023.114643
38. Chatterjee N, Bivona TG. Polytherapy and targeted cancer drug resistance. Trends Cancer. (2019) 5:170. doi: 10.1016/j.trecan.2019.02.003
39. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. (2018) 15:325. doi: 10.1038/nrclinonc.2018.29
40. Lopes-Coelho F, Martins F, Pereira SA, Serpa J. Anti-angiogenic therapy: current challenges and future perspectives. Int J Mol Sci. (2021) 22:3765. doi: 10.3390/ijms22073765
41. Cao Z, Liu Y, Wang Y, Leng P. Research progress on the role of PDGF/PDGFR in type 2 diabetes. Biomedicine Pharmacotherapy. (2023) 164:114983. doi: 10.1016/j.biopha.2023.114983
42. Zou X, Tang XY, Qu ZY, Sun ZW, Ji ZF, Li YJ, et al. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int J Biol Macromolecules. (2022) 202:539–57. doi: 10.1016/j.ijbiomac.2022.01.113
43. Noskovičová N, Petřek M, Eickelberg O, Heinzelmann K. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Respir Cell Mol Biol. (2015) 52:263–84. doi: 10.1165/rcmb.2014-0294TR
44. McDermott U, Ames RY, Iafrate AJ, Maheswaran S, Stubbs H, Greninger P, et al. Ligand-dependent PDGF receptor-alpha activation sensitizes rare lung cancer and sarcoma cells to PDGF receptor kinase inhibitors. Cancer Res. (2009) 69:3937. doi: 10.1158/0008-5472.CAN-08-4327
45. Bahlawane C, Eulenfeld R, Wiesinger MY, Wang J, Muller A, Girod A, et al. Constitutive activation of oncogenic PDGFRα-mutant proteins occurring in GIST patients induces receptor mislocalisation and alters PDGFRα signalling characteristics. Cell Communication Signaling : CCS. (2015) 13:21. doi: 10.1186/s12964-015-0096-8
46. Bottrell A, Meng YH, Najy AJ, Newton Hurst J, Kim S, Kim CJ, et al. An oncogenic activity of PDGF-C and its splice variant in human breast cancer. Growth factors (Chur Switzerland). (2019) 37:131. doi: 10.1080/08977194.2019.1662415
47. Cadamuro M, Brivio S, Mertens J, Vismara M, Monscek A, Milani A, et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. (2018) 70:700. doi: 10.1016/j.jhep.2018.12.004
48. Ai JY, Liu CF, Zhang W, Rao GW. Current status of drugs targeting PDGF/PDGFR. Drug Discovery Today. (2024) 29:103989. doi: 10.1016/j.drudis.2024.103989
49. Raica M, Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals. (2010) 3:572–99. doi: 10.3390/ph3030572
51. Xie C, Wan X, Quan H, Zheng M, Fu L, Li Y, et al. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci. (2018) 109:1207–19. doi: 10.1111/cas.13536
52. Shen G, Zheng F, Ren D, Du F, Dong Q, Wang Z, et al. Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol Oncol. (2018) 11:120. doi: 10.1186/s13045-018-0664-7
53. Gao Y, Liu P, Shi R. Anlotinib as a molecular targeted therapy for tumors. Oncol Lett. (2020) 20:1001–14. doi: 10.3892/ol.2020.11685
54. Cowman SJ, Koh MY. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer. (2022) 8:28–42. doi: 10.1016/j.trecan.2021.10.004
55. Fallah J, Rini BI. HIF inhibitors: status of current clinical development. Curr Oncol Rep. (2019) 21:6. doi: 10.1007/s11912-019-0752-z
56. Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M, et al. Up-down regulation of HIF-1α in cancer progression. Gene. (2021) 798:145796. doi: 10.1016/j.gene.2021.145796
57. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci. (2021) 22:5703. doi: 10.3390/ijms22115703
58. Manuelli V, Pecorari C, Filomeni G, Zito E. Regulation of redox signaling in HIF-1-dependent tumor angiogenesis. FEBS J. (2022) 289:5413–25. doi: 10.1111/febs.16110
59. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Research : CR. (2020) 39:204. doi: 10.1186/s13046-020-01709-5
60. Magar AG, Morya VK, Kwak MK, Oh JU, Noh KC. A molecular perspective on HIF-1α and angiogenic stimulator networks and their role in solid tumors: an update. Int J Mol Sci. (2024) 25:3313. doi: 10.3390/ijms25063313
61. Xiong Q, Liu B, Ding M, Zhou J, Yang C, Chen Y. Hypoxia and cancer related pathology. Cancer Lett. (2020) 486:1–7. doi: 10.1016/j.canlet.2020.05.002
62. Shen J, Han L, Xue Y, Li C, Jia H, Zhu K. Ropivacaine inhibits lung cancer cell Malignancy through downregulation of cellular signaling including HIF-1α In vitro. Front Pharmacol. (2022) 12:806954. doi: 10.3389/fphar.2021.806954
63. Zheng S, Mo J, Zhang J, Chen Y. HIF−1α inhibits ferroptosis and promotes Malignant progression in non−small cell lung cancer by activating the Hippo−YAP signalling pathway. Oncol Lett. (2023) 25:90. doi: 10.3892/ol.2023.13676
64. Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discovery. (2019) 14:667–82. doi: 10.1080/17460441.2019.1613370
65. Zhu X, Qiu C, Wang Y, Jiang Y, Chen Y, Fan L, et al. FGFR1 SUMOylation coordinates endothelial angiogenic signaling in angiogenesis. Proc Natl Acad Sci United States America. (2022) 119:e2202631119. doi: 10.1073/pnas.2202631119
66. PubMed central link . Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10435562/.
67. Li X, Kumar A, Zhang F, Lee C, Tang Z. Complicated life, complicated VEGF-B. Trends Mol Med. (2012) 18:119–27. doi: 10.1016/j.molmed.2011.11.006
68. Sadeghi MS, Lotfi M, Soltani N, Farmani E, Fernandez JH, Akhlaghitetehrani S, et al. Recent advances on high-efficiency of microRNAs in different types of lung cancer: a comprehensive review. Cancer Cell Int. (2023) 23:284. doi: 10.1186/s12935-023-03133-z
69. Ma Z, Chen G, Chen Y, Guo Z, Chai H, Tang Y, et al. MiR-937-3p promotes metastasis and angiogenesis and is activated by MYC in lung adenocarcinoma. Cancer Cell Int. (2022) 22:31. doi: 10.1186/s12935-022-02453-w
70. Albini A, Noonan DM, Corradino P, Magnoni F, Corso G. The past and future of angiogenesis as a target for cancer therapy and prevention. Cancer Prev Res. (2024) 17:289–303. doi: 10.1158/1940-6207.CAPR-24-0085
71. Pang ZQ, Wang JS, Wang JF, Wang YX, Ji B, Xu YD, et al. JAM3: A prognostic biomarker for bladder cancer via epithelial-mesenchymal transition regulation. Biomol Biomed. (2024) 24:897–911. doi: 10.17305/bb.2024.9979
72. Huang Z, Zhang Z, Zhou C, Liu L, Huang C. Epithelial–mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities. MedComm. (2022) 3:e144. doi: 10.1002/mco2.144
73. Babaei G, Aziz SGG, Jaghi NZZ. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomedicine Pharmacotherapy. (2021) 133:110909. doi: 10.1016/j.biopha.2020.110909
74. Wang X, Eichhorn PJA, Thiery JP. TGF-β, EMT, and resistance to anti-cancer treatment. Semin Cancer Biol. (2023) 97:1–11. doi: 10.1016/j.semcancer.2023.10.004
75. Hill CS. Transcriptional control by the SMADs. Cold Spring Harbor Perspect Biol. (2016) 8:a022079. doi: 10.1101/cshperspect.a022079
76. Derynck R, Budi EH. Specificity, versatility and control of TGF-β family signaling. Sci Signaling. (2019) 12:eaav5183. doi: 10.1126/scisignal.aav5183
77. Zhang YE, Stuelten CH. Alternative splicing in EMT and TGF-β signaling during cancer progression. Semin Cancer Biol. (2024) 101:1–11. doi: 10.1016/j.semcancer.2024.04.001
78. Kuburich NA, Sabapathy T, Demestichas BR, Maddela JJ, Den Hollander P, Mani SA. Proactive and reactive roles of TGF-β in cancer. Semin Cancer Biol. (2023) 95:120–39. doi: 10.1016/j.semcancer.2023.08.002
79. Zhong PC, Liu ZW, Xing QC, Chen J, Yang RP. Neferine inhibits the development of lung cancer cells by downregulating TGF-β to regulate MST1/ROS-induced pyroptosis. Kaohsiung J Med Sci. (2023) 39:1106–18. doi: 10.1002/kjm2.12752
80. Aftabi S, Barzegar Behrooz A, Cordani M, Rahiman N, Sadeghdoust M, Aligolighasemabadi F, et al. Therapeutic targeting of TGF-β in lung cancer. FEBS J. (2024). doi: 10.1111/febs.17234
81. Batlle E, Massagué J. Transforming grown factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
82. Khan AQ, Hasan A, Mir SS, Rashid K, Uddin S, Steinhoff M. Exploiting transcription factors to target EMT and cancer stem cells for tumor modulation and therapy. Semin Cancer Biol. (2024) 100:1–16. doi: 10.1016/j.semcancer.2024.03.002
83. García de Herreros A. Dual role of Snail1 as transcriptional repressor and activator. Biochim Biophys Acta (BBA) - Rev Cancer. (2024) 1879:189037. doi: 10.1016/j.bbcan.2023.189037
84. Lai CY, Yeh DW, Lu CH, Liu YL, Chuang YC, Ruan JW, et al. Epigenetic silencing of ubiquitin specific protease 4 by snail1 contributes to macrophage-dependent inflammation and therapeutic resistance in lung cancer. Cancers. (2020) 12:148. doi: 10.3390/cancers12010148
85. Kaufhold S, Bonavida B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Research : CR. (2014) 33:62. doi: 10.1186/s13046-014-0062-0
86. Shah P, Gau Y, Sabnis G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. (2014) 143:99–111. doi: 10.1007/s10549-013-2784-7
87. Lu J, Fei F, Wu C, Mei J, Xu J, Lu P. ZEB1: Catalyst of immune escape during tumor metastasis. Biomedicine Pharmacotherapy. (2022) 153:113490. doi: 10.1016/j.biopha.2022.113490
88. Ren J, Crowley SD. Twist1: A double-edged sword in kidney diseases. Kidney Dis. (2020) 6:247. doi: 10.1159/000505188
89. Casas E, Kim J, Bendesky A, Ogno-Machado L, Solfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial-mesenchymal transition and metastasis. Cancer Res. (2011) 71:245. doi: 10.1158/0008-5472.CAN-10-2330
90. Wang Y, Shi L, Li J, Wang H, Yang H. Involvement of twist in NNK exposure-promoted lung cancer cell migration and invasion. Toxicol Vitro. (2020) 63:104740. doi: 10.1016/j.tiv.2019.104740
91. Meng Q, Liu M, Cheng R. LINC00461/miR-4478/E2F1 feedback loop promotes non-small cell lung cancer cell proliferation and migration. Bioscience Rep. (2020) 40:BSR20191345. doi: 10.1042/BSR20191345
92. Wang T, Chen X, Qiao W, Kong L, Sun D, Li Z. Transcription factor E2F1 promotes EMT by regulating ZEB2 in small cell lung cancer. BMC Cancer. (2017) 17:719. doi: 10.1186/s12885-017-3701-y
93. Vizcaíno C, Mansilla S, Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacology & Therapeutics (2015) 152:111–24. doi: 10.13039/501100003339
94. Zhang H, Zhang G, Zhang J, Xiao M, Sui S, Wu S, et al. Transcription factor SP1 and oncoprotein PPP1R13L regulate nicotine-induced epithelial-mesenchymal transition in lung adenocarcinoma via a feedback loop. Biochem Pharmacol. (2022) 206:115344. doi: 10.1016/j.bcp.2022.115344
95. Yang X, Wang C. SGPP2 is activated by SP1 and promotes lung adenocarcinoma progression. Anti-Cancer Drugs (2024) 35:943. doi: 10.1097/CAD.0000000000001648
Keywords: lung cancer, angiogenesis, EMT, immunotherapy, TME
Citation: Yan T and Shi J (2024) Angiogenesis and EMT regulators in the tumor microenvironment in lung cancer and immunotherapy. Front. Immunol. 15:1509195. doi: 10.3389/fimmu.2024.1509195
Received: 10 October 2024; Accepted: 28 November 2024;
Published: 16 December 2024.
Edited by:
Minghua Ren, First Affiliated Hospital of Harbin Medical University, ChinaReviewed by:
Zhijia Xia, Ludwig Maximilian University of Munich, GermanyCopyright © 2024 Yan and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiahai Shi, aGFwcHlzamgxNjdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.