Abstract
Pathogenic bacteria trigger complex molecular interactions in hosts that are characterized mainly by an increase in reactive oxygen species (ROS) as well as an inflammation-associated response. To counteract oxidative damage, cells respond through protective mechanisms to promote resistance and avoid tissue damage and infection; among these cellular mechanisms the activation or inhibition of the nuclear factor E2-related factor 2 (Nrf2) is frequently observed. The transcription factor Nrf2 is considered the master regulator of several hundred cytoprotective and antioxidant genes. Under normal conditions, the Keap1/Nrf2 signaling protects the cellular environment by sensing deleterious oxygen radicals and inducing the expression of genes coding for proteins intended to neutralize the harmful effects of ROS. However, bacteria have developed strategies to harness Nrf2 activity to their own benefit, complicating the host response. This review is aimed to present the most recent information and probable mechanisms employed by a variety of bacteria to modulate the Keap1/Nrf2 activity in order to survive in the infected tissue. Particularly, those utilized by the Gram-positive bacteria Staphylococcus aureus, Streptococcus pneumoniae, Listeria monocytogenes, and Mycobacterium tuberculosis as well as by the Gram-negative bacteria Escherichia coli, Helicobacter pylori, Legionella pneumophila, Pseudomonas aeruginosa and Salmonella typhimurium. We also discuss and highlight the beneficial impact of the Keap1/Nrf2 antioxidant and anti-inflammatory role in bacterial clearance.
Introduction
The innate immune response is the first line of defense against infectious agents such as parasites, viruses, fungi, and bacteria. It involves a complex network of molecular interactions and cellular defense mechanisms between host and pathogen, intended not only to eliminate the pathogen (resistance) but also to limit tissue damage and infection (tolerance) without directly targeting the pathogen. Under normal conditions, these complex mechanisms give rise to a successful resolution of the infection and a recovery of the tissue homeostasis (1). Among the different mechanisms currently studied and known to contribute to immune tolerance associated with infections caused by bacterial pathogens, the activation of nuclear transcription factor erythroid 2-related factor 2 (Nrf2) has been considered of central importance (2, 3). Nrf2 is regarded as the antioxidant and anti-inflammatory master regulator that protects cells from tissue damage caused by oxidative stress via an antioxidant defense response (4). Although the importance of Nrf2 as one of the multiple responses to infection is accepted worldwide, many questions remain unanswered, especially those related to oxidative stress and the molecular mechanisms involved in such regulation. In particular, one of these questions concerns how pathogenic Gram-positive and Gram-negative bacteria manipulate the Keap1/Nrf2 signaling pathway and how cells respond to limit bacterial survival and replication. Understanding these molecular mechanisms should be fundamental to design new therapeutic strategies to regulate and control infections, reducing tissue damage and its inflammatory consequences.
The main goal of this review is to present new findings that strongly support the regulatory role of the transcription factor Nrf2 during bacterial infections. The first section includes an overview of the Keap1/Nrf2 relationship to immune modulation and inflammation, and a detailed description of the signaling pathway mechanism. The second and third parts are devoted to concisely describe the molecular mechanisms employed by the Gram-positive bacteria Staphylococcus aureus, Streptococcus pneumoniae, Listeria monocytogenes, and Mycobacterium tuberculosis and by the Gram-negative bacteria Escherichia coli, Helicobacter pylori, Legionella pneumophila, Pseudomonas aeruginosa, and Salmonella typhimurium in the activation or inhibition of the Keap1/Nrf2 signaling.
The role of Keap1/Nrf2 in the inflammatory response
Nrf2 activation is particularly relevant in inflammation, which is the most essential process within the innate immune response. Inflammation started with the recognition of pathogen-associated molecular patterns (PAMPs) or cell damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs) such as transmembrane Toll-like receptors (TLRs) and intracellular nucleotide oligomerization domain-like receptors (NODs or NLRs). These PRRs promote the expression of inflammatory mediators such as chemokines, cytokines, eicosanoids, proteases, and reactive oxygen species (ROS) (5). The most relevant ROS include the superoxide anion, hydrogen peroxide, and hydroxyl radicals formed because of the partial reduction of oxygen (6). Although low levels of ROS are essential to eliminate pathogens, an excess can induce the generation of oxidative stress as a result of an imbalance between oxidizing and antioxidant molecules. Since ROS are very unstable and may quickly react and damage biological macromolecules including DNA, proteins, and lipids, cells must maintain tight control over ROS to keep cellular homeostasis. Structure alteration of these biomolecules caused by ROS may lead to the development of chronic and degenerative diseases, including Alzheimer’s, Parkinson’s, rheumatoid arthritis, premature aging, cancer, cardiovascular and pulmonary disorders, as well as viral, parasitic and bacterial infections (7–10).
During an infection process, cells of the immune system, including lymphocytes, dendritic cells, macrophages, and neutrophils sense pathogenic microorganisms that are subsequently engulfed and eliminated by the formation of phagosomal structures. Once phagosomes mature by fusion with endosomes and lysosomes, a ROS oxidative burst develops (11). A sustained increase in ROS may be detrimental and even lead to cell death by apoptosis. In addition, high concentrations of ROS activate signaling pathways involved in the production of free radicals and inflammation by activation of the transcription factor nuclear factor kappa B (NF-κB) that induces the expression of the proinflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-1β, and many others (12, 13). In this context, activation of antioxidant and anti-inflammatory genes is of vital importance to counteract the inflammation and maintain redox homeostasis. Among the large battery of biomolecules that contribute to restrain the deleterious effects of ROS are worth to mention reduced glutathione (GSH), superoxide dismutase (SOD), catalase (CAT) and phase II detoxifying enzymes such as heme oxygenase-1 (HO-1), NADPH: Quinone Oxidoreductase 1 (NQO-1), Glutathione S-Transferase (GST), thioredoxin and peroxiredoxin systems, NADPH generation systems, and iron metabolism (14). Notably, Nrf2 regulates the expression of genes encoding most of these antioxidant and detoxifying enzymes.
The Nrf2 signaling pathway
Nrf2 belongs to the cap ‘n’ collar (CNC) subfamily of transcription factors with an essential leucine zipper (bZip) located at its C-terminal region (15). Structurally, it comprises seven highly conserved domains called Neh (Nrf2-ECH homology). Under normal physiological conditions, Nrf2 activity is regulated by its cytoplasmic interactions with the two Kelch domains of the homodimeric Kelch-like ECH-associated protein Keap1. The homodimeric Keap1 interacts with the high-affinity (ETGE) and low-affinity (DLG) motifs of the Neh2 domain of Nrf2, which allows Keap1 to function as an adaptor of the RING box protein 1 (RBX1)-associated ubiquitin E2 ligase Cullin-3 (CUL3). The complex Keap1-RBX1-CUL3 ubiquitinates Nrf2 at Lys residues located between the DLG and ETGE motifs, priming its degradation by the proteasome 26S (Figure 1A) (16, 17). However, under oxidative or electrophilic conditions, the Cys thiol groups of Cys158 present in the Keap1 Broad complex Tramtrack and Bric-a-Brac (BTB) domain or that of Cys288 at the intervening region (IVR) are oxidized. This induces a mechanism of dissociation between Nrf2 and Keap1 that is different from the “Hinge-Latch” mechanism proposed by Horie et al. (2021) for protein-protein interaction inhibitors, thus facilitating the cytoplasmic accumulation of Nrf2 and its subsequent translocation to the nucleus (18). Once in the nucleus, Nrf2 binds to small aponeurotic muscle fibrosarcoma (sMaf) proteins and induces the expression of antioxidant response element (ARE)-driven genes encoding antioxidant and detoxifying enzymes that neutralize ROS, electrophiles, hydroperoxides, carbonyls, quinones, and exogen stimuli such as xenobiotics and UV light thereby restoring the redox balance (Figure 1B) (19). Recovery from oxidative stress is achieved when Keap1 translocates to the nucleus and promotes Nrf2 nuclear export. This mechanism restores normal physiological levels of Nrf2 and prevents the antioxidant and detoxifying response from being unnecessarily prolonged (Figure 1C).
Figure 1

The NRF2/KEAP1 signaling pathway adapted from [17]. (A) Under basal conditions, Nrf2 remains inactive in the cytoplasm bound to its regulatory protein Keap1. This protein is associated with the CUL3-RBX1 complex, allowing the transfer of ubiquitin (Ub) to the Lys residues located between the DLG and ETGE motifs of the Nrf2 Neh2 domain. Then, degradation of Nrf2 occurs by ubiquitination and recognition by the proteasome 26S. (B) During oxidative stress, Cys residues at KEAP1 are oxidized at their BTB and/or IVR domains, inducing dissociation between Nrf2 and Keap1. Nrf2 is released to the cytoplasm and, after accumulation, it is translocated to the nucleus where it binds to the small aponeurotic muscle fibrosarcoma (sMaf) proteins and induces the expression of antioxidant and detoxifying genes coding for enzymes that neutralize ROS and restore redox balance. (C) To restore redox balance, Keap1 is translocated to the nucleus and translocates Nrf2 back to the cytoplasm. This controls the time of the antioxidant and detoxifying response. ARE, antioxidant response element; BTB, Broad complex Tramtrack and Bric-a-Brac; Cul3, Cullin 3 protein; DLG, low-affinity motif of the Nrf2 Neh2 domain; E3, E3 ubiquitin ligase; ETGE, high-affinity motif of the Nrf2 Neh2 domain; GCLM, Glutamate-cysteine ligase modifier subunit; GST, Glutathione S-Transferase; HO-1, Heme Oxygenase-1; Keap1, Kelch-like ECH-associated protein 1; NQO-1, NADPH: Quinone Oxidoreductase 1; Nrf2, nuclear transcription factor erythroid 2-related factor 2; RBX1, RING box protein-1; sMaF, small aponeurotic muscle fibrosarcoma proteins; Ub, ubiquitin.
Although Nrf2 is activated mainly by high ROS concentrations, several non-canonical mechanisms that regulate Nrf2 activity have also been described. One of them is characterized by phosphorylation of Nrf2 at Ser/Thr residues carried out by protein kinases such as ERK, p38, PKC, JNK, MSK1/2, and PI3K (20, 21). Another mechanism involves formation of a cytoplasmic macromolecular complex constituted by the enzyme glycogen synthase kinase-3β (GSK3β), Axin-1 and Cullin1-associated E3 ubiquitin ligase and beta-transducin repeats-containing proteins (β-TrCP), whose function is to remove low concentrations of ROS produced by normal cellular metabolism (22, 23). Interestingly, this mechanism also controls Nrf2 activity by promoting its degradation in a Keap1-independent manner, through a process that requires phosphorylation at Ser residues of the Neh6 domain (24). A third mechanism of Nrf2 activation involves the autophagy signaling pathway p62/Sequestosome 1 (SQSTM1; hereinafter referred to as p62) protein-dependent degradation of Keap1, in which many proteins are digested (4, 25).
Pharmacological activation of Nrf2 can be achieved through various compounds with electrophilic properties capable of covalently modifying cysteine (Cys) residues in Keap1. This chemical modification induces conformational changes that prevent the ubiquitination and subsequent proteasomal degradation of Nrf2 (26). Examples of such compounds include sulforaphane, chalcone, and dimethyl fumarate (DMF) (26–28). Alternatively, non-electrophilic compounds can activate this signaling pathway by reversibly inhibiting the Nrf2-Keap1 protein-protein interaction (PPI). These include compound 7, diaryl-triazole-derived compounds (27, 29), Nrf2 peptides containing ETGE or DLG motifs, p62-derived peptides, compounds derived from tetrahydroisoquinoline (THIQ), indoline, or related to 1,4-diaminonaphthalene or iminocoumarin-benzothiazole, among many others (27). Interestingly, the selective activation of Nrf2 by various chemical molecules has been shown to induce a cytoprotective effect against bacterial infections, as will be discussed in the following sections.
Gram positive bacteria
Staphylococcus aureus
S. aureus is an intracellular bacterium that causes a wide range of infections in humans, ranging from mild skin to severe and life-threatening conditions such as pneumonia, sepsis, and endocarditis. The interaction between S. aureus and the host immune system involves various molecular and cellular pathways, including the Keap1/Nrf2/ARE. Although this bacterium may activate the production of ROS that ultimately could lead to Nrf2 activation (Figure 2-1), some studies have proposed that S. aureus may have developed mechanisms to manipulate the host immune responses for its survival and proliferation, including the modulation of the Nrf2 activity (30). An interesting model to investigate these type of mechanisms is bovine mastitis where a weak immune response is often observed during S. aureus infections. In primary bovine mammary epithelial cells (pbMECs) the presence of S. aureus was unable to either induce changes in Nrf2 activity or induce the expression of the Nrf2-dependent genes NQO1 and GCLM even when ROS concentration in the infected cells is high (31). The idea that S. aureus is able to manipulate the Nrf2 pathway arose from the observation that macrophages (Raw 264.7 cells) expressed HO-1 when stimulated with peptidoglycan (PGN), suggesting the activation of Nrf2 and implicating that the whole bacteria is needed to repress Nrf2 activation as it was observed in pbMECs (32). Activation of the MAPK/p38 signaling pathway in THP-1 macrophages infected with S. aureus triggers the production of the proinflammatory cytokines IL-6, IL-1β, and TNF-α (Figure 2-2). Remarkably, S. aureus took advantage of these cytokines to promote its own intracellular survival (33, 34). An even more surprising strategy is the sequestration of the p62/SQTM1-Keap1/Nrf2 signaling pathway, which normally protects cells from oxidative stress. In this way, S. aureus suppresses the macrophage´s antioxidant response and builds up a favorable environment for their replication inside cells (Figure 2-3). Consequently, in the initial stage of the infection this bacterium avoids elimination and is able to persists for longer times within host cells. A possible mechanism explaining this behavior was elucidated in pbMECs infected with S. aureus. Such study showed that Keap1 was not degraded despite an increase in p62 protein levels (31), which suggests that S. aureus can manipulate p62 during an early phase of infection by reducing the phosphorylation levels of p62 at Ser349. It was later demonstrated that p62 phosphorylation affects the Keap1 inactivation machinery, resulting in repression of Nrf2 without necessarily compromising intracellular Keap1 levels. These data were complemented with results obtained from THP-1 macrophages infected with S. aureus and subjected to repetitive magnetic stimulation (rMS). In this model, there was a decreased phosphorylation of MAPK/p38 and an increased phosphorylation of p62 at Ser349, which in turn increased its binding affinity to Keap1. The Keap1-p62 complex was degraded by autophagy, allowing the release of Nrf2 that can freely translocate to the nucleus (Figure 2-4) (35). Interestingly, NF-κB activity, that is needed to develop a proper immune response and pro-inflammatory cytokine production, is only achieved when Nrf2 was overexpressed in A549 cells, indicating that Nrf2 function is a necessary previous step for a proper NF-κB activation (36). Although a detailed mechanism for this process has not yet been described it is worth mentioning that Keap1 was able to downregulate NF-κB through its interaction with IKKβ. This event ultimately led to IκBα stabilization, suggesting that S. aureus manipulates both pathways by preventing Keap1 degradation (Figure 2-5).
Figure 2
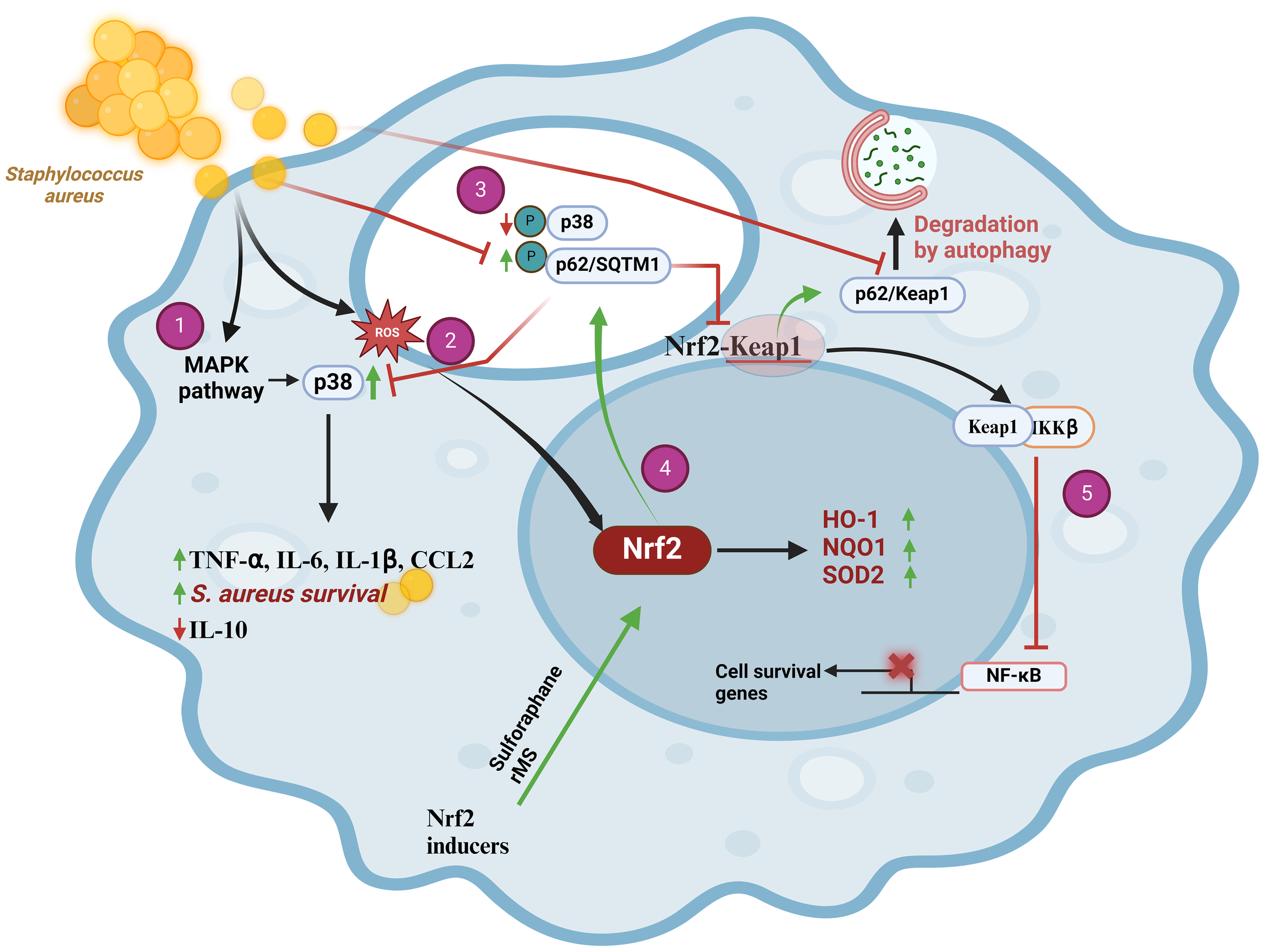
Staphylococcus aureus prevents Nrf2 pathway activation in THP-1 macrophages through upregulation of MAPK/p38 and downregulation of p62/SQTM1, scheme modified from [35]. (1) During the initial phase of infection, S. aureus causes an increase in ROS that activates Nrf2. (2) Upon internalization, S. aureus induces the expression of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, and CCL2) by activating the MAPK signaling pathway. This causes p38 phosphorylation that promotes bacterial cell survival and decreases the expression of the anti-inflammatory cytokine IL-10. (3) S. aureus manages to suppress the cellular antioxidant response by sequestering the p62/SQSTM1-Keap1/Nrf2 signaling pathways. This bacterium causes a decrease in p62/SQTM1 phosphorylation at Ser349, which diminishes its affinity for the Keap1 protein, thereby preventing formation of the p62-Keap1 complex and its degradation through the autophagy pathway. The final outcome of this mechanism is the repression of Nrf2. (4) Selective activation of Nrf2 decreased MAPK/p38 phosphorylation that increases p62 phosphorylation at Ser349 and its affinity for Keap1. Then, Keap1 is degraded via autophagy and Nrf2 translocates to nucleus to activate the antioxidant response, which could aid in S. aureus clearance. (5) Keap1 negatively regulates NF-κB by interacting with IKKβ. Ultimately, this stabilizes IκBα. CCL2, chemokine (C-C motif) ligand 2; HO-1, Heme Oxygenase-1; IKKβ, inhibitory kappa B kinase beta; IL-10, Interleukin 10; IL-6, interleukin-6; IL-1β, interleukin-1beta; Keap1, Kelch-like ECH-associated protein 1; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; NQO-1 NADPH, Quinone Oxidoreductase 1; Nrf2, nuclear transcription factor erythroid 2-related factor 2; p38, p38 mitogen-activated protein kinases; p62/SQSTM1, p62 protein, also called sequestosome 1; ROS, reactive oxygen species; SOD, superoxide dismutase.
Roles for the Keap1/Nrf2/ARE pathway in S. aureus infections have also been reported in a pneumonia mouse model, where Nrf2 promoted the activation of mitochondrial biogenesis. These events resulted in a limitation of acute lung injury and pro-inflammatory cytokine production that rescued mice from lethal sepsis (37). According to this study, when Nrf2-/- mice were infected with S. aureus, a delayed expression of HO-1 and superoxide dismutase 2 (SOD2) was accompanied with an overexpression of TNF-α, IL-1β;, and chemokine ligand 2 (CCL2; from the CC motif subfamily) in the lungs as compared to WT mice. Moreover, in the Nrf2-/- mice lungs the level of IL-10 expression was lower, reinforcing the idea that the absence of Nrf2 promotes a pro-inflammatory environment with more intense infiltration of PMN cells, edema, and loss of alveolar membrane integrity. Of note, in contrast to the mastitis model, the S. aureus lung infection is associated with downregulation of p62 levels, leading to the autophagy reduction of damaged mitochondrial proteins that are essential for tissue repair.
It is clear that Nrf2 activation in S. aureus infected tissues has a prominent role as a cytoprotective factor and aids to develop a proper immune response. This affirmation has been strengthened by data obtained in studies analyzing the sulforaphane (SFN, an activator of Nrf2) effects in cases of antibiotic resistant to S. aureus infections. SFN reduced the internalization and survival of S. aureus although both NQO1 and HO-1 expression was not modified (30). These data support the idea that S. aureus is able to harness the Nrf2 signaling to extend its survival inside cells.
Streptococcus pneumoniae
Extracellular pathogen S. pneumoniae is a leading cause of community-spread pneumonia. Recently, different studies have pointed out the role of Nrf2 activity during infections caused by this bacterium. In 2020 Maurice et al. showed an increase in the expression of anti-oxidant proteins like Nrf2, in response to mitochondrial-derived oxidative injury in a model of human primary bronchial epithelial cells infected with S. pneumoniae (38). In a subsequent study and using a Nrf2-/- mouse model of pneumonia, this bacterium caused epithelial barrier dysfunction accompanied by enhanced neutrophil recruitment to the alveolar space, lung injury, and leukocytosis as well as a high mortality rate in the knock-outs relative to the wild-type animals, suggesting that Nrf2 activation exhibits a protective mechanism during S. pneumoniae infections (39). These results were strengthened in several models of pneumonia and keratitis, in which the Nrf2 activators, tert-butylhydroquinone (tBHQ) and resveratrol, down-regulated S. pneumoniae inflammation, lung injury, and promoted catalase function that counteracted the H2O2 secreted by this bacterium (40, 41). Infection of human corneal epithelial cells by S. pneumoniae showed that tBHQ down-regulated ROS production, protected cells from cytotoxic effects, and induced Nrf2-dependent autophagy to enhance bacterial clearance. Furthermore, inhibition of IKKβ; activity by free Keap1 contributed to reduce the inflammatory response mediated by NF-κB (Figure 3A). Notably, tBHQ also promoted LL-37 antimicrobial peptide expression although no relation to Nrf2 was established (41).
Figure 3
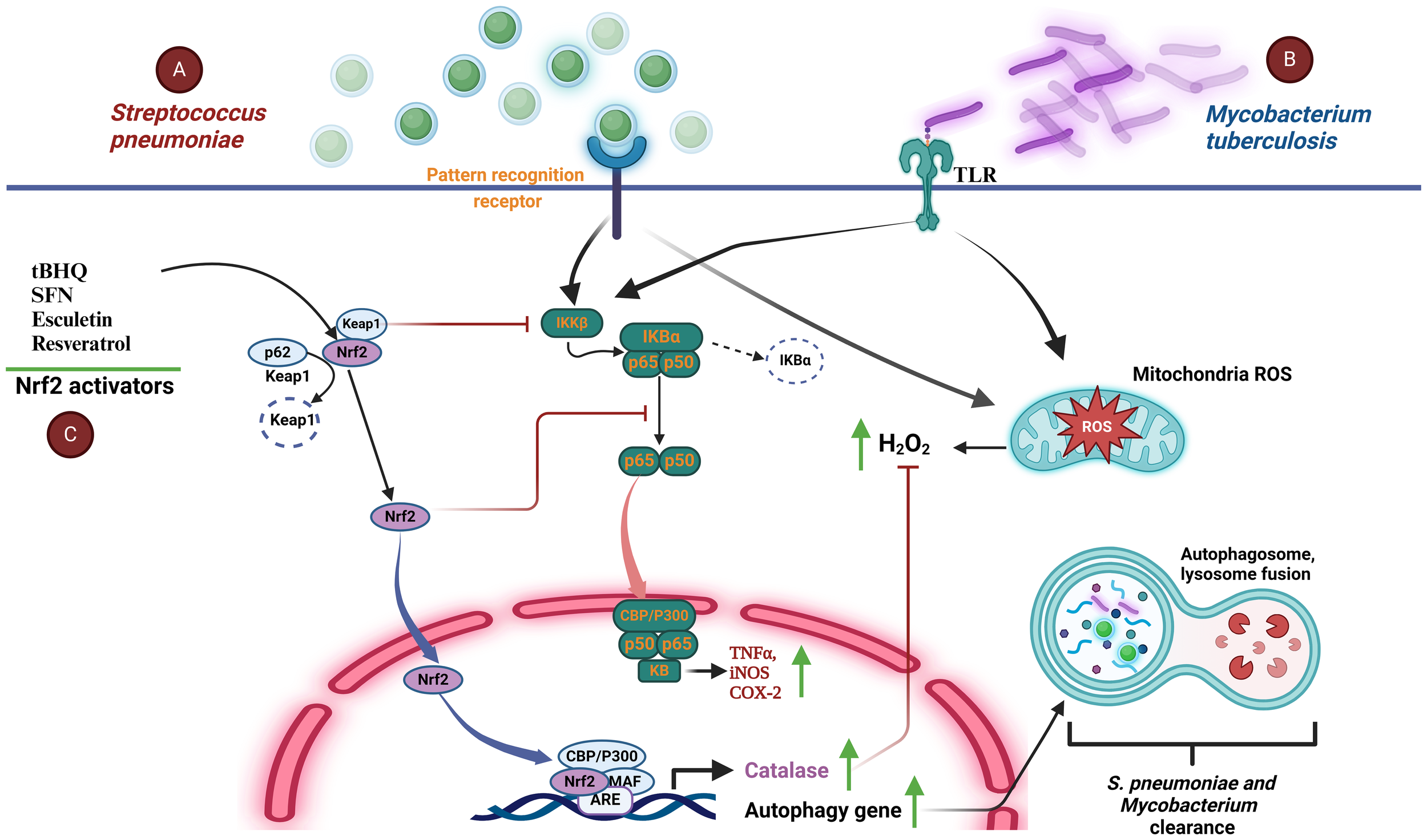
Activation of Nrf2 in S. pneumoniae and M. tuberculosis infections. (A) The inflammatory and redox response to S. pneumoniae infection (green spheres) in lung epithelial cells is initiated by their interaction with pattern recognition receptors (PRRs) that stimulate the activation of the inflammatory response mediated by NF-κB, which translocates to the nucleus and promotes the expression of TNF-α, iNOS, and COX-2. Simultaneously, S. pneumoniae induces the generation of mitochondrial reactive species (i.e. H2O2). An increment of H2O2 activates the transcription factor Nrf2. This in turn promotes expression of antioxidant and autophagy genes, allowing elimination of H2O2, promoting bacterial phagocytosis, and reducing the inflammatory response by blocking the nuclear translocation of NF-κB. (B)M. tuberculosis (purple rods) uses a mechanism very similar to that employed by S. pneumoniae because it interacts specifically with host cell TLRs, activating NF-κB-mediated inflammation. At the same time, the increase in H2O2 upon infection activates Nrf2, which promotes the expression of antioxidant enzymes thereby re-establishing cellular homeostasis, reducing inflammation, and favoring bacterial clearance through the autophagosome pathway. (C) Nrf2 activation by molecules such as SFN, TBHQ, and resveratrol, among others, restores the host redox imbalance, tissue damage and inflammation. These compounds are also able to regulate bacterial persistence and burden by inducing the expression of genes targeting autophagy. ARE, antioxidant response element; CBP, creb-binding protein; COX-2, Cyclooxygenase-2; H2O2, hydrogen peroxide; IκBα, inhibitor of nuclear factor kappa b; IKKβ, inhibitory kappa kinase beta; Keap1, kelch-like ECH-associated protein 1; NF-κB, factor nuclear factor kappa b; Nrf2, nuclear transcription factor erythroid 2-related factor 2; P300, e1a binding protein p300; PRRs, pattern recognition receptors; ROS, reactive oxygen species; SFN, sulforaphane; sMaF, small aponeurotic muscle fibrosarcoma proteins; TLRs, toll-like receptors; TNF-α, tumor necrosis factor-α; tBHQ, tert-butylhydroquinone; iNOS, inducible nitric oxide synthase.
The importance of the genes transcribed under the Nrf2 control in infections caused by S. pneumoniae was also observed in macrophages isolated from chronic obstructive pulmonary disease (COPD) patients. These macrophages showed a defect in S. pneumoniae phagocytosis that was related to impaired transcriptional response, including the Nrf2 pathway. Phagocytosis capacity was recovered by stimulation of COPD-derived macrophages with the Nrf2 agonists SFN and compound-7 (42). Moreover, when Raw 264.7 macrophages were stimulated with lipoteichoic acid (LTA) from Gram-positive bacterial cell wall, esculetin (a compound from Dipteryx odorata) down-regulated nitric oxide synthase (iNOS), nitric oxide (NO) and the NF-κB pathway while promoting Nrf2 activation, which mitigated LTA-induced inflammation by NF-κB activity inhibition (43).
Nrf2 also regulates gene expression of the NADPH-generating enzyme, malic enzyme 1 (ME1) (44). Malic enzyme 1 is essential in fatty acid synthesis, catalyzing the reversible decarboxylation of malate to pyruvate while regenerating NADPH from NADP (45). Reduced ME1 expression in alveolar macrophages (AM) and peripheral blood monocyte-derived macrophages (MDM) from patients with chronic obstructive pulmonary disease (COPD) has been linked to defects in the phagocytosis process of bacteria such as Haemophilus influenzae and Streptococcus pneumoniae (42, 46). Macrophages exhibited reduced transcriptional responses to opsonized bacteria, including Nrf2-mediated oxidative stress responses, and have defects in efferocytosis, phagocytosis, glycolytic reserve, and metabolic plasticity. The altered responses were reversed by treatment with Nrf2 agonists such as sulforaphane and compound 7, capable of rescuing ME1 expression, thus demonstrating that this enzyme can become a new therapeutic target against these bacterial infections through Keap1-Nrf2 PPI inhibitors (42, 46).
Altogether these data demonstrate that activation of Nrf2 is beneficial to the host during S. pneumoniae infection and highlights that activation of the Nrf2 pathway may be a novel strategy to counteract the damage caused by this bacterium.
Mycobacterium tuberculosis
Infections caused by intracellular pathogen M. tuberculosis stand as a prominent global cause of mortality, posing a significant threat to public health. During the course of infection, M. tuberculosis induces a disruption in redox balance and compromises the efficacy of protective immunity and emerging evidence suggests that Nrf2 plays a pivotal role in the host response to mycobacterial infections (47, 48). On one hand, Nrf2 activation has been associated with increased antimicrobial activities, promoting the clearance of mycobacteria. This involves the upregulation of autophagy genes, inflammation, and immune modulation. On the other hand, persistent activation of Nrf2 may lead to an immunosuppressive environment, facilitating mycobacterial survival and exacerbating chronic infections (Figure 3B) (49). Additionally, RNA-seq and immunoblotting data revealed an elevation in Nrf2 gene expression following THP-1 macrophages infected by M. tuberculosis (49). However, longer M. tuberculosis exposure caused a decline in Nrf2 protein levels while the expression of Keap1 protein, a negative regulator of Nrf2, remained consistently low. Furthermore, SFN (sulforaphane) induced a pro-inflammatory cytokines reduction, phagocytosis, host cell apoptosis, ROS increase, and autophagy of THP-1 infected with M. tuberculosis. Notably, SFN-activated Nrf2 stimulated an enhancement in the THP-1 capacity to eliminate bacteria, possibly through the modulation of protective immunity mediated by Nrf2. It seems that Nrf2 acts like a two-sided weapon against mycobacterial infections (Figure 3C). That is, while Nrf2 activity may help clearing M. tuberculosis in short term infections, in longer term cases a reduction of Nrf2 levels is observed. Understanding the complex mechanistic behavior of Nrf2 regulation in M. tuberculosis infected tissues may guide to novel therapeutic strategies to combat these infections.
Listeria monocytogenes
L. monocytogenes is a motile, rod-shaped, and intracellular facultative anaerobic bacterium causative of the foodborne illness known as listeriosis. L. monocytogenes is unique among the pathogenic bacteria because it is able to survive and proliferate at low temperatures (50). This bacterium disrupts the Nrf2 pathway to evade the host immune response through as-yet-unknown mechanism that involves the activation of the immunoglobulin superfamily T-cell membrane protein mucin-3 (Tim-3). Tim-3 has been recognized as an immune checkpoint inhibitor, and its dysregulation has been associated with T-cell tolerance and various immune disorders, including tumors and tolerance to infections (51). However, a precise role of Tim-3 in innate immunity has so far remained unclear. A recent report revealed that Tim-3 hinders the phagocytic activity of macrophages against L. monocytogenes by inhibiting the Nrf2 signaling pathway, whose consequence is an increase in bacterial burden. It is likely, as it is observed in some type of cancers, that Tim-3 signaling pathway facilitates Nrf2 degradation through the activation of the tripartite motif (TRIM) family proteins known to induce E3 ligase-dependent but Keap1-independent ubiquitination giving rise to a reduction of Nrf2 nuclear translocation (Figure 4) (52, 53). Interestingly, downregulation of two Nrf2-dependent genes CD36 and HO-1 were identified as responsible for L. monocytogenes phagocytosis and increased bacterial survival. Restoration of CD36 and HO-1 expression in macrophages reduced bacterial burden and severity of infection (52). These data indicate that downregulation of the Keap1/Nrf2 pathway increases bacterial survival, which suggests that activation of Nrf2 may be a useful strategy to improve L. monocytogenes clearance from infected tissues.
Figure 4
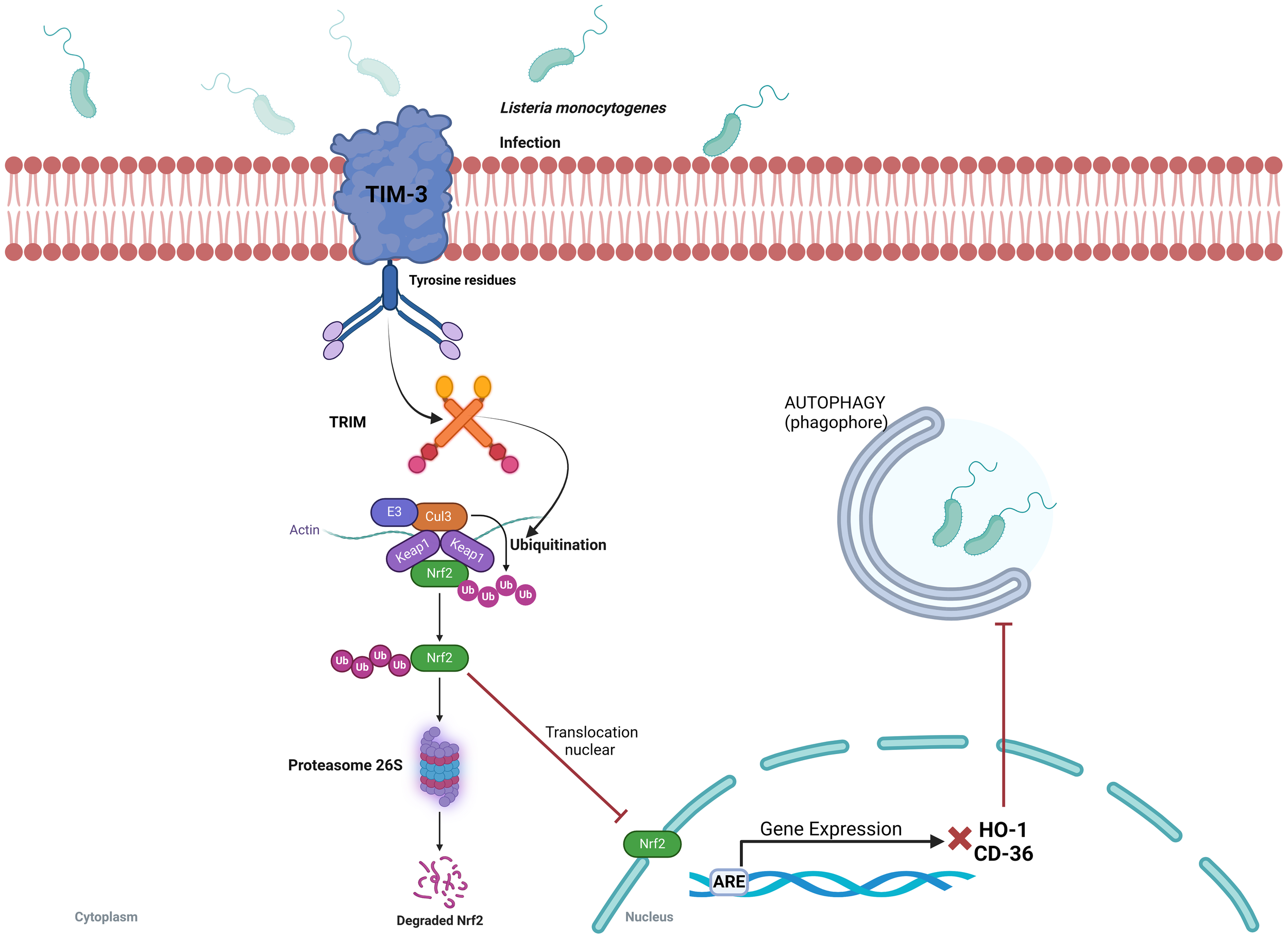
Listeria monocytogenes evades the host immune response by Tim-3-dependent inhibition of the Keap1/Nrf2 signaling pathway. Interaction of L. monocytogenes with Tim-3 (the macrophage type I membrane protein) induces ubiquitination and degradation of the downstream transcription factor Nrf2. A decrease in Nrf2 levels reduces the expression of the effector genes CD36 and HO-1, which decreases the phagocytic capacity of macrophages and favors bacterial survival. Restoration of Nrf2 levels reverses these effects, suggesting that Nrf2 activation may enhance bacterial clearance from infected tissues. ARE, antioxidant response element; CD-36, platelet glycoprotein 4; Cul3, Cullin 3 protein; E3, E3 ubiquitin ligase; HO-1, Heme oxygenase-1; Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear transcription factor erythroid 2-related factor 2; TRIM, tripartite motif family proteins; Ub, ubiquitin.
Gram negative bacteria
Escherichia coli
E. coli is a bacterium susceptible to natural and random genetic alteration. Most of the pathogenic E. coli strains are extracellular, although some, such as uropathogenic E. coli, can become intracellular under certain conditions (54). Hence, it is common to find variants within the same species that are divided into those causing diarrheal and extra intestinal infections. These variants can be facultative or obligate pathogens causing infections that range from moderate to lethal cases (55). Also, normal intestinal microbiota of both humans and animals contains non-pathogenic variants of this bacterium (55). To date, nine pathotypes have been isolated from humans suffering diarrheal and extra intestinal disease (56). However, urinary tract infections (UTIs) caused by uropathogenic E. coli (UPEC) are the most common variant worldwide, with an estimate of about 150 million cases per year (57).
Several studies have shown that E. coli infections are associated with ROS oxidative stress (58–61). That is why UTI-UPEC are commonly accompanied by an increase in lipid peroxidation and a decrease in the activity of the Nrf2-dependent antioxidant enzymes such as catalase (CAT) and superoxide dismutase (SOD) (62). Furthermore, in a mouse model of acute lung injury (ALI) caused by E. coli, it was found an increase in Keap1, low expression of the antioxidant enzymes NQO1and HO-1, and low levels of Nrf2 (63). Reversion of these effects was observed in mice treated with the anti-inflammatory isoquinoline alkaloid sinomenine (SIN) (64). These mice presented a down-regulation of Keap1 expression, an increase in the levels of Nrf2, and high levels of the antioxidant enzymes NQO1 and HO-1. SIN also down-regulated the expression of the pro-inflammatory mediators IL-6, IL-1β, and TNF-α that characterize acute inflammation in E. coli-ALI through a mechanism that inhibits the NF-κB nuclear translocation by blocking phosphorylation of IκBα. It seems that SIN significantly ameliorated E. coli-induced histopathological changes by reducing lung injury in mice in an Nrf2-dependent manner (63). Nrf2 also induced overexpression of NQO1 in response to LPS during infections caused by E. coli in lung tissues and THP-1-derived macrophages (63–66). In this case it was observed a reduction of the NF-κB signaling activity that resulted in lowering the expression of the pro-inflammatory genes IL-6 and IL-1β (63, 65–67). In another study, activation of Nrf2 nuclear translocation after p62 binding to Keap1 in urothelial cells infected with E. coli enabled bacterial exocytosis (68). Once in the nucleus, Nrf2 interacted with the promoter region of the RAB27B gene thereby regulating the expression of this protein (68). An important aspect of this work was that increased expression of NQO1, glutamate-cysteine ligase catalytic subunit (GCLC), and HO-1 was required to mitigate the effects caused by ROS. It was also necessary a constant increase in Nrf2 expression to reduce the bacterial load of intracellular E. coli in bladder epithelial or urothelial cells (68).
From these results it is evident that modulation of Nrf2 activity in UTI caused by E. coli is undoubtedly of paramount importance. Nevertheless, more studies are needed to provide a deep insight of these mechanisms in order to design possible therapeutic strategies that help to control this type of infections.
Helicobacter pylori
H. pylori is generally viewed as an extracellular pathogen, but also exhibit intracellular behavior under certain conditions and is considered a major risk factor in the development of peptic ulcers and gastric cancer. After infection, several virulence factors trigger a transformation process in gastric cells, causing intestinal injury that leads to the appearance of peptic ulcers, atrophic gastritis, intestinal metaplasia, dysplasia, and adenocarcinoma (69). It is estimated that more than 50% of the world’s population is infected by this bacterium, although only about 10% of infected patients develop gastric cancer, suggesting the existence of defense mechanisms by the host against H. pylori (70). The inflammatory response initiated by H. pylori is closely related to secretion of virulence factors such as cytotoxin-associated gene A (CagA), type IV secretion system (T4SS) (71), vacuolating cytotoxin A (VacA), ureases, adhesins, peptidoglycan outer membrane proteins (i.e., BabA, SabA, OipA, HtrA, DupA, IceA), arginase, γ-glutamyl transpeptidase (GGT), catalase, and SOD (72).
An important toxin secreted by H. pylori is VacA, which has the ability to induce formation of vacuoles, induce apoptosis, improve colonization capacity in the stomach, contribute to the pathogenesis of gastric adenocarcinoma, peptic ulcer, and autophagy (73). VacA-activated autophagy is believed to contribute to limiting toxin damage to the host cell (74) and induce oxidative stress, which results in membrane pores formation with subsequent loss of mitochondrial activity (75). The importance of oxidative stress has been widely discussed in the context of gastric cancer (GC) (76), which suggests a highly relevant role of Nrf2. In this scenario, polymorphisms present in the Nrf2 promoter are known to be associated with the development of gastric mucosal inflammation, either independently or associated with H. pylori infection (77). By using three different gastric epithelial cell lines (non-cancerous HFE-145, diffuse gastric cancer AGS, and intestinal GC subtype MKN74) Bacon et al. (2022) were able to demonstrate that Nrf2 signaling pathway was differentially regulated depending on the stage of H. pylori infection. Moreover, VacA-dependent downregulation of Nrf2 was observed after 24 h of infection, which resulted in an increase in epithelial-to-mesenchymal transition (EMT) an important stage in the development of GC (78). These results suggest that Nrf2 is crucial for maintaining a normal epithelial gastric cell phenotype.
Regulation of Nrf2 during H. pylori infection could also be related to an increase in ROS production, which in turn triggers a p62 -Keap1/Nrf2/HO-1 selective autophagy. This process is regulated by the ubiquitination of p62 and by sequential phosphorylation reactions (79). Two important facts are worth to mention on the Keap1/Nrf2/HO-1 signaling axis participation in the autophagy process activated by H. pylori in human gastric adenocarcinoma (HGA) that allows oxidative stress-damaged epithelial cells to be eliminated (70): 1) There was a nuclear translocation of Nrf2 upon co-incubation of HGA cells with H. pylori for a period of 3 h and 2) Nrf2 induced the expression of HO-1 whose byproduct is carbon monoxide (CO), a molecule considered antioxidant and anti-inflammatory. Therefore, in H. pylori infections the Keap1/Nrf2 signaling pathway has been considered as one of the cellular defense mechanisms capable of regulating the autophagy process by modulating the p62 protein. Although the molecular mechanisms in H. pylori infections are yet to be fully elucidated, therapeutic approaches targeting Nrf2 seem quite attractive.
Legionella pneumophila
L. pneumophila is an intracellular pathogen considered one of the main causative agents of acquired pneumonia in the European community (80). This bacterium is widely distributed in the environment, causing sporadic outbreaks of opportunistic infections, particularly in young children and the elderly immunocompromised individuals (81). L. pneumophila causes two different types of pathologies: 1) Legionnaires’ disease, which includes lung infection symptoms such as cough, difficulty of breathing, fever, and headache and 2) Pontiac fever, which displays flu-like symptoms but does not involve pneumonia. During the infection process, L. pneumophila interacts with several immunomodulatory molecules on the cell surface of infected cells, such as TLRs on the plasma membrane and NLRs in the cytoplasm (82, 83). Activation of these receptors induces an inflammatory process due to excessive production of pro-inflammatory cytokines that may contribute to pneumonia symptoms (83). L. pneumophila alters the cellular redox metabolism, which in turns activates the Keap1/Nrf2-dependent antioxidant signaling pathway (84). It is believed that MitF (mitochondrial fragmentation factor), an effector injected by the type IV secretion system of L. pneumophila to the alveolar macrophages, is one of the causative agents associated with increased ROS production and mitochondrial fragmentation (85, 86). MitF is a protein produced by mitochondrial accumulation of dynamin-1-like protein (DNM1L) a GTPase essential for the regulation of mitochondrial fission and activator of Ran GTPase (Ras-related nuclear protein) and translocation of RNA and proteins through the nuclear pore complex (85). Furthermore, cellular metabolism and autophagy, being closely related processes to redox signaling, link the p62 autophagic system to Nrf2 activation by inhibiting the interaction with Keap1. This mechanism involves phosphorylation of the p62 Keap1-interacting region (KIR) at Ser351, which increases the affinity of p62 to Keap1 preventing the interaction between Keap1 and Nrf2 (84, 87). However, during the early stages of L. pneumophila infection (1-2 h post-infection, hpi) to A549 lung cells, ubiquitination of p62 at residues Ser361/365/366, near the p62-KIR, by ubiquitin-linked phosphoribosyl (PR-Ub) prevents its binding to Keap1. Consequently, Keap1 is free to interact with Nrf2, leading to its rapid degradation by the proteasome 26S. This mechanism prevents premature activation of the Nrf2-dependent redox response. In contrast, at 4 hpi, there is a significant decrease in PR-Ub-p62 levels, which promotes Nrf2-dependent transcriptional activation of target genes because phosphorylated p62 at Ser351 binds Keap1 and blocks its interaction with Nrf2 (Figure 5) (84, 88).
Figure 5
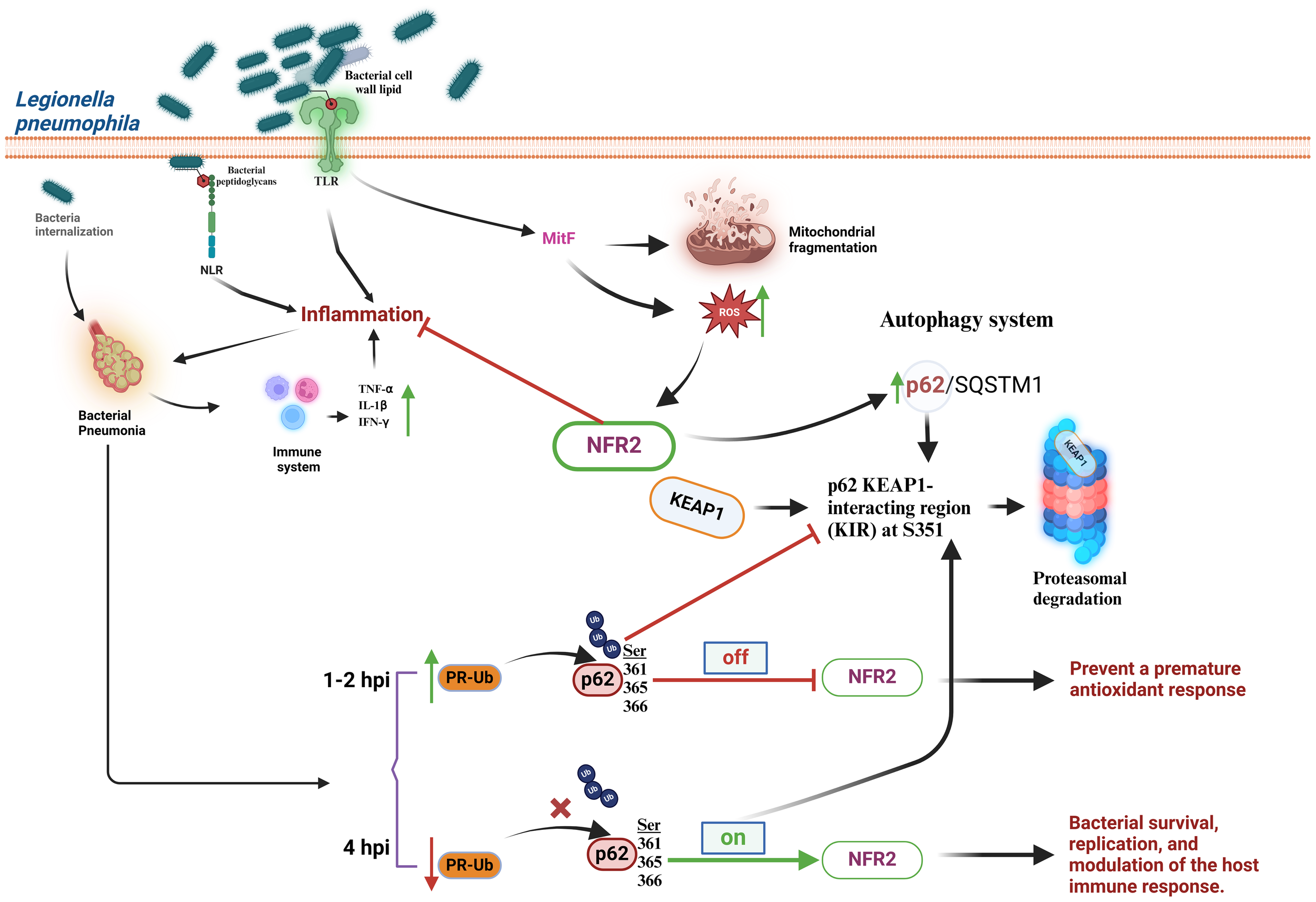
Legionella pneumophila manipulates the cellular antioxidant response at initial and later stages of infection. L. pneumophila interacts with TLRs or NLRs, triggering the synthesis of the pro-inflammatory cytokines TNF-α, IL-1β, and IFN-γ, increase in ROS production, activation of Nrf2, and mitochondrial fragmentation. Nrf2 may also be linked to an increase in the p62/SQSTM1 autophagic system by promoting the interaction of the Keap1-interacting region of p62 (KIR) with Keap1. At early stages (1-2 h post-infection, hpi) (Off) L. pneumophila blocks activation of Nrf2 in a process dependent upon the activity of ubiquitin-linked phosphoribosyl (PR-Ub). At later stages (≥ 4 hpi) (On) a reduction of p62 ubiquitination is observed because the PR-Ub levels decrease. This allows p62 to interact with Keap1 and block Nrf2 degradation. IL-β, interleukin-1beta; IFN-γ, interferon gamma; Keap1, Kelch-like ECH-associated protein 1; KIR, the p62 KEAP1-interacting region; MitF, mitochondrial fragmentation factor; NLRs, Nod-like receptors; Nrf2, nuclear transcription factor erythroid 2-related factor 2; p62/SQSTM1, p62 protein, also called sequestosome 1; PR-Ub, ubiquitin-linked phosphoribosyl; ROS, reactive oxygen species; TLRs, Toll-like receptors; TNF-α, tumor necrosis factor-α.
Interestingly, the interplay between L. pneumophila infection and the host cell redox response seems to be a carefully orchestrated battlefield. This bacterium strategically inhibits Nrf2 activation at early stages of infection by manipulating p62 ubiquitination to prevent an antioxidant response. Such a delay may be beneficial for bacterial survival and replication. In contrast, at later stages L. pneumophila induces activation of Nrf2, to exploit, perhaps, the antioxidant mechanisms for its own benefit.
Pseudomonas aeruginosa
P. aeruginosa is an extracellular pathogen, and facultative opportunistic aerobic bacterium typically found in soil or water sources that is associated with hospital-acquired pneumonia and chronic lung infections in cystic fibrosis patients (89, 90). The ability of P. aeruginosa to cause infections stems from two key factors: 1) Formation of biofilms that resist components of the host immune system and 2) secretion of virulence factors such as exoproteases, phospholipases, hemolysins, and phenazines that facilitate colonization in the pulmonary epithelium (91–93). One of the main virulence factors of P. aeruginosa-induced pulmonary disease is the phenazine pyocyanin (1-hydroxy-5-methyl-phenazine, PCN) because of its ability to induce ROS response. ROS production may accelerate bacterial invasion, alter the immune response, and damage lung epithelial cells (94, 95). PCN undergoes reduction by NADPH, allowing it to react with molecular oxygen and generate superoxide radicals and hydrogen peroxide (94, 96).
Several studies have shown that PCN may be an activator of Nrf2 (97–99). Evidence of Nrf2 accumulation and nuclear translocation was demonstrated to occur in A549 lung cells stimulated with PCN, leading to the expression of the antioxidant Nrf2-dependent genes NQO1 and γ-glutamylcysteine synthetase heavy subunit (γ-GCSH) (98, 100). A proposed mechanism to explain these data suggests that PCN-triggered ROS production causes the expression of Nrf2-dependent genes (HO-1, catalase, and SOD) and TNF-α, IL-6, and IL-1β; that activate the epidermal growth factor receptor (EGFR) either by inducing its expression (TNF-α) (101), by co-regulation of their signaling components (IL-6) (102) or by direct binding (IL-1β;) (103). ROS also induces the expression of ligands such as transforming growth factor alpha (TGF-α) and heparin-bound epidermal growth factor (HB-EGF) that activate this receptor (104). EGFR activates phosphatidylinositol-3-kinase (PI3K), which phosphorylates protein kinase B (Akt) and mitogen-activated protein kinase kinase kinase complex (MAPKKK, MEK1/2) and extracellular signal-regulated MAPK subfamily proteins ERK1/2 (Figure 6). These kinases are thought to phosphorylate Nrf2 at specific Ser and Thr residues (Ser215, Ser408, Ser558, Ser577, and Thr559), causing its dissociation from Keap1 and nuclear translocation (101, 105). Redox regulation in lung cells might also involve PCN-induced Nrf2 activation by high mobility group nucleosome-binding protein 2 (HMGN2). These chromatin-associated, non-histone nuclear proteins, play a role in gene transcription, replication, DNA repair, and cell differentiation (98, 106, 107). Nuclear translocation of Nrf2 is among the most important mechanism activated by HMGN2 along with the expression of antioxidant genes, scavenging of ROS, actin rearrangement, and phosphorylation of ERK1/2 and JNK, while regulating P. aeruginosa invasion (98).
Figure 6
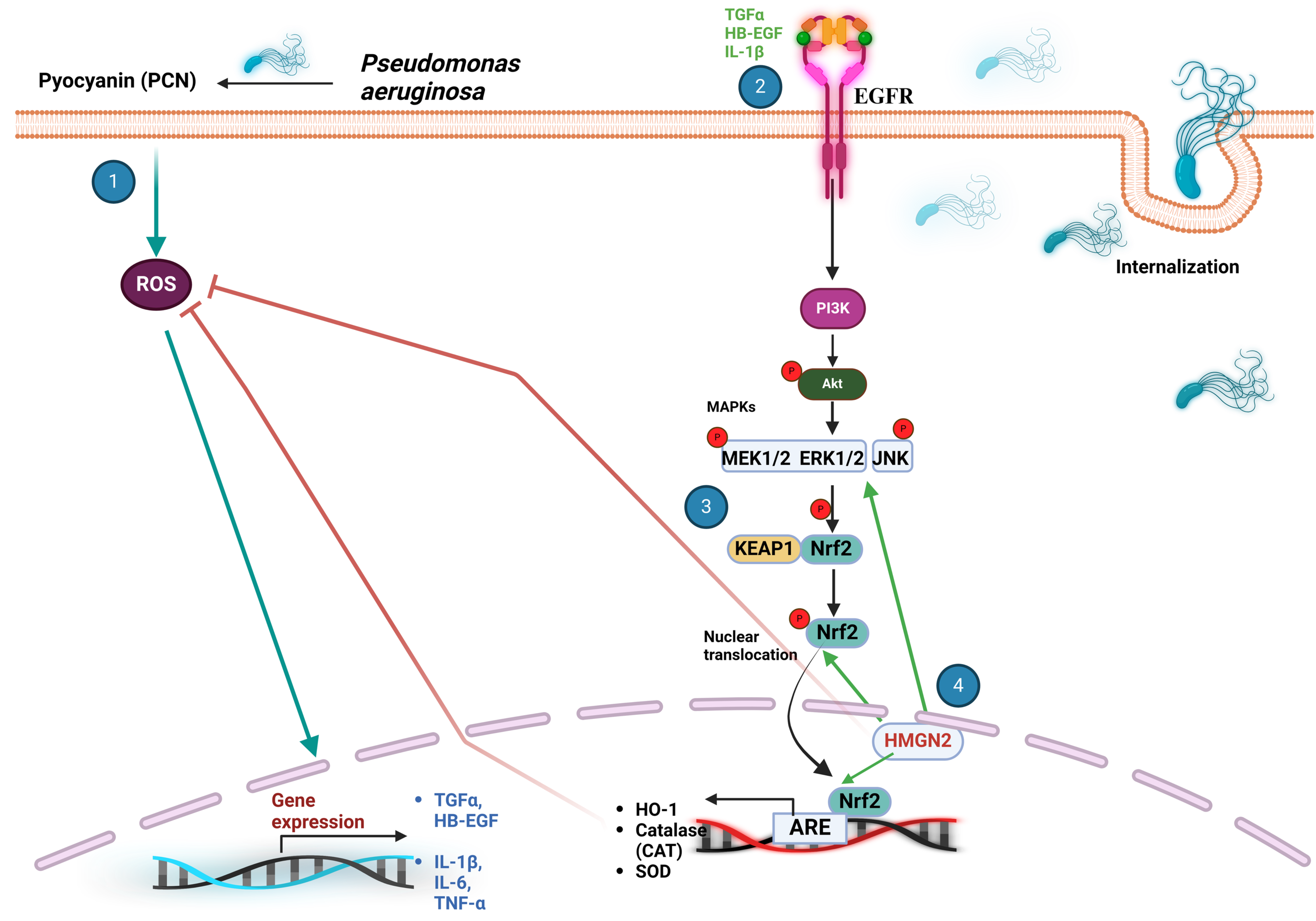
The Keap1/Nrf2 signaling pathway plays a crucial role in the cellular response to P. aeruginosa infection. (1) Pyocyanin (PCN) induces an increment in ROS, leading to the expression of TNF-α, IL-6, IL-1β;, TGF-α and HB-EGF that modulate the activity of EGFR. (2) EGFR triggers a downstream signaling cascade that activates PI3K, Akt, the MAPKKK MEK1/2, ERK1/2, and JNK. (3) These protein kinases phosphorylate Nrf2 at Ser215, Ser408, Ser558, Ser577 and Thr559, promoting its nuclear translocation. (4) PCN also activates Nrf2 via HMGN2 that assists ROS scavenging through antioxidant Nrf2-dependent gene expression, actin rearrangement, and activation of ERK1/2 and JNK. ARE, antioxidant response element; Akt, protein kinase B; CAT, catalase; EGFR, epidermal growth factor receptor; HB-EGF, heparin-bound epidermal growth factor; HMGN2, high-mobility group nucleosome-binding protein 2; IL-6 and IL-β, interleukin-6 and interleukin-1beta; JNK, c-Jun N-terminal kinase; Keap1, Kelch-like ECH-associated protein 1; MAPK, mitogen-activated protein kinase; Nrf2, nuclear transcription factor erythroid 2-related factor 2; PCN, pyocyanin; PI3K, phosphatidylinositol-3-kinase; ROS, reactive oxygen species; SOD, superoxide dismutase; TGF-α, tissue growth factor alpha; TNF-α, tumor necrosis factor-α.
Despite all these important advances, the exact mechanism of Nrf2 activation by P. aeruginosa still remains under investigation. Involvement of canonical and non-canonical Keap1/Nrf2 pathways suggest new possibilities for targeted drug design to combat this and other pathogenic bacteria.
Salmonella typhimurium
S. typhimurium, a rod-shaped and intracellular bacterium, is a major cause of foodborne illness worldwide. It infects both humans and animals, leading to the development of diseases like chronic gastroenteritis (108, 109). During the course of infection, S. typhimurium secretes adhesion and virulence factors that are injected to the host intracellular environment by the type III secretion system (T3SS) (110). Notably, the cytotoxic protein SpvB, secreted via T3SS, modulates the inflammatory response by directly targeting the NF-κB signaling pathway. SpvB prevents IKKβ phosphorylation, thereby hindering the subsequent phosphorylation and degradation of IκBα. Ultimately, this represses NF-κB nuclear translocation and pro-inflammatory gene expression, dampening the host’s immune response and promoting bacterial survival and replication (111).
In addition to manipulating inflammation via NF-κB, S. typhimurium also induces oxidative stress, which in turn activates the Keap1/Nrf2 pathway. This pathway leads to the expression of genes involved in defense and detoxification, potentially protecting host cells from oxidative damage (112, 113). Recent studies have demonstrated that esculetin (EST), a natural coumarin found in various dietary foods and herbs, can activate Nrf2. This activation effectively inhibits the proliferation and expansion of multidrug-resistant (MDR) Salmonella Typhimurium. Notably, EST significantly downregulates the expression of the type 3 secretion system-1 (T3SS-1) in both intestinal epithelial cells (IEC-6) and a murine model (114). These findings suggest that dietary coumarins or a targeted plant-based diet may offer a promising strategy to counteract MDR bacterial-induced enteric diseases through Nrf2 activation.
However, confirming data on targeting the Nrf2 pathway by S. typhimurium are limited. For instance, in bone marrow-derived mouse macrophages (BMDM) infected with this bacterium, type I interferon signaling (IFN-I) disrupts Nrf2 responses to oxidative stress, which results in mitochondrial dysfunction and subsequent cell death by necrosis. IFN-I induces a positive regulation of the mitochondrial phosphatase Pgam5 that represses the Nrf2-dependent transcription of antioxidant genes (113). Conversely, mitochondrial damage caused by ROS promotes autophagy of p62, a protein that competes with the Nrf2 binding to Keap1. This further reduces the antioxidant activity of Nrf2 during Salmonella infection (113, 115). In a different study in RAW264.7 mouse macrophages infected with S. typhimurium the SpvB protein disrupted iron metabolism facilitating bacterial survival and replication (116). This work also revealed that the SpvB C-terminal domain targets Nrf2 for degradation via the proteasomal pathway and prevents transcription of ferroportin (FPN), a protein responsible for iron export from macrophages. Consequently, iron accumulates intracellularly, leading to severe serum hypoferremia, cellular injury, and acute inflammatory processes. Additionally, Deng et al. (2022) were able to demonstrate that NLRP6, a member of the nucleotide-oligomerization domain-like receptors (NLR) family, regulating host iron metabolism that perturbs host resistance to bacterial infection in both macrophages and enterocytes through increasing the unique iron exporter ferroportin-mediated iron efflux in a Nrf2-dependent manner during S. typhimurium infection (117). In this investigation, it was observed that NLRP6 negatively regulates the activation of Nrf2 by binding to AKT, reducing the dissociation of the Nrf2-Keap1 complex, and the nuclear translocation of Nrf2. Consequently, there is a decrease in the fpn gene, which codes for ferroportin (FPN), and a reduction in iron export, as well as an increase in intracellular iron levels that promote bacterial survival. In the absence of NLRP6, AKT phosphorylates p62 at Ser351, mediated by the mammalian target of rapamycin complex 1 (mTOR). This phosphorylation negatively regulates the transcription of Keap1 by promoting the phosphorylation of the transcription factor Forkhead box class O 3a (FOXO3A) while simultaneously activating the nuclear translocation of Nrf2 (117). All these data have allowed us to consider NLRP6 as a new potential therapeutic target to limit bacterial iron acquisition during S. typhimurium infections through the regulation of Nrf2.
Overall, the interplay between Salmonella and the Nrf2 pathway is complex. It is evident that more research is needed to fully understand the role of Nrf2 during Salmonella infection. This knowledge could be crucial for developing targeted therapies that modulate the Nrf2 pathway for the treatment of Salmonella-associated diseases.
Concluding remarks and future perspectives
Harnessing the Nrf2 signaling pathway seems to be a recurrent strategy exploited by a variety of bacteria to facilitate their invasion, survival, and proliferation. In general, the inhibition of the Nrf2-associated antioxidant and anti-inflammatory genes seems to be beneficial in most of the cases discussed here. Nonetheless, the intricate interplay between the microorganism and the host is more complex as observed in infection caused by S. pneumoniae and S. typhimurium. In these cases, bacterial-mediated activation of the Nrf2 pathway paradoxically benefits the microorganism. A notable finding is the temporal variability in the modulation of the Nrf2 signaling pathway by diverse bacterial species. Both upregulation and downregulation of Nrf2 have been observed at different post-infection time points, highlighting the importance of considering temporal dynamics in future scientific endeavors.
In this context, what seems to be emerging as a common strategy is the fine-tuning timing that bacteria utilize to manipulate various canonical and non-canonical mechanisms to regulate the Nrf2 activity to their own benefit. Understanding not only the particular interactions that each infectious microorganism uses to control the antioxidant response but also the time evolution of such manipulation may delineate the development of novel therapies to tackle these infections.
On the other hand, recent research has demonstrated the participation of Nrf2 as a key regulator of mitochondrial function, which opens new perspectives as therapeutic strategies for the management of bacterial infections. Given the diversity of bacterial virulence factors, many bacteria alter mitochondrial function for their benefit (118). Interestingly, many of these virulence factors specifically target cellular processes that modulate host mitochondrial pathways, such as apoptosis, mitochondrial fusion and fission, energy production, mitochondrial dynamics, mtDNA damage, biogenesis, mitophagy, and the mitochondrial unfolded protein response (118–120). For instance, Escherichia coli secretes the T3SS effectors Map and EspF, deregulating mitochondrial morphology and promoting calcium release-mediated apoptosis (121–123). In contrast, S. flexneri and S. typhimurium secrete the T3SS effectors IpaD and SipD, inducing mitochondrial depolarization, activation of the non-canonical intrinsic apoptosis pathway, and autophagy-mediated type II programmed cell death, respectively (118, 123). Other bacteria, such as S. pneumoniae, M. tuberculosis, and P. aeruginosa, cause the release of mitochondrial genetic material (mtDNA), leading to acute lung injury in mouse models (124–126). Additionally, bacteria like L. monocytogenes and S. aureus suppress mitochondrial ROS, necessary for the elimination of the bacterial agent, and alters mitochondrial biogenesis and the mitophagy process (118, 126).
Antioxidant therapies carried out by activating Nrf2 with molecules such as bardoxolone methyl and rosmarinic acid have been shown to help restore mitochondrial function, leading to increased bacterial clearance and resolution of inflammation through enhanced antioxidant, mitophagy and mitochondrial biogenesis pathways, demonstrating the therapeutic potential of Nrf2 on bacterial infections (127, 128).
Statements
Author contributions
MR: Conceptualization, Investigation, Writing – original draft. OS: Conceptualization, Writing – original draft. JP: Writing – review & editing. VB: Conceptualization, Formal analysis, Funding acquisition, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Coordinación de la Investigación Científica, Universidad Michoacana de San Nicolás de Hidalgo, Research Program 2024 to VB. MR is a recipient of a doctoral scholarship number 814880 from Consejo Nacional de Humanidades Ciencia Tecnología e Innovación (CONAHCYT, México).
Acknowledgments
Authors would like to thank BioRender: Scientific Image and Illustration Software (https://www.biorender.com/). Each Figure was created under the Confirmation of Publication and License Rights –Open Access- to MR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Schneider DS Ayres JS . Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. (2008) 8:889–95. doi: 10.1038/nri2432
2
Soares MP Ribeiro AM . Nrf2 as a master regulator of tissue damage control and disease tolerance to infection. Biochem Soc Trans. (2015) 43:663–8. doi: 10.1042/BST20150054
3
Klemm P Rajendiran A Fragoulis A Wruck C Schippers A Wagner N et al . Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur J Immunol. (2020) 50:515–24. doi: 10.1002/eji.201948285
4
Vomund S Schäfer A Parnham MJ Brüne B von Knethen A . Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. (2017) 18:2772. doi: 10.3390/ijms18122772
5
Fitzgerald KA Kagan JC . Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
6
Collin F . Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci. (2019) 20:2407. doi: 10.3390/ijms20102407
7
Niedzielska E Smaga I Gawlik M Moniczewski A Stankowicz P Pera J et al . Oxidative stress in neurodegenerative diseases. Mol Neurobiol. (2016) 53:4094–125. doi: 10.1007/s12035-015-9337-5
8
Butterfield DA Halliwell B . Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. (2019) 20:148–60. doi: 10.1038/s41583-019-0132-6
9
Puspita L Chung YS Shim JW . Oxidative stress and cellular pathologies in Parkinson’s disease. Mol brain. (2017) 10:1–12. doi: 10.1186/s13041-017-0340-9
10
Ivanov VA Bartosch B Isaguliants GM . Oxidative stress in infection and consequent disease. Oxid Med Cell Longev. (2017) 2017:3496043. doi: 10.1155/2017/3496043
11
Deramaudt TB Dill C . Regulation of oxidative stress by Nrf2 in the pathophysiology of infectious diseases. Med Mal. Infect. (2013) 43:100–7. doi: 10.1016/j.medmal.2013.02.004
12
Redza-Dutordoir M Averill-Bates DA . Activation of apoptosis signalling pathways by reactive oxygen species. BBA. (2016) 1863:2977–92. doi: 10.1016/j.bbamcr.2016.09.012
13
Mittal M Mohammad RS Tran K Reddy PS Malik AB . Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
14
Ngo V Duennwald LM . Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease. Antioxidants. (2022) 11:2345. doi: 10.3390/antiox11122345
15
Dodson M Cholanians BA Schmidlin JC Chapman E Zhang DD . Modulating Nrf2 in disease: timing is everything. Annu Rev Pharmacol Toxicol. (2019) 59:555–75. doi: 10.1146/annurev-pharmtox-010818-021856
16
Cores A Piquero M Villacampa M León R Menéndez JC . Nrf2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules. (2020) 10:1–38. doi: 10.3390/biom10060904
17
Bono S Corbo M . Impaired antioxidant Keap1-Nrf2 system in amyotrophic lateral sclerosis: Nrf2 activation as a potential therapeutic strategy. Mol Neurodegener. (2021) 16:1–26. doi: 10.1186/s13024-021-00479-8
18
Horie Y Suzuki T Inoue J Iso T Wells G Moore WT et al . Molecular basis for the disruption of Keap1–Nrf2 interaction via Hinge & Latch mechanism. Commun Biol. (2021) 4:576. doi: 10.1038/s42003-021-02100-6
19
Tonelli C Chio IIC Tuveson DA . Transcriptional regulation by nrf2. Antioxid Redox Signal. (2018) 29:1727–45. doi: 10.1089/ars.2017.7342
20
Müller S Behnen M Bieber K Möller S Hellberg L Witte M et al . Dimethylfumarate impairs neutrophil functions. J Invest. Dermatol. (2016) 136:117–26. doi: 10.1038/JID.2015.361
21
Shaw P Chattopadhyay A . Nrf2–ARE signaling in cellular protection: mechanism of action and the regulatory mechanisms. J Cell Physiol. (2020) 235:3119–30. doi: 10.1002/jcp.29219
22
Chowdhry S Zhang Y McMahon M Sutherland C Cuadrado A Hayes JD . Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. (2013) 32:3765–81. doi: 10.1038/onc.2012.388
23
Cuadrado A Manda G Hassan A Alcaraz MJ Barbas C Daiber A et al . Transcription factor Nrf2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. (2018) 70:348–83. doi: 10.1124/pr.117.014753
24
Rada P Rojo AI Chowdhry S McMahon M Hayes JD Cuadrado A . SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. (2011) 31:1121–33. doi: 10.1128/MCB.01204-10
25
Zhang W Feng C Jiang H . Novel target for treating Alzheimer’s diseases: crosstalk between the Nrf2 pathway and autophagy. Ageing Res Rev. (2021) 65:101207. doi: 10.1016/j.arr.2020.101207
26
Esteras N Abramov AY . Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic Biol Med. (2022) 189:136–53. doi: 10.1016/j.freeradbiomed.2022.07.013
27
Kumar V Kumar S Hassan M Wu H Thimmulappa RK Kumar A et al . Novel chalcone derivatives as potent Nrf2 activators in mice and human lung epithelial cells. J Med Chem. (2011) 54:4147–59. doi: 10.1021/jm2002348
28
Muri J Wolleb H Broz P Carreira EM Kopf M . Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1β production and inflammatory apoptosis at high dose. Redox Biol. (2020) 36:101647. doi: 10.1016/j.redox.2020.101647
29
Wu H Wu J Jiang J Qian Z Yang S Sun Y et al . Compound 7 regulates microglia polarization and attenuates radiation-induced myelopathy via the Nrf2 signaling pathway in vivo and in vitro studies. Mol Med. (2024) 30:198. doi: 10.1186/s10020-024-00951-3
30
Bonay M Deramaudt T . Use of Nrf2 activators for the treatment of Staphylococcus aureus infections. Patent No WO2020094767A. Paris France patent (2020). Available at: https://patents.google.com/patent/WO2020094767A1/en (accessed May 24, 2024).
31
Ying YT Yang J Tan X Liu R Zhuang Y Xu JX et al . Escherichia coli and Staphylococcus aureus differentially regulate Nrf2 pathway in bovine mammary epithelial cells: Relation to distinct innate immune response. Cells. (2021) 10:3426. doi: 10.3390/cells10123426
32
Yin S Cao W . Toll-like receptor signaling induces Nrf2 pathway activation through p62-triggered Keap1 degradation. Mol Cell Biol. (2015) 35:2673–83. doi: 10.1128/MCB.00105-15
33
Oviedo-Boyso J Barriga-Rivera JG Valdez-Alarcón JJ Bravo-Patiño A Cárabez-Trejo A Cajero-Juárez M et al . Internalization of Staphylococcus aureus by bovine endothelial cells is associated with the activity state of NF-kappaB and modulated by the pro-inflammatory cytokines TNF-alpha and IL-1beta. Scand J Immunol. (2008) 67:169–76. doi: 10.1111/j.1365-3083.2007.02056.x
34
Deramaudt TB Ali M Vinit S Bonay M . Sulforaphane reduces intracellular survival of Staphylococcus aureus in macrophages through inhibition of JNK and p38 MAPK induced inflammation. Int J Molec Med. (2020) 45:1927–41. doi: 10.3892/ijmm.2020.4563
35
Deramaudt TB Chehaitly A Charrière T Arnaud J Bonay M . High-Frequency repetitive magnetic stimulation activates bactericidal activity of macrophages via modulation of p62/Keap1/Nrf2 and p38 MAPK pathways. Antioxidants. (2023) 12:1695. doi: 10.3390/antiox12091695
36
Jin SH Sun JJ Liu G Shen LJ Weng Y Li JY et al . Nrf2/PHB2 alleviates mitochondrial damage and protects against Staphylococcus aureus-induced acute lung injury. MedComm. (2023) 4:e448. doi: 10.1002/mco2.448
37
MacGarvey NC Suliman HB Bartz RR Fu P Withers CM Welty-Wolf KE et al . Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med. (2012) 185:851–61. doi: 10.1164/rccm.201106-1152OC
38
Maurice N Bedi B Yuan Z Koval M Hart C Sadikot R . Streptococcus pneumoniae-induced epithelial barrier dysfunction is mediated by mitochondrial reactive oxygen species generation and attenuated by a mitochondrially-targeted antioxidant D102. Bacterial infections: immunity and basic mechanisms. Am Thorac Soc. (2020) 201:A7733–3. doi: 10.1164/ajrccm-conference.2020.201.1
39
Gomez JC Dang H Martin JR Doerschuk CM . Nrf2 modulates host defense during Streptococcus pneumoniae pneumonia in mice. J Immunol. (2016) 197:2864–79. doi: 10.4049/jimmunol.1600043
40
Zahlten J Kim YJ Doehn JM Pribyl T Hocke AC García P et al . Streptococcus pneumoniae-induced oxidative stress in lung epithelial cells depends on pneumococcal autolysis and is reversible by resveratrol. J Infect Dis. (2015) 211:1822–30. doi: 10.1093/infdis/jiu806
41
Sharma P Roy S . Streptococcus pneumoniae exerts oxidative stress, subverts antioxidant signaling and autophagy in human corneal epithelial cells that is alleviated by tert-Butylhydroquinone. Med Microbiol Immunol. (2022) 211:119–32. doi: 10.1007/s00430-022-00731-y
42
Bewley MA Budd RC Ryan E Cole J Collini P Marshall J et al . Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med. (2018) 198:739–50. doi: 10.1164/rccm.201705-0903OC
43
Jayakumar T Huang CJ Yen TL Hsia CW Sheu JR Bhavan PS et al . Activation of Nrf2 by Esculetin mitigates inflammatory responses through suppression of NF-κB signaling cascade in RAW 264.7 cells. Molecules. (2022) 27:5143. doi: 10.3390/molecules27165143
44
Dinkova-Kostova AT Abramov AY . The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. (2015) 88:179–88. doi: 10.1016/j.freeradbiomed.2015.04.036
45
Simmen FA Alhallak I Simmen RCM . Malic enzyme 1 (ME1) in the biology of cancer: it is not just intermediary metabolism. J Mol Endocrinol. (2020) 65:R77–90. doi: 10.1530/JME-20-0176
46
Ryan EM Sadiku P Coelho P Watts ER Zhang A Howden AJM et al . NRF2 activation reprograms defects in oxidative metabolism to restore macrophage function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2023) 207:998–1011. doi: 10.1164/rccm.202203-0482OC
47
Rothchild AC Olson GS Nemeth J Amon LM Mai D Gold ES et al . Alveolar macrophages generate a noncanonical NRF2-driven transcriptional respons to Mycobacterium tuberculosis in vivo. Sci Immunol. (2019) 4:eaaw6693. doi: 10.1126/sciimmunol.aaw6693
48
Matsuyama M Nonaka M Nakajima M Morishima Y Ishii Y Hizawa N . The role of NRF2 in mycobacterial infection. Antioxidants. (2021) 10:1861. doi: 10.3390/antiox10121861
49
Zhou J Fang F Qi J Li T Zhang L Liu H et al . Activation of Nrf2 modulates protective immunity against Mycobacterium tuberculosis infection in THP1-derived macrophages. Free. Radic Biol Med. (2022) 193:177–89. doi: 10.1016/j.freeradbiomed.2022.10.274
50
Osek J Lachtara B Wieczorek K . Listeria monocytogenes - How this pathogen survives in food-production environments? Front Microbiol. (2022) 13:866462. doi: 10.3389/fmicb.2022.866462
51
Wu FH Yuan Y Li D Lei Z Song CW Liu YY et al . Endothelial cell-expressed Tim-3 facilitates metastasis of melanoma cells by activating the NF-kappaB pathway. Oncol Rep. (2010) 24:693–9. doi: 10.3892/or_00000909
52
Wang Z Sun D Chen G Li G Dou S Wang R et al . Tim-3 inhibits macrophage control of Listeria monocytogenes by inhibiting Nrf2. Sci Rep. (2017) 7:42095. doi: 10.1038/srep42095
53
Liu W Zhao Y Wang G Feng S Ge X Ye W et al . TRIM22 inhibits osteosarcoma progression through destabilizing Nrf2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. (2022) 53:102344. doi: 10.1016/j.redox.2022.102344
54
Braz VS Melchior K Moreira CG . Escherichia coli as a multifaceted pathogenic and versatile bacterium. Front Cell Infect Microbiol. (2020) 10:548492. doi: 10.3389/fcimb.2020.548492
55
Köhler CD Dobrindt U . What defines extraintestinal pathogenic Escherichia coli? Int J Med Microbiol. (2011) 301:642–7. doi: 10.1016/j.ijmm.2011.09.006
56
Nash JH Villegas A Kropinski AM Aguilar-Valenzuela R Konczy P Mascarenhas M et al . Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics. (2010) 11:667. doi: 10.1186/1471-2164-11-667
57
Terlizzi ME Gribaudo G Maffei ME . UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front Microbiol. (2017) 8:1566. doi: 10.3389/fmicb.2017.01566
58
Blomgran R Zheng L Stendahl O . Uropathogenic Escherichia coli triggers oxygen-dependent apoptosis in human neutrophils through the cooperative effect of type 1 fimbriae and lipopolysaccharide. Infect Immun. (2004) 72:4570–8. doi: 10.1128/IAI.72.8.4570-4578.2004
59
Aubron C Glodt J Matar C Huet O Borderie D Dobrindt U et al . Variation in endogenous oxidative stress in Escherichia coli natural isolates during growth in urine. BMC Microbiol. (2012) 12:120. doi: 10.1186/1471-2180-12-120
60
Baronetti JL Villegas NA Aiassa V Paraje MG Albesa I . Hemolysin from Escherichia coli induces oxidative stress in blood. Toxicon. (2013) 70:15–20. doi: 10.1016/j.toxicon.2013.03.014
61
Mendoza-Chamizo B Løbner-Olesen A Charbon G . Coping with reactive oxygen species to ensure genome stability in Escherichia coli. Genes. (2018) 9:565. doi: 10.3390/genes9110565
62
Kurutas EB Ciragil P Gul M Kilinc M . The effects of oxidative stress in urinary tract infection. Mediators. Inflamm. (2005) 2005:242–4. doi: 10.1155/MI.2005.242
63
Liu S Chen Q Liu J Yang X Zhang Y Huang F . Sinomenine protects against E. coli-induced acute lung injury in mice through Nrf2-NF-κB pathway. Biomed Pharmacother. (2018) 107:696–702. doi: 10.1016/j.biopha.2018.08.048
64
Hou W Huang L Huang H Liu S Dai W Tang J et al . Bioactivities and mechanisms of action of Sinomenine and its derivatives: a comprehensive review. Molecules. (2024) 29:540. doi: 10.3390/molecules29020540
65
Ali M Bonay M Vanhee V Vinit S Deramaudt TB . Comparative effectiveness of 4 natural and chemical activators of Nrf2 on inflammation, oxidative stress, macrophage polarization, and bactericidal activity in an in vitro macrophage infection model. PloS One. (2020) 15:e0234484. doi: 10.1371/journal.pone.0234484
66
Kimura A Kitajima M Nishida K Serada S Fujimoto M Naka T et al . NQO1 inhibits the TLR-dependent production of selective cytokines by promoting IκB-ζ degradation. J Exp Med. (2018) 215:2197–209. doi: 10.1084/jem.20172024
67
Kobayashi EH Suzuki T Funayama R Nagashima T Hayashi M Sekine H et al . Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:11624. doi: 10.1038/ncomms11624
68
Joshi CS Mora A Felder PA Mysorekar IU . Nrf2 promotes urothelial cell response to bacterial infection by regulating reactive oxygen species and RAB27B expression. Cell Rep. (2021) 37:109856. doi: 10.1016/j.celrep.2021.109856
69
Díaz P Valenzuela VM Bravo J Quest AFG . Helicobacter pylori and gastric cancer: adaptive cellular mechanisms involved in disease progression. Front Microbiol. (2018) 9:5. doi: 10.3389/fmicb.2018.00005
70
Paik JY Lee HG Piao JY Kim SJ Kim DH Na HK et al . Helicobacter pylori infection promotes autophagy through Nrf2-mediated heme oxygenase upregulation in human gastric cancer cells. Biochem Pharmacol. (2019) 162:89–97. doi: 10.1016/j.bcp.2019.02.003
71
Tegtmeyer N Wessler S Necchi V Rohde M Harrer A Rau TT et al . Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host. Microbe. (2017) 22:552–560.e5. doi: 10.1016/j.chom.2017.09.005
72
Chang WL Yeh YC Sheu BS . The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J Biomed Sci. (2018) 25:68. doi: 10.1186/s12929-018-0466-9
73
Foegeding NJ Caston RR McClain MS Ohi MD Cover TL . An overview of Helicobacter pylori VacA toxin biology. Toxins. (2016) 8:173. doi: 10.3390/toxins8060173
74
Yahiro K Satoh M Nakano M Hisatsune J Isomoto H Sap J et al . Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem. (2012) 287:31104–15. doi: 10.1074/jbc.M112.387498
75
Han L Shu X Wang J . Helicobacter pylori-mediated oxidative stress and gastric diseases: a review. Front Microbiol. (2022) 13:811258. doi: 10.3389/fmicb.2022.811258
76
Liu Y Shi Y Han R Liu C Qin X Li P et al . Signaling pathways of oxidative stress response: the potential therapeutic targets in gastric cancer. Front Immunol. (2023) 14:1139589. doi: 10.3389/fimmu.2023.1139589
77
Arisawa T Tahara T Shibata T Nagasaka M Nakamura M Kamiya Y et al . The relationship between Helicobacter pylori infection and promoter polymorphism of the Nrf2 gene in chronic gastritis. Int J Mol Med. (2007) 19:143–8. doi: 10.3892/ijmm.19.1.143
78
Bacon S Seeneevassen L Fratacci A Rose F Tiffon C Sifré E et al . Nrf2 downregulation contributes to Epithelial-to-Mesenchymal transition in Helicobacter pylori-infected cells. Cancers. (2022) 14:4316. doi: 10.3390/cancers14174316
79
Katsuragi Y Ichimura Y Komatsu M . Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr Opin Toxicol. (2016) 1:54–61. doi: 10.1016/j.cotox.2016.09.005
80
Lupia T Corcione S Shbaklo N Rizzello B De Benedetto I Concialdi E et al . Legionella pneumophila infections during a 7-Year retrospective analysis (2016-2022): epidemiological, clinical features and outcomes in patients with Legionnaires’ disease. Microorganisms. (2023) 11:498. doi: 10.3390/microorganisms11020498
81
Friedman H Yamamoto Y Klein TW . Legionella pneumophila pathogenesis and immunity. Semin Pediatr Infect Dis. (2002) 13:273–9. doi: 10.1053/spid.2002.127206
82
Palusinska-Szysz M Janczarek M . Innate immunity to Legionella and toll-like receptors - review. Folia. Microbiol. (2010) 55:508–14. doi: 10.1007/s12223-010-0084-8
83
Grigoryeva LS Cianciotto NP . Human macrophages utilize a wide range of pathogen recognition receptors to recognize Legionella pneumophila, including Toll-like Receptor 4 engaging Legionella lipopolysaccharide and the Toll-like Receptor 3 nucleic-acid sensor. PloS pathogens. (2021) 17:e1009781. doi: 10.1371/journal.ppat.1009781
84
Mukherjee R Hoffmann M Gonzalez A Bhattacharya A Kuncha SK Rathore R et al . Serine ubiquitination of p62 regulates Nrf2 dependent redox homeostasis. BioRxiv. (2024) 06:17. doi: 10.1080/15548627.2024.2404375
85
Escoll P Song OR Viana F Steiner B Lagache T Olivo-Marin JC et al . Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host. Microbe. (2017) 22:302–316.e7. doi: 10.1016/j.chom.2017.07.020
86
García-Rodríguez FJ Buchrieser C Escoll P . Legionella and mitochondria, an intriguing relationship. Int Rev Cell Mol Biol. (2023) 374:37–81. doi: 10.1016/bs.ircmb.2022.10.001
87
Kageyama S Saito T Obata M Koide RH Ichimura Y Komatsu M . Negative regulation of the Keap1-Nrf2 pathway by a p62/Sqstm1 splicing variant. Mol Cell Biol. (2018) 38:e00642–17. doi: 10.1128/MCB.00642-17
88
Park JY Sohn HY Koh YH Jo C . Curcumin activates Nrf2 through PKCδ-mediated p62 phosphorylation at Ser351. Sci Rep. (2021) 11:8430. doi: 10.1038/s41598-021-87225-8
89
Debnath SK Debnath M Srivastava R . Opportunistic etiological agents causing lung infections: emerging need to transform lung-targeted delivery. Heliyon. (2022) 8:e12620. doi: 10.1016/j.heliyon.2022.e12620
90
Wood SJ Kuzel TM Shafikhani SH . Pseudomonas aeruginosa: infections, animal modeling, and therapeutics. Cells. (2023) 12:199. doi: 10.3390/cells12010199
91
Strempel N Neidig A Nusser M Geffers R Vieillard J LesouHaitier O et al . Human host defense peptide LL-37 stimulates virulence factor production and adaptive resistance in Pseudomonas aeruginosa. PloS One. (2013) 8:e82240. doi: 10.1371/journal.pone.0082240
92
Thi MTT Wibowo D Rehm BHA . Pseudomonas aeruginosa biofilms. Int J Mol Sci. (2020) 21:8671. doi: 10.3390/ijms21228671
93
Qin S Xiao W Zhou C Pu Q Deng X Lan L et al . Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduction Targeting Ther. (2022) 7:199. doi: 10.1038/s41392-022-01056-1
94
Hall S McDermott C Anoopkumar-Dukie S McFarland AJ Forbes A Perkins AV et al . Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins. (2016) 8:236. doi: 10.3390/toxins8080236
95
Chadha J Harjai K Chhibber S . Revisiting the virulence hallmarks of Pseudomonas aeruginosa: a chronicle through the perspective of quorum sensing. Environ Microbiol. (2022) 24:2630–56. doi: 10.1111/1462-2920.15784
96
Reszka KJ O’Malley Y McCormick ML Denning GM Britigan BE . Oxidation of pyocyanin, a cytotoxic product from Pseudomonas aeruginosa, by microperoxidase 11 and hydrogen peroxide. Free Radic Biol Med. (2004) 36:1448–59. doi: 10.1016/j.freeradbiomed.2004.03.011
97
Hojo T Maishi N Towfik AM Akiyama K Ohga N Shindoh M et al . ROS enhance angiogenic properties via regulation of Nrf2 in tumor endothelial cells. Oncotarget. (2017) 8:45484–95. doi: 10.18632/oncotarget.17567
98
Liu K Wang X Sha K Zhang F Xiong F Wang X et al . Nuclear protein HMGN2 attenuates pyocyanin-induced oxidative stress via Nrf2 signaling and inhibits Pseudomonas aeruginosa internalization in A549 cells. Free Radic Biol Med. (2017) 108:404–17. doi: 10.1016/j.freeradbiomed.2017.04.007
99
Zhu H Cao W Huang Y Karrow NA Yang Z . Involvement of pyocyanin in promoting LPS-induced apoptosis, inflammation, and oxidative stress in bovine mammary epithelium cells. Agriculture. (2023) 13:2192. doi: 10.3390/agriculture13122192
100
Xu Y Duan C Kuang Z Hao Y Jeffries JL Lau GW . Pseudomonas aeruginosa pyocyanin activates Nrf2-ARE-mediated transcriptional response via the ROS-EGFR-PI3K-AKT/MEK-ERK MAP kinase signaling in pulmonary epithelial cells. PloS One. (2013) 8:e72528. doi: 10.1371/journal.pone.0072528
101
Yoo J Rodriguez-Perez CE Nie W Edwards RA Sinnett-Smith J Rozengurt E . TNF-α induces upregulation of EGFR expression and signaling in human colonic myofibroblasts. Am J Physiol Gastrointest. Liver. Physiol. (2012) 302:G805–14. doi: 10.1152/ajpgi.00522.2011
102
Ray K Ujvari B Ramana V Donald J . Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer. Growth Factor. Rev. (2018) 41:18–27. doi: 10.1016/j.cytogfr.2018.04.002
103
Sanchez-Guerrero E Chen E Kockx M An SW Chong BH Khachigian LM . IL-1beta signals through the EGF receptor and activates Egr-1 through MMP-ADAM. PloS One. (2012) 7:e39811. doi: 10.1371/journal.pone.0039811
104
Rada B Gardina P Myers TG Leto TL . Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucosal Immunol. (2011) 4:158–71. doi: 10.1038/mi.2010.62
105
Sun Z Huang Z Zhang DD . Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PloS One. (2009) 4:e6588. doi: 10.1371/journal.pone.0006588
106
Liang G Xu E Yang C Zhang C Sheng X Zhou X . Nucleosome-binding protein HMGN2 exhibits antitumor activity in human SaO2 and U2-OS osteosarcoma cell lines. Oncol Rep. (2015) 33:1300–6. doi: 10.3892/or.2014.3689
107
Geng F Liu Z Chen X Chen H Liu Y Yang J et al . High mobility group nucleosomal binding 2 reduces integrin α5/β1-mediated adhesion of Klebsiella pneumoniae on human pulmonary epithelial cells via nuclear factor I. Microbiol Immunol. (2020) 64:825–34. doi: 10.1111/1348-0421.12855
108
Wang L Yan J Niu H Huang R Wu S . Autophagy and ubiquitination in Salmonella infection and the related inflammatory responses. Front Cell Infect Microbiol. (2018) 8:78. doi: 10.3389/fcimb.2018.00078
109
Eng SK Pusparajah P Ab Mutalib NS Ser HL Chan KG Lee LH . Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front Life Sci. (2015) 8:284–93. doi: 10.1080/21553769.2015.1051243
110
Jajere SM . A review of Salmonella enterica with particular focus on the pathogenicity and virulence factors, host specificity and antimicrobial resistance including multidrug resistance. Vet World. (2019) 12:504–21. doi: 10.14202/vetworld.2019.504-521
111
Yang S Deng Q Sun L Zhu Y Dong K Wu S et al . Salmonella effector SpvB inhibits NF-κB activity via KEAP1-mediated downregulation of IKKβ. Front Cell Infect Microbiol. (2021) 11:641412. doi: 10.3389/fcimb.2021.641412
112
Kim JS Liu L Davenport B Kant S Morrison TE Vazquez-Torres A . Oxidative stress activates transcription of Salmonella pathogenicity island-2 genes in macrophages. J Biol Chem. (2022) 298:102130. doi: 10.1016/j.jbc.2022.102130
113
Hos NJ Ganesan R Gutiérrez S Hos D Klimek J Abdullah Z et al . Type I interferon enhances necroptosis of Salmonella Typhimurium–infected macrophages by impairing antioxidative stress responses. J Cell Biol. (2017) 216:4107–21. doi: 10.1083/jcb.201701107
114
Xu W Ding W Jia L Zhu K Luo Q . Esculetin combats multidrug-resistant Salmonella infection and ameliorates intestinal dysfunction via the Nrf2 pathway. Antioxidants. (2024) 13:1170. doi: 10.3390/antiox13101170
115
Komatsu M Kurokawa H Waguri S Taguchi K Kobayashi A Ichimura Y et al . The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. (2010) 12:213–23. doi: 10.1038/ncb2021
116
Yang S Deng Q Sun L Dong K Li Y Wu S et al . Salmonella effector SpvB interferes with intracellular iron homeostasis via regulation of transcription factor Nrf2. FASEB J. (2019) 33:13450–64. doi: 10.1096/fj.201900883RR
117
Deng Q Yang S Sun L Huang K Dong K Zhu Y et al . A detrimental role of NLRP6 in host iron metabolism during Salmonella infection. Redox Biol. (2022) 49:102217. doi: 10.1016/j.redox.2021.102217
118
Maurice NM Sadikot RT . Mitochondrial dysfunction in bacterial infections. Pathogens. (2023) 12:1005. doi: 10.3390/pathogens12081005
119
Harishankar A Viswanathan VK . Attaching and effacing pathogens modulate host mitochondrial structure and function. Int Rev Cell Mol Biol. (2023) 377:65–86. doi: 10.1016/bs.ircmb.2023.03.001
120
Tiku V Tan MW Dikic I . Mitochondrial functions in infection and immunity. Trends Cell Biol. (2020) 30:263–75. doi: 10.1016/j.tcb.2020.01.006
121
Papatheodorou P Domańska G Oxle M Mathieu J Selchow O Kenny B et al . The enteropathogenic Escherichia coli (EPEC) Map effector is imported into the mitochondrial matrix by the TOM/Hsp70 system and alters organelle morphology. Cell Microbiol. (2006) 8:677–89. doi: 10.1111/j.1462-5822.2005.00660.x
122
Singh AP Sharma S Pagarware K Siraji RA Ansari I Mandal A et al . Enteropathogenic E. coli effectors EspF and Map independently disrupt tight junctions through distinct mechanisms involving transcriptional and post-transcriptional regulation. Sci Rep. (2018) 8:3719. doi: 10.1038/s41598-018-22017-1
123
Nandi I Aroeti L Ramachandran RP Kassa EG Zlotkin-Rivkin E Aroeti B . Type III secreted effectors that target mitochondria. Cell Microbiol. (2021) 23:e13352. doi: 10.1111/cmi.13352
124
Newman LE Shadel GS . Mitochondrial DNA release in innate immune signaling. Annu Rev Biochem. (2023) 92:299–332. doi: 10.1146/annurev-biochem-032620-104401
125
Wang B Zhou C Wu Q Lin P Pu Q Qin S et al . cGAS modulates cytokine secretion and bacterial burdens by altering the release of mitochondrial DNA in pseudomonas pulmonary infection. Immunology. (2022) 166:408–23. doi: 10.1111/imm.13482
126
Mukherjee A Ghosh KK Chakrabortty S Gulyás B Padmanabhan P Ball WB . Mitochondrial reactive oxygen species in infection and immunity. Biomolecules. (2024) 14:670. doi: 10.3390/biom14060670
127
Kalvala AK Kumar R Sherkhane B Gundu C Arruri VK Kumar A . Bardoxolone methyl ameliorates hyperglycemia induced mitochondrial dysfunction by activating the keap1-Nrf2-ARE pathway in experimental diabetic neuropathy. Mol Neurobiol. (2020) 57:3616–31. doi: 10.1007/s12035-020-01989-0
128
Zhang W Cheng C Sha Z Chen C Yu C Lv N et al . Rosmarinic acid prevents refractory bacterial pneumonia through regulating Keap1/Nrf2-mediated autophagic pathway and mitochondrial oxidative stress. Free Radic Biol Med. (2021) 168:247–57. doi: 10.1016/j.freeradbiomed.2021.03.038
Summary
Keywords
KEAP1, Nrf2, bacteria, infection, inflammatory mechanisms, oxidative stress
Citation
Romero-Durán MA, Silva-García O, Perez-Aguilar JM and Baizabal-Aguirre VM (2024) Mechanisms of Keap1/Nrf2 modulation in bacterial infections: implications in persistence and clearance. Front. Immunol. 15:1508787. doi: 10.3389/fimmu.2024.1508787
Received
09 October 2024
Accepted
06 December 2024
Published
23 December 2024
Volume
15 - 2024
Edited by
Markus M. Heimesaat, Charité University Medicine Berlin, Germany
Reviewed by
Prathyusha Bagam, National Center for Toxicological Research (FDA), United States
Therese Deramaudt, Université de Versailles Saint-Quentin-en-Yvelines, France
Updates
Copyright
© 2024 Romero-Durán, Silva-García, Perez-Aguilar and Baizabal-Aguirre.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Víctor Manuel Baizabal-Aguirre, victor.baizabal@umich.mx
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.