 Xianqiang Liu1,2†‡
Xianqiang Liu1,2†‡ Xiaowei Xi3†
Xiaowei Xi3† Shengshan Xu1*†
Shengshan Xu1*† Hongyu Chu4†Penghui Hu5Dong Li6Bin Zhang7,8Hejie Liu1
Hongyu Chu4†Penghui Hu5Dong Li6Bin Zhang7,8Hejie Liu1 Tianxiao Jiang9*
Tianxiao Jiang9* Zhuming Lu1*
Zhuming Lu1*- 1Department of Thoracic Surgery, Jiangmen Central Hospital, Jiangmen, Guangdong, China
- 2Graduate School, Medical School of Chinese PLA, Beijing, China
- 3Technical University of Munich (TUM) School of Medicine and Health, Munich, Germany
- 4Department of Gastrointestinal, Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Changchun, China
- 5Scientific Research and Education Department, Jiangmen Central Hospital, Jiangmen, Guangdong, China
- 6Department of Intensive Care Unit and Clinical Experimental Center, Jiangmen Central Hospital, Jiangmen, China
- 7Department of Cardiovascular Disease and Clinical Experimental Center, Jiangmen Central Hospital, Jiangmen, China
- 8Department of Cardiology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
- 9Department of General, Visceral, and Transplant Surgery, Ludwig-Maximilians-University Munich, Munich, Germany
Lung cancer continues to be a major contributor to cancer-related deaths globally. Recent advances in immunotherapy have introduced promising treatments targeting T cell functionality. Central to the efficacy of these therapies is the role of T cells, which are often rendered dysfunctional due to continuous antigenic stimulation in the tumor microenvironment–a condition referred to as T cell exhaustion. This review addresses the critical challenge of T cell exhaustion in non-small cell lung cancer (NSCLC), offering a detailed examination of its molecular underpinnings and the resultant therapeutic ineffectiveness. We synthesize current knowledge on the drivers of T cell exhaustion, evaluate emerging strategies for its reversal, and explore the potential impact of these insights for enhancing the clinical efficacy of immunotherapies. By consolidating reported clinical trials and preclinical studies, this article highlights innovative approaches to modulate immune responses and improve patient outcomes, thus providing a roadmap for future research and therapeutic development in lung cancer immunotherapy.
1 Introduction
Lung cancer continues to pose a significant global health issue, consistently ranking as the second most diagnosed malignancy as of 2020 and the leading contributor to cancer-related mortality. This disease is primarily divided into two subtypes: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) (1). SCLC, representing about 15% of all lung cancer instances, characterized by high aggressiveness and low survival rates, with a five-year survival rate of 7% (2). NSCLC comprises about 85% of cases and has a somewhat higher five-year survival rate of around 25% (3). Despite advancements in treatment, about half of all lung cancer patients are diagnosed with advanced stages of the disease, undergoing treatment regimens predominantly based on platinum-based chemotherapy (4). Even with the integration of chemotherapy and targeted therapies or other treatment modalities, the average five-year survival rate languishes at approximately 6%, underscoring a dire need for more effective therapeutic options (5).
In recent years, the emergence of immunotherapy has diversified the treatment modalities for lung cancer. T cells are vital for the effectiveness of immunotherapies, but their function can be significantly hindered by continuous antigen exposure from chronic infections or tumors, leading to T cell exhaustion. This state is marked by diminished production of effector cytokines such as IL-2 and IFN-γ, reduced proliferative capacity, and compromised cytotoxic and pro-inflammatory functions (6–8). The exacerbated expression of multiple immune checkpoints like PD-1, CTLA-4, LAG-3, TIGIT, BTLA, and TIM-3 further illustrates the impaired functionality of T cells under such conditions (9, 10). Consequently, many patients receiving immunotherapies like PD-1 or PD-L1 inhibitors do not maintain strong anti-tumor effects (11), propelling ongoing research to focus on methods to reverse T cell exhaustion (12). This review seeks to delve into emerging strategies to rejuvenate T cell activity, potentially counteracting the negative impact of continuous antigenic stimulation and fostering a more effective pathway for lung cancer treatment.
2 Inhibitory receptors involved in T cell exhaustion in lung cancer
Elevated expression of multiple inhibitory receptors has been well-documented in lung cancer patients, signifying a state of T cell exhaustion (13). Further complicating the treatment landscape, resistance to PD-1 inhibitors in lung cancer has been correlated with upregulation of other immune checkpoints such as TIM-3 and LAG-3 in mouse models (14).
2.1 PD-1
Programmed death receptor-1 (PD-1), a member of the CD28 transmembrane protein receptor family, plays a critical role in the immune system’s ability to respond to tumor cells. PD-1 suppresses the anti-cancer immune activity of T cells through its interaction with ligands PD-L1 or PD-L2 (15, 16), leading to downregulation of the T cell receptor and subsequent inhibition of T cell activation and cytokine release (17, 18). Notably, interferon-gamma activates the PI3K/AKT and JAK/STAT3 pathways in NSCLC, which enhances PD-L1 expression, a mechanism indicating IFN-γ induced immune suppression (19). Innovative treatments, such as vaccines using plasmacytoid dendritic cells in conjunction with anti-PD-1 therapies, have proved promise in amplifying the tumor-specific CD8+ T cell response in lung cancer (20). Other studies indicate that the miR-3127-5p/p-STAT3 axis (21), the EZH2-HIF-1α axis (22), KLF12 (23), and POU2F1 (24) are also involved in controlling PD-L1 expression, ultimately affecting T cell exhaustion. Therefore, a deeper understanding of the mechanisms that regulate PD-L1 expression could lead to improved strategies for enhancing the effectiveness of PD-L1/PD-1 inhibitors in lung cancer treatment. For a detailed summary of clinical trials employing PD-1 inhibitors in lung cancer, refer to Supplementary Table 1, which catalogues and summarizes these trials.
2.2 CTLA-4
As another inhibitory member of the CD28 family, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), mainly found on T cells, CTLA-4 competitively binds with the ligands CD80/CD86, curtailing CD28 signaling and subsequently reducing IL-2 production and T cell-APC contact time-crucially weakening T cell responses (25). This receptor also triggers the PI3-K pathway, diminishing T cell effectiveness further. The enhancement of PD-1/PD-L1 and CTLA-4 expression via the EGFR pathway suggests a potent avenue for immune suppression in lung cancer, exacerbated by ERK and NF-κB pathways (26). In a Phase Ib/II study (NCT04646330), researchers assessed the combined use of cadonilimab (a PD-1/CTLA-4 bispecific antibody) with anlotinib as a primary treatment for advanced NSCLC, revealing the regimen’s safety and potential efficacy at 10 mg/kg every three weeks. Employing anti-PD-1/PD-L1 and anti-CTLA-4 drugs targets different stages of the cancer immunity cycle; their combined use can produce a synergistic effect, helping to overcome resistance to monotherapy (27, 28). Additionally, Supplementary Table 2 provides an overview of clinical trials investigating CTLA-4 inhibitors in lung cancer.
2.3 TIM-3
T cell immunoglobulin and mucin-domain containing-3 (TIM-3), a member of the immunoglobulin superfamily, was first identified on CD4+ Th1 and CD8+ Tc1 cells as a surface marker that binds to galectin-9. This interaction negatively regulates the function of these cells (29, 30). TIM-3 is also found on a variety of cells within the tumor microenvironment (TME), such as NK cells, dendritic cells (DCs), and tumor cells, which underscores its pivotal role in disease progression in lung cancer (30, 31). High levels of TIM-3 on NK cells and tumor-associated macrophages are related to advanced disease stages and reduced survival rates in lung cancer patients (32, 33). Therapeutically, combining anti-TIM-3 with anti-PD-1 therapies has been effective in reviving the effector functions of CD8 T cells in vitro, indicating that TIM-3 plays a significant role not only as a biomarker of resistance but also in the mechanisms of resistance itself. A Phase I/II trial (NCT02608268) evaluating MGB453 (an anti-TIM-3 drug) combined with PDR001 (an anti-PD-1 therapy) demonstrated good safety and antitumor activity, highlighting the clinical potential of targeting TIM-3 in advanced NSCLC (34). Ongoing clinical trials continue to assess the efficacy of several novel TIM-3 inhibitors, including AZD7789, INCAGN02390, BMS-986258, cobolimab, and sabatolimab (MBG453) (35).
2.4 LAG3
Lymphocyte activation gene-3 (LAG-3) exhibits structural similarities to CD4 in its extracellular domain, yet its intracellular domain markedly differs, indicating unique functional attributes (36). LAG-3 more strongly binds MHC-II compared to CD4, effectively interfering with CD4’s interaction with MHC-II, ultimately suppressing T cell activation (37). Unlike naïve T cells, LAG-3 is expressed on CD4+ and CD8+ T cells after antigen exposure, serving to inhibit their proliferation (38, 39). In the TME, constant antigen exposure heightens LAG-3 expression, contributing to T cell exhaustion and impaired tumor cell elimination. T cells that express both LAG-3 and PD-1 exhibit a greater degree of exhaustion than those expressing PD-1 alone, making LAG-3 a potential target for rescuing dysfunctional T cells in lung cancer. A Phase II study (NCT03365791) evaluating LAG525 combined with PDR001 in advanced solid tumors showed promising anti-tumor effects, achieving a 24-week clinical benefit rate of 27% in SCLC cases. Additionally, Xencor’s bispecific antibody XmAb®22841 targets both CTLA-4 and LAG-3, boosting T cell activation and proliferation (40). A Phase I study (NCT03849469) is currently exploring the optimal dose of XmAb22841, alone or with pembrolizumab, for treating advanced solid tumors, including SCLC (41).
2.5 TIGIT
The T cell immunoreceptor with Ig and ITIM domains (TIGIT) is a recently identified immune checkpoint receptor primarily found on T cells and NK cells, binding to the ligand CD155. TIGIT serves as a marker for T cell exhaustion, effectively identifying exhausted T cells across different stages of differentiation and fostering the exhaustion of CD8 T cells and NK cells under chronic conditions (42–44). Furthermore, research by Yang et al. found that the positivity rate of CD155 in the immunohistochemistry results of squamous lung cancer is significantly higher than that of PD-L1, indicating a crucial role for the TIGIT/CD155 axis in the development and progression of this cancer type (45). Additionally, high TIGIT levels are linked to greater severity in lung adenocarcinoma, and patients with overexpressed CD155 in lung adenocarcinoma and SCLC have shorter progression-free survival and overall survival (46–48). Studies show that antagonistic antibodies against TIGIT, when used with PD-1 inhibitors, effectively curb lung cancer growth in immunocompetent mice (49). Ongoing clinical trials, such as NCT02964013 (MK-7684-001), are assessing the efficacy of the anti-TIGIT antibody vibostolimab in combination with pembrolizumab, demonstrating promising antitumor effects particularly in patients new to anti-PD-1/PD-L1 therapy and those with PD-L1 positivity (50).
2.6 BTLA
The B and T lymphocyte attenuator (BTLA) belongs to the immunoglobulin superfamily and is primarily found on the surfaces of T cells, B cells, NK cells, and DCs (51, 52). As an inhibitory receptor, BTLA limits the activation, proliferation, and production of pro-inflammatory cytokines (IFN-γ, IL-2, and IL-10) in T and B cells through its interaction with the ligand HVEM (53). In lung cancer, BTLA plays a particularly important role, co-participating in T cell exhaustion with other inhibitory receptors like PD-1 and CTLA-4 (54, 55). Within the TME, elevated BTLA levels result in reduced T cell efficacy, affecting their immune response to tumors (56). Studies by Mittal et al. and Thommen et al. demonstrate that BTLA, along with markers like PD-1 and TIM-3, is upregulated in T cells post-tumor implantation in mice and in infiltrating CD8 T cells in advanced lung cancer patients, underscoring its role in T cell dysfunction during tumor progression (57, 58). Clinical studies suggest that high expression of BTLA is linked to unfavorable outcomes in NSCLC and is viewed as a new target for immunotherapy, potentially by combining other therapeutic strategies (such as PD-1 blockade) to restore T cell function (59).
2.7 IDO
Indoleamine 2,3-dioxygenase (IDO) is an intracellular enzyme that catalyzes the conversion of the essential amino acid L-tryptophan into N-formylkynurenine (60). By diminishing local tryptophan levels and augmenting the production of immunoregulatory metabolites, IDO exhibits its immunosuppressive effects (61). These metabolites not only inhibit T lymphocyte proliferation but also promote apoptosis and drive the transformation of naïve T cells into regulatory T cells. Additionally, overexpression of IDO in DCs affects their maturation, reducing antigen presentation and decreasing co-stimulatory molecule expression (60). Increasing evidence links IDO overexpression with adverse outcomes in several cancers (62, 63). IDO is considered a mechanism of resistance that might hinder the effectiveness of checkpoint inhibitor therapies. In a mouse model of lung cancer, silencing IDO1 inhibited tumor growth by reversing T cell exhaustion. Furthermore, blocking the T cell exhaustion induced by IDO1 can enhance the performance of PD-1 inhibitors in lung cancer treatment (64). Therefore, IDO as a potential immunosuppressant is worth further exploration. Table 1 provides a comprehensive overview of the clinical trials involving immune checkpoint modulators, such as TIM-3, LAG-3, TIGIT, BTLA, and IDO, in the context of lung cancer.
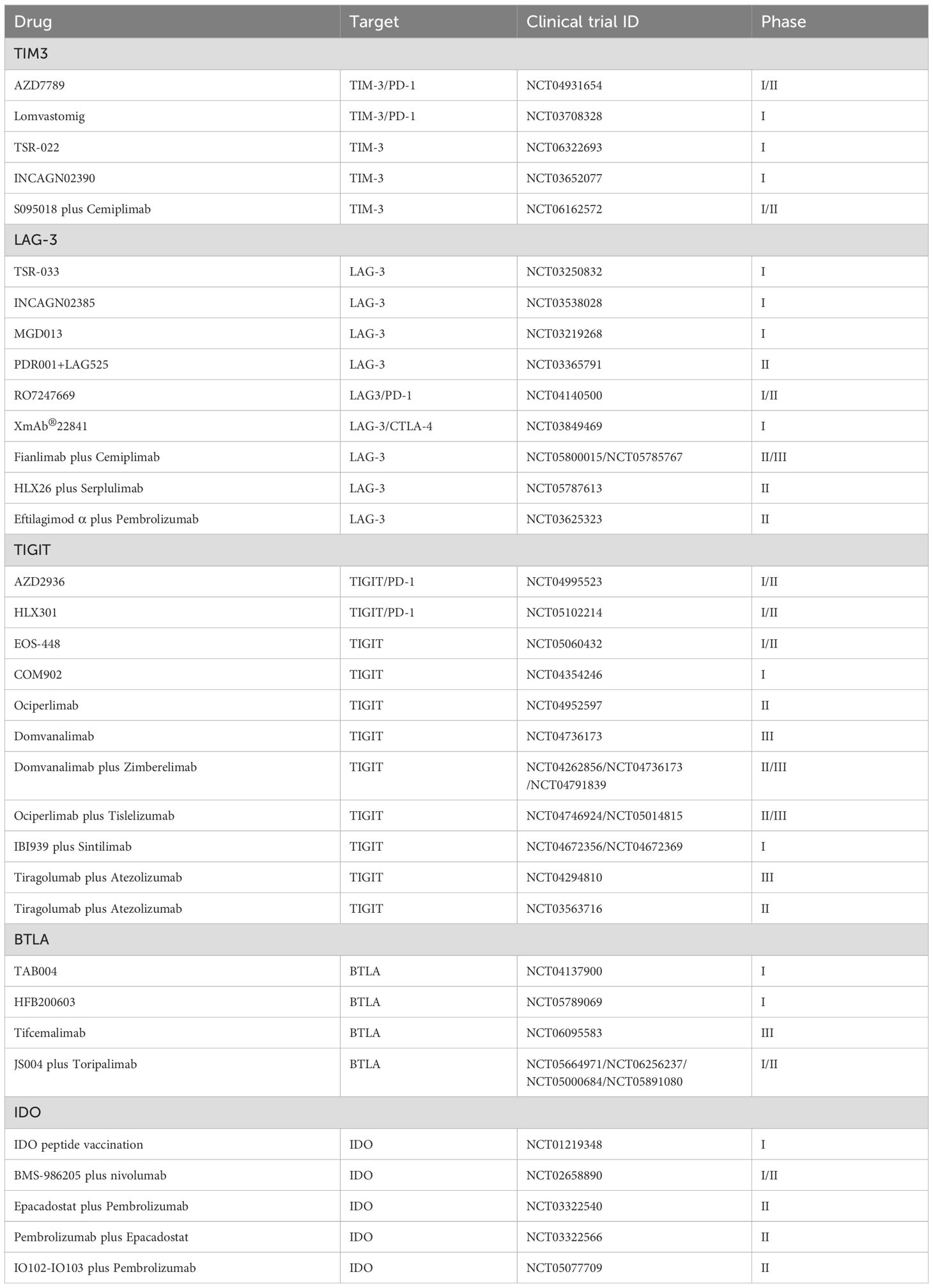
Table 1. Clinical trials of immune checkpoint modulators (TIM3, LAG-3, TIGIT, BTLA, and IDO) in lung cancer.
3 The impact of cytokines on T cell exhaustion in lung cancer
3.1 IL-10
IL-10, a key anti-inflammatory cytokine, is secreted by various immune cells, such as DCs, B cells, CD8 T cells, and non-regulatory CD4 T cells. It plays a critical role in maintaining self-tolerance and protecting against tissue damage in inflamed conditions by inhibiting Th17 cell-mediated inflammation and reducing levels of TNF-α and IL-6 (65, 66). IL-10 exhibits a complex, dual role in antitumor immunity, with its effects shaped by various factors, including cancer type, cell type, TME conditions, and IL-10 concentration. In models of breast cancer, cutaneous squamous cell carcinoma, and lymphoma, IL-10 deficiency correlates with increased tumor burden and impaired immune surveillance, along with elevated rates of CD8+ T cell exhaustion (67, 68). Conversely, in colorectal cancer, bladder cancer, and melanoma models, IL-10 has been shown to promote CD8+ T cell exhaustion, thereby weakening antitumor immunity (69–72). Additionally, in non-small cell lung cancer, IL-10, in conjunction with Tregs and IL-35, is believed to further exacerbate T cell exhaustion, suppressing antitumor responses (70).
The concentration of IL-10 plays a critical role in determining its antitumor effects. High levels of IL-10 can stimulate CD8+ T cell proliferation and enhance cytotoxicity, while low levels tend to exert immunosuppressive effects, diminishing the antitumor immune response (68). In chronic inflammatory settings, IL-10’s anti-inflammatory properties may help maintain CD8+ T cell antitumor functions (73, 74). Furthermore, IL-10 influences CD8+ T cell functionality through metabolic reprogramming, particularly by inducing oxidative phosphorylation (OXPHOS) pathways (75, 76). This upregulation of OXPHOS can partially reinvigorate exhausted T cells, enhancing their proliferative capacity, effector functions, and overall antitumor immunity (76). IL-10’s effects also vary among different cell types. For example, in B cells, IL-10 promotes proliferation, stimulates immunoglobulin secretion and isotype switching, and enhances tumor-killing capabilities (77, 78). Future research should focus on the role of IL-10 in relation to exhausted CD8+ T cells in non-small cell lung cancer, as well as the key factors influencing this dynamic, to improve the efficacy of lung cancer immunotherapies.
3.2 IFNα/β
IFNα/β are pro-inflammatory cytokines with various anti-tumor activities, such as direct tumor cell eradication and the stimulation of immune cells like DCs and CD8 T cells (79–81). Studies suggest that IFNα/β can trigger the expression of IL-10, PD-L1, and other inhibitory regulators, potentially leading to T cell exhaustion through the influence on the transcription factor Tcf1 (82, 83). To date, IFNα/β has been approved for treating multiple malignancies, such as renal cell carcinoma and melanoma. Overall, the efficacy of combining PD-1/PD-L1 inhibitors with IFNα/β therapy is influenced by various factors, including the type and dosage of IFNα/β, timing and duration of treatment, patient immune status, and cancer type. Thus, understanding the mechanisms of action of IFNα/β and its interaction with PD-1/PD-L1 combined therapy is crucial for optimizing cancer treatment strategies.
3.3 TGF-β
TGF-β, a versatile cytokine, plays a critical role in regulating cell growth and differentiation. It also fosters T cell exhaustion; notably, TGF-β’s stimulation of exhausted T cell precursors leads to the inhibition of mTOR signaling, which in turn produces inhibitory cytokines that dampen the immune response (84). Furthermore, TGF-β reduces or suppresses immune cell activation through triggering SMAD transcription regulators downstream. Tumor cells release TGF-β, which directly increases PD-1 transcription in these cells, contributing to T cell exhaustion. Consequently, blocking TGF-β signaling could directly bolster anti-tumor immunity (85). The significant link between TGF-β signaling and impaired immune responses in lung cancer underscores the need to focus on this pathway when developing new therapeutic strategies. Extensive research is required to unravel its full mechanisms and evaluate the effectiveness of TGF-β inhibitors in treating lung cancer.
3.4 IL-2
IL-2, commonly referred to as T cell growth factor, is integral to T cell proliferation. At initial tumor growth stages, IL-2-driven BLIMP1 expression is vital for the development and differentiation of CD8 T cells. Recent findings suggest that IL-2 signaling can alter the differentiation trajectory of exhausted CD8 T cell precursors, potentially reversing T cell exhaustion (86). However, studies indicate that IL-2 can enhance 5-HTP production through the STAT5-TPH1 pathway, which activates the aryl hydrocarbon receptor and may lead to CD8 T cell exhaustion (87). Thus, IL-2 serves a dual function: it both contributes to and could potentially reverse the exhausted T cell phenotype. Currently, combining IL-2 with PD-1 inhibitors has demonstrated promising synergistic effects in reactivating exhausted CD8 T cells (88, 89). Ongoing research is crucial to fully ascertain the therapeutic impact of IL-2 in lung cancer treatment.
4 Non-cytokine mediators of T cell exhaustion
Beyond cytokines, non-cytokine factors such as metabolites and metabolic fuel sources like adenosine, cholesterol, and fatty acids, also contribute to T cell exhaustion (90, 91).
4.1 Prostaglandin E2
The arachidonic acid pathway, involved in immune suppression across various cancer types, includes the enzyme microsomal prostaglandin E2 synthase-1, downstream of cyclooxygenase 2, which limits the body’s anti-tumor immunity. Prostaglandin E2 (PGE2), known for its immunosuppressive effects, can suppress the activation and proliferation of immune cells such as NK cells and B cells via the PGE2-EP signaling pathway (92–94). Recent research in lung cancer has shown that PGE2 promotes immune tolerance by upregulating PD-1 on infiltrating CD8 T cells, thereby contributing to their exhaustion (95). Consequently, targeting the PGE2-EP signaling pathway presents a promising approach to counteract T cell exhaustion in lung cancer therapy.
4.2 Adenosine
Adenosine (Ado) is produced from extracellular ATP through the actions of ectonucleotidases CD39 and CD73 on cell surfaces (96). Persistent high levels of Ado can contribute to an immunosuppressive microenvironment, although the precise mechanisms remain unclear (97). Ado impairs the activation of CD8 T cells in the TME and disrupts their tumor recognition capabilities, mainly through the A2AR/PKA/mTORC1 signaling pathway (98). Additionally, Ado further dampens tumor immunity by decreasing the infiltration of immune cells into tumors (99). Due to poor vascular development in tumors, the TME often exists in a hypoxic state, facilitate Ado production and activate immunosuppressive A2A and A2B receptors. Shifts in tumor metabolism towards glycolysis and the resulting increase in lactate also lower pH levels, promoting M2 polarization and inhibiting T cell activation (100, 101). Additionally, studies by Maj and colleagues have shown that during checkpoint therapy, the apoptosis of cancer cells and Tregs leads to an increase in ATP release, which is subsequently transformed into adenosine by the enzymes CD39 and CD73. The increased Ado in the TME counteracts the effects of checkpoint therapy, further suppressing anti-tumor immune responses (102). This suggests that Ado may be a potential target for advanced immunotherapies. Currently, a phase I clinical trial involving CPI-444 (an A2AR antagonist) combined with the PD-1 inhibitor nivolumab for treating non-small cell lung cancer is underway (NCT0265582).
4.3 Cholesterol
Cholesterol, a vital component of cell membranes, affects membrane fluidity as well as gene expression and metabolism, and impacting anti-tumor immunity (103). Its role in T cell activation is debated; some studies indicate it may suppress TCR signaling either by binding to the transmembrane region of TCRβ or by interfering with TCR oligomerization (104). In the TME, cholesterol is known to promote the expression of immunosuppressive receptors, leading to CD8 T cell exhaustion. Additionally, research has found that cholesterol levels in tumor-infiltrating CD8 T cells correlate with exhaustion status and the presence of immune checkpoints such as PD-1, TIM-3, and LAG-3 (104, 105). Statins, the primary compounds inhibiting cholesterol synthesis, are highly safe, with atorvastatin reported to downregulate the expression of inhibitory receptors like LAG-3, PD-1, TIM-3, and CTLA-4 in T cells, indirectly indicating the relationship between cholesterol and immune checkpoint expression (106). Moreover, statins have been found to reduce cancer-related mortality by 15% (107). Therefore, cholesterol-lowering medications may be combined with immunotherapy for cancer patients, offering new possibilities for treatment.
4.4 Clinical implications of T cell exhaustion in lung cancer immunotherapy
Understanding T cell exhaustion is crucial for monitoring and treating lung cancer. Figure 1 provides an overview of the pathways of T cell exhaustion and therapeutic interventions in lung cancer currently under study. Exhausted T cells are characterized by the expression of inhibitory receptors such as PD-1 and TIM-3, with their co-expression often signaling severe exhaustion and correlating with a poorer prognosis in lung cancer. In the TME, the presence of these markers on CD8 T cells can predict the response to immune checkpoint inhibitor therapy. Utilizing transcriptomics and multiplex staining techniques to identify patients who may benefit from ICI therapy is increasingly valuable in clinical research. Approaches to reverse T cell exhaustion and enhance anti-tumor immunity are promising, yet obstacles like immunosuppressive cytokines, certain immune cells, and elevated inhibitory receptors in the TME all play roles in promoting T cell exhaustion, which in turn dampens anti-tumor responses. Studies show that blocking inhibitory receptors and cytokines can effectively reverse T cell exhaustion in cancer patients, particularly in those with advanced disease, enhancing anti-tumor immunity. Clinical application of anti-PD-1 antibodies has been successful, and blocking PD-1 or PD-L1 improves the functionality of exhausted CD8 T cells (9, 108, 109), indicating that T cell exhaustion is not irreversible. Preclinical studies confirm that anti-PD-1 immune checkpoint inhibitors can effectively ameliorate T cell exhaustion and are more effective when combined with other immune checkpoint inhibitors (110); This has led to FDA approval for combination therapies like nivolumab plus ipilimumab (111). While single-agent CTLA-4 or PD-1 blockade has significantly extended survival in some cancer patients, most do not respond. Therefore, strategies against T cell exhaustion are becoming a focus for enhancing the effectiveness of anti-tumor therapies, especially in enhancing the efficacy and durability of treatments like CAR-T cell therapies.
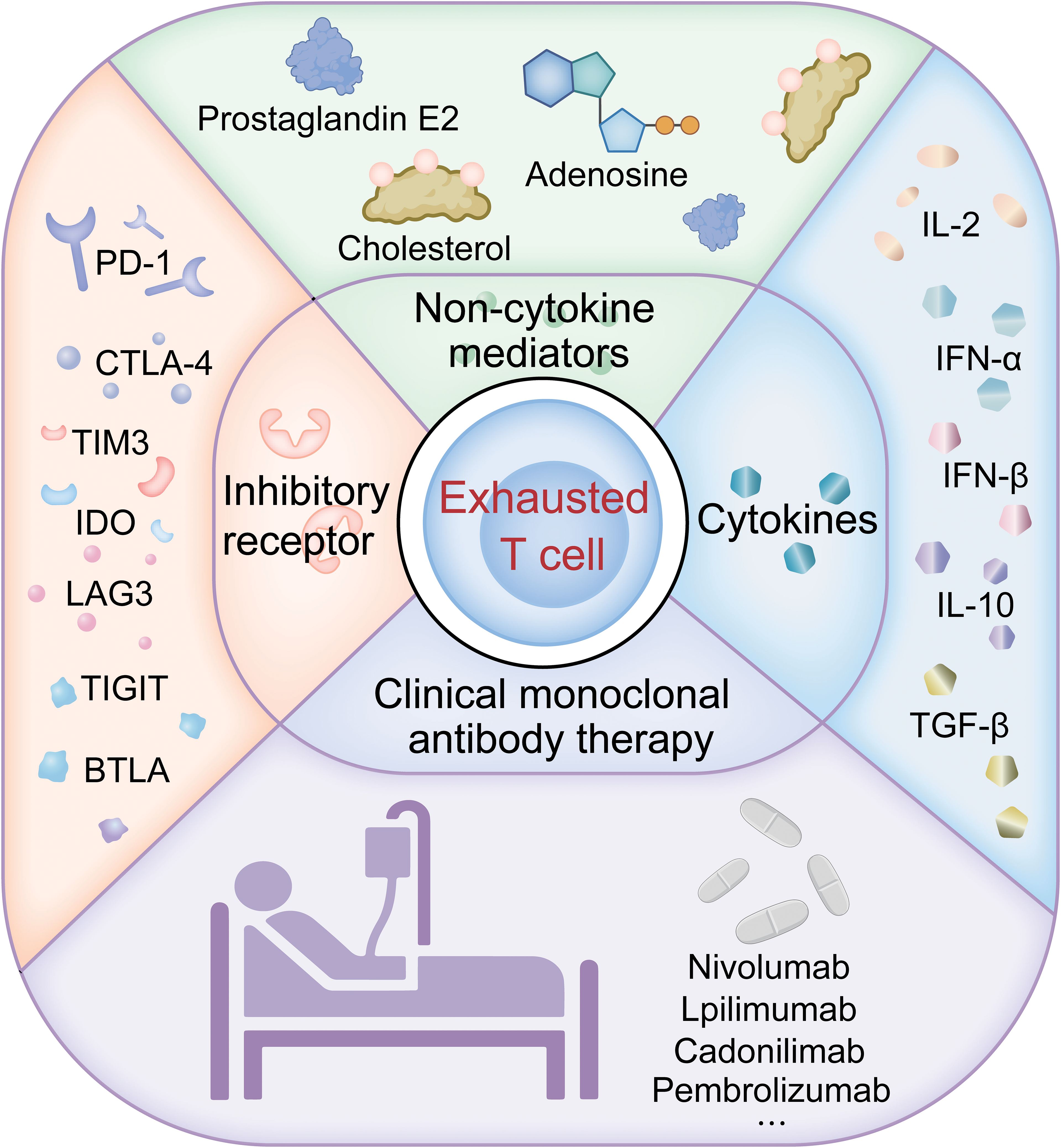
Figure 1. Pathways of T cell exhaustion and therapeutic interventions in lung cancer.
5 Discussion and conclusion
Recent advances in immunotherapy have expanded treatment options for lung cancer, with T cell exhaustion emerging as a key area of interest in the disease’s pathogenesis, progression, and therapy. While the precise mechanisms behind T cell exhaustion continue to be studied, further research and clinical trials are essential to establish clear findings. Additionally, the exploration of T cell dysfunction presents a new avenue in immunotherapy. A thorough understanding of T cell exhaustion and its underlying mechanisms is critical to comprehending the immune dynamics in lung cancer. This knowledge is fundamental to crafting innovative treatment approaches for this challenging disease.
Author contributions
XL: Writing – original draft, Writing – review & editing. XX: Writing – review & editing. SX: Writing – review & editing, Writing – original draft. HC: Writing – original draft, Writing – review & editing. PH: Writing – review & editing. DL: Writing – review & editing. BZ: Writing – review & editing. HL: Writing – review & editing. TJ: Writing – review & editing. ZL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Technology Project of Jiangmen (Nos. 2220002000183, 2320002000933), the Medical Science Foundation of Jiangmen Central Hospital (J202401), and the Postdoctoral Project of the International Training Program for Guangdong Outstanding Young Research Talents and the General Project of China Postdoctoral Science Foundation (No. 2024M761177).
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1507501/full#supplementary-material
References
1. Khanmohammadi A, Aghaie A, Vahedi E, Qazvini A, Ghanei M, Afkhami A, et al. Electrochemical biosensors for the detection of lung cancer biomarkers: A review. Talanta. (2020) 206:120251. doi: 10.1016/j.talanta.2019.120251
2. Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. (2017) 17:725–37. doi: 10.1038/nrc.2017.87
3. Arbour KC, Riely GJ. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: A review. JAMA. (2019) 322:764–74. doi: 10.1001/jama.2019.11058
4. Qu Y, Emoto K, Eguchi T, Aly RG, Zheng H, Chaft JE, et al. Pathologic assessment after neoadjuvant chemotherapy for Nsclc: importance and implications of distinguishing adenocarcinoma from squamous cell carcinoma. J Thorac Oncol. (2019) 14:482–93. doi: 10.1016/j.jtho.2018.11.017
5. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA: A Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
6. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
7. Gehring AJ, Ho ZZ, Tan AT, Aung MO, Lee KH, Tan KC, et al. Profile of tumor antigen-specific cd8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology. (2009) 137:682–90. doi: 10.1053/j.gastro.2009.04.045
8. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen–specific Cd8 T cells infiltrating the tumor express high levels of Pd-1 and are functionally impaired. Blood. (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
10. Franco F, Jaccard A, Romero P, Yu Y-R, Ho P-C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab. (2020) 2:1001–12. doi: 10.1038/s42255-020-00280-9
11. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer immunotherapy targeting Pd-1 and Gitr can rescue Cd8(+) T cell dysfunction and maintain memory phenotype. Sci Immunol. (2018) 3(29). doi: 10.1126/sciimmunol.aat7061
12. Zhu Y, Tan H, Wang J, Zhuang H, Zhao H, Lu X. Molecular insight into T cell exhaustion in hepatocellular carcinoma. Pharmacol Res. (2024) 203:107161. doi: 10.1016/j.phrs.2024.107161
13. Philip M, Schietinger A. Cd8+ T cell differentiation and dysfunction in cancer. Nat Rev Immunol. (2022) 22:209–23. doi: 10.1038/s41577-021-00574-3
14. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic Pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
15. Dolina JS, Van Braeckel-Budimir N, Thomas GD, Salek-Ardakani S. Cd8(+) T cell exhaustion in cancer. Front Immunol. (2021) 12:715234. doi: 10.3389/fimmu.2021.715234
16. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. (2021) 184:5309–37. doi: 10.1016/j.cell.2021.09.020
17. Constantinidou A, Alifieris C, Trafalis DT. Targeting programmed cell death -1 (Pd-1) and ligand (Pd-L1): A new era in cancer active immunotherapy. Pharmacol Ther. (2019) 194:84–106. doi: 10.1016/j.pharmthera.2018.09.008
18. Dermani FK, Samadi P, Rahmani G, Kohlan AK, Najafi R. Pd-1/Pd-L1 immune checkpoint: potential target for cancer therapy. J Cell Physiol. (2019) 234:1313–25. doi: 10.1002/jcp.27172
19. Zhang J, Liu S, Chen X, Xu X, Xu F. Non-immune cell components in tumor microenvironment influencing lung cancer immunotherapy. Biomedicine Pharmacotherapy. (2023) 166:115336. doi: 10.1016/j.biopha.2023.115336
20. Hannani D, Leplus E, Laurin D, Caulier B, Aspord C, Madelon N, et al. A new plasmacytoid dendritic cell-based vaccine in combination with anti-Pd-1 expands the tumor-specific Cd8+ T cells of lung cancer patients. Int J Mol Sci. (2023) 24(3). doi: 10.3390/ijms24031897
21. Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The Mir-3127-5p/P-Stat3 axis up-regulates Pd-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. (2018) 22:3847–56. doi: 10.1111/jcmm.13657
22. Zhao Y, Wang XX, Wu W, Long H, Huang J, Wang Z, et al. Ezh2 regulates Pd-L1 expression via Hif-1α in non-small cell lung cancer cells. Biochem Biophys Res Commun. (2019) 517:201–9. doi: 10.1016/j.bbrc.2019.07.039
23. Pan X, Zhang W, Wang L, Guo H, Zheng M, Wu H, et al. Klf12 transcriptionally regulates Pd-L1 expression in non-small cell lung cancer. Mol Oncol. (2023) 17:2659–74. doi: 10.1002/1878-0261.13512
24. Li F, Wang T, Huang Y. Pou2f1 induces the immune escape in lung cancer by up-regulating Pd-L1. Am J Trans Res. (2021) 13:672–83.
25. Rudd CE, Taylor A, Schneider H. Cd28 and Ctla-4 coreceptor expression and signal transduction. Immunol Rev. (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
26. Liang H, Liu X, Wang M. Immunotherapy combined with epidermal growth factor receptor-tyrosine kinase inhibitors in non-small-cell lung cancer treatment. OncoTargets Ther. (2018) 11:6189–96. doi: 10.2147/ott.S178497
27. Feng Y, Roy A, Masson E, Chen TT, Humphrey R, Weber JS. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin Cancer Res. (2013) 19:3977–86. doi: 10.1158/1078-0432.Ccr-12-3243
28. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-Ctla-4 therapy broadens the melanoma-reactive Cd8+ T cell response. Sci Trans Med. (2014) 6:254ra128. doi: 10.1126/scitranslmed.3008918
29. Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The Tim gene family regulates autoimmune and allergic diseases. Trends Mol Med. (2005) 11:362–9. doi: 10.1016/j.molmed.2005.06.008
30. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
31. Jiang X, Yu J, Shi Q, Xiao Y, Wang W, Chen G, et al. Tim-3 promotes intestinal homeostasis in Dss colitis by inhibiting M1 polarization of macrophages. Clin Immunol (Orlando Fla). (2015) 160:328–35. doi: 10.1016/j.clim.2015.07.008
32. Xu LY, Chen DD, He JY, Lu CC, Liu XG, Le HB, et al. Tim-3 expression by peripheral natural killer cells and natural killer T cells increases in patients with lung cancer–reduction after surgical resection. Asian Pacific J Cancer prevention: APJCP. (2014) 15:9945–8. doi: 10.7314/apjcp.2014.15.22.9945
33. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of Pd-1, Lag-3, and Tim-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. (2019) 25:4663–73. doi: 10.1158/1078-0432.Ccr-18-4142
34. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an anti-Tim-3 antibody, alone and in combination with spartalizumab, an anti-Pd-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.Ccr-20-4746
35. Roy D, Gilmour C, Patnaik S, Wang LL. Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front Immunol. (2023) 14:1264327. doi: 10.3389/fimmu.2023.1264327
36. Crise B, Rose JK. Identification of palmitoylation sites on Cd4, the human immunodeficiency virus receptor. J Biol Chem. (1992) 267:13593–7. doi: 10.1016/S0021-9258(18)42253-3
37. Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DA. Phenotypic analysis of the murine Cd4-related glycoprotein, Cd223 (Lag-3). Eur J Immunol. (2002) 32:2255–63. doi: 10.1002/1521-4141(200208)32:8<2255::Aid-immu2255>3.0.Co;2-a
38. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. Lag-3, a novel lymphocyte activation gene closely related to Cd4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
39. Workman CJ, Cauley LS, Kim I-J, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (Cd223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. (2004) 172:5450–5. doi: 10.4049/jimmunol.172.9.5450
40. Uboha NV, Milhem MM, Kovacs C, Amin A, Magley A, Purkayastha DD, et al. Phase ii study of spartalizumab (Pdr001) and lag525 in advanced solid tumors and hematologic Malignancies. Am Soc Clin Oncol. (2019) 37(15_suppl):2553-. doi: 10.1200/JCO.2019.37.15_suppl.2553
41. Jacob S, Daud A. Phase ib/ii study of xmab23104 (Pd1 X icos) and xmab22841 (Ctla-4 X lag3) combination in metastatic melanoma refractory to prior immune checkpoint inhibitor therapy with and without cns disease. J Clin Oncol. (2023) 41:TPS9595–TPS. doi: 10.1200/JCO.2023.41.16_suppl.TPS9595
42. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of tigit with pvr and pvrl2 inhibits human Nk cell cytotoxicity. Proc Natl Acad Sci U S A. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
43. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and itim domain (Tigit) receptor/poliovirus receptor (Pvr) ligand engagement suppresses interferon-Γ Production of natural killer cells via B-arrestin 2-mediated negative signaling*. J Biol Chem. (2014) 289:17647–57. doi: 10.1074/jbc.M114.572420
44. Johnston Robert J, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor tigit regulates antitumor and antiviral Cd8+ T cell effector function. Cancer Cell. (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
45. Yang Z, Peng Y, Xu J, Chen P, Zhao Z, Cai Q, et al. Pvr/Tigit and Pd-L1/Pd-1 expression predicts survival and enlightens combined immunotherapy in lung squamous cell carcinoma. Trans Oncol. (2022) 24:101501. doi: 10.1016/j.tranon.2022.101501
46. Xiao K, Xiao K, Li K, Xue P, Zhu S. Prognostic role of Tigit expression in patients with solid tumors: A meta-analysis. J Immunol Res. (2021) 2021:5440572. doi: 10.1155/2021/5440572
47. Xu Y, Cui G, Jiang Z, Li N, Zhang X. Survival analysis with regard to Pd-L1 and Cd155 expression in human small cell lung cancer and a comparison with associated receptors. Oncol Lett. (2019) 17:2960–8. doi: 10.3892/ol.2019.9910
48. Sun Y, Luo J, Chen Y, Cui J, Lei Y, Cui Y, et al. Combined evaluation of the expression status of Cd155 and Tigit plays an important role in the prognosis of luad (Lung adenocarcinoma). Int Immunopharmacol. (2020) 80:106198. doi: 10.1016/j.intimp.2020.106198
49. Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome profiling identifies Tigit as a marker of T-cell exhaustion in liver cancer. Hepatology. (2021) 73:1399–418. doi: 10.1002/hep.31466
50. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-tigit antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
51. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. Btla is a lymphocyte inhibitory receptor with similarities to Ctla-4 and Pd-1. Nat Immunol. (2003) 4:670–9. doi: 10.1038/ni944
52. Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol. (2022) 86:137–50. doi: 10.1016/j.semcancer.2022.03.014
53. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. (2005) 6:90–8. doi: 10.1038/ni1144
54. Shui JW, Steinberg MW, Kronenberg M. Regulation of inflammation, autoimmunity, and infection immunity by Hvem-Btla signaling. J leukocyte Biol. (2011) 89:517–23. doi: 10.1189/jlb.0910528
55. Wang XF, Chen YJ, Wang Q, Ge Y, Dai Q, Yang KF, et al. Distinct expression and inhibitory function of B and T lymphocyte attenuator on human T cells. Tissue Antigens. (2007) 69:145–53. doi: 10.1111/j.1399-0039.2006.00710.x
56. Ren S, Tian Q, Amar N, Yu H, Rivard CJ, Caldwell C, et al. The immune checkpoint, hvem may contribute to immune escape in non-small cell lung cancer lacking Pd-L1 expression. Lung Cancer (Amsterdam Netherlands). (2018) 125:115–20. doi: 10.1016/j.lungcan.2018.09.004
57. Mittal R, Chen CW, Lyons JD, Margoles LM, Liang Z, Coopersmith CM, et al. Murine lung cancer induces generalized T-cell exhaustion. J Surg Res. (2015) 195:541–9. doi: 10.1016/j.jss.2015.02.004
58. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. (2015) 3:1344–55. doi: 10.1158/2326-6066.Cir-15-0097
59. Li X, Xu Z, Cui G, Yu L, Zhang X. Btla expression in stage I-iii non-small-cell lung cancer and its correlation with Pd-1/Pd-L1 and clinical outcomes. OncoTargets Ther. (2020) 13:215–24. doi: 10.2147/ott.S232234
60. Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2, 3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunology Immunotherapy. (2018) 67:1305–15. doi: 10.1007/s00262-018-2190-4
61. Lim J-Y, Lee S-E, Park G, Choi EY, Min C-K. Inhibition of indoleamine 2, 3-dioxygenase by stereoisomers of 1-methyl tryptophan in an experimental graft-versus-tumor model. Exp Hematol. (2014) 42:862–6.e3. doi: 10.1016/j.exphem.2014.06.006
62. Asghar K, Farooq A, Zulfiqar B, Rashid MU. Indoleamine 2,3-dioxygenase: as a potential prognostic marker and immunotherapeutic target for hepatocellular carcinoma. World J Gastroenterol. (2017) 23:2286–93. doi: 10.3748/wjg.v23.i13.2286
63. Schalper KA, Carvajal-Hausdorf D, McLaughlin J, Altan M, Velcheti V, Gaule P, et al. Differential expression and significance of Pd-L1, Ido-1, and B7-H4 in human lung cancer. Clin Cancer Res. (2017) 23:370–8. doi: 10.1158/1078-0432.Ccr-16-0150
64. Shang K, Wang Z, Hu Y, Huang Y, Yuan K, Yu Y. Gene silencing of indoleamine 2,3-dioxygenase 1 inhibits lung cancer growth by suppressing T-cell exhaustion. Oncol Lett. (2020) 19:3827–38. doi: 10.3892/ol.2020.11477
65. Yang Y, Liu F, Liu W, Ma M, Gao J, Lu Y, et al. Analysis of single-cell rnaseq identifies transitional states of T cells associated with hepatocellular carcinoma. Clin Trans Med. (2020) 10:e133. doi: 10.1002/ctm2.133
66. Wilson EB, Brooks DG. The role of Il-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol. (2011) 350:39–65. doi: 10.1007/82_2010_96
67. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. Il-10 elicits Ifnγ-dependent tumor immune surveillance. Cancer Cell. (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
68. Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B-cell lymphoma caused by Il-10r deficiency. Blood. (2013) 122:3713–22. doi: 10.1182/blood-2013-06-508267
69. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage Il-10 blocks Cd8+ T cell-dependent responses to chemotherapy by suppressing Il-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
70. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of Il-10(+) and Il-35(+) T(Reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
71. Xu Y, Zeng H, Jin K, Liu Z, Zhu Y, Xu L, et al. Immunosuppressive tumor-associated macrophages expressing interlukin-10 conferred poor prognosis and therapeutic vulnerability in patients with muscle-invasive bladder cancer. J immunotherapy Cancer. (2022) 10(3). doi: 10.1136/jitc-2021-003416
72. Shiri AM, Zhang T, Bedke T, Zazara DE, Zhao L, Lücke J, et al. Il-10 dampens antitumor immunity and promotes liver metastasis via Pd-L1 induction. J Hepatol. (2024) 80:634–44. doi: 10.1016/j.jhep.2023.12.015
73. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
74. Huang Y, Zou K, Jiang H, Li Z. The complex role of Il-10 in Malignant ascites: A review. Cancer immunology immunotherapy: CII. (2024) 73:32. doi: 10.1007/s00262-023-03616-y
75. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor Pd-1 are an early driver of Cd8+ T cell exhaustion. Immunity. (2016) 45:358–73.
76. Guo Y, Xie Y-Q, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted Cd8+ T cells by Il-10 enhances anti-tumor immunity. Nat Immunol. (2021) 22:746–56. doi: 10.1038/s41590-021-00940-2
77. Commins S, Steinke JW, Borish L. The extended Il-10 superfamily: Il-10, Il-19, Il-20, Il-22, Il-24, Il-26, Il-28, and Il-29. J Allergy Clin Immunol. (2008) 121:1108–11. doi: 10.1016/j.jaci.2008.02.026
78. Banchereau J, Brière F, Liu YJ, Rousset F. Molecular control of B lymphocyte growth and differentiation. Stem Cells (Dayton Ohio). (1994) 12:278–88. doi: 10.1002/stem.5530120304
79. Shi W, Yao X, Fu Y, Wang Y. Interferon-A and its effects on cancer cell apoptosis. Oncol Lett. (2022) 24:235. doi: 10.3892/ol.2022.13355
80. Fujimura T, Hidaka T, Kambayashi Y, Furudate S, Kakizaki A, Tono H, et al. Phase I study of nivolumab combined with Ifn-B for patients with advanced melanoma. Oncotarget. (2017) 8:71181–7. doi: 10.18632/oncotarget.17090
81. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
82. Marshall HD, Urban SL, Welsh RM. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J Virol. (2011) 85:5929–39. doi: 10.1128/jvi.02516-10
83. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The tcf1-bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. (2016) 1(6). doi: 10.1126/sciimmunol.aai8593
84. Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol. (2020) 21:1022–33. doi: 10.1038/s41590-020-0725-2
85. Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. Tgfβ1-mediated Smad3 enhances Pd-1 expression on antigen-specific T cells in cancer. Cancer Discovery. (2016) 6:1366–81. doi: 10.1158/2159-8290.CD-15-1347
86. Hashimoto M, Araki K, Cardenas MA, Li P, Jadhav RR, Kissick HT, et al. Pd-1 combination therapy with Il-2 modifies Cd8(+) T cell exhaustion program. Nature. (2022) 610:173–81. doi: 10.1038/s41586-022-05257-0
87. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. Il-2 regulates tumor-reactive Cd8+ T cell exhaustion by activating the Aryl hydrocarbon receptor. Nat Immunol. (2021) 22:358–69. doi: 10.1038/s41590-020-00850-9
88. Zhu W, Li Y, Han M, Jiang J. Regulatory mechanisms and reversal of Cd8+ T cell exhaustion: A literature review. Biology. (2023) 12:541. doi: 10.3390/biology12040541
89. Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. (2021) 20:1–27. doi: 10.1186/s12943-021-01464-x
90. Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol (Baltimore Md: 1950). (2014) 192:136–44. doi: 10.4049/jimmunol.1301158
91. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. (2010) 236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x
92. Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. (2002) 23:144–50. doi: 10.1016/s1471-4906(01)02154-8
93. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. (2015) 162:1257–70. doi: 10.1016/j.cell.2015.08.015
94. Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. (2006) 116:1391–9. doi: 10.1172/jci27540
95. Wang J, Zhang L, Kang D, Yang D, Tang Y. Activation of Pge2/Ep2 and Pge2/Ep4 signaling pathways positively regulate the level of Pd-1 in infiltrating Cd8(+) T cells in patients with lung cancer. Oncol Lett. (2018) 15:552–8. doi: 10.3892/ol.2017.7279
96. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express Cd39 and Cd73, which suppress T cells through adenosine production. J Immunol (Baltimore Md: 1950). (2011) 187:676–83. doi: 10.4049/jimmunol.1003884
97. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A(2a) adenosine receptor gene deletion or synthetic a(2a) antagonist liberate tumor-reactive Cd8(+) T cells from tumor-induced immunosuppression. J Immunol (Baltimore Md: 1950). (2018) 201:782–91. doi: 10.4049/jimmunol.1700850
98. Mastelic-Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating Cd8(+) T cells. J immunotherapy Cancer. (2019) 7:257. doi: 10.1186/s40425-019-0719-5
99. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
100. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. (2017) 17:577–93. doi: 10.1038/nrc.2017.77
101. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
102. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and Pd-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
103. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. (2008) 9:125–38. doi: 10.1038/nrm2336
104. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces Cd8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–56.e5. doi: 10.1016/j.cmet.2019.04.002
105. Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, et al. Cholesterol negatively regulates Il-9-producing Cd8(+) T cell differentiation and antitumor activity. J Exp Med. (2018) 215:1555–69. doi: 10.1084/jem.20171576
106. Law M, Rudnicka AR. Statin safety: A systematic review. Am J Cardiol. (2006) 97:52c–60c. doi: 10.1016/j.amjcard.2005.12.010
107. Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. New Engl J Med. (2012) 367:1792–802. doi: 10.1056/NEJMoa1201735
108. Takamori S, Toyokawa G, Takada K, Shoji F, Okamoto T, Maehara Y. Combination therapy of radiotherapy and anti-Pd-1/Pd-L1 treatment in non-small-cell lung cancer: A mini-review. Clin Lung Cancer. (2018) 19:12–6. doi: 10.1016/j.cllc.2017.06.015
109. McLane LM, Abdel-Hakeem MS, Wherry EJ. Cd8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
110. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against Immune Checkpoint Molecules Restore functions of Tumor-Infiltrating T Cells in Hepatocellular carcinomas. Gastroenterology. (2017) 153:1107–19.e10. doi: 10.1053/j.gastro.2017.06.017
Keywords: T cell exhaustion, immunotherapy, non-small cell lung cancer, cytokines, immune checkpoint inhibitors
Citation: Liu X, Xi X, Xu S, Chu H, Hu P, Li D, Zhang B, Liu H, Jiang T and Lu Z (2024) Targeting T cell exhaustion: emerging strategies in non-small cell lung cancer. Front. Immunol. 15:1507501. doi: 10.3389/fimmu.2024.1507501
Received: 07 October 2024; Accepted: 22 November 2024;
Published: 12 December 2024.
Edited by:
Xiangyu Chen, Chongqing Medical University, ChinaReviewed by:
Sheefa Mirza, University of the Witwatersrand, South AfricaCopyright © 2024 Liu, Xi, Xu, Chu, Hu, Li, Zhang, Liu, Jiang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengshan Xu, eHVzaGVuZ3NoYW45N0AxNjMuY29t; Tianxiao Jiang, dGlhbnhpYW8uamlhbmdAbWVkLnVuaS1tdWVuY2hlbi5kZQ==; Zhuming Lu, bHptMjE5QGpudS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
‡ORCID: Xianqiang Liu, orcid.org/0009-0006-7094-2177