 Zhumin Cao
Zhumin Cao Ke Tian1†
Ke Tian1† Xiaowei Tang
Xiaowei Tang- 1Department of Hepatobiliary Surgery, The Seventh People’s Hospital of Chongqing, Chongqing, China
- 2Department of Hepatobiliary Surgery, District Traditional Chinese Medicine Hospital, Chongqing, China
The significant identification of Beclin-1’s function in regulating autophagy flow signified a significant progression in our understanding of cellular operations. Beclin-1 acts as a scaffold for forming the PI3KC3 complex, controlling autophagy and cellular trafficking processes in a complicated way. This intricate protein has garnered considerable attention due to its substantial impact on the development of tumors. Strong evidence indicates Beclin-1 plays a critical role in controlling autophagy in various human cancer types and its intricate connection with apoptosis and ferroptosis. The potential of Beclin-1 as a viable target for cancer therapy is highlighted by its associations with key autophagy regulators such as AMPK, mTOR, and ATGs. Beclin-1 controls the growth and dissemination of tumors by autophagy. It also affects how tumors react to therapies such as chemotherapy and radiation therapy. The role of Beclin-1 in autophagy can influence apoptosis, depending on whether it supports cell survival or leads to cell death. Beclin-1 plays a crucial role in ferroptosis by increasing ATG5 levels, which in turn promotes autophagy-triggered ferroptosis. Finally, we analyzed the possible function of Beclin-1 in tumor immunology and drug sensitivity in cancers. In general, Beclin-1 has a significant impact on regulating autophagy, offering various potentials for medical intervention and altering our understanding of cancer biology.
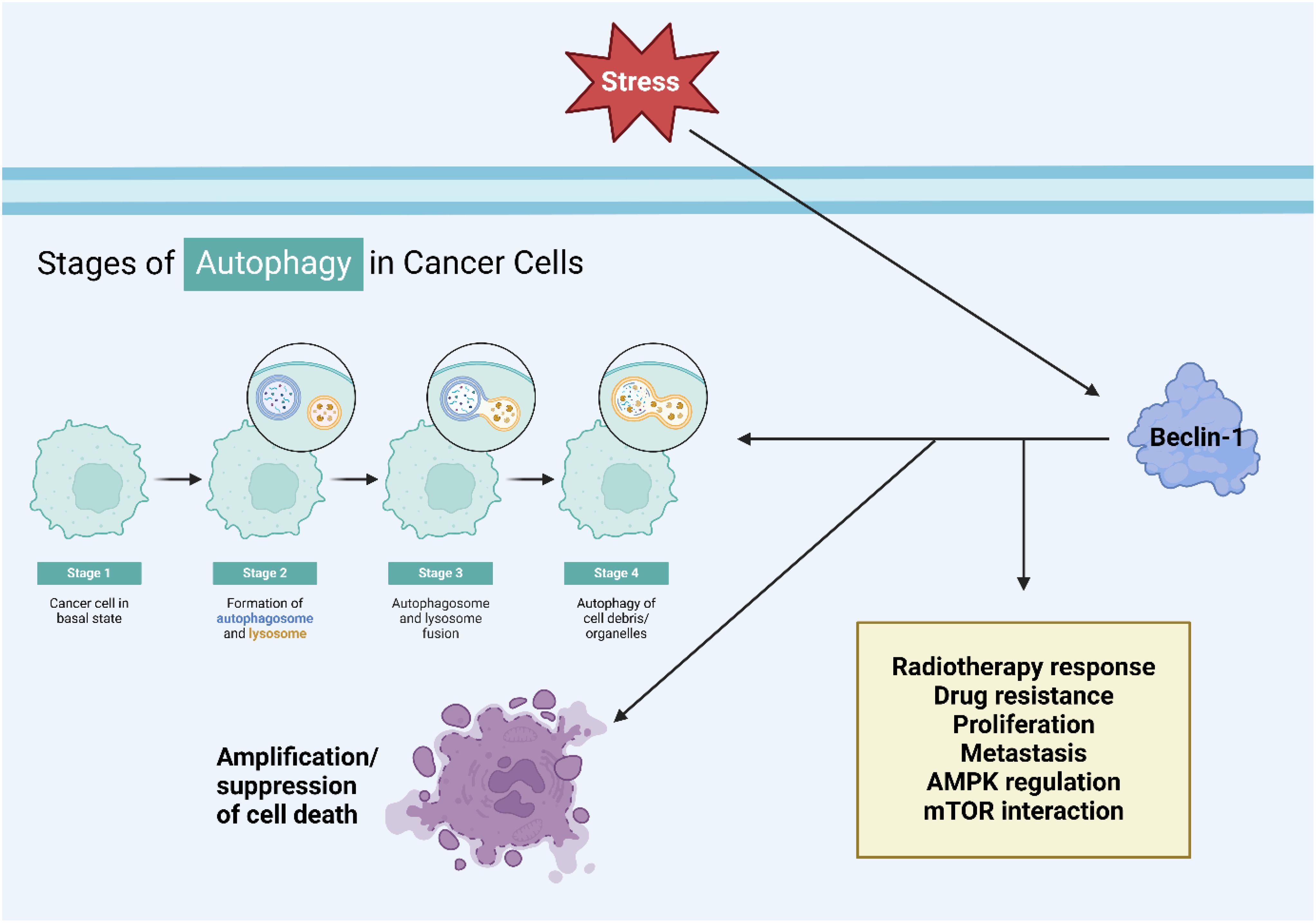
Graphical Abstract.
1 Introduction
Growing proof has shown that cell death is an essential aspect of cellular existence. Nevertheless, the sophisticated biological studies have showcased the genetically encoded processes that rid the body of unnecessary, permanently damaged, or possibly dangerous cells (1–5). It should be pointed out that regulated cell death (RCD) is not limited to multicellular organisms. Although RCD unquestionably aids in maintaining balance within organisms in normal and abnormal situations, it is also present in simpler forms in single-celled eukaryotes like Dictyostelium discoideum and different yeast species that may gather in colonies, as well as in specific prokaryotes like Escherichia coli. RCD relies on certain molecular machinery, indicating that it can be modified (accelerated or decelerated) through genetic or pharmacological interventions. This is in stark opposition to unintentional cell demise, which is the swift and disastrous death of cells that have been subjected to intense physical, chemical, or mechanical injuries (6–18).
Despite the high level of similarity in the chemical reactions involved, RCD plays a role in two distinct scenarios. RCD can occur spontaneously, serving as a natural part of the body’s growth or tissue renewal processes (7, 19). The term commonly used for these completely natural types of RCD is programmed cell death (PCD). On the other hand, RCD can be activated by changes in the intracellular or extracellular surroundings that are too extreme or extended for the body’s adaptive processes to handle, hindering the restoration of cellular balance. Similarly to adaptive stress responses, stress-induced programmed cell death is an approach used to preserve biological equilibrium (6). Unlike adaptive stress responses, which support cellular and overall homeostasis, programmed cell death (RCD) targets the organism or colony directly, independent of cellular homeostasis (6). This process of maintaining balance in the body involves not just getting rid of harmful or unnecessary cells, but also allows dying cells to release chemicals that alert the organism or colony of potential dangers. DAMPs or alarmins are common terms used to describe molecular patterns associated with damage, serving as warning signals (20–23).
Macroscopic morphological alterations indicate the occurrence of cell demise. In the past, these morphotypes were used to classify cell death into three different types and determine the mechanisms responsible for clearing dead cells and their remnants: (1) apoptosis, characterized by cytoplasmic shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blebbing, leading to the formation of apoptotic bodies that are engulfed and degraded by neighboring cells; (2) autophagy, showing cytoplasmic vacuolization and eventual lysosomal degradation after phagocytic uptake; and (3) necrosis, which lacks specific features of apoptosis and autophagy, ending with the removal of cell corpses without phagocytic or lysosomal involvement (24, 25). Nevertheless, despite various limitations and constraints, this morphological classification remains commonly utilized. Starting in 2005, the Nomenclature Committee on Cell Death (NCDD) convened consistently for four reasons: (1) to address problems with a morphology-centered naming of cell death; (2) to accurately delineate key cell death processes using genetic, biochemical, pharmacological, and functional criteria instead of morphology; (3) to distinguish between causes and correlations of cell death; and (4) to set agreed-upon standards for recognizing dead cells with irreversible plasma membrane permeabilization or complete cellular fragmentation as signs of cell death (18, 26–29).
The disruption of cell death processes is a crucial element involved in many diseases, especially cancer. Different pathways of cell death, such as autophagy and apoptosis, interact in intricate ways, impacting the advancement of cancer and how it responds to treatment. This review examines the different functions of autophagy in human cancers, emphasizing its significance as both a protective and detrimental element in tumor biology. Among the numerous regulators of autophagy that have been identified, Beclin-1 is acknowledged as one of the most important. Beclin-1, a vital regulatory protein, is necessary for initiating autophagy and impacts the fine balance between cell survival and death. The present review will comprehensively investigate how Beclin-1 controls autophagy in human cancers, assessing its effects on tumor advancement and drug resistance. Examining Beclin-1’s role reveals cancer cells’ reliance on autophagy for survival. This review explores Beclin-1’s impact on cell death pathways. The connection between these processes is essential in deciding the outcome of cancer cells following medical treatments. Exploring how autophagy affects cancer therapies could lead to new strategies. This review will delve into Beclin-1’s vital role in controlling autophagy, its influence on cancer growth, drug resistance, and interactions with cell death mechanisms. By revealing these connections, we can offer fresh perspectives to enhance the creation of groundbreaking healing strategies for improved treatment of human cancers.
2 An overview of cell death
PCD could potentially regulate the equilibrium between cell death and normal cell survival (30). PCD is important in deciding the ultimate destiny of cancer cells when cellular balance is disturbed (31, 32). Noteworthy is the fact that the three main forms of PCD include apoptosis, autophagy, and programmed necrosis. These three forms can be distinguished from each other by their morphological differences. Kerr and colleagues were the first to introduce apoptosis, also referred to as type I PCD. Apoptosis involves specific changes in dying cells, including cell shrinkage, nucleus condensation, membrane blebbing, and detachment from neighboring cells or matrix. The breaking of chromosomal DNA into fragments between nucleosomes, the movement of phosphatidylserine to the outside of the cell membrane, and various internal substrate cleavages due to specific proteolysis are all instances of biochemical changes (33–38). Autophagy, or type II PCD, is a conserved degradation process in various species. It involves the formation of autophagosomes, enclosed by a double membrane, containing macromolecules and organelles for recycling (39–42). Overall, autophagy is crucial in times of famine or stress caused by a lack of growth factors (43), and it is key in preserving cell homeostasis. However, a growing body of evidence indicates that autophagy in cells could experience autolysis, leading to cell death as a response to intense stress. This phenomenon is separate from apoptosis and programmed necrosis (34, 44). Autophagy, acting as “the Janus role,” controls various physiological processes like starvation, differentiation, survival, and cell death (45). Programmed necrosis is a type of programmed cell death that happens alongside apoptosis and autophagy. It is defined by the swelling of cells, dysfunction of organelles, and breakdown of living cells (46–48). Hence, PCD could have a crucial impact on maintaining tissue balance and eliminating unhealthy cells, which is important for cancerous tissues (34).
Typically, apoptosis and autophagy-mediated cell death are considered necessary processes of regulated cell death (49). These processes can lead to organelle malfunction or cell death under conditions of cellular stress. Additionally, they have a crucial role in regulating cancer cell death and specific cancer treatments. Apoptosis plays a crucial role in controlling cell numbers and preserving overall organismal balance (50). Apoptosis is linked to numerous morphological characteristics. These features consist of cell shrinkage, chromatin condensation, membrane blebbing, fragmentation of DNA, and the formation of apoptotic bodies (51, 52). Apoptosis occurs via extrinsic pathway (death receptor activation) and intrinsic pathway (mitochondria control). Ligands like tumor necrosis factor α (TNFα), Fas ligand, and TRAIL trigger extrinsic pathway by binding death receptors on cell surfaces. This activation triggers caspase-8, initiating the final stage of cell death known as apoptosis (53). Proteins in the B-cell lymphoma 2 (Bcl-2) family typically have a function in regulating the intrinsic pathway when there is permanent cellular harm. These proteins control the release of Cytochrome c (Cyt-C) and SMAC/DIABLO, which is a direct IAP-binding protein with low pI, from the mitochondria. The joining of Cyt-C and Apaf-1 proteins triggers caspase 9 activation, causing cancer cell apoptosis (54, 55). Autophagy is an essential phagocytic process for ensuring cell function and balance. Lysosomal fusion can decompose toxic proteins or organelles (56). Autophagy plays two distinct roles in tumor growth, showing that it can either aid or hinder cancer progression, based on the specific tumor subtype and its mutation profile (57). In the pre-cancer stage, inhibiting autophagy leads to the accumulation of ROS and genomic dysfunction. When these two elements are merged, they create pressure on the endoplasmic reticulum (ER) to expand and cause DNA harm, ultimately resulting in tumor development. However, autophagy can provide energy and nutrients to tumors when triggered by a lack of food or oxidative stress (58, 59). Autophagy-related genes, including Beclin-1, light chain 3 (LC3), p62, forkhead box O (FoxO), and Unc-51-like kinase 1 (ULK1), regulate the autophagy process. ULK1 specifically plays a key role in initiating and controlling autophagy, increasing the chances of cancer cell survival (60). Furthermore, autophagy-associated signaling pathways, such as the phosphatidylinositol 3 kinase complex 1 (PI3KC1)-protein kinase B (Akt)-mammalian target of rapamycin complex 1 (mTORC1) pathway, the Ras-Raf-mitogen activated protein kinases (MAPKs) pathway, and the nuclear factor kappa-B (NF-κB) pathway, play a crucial role in fighting against tumor advancement and spread of metastasis. Autophagy and apoptosis are the main mechanisms that control cell destiny and uphold cellular balance within the cell. Alternatively, there is a distinct relationship between apoptosis and autophagy that can result in cell death via methods that are separate or supportive (61). The controlled management of apoptosis and autophagy has proven the therapeutic effectiveness of small-molecule compounds in the development of cancer treatments (62). For instance, studies have demonstrated that Ampelopsin (Amp) triggers apoptosis and autophagy-related cell death in glioma cells by stimulating ROS production and activating c-Jun N-terminal kinase (JNK) (63). Galectin-1, a member of the galactose lectins family, carries out a range of biological functions. This protein, present in many tumor structures, regulates the proliferation, migration, and growth of tumor cells (64). Table 1 outlines the pathways of cell death involved in cancer.
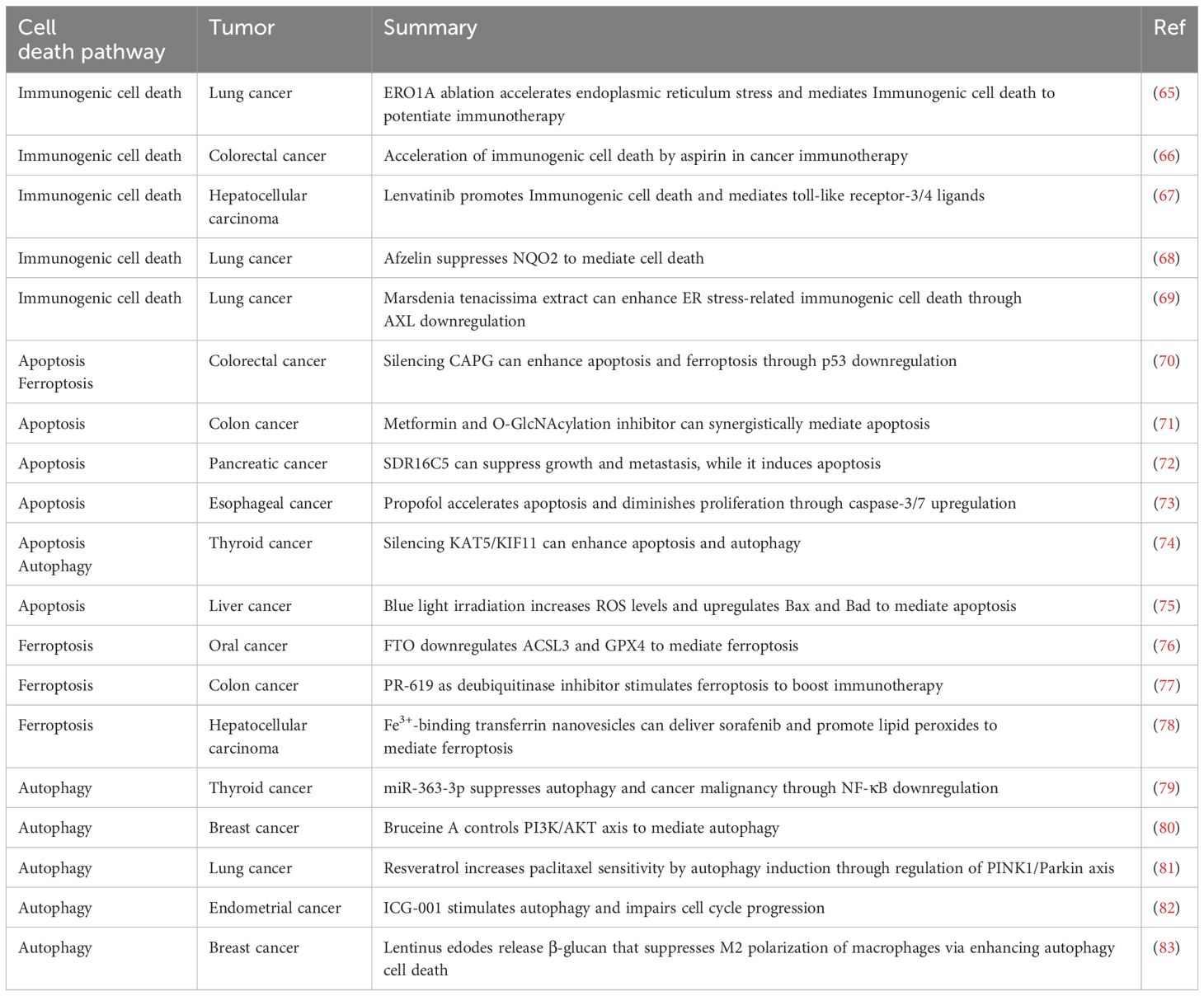
Table 1. Cell death pathways in cancer.
Researchers developed chitosan nanoparticles that respond to dual pH to enhance drug delivery and accumulation in MDR breast cancer cells, named DCCA/DOX-NPs. These nanoparticles exhibited increased cellular absorption, heightened toxicity, and enhanced antitumor effectiveness in living organisms by overcoming resistance mechanisms (84). In hepatocellular carcinoma, sorafenib and its derivative SC-59 both trigger autophagy through SHP-1-STAT3-Mcl-1-Beclin 1 pathway, with SC-59 showing greater efficacy in decreasing cancer cell viability and tumor growth (85). The use of Adenovirus vector to express XAF1 in gastric cancer cells promotes autophagy through increasing Beclin-1 levels and inhibiting the Akt/p70S6K pathway, leading to enhanced apoptosis simultaneously (86). Rhus coriaria extract (RCE) exhibits notable effectiveness in fighting colorectal cancer cells by diminishing cell viability and colony formation, triggering Beclin-1-independent autophagy and caspase-7-dependent apoptosis, impacting the AKT/mTOR pathway, and influencing crucial proteins related to proteasome-mediated breakdown (87). Tanshinone IIA (TAN) slows the growth of oral squamous cell carcinoma (OSCC) by inducing cell death and self-cleaning mechanisms through specific pathways, while also decreasing the activity of other pathways, with its effectiveness being linked to Beclin-1 levels (88). Cisplatin enhances autophagy and apoptosis in A549 human lung cancer cells by increasing Beclin 1 and Atg5, while blocking these autophagy-related proteins worsens apoptotic cell death (89). Autophagy may promote programmed cell death in cancer cells exposed to cobalt chloride (CoCl2)-induced hypoxia through an ATG5-dependent mechanism, which includes crosstalk with endoplasmic reticulum stress and mitochondrial pathways, and identifies two separate autophagy-related routes that could aid in the creation of new anti-cancer treatments (90). Thus, Beclin-1 controls cell death in human cancers (91–93).
3 Autophagy flux
Autophagy, frequently referred to as the self-recycling process, involves complex lysosome-dependent pathways in eukaryotic cells to control protein, lipid, and organelle levels. Autophagy originates from the Greek terms “auto” and “phagein,” denoting “self-consumption.” When the body experiences a lack of nutrition or energy, it initiates autophagy binge to compensate. Autophagy can also be triggered by various cellular stressors, including the buildup of harmful protein clusters that are specifically broken down through autophagic processes. In the same way, lysosomes can engulf cellular organelles like LDs through autophagy processes to create metabolites. Hence, autophagy serves as an important control mechanism for a range of cellular and tissue functions, such as growth, coping with stress, immune reactions, and metabolic processes. Furthermore, there are three forms of autophagy: macroautophagy/autophagy, microautophagy, and chaperone-mediated autophagy (CMA) (94, 95).
Autophagy dysfunction is associated with illnesses such as neurodegeneration, macular degeneration, Crohn’s disease, and cancer. It plays crucial roles in controlling cellular growth, metastasis, and treatment resistance in cancer. As a result, autophagy has garnered significant attention in cancer treatment, with researchers focusing on regulating it using nanoparticles and anti-cancer medications (96–100). Additionally, aging is linked to malfunctions in the autophagy process. Autophagy is divided into three types: (1) Microautophagy involves the direct ingestion of cellular material by lysosomes. (2) Chaperones selectively recognize substrate proteins, then unfold and translocate them to lysosomes through a lysosomal receptor, and (3) Macroautophagy involves non-selectively enclosing bulk cytoplasm in double-membraned vesicles, which later fuse with lysosomes for degradation. Additional information on autophagy can be found in several scientific reviews (101–104). Many eukaryotic organisms have over 90% of their cells participating in basic macroautophagy processes. Macroautophagy starts with a precursor structure, known as the phagophore, that eventually transforms into an autophagosome. The recently created autophagosome combines with lysosomes, which then expose the internal cytosolic material to enzymes that degrade it in an acidic setting. Autophagosomes gather the lipids they need for formation from different organelles within the cell including the Golgi complex, mitochondria, recycling endosomes, the ER, and even the plasma membrane. Proteins called autophagy-related (ATG) proteins supply these lipids in vesicular form (105–109). Yoshinori Ohsumi, Daniel Klionsky, and Michael Thumm were the first to discover and characterize ATG genes (109–111). Activation of the ULK1/Atg1 kinase complex is necessary to initiate the process of autophagosome formation in autophagy. The ULK1 kinase complex primarily incorporates signals from two kinases: mTORC1 and PRKA/AMPK, both of which are upstream molecular signals. These complexes are vital in controlling the initiation of the autophagy process (112–114). Another part of creating a phagophore from scratch includes its growth in particular ER subdomains referred to as omegasomes. The omegasomes need the ULK1 kinase complex to be activated in order to form (115). The second group acts as a nursery for the growth of autophagosome (116).
The ULK1 complex phosphorylates and activates a different protein kinase complex, called the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (BECLIN1/Vps30/Atg6-PIK3R4/Vps15-ATG14-PIK3C3/Vps34-AMBRA1). When Beclin-1/Vps30/Atg6 combines with PIK3R4/VPS15 and ATG14, PIK3C3/VPS34 is triggered to produce PtdIns3P. Proteins such as DFCP1 (zinc finger FYVE-type containing 1) (115) and phosphoinositide interacting (WIPI) are connected through binding proteins to participate in cellular processes (117). WIPI2 and WIPI1 collaborate to bring in the ATG12-ATG5 complex, together with ATG16L1 (118). ATG16 combines with the ATG12-ATG5 complex to initiate the formation of phagophores (119). The elongation complex (WDR45B/WIPI3 and WDR45/WIPI4) and WDR45, specifically interact with ATG2 (118, 120). The ATG12-ATG5-ATG16L1 conjugate acts as an E3-like ligase (109), to attach MAP1LC3 from the LC3 protein family to phosphatidylethanolamine (119). The LC3 protein family is made up of seven members, with four in the MAP1LC3 group (LC3A, LC3B, LC3C, and LC3D) and the other three in the GABA Type A receptor-associated protein groups (GABARAP, GABARAPL1, and GABARAPL2), representing GABA Type A receptor-associated protein and GABA type A receptor associated protein like 1 and 2, respectively. In terms of function, LC3 proteins play a role in selecting cargo, elongating, and closing phagophores (119, 120), resulting in the development of fully mature autophagosomes that merge with lysosomes to form autolysosomes (121).
4 Evolution of autophagy genes
The origins of all three domains of life - bacteria, archaea (prokaryotes), and eukaryotes - can be traced back to the last eukaryotic common ancestor (LECA). Thaumarchaeota is one of the primary branches of archaea, followed by Asgard, and then Crenarchaeota, Korarchaeota, and Aigarchaeota (TACK) (122). Prokaryotes lack the autophagy pathway found in eukaryotic cells. This system relies on the existence of intracellular membrane compartments. The diverse functions seen within eukaryotic lineages are probably due to the presence of most, if not all, core autophagy related (ATG) proteins in the Last Eukaryotic Common Ancestor (LECA). Not every subgroup participates in autophagy, but it is likely that early on in the LECA era, two ATG protein families - Atg1/ULK and PROPPINs - which interact with polyphosphoinositides, had already branched out into various subgroups. Plants and vertebrates experienced numerous duplication events. In the usual ATG conjugation systems, ATG12 is bonded to ATG5 through a covalent attachment. Nevertheless, essential components required for this conjugation are absent in Toxoplasma and Plasmodium, which are part of the Alveolata within the SAR supergroup of Stramenopiles, Alveolata, and Rhizaria, as well as in Komagataella, a yeast genus. Specifically, the E2-like enzyme ATG10 and the C-terminal glycine of ATG12 are not present. Therefore, these organisms depend on noncovalent connections involving ATG12 and ATG5 instead (123, 124). The noncovalent form is considered adaptable because it does not depend on enzymes or ATP. Eukaryotic organisms might have experienced as many as sixteen transitions from covalent to noncovalent bonds.
Saccharomyces cerevisiae, frequently utilized in studies on budding yeast, is commonly acknowledged as the gold standard in autophagy investigations. This reputation is because of its crucial involvement in breakthroughs like the discovery of the ATG gene. Research indicates autophagy in S. cerevisiae is different from other species.
In different kinds of organisms, Atg1 complex consists of Atg29 and Atg31 in addition to Atg17. Nevertheless, the S. cerevisiae Atg1 complex does not have ATG101 like other species (125), preventing the stabilization of ATG13 and the formation of a complex. It is not clear if the loss of ATG101 is related to the acquisition of Atg29 and Atg31. S. cerevisiae does not have VMP1, a crucial protein for autophagy in various other organisms such as metazoa, Dictyostelium, and potentially green algae. VMP1 is located in the ER and is situated after the hisT gene in the Escherichia coli DNA gene A superfamily. Crucially, S. cerevisiae employs a distinctive biosynthetic mechanism called the cytoplasm-to-vacuole targeting (Cvt) pathway for delivering vacuolar hydrolases to the vacuole. Furthermore, Schizosaccharomyces pombe also contains an alternative metabolic pathway called the Nbr1-mediated vacuolar targeting (NVT) pathway.
Furthermore, proteins associated with specific autophagy have been discovered to increase in gene families. NBR1 (Atg19 in S. cerevisiae) is found in various eukaryotic organisms, and SQSTM1 likely evolved from duplication of NBR1 and loss of NBR1 domains. The presence of OPTN and CALCOCO families in the majority of metazoan species indicates that they underwent expansion in vertebrate evolutionary lineages. While most ATG proteins are found only in eukaryotic organisms, certain ones may have originated in prokaryotes. Many functional complexes in the autophagy pathway have proteins in common with distant prokaryote counterparts. Various instances of this phenomenon are demonstrated, including the DedA superfamily proteins (TMEM41B and VMP1), the Hop1, Rev7 and Mad2 (HORMA)-domain-containing proteins (ATG13 and ATG101), transmembrane segment of ATG9, chorein-N domain at the N termini of lipid transfer proteins (ATG2), and ubiquitin-like ATG conjugation systems. This implies that autophagy developed partially through the utilization of genes already present. Prokaryotes are the ancestors of the endosomal sorting complex required for transport (ESCRT) proteins, crucial for micro- and macroautophagy. Even though ESCRT-I, -II, and -III function sequentially in eukaryotic cells during membrane fission, bacterial and archaeal proteins like PspA/Vipp1 and CdvB appeared earlier in evolution. Although the Asgard archaea group is believed to be the initial habitat of the ESCRT-I and -II proteins, they were introduced at a later time. Therefore, it is believed that eukaryotic organisms evolved from the Asgard group, which had a complete ESCRT system (although lacking ESCRT-0, found only in Opisthokonta) (123, 126–129).
5 Autophagy in cancer drug resistance
Inhibiting autophagy may enhance tumor cells’ responsiveness to regular medications or combat the resistance they have built up against chemotherapy (130–132). In the upcoming section, we will explore the latest and most relevant findings on combining various anticancer drugs with autophagy inhibitors and activators. Combining CQ and HCQ with medicines like 5-fluorouracil (133), cisplatin (134), and temozolomide (135, 136), enhances their cytotoxicity. Additionally, the use of both CQ and trastuzumab together was able to block tumor growth by over 90% in a HER2-positive breast cancer tumor xenograft that had total resistance to trastuzumab (137). Furthermore, different autophagy blockers have demonstrated positive outcomes, such as verteporfin’s improvement of gemcitabine’s effectiveness in an in vitro model of pancreatic cancer (138). SBI-0206965 was able to overcome resistance to cisplatin in NSCLC cells (139) as well as resistance to cabozantinib in metastatic colorectal cancer (140). The combination of celecoxib, a specific inhibitor of cyclooxygenase-2, with CQ and SAR405 led to an enhancement in cell death (141). Furthermore, research demonstrated that 3MA increased the level of cell demise caused by bortezomib in glioblastoma cells (142). Furthermore, the use of CQ analog lys05 with the second generation tyrosine kinase inhibitor, nilotinib, has shown to have an extra effect on reducing the amount of leukemia stem cells in mouse models of chronic myelogenous leukemia (143). By inhibiting autophagy, UAMC-2526 can improve the effectiveness of oxaliplatin in a colorectal cancer mouse model. In addition, UAMC-2526 treatment led to a more differentiated phenotype in tumors (144). Research has investigated using both autophagy activators and inhibitors together; for instance, CQ and HCQ have been shown to boost the impact of mTOR inhibitors like temsirolimus (145) or everolimus (146), in colorectal cancer (147), melanoma (145), and neuroendocrine neoplasms (146). This indicates that blocking autophagy can be utilized to address resistance to mTOR inhibitors. In addition, research has shown that CQ can improve the cancer-fighting abilities of vorinostat (148). Moreover, the synergistic effects of combining everolimus with SAR405 have been shown in studies (149, 150). In light of these observations, it can be inferred that suppressing autophagy proves to be a more efficient approach to treatment. This happens due to the fact that autophagy has a protective function in the models being studied. Autophagy inducers, however, can also help overcome chemoresistance, although to a lesser extent. Studies have demonstrated that Temsirolimus can enhance the efficacy of gemcitabine and cisplatin in bladder cancer cell lines (151). Furthermore, studies have demonstrated that it can decrease the susceptibility of colon cancer cells to cetuximab (152). In the same way, curcumin has been found to enhance the effectiveness of gefitinib in primary gefitinib-resistant small-cell lung cancer cells through autophagy-dependent synergism (153). However, studies conducted before clinical trials have revealed that controlling autophagy can lead to conflicting outcomes under specific circumstances. Lab research shows mTOR + ULK1 inhibitor combo induces A549 cell apoptosis (154, 155). SBI-0206965 sensitizes neuroblastoma cells to TRAIL, not mTOR inhibitors. This implies that autophagy does not have a protective function within this model (155). At this point, it is essential to utilize molecular markers that can anticipate how tumors will react to autophagy modulators. One illustration of such an indicator is the BRAF V600E mutation, linked to protective autophagy (156).
5-FU, antimetabolite chemo used long term for solid tumors like head/neck, breast, GI, and pancreatic cancer (157, 158). In S phase, halts thymidylate synthetase, decreasing thymidylate for DNA replication, affecting cell cycle progression (159). Nevertheless, the effectiveness of 5-FU is limited by the promotion of protective autophagy as an unintended consequence in different cancer types through multiple pathways: (1) augmentation of Beclin-1 expression, aiding in the transformation of LC3I to LC3II (160); (2) JNK-triggered protective autophagy and activation of Bcl-2 phosphorylation, resulting in resilience to 5-FU; and (3) increased autophagy flow when 5-FU is present (161). Despite its purpose of damaging DNA in the guanine residue, TMZ, an alkylating medication utilized for treating glioma (162), often shows restricted effectiveness. This is partly because it triggers protective autophagy, which may play a role in building resistance (163, 164). Following temozolomide treatment, various mechanisms have been demonstrated to trigger protective autophagy. Some of these mechanisms involve ATM increasing the AMPK-ULK1 pathway (165), generating ROS, activating the signal-regulated kinase (ERK) pathway (166), and creating mitochondrial and ER stress (167). Cisplatin-based chemotherapeutic regimens are standard treatments for solid tumors due to their DNA damage and mitochondria apoptosis effects, although drug resistance can limit their effectiveness (168, 169).
Autophagy plays a significant role in various pathways of cisplatin-induced chemoresistance, specifically in ovarian cancer, where it is triggered by the activation of the ERK pathway (170), and the upregulation of Beclin-1 (171). Cisplatin induces protective autophagy in esophageal cancer by increasing Beclin-1 levels, converting LC3-I to LC3-II (172), and upregulating ATG7 expression (173). Consequently, inhibiting autophagy along with cisplatin treatment results in increased cell death in esophageal cancer (174).
6 Major regulators of autophagy machinery
6.1 Beclin-1
Beclin-1 is a new protein containing only the BH3 domain of Bcl-2 (175). Unlike other animals lacking autophagy genes, which die in early embryo development (before E7.5) because of issues with proamniotic canal closure, Beclin-1 null mice have a significantly more severe embryonic phenotype (176). Beclin-1 located in cytoplasm, ER, mitochondria, and perinuclear membranes. Found in various human and mouse tissues. Also seen in different regions of human colon cancer tissue via immunohistochemistry (177). Beclin-1 is comprised of three specific structural regions: a BH3 domain at the N-terminal (amino acids 114-123), a coiled-coil domain (CCD) (amino acids 144-269), and an evolutionarily conserved domain (ECD) (amino acids 244-337). ECD domain important for Beclin-1 to regulate autophagy, prevent cancer.
The short leucine-rich amino acid sequence makes the nuclear export signal (NES) of Beclin-1 effective. The Beclin-1 NES mutation hinders its capacity to induce autophagy during food scarcity and to inhibit cancer progression. Twelve Bcl-2 family members, known for their ability to prevent cell death, bind to the BH3 domain of Beclin-1. Activators such as activating molecule in Beclin-1-regulated autophagy (Ambra1), UV radiation resistance-associated gene (UVRAG), and Atg14L interact with the CCD, while both the ECD and CCD are connected to PI3KC3/Vps34 (Figure 1, Table 2) (191, 192). Beclin-1 not only controls autophagy, but also regulates mitophagy, a specific form of autophagy (193).
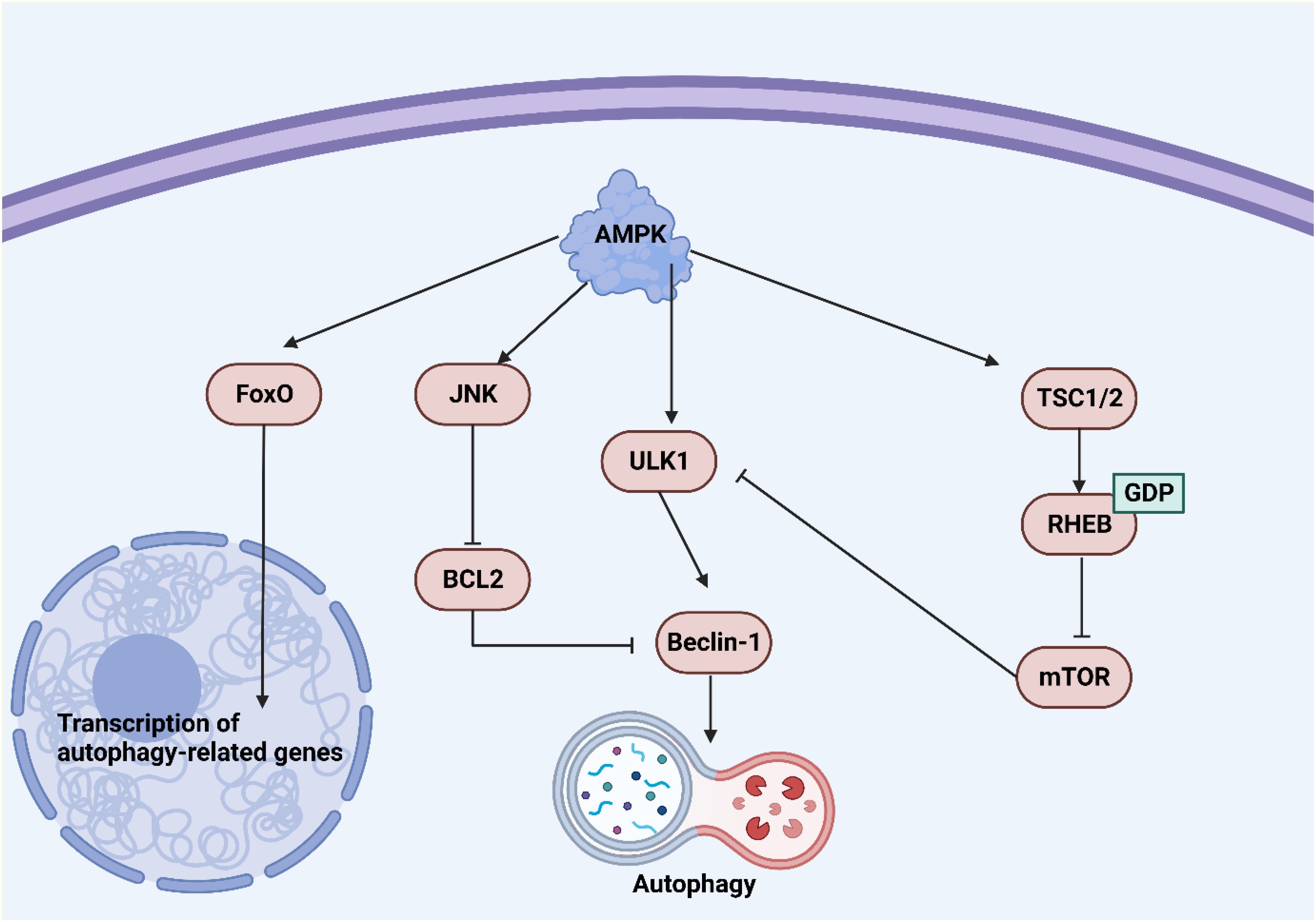
Figure 1. An overview of Beclin-1 in autophagy. Upregulation of Beclin-1 can induce autophagy. AMPK upregulates ULK1 to increase Beclin-1 levels for autophagy induction. On the other hand, an increase in mTOR levels can suppress ULK1 to reduce Beclin-1 levels in autophagy suppression.
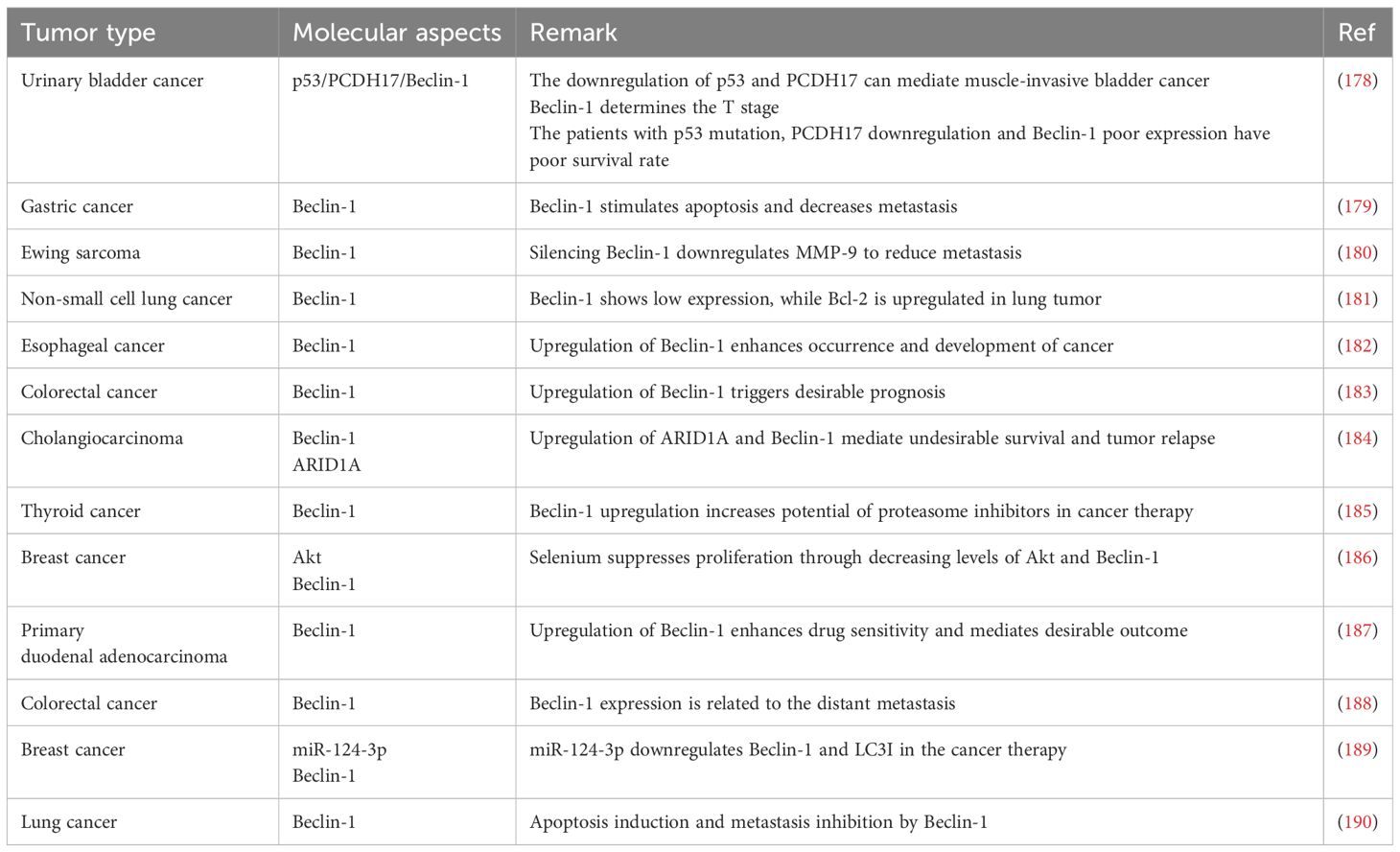
Table 2. Beclin-1-mediated regulation of cancer progression.
Beclin-1 is crucial for maintaining cellular balance and controlling autophagy, acting as a key center that combines different cellular cues. Its dual role involves promoting autophagy cell death and also inhibiting apoptosis by interacting with the Bcl-2 protein family. Maintaining a delicate balance between these processes is essential for how cells respond to stress and keep tissues healthy. Beclin-1 plays a crucial role in autophagy and is involved in various cellular processes like inflammation and immune responses. Its significance extends beyond autophagy regulation and dysfunction is linked to cancer. Targeting Beclin-1 for treatments may offer new approaches in cancer therapy, making it a potential biomarker and therapeutic target.
6.2 mTOR
Cell growth and metabolism are influenced by hormonal, nutrient, and energy signals via signaling pathways and transcription factors. mTOR, a crucial kinase, regulates cellular metabolism and responds to environmental cues, with its overactivity associated with diseases. mTOR interacts with multiple proteins to create two complex entities known as mTOR complex 1 (mTORC1) and mTORC2 (194). A fundamental list of individuals found in both mTORC1 and mTORC2 consists of the Tti1/Tel2 complex, mLST8, and DEPTOR, all functioning in both groups (195–198). RICTOR, mSin1, and PROCTOR1/2 are unique to mTORC2, whereas RAPTOR and PRAS40 are dedicated solely to mTORC1 (199–209). Two kinase complexes can activate signal cascades and control cellular activities independently based on various substrate preferences. mTOR creates two distinct signaling complexes called mTORC1 and mTORC2. One function of mTORC1 is to integrate signals from growth factors and nutrients to promote increased anabolic metabolism, including elevated protein and lipid synthesis, and inhibition of autophagy or lysosome production. The TSC complex prevents mTORC1 activation by stimulating Rheb GTPase, a requirement for mTORC1 activity. The compound is blocked by signaling pathways triggered by growth factors like PI3K/AKT or Ras/Erc, or stimulated by cellular stressors that involve mTORC1. During stressful situations like low energy or oxygen levels, AMPK activates and also inhibits the TSC complex by converting ATP to AMP. This inhibition also includes AMPK’s phosphorylation of RAPTOR, which directly impacts mTORC1. Moreover, Rag GTPases move mTORC1 to the lysosomal surface upon amino acid stimulation, where Rheb GTPase subsequently triggers its activation. On the other hand, mTORC2 is activated by growth factor/RTK/PI3K signaling and has important functions in cell metabolism, the cytoskeleton, and survival through AGC family kinases. Activation of mTORC1 has the ability to block IRS and enhance GRB10 functionality, leading to the suppression of RTK/PI3K/AKT signaling. An mTORC1 effector, S6K, additionally suppresses IRS and indirectly impacts mTORC2 via inhibitory phosphorylation (Figure 2) (210).
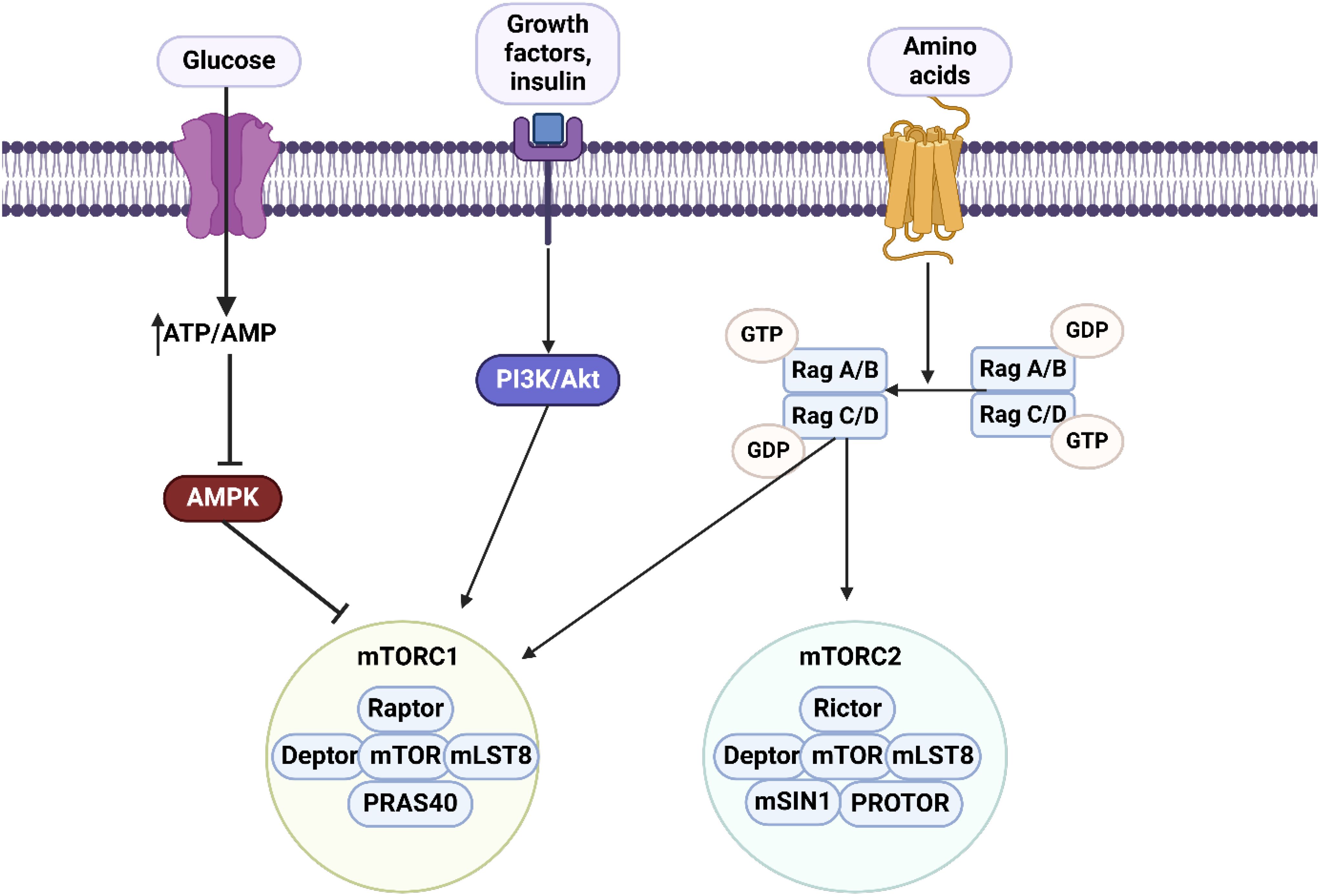
Figure 2. An overview of mTOR axis. There are two complexes including mTORC1 comprised of Raptor, Deptor, mTOR, mLST8 and PRAS40, and mTORC2 comprised of Rictor, Deptor, mTOR, mLST8, mSIN1 and Protor. The upstream regulator of mTORC1 is AMPK which suppresses the mTORC1 axis. Lack of glucose and an increase in ATP/AMP levels can regulate the AMPK/mTORC1 axis. Changes in amino acid levels can affect both mTORC1 and mTORC2 levels. Moreover, PI3K/Akt axis is considered as the upstream regulator of mTORC1. Therefore, most of the molecular pathways are related to mTORC1.
6.3 AMPK
AMPK is believed to be made up of three parts - α, β, and γ subunits - with slight structural differences seen in various species (211). The α subunit of AMPK has a Ser/Thr kinase domain at the start of the protein. Phosphorylation at Thr-172 is crucial for regulating its activity in humans. LKB1, TAK1, and CaMKKβ are upstream kinases targeting Thr-172. Circac1 also plays a role in controlling AMPK function (212). Thr-172. Circac1 is also involved in controlling AMPK function (212). Moreover, AMPK also includes multiple allosteric activation sites. These consist of the self-inhibiting domain (AID) on the α subunit, as well as domains for interacting with the β and γ subunits. The main functions of the β-regulatory subunit are found in two key areas: the carbohydrate binding region (CBM) which helps with energy supply, and the end area which links to the α and γ subunits. The γ regulatory subunit contains multiple cystathionine-β-synthase (CBS) tandem repeats that make up the Bateman domain, which is essential for detecting energy levels and interacting with ATP and AMP (213). Nutrient depletion will result in elevated AMP/ATP ratio and lowered ATP levels. It was previously believed that activation of AMPK γ subunit required extremely high levels of 5′-AMP when AMP was present in millimolar concentrations (214). AMPK Thr-172 can be targeted by a very high concentration of AMP through calcium ion-dependent CaMKKβ and AMP through the AMP-dependent LKB-1. Before, it was thought that AMPK only controlled bioenergetic metabolism, but now it is understood that it also protects the nervous system, lengthens telomeres, triggers the tumor suppressor p53, and improves mitophagy and synthesis. It has been experimentally demonstrated that boosting the AMPK signaling pathway in the body-wall muscles of nematodes can extend their average lifespan. Moreover, the longevity of the nematode is prolonged by neuronal TORC1 through the reduction of mitochondrial activity by activating AMPK (215). Alpha-ketoglutarate (AKG) prolongs the lifespan of fruit flies by blocking the mTOR pathway and boosting the AMPK signaling pathway when glucose is transformed into blood sugar in periods of high energy, ultimately raising insulin levels. Insulin indicates a surplus of nutrients, causing cells to absorb and use more resources by attaching to receptors on the cell surface and initiating the PI3K-AKT pathway through IRS1. The AKT-TSC1/2-RheB-mTORC1 pathway is known to be the route through which insulin inhibits AMPK activity and stimulates mTORC1 (216).
6.4 ULK1
Only one autophagy-linked kinase has been identified in S. cerevisiae, which is known as Atg1 and is a conserved serine/threonine kinase (217, 218). Atg1 loss leads to early termination of yeast autophagy (218). In mammals, Atg1 is similar to ULK1 and ULK2, which are kinases that resemble uncoordinated-51. ULK1 and ULK2 work together; the absence of ULK1 results in a slight phenotype in mice (219). In mammals, Papinski and Kraft (2016) (220), discovered that the start of autophagy is triggered by ULK1 combining with FIP200, ATG13, and ATG101. ULK1 activity is regulated by AMPK and MTORC1 as energy and nutrient sensors. In environments with abundant nutrients, MTORC1 suppresses autophagy, but fasting or rapalog treatment inhibits MTORC1, causing an increase in ULK1 kinase activity in mammalian cells (221). Studies indicate that MTORC1 might regulate the phosphorylation of two proteins in the initiation complex, ULK1 and ATG13 (112). The discovery was made that MTORC1 phosphorylation on Ser757 can inhibit the activity of ULK1 and interfere with its interaction with AMPK (222). AMPK phosphorylation initiates autophagy by activating ULK1, in contrast to MTORC1 phosphorylation. During amino acid deprivation, AMPK adds phosphate groups to ULK1, assisting in the degradation of mitochondria via mitophagy (223). AMPK phosphorylates ULK1 at Ser555, regulating hypoxia-triggered autophagic breakdown of mitochondri (224). ULK1 phosphorylates autophagy regulators. ULK1 phosphorylates its binding partners ATG13, FIP200, and ATG101. The PI3KC3 complex 1, which is specific to autophagy, consists of Beclin-1, Vps34/PIK3C3, and Ambra, all phosphorylated by ULK1 (220). Phosphorylation of FUNDC1 by ULK1 is crucial for promoting the proper association of the mitophagy adapter LC3 with FUNDC1, which helps advance mitophagy (225). Additionally, ULK1 helps maintain autophagy flow by triggering a feedback inhibition of the MTORC1 complex via phosphorylating Raptor, ultimately leading to its inhibition (226). AMPK phosphorylation accompanies ULK1-regulated autophagy, leading to the formation of a negative feedback loop (Figure 3) (227).
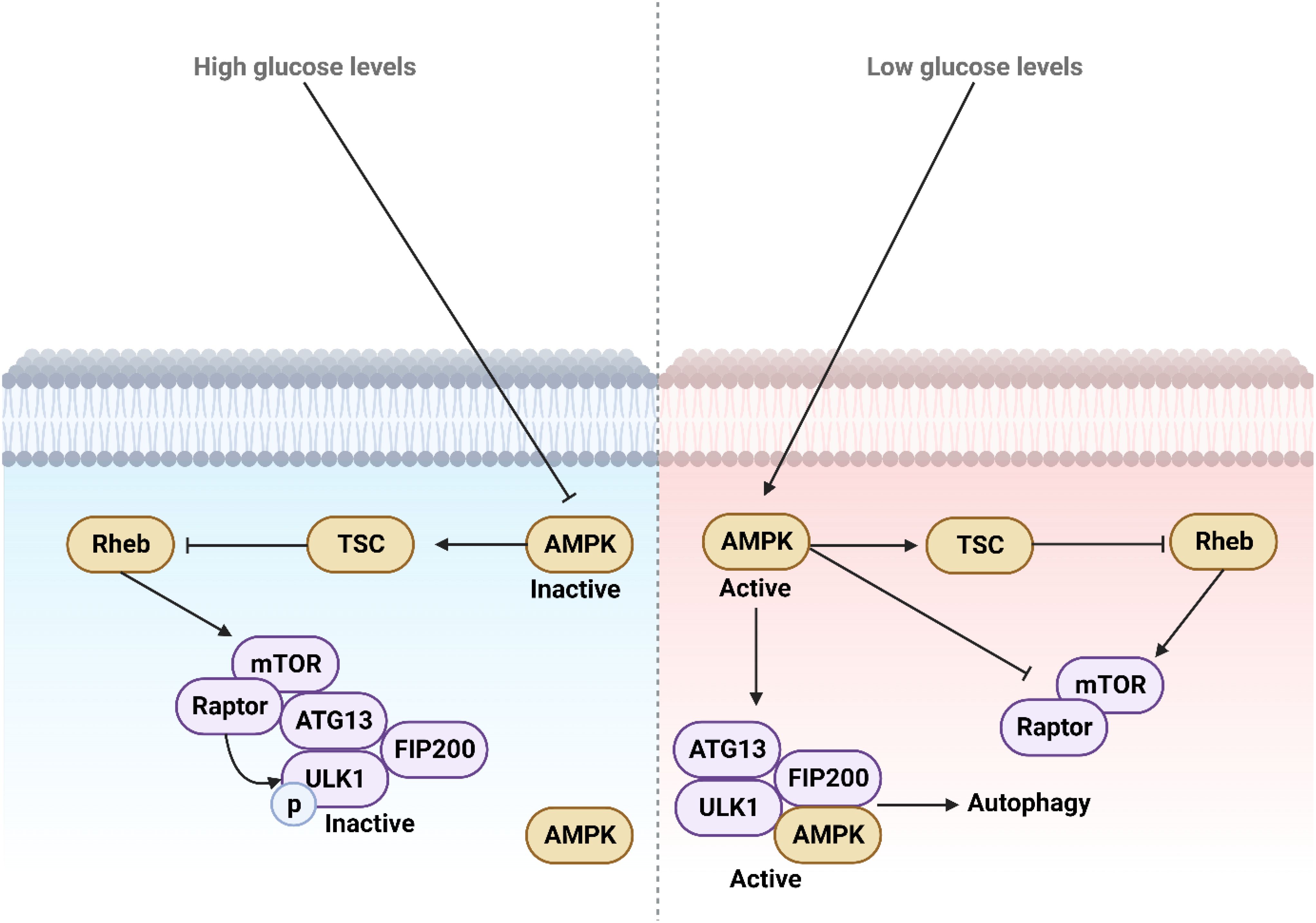
Figure 3. The function and regulation of ULK1 during low and high glucose levels. High glucose levels deactivate AMPK by suppressing its activity, whereas low glucose levels activate and upregulate AMPK, which in turn mediates autophagy. This process provides necessary energy through the degradation of macromolecules and aged organelles.
6.5 Autophagy-related genes
Essential for autophagosome formation are core ATG genes (228). They are made up of 20 genes or gene families. These genes are responsible for producing six different proteins: ATG1 (known as ULK in mammals), ATG9 vesicle, ATG14-containing PtdIns 3-kinase complex, ATG2-ATG18 complex, ATG12 conjugation system, and ATG8 conjugation system make up the initial six complexes (107, 229). ATG8(s) and their counterparts in mammals are known as microtubule-associated protein 1 LC3 and GABARAP, respectively, unless the need arises to specifically name each ATG8. ATG8 and ATG12 systems serve a role similar to ubiquitin through covalent conjugation mechanisms (230–232). ATP-driven mechanism triggers the starting point of activating ATG12’s C-terminal glycine by E1-like enzyme ATG7 in ubiquitin-like protein. After that, thioester intermediates are gradually formed by ATG12, ATG7, and E2-like enzyme ATG10. The final stage involves the formation of an isopeptide bond between ATG12 and the lysine residue in ATG5. Two pairs of ATG12-ATG5 conjugates unite within a compound containing ATG16(L) dimer. The ATG8 system begins with the production of ATG8, a protein that is comparable to ubiquitin, in its proform state. Enzymes belonging to the ATG4 family cut the C-terminal region of this proform to expose a glycine residue. Afterwards, the ATG8 that has been processed is activated by ATG7 and ATG12 enzymes, with the help of E2-like enzyme ATG3, and then attaches to the PE head group. The membranes of autophagy contain ATG8-PE. Although ATG12 conjugation remains unchanged, ATG8-PE can be re-conjugated by ATG4. Remarkably, by interacting with ATG3, ATG12 enhances the ATG8-PE conjugation through ATG12-ATG5 conjugate functioning as an E3-like enzyme (233–236). ATG16L1’s membrane binding dictates where ATG8 lipidation occurs, despite not being essential for the process (237–239). ATG8 lipidation can occur on non-autophagy membranes, but it can be reversed through deconjugation carried out by ATG4, a process regulated by ATG1 and ULK1 An instance would be LC3-I representing modified ATG8, while LC3-II represents modified ATG8-PE. The articles that discussed the discovery of these two systems were part of the four “key publications” for Dr. Ohsumi’s 2016 Nobel Prize, as stated in references (230, 231). These publications significantly broadened the scope of research on autophagy. Even though ATG8 is the most commonly used marker for autophagosomes, ATG5 and ATG7 have been heavily utilized in knockout mouse research (Figure 4).
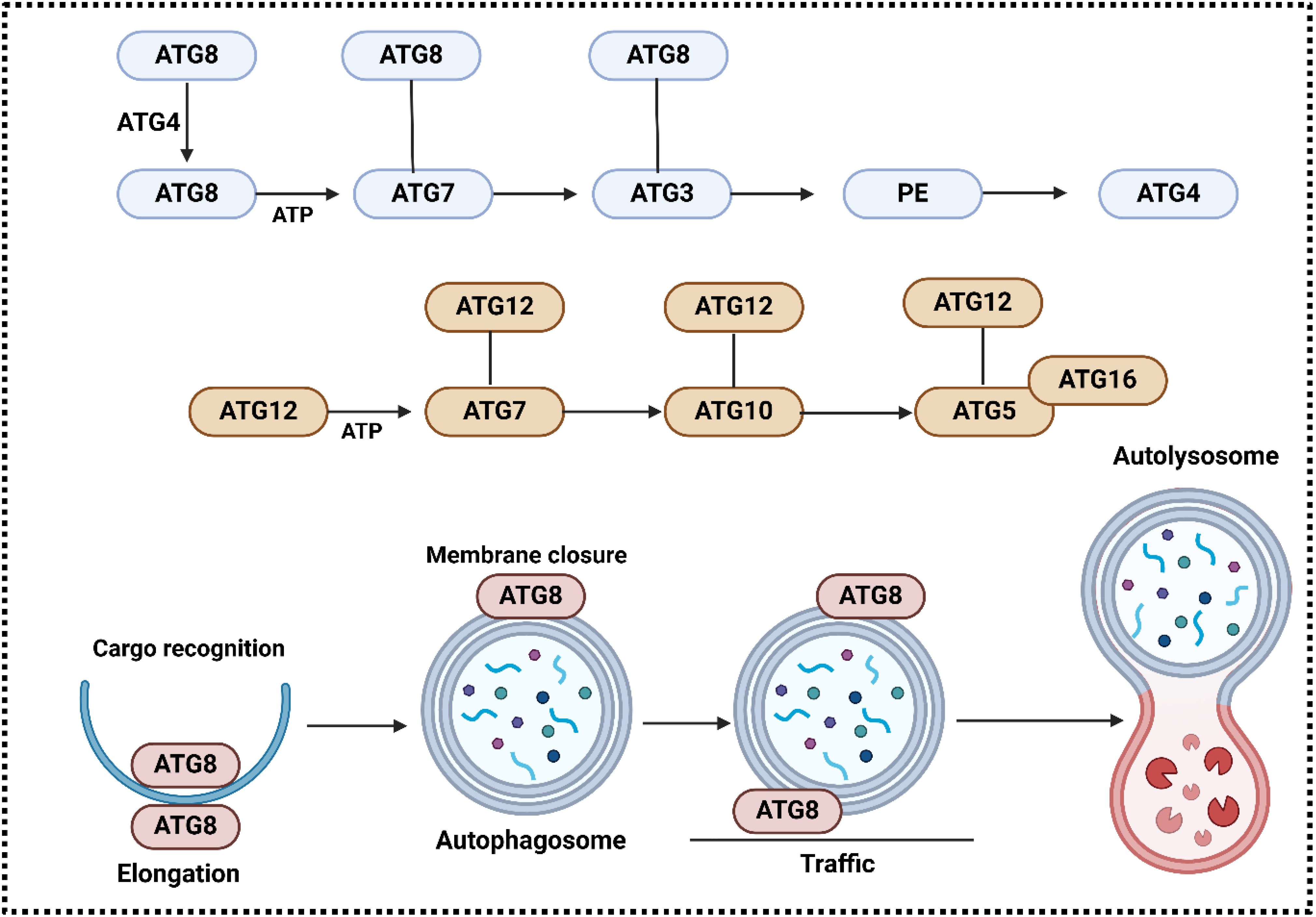
Figure 4. The involvement of ATG system in the regulation of autophagy. ATG7 (E1-like) activates ATG12 in an ATP-dependent way, and a high-energy thioester bond couples its C-terminal glycine to the cysteine of ATG7 active site. Another thioester intermediate is formed when ATG12 is transferred to ATG10 (which is E2-like). An isopeptide bond binds ATG12 to the acceptor lysine in ATG5. Connected to ATG16(L) is ATG5. The ATG12-ATG5-ATG16 complex is formed when ATG16(L) forms a dimer. Following proteolytic processing by ATG4 enzymes, the ATG8 system proceeds to activate the ATG8 protein(s), which in turn produce a thioester with the E2-like enzyme ATG3. In the end, phosphatidylethanolamine (PE) is bound to ATG8. autophagy membranes contain the conjugated ATG8 form, which is recycled by deconjugation by ATG4(s). Conjugation of ATG8 with PE is enhanced by ATG12-ATG5 (red arrow). It is suggested that ATG8 (LC3 and GABARAP) serves various purposes during autophagy, including as elongating the membrane, recognizing cargo, closing the edge, moving autophagosomes, and connecting them to lysosomes (228).
7 Non-autophagy functions of Beclin-1
Current research shows that Beclin-1 plays a role in processes beyond autophagy in addition to its known function as an autophagy effecter (240). Beclin-1 does not exhibit a pro-apoptotic function as it is solely a BH3 protein. It is interesting that Beclin-1’s control over autophagy and apoptosis is given through caspase-mediated cleavage (191). Caspases can cause Beclin-1 to be divided, resulting in cleaved N and C-terminal fragments that are no longer capable of inducing autophagy (241–243). Unlike the N-terminal Beclin-1 fragment containing the BH3 domain, only the C-terminal fragment is able to make cells more sensitive to signals that trigger apoptosis. Beclin-1-C must translocate to the mitochondria surface in order to induce the release of cytochrome c (241). Beclin-1-C enhances Bax translocation to mitochondria, leading to elevated apoptosis and reduced autophagy (244). Beclin-1 controls cell death and self-cannibalization, implying possible treatment benefits. It plays a role in multiple pathways of controlled cell death such as apoptosis, ferroptosis, and necroptosis. Certainly, new functions of Beclin-1 apart from autophagy have just become evident. Phosphorylation of Ser90, Ser93, and Ser96 in Beclin-1 by active AMPK leads to the formation of Beclin-1-SLC7A11 complex, which directly inhibits system Xc− activity and ultimately promotes ferroptosis (245). Another newly discovered, adversarial component of the necrosome complex is Beclin-1.Beclin-1 serves as a blocker of necroptosis by linking with phosphorylated MLKL in the necrosome complex, preventing MLKL from forming oligomers (246). Beclin-1 is involved in ferroptosis and necroptosis, and could be targeted for treatment in associated disorders.
In addition to autophagy, Beclin-1 has been linked to DNA damage responses that do not depend on autophagy, whether they need it or not. Activation of nonhomologous end joining (NHEJ) for DNA double-strand break repair necessitates a decrease in Beclin-1-UVRAG activity to uphold genome stability (247). Nuclear Beclin-1 and DNA topoisomerase IIβ are required for effective repair of DNA double-strand breaks caused by ionizing radiation in both autophagy-independent and autophagy-dependent DNA damage responses (248). Furthermore, Beclin-1 plays a role in tumor spreading, blood vessel formation, and immune system regulation through its actions that are not related to autophagy (249). In models of mouse melanoma tumors, animals with Beclin-1 (+/−) showed a more aggressive tumor growth and increased angiogenic activity, compared to mice with Beclin-1 wild-type, by boosting the protein expression and stability of hypoxia-inducible factor-2α (HIF-2α) (250). Increased levels of Beclin-1 lead to a decrease in angiogenesis in laboratory settings by lowering the production of matrix metalloprotease 9 and vascular endothelial growth factor (251). Beclin-1 also plays a role in regulating the immune system (252, 253). An example is how the lack of Beclin-1 is associated with the abnormal activation of the neutrophil MEKK3/p38 signaling pathway, increased B cell movement through the Cxcl9-Cxcr3 axis, and the conversion of precursor B cells into malignant ones (253).
8 Beclin-1 and ATG interaction in cancer
Beclin-1 and ATGs regulate autophagy and cancer progression. Understanding their interaction may lead to innovative cancer treatments. CUL3 E3 ligase promotes Beclin-1 degradation. This communication not just decreases autophagy function by blocking Beclin-1 but also includes KLHL38 as the adapter for substrates in the ubiquitination process controlled by the CUL3 E3 ligase complex. Moreover, elevated CUL3 levels are linked to a negative outlook in cases of breast and ovarian cancers, indicating its involvement in facilitating the growth of tumor cells in these scenarios (254). Additionally, Beclin-1 and ATGs may serve as predictive indicators in cancer. Out of 90 cancer cases, 55 showed positive Beclin-1 staining (61.1%) and 52 showed positive Atg5 staining (57.8%). Beclin-1 and Atg5 were both expressed in 40 tumors, but not in 23 tumors. Beclin-1 expression was associated with lymph node metastasis and tumor grade, whereas Atg5 expression was associated with tumor grade, clinical stage, tumor size, and lymph node metastasis. Beclin-1 patients showed increased 3-year overall survival (OS) rates and improved time to recurrence (TTR). However, there were no significant differences in survival between groups with positive and negative Atg5. Patients who displayed positive levels of both Beclin-1 and Atg5 had notably improved 3-year OS and TTR compared to those who had negative levels of both (p=0.022 and p=0.026, respectively) (255). Hence, ATGs and Beclin-1 serve as dependable prognostic indicators in cancer (256).
ATGs have the ability to also control Beclin-1 in a meaningful way. In Ca Ski cells, RCE-4 disrupted the formation of the Bcl-2-Beclin 1 complex through various pathways, with ATG 4B proteins playing an important role. It was shown that using an ATG 4B siRNA plasmid led to a significant increase in Ca Ski cell sensitivity by reducing ATG 4B expression, as proven by RCE-4 (257). The regulation of Beclin-1 and ATG2 is essential in the autophagy mechanisms related to cancer treatment. Exposure to cisplatin in A549 cells increased indicators of both autophagy and apoptosis, including conversion of LC3B-I/II, LC3B puncta, and formation of autophagosomes. Cisplatin also raised the mRNA and protein quantities of autophagy proteins Beclin-1 and Atg5. Nonetheless, it did not cause a substantial change in the levels of expression of serine/threonine-protein kinase ULK1, Atg3, Atg7, Atg12, and sequestosome-1. Additionally, silencing Atg5 and Beclin-1 with small interfering RNA (siRNA) led to decreased autophagy reactions to cisplatin, heightened caspase-3 cleavage, and diminished cell viability (89).
Although inhibiting apoptosis by silencing BAX and BAK, obatoclax still induced acute toxicity and loss of clonogenicity through activation of mitochondrial apoptosis via the BCL-2 antagonist killer/BAX pathway. Obatoclax greatly decreased cell viability while also keeping the integrity of the plasma membrane. This therapy also led to a decrease in 6 kinase S phosphorylation, ongoing LC3 autophagy processing, and significant vacuolation of the ultrastructure, which is unusual for autophagy. Unexpectedly, knocking down Beclin-1 did not affect the processing of LC3. Even though the suppression of autophagy-related protein 7 (Atg7) stopped LC3 processing, it did not impact the decline in clonogenicity or structural changes caused by obatoclax. Interestingly, inhibiting Atg7 with siRNA did not stop cell death in BAX/BAK mutant mouse embryonic fibroblasts caused by obatoclax. In line with apoptosis, cells that were not affected by obatoclax exhibited decreased LC3 processing and survival (258).
Blocking autophagy increases how well tumor cells react to cisplatin treatment. Cisplatin-sensitive FaDu cells have the potential to generate cystatin-resistant FaDu cells in a stable manner. In cisplatin-resistant FaDu cells, there is an increased level of expression of autophagy-related genes and proteins, such as Beclin-1, when compared to the control group. Treating the FaDu cells with an autophagy inhibitor after 24 hours led to a significant decrease in cell survival, higher rates of apoptosis, and notable impact on cell cycle, resulting in G1 phase arrest. There was a notable decrease in the quantity of autophagy vacuoles in the group treated with 3-MA. After being treated with 3-MA, p62 expression rose due to the interruption of autophagy flux. Furthermore, there was a notable decrease in the levels of Beclin-1, LC3-I, LC3-II, and Atg-5 proteins (259).
9 Beclin-1 and AMPK crosstalk
The relationship between Beclin-1 and AMPK is influenced by the curcumin analog ZYX01. ZYX01 triggers cell death in A549 cells through autophagy in a way that is influenced by both the dose and timing. ZYX01 treatment resulted in alterations in the LC3-II/LC3-I ratio, Beclin-1 expression, and p62 levels as indicated by Western blot analysis. Exposure to ZYX01 verified the activation of the AMPK/ULK1/Beclin-1 signaling pathway in A549 cells. ZYX01’s effect on A549 cell migration was evaluated through wound healing and transwell tests (260). Allyl Isothiocyanate (AITC) plays a role in controlling Beclin-1 in prostate cancer. AITC increases the amount of LC3-II protein in RV1 and PC3 cells over time, promoting autophagy. AITC does not impact PrEC. Blocking of autophagy in cells treated with AITC resulted in decreased viability and increased apoptosis, indicating a potential protective role of autophagy. Cells were exposed to varying AITC concentrations and autophagy marker levels were assessed at different time intervals. It was discovered that cells exposed to AITC showed activation of numerous pathways. Phosphorylated mTOR, ERK, AMPK, JNK, and p38 were specifically phosphorylated in the cells treated with AITC, with ERK, AMPK, and JNK being activated. AITC did not change the levels of Akt and its associated proteins. Nevertheless, despite prior treatment with targeted inhibitors like rapamycin, LY294002, and PI-103, autophagy induction persisted to rise as AITC triggered the ERK, AMPK, and JNK signaling pathways. AITC also induced an increase in the expression of Beclin-1. The study showed that using its specific siRNA to block Beclin-1 decreases the level of autophagy caused by AITC, suggesting that Beclin-1 is essential for AITC-induced autophagy (261). LETM1 controls both autophagy and apoptosis in liver cancer, with elevated levels found in hepatocellular carcinoma tissue, cell lines, and linked to a negative outlook. Depletion of LETM1 triggers apoptosis, autophagy, and hindrance of growth in liver cancer cell lines. AMPK phosphorylation of Bcl-2 after LETM1 depletion caused disruption in the Beclin-1/Bcl-2 complex (262).
10 Beclin-1 and mTOR interaction in autophagy regulation
In several experiments, the levels of expression for mTOR and Beclin-1 are evaluated separately as well as in relation to their interaction. Xie-Bai-San (XBS) was discovered to regulate both mTOR and Beclin-1. XBS inhibits gefitinib-induced autophagy in NSCLC cells, promoting cell death and hindering growth. It enhances p-mTOR and Bcl-2 levels while decreasing Beclin-1 levels, without affecting autophagosome-lysosome fusion or lysosome activity. XBS improves Bcl-2 and Beclin-1 communication, making it an effective treatment for gefitinib-resistant non-small cell lung cancer (NSCLC). Increased expression of Beclin-1 in NSCLC cells leads to enhanced cell growth and reduces cell death caused by XBS (263). Another element that controls autophagy in liver cancer is Oroxylin A. Oroxylin A was utilized to trigger Beclin 1-facilitated macroautophagy in the HepG2 cell line of human hepatocellular carcinoma. Within 12 hours, LC3-I in cells exposed to 80 μM oroxylin A changes to LC3-II and binds to the autophagosomal membrane, becoming water-insoluble. Nonetheless, the breakdown of autophagosomes by lysosomes/vacuoles is hindered after 24 hours as a result of cell death triggered by oroxylin A. Moreover, oroxylin A substantially inhibits the PI3K-PTEN-Akt-mTOR signaling pathway. Utilizing siBeclin 1 with 3-methyladenine (3-MA), which hinders autophagy, and enhancing autophagy-related genes like Atg5 and Atg7, can aid in establishing if autophagy granule protein (AGP) serves as a PCD mechanism (264).
LTX-315, a polypeptide, controls the Beclin-1/PI3K/mTOR axis to adjust autophagy and drug responsiveness in ovarian cancer. DDP-resistant ovarian cancer cell models were created, and LTX-315 treatment decreased the IC50 of DDP. The use of LTX-315 halted the advancement of cell cycle, raised apoptosis rates, and boosted levels of cleaved caspase-3, cleaved PARP, and Bax in ovarian cancer cells. Additionally, the use of LTX-315 decreased levels of Bcl-2 and impeded cellular movement and penetration. It also raised Beclin-1 levels and changed the levels of phosphorylated Akt (p-Akt) and phosphorylated mTOR (p-mTOR). However, 3-MA managed to partially mitigate the effects of LTX-315 on OC cells (265). It is understood that mTOR plays a role in controlling tumor growth and spread by regulating Beclin-1 (266).
The exact function of Interleukin-7 (IL-7) in controlling tumor cell autophagy, lymphangiogenesis, growth, and cell death is not completely clear; studies are centered on mTOR and Beclin-1. IL-7 was discovered to lower Beclin-1 levels and activate the PI3 K/Akt/mTOR pathway in lung cancer cells. Furthermore, the levels of Beclin-1 and mTOR are closely linked to the clinical stage and outcome of NSCLC patients. In cases of lung cancer, IL-7R, mTOR, and tumor stage are important predictors of prognosis (267).
Prodigiosin (PG), found in gram-negative bacteria, is a strong inducer of cell death and a red pigment that does not dissolve. It has shown effectiveness against lung cancer by inhibiting half of the maximum tumor growth at a dose of 10 μM in cells that are both sensitive and resistant to doxorubicin, suggesting comparable toxic effects. Autophagy was indicated in both cell types due to a slight rise in the sub-G1 phase and an increase in the levels of microtubule-associated proteins 1A/1B light chain 3B-phosphatidylethanolamine conjugate (LC3-II). Moreover, an increase in cleaved-poly ADP ribose polymerase (cleaved-PARP) suggested a subset susceptible to cell death. Impeding the PI3K-p85/Akt/mTOR signaling pathways hindered autophagy initiation in both cell types. Nonetheless, PG-induced autophagy was associated with the decrease of Beclin-1/PI3K-Class III, indicating the triggering of alternative autophagy mechanisms. While the growth of tumors in the animals’ tracheas increased during PG treatment, the therapy effectively decreased their size, validating PG’s effectiveness against both Dox-S and Dox-R lung cancers (92). Piceatannol’s inhibition of Beclin-1 may hinder the advancement of gastric cancer. Piceatannol boosts protein binding of UV radiation resistance-associated genes and supports Beclin-1-dependent autophagy signaling while also interfering with Beclin-1 and Bcl-2 interactions. Everolimus, a type of mTOR inhibitor, stimulates autophagy to enhance the anticancer effects of piceatannol (268).
11 Beclin-1-mediated autophagy regulation in cancer drug resistance and radioresistance
Chemotherapy and radiotherapy are the primary conventional treatments for cancer. These treatment methods are commonly used to control cell death and genetic damage in cancer cells hindering their advancement. Nonetheless, if the tumor cells spread and migrate to different parts of the body, it becomes challenging to completely eliminate all of them, leading to potential tumor relapse and recurrence. Yet, a major obstacle in cancer treatment currently lies in tumor cells’ capacity to become resistant to cancer medications. The resistance frequently occurs due to the dysfunction of molecular pathways and drug efflux transporters, among other reasons, leading to a difficult challenge for researchers (269, 270). The current portion concentrates on Beclin-1-mediated autophagy regulation’s involvement in cancer drug resistance and radioresistance. Hypoxia in glioblastoma (GBM) can lead to drug resistance. It has been shown that HIF1A-triggered autophagy plays a crucial role in the radioresistance of GBM. Suppressing HIF1A increased the radiosensitivity of GBM in both lab settings and living organisms. One possible explanation for this impact is the decrease in Beclin-1 caused by the suppression of HIF1A, which could provide protection for cells. Another option is that the blocking effect of 3-MA is partly influenced by Beclin-1; therefore, boosting Beclin-1 levels could offset the sensitivity caused by 3-MA. Moreover, there could be undiscovered resistance pathways that help cells sustain their radioresistance. Hence, the autophagy controlled by HIF1A and Beclin-1 is essential in the radioresistance seen in GBM (271). miR-216a has also been proven to control Beclin-1, impacting radiosensitivity. MiR-216a levels were noticeably decreased compared to controls, while autophagy activity was elevated. The presence of miR-216a inhibited autophagy and the key autophagy factor Beclin-1 by directly interacting with the 3’-UTR of Beclin-1. Moreover, increased levels of miR-216a in radio-resistant pancreatic cancer cells resulted in improved apoptosis under irradiated conditions, along with reduced cell proliferation and colony formation abilities. Increasing the levels of Beclin-1 reversed the impacts of reducing miR-216a. In general, miR-216a increases xenograft tumors’ responsiveness to radiotherapy and prevents radiation-triggered autophagy by controlling Beclin-1 (272).
Nevertheless, the majority of research has concentrated on the role of Beclin-1-mediated autophagy in chemoresistance. EGFR has the ability to control Beclin-1 in order to inhibit autophagy and manage chemoresistance. When there is an active EGFR signaling pathway, Beclin-1 may be contained, resulting in its tyrosine phosphorylation, increased inhibitory binding, and the interference of its linked VPS34 kinase function. In NSCLC cells with sensitive EGFR mutations, EGFR kinase inhibitors can trigger autophagy by disrupting tyrosine phosphorylation of Beclin-1, leading to enhanced tumor growth and resistance to TKI treatment (273).
Interplay between non-coding RNAs can influence drug sensitivity by regulating Beclin-1. Increased levels of PVT1 were linked to unfavorable outcomes in NSCLC patients (*P < 0.05). Overexpressing PVT1 negatively affected A549 cells, while reducing PVT1 intensified cisplatin’s effects on cell viability and apoptosis in A549/DDP cells. PVT1 upregulation promoted autophagy and tumor growth in NSCLC cells, with miR-216b interacting with PVT1 and Beclin-1. Beclin-1 reversed miR-216b’s impact on autophagy and apoptosis, and PVT1 and miR-216b communication controlled Beclin-1 expression. (*P < 0.05, #P < 0.05) (274).
In human cancers, increasing drug sensitivity is possible by inhibiting protective autophagy. In particular, miR-30a hinders Beclin-1, decreasing autophagy and enhancing the susceptibility of gastrointestinal tumors to imatinib (275). Low miR-30a linked to osteosarcoma drug resistance, upregulating Beclin-1 for increased survival autophagy (276). Research showed that reducing miR-30a-5p levels in lung cancer can increase Beclin-1 expression, causing drug resistance. Similar effects were seen in colon cancer (277). Enhancing sensitivity to oxaliplatin in colon cancer can be achieved by suppressing the Beclin-1/autophagy axis through the restoration of miR-409-3p expression (278).
12 Beclin-1-mediated autophagy regulation and apoptosis
In apoptosis, proteins like caspases 3, 6, and 7 destroy cells by cutting numerous proteins (279, 280). The development of pores in target cells by perforin can initiate apoptosis, along with the release of granzymes from cytotoxic granules of T-cells and Natural Killer (NK) cells (281). DNA in the cell nucleus is usually packed in a dormant state due to the fact that the vast length of DNA would not be able to fit inside the cell if it was completely stretched out. This method consists of coiling DNA in a counter-clockwise direction around protein clusters to create nucleosomes. Nonetheless, somatic topoisomerase 2 enzyme has the ability to interfere with this arrangement by turning the DNA coils in a different direction. This process causes the DNA to resemble a ladder pattern when viewed on an agarose gel (282, 283). This occurrence does not only happen in nuclear apoptosis but also when cells are preparing and experiencing apoptosis. In this procedure, the nuclear membrane forms blebs, necessitating the breaking down of lamins by caspase 6. Moreover, a significant indicator of apoptosis, especially the activation of caspase 3, is the cleavage of PARP, which takes place during cell death (284). Serum M30, a form of cleaved cytokeratin 18 by caspase 3, is detected in patients with tumor response to treatment, signifying cell death (285, 286). Western blotting and the TUNEL assay are frequently utilized methods to identify cleaved caspases and fragmented DNA in tissue samples, showing presence of Serum M30, a caspase 3-cleaved cytokeratin 18, in patients with successful tumor responses. Immunohistochemistry detects granzyme B in cytolytic T cells inducing cell death (287). Flow cytometry is able to detect apoptosis in cultured cells by assessing different indicators like cleaved caspases (288), phosphatidylserine externalization, or sub-G1 DNA content. Furthermore, caspase enzyme functioning and assessments done on cells, along with imaging tests on humans, can also identify caspase activation (289, 290). The identification of apoptosis has made preclinical studies in drug development easier, utilizing PET scans to show markers like Granzyme B (291) as signals of drug effectiveness and impact in clinical specimens. Using standardized terminology for cell death enables the categorization of different forms of cell death and the use of suitable techniques for identifying these mechanisms (5).
Beclin-1 plays a crucial role in linking autophagy and apoptosis in the control of cancer advancement. It acts as a central regulator connecting these pathways. The pairing of bortezomib and mitomycin C mainly led to cell death and cytotoxicity, with little involvement of autophagy, either through additive or synergistic effects. This specific therapy resulted in Akt being inactivated, leading to the dephosphorylation of Beclin-1 at Ser 234/295. Dephosphorylating Beclin-1 inhibited autophagy and caused a pro-apoptotic effect, leading to the cleavage of Beclin-1 and disruption of the R-BiP/Beclin-1/p62 complex. Significantly, this combination greatly diminished autophagy, inactivated Akt, and triggered Beclin-1 cleavage in vivo, leading to a substantial decrease in the growth of intraperitoneal xenografted LS174T tumors in mice. Additionally, in both laboratory and live settings, LS174T xenografted tumors containing a mutated caspase 8 cleavage site of Beclin-1 showed significant resistance to the anti-tumor effects of the combined treatment (292).
Apoptosis caused by radiotherapy (RT) is affected by autophagy mediated by Beclin-1. Both apoptosis and autophagy are mechanisms that RT activates to cause death in thyroid cancer cells. Beclin-1 and LC3, proteins related to autophagy, saw an increase after exposure to radiation. Furthermore, blocking autophagy with 3MA and Beclin-1 siRNA increased radiation-induced cell death and led to higher levels of p53 expression (293).
The relationship between apoptosis and autophagy is significant in prostate tumors, with autophagy-like changes seen in miR-139-treated cancer cells. Although miR-139 antagomir successfully blocked the conversion of LC3-I to LC3-II through autophagy, miR-139 promoted this process. Confocal microscopy data additionally validated the elevated levels of LC3-II. MiR-139 controlled the regulation of two important molecules, mTOR and Beclin-1, in autophagy. After miR-139 treatment, there was an increase in the accumulation of cargo receptor protein p62 which is typically degraded during autophagy (294).
The primary and most effective component in the water extract of Scutellaria baicalensis Georgi’s root, known as Baicalein (BA), can effectively overcome cephalotin resistance. This discovery establishes a theoretical foundation for utilizing BA in clinical settings to combat cephalosporin-resistant strains and endorses its capacity as a promising alternative treatment option. BA was discovered to block cell growth and induce cell death in ovarian cancer cells. BA therapy led to elevated levels of vacuoles stained with acridine orange, puncta of GFP-LC3, and expression of LC3-II. Additionally, both the HEY and A2780 ovarian cancer cell lines showed increased cleavage of poly (ADP-ribose) polymerase (PARP) and decreased cell viability when treated with both chloroquine and BA. This implies that BA may induce cyto-protective autophagy in these cells. Reducing Beclin-1 rates in HT29 cells resulted in a decline in BA-induced LC3-II lipidation. BA treatment led to elevated rates of AKT phosphorylation at Ser473 and extracellular signal-regulated kinase (ERK) phosphorylation at Thr202/Thr204 (295), illustrating an association among autophagy, apoptosis, and the role of Beclin-1 (93, 242, 296–300).
13 Beclin-1-mediated autophagy regulation and ferroptosis
Lipid peroxidation, which is influenced by the amount of iron in cells, is the primary cause of ferroptosis. Regulation of ferroptosis is closely associated with different aspects of iron metabolism, such as iron absorption, transfer, retention, and use. The activation of lipid metabolic pathway enzymes, including LACS4, LPLAT5, LOX, and NOX, also impacts this process by promoting ferroptosis and lipid peroxidation. Furthermore, the cystine-glutamate antiporter (system xc−) is crucial in the classic pathway that prevents ferroptosis by aiding in the synthesis of glutathione (GSH), a necessary cofactor for glutathione peroxidase 4 (GPX4) to convert phospholipid hydroperoxides into alcohols. The system involving ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 (CoQ10) also plays a role in controlling phospholipid peroxidation. Iron metabolism affects ferroptosis by two main routes of non-heme iron intake in cells: uptake of TF-bound iron through TFR1 and uptake of non-TF-bound iron through ZIP14. The Fenton reaction, triggered by haem degradation and ferritinophagy mediated by nuclear receptor coactivator 4 (NCOA4), can result in heightened vulnerability to ferroptosis (301–308).
Beclin-1, an important controller of autophagy, plays a role in connections with ferroptosis and its related pathways, molecules, and proteins (309). Proteins that control various cellular functions, such as autophagy and cell death, have been identified through proteomic studies of the Beclin-1 interactome (191). Erastin triggers the formation of Beclin-1-SLC7A11 complexes, leading to ferroptosis activation. These complexes are said to not be necessary for cell death and do not contribute to ferroptosis triggered by type 2 activators (RSL3 and FIN56) (310). Moreover, by phosphorylating Beclin-1 at Ser90/93/96, PRKAA/AMPKα boosts the assembly of the Beclin-1-SLC7A11 complex, inhibits system xc−, and stimulates the resulting ferroptotic demise of cancer cells (311). Moreover, by phosphorylating Beclin-1 at Ser90/93/96, PRKAA/AMPKα boosts the assembly of the Beclin-1-SLC7A11 complex, inhibits system xc−, and stimulates the resulting ferroptotic demise of cancer cells (311). The Tat-Beclin-1 peptide boosts the erastin reaction in a laboratory setting or within a living organism (311). ELAVL1, present in normal livers, also produces Beclin-1 in stellate cells leading to an elevated chance of ferroptosis (312). This information backs the claim that Beclin-1 directly inhibits system xc− to facilitate the beginning of ferroptosis. Still unknown whether OTUB1 affects the stability of SLC7A11 by influencing the interaction between Beclin-1 and SLC7A11 (313).
Nanoparticles have been designed to regulate this process since the finding of autophagy-induced ferroptosis in treating cancer. Ultrasmall iron oxide nanostructures have been created with excellent water solubility to induce ferroptosis in glioblastoma. These nanoparticles also decrease the levels of factors that protect against ferroptosis.
Suppression of autophagy reduces the ability of these nanoparticles to induce ferroptosis, indicating a potential interaction between autophagy and ferroptosis. Noticeably, tiny iron oxide structures enhance Beclin-1 production, leading to increased ATG5 levels, promoting autophagy-triggered ferroptosis in the treatment of glioblastoma (Table 3) (310).

Table 3. Beclin-1-mediated autophagy regulation in cancer.
14 Beclin-1 modulators in cancer therapy
Quercetin suppressed HeLa cell proliferation and triggered autophagy in a manner that was dependent on the dosage used (319). It decreased cell growth, elevated Beclin-1 and LC3-I/II levels, and lowered S6K1 phosphorylation levels (320). Examination of omics data in LGG discovered that ZFP36L2 and RAB13 are involved in regulating autophagy by enhancing Beclin-1 and other crucial autophagy elements. The presence of Gallic acid, which can block RAB13, has been shown to reduce autophagy and induce cell death in SW1088 cells (62). The collaboration of nobiletin and vorinostat in small cell lung cancer (SCLC) causes autophagy and apoptosis by interfering with the BCL-2 and Beclin-1 complex, releasing Beclin-1 to initiate autophagy and inhibiting PI3K-AKT-mTOR signaling, suggesting nobiletin’s potential as a BH3 mimetic for SCLC combination treatment (321). Resveratrol increases Beclin-1, LC3-II, and p53 expression in A549 NSCLC cells, inducing apoptosis and autophagy, reducing cell survival via p53 pathway activation. Autophagy inhibition partly reverses this effect (322). Resveratrol alleviates doxorubicin-induced heart damage by inhibiting S6K1 and reducing autophagy. Beclin-1 overexpression counteracts Resveratrol’s protective effects (323). Resveratrol induces apoptosis in MCF-7 breast cancer cells through caspase-dependent and -independent pathways. This unconventional type of autophagy cell death is significant in caspase-3-deficient cells (324). Resveratrol induces cell death in MCF-7 breast cancer cells through caspase-dependent and caspase-independent pathways, triggering macroautophagy independently of Beclin 1. This atypical form of autophagic cell death is especially important in cells that do not have caspase-3 (325). Resveratrol induces a defensive autophagy mechanism that boosts cancer cell demise by raising caspase activation and cell death in the absence of autophagy; this mechanism includes Beclin-1 binding with p53 and results in impaired mitochondrial function marked by decreased mtDNA content (326). Resveratrol enhances cell death in ovarian cancer cells by blocking STAT3 signaling, leading to higher autophagosome production and elimination of mitochondria, ultimately leading to inhibition of growth and cell death (327). Resveratrol prevents the spread of breast cancer and its growth in other areas by reversing TGF-β1-induced EMT and boosting autophagy through the SIRT3/AMPK pathway (328). Therefore, increasing research indicates the significance of pharmacological compounds in modulating Beclin-1 in human cancer and overseeing autophagy (153, 260, 329–332). Figure 5 provides a comprehensive view of Beclin-1’s regulation of autophagy in human cancer.
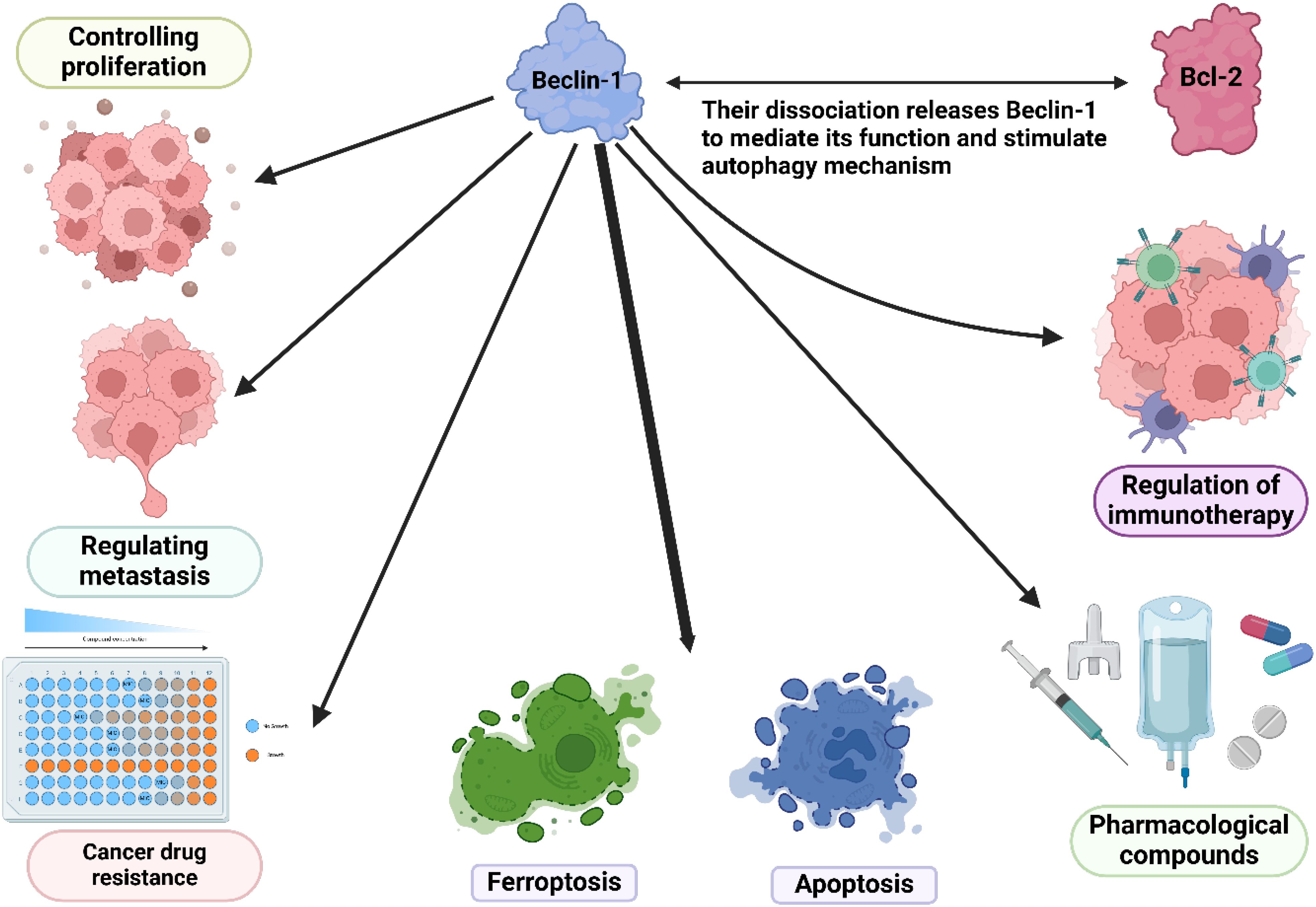
Figure 5. The overall function of Beclin-1 in the regulation of cancer hallmarks and cell death mechanisms.
15 The multifaceted role of Beclin-1 in tumor immunology and drug sensitivity
Beclin-1 is a key regulatory factor in the autophagy process, and research advancements regarding its role in the tumor immune microenvironment, immunotherapy, and drug sensitivity indicate its potential application value in cancer treatment (333, 334). In the tumor immune microenvironment, Beclin-1 influences the interaction between tumor cells and immune cells by regulating autophagy. Specifically, the expression of Beclin-1 can alter the polarization state of tumor-associated macrophages (TAMs), enhancing their immunosuppressive function and thus promoting tumor development (252, 335). Additionally, Beclin-1 affects the antigen presentation capability of tumor cells, altering how tumor cells are recognized and attacked by the immune system, which allows tumor cells to partially evade immune surveillance. Regarding immunotherapy, studies have found that Beclin-1 may enhance the efficacy of immune checkpoint inhibitors, making it a potential target for combination therapy. By combining Beclin-1 modulators with immunotherapeutic agents, it is possible to significantly improve anti-tumor immune responses (336). Furthermore, the expression level of Beclin-1 may serve as a biomarker for evaluating patient responses to immunotherapy, aiding clinicians in developing more targeted personalized treatment plans (337). In terms of drug sensitivity, Beclin-1 also plays an important role in the sensitivity of tumor cells to chemotherapy. Research indicates that the regulation of autophagy is closely linked to the survival and death of tumor cells, with the overexpression of Beclin-1 being closely associated with chemotherapy resistance (336, 338). Therefore, based on the function of Beclin-1, researchers are exploring drugs that target autophagy regulation to enhance the sensitivity of tumor cells to chemotherapy and targeted therapy. Overall, the complex roles of Beclin-1 in the tumor immune microenvironment, immunotherapy, and drug sensitivity make it an important focus of cancer treatment research. Future studies need to further investigate the specific mechanisms of Beclin-1 to develop more effective therapeutic strategies, particularly in personalized and combination therapies, which will help improve the survival rates and quality of life for cancer patients.
16 Conclusion
Autophagy is one of the most frequently disrupted processes in cancer. Autophagy dysregulation has been noted in both solid and hematological cancers. Additionally, autophagy is a versatile process that controls different characteristics of cancer. Autophagy in clinical trials led to new cancer drugs. It plays roles in both cell survival and death. Hence, dual blocking and stimulation of autophagy are significant. Even though several autophagy regulators like mTOR, ULK1, and AMPK have been identified, Beclin-1 is considered a crucial controller of autophagy in cancer. The present study shows that controlling autophagy through Beclin-1 can impact cell growth, resistance to drugs, and response to radiotherapy. In addition, Beclin-1 regulates autophagy in human cancers by interacting with ATGs, mTOR, and AMPK. Hence, manipulating Beclin-1 can impact regulators both before and after it in the signaling pathway. Additionally, there have been reports stating that Beclin-1 controls autophagy, which can potentially affect apoptosis and ferroptosis in human cancer. Hence, purposeful adjustment of Beclin-1 and autophagy could greatly increase cell mortality in human cancers. This evaluation can be viewed from various angles. In terms of fundamental research, the dysregulation of Beclin-1 has been extensively documented in numerous studies. Nevertheless, Beclin-1 poses difficulties because of its dual function. While its role in cancer is intricate, Beclin-1 is crucial clinically, as its levels can impact how cancer patients respond to chemotherapy. Furthermore, Beclin-1 has the potential to influence survival, prognosis, growth, and tumor recurrence. Hence, regulating the Beclin-1 expression can result in the creation of innovative treatments. Beclin-1 plays a crucial role in regulating autophagy, impacting immunogenic cell death, necroptosis, and ferroptosis. This is important for timely detection and outcome prediction in cancer patients, with potential implications for cancer treatment (21, 339–343). Hence, it is recommended that upcoming research assess the relationship between the Beclin-1/autophagy axis and immunogenic cell death in treating human cancers. Moreover, addressing cancer is greatly challenged by drug resistance and radioresistance (344–347). Thus, the utilization of immunotherapy and checkpoint inhibitors has risen for the removal of tumors (348–352). Current research studies are limited because they fail to consider the impact of the Beclin-1/autophagy axis on immune interactions and how it affects cancer cells’ reactions to immune checkpoint inhibitors. Autophagy offers significant clinical insights in addition to Beclin-1’s basic and clinical relevance. Autophagy genes as well as non-coding RNAs play a crucial role in evaluating cancer diagnosis and treatment.
Beclin-1 is essential for regulating various cellular processes like autophagy, apoptosis, and ferroptosis. Beclin-1, an essential initiator of autophagy, interacts with VPS34, a type III phosphatidylinositol 3-kinase, to start the process of autophagy vesicle formation. A chemical creates PI3P, drawing in autophagy proteins to create phagophores, which then transform into autophagosomes. Beclin-1 aids in autophagosome development and breakdown, merging them with lysosomes for material degradation. Beclin-1 interactions with ATG proteins and LC3 enhance autophagy, emphasizing its role in cellular balance. When it comes to apoptosis, Beclin-1 shows a complicated connection and can often function as a pro-apoptotic factor in specific situations. Beclin-1 impacts programmed cell death by interacting with caspases, like regulating caspase-3, crucial for cell death. It binds to Bcl-2 proteins, trapping anti-apoptotic ones and inducing apoptosis. Also, it affects mitochondria function, releasing pro-apoptotic factors, triggering apoptosis. Beclin-1 can determine cell fate through supporting survival via autophagy or aiding apoptosis under stress. It also regulates ferroptosis, dependent on iron and lipid peroxides. Autophagy breaks down damaged lipids, preventing lipid peroxidation and reducing ferroptotic cell death. Furthermore, Beclin-1 can help regulate the levels of reactive oxygen species (ROS) by promoting the breakdown of impaired organelles, consequently reducing the oxidative stress linked to ferroptosis. Beclin-1’s interactions with GPX4 influence cell susceptibility to ferroptosis. Beclin-1, a flexible protein, controls autophagy, apoptosis, and ferroptosis processes, making it a possible treatment focus in illnesses. Utilizing Beclin-1 in treatments shows promise but requires caution due to potential challenges. Beclin-1 regulates autophagy and is vital for cellular balance, stress response, and cell death. Enhancing autophagy by improving Beclin-1 function is being studied to overcome blockages. Therapies targeting Beclin-1 levels can aid in removing harmful substances and supporting cell survival by restoring autophagy. However, Beclin-1’s involvement in autophagy and apoptosis complicates its therapeutic use. While increasing autophagy with Beclin-1 can help in some cases, it might promote cancer growth by supporting cell survival. Changing Beclin-1 levels could disrupt the balance between autophagy and apoptosis, leading to issues like increased cell death or reduced response to chemotherapy. Using Beclin-1 modulators may also trigger inflammatory responses, complicating treatment. Disruption of Beclin-1 can alter cytokine production and immune cell function, impacting inflammatory conditions and infection defense. Understanding Beclin-1’s roles and potential side effects is crucial for developing treatments across diseases. Future research should identify patient groups benefiting from Beclin-1 adjustments and develop targeted delivery methods to enhance treatment efficacy and minimize side effects. Further investigation is essential to understand Beclin-1’s intricate role in cancer by studying its interactions with pathways such as autophagy. Autophagy is interconnected with signaling pathways controlling cell survival, growth, and death, like the PI3K pathway crucial for autophagy initiation. Disrupted PI3K pathway functioning can impact autophagy levels, influencing cancer development and treatment response. Beclin-1’s interaction with the Bcl-2 protein family highlights the balance between cell survival and death. Understanding how Beclin-1 integrates signals from these pathways can uncover how cancer cells react to stress and treatment evasion. Cancer cells change metabolism to survive, possibly through Beclin-1 affecting metabolic reprogramming by regulating autophagy, which links to glucose and oxidative stress. Research must also focus on autophagy’s connection with immune signaling pathways, impacting tumor immunogenicity and response to immunotherapies. Fully grasping Beclin-1’s involvement in cancer requires exploring its links to cellular pathways for new treatment strategies. Future studies should elucidate these relationships for advancements in cancer treatment and patient outcomes.
Author contributions
ZC: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. KT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. YR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. HZ: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. Z’MC: Conceptualization, Investigation, Methodology, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. YD: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. XT: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. LZ: Conceptualization, Investigation, Data curation, Software, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Figures have been created with Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ULK1, Unc-51 Like Autophagy Activating Kinase 1; mTOR, Mammalian Target of Rapamycin; AMPK, AMP-Activated Protein Kinase; Autophagy Related; ATG, Autophagy Related Gene; PIK3C3, Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3; VPS34, Vacuolar Protein Sorting 34; AMBRA1, Autophagy and Beclin 1 Regulator 1; ATG14, Autophagy Related 14; ATG16L1, Autophagy Related 16 Like 1; LC3, Microtubule Associated Protein 1 Light Chain 3; GABARAP, GABA Type A Receptor Associated Protein; ATG12, Autophagy Related 12; ATG5, Autophagy Related 5; ATG7, Autophagy Related 7; ATG3, Autophagy Related 3; ATG4, Autophagy Related 4; WIPI, WD Repeat Domain, Phosphoinositide Interacting; DFCP1, Zinc Finger; FYVE-Type Containing 1.
References
1. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery. (2016) 15:348–66. doi: 10.1038/nrd.2015.6
2. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
3. Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. (2015) 16:329–44. doi: 10.1038/nrm3999
4. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
5. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death differentiation. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
6. Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G. Regulated cell death and adaptive stress responses. Cell Mol Life sciences: CMLS. (2016) 73:2405–10. doi: 10.1007/s00018-016-2209-y
7. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. (2011) 147:742–58. doi: 10.1016/j.cell.2011.10.033
8. Galluzzi L, López-Soto A, Kumar S, Kroemer G. Caspases connect cell-death signaling to organismal homeostasis. Immunity. (2016) 44:221–31. doi: 10.1016/j.immuni.2016.01.020
9. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. (2017) 17:151–64. doi: 10.1038/nri.2016.147
10. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. (2017) 17:333–40. doi: 10.1038/nri.2016.153
11. Cornillon S, Foa C, Davoust J, Buonavista N, Gross JD, Golstein P. Programmed cell death in Dictyostelium. J Cell Sci. (1994) 107:2691–704. doi: 10.1242/jcs.107.10.2691
12. Olie RA, Durrieu F, Cornillon S, Loughran G, Gross J, Earnshaw WC, et al. Apparent caspase independence of programmed cell death in Dictyostelium. Curr biology: CB. (1998) 8:955–8. doi: 10.1016/S0960-9822(98)70395-1
13. Cornillon S, Olie RA, Golstein P. An insertional mutagenesis approach to Dictyostelium cell death. Cell Death differentiation. (1998) 5:416–25. doi: 10.1038/sj.cdd.4400361
14. Madeo F, Fröhlich E, Fröhlich KU. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. (1997) 139:729–34. doi: 10.1083/jcb.139.3.729
15. Eisenberg T, Büttner S, Kroemer G, Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis: an Int J programmed Cell Death. (2007) 12:1011–23. doi: 10.1007/s10495-007-0758-0
16. Büttner S, Eisenberg T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol. (2006) 175:521–5. doi: 10.1083/jcb.200608098
17. Green DR, Fitzgerald P. Just so stories about the evolution of apoptosis. Curr biology: CB. (2016) 26:R620–r627. doi: 10.1016/j.cub.2016.05.023
18. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death differentiation. (2015) 22:58–73. doi: 10.1038/cdd.2014.137
19. Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. (2009) 43:493–523. doi: 10.1146/annurev.genet.42.110807.091533
20. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. (2017) 17:363–75. doi: 10.1038/nri.2017.21
21. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. (2012) 12:860–75. doi: 10.1038/nrc3380
22. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. (2012) 13:780–8. doi: 10.1038/nrm3479
23. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Sci (New York NY). (2010) 330:362–6. doi: 10.1126/science.1195491
24. Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. (1973) 7:253–66. doi: 10.1002/tera.1420070306
25. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death differentiation. (2007) 14:1237–43. doi: 10.1038/sj.cdd.4402148
26. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death differentiation. (2005) 12 Suppl 2:1463–7. doi: 10.1038/sj.cdd.4401724
27. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death differentiation. (2009) 16:3–11. doi: 10.1038/cdd.2008.150
28. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death differentiation. (2012) 19:107–20. doi: 10.1038/cdd.2011.96
29. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death differentiation. (2009) 16:1093–107. doi: 10.1038/cdd.2009.44
30. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell proliferation. (2012) 45:487–98. doi: 10.1111/j.1365-2184.2012.00845.x
31. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
32. Laubenbacher R, Hower V, Jarrah A, Torti SV, Shulaev V, Mendes P, et al. A systems biology view of cancer. Biochim Biophys Acta. (2009) 1796:129–39. doi: 10.1016/j.bbcan.2009.06.001
33. Tan ML, Ooi JP, Ismail N, Moad AI, Muhammad TS. Programmed cell death pathways and current antitumor targets. Pharm Res. (2009) 26:1547–60. doi: 10.1007/s11095-009-9895-1
34. Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. (2010) 35:556–64. doi: 10.1016/j.tibs.2010.04.008
35. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
36. Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. (2008) 103:343–51. doi: 10.1161/CIRCRESAHA.108.175448
37. Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. (1995) 82:349–52. doi: 10.1016/0092-8674(95)90422-0
38. Cohen GM, Sun XM, Fearnhead H, MacFarlane M, Brown DG, Snowden RT, et al. Formation of large molecular weight fragments of DNA is a key committed step of apoptosis in thymocytes. J Immunol (Baltimore Md: 1950). (1994) 153:507–16. doi: 10.4049/jimmunol.153.2.507
39. Liu JJ, Lin M, Yu JY, Liu B, Bao JK. Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett. (2011) 300:105–14. doi: 10.1016/j.canlet.2010.10.001
40. Huett A, Goel G, Xavier RJ. A systems biology viewpoint on autophagy in health and disease. Curr Opin Gastroenterol. (2010) 26:302–9. doi: 10.1097/MOG.0b013e32833ae2ed
41. Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta. (2010) 1806:220–9. doi: 10.1016/j.bbcan.2010.07.003
42. Li ZY, Yang Y, Ming M, Liu B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem Biophys Res Commun. (2011) 414:5–8. doi: 10.1016/j.bbrc.2011.09.046
43. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
44. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death differentiation. (2009) 16:966–75. doi: 10.1038/cdd.2009.33
45. Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta pharmacologica Sin. (2010) 31:1154–64. doi: 10.1038/aps.2010.118
46. Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev oncology/hematology. (2012) 82:249–58. doi: 10.1016/j.critrevonc.2011.08.004
47. McCall K. Genetic control of necrosis - another type of programmed cell death. Curr Opin Cell Biol. (2010) 22:882–8. doi: 10.1016/j.ceb.2010.09.002
48. Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. (2001) 2:589–98. doi: 10.1038/35085008
49. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduction Targeted Ther. (2022) 7:286. doi: 10.1038/s41392-022-01110-y
50. Fairlie WD, Tran S, Lee EF. Crosstalk between apoptosis and autophagy signaling pathways. Int Rev Cell Mol Biol. (2020) 352:115–58. doi: 10.1016/bs.ircmb.2020.01.003
51. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
52. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
53. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging. (2016) 8:603–19. doi: 10.18632/aging.100934
54. Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun. (2017) 8:16078. doi: 10.1038/ncomms16078
55. Derakhshan A, Chen Z, Van Waes C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer research: an Off J Am Assoc Cancer Res. (2017) 23:1379–87. doi: 10.1158/1078-0432.CCR-16-2172
56. Xiang H, Zhang J, Lin C, Zhang L, Liu B, Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. (2020) 10:569–81. doi: 10.1016/j.apsb.2019.10.003
57. Cui J, Shen H-M, Lim LHK. The role of autophagy in liver cancer: crosstalk in signaling pathways and potential therapeutic targets. Pharmaceuticals. (2020) 13:432. doi: 10.3390/ph13120432
58. Mokarram P, Albokashy M, Zarghooni M, Moosavi MA, Sepehri Z, Chen QM, et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy. (2017) 13:781–819. doi: 10.1080/15548627.2017.1290751
59. Zhang J, Wang G, Zhou Y, Chen Y, Ouyang L, Liu B. Mechanisms of autophagy and relevant small-molecule compounds for targeted cancer therapy. Cell Mol Life sciences: CMLS. (2018) 75:1803–26. doi: 10.1007/s00018-018-2759-2
60. Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. (2017) 10:67. doi: 10.1186/s13045-017-0436-9
61. Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol. (2017) 232:2977–84. doi: 10.1002/jcp.v232.11
62. Su W, Liao M, Tan H, Chen Y, Zhao R, Jin W, et al. Identification of autophagic target RAB13 with small-molecule inhibitor in low-grade glioma via integrated multi-omics approaches coupled with virtual screening of traditional Chinese medicine databases. Cell Prolif. (2021) 54:e13135. doi: 10.1111/cpr.v54.12
63. Guo Z, Guozhang H, Wang H, Li Z, Liu N. Ampelopsin inhibits human glioma through inducing apoptosis and autophagy dependent on ROS generation and JNK pathway. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2019) 116:108524. doi: 10.1016/j.biopha.2018.12.136
64. Sethi A, Sanam S, Alvala R, Alvala M. An updated patent review of galectin-1 and galectin-3 inhibitors and their potential therapeutic applications (2016-present). Expert Opin Ther patents. (2021) 31:709–21. doi: 10.1080/13543776.2021.1903430
65. Liu L, Li S, Qu Y, Bai H, Pan X, Wang J, et al. Ablation of ERO1A induces lethal endoplasmic reticulum stress responses and immunogenic cell death to activate anti-tumor immunity. Cell Rep Med. (2023) 4:101206. doi: 10.1016/j.xcrm.2023.101206
66. Lei J, Zhou Z, Fang J, Sun Z, He M, He B, et al. Aspirin induces immunogenic cell death and enhances cancer immunotherapy in colorectal cancer. Int Immunopharmacol. (2023) 121:110350. doi: 10.1016/j.intimp.2023.110350
67. Zhou C, Yang ZF, Sun BY, Yi Y, Wang Z, Zhou J, et al. Lenvatinib induces immunogenic cell death and triggers toll-Like receptor-3/4 ligands in hepatocellular carcinoma. J hepatocellular carcinoma. (2023) 10:697–712. doi: 10.2147/JHC.S401639
68. Xia L, Xu X, Li M, Zhang X, Cao F. Afzelin induces immunogenic cell death against lung cancer by targeting NQO2. BMC complementary Med therapies. (2023) 23:381. doi: 10.1186/s12906-023-04221-3
69. Yuan Y, Guo Y, Guo ZW, Hao HF, Jiao YN, Deng XX, et al. Marsdenia tenacissima extract induces endoplasmic reticulum stress-associated immunogenic cell death in non-small cell lung cancer cells through targeting AXL. J ethnopharmacology. (2023) 314:116620. doi: 10.1016/j.jep.2023.116620
70. Zhao Y, Ma R, Wang C, Hu R, Wu W, Sun X, et al. CAPG interference induces apoptosis and ferroptosis in colorectal cancer cells through the P53 pathway. Mol Cell Probes. (2023) 71:101919. doi: 10.1016/j.mcp.2023.101919
71. Lee DE, Lee GY, Lee HM, Choi SY, Lee SJ, Kwon OS. Synergistic apoptosis by combination of metformin and an O-GlcNAcylation inhibitor in colon cancer cells. Cancer Cell Int. (2023) 23:108. doi: 10.1186/s12935-023-02954-2
72. Hong K, Yang Q, Yin H, Zhang J, Yu B. SDR16C5 promotes proliferation and migration and inhibits apoptosis in pancreatic cancer. Open Life Sci. (2023) 18:20220630. doi: 10.1515/biol-2022-0630
73. Zhou R, Konishi Y, Zhang A, Nishiwaki K. Propofol elicits apoptosis and attenuates cell growth in esophageal cancer cell lines. Nagoya J Med Sci. (2023) 85:579–91. doi: 10.18999/nagjms.85.3.579
74. Fu L, Zhao L, Liao C, Wang P, Gu Y, Li S, et al. Knockdown of KAT5/KIF11 induces autophagy and promotes apoptosis in anaplastic thyroid cancer cells. Exp Ther Med. (2023) 25:247. doi: 10.3892/etm.2023.11946
75. Teng Y, Li Z, Liu J, Teng L, Li H. Proliferation inhibition and apoptosis of liver cancer cells treated by blue light irradiation. Med Oncol (Northwood London England). (2023) 40:227. doi: 10.1007/s12032-023-02096-5
76. Wang Z, Li H, Cai H, Liang J, Jiang Y, Song F, et al. FTO sensitizes oral squamous cell carcinoma to ferroptosis via suppressing ACSL3 and GPX4. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms242216339
77. Wu J, Liu C, Wang T, Liu H, Wei B. Deubiquitinase inhibitor PR-619 potentiates colon cancer immunotherapy by inducing ferroptosis. Immunology. (2023) 170:439–51. doi: 10.1111/imm.v170.3
78. Xiao Y, Xu Z, Cheng Y, Huang R, Xie Y, Tsai HI, et al. Fe(3+)-binding transferrin nanovesicles encapsulating sorafenib induce ferroptosis in hepatocellular carcinoma. Biomaterials Res. (2023) 27:63. doi: 10.1186/s40824-023-00401-x
79. Zhang J, Ren G, Huang T, Sang Y, Zhong Y, Yi Y. miRNA-363-3p hinders proliferation, migration, invasion and autophagy of thyroid cancer cells by controlling SYT1 transcription to affect NF-κB. Endocrine Metab Immune Disord Drug Targets. (2024) 24:153–62. doi: 10.2174/1871530323666230504112553
80. Gao Z, Zhang Y, Shen W, Liu X, Wei Y, Li L, et al. Bruceine A inhibited breast cancer proliferation and metastasis by inducing autophagy via targeting PI3K-AKT signaling pathway. Chem Biol Drug design. (2024) 103:e14398. doi: 10.1111/cbdd.v103.1
81. Kong F, Zhang L, Zhao X, Zhao L, Wang P, Zhang R, et al. Resveratrol augments paclitaxel sensitivity by modulating miR-671-5p/STOML2/PINK1/Parkin-mediated autophagy signaling in A549 cell. J Biochem Mol Toxicol. (2024) 38:e23557. doi: 10.1002/jbt.23557
82. Hsin IL, Wu PJ, Tang SC, Ou CC, Chang HY, Shen HP, et al. [amp]]beta;-catenin inhibitor ICG-001 suppress cell cycle progression and induce autophagy in endometrial cancer cells. J Cell Physiol. (2023) 238:2440–50. doi: 10.1002/jcp.v238.10
83. Zhu F, Zhang Q, Feng J, Zhang X, Li T, Liu S, et al. [amp]]beta;-Glucan produced by Lentinus edodes suppresses breast cancer progression via the inhibition of macrophage M2 polarization by integrating autophagy and inflammatory signals. Immunity Inflammation Dis. (2023) 11:e876. doi: 10.1002/iid3.876
84. Xiao W, Dai B, Zhu Y, Ye D. Norcantharidin induces autophagy-related prostate cancer cell death through Beclin-1 upregulation by miR-129-5p suppression. Tumour biology: J Int Soc Oncodevelopmental Biol Med. (2015). doi: 10.1007/s13277-015-4488-6
85. Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. (2013) 4:e485. doi: 10.1038/cddis.2013.18
86. Sun PH, Zhu LM, Qiao MM, Zhang YP, Jiang SH, Wu YL, et al. The XAF1 tumor suppressor induces autophagic cell death via upregulation of Beclin-1 and inhibition of Akt pathway. Cancer Lett. (2011) 310:170–80. doi: 10.1158/1538-7445.AM2011-170
87. Athamneh K, Hasasna HE, Samri HA, Attoub S, Arafat K, Benhalilou N, et al. Rhus coriaria increases protein ubiquitination, proteasomal degradation and triggers non-canonical Beclin-1-independent autophagy and apoptotic cell death in colon cancer cells. Sci Rep. (2017) 7:11633. doi: 10.1038/s41598-017-11202-3
88. Qiu Y, Li C, Wang Q, Zeng X, Ji P. Tanshinone IIA induces cell death via Beclin-1-dependent autophagy in oral squamous cell carcinoma SCC-9 cell line. Cancer Med. (2018) 7:397–407. doi: 10.1002/cam4.2018.7.issue-2
89. Chen J, Zhang L, Zhou H, Wang W, Luo Y, Yang H, et al. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol Med Rep. (2018) 17:6859–65. doi: 10.3892/mmr.2018.8686
90. Sun L, Liu N, Liu SS, Xia WY, Liu MY, Li LF, et al. Beclin-1-independent autophagy mediates programmed cancer cell death through interplays with endoplasmic reticulum and/or mitochondria in colbat chloride-induced hypoxia. Am J Cancer Res. (2015) 5:2626–42.
91. Zeng C, Zhang Z, Luo W, Wang L, Zhou H, Nie C. JNK initiates Beclin-1 dependent autophagic cell death against Akt activation. Exp Cell Res. (2022) 414:113105. doi: 10.1016/j.yexcr.2022.113105
92. Chiu WJ, Lin SR, Chen YH, Tsai MJ, Leong MK, Weng CF. Prodigiosin-emerged PI3K/beclin-1-independent pathway elicits autophagic cell death in doxorubicin-sensitive and -resistant lung cancer. J Clin Med. (2018) 7. doi: 10.3390/jcm7100321
93. Kim SY, Song X, Zhang L, Bartlett DL, Lee YJ. Role of Bcl-xL/Beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death. Biochem Pharmacol. (2014) 88:178–88. doi: 10.1016/j.bcp.2014.01.027
94. Filali-Mouncef Y, Hunter C, Roccio F, Zagkou S, Dupont N, Primard C, et al. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy. (2022) 18:50–72. doi: 10.1080/15548627.2021.1895658
95. Wu N, Zheng W, Zhou Y, Tian Y, Tang M, Feng X, et al. Autophagy in aging-related diseases and cancer: Principles, regulatory mechanisms and therapeutic potential. Ageing Res Rev. (2024) 100:102428. doi: 10.1016/j.arr.2024.102428
96. Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S. Autophagy regulation using luteolin: new insight into its anti-tumor activity. Cancer Cell Int. (2020) 20:537. doi: 10.1186/s12935-020-01634-9
97. Qin Y, Ashrafizadeh M, Mongiardini V, Grimaldi B, Crea F, Rietdorf K, et al. Autophagy and cancer drug resistance in dialogue: Pre-clinical and clinical evidence. Cancer Lett. (2023) 570:216307. doi: 10.1016/j.canlet.2023.216307
98. Ashrafizadeh M, Zhang W, Zou R, Sethi G, Klionsky DJ, Zhang X. A bioinformatics analysis, pre-clinical and clinical conception of autophagy in pancreatic cancer: Complexity and simplicity in crosstalk. Pharmacol Res. (2023) 194:106822. doi: 10.1016/j.phrs.2023.106822
99. Ashrafizadeh M, Paskeh MDA, Mirzaei S, Gholami MH, Zarrabi A, Hashemi F, et al. Targeting autophagy in prostate cancer: preclinical and clinical evidence for therapeutic response. J Exp Clin Cancer Res. (2022) 41:105. doi: 10.1186/s13046-022-02293-6
100. Paskeh MDA, Entezari M, Clark C, Zabolian A, Ranjbar E, Farahani MV, et al. Targeted regulation of autophagy using nanoparticles: New insight into cancer therapy. Biochim Biophys Acta (BBA) - Mol Basis Dis. (2022) 1868:166326. doi: 10.1016/j.bbadis.2021.166326
101. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol. (2021) 17:647–61. doi: 10.1038/s41574-021-00551-9
102. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
103. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. (2018) 14:221–32. doi: 10.1080/15548627.2017.1389823
104. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol. (2020) 16:489–508. doi: 10.1038/s41581-020-0309-2
105. Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. (2017) 42:28–41. doi: 10.1016/j.tibs.2016.09.008
106. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. (2018) 14:207–15. doi: 10.1080/15548627.2017.1378838
107. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
108. Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. (2014) 21:336–45. doi: 10.1038/nsmb.2787
109. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. (1993) 333:169–74. doi: 10.1016/0014-5793(93)80398-E
110. Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. (1994) 349:275–80. doi: 10.1016/0014-5793(94)00672-5
111. Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. (1995) 131:591–602. doi: 10.1083/jcb.131.3.591
112. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. (2009) 20:1981–91. doi: 10.1091/mbc.e08-12-1248
113. Nazio F, Cecconi F. mTOR, AMBRA1, and autophagy: an intricate relationship. Cell Cycle (Georgetown Tex). (2013) 12:2524–5. doi: 10.4161/cc.25835
114. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. (2009) 20:1992–2003. doi: 10.1091/mbc.e08-12-1249
115. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. (2008) 182:685–701. doi: 10.1083/jcb.200803137
116. Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. (2013) 55:17–27. doi: 10.1042/bse0550017
117. Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem. (2018) 293:5386–95. doi: 10.1074/jbc.R117.810366
118. Bakula D, Müller AJ, Zuleger T, Takacs Z, Franz-Wachtel M, Thost AK, et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. (2017) 8:15637. doi: 10.1038/ncomms15637
119. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. (2014) 55:238–52. doi: 10.1016/j.molcel.2014.05.021
120. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. (2010) 466:68–76. doi: 10.1038/nature09204
121. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
122. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet. (2023) 24:382–400. doi: 10.1038/s41576-022-00562-w
123. Zhang S, Yazaki E, Sakamoto H, Yamamoto H, Mizushima N. Evolutionary diversification of the autophagy-related ubiquitin-like conjugation systems. Autophagy. (2022) 18:2969–84. doi: 10.1080/15548627.2022.2059168
124. Pang Y, Yamamoto H, Sakamoto H, Oku M, Mutungi JK, Sahani MH, et al. Evolution from covalent conjugation to non-covalent interaction in the ubiquitin-like ATG12 system. Nat Struct Mol Biol. (2019) 26:289–96. doi: 10.1038/s41594-019-0204-3
125. Noda NN, Mizushima N. Atg101: not just an accessory subunit in the autophagy-initiation complex. Cell structure Funct. (2016) 41:13–20. doi: 10.1247/csf.15013
126. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. (2010) 12:836–41. doi: 10.1038/ncb0910-836
127. Levine TP. Remote homology searches identify bacterial homologues of eukaryotic lipid transfer proteins, including Chorein-N domains in TamB and AsmA and Mdm31p. BMC Mol Cell Biol. (2019) 20:43. doi: 10.1186/s12860-019-0226-z
128. Liu J, Tassinari M, Souza DP, Naskar S, Noel JK, Bohuszewicz O, et al. Bacterial Vipp1 and PspA are members of the ancient ESCRT-III membrane-remodeling superfamily. Cell. (2021) 184:3660–3673.e3618. doi: 10.1016/j.cell.2021.05.041
129. Moon CW, Kim Y, Hyun JK. Active electrochemical high-contrast gratings as on/off switchable and color tunable pixels. Nat Commun. (2022) 13:3391. doi: 10.1038/s41467-022-31083-z
130. Bhat P, Kriel J, Priya BS, Shivananju NS, Loos B. Modulating autophagy in cancer therapy: advancements and challenges for cancer cell death sensitization. Biochem Pharmacol. (2018) 147:170–82. doi: 10.1016/j.bcp.2017.11.021
131. Pérez-Hernández M, Arias A, Martínez-García D, Pérez-Tomás R, Quesada R, Soto-Cerrato V. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers. (2019) 11:1599. doi: 10.3390/cancers11101599
132. Yang Y, Liu L, Tian Y, Gu M, Wang Y, Ashrafizadeh M, et al. Autophagy-driven regulation of cisplatin response in human cancers: Exploring molecular and cell death dynamics. Cancer Lett. (2024) 587:216659. doi: 10.1016/j.canlet.2024.216659
133. Choi JH, Yoon JS, Won YW, Park BB, Lee YY. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. Apmis. (2012) 120:597–604. doi: 10.1111/j.1600-0463.2012.02876.x
134. Qin L, Xu T, Xia L, Wang X, Zhang X, Zhang X, et al. Chloroquine enhances the efficacy of cisplatin by suppressing autophagy in human adrenocortical carcinoma treatment. Drug design Dev Ther. 2016:1035–45. doi: 10.2147/DDDT.S101701
135. Gonçalves RM, Agnes JP, Delgobo M, de Souza PO, Thomé MP, Heimfarth L, et al. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem Pharmacol. (2019) 163:440–50. doi: 10.1016/j.bcp.2019.03.015
136. Hori YS, Hosoda R, Akiyama Y, Sebori R, Wanibuchi M, Mikami T, et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J neuro-oncology. (2015) 122:11–20. doi: 10.1007/s11060-014-1686-9
137. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E, et al. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci Rep. (2013) 3:2469. doi: 10.1038/srep02469
138. Thelen M, Wymann MP, Langen H. Wortmannin binds specifically to 1-phosphatidylinositol 3-kinase while inhibiting guanine nucleotide-binding protein-coupled receptor signaling in neutrophil leukocytes. Proc Natl Acad Sci. (1994) 91:4960–4. doi: 10.1073/pnas.91.11.4960
139. Tang F, Hu P, Yang Z, Xue C, Gong J, Sun S, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep. (2017) 37:3449–58. doi: 10.3892/or.2017.5635
140. Scott AJ, Arcaroli JJ, Bagby SM, Yahn R, Huber KM, Serkova NJ, et al. Cabozantinib exhibits potent antitumor activity in colorectal cancer patient-Derived tumor xenograft models via autophagy and signaling mechanisms. Mol Cancer Ther. (2018) 17:2112–22. doi: 10.1158/1535-7163.MCT-17-0131
141. Zhou P, Li Y, Li B, Zhang M, Xu C, Liu F, et al. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle (Georgetown Tex). (2018) 17:997–1006. doi: 10.1080/15384101.2018.1467677
142. Zhang X, Li W, Wang C, Leng X, Lian S, Feng J, et al. Inhibition of autophagy enhances apoptosis induced by proteasome inhibitor bortezomib in human glioblastoma U87 and U251 cells. Mol Cell Biochem. (2014) 385:265–75. doi: 10.1007/s11010-013-1835-z
143. Baquero P, Dawson A, Mukhopadhyay A, Kuntz EM, Mitchell R, Olivares O, et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia. (2019) 33:981–94. doi: 10.1038/s41375-018-0252-4
144. Kurdi A, Cleenewerck M, Vangestel C, Lyssens S, Declercq W, Timmermans JP, et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem Pharmacol. (2017) 138:150–62. doi: 10.1016/j.bcp.2017.06.119
145. Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PloS One. (2013) 8:e55096. doi: 10.1371/journal.pone.0055096
146. Avniel-Polak S, Leibowitz G, Doviner V, Gross DJ, Grozinsky-Glasberg S. Combining chloroquine with RAD001 inhibits tumor growth in a NEN mouse model. Endocrine-Related Cancer. (2018) 25:677–86. doi: 10.1530/ERC-18-0121
147. Kaneko M, Nozawa H, Hiyoshi M, Tada N, Murono K, Nirei T, et al. Temsirolimus and chloroquine cooperatively exhibit a potent antitumor effect against colorectal cancer cells. J Cancer Res Clin Oncol. (2014) 140:769–81. doi: 10.1007/s00432-014-1628-0
148. Carew JS, Medina EC, Esquivel JA II, Mahalingam D, Swords R, Kelly K, et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J Cell Mol Med. (2010) 14:2448–59. doi: 10.1111/j.1582-4934.2009.00832.x
149. Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. (2014) 10:1013–9. doi: 10.1038/nchembio.1681
150. Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. (2015) 11:725–6. doi: 10.1080/15548627.2015.1033601
151. Pinto-Leite R, Arantes-Rodrigues R, Ferreira R, Palmeira C, Colaço A. Moreira da Silva V et al: Temsirolimus improves cytotoxic efficacy of cisplatin and gemcitabine against urinary bladder cancer cell lines. Urologic Oncol. (2014) 32:41.e11–22. doi: 10.1016/j.urolonc.2013.04.012
152. Wang HW, Yang SH, Huang GD, Lin JK, Chen WS, Jiang JK, et al. Temsirolimus enhances the efficacy of cetuximab in colon cancer through a CIP2A-dependent mechanism. J Cancer Res Clin Oncol. (2014) 140:561–71. doi: 10.1007/s00432-014-1596-4
153. Chen P, Huang HP, Wang Y, Jin J, Long WG, Chen K, et al. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J Exp Clin Cancer research: CR. (2019) 38:254. doi: 10.1186/s13046-019-1234-8
154. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. (2015) 59:285–97. doi: 10.1016/j.molcel.2015.05.031
155. Dower CM, Bhat N, Gebru MT, Chen L, Wills CA, Miller BA, et al. Targeted inhibition of ULK1 promotes apoptosis and suppresses tumor growth and metastasis in neuroblastoma. Mol Cancer Ther. (2018) 17:2365–76. doi: 10.1158/1535-7163.MCT-18-0176
156. Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. (2014) 124:1406–17. doi: 10.1172/JCI70454
157. Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. (2018) 37:749–66. doi: 10.1007/s10555-018-9727-z
158. Malet-Martino M, Jolimaitre P, Martino R. The prodrugs of 5-fluorouracil. Curr medicinal Chem Anti-cancer Agents. (2002) 2:267–310. doi: 10.2174/1568011023354146
159. Walko CM, Lindley C. Capecitabine: a review. Clin Ther. (2005) 27:23–44. doi: 10.1016/j.clinthera.2005.01.005
160. Park JM, Huang S, Wu TT, Foster NR, Sinicrope FA. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther. (2013) 14:100–7. doi: 10.4161/cbt.22954
161. Liang X, Tang J, Liang Y, Jin R, Cai X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell bioscience. (2014) 4:10. doi: 10.1186/2045-3701-4-10
162. Zhu W, Zhou L, Qian JQ, Qiu TZ, Shu YQ, Liu P. Temozolomide for treatment of brain metastases: A review of 21 clinical trials. World J Clin Oncol. (2014) 5:19–27. doi: 10.5306/wjco.v5.i1.19
163. Yan Y, Xu Z, Dai S, Qian L, Sun L, Gong Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J Exp Clin Cancer Res. (2016) 35:1–14. doi: 10.1186/s13046-016-0303-5
164. Würstle S, Schneider F, Ringel F, Gempt J, Lämmer F, Delbridge C, et al. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncol Lett. (2017) 14:322–8. doi: 10.3892/ol.2017.6107
165. Zou Y, Wang Q, Li B, Xie B, Wang W. Temozolomide induces autophagy via ATM−AMPK−ULK1 pathways in glioma. Mol Med Rep. (2014) 10:411–6. doi: 10.3892/mmr.2014.2151
166. Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH, Lin YF, et al. Resveratrol enhances the therapeutic effect of temozolomide against Malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radical Biol Med. (2012) 52:377–91. doi: 10.1016/j.freeradbiomed.2011.10.487
167. Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, Chen TH, et al. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PloS One. (2012) 7:e38706. doi: 10.1371/journal.pone.0038706
168. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. (2012) 31:1869–83. doi: 10.1038/onc.2011.384
169. Ferreira JA, Peixoto A, Neves M, Gaiteiro C, Reis CA, Assaraf YG, et al. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug resistance updates: Rev commentaries antimicrobial Anticancer chemotherapy. (2016) 24:34–54. doi: 10.1016/j.drup.2015.11.003
170. Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. (2014) 289:17163–73. doi: 10.1074/jbc.M114.558288
171. Bao L, Jaramillo MC, Zhang Z, Zheng Y, Yao M, Zhang DD, et al. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. (2015) 11:91–8. doi: 10.3892/mmr.2014.2671
172. Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol (Northwood London England). (2011) 28:105–11. doi: 10.1007/s12032-009-9397-3
173. Zhu L, Du H, Shi M, Chen Z, Hang J. ATG7 deficiency promote apoptotic death induced by Cisplatin in human esophageal squamous cell carcinoma cells. Bull du Cancer. (2013) 100:E15–21. doi: 10.1684/bdc.2013.1749
174. Cheng CY, Liu JC, Wang JJ, Li YH, Pan J, Zhang YR. Autophagy inhibition increased the anti-tumor effect of cisplatin on drug-resistant esophageal cancer cells. J Biol regulators homeostatic Agents. (2017) 31:645–52.
175. Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. (2007) 282:13123–32. doi: 10.1074/jbc.M700492200
176. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci United States America. (2003) 100:15077–82. doi: 10.1073/pnas.2436255100
177. Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, et al. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy. (2009) 5:303–6. doi: 10.4161/auto.5.3.7491
178. Chen L, Liu Y, Zhang Q, Zhang M, Han X, Li Q, et al. p53/PCDH17/Beclin-1 Proteins as Prognostic Predictors for Urinary Bladder Cancer. J Cancer. (2019) 10:6207–16. doi: 10.7150/jca.37335
179. Wang Y, Xie J, Wang H, Huang H, Xie P. Beclin-1 suppresses gastric cancer progression by promoting apoptosis and reducing cell migration. Oncol Lett. (2017) 14:6857–62. doi: 10.3892/ol.2017.7046
180. Ye C, Yu X, Liu X, Zhan P, Nie T, Guo R, et al. Beclin-1 knockdown decreases proliferation, invasion and migration of Ewing sarcoma SK-ES-1 cells via inhibition of MMP-9. Oncol Lett. (2018) 15:3221–5. doi: 10.3892/ol.2017.7667
181. Du H, Chen L, Luo F, Chen X, Li Y, Cheng Q. Beclin-1 expression is associated with prognosis in a Bcl-2-dependent manner in non-small cell lung cancer. Oncol Lett. (2020) 20:9. doi: 10.3892/ol.2020.11870
182. Du H, Che J, Shi M, Zhu L, Hang JB, Chen Z, et al. Beclin 1 expression is associated with the occurrence and development of esophageal squamous cell carcinoma. Oncol Lett. (2017) 14:6823–8. doi: 10.3892/ol.2017.7015
183. Yang Z, Ghoorun RA, Fan X, Wu P, Bai Y, Li J, et al. High expression of Beclin-1 predicts favorable prognosis for patients with colorectal cancer. Clinics Res Hepatol Gastroenterol. (2015) 39:98–106. doi: 10.1016/j.clinre.2014.06.014
184. Bi C, Liu M, Rong W, Wu F, Zhang Y, Lin S, et al. High Beclin-1 and ARID1A expression corelates with poor survival and high recurrence in intrahepatic cholangiocarcinoma: a histopathological retrospective study. BMC Cancer. (2019) 19:213. doi: 10.1186/s12885-019-5429-3
185. Zhang HY, Du ZX, Meng X, Zong ZH, Wang HQ. Beclin 1 enhances proteasome inhibition-mediated cytotoxicity of thyroid cancer cells in macroautophagy-independent manner. J Clin Endocrinol Metab. (2013) 98:E217–226. doi: 10.1210/jc.2012-2679
186. Woo J, Kim JB, Cho T, Yoo EH, Moon BI, Kwon H, et al. Selenium inhibits growth of trastuzumab-resistant human breast cancer cells via downregulation of Akt and beclin-1. PloS One. (2021) 16:e0257298. doi: 10.1371/journal.pone.0257298
187. Wu XY, Chen J, Cao QH, Dong M, Lin Q, Fan XJ, et al. Beclin 1 activation enhances chemosensitivity and predicts a favorable outcome for primary duodenal adenocarcinoma. Tumour biology: J Int Soc Oncodevelopmental Biol Med. (2013) 34:713–22. doi: 10.1007/s13277-012-0599-5
188. Zhang MY, Gou WF, Zhao S, Mao XY, Zheng ZH, Takano Y, et al. Beclin 1 expression is closely linked to colorectal carcinogenesis and distant metastasis of colorectal carcinoma. Int J Mol Sci. (2014) 15:14372–85. doi: 10.3390/ijms150814372
189. Zhang F, Wang B, Long H, Yu J, Li F, Hou H, et al. Decreased miR-124-3p expression prompted breast cancer cell progression mainly by targeting beclin-1. Clin Lab. (2016) 62:1139–45. doi: 10.7754/Clin.Lab.2015.151111
190. Wang W, Fan H, Li X, Wu G, Zhao W, Zhang G, et al. Beclin 1 promotes apoptosis and decreases invasion by upregulating the expression of ECRG4 in A549 human lung adenocarcinoma cells. Mol Med Rep. (2016) 14:355–60. doi: 10.3892/mmr.2016.5219
191. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death differentiation. (2011) 18:571–80. doi: 10.1038/cdd.2010.191
192. Liang XH, Yu J, Brown K, Levine B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. (2001) 61:3443–9.
193. Zeng X, Sun J, Li F, Peng L, Zhang C, Jiang X, et al. Beclin 1 haploinsufficiency ameliorates high-Fat diet-Induced myocardial injury via inhibiting alternative mitophagy. Antioxidants Redox Signaling. (2024). doi: 10.1089/ars.2023.0399
194. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
195. Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. (2003) 11:895–904. doi: 10.1016/S1097-2765(03)00114-X
196. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. (2004) 6:1122–8. doi: 10.1038/ncb1183
197. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. (2009) 137:873–86. doi: 10.1016/j.cell.2009.03.046
198. Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. (2010) 285:20109–16. doi: 10.1074/jbc.M110.121699
199. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. (2002) 110:177–89. doi: 10.1016/S0092-8674(02)00833-4
200. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR. Erdjument-Bromage H et al: mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. (2002) 110:163–75. doi: 10.1016/S0092-8674(02)00808-5
201. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. (2007) 25:903–15. doi: 10.1016/j.molcel.2007.03.003
202. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. (2007) 9:316–23. doi: 10.1038/ncb1547
203. Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jenö P, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PloS One. (2007) 2:e1217. doi: 10.1371/journal.pone.0001217
204. Wang L, Harris TE, Roth RA, Lawrence JC Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. (2007) 282:20036–44. doi: 10.1074/jbc.M702376200
205. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR. Erdjument-Bromage H et al: Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr biology: CB. (2004) 14:1296–302. doi: 10.1016/j.cub.2004.06.054
206. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. (2006) 127:125–37. doi: 10.1016/j.cell.2006.08.033
207. Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr biology: CB. (2006) 16:1865–70. doi: 10.1016/j.cub.2006.08.001
208. Pearce LR, Huang X, Boudeau J, Pawłowski R, Wullschleger S, Deak M, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. (2007) 405:513–22. doi: 10.1042/BJ20070540
209. Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. (2006) 20:2820–32. doi: 10.1101/gad.1461206
210. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. (2015) 125:25–32. doi: 10.1172/JCI73939
211. Yan Y, Zhou XE, Xu HE, Melcher K. Structure and physiological regulation of AMPK. Int J Mol Sci. (2018) 19:3534. doi: 10.3390/ijms19113534
212. Li Q, Wang Y, Wu S, Zhou Z, Ding X, Shi R, et al. CircACC1 regulates assembly and activation of AMPK complex under metabolic stress. Cell Metab. (2019) 30:157–173. e157. doi: 10.1016/j.cmet.2019.05.009
213. Lin S-C, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. (2018) 27:299–313. doi: 10.1016/j.cmet.2017.10.009
214. Hardie DG. Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface. (2018) 15:20170774. doi: 10.1098/rsif.2017.0774
215. Zhang Y, Lanjuin A, Chowdhury SR, Mistry M, Silva-García CG, Weir HJ, et al. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. Elife. (2019) 8:e49158. doi: 10.7554/eLife.49158
216. Ge Y, Zhou M, Chen C, Wu X, Wang X. Role of AMPK mediated pathways in autophagy and aging. Biochimie. (2022) 195:100–13. doi: 10.1016/j.biochi.2021.11.008
217. Rajak S, Raza S, Sinha RA. ULK1 signaling in the liver: autophagy dependent and independent actions. Front Cell Dev Biol. (2022) 10:836021. doi: 10.3389/fcell.2022.836021
218. Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. (1997) 192:245–50. doi: 10.1016/S0378-1119(97)00084-X
219. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. (2008) 112:1493–502. doi: 10.1182/blood-2008-02-137398
220. Papinski D, Kraft C. Regulation of autophagy by signaling through the atg1/ULK1 complex. J Mol Biol. (2016) 428:1725–41. doi: 10.1016/j.jmb.2016.03.030
221. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. (2004) 36:585–95. doi: 10.1038/ng1362
222. Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci United States America. (2011) 108:4788–93. doi: 10.1073/pnas.1100844108
223. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Sci (New York NY). (2011) 331:456–61. doi: 10.1126/science.1196371
224. Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. (2015) 589:1847–54. doi: 10.1016/j.febslet.2015.05.020
225. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. (2014) 15:566–75. doi: 10.1002/embr.201438501
226. Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. (2011) 7:737–47. doi: 10.4161/auto.7.7.15491
227. Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. (2011) 7:696–706. doi: 10.4161/auto.7.7.15451
228. Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. (2020) 63:1–10. doi: 10.1016/j.ceb.2019.12.001
229. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. (2009) 10:458–67. doi: 10.1038/nrm2708
230. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. (1998) 395:395–8. doi: 10.1038/26506
231. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. (2000) 408:488–92. doi: 10.1038/35044114
232. Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Seminars Cell Develop Biol. (2004) 15(2):231–6.
233. Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. (2001) 20:5971–81. doi: 10.1093/emboj/20.21.5971
234. Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. (2001) 152:657–68. doi: 10.1083/jcb.152.4.657
235. Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12~ ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. (2013) 20:59–66. doi: 10.1038/nsmb.2431
236. Zheng Y, Qiu Y, Grace CR, Liu X, Klionsky DJ, Schulman BA. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat Commun. (2019) 10:3600. doi: 10.1038/s41467-019-11435-y
237. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. (2007) 282:37298–302. doi: 10.1074/jbc.C700195200
238. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. (2008) 19:2092–100. doi: 10.1091/mbc.e07-12-1257
239. Lystad AH, Carlsson SR, de la Ballina LR, Kauffman KJ, Nag S, Yoshimori T, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. (2019) 21:372–83. doi: 10.1038/s41556-019-0274-9
240. Ye J, Zhang J, Zhu Y, Wang L, Jiang X, Liu B, et al. Targeting autophagy and beyond: Deconvoluting the complexity of Beclin-1 from biological function to cancer therapy. Acta Pharm Sin B. (2023) 13:4688–714. doi: 10.1016/j.apsb.2023.08.008
241. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. (2010) 1:e18. doi: 10.1038/cddis.2009.16
242. Li H, Wang P, Yu J, Zhang L. Cleaving Beclin 1 to suppress autophagy in chemotherapy-induced apoptosis. Autophagy. (2011) 7:1239–41. doi: 10.4161/auto.7.10.16490
243. Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang X, et al. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. (2010) 1:468–77. doi: 10.1007/s13238-010-0048-4
244. Siddiqui MA, Mukherjee S, Manivannan P, Malathi K. RNase L cleavage products promote switch from autophagy to apoptosis by caspase-mediated cleavage of beclin-1. Int J Mol Sci. (2015) 16:17611–36. doi: 10.3390/ijms160817611
245. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr biology: CB. (2018) 28:2388–2399.e2385. doi: 10.1016/j.cub.2018.05.094
246. Seo J, Seong D, Nam YW, Hwang CH, Lee SR, Lee CS, et al. Beclin 1 functions as a negative modulator of MLKL oligomerisation by integrating into the necrosome complex. Cell Death differentiation. (2020) 27:3065–81. doi: 10.1038/s41418-020-0561-9
247. Park JM, Tougeron D, Huang S, Okamoto K, Sinicrope FA. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PloS One. (2014) 9:e100819. doi: 10.1371/journal.pone.0100819
248. Xu F, Fang Y, Yan L, Xu L, Zhang S, Cao Y, et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci Rep. (2017) 7:45385. doi: 10.1038/srep45385
249. Wijshake T, Zou Z, Chen B, Zhong L, Xiao G, Xie Y, et al. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc Natl Acad Sci United States America. (2021) 118. doi: 10.1073/pnas.2020478118
250. Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy. (2011) 7:829–39. doi: 10.4161/auto.7.8.15598
251. Sun Y, Liu JH, Sui YX, Jin L, Yang Y, Lin SM, et al. Beclin1 overexpression inhibitis proliferation, invasion and migration of CaSki cervical cancer cells. Asian Pacific J Cancer prevention: APJCP. (2011) 12:1269–73.
252. Su YL, Kortylewski M. Beclin-1 as a neutrophil-specific immune checkpoint. J Clin Invest. (2019) 129:5079–81. doi: 10.1172/JCI132534
253. Tan P, He L, Xing C, Mao J, Yu X, Zhu M, et al. Myeloid loss of Beclin 1 promotes PD-L1hi precursor B cell lymphoma development. J Clin Invest. (2019) 129:5261–77. doi: 10.1172/JCI127721
254. Li X, Yang KB, Chen W, Mai J, Wu XQ, Sun T, et al. CUL3 (cullin 3)-mediated ubiquitination and degradation of BECN1 (beclin 1) inhibit autophagy and promote tumor progression. Autophagy. (2021) 17:4323–40. doi: 10.1080/15548627.2021.1912270
255. Tang JY, Fang YY, Hsi E, Huang YC, Hsu NC, Yang WC, et al. Immunopositivity of Beclin-1 and ATG5 as indicators of survival and disease recurrence in oral squamous cell carcinoma. Anticancer Res. (2013) 33:5611–6.
256. Cao QH, Liu F, Yang ZL, Fu XH, Yang ZH, Liu Q, et al. Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am J Trans Res. (2016) 8:3831–47.
257. You FF, Zhang J, Cheng F, Zou K, Zhang XQ, Chen JF. ATG 4B serves a crucial role in RCE-4-induced inhibition of the bcl-2-beclin 1 complex in cervical cancer ca ski cells. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222212302
258. McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O'Hagan B, et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis. (2010) 1:e108. doi: 10.1038/cddis.2010.86
259. Zhang J, Mao W, Liu Y, Ding J, Wang J, Yu Z, et al. 3-MA enhanced chemosensitivity in cisplatin resistant hypopharyngeal squamous carcinoma cells via inhibiting beclin -1 mediated autophagy. Curr Pharm design. (2021) 27:996–1005. doi: 10.2174/1381612826666201221150431
260. Zhou GZ, Wang QQ, Wang PB, Wang ZC, Sun GC. One novel curcumin derivative ZYX01 induces autophagy of human non-small lung cancer cells A549 through AMPK/ULK1/Beclin-1 signaling pathway. Cell Mol Biol (Noisy-le-Grand France). (2019) 65:1–6. doi: 10.14715/cmb/2019.65.2.1
261. Chen HE, Lin JF, Tsai TF, Lin YC, Chou KY, Hwang TI. Allyl isothiocyanate induces autophagy through the up-regulation of beclin-1 in human prostate cancer cells. Am J Chin Med. 2018:1–19. doi: 10.1142/S0192415X18500830
262. Zhou B, Yang C, Yan X, Shi Z, Xiao H, Wei X, et al. LETM1 knockdown promotes autophagy and apoptosis through AMP-activated protein kinase phosphorylation-mediated beclin-1/bcl-2 complex dissociation in hepatocellular carcinoma. Front Oncol. (2020) 10:606790. doi: 10.3389/fonc.2020.606790
263. Ma C, Zhang X, Mo X, Yu Y, Xiao Z, Wu J, et al. Xie-Bai-San increases NSCLC cells sensitivity to gefitinib by inhibiting Beclin-1 mediated autophagosome formation. Phytomedicine: Int J phytotherapy phytopharmacology. (2024) 125:155351. doi: 10.1016/j.phymed.2024.155351
264. Zou M, Lu N, Hu C, Liu W, Sun Y, Wang X, et al. Beclin 1-mediated autophagy in hepatocellular carcinoma cells: implication in anticancer efficiency of oroxylin A via inhibition of mTOR signaling. Cell signalling. (2012) 24:1722–32. doi: 10.1016/j.cellsig.2012.04.009
265. Ma J, Liu L, Ling Y, Zheng J. Polypeptide LTX-315 reverses the cisplatin chemoresistance of ovarian cancer cells via regulating Beclin-1/PI3K/mTOR signaling pathway. J Biochem Mol Toxicol. (2021) 35:e22853. doi: 10.1002/jbt.22853
266. Nicco C, Thomas M, Guillermet J, Havard M, Laurent-Tchenio F, Doridot L, et al. Mechanistic target of rapamycin (mTOR) regulates self-sustained quiescence, tumor indolence, and late clinical metastasis in a Beclin-1-dependent manner. Cell Cycle (Georgetown Tex). (2023) 22:542–64. doi: 10.1080/15384101.2022.2123187
267. Jian M, Yunjia Z, Zhiying D, Yanduo J, Guocheng J. Interleukin 7 receptor activates PI3K/Akt/mTOR signaling pathway via downregulation of Beclin-1 in lung cancer. Mol carcinogenesis. (2019) 58:358–65. doi: 10.1002/mc.22933
268. Huangfu L, Wang X, Tian S, Chen J, Wang X, Fan B, et al. Piceatannol enhances Beclin-1 activity to suppress tumor progression and its combination therapy strategy with everolimus in gastric cancer. Sci China Life Sci. (2023) 66:298–312. doi: 10.1007/s11427-022-2185-9
269. Ashrafizaveh S, Ashrafizadeh M, Zarrabi A, Husmandi K, Zabolian A, Shahinozzaman M, et al. Long non-coding RNAs in the doxorubicin resistance of cancer cells. Cancer Lett. (2021) 508:104–14. doi: 10.1016/j.canlet.2021.03.018
270. Mirzaei S, Gholami MH, Hashemi F, Zabolian A, Farahani MV, Hushmandi K, et al. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discovery Today. (2022) 27:436–55. doi: 10.1016/j.drudis.2021.09.020
271. Wei J, Zhu K, Yang Z, Zhou Y, Xia Z, Ren J, et al. Hypoxia-Induced autophagy is involved in radioresistance via HIF1A-Associated beclin-1 in glioblastoma multiforme. Heliyon. (2023) 9:e12820. doi: 10.1016/j.heliyon.2023.e12820
272. Zhang X, Shi H, Lin S, Ba M, Cui S. MicroRNA-216a enhances the radiosensitivity of pancreatic cancer cells by inhibiting beclin-1-mediated autophagy. Oncol Rep. (2015) 34:1557–64. doi: 10.3892/or.2015.4078
273. Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. (2013) 154:1269–84. doi: 10.1016/j.cell.2013.08.015
274. Chen L, Han X, Hu Z, Chen L. The PVT1/miR-216b/Beclin-1 regulates cisplatin sensitivity of NSCLC cells via modulating autophagy and apoptosis. Cancer chemotherapy Pharmacol. (2019) 83:921–31. doi: 10.1007/s00280-019-03808-3
275. Chen W, Li Z, Liu H, Jiang S, Wang G, Sun L, et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. (2020) 11:198. doi: 10.1038/s41419-020-2390-7
276. Xu R, Liu S, Chen H, Lao L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol Rep. (2016) 35:1757–63. doi: 10.3892/or.2015.4497
277. Yang X, Bai F, Xu Y, Chen Y, Chen L. Intensified beclin-1 mediated by low expression of mir-30a-5p promotes chemoresistance in human small cell lung cancer. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2017) 43:1126–39. doi: 10.1159/000481754
278. Tan S, Shi H, Ba M, Lin S, Tang H, Zeng X, et al. miR-409-3p sensitizes colon cancer cells to oxaliplatin by inhibiting Beclin-1-mediated autophagy. Int J Mol Med. (2016) 37:1030–8. doi: 10.3892/ijmm.2016.2492
279. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. (2020) 17:395–417. doi: 10.1038/s41571-020-0341-y
280. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harbor Perspect Biol. (2013) 5:a008656. doi: 10.1101/cshperspect.a008656
281. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
282. Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res. (2000) 45:528–37. doi: 10.1016/S0008-6363(99)00384-3
283. Elmore S. Apoptosis: a review of programmed cell death. Toxicologic Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
284. Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. (1994) 371:346–7. doi: 10.1038/371346a0
285. Olofsson MH, Ueno T, Pan Y, Xu R, Cai F, van der Kuip H, et al. Cytokeratin-18 is a useful serum biomarker for early determination of response of breast carcinomas to chemotherapy. Clin Cancer research: an Off J Am Assoc Cancer Res. (2007) 13:3198–206. doi: 10.1158/1078-0432.CCR-07-0009
286. Ulukaya E, Karaagac E, Ari F, Oral AY, Adim SB, Tokullugil AH, et al. Chemotherapy increases caspase-cleaved cytokeratin 18 in the serum of breast cancer patients. Radiol Oncol. (2011) 45:116–22. doi: 10.2478/v10019-011-0006-7
287. Hameed A, Truong LD, Price V, Kruhenbuhl O, Tschopp J. Immunohistochemical localization of granzyme B antigen in cytotoxic cells in human tissues. Am J Pathol. (1991) 138:1069–75.
288. Wlodkowic D, Skommer J, Darzynkiewicz Z. Flow cytometry-based apoptosis detection. Methods Mol Biol (Clifton NJ). (2009) 559:19–32.
289. Liu JJ, Wang W, Dicker DT, El-Deiry WS. Bioluminescent imaging of TRAIL-induced apoptosis through detection of caspase activation following cleavage of DEVD-aminoluciferin. Cancer Biol Ther. (2005) 4:885–92. doi: 10.4161/cbt.4.8.2133
290. Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, et al. Imaging of apoptosis (programmed cell death) with 99mTc annexin V. J Nucl medicine: Off publication Soc Nucl Med. (1999) 40:184–91.
291. Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K, et al. Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res. (2017) 77:2318–27. doi: 10.1158/0008-5472.CAN-16-3346
292. Song X, Lee DH, Dilly AK, Lee YS, Choudry HA, Kwon YT, et al. Crosstalk between apoptosis and autophagy is regulated by the arginylated biP/Beclin-1/p62 complex. Mol Cancer research: MCR. (2018) 16:1077–91. doi: 10.1158/1541-7786.MCR-17-0685
293. Gao P, Hao F, Dong X, Qiu Y. The role of autophagy and Beclin-1 in radiotherapy-induced apoptosis in thyroid carcinoma cells. Int J Clin Exp Pathol. (2019) 12:885–92.
294. Nam RK, Benatar T, Amemiya Y, Sherman C, Seth A. Mir-139 regulates autophagy in prostate cancer cells through beclin-1 and mTOR signaling proteins. Anticancer Res. (2020) 40:6649–63. doi: 10.21873/anticanres.14689
295. Wang YF, Xu YL, Tang ZH, Li T, Zhang LL, Chen X, et al. Baicalein induces beclin 1- and extracellular signal-Regulated kinase-Dependent autophagy in ovarian cancer cells. Am J Chin Med. (2017) 45:123–36. doi: 10.1142/S0192415X17500094
296. Liu L, Zhao WM, Yang XH, Sun ZQ, Jin HZ, Lei C, et al. Effect of inhibiting Beclin-1 expression on autophagy, proliferation and apoptosis in colorectal cancer. Oncol Lett. (2017) 14:4319–24. doi: 10.3892/ol.2017.6687
297. Lin F, Ghislat G, Luo S, Renna M, Siddiqi F, Rubinsztein DC. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFκB activation. Hum Mol Genet. (2015) 24:2899–913. doi: 10.1093/hmg/ddv052
298. Lian J, Karnak D, Xu L. The Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy. (2010) 6:1201–3. doi: 10.4161/auto.6.8.13549
299. Hasan A, Haque E, Hameed R, Maier PN, Irfan S, Kamil M, et al. Hsp90 inhibitor gedunin causes apoptosis in A549 lung cancer cells by disrupting Hsp90:Beclin-1:Bcl-2 interaction and downregulating autophagy. Life Sci. (2020) 256:118000. doi: 10.1016/j.lfs.2020.118000
300. Yu FS, Yu CS, Chen JC, Yang JL, Lu HF, Chang SJ, et al. Tetrandrine induces apoptosis Via caspase-8, -9, and -3 and poly (ADP ribose) polymerase dependent pathways and autophagy through beclin-1/ LC3-I, II signaling pathways in human oral cancer HSC-3 cells. Environ Toxicol. (2016) 31:395–406. doi: 10.1002/tox.22053
301. Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. (2022) 15:174. doi: 10.1186/s13045-022-01392-3
302. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
303. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/j.cell.2022.06.003
304. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
305. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
306. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. (2020) 5:108. doi: 10.1038/s41392-020-00216-5
307. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:34. doi: 10.1186/s13045-019-0720-y
308. Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (London England). (2022) 42:88–116. doi: 10.1002/cac2.12250
309. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
310. Wen J, Chen H, Ren Z, Zhang P, Chen J, Jiang S. Ultrasmall iron oxide nanoparticles induced ferroptosis via Beclin1/ATG5-dependent autophagy pathway. Nano convergence. (2021) 8:10. doi: 10.1186/s40580-021-00260-z
311. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc–activity. Curr Biol. (2018) 28:2388–2399. e2385. doi: 10.1016/j.cub.2018.05.094
312. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. (2018) 14:2083–103. doi: 10.1080/15548627.2018.1503146
313. Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. (2019) 79:1913–24. doi: 10.1158/0008-5472.CAN-18-3037
314. Wang C, Liu T, Wang J, Cheng C, Zhang Z, Zhang J, et al. CircHIPK3 negatively regulates autophagy by blocking VCP binding to the Beclin 1 complex in bladder cancer. Discover Oncol. (2023) 14:86. doi: 10.1007/s12672-023-00689-0
315. Ünal TD, Hamurcu Z, Delibaşı N, Çınar V, Güler A, Gökçe S, et al. Thymoquinone inhibits proliferation and migration of MDA-MB-231 triple negative breast cancer cells by suppressing autophagy, beclin-1 and LC3. Anti-cancer Agents medicinal Chem. (2021) 21:355–64. doi: 10.2174/1871520620666200807221047
316. Chen Z, Liu L, Liu Y, Wang S, Zhang S, Dong R, et al. Hydroxysafflor yellow A induces autophagy in human liver cancer cells by regulating Beclin 1 and ERK expression. Exp Ther Med. (2020) 19:2989–96. doi: 10.3892/etm.2020.8552
317. Huang Z, Fang W, Liu W, Wang L, Liu B, Liu S, et al. Aspirin induces Beclin-1-dependent autophagy of human hepatocellular carcinoma cell. Eur J Pharmacol. (2018) 823:58–64. doi: 10.1016/j.ejphar.2018.01.031
318. Li J, Wang Y, Luo Y, Liu Y, Yi Y, Li J, et al. USP5-Beclin 1 axis overrides p53-dependent senescence and drives Kras-induced tumorigenicity. Nat Commun. (2022) 13:7799. doi: 10.1038/s41467-022-35557-y
319. Wang Y, Zhang W, Lv Q, Zhang J, Zhu D. The critical role of quercetin in autophagy and apoptosis in HeLa cells. Tumour biology: J Int Soc Oncodevelopmental Biol Med. (2016) 37:925–9. doi: 10.1007/s13277-015-3890-4
320. Wang K, Liu R, Li J, Mao J, Lei Y, Wu J, et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy. (2011) 7:966–78.
321. Li YQ, Fan F, Wang YR, Li LY, Cao YJ, Gu SM, et al. The novel small molecule BH3 mimetic nobiletin synergizes with vorinostat to induce apoptosis and autophagy in small cell lung cancer. Biochem Pharmacol. (2023) 216:115807. doi: 10.1016/j.bcp.2023.115807
322. Fan Y, Li J, Yang Y, Zhao X, Liu Y, Jiang Y, et al. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53−dependent pathway: Integrated bioinformatics analysis and experimental validation. Int J Oncol. (2020) 57:925–38. doi: 10.3892/ijo.2020.5107
323. Xu X, Chen K, Kobayashi S, Timm D, Liang Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J Pharmacol Exp Ther. (2012) 341:183–95. doi: 10.1124/jpet.111.189589
324. Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death differentiation. (2008) 15:1318–29. doi: 10.1038/cdd.2008.51
325. Esposito A, Ferraresi A, Salwa A, Vidoni C, Dhanasekaran DN, Isidoro C. Resveratrol contrasts IL-6 pro-growth effects and promotes autophagy-mediated cancer cell dormancy in 3D ovarian cancer: role of miR-1305 and of its target ARH-I. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14092142
326. Prabhu V, Srivastava P, Yadav N, Amadori M, Schneider A, Seshadri A, et al. Resveratrol depletes mitochondrial DNA and inhibition of autophagy enhances resveratrol-induced caspase activation. Mitochondrion. (2013) 13:493–9. doi: 10.1016/j.mito.2012.10.010
327. Zhong LX, Zhang Y, Wu ML, Liu YN, Zhang P, Chen XY, et al. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discovery. (2016) 2:15071. doi: 10.1038/cddiscovery.2015.71
328. Wang J, Huang P, Pan X, Xia C, Zhang H, Zhao H, et al. Resveratrol reverses TGF-β1-mediated invasion and metastasis of breast cancer cells via the SIRT3/AMPK/autophagy signal axis. Phytotherapy research: PTR. (2023) 37:211–30. doi: 10.1002/ptr.v37.1
329. Zhou GZ, Shi YY, Wei LL, Sun GC. Autophagy induction and antiproliferative effect of a novel curcumin derivative MOMI-1 on the human lung cancer cells A549. J Biochem Mol Toxicol. (2019) 33:e22280. doi: 10.1002/jbt.2019.33.issue-4
330. Zhang L, Xu S, Cheng X, Wu J, Wu L, Wang Y, et al. Curcumin induces autophagic cell death in human thyroid cancer cells. Toxicol vitro: an Int J published Assoc BIBRA. (2022) 78:105254. doi: 10.1016/j.tiv.2021.105254
331. Zheng X, Liu J, Hu W, Jiang B, Zhou X, Zhang M, et al. Curcumin induces autophagy-mediated ferroptosis by targeting the PI3K/AKT/mTOR signaling pathway in gastric cancer. Turkish J gastroenterology: Off J Turkish Soc Gastroenterol. (2024) 35:625–33.
332. Akkoç Y, Berrak Ö, Arısan ED, Obakan P, Çoker-Gürkan A, Palavan-Ünsal N. Inhibition of PI3K signaling triggered apoptotic potential of curcumin which is hindered by Bcl-2 through activation of autophagy in MCF-7 cells. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2015) 71:161–71. doi: 10.1016/j.biopha.2015.02.029
333. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
334. Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. (2014) 15:228–38. doi: 10.1016/j.chom.2014.01.009
335. Liu H, He Z, Simon HU. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol. (2013) 23:352–60. doi: 10.1016/j.semcancer.2013.06.008
336. Frias A, Di Leo L, Antoranz A, Nazerai L, Carretta M, Bodemeyer V, et al. Ambra1 modulates the tumor immune microenvironment and response to PD-1 blockade in melanoma. J immunotherapy Cancer. (2023) 11. doi: 10.1136/jitc-2022-006389
337. Cai W, Hu M, Li C, Wu R, Lu D, Xie C, et al. FOXP3+ macrophage represses acute ischemic stroke-induced neural inflammation. Autophagy. (2023) 19:1144–63. doi: 10.1080/15548627.2022.2116833
338. Toton E, Lisiak N, Sawicka P, Rybczynska M. Beclin-1 and its role as a target for anticancer therapy. J Physiol pharmacology: an Off J Polish Physiol Soc. (2014) 65:459–67.
339. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
340. Duan X, Chan C, Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angewandte Chemie (International ed English). (2019) 58:670–80. doi: 10.1002/anie.201804882
341. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
342. Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
343. Ruan H, Leibowitz BJ, Zhang L, Yu J. Immunogenic cell death in colon cancer prevention and therapy. Mol carcinogenesis. (2020) 59:783–93. doi: 10.1002/mc.23183
344. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. (2017) 170:548–563.e516. doi: 10.1016/j.cell.2017.07.008
345. Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in pancreatic cancer. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20184504
346. Olivares-Urbano MA, Griñán-Lisón C, Marchal JA, Núñez MI. CSC radioresistance: A therapeutic challenge to improve radiotherapy effectiveness in cancer. Cells. (2020) 9. doi: 10.3390/cells9071651
347. Bai X, Ni J, Beretov J, Wang S, Dong X, Graham P, et al. THOC2 and THOC5 regulate stemness and radioresistance in triple-negative breast cancer. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2021) 8:e2102658. doi: 10.1002/advs.202102658
348. Baxevanis CN, Perez SA, Papamichail M. Cancer immunotherapy. Crit Rev Clin Lab Sci. (2009) 46:167–89. doi: 10.1080/10408360902937809
349. Wang G. Cancer immunotherapy and immunomodulation. Curr medicinal Chem. (2019) 26:2988–9. doi: 10.2174/092986732617190820123515
350. Cha JH, Chan LC, Song MS, Hung MC. New approaches on cancer immunotherapy. Cold Spring Harbor Perspect Med. (2020) 10. doi: 10.1101/cshperspect.a036863
351. Greten TF, Schwabe R, Bardeesy N, Ma L, Goyal L, Kelley RK, et al. Immunology and immunotherapy of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. (2023) 20:349–65. doi: 10.1038/s41575-022-00741-4
Keywords: autophagy, Beclin-1, mTOR, autophagy flux, apoptosis and ferroptosis, immunotherapy
Citation: Cao Z, Tian K, Ran Y, Zhou H, Zhou L, Ding Y and Tang X (2024) Beclin-1: a therapeutic target at the intersection of autophagy, immunotherapy, and cancer treatment. Front. Immunol. 15:1506426. doi: 10.3389/fimmu.2024.1506426
Received: 05 October 2024; Accepted: 01 November 2024;
Published: 22 November 2024.
Edited by:
Zhiwen Luo, Fudan University, ChinaReviewed by:
Kaige Chen, Wake Forest University, United StatesShangke Huang, Southwest Medical University, China
Copyright © 2024 Cao, Tian, Ran, Zhou, Zhou, Ding and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Tang, Z2R3a3R4d0AxNjMuY29t; Yana Ding, MTg5OTYxNDIwMjFAMTYzLmNvbQ==
†These authors have contributed equally to this work