 Alexander B. Sigalov
Alexander B. Sigalov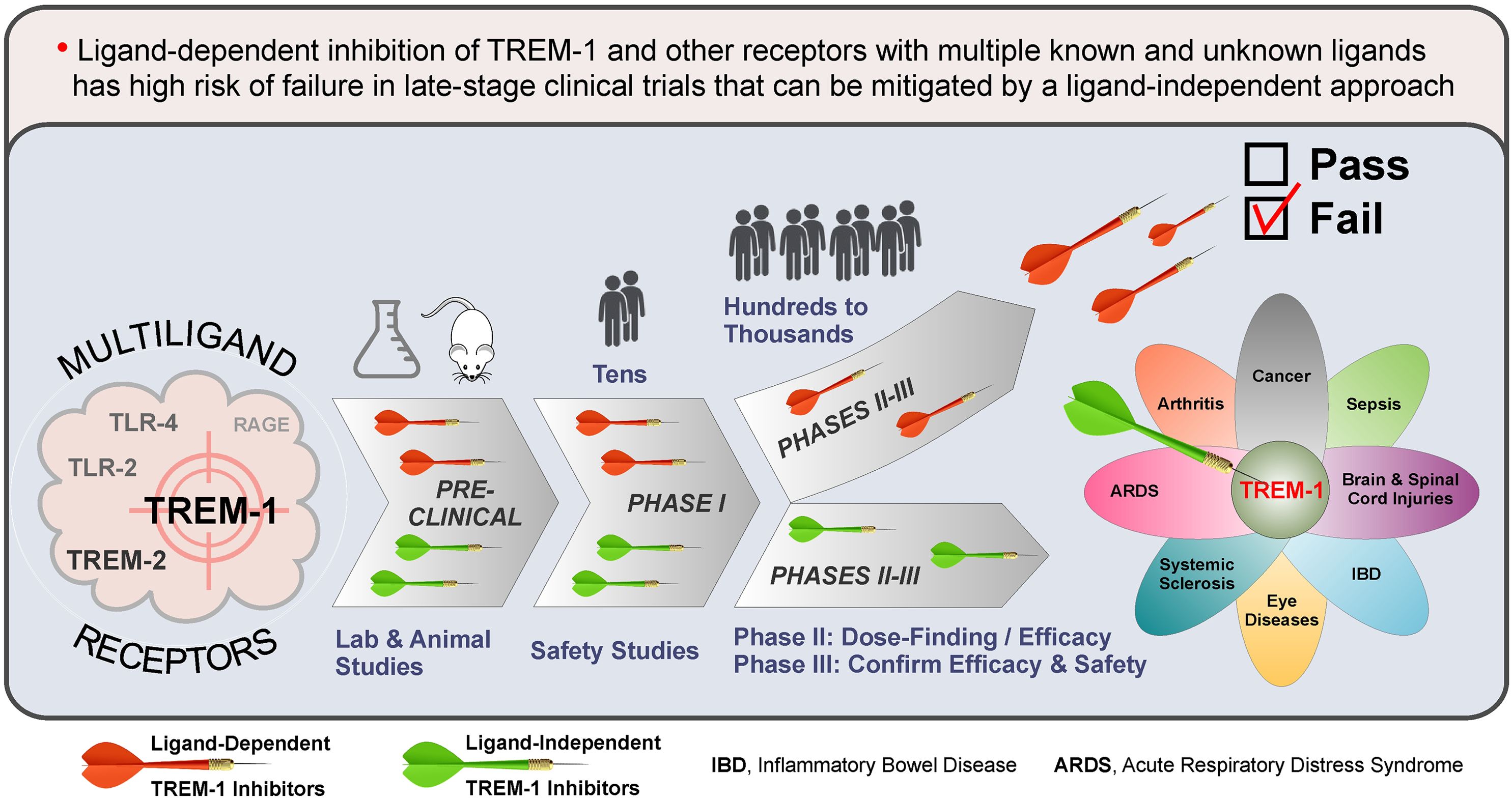
Graphical Abstract.
1 Introduction
Triggering receptor expressed on myeloid cells 1 (TREM-1) plays a pivotal role in human health and disease (1–6). Despite promising preclinical data and safety in humans, the first clinical inhibitor of TREM-1 (peptide LR12 or nangibotide) failed to reach significance for the primary endpoint in ASTONISH phase IIb sepsis trial (7, 8). Here, based on the multiplicity of TREM-1 ligands (9–11), I hypothesize that the ligand-dependent mechanism of action of LR12 rather than TREM-1 inhibition per se is the reason of failure. Similar reasoning can explain why an antagonistic monoclonal antibody (mAb) against TREM-2, another multiligand receptor, failed recently in phase Ia/b oncology trials due to lack of efficacy, despite success in animal models (12).
This Opinion focuses on the problems inherent in current, ligand-dependent approaches to pharmacological inhibition of TREM-1, TREM-2 and other multiligand receptors, and suggests solutions to these problems by using ligand-independent strategies that will minimize the risk of failure in the clinic.
2 Inhibitors for multiligand receptors
Multiligand receptors are those that specifically interact with multiple ligands (13). Examples are TREM-1 and TREM-2 (5, 14, 15), toll-like receptors (TLR) (16, 17), receptor for advanced glycation end products (RAGE) (18–21), CD36 (22–24) and scavenger receptors (SR) (25–28). Despite numerous preclinical findings supporting a therapeutic value of blocking these receptors, no approved agents are currently available for their specific inhibition.
2.1 Poor preclinical to clinical translatability
2.1.1 TREM-1
Discovered in 2000 (14), TREM-1, mainly expressed on neutrophils, monocytes, and macrophages, is a key player in the pathogenesis of inflammatory diseases (1–6). TREM-1 is upregulated upon inflammation (5, 29–32) and functions as an inflammation amplifier (5, 14, 33–37) by mediating release of proinflammatory cytokines (5, 38, 39) (Figure 1A).

Figure 1. TREM-1: receptor assembly, signaling and mechanisms of ligand-dependent and ligand-independent inhibition (A) TREM-1/DAP-12 receptor complex assembly and signaling are depicted. TREM-1 and DAP-12 are bound together by electrostatic interactions between their basic and acidic amino acids in the cell membrane. Binding to the known and unknown natural human ligands of TREM-1 results in dimerization/multimerization of the TREM-1/DAP-12 complex followed by homooligomerization of cytoplasmic DAP-12 domains that triggers the receptor. (B) Ligand-dependent inhibitors attempt to block interaction of TREM-1 with its multiple ligands by binding with either the ligands (decoy peptides) or TREM-1 (anti-TREM-1 blocking antibodies, peptides and small molecules). (C) Ligand-independent peptide inhibitor GF9 (SCHOOL mechanism-based peptide) disrupts the interactions between TREM-1 and DAP-12 in the cell membrane that upon binding to the ligands, results in dimerization/multimerization of only TREM-1 but not DAP-12, therefore completely blocking the transmembrane signaling. GF9 can reach its site of action in the cell membrane from not only outside but also inside the cell enabling targeted intracellular delivery of GF9 peptide sequence-based TREM-1 inhibitor to certain types of TREM-1-expressing cells. eCIRP, extracellular cold-inducible RNA-binding protein; HMGB1, high mobility group box 1; Hsp70, heat shock protein 70 kDa; PGLYRP1, peptidoglycan recognition protein 1; SCHOOL, signaling chain homooligomerization.
Therapeutic effect of TREM-1 blockade was demonstrated in animal models of sepsis (40, 41), cancer (42–44), acute respiratory distress syndrome (45–47), inflammatory bowel disease (32, 48), rheumatoid arthritis (RA) (49–51), and other inflammation-associated pathologies (52–75).
However, despite promising preclinical efficacy and proven safety in humans (76–82), the first clinical TREM-1 blocker, LR12, failed in the clinic (7, 8). Thus, after more than two decades of intensive research, there are still no approved TREM-1 inhibitors.
2.1.2 TREM-2
First reported in 2001 (83), TREM-2 is expressed on human monocyte-derived dendritic cells, microglia, osteoclasts and tissue macrophage subsets (5, 15). In contrast to TREM-1 that amplifies inflammation (2, 5, 37), TREM-2 acts as a negative (66, 84–88) or positive (51, 89–92) regulator of inflammation.
TREM-2 is overexpressed on tumor-associated macrophages (TAMs) and plays a role in tumorigenesis (5, 93–97). Recent studies (98) resulted in the development of the first anti-TREM-2 antagonistic mAb, PY314, that moved in 2020 into a clinical trial on patients with solid tumors (NCT04691375) (12, 99). However, the limited anti-tumor effect of PY314 observed (12) led to the termination of the trial. Currently, there are no clinical inhibitors of this emerging cancer target.
2.1.3 Other multiligand receptors
TLRs. Discovered decades ago (100), TLRs play a role in human health and disease (101) and are emerging targets with wide applicability (102, 103), including the treatment of autoimmune and infectious diseases, and cancer (104, 105). Various TLR-inhibitory therapeutics showed efficacy in preclinical studies but failed at different stages of clinical testing (103, 106) and none have been approved for clinical uses (106).
RAGE. First reported in 1992 (107), RAGE is involved in the pathogenesis of inflammation (108–110). Despite decades of interest and research, many RAGE inhibitors that yielded promising preclinical findings failed to translate to humans (108, 109, 111–113). There are no approved therapeutics based on the RAGE antagonists as yet.
Scavenger Receptors. CD36 is a multiligand receptor (22, 114, 115) that was first identified in 1977 (116). CD36 is an emerging target in cancer (117, 118) but most of CD36-targeting drug candidates that demonstrated efficacy in preclinical studies failed in humans because of severe adverse events and unsatisfactory efficacy (114). SR-BI, first isolated in 1993 (119), is a multiligand receptor that binds to diverse ligands (120, 121), including lipopolysaccharide (LPS) (122). Despite recognition of SR-BI as an important therapeutic target (123) and promising preclinical findings (123–125), none of the SR-BI inhibitors tested was successful in humans. First described in 1979 (126), SR-A (or CD204) is a multiligand receptor (127) that plays key roles in innate immunity and inflammation (128). Several SR-A inhibitors were reported (129–132), but none is specific for SR-A (132). Thus, there are currently no clinically available inhibitors of CD36, SR-BI and SR-A.
2.1.4 Summary
In summary, despite drug discovery interest and numerous preclinical and clinical studies over several decades, no clinical inhibitors of TREM-1, TREM-2 and other multiligand receptors have been developed to date. I hypothesize that a ligand-dependent mechanism of action of most of the inhibitors tested was and remains a major driver for their failures in humans and that this can be overcome by using a ligand-independent inhibition strategy.
2.2 Ligand-dependent mechanism of action: excellence in preclinical, failure in late clinical?
TREM-1 is noncovalently bound in the membrane with its signaling partner, DAP-12, and belongs to a family of multichain immune recognition receptors (MIRRs) (133). According to the Signaling Chain HOmoOLigomerization (SCHOOL) model of MIRR signaling (134, 135), binding of TREM-1 to its ligand(s) results to dimerization/multimerization of the TREM-1/DAP-12 complex followed by homooligomerization of DAP-12 that triggers the receptor (Figure 1A).
Since 2015 when the first ligand for TREM-1, PGLYRP1, was identified (9), at least, four other potential ligands have been reported: actin, eCIRP, HMGB1, and Hsp70 (10, 11). It is likely that still more remain unidentified and uncharacterized at present.
Most of current strategies for inhibition of TREM-1 use ligand-dependent inhibitors such as decoy receptor/peptides or anti-TREM-1 antagonistic mAbs (Figure 1B). Mechanistically, they attempt to block the interactions of TREM-1 with its multiple known and unknown ligands by binding either to ligands (e.g., decoy peptides LP17 and LR12 (81, 136) or to TREM-1 (e.g., an anti-TREM-1 blocking mAB (48); human eCIRP-derived peptide M3 (70, 73, 137, 138); PGLYRP1-derived peptide N1 (139, 140) and small molecule VJDT (141, 142). Despite the efficacy of these agents in animal models of sepsis (41) and other inflammatory diseases (48, 55, 67, 70, 74, 137–139, 141, 143), only one of them, LR12, has entered clinical trials but failed in phase IIb (7, 8, 41).
Similarly, a humanized anti-TREM-2 mAb PY314 exhibited potent anti-tumor activity in mouse cancer models (144), but was ineffective in the clinical setting (12). Notably, the majority of inhibitors for other multiligand receptors that succeeded in animals but failed in humans (106, 109, 113, 114, 145) were also ligand-dependent.
My hypothesis is that traditional approaches aiming to develop drugs that interfere with the interactions of TREM-1, TREM-2 and other multiligand receptors with their multiple known and unknown ligands all bear a high risk of failure not only in preclinical but also in clinical efficacy testing. This is illustrated on the example of TREM-1 and based on the following major considerations.
First of all, in preclinical testing, potential TREM-1 inhibitors are not and practically cannot be evaluated for their efficacy in blocking interactions between TREM-1 and all its multiple ligands of human origin. Thus, a clinical efficacy trial is the first setting where TREM-1 inhibitory drug candidates compete with known and unknown human ligands or TREM-1 by interaction either with ligands (decoy receptor/peptides) or receptor (anti-TREM-1 blocking antibodies) (Figure 1B). The diverse roles played by different ligands of TREM-1 in the pathogenesis of inflammatory diseases and the likely existence of not yet discovered ligands of TREM-1 add more uncertainty to the outcome of clinical efficacy trials. In addition, as demonstrated in sepsis, the expression levels and timing of different TREM-1 ligands may vary depending on the stage of sepsis and type of infection (146–148), further increasing risk of failure of ligand-dependent TREM-1 inhibitors.
Second, decoy peptide-based inhibitors (e.g., LR12) may have off-target effects due to ligand promiscuity. For example, the biological function of HMGB1 is mediated by not only TREM-1 but also other receptors such as TLRs and RAGE, which are expressed on different cells (149). Thus, by binding to HMGB1, LR12 can affect other signaling pathways.
Third, ligand-dependent inhibitors of TREM-1 that bind to ligands (LP17 and LR12) or receptor (anti-TREM-1 blocking mAbs, peptides M3 and N1, and a small molecule VJDT) (Figure 1B), all are “pan-TREM-1” inhibitors, i.e., they inhibit TREM-1 on all TREM-1-expressing cells. However, these cells may play different roles in the pathogenesis of infectious and non-infectious inflammatory diseases. For example, in mice with Pseudomonas aeruginosa-induced pneumonia, TREM-1/3 deficiency in neutrophils causes increased mortality due to the failure to clear lung bacteria (150). Increased bacterial growth and dissemination and decreased survival were also observed in TREM-1/3-deficient mice with Klebsiella-derived pneumosepsis (151) and pneumococcal pneumonia (152).
In summary, for ligand-dependent inhibitors of TREM-1, TREM-2 and other multiligand receptors, promising preclinical and safety studies may not translate to clinical efficacy. This risk can be mitigated by using the inhibitors that employ ligand-independent mechanisms of action.
2.3 Ligand-independent mechanism of action: minimizing risk of drug failure
The interactions in the membrane between ligand-recognizing and signal-transducing MIRR subunits can be targeted by short synthetic peptides with receptor-specific sequences (153–155). TREM-1 inhibitory peptide GF9, designed to disrupt the interactions between TREM-1 and DAP-12, thus preventing formation of DAP-12 homooligomers (Figure 1C), ameliorates various inflammation-associated diseases in animal models (51, 65, 71, 153, 156–158). Peptide IA9, designed for ligand-independent inhibition of TREM-2, suppresses joint inflammation and damage in experimental RA (51).
In addition to their ligand-independent mechanism of action, another chief advantage of GF9 and IA9 is that their site of action in the membrane enables the development of targeted therapies. As illustrated for TREM-1 (Figure 1C), when systemically administered, the GF9 peptide inserts into the membrane of any cell and inhibits TREM-1 wherever it is expressed, acting thus as a pan-TREM-1 inhibitor. When delivered intracellularly to TREM-1-expressing cells of interest (e.g., macrophages including TAMs), GF9 exerts its therapeutic action by inserting into the membrane from inside the cell. This key feature allowed to develop macrophage-targeted formulations of GF9 sequence-based inhibitors effective in suppressing TREM-1-mediated macrophage activation and ameliorating inflammatory diseases in several animal models (51, 71, 153). Following a similar strategy, a macrophage-targeted formulation of IA9 sequence-based TREM-2 inhibitor was designed and demonstrated to ameliorate arthritis in mice (51).
Thus, the use of ligand-independent inhibitors of TREM-1 and TREM-2 not only addresses the multiplicity and promiscuity of their multiple known and unknown ligands, but also enables targeted delivery of these inhibitors to block TREM-1 and TREM-2 on selected cell types. This addresses the potential for different cell types playing different roles in the pathogenesis of many diseases.
Ligand-independent inhibition strategies have been also considered for other multiligand receptors. TLR2 inhibition via blockage of TLR2 dimerization has been recently reported (159). An anti-human TLR4 mAb, NI-0101, that blocks TLR4 dimerization (160) showed efficacy in healthy volunteers receiving LPS (161). Several small molecules that inhibit RAGE by blocking the interaction of the RAGE cytoplasmic domain with DIAPH1 are currently in development (111, 162, 163).
3 Conclusion and perspectives
Despite growing interest in therapeutic targeting TREM-1 and TREM-2 over the past two decades (2, 5, 15, 40, 42, 43, 66, 95, 96, 98, 164–168), promising preclinical findings for the first clinical inhibitors of TREM-1 (LR12) and TREM-2 (an anti-TREM-2 antibody PY314) did not translate into clinical efficacy in sepsis (7, 8) and oncology (12) patients, respectively.
To my knowledge, this Opinion is the first attempt to analyze poor preclinical to clinical translatability of inhibitors for TREM-1 and TREM-2 from the angle of multiplicity of their known and unknown ligands and extend this analysis to other multiligand receptors. In summary, deploying a ligand-independent mechanism of action for the pharmacological inhibition of TREM-1 and TREM-2 could bridge the efficacy gap between preclinical and clinical testing observed for ligand-dependent inhibitors as well as mitigate the risk of drug failure. Importantly, the existing preclinical development path may not reveal any difference between ligand-dependent and ligand-independent inhibitors of TREM-1 and TREM-2 since in animal models, these inhibitors do not compete with various TREM-1 and TREM-2 ligands of human origin.
In conclusion, ligand-independent inhibition is a promising alternative strategy to target TREM-1 and TREM-2 in human disease. By addressing the multiplicity and promiscuity of TREM-1 and TREM-2 ligands as well as the differential roles played by these receptors expressed on different cells, this approach inherently mitigates the risk of failure in the clinic, which can save time and resources and substantially increase the odds of success in developing of novel drugs targeting the TREM-1 and TREM-2 signaling pathways.
Author contributions
AS: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by grants R44CA217400 from the National Cancer Institute of (NCI), R44EY034015 from the National Eye Institute (NEI), R43GM128369 from the National Institute of General Medical Sciences (NIGMS), R43HL165734 from the National Heart, Lung, and Blood Institute (NHLBI). NCI, NEI, NIGMS, and NHLBI are components of the National Institutes of Health (NIH).
Acknowledgments
I thank Dr. Zu Shen for his critical reading of the manuscript.
Conflict of interest
The author AS is employed by SignaBlok, Inc.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roe K, Gibot S and Verma S. Triggering receptor expressed on myeloid cells-1 (TREM-1): a new player in antiviral immunity? Front Microbiol. (2014) 5:627. doi: 10.3389/fmicb.2014.00627
2. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S and Dessing MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther. (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
3. Pelham CJ, Agrawal DK. Emerging roles for triggering receptor expressed on myeloid cells receptor family signaling in inflammatory diseases. Expert Rev Clin Immunol. (2014) 10:243–56. doi: 10.1586/1744666X.2014.866519
4. Nguyen AH, Berim IG and Agrawal DK. Chronic inflammation and cancer: emerging roles of triggering receptors expressed on myeloid cells. Expert Rev Clin Immunol. (2015) 11:849–57. doi: 10.1586/1744666X.2015.1043893
5. Colonna M. The biology of TREM receptors. Nat Rev Immunol. (2023) 23:580–94. doi: 10.1038/s41577-023-00837-1
6. Gao S, Yi Y, Xia G, Yu C, Ye C, Tu F, et al. The characteristics and pivotal roles of triggering receptor expressed on myeloid cells-1 in autoimmune diseases. Autoimmun Rev. (2019) 18:25–35. S1568-9972(18)30257-X doi: 10.1016/j.autrev.2018.07.008
7. Francois B, Lambden S, Fivez T, Gibot S, Derive M, Grouin JM, et al. Prospective evaluation of the efficacy, safety, and optimal biomarker enrichment strategy for nangibotide, a TREM-1 inhibitor, in patients with septic shock (ASTONISH): a double-blind, randomised, controlled, phase 2b trial. Lancet Respir Med. (2023) 11:894–904. doi: 10.1016/S2213-2600(23)00158-3
8. Neupane M, Kadri SS. Nangibotide for precision immunomodulation in septic shock and COVID-19. Lancet Respir Med. (2023) 11:855–7. S2213-2600(23)00220-5 doi: 10.1016/S2213-2600(23)00220-5
9. Read CB, Kuijper JL, Hjorth SA, Heipel MD, Tang X, Fleetwood AJ, et al. Cutting Edge: identification of neutrophil PGLYRP1 as a ligand for TREM-1. J Immunol. (2015) 194:1417–21.jimmunol.1402303 doi: 10.4049/jimmunol.1402303
10. Singh H, Rai V, Nooti SK and Agrawal DK. Novel ligands and modulators of triggering receptor expressed on myeloid cells receptor family: 2015-2020 updates. Expert Opin Ther Pat. (2021) 31:549–61. doi: 10.1080/13543776.2021.1883587
11. de Oliveira Matos A, dos Santos Dantas PH, Colmenares MTC, Sartori GR, Silva-Sales M, Da Silva JHM, et al. The CDR3 region as the major driver of TREM-1 interaction with its ligands, an in silico characterization. Comput Struct Biotechnol J. (2023) 21:2579–90. doi: 10.1016/j.csbj.2023.04.008
12. Beckermann KE, Patnaik A, Winer I, Tan W, Bashir B, Kyriakopoulos CE, et al. A phase 1b open-label study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of py314 in combination with pembrolizumab in patients with advanced renal cell carcinoma. Invest New Drugs. (2024) 42:179–84. doi: 10.1007/s10637-024-01419-1
13. Krieger M, Stern DM. Series introduction: multiligand receptors and human disease. J Clin Invest. (2001) 108:645–7. doi: 10.1172/JCI13932
14. Bouchon A, Dietrich J and Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. (2000) 164:4991–5. doi: 10.4049/jimmunol.164.10.4991
15. Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. (2003) 3:445–53. doi: 10.1038/nri1106
16. Sabroe I, Read RC, Whyte MK, Dockrell DH, Vogel SN and Dower SK. Toll-like receptors in health and disease: complex questions remain. J Immunol. (2003) 171:1630–5. doi: 10.4049/jimmunol.171.4.1630
17. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. (2004) 4:499–511. doi: 10.1038/nri1391
18. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. (2018) 69:349–64. doi: 10.1146/annurev-med-041316-085215
19. Rouhiainen A, Kuja-Panula J, Tumova S and Rauvala H. RAGE-mediated cell signaling. Methods Mol Biol. (2013) 963:239–63. doi: 10.1007/978-1-62703-230-8_15
20. Rauvala H, Rouhiainen A. RAGE as a receptor of HMGB1 (Amphoterin): roles in health and disease. Curr Mol Med. (2007) 7:725–34. doi: 10.2174/156652407783220750
21. Yan SD, Stern D and Schmidt AM. What’s the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest. (1997) 27:179–81. doi: 10.1046/j.1365-2362.1996.00072.x
22. Chen Y, Zhang J, Cui W and Silverstein RL. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J Exp Med. (2022) 219:e20211314. doi: 10.1084/jem.20211314
23. Karunakaran U, Elumalai S, Moon JS and Won KC. CD36 signal transduction in metabolic diseases: novel insights and therapeutic targeting. Cells. (2021) 10:1833. doi: 10.3390/cells10071833
24. Daviet L, McGregor JL. Vascular biology of CD36: roles of this new adhesion molecule family in different disease states. Thromb Haemost. (1997) 78:65–9.
25. Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu Rev Biochem. (1994) 63:601–37. doi: 10.1146/annurev.bi.63.070194.003125
26. Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. (2001) 108:793–7. doi: 10.1172/JCI14011
27. Xie Y, Jia Y, Li Z and Hu F. Scavenger receptor A in immunity and autoimmune diseases: Compelling evidence for targeted therapy. Expert Opin Ther Targets. (2022) 26:461–77. doi: 10.1080/14728222.2022.2072729
28. Yu L, Dai Y and Mineo C. Novel functions of endothelial scavenger receptor class B type I. Curr Atheroscler Rep. (2021) 23:6. doi: 10.1007/s11883-020-00903-2
29. Gonzalez-Roldan N, Ferat-Osorio E, Aduna-Vicente R, Wong-Baeza I, Esquivel-Callejas N, Astudillo-de la Vega H, et al. Expression of triggering receptor on myeloid cell 1 and histocompatibility complex molecules in sepsis and major abdominal surgery. World J Gastroenterol. (2005) 11:7473–9. doi: 10.3748/wjg.v11.i47.7473
30. Koussoulas V, Vassiliou S, Demonakou M, Tassias G, Giamarellos-Bourboulis EJ, Mouktaroudi M, et al. Soluble triggering receptor expressed on myeloid cells (sTREM-1): a new mediator involved in the pathogenesis of peptic ulcer disease. Eur J Gastroenterol Hepatol. (2006) 18:375–9. doi: 10.1097/00042737-200604000-00010
31. Wang DY, Qin RY, Liu ZR, Gupta MK and Chang Q. Expression of TREM-1 mRNA in acute pancreatitis. World J Gastroenterol. (2004) 10:2744–6. doi: 10.3748/wjg.v10.i18.2744
32. Schenk M, Bouchon A, Seibold F and Mueller C. TREM-1–expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest. (2007) 117:3097–106. doi: 10.1172/JCI30602
33. Bouchon A, Facchetti F, Weigand MA and Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. (2001) 410:1103–7. doi: 10.1038/35074114
34. Bleharski JR, Kiessler V, Buonsanti C, Sieling PA, Stenger S, Colonna M, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol. (2003) 170:3812–8. doi: 10.4049/jimmunol.170.7.3812
35. Tessarz AS, Cerwenka A. The TREM-1/DAP12 pathway. Immunol Lett. (2008) 116:111–6. doi: 10.1016/j.imlet.2007.11.021
36. Klesney-Tait J, Turnbull IR and Colonna M. The TREM receptor family and signal integration. Nat Immunol. (2006) 7:1266–73. doi: 10.1038/ni1411
37. Colonna M, Facchetti F. TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J Infect Dis. (2003) 187 Suppl 2:S397–401. doi: 10.1086/jid.2003.187.issue-s2
38. Kuai J, Gregory B, Hill A, Pittman DD, Feldman JL, Brown T, et al. TREM-1 expression is increased in the synovium of rheumatoid arthritis patients and induces the expression of pro-inflammatory cytokines. Rheumatol (Oxford). (2009) 48:1352–8. doi: 10.1093/rheumatology/kep235
39. Dower K, Ellis DK, Saraf K, Jelinsky SA and Lin LL. Innate immune responses to TREM-1 activation: overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J Immunol. (2008) 180:3520–34. doi: 10.4049/jimmunol.180.5.3520
40. Siskind S, Brenner M and Wang P. TREM-1 modulation strategies for sepsis. Front Immunol. (2022) 13:907387. doi: 10.3389/fimmu.2022.907387
41. Theobald V, Schmitt FCF, Middel CS, Gaissmaier L, Brenner T and Weigand MA. Triggering receptor expressed on myeloid cells-1 in sepsis, and current insights into clinical studies. Crit Care. (2024) 28:17. doi: 10.1186/s13054-024-04798-2
42. Muller M, Haghnejad V, Lopez A, Tiotiu A, Renaud S, Derive M, et al. Triggering receptors expressed on myeloid cells 1: our new partner in human oncology? Front Oncol. (2022) 12:927440. doi: 10.3389/fonc.2022.927440
43. Li C, Cai C, Xu D, Chen X and Song J. TREM1: Activation, signaling, cancer and therapy. Pharmacol Res. (2024) 204:107212. doi: 10.1016/j.phrs.2024.107212
44. Bosco MC, Raggi F and Varesio L. Therapeutic potential of targeting TREM-1 in inflammatory diseases and cancer. Curr Pharm Des. (2016) 22:6209–33. doi: 10.2174/1381612822666160826110539
45. Yuan Z, Syed M, Panchal D, Joo M, Bedi C, Lim S, et al. TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L426–38. doi: 10.1152/ajplung.00195.2015
46. Sadikot RT, Kolanjiyil AV, Kleinstreuer C, Rubinstein I. Nanomedicine for treatment of acute lung injury and acute respiratory distress syndrome. BioMed Hub. (2017) 2:1–12. doi: 10.1159/000477086
47. Shi R, Zhang J, Peng Z, Yuan S, Gao S, Chen L, et al. Expression level of 12-amino acid triggering receptor on myeloid cells-like transcript 1 derived peptide alleviates lipopolysaccharide-induced acute lung injury in mice. Int J Mol Med. (2018) 41:2159–68. doi: 10.3892/ijmm.2018.3443
48. Brynjolfsson SF, Magnusson MK, Kong PL, Jensen T, Kuijper JL, Hakansson K, et al. An antibody against triggering receptor expressed on myeloid cells 1 (TREM-1) dampens proinflammatory cytokine secretion by lamina propria cells from patients with IBD. Inflammation Bowel Dis. (2016) 22:1803–11. doi: 10.1097/MIB.0000000000000822
49. Murakami Y, Akahoshi T, Aoki N, Toyomoto M, Miyasaka N and Kohsaka H. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. (2009) 60:1615–23. doi: 10.1002/art.24554
50. Shen ZT, Sigalov AB. Rationally designed ligand-independent peptide inhibitors of TREM-1 ameliorate collagen-induced arthritis. J Cell Mol Med. (2017) 21:2524–34. doi: 10.1111/jcmm.13173
51. Sigalov AB. Inhibition of TREM-2 markedly suppresses joint inflammation and damage in experimental arthritis. Int J Mol Sci. (2022) 23:8857. doi: 10.3390/ijms23168857
52. Rojas MA, Shen ZT, Caldwell RB and Sigalov AB. Blockade of TREM-1 prevents vitreoretinal neovascularization in mice with oxygen-induced retinopathy. Biochim Biophys Acta. (2018) 1864:2761–8. doi: 10.1016/j.bbadis.2018.05.001
53. He Y, Yang Q, Wang X, Jia A, Xie W and Zhou J. Inhibition of triggering receptor expressed on myeloid cell-1 alleviates acute gouty inflammation. Mediators Inflammation. (2019) 2019:5647074. doi: 10.1155/2019/5647074
54. Potteaux S, Joffre J, Esposito B, Tedgui A, Gibot S, Mallat Z, et al. Exploration and modulation of TREM-1 in experimental atherosclerosis. Arterioscler Thromb Vasc Biol. (2013) 33:A135. doi: 10.1161/atvb.33.suppl_1.A135
55. Joffre J, Potteaux S, Zeboudj L, Loyer X, Boufenzer A, Laurans L, et al. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J Am Coll Cardiol. (2016) 68:2776–93. doi: 10.1016/j.jacc.2016.10.015
56. Luo L, Zhou Q, Chen XJ, Qin SM, Ma WL and Shi HZ. Effects of the TREM-1 pathway modulation during empyema in rats. Chin Med J (Engl). (2010) 123:1561–5.
57. Sun XG, Duan H, Jing G, Wang G, Hou Y and Zhang M. Inhibition of TREM-1 attenuates early brain injury after subarachnoid hemorrhage via downregulation of p38MAPK/MMP-9 and preservation of ZO-1. Neuroscience. (2019) 406:369–75. doi: 10.1016/j.neuroscience.2019.03.032
58. Zhao T, Zhou Y, Zhang D, Han D, Ma J, Li S, et al. Inhibition of TREM-1 alleviates neuroinflammation by modulating microglial polarization via SYK/p38MAPK signaling pathway after traumatic brain injury. Brain Res. (2024) 1834:148907. doi: 10.1016/j.brainres.2024.148907
59. Xie Y, He W, Ma L, Ren R, Yang S and Lu Q. Endothelial TREM-1 receptor regulates the blood-brain barrier integrity after intracerebral hemorrhage in mice via SYK/beta-catenin signaling. CNS Neurosci Ther. (2023) 29:3228–38. doi: 10.1111/cns.14255
60. Li Z, Wu F, Xu D, Zhi Z and Xu G. Inhibition of TREM1 reduces inflammation and oxidative stress after spinal cord injury (SCI) associated with HO-1 expressions. Biomedicine Pharmacotherapy. (2019) 109:2014–21. doi: 10.1016/j.biopha.2018.08.159
61. Wu X, Zeng H, Xu C, Chen H, Fan L, Zhou H, et al. TREM1 regulates neuroinflammatory injury by modulate proinflammatory subtype transition of microglia and formation of neutrophil extracellular traps via interaction with SYK in experimental subarachnoid hemorrhage. Front Immunol. (2021) 12:766178. doi: 10.3389/fimmu.2021.766178
62. Li F, Qin N, Yu Y, Dong R, Li X, Gong S, et al. TREM-1 inhibition or ondansetron administration ameliorates NLRP3 inflammasome and pyroptosis in traumatic brain injury-induced acute lung injury. Arch Med Sci. (2024) 20:984–96. doi: 10.5114/aoms/174264
63. Feng CW, Chen NF, Sung CS, Kuo HM, Yang SN, Chen CL, et al. Therapeutic effect of modulating TREM-1 via anti-inflammation and autophagy in parkinson’s disease. Front Neurosci. (2019) 13:769. doi: 10.3389/fnins.2019.00769
64. Tornai D, Furi I, Shen ZT, Sigalov AB, Coban S and Szabo G. Inhibition of triggering receptor expressed on myeloid cells 1 ameliorates inflammation and macrophage and neutrophil activation in alcoholic liver disease in mice. Hepatol Commun. (2019) 3:99–115. doi: 10.1002/hep4.1269
65. Giraud J, Chalopin D, Ramel E, Boyer T, Zouine A, Derieppe MA, et al. THBS1(+) myeloid cells expand in SLD hepatocellular carcinoma and contribute to immunosuppression and unfavorable prognosis through TREM1. Cell Rep. (2024) 43:113773. doi: 10.1016/j.celrep.2024.113773
66. Sun H, Feng J and Tang L. Function of TREM1 and TREM2 in liver-related diseases. Cells. (2020) 9:2626. doi: 10.3390/cells9122626
67. Wang Y, Xie X, Liu H and Jiang H. LR12 promotes liver repair by improving the resolution of inflammation and liver regeneration in mice with thioacetamide- (TAA-) induced acute liver failure. Mediators Inflammation. (2021) 2021:2327721. doi: 10.1155/2021/2327721
68. Nguyen-Lefebvre AT, Ajith A, Portik-Dobos V, Horuzsko DD, Arbab AS, Dzutsev A, et al. The innate immune receptor TREM-1 promotes liver injury and fibrosis. J Clin Invest. (2018) 128:4870–83. doi: 10.1172/JCI98156
69. Tammaro A, Kers J, Emal D, Stroo I, Teske GJD, Butter LM, et al. Effect of TREM-1 blockade and single nucleotide variants in experimental renal injury and kidney transplantation. Sci Rep. (2016) 6:38275. doi: 10.1038/srep38275
70. Siskind S, Royster W, Brenner M and Wang P. A novel eCIRP/TREM-1 pathway inhibitor attenuates acute kidney injury. Surgery. (2022) 172:639–47. doi: 10.1016/j.surg.2022.02.003
71. Gallop D, Scanlon KM, Ardanuy J, Sigalov AB, Carbonetti NH and Skerry C. Triggering receptor expressed on myeloid cells-1 (TREM-1) contributes to bordetella pertussis inflammatory pathology. Infect Immun. (2021) 89:e0012621. doi: 10.1128/IAI.00126-21
72. Xiong JB, Duan JX, Jiang N, Zhang CY, Zhong WJ, Yang JT, et al. TREM-1 exacerbates bleomycin-induced pulmonary fibrosis by aggravating alveolar epithelial cell senescence in mice. Int Immunopharmacol. (2022) 113:109339. doi: 10.1016/j.intimp.2022.109339
73. Denning NL, Aziz M, Murao A, Gurien SD, Ochani M, Prince JM, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight. (2020) 5:e134172. doi: 10.1172/jci.insight.134172
74. Lemarie J, Boufenzer A, Popovic B, Tran N, Groubatch F, Derive M, et al. Pharmacological inhibition of the triggering receptor expressed on myeloid cells-1 limits reperfusion injury in a porcine model of myocardial infarction. ESC Heart Fail. (2015) 2:90–9. doi: 10.1002/ehf2.12029
75. Jolly L, Lemarie J, Carrasco K, Popovic B, Derive M, Boufenzer A, et al. Triggering Receptor Expressed on Myeloid cells-1: a new player in platelet aggregation. Thromb Haemost. (2017) 117:1772–81. doi: 10.1160/TH17-03-0156
76. Gibot S, Buonsanti C, Massin F, Romano M, Kolopp-Sarda MN, Benigni F, et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun. (2006) 74:2823–30. doi: 10.1128/IAI.74.5.2823-2830.2006
77. Gibot S, Massin F, Marcou M, Taylor V, Stidwill R, Wilson P, et al. TREM-1 promotes survival during septic shock in mice. Eur J Immunol. (2007) 37:456–66. doi: 10.1002/eji.200636387
78. Cuvier V, Lorch U, Witte S, Olivier A, Gibot S, Delor I, et al. A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition. Br J Clin Pharmacol. (2018) 84:2270–9. doi: 10.1111/bcp.13668
79. Francois B, Wittebole X, Mira JP, Dugernier T, Gibot S, Derive M, et al. P1 Safety and pharmacodynamic activity of a novel TREM-1 pathway inhibitory peptide in septic shock patients: phase IIa clinical trial results. Intensive Care Med Exp. (2018) 6. doi: 10.1186/s40635-018-0196-z
80. Francois B, Wittebole X, Ferrer R, Mira JP, Dugernier T, Gibot S, et al. Nangibotide in patients with septic shock: a Phase 2a randomized controlled clinical trial. Intensive Care Med. (2020) 46:1425–37. doi: 10.1007/s00134-020-06109-z
81. Derive M, Boufenzer A, Bouazza Y, Groubatch F, Alauzet C, Barraud D, et al. Effects of a TREM-like transcript 1-derived peptide during hypodynamic septic shock in pigs. Shock. (2013) 39:176–82. doi: 10.1097/SHK.0b013e31827bcdfb
82. Derive M, Boufenzer A and Gibot S. Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate. Anesthesiology. (2014) 120:935–42. doi: 10.1097/ALN.0000000000000078
83. Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. (2001) 194:1111–22. doi: 10.1084/jem.194.8.1111
84. Gao X, Dong Y, Liu Z and Niu B. Silencing of triggering receptor expressed on myeloid cells-2 enhances the inflammatory responses of alveolar macrophages to lipopolysaccharide. Mol Med Rep. (2013) 7:921–6. doi: 10.3892/mmr.2013.1268
85. Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. (2006) 177:3520–4. doi: 10.4049/jimmunol.177.6.3520
86. Li T, Lu L, Pember E, Li X, Zhang B and Zhu Z. New insights into neuroinflammation involved in pathogenic mechanism of alzheimer’s disease and its potential for therapeutic intervention. Cells. (2022) 11:1925. doi: 10.3390/cells11121925
87. Deczkowska A, Weiner A and Amit I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell. (2020) 181:1207–17. doi: 10.1016/j.cell.2020.05.003
88. Rai V, Dietz NE, Dilisio MF, Radwan MM and Agrawal DK. Vitamin D attenuates inflammation, fatty infiltration, and cartilage loss in the knee of hyperlipidemic microswine. Arthritis Res Ther. (2016) 18:203. doi: 10.1186/s13075-016-1099-6
89. Wang M, Gao X, Zhao K, Chen H, Xu M and Wang K. Effect of TREM2 on release of inflammatory factor from LPS-stimulated microglia and its possible mechanism. Ann Clin Lab Sci. (2019) 49:249–56.
90. Suchankova M, Bucova M, Tibenska E, Tedlova E, Demian J, Majer I, et al. Triggering receptor expressed on myeloid cells-1 and 2 in bronchoalveolar lavage fluid in pulmonary sarcoidosis. Respirology. (2013) 18:455–62. doi: 10.1111/resp.12028
91. Sharif O, Gawish R, Warszawska JM, Martins R, Lakovits K, Hladik A, et al. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumonia. PloS Pathog. (2014) 10:e1004167. doi: 10.1371/journal.ppat.1004167PPATHOGENS-D-13-02922
92. Genua M, Rutella S, Correale C and Danese S. The triggering receptor expressed on myeloid cells (TREM) in inflammatory bowel disease pathogenesis. J Transl Med. (2014) 12:293. doi: 10.1186/s12967-014-0293-zs12967-014-0293-z
93. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. (2020) 182:886–900.e17. doi: 10.1016/j.cell.2020.07.013
94. Zheng W, Yu W, Hua R, He J, Wu N, Tian S, et al. Systematic analysis of TREM2 and its carcinogenesis in pancreatic cancer. Transl Cancer Res. (2024) 13:3200–16. doi: 10.21037/tcr-24-201
95. Zheng P, Tan Y, Liu Q, Wu C, Kang J, Liang S, et al. Deciphering the molecular and clinical characteristics of TREM2, HCST, and TYROBP in cancer immunity: A comprehensive pan-cancer study. Heliyon. (2024) 10:e26993. doi: 10.1016/j.heliyon.2024.e26993
96. Lei X, Gou YN, Hao JY and Huang XJ. Mechanisms of TREM2 mediated immunosuppression and regulation of cancer progression. Front Oncol. (2024) 14:1375729. doi: 10.3389/fonc.2024.1375729
97. Nasir I, McGuinness C, Poh AR, Ernst M, Darcy PK and Britt KL. Tumor macrophage functional heterogeneity can inform the development of novel cancer therapies. Trends Immunol. (2023) 44:971–85. doi: 10.1016/j.it.2023.10.007
98. Molgora M, Liu YA, Colonna M and Cella M. TREM2: A new player in the tumor microenvironment. Semin Immunol. (2023) 67:101739. doi: 10.1016/j.smim.2023.101739
99. Yeku OO, Barve M, Tan WW, Wang JS, Patnaik A, Lorusso P, et al. 126P Evaluation of myeloid targeting agents, PY159 and PY314, in two dose expansion phase Ib trials in platinum-resistant ovarian cancer. Immuno-Oncology Technol. (2023) 20:100598. doi: 10.1016/j.iotech.2023.100598
100. O’Neill LA, Golenbock D and Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. (2013) 13:453–60. doi: 10.1038/nri3446
101. Wang K, Huang H, Zhan Q, Ding H and Li Y. Toll-like receptors in health and disease. MedComm (2020). (2024) 5:e549. doi: 10.1002/mco2.549
102. Lepper PM, Triantafilou M, O’Neill LA, Novak N, Wagner H, Parker AE, et al. Modulation of toll-like receptor signalling as a new therapeutic principle. Mediators Inflammation. (2010) 2010:705612. doi: 10.1155/2010/705612
103. Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics. Nat Rev Drug Discovery. (2010) 9:293–307. doi: 10.1038/nrd3203
104. Farooq M, Batool M, Kim MS and Choi S. Toll-like receptors as a therapeutic target in the era of immunotherapies. Front Cell Dev Biol. (2021) 9:756315. doi: 10.3389/fcell.2021.756315
105. El-Zayat SR, Sibaii H and Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Centre. (2019) 43:187. doi: 10.1186/s42269-019-0227-2
106. Gao W, Xiong Y, Li Q and Yang H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: A journey from molecular to nano therapeutics. Front Physiol. (2017) 8:508. doi: 10.3389/fphys.2017.00508
107. Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. (1992) 267:14987–97.S0021-9258(18)42137-0 doi: 10.1016/S0021-9258(18)42137-0
108. Dong H, Zhang Y, Huang Y and Deng H. Pathophysiology of RAGE in inflammatory diseases. Front Immunol. (2022) 13:931473. doi: 10.3389/fimmu.2022.931473
109. Reddy VP, Aryal P and Soni P. RAGE inhibitors in neurodegenerative diseases. Biomedicines. (2023) 11:1131. doi: 10.3390/biomedicines11041131
110. Taguchi K, Fukami K. RAGE signaling regulates the progression of diabetic complications. Front Pharmacol. (2023) 14:1128872. doi: 10.3389/fphar.2023.1128872
111. Egana-Gorrono L, Lopez-Diez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, et al. Receptor for advanced glycation end products (RAGE) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: insights from human subjects and animal models. Front Cardiovasc Med. (2020) 7:37. doi: 10.3389/fcvm.2020.00037
112. Magna M, Hwang GH, McIntosh A, Drews-Elger K, Takabatake M, Ikeda A, et al. RAGE inhibitor TTP488 (Azeliragon) suppresses metastasis in triple-negative breast cancer. NPJ Breast Cancer. (2023) 9:59. doi: 10.1038/s41523-023-00564-9
113. Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I and Altstiel L. Development of azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild alzheimer’s disease. J Prev Alzheimers Dis. (2018) 5:149–54. doi: 10.14283/jpad.2018.18
114. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. (2019) 9:4893–908. doi: 10.7150/thno.36037
115. Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. (2001) 108:785–91. doi: 10.1172/JCI14006
116. Clemetson KJ, Pfueller SL, Luscher EF and Jenkins CS. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. Biochim Biophys Acta. (1977) 464:493–508. doi: 10.1016/0005-2736(77)90025-6
117. Ruan C, Meng Y and Song H. CD36: an emerging therapeutic target for cancer and its molecular mechanisms. J Cancer Res Clin Oncol. (2022) 148:1551–8. doi: 10.1007/s00432-022-03957-8
118. Jiang M, Karsenberg R, Bianchi F, van den Bogaart G. CD36 as a double-edged sword in cancer. Immunol Lett. (2024) 265:7–15. doi: 10.1016/j.imlet.2023.12.002
119. Calvo D, Vega MA. Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J Biol Chem. (1993) 268:18929–35.S0021-9258(17)46716-0 doi: 10.1016/S0021-9258(17)46716-0
120. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH and Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. (1996) 271:518–20. doi: 10.1126/science.271.5248.518
121. Rigotti A, Miettinen HE and Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. (2003) 24:357–87. doi: 10.1210/er.2001-0037
122. Morin EE, Guo L, Schwendeman A and Li XA. HDL in sepsis - risk factor and therapeutic approach. Front Pharmacol. (2015) 6:244. doi: 10.3389/fphar.2015.00244
123. Hoekstra M, Sorci-Thomas M. Rediscovering scavenger receptor type BI: surprising new roles for the HDL receptor. Curr Opin Lipidol. (2017) 28:255–60. doi: 10.1097/MOL.0000000000000413
124. Faloon PW, Dockendorff C, Germain A, Yu M, Nag PP, Youngsaye W, et al. A small molecule inhibitor of scavenger receptor BI-mediated lipid uptake-probe. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). (2010) 2.
125. Faloon PW, Dockendorff C, Youngsaye W, Yu M, Nag PP, Lewis TA, et al. A small molecule inhibitor of scavenger receptor BI-mediated lipid uptake-probe. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). (2010) 1.
126. Goldstein JL, Ho YK, Basu SK and Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U.S.A. (1979) 76:333–7. doi: 10.1073/pnas.76.1.333
127. Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse’s tale. J Clin Invest. (2001) 108:649–54. doi: 10.1172/JCI13903
128. Kelley JL, Ozment TR, Li C, Schweitzer JB, Williams DL. Scavenger receptor-A (CD204): a two-edged sword in health and disease. Crit Rev Immunol. (2014) 34:241–61. doi: 10.1615/critrevimmunol.2014010267
129. Neyen C, Mukhopadhyay S, Gordon S and Hagemann T. An apolipoprotein A-I mimetic targets scavenger receptor A on tumor-associated macrophages: A prospective anticancer treatment? Oncoimmunology. (2013) 2:e24461. doi: 10.4161/onci.24461
130. Neyen C, Pluddemann A, Mukhopadhyay S, Maniati E, Bossard M, Gordon S, et al. Macrophage scavenger receptor a promotes tumor progression in murine models of ovarian and pancreatic cancer. J Immunol. (2013) 190:3798–805. doi: 10.4049/jimmunol.1203194
131. Hu F, Jiang X, Guo C, Li Y, Chen S, Zhang W, et al. Scavenger receptor-A is a biomarker and effector of rheumatoid arthritis: A large-scale multicenter study. Nat Commun. (2020) 11:1911. doi: 10.1038/s41467-020-15700-3
132. Watson DC, Bayik D, Srivatsan A, Bergamaschi C, Valentin A, Niu G, et al. Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials. (2016) 105:195–205. doi: 10.1016/j.biomaterials.2016.07.003
133. Keegan AD, Paul WE. Multichain immune recognition receptors: similarities in structure and signaling pathways. Immunol Today. (1992) 13:63–8. doi: 10.1016/0167-5699(92)90136-U
134. Sigalov AB. Multichain immune recognition receptor signaling: different players, same game? Trends Immunol. (2004) 25:583–9. doi: 10.1016/j.it.2004.08.009
135. Sigalov AB. The SCHOOL of nature: I. Transmembrane signaling. Self Nonself. (2010) 1:4–39. doi: 10.4161/self.1.1.108321938-2030-1-1-3
136. Gibot S, Alauzet C, Massin F, Sennoune N, Faure GC, Bene MC, et al. Modulation of the triggering receptor expressed on myeloid cells-1 pathway during pneumonia in rats. J Infect Dis. (2006) 194:975–83. doi: 10.1086/506950
137. Denning NL, Aziz M, Diao L, Prince JM and Wang P. Targeting the eCIRP/TREM-1 interaction with a small molecule inhibitor improves cardiac dysfunction in neonatal sepsis. Mol Med. (2020) 26:121. doi: 10.1186/s10020-020-00243-6
138. Denning NL, Aziz M, Ochani M, Prince JM and Wang P. Inhibition of a triggering receptor expressed on myeloid cells-1 (TREM-1) with an extracellular cold-inducible RNA-binding protein (eCIRP)-derived peptide protects mice from intestinal ischemia-reperfusion injury. Surgery. (2020) 168:478–85. doi: 10.1016/j.surg.2020.04.010
139. Sharapova TN, Romanova EA, Chernov AS, Minakov AN, Kazakov VA, Kudriaeva AA, et al. Protein PGLYRP1/tag7 peptides decrease the proinflammatory response in human blood cells and mouse model of diffuse alveolar damage of lung through blockage of the TREM-1 and TNFR1 receptors. Int J Mol Sci. (2021) 22:11213. doi: 10.3390/ijms222011213
140. Yurkina DM, Romanova EA, Feoktistov AV, Soshnikova NV, Tvorogova AV, Yashin DV, et al. The interaction of HMGB1 with the proinflammatory TREM-1 receptor generates cytotoxic lymphocytes active against HLA-negative tumor cells. Int J Mol Sci. (2024) 25:627. doi: 10.3390/ijms25010627
141. Ajith A, Mamouni K, Horuzsko DD, Musa A, Dzutsev AK, Fang JR, et al. Targeting TREM1 augments antitumor T cell immunity by inhibiting myeloid-derived suppressor cells and restraining anti-PD-1 resistance. J Clin Invest. (2023) 133:e167951. doi: 10.1172/JCI167951
142. Ren L, Qiao GL, Zhang SX, Zhang ZM and Lv SX. Pharmacological inhibition or silencing of TREM1 restrains HCC cell metastasis by inactivating TLR/PI3K/AKT signaling. Cell Biochem Biophys. (2024) 82:2673–85. doi: 10.1007/s12013-024-01377-8
143. Kokten T, Gibot S, Lepage P, D’Alessio S, Hablot J, Ndiaye NC, et al. TREM-1 inhibition restores impaired autophagy activity and reduces colitis in mice. J Crohns Colitis. (2018) 12:230–44. doi: 10.1093/ecco-jcc/jjx129
144. Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T, et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. (2021) 37:109844. doi: 10.1016/j.celrep.2021.109844
145. Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M and Mjalli AM. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. (2014) 14:12. doi: 10.1186/1471-2377-14-12
146. Kolte A, Taudien S, Giamarellos-Bourboulis E, Sponholz C, Kurzai O, Bauer M, et al. Understanding patients’ genetic variants for bacterial induced sepsis to identify potential drug targets. Infection. (2015) 43:S36–S7.
147. Zhou Y, Dong H, Zhong Y, Huang J, Lv J and Li J. The cold-inducible RNA-binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PloS One. (2015) 10:e0137721. doi: 10.1371/journal.pone.0137721
148. Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD and Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. (2017) 7:5850. doi: 10.1038/s41598-017-06205-z
149. Khambu B, Yan S, Huda N and Yin XM. Role of high-mobility group box-1 in liver pathogenesis. Int J Mol Sci. (2019) 20:5314. doi: 10.3390/ijms20215314
150. Klesney-Tait J, Keck K, Li X, Gilfillan S, Otero K, Baruah S, et al. Transepithelial migration of neutrophils into the lung requires TREM-1. J Clin Invest. (2013) 123:138–49. doi: 10.1172/JCI64181
151. Hommes TJ, Dessing MC, Veer C, Florquin S, Colonna M, de Vos AF, et al. Role of triggering receptor expressed on myeloid cells-1/3 in Klebsiella-derived pneumosepsis. Am J Respir Cell Mol Biol. (2015) 53:647–55. doi: 10.1165/rcmb.2014-0485OC
152. Hommes TJ, Hoogendijk AJ, Dessing MC, Van’t Veer C, Florquin S, Colonna M, et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) improves host defence in pneumococcal pneumonia. J Pathol. (2014) 233:357–67. doi: 10.1002/path.4361
153. Sigalov AB. SCHOOL of nature: ligand-independent immunomodulatory peptides. Drug Discovery Today. (2020) 25:1298–306. doi: 10.1016/j.drudis.2020.05.005
154. Sigalov AB. Immune cell signaling: a novel mechanistic model reveals new therapeutic targets. Trends Pharmacol Sci. (2006) 27:518–24. doi: 10.1016/j.tips.2006.08.004
155. Sigalov AB. The SCHOOL of nature: III. From mechanistic understanding to novel therapies. Self Nonself. (2010) 1:192–224. doi: 10.4161/self.1.3.12794
156. Wei B, Ma Y. Synergistic effect of GF9 and streptomycin on relieving gram-negative bacteria-induced sepsis. Front Bioeng Biotechnol. (2022) 10:973588. doi: 10.3389/fbioe.2022.973588
157. Hu L, Bai G, Xu Q, Zhao G, Jiang N, Yao H, et al. Candidalysin amplifies the immune inflammatory response in Candida albicans keratitis through the TREM-1/DAP12 pathway. Int Immunopharmacol. (2023) 119:110195. doi: 10.1016/j.intimp.2023.110195
158. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology. (2019) 70:198–214. doi: 10.1002/hep.30593
159. Ahmad B, Choi S. Unraveling the tomaralimab epitope on the toll-like receptor 2 via molecular dynamics and deep learning. ACS Omega. (2022) 7:28226–37. doi: 10.1021/acsomega.2c02559
160. Daubeuf B, Giovannoni L, Salgado-Pires S, Elson G, Ferlin W, Kosco-Vilbois M, et al. O033 Ligand-independent blockade of TLR4 activation represents a promising strategy to protect from inflammatory disease pathogenesis in islet-transplantation. Cytokine. (2012) 59:513. doi: 10.1016/j.cyto.2012.06.068
161. Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf K, Reijers J, et al. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin Pharmacol Ther. (2017) 101:200–8. doi: 10.1002/cpt.522
162. Singh H, Agrawal DK. Therapeutic potential of targeting the receptor for advanced glycation end products (RAGE) by small molecule inhibitors. Drug Dev Res. (2022) 83:1257–69. doi: 10.1002/ddr.21971
163. Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, et al. Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci Rep. (2016) 6:22450. doi: 10.1038/srep22450
164. Fan R, Cheng Z, Huang Z, Yang Y, Sun N, Hu B, et al. TREM-1, TREM-2 and their association with disease severity in patients with COVID-19. Ann Med. (2023) 55:2269558. doi: 10.1080/07853890.2023.2269558
165. Saadipour K. TREM1: A potential therapeutic target for alzheimer’s disease. Neurotox Res. (2017) 32:14–6. doi: 10.1007/s12640-017-9716-y
166. Roselli F, Huber-Lang M. TREM1-ors shake the brain and gut after stroke. Nat Immunol. (2019) 20:950–2. doi: 10.1038/s41590-019-0443-9
167. Yao Y, Yang L, Zhang Z, Wang B, Feng B and Liu Z. Identification of targets for subsequent treatment of crohn’s disease patients after failure of anti-TNF therapy. J Inflammation Res. (2023) 16:4617–31. doi: 10.2147/JIR.S422881
Keywords: TREM-1, TREM-2, inflammation, clinical trial failure, multiligand receptors, antagonists & inhibitors, ligand-independent inhibition, drug discovery & development
Citation: Sigalov AB (2024) TREM-1 and TREM-2 as therapeutic targets: clinical challenges and perspectives. Front. Immunol. 15:1498993. doi: 10.3389/fimmu.2024.1498993
Received: 20 September 2024; Accepted: 30 November 2024;
Published: 16 December 2024.
Edited by:
Alexandru Movila, Indiana University, United StatesReviewed by:
Tsuguno Yamaguchi, Lion Corporation, JapanCopyright © 2024 Sigalov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander B. Sigalov, c2lnYWxvdkBzaWduYWJsb2suY29t