 Xiaohong Duan1,2,3†
Xiaohong Duan1,2,3† Zhiyao Xing
Zhiyao Xing Shan Qin
Shan Qin Xuejing Zhao
Xuejing Zhao Yanhua Gong
Yanhua Gong Xueren Li
Xueren Li- 1School of Disaster and Emergency Medicine, Faculty of Medicine, Tianjin University, Tianjin, China
- 2Institute of Disaster and Emergency Medicine, Faculty of Medicine, Tianjin University, Tianjin, China
- 3Medical School, Faculty of Medicine, Tianjin University, Tianjin, China
- 4Tianjin University and Health-Biotech United Group Joint Laboratory of Innovative Drug Development and Translational Medicine, School of Pharmaceutical Science and Technology, Faculty of Medicine, Tianjin University, Tianjin, China
- 5Department of Respiratory Medicine, Jinnan Hospital, Tianjin University, Tianjin, China
- 6Department of Respiratory Medicine, Tianjin Jinnan Hospital, Tianjin, China
- 7The Province and Ministry Co-sponsored Collaborative Innovation Center for Medical Epigenetics, Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
Histones play crucial roles in both promoting and repressing gene expression, primarily regulated through post-translational modifications (PTMs) at specific amino acid residues. Histone PTMs, including methylation, acetylation, ubiquitination, phosphorylation, lactylation, butyrylation, and propionylation, act as important epigenetic markers. These modifications influence not only chromatin compaction but also gene expression. Their importance extends to the treatment and prevention of various human diseases, particularly cancer, due to their involvement in key cellular processes. Abnormal histone modifications and the enzymes responsible for these alterations often serve as critical drivers in tumor cell proliferation, invasion, apoptosis, and stemness. This review introduces key histone PTMs and the enzymes responsible for these modifications, examining their impact on tumorigenesis and cancer progression. Furthermore, it explores therapeutic strategies targeting histone PTMs and offers recommendations for identifying new potential therapeutic targets.
1 Introduction
The regulation of chromatin structure, nucleosome positioning, and gene transcription is primarily controlled by histone proteins. The nucleosome, the fundamental unit of chromatin, consists of a central histone octamer, around which approximately 1.75 left-handed superhelical turns of DNA are wrapped (1, 2). Each nucleosome is made up of two identical subunits, and each subunit contains four core histones: H2A, H2B, H3, and H4. Additionally, histone H1, which acts as a linker, is not part of the nucleosome itself but plays a crucial role in stabilizing the DNA between nucleosomes (3). These histones undergo various forms of post-translational modifications (PTMs), which serve as epigenetic markers that influence their interaction with DNA. Under normal physiological conditions, these histone PTMs are essential for maintaining nucleosome structure and functioning as regulatory mechanisms. They play vital roles in key cellular processes, including DNA replication, gene expression, DNA damage repair, and chromatin organization (4).
At least eleven types of post-translational modifications (PTMs) have been identified on histones, including methylation, acetylation, propionylation, butyrylation, formylation, ubiquitylation, phosphorylation, sumoylation, citrullination, proline isomerization, and ADP ribosylation, occurring at more than 60 different amino acid residues (5) (Figure 1). These modifications can occur in various combinations, contributing to a wide range of biological functions. Upon histone modification, the chromatin structure is altered, which subsequently influences the interaction between histones and DNA, thereby regulating gene transcription.
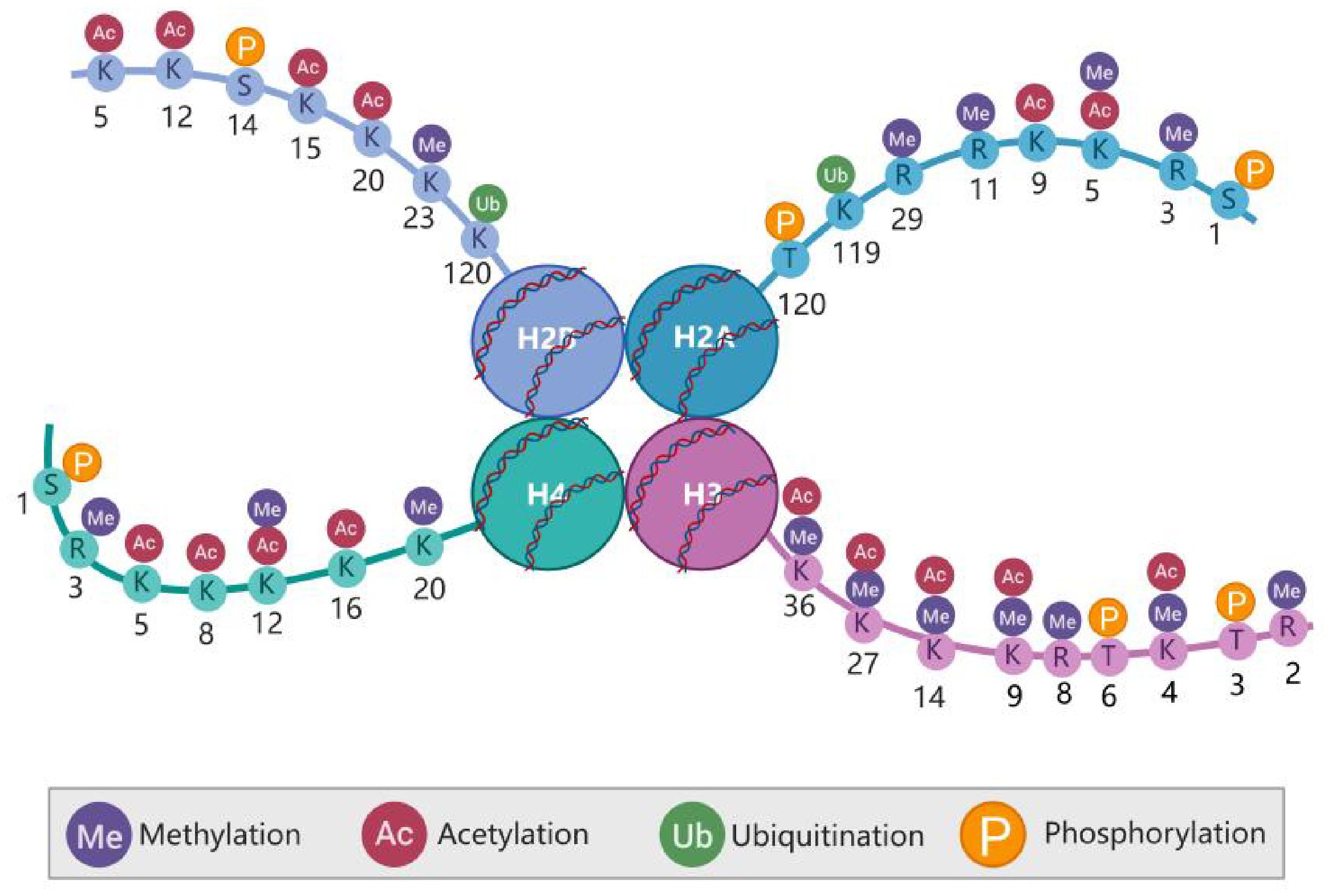
Figure 1. Post-translational modifications (PTMs) of the histone amino terminus. Histones in nucleosomes (two each of H2A. H2B. H3. and H4), Histone tails are subject to various PTMs that affect not only the overall compression of chromatin but also gene expression. Created in BioRender. Xing, Z. (2024) https://BioRender.com/l06b379.
Most histone post-translational modifications (PTMs) are localized within the N-terminal tail domain of core histones, although some crucial PTMs involved in histone–DNA and histone–histone interactions also occur in the globular domain of core histones (6–8). The structural domains at the ends of histone tails are positively charged and interact with negatively charged DNA. Much of the N-terminal part of the histone tail does not participate in nucleosome assembly and protrudes from the core structure, making it more suitable for interactions with the surrounding environment and, thus, more susceptible to PTMs (9). Compared with those of H2A and H2B, the histone tails of H3 and H4 are particularly vulnerable to PTMs. PTMs on histone tails are typically recognized by “reader” or effector proteins, which, in turn, regulate chromatin function (Figure 2). Histone methylation and acetylation predominantly occur on the N-terminal tail and are key regulators of gene transcription. PTMs in the globular domain of histones can disrupt histone–histone interactions, destabilize nucleosomes or alter histone-DNA interactions, impacting nucleosome dynamics and chromatin function, often without the need for effector proteins.
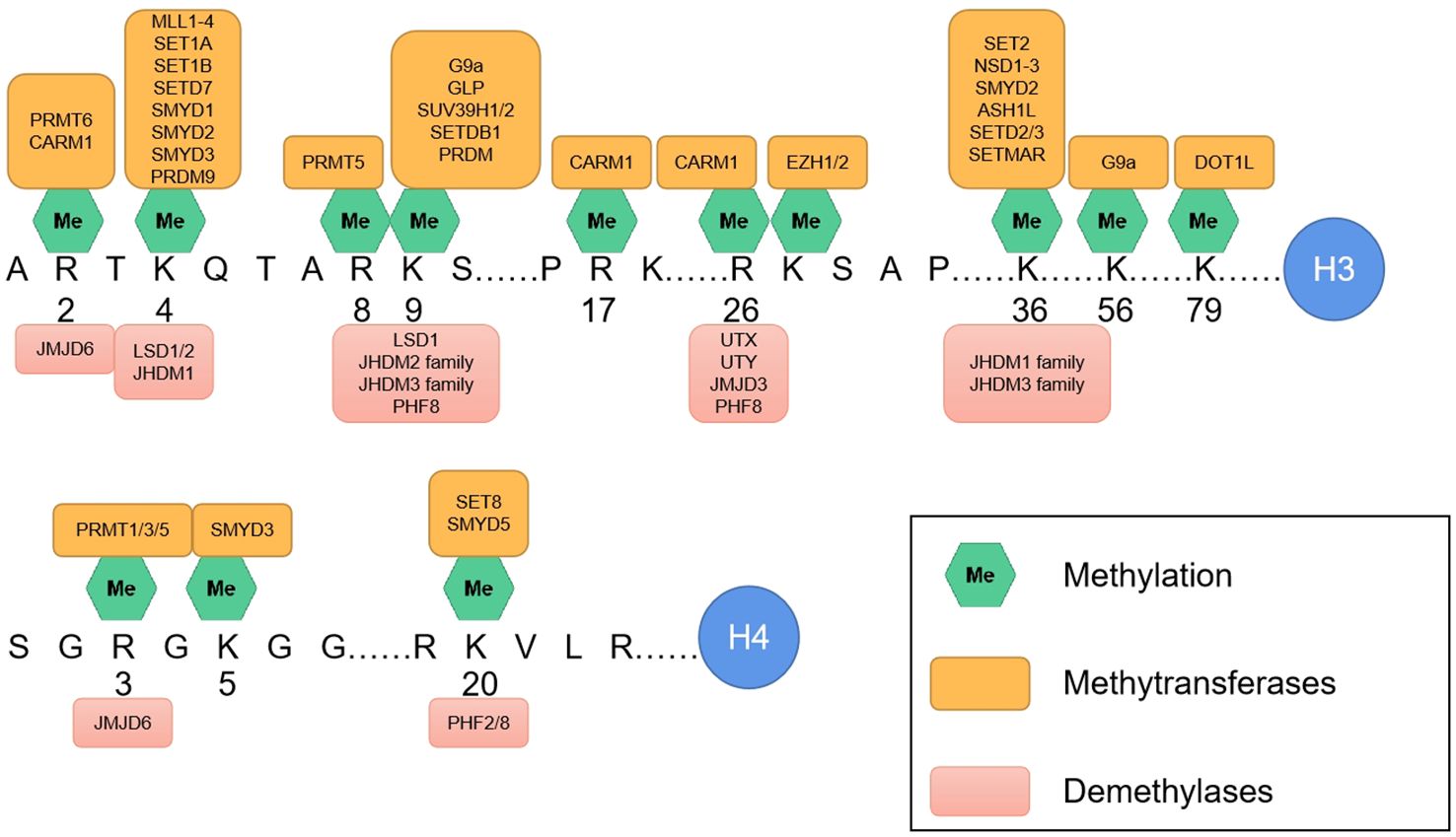
Figure 2. The main methylation sites on the amino termini of H3 and H4. Along with the associated methyltransferases (above) and demethylases (below).
Mounting evidence indicates that histone post-translational modifications (PTMs) play essential roles in a variety of biological processes, including cell differentiation and organismal development. The dysregulation of histone PTMs under pathological conditions is closely associated with the onset and progression of major human diseases, especially cancer. Enzymes such as histone methyltransferases, demethylases, acetyltransferases, and deacetylases regulate gene expression through these modifications. For instance, histone methylation typically results in gene silencing, while acetylation activates gene transcription; in contrast, demethylation and deacetylation generally reverse these effects. The abnormal expression of these modifying enzymes is a key factor contributing to tumor development and progression. Thus, understanding the biological roles of histone PTMs is critical for elucidating their pathophysiology. This review emphasizes the role of histone PTMs in cancer and explores the mechanisms underlying abnormal modification events. Additionally, this review discusses the potential of developing therapeutic drugs that target histone-modifying enzymes, offering new directions for identifying novel therapeutic targets and strategies for cancer treatment.
2 Histone modifications related to cancer
2.1 Histone methylation
2.1.1 Histone methylation in cancer
Histone methylation predominantly occurs on lysine and arginine residues and represents a critical post-translational modification catalyzed by histone methyltransferase enzymes. This modification can either activate or repress transcription, depending on the specific sites involved. For example, methylation at H3K4, H3K36, and H3K79 is generally linked to transcriptional activation, whereas methylation at H3K9, H3K27, and H4K20 is typically associated with transcriptional repression (10). The impact of methylation also varies on the basis of degree of methylation. For example, monomethylation of H4K20 (H4K20me1) is observed in active gene bodies, whereas trimethylation of H4K20 (H4K20me3) is associated with gene repression and chromatin compaction (11, 12). Additionally, the position of the methylated lysine residues relative to the DNA sequence plays a crucial role in gene regulation. For example, H3K9me3 at the promoter region is linked to gene silencing, whereas H3K9me3 within the gene body is often found in inducible genes. Since this modification is electrically neutral and chemically inert, it relies on other proteins with binding motifs to exert its function. Proper histone methylation is essential for genomic programming during development. However, during tumorigenesis, dysregulated histone methylation promotes tumor cell proliferation, migration, and invasion, ultimately contributing to tumor progression and poor prognosis (Table 1).
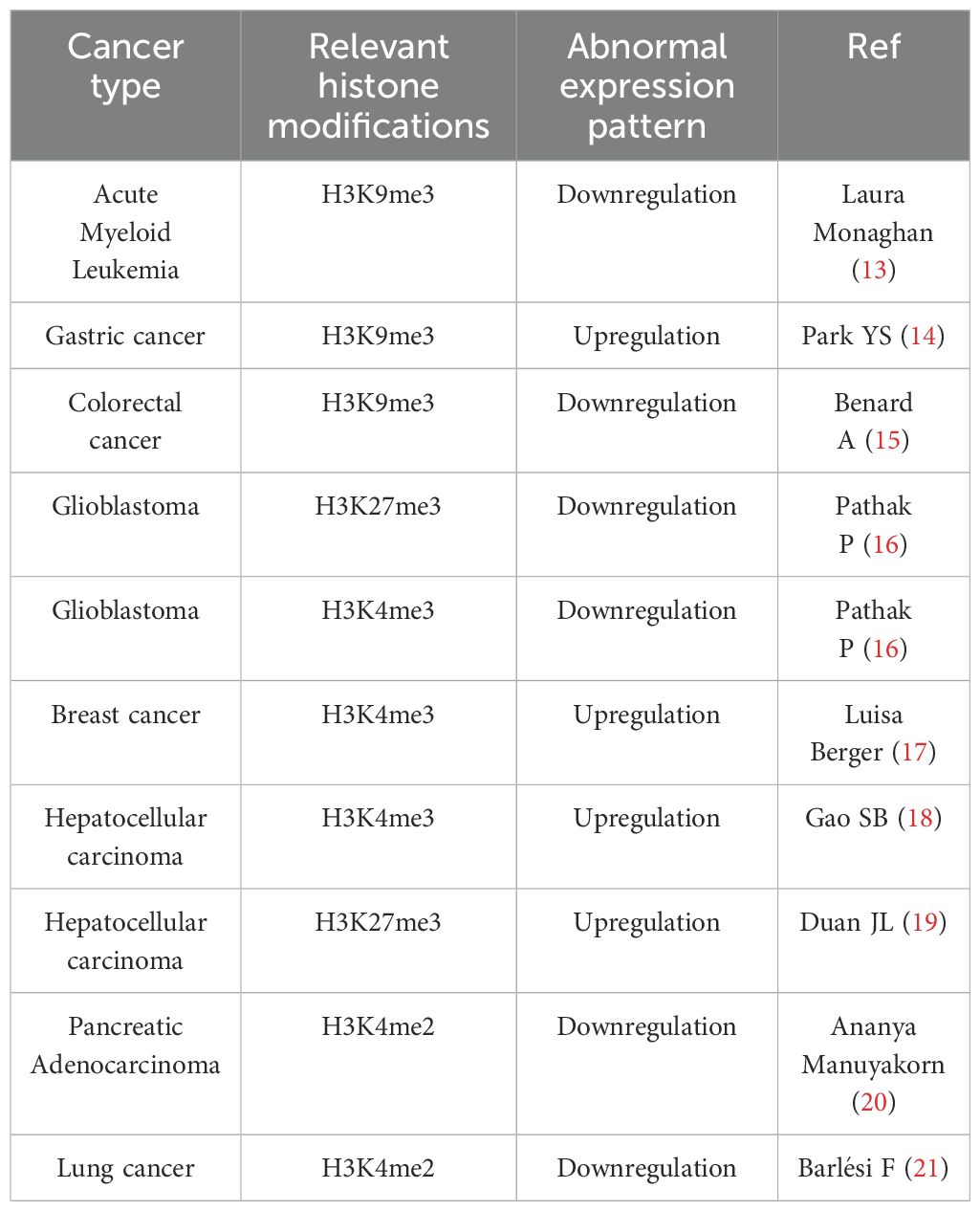
Table 1. Abnormal histone methylation in cancer.
2.1.1.1 H3K9me3
The expression levels of numerous histone modification markers are closely linked to the prognosis of various human cancers. Among these, H3K9me3 is a key histone modification marker that plays a significant role in tumor development and patient outcomes. H3K9me3 is generally associated with gene transcriptional silencing and influences cancer progression in multiple ways. On the one hand, H3K9me3 contributes to the abnormal silencing of tumor suppressor genes, thereby promoting tumor progression and leading to poorer patient prognosis. For instance, in HCT116 cells, the promoter and adjacent 3’ regions of the tumor suppressor gene DCC are enriched with the repressive H3K9me3 marker, which inhibits DCC transcription and promotes colorectal cancer development (22). Elevated H3K9me3 levels are also prognostic markers in cancers such as acute myeloid leukemia, gastric adenocarcinoma, salivary carcinoma, and bladder cancer (23–26). On the other hand, H3K9me3 helps to repress the aberrant expression of oncogenes and regulates the silencing of repetitive sequences in the genome. Studies have shown that higher H3K9me3 immunostaining scores are inversely correlated with disease recurrence, particularly distant metastasis, and improved disease-free survival in patients with non-small cell lung cancer (27). Furthermore, reduced levels of both H3K9me3 and H4K20me3 are associated with shorter survival times and increased tumor recurrence rates in patients with early-stage colon cancer (15).
2.1.1.2 H3K4me3
H3 lysine 4 (H3K4) methylation is among the most extensively studied histone modifications due to its strong association with gene expression and cancer development. This methylation is catalyzed by the SET1/COMPASS complex, which comprises several lysine methyltransferases and essential subunits, including six catalytic members: SETD1A, SETD1B, and MLL1-4 (28). H3K4me3 is typically found at transcription start sites (TSSs) and is believed to enhance transcription by recruiting PHD finger-containing proteins, such as TATA-box binding protein-associated factor 3 (TAF3), which play critical roles in transcription initiation (29). Additionally, H3K4me3 at promoter regions can counterbalance repressive histone modifications such as H3K9me3 and H3K27me3, helping to activate gene transcription (30–32). H3K4me3 is a hallmark of actively transcribed genes and has been implicated in promoting changes in gene expression and advancing tumor progression. Recent research has indicated that H3K4me3 actively participates in driving the progression of several cancers, including lung cancer, liver cancer, multiple myeloma, and prostate cancer (33–37). Notably, in gastric cancer (GC) patients, H3K4me3 is significantly upregulated at the TM4SF1-AS1 locus, promoting the expression of TM4SF1-AS1, which in turn inhibits apoptosis in gastric cancer cells (38). While most expressed genes present H3K4me3 restricted to the promoter and 5’ regions of the gene body, a subset of genes exhibit broader H3K4me3 regions (39). These broad domains often identify genes involved in crucial functions such as cell identity, tumor suppression, and disease-related processes (32, 40). For example, highly metastatic triple-negative breast cancer cells show higher expression levels of oncogenes linked to broad H3K4me3 domains compared to normal breast epithelial cells and less malignant breast cancer cell lines (39).
2.1.1.3 H3K27me3
H3K27 methylation is catalyzed by polycomb repressive complex 2 (PRC2), a key regulator whose subunits can recognize the H3 tail for complex binding (41). The primary function of PRC2 is to deposit methyl groups onto the lysine 27 residue of histone H3 (H3K27), resulting in gene repression (42). This trimethylation of H3K27 is mediated by the histone methyltransferase enhancer zeste homolog 2 (EZH2), an essential component of PRC2 (43). H3K27me3 plays a critical role in cell differentiation, and studies have demonstrated that the disruption of PRC2 impairs the differentiation of embryonic stem cells (44).
In addition to its pivotal role in development, alterations in H3K27me3 are observed in various cancer types, where H3K27me3 contributes to different stages of tumor initiation, progression, and metastasis. Both the upregulation and downregulation of H3K27me3 have been implicated in numerous cancers. For example, in ovarian cancer, overexpression of EZH2 leads to increased H3K27me3 levels, which suppresses the expression of genes such as E-cadherin, TIMP2, and TIMP3, all of which are involved in cell migration. Their repression facilitates tumor metastasis (45). EZH2 also modulates the expression of DAB2IP by trimethylating H3K27 at the DAB2IP promoter. DAB2IP, known for its Ras-GTPase activity, suppresses cancer stem cell phenotype in several cancers, and its repression by EZH2 promotes cancer cell stemness (46, 47).
Conversely, the loss of H3K27me3 can also contribute to tumor development. In such cases, reduced H3K27me3 may lead to the activation of tumor suppressor genes, accelerating tumor progression. In diffuse midline gliomas, for example, the loss of H3K27 trimethylation is a primary driver of tumor growth (48). This reduction in H3K27 methylation promotes glial cell stemness and silences tumor suppressor genes (42). Thus, dynamic changes in H3K27me3 play multifaceted and complex roles at various stages of tumor initiation and progression.
2.1.2 Histone methyltransferases in cancer
Histone methyltransferases (HMTs) are categorized into two main subfamilies: histone lysine methyltransferases (HKMs) and histone arginine methyltransferases (HRMs) (49). Lysine methyltransferases (KMTs) can be further divided into two families based on the sequence of their catalytic domain: those with the SET domain (located in the histone tails) and those with non-SET domains (located in the histone core) (50–52). All HKMTs, except those in the Dot1 family, contain a conserved enzymatic SET [SU(VAR)3-9, E(Z), and TRX] domain, which was initially discovered in su(var)3-9, enhancer-of-zeste, and trithorax proteins (52). The SET domain includes enzymes such as SUV39H1/2, G9a, EZH2, GLP, and SETDB1 (53).
In the histone arginine methyltransferase (HRM) subfamily, protein arginine methyltransferases (PRMTs) methylate arginine residues in histones through mono-, symmetric-, or asymmetric-dimethylation (54). Currently, nine PRMTs have been identified in mammals, all of which possess four conserved motifs. Mammalian PRMTs are classified into two groups on the basis of the type and position of methylation. Class I includes PRMT1, 3, 4, 6, and 8, which catalyze monomethylation and asymmetric dimethylation of arginine. Class II comprises PRMT5 and PRMT7, which catalyze monomethylation and symmetric dimethylation of arginine (55–57).
The dysregulated expression of histone methyltransferases (HMTs) can result in aberrant histone methylation of cancer-related genes, thereby contributing to tumor development. The histone-lysine N-methyltransferase (KMT2) family plays crucial roles in regulating transcription. Mutations in the KMT2 family are among the most frequently observed genetic aberrations in various cancers, including hematological malignancies and solid tumors such as colorectal, lung, endometrial, breast, bladder, and brain cancers (58–63). Members of the PRMT family have also gained importance in the study of different cancer types. The overexpression and dysregulation of PRMT4, PRMT5, and PRMT7 are known to drive the progression of several hematological malignancies and solid tumors, making them effective therapeutic targets (64–66). Therefore, PRMT family members can serve as effective targets for cancer therapy.
Aberrant expression of HMTs disrupts the transcriptional regulation of genes linked to disease, including oncogenes and tumor suppressor genes, which can promote tumorigenesis. HMTs are associated with chemotherapy resistance and immune evasion, highlighting their potential as therapeutic targets. Chemically targeting these enzymes represents a promising approach for the development of novel cancer therapies.
2.1.2.1 SETD1A
SETD1A is a histone lysine methyltransferase that contains the SET domain and is part of the SET1/COMPASS complex family, alongside its paralog SETD1B (28, 67). SETD1A specifically methylates H3K4, a modification crucial for the transcriptional activation of genes that regulate the self-renewal and differentiation of embryonic stem cells (68). Additionally, SETD1A plays a significant role in maintaining mitosis and cell proliferation. Research has shown that SETD1A is upregulated in various cancers, and its overexpression is linked to accelerated tumor cell proliferation and invasion, which is often correlated with poor prognosis.
In lung cancer, high levels of SETD1A expression drive the deposition of H3K4me3 on the promoters of key oncogenes such as MYC, GLI1, FOXM1, and DNMT1. This promotes the transcription of these oncogenes, thereby facilitating the onset and progression of lung cancer (33). Furthermore, studies indicate that SETD1A contributes to the development of gastric cancer by increasing H3K4me3 levels at hypoxia response elements in the promoters of HK2 and PFK2. This, in turn, enhances the transactivation of HIF1α and upregulates the expression of its target genes, ultimately leading to increased glycolysis in gastric cancer cells (69). Similarly, SETD1A is overexpressed in pancreatic ductal adenocarcinoma (PDAC) and is associated with poor patient prognosis. It binds to the promoter of the oncogenic protein ATP-dependent DNA helicase gene RUVBL1, increasing H3K4me3 levels and promoting transcriptional regulation of the gene, which plays a pivotal role in PDAC cell proliferation and motility (70). These findings suggest that SETD1A may serve as a potential predictive marker for various cancer types.
2.1.2.2 KMT2D
Lysine-specific methyltransferase (KMT2D), also known as myeloid/lymphoid or mixed-lineage leukemia 2 (MLL2), is a key histone methyltransferase essential for regulating gene transcription. It specifically targets histone H3 lysine 4 (H3K4), whose methylation serves as a marker for gene activation. KMT2D, along with SETD1A, is a member of the SET/MLL (mixed lineage leukemia) methyltransferase family, which is conserved across species, from yeast to mammals. In humans, this family includes six H3K4 methyltransferases (HMTs): MLL1 (MLL/KMT2A), MLL2 (KMT2B), MLL3 (KMT2C), MLL4 (KMT2D), SETD1A (KMT2F), and SETD1B (KMT2G) (58, 71).
In recent years, KMT2D has emerged as one of the most frequently mutated genes in various cancers and human diseases, including lymphoma, medulloblastoma, and gastric cancer (72–74). Mutations in KMT2D often result in a loss of function, suggesting its role as a tumor suppressor in various tissues. The absence of KMT2D affects the proliferation and migration of colorectal cancer cell lines. KMT2D regulates H3K4 monomethylation and is associated with enhancer elements in the HCT116 cell line. The SET domain of the enzyme is critical for maintaining effective H3K4 monomethylation, and its activity is directly involved in regulating H3K4me1, which is essential for sustaining tumor cell proliferation.
Moreover, KMT2D plays a role in addressing tumor resistance. For example, drugs targeting the PI3K signaling pathway are effective in some breast cancer patients; however, estrogen receptor (ER)-positive breast cancer patients often develop resistance to these therapies. Toska et al. reported that PI3K inhibition activates KMT2D and that H3K4 methylation catalyzed by KMT2D leads to a more open chromatin state, facilitating estrogen receptor-dependent transcription (75). As a result, researchers have suggested that combination therapy consisting of PI3K inhibitors and KMT2D inhibitors may be more effective than PI3K inhibitors alone.
2.1.2.3 PRMT1
PRMT1 is the predominant type 1 protein arginine methyltransferase (PRMT), accounting for more than 85% of arginine methylation in mammals, with histone H4 as its primary methylation target. Arginine dimethylation of histone H4 (H4R3me2a) enhances histone acetylation, chromatin accessibility, and transcriptional activation. Recent studies have underscored the critical role of arginine methylation in various human diseases, particularly cancer. Elevated levels of PRMT1 are linked to poor prognosis in many cancer types.
Research conducted by Ku et al. demonstrated that high PRMT1 levels are associated with unfavorable outcomes in both human and mouse pancreatic cancer patients (76). PRMT1 activity is essential for regulating chromatin accessibility and controlling the expression of key glycolytic genes, such as GLUT1 and HK2. Furthermore, inhibiting PRMT1 can disrupt KRAS-driven glycolysis in pancreatic ductal adenocarcinoma (PDAC), thereby affecting tumor metabolism.
PRMT1 also acts in synergy with SMARCA4, an ATPase subunit of the SWI/SNF chromatin remodeling complex, to drive the progression of colorectal cancer (CRC) (77). Mechanistically, it was shown that H4R3me2a directly recruits SMARCA4, enhancing the proliferative, colony-forming, and migratory capacities of CRC cells by activating EGFR signaling pathways. PRMT1 is further involved in numerous interactions with transcription factors and gene promoters. The overexpression and aberrant splicing of PRMT1 are directly implicated in the development of several cancers, including breast, lung, and bladder cancers and leukemia (78–81).
2.1.3 Histone demethylases in cancer
Histone methylation is a reversible modification that is dynamically regulated by the interplay between histone methyltransferases and histone demethylases. Histone demethylation is catalyzed by histone demethylases (HDMs), which remove methyl groups from specific amino acids on the N-terminal tails of histones. These enzymes primarily target lysine (K) residues on histone H3, such as K4, K9, K27, and K36, and are also known as lysine demethylases (KDMs). Currently, two evolutionarily conserved families of histone demethylases have been identified: lysine-specific demethylases (LSDs) and demethylases containing the Jumonji C (JmjC) domain (JHDM). These two families utilize different mechanisms to remove methyl groups (82, 83).
The LSD family includes LSD1 and LSD2, which demethylate mono- and dimethylated lysine residues through a flavin adenine dinucleotide (FAD)-dependent amine oxidase reaction. LSD1, also known as KDM1A or AOF2, was the first discovered histone lysine demethylase. It removes mono- and dimethyl groups from lysine 4 (H3K4me1/2) or lysine 9 (H3K9me1/2) of histone H3, serving as either repressors or activators of gene expression (84). However, owing to its reliance on FAD and protonated nitrogen, LSD1 can only demethylate mono- or dimethylated lysines and is ineffective against trimethylated lysines (82, 85). The catalytic mechanism of LSD1 limits its ability to demethylate trimethylated lysine, a widely observed modification. This has led researchers to propose that other catalytic mechanisms may exist for histone lysine demethylation.
Zhang et al. discovered that F-box and leucine-rich repeat protein 11 (FBX11) possesses histone demethylase activity and contains a JmjC domain, classifying it as JHDM1A (JmjC domain-containing histone demethylase 1A) (86). JmjC domain-containing proteins exhibit hydroxylase activity, which enables their demethylation function. JHDM1A specifically removes dimethylation marks from H3K36me2 on histone H3 in the presence of divalent iron ions and α-ketoglutarate. The JHDM1A protein comprises several domains: a JmjC domain, an F-box domain, a PHD, a zinc finger domain, and three leucine-rich repeat regions, with the JmjC domain serving as the catalytic domain. On the basis of sequence information, proteins containing the JmjC domain are classified into seven families: JHDM1, JHDM2, JHDM3, JARID1, UTX/UTY, PHF8, and those containing only the JmjC domain. The JmjC family has 30 members, nearly 20 of which have demonstrated histone demethylase activity. Unlike LSDs, JmjC domain-containing proteins do not require a hydrogen donor, allowing them to demethylate all three methylation states of lysine residues.
Proteomic analyses have shown that histone arginine methylation is also a reversible modification. However, the “arginine demethylases” (RDMs), which directly remove methyl groups from arginine, remain poorly characterized. The most studied candidates are peptidylarginine deiminase 4 (PADI4) and Jumonji domain-containing protein 6 (Jmjd6) (87, 88). PADIs are Ca2+-dependent enzymes that catalyze the conversion of arginine to citrulline in proteins. They can also convert monomethylated arginine (MMA) to citrulline, affecting histone H3 and H4 modifications. Additionally, Chang et al. identified Jmjd6 as a histone arginine demethylase that specifically demethylates histone H3 at arginine 2 (H3R2) and histone H4 at arginine 3 (H4R3).
These histone demethylases can target both histone and non-histone substrates and are involved in various biological processes, including development and metabolic diseases such as diabetes and cancer. Abnormal expression of histone demethylases is linked to tumorigenesis, cancer progression, and drug resistance (Table 2). For example, LSD1 is abnormally expressed in several cancers and has been shown to inhibit cancer cell differentiation while promoting proliferation, metastasis, and invasion. LSD1 overexpression is associated with poor prognosis in conditions such as non-small cell lung cancer, neuroblastoma, pancreatic cancer, prostate cancer, and breast cancer (101–105). Inhibiting LSD1 activity may help reduce or halt the growth of these tumors.

Table 2. The mechanism of KDMs in cancer.
2.1.3.1 JMJD1A
JMJD1A, also known as lysine demethylase 3A (KDM3A), is a member of the Jumonji C (JmjC) family of histone demethylases. JMJD1A plays a key role in promoting the expression and activity of various transcription factors through the demethylation of H3K9, thereby regulating critical biological processes such as spermatogenesis, stem cell activity, and sex determination. Notably, JMJD1A is upregulated in multiple malignant tumors, including neuroblastoma, breast cancer, cervical cancer, non-small cell lung cancer, liver cancer, and gastric cancer (106–111).
In prostate cancer, JMJD1A plays several important roles (112). First, the interaction between JMJD1A and the androgen receptor (AR) promotes AR chromatin binding by demethylating H3K9 on AR target genes. Additionally, JMJD1A regulates the oncogene c-Myc through three distinct mechanisms: (1) JMJD1A induces H3K9 demethylation at the c-Myc enhancer, thereby promoting c-Myc transcription (113); (2) JMJD1A acts as a coactivator of c-Myc by interacting with it and enhancing c-Myc recruitment to chromatin via H3K9 demethylation; and (3) JMJD1A interacts with the protein HUWE1, which is involved in the ubiquitination and degradation of c-Myc (114). By inhibiting c-Myc degradation, JMJD1A enhances the stability of c-Myc.
Furthermore, JMJD1A has been implicated in promoting the progression of urinary bladder cancer (115). It does so by enhancing glycolysis through the coactivation of HIF-1α, contributing to cancer cell growth and survival. These findings highlight JMJD1A as a potential therapeutic target in various cancers.
2.1.3.2 UTX/KDM6A
Lysine-specific demethylase 6A (UTX), encoded by the KDM6A gene, is a key component of the COMPASS complex, which plays a critical role in gene activation. UTX functions as part of a transcriptional activation complex that includes MLL2/MLL3 (H3K4 methyltransferases) and P300/CBP histone acetyltransferases (116). This collaborative mechanism enables the removal of the repressive histone mark H3K27me3 and the deposition of activation-associated marks such as H3K27 acetylation and H3K4 methylation, facilitating transcriptional activation.
Mutations in KDM6A are frequently observed in various cancer types, particularly in primary multiple myeloma (MM) and certain types of T-cell leukemia (117–119). Loss of UTX in MM promotes tumor cell proliferation, clonogenicity, and adhesion. Moreover, UTX-mutant MM cells exhibit increased sensitivity to EZH2 inhibition both in vitro and in vivo, which correlates with reduced levels of IRF4 and c-MYC, as well as the activation of IRF4 repressors specific to germinal center B cells, such as BCL6 and IRF1.
Interestingly, UTX can either suppress or promote cancer development through interactions with transcription factors. For example, in breast cancer, UTX regulates the oncogenic functions of estrogen receptor α (ERα) (120). In bladder cancer, UTX functions as a tumor suppressor by localizing to enhancers and regulating key genes involved in bladder differentiation through a catalytic-independent mechanism (121). UTX also attenuates the transcriptional and phenotypic effects of aberrant fibroblast growth factor receptor 3 (FGFR3) activation. These findings suggest that the concurrent loss of UTX function and FGFR3 mutations synergistically drive tumorigenesis in bladder cancer. UTX/KDM6A is encoded on the X chromosome and escapes X inactivation in females. In females, UTX lesions are often homozygous, whereas in males, these mutations are frequently accompanied by the loss of the paralog UTY, further supporting its role as a tumor suppressor.
2.1.3.3 JMJD2
The JMJD2A-D proteins, now commonly referred to as KDM4A-D (lysine demethylases 4 A-D), belong to one of the largest subfamilies of JMJD proteins. These enzymes have garnered significant attention for their ability to recognize and demethylate dimethylated and trimethylated histones, specifically H3K9 and H3K36, as well as trimethylated H1.4K26. The KDM4 family consists of three ∼130-kDa proteins (KDM4A-C) and a smaller member, KDM4D/JMJD2D, which lacks the double PHD and Tudor domains found in the other KDM4 proteins (122). These domains function as epigenome readers, and their absence in KDM4D results in different substrate specificities than those of the other family members.
Various studies have shown that KDM4A/JMJD2A, KDM4B/JMJD2B, and/or KDM4C/JMJD2C are overexpressed in breast cancer, colorectal cancer, lung cancer, prostate cancer, and other tumors and are essential for the efficient growth of cancer cells (122, 123). KDM4A/JMJD2A is the most extensively studied member of the KDM4 family, and it can demethylate H3K9 and H3K36, thereby regulating gene transcription. In breast and prostate cancers, KDM4A forms a complex with estrogen and androgen receptors and activates downstream target genes. Therefore, depletion of KDM4A in ER-positive T47D breast cancer cells reduces the expression of ER target genes (such as the oncogenes c-Jun and Cyclin D1) and leads to decreased cell growth (124). Like in breast cancer cells, the knockdown of KDM4A in multiple colorectal cancer cell lines led to reduced cell proliferation, further increased apoptosis, and delayed G2−M phase progression of the cell cycle (125).
KDM4B and KDM4C, which share structural similarities with KDM4A, exhibit the same target specificity and similar enzymatic activities in vitro (126, 127). The expression of KDM4B and KDM4C is elevated in breast tumors. Functionally, KDM4C overexpression has been linked to the development of ER-negative breast cancers, while KDM4B contributes to the tumorigenic transformation of ER-positive breast cancer cells (128–131). Furthermore, KDM4B overexpression has been observed in gastric, bladder, lung, and colorectal cancers, where it is essential for the proliferation, colony formation, invasion, and survival of cancer cells (132, 133).
KDM4D, which is distinct from other KDM4 proteins due to its lack of PHD and Tudor domains, also acts as a coactivator of the androgen receptor, similar to KDM4A and KDM4C, thereby promoting tumor progression. Additionally, KDM4D plays a role in mediating inflammatory responses triggered by cytokines such as TNF-α, potentially influencing tumorigenesis within the tumor microenvironment and immune cells (134).
2.1.3.4 JMJD3/KDM6B
Lysine-specific demethylase 6B (KDM6B, also known as JMJD3) is a member of the UTX/UTY JmjC domain protein subfamily that demethylates H3K27 residues, including both the trimethylated and dimethylated forms, in conjunction with UTX (135). KDM6B promotes gene transcription by removing H3K27me3 marks from the promoters of its target genes, thereby preventing the binding of polycomb repressive complex 2 (PRC2), or by recruiting coactivators and mediating their interactions with transcription factors (TFs), which can be dependent on or independent of its demethylase activity (136, 137). KDM6B is a crucial histone demethylase involved in various biological and pathological processes, including development, inflammation, aging, and cancer.
KDM6B functions as both a tumor suppressor and an oncogene, depending on the cellular context. In certain cancer types, such as neuroblastoma, hepatocellular carcinoma, lung adenocarcinoma, and endometrial cancer, KDM6B functions as a tumor suppressor, and its expression is associated with improved survival rates and prognoses (138–141). For example, in non-small cell lung cancer (NSCLC), KDM6B expression is reduced, and when KDM6B is overexpressed, it restricts cell proliferation and migration while inducing apoptosis. This is achieved by inhibiting the phosphorylation of FOXO1, leading to its accumulation in the nucleus (142). In acute myeloid leukemia (AML), particularly the M2 and M3 subtypes, KDM6B overexpression reduces the number of leukemia stem cells, promotes bone marrow differentiation, and induces cellular senescence by upregulating the expression of C/EBPβ and its target genes (143).
Despite its tumor-suppressive functions, KDM6B can also act as an oncoprotein in certain contexts, promoting tumor progression. In prostate cancer cells, KDM6B is highly expressed, with levels increasing as the disease progresses, particularly in metastatic cases. Additionally, in estrogen receptor α (ERα)-dependent breast cancer, KDM6B forms a complex with ERα and binds to ERα sites in the BCL2 enhancer region, leading to BCL2 overexpression following estrogen treatment (144). KDM6B also induces the expression of SNAI1, promoting epithelial-mesenchymal transition (EMT) and contributing to metastasis in patients (145). In esophageal squamous cell carcinoma (ESCC), the RAS/MEK pathway induces KDM6B overexpression, which is associated with disease stage and patient survival (146). Thus, KDM6B plays dual roles in cancer, acting as either a tumor suppressor or an oncogene, depending on the specific cancer type and context.
2.2 Histone acetylation
Acetylation, one of the earliest discovered histone modifications affecting transcriptional regulation, introduces negative charges to lysine residues located in the N-terminal histone tails protruding from the nucleosome (147, 148). These negative charges repel negatively charged DNA, resulting in a more relaxed chromatin structure that allows transcription factors to bind more easily, thereby significantly increasing gene expression (149). The dynamic balance of histone acetylation is regulated by two classes of enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs) (150). HDACs can remove acetyl groups from histones, leading to a more compact chromatin structure and the suppression of gene transcription. In contrast, HATs add acetyl groups to histones, resulting in a more relaxed chromatin structure and promoting gene transcription. The dynamic balance between these two classes of enzymes determines the level of histone acetylation at specific gene loci, thereby influencing the expression of the corresponding genes (151).
Histone acetylation plays a vital role in regulating various cellular processes, such as the cell cycle, cell proliferation, apoptosis, differentiation, DNA replication and repair, nuclear transport, and neuronal inhibition (152). Imbalances in histone acetylation are closely associated with tumor development and cancer progression, as aberrations in acetylation can disrupt the regulation of gene expression involved in these key processes.
In various cancers, abnormal changes in H3K27ac modification are often linked to the dysregulation of tumor-related genes, affecting critical cellular processes such as proliferation, differentiation, and apoptosis. Several studies have highlighted the potential of H3K27ac modification changes as biomarkers for cancer diagnosis and prognosis, aiding in identifying the onset and progression of malignancies. In pancreatic ductal adenocarcinoma (PDAC), a highly invasive tumor, the pro-apoptotic protein NOXA serves as a marker of the invasive subtype. Research has demonstrated that inhibition of the transcription factor RUNX1 leads to enrichment of H3K27ac, which, in turn, activates the proximal NOXA promoter region (153). This drug-induced enrichment of H3K27ac triggers NOXA-dependent cell death. Similarly, in cervical cancer cells, the long non-coding RNA EGFR-AS1 promotes migration and invasion while inhibiting apoptosis. Studies have shown that CBP interacts with the promoter of EGFR-AS1, activating H3K27ac and subsequently leading to the upregulation of EGFR-AS1, which influences the WNT pathway through ACTN4, promoting cervical cancer cell growth (153).
In summary, H3K27ac plays a crucial role in cancer development and progression. Given its significance as an epigenetic marker, targeting the mechanisms that regulate H3K27ac, such as CBP/p300, has emerged as a promising therapeutic strategy for treating various cancers.
2.2.1 Histone deacetylases in cancer
Lysine acetylation on histone tails is a highly dynamic process crucial for regulating chromatin structure, transcription, and DNA repair. This process is controlled by two enzyme families: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (154). HATs catalyze the transfer of an acetyl group from acetyl-CoA to the ϵ-amino group of lysine residues on histones, leading to a more relaxed chromatin structure that promotes transcription. In contrast, HDACs remove the acetyl group from histones, resulting in a more compact chromatin conformation that reduces transcription factor accessibility. This dynamic regulation of histone acetylation plays a key role in gene expression and chromatin organization.
HDACs, in particular, serve as critical transcriptional corepressors in a variety of physiological and pathological systems. In mammals, there are 18 HDACs, which are classified into four major classes (155, 156). Class I HDACs (HDACs 1, 2, 3, and 8) are ubiquitously expressed in human cells and are primarily localized in the nucleus. Class II HDACs (HDACs 4, 5, 6, 7, 9, and 10) exhibit tissue-specific expression and shuttle between the nucleus and cytoplasm. Compared with other HDAC classes, class III HDACs, also known as sirtuins (SIRT1–7), are NAD+-dependent and have a unique catalytic mechanism. Finally, Class IV contains only one member, HDAC11, which has been found to deacetylate various histone sites, resulting in low substrate specificity and functional redundancy in certain contexts.
Mutations and abnormal expression of HDACs are frequently observed in human diseases, especially cancer, where the dysregulation of histone acetylation contributes to oncogenesis. As a result, HDACs have emerged as significant therapeutic targets in cancer, with their inhibition leading to increased histone acetylation and potential restoration of normal gene expression patterns in tumor cells. Consequently, the overall pattern of histone acetylation becomes dysregulated in cancer, further driving disease progression.
2.2.1.1 Class I HDACs
All members of the class I subfamily of HDACs are dysregulated in many types of cancer. The overexpression of HDAC1 has been observed in patients with breast cancer, prostate cancer, gastric cancer, and pancreatic cancer, and its upregulation is correlated with poor prognosis (157–160). Specifically, HDAC1 is highly expressed in glioblastoma (GBM) tissues, where it promotes the invasion and migration of GBM cells by regulating the epithelial-mesenchymal transition (EMT) process (161). Moreover, HDAC1 upregulation has been identified as a key factor in the development of drug resistance in ovarian cancer. Enhancing c-Myc-dependent miR-34a expression to target HDAC1 may offer a promising strategy to improve the efficacy of cisplatin treatment in these patients (162).
In human lung cancer cell lines, HDAC2 inactivation leads to apoptosis via the activation of p53 and Bax (163). Conversely, in colorectal cancer cells, the loss or knockdown of HDAC2 induces EMT and lung metastasis by upregulating the long non-coding RNA H19 (lncRNA H19) (164). Additionally, frequent mutations in the CREBBP gene in B-cell lymphomas drive tumorigenesis in vivo through the involvement of HDAC3 (165). HDAC3 also plays a crucial role in the development of non-small cell lung cancer (NSCLC) driven by the KL and KP genotypes (166). Furthermore, knockdown of HDAC8 has been shown to inhibit cell proliferation in lung cancer, colorectal cancer, and cervical cancer cell lines, highlighting its potential as a therapeutic target across multiple cancers (167–169).
2.2.1.2 Class II HDACs
Class II HDACs are divided into two subfamilies: Class IIa (HDAC4, 5, 7, 9) and Class IIb (HDAC6 and 10) (154). Some members of the Class IIa subfamily play dual roles in cancer. HDAC4, for example, is upregulated in breast cancer patients, yet its inhibition or downregulation can also affect cancer progression. Interestingly, homozygous deletion of HDAC4 has been observed in melanoma cell lines, indicating that HDAC4 can function both as an oncogenic factor and as a tumor suppressor depending on the context (170). Similarly, HDAC7 demonstrates dual roles in tumor biology. High expression of HDAC7 and HDAC9 has been linked to poor prognosis in children with acute lymphoblastic leukemia (ALL), whereas Skov et al. reported significant downregulation of HDAC7 in myeloproliferative tumors (171, 172). These findings suggest that Class IIa HDACs may serve as both proliferation-promoting and tumor-suppressive factors, depending on the cellular environment. In addition, Peixoto et al. reported that HDAC5 is essential for the replication fork process in cancer cells, as it is capable of maintaining and assembling the structure of heterochromatin around centromeres in cancer cells (173).
Among the Class IIb HDACs, HDAC6 has been extensively studied for its role in tumorigenesis. Elevated HDAC6 expression is positively correlated with cancer progression in oral squamous cell carcinoma, and estrogen stimulation increases HDAC6 gene expression in MCF-7 breast cancer cells (174). Moreover, acute myeloid leukemia samples and leukemia cell lines (such as HL60, K562, and KG1a) present elevated levels of HDAC6 expression (175). HDAC10, another member of Class IIb, has been implicated in gastric cancer development. It plays a crucial role in regulating the production of reactive oxygen species (ROS) in gastric cancer cells. Inhibition of HDAC10 leads to ROS accumulation, which triggers apoptosis in these cells, suggesting its potential as a therapeutic target (176).
2.2.1.3 Class III HDACs
Sirtuins, Class III HDACs, play both oncogenic and tumor-suppressive roles in cancer, depending on the context. Various sirtuins have been found to be aberrantly expressed in multiple types of cancers. The most studied of these genes is SIRT1, which is widely recognized as a key epigenetic regulator involved in numerous biological processes (177). SIRT1 is responsible for deacetylating histone H1 lysine 26 (H1K26ac), histone H3 lysine 9 (H3K9ac), and histone H4 lysine 16 (H4K16ac) (178). Compared with that in normal tissues, the overexpression of SIRT1 has been reported in several cancers, including prostate cancer, colorectal cancer, leukemia, and melanoma. However, SIRT1 downregulation has also been observed in other types of tumors, such as breast cancer and hepatocellular carcinoma (179, 180), reflecting its multifaceted effects that depend on its subcellular localization and roles in different tissues.
On the other hand, SIRT2 primarily acts as a tumor suppressor. Studies have shown that a deficiency in SIRT2 impairs the mitotic checkpoint, leading to genomic instability and tumorigenesis (181). Furthermore, a significant decrease in SIRT2 expression has been reported in patients with gliomas (182). In cholangiocarcinoma, SIRT3 plays a tumor-suppressive role by downregulating the HIF1α/PDK1/PDHA1 pathway, leading to tumor regression (183). SIRT3 also inhibits renal cancer tumorigenesis by blocking mitochondrial biogenesis and inducing ferroptosis. In gallbladder cancer, SIRT3 suppresses tumors by inhibiting AKT-dependent mitochondrial metabolism and epithelial−mesenchymal transition (EMT) (184). However, in cervical cancer, SIRT3 overexpression promotes cancer cell progression by reprogramming fatty acid synthesis.
SIRT7, another member of this class, is a promoter-associated, highly selective H3K18Ac deacetylase. It plays a role in mediating transcriptional repression and stabilizing the cancer cell phenotype, further underscoring the diverse and complex roles of sirtuins in cancer.
2.2.1.4 Class IV HDACs
HDAC11, a newly discovered member of the HDAC family, is encoded by a gene located on human chromosome 3q25.1 and is found in both the nucleus and cytoplasm (185). Its role in cancer appears to be cancer type specific. HDAC11 is highly expressed in lung adenocarcinoma and squamous cell carcinoma, where it is associated with poor patient prognosis (186, 187). In these cancers, HDAC11 promotes tumor cell migration, stemness, and drug resistance. In contrast, in basal-like breast cancer (BLBC), HDAC11 expression is downregulated, and overexpression of HDAC11 can suppress in vitro invasion and in vivo metastasis in xenograft breast cancer models, such as the SUM1315 and BT549 cell lines (128, 188).
The role of HDAC11 in hepatocellular carcinoma (HCC) metastasis is complex and may depend on the specific cancer cell lines and preclinical models used. Wang et al. reported that the downregulation of HDAC11 significantly reduced the migration and invasion abilities of highly metastatic MHCC97H HCC cells (189). However, Zhu et al. reported that while the knockdown of HDAC11 inhibited the proliferation of HepG2 cells, it did not affect invasion or migration (190).
In addition to its role in metastasis, HDAC11 is implicated in maintaining stemness and promoting tumor development in HCC. Downregulation of HDAC11 significantly suppresses glycolysis in HCC cancer stem cells and inhibits the stem cell-like properties of these cells by modulating their glycolytic levels (191). These findings highlight the context-dependent functions of HDAC11 across different cancer types.
2.3 Histone ubiquitination
All core histone proteins can undergo ubiquitination, but H2A and H2B are the most ubiquitinated and commonly modified histones in the nucleus (192, 193). Histone H2A was the first identified ubiquitination substrate, with lysine 119 (H2AK119ub) serving as the single ubiquitination site, catalyzed by the polycomb repressive complex 1 (PRC1) E3 ligase (194–196). H2AK119ub1 is highly enriched in the promoter regions of polycomb target genes and functions as a transcriptional repressor through various mechanisms. Histone H2A ubiquitination (H2Aub) plays a key role in several biological processes, including gene transcription and DNA damage repair (197, 198). Additionally, H2Aub promotes the binding of histone H1 to the nucleosome, stabilizing the nucleosome by preventing the dissociation of DNA from it (199).
Similarly, the ubiquitination site for histone H2B in mammals is typically located at lysine 120 (H2BK120ub) (200). In vivo, the ubiquitination of H2B (H2BK123ub) is catalyzed by the E2 transferase Rad6 and the E3 ligase Bre1. H2Bub rapidly accumulates at double-strand break (DSB) sites, where it plays a pivotal role in DSB repair (201, 202). Studies indicate that H2BK123ub promotes the methylation of histone H3 at lysines 4, 46, and 79 (H3K4, H3K46, H3K79) and that methylation at H3K46 and H3K79 is essential for effective DSB repair (203). These dynamic post-translational modifications regulate gene transcription and DNA repair through several mechanisms, including histone−DNA interaction regulation, nucleosome stability, histone eviction, chromatin compaction, histone cross-talk, and the recruitment of effector proteins.
In cancer, the mechanisms that regulate histone ubiquitination are frequently disrupted. Abnormal histone ubiquitination can drive tumorigenesis by altering the expression of tumor suppressors and oncogenes, misregulating cell differentiation, and promoting the proliferation of cancer cells. Compared with that in normal tissues, H2AK119ub1 expression is generally reduced in prostate cancer tissues. A global loss of H2BK120ub has been observed in patients with triple-negative breast cancer, gastric cancer, and colorectal cancer. Depletion of H2BK120ub significantly decreases p53 expression while simultaneously promoting c-MYC expression (204–207). Furthermore, DNA and RNA sequencing data revealed that genes encoding histone E3 ubiquitin ligases are frequently altered in cancer.
The polycomb E3 ubiquitin ligase subunits RING1A, RING1B, and BMI1, along with H2AK119ub1, help maintain the adult stem cell pool, and they may also contribute to the maintenance of cancer stem cells (208). In leukemia cells, BMI1 promotes cancer cell self-renewal by mediating the repression of key tumor suppressor genes (including the INK4A/ARF locus) through H2AK119ub1 (209). In addition to promoting acute leukemia, BMI1 promotes the proliferation of cancer cells in various solid tumors, including gastric cancer, pancreatic cancer, and epithelial ovarian cancer, by catalyzing H2A ubiquitination at lysine 119 (H2AK119ub) (210–212).
Given these roles, targeting abnormal histone ubiquitination represents a viable strategy for reprogramming transcription in cancer cells. The development of inhibitors that target aberrant histone ubiquitination sites or E3 ubiquitin ligases to block cancer cell proliferation and induce cell death is a promising avenue in histone ubiquitination-targeted cancer therapies.
2.4 Histone lactylation
Lactylation, a newly discovered post-translational modification (PTM) of histones, is closely related to the glycolytic metabolite lactate. Therefore, histone lactylation plays a significant role in cellular metabolic reprogramming. In 2019, Zhang et al. first reported a novel post-translational modification (PTM) of histones induced by lactate-lactylation (213). They discovered that lysine residues in histone tails can undergo lactylation. Subsequent studies confirmed the widespread existence of lactylation in various cancers and its close association with processes such as the development of malignancies. In hepatocellular carcinoma (HCC), researchers have reported that histone lactylation activates the transcription of ESM1 in HCC cells and that ESM1 is highly expressed in HCC, where it plays a carcinogenic role. Histone lactylation promotes the malignant phenotype of cells, tumor growth, and metastasis by increasing the expression of ESM1 in HCC. These findings may provide new therapeutic targets for the treatment of HCC (214). Yang et al. reported that the level of histone lactylation is associated with poor prognosis in patients with clear cell renal cell carcinoma (ccRCC). Mechanistic studies have shown that histone lactylation can promote the progression of ccRCC by activating the transcription of platelet-derived growth factor receptor β (PDGFRβ). Targeting histone lactylation can inhibit the proliferation and metastasis of ccRCC cells in vivo (215). In addition to the pro-cancer effects, histone lactylation can also have tumor-suppressive effects.
According to a study by Jiang et al. on non-small cell lung cancer (NSCLC), increased histone lactylation can lead to decreased levels of hexokinase 1 (HK-1) and pyruvate kinase M (PKM) in glycolysis, as well as increased levels of succinate dehydrogenase (SDHA) and isocitrate dehydrogenase 3γ (IDH3γ) in the tricarboxylic acid (TCA) cycle. This further results in the inhibition of tumor cell glycolysis, as well as reduced cell proliferation and migration abilities (216). In recent years, there have been several advancements in research on the roles of lactylation-related enzymes in tumors. Jin et al. reported that the delactylase SIRT3 can suppress the proliferation of liver cancer cells by regulating the lactylation of Cyclin E2 (217). In future research, studying histone lactylation modifications and their regulatory sites may lead to the identification of effective therapeutic targets for cancer treatment.
3 Clinical therapeutics
The previous discussion highlighted numerous abnormal histone modification sites and alterations in the activity of histone-modifying enzymes in cancer. These aberrant modifications and changes in the expression of modifying enzymes can serve as valuable biomarkers for the accurate screening, detection, diagnosis, and prognosis of tumors. Since different enzymes catalyze modifications at specific histone sites, targeting these histone-modifying enzymes with oncogenic potential represents a promising strategy in cancer therapy.
3.1 Epigenetic markers for prognosis
Epigenetic changes, particularly histone modifications observed in the early stages of tumor development and cancer progression, have been proposed as biomarkers for early cancer detection, prognosis, and treatment response. In most breast cancer patients, low or absent expression of H4K16ac has been identified, making H4K16ac an early marker of the disease (218). During epithelial−mesenchymal transition (EMT), the loss of H4K16ac in mesenchymal cells can distinguish between epithelial and mesenchymal phenotypes. Additionally, low levels of H4R3me2, H3K4me2, and H4K20me3 are associated with poor prognosis in patients with breast cancer (219). In gastric cancer, Jang et al. reported high expression of H3K9me3, which is positively correlated with tumor stage. Abnormal expression of KDM5B also promotes gastric cancer metastasis by regulating various signaling pathways, contributing to poor prognosis. In ovarian cancer, overexpression of HDAC3 and loss of H3K27me3 are linked to prognosis and disease progression. Furthermore, in glioblastoma (GBM), the nuclear expression levels of lysine methyltransferases, such as SETDB1, KMT5B, Suv-39h1, and EZH2, are elevated and associated with advanced histological cancer grades. Overall, changes in histone post-translational modifications have demonstrated clinical utility and are increasingly recognized as promising biomarkers for early cancer detection and diagnosis.
3.2 New targets and therapy
3.2.1 HMT inhibitors
Increasing evidence suggests that histone methyltransferases (HMTs) may serve as potential therapeutic targets for cancer treatment. As a result, the development of HMT inhibitors has gained considerable attention over the past decade. Most of these inhibitors target enzymes such as EZH2 and PRMT5 (220). To date, the U.S. FDA has approved two small-molecule inhibitors: tazemetostat (EPZ-6438) and valemetostat (DS-3201b), which target EZH2 or both EZH1 and EZH2 for cancer therapy. In addition, multiple EZH2 inhibitors (EZH2is), such as CPI-1205, SHR2554, and PF-06821497, are currently in various stages of clinical trials. PRMT5, another promising therapeutic target, is also being explored for cancer treatment. GSK3326595, a PRMT5 inhibitor, is in phase I trials for non-Hodgkin lymphoma, while other PRMT5 inhibitors, such as the PRT543 series, are in phase I trials for relapsed/refractory advanced solid tumors, and the PRT811 series is in phase I trials for relapsed glioma, advanced solid tumors, and CNS lymphoma. Other HMT inhibitors, such as the DOT1L inhibitor EPZ-5676, which is undergoing clinical trials for AML, ALL, and CML, have made significant progress in recent years.
While substantial progress has been made in the development of HMT inhibitors, the emergence of resistance with prolonged use highlights the limitations of monotherapy in treating solid tumors. Studies have shown that combining HMT inhibitors, such as EZH2 inhibitors, with immunotherapy, targeted therapy, chemotherapy, and endocrine therapy can result in synergistic anti-tumor effects, improving treatment outcomes. However, owing to the high homology between EZH1 and EZH2, EZH2 inhibitors often inhibit both, presenting challenges for the development of highly specific EZH2 inhibitors. Adachi et al. reported the discovery of two novel orally bioavailable dual-target EZH1/2 inhibitors, OR-S1 (compound 1) and OR-S2 (compound 2). These compounds exhibit potent and selective inhibition of both EZH2 and EZH1, effectively reducing H3K27me3 levels, and have demonstrated significant anti-tumor activity against diffuse large B-cell lymphoma cells with gain-of-function mutations. Optimizing the therapeutic efficacy of these inhibitors and minimizing their toxicity and side effects will be the focus of future research.
Inhibition of EZH2 can boost tumor immunotherapy through various mechanisms. Resistance to immune checkpoint inhibitors (ICIs) is currently a significant challenge in cancer immunotherapy. DuCote et al. reported that EZH2 inhibitors could increase ICI responses in patients undergoing treatment for lung squamous cell carcinoma (LSCC) (221). In LSCC, tumors can evade the immune system through the expression of PD-L1, thereby suppressing T-cell activation. Additionally, these tumors secrete high levels of CXCL1/2/3, which recruit T-cell-suppressive neutrophils and express high levels of arginase, further driving T-cell suppression. Under EZH2 inhibition, tumors upregulate the antigen presentation mechanisms of MHC I and MHC II and shift from the expression of CXCL1/2/3 to the expression of the T-cell-promoting cytokines CXCL9/10/11 and the anti-inflammatory molecule ALOX15. The combined use of anti-PD1 therapy and EZH2 inhibitors (such as GSK126 or EPZ6438) has also shown significant tumor-suppressive effects in LSCC patients. Furthermore, the EZH2 inhibitor tazemetostat has recently received FDA approval and is currently in clinical trials for the use of ICI therapy in urothelial carcinoma (NCT03854474). These studies suggest that EZH2 inhibition combined with ICI treatment may be an effective treatment strategy for ICI-resistant solid tumors.
3.2.2 HDM inhibitors
The targeting of demethylases is an emerging approach for the treatment of various cancers, and numerous demethylase inhibitors have been reported, some of which are currently undergoing clinical evaluation for cancer therapy. KDM inhibitors can be classified into two categories: KDM1 and JmjC family histone lysine demethylase inhibitors. Trans-2-phenylcyclopropylamine (TCP) and its derivatives can inhibit KDM1A and KDM1B (84, 222). Currently, six TCP-based KDM1A inhibitors have been developed and are in clinical trials, including TCP, ORY-1001, ORY-2001, GSK-2879552, INCB059872, and IMG-7289, which covalently bind to the FAD domain within KDM1A (84, 223). ORY-1001 inhibits the proliferation of TNBC cells and induces apoptosis by inactivating AR (224). The TCP derivatives (NCL-1, NCD-38, MC_2580, and DDP_38003) also exhibit anti-tumor effects as KDM1 inhibitors. NCL-1 and NCD-38 inhibit KDM1A, leading to reduced viability and increased apoptosis of GSCs, with minimal effects on differentiated cells (90) Maes et al. emphasized the therapeutic potential of combining ORY-1001 with checkpoint inhibitors for the treatment of melanoma. After cotreatment with ORY-1001 and anti-PD1 antibodies, significant tumor growth inhibition (TGI) was achieved, which was 54% higher than that in the anti-PD1 antibody treatment group (225).
JmjC family inhibitors are divided into broad-spectrum and subfamily-specific inhibitors. IOX1 is a broad-spectrum inhibitor that targets several subfamilies, including KDM2, KDM3, KDM4, KDM5, KDM6, and KDM7 (226). Liu et al. reported that IOX1 inhibits the expression of P-glycoprotein (P-gp) in cancer cells through the JMJD1A/β-catenin/P-gp pathway, thereby reducing the expression level of PD-L1. Moreover, IOX1 significantly enhances immunogenic cell death (ICD) induced by doxorubicin (DOX), promotes T-cell infiltration, and markedly suppresses tumor growth in preclinical models. These results suggest that the combination of IOX1 with immune checkpoint inhibitors may represent a promising strategy for cancer treatment (227). On the other hand, SD49-7 is a small-molecule inhibitor specific to KDM4A and KDM4C. Inhibiting these demethylases disrupts the expression of MDM2, activates p21, and suppresses the stemness of leukemia cells, leading to increased cell apoptosis (228). PBIT is a potent, selective inhibitor of KDM5B that reduces the expression of JARID1B, which results in decreased levels of cancer stem cell (CSC) and epithelial-mesenchymal transition (EMT) markers in cisplatin-resistant non-small cell lung cancer (229). PBIT also enhances the sensitivity of cancer cells to radiation therapy. These small-molecule KDM inhibitors, which exhibit drug-like specificity and selectivity, can be used either as standalone therapies or in combination with other immunotherapies and chromatin-targeting agents, thereby offering more viable treatment options for cancer.
3.2.3 HAT inhibitors
Among the existing small-molecule histone acetyltransferase (HAT) inhibitors, many studies have focused on compounds that target p300/CBP, which play key roles in acetylating histone H3 at lysines 18 and 27 (H3K18, H3K27) to facilitate gene activation critical for cell growth and differentiation (230). In addition to the enzymatic HAT domain, p300/CBP contains several other functional domains, such as three cysteine-histidine-rich domains (CH1, CH2, and CH3), a KIX domain, a bromodomain, and a steroid receptor coactivator interaction domain (SRC-1 interaction domain). Inhibitors targeting p300/CBP are designed primarily to exploit these domain characteristics.
One promising inhibitor is CCS1477, a CBP/p300 bromodomain inhibitor developed by CellCentric, which is currently in phase 1b/2a clinical trials for treating hematological malignancies and late-stage castration-resistant prostate cancer. A-485 is another potent and highly selective p300/CBP inhibitor that has shown anti-tumor activity in prostate cancer cell lines (231). Additionally, Ding et al. identified compound 13f, a novel p300/CBP HAT inhibitor, which demonstrated significant anti-tumor effects in an ovarian cancer xenograft mouse model (232).
Natural compounds have also shown promise. Garcinol, a natural inhibitor of p300/CBP and PCAF, has anti-tumor effects on several cancer cell lines, including hepatocellular carcinoma, gastric cancer, and triple-negative breast cancer (233). WM-3835 selectively inhibits HBO1, another member of the HAT family, and effectively suppresses mouse osteosarcoma (OS) tumor cell growth (234). Additionally, the small molecule NBP targets KAT7 to inhibit PD-L1 expression and weaken the PD-1/PD-L1 axis, reducing T-cell apoptosis to alleviate lung cancer progression. It may serve as a potential therapeutic strategy for immunotherapy in lung cancer (235).
Despite these advances, the development of targeted drugs for HAT inhibition remains in its early stages. Future research should focus on identifying and developing inhibitors for other members of the HAT family, offering new and effective strategies for anti-tumor therapy.
3.2.4 HDAC inhibitors
Histone deacetylase (HDAC) inhibitors play crucial roles in restoring the balance of acetylation and deacetylation of lysine residues of histones and nonhistone proteins and are used to treat several diseases, including cancer (Table 3). Four HDAC inhibitors (HDACis), vorinostat (SAHA), belinostat (PXD101), panobinostat (LBH589), and romidepsin (FK228), have been approved by the US FDA for the treatment of cutaneous T-cell lymphoma (CTCL), relapsed or refractory peripheral T-cell lymphoma (PTCL), and multiple myeloma (MM), whereas chidamide (CS055) has also been approved by China’s NMPA for the treatment of relapsed or refractory peripheral T-cell lymphoma (249). Additionally, several HDAC inhibitors are in clinical trial stages.
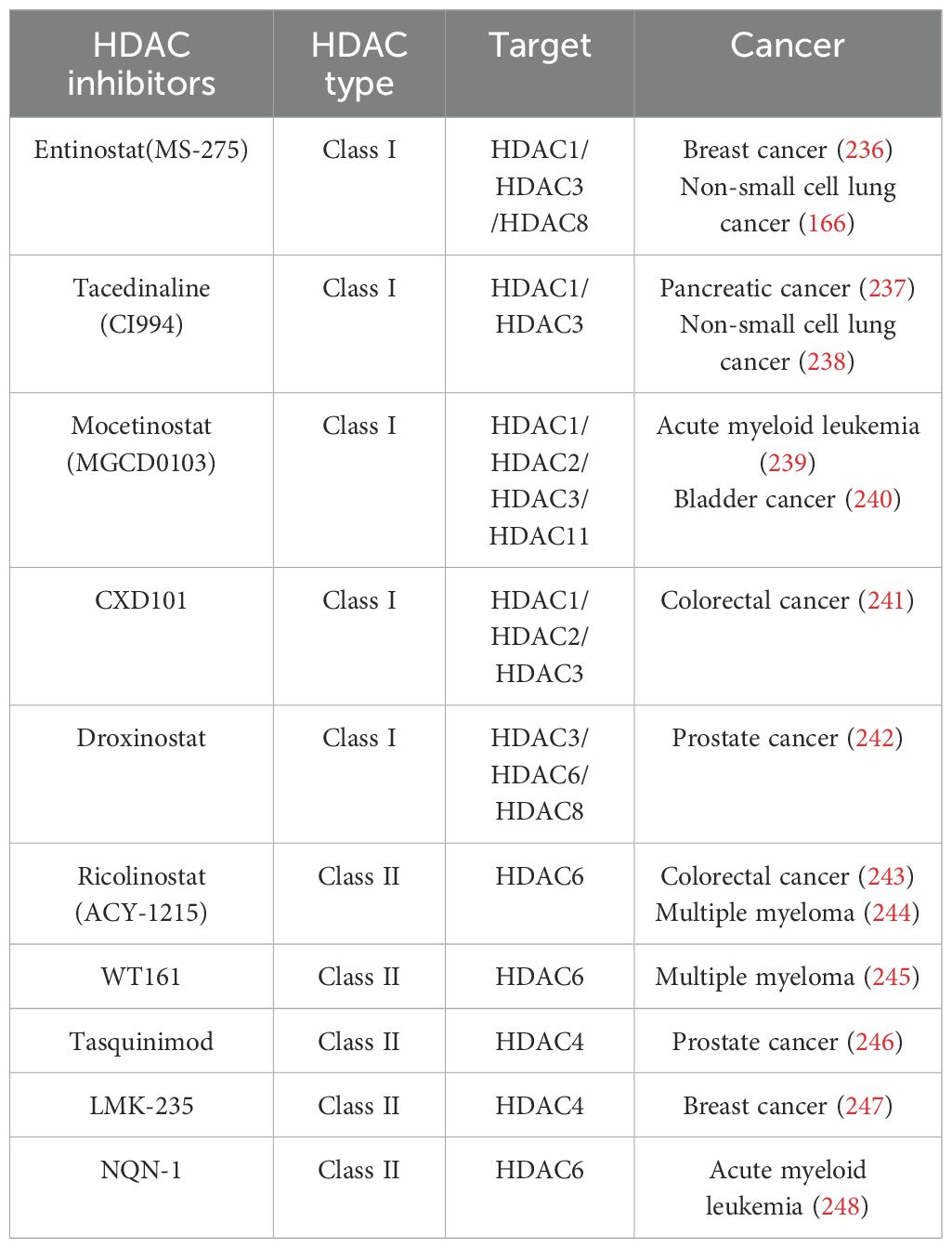
Table 3. HDACis in cancer therapy.
The majority of HDACi structures contain three pharmacophoric elements: a cap structure (Cap region) that interacts with amino acid residues at the entrance edge of the HDAC active site; a zinc-binding group (ZBG) that chelates the catalytic zinc ion located at the bottom of the active site pocket; and a linker region that connects the Cap region and ZBG, which interacts with the hydrophobic channel of the active site (250–252). The chemical structures of HDAC inhibitors can be divided into four main categories: hydroxamic acids (such as vorinostat), benzamide derivatives (such as chidamide), cyclic peptides (such as romidepsin), and fatty acids (such as 2-propylpentanoic acid).
Mocetinostat (MGCD0103) is a novel benzamide-class HDAC inhibitor that can inhibit HDAC1, 2, 3, and 11. It is currently being researched for diseases such as acute myeloid leukemia, bladder cancer, and non-small cell lung cancer and has entered phase II clinical trials (253). Largazole is a cyclic depsipeptide natural product that shows good activity and selectivity for HDAC1. It inhibits the proliferation of the human breast cancer cell line MDA-MB-231 and the human osteosarcoma cell line U2OS but has no effect on normal cells (254). AR-42 is an orally effective HDAC inhibitor that is currently in clinical trials for the treatment of multiple myeloma, leukemia, and lymphoma (255). CAY10603 is a potent HDAC6 inhibitor that effectively inhibits the activity of HDAC6. It inhibits the proliferation of Burkitt’s lymphoma cell lines and induces caspase-dependent apoptosis (256).
The combination of HDACis and immunotherapy has made great progress in cancer immunotherapy. The activation of the immune response by HDACis could prevent cancer relapse. Entinostat (ENT; Class I HDAC inhibitor) has shown significant efficacy in tumor immunotherapy when combined with anti-PD-1/anti-CTLA-4 antibodies, functioning by reducing the number of tumor-infiltrating G-MDSCs (257). Owing to the typical downregulation of MHC class I expression caused by epigenetic mechanisms in cancer, HDAC inhibitors can upregulate MHC class I expression in various types of cancer. Romidepsin (HDAC1/2 inhibitor), valproic acid (Class I HDAC inhibitor), or RGFP966 (HDAC3 inhibitor) can upregulate MHC class I expression and the expression of costimulatory molecules such as CD80 and CD86 in B-cell lymphomas (258). In HER2+ breast cancer, the combination of palbociclib and trastuzumab (anti-HER2) achieves anti-tumor efficacy by stimulating the release of CXCR3-reactive chemokines and increasing the recruitment of tumor-associated natural killer (NK) cells (259). In addition, HDAC inhibitors (HDACis) can modulate the effector functions of activated immune cells and potentiate the efficacy of immunotherapeutic strategies against established solid tumors. The HDAC inhibitor MS-275 can enhance the lymphocyte depletion induced by oncolytic virus vectors, leading to the selective exhaustion of conventional lymphocytes and regulatory T cells (Tregs) while allowing the expansion of antigen-specific secondary responses (260). HDAC inhibitors may participate in the anticancer immune response by directly altering the immunogenicity of tumor cells and potentially rescuing the functional activity of exhausted CD8+ T cells (261).
Currently, the main applications of HDAC inhibitors (HDACis) are limited to the treatment of peripheral T-cell lymphoma and cutaneous T-cell lymphoma, whereas the development of drugs for solid tumors is relatively limited. Additionally, clinical trials of HDAC inhibitors have revealed numerous adverse reactions in patients, including thrombocytopenia-induced bleeding, neutropenia-induced susceptibility to infections, anemia due to hemoglobin reduction, arrhythmias, myocardial hypertrophy, and neurotoxicity, which also pose major limitations in the development of HDAC inhibitors (262). Furthermore, most HDACis are broad-spectrum inhibitors, making the development of efficient inhibitors with cell selectivity and isoform specificity a significant challenge.
4 Summary and future perspectives
Epigenetic modifications have become key regulatory factors and drivers in the occurrence and development of cancer in recent years. The processes of DNA repair, replication, transcription, translation, and posttranscriptional and post-translational regulation are all under the control of epigenetics. Therefore, abnormal expression patterns or epigenomic alterations can lead to dysregulation, ultimately resulting in cancer. Changes in the epigenetic landscape of cancer can affect the expression of genes involved in cellular metabolism, primarily through the dysregulation of metabolic signaling pathways caused by abnormal DNA methylation, histone modifications, and non-coding RNAs.
Histone post-translational modifications (PTMs) do not alter the DNA sequence but can change the expression and functional levels, providing new explanations for many biological activities. In addition to common modifications such as methylation, acetylation, ubiquitination, and phosphorylation, in recent years, histone butyrylation, lactylation, propionylation, and isonicotinamide have gradually become new research hotspots. Most PTMs are reversible and therefore can regulate the functionality of the proteome in a cell type-specific manner to modulate gene expression. Abnormalities in histone post-translational modifications often affect key molecular regulatory mechanisms involved in the progression of tumors.
In this review, we elaborate on the main types of histone PTMs and their functions in different categories of cancers. We subsequently summarize the key enzymes that influence histone modifications and their inhibitors, providing feasible targets for cancer treatment research. Furthermore, the crosstalk between different histone PTMs is also an interesting research direction. Methylation of H3K4 can increase the acetylation activity of HATs on the H3 tail. Therefore, the impact of histone modification enzymes on cancer development is certainly not singular.
As the role of histone PTMs in the cancer field has gained increasing attention, targeted drugs against histone modification enzymes have also become a research focus in this area. As mentioned previously, HDAC inhibitors (HDACis) are currently the most widely used HDACis in clinical practice. While various HDAC inhibitors that target different HDAC classes have been approved, these inhibitors are currently limited to the treatment of hematological cancers, with few HDAC inhibitors that target solid tumors. Therefore, developing more HDAC inhibitors that target solid tumors is also a future research focus for researchers. Furthermore, since the therapeutic efficacy of HDAC inhibitors as single agents is limited, there is a need to develop dual-target HDAC drugs. Dual-target HDAC inhibitors can act on multiple signaling pathways involved in tumor development, thereby more comprehensively regulating epigenetic modifications and gene expression in cells. For example, researchers at China Pharmaceutical University have recently developed a series of innovative dual PD-L1/HDAC6 inhibitors, C1-C6 (patent number: CN113387840A). In the CT26 syngeneic colon tumor model, the tumor inhibition effect of C5 exceeded the efficacy of BMS202 (a PD-L1 inhibitor) or SAHA (an approved HDAC inhibitor) monotherapy.
Histone modification enzyme inhibitors are also a hot topic in the clinical treatment of cancer immunotherapy. Immunotherapy works by activating the immune system to fight cancer cells, achieving good efficacy in some patients. However, in many patients, tumor cells evade detection by the immune system because of their low mutational burden, resulting in “cold tumors.” Additionally, cancer cells can develop various resistance mechanisms to immune checkpoint inhibitors, such as reduced antigen presentation, modulation of immune cell recruitment, secretion of immunosuppressive factors, decreased co-stimulatory molecules, and induction of T cell apoptosis. These factors greatly limit the effectiveness of immunotherapy in cancer patients. Currently, multiple studies have shown that histone modification enzyme inhibitors, important epigenetic modifiers, can not only enhance anti-tumor immunity by inhibiting the expression of immune checkpoint molecules (ICMs) but also be used in combination with immune checkpoint inhibitors (ICIs) to enhance the tumor response to immunotherapy by increasing ICM expression. A study revealed that the combination of HDAC inhibitors (HDACis) and immune checkpoint inhibitors (ICIs) altered the infiltration and function of innate immune cells, leading to a more robust adaptive immune response via the inhibition of myeloid-derived suppressor cells (MDSCs) and immune-resistant breast tumors (263). Many drugs targeting dysregulated epigenetic regulators have entered clinical use for the treatment of hematological malignancies. Increasing compelling evidence suggests that epigenetic therapies have the potential to transform immunosuppressive (“cold”) tumors into immunopermissive (“hot”) tumors. The combination of epigenetic therapies and immunotherapy can consolidate anti-tumor immune responses, reprogram the immunosuppressive TME, and improve treatment outcomes.
In future cancer treatment, the ability of HDAC to synergize with other therapies makes it a promising candidate for cancer immunotherapy. In future tumor treatment strategies centered on histone PTMs, only by clearly elucidating the specific mechanisms of action of histone PTMs in tumors will it be possible to conduct research on targeted anticancer drugs based on the molecular mechanisms of PTMs and thus promote cancer treatment.
Author contributions
XD: Writing – original draft, Writing – review & editing. ZX: Writing – original draft, Writing – review & editing. LQ: Writing – original draft, Writing – review & editing. SQ: Writing – original draft. XZ: Writing – original draft, Writing – review & editing. YG: Writing – original draft, Writing – review & editing. XL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Tianjin Health science and technology project (TJWJ2024ZD012) granted from Tianjin Health Commission and entrusted Tianjin Jinnan Hospital to undertake.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. (2013) 58:439–45. doi: 10.1038/jhg.2013.66
2. Lai WKM, Pugh BF. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol. (2017) 18:548–62. doi: 10.1038/nrm.2017.47
3. Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. (2006) 14:17–25. doi: 10.1007/s10577-005-1024-3
4. Wang M, Lin H. Understanding the function of mammalian sirtuins and protein lysine acylation. Annu Rev Biochem. (2021) 90:245–85. doi: 10.1146/annurev-biochem-082520-125411
5. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. (2011) 146:1016–28. doi: 10.1016/j.cell.2011.08.008
6. Millán-Zambrano G, Burton A, Bannister AJ, Schneider R. Histone post-translational modifications - cause and consequence of genome function. Nat Rev Genet. (2022) 23:563–80. doi: 10.1038/s41576-022-00468-7
7. Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. (2004) 11:1037–43. doi: 10.1038/nsmb851
8. Garcia BA, Shabanowitz J, Hunt DF. Characterization of histones and their post-translational modifications by mass spectrometry. Curr Opin Chem Biol. (2007) 11:66–73. doi: 10.1016/j.cbpa.2006.11.022
9. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. (2000) 403:41–5. doi: 10.1038/47412
10. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006
11. Lv X, Han Z, Chen H, Yang B, Yang X, Xia Y, et al. A positive role for polycomb in transcriptional regulation via H4K20me1. Cell Res. (2017) 27:594. doi: 10.1038/cr.2017.50
12. Agredo A, Kasinski AL. Histone 4 lysine 20 tri-methylation: a key epigenetic regulator in chromatin structure and disease. Front Genet. (2023) 14:1243395. doi: 10.3389/fgene.2023.1243395
13. Monaghan L, Massett ME, Bunschoten RP, Hoose A, Pirvan PA, Liskamp RMJ, et al. The emerging role of H3K9me3 as a potential therapeutic target in acute myeloid leukemia. Front Oncol. (2019) 9:705. doi: 10.3389/fonc.2019.00705
14. Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol. (2008) 15:1968–76. doi: 10.1245/s10434-008-9927-9
15. Benard A, Goossens-Beumer IJ, van Hoesel AQ, de Graaf W, Horati H, Putter H, et al. Histone trimethylation at H3K4, H3K9 and H4K20 correlates with patient survival and tumor recurrence in early-stage colon cancer. BMC Cancer. (2014) 14:531. doi: 10.1186/1471-2407-14-531
16. Pathak P, Jha P, Purkait S, Sharma V, Suri V, Sharma MC, et al. Altered global histone-trimethylation code and H3F3A-ATRX mutation in pediatric GBM. J Neurooncol. (2015) 121:489–97. doi: 10.1007/s11060-014-1675-z
17. Berger L, Kolben T, Meister S, Kolben TM, Schmoeckel E, Mayr D, et al. Expression of H3K4me3 and H3K9ac in breast cancer. J Cancer Res Clin Oncol. (2020) 146:2017–27. doi: 10.1007/s00432-020-03265-z
18. Gao SB, Xu B, Ding LH, Zheng QL, Zhang L, Zheng QF, et al. The functional and mechanistic relatedness of EZH2 and menin in hepatocellular carcinoma. J Hepatol. (2014) 61:832–9. doi: 10.1016/j.jhep.2014.05.015
19. Duan JL, Nie RC, Xiang ZC, Chen JW, Deng MH, Liang H, et al. Prognostic model for the risk stratification of early and late recurrence in hepatitis B virus-related small hepatocellular carcinoma patients with global histone modifications. J Hepatocell Carcinoma. (2021) 8:493–505. doi: 10.2147/jhc.S309451
20. Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. (2010) 28:1358–65. doi: 10.1200/jco.2009.24.5639
21. Barlési F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. (2007) 25:4358–64. doi: 10.1200/jco.2007.11.2599
22. Derks S, Bosch LJW, Niessen HEC, Moerkerk PTM, van den Bosch SM, Carvalho B, et al. Promoter CpG island hypermethylation- and H3K9me3 and H3K27me3-mediated epigenetic silencing targets the deleted in colon cancer (DCC) gene in colorectal carcinogenesis without affecting neighboring genes on chromosomal region 18q21. Carcinogenesis. (2009) 30:1041–8. doi: 10.1093/carcin/bgp073
23. Müller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N, et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood. (2010) 116:3564–71. doi: 10.1182/blood-2009-09-240978
24. Okabe A, Huang KK, Matsusaka K, Fukuyo M, Xing M, Ong X, et al. Cross-species chromatin interactions drive transcriptional rewiring in Epstein-Barr virus-positive gastric adenocarcinoma. Nat Genet. (2020) 52:919–30. doi: 10.1038/s41588-020-0665-7
25. Xia R, Zhou R, Tian Z, Zhang C, Wang L, Hu Y, et al. High expression of H3K9me3 is a strong predictor of poor survival in patients with salivary adenoid cystic carcinoma. Arch Pathol Lab Med. (2013) 137:1761–9. doi: 10.5858/arpa.2012-0704-OA
26. Ellinger J, Bachmann A, Göke F, Behbahani TE, Baumann C, Heukamp LC, et al. Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urol Int. (2014) 93:113–8. doi: 10.1159/000355467
27. Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int. (2012) 62:182–90. doi: 10.1111/j.1440-1827.2011.02776.x
28. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. (2012) 81:65–95. doi: 10.1146/annurev-biochem-051710-134100
29. Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. (2013) 152:1021–36. doi: 10.1016/j.cell.2013.01.052
30. Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. (2010) 142:967–80. doi: 10.1016/j.cell.2010.08.020
31. Sankar A, Lerdrup M, Manaf A, Johansen JV, Gonzalez JM, Borup R, et al. KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat Cell Biol. (2020) 22:380–8. doi: 10.1038/s41556-020-0494-z
32. Liu X, Wang C, Liu W, Li J, Li C, Kou X, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. (2016) 537:558–62. doi: 10.1038/nature19362
33. Du M, Gong P, Zhang Y, Liu Y, Liu X, Zhang F, et al. Histone methyltransferase SETD1A participates in lung cancer progression. Thorac Cancer. (2021) 12:2247–57. doi: 10.1111/1759-7714.14065
34. Phoyen S, Sanpavat A, Ma-On C, Stein U, Hirankarn N, Tangkijvanich P, et al. H4K20me3 upregulated by reactive oxygen species is associated with tumor progression and poor prognosis in patients with hepatocellular carcinoma. Heliyon. (2023) 9:e22589. doi: 10.1016/j.heliyon.2023.e22589
35. Alaterre E, Ovejero S, Herviou L, de Boussac H, Papadopoulos G, Kulis M, et al. Comprehensive characterization of the epigenetic landscape in Multiple Myeloma. Theranostics. (2022) 12:1715–29. doi: 10.7150/thno.54453
36. Wang Q, Trevino LS, Wong RL, Medvedovic M, Chen J, Ho SM, et al. Reprogramming of the epigenome by MLL1 links early-life environmental exposures to prostate cancer risk. Mol Endocrinol. (2016) 30:856–71. doi: 10.1210/me.2015-1310
37. Zhao F, Chen Y, Zeng L, Li R, Zeng R, Wen L, et al. Role of triptolide in cell proliferation, cell cycle arrest, apoptosis and histone methylation in multiple myeloma U266 cells. Eur J Pharmacol. (2010) 646:1–11. doi: 10.1016/j.ejphar.2010.05.034
38. Kitajima H, Maruyama R, Niinuma T, Yamamoto E, Takasawa A, Takasawa K, et al. TM4SF1-AS1 inhibits apoptosis by promoting stress granule formation in cancer cells. Cell Death Dis. (2023) 14:424. doi: 10.1038/s41419-023-05953-3
39. López C, Barnon MT, Beacon TH, Nardocci G, Davie JR. The key role of differential broad H3K4me3 and H3K4ac domains in breast cancer. Gene. (2022) 826:146463. doi: 10.1016/j.gene.2022.146463
40. Beacon TH, Delcuve GP, López C, Nardocci G, Kovalchuk I, van Wijnen AJ, et al. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin Epigenetics. (2021) 13:138. doi: 10.1186/s13148-021-01126-1
41. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. (2011) 469:343–9. doi: 10.1038/nature09784
42. Day CA, Hinchcliffe EH, Robinson JP. H3K27me3 in diffuse midline glioma and epithelial ovarian cancer: opposing epigenetic changes leading to the same poor outcomes. Cells. (2022) 11(21):3376. doi: 10.3390/cells11213376
43. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. (2008) 32:503–18. doi: 10.1016/j.molcel.2008.11.004
44. Liefke R, Shi Y. The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discovery. (2015) 1:15008. doi: 10.1038/celldisc.2015.8
45. Yi X, Guo J, Guo J, Sun S, Yang P, Wang J, et al. EZH2-mediated epigenetic silencing of TIMP2 promotes ovarian cancer migration and invasion. Sci Rep. (2017) 7:3568. doi: 10.1038/s41598-017-03362-z
46. Zong X, Wang W, Ozes A, Fang F, Sandusky GE, Nephew KP. EZH2-mediated downregulation of the tumor suppressor DAB2IP maintains ovarian cancer stem cells. Cancer Res. (2020) 80:4371–85. doi: 10.1158/0008-5472.Can-20-0458
47. Yun EJ, Baek ST, Xie D, Tseng SF, Dobin T, Hernandez E, et al. DAB2IP regulates cancer stem cell phenotypes through modulating stem cell factor receptor and ZEB1. Oncogene. (2015) 34:2741–52. doi: 10.1038/onc.2014.215
48. Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. (2020) 139:1109–13. doi: 10.1007/s00401-020-02142-w
49. Wang MY, Liow P, Guzman MIT, Qi J. Exploring methods of targeting histone methyltransferases and their applications in cancer therapeutics. ACS Chem Biol. (2022) 17:744–55. doi: 10.1021/acschembio.2c00062
50. McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther. (2015) 150:1–22. doi: 10.1016/j.pharmthera.2015.01.002
51. Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol. (2016) 9:49. doi: 10.1186/s13045-016-0279-9
52. Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. (2019) 26:880–9. doi: 10.1038/s41594-019-0298-7
53. Saha N, Muntean AG. Insight into the multi-faceted role of the SUV family of H3K9 methyltransferases in carcinogenesis and cancer progression. Biochim Biophys Acta Rev Cancer. (2021) 1875:188498. doi: 10.1016/j.bbcan.2020.188498
54. Jarrold J, Davies CC. PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol Med. (2019) 25:993–1009. doi: 10.1016/j.molmed.2019.05.007
55. Ning J, Chen L, Xiao G, Zeng Y, Shi W, Tanzhu G, et al. The protein arginine methyltransferase family (PRMTs) regulates metastases in various tumors: From experimental study to clinical application. BioMed Pharmacother. (2023) 167:115456. doi: 10.1016/j.biopha.2023.115456
56. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. (2019) 20:642–57. doi: 10.1038/s41580-019-0155-x
57. Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci. (2009) 66:2109–21. doi: 10.1007/s00018-009-0010-x
58. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. (2015) 15:334–46. doi: 10.1038/nrc3929
59. Kumari N, Singh RK, Mishra SK, Krishnani N, Mohindra S, R L. Identification of PI3K-AKT signaling as the dominant altered pathway in intestinal type ampullary cancers through whole-exome sequencing. J Pathol Transl Med. (2021) 55:192–201. doi: 10.4132/jptm.2021.01.23
60. Bosgana P, Nikou S, Dimitrakopoulos FI, Logotheti S, Tzelepi V, Kalophonos C, et al. H3K4 methylation status and lysine specific methyltransferase KMT2C expression correlate with prognosis in lung adenocarcinoma. Curr Mol Pharmacol. (2021) 14:1028–36. doi: 10.2174/1874467213999200831130739
61. Cai WL, Chen JF, Chen H, Wingrove E, Kurley SJ, Chan LH, et al. Human WDR5 promotes breast cancer growth and metastasis via KMT2-independent translation regulation. Elife. (2022) 11:e78163. doi: 10.7554/eLife.78163
62. Laukhtina E, Lemberger U, Bruchbacher A, Ilijazi D, Korn S, Berndl F, et al. Expression analysis and mutational status of histone methyltransferase KMT2D at different upper tract urothelial carcinoma locations. J Pers Med. (2021) 11(11):1147. doi: 10.3390/jpm11111147
63. Seehawer M, Li Z, Nishida J, Foidart P, Reiter AH, Rojas-Jimenez E, et al. Loss of Kmt2c or Kmt2d drives brain metastasis via KDM6A-dependent upregulation of MMP3. Nat Cell Biol. (2024) 26:1165–75. doi: 10.1038/s41556-024-01446-3
64. Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. (2016) 30:179–80. doi: 10.1016/j.ccell.2016.06.013
65. Jing P, Zhao N, Ye M, Zhang Y, Zhang Z, Sun J, et al. Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Lett. (2018) 427:38–48. doi: 10.1016/j.canlet.2018.04.019
66. Cheng D, He Z, Zheng L, Xie D, Dong S, Zhang P. PRMT7 contributes to the metastasis phenotype in human non-small-cell lung cancer cells possibly through the interaction with HSPA5 and EEF2. Onco Targets Ther. (2018) 11:4869–76. doi: 10.2147/ott.S166412
67. Sze CC, Ozark PA, Cao K, Ugarenko M, Das S, Wang L, et al. Coordinated regulation of cellular identity-associated H3K4me3 breadth by the COMPASS family. Sci Adv. (2020) 6(26):eaaz4764. doi: 10.1126/sciadv.aaz4764
68. Sze CC, Cao K, Collings CK, Marshall SA, Rendleman EJ, Ozark PA, et al. Histone H3K4 methylation-dependent and -independent functions of Set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. (2017) 31:1732–7. doi: 10.1101/gad.303768.117
69. Wu J, Chai H, Xu X, Yu J, Gu Y. Histone methyltransferase SETD1A interacts with HIF1α to enhance glycolysis and promote cancer progression in gastric cancer. Mol Oncol. (2020) 14:1397–409. doi: 10.1002/1878-0261.12689
70. Ishii T, Akiyama Y, Shimada S, Kabashima A, Asano D, Watanabe S, et al. Identification of a novel target of SETD1A histone methyltransferase and the clinical significance in pancreatic cancer. Cancer Science. (2023) 114:463–76. doi: 10.1111/cas.15615
71. Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. (2013) 4:2144–53. doi: 10.18632/oncotarget.1555
72. Xu J, Zhong A, Zhang S, Chen M, Zhang L, Hang X, et al. KMT2D deficiency promotes myeloid leukemias which is vulnerable to ribosome biogenesis inhibition. Adv Sci (Weinh). (2023) 10:e2206098. doi: 10.1002/advs.202206098
73. Sanghrajka RM, Koche R, Medrano H, El Nagar S, Stephen DN, Lao Z, et al. KMT2D suppresses Sonic hedgehog-driven medulloblastoma progression and metastasis. iScience. (2023) 26:107831. doi: 10.1016/j.isci.2023.107831
74. Li Q, Wu R, Wu F, Chen Q. KMT2D promotes proliferation of gastric cancer cells: evidence from ctDNA sequencing. J Clin Lab Anal. (2021) 35:e23721. doi: 10.1002/jcla.23721
75. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. (2017) 355:1324–30. doi: 10.1126/science.aah6893
76. Ku B, Eisenbarth D, Baek S, Jeong TK, Kang JG, Hwang D, et al. PRMT1 promotes pancreatic cancer development and resistance to chemotherapy. Cell Rep Med. (2024) 5:101461. doi: 10.1016/j.xcrm.2024.101461
77. Yao B, Gui T, Zeng X, Deng Y, Wang Z, Wang Y, et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. (2021) 13:58. doi: 10.1186/s13073-021-00871-5
78. Li Z, Wang D, Lu J, Huang B, Wang Y, Dong M, et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. (2020) 27:3226–42. doi: 10.1038/s41418-020-00615-9
79. He L, Hu Z, Sun Y, Zhang M, Zhu H, Jiang L, et al. PRMT1 is critical to FEN1 expression and drug resistance in lung cancer cells. DNA Repair (Amst). (2020) 95:102953. doi: 10.1016/j.dnarep.2020.102953
80. Deng X, Von Keudell G, Suzuki T, Dohmae N, Nakakido M, Piao L, et al. PRMT1 promotes mitosis of cancer cells through arginine methylation of INCENP. Oncotarget. (2015) 6:35173–82. doi: 10.18632/oncotarget.6050
81. Zhu Y, He X, Lin YC, Dong H, Zhang L, Chen X, et al. Targeting PRMT1-mediated FLT3 methylation disrupts maintenance of MLL-rearranged acute lymphoblastic leukemia. Blood. (2019) 134:1257–68. doi: 10.1182/blood.2019002457
82. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
83. Hoffmann I, Roatsch M, Schmitt ML, Carlino L, Pippel M, Sippl W, et al. The role of histone demethylases in cancer therapy. Mol Oncol. (2012) 6:683–703. doi: 10.1016/j.molonc.2012.07.004
84. Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol. (2019) 12:129. doi: 10.1186/s13045-019-0811-9
85. Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry. (2007) 46:8058–65. doi: 10.1021/bi700664y
86. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. (2006) 439:811–6. doi: 10.1038/nature04433
87. Zhang J, Jing L, Li M, He L, Guo Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol Med Rep. (2019) 19:3963–71. doi: 10.3892/mmr.2019.10111
88. Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. (2007) 318:444–7. doi: 10.1126/science.1145801
89. Lu Y, Liu Y, Oeck S, Glazer PM. Hypoxia promotes resistance to EGFR inhibition in NSCLC cells via the histone demethylases, LSD1 and PLU-1. Mol Cancer Res. (2018) 16:1458–69. doi: 10.1158/1541-7786.Mcr-17-0637
90. Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene. (2017) 36:2423–34. doi: 10.1038/onc.2016.395
91. Lin Q, Wu Z, Yue X, Yu X, Wang Z, Song X, et al. ZHX2 restricts hepatocellular carcinoma by suppressing stem cell-like traits through KDM2A-mediated H3K36 demethylation. EBioMedicine. (2020) 53:102676. doi: 10.1016/j.ebiom.2020.102676
92. Ramadoss S, Sen S, Ramachandran I, Roy S, Chaudhuri G, Farias-Eisner R. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene. (2017) 36:1537–45. doi: 10.1038/onc.2016.320
93. Tang DE, Dai Y, He JX, Lin LW, Leng QX, Geng XY, et al. Targeting the KDM4B-AR-c-Myc axis promotes sensitivity to androgen receptor-targeted therapy in advanced prostate cancer. J Pathol. (2020) 252:101–13. doi: 10.1002/path.5495
94. Feng T, Wang Y, Lang Y, Zhang Y. KDM5A promotes proliferation and EMT in ovarian cancer and closely correlates with PTX resistance. Mol Med Rep. (2017) 16:3573–80. doi: 10.3892/mmr.2017.6960
95. Xu L, Wu H, Hu X. Histone demethylase KDM5A enhances cell proliferation, induces EMT in lung adenocarcinoma cells, and have a strong causal association with paclitaxel resistance. Acta Biochim Pol. (2021) 68:593–602. doi: 10.18388/abp.2020_5437
96. Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. (2018) 34:939–53.e9. doi: 10.1016/j.ccell.2018.10.014
97. Kuo KT, Huang WC, Bamodu OA, Lee WH, Wang CH, Hsiao M, et al. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin Epigenetics. (2018) 10:107. doi: 10.1186/s13148-018-0533-9
98. Liu J, Nie C. KDM5B regulates the PTEN/PI3K/Akt pathway to increase sorafenib-resistance in hepatocellular carcinoma. Anticancer Drugs. (2022) 33:840–9. doi: 10.1097/cad.0000000000001329
99. He C, Sun J, Liu C, Jiang Y, Hao Y. Elevated H3K27me3 levels sensitize osteosarcoma to cisplatin. Clin Epigenetics. (2019) 11:8. doi: 10.1186/s13148-018-0605-x
100. Wang W, Lim KG, Feng M, Bao Y, Lee PL, Cai Y, et al. KDM6B counteracts EZH2-mediated suppression of IGFBP5 to confer resistance to PI3K/AKT inhibitor treatment in breast cancer. Mol Cancer Ther. (2018) 17:1973–83. doi: 10.1158/1535-7163.Mct-17-0802
101. Xu S, Zheng L, Kang L, Xu H, Gao L. microRNA-let-7e in serum-derived exosomes inhibits the metastasis of non-small-cell lung cancer in a SUV39H2/LSD1/CDH1-dependent manner. Cancer Gene Ther. (2021) 28:250–64. doi: 10.1038/s41417-020-00216-1
102. Chen K, Cai Y, Cheng C, Zhang J, Lv F, Xu G, et al. MYT1 attenuates neuroblastoma cell differentiation by interacting with the LSD1/CoREST complex. Oncogene. (2020) 39:4212–26. doi: 10.1038/s41388-020-1268-6
103. Qin Y, Zhu W, Xu W, Zhang B, Shi S, Ji S, et al. LSD1 sustains pancreatic cancer growth via maintaining HIF1α-dependent glycolytic process. Cancer Lett. (2014) 347:225–32. doi: 10.1016/j.canlet.2014.02.013
104. Kumaraswamy A, Duan Z, Flores D, Zhang C, Sehrawat A, Hu YM, et al. LSD1 promotes prostate cancer reprogramming by repressing TP53 signaling independently of its demethylase function. JCI Insight. (2023) 8(15):e167440. doi: 10.1172/jci.insight.167440
105. Shen DD, Pang JR, Bi YP, Zhao LF, Li YR, Zhao LJ, et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol Cancer. (2022) 21:75. doi: 10.1186/s12943-022-01557-1
106. Tee AE, Ling D, Nelson C, Atmadibrata B, Dinger ME, Xu N, et al. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. (2014) 5:1793–804. doi: 10.18632/oncotarget.1785
107. Zhao QY, Lei PJ, Zhang X, Zheng JY, Wang HY, Zhao J, et al. Global histone modification profiling reveals the epigenomic dynamics during Malignant transformation in a four-stage breast cancer model. Clin Epigenetics. (2016) 8:34. doi: 10.1186/s13148-016-0201-x
108. Liu J, Zhu M, Xia X, Huang Y, Zhang Q, Wang X. Jumonji domain-containing protein 1A promotes cell growth and progression via transactivation of c-Myc expression and predicts a poor prognosis in cervical cancer. Oncotarget. (2016) 7:85151–62. doi: 10.18632/oncotarget.13208
109. Zhan M, Wen F, Liu L, Chen Z, Wei H, Zhou H. JMJD1A promotes tumorigenesis and forms a feedback loop with EZH2/let-7c in NSCLC cells. Tumour Biol. (2016) 37:11237–47. doi: 10.1007/s13277-016-4999-9
110. Feng B, Zhu Y, Su Z, Tang L, Sun C, Li C, et al. Basil polysaccharide attenuates hepatocellular carcinoma metastasis in rat by suppressing H3K9me2 histone methylation under hepatic artery ligation-induced hypoxia. Int J Biol Macromol. (2018) 107:2171–9. doi: 10.1016/j.ijbiomac.2017.10.088
111. Ning K, Shao Y, He Y, Wang F, Cui X, Liu F, et al. Histone demethylase Jumonji domain-containing 1A inhibits proliferation and progression of gastric cancer by upregulating runt-related transcription factor 3. Cancer Sci. (2020) 111:3679–92. doi: 10.1111/cas.14594
112. Jeon H-Y, Ryu H, Pornour M, Qi J. Histone demethylase JMJD1A in cancer progression and therapeutic resistance. Mol Carcinogenesis. (2022) 61:392–6. doi: 10.1002/mc.23390
113. Fan L, Peng G, Sahgal N, Fazli L, Gleave M, Zhang Y, et al. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene. (2016) 35:2441–52. doi: 10.1038/onc.2015.309
114. Fan L, Xu S, Zhang F, Cui X, Fazli L, Gleave M, et al. Histone demethylase JMJD1A promotes expression of DNA repair factors and radio-resistance of prostate cancer cells. Cell Death Dis. (2020) 11:214. doi: 10.1038/s41419-020-2405-4
115. Wan W, Peng K, Li M, Qin L, Tong Z, Yan J, et al. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene. (2017) 36:3868–77. doi: 10.1038/onc.2017.13
116. Tran N, Broun A, Ge K. Lysine demethylase KDM6A in differentiation, development, and cancer. Mol Cell Biol. (2020) 40(20):e00341-20. doi: 10.1128/mcb.00341-20
117. Ezponda T, Dupéré-Richer D, Will CM, Small EC, Varghese N, Patel T, et al. UTX/KDM6A loss enhances the Malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. (2017) 21:628–40. doi: 10.1016/j.celrep.2017.09.078
118. Levinson AL, Tjoa K, Huang B, Meyer LK, Kim MO, Brady SW, et al. Opposing effects of KDM6A and JDP2 on glucocorticoid sensitivity in T-ALL. Blood Adv. (2023) 7:3479–84. doi: 10.1182/bloodadvances.2021006881
119. Schulz WA, Lang A, Koch J, Greife A. The histone demethylase UTX/KDM6A in cancer: Progress and puzzles. Int J Cancer. (2019) 145:614–20. doi: 10.1002/ijc.32116
120. Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. (2014) 74:1705–17. doi: 10.1158/0008-5472.Can-13-1896
121. Barrows D, Feng L, Carroll TS, Allis CD. Loss of UTX/KDM6A and the activation of FGFR3 converge to regulate differentiation gene-expression programs in bladder cancer. Proc Natl Acad Sci U S A. (2020) 117:25732–41. doi: 10.1073/pnas.2008017117
122. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. (2013) 73:2936–42. doi: 10.1158/0008-5472.Can-12-4300
123. Li BX, Luo CL, Li H, Yang P, Zhang MC, Xu HM, et al. Effects of siRNA-mediated knockdown of jumonji domain containing 2A on proliferation, migration and invasion of the human breast cancer cell line MCF-7. Exp Ther Med. (2012) 4:755–61. doi: 10.3892/etm.2012.662
124. Li BX, Zhang MC, Luo CL, Yang P, Li H, Xu HM, et al. Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. J Exp Clin Cancer Res. (2011) 30:90. doi: 10.1186/1756-9966-30-90
125. Kim TD, Shin S, Berry WL, Oh S, Janknecht R. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J Cell Biochem. (2012) 113:1368–76. doi: 10.1002/jcb.24009
126. Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C, et al. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J Biol Chem. (2011) 286:41616–25. doi: 10.1074/jbc.M111.283689
127. Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, et al. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem. (2009) 284:8395–405. doi: 10.1074/jbc.M807818200
128. Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, et al. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. (2009) 28:4491–500. doi: 10.1038/onc.2009.297
129. Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PloS One. (2011) 6:e17830. doi: 10.1371/journal.pone.0017830
130. Berdel B, Nieminen K, Soini Y, Tengström M, Malinen M, Kosma VM, et al. Histone demethylase GASC1–a potential prognostic and predictive marker in invasive breast cancer. BMC Cancer. (2012) 12:516. doi: 10.1186/1471-2407-12-516
131. Slee RB, Steiner CM, Herbert BS, Vance GH, Hickey RJ, Schwarz T, et al. Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene. (2012) 31:3244–53. doi: 10.1038/onc.2011.502
132. Jing JC, Feng Z, Chen ZH, Ji BN, Hong J, Tang N, et al. KDM4B promotes gastric cancer metastasis by regulating miR-125b-mediated activation of Wnt signaling. J Cell Biochem. (2019) 120:7897–906. doi: 10.1002/jcb.28065
133. Toyokawa G, Cho HS, Iwai Y, Yoshimatsu M, Takawa M, Hayami S, et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev Res (Phila). (2011) 4:2051–61. doi: 10.1158/1940-6207.Capr-11-0290
134. Zhu Y, van Essen D, Saccani S. Cell-type-specific control of enhancer activity by H3K9 trimethylation. Mol Cell. (2012) 46:408–23. doi: 10.1016/j.molcel.2012.05.011
135. Lagunas-Rangel FA. KDM6B (JMJD3) and its dual role in cancer. Biochimie. (2021) 184:63–71. doi: 10.1016/j.biochi.2021.02.005
136. Burchfield JS, Li Q, Wang HY, Wang RF. JMJD3 as an epigenetic regulator in development and disease. Int J Biochem Cell Biol. (2015) 67:148–57. doi: 10.1016/j.biocel.2015.07.006
137. Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. JMJD3 is a histone H3K27 demethylase. Cell Res. (2007) 17:850–7. doi: 10.1038/cr.2007.83
138. Yang L, Zha Y, Ding J, Ye B, Liu M, Yan C, et al. Histone demethylase KDM6B has an anti-tumorigenic function in neuroblastoma by promoting differentiation. Oncogenesis. (2019) 8:3. doi: 10.1038/s41389-018-0112-0
139. Tang B, Qi G, Tang F, Yuan S, Wang Z, Liang X, et al. Aberrant JMJD3 expression upregulates slug to promote migration, invasion, and stem cell-like behaviors in hepatocellular carcinoma. Cancer Res. (2016) 76:6520–32. doi: 10.1158/0008-5472.Can-15-3029
140. Chang S, Yim S, Park H. The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp Mol Med. (2019) 51:1–17. doi: 10.1038/s12276-019-0230-6
141. Chang WH, Forde D, Lai AG. Dual prognostic role of 2-oxoglutarate-dependent oxygenases in ten cancer types: implications for cell cycle regulation and cell adhesion maintenance. Cancer Commun (Lond). (2019) 39:23. doi: 10.1186/s40880-019-0369-5
142. Jones SE, Olsen L, Gajhede M. Structural basis of histone demethylase KDM6B histone 3 lysine 27 specificity. Biochemistry. (2018) 57:585–92. doi: 10.1021/acs.biochem.7b01152
143. Yu S-H, Zhu K-Y, Chen J, Liu X-Z, Xu P-F, Zhang W, et al. JMJD3 facilitates C/EBPβ-centered transcriptional program to exert oncorepressor activity in AML. Nat Commun. (2018) 9:3369. doi: 10.1038/s41467-018-05548-z
144. Svotelis A, Bianco S, Madore J, Huppé G, Nordell-Markovits A, Mes-Masson AM, et al. H3K27 demethylation by JMJD3 at a poised enhancer of anti&x2010;apoptotic gene BCL2 determines ER&x3b1; ligand dependency. EMBO J. (2011) 30:3947–61. doi: 10.1038/emboj.2011.284
145. Ramadoss S, Chen X, Wang C-Y. Histone demethylase KDM6B promotes epithelial-mesenchymal transition*. J Biol Chem. (2012) 287:44508–17. doi: 10.1074/jbc.M112.424903
146. Li SH, Lu HI, Chen YH, Lo CM, Huang WT, Tien WY, et al. JMJD3 expression is an independent prognosticator in patients with esophageal squamous cell carcinoma. Surgery. (2019) 165:946–52. doi: 10.1016/j.surg.2018.11.015
147. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc Natl Acad Sci U S A. (1964) 51:786–94. doi: 10.1073/pnas.51.5.786
148. Avvakumov N, Côté J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. (2007) 26:5395–407. doi: 10.1038/sj.onc.1210608
149. Alaskhar Alhamwe B, Khalaila R, Wolf J, von Bülow V, Harb H, Alhamdan F, et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin Immunol. (2018) 14:39. doi: 10.1186/s13223-018-0259-4
150. Zaib S, Rana N, Khan I. Histone modifications and their role in epigenetics of cancer. Curr Med Chem. (2022) 29:2399–411. doi: 10.2174/0929867328666211108105214
151. Shen Y, Wei W, Zhou DX. Histone acetylation enzymes coordinate metabolism and gene expression. Trends Plant Sci. (2015) 20:614–21. doi: 10.1016/j.tplants.2015.07.005
152. Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell Mol Life Sci. (2001) 58:728–36. doi: 10.1007/pl00000896
153. Doffo J, Bamopoulos SA, Köse H, Orben F, Zang C, Pons M, et al. NOXA expression drives synthetic lethality to RUNX1 inhibition in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2022) 119(9):e2105691119. doi: 10.1073/pnas.2105691119
154. Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. (2012) 6:579–89. doi: 10.1016/j.molonc.2012.07.003
155. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
156. Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. (2010) 10:454–60. doi: 10.1016/j.coph.2010.04.004
157. Injinari N, Amini-Farsani Z, Yadollahi-Farsani M, Teimori H. Apoptotic effects of valproic acid on miR-34a, miR-520h and HDAC1 gene in breast cancer. Life Sci. (2021) 269:119027. doi: 10.1016/j.lfs.2021.119027
158. Ma JB, Bai JY, Zhang HB, Jia J, Shi Q, Yang C, et al. KLF5 inhibits STAT3 activity and tumor metastasis in prostate cancer by suppressing IGF1 transcription cooperatively with HDAC1. Cell Death Dis. (2020) 11:466. doi: 10.1038/s41419-020-2671-1
159. Yu Z, Zeng J, Liu H, Wang T, Yu Z, Chen J. Role of HDAC1 in the progression of gastric cancer and the correlation with lncRNAs. Oncol Lett. (2019) 17:3296–304. doi: 10.3892/ol.2019.9962
160. Wright CA, Gordon ER, Cooper SJ. Genomic analysis reveals HDAC1 regulates clinically relevant transcriptional programs in Pancreatic cancer. BMC Cancer. (2023) 23:1137. doi: 10.1186/s12885-023-11645-0
161. Cheng Z, Li S, Yuan J, Li Y, Cheng S, Huang S, et al. HDAC1 mediates epithelial-mesenchymal transition and promotes cancer cell invasion in glioblastoma. Pathol Res Pract. (2023) 246:154481. doi: 10.1016/j.prp.2023.154481
162. Liu X, Yu Y, Zhang J, Lu C, Wang L, Liu P, et al. HDAC1 silencing in ovarian cancer enhances the chemotherapy response. Cell Physiol Biochem. (2018) 48:1505–18. doi: 10.1159/000492260
163. Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Xie HJ, et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J Cell Biochem. (2012) 113:2167–77. doi: 10.1002/jcb.24090
164. Hu X-t, Xing W, Zhao R-s, Tan Y, Wu X-f, Ao L-q, et al. HDAC2 inhibits EMT-mediated cancer metastasis by downregulating the long noncoding RNA H19 in colorectal cancer. J Exp Clin Cancer Res. (2020) 39:270. doi: 10.1186/s13046-020-01783-9
165. Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, et al. CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discovery. (2017) 7:38–53. doi: 10.1158/2159-8290.Cd-16-0975
166. Eichner LJ, Curtis SD, Brun SN, McGuire CK, Gushterova I, Baumgart JT, et al. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non–small cell lung cancer. Sci Adv. (2023) 9:eadd3243. doi: 10.1126/sciadv.add3243
167. Li Y, Liang R, Sun M, Li Z, Sheng H, Wang J, et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. (2020) 478:82–92. doi: 10.1016/j.canlet.2020.03.007
168. Li Y, Li X, Guo B. Chemopreventive agent 3,3’-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. (2010) 70:646–54. doi: 10.1158/0008-5472.Can-09-1924
169. Vanaja GR, Ramulu HG, Kalle AM. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun Signal. (2018) 16:20. doi: 10.1186/s12964-018-0231-4
170. Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. (2007) 67:2632–42. doi: 10.1158/0008-5472.Can-06-4152
171. Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. (2010) 150:665–73. doi: 10.1111/j.1365-2141.2010.08301.x
172. Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, et al. Increased gene expression of histone deacetylases in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Lymphoma. (2012) 53:123–9. doi: 10.3109/10428194.2011.597905
173. Peixoto P, Castronovo V, Matheus N, Polese C, Peulen O, Gonzalez A, et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. (2012) 19:1239–52. doi: 10.1038/cdd.2012.3
174. Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, et al. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. (2004) 10:6962–8. doi: 10.1158/1078-0432.Ccr-04-0455
175. Bulut I, Lee A, Cevatemre B, Ruzic D, Belle R, Kawamura A, et al. Dual LSD1 and HDAC6 inhibition induces doxorubicin sensitivity in acute myeloid leukemia cells. Cancers (Basel). (2022) 14(23):6014. doi: 10.3390/cancers14236014
176. Lee JH, Jeong EG, Choi MC, Kim SH, Park JH, Song SH, et al. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells. (2010) 30:107–12. doi: 10.1007/s10059-010-0094-z
177. Guarente L, Picard F. Calorie restriction–the SIR2 connection. Cell. (2005) 120:473–82. doi: 10.1016/j.cell.2005.01.029
178. Alves-Fernandes DK, Jasiulionis MG. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int J Mol Sci. (2019) 20(13):3153. doi: 10.3390/ijms20133153
179. Liarte S, Alonso-Romero JL, Nicolás FJ. SIRT1 and estrogen signaling cooperation for breast cancer onset and progression. Front Endocrinol (Lausanne). (2018) 9:552. doi: 10.3389/fendo.2018.00552
180. Farcas M, Gavrea AA, Gulei D, Ionescu C, Irimie A, Catana CS, et al. SIRT1 in the development and treatment of hepatocellular carcinoma. Front Nutr. (2019) 6:148. doi: 10.3389/fnut.2019.00148
181. Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. (2003) 23:3173–85. doi: 10.1128/mcb.23.9.3173-3185.2003
182. Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, et al. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun. (2003) 309:558–66. doi: 10.1016/j.bbrc.2003.08.029
183. Xu L, Li Y, Zhou L, Dorfman RG, Liu L, Cai R, et al. SIRT3 elicited an anti-Warburg effect through HIF1α/PDK1/PDHA1 to inhibit cholangiocarcinoma tumorigenesis. Cancer Med. (2019) 8:2380–91. doi: 10.1002/cam4.2089
184. Liu L, Li Y, Cao D, Qiu S, Li Y, Jiang C, et al. SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett. (2021) 510:93–104. doi: 10.1016/j.canlet.2021.04.007
185. Liu Y, Tong X, Hu W, Chen D. HDAC11: A novel target for improved cancer therapy. BioMed Pharmacother. (2023) 166:115418. doi: 10.1016/j.biopha.2023.115418
186. Bora-Singhal N, Mohankumar D, Saha B, Colin CM, Lee JY, Martin MW, et al. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci Rep. (2020) 10:4722. doi: 10.1038/s41598-020-61295-6
187. He Y, Zheng CC, Yang J, Li SJ, Xu TY, Wei X, et al. Lysine butyrylation of HSP90 regulated by KAT8 and HDAC11 confers chemoresistance. Cell Discovery. (2023) 9:74. doi: 10.1038/s41421-023-00570-y
188. Yi Z, Wenwen L, Kun W, Jian S. Overexpression of histone deacetylase 11 suppresses basal-like breast cancer cell invasion and metastasis. Nan Fang Yi Ke Da Xue Xue Bao. (2019) 39:751–9. doi: 10.12122/j.issn.1673-4254.2019.07.01
189. Wang W, Ding B, Lou W, Lin S. Promoter hypomethylation and miR-145-5p downregulation- mediated HDAC11 overexpression promotes sorafenib resistance and metastasis of hepatocellular carcinoma cells. Front Cell Dev Biol. (2020) 8:724. doi: 10.3389/fcell.2020.00724
190. Zhu W, Zhang X, Yu M, Zhang Y, Li S, Yu C. Profiles of acetylation regulation genes contribute to Malignant progression and have a clinical prognostic impact on liver cancer. Dis Markers. (2022) 2022:1724301. doi: 10.1155/2022/1724301
191. Bi L, Ren Y, Feng M, Meng P, Wang Q, Chen W, et al. HDAC11 regulates glycolysis through the LKB1/AMPK signaling pathway to maintain hepatocellular carcinoma stemness. Cancer Res. (2021) 81:2015–28. doi: 10.1158/0008-5472.Can-20-3044
192. Tweedie-Cullen RY, Reck JM, Mansuy IM. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J Proteome Res. (2009) 8:4966–82. doi: 10.1021/pr9003739
193. West MH, Bonner WM. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res. (1980) 8:4671–80. doi: 10.1093/nar/8.20.4671
194. de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. (2004) 7:663–76. doi: 10.1016/j.devcel.2004.10.005
195. Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. (2005) 20:845–54. doi: 10.1016/j.molcel.2005.12.002
196. Barbour H, Daou S, Hendzel M, Affar EB. Polycomb group-mediated histone H2A monoubiquitination in epigenome regulation and nuclear processes. Nat Commun. (2020) 11:5947. doi: 10.1038/s41467-020-19722-9
197. Zhou W, Wang X, Rosenfeld MG. Histone H2A ubiquitination in transcriptional regulation and DNA damage repair. Int J Biochem Cell Biol. (2009) 41:12–5. doi: 10.1016/j.biocel.2008.09.016
198. Marsh DJ, Ma Y, Dickson KA. Histone monoubiquitination in chromatin remodelling: focus on the histone H2B interactome and cancer. Cancers (Basel). (2020) 12(11):3462. doi: 10.3390/cancers12113462
199. Xiao X, Liu C, Pei Y, Wang YZ, Kong J, Lu K, et al. Histone H2A ubiquitination reinforces mechanical stability and asymmetry at the single-nucleosome level. J Am Chem Soc. (2020) 142:3340–5. doi: 10.1021/jacs.9b12448
200. Wu L, Zee BM, Wang Y, Garcia BA, Dou Y. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol Cell. (2011) 43:132–44. doi: 10.1016/j.molcel.2011.05.015
201. Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell. (2011) 41:529–42. doi: 10.1016/j.molcel.2011.02.015
202. Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell. (2011) 41:515–28. doi: 10.1016/j.molcel.2011.02.002
203. Schulze JM, Hentrich T, Nakanishi S, Gupta A, Emberly E, Shilatifard A, et al. Splitting the task: Ubp8 and Ubp10 deubiquitinate different cellular pools of H2BK123. Genes Dev. (2011) 25:2242–7. doi: 10.1101/gad.177220.111
204. Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. (2011) 71:5739–53. doi: 10.1158/0008-5472.Can-11-1896
205. Wang Z, Zhu L, Guo T, Wang Y, Yang J. Decreased H2B monoubiquitination and overexpression of ubiquitin-specific protease enzyme 22 in Malignant colon carcinoma. Hum Pathol. (2015) 46:1006–14. doi: 10.1016/j.humpath.2015.04.001
206. Wang ZJ, Yang JL, Wang YP, Lou JY, Chen J, Liu C, et al. Decreased histone H2B monoubiquitination in Malignant gastric carcinoma. World J Gastroenterol. (2013) 19:8099–107. doi: 10.3748/wjg.v19.i44.8099
207. Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. (2008) 22:2664–76. doi: 10.1101/gad.1703008
208. Jeusset LM, McManus KJ. Developing targeted therapies that exploit aberrant histone ubiquitination in cancer. Cells. (2019) 8(2):165. doi: 10.3390/cells8020165
209. Saudy NS, Fawzy IM, Azmy E, Goda EF, Eneen A, Abdul Salam EM. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol Dis. (2014) 53:194–8. doi: 10.1016/j.bcmd.2014.07.002
210. Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. (2014) 20:29–36. doi: 10.1038/nm.3418
211. Proctor E, Waghray M, Lee CJ, Heidt DG, Yalamanchili M, Li C, et al. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PloS One. (2013) 8:e55820. doi: 10.1371/journal.pone.0055820
212. Zhang F, Sui L, Xin T. Correlations of BMI-1 expression and telomerase activity in ovarian cancer tissues. Exp Oncol. (2008) 30:70–4.
213. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
214. Zhao P, Qiao C, Wang J, Zhou Y, Zhang C. Histone lactylation facilitates hepatocellular carcinoma progression by upregulating endothelial cell-specific molecule 1 expression. Mol Carcinog. (2024) 63(11):2078–89. doi: 10.1002/mc.23794
215. Yang J, Luo L, Zhao C, Li X, Wang Z, Zeng Z, et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ Signaling drives clear cell renal cell carcinoma progression. Int J Biol Sci. (2022) 18:3470–83. doi: 10.7150/ijbs.73398
216. Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front Oncol. (2021) 11:647559. doi: 10.3389/fonc.2021.647559
217. Jin J, Bai L, Wang D, Ding W, Cao Z, Yan P, et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. (2023) 24:e56052. doi: 10.15252/embr.202256052
218. Wang B, Zhou M, Gan XL, Ren YX, Yang YZ, Weng ZJ, et al. Combined low levels of H4K16ac and H4K20me3 predicts poor prognosis in breast cancer. Int J Clin Oncol. (2023) 28:1147–57. doi: 10.1007/s10147-023-02378-y
219. Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. (2009) 69:3802–9. doi: 10.1158/0008-5472.Can-08-3907
220. Li D, Peng X, Hu Z, Li S, Chen J, Pan W. Small molecules targeting selected histone methyltransferases (HMTs) for cancer treatment: Current progress and novel strategies. Eur J Med Chem. (2024) 264:115982. doi: 10.1016/j.ejmech.2023.115982
221. DuCote TJ, Song X, Naughton KJ, Chen F, Plaugher DR, Childress AR, et al. EZH2 inhibition promotes tumor immunogenicity in lung squamous cell carcinomas. Cancer Res Commun. (2024) 4:388–403. doi: 10.1158/2767-9764.Crc-23-0399
222. Zheng YC, Yu B, Jiang GZ, Feng XJ, He PX, Chu XY, et al. Irreversible LSD1 inhibitors: application of tranylcypromine and its derivatives in cancer treatment. Curr Top Med Chem. (2016) 16:2179–88. doi: 10.2174/1568026616666160216154042
223. Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. (2015) 28:57–69. doi: 10.1016/j.ccell.2015.06.002
224. Wang T, Zhang F, Sun F. ORY-1001, a KDM1A inhibitor, inhibits proliferation, and promotes apoptosis of triple negative breast cancer cells by inactivating androgen receptor. Drug Dev Res. (2022) 83:208–16. doi: 10.1002/ddr.21860
225. Maes T, Mascaró C, Sacilotto N, Lufino MMP, Ciceri F. Targeting KDM1A with iadademstat in combination with immunotherapy in an in vivo model of melanoma. J Clin Oncol. (2019) 37:e14248–e. doi: 10.1200/JCO.2019.37.15_suppl.e14248
226. Macedo-Silva C, Miranda-Gonçalves V, Lameirinhas A, Lencart J, Pereira A, Lobo J, et al. JmjC-KDMs KDM3A and KDM6B modulate radioresistance under hypoxic conditions in esophageal squamous cell carcinoma. Cell Death Dis. (2020) 11:1068. doi: 10.1038/s41419-020-03279-y
227. Liu J, Zhao Z, Qiu N, Zhou Q, Wang G, Jiang H, et al. Co-delivery of IOX1 and doxorubicin for antibody-independent cancer chemo-immunotherapy. Nat Commun. (2021) 12:2425. doi: 10.1038/s41467-021-22407-6
228. Li Y, Wang C, Gao H, Gu J, Zhang Y, Zhang Y, et al. KDM4 inhibitor SD49-7 attenuates leukemia stem cell via KDM4A/MDM2/p21(CIP1) axis. Theranostics. (2022) 12:4922–34. doi: 10.7150/thno.71460
229. Das A, Bortner J, Desai D, Amin S, El-Bayoumy K. The selenium analog of the chemopreventive compound S,S’-(1,4-phenylenebis[1,2-ethanediyl])bisisothiourea is a remarkable inducer of apoptosis and inhibitor of cell growth in human non-small cell lung cancer. Chem Biol Interact. (2009) 180:158–64. doi: 10.1016/j.cbi.2009.03.003
230. He Z-X, Wei B-F, Zhang X, Gong Y-P, Ma L-Y, Zhao W. Current development of CBP/p300 inhibitors in the last decade. Eur J Medicinal Chem. (2021) 209:112861. doi: 10.1016/j.ejmech.2020.112861
231. Feng L, Yu S, Wang H, Yang S, Li X, Dai H, et al. Synthesis and biological evaluation of spirocyclic chromane derivatives as a potential treatment of prostate cancer. Molecules. (2021) 26(11):3162. doi: 10.3390/molecules26113162
232. Ding H, Pei Y, Li Y, Xu W, Mei L, Hou Z, et al. Design, synthesis and biological evaluation of a novel spiro oxazolidinedione as potent p300/CBP HAT inhibitor for the treatment of ovarian cancer. Bioorg Med Chem. (2021) 52:116512. doi: 10.1016/j.bmc.2021.116512
233. Kopytko P, Piotrowska K, Janisiak J, Tarnowski M. Garcinol—A natural histone acetyltransferase inhibitor and new anti-cancer epigenetic drug. Int J Mol Sci. (2021) 22:2828. doi: 10.3390/ijms22062828
234. Gao YY, Ling ZY, Zhu YR, Shi C, Wang Y, Zhang XY, et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics. (2021) 11:4599–615. doi: 10.7150/thno.55655
235. Jiang Q, Zhang N, Li X, Hou W, Zhao XQ, Liu L. Dl-3-N-butylphthalide presents anti-cancer activity in lung cancer by targeting PD-1/PD-L1 signaling. Cancer Manag Res. (2021) 13:8513–24. doi: 10.2147/cmar.S333416
236. Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther. (2010) 9:3254–66. doi: 10.1158/1535-7163.Mct-10-0582
237. Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. (2006) 17:1096–102. doi: 10.1093/annonc/mdl081
238. Gridelli C, Rossi A, Maione P. The potential role of histone deacetylase inhibitors in the treatment of non-small-cell lung cancer. Crit Rev Oncol Hematol. (2008) 68:29–36. doi: 10.1016/j.critrevonc.2008.03.002
239. Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H, et al. The combination of a histone deacetylase inhibitor with the BH3-mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Autophagy. (2010) 6:976–8. doi: 10.4161/auto.6.7.13117
240. Groselj B, Ruan JL, Scott H, Gorrill J, Nicholson J, Kelly J, et al. Radiosensitization in vivo by histone deacetylase inhibition with no increase in early normal tissue radiation toxicity. Mol Cancer Ther. (2018) 17:381–92. doi: 10.1158/1535-7163.Mct-17-0011
241. Blaszczak W, Liu G, Zhu H, Barczak W, Shrestha A, Albayrak G, et al. Immune modulation underpins the anti-cancer activity of HDAC inhibitors. Mol Oncol. (2021) 15:3280–98. doi: 10.1002/1878-0261.12953
242. McCourt C, Maxwell P, Mazzucchelli R, Montironi R, Scarpelli M, Salto-Tellez M, et al. Elevation of c-FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin Cancer Res. (2012) 18:3822–33. doi: 10.1158/1078-0432.Ccr-11-3277
243. Wen Y, Ye S, Li Z, Zhang X, Liu C, Wu Y, et al. HDAC6 inhibitor ACY-1215 enhances STAT1 acetylation to block PD-L1 for colorectal cancer immunotherapy. Cancer Immunol Immunother. (2024) 73:7. doi: 10.1007/s00262-023-03624-y
244. Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br J Haematol. (2015) 169:423–34. doi: 10.1111/bjh.13315
245. Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, West N, et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc Natl Acad Sci U S A. (2016) 113:13162–7. doi: 10.1073/pnas.1608067113
246. Olsson A, Björk A, Vallon-Christersson J, Isaacs JT, Leanderson T. Tasquinimod (ABR-215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer. (2010) 9:107. doi: 10.1186/1476-4598-9-107
247. Oltra SS, Cejalvo JM, Tormo E, Albanell M, Ferrer A, Nacher M, et al. HDAC5 inhibitors as a potential treatment in breast cancer affecting very young women. Cancers (Basel). (2020) 12(2):412. doi: 10.3390/cancers12020412
248. Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol. (2012) 7:331–9. doi: 10.1021/cb200134p
249. Sultana F, Manasa Lakshmi K, Shaik Pasha S, Bonam Reddy S, Kamal A. Zinc dependent histone deacetylase inhibitors in cancer therapeutics: recent update. Curr Medicinal Chem. (2019) 26:7212–80. doi: 10.2174/0929867325666180530094120
250. Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Medicinal Chem. (2003) 46:5097–116. doi: 10.1021/jm0303094
251. Marson MC. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anti-Cancer Agents Medicinal Chem. (2009) 9:661–92. doi: 10.2174/187152009788679976
252. Luan Y, Li J, Bernatchez JA, Li R. Kinase and histone deacetylase hybrid inhibitors for cancer therapy. J Medicinal Chem. (2019) 62:3171–83. doi: 10.1021/acs.jmedchem.8b00189
253. Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget M-C, Kalita A, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. (2008) 7:759–68. doi: 10.1158/1535-7163.Mct-07-2026
254. Poli G, Di Fabio R, Ferrante L, Summa V, Botta M. Largazole analogues as histone deacetylase inhibitors and anticancer agents: an overview of structure–activity relationships. ChemMedChem. (2017) 12:1917–26. doi: 10.1002/cmdc.201700563
255. Tng J, Lim J, Wu KC, Lucke AJ, Xu W, Reid RC, et al. Achiral derivatives of hydroxamate AR-42 potently inhibit class I HDAC enzymes and cancer cell proliferation. J Med Chem. (2020) 63:5956–71. doi: 10.1021/acs.jmedchem.0c00230
256. Ma XJ, Xu G, Li ZJ, Chen F, Wu D, Miao JN, et al. HDAC-selective inhibitor cay10603 has single anti-tumour effect in burkitt’s lymphoma cells by impeding the cell cycle. Curr Med Sci. (2019) 39:228–36. doi: 10.1007/s11596-019-2024-4
257. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. (2014) 111:11774–9. doi: 10.1073/pnas.1410626111
258. Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C. HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol Cancer Ther. (2019) 18:900–8. doi: 10.1158/1535-7163.Mct-18-1068
259. Medon M, Vidacs E, Vervoort SJ, Li J, Jenkins MR, Ramsbottom KM, et al. HDAC inhibitor panobinostat engages host innate immune defenses to promote the tumoricidal effects of trastuzumab in HER2(+) tumors. Cancer Res. (2017) 77:2594–606. doi: 10.1158/0008-5472.Can-16-2247
260. Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. (2013) 21:887–94. doi: 10.1038/mt.2012.265
261. Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective CD8+ T cells from chronic viral infection. Mol Ther. (2014) 22:1698–706. doi: 10.1038/mt.2014.91
262. Li G, Tian Y, Zhu WG. The roles of histone deacetylases and their inhibitors in cancer therapy. Front Cell Dev Biol. (2020) 8:576946. doi: 10.3389/fcell.2020.576946
Keywords: histone PTMs, methylation, acetylation, HMTs, KDMs, HDAC, cancer
Citation: Duan X, Xing Z, Qiao L, Qin S, Zhao X, Gong Y and Li X (2024) The role of histone post-translational modifications in cancer and cancer immunity: functions, mechanisms and therapeutic implications. Front. Immunol. 15:1495221. doi: 10.3389/fimmu.2024.1495221
Received: 12 September 2024; Accepted: 18 October 2024;
Published: 15 November 2024.
Edited by:
Xiangpeng Dai, Jilin University, ChinaReviewed by:
Zhiwei Wang, Wenzhou Medical University, ChinaRuize Gao, Luxembourg Institute of Health, Luxembourg
Copyright © 2024 Duan, Xing, Qiao, Qin, Zhao, Gong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhua Gong, Z29uZ3lhbmh1YUB0anUuZWR1LmNu; Xueren Li, MTM4MjA5MzE4NDdAMTYzLmNvbQ==
†These authors share first authorship