 Xiaoke Wu
Xiaoke Wu Shaokuan Fang
Shaokuan Fang- Department of Neurology, Neuroscience Centre, The First Hospital of Jilin University, Changchun, China
Oncolytic viruses are either naturally occurring or genetically engineered viruses that can activate immune cells and selectively replicate in and destroy cancer cells without damaging healthy tissues. Oncolytic virus therapy (OVT) represents an emerging treatment approach for cancer. In this review, we outline the properties of oncolytic viruses and then offer an overview of the immune cells and tumor microenvironment (TME) across various OVTs. A thorough understanding of the immunological mechanisms involved in OVTs could lead to the identification of novel and more effective therapeutic targets for cancer treatment.
1 Introduction
Treatment options for tumors have expanded significantly in recent years, driven by an enhanced understanding of the immunologic mechanisms underlying these diseases (1, 2). These options now include traditional surgical treatments and chemoradiotherapy, as well as innovative approaches like immune checkpoint inhibitors, chimeric antigen receptor T-cells, adoptive cell immunotherapy, monoclonal antibodies, nanoantibodies, and other forms of immunotherapy (3–7). While surgical resection remains the most effective treatment for cancer and can substantially relieve symptoms, it is often not viable for patients in advanced stages of the disease. Moreover, surgery often leads to distant metastasis and local recurrence post-operatively (8). Chemoradiotherapy can decelerate cancer growth and prolong survival, however, it poses severe risks due to its damaging effects on normal cells while targeting cancer cells (9, 10). Immunotherapies, despite their potential, benefit only a select group of patients due to various immunosuppressive factors such as immune system suppression, deficiencies in cytokine types, reduced activity of tumor-infiltrating lymphocytes, impaired function of antigen-presenting cells, and weakened effector T-cell activity (11–13). Oncolytic viruses have emerged as one of the most promising treatments to address these challenges (14). These viruses are capable of selectively replicating within tumor cells, delivering multiple eukaryotic transgene payloads, inducing immunogenic cell death, enhancing antitumor immunity, and exhibiting a safety profile that generally does not overlap with other cancer treatments (15, 16). Recent clinical trials of oncolytic virus therapy (OVT) have shown minimal severe adverse reactions, with only local injection site reactions and low-grade systemic symptoms noted (17–20). In 2005, Chinese regulators approved the first genetically modified oncolytic adenovirus for the treatment of nasopharyngeal carcinoma in combination with chemotherapy, marking the world’s first oncolytic virus approved for clinical cancer treatment (21).
Oncolytic viruses, either genetically modified or naturally occurring, have the capability to activate immune cells that specifically target and destroy cancer cells while sparing healthy cells (22). These viruses selectively infect tumor cells harboring mutations associated with malignancies (23), and often have modifications in or deletions of certain viral genes essential for replication in normal cells but not in tumor cells (24). This selective infection is facilitated by the viruses’ ability to bind specifically to certain receptors on tumor cells, enhanced by altering the virus’s tropism (25). The viral genes are controlled by tumor-specific and/or tissue-specific gene promoters, enhancing the safety by confining viral replication to tumor cells (26, 27). Tumor cells frequently exhibit dysfunctional immune response signaling pathways, which support the virus’s replication and proliferation within them (28, 29). Oncolytic viruses inhibit the production of host cellular products while enhancing viral product synthesis within the tumor cells. When oncolytic viruses lyse tumor cells, they release large quantities of tumor antigens that are then processed by antigen-presenting cells, triggering T-cells to target both infected and uninfected tumor cells (30, 31). Upon infecting tumor cells, the viruses can either directly affect immune cells or stimulate the tumor cells to produce more cytokines, thereby enhancing the immune system to eliminate any remaining tumor cells by phagocytosing them (32). Oncolytic viruses induce various cell death pathways in cancer cells, including necroptosis, pyroptosis, apoptosis, and autophagy (Figure 1) (33–36). For example, the N-terminal gasdermin domain (GSDMNT), when delivered into tumor cells via a recombinant adeno-associated virus, induces pyroptosis, offering significant therapeutic potential (34). Similarly, the oncolytic vaccinia virus, when armed with the aphrocallistes vastus lectin gene, can alter the metabolism of hepatocellular carcinoma cells, increase reactive oxygen species (ROS) production, and promote apoptosis, all contributing to its antitumor effects (37). OVT works by killing cancer cells using viruses that have a selective replication function (1, 38). There are variations among oncolytic viruses in terms of how they interact with immune cells and the tumor microenvironment (TME) during treatment (39, 40). In this paper, we provide a detailed overview of the immune cells, TME, and oncolytic viruses, and discuss the variations in TME and immune responses observed with current OVTs.
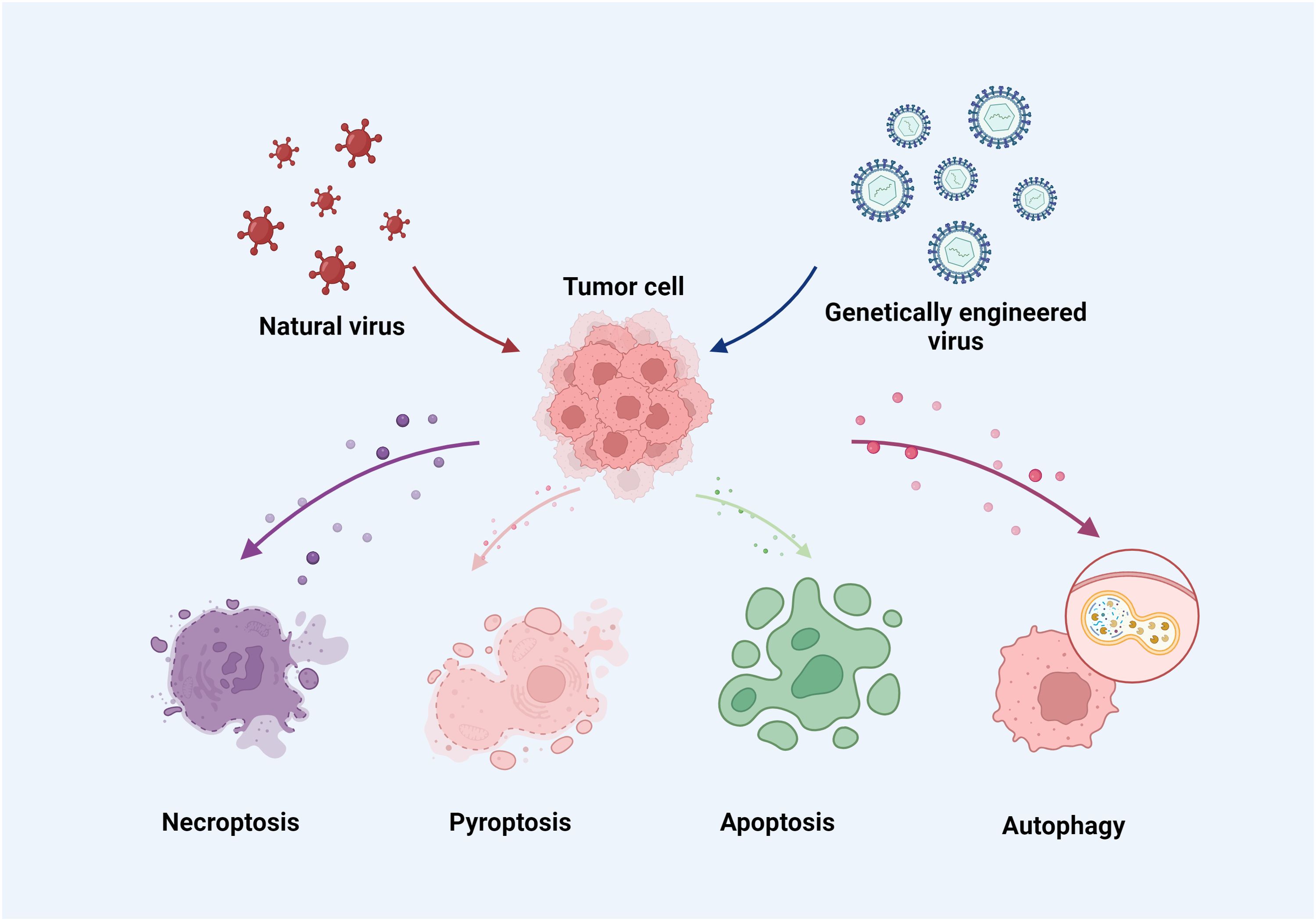
Figure 1. Oncolytic viruses induce necroptosis, pyroptosis, apoptosis and autophagy in cancer cells.
2 Origins and characteristics of oncolytic viruses
For nearly a century, researchers have been studying viruses. The first viruses identified were the foot-and-mouth disease virus from animals and the contagium vivium fluidum from plants, both discovered in 1898 (41, 42). Additionally, the yellow fever virus was first identified in 1901 and associated with human disease (43, 44). Advances in technology and an enhanced understanding of virus morphology and virulence have enabled ex vivo virus replication. This has facilitated the linking of several diseases, including rabies and influenza, to specific viruses (45, 46). Subsequent research has provided a detailed understanding of viral species, including their structure and biological characteristics (47). Simultaneously, researchers discovered that the virus may be utilized to cure tumors in addition to causing infectious diseases (48, 49).
Studies have documented cases of tumor regression following viral infection (49, 50). However, these remissions were typically short-lived, generally lasting only one to two months. In 1922, Levaditi and colleagues observed that the vaccinia virus could inhibit various cancers in mice and rats (51). In 1950, Pack reported remissions in patients with metastatic melanoma who were treated with the rabies virus. Additionally, patients with hematological malignancies such as leukemia or lymphoma experienced remission following infections with chickenpox or influenza (52, 53). Concurrent regressions of leukemia, Hodgkin’s disease, and Burkitt’s lymphoma have also been noted during measles infections, suggesting that under the right circumstances, certain viruses can target malignancies effectively without endangering the patient (54, 55). For instance, in a study of juvenile diffuse intrinsic pontine glioma treated with the oncolytic DNX-2401 virus, magnetic resonance imaging showed a reduction in tumor volume in 75% of the cases, and 66.7% of the patients achieved stable disease (56). Furthermore, a study involving 19 patients with recurrent glioblastoma treated with the oncolytic virus DNX-2401 demonstrated its safety and feasibility; notably, one patient achieved complete regression and was still alive eight years later (57).
Various viruses have been evaluated for their oncolytic properties in human tumor cell lines prior to their progressive utilization in clinical trials and other therapeutic contexts (58). In models of KRAS-mutated colorectal cancer, such as HCT116 colon cancer cells and patient-derived peripheral blood mononuclear cells, the oncolytic reovirus (pelareorep) exploits host autophagic machinery to enhance its proliferation and achieve selective oncolysis (59). In a phase 1 dose-escalation study, patients with newly diagnosed high-grade glioma who were treated with neural stem cell-delivered engineered oncolytic adenovirus showed promising safety and efficacy (60). Originally, hepatitis viruses were used to treat hematological malignancies and leukemia symptoms remitted in patients infected with adenovirus or EB virus (61). Subsequently, Alice Moore solidified the existence of an oncolytic virus in 1950 through in vivo tumor models and clinical and preclinical research (62). Oncolytic viruses, defined as naturally occurring or genetically modified viruses, selectively replicate in tumor cells, destroying them without harming healthy tissues and can also stimulate systemic or localized anti-tumor immunity (14, 63). These viruses target specific cell surface receptors overly expressed by cancer cells and naturally prefer cell surface proteins, enabling them to bind these receptors and penetrate the cells (64, 65). Oncolytic viruses are divided into two main categories: RNA and DNA viruses, including single-stranded (ssRNA and ssDNA) and double-stranded (dsRNA and dsDNA) viruses (Figure 2) (66).The most common RNA viruses used are measles and coxsackie virus group B, while commonly used DNA viruses include adenovirus, herpes simplex virus 1, and vaccinia virus. Except for vaccinia, DNA viruses have longer replication cycles than RNA viruses and replicate in the nuclei of infected cells (67, 68). Oncolytic viruses can also be classified into genetically engineered and naturally occurring strains. Engineered strains exhibit decreased pathogenicity, enhanced tumor expression, and increased lethality compared to wild-type strains (69). Mutations in the presence or lack of certain viral genes that are necessary for the virus to multiply in healthy cells but not in tumor cells (24). Genetically engineered oncolytic viruses are also designed to improve targeting selectivity for tumor cells and host cells with cancer-related mutations (70). Oncolytic viruses are intended to identify tumor-upregulated receptors, allowing the virus for an improved fidelity (71).To increase safety and limit viral replication to tumor cells, their principal genes are cloned into tumor-specific or tissue-specific promoters (26, 27). Besides their direct oncolytic activity of preferentially replicating within and destroying cancer cells, oncolytic viruses are highly effective in triggering immune responses against both the tumor cells and themselves. They are capable of inducing both local and systemic anti-tumor immunity, utilizing immune evasion strategies employed by cancer cells (72).
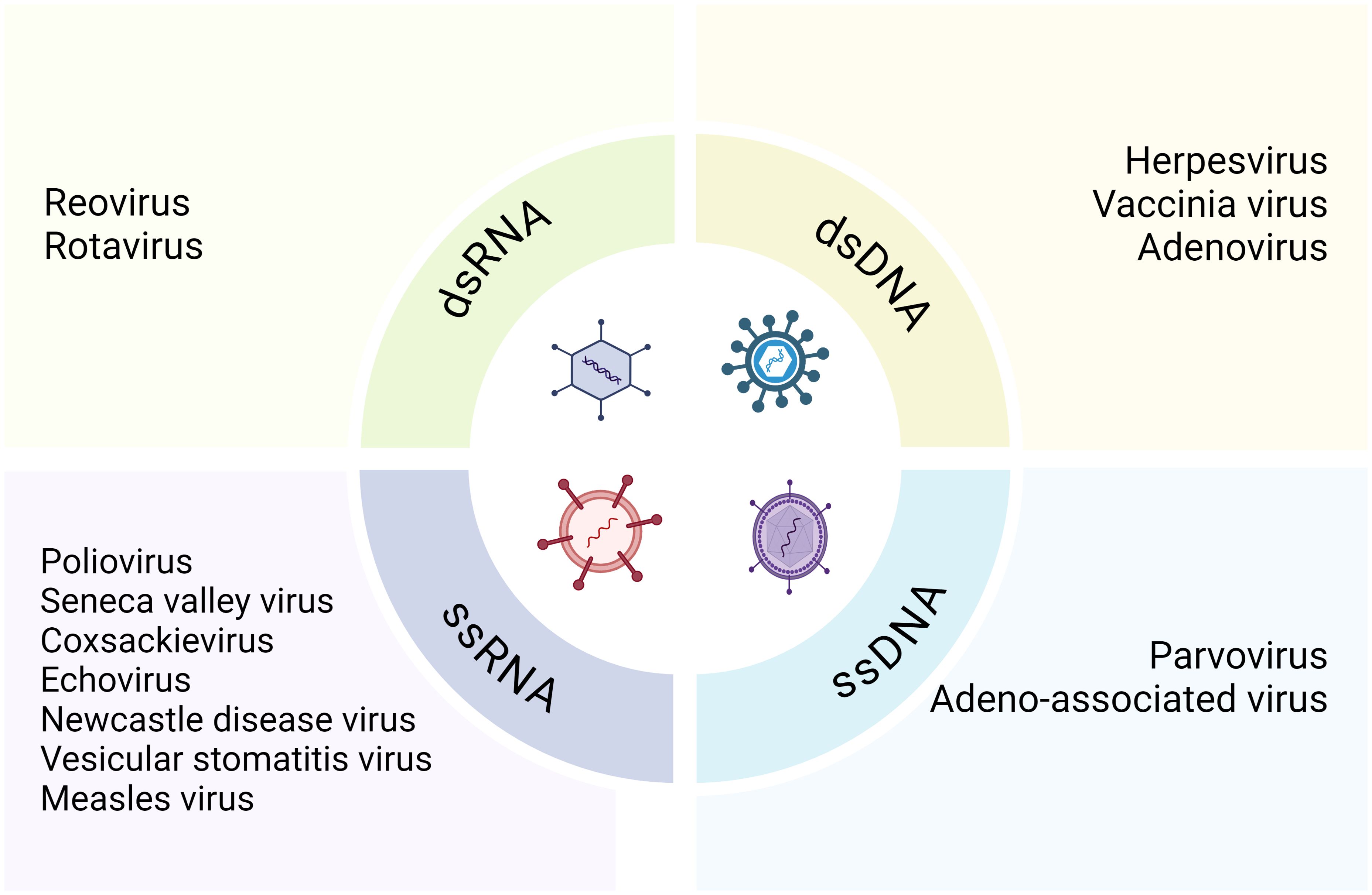
Figure 2. Classification of the oncolytic viruses.
3 Tumor-associated immune cells and immune microenvironment
3.1 Tumor-associated immune microenvironment
Since the 1970s, there has been accumulating evidence that TME plays a crucial role in tumor development, either by facilitating or hindering it (73, 74). Additionally, Stephen Paget’s “seed and soil theory,” introduced in 1989, conceptualized the interplay between cancer cells and the TME (75, 76).There is increasing evidence that tumor progression requires the recruitment and reprogramming of adjacent normal cells. The TME consists of a complex network of exosomes, the extracellular matrix (ECM), and stromal cells. During the occurrence and progression of cancer, tumor cells interact with surrounding cells to influence the cancer’s spread, proliferation, immune evasion, and chemoresistance within the TME; concurrently, these components undergo dynamic changes (77, 78). Different tumor locations and types exhibit specific TMEs, notable for their heterogeneity and dynamic changes. In colorectal cancer, stratifying patients based on the interindividual variability of the TME revealed differences in the immune evasion tactics employed by cancer cells among various TME subtypes (79). A cell-level analysis in prostate cancer samples studied the cell states associated with tumorigenesis, focusing on epithelial cell subsets, stromal cells, and the TME. This analysis found that ERG- cells show heterogeneity with luminal epithelial cells and differ from ERG+ tumor cells, potentially inducing a characteristic TME response (80).
The TME is comprised of stromal cells including cancer-associated fibroblasts (CAFs), macrophages, dendritic cells (DCs), natural killer (NK) cells, neutrophils, lymphocytes, endothelial cells, as well as chemokines, matrix metalloproteinases (MMPs), integrins, and other secreted molecules (Figure 3) (81). The TME contains various signaling molecules and pathways that contribute to immune suppression and angiogenic responses (82–84). The methyltransferase-like 3 associated with RNA N6-methyladenosine (m6A) modification is strongly activated by lactate accumulation in the TME, which enhances m6A modification in tumor-infiltrating myeloid cells (TIMs) through H3K18 lactylation. This activation of the m6A-YTHDF1/JAK1/STAT3 axis is associated with poor prognosis in colon cancer and increases the immunosuppressive capabilities of TIMs (83). Hypoxia in the TME can induce high levels of diacylglycerol kinase gamma in tumor vascular endothelial cells, which in turn can promote tumor angiogenesis and immune evasion in hepatocellular carcinoma through the ZEB2/TGF-β1 axis (84). The Food and Drug Administration’s (FDA) approval of antiangiogenic drugs and, more recently, immunological checkpoint inhibitors has reignited interest in understanding the role of the TME (85). Angiogenesis primarily depends on vascular endothelial growth factor (VEGF). Antiangiogenic drugs are those that inhibit tumor angiogenesis by targeting VEGF, its receptors, and other related molecules (86). The first antiangiogenic targeted medication to target VEGF was bevacizumab, which the FDA approved for application in 2004 (87). Clinical trials have shown that recombinant poliovirus therapy for gliomas, when combined with bevacizumab, had synergistic effects by reducing the local inflammatory response (88). Immune checkpoint inhibitors, including monoclonal antibodies targeting programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), counteract the mechanisms tumors use to evade immune surveillance. The first monoclonal antibody targeting CTLA-4, ipilimumab, was approved by the FDA in 2011 for treating melanoma (89). In advanced melanoma patients, combining ipilimumab with a modified oncolytic herpes simplex virus type 1 (HSV-1) demonstrated enhanced anticancer efficacy without additional harm, and these improved response rates persisted at the 5-year follow-up (90).
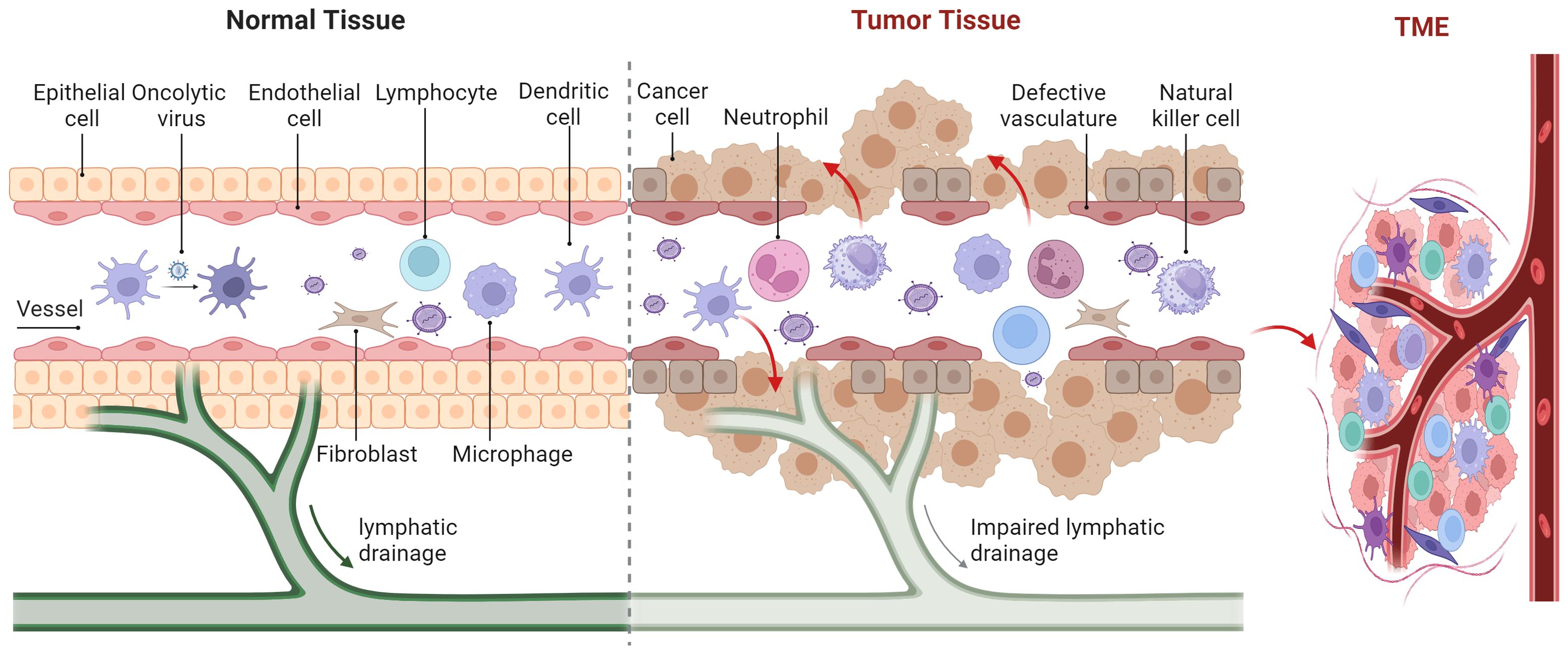
Figure 3. Various cells and oncolytic viruses in the tumor microenvironment.
3.2 Tumor-associated immune cells
3.2.1 Cancer-associated fibroblasts
The TME significantly influences the survival, proliferation, migration, and even dormancy of cancer cells (91, 92). It has been established that cancer-associated fibroblasts (CAFs) play a multifaceted role in promoting tumor growth within the TME. CAFs secrete inflammatory ligands, growth factors, and extracellular matrix (ECM) proteins that support tumor growth, contribute to immune exclusion, and foster resistance to treatment (91). As a principal component of the stromal cells, CAFs are recognized for their tumor-promoting properties. However, evidence that CAFs activate the Hedgehog signaling pathway indicates that under certain conditions, CAFs might also exhibit tumor-suppressing functions (93, 94).
Studies have demonstrated that CAFs promote tumor growth through various mechanisms, such as secreting ECM proteins, inducing inflammation and neovascularization, enhancing angiogenesis, increasing the prevalence of tumor-initiating cells, and altering cancer cell signaling (95). Under specific cellular conditions, the multifunctional cytokine TGF-β can both promote and inhibit tumor growth. In pre-menopausal breast cancer, knocking down the TGF-β receptor in cancer-associated fibroblasts (CAFs) has been identified as a predictive factor that can increase the growth, proliferation, and clonogenic survival of breast cancer cells (96). In pancreatic ductal adenocarcinoma, a population of predominantly quiescent CAFs is observed among long-term survivors; those with activated CAFs are highly likely to benefit from therapeutic interventions targeting CAFs (97). In cases of nasopharyngeal carcinoma, CAFs and tumor cells can enhance neoangiogenesis by recruiting endothelial progenitor cells from the bone marrow into the tumor stroma, a process that relies on VEGF and stroma-derived factor-1. Thus, molecules related to angiogenesis may offer viable therapeutic targets in nasopharyngeal carcinom (98). In head and neck cancers (HNCs), fibroblast activation protein (FAP) is expressed by CAFs within the tumor microenvironment. Radiolabeled inhibitors targeting FAP administered to patients with HNCs have demonstrated localized uptake in tumor lesions, indicating activity above background levels (99).
3.2.2 Macrophages
Macrophages are both ubiquitous and specialized across different tissues, playing roles in tissue development, coagulation, inflammation, and every stage of wound healing (100). Macrophages are categorized into two types: immune-suppressive M2 (alternatively activated) and inflammatory M1 (classically activated) (101, 102). Macrophages are essential for immunological homeostasis because they not only play a crucial role in wound healing and tissue repair but also control immune responses through pathogen phagocytosis and antigen presentation. The tumor-promoting actions of immune-suppressive M2 macrophages are enhanced by the TME. A high level of macrophage infiltration in tumors is generally associated with a poor prognosis (103, 104). In the TME, macrophages influence epithelial cell motility, which can be exploited by tumor cells to facilitate their migration and invasion (105).
In prostate cancer, interactions between tumor cells and macrophages are known to facilitate tumor development, although the precise mechanisms remain unclear. It has been found that high-mobility group box 1 activates macrophages, which then produce IL-6. This, in turn, promotes prostate cancer progression, resistance to androgen deprivation therapy (ADT), and gankyrin expression via the STAT3 signaling pathway, creating a self-reinforcing loop. Interestingly, inhibiting the interactions within this loop in a tumor xenograft model has shown to prevent ADT resistance (106). Another study found that patients with metastatic colorectal cancer undergoing chemotherapy that included bevacizumab experienced significantly improved clinical outcomes when they had genetic variations affecting macrophage-related functions. Additionally, alterations in genes related to macrophages may predict the outcome of bevacizumab treatment depending on the KRAS status (107).
3.2.3 Dendritic cells
The innate and adaptive immune systems are bridged by DCs, which are the most potent antigen-presenting cells and are crucial for initiating the adaptive immune response (108). DCs are classified into various subtypes, such as classical DCs, plasmacytoid DCs, and monocyte-derived inflammatory DCs, based on their functional attributes. The cross-priming of tumor-specific T cells by DCs is vital for initiating and sustaining anti-tumor immunity, a process that involves dynamic interactions between different DC subtypes and the tumor (109). The specific type or subtype of the tumor, along with its unique TME, significantly influences the composition and function of DCs. DCs within the tumor are associated with improved patient survival and are capable of inducing T cell responses that enhance protective immunity and decelerate cancer progression (110, 111). Typically, tumors manipulate their environment to ensure their survival, encountering immune-suppressive agents such as VEGF and IL-10 in the TME. These factors hinder the maturation of DCs into immunogenic cells, promoting instead the development of a tolerogenic phenotype in DCs (112, 113).
For some melanoma patients, immunization with mature monocyte-derived DCs)that were loaded with tumor antigens led to tumor regression. Additional refinement of the DC immunization protocol is required to determine which factors contribute to better clinical outcomes and enhanced anti-tumor responses (114). DC vaccines that were transfected with personalized tumor-associated antigen mRNA triggered specific CD4+ and CD8+ T cell responses in patients with advanced lung cancer or glioblastoma multiforme (GBM), and these responses were associated with favorable overall survival without significant autoimmune side effects (115). In a phase I trial, the safety and specificity of immune responses to tumor antigens were evaluated following in situ vaccination with autologous DCs transduced with an adenoviral vector expressing the CCL21 gene in patients with advanced non-small cell lung carcinoma (116).
3.2.4 Neutrophils
Neutrophils, constituting up to 70% of circulating leukocytes, serve as the primary line of defense against infections. Typically, they have a short lifespan, remaining in circulation for up to five days. When tissue is infected or damaged, epithelial cells emit chemokines that direct neutrophils to the affected site. Upon arrival, neutrophils deploy extracellular traps (NETs), release inflammatory cytokines, and engulf invading pathogens (117, 118). These NETs, which carry toxins and antimicrobial peptides on a chromatin backbone, serve as an additional mechanism of attack, albeit at the cost of the neutrophil’s own survival (119). The phenotype of neutrophils within the TME varies depending on the type of tumor and the stage of the disease. Initially, during the early stages of tumor development, neutrophils exhibit an inflammatory behavior, but as the tumor progresses, they adopt an immunosuppressive role. Neutrophils manage inflammation by producing ROS and nitrogen species. They also support angiogenesis, tumor invasion, and the remodeling of the ECM by releasing neutrophil elastase and MMPs in the TME. These proteases break down pro-inflammatory cytokines and restructure the TME, fostering tumor growth and metastasis (120, 121).
In the early stages of lung cancer, tumor-associated neutrophils do not suppress the immune system, instead, they enhance T cell responses. This activity leads to a marked increase in costimulatory molecules on the surface of neutrophils, which in turn fosters T cell proliferation in a positive feedback loop (122). Moreover, prior studies have shown that some patients with non-small cell lung cancer exhibit significant anticancer effects after undergoing salvage chemotherapy following PD-1 inhibition. For patients who did not respond to salvage chemotherapy, both the neutrophil-to-lymphocyte ratio (NLR) and the absolute neutrophil count (ANC) increased over the course of treatment with nivolumab. An inverse relationship was observed between the response to the drug and NLR or ANC at four to six weeks (123).
3.2.5 Natural killer cells and natural killer T cells
NK cells are innate lymphoid cells recognized for their cytotoxic capabilities (124). These cells target tumor cells to inhibit the formation of primary tumors through apoptosis induced by death receptors and cytotoxic mechanisms mediated by perforin and granzyme. While NK cells are effective at eliminating circulating tumor cells, they struggle to kill cells within the TME. Tumors deploy various tactics to evade destruction by NK cells, such as surrounding themselves with collagen to trigger inhibitory NK receptors and using platelets as shields to prevent NK cell detection (125, 126). Additionally, many cytokines commonly present in the TME can effectively dampen NK cell effector functions. Natural killer T cells (NKTs) are innate-like T lymphocytes that bind to CD1d and, like conventional T cells, possess a T cell receptor and respond quickly to antigen exposure. These cells are also prevalent within the TME (127). To be more precise, NKTIs may be further classified into subtypes such as Th1-like, Th2-like, Th17-like, regulatory T-like (Treg-like), and T follicular helper (TFH)-like for type I NKTs; whereas type II NKTs can be classified as Th1-like and Th2-like. It has been observed that NKTs can switch roles in the environment, toggling between immune-suppressive and inflammatory roles. Type I NKTs generally exhibit anti-tumor properties, whereas type II NKTs tend to support tumor growth (128, 129).
In ovarian cancer patients, including those from whom cells were isolated from ascites, NK cells demonstrated effective killing of autologous tumor-associated macrophages (TAMs) that expressed low, non-protective levels of HLA class I molecules when appropriately stimulated within the complex TME (130). For achieving pathological complete responses in patients with HER2-positive breast cancer, maintaining functional T cell responses to specific antigens and enhancing NK cell efficacy during neoadjuvant chemotherapy appears crucial (131). Hypoxic TAMs, induced by the tumor, secrete chemokine ligand-21 (CCL21), which attracts and suppresses NKTs. CCL21 further impairs NKT survival and function, inhibiting NKT migration in vitro toward tumor-conditioned hypoxic monocytes and preventing their localization to neuroblastoma grafts in mice (132). Patients with asymptomatic myeloma who underwent combination therapy exhibited signs of NK cell activation and an activation-induced reduction in detectable iNKT cells, with the therapy proving effective in driving tumor regression and synergistically activating various innate immune cells (133).
3.2.6 Innate lymphoid cells
Five distinct cell types comprise the innate lymphoid cells (ILCs), including NK cells) (134, 135). Unlike other ILCs, which primarily produce cytokines in reaction to different stimuli, NK cells exhibit the highest cytotoxic activity within the ILC group. These cells are a crucial part of the tumor microenvironment TME (136). ILCs originate from the same lymphoid progenitor as B and T cells but are categorized as innate immune cells due to their lack of B and T cell receptors. They play a role in T cell polarization by presenting antigens and secreting cytokines (137).
Prior to transplantation, the presence of acute leukemia patient ILCs and donor ILCs expressing specific markers was associated with a reduced risk of acute graft-versus-host disease (GVHD) and therapy-induced mucositis. Consequently, the dynamics of ILC recovery and its interaction with treatment-related tissue damage influence the development of GVHD (138). ILCs exhibit a dual role in cancer contexts, showing either pro-tumor or anti-tumor effects depending on the ILC subset and the type of cancer involved. ILC1s, in particular, are an early source of IFN-γ and are generally associated with anti-tumor activity through mechanisms like macrophage activation, Th1 polarization, and enhancement of major histocompatibility complex molecules (139). In cases of metastatic colorectal cancer, the overall frequency of ILCs was markedly increased compared to healthy donors and showed an inverse relationship with Th1 immune responses (140).
4 Oncolytic viruses mediate the immune cells and the immune microenvironment
Immunosuppression in the tumor microenvironment, exacerbated by immune checkpoint inhibitors like PD-1/PD-L, diminishes effective neoantigen presentation and hinders anti-cancer T cell responses (141). An engineered oncolytic virus designed to express PD-L1 inhibitors can initiate tumor neoantigen-specific T cell responses. This approach has shown promise in enhancing endogenous T cell reactions against tumor neoantigens in patients with advanced cancers, leading to durable outcomes, including complete responses in various cancer types such as melanoma, and metastatic lung, kidney, and bladder cancers (142, 143).
Oncolytic viruses, capable of inducing anti-tumor immunity both locally and systemically, are either injected directly into the tumor or administered systemically. Once these viruses infect tumor cells, they multiply, often triggering immunogenic cell death, and spread throughout the tumor, stimulating an inflammatory response that can be modulated by macrophages and NK cells of the innate immune system (144). Armed oncolytic viruses also express immunomodulatory transgenes, enhancing immune reactions against the tumor. The immunogenic cell death and inflammation attract DCs to the tumor site, where they initiate a comprehensive immune attack on the tumor by activating T and B cells (145).
Oncolytic viruses can counteract immune evasion strategies utilized by cancer cells. These strategies often involve immuno-inhibitory receptors on tumor cells and in the surrounding microenvironment, which deactivate immune effector cells and promote the secretion of IL-10 and TGF-β. These factors can attract immunosuppressive cells, such as myeloid-derived suppressor cells and tumor-associated macrophages, to the tumor site (146, 147). Oncolytic viruses disrupt this suppressive environment through various mechanisms that alter the cytokine landscape and the composition of immune cells within the TME (148).
Furthermore, several oncolytic viruses possess the capability to express therapeutic genes or alter the function of tumor-associated endothelial cells, enhancing the recruitment of T cells into TMEs that are otherwise immune-deserted or immune-excluded.
5 Promisting oncolytic viruses in tumor therapy
5.1 Nervous system tumor
Gliomas account for approximately 81% of all central nervous system (CNS) tumors, making them the most common type of malignant brain tumor (149). These tumors are notorious for their aggressive behavior, rapid growth, therapy resistance, and generally poor prognosis. Several non-neurotoxic viruses such as parvovirus, myxoma virus, M1 virus, and Seneca Valley virus are used in treatments as they generally do not require additional modifications for safety (150). The CNS is considered immune-privileged due to the blood-brain barrier (BBB), comprised of astrocytes, pericytes, and vascular endothelial cells forming tight junctions, which typically restricts access of peripheral immune cells to the brain. Overcoming this barrier involves strategies such as direct injection into CNS tumors or using external reservoirs that interface with brain tumor sites. Notably, parvovirus can naturally cross the BBB, facilitating the entry of oncolytic viruses into the bloodstream (151). In GBM clinical studies, parvovirus H-1PV has been utilized. However, there have been relatively few in vivo studies on the distribution of oncolytic viruses across the CNS to evaluate viral penetration effectively (152, 153).
5.2 Digestive system tumor
Digestive system cancers (DSC), including colorectal cancer (CRC), gastric cancer, hepatocellular carcinoma, pancreatic cancer (PC), and esophageal squamous cell carcinoma, are a leading cause of cancer-related deaths worldwide. Patients with advanced DSC generally have a poor prognosis. Peritoneal metastases, which often occur due to the spread of tumor cells within the peritoneal cavity, commonly triggered by CRC, represent the second most frequent type of CRC metastasis following those in the lung and liver. Approximately 25% of CRC patients develop metastatic disease, and peritoneal metastases are associated with particularly poor outcomes and are a major cause of mortality (154). According to a study, a tumor-lysing vaccinia virus that carries GM-CSF was found to successfully prevent CRC from spreading to the peritoneum by selectively infecting and lysing peritoneal tumor cells, as well as by activating peritoneal DCs and CD8 T cells to restore peritoneal anti-cancer immunity. Various oncolytic viruses, such as vaccine viruses, reoviruses, HSV, adenovirus, oncolytic measles virus, and of virus, are being explored for treating CRC (155). In pancreatic ductal adenocarcinoma, pelareorep, an intravenously administered oncolytic reovirus, has been shown to induce a T-cell-inflamed phenotype in tumors. This treatment has led to T-cell infiltration, increased PD-L1 expression, and active reovirus replication in the tumors of treated patients (59). For gastric cancer, HSV-based OVT has shown promise. The recombinant vaccinia virus used in these treatments has proven safe in vivo and demonstrates enhanced replication in tumor cells is an exciting treatment used in OVT for patients with gastric cancer. The recombinant vaccinia virus was safe in vivo and had a greater capacity for reproduction in tumor cells (156).
5.3 Urogenital system cancer
Prostate cancer is the second most common malignant tumor globally, with an incidence rate of 13.5% and a mortality rate of 6.7%. Current treatments include surgery, hormone therapy, chemotherapy, and radiation therapy (157). However, these treatments often fall short, especially in cases of advanced metastatic prostate cancer. Oncolytic viruses offer a promising alternative due to their high selectivity, efficiency, and low toxicity. These viruses are limited in their ability to replicate in healthy cells but can proliferate within tumor cells, inducing apoptosis and promoting viral growth. The released progeny viruses can then infect adjacent tumor cells, leading to tumor destruction. A phase II study revealed that bladder cancer patients treated with the intravesical oncolytic virus CG0070 experienced a 47% complete response rate at six months (158). Additionally, the bluetongue virus has been shown to infect and selectively lyse human hepatic and prostate cancer cells while significantly increasing the proportion of apoptotic renal cancer cells (159).
5.4 Potential clinical applications
There is an increasing body of research on oncolytic viruses that has enhanced our understanding of their role in tumor therapy. Some oncolytic viral therapies, identified as potential prognostic markers and therapeutic targets, have already been approved for clinical use or are advancing into clinical trials for tumor treatment. Currently, single-method treatments for some types of tumors have proven inadequate for achieving optimal results. Oncolytic viruses can inhibit DNA damage repair proteins, thereby minimizing the DNA damage caused by chemoradiation when used in combination with it. Furthermore, chemoradiation can promote tumor cell death, which in turn supports the replication and dissemination of oncolytic viruses. Additionally, combining immunotherapy drugs with oncolytic viruses enhances their synergistic effect, leading to more potent and sustained therapeutic outcomes. Due to their extensive mechanisms of action, oncolytic viruses are vital for intercellular communication and therapeutic applications. It has been observed that insufficient tumor cell tropism and transduction can make the therapeutic virus ineffective when administered systemically in clinical settings. To counter this, modifications to the viral surface have been employed to enhance tumor cell targeting. One such modification is the insertion of an RGD motif into the HI loop of the adenovirus fiber knob domain, which has significantly improved the virus’s infection efficiency and anti-tumor efficacy, especially in CAR-negative tumor models. Additionally, using different virus types, such as Human adenovirus (HAdV)-G52, which binds to polysialic acid on target cells, can offer another layer of targeting specificity. HAdV-G52 might specifically infect cancers, such as brain and lung cancers, that express high levels of polysialic acid, although adjustments are necessary to mitigate any potential neurotropism. Through various modifications, oncolytic viruses can be tailored to enhance their safety, infection capability through tumor cellular receptors, tumor-targeting selectivity, and replication efficiency within tumor cell cytoplasm. These modifications can aid to improve viruses’ immunostimulatory capacity and tissue tropism while maintaining safety and antitumor efficiency.
6 Conclusion
There are several challenges with the oncolytic viral therapy approach. Research on oncolytic viruses is still in its infancy. Many oncolytic viruses are currently being explored both theoretically and experimentally, and their potential therapeutic value remains uncertain. Additionally, there are no specific diagnostic criteria to identify patients who might benefit from oncolytic viral therapies.
In conclusion, the function of oncolytic viruses in tumor therapy and the tumor microenvironment is gradually becoming apparent. As either naturally occurring or genetically engineered agents, oncolytic viruses achieve their therapeutic effects by influencing immune cells, tumor cells, and the tumor microenvironment. This review has discussed the history and characteristics of the tumor microenvironment, immune cells, and oncolytic viruses, as well as the potential therapeutic efficacy of oncolytic viruses. These viruses activate various pathways to induce tumor cell death. A significant challenge in cancer treatment is that many oncolytic virus therapies are still in the preclinical trial stage. While a few oncolytic viruses have been approved for clinical use, transitioning these therapies into clinical applications remains a substantial hurdle. Consequently, understanding how oncolytic viruses modify the tumor microenvironment in different tumors will lead to the enhancement and development of oncolytic virus treatments for cancer. Moving forward, continued research into oncolytic viruses is essential.
Author contributions
XW: Data curation, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. SF: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 82371304).
Acknowledgments
We would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript. All illustrations were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
OVT: Oncolytic virus therapy
TME: Tumor microenvironment
ROS: Reactive oxygen species
ECM: Extracellular matrix
CAF: Cancer-associated fibroblast
DC: Dendritic cell
NK: Natural killer cell
NKT: Natural killer T cell
MMP: Matrix metalloproteinase
m6A: RNA N6-methyladenosine
TIM: Tumor-infiltrating myeloid cells
FDA: Food and Drug Administration
VEGF: Vascular endothelial growth factor
PD-1: Programmed death-1
PD-L1: Programmed death-ligand 1
CTLA-4: Cytotoxic T-lymphocyte-associated protein 4
HSV-1: Herpes simplex virus type 1
FAP: Fibroblast activation protein
HNC: Head and neck cancer
ADT: Androgen deprivation therapy
CCL21: Chemokine ligand-21
TAM: Tumor-associated macrophages
NLR: Neutrophil-to-lymphocyte ratio
ANC: Absolute neutrophil count
ILC: Innate lymphoid cells
GVHD: Graft-versus-host disease
CNS: Central nervous system
BBB: Blood-brain barrier
DSC: Digestive system tumor
PC: Pancreatic cancer
CRC: Colorectal cancer
HAdV: Human adenovirus
References
1. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci (2016) 107:1373–9. doi: 10.1111/cas.13027
2. Fu LQ, Wang SB, Cai MH, Wang XJ, Chen JY, Tong XM, et al. Recent advances in oncolytic virus-based cancer therapy. Virus Res (2019) 270:197675. doi: 10.1016/j.virusres.2019.197675
3. Rajewsky K. The advent and rise of monoclonal antibodies. Nature (2019) 575:47–9. doi: 10.1038/d41586-019-02840-w
4. Liu M, Li L, Jin D, Liu Y. Nanobody-a versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip Rev Nanomed Nanobiotechnol (2021) 13:e1697. doi: 10.1002/wnan.1697
5. Lee DA. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol Rev (2019) 290:85–99. doi: 10.1111/imr.12793
6. Castellanos-Rueda R, Di Roberto RB, Schlatter FS, Reddy ST. Leveraging single-cell sequencing for chimeric antigen receptor t cell therapies. Trends Biotechnol (2021) 39:1308–20. doi: 10.1016/j.tibtech.2021.03.005
7. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (2021) 398:1002–14. doi: 10.1016/s0140-6736(21)01206-x
8. Chen Z, Zhang P, Xu Y, Yan J, Liu Z, Lau WB, et al. Surgical stress and cancer progression: The twisted tango. Mol Cancer (2019) 18:132. doi: 10.1186/s12943-019-1058-3
9. Levy A, Le Péchoux C, Faivre-Finn C. Twice-daily chemoradiotherapy in limited-stage small-cell lung cancer. Lancet Oncol (2021) 22:e220. doi: 10.1016/s1470-2045(21)00255-2
10. Fokas E, Schlenska-Lange A, Polat B, Klautke G, Grabenbauer GG, Fietkau R, et al. Chemoradiotherapy plus induction or consolidation chemotherapy as total neoadjuvant therapy for patients with locally advanced rectal cancer: Long-term results of the cao/aro/aio-12 randomized clinical trial. JAMA Oncol (2022) 8:e215445. doi: 10.1001/jamaoncol.2021.5445
11. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discovery (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
12. Cha JH, Chan LC, Song MS, Hung MC. New approaches on cancer immunotherapy. Cold Spring Harb Perspect Med (2020) 10. doi: 10.1101/cshperspect.a036863
13. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin (2020) 70:86–104. doi: 10.3322/caac.21596
14. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug Discovery (2015) 14:642–62. doi: 10.1038/nrd4663
15. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: Progress and challenges. Nat Rev Clin Oncol (2023) 20:160–77. doi: 10.1038/s41571-022-00719-w
16. Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: A review. JAMA Oncol (2017) 3:841–9. doi: 10.1001/jamaoncol.2016.2064
17. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2020-001486
18. Bahreyni A, Mohamud Y, Luo H. Oncolytic virus-based combination therapy in breast cancer. Cancer Lett (2024) 585:216634. doi: 10.1016/j.canlet.2024.216634
19. Sakhi H, Arabi M, Ghaemi A, Movafagh A, Sheikhpour M. Oncolytic viruses in lung cancer treatment: A review article. Immunotherapy (2024) 16:75–97. doi: 10.2217/imt-2023-0124
20. Galanis E, Dooley KE, Keith Anderson S, Kurokawa CB, Carrero XW, Uhm JH, et al. Carcinoembryonic antigen-expressing oncolytic measles virus derivative in recurrent glioblastoma: A phase 1 trial. Nat Commun (2024) 15:493. doi: 10.1038/s41467-023-43076-7
21. Liang M. Oncorine, the world first oncolytic virus medicine and its update in china. Curr Cancer Drug Targets (2018) 18:171–6. doi: 10.2174/1568009618666171129221503
22. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30:658–70. doi: 10.1038/nbt.2287
23. Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer (2023) 9:122–39. doi: 10.1016/j.trecan.2022.10.003
24. Chen X, Liu J, Li Y, Zeng Y, Wang F, Cheng Z, et al. Idh1 mutation impairs antiviral response and potentiates oncolytic virotherapy in glioma. Nat Commun (2023) 14:6781. doi: 10.1038/s41467-023-42545-3
25. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus g47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat Med (2022) 28:1630–9. doi: 10.1038/s41591-022-01897-x
26. Dai W, Tian R, Yu L, Bian S, Chen Y, Yin B, et al. Overcoming therapeutic resistance in oncolytic herpes virotherapy by targeting igf2bp3-induced netosis in malignant glioma. Nat Commun (2024) 15:131. doi: 10.1038/s41467-023-44576-2
27. Fukuhara H, Takeshima Y, Todo T. Triple-mutated oncolytic herpes virus for treating both fast- and slow-growing tumors. Cancer Sci (2021) 112:3293–301. doi: 10.1111/cas.14981
28. Keshavarz M, Mohammad Miri S, Behboudi E, Arjeini Y, Dianat-Moghadam H, Ghaemi A. Oncolytic virus delivery modulated immune responses toward cancer therapy: Challenges and perspectives. Int Immunopharmacol (2022) 108:108882. doi: 10.1016/j.intimp.2022.108882
29. Ma W, He H, Wang H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol (2018) 19:40. doi: 10.1186/s12865-018-0281-9
30. Bai Y, Hui P, Du X, Su X. Updates to the antitumor mechanism of oncolytic virus. Thorac Cancer (2019) 10:1031–5. doi: 10.1111/1759-7714.13043
31. Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: An underappreciated aspect of oncolytic virotherapies. Trends Immunol (2018) 39:209–21. doi: 10.1016/j.it.2017.11.006
32. Lin C, Teng W, Tian Y, Li S, Xia N, Huang C. Immune landscape and response to oncolytic virus-based immunotherapy. Front Med (2024) 18:411–29. doi: 10.1007/s11684-023-1048-0
33. Aspirin AP, de Los Reyes VA, Kim Y. Polytherapeutic strategies with oncolytic virus-bortezomib and adjuvant nk cells in cancer treatment. J R Soc Interface (2021) 18:20200669. doi: 10.1098/rsif.2020.0669
34. Lu Y, He W, Huang X, He Y, Gou X, Liu X, et al. Strategies to package recombinant adeno-associated virus expressing the n-terminal gasdermin domain for tumor treatment. Nat Commun (2021) 12:7155. doi: 10.1038/s41467-021-27407-0
35. Garant KA, Shmulevitz M, Pan L, Daigle RM, Ahn DG, Gujar SA, et al. Oncolytic reovirus induces intracellular redistribution of ras to promote apoptosis and progeny virus release. Oncogene (2016) 35:771–82. doi: 10.1038/onc.2015.136
36. Lin J, Sun S, Zhao K, Gao F, Wang R, Li Q, et al. Oncolytic parapoxvirus induces gasdermin e-mediated pyroptosis and activates antitumor immunity. Nat Commun (2023) 14:224. doi: 10.1038/s41467-023-35917-2
37. Zhang Y, Zhu Y, Jiang G, Chen K, Zhang G, Chen K, et al. Ros induced by aphrocallistes vastus lectin enhance oncolytic vaccinia virus replication and induce apoptosis in hepatocellular carcinoma cells. Mar Drugs (2024) 22. doi: 10.3390/md22070307
38. Martin NT, Bell JC. Oncolytic virus combination therapy: Killing one bird with two stones. Mol Ther (2018) 26:1414–22. doi: 10.1016/j.ymthe.2018.04.001
39. Meisen WH, Wohleb ES, Jaime-Ramirez AC, Bolyard C, Yoo JY, Russell L, et al. The impact of macrophage- and microglia-secreted tnfα on oncolytic hsv-1 therapy in the glioblastoma tumor microenvironment. Clin Cancer Res (2015) 21:3274–85. doi: 10.1158/1078-0432.Ccr-14-3118
40. Xu B, Tian L, Chen J, Wang J, Ma R, Dong W, et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat Commun (2021) 12:5908. doi: 10.1038/s41467-021-26003-6
41. Bos L. Beijerinck's work on tobacco mosaic virus: Historical context and legacy. Philos Trans R Soc Lond B Biol Sci (1999) 354:675–85. doi: 10.1098/rstb.1999.0420
42. Arzt J, Juleff N, Zhang Z, Rodriguez LL. The pathogenesis of foot-and-mouth disease i: Viral pathways in cattle. Transbound Emerg Dis (2011) 58:291–304. doi: 10.1111/j.1865-1682.2011.01204.x
43. Chaves-Carballo E. Clara maass, yellow fever and human experimentation. Mil Med (2013) 178:557–62. doi: 10.7205/milmed-d-12-00430
44. Clements AN, Harbach RE. History of the discovery of the mode of transmission of yellow fever virus. J Vector Ecol (2017) 42:208–22. doi: 10.1111/jvec.12261
45. Bloom K, van den Berg F, Arbuthnot P. Self-amplifying rna vaccines for infectious diseases. Gene Ther (2021) 28:117–29. doi: 10.1038/s41434-020-00204-y
46. Beaman MH. Community-acquired acute meningitis and encephalitis: A narrative review. Med J Aust (2018) 209:449–54. doi: 10.5694/mja17.01073
47. Sinha A, Singh AK, Kadni TS, Mullick J, Sahu A. Virus-encoded complement regulators: Current status. Viruses (2021) 13. doi: 10.3390/v13020208
48. Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther (2002) 9:967–78. doi: 10.1038/sj.cgt.7700537
49. Dirven CM, van Beusechem VW, Lamfers ML, Grill J, Gerritsen WR, Vandertop WP. Oncolytic adenoviruses for treatment of brain tumours. Expert Opin Biol Ther (2002) 2:943–52. doi: 10.1517/14712598.2.8.943
50. Hermine O, Lefrère F, Bronowicki JP, Mariette X, Jondeau K, Eclache-Saudreau V, et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis c virus infection. N Engl J Med (2002) 347:89–94. doi: 10.1056/NEJMoa013376
51. Eisenberg T, Glaeser SP, Ewers C, Semmler T, Nicklas W, Rau J, et al. Streptobacillus notomytis sp. Nov., isolated from a spinifex hopping mouse (notomys alexis thomas, 1922), and emended description of streptobacillus levaditi et al. 1925, eisenberg et al. 2015 emend. Int J Syst Evol Microbiol (2015) 65:4823–9. doi: 10.1099/ijsem.0.000654
52. Addou S, Sarkozy C, Lazarovici J, Champiat S, Stamatoullas A, Jardin F, et al. Relapsed and refractory classical hodgkin lymphoma: Could virotherapy help solve the equation? Hum Vaccin Immunother (2021) 17:3502–10. doi: 10.1080/21645515.2021.1924521
53. Krugmann J, Tzankov A, Gschwendtner A, Fischhofer M, Greil R, Fend F, et al. Longer failure-free survival interval of epstein-barr virus-associated classical hodgkin's lymphoma: A single-institution study. Mod Pathol (2003) 16:566–73. doi: 10.1097/01.Mp.0000071843.09960.Bf
54. Pidelaserra-Martí G, Engeland CE. Mechanisms of measles virus oncolytic immunotherapy. Cytokine Growth Factor Rev (2020) 56:28–38. doi: 10.1016/j.cytogfr.2020.07.009
55. Pasquinucci G. Possible effect of measles on leukaemia. Lancet (1971) 1:136. doi: 10.1016/s0140-6736(71)90869-5
56. Gállego Pérez-Larraya J, Garcia-Moure M, Labiano S, Patiño-García A, Dobbs J, Gonzalez-Huarriz M, et al. Oncolytic dnx-2401 virus for pediatric diffuse intrinsic pontine glioma. N Engl J Med (2022) 386:2471–81. doi: 10.1056/NEJMoa2202028
57. van Putten EHP, Kleijn A, van Beusechem VW, Noske D, Lamers CHJ, de Goede AL, et al. Convection enhanced delivery of the oncolytic adenovirus delta24-rgd in patients with recurrent gbm: A phase i clinical trial including correlative studies. Clin Cancer Res (2022) 28:1572–85. doi: 10.1158/1078-0432.Ccr-21-3324
58. Ricordel M, Foloppe J, Pichon C, Findeli A, Tosch C, Cordier P, et al. Oncolytic properties of non-vaccinia poxviruses. Oncotarget (2018) 9:35891–906. doi: 10.18632/oncotarget.26288
59. Jiffry J, Thavornwatanayong T, Rao D, Fogel EJ, Saytoo D, Nahata R, et al. Oncolytic reovirus (pelareorep) induces autophagy in kras-mutated colorectal cancer. Clin Cancer Res (2021) 27:865–76. doi: 10.1158/1078-0432.Ccr-20-2385
60. Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol (2021) 22:1103–14. doi: 10.1016/s1470-2045(21)00245-x
61. Wang CR, Tsai HW. Human hepatitis viruses-associated cutaneous and systemic vasculitis. World J Gastroenterol (2021) 27:19–36. doi: 10.3748/wjg.v27.i1.19
62. Cabasso VJ, Moore IF, Cox HR. In vitro and in vivo action of aureomycin on vaccinia virus in the chick embryo and in the rabbit. J Infect Dis (1952) 91:79–85. doi: 10.1093/infdis/91.1.79
63. Ylösmäki E, Cerullo V. Design and application of oncolytic viruses for cancer immunotherapy. Curr Opin Biotechnol (2020) 65:25–36. doi: 10.1016/j.copbio.2019.11.016
64. Kelly E, Russell SJ. History of oncolytic viruses: Genesis to genetic engineering. Mol Ther (2007) 15:651–9. doi: 10.1038/sj.mt.6300108
65. Sinkovics JG, Horvath JC. Natural and genetically engineered viral agents for oncolysis and gene therapy of human cancers. Arch Immunol Ther Exp (Warsz) (2008) 56. doi: 10.1007/s00005-008-0047-9
66. Melcher A, Harrington K, Vile R. Oncolytic virotherapy as immunotherapy. Science (2021) 374:1325–6. doi: 10.1126/science.abk3436
67. Rosario K, Fierer N, Miller S, Luongo J, Breitbart M. Diversity of DNA and rna viruses in indoor air as assessed via metagenomic sequencing. Environ Sci Technol (2018) 52:1014–27. doi: 10.1021/acs.est.7b04203
68. Stedman KM. Deep recombination: Rna and ssdna virus genes in DNA virus and host genomes. Annu Rev Virol (2015) 2:203–17. doi: 10.1146/annurev-virology-100114-055127
69. Lin D, Shen Y, Liang T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct Target Ther (2023) 8:156. doi: 10.1038/s41392-023-01407-6
70. Yamamoto Y, Nagasato M, Yoshida T, Aoki K. Recent advances in genetic modification of adenovirus vectors for cancer treatment. Cancer Sci (2017) 108:831–7. doi: 10.1111/cas.13228
71. Mathis JM, Stoff-Khalili MA, Curiel DT. Oncolytic adenoviruses - selective retargeting to tumor cells. Oncogene (2005) 24:7775–91. doi: 10.1038/sj.onc.1209044
72. De Munck J, Binks A, McNeish IA, Aerts JL. Oncolytic virus-induced cell death and immunity: A match made in heaven? J Leukoc Biol (2017) 102:631–43. doi: 10.1189/jlb.5RU0117-040R
73. Preisler HD, Bjornsson S, Mori M, Barcos M. Granulocyte differentiation by friend leukemia cells. Cell Differ (1975) 4:273–83. doi: 10.1016/0045-6039(75)90012-3
74. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
75. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8:98–101.
76. Mendoza M, Khanna C. Revisiting the seed and soil in cancer metastasis. Int J Biochem Cell Biol (2009) 41:1452–62. doi: 10.1016/j.biocel.2009.01.015
77. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.Can-18-3962
78. DeBerardinis RJ. Tumor microenvironment, metabolism, and immunotherapy. N Engl J Med (2020) 382:869–71. doi: 10.1056/NEJMcibr1914890
79. Chu X, Li X, Zhang Y, Dang G, Miao Y, Xu W, et al. Integrative single-cell analysis of human colorectal cancer reveals patient stratification with distinct immune evasion mechanisms. Nat Cancer (2024) 5:1409–26. doi: 10.1038/s43018-024-00807-z
80. Song H, Weinstein HNW, Allegakoen P, Wadsworth MH 2nd, Xie J, Yang H, et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Nat Commun (2022) 13:141. doi: 10.1038/s41467-021-27322-4
82. Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D, et al. Stromal hif2 regulates immune suppression in the pancreatic cancer microenvironment. Gastroenterology (2022) 162:2018–31. doi: 10.1053/j.gastro.2022.02.024
83. Xiong J, He J, Zhu J, Pan J, Liao W, Ye H, et al. Lactylation-driven mettl3-mediated rna m(6)a modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell (2022) 82:1660–77.e10. doi: 10.1016/j.molcel.2022.02.033
84. Zhang L, Xu J, Zhou S, Yao F, Zhang R, You W, et al. Endothelial dgkg promotes tumor angiogenesis and immune evasion in hepatocellular carcinoma. J Hepatol (2024) 80:82–98. doi: 10.1016/j.jhep.2023.10.006
85. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat Rev Cancer (2019) 19:9–31. doi: 10.1038/s41568-018-0081-9
86. Patel SA, Nilsson MB, Le X, Cascone T, Jain RK, Heymach JV. Molecular mechanisms and future implications of vegf/vegfr in cancer therapy. Clin Cancer Res (2023) 29:30–9. doi: 10.1158/1078-0432.Ccr-22-1366
87. Ofek P, Tiram G, Satchi-Fainaro R. Angiogenesis regulation by nanocarriers bearing rna interference. Adv Drug Delivery Rev (2017) 119:3–19. doi: 10.1016/j.addr.2017.01.008
88. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
89. May KF Jr., Gulley JL, Drake CG, Dranoff G, Kantoff PW. Prostate cancer immunotherapy. Clin Cancer Res (2011) 17:5233–8. doi: 10.1158/1078-0432.Ccr-10-3402
90. Chesney JA, Puzanov I, Collichio FA, Singh P, Milhem MM, Glaspy J, et al. Talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone for advanced melanoma: 5-year final analysis of a multicenter, randomized, open-label, phase ii trial. J Immunother Cancer (2023) 11. doi: 10.1136/jitc-2022-006270
91. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
92. Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett (2015) 368:7–13. doi: 10.1016/j.canlet.2015.07.039
93. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer (2020) 146:895–905. doi: 10.1002/ijc.32193
94. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther (2021) 6:218. doi: 10.1038/s41392-021-00641-0
95. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J Hematol Oncol (2019) 12:86. doi: 10.1186/s13045-019-0770-1
96. Busch S, Acar A, Magnusson Y, Gregersson P, Rydén L, Landberg G. Tgf-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene (2015) 34:27–38. doi: 10.1038/onc.2013.527
97. Sadozai H, Acharjee A, Eppenberger-Castori S, Gloor B, Gruber T, Schenk M, et al. Distinct stromal and immune features collectively contribute to long-term survival in pancreatic cancer. Front Immunol (2021) 12:643529. doi: 10.3389/fimmu.2021.643529
98. Wang S, Ma N, Kawanishi S, Hiraku Y, Oikawa S, Xie Y, et al. Relationships of alpha-sma-positive fibroblasts and sdf-1-positive tumor cells with neoangiogenesis in nasopharyngeal carcinoma. BioMed Res Int (2014) 2014:507353. doi: 10.1155/2014/507353
99. Zhao L, Pang Y, Chen S, Chen J, Li Y, Yu Y, et al. Prognostic value of fibroblast activation protein expressing tumor volume calculated from [(68) ga]ga-fapi pet/ct in patients with esophageal squamous cell carcinoma. Eur J Nucl Med Mol Imaging (2023) 50:593–601. doi: 10.1007/s00259-022-05989-1
100. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
101. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
102. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer (2019) 18:177. doi: 10.1186/s12943-019-1102-3
103. Baradaran A, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Shadbad MA, Khosravi N, et al. The cross-talk between tumor-associated macrophages and tumor endothelium: Recent advances in macrophage-based cancer immunotherapy. BioMed Pharmacother (2022) 146:112588. doi: 10.1016/j.biopha.2021.112588
104. Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med (2011) 9:216. doi: 10.1186/1479-5876-9-216
105. Wang H, Yung MMH, Ngan HYS, Chan KKL, Chan DW. The impact of the tumor microenvironment on macrophage polarization in cancer metastatic progression. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22126560
106. Peng G, Wang C, Wang H, Qu M, Dong K, Yu Y, et al. Gankyrin-mediated interaction between cancer cells and tumor-associated macrophages facilitates prostate cancer progression and androgen deprivation therapy resistance. Oncoimmunology (2023) 12:2173422. doi: 10.1080/2162402x.2023.2173422
107. Sunakawa Y, Stintzing S, Cao S, Heinemann V, Cremolini C, Falcone A, et al. Variations in genes regulating tumor-associated macrophages (tams) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: Results from tribe and fire3 trials. Ann Oncol (2015) 26:2450–6. doi: 10.1093/annonc/mdv474
108. Gardner A, de Mingo Pulido Á, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol (2020) 11:924. doi: 10.3389/fimmu.2020.00924
109. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
110. Strioga M, Schijns V, Powell DJ Jr., Pasukoniene V, Dobrovolskiene N, Michalek J. Dendritic cells and their role in tumor immunosurveillance. Innate Immun (2013) 19:98–111. doi: 10.1177/1753425912449549
111. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12:265–77. doi: 10.1038/nrc3258
112. Seyfizadeh N, Muthuswamy R, Mitchell DA, Nierkens S, Seyfizadeh N. Migration of dendritic cells to the lymph nodes and its enhancement to drive anti-tumor responses. Crit Rev Oncol Hematol (2016) 107:100–10. doi: 10.1016/j.critrevonc.2016.09.002
113. Kalinski P, Muthuswamy R, Urban J. Dendritic cells in cancer immunotherapy: Vaccines and combination immunotherapies. Expert Rev Vaccines (2013) 12:285–95. doi: 10.1586/erv.13.22
114. Kim DS, Kim DH, Goo B, Cho YH, Park JM, Lee TH, et al. Immunotherapy of malignant melanoma with tumor lysate-pulsed autologous monocyte-derived dendritic cells. Yonsei Med J (2011) 52:990–8. doi: 10.3349/ymj.2011.52.6.990
115. Wang QT, Nie Y, Sun SN, Lin T, Han RJ, Jiang J, et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother (2020) 69:1375–87. doi: 10.1007/s00262-020-02496-w
116. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase i trial of intratumoral injection of ccl21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and cd8(+) t-cell infiltration. Clin Cancer Res (2017) 23:4556–68. doi: 10.1158/1078-0432.Ccr-16-2821
117. Wu L, Zhang XH. Tumor-associated neutrophils and macrophages-heterogenous but not chaotic. Front Immunol (2020) 11:553967. doi: 10.3389/fimmu.2020.553967
118. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of neutrophil extracellular traps (nets) in tumor progression and metastasis. Front Immunol (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
119. Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A, et al. Il8, neutrophils, and nets in a collusion against cancer immunity and immunotherapy. Clin Cancer Res (2021) 27:2383–93. doi: 10.1158/1078-0432.Ccr-20-1319
120. Swierczak A, Mouchemore KA, Hamilton JA, Anderson RL. Neutrophils: Important contributors to tumor progression and metastasis. Cancer Metastasis Rev (2015) 34:735–51. doi: 10.1007/s10555-015-9594-9
121. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: Neutral no more. Nat Rev Cancer (2016) 16:431–46. doi: 10.1038/nrc.2016.52
122. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate t cell responses in early-stage human lung cancer. J Clin Invest (2014) 124:5466–80. doi: 10.1172/jci77053
123. Soda H, Ogawara D, Fukuda Y, Tomono H, Okuno D, Koga S, et al. Dynamics of blood neutrophil-related indices during nivolumab treatment may be associated with response to salvage chemotherapy for non-small cell lung cancer: A hypothesis-generating study. Thorac Cancer (2019) 10:341–6. doi: 10.1111/1759-7714.12952
124. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19:120. doi: 10.1186/s12943-020-01238-x
125. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. Nk cell metabolism and tumor microenvironment. Front Immunol (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
126. Zhang C, Hu Y, Shi C. Targeting natural killer cells for tumor immunotherapy. Front Immunol (2020) 11:60. doi: 10.3389/fimmu.2020.00060
127. Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, et al. Cxcr6 inhibits hepatocarcinogenesis by promoting natural killer t- and cd4(+) t-cell-dependent control of senescence. Gastroenterology (2019) 156:1877–89.e4. doi: 10.1053/j.gastro.2019.01.247
128. Wang H, Yin S. Natural killer t cells in liver injury, inflammation and cancer. Expert Rev Gastroenterol Hepatol (2015) 9:1077–85. doi: 10.1586/17474124.2015.1056738
129. Singh AK, Gaur P, Das SN. Natural killer t cell anergy, co-stimulatory molecules and immunotherapeutic interventions. Hum Immunol (2014) 75:250–60. doi: 10.1016/j.humimm.2013.12.004
130. Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, et al. Tlr activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of nk cells. Eur J Immunol (2014) 44:1814–22. doi: 10.1002/eji.201344130
131. Muraro E, Comaro E, Talamini R, Turchet E, Miolo G, Scalone S, et al. Improved natural killer cell activity and retained anti-tumor cd8(+) t cell responses contribute to the induction of a pathological complete response in her2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J Transl Med (2015) 13:204. doi: 10.1186/s12967-015-0567-0
132. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. Il-15 protects nkt cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest (2012) 122:2221–33. doi: 10.1172/jci59535
133. Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, et al. Clinical regressions and broad immune activation following combination therapy targeting human nkt cells in myeloma. Blood (2013) 121:423–30. doi: 10.1182/blood-2012-06-435503
134. Yuan X, Rasul F, Nashan B, Sun C. Innate lymphoid cells and cancer: Role in tumor progression and inhibition. Eur J Immunol (2021) 51:2188–205. doi: 10.1002/eji.202049033
135. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector nk cells into type 1 innate lymphoid cells. Nat Immunol (2017) 18:1004–15. doi: 10.1038/ni.3800
136. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
137. Bottino C, Dondero A, Castriconi R. Inhibitory axes impacting on the activity and fate of innate lymphoid cells. Mol Aspects Med (2021) 80:100985. doi: 10.1016/j.mam.2021.100985
138. Munneke JM, Björklund AT, Mjösberg JM, Garming-Legert K, Bernink JH, Blom B, et al. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood (2014) 124:812–21. doi: 10.1182/blood-2013-11-536888
139. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev (2008) 226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x
140. Loyon R, Jary M, Salomé B, Gomez-Cadena A, Galaine J, Kroemer M, et al. Peripheral innate lymphoid cells are increased in first line metastatic colorectal carcinoma patients: A negative correlation with th1 immune responses. Front Immunol (2019) 10:2121. doi: 10.3389/fimmu.2019.02121
141. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-pd-1 immunotherapy. Cell (2017) 170:1109–19.e10. doi: 10.1016/j.cell.2017.08.027
142. Chen CY, Hutzen B, Wedekind MF, Cripe TP. Oncolytic virus and pd-1/pd-l1 blockade combination therapy. Oncolytic Virother (2018) 7:65–77. doi: 10.2147/ov.S145532
143. Feola S, Capasso C, Fusciello M, Martins B, Tähtinen S, Medeot M, et al. Oncolytic vaccines increase the response to pd-l1 blockade in immunogenic and poorly immunogenic tumors. Oncoimmunology (2018) 7:e1457596. doi: 10.1080/2162402x.2018.1457596
144. Oh CM, Chon HJ, Kim C. Combination immunotherapy using oncolytic virus for the treatment of advanced solid tumors. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21207743
145. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
146. Bollino D, Colunga A, Li B, Aurelian L. Δpk oncolytic activity includes modulation of the tumour cell milieu. J Gen Virol (2016) 97:496–508. doi: 10.1099/jgv.0.000353
147. Chen D, Huang L, Zhou H, Zhang Y. Combining il-10 and oncolytic adenovirus demonstrates enhanced antitumor efficacy through cd8(+) t cells. Front Immunol (2021) 12:615089. doi: 10.3389/fimmu.2021.615089
148. Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett (2007) 254:178–216. doi: 10.1016/j.canlet.2007.02.002
149. Morgan LL. The epidemiology of glioma in adults: A "state of the science" review. Neuro Oncol (2015) 17:623–4. doi: 10.1093/neuonc/nou358
150. Zhang Q, Liu F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis (2020) 11:485. doi: 10.1038/s41419-020-2696-5
151. Reddy PS, Burroughs KD, Hales LM, Ganesh S, Jones BH, Idamakanti N, et al. Seneca valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J Natl Cancer Inst (2007) 99:1623–33. doi: 10.1093/jnci/djm198
152. Zeng J, Li X, Sander M, Zhang H, Yan G, Lin Y. Oncolytic viro-immunotherapy: An emerging option in the treatment of gliomas. Front Immunol (2021) 12:721830. doi: 10.3389/fimmu.2021.721830
153. Jiang H, Alonso MM, Gomez-Manzano C, Piao Y, Fueyo J. Oncolytic viruses and DNA-repair machinery: Overcoming chemoresistance of gliomas. Expert Rev Anticancer Ther (2006) 6:1585–92. doi: 10.1586/14737140.6.11.1585
154. Kana SI, Essani K. Immuno-oncolytic viruses: Emerging options in the treatment of colorectal cancer. Mol Diagn Ther (2021) 25:301–13. doi: 10.1007/s40291-021-00517-7
155. Chen D, Wang R, Long M, Li W, Xiao B, Deng H, et al. Identification of in vitro and in vivo oncolytic effect in colorectal cancer cells by orf virus strain na1/11. Oncol Rep (2021) 45:535–46. doi: 10.3892/or.2020.7885
156. Moehler M, Delic M, Goepfert K, Aust D, Grabsch HI, Halama N, et al. Immunotherapy in gastrointestinal cancer: Recent results, current studies and future perspectives. Eur J Cancer (2016) 59:160–70. doi: 10.1016/j.ejca.2016.02.020
157. Lee P, Gujar S. Potentiating prostate cancer immunotherapy with oncolytic viruses. Nat Rev Urol (2018) 15:235–50. doi: 10.1038/nrurol.2018.10
158. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL 3rd, et al. An open label, single-arm, phase ii multicenter study of the safety and efficacy of cg0070 oncolytic vector regimen in patients with bcg-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol Oncol (2018) 36:440–7. doi: 10.1016/j.urolonc.2017.07.005
Keywords: oncolytic virus, tumor, treatment, immune cell, tumor microenvironment
Citation: Wu X and Fang S (2024) Comparison of differences in immune cells and immune microenvironment among different kinds of oncolytic virus treatments. Front. Immunol. 15:1494887. doi: 10.3389/fimmu.2024.1494887
Received: 11 September 2024; Accepted: 24 October 2024;
Published: 11 November 2024.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesCopyright © 2024 Wu and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaokuan Fang, amx1bmV1QDE2My5jb20=