 Xiaocao Ma1,2†
Xiaocao Ma1,2† Tingting Miao
Tingting Miao- 1Department of Nuclear Medicine, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, China
- 2Jiangxi Province Key Laboratory of Immunology and Inflammation, Jiangxi Provincial Clinical Research Center for Laboratory Medicine, Department of Clinical Laboratory, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, Jiangxi, China
- 3Department of Radiology, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, China
- 4Intelligent Medical Imaging of Jiangxi Key Laboratory, Nanchang, China
Macrophages, the predominant immune cells in the liver, are essential for maintaining hepatic homeostasis and responding to liver injury caused by external stressors. The hepatic macrophage population is highly heterogeneous and plastic, mainly comprised of hepatic resident kuffer cells (KCs), monocyte-derived macrophages (MoMφs), lipid-associated macrophages (LAMs), and liver capsular macrophages (LCMs). KCs, a population of resident macrophages, are localized in the liver and can self-renew through in situ proliferation. However, MoMφs in the liver are recruited from the periphery circulation. LAMs are a self-renewing subgroup of liver macrophages near the bile duct. While LCMs are located in the liver capsule and derived from peripheral monocytes. LAMs and LCMs are also involved in liver damage induced by various factors. Hepatic macrophages exhibit distinct phenotypes and functions depending on the specific microenvironment in the liver. KCs are critical for initiating inflammatory responses after sensing tissue damage, while the MoMφs infiltrated in the liver are implicated in both the progression and resolution of chronic hepatic inflammation and fibrosis. The regulatory function of liver macrophages in hepatic fibrosis has attracted significant interest in current research. Numerous literatures have documented that the MoMφs in the liver have a dual impact on the progression and resolution of liver fibrosis. The MoMφs in the liver can be categorized into two subtypes based on their Ly-6C expression level: inflammatory macrophages with high Ly-6C expression (referred to as Ly-6Chi subgroup macrophages) and reparative macrophages with low Ly-6C expression (referred to as Ly-6Clo subgroup macrophages). Ly-6Chi subgroup macrophages are conducive to the occurrence and progression of liver fibrosis, while Ly-6Clo subgroup macrophages are associated with the degradation of extracellular matrix (ECM) and regression of liver fibrosis. Given this, liver macrophages play a pivotal role in the occurrence, progression, and regression of liver fibrosis. Based on these studies, treatment therapies targeting liver macrophages are also being studied gradually. This review aims to summarize researches on the composition and origin of liver macrophages, the macrophage heterogeneity in the progression and regression of liver fibrosis, and anti-fibrosis therapeutic strategies targeting macrophages in the liver.
1 Introduction
Chronic liver disease arises from exposure to diverse pathogenic factors for the long term in the liver, which largely affects the normal physiological functions of the liver. At present, chronic liver disease is a serious public health issue, with approximately 2 million deaths per year on a global scale (1, 2). The reparative response of the liver following inflammatory insults, induced by a variety of exogenous factors (e.g. infection, alcohol consumption, high-fat diet, etc.) or endogenous factors (e.g. reactive oxygen species (ROS), inflammasome activation, etc.), frequently leads to hepatic fibrosis (3). Liver fibrosis is primarily characterized by excessive deposition of ECM components, notably collagen fibers and some other fibrins, such as elastin, in the liver sinusoidal space (4). Prolonged and severe liver fibrosis can trigger liver scar formation via ECM accumulation, and ultimately further progressing into liver cirrhosis, liver cancer, and even liver failure (3, 5). According to previous studies, liver injury caused by various etiologies like chronic viral hepatitis and nonalcoholic fatty liver disease (NAFLD) can progress into liver fibrosis. However, early liver fibrosis can be reversed through effective anti-viral therapy and lifestyle adjustments, indicating that liver fibrosis is a reversible dynamic balance process (6–9). Therefore, elucidating the pathogenesis of liver fibrosis and developing targeted therapeutic drugs hold significant clinical importance and research value.
The progression of liver fibrosis is always in a dynamic balance between fibrogenesis and fibrolysis (10). Hepatic stellate cells (HSCs) are responsible for the secretion of ECM components, specifically collagen, and play a crucial role in the pathogenesis of liver fibrosis. Additionally, macrophages also play a pivotal role in the initiation and progression of liver fibrosis (11). Macrophages are widely distributed in multiple organs and tissues in the body, they can be divided into resident macrophages and monocyte-derived macrophages within the liver sinusoids (12, 13). Interestingly, liver macrophages are a population of immune cells characterized by highly heterogeneous and plastic, and these macrophages could exhibit distinct cellular phenotypes depending on microenvironmental signals (14). Liver macrophages play crucial roles in a range of physiological and pathological processes, including the maintenance of tissue homeostasis, defense against pathogens, and tissue repairment (15).
In recent years, a substantial amount of literature has reported that macrophages claimed great importance in hepatic inflammation and fibrosis, and macrophage infiltration is a common feature of liver fibrosis caused by multiple chronic liver injuries (16). In addition, macrophages exhibit a dual functionality in the progression and regression phases of liver fibrosis due to high cell heterogeneity (17). Therefore, it is urgent to explore the heterogeneity of macrophages in different disease states and develop approaches for inhibiting or reversing liver fibrosis, which not only enhances our comprehension of hepatic macrophages from an immunological perspective, but also contributes to developing effective drugs to treat liver fibrosis targeting macrophage in future studies. This review aims to conclude the composition and origin of hepatic macrophages, as well as the heterogeneity of macrophages in the progression of liver fibrosis. Subsequently, potential anti-fibrotic strategies targeting macrophages are discussed. This data provides a basis for further studying the mechanism of macrophage regulation and developing therapies for liver fibrosis through regulating macrophage heterogeneity.
2 The composition and origin of liver Macrophages
Macrophages account for the largest proportion in the liver among all solid organs in the body. Macrophages are crucial in maintaining immune homeostasis, disease progression and outcome (18). According to recent literatures, macrophages are characterized by a high degree of heterogeneity (19). In the past, macrophages were simply classified into pro-inflammatory M1-like macrophages and anti-inflammatory M2-like macrophages. However, this traditional classification method fails to fully display the functional heterogeneity of liver macrophages (19, 20). In the past few years, liver macrophages have been more accurately categorized as KCs, MoMφs, LAMs, and LCMs based on their origin, function and associated surface markers (21). As for mouse KCs, apart from F4/80hiCD11bint, Clec4F(C-type lectin domain family 4 member F)and Tim-4(T cell immunoglobulin and mucin domain-containing protein-4) are used as specific cell-surface marker. And mouse KCs are characterized by F4/80hiCD11bintClec4F+Tim4+, whereas mouse MoMφs are identified by F4/80intCD11bhiClec4F-Tim4- (22, 23). Furthermore, Clec4F was identified as a specific marker of resident KCs, but it is expressed relatively late in the development of KCs, making it difficult to identify cells that are developing into KCs (24). Clec2, encoded by the Clec1b gene, is an early marker for KCs and continues to be expressed throughout their lifespan. Therefore, Clec2 is useful for identifying monocyte-derived KCs before they express Clec4F (24, 25). Therefore, KCs and MoMφs in the mouse liver can be effectively distinguished based on their surface marker profiles (26, 27). Human liver macrophages are mainly composed of KCs and MoMϕs, but the macrophage markers are different from those in mice. Human KCs are usually characterized as CD14+CD68+, especially CD68, a recognized marker of human KCs (27, 28). While human MoMϕs are usually recognized as CD14+CCR2+ (17). In addition, the liver also contains LAMs and LCMs (24, 29). The surface markers of human LAMs are CD14, CD9, TREM2, and GPNMB (30, 31). No definite surface markers have been identified in the mouse LAMs (24). While LCMs express general macrophage markers such as F4/80, CD64, and CX3CR1 (24, 29).
Under liver homeostatic conditions, resident KCs which originated from fetal yolk sac erythromyeloid progenitors and are mainly located in the liver sinusoids, constitute the primary macrophage population, and the lifespan of KCs in the liver is relatively brief (32). KCs can sustain their population through continuous self-renewal (19). KCs are highly efficient phagocytes that constitute the first line of host defense against pathogens, thereby maintaining hepatic homeostasis (33). In addition, KCs express a variety of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), nucleotide oligomerization domain-like receptors (NODs), and retinol-inducible gene I (RIG-I), which contributes to the effective identification and elimination of foreign pathogens (17, 34) (Figure 1). Simultaneously, KCs are involved in many metabolic pathways, including removing damaged cells, erythrocyte-derived hemoglobin-containing vesicles, and metabolic waste products through scavenger receptors (35, 36). Moreover, KCs can regulate cholesterol metabolism and contribute to maintaining cholesterol homeostasis in the body (37). Meanwhile, KCs can also recycle iron by inducing phagocytosis of damaged erythrocytes, which can participate in maintaining the iron metabolism balance (38). Additionally, KCs play a significant role in mediating immune tolerance (39). Interestingly, KCs can not only act as guardians to maintain liver homeostasis, but also release damage-associated molecular pattern (DAMP)after sensing liver injury, and the DAMP can activate KCs to secrete TNF-α and IL-1β, further causing damage to hepatocytes (40, 41) (Figure 1). The number of KCs and monocyte-derived macrophages maintains a dynamic balance in the liver, when the KCs are exhausted or depleted due to various internal or external factors, myeloid-derived monocytes will replenish self-renewing KCs to respond to liver damage (42).
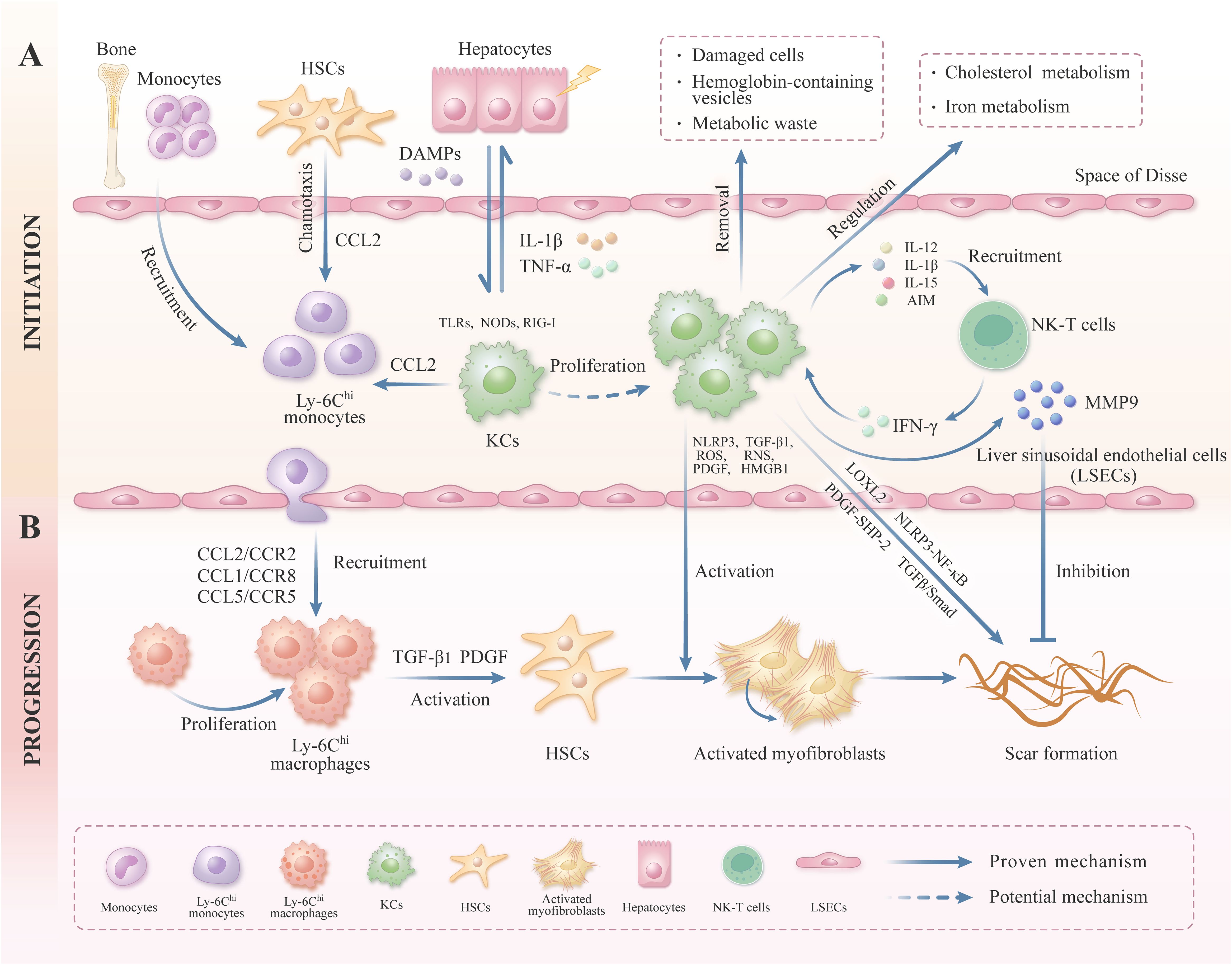
Figure 1. The role of liver macrophage in the initiation and progression of liver fibrosis. This figure provides an overview of the heterogeneity and plasticity of liver macrophages. (A) KCs, located in the hepatic sinus endothelium, can be activated by DAMPs after suffering from liver injury, and activated KCs can release macrophage chemokine CCL2 to recruit the Ly6Chi subgroup monocytes to the liver. KCs express a variety of PRRs, including TLRs, NODs, and RIG-I, which facilitate the effective identification and removal of foreign pathogens. KCs can also secrete inflammatory cytokines (e.g.IL-1β and TNF-α) to aggravate the damage of hepatocytes. KCs can secrete IL-12, IL-15, IL-1β, and AIM, which further recruit and activate NK-T cells. In turn, NK-T cells can produce inflammatory cytokines, including IFN-γ, thereby modulating the function of KCs and impacting the progression of liver fibrosis. In addition, KCs can regulate a variety of metabolic pathways, including the cholesterol and iron metabolism. Besides, KCs can remove damaged cells and metabolic waste to maintain the metabolic balance. (B) Under the chemotaxis axis, Ly-6Chi subgroup monocytes can be recruited to the liver, where they differentiate into proliferating Ly-6Chi subgroup macrophages, and these macrophages can secrete TGF-β1 and PDGF to activate HSCs. In addition, KCs can also release NLRP3, TGFβ, PDGF, ROS, and RNS, then induce the quiescent HSCs activated into myofibroblasts, ultimately secreting ECM and inducing liver fibrosis. KCs can also secrete MMP9, which is conducive to the degradation of liver ECM. In addition, KCs can promote collagen cross-linking and scar formation by modulating the expression of LOXL2.
Compared with KCs, MoMφs derived primarily from peripherally circulating monocytes, constitute only a small percentage of the macrophage pool in healthy livers (43). When the liver was damaged, monocytes derived from the peripheral circulation are quickly recruited to the liver via the CCL2/CCR2 (CC chemokine ligand-2/CC chemokine receptor-2), CCL5/CCR5, and CCL1/CCR8 chemotactic axes, and then trans-differentiated into mature macrophages (17, 44–46) (Figure 1). The MoMφs, after being continuously recruited to the liver, dominate the macrophage population and play a crucial role in regulating liver damage and repair (17). Interestingly, MoMφs are highly heterogeneous, composed of a spectrum of functional plastic cells, these MoMφs can undergo constant variation under a dynamically changing micro-environment in the liver (14). MoMφs can be further classified into pro-inflammatory Ly-6Chi subgroup macrophages and anti-inflammatory Ly-6Clo subgroup macrophages according to the surface markers and functional characteristics, the former exhibits pro-inflammatory and pro-fibrotic properties, and the latter displays anti-inflammatory and anti-fibrotic properties (17, 47). Under specific conditions, pro-inflammatory Ly-6Chi subgroup macrophages can trans-differentiate into reparative Ly-6Clo subgroup macrophages via phagocytosis (48) (Figure 2). It has been reported that a large number of Ly-6Chi subgroup macrophages can infiltrate into the liver and exert pro-inflammatory and pro-fibrotic functions during the progression stage of carbon tetrachloride (CCL4)-induced liver fibrosis, and Ly-6Chi subgroup macrophages can transform into Ly-6Clo subgroup macrophages after CCL4 removal, leading to collagen degradation and gradual liver repair (26). Therefore, the MoMφs in the liver are highly plastic and heterogenous cells.
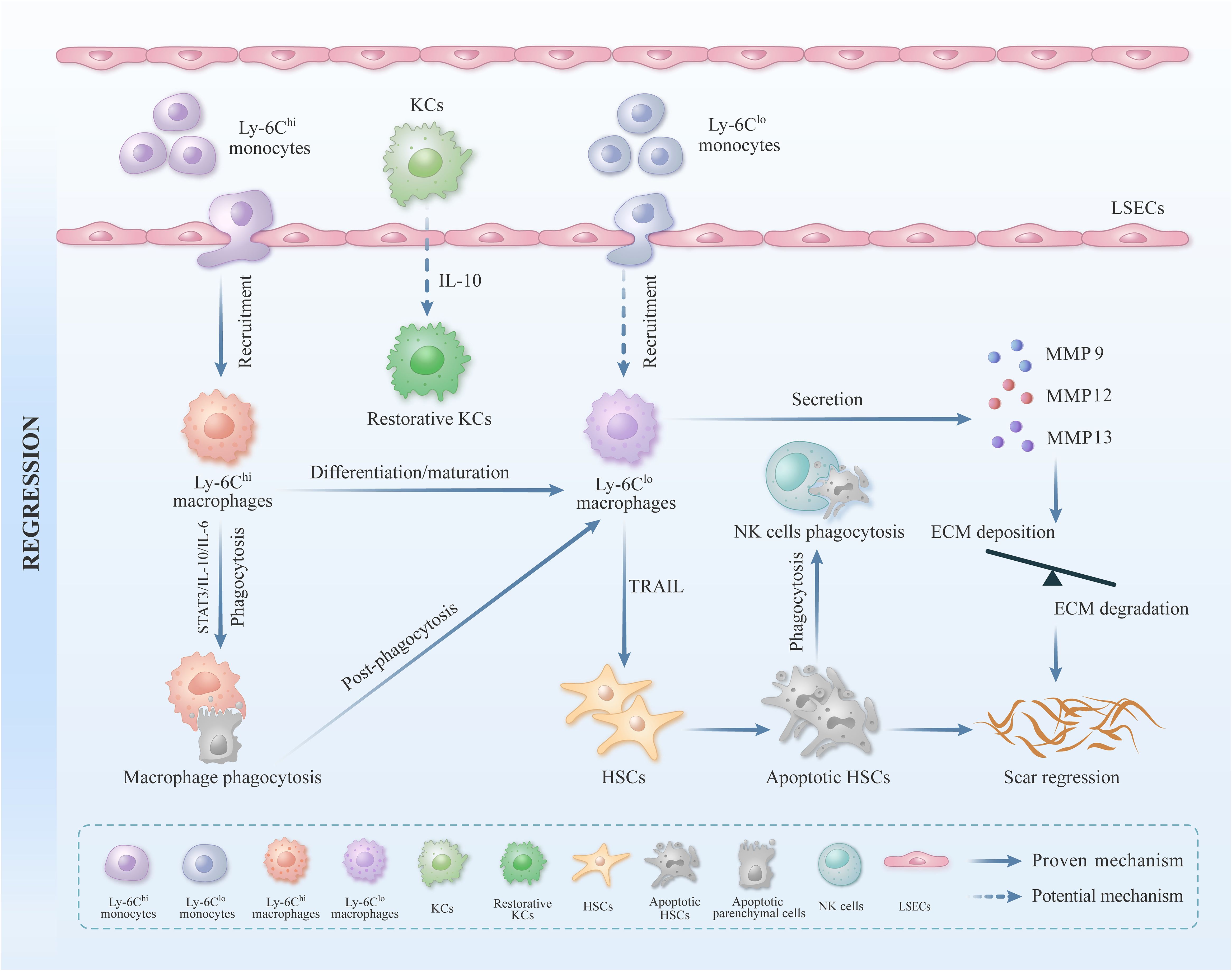
Figure 2. The role of liver macrophages in liver fibrosis repair. Apart from KCs, MoMφs play a crucial regulatory role in liver inflammation and fibrosis. When the liver suffers from external damage, a substantial influx of peripheral monocytes can be recruited to the liver and subsequently differentiate into mature Ly-6Chi subgroup macrophages. These Ly-6Chi subgroup macrophages exhibit pro-inflammatory and pro-fibrotic characteristics. In the stage of liver injury repair, Ly-6Chi subgroup macrophages can be transformed into Ly-6Clo subgroup macrophages by activating STAT3/IL-10/IL-6 signaling pathway, and then engulfing hepatocyte fragments. Ly-6Clo subgroup macrophages can promote ECM degradation by secreting MMP9, MMP12, and MMP13. Ly-6Clo subgroup macrophages can also induce the apoptosis of activated HSCs by expressing TRAIL, thus inhibiting the formation of liver collagen and accelerating the repair process of liver fibrosis.
Furthermore, LAMs and LCMs are also two critical subgroups macrophages in the liver. LAMs are mainly situated near the hepatobiliary ducts, and their quantity is relatively low under physiological conditions. In contrast, the population of LAMs increases and predominantly accumulates in adipose and inflammatory tissues in pathological conditions like liver injury (24, 49). LAMs originate from precursor cells derived from peripheral monocytes and possess the capacity for self-renewal. The precursor cells of LAMs express CCR2 and CX3CR1, which facilitate the migration and recruitment of monocytes (50). Research has reported that when KCs are exhausted, LAMs can transform into KCs to replenish the KCs pool in the liver (25). LAMs possess an abundance of intracellular lipid structures and lysosomes, which are indispensable for lipid metabolism, ECM remodeling, and the clearance of apoptotic hepatocytes (49, 51) (Figure 3). Numerous studies have investigated the role of LAMs in liver inflammatory injury and tissue remodeling. It was reported that LAMs deficiency in the liver can aggravate liver inflammation and fibrosis induced by nonalcoholic steatohepatitis (NASH) in mice, indicating that LAMs can inhibit the progression of inflammation and liver fibrosis (49, 52). As for LCMs, they originate from peripheral circulating monocytes via CCR2 and CX3CR1 and have elongated dendrites on their cell membrane surface. These dendritic structures facilitate intercellular cross-linking on the liver capsule surface, forming an intricate cellular network. This network plays a crucial role in immune surveillance by sensing and limiting the dissemination of bacteria in the liver capsule (24, 53). In addition, LCMs also contribute to regulating liver inflammation (54) (Figure 3).
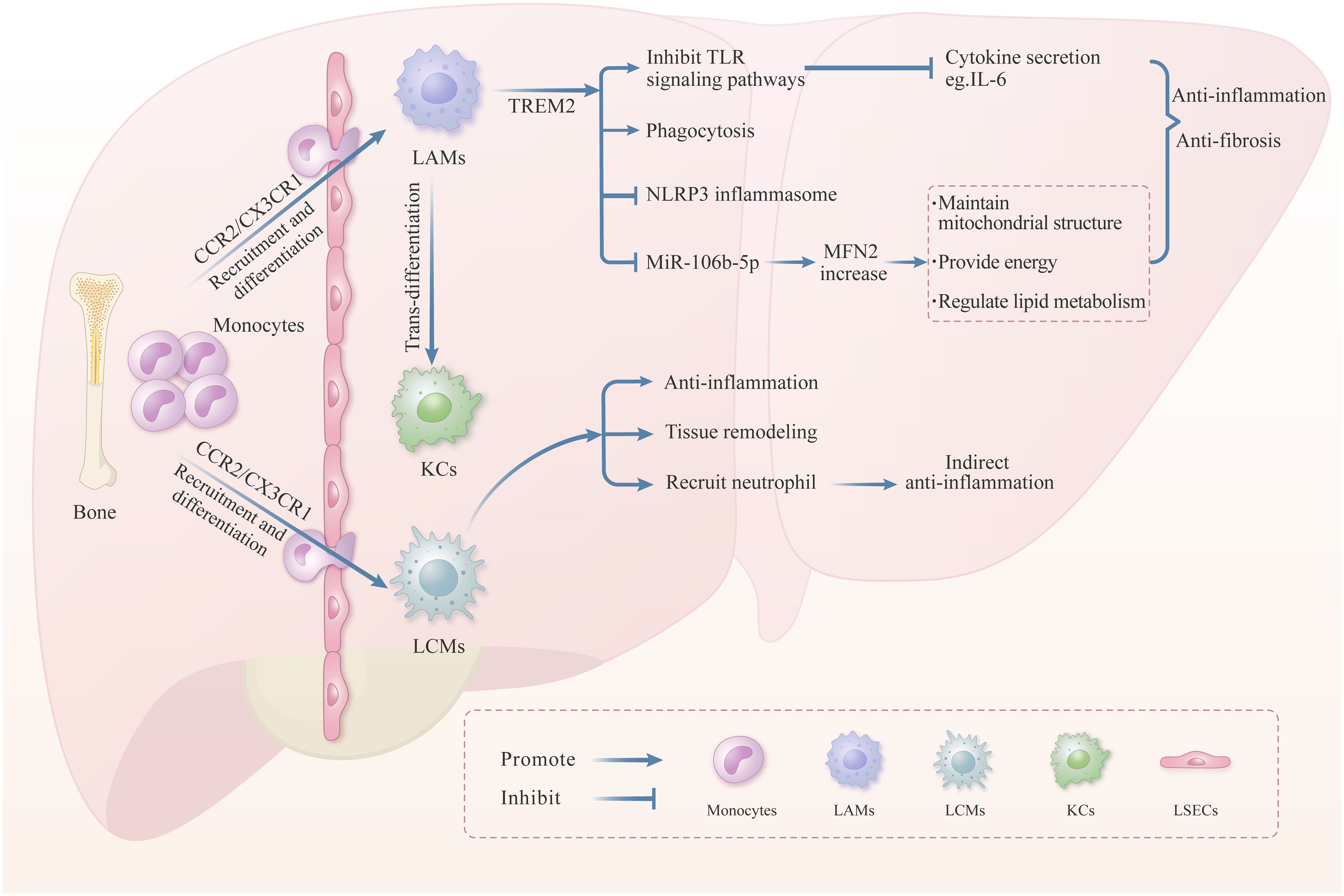
Figure 3. The role of LAMs and LCMs in liver injury. In liver pathological conditions, LAMs precursor cells are recruited to the liver via chemotaxis and then differentiate into LAMs. The transmembrane receptor TREM2, on the surface of LAMs, plays a vital role in regulating the LAMs function. Numerous literatures have reported that TREM2 can promote anti-inflammation and anti-fibrosis through inhibiting the TLRs signaling pathway, promoting phagocytosis, and inhibiting the NLRP3 inflammasome. Moreover, TREM2 can inhibit the production of MiR-106b-5p in the exosomes derived from macrophage, thereby enhancing the MFN2 expression to maintain mitochondrial structure and lipid metabolism balance. Besides, LAMs can transform into KCs when the KCs were exhausted. LCMs located in the liver capsule also play a significant role in liver inflammation and tissue remodeling. LAMs are recruited from peripheral monocytes, and mitigate the dissemination of intraperitoneal bacteria by recruiting neutrophils in the liver, thereby inhibiting the liver inflammation indirectly.
Human monocytes in the peripheral can also be categorized into distinct subsets with unique phenotypic and functional characteristics (55). The literature indicates that human peripheral blood monocytes can be categorized into classical monocytes (identified as CD14++CD16- or CD14+CD16-), intermediate monocytes (identified as CD14++CD16+ or CD14+CD16+), and non-classical monocytes (identified as CD14-CD16++ or CD14-CD16+) based on the expression of surface marker (55, 56). To a certain extent, the gene expression profiles of CD14++CD16- monocytes and CD14+CD16++ monocytes in human peripheral blood respectively exhibit similarities to Ly-6Chi subgroup monocytes and Ly-6Clo subgroup monocytes in mouse peripheral blood in previous studies (56, 57). Based on the above literature, the phenotype and function of macrophages are modulated by specific liver micro-environment, ultimately determining whether they exacerbate or ameliorate liver damage. These findings explain why liver macrophages perform different and sometimes even opposite functions during hepatic diseases (58).
3 The function of KCs in liver fibrosis
KCs, situated in the periportal area of the hepatic sinusoids, constitute an important component of the innate immune system within the body. KCs maintain their population mainly through proliferation and act as scavengers to maintain hepatic homeostasis (17). Monitoring infectious and non-infectious insults in the liver is a pivotal role of KCs (59, 60). KCs can modulate the occurrence and progression of liver fibrosis by regulating NOD-like receptor protein 3 (NLRP3) inflammasome, PRR, transforming growth factor-β (TGF-β) signaling pathway, and platelet-derived growth factor (PDGF) signaling pathway (61–64) (Figure 1). For example, the KCs-derived NLRP3 inflammasome can promote the occurrence of liver fibrosis by activating the nuclear factor kappa-B (NF-κB) signal pathway in Schistosoma japonicum-infected mice (61). KCs can also release pro-inflammatory cytokines, including TNF-α and IL-1β, and recruit peripheral mononuclear macrophages to the liver through the CCL2/CCR2 axis, further aggravating liver fibrosis (17). In addition, KCs can also stimulate quiescent HSCs transformed into activated HSCs through various mechanisms, leading to increased production and secretion of ECM (65, 66). For example, TGF-β, a pro-fibrotic cytokine secreted by KCs, can activate HSCs by activating the TGF-β/Smad signaling pathway, further promoting ECM deposition (67, 68). PDGF, a potent proliferative cytokine secreted by KCs, can induce tyrosine phosphorylation and activate the downstream signaling molecule SHP-2 (Src homology region 2 domain-containing protein tyrosine phosphatase-2) by binding to the cell-surface receptors on HSCs, and then exacerbate liver fibrosis in mice (69–71). It is well established that the activated KCs can also produce ROS/reactive nitrogen species (RNS) to stimulate the activation and proliferation of HSCs, ultimately increasing ECM deposition in the liver (66). Furthermore, it has been demonstrated that HMGB1 (High-mobility group box-1), produced by hepatic cells like KCs and hepatocytes, can activate HSCs to express collagen I via its receptor RAGE (the Receptor for Advanced Glycation End-products), thereby exacerbating liver inflammation and fibrosis (72). Some studies have further confirmed that KCs depletion can not only inhibit the production of IL-1β and TNF-α but also inhibit the activation of HSC, thereby alleviating liver fibrosis in a bile duct ligation (BDL)-induced liver fibrosis mouse model (73). Besides, when viruses like hepatitis B (HBV) infect the liver, KCs can detect the danger signals and recruit circulating monocytes to the liver, where these monocytes differentiate into MoMφs, ultimately regulating anti-viral immunity. Furthermore, these macrophages can also release TGF-β1 to promote the progression of viral hepatitis to liver fibrosis (74, 75). In addition, KCs can also directly regulate ECM remodeling and exhibit significant cellular heterogeneity, such as KCs can promote collagen cross-linking and scar formation by modulating the expression of lysyl oxidase-like protein 2 (LOXL2). Conversely, KCs can also secret matrix metalloproteinase (MMP), such as MMP9, to degrade collagen under certain circumstances (32).
KCs can regulate the development and progression of liver fibrosis through the mechanisms above. Additionally, KCs can also interact with various immune cells such as natural killer T (NK-T)cells, consequently impacting the development of liver fibrosis (Figure 1). KCs can secrete IL-12, IL-15, IL-1β, and apoptosis inhibitors of macrophage (AIM) to recruit and activate NK-T cells. In response, NK-T cells can produce pro-inflammatory cytokines such as interferon-gamma (IFN-γ) to modulate the function of KCs, influencing the progression of liver fibrosis (76) (Figure 1). Furthermore, KCs can release chemical mediators to recruit neutrophils to the liver; whereupon neutrophils can release inflammatory mediators such as ROS to participate in liver repair (77, 78). In conclusion, KCs play a significant role in the regulation of liver fibrosis through various direct and indirect mechanisms. Nevertheless, further research is needed to determine whether KCs can be converted into reparative KCs and enhance the hepatic regenerative response during liver injury repair (Figure 2).
4 The role of MoMφs in liver fibrosis
Except for KCs, MoMφs are also essential regulatory cells for liver inflammation and fibrosis. When the liver suffers from injury, KCs are capable of sensing liver damage and releasing inflammatory cytokines, such as TNF-α and IL-1β. Simultaneously, a large number of peripherally derived monocytes can recruit to the liver and differentiate into mature MoMφs (17) (Figure 1). In recent years, MoMφs have drawn significant attention due to their high heterogeneity and plasticity. In the initial phase of chronic persistent liver fibrosis, MoMφs are predominant in the liver, and they can activate HSCs by secreting TGF-β1 and PDGF, which further exacerbates liver fibrosis (79, 80) (Figure 1). During the regression phase of liver fibrosis, MoMφs can participate in ECM degradation by secreting MMPs such as MMP9, MMP12, and MMP13, which facilitates the regression of liver fibrosis (81) (Figure 2). MoMφs can exhibit different cell phenotypes based on the local environmental cue to maintain liver homeostasis or affect disease progression (82). For example, macrophages can secrete cytokines, chemokines, and ROS to modulate liver inflammation and fibrosis in both alcoholic liver disease (ALD) and NAFLD (83, 84). In ALD, ethanol and its metabolite acetaldehyde can activate KCs and then lead to the release of pro-inflammatory mediators such as IL-1β and TNF-α, which results in an acute pro-inflammatory response, simultaneously accompanied by a small number of MoMφs infiltration (21). In NAFLD, the chronic low-grade inflammation induced by lipid accumulation and metabolic dysregulation can lead to substantial MoMφs infiltration in the liver. In the initial stage of NAFLD, macrophages display a pro-inflammatory and phagocytic phenotype that facilitates the clearance of lipid-overloaded hepatocytes. Conversely, in the advanced stages of NAFLD, there is a predominance of M2 macrophages characterized by an anti-inflammatory phenotype. Consequently, pathogenic and reparative macrophages within the liver microenvironment undergo dynamic regulation in NAFLD (84).
Numerous studies have indicated that the conventional M1/M2 binary classification method, which distinguishes between classically activated and alternatively activated macrophages, this classification is insufficient to capture the full complexity of macrophages (19, 85). In recent years, more researchers have categorized MoMφs into pro-inflammatory Ly-6Chi subgroup macrophages and reparative Ly-6Clo subgroup macrophages based on the expression of the cell surface glycoprotein Ly-6C (21). In the context of liver injury, pro-inflammatory Ly-6Chi subgroup macrophages are primarily recruited to the liver through chemokines, including CCL1, CCL2, and CX3CL1, subsequently exacerbating liver inflammation and fibrosis (21, 47) (Figure 1). Previous studies have indicated that selective depletion of hepatic Ly-6Chi subgroup macrophages can impede HSCs activation and reduce ECM deposition during the initial stage of liver fibrosis, while selective depletion of hepatic Ly-6Clo subgroup macrophages will be detrimental in the repair phase of the liver fibrosis (26, 86). Baeck. C et al. demonstrated that the administration of the CCL2 inhibitor mNOX-E36 could effectively impede the recruitment of Ly-6Chi subgroup macrophages to the liver, thereby reducing the degree of liver fibrosis. The above findings indicate that Ly-6Chi subgroup macrophages predominantly exert a pro-inflammatory and pro-fibrotic effect (87). During the repair stage of liver injury, it has been observed that Ly-6Chi subgroup macrophages can transform into Ly-6Clo subgroup macrophages under specific circumstances in vivo. In vitro studies have demonstrated that this process was mainly accomplished by engulfing apoptotic hepatocyte fragments. Recent research has further indicated that the activation of the STAT3/IL-10/IL-6 signaling pathway plays a crucial role in mediating phagocytosis and orchestrating the phenotypic conversion of these two subgroups of macrophage (88, 89) (Figure 2).
In addition, the phenotypic switch from Ly-6Chi subgroup macrophages to Ly-6Clo subgroup macrophages is influenced by the interaction between macrophages and other immune cells. For instance, Ly-6Chi subgroup macrophages can promote neutrophils generating ROS and promote CD4+T cells generating IL-4. These processes can facilitate the phenotype switch of two subgroups of macrophages (90–92). The phenotypic conversion of Ly-6Chi subgroup macrophages to Ly-6Clo subgroup macrophages plays a significant role in facilitating the regression of liver fibrosis induced by multiple factors in mice (93). Additionally, the phenotypic transformation of two subgroups of macrophages in the liver marks the shift from the pro-inflammation initiation phase to the anti-inflammatory and resolution phase (94, 95). In summary, the phenotypic transformation of liver macrophage subsets plays a crucial role in the reversal of liver fibrosis. Further research is warranted to determine whether Ly-6Clo subgroup monocytes in peripheral blood can be recruited to the liver during the repair phase of liver injury, where they may differentiate and mature into Ly-6Clo subgroup macrophages, thereby potentially facilitating the resolution of liver fibrosis (Figure 2).
Previous studies demonstrated that collagen degradation was weakened when Ly-6Clo subgroup macrophages were knocked out in the repair stage of the CCL4-induced liver fibrosis mouse model, which strongly proves that Ly-6Clo subgroup macrophages play a crucial role in degrading liver collagen (26). Ly-6Clo subgroup macrophages primarily repair liver fibrosis by upregulating MMPs (MMP9, MMP12, and MMP13), which facilitates the degradation of ECM. These cells can also increase the expression of some growth factors like hepatocyte growth factor and insulin-like growth factor for hepatocyte repairment. Furthermore, Ly-6Clo subgroup macrophages can upregulate phagocytosis-related genes like MARCO (macrophage receptor with collagenous structure), which in turn fuel themselves to engulf apoptotic cells (26, 88, 91, 95–97). It has been reported that the activation of HSCs played a pivotal role in the occurrence and development of liver fibrosis (3). Ly-6Clo subgroup macrophages can induce the apoptosis of activated HSCs through expressing TRAIL (TNF-related apoptosis-inducing ligand), subsequently rendering them susceptible to cytotoxic NK cells, besides, Ly-6Clo subgroup macrophages can also revert the activated HSCs back to a quiescent state (98, 99) (Figure 2). In conclusion, the aforementioned studies have reveal the significant contribution of Ly-6Clo subgroup macrophages in the process of repairing chronic liver fibrosis.
5 The role of LAMs and LCMs in liver fibrosis
The number of LAMs is relatively tiny in normal liver. However, during pathological conditions such as hepatic steatosis, liver inflammation, and liver fibrosis, a large number of peripheral monocyte-derived LAMs precursor cells are recruited to the liver by the CCL2/CCR2 axis and then differentiate into LAMs (24, 50). Studies indicated that knocking out CCR2 can lead to an absence of LAMs in the liver, which aggravates liver inflammation and fibrosis induced by NASH in mice. Therefore, LAMs could inhibit the progression of liver inflammation and fibrosis (50). TREM2, a transmembrane receptor of the immunoglobulin superfamily, has been identified on the surface of LAMs and is pivotal for modulating their function (100). TREM2 can not only recognize lipids and apolipoproteins but also combine with phospholipid molecules on the surface of apoptotic cells, thereby regulating processes including lipid metabolism and cellular phagocytosis in the liver (101). Furthermore, TREM2 can also regulate liver inflammation and tissue repair (49). In murine models of NAFLD, exosomes derived from TREM2-deficient macrophages exhibit elevated levels of miR-106b-5p, which leads to the downregulation of mitochondrial fusion protein 2 (MFN2), thereby damaging mitochondrial architecture and accelerating NAFLD (102) (Figure 3). Some studies have also indicated that macrophage-specific knockout of TREM2 would impair its phagocytic capacity for apoptotic hepatocytes, further aggravating the progression of NASH (52). Other studies have demonstrated that mice deficient in TREM2 exhibit aggravated hepatic inflammation in liver injury models induced by CCl4 and acetaminophen (APAP), and the mechanism is related to enhanced TLR signal pathways and subsequent pro-inflammatory cytokines secretion (103). The latest research shows that TREM2+LAMs can inhibit NLRP3 activation and pro-inflammatory cytokine secretion, then degrade the ECM in the liver, thereby accelerating the regression of liver fibrosis (104) (Figure 3). The above studies indicate that LAMs are involved in regulating liver lipid metabolism and cellular phagocytosis through their surface receptor TREM2, and play an essential role in inhibiting liver inflammation and facilitating the repair of liver injury (Figure 3). Future research may concentrate on further elucidating the functions of LAMs and developing effective therapeutic approaches for liver fibrosis based on the relevant target.
LCMs also play an essential regulatory role in liver injury (Figure 3). Research indicates that LCMs can mitigate the dissemination of intraperitoneal bacteria in the liver, which was achieved by recruiting neutrophils in response to bacterial detection, consequently attenuating hepatic inflammation (53). In a murine model of liver injury induced by 5-chloro-2-(2,4-dichlorophenoxy), LCMs exhibited a shift from M1 to M2 macrophage phenotypes, suggesting their potential involvement in regulating liver tissue remodeling and fibrosis by releasing anti-inflammatory or pro-resolving mediators (105). In the advanced stages of the NAFLD mouse model, the number of liver LCMs increased, accompanied by morphological alterations, which suggests that LCMs may have a regulatory function in the progression of advanced-stage NAFLD (54). The literature above indicates that LCMs constitute a significant subpopulation of macrophages involved in the regulation of liver injury (Figure 3). However, little evidence addresses the role of LCMs in liver fibrosis, further research is warranted to elucidate the function of LCMs in this context.
6 Potential therapeutic strategies to target macrophages for liver fibrosis
6.1 The regulation of intestinal microbiota
Increased bacterial ectopy is one of the important signs of chronic liver diseases, as pathogen-associated molecular patterns (PAMPs) can not only cause typical infectious complications but also activate TLRs like TLR4 on macrophages, thereby causing liver inflammation and fibrosis (106, 107). A great number of bacterial products that originate from the hepatic portal vein can activate the TLR4 receptor, further activating HSCs and recruiting peripheral inflammatory macrophages to the liver, ultimately leading to liver fibrosis (21, 106, 108). Some studies have shown that the endotoxins produced by gram-negative bacteria can induce hepatic fibrogenesis. The transfer of fecal microbiota from healthy donors to individuals with chronic liver diseases, known as fecal microbiota transplantation (FMT), has emerged as a potential novel therapeutic approach, which further reinforces the close link between gut microbiota and liver fibrosis (109, 110). Clinical studies have confirmed that intestinal flora imbalance can drive the progression of liver fibrosis by affecting macrophages, while the antibiotic rifaximin has been found to alleviate alcoholic liver fibrosis by modulating intestinal microflora (108, 111). The above studies indicate that rifaximin has the potential to ameliorate liver fibrosis by targeting macrophages. In addition, the combination therapy of vancomycin, gentamicin, and meropenem shows promise in improving intestinal flora and treating liver disease, which is currently being explored in clinical trials(NCT03157388) (112). It has also been reported that supplementation with probiotic Lacticaseibacillus rhamnosus can potentially mitigate liver fibrosis by inhibiting bile acid synthesis and enhancing bile acid excretion (113). The above studies support the existence of cross-talk between intestinal flora and liver diseases, specifically liver fibrosis. Consequently, modulating intestinal flora may represent a novel approach for targeting macrophages in the treatment of liver fibrosis or other liver diseases.
6.2 The inhibition of IL-1β signaling pathway
Numerous studies have confirmed a correlation between the NLRP3 inflammasome and liver fibrogenesis (130, 131). The studies demonstrated that NLRP3 inflammasome activation can induce the KCs activation and then release inflammatory cytokines such as IL-1β during liver injury, which plays a crucial role in the progression of liver fibrogenesis (61, 131). IL-1β, a potent pro-inflammatory cytokine, exerts a pro-inflammatory role through interaction with the interleukin-1 receptor (IL-1R1) in an autocrine or paracrine manner, thereby driving the occurrence of liver fibrosis (132, 133). It has been shown that a naturally existing IL-1R1 antagonist (IL-1Ra)can block the biological interaction between IL-1β and its cell surface receptors. Anakinra, a specific antagonist of the IL-1β receptor, can ameliorate liver fibrosis caused by various etiological factors (114–116). The aforementioned studies suggest that blocking the IL-1β receptor may be a promising strategy for the treatment of liver diseases.
6.3 The inhibition of the chemotactic axis in macrophages
In the progress of liver fibrosis, peripheral pro-inflammatory mononuclear MoMφs can be attracted to the liver depending on CCL2/CCR2, CCL5/CCR5, and CCL1/CCR8 chemokines axis and aggravate liver fibrosis (44, 134). In this context, inhibiting the recruitment of pro-inflammatory MoMφs to the liver by interfering with the chemotactic axis is a significant therapeutic strategy. mNOX-E36, a CCL2 inhibitor, can suppress the recruitment of pro-inflammatory MoMφs to the liver in murine models of CCL4-induced liver fibrosis and methionine-choline-deficient diet (MCD)-induced non-alcoholic steatohepatitis, therefore, the balance of macrophage subgroups can shift to a state that was dominated by reparative MoMφs, ultimately contributing to the fibrosis regression (87, 117, 135). In recent years, numerous studies have extensively reported that Cenicriviroc (CVC) functions as a dual antagonist of CCR2/CCR5 (118). The usage of CVC has been shown to effectively block the recruitment of pro-inflammatory mononuclear MoMφs to the liver that is mediated by the CCL2/CCR2 and CCL5/CCR5 axes, which consequently inhibits both hepatic inflammation and fibrosis (136, 137). In a randomized, double-blind, placebo-controlled trial, Friedman et al. found that hepatic inflammation and fibrosis were significantly improved following a one-year treatment with CVC in NASH (non-alcoholic fatty liver disease activity score NAS ≥ 4) or liver fibrosis (stages I-III) patients (119). Furthermore, the anti-fibrotic effect of CVC treatment can be maintained in the second year (120). Besides, propagermanium (a CCR2 inhibitor) and maraviroc (a CCR5 inhibitor) are also considered as relative chemotactic axis inhibitors, which all promote the amelioration of NASH in murine models (121, 122). It has been reported that CCR8 knockout can inhibit the recruitment of MoMφs derived from peripheral monocytes to the fibrotic liver, thereby alleviating experimental liver inflammation and fibrosis induced by either CCL4 or BDL in mice (45). Furthermore targeting the CCL1/CCR8 chemotactic axis can also alleviate inflammatory and fibrotic damage in the lung and peritoneum (138, 139). The above data indicates that the CCL1/CCR8 chemotactic axis is important for driving inflammation and fibrosis in liver or other organs. However, no clinical trials have explored whether blocking the CCL1/CCR8 chemotactic axis can inhibit liver inflammation and fibrosis. Therefore, targeting the CCL1/CCR8 chemotactic axis represents a promising potential approach for clinical intervention in liver inflammation and fibrosis.
These studies show that the chemotaxis effects mediated by chemokines and their receptors initiate the recruitment of peripheral mononuclear MoMφs to the damaged liver. Hence, the inhibition of CCL2/CCR2, CCL5/CCR5, and CCL1/CCR8 chemotaxis shows potential efficacy to repair liver fibrosis.
6.4 Galectin-3 antagonist
Galectin-3 (Gal-3) is a β-galactoside-binding lectin that can be produced from hepatic macrophages (81, 140). Gal-3 exerts multiple regulatory effects on the innate immune and adaptive inflammatory response (141). It has been reported that Gal-3 is involved in the development of fibrosis in various tissues by influencing phenotypic transformation and migration of macrophages. Moreover, Gal-3 can also function as a pro-inflammatory and pro-fibrotic mediator in the liver (142, 143). Jiang et al. reported that Gal-3-knockout could inhibit the HSCs activation and effectively alleviate liver fibrosis while Gal-3 overexpression could reverse it in mice after BDL (144). In addition, it was also observed that Gal-3 inhibition can also suppress liver inflammation and liver fibrosis in mouse models of NASH (123). The above studies have shown that the Gal-3, secreted by macrophages, could contribute to hepatic fibrogenesis, this notion may suggest that Gal-3 could serve as a potential therapeutic target for the patients with liver fibrosis. As previously reported, GR-MD-02, a Gal-3 inhibitor, is a complex carbohydrate compound extracted from natural plants. In a phase III clinical trial for NASH-induced advanced liver fibrosis, GR-MD-02 has been shown high safety and tolerability on the human body, as well as the potential to ameliorate liver fibrosis (123). GB1211, another Gal-3 inhibitor, also has been confirmed to have favorable safety and tolerability in phase I clinical trial, which indicates that GB1211 warrants further clinical trial research for anti-fibrotic treatments (124). In summary, the above studies suggest that targeted inhibition of macrophage-derived Gal-3 is conducive to the repairment of liver fibrosis.
6.5 PPARs agonist
Peroxisome proliferator-activated receptors (PPARs) are a group of key nuclear transcription factors that play important roles in regulating lipid metabolism, cell differentiation, and maintaining liver homeostasis (145). In mammals, PPARs consist of three isoforms: PPAR-α, PPAR-δ (also referred to as PPAR-β), and PPAR-γ (146). The liver is an organ that mainly regulates systemic metabolism and maintains energy homeostasis, with PPARα governing lipid metabolism in the liver, and the dysregulation of lipid metabolism may lead to steatohepatitis or even liver fibrosis (147). PPAR-δ has been shown to inhibit liver steatosis and block the progression of steatohepatitis by upregulating low-density lipoprotein receptor(LDL-R) (148). Additionally, PPARγ can inhibit NF-κB activity through directly binding to its p65 subunit, thereby improving steatohepatitis (149). Research has reported that PPARs activation can inhibit the advancement of liver fibrosis by regulating the inflammatory responses in macrophages. Lanifibranor, a PPARs agonist with broad-spectrum activity, can attenuate the inflammatory response induced by palmitic acid on human monocytes and mouse bone marrow-derived macrophages in vitro experiments (125). In vivo animal experiments also demonstrated that Lanifibranor has the potential to ameliorate liver inflammation and fibrosis induced by NAFLD (125, 150). In addition, in a phase IIb randomized controlled clinical trial, it has been corroborated that lanifibranor treatment for six months can alleviate liver fibrosis induced by NASH. Building on this evidence, phase III clinical trials are currently being explored (151). The above literature confirmed that modulating the activation of PPARs to control the inflammatory response of macrophages is a promising therapeutic approach for liver inflammation and fibrosis.
6.6 The Farnitol X receptor agonist
The farnitol X receptor (FXR) agonist is a nuclear receptor that plays a crucial role in regulating bile acid and lipid homeostasis, enhancing cholesterol transport in macrophages, and serving as a key regulator in hepatic steatosis, inflammation, and fibrosis (152, 153). FXR activation can not only decrease the synthesis of bile acids, but also suppress the production of pro-inflammatory cytokines from hepatic infiltrating inflammatory cells, thereby preventing the progression of liver fibrosis (154). Obeccholic acid, an FXR agonist, is beneficial to lipid metabolism and glucose metabolism, and is considered as a leading candidate for the treatment of liver fibrosis induced by NASH (126). These studies suggest that FXR may be an important therapeutic approach to improve the outcome of liver fibrosis by affecting liver macrophages.
6.7 Splenectomy
Advanced liver fibrosis and cirrhosis often have multiple complications, including splenomegaly and hypersplenism (127). It has been reported that advanced liver fibrosis and cirrhosis are often accompanied by thrombocytopenia, which results from the destruction of circulating platelets during secondary portal hypertension or hypersplenism (155). Animal experimental studies have demonstrated that thrombocytopenia can further aggravate liver fibrosis induced by BDL and CCL4 in mice. The underlying mechanism involves the upregulation of the pro-fibrotic cytokine TGF-β1 and the downregulation of MMPs in the liver. Consequently, therapies targeting thrombocytopenia may serve as an effective strategy for repairing advanced liver fibrosis and cirrhosis (155, 156). Recent research has indicated that splenectomy is beneficial for liver repair and regeneration. Mechanistically, splenectomy has been shown to alleviate advanced liver fibrosis by ameliorating thrombocytopenia, and splenectomy can also facilitate the recruitment of Ly-6Chi subgroup macrophages into the liver and differentiation into Ly-6Clo subgroup macrophages, thereby accelerating the regression of hepatic fibrosis induced by thioacetamide (TAA) and concanavalin A (ConA) in mice (127, 128). In addition, advanced liver fibrosis and cirrhosis are frequently associated with gut microflora dysbiosis. However, splenectomy can ameliorate liver fibrosis and cirrhosis by restoring gut barrier function and maintaining gut microbiota balance by inhibiting the TLR4/NLRP3 signaling pathway (128, 129). The above reports enhance our understanding of the mechanism by which splenectomy reverses advanced liver fibrosis and cirrhosis. Although splenectomy for treating liver cirrhosis is available in clinical practice, many patients with liver cirrhosis have contraindications for splenectomy. Therefore, it is critical to explore non-surgical alternatives for the treatment of liver fibrosis and cirrhosis.
6.8 Other treatment strategies
Except for the above therapies, some other interventions such as macrophage adoptive transfer, and drug-directed delivery approaches can also impact the progress and outcome of liver fibrosis by specifically targeting macrophages. Thomas et al. report that the adoptive transfer of anti-inflammatory macrophages can alleviate CCL4-induced liver fibrosis in mice (157). In addition, it was reported that liposomes can be routinely used as carrier materials for efficiently delivering drug molecules to pathological sites. For example, dexamethasone (Dex)-loaded liposomes have the potential to induce anti-inflammatory polarization of hepatic macrophages. In the context of experimental chronic liver damage, the administration of dex-loaded liposomes has been shown to markedly alleviate liver injury and fibrosis (158). Moreover, the phagocytosis function of macrophages can promote the inflammatory macrophages transforming into reparative macrophages in a well-characterized murine model of CCl4-induced liver fibrosis, leading to accelerated degradation of liver fibrosis (26). Overall, these studies suggest that the targeted regulation of macrophage activity and function may be a promising approach for treating liver fibrosis.
Liver macrophages play pivotal roles in the liver fibrosis during its initiation, progression, and regression. Therefore, targeting these macrophages is essential for antifibrotic therapeutic strategies. A variety of therapeutic approaches and related mechanisms on the above are summarized and listed in Table 1.
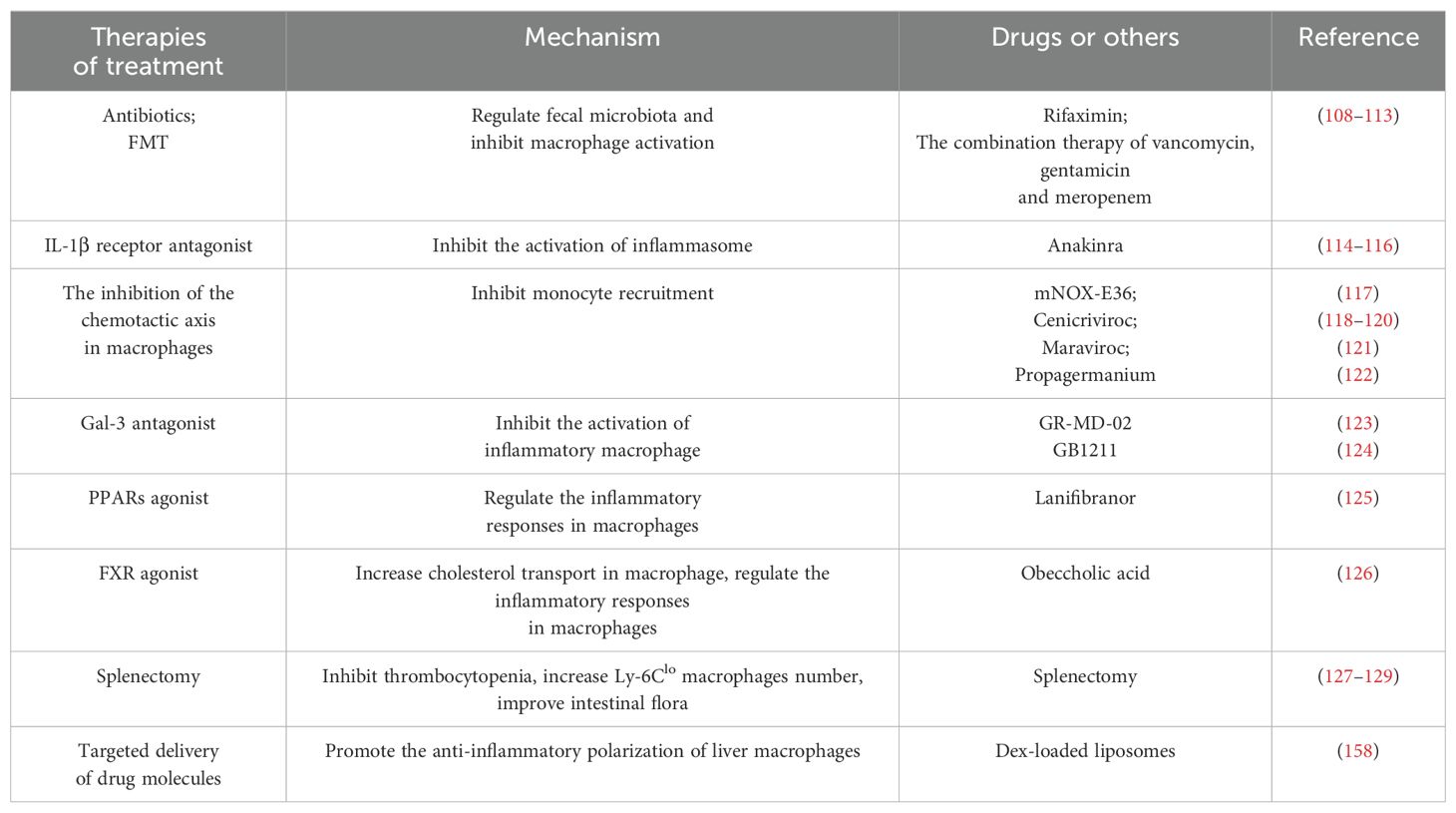
Table 1. Therapeutic strategies of anti-liver fibrosis targeting macrophages.
6 Conclusion and perspectives
Liver macrophages play an essential role in regulating hepatic homeostasis, maintaining immune tolerance, and influencing the outcome of liver diseases. Liver macrophages are a highly heterogeneous cell population that composed of KCs, MoMφs, LAMs, and LCMs. These macrophages perform diverse regulatory functions in various liver microenvironments. Under the condition of various liver injury, KCs are capable of modulating the occurrence and development of liver fibrosis through multiple pathways such as inflammasome. In addition, monocytes in the peripheral circulation can be recruited to the injured liver via chemokine receptor-ligand axis and subsequently differentiated into mature macrophages. Latest reports have highlighted the dual functions of hepatic MoMφs in the development and progression of liver fibrosis. In the progressive stage of liver fibrosis, the hepatic MoMφs are predominantly comprised of pro-inflammatory Ly-6Chi subgroup macrophages, which primarily exerts pro-inflammatory and pro-fibrotic function. In the resolution stage of liver fibrosis, the hepatic MoMφs are predominantly composed of anti-inflammatory Ly-6Clo subgroup macrophages, which mainly exhibit both anti-inflammatory and anti-fibrotic properties. Furthermore, LAMs play a crucial role in mitigating liver inflammation and facilitating the repair of liver injury. Similarly, LCMs are also involved in the regulation of liver injury. In recent years, many literatures have extensively documented the heterogeneity of macrophages in the liver under both homeostatic conditions and a variety of disease states. Liver macrophages could be a promising therapeutic target for various liver diseases. Strategies aimed at decreasing the influx of inflammatory monocyte-derived macrophages to the liver and promoting their transition to a reparative phenotype are the therapeutic focus. Several potential treatments targeting liver macrophages are currently undergoing clinical trials. Further investigation of the phenotype and functions of macrophages in different stages and developing effective therapeutic approaches is needed to reverse liver fibrosis.
Author contributions
XM: Writing – original draft, Writing – review & editing, Conceptualization. JQ: Writing – original draft, Writing – review & editing. SZ: Writing – original draft, Writing – review & editing. LT: Writing – original draft, Writing – review & editing. TM: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Jiangxi Province (20232BAB216089), the Fund Incubation Plan Project of the Second Affiliated Hospital of Nanchang University (2022YNFY12034), the National Natural Science Foundation of China (32460184).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Devarbhavi H, Asrani SK, Arab JP, Nartey YA, Pose E, Kamath PS. Global burden of liver disease: 2023 update. J Hepatol. (2023) 79:516–37. doi: 10.1016/j.jhep.2023.03.017
2. Ginès P, Castera L, Lammert F, Graupera I, Serra-Burriel M, Allen AM, et al. Population screening for liver fibrosis: toward early diagnosis and intervention for chronic liver diseases. Hepatology. (2022) 75:219–28. doi: 10.1002/hep.32163
3. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. (2023) 20:633–46. doi: 10.1038/s41575-023-00807-x
4. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
5. Dioguardi Burgio M, Grégory J, Ronot M, Sartoris R, Chatellier G, Vilgrain V. 2d-shear wave elastography: number of acquisitions can be reduced according to clinical setting. Insights Imaging. (2021) 12:145. doi: 10.1186/s13244-021-01090-7
6. Lin MH, Li HQ, Zhu L, Su HY, Peng LS, Wang CY, et al. Liver fibrosis in the natural course of chronic hepatitis B viral infection: A systematic review with meta-analysis. Dig Dis Sci. (2022) 67:2608–26. doi: 10.1007/s10620-021-07009-y
7. Simon TG, Roelstraete B, Khalili H, Hagström H, Ludvigsson JF. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: results from a nationwide cohort. Gut. (2021) 70:1375–82. doi: 10.1136/gutjnl-2020-322786
8. Pouwels S, Sakran N, Graham Y, Leal A, Pintar T, Yang W, et al. Non-alcoholic fatty liver disease (Nafld): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr Disord. (2022) 22:63. doi: 10.1186/s12902-022-00980-1
9. Li W, Zhou C, Fu Y, Chen T, Liu X, Zhang Z, et al. Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats. Acta Pharm Sin B. (2020) 10:693–710. doi: 10.1016/j.apsb.2019.07.003
10. Dhar D, Baglieri J, Kisseleva T, Brenner DA. Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med (Maywood). (2020) 245:96–108. doi: 10.1177/1535370219898141
11. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. (2021) 18:151–66. doi: 10.1038/s41575-020-00372-7
12. Fan XG, Pei SY, Zhou D, Zhou PC, Huang Y, Hu XW, et al. Melittin ameliorates inflammation in mouse acute liver failure via inhibition of pkm2-mediated warburg effect. Acta Pharmacol Sin. (2021) 42:1256–66. doi: 10.1038/s41401-020-00516-0
13. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic reprogramming in macrophage responses. biomark Res. (2021) 9:1. doi: 10.1186/s40364-020-00251-y
14. Lopez-Yrigoyen M, Yang CT, Fidanza A, Cassetta L, Taylor AH, McCahill A, et al. Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nat Commun. (2019) 10:881. doi: 10.1038/s41467-019-08705-0
15. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
16. Hara-Chikuma M, Tanaka M, Verkman AS, Yasui M. Inhibition of aquaporin-3 in macrophages by a monoclonal antibody as potential therapy for liver injury. Nat Commun. (2020) 11:5666. doi: 10.1038/s41467-020-19491-5
17. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
18. Arvanitakis K, Koletsa T, Mitroulis I, Germanidis G. Tumor-associated macrophages in hepatocellular carcinoma pathogenesis, prognosis and therapy. Cancers (Basel). (2022) 14:226. doi: 10.3390/cancers14010226
19. Barreby E, Chen P, Aouadi M. Macrophage functional diversity in nafld - more than inflammation. Nat Rev Endocrinol. (2022) 18:461–72. doi: 10.1038/s41574-022-00675-6
20. Jin GL, Liu HP, Huang YX, Zeng QQ, Chen JX, Lan XB, et al. Koumine regulates macrophage M1/M2 polarization via tspo, alleviating sepsis-associated liver injury in mice. Phytomedicine. (2022) 107:154484. doi: 10.1016/j.phymed.2022.154484
21. Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol. (2019) 10:3112. doi: 10.3389/fimmu.2019.03112
22. Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A, et al. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol. (2017) 322:74–83. doi: 10.1016/j.cellimm.2017.10.006
23. De Muynck K, Vanderborght B, De Ponti FF, Gijbels E, Van Welden S, Guilliams M, et al. Kupffer cells contested as early drivers in the pathogenesis of primary sclerosing cholangitis. Am J Pathol. (2023) 193:366–79. doi: 10.1016/j.ajpath.2022.12.008
24. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. (2022) 55:1515–29. doi: 10.1016/j.immuni.2022.08.002
25. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. (2020) 53:627–40.e5. doi: 10.1016/j.immuni.2020.06.003
26. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential ly-6c expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U.S.A. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
27. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
28. Elchaninov AV, Fatkhudinov TK, Vishnyakova PA, Lokhonina AV, Sukhikh GT. Phenotypical and functional polymorphism of liver resident macrophages. Cells. (2019) 8:1032. doi: 10.3390/cells8091032
29. Elchaninov A, Vishnyakova P, Menyailo E, Sukhikh G, Fatkhudinov T. An eye on kupffer cells: development, phenotype and the macrophage niche. Int J Mol Sci. (2022) 23:9868. doi: 10.3390/ijms23179868
30. Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Miličević N, Hoebinger C, et al. Soluble trem2 levels reflect the recruitment and expansion of trem2(+) macrophages that localize to fibrotic areas and limit nash. J Hepatol. (2022) 77:1373–85. doi: 10.1016/j.jhep.2022.06.004
31. Bianco V, Kratky D. Glycoprotein non-metastatic protein B (Gpnmb): the missing link between lysosomes and obesity. Exp Clin Endocrinol Diabetes. (2023) 131:639–45. doi: 10.1055/a-2192-0101
32. Li W, Chang N, Li L. Heterogeneity and function of kupffer cells in liver injury. Front Immunol. (2022) 13940867:940867. doi: 10.3389/fimmu.2022.940867
33. Yang Zhou Innate Immunity J. and early liver inflammation. Front Immunol. (2023) 141175147:1175147. doi: 10.3389/fimmu.2023.1175147
34. Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, Kubes P. Crig functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe. (2016) 20:99–106. doi: 10.1016/j.chom.2016.06.002
35. Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of kupffer cells regulates metabolism through the expression of cd36. Immunity. (2021) 54:2101–16.e6. doi: 10.1016/j.immuni.2021.08.006
36. Lee Y, Kim JH. The emerging roles of extracellular vesicles as intercellular messengers in liver physiology and pathology. Clin Mol Hepatol. (2022) 28:706–24. doi: 10.3350/cmh.2021.0390
37. Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. (2023) 23:563–79. doi: 10.1038/s41577-023-00848-y
38. Arsiwala T, Vogt AS, Barton AE, Manolova V, Funk F, Flühmann B, et al. Kupffer cells and blood monocytes orchestrate the clearance of iron-carbohydrate nanoparticles from serum. Int J Mol Sci. (2022) 23:2666. doi: 10.3390/ijms23052666
39. Zheng W, Yang L, Jiang S, Chen M, Li J, Liu Z, et al. Role of kupffer cells in tolerance induction after liver transplantation. Front Cell Dev Biol. (2023) 11:1179077. doi: 10.3389/fcell.2023.1179077
40. Hou X, Liu Q, Gao Y, Yong L, Xie H, Li W, et al. Mesencephalic astrocyte-derived neurotrophic factor reprograms macrophages to ameliorate acetaminophen-induced acute liver injury via P38 mapk pathway. Cell Death Dis. (2022) 13:100. doi: 10.1038/s41419-022-04555-9
41. Li W, Yang Y, Yang L, Chang N, Li L. Monocyte-derived kupffer cells dominate in the kupffer cell pool during liver injury. Cell Rep. (2023) 42:113164. doi: 10.1016/j.celrep.2023.113164
42. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. Alk1 signaling is required for the homeostasis of kupffer cells and prevention of bacterial infection. J Clin Invest. (2022) 132:e150489. doi: 10.1172/jci150489
43. Shan Z, Ju C. Hepatic macrophages in liver injury. Front Immunol. (2020) 11322:322. doi: 10.3389/fimmu.2020.00322
44. Tian X, Wang Y, Lu Y, Wang W, Du J, Chen S, et al. Conditional depletion of macrophages ameliorates cholestatic liver injury and fibrosis via lncrna-H19. Cell Death Dis. (2021) 12:646. doi: 10.1038/s41419-021-03931-1
45. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Rüsseler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology. (2012) 55:898–909. doi: 10.1002/hep.24764
46. Puengel T, Lefere S, Hundertmark J, Kohlhepp M, Penners C, Van de Velde F, et al. Combined therapy with a ccr2/ccr5 antagonist and fgf21 analogue synergizes in ameliorating steatohepatitis and fibrosis. Int J Mol Sci. (2022) 23:6696. doi: 10.3390/ijms23126696
47. Li YH, Zhang Y, Pan G, Xiang LX, Luo DC, Shao JZ. Occurrences and functions of ly6c(Hi) and ly6c(Lo) macrophages in health and disease. Front Immunol. (2022) 13:901672. doi: 10.3389/fimmu.2022.901672
48. Zhao X, Chen J, Sun H, Zhang Y, Zou D. New insights into fibrosis from the ecm degradation perspective: the macrophage-mmp-ecm interaction. Cell Biosci. (2022) 12:117. doi: 10.1186/s13578-022-00856-w
49. Xu R, Vujić N, Bianco V, Reinisch I, Kratky D, Krstic J, et al. Lipid-associated macrophages between aggravation and alleviation of metabolic diseases. Trends Endocrinol Metab. (2024) 35:981–95. doi: 10.1016/j.tem.2024.04.009
50. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in nash. Cell Rep. (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
51. Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. (2013) 18:816–30. doi: 10.1016/j.cmet.2013.11.001
52. Wang X, He Q, Zhou C, Xu Y, Liu D, Fujiwara N, et al. Prolonged hypernutrition impairs trem2-dependent efferocytosis to license chronic liver inflammation and nash development. Immunity. (2023) 56:58–77.e11. doi: 10.1016/j.immuni.2022.11.013
53. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity. (2017) 47:374–88.e6. doi: 10.1016/j.immuni.2017.07.018
54. Kim ES, Lee JM, Kwak JY, Lee HW, Lee IJ, Kim HM. Multicolor two-photon microscopy imaging of lipid droplets and liver capsule macrophages in vivo. Anal Chem. (2024) 96:8467–73. doi: 10.1021/acs.analchem.4c00228
55. Ożańska A, Szymczak D, Rybka J. Pattern of human monocyte subpopulations in health and disease. Scand J Immunol. (2020) 92:e12883. doi: 10.1111/sji.12883
56. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
57. Brempelis KJ, Crispe IN. Infiltrating monocytes in liver injury and repair. Clin Transl Immunol. (2016) 5:e113. doi: 10.1038/cti.2016.62
58. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
59. Musrati MA, De Baetselier P, Movahedi K, Van Ginderachter JA. Ontogeny, functions and reprogramming of kupffer cells upon infectious disease. Front Immunol. (2023) 14:1238452. doi: 10.3389/fimmu.2023.1238452
60. Kermanizadeh A, Berthing T, Guzniczak E, Wheeldon M, Whyte G, Vogel U, et al. Assessment of nanomaterial-induced hepatotoxicity using a 3d human primary multi-cellular microtissue exposed repeatedly over 21 Days - the suitability of the in vitro system as an in vivo surrogate. Part Fibre Toxicol. (2019) 16:42. doi: 10.1186/s12989-019-0326-0
61. Zhang WJ, Fang ZM, Liu WQ. Nlrp3 inflammasome activation from kupffer cells is involved in liver fibrosis of schistosoma japonicum-infected mice via nf-Kb. Parasit Vectors. (2019) 12:29. doi: 10.1186/s13071-018-3223-8
62. Zhang L, Bansal MB. Role of kupffer cells in driving hepatic inflammation and fibrosis in hiv infection. Front Immunol. (2020) 11:1086. doi: 10.3389/fimmu.2020.01086
63. Feng Y, Dong H, Sun B, Hu Y, Yang Y, Jia Y, et al. Mettl3/mettl14 transactivation and M(6)a-dependent tgf-B1 translation in activated kupffer cells. Cell Mol Gastroenterol Hepatol. (2021) 12:839–56. doi: 10.1016/j.jcmgh.2021.05.007
64. Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. (2020) 9:875. doi: 10.3390/cells9040875
65. Maia J, Otake AH, Poças J, Carvalho AS, Beck HC, Magalhães A, et al. Transcriptome reprogramming of cd11b(+) bone marrow cells by pancreatic cancer extracellular vesicles. Front Cell Dev Biol. (2020) 8:592518. doi: 10.3389/fcell.2020.592518
66. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
67. Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X, et al. Cxcl6-egfr-induced kupffer cells secrete tgf-B1 promoting hepatic stellate cell activation via the smad2/brd4/C-myc/ezh2 pathway in liver fibrosis. J Cell Mol Med. (2018) 22:5050–61. doi: 10.1111/jcmm.13787
68. Liu J, Kong D, Qiu J, Xie Y, Lu Z, Zhou C, et al. Praziquantel ameliorates ccl(4) -induced liver fibrosis in mice by inhibiting tgf-B/smad signalling via up-regulating smad7 in hepatic stellate cells. Br J Pharmacol. (2019) 176:4666–80. doi: 10.1111/bph.14831
69. Bazenet CE, Gelderloos JA, Kazlauskas A. Phosphorylation of tyrosine 720 in the platelet-derived growth factor alpha receptor is required for binding of grb2 and shp-2 but not for activation of ras or cell proliferation. Mol Cell Biol. (1996) 16:6926–36. doi: 10.1128/mcb.16.12.6926
70. Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. (2020) 73:1144–54. doi: 10.1016/j.jhep.2020.04.044
71. Zehender A, Huang J, Györfi AH, Matei AE, Trinh-Minh T, Xu X, et al. The tyrosine phosphatase shp2 controls tgfβ-induced stat3 signaling to regulate fibroblast activation and fibrosis. Nat Commun. (2018) 9:3259. doi: 10.1038/s41467-018-05768-3
72. Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N, et al. High mobility group box-1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice. Hepatology. (2018) 68:2380–404. doi: 10.1002/hep.30093
73. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. (2013) 58:1461–73. doi: 10.1002/hep.26429
74. Boltjes A, van Montfoort N, Biesta PJ, Op den Brouw ML, Kwekkeboom J, van der Laan LJ, et al. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J Infect Dis. (2015) 211:1268–78. doi: 10.1093/infdis/jiu599
75. Movita D, van de Garde MD, Biesta P, Kreefft K, Haagmans B, Zuniga E, et al. Inflammatory Monocytes Recruited to the Liver within 24 Hours after Virus-Induced Inflammation Resemble Kupffer Cells but Are Functionally Distinct. J Virol. (2015) 89:4809–17. doi: 10.1128/jvi.03733-14
76. Gu X, Chu Q, Ma X, Wang J, Chen C, Guan J, et al. New insights into inkt cells and their roles in liver diseases. Front Immunol. (2022) 13:1035950. doi: 10.3389/fimmu.2022.1035950
77. Yaghoubi A, Azarpira N, Karbalay-Doust S, Daneshi S, Vojdani Z, Talaei-Khozani T. Prednisolone and mesenchymal stem cell preloading protect liver cell migration and mitigate extracellular matrix modification in transplanted decellularized rat liver. Stem Cell Res Ther. (2022) 13:36. doi: 10.1186/s13287-022-02711-8
78. Liu C, Zhu Y, Lu Z, Guo W, Tumen B, He Y, et al. Cadmium induces acute liver injury by inhibiting nrf2 and the role of nf-Kb, nlrp3, and mapks signaling pathway. Int J Environ Res Public Health. (2019) 17:138. doi: 10.3390/ijerph17010138
79. Gao CC, Bai J, Han H, Qin HY. The versatility of macrophage heterogeneity in liver fibrosis. Front Immunol. (2022) 13:968879. doi: 10.3389/fimmu.2022.968879
80. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
81. Wang Z, Du K, Jin N, Tang B, Zhang W. Macrophage in liver fibrosis: identities and mechanisms. Int Immunopharmacol. (2023) 120:110357. doi: 10.1016/j.intimp.2023.110357
82. Zwicker C, Bujko A, Scott CL. Hepatic macrophage responses in inflammation, a function of plasticity, heterogeneity or both? Front Immunol. (2021) 12:690813. doi: 10.3389/fimmu.2021.690813
83. Sun J, Zhao P, Shi Y, Li Y. Recent insight into the role of macrophage in alcohol-associated liver disease: A mini-review. Front Cell Dev Biol. (2023) 111:1292016. doi: 10.3389/fcell.2023.1292016
84. Vonderlin J, Chavakis T, Sieweke M, Tacke F. The multifaceted roles of macrophages in nafld pathogenesis. Cell Mol Gastroenterol Hepatol. (2023) 15:1311–24. doi: 10.1016/j.jcmgh.2023.03.002
85. Laban H, Weigert A, Zink J, Elgheznawy A, Schürmann C, Günther L, et al. Vasp regulates leukocyte infiltration, polarization, and vascular repair after ischemia. J Cell Biol. (2018) 217:1503–19. doi: 10.1083/jcb.201702048
86. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. (2005) 115:56–65. doi: 10.1172/jci22675
87. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (Monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing ly-6c(+) macrophage infiltration in mice. Hepatology. (2014) 59:1060–72. doi: 10.1002/hep.26783
88. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. (2014) 96:657–65. doi: 10.1189/jlb.6A0114-004RR
89. Campana L, Starkey Lewis PJ, Pellicoro A, Aucott RL, Man J, O’Duibhir E, et al. The stat3-il-10-il-6 pathway is a novel regulator of macrophage efferocytosis and phenotypic conversion in sterile liver injury. J Immunol. (2018) 200:1169–87. doi: 10.4049/jimmunol.1701247
90. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ros to orchestrate liver repair. Nat Commun. (2019) 10:1076. doi: 10.1038/s41467-019-09046-8
91. Graubardt N, Vugman M, Mouhadeb O, Caliari G, Pasmanik-Chor M, Reuveni D, et al. Ly6c(Hi) monocytes and their macrophage descendants regulate neutrophil function and clearance in acetaminophen-induced liver injury. Front Immunol. (2017) 8:626. doi: 10.3389/fimmu.2017.00626
92. Girgis NM, Gundra UM, Ward LN, Cabrera M, Frevert U, Loke P. Ly6c(High) monocytes become alternatively activated macrophages in schistosome granulomas with help from cd4+ Cells. PloS Pathog. (2014) 10:e1004080. doi: 10.1371/journal.ppat.1004080
93. Li YH, Shen S, Shao T, Jin MT, Fan DD, Lin AF, et al. Mesenchymal stem cells attenuate liver fibrosis by targeting ly6c(Hi/lo) macrophages through activating the cytokine-paracrine and apoptotic pathways. Cell Death Discovery. (2021) 7:239. doi: 10.1038/s41420-021-00584-z
94. Mabire M, Hegde P, Hammoutene A, Wan J, Caër C, Sayegh RA, et al. Mait cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat Commun. (2023) 14:1830. doi: 10.1038/s41467-023-37453-5
95. Liu L, Wang P, Wang YS, Zhang YN, Li C, Yang ZY, et al. Mir-130a-3p alleviates liver fibrosis by suppressing hscs activation and skewing macrophage to ly6c(Lo) phenotype. Front Immunol. (2021) 12:696069. doi: 10.3389/fimmu.2021.696069
96. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of ccr2+ Monocytes at a site of sterile injury. J Exp Med. (2015) 212:447–56. doi: 10.1084/jem.20141539
97. Starkey Lewis P, Campana L, Aleksieva N, Cartwright JA, Mackinnon A, O’Duibhir E, et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J Hepatol. (2020) 73:349–60. doi: 10.1016/j.jhep.2020.02.031
98. Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. (2015) 63:1038–9. doi: 10.1016/j.jhep.2015.03.039
99. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. (2015) 61:1066–79. doi: 10.1002/hep.27332
100. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
101. Colonna M. The biology of trem receptors. Nat Rev Immunol. (2023) 23:580–94. doi: 10.1038/s41577-023-00837-1
102. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. Trem2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest. (2021) 131:e135197. doi: 10.1172/jci135197
103. Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, et al. Non-parenchymal trem-2 protects the liver from immune-mediated hepatocellular damage. Gut. (2019) 68:533–46. doi: 10.1136/gutjnl-2017-314107
104. Ganguly S, Rosenthal SB, Ishizuka K, Troutman TD, Rohm TV, Khader N, et al. Lipid-associated macrophages’ Promotion of fibrosis resolution during mash regression requires trem2. Proc Natl Acad Sci U.S.A. (2024) 121:e2405746121. doi: 10.1073/pnas.2405746121
105. Bai YM, Yang F, Luo P, Xie LL, Chen JH, Guan YD, et al. Single-cell transcriptomic dissection of the cellular and molecular events underlying the triclosan-induced liver fibrosis in mice. Mil Med Res. (2023) 10:7. doi: 10.1186/s40779-023-00441-3
106. Villard A, Boursier J, Andriantsitohaina R. Bacterial and eukaryotic extracellular vesicles and nonalcoholic fatty liver disease: new players in the gut-liver axis? Am J Physiol Gastrointest Liver Physiol. (2021) 320:G485–g95. doi: 10.1152/ajpgi.00362.2020
107. Maslennikov R, Poluektova E, Zolnikova O, Sedova A, Kurbatova A, Shulpekova Y, et al. Gut microbiota and bacterial translocation in the pathogenesis of liver fibrosis. Int J Mol Sci. (2023) 24:16502. doi: 10.3390/ijms242216502
108. Gandhi CR. Pro- and anti-fibrogenic functions of gram-negative bacterial lipopolysaccharide in the liver. Front Med (Lausanne). (2020) 7130:130. doi: 10.3389/fmed.2020.00130
109. De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. (2014) 59:1738–49. doi: 10.1002/hep.26695
110. Ferrere G, Wrzosek L, Cailleux F, Turpin W, Puchois V, Spatz M, et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J Hepatol. (2017) 66:806–15. doi: 10.1016/j.jhep.2016.11.008
111. Madsen BS, Trebicka J, Thiele M, Israelsen M, Arumugan M, Havelund T, et al. Antifibrotic and molecular aspects of rifaximin in alcoholic liver disease: study protocol for a randomized controlled trial. Trials. (2018) 19:143. doi: 10.1186/s13063-018-2523-9
112. Ohashi K, Pimienta M, Seki E. Alcoholic liver disease: A current molecular and clinical perspective. Liver Res. (2018) 2:161–72. doi: 10.1016/j.livres.2018.11.002
113. Liu Y, Chen K, Li F, Gu Z, Liu Q, He L, et al. Probiotic lactobacillus rhamnosus gg prevents liver fibrosis through inhibiting hepatic bile acid synthesis and enhancing bile acid excretion in mice. Hepatology. (2020) 71:2050–66. doi: 10.1002/hep.30975
114. Colantuoni M, Jofra Hernandez R, Pettinato E, Basso-Ricci L, Magnani L, Andolfi G, et al. Constitutive il-1ra production by modified immune cells protects against il-1-mediated inflammatory disorders. Sci Transl Med. (2023) 15:eade3856. doi: 10.1126/scitranslmed.ade3856
115. Puccetti M, Pariano M, Stincardini C, Wojtylo P, Schoubben A, Nunzi E, et al. Pulmonary drug delivery technology enables anakinra repurposing in cystic fibrosis. J Control Release. (2023) 353:1023–36. doi: 10.1016/j.jconrel.2022.11.043
116. Tu W, Gawrieh S, Dasarathy S, Mitchell MC, Simonetto DA, Patidar KR, et al. Design of a multicenter randomized clinical trial for treatment of alcohol-associated hepatitis. Contemp Clin Trials Commun. (2023) 32:101074. doi: 10.1016/j.conctc.2023.101074
117. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine ccl2 (Mcp-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
118. Kwiatkowski K, Pawlik K, Ciapała K, Piotrowska A, Makuch W, Mika J. Bidirectional action of cenicriviroc, a ccr2/ccr5 antagonist, results in alleviation of pain-related behaviors and potentiation of opioid analgesia in rats with peripheral neuropathy. Front Immunol. (2020) 11:615327. doi: 10.3389/fimmu.2020.615327
119. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
120. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b centaur study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
121. Pérez-Martínez L, Pérez-Matute P, Aguilera-Lizarraga J, Rubio-Mediavilla S, Narro J, Recio E, et al. Maraviroc, a ccr5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (Nafld). J Antimicrob Chemother. (2014) 69:1903–10. doi: 10.1093/jac/dku071
122. Mulder P, van den Hoek AM, Kleemann R. The ccr2 inhibitor propagermanium attenuates diet-induced insulin resistance, adipose tissue inflammation and non-alcoholic steatohepatitis. PloS One. (2017) 12:e0169740. doi: 10.1371/journal.pone.0169740
123. Al Attar A, Antaramian A, Noureddin M. Review of galectin-3 inhibitors in the treatment of nonalcoholic steatohepatitis. Expert Rev Clin Pharmacol. (2021) 14:457–64. doi: 10.1080/17512433.2021.1894127
124. Barde PJ, Viswanadha S, Veeraraghavan S, Vakkalanka SV, Nair A. A first-in-human study to evaluate the safety, tolerability and pharmacokinetics of rp3128, an oral calcium release-activated calcium (Crac) channel modulator in healthy volunteers. J Clin Pharm Ther. (2021) 46:677–87. doi: 10.1111/jcpt.13322
125. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-ppar agonists on experimental steatohepatitis and hepatic macrophages(☆). J Hepatol. (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
126. Fu J, Zhang P, Sun Z, Lu G, Cao Q, Chen Y, et al. A combined nanotherapeutic approach targeting farnesoid X receptor, ferroptosis, and fibrosis for nonalcoholic steatohepatitis treatment. Acta Pharm Sin B. (2024) 14:2228–46. doi: 10.1016/j.apsb.2024.02.017
127. Yada A, Iimuro Y, Uyama N, Uda Y, Okada T, Fujimoto J. Splenectomy attenuates murine liver fibrosis with hypersplenism stimulating hepatic accumulation of ly-6c(Lo) macrophages. J Hepatol. (2015) 63:905–16. doi: 10.1016/j.jhep.2015.05.010
128. Elchaninov A, Vishnyakova P, Sukhikh G, Fatkhudinov Spleen T. Reparative regeneration and influence on liver. Life (Basel). (2022) 12:626. doi: 10.3390/life12050626
129. Jin Y, Shi M, Feng J, Zhang Z, Zhao B, Li Q, et al. Splenectomy ameliorates liver cirrhosis by restoring the gut microbiota balance. Cell Mol Life Sci. (2024) 81:32. doi: 10.1007/s00018-023-05055-5
130. Brahadeeswaran S, Dasgupta T, Manickam V, Saraswathi V, Tamizhselvi R. Nlrp3: A new therapeutic target in alcoholic liver disease. Front Immunol. (2023) 14:1215333. doi: 10.3389/fimmu.2023.1215333
131. Charan HV, Dwivedi DK, Khan S, Jena G. Mechanisms of nlrp3 inflammasome-mediated hepatic stellate cell activation: therapeutic potential for liver fibrosis. Genes Dis. (2023) 10:480–94. doi: 10.1016/j.gendis.2021.12.006
132. Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev. (2018) 281:197–232. doi: 10.1111/imr.12606
133. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. (2021) 74:156–67. doi: 10.1016/j.jhep.2020.07.041
134. Lee CM, Peng HH, Yang P, Liou JT, Liao CC, Day YJ. C-C chemokine ligand-5 is critical for facilitating macrophage infiltration in the early phase of liver ischemia/reperfusion injury. Sci Rep. (2017) 7:3698. doi: 10.1038/s41598-017-03956-7
135. Oberthür D, Achenbach J, Gabdulkhakov A, Buchner K, Maasch C, Falke S, et al. Crystal structure of a mirror-image L-rna aptamer (Spiegelmer) in complex with the natural L-protein target ccl2. Nat Commun. (2015) 6:6923. doi: 10.1038/ncomms7923
136. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of ccr2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology. (2019) 69:1105–21. doi: 10.1002/hep.30249
137. Guo Y, Zhao C, Dai W, Wang B, Lai E, Xiao Y, et al. C-C motif chemokine receptor 2 inhibition reduces liver fibrosis by restoring the immune cell landscape. Int J Biol Sci. (2023) 19:2572–87. doi: 10.7150/ijbs.83530
138. Liu SS, Liu C, Lv XX, Cui B, Yan J, Li YX, et al. The chemokine ccl1 triggers an amfr-spry1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity. (2021) 54:2042–56.e8. doi: 10.1016/j.immuni.2021.06.008
139. Lee Y, Lee J, Park M, Seo A, Kim KH, Kim S, et al. Inflammatory chemokine (C-C motif) ligand 8 inhibition ameliorates peritoneal fibrosis. FASEB J. (2023) 37:e22632. doi: 10.1096/fj.202200784R
140. Merino-Merino A, Gonzalez-Bernal J, Fernandez-Zoppino D, Saez-Maleta R, Perez-Rivera JA. The role of galectin-3 and st2 in cardiology: A short review. Biomolecules. (2021) 11:1167. doi: 10.3390/biom11081167
141. Díaz-Alvarez L, Ortega E. The many roles of galectin-3, a multifaceted molecule, in innate immune responses against pathogens. Mediators Inflammation. (2017) 2017:9247574. doi: 10.1155/2017/9247574
142. Mackinnon AC, Tonev D, Jacoby B, Pinzani M, Slack RJ. Galectin-3: therapeutic targeting in liver disease. Expert Opin Ther Targets. (2023) 27:779–91. doi: 10.1080/14728222.2023.2258280
143. Ezhilarasan D. Unraveling the pathophysiologic role of galectin-3 in chronically injured liver. J Cell Physiol. (2023) 238:673–86. doi: 10.1002/jcp.30956
144. Jiang JX, Chen X, Hsu DK, Baghy K, Serizawa N, Scott F, et al. Galectin-3 modulates phagocytosis-induced stellate cell activation and liver fibrosis in vivo. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G439–46. doi: 10.1152/ajpgi.00257.2011
145. Qiu YY, Zhang J, Zeng FY, Zhu YZ. Roles of the peroxisome proliferator-activated receptors (Ppars) in the pathogenesis of nonalcoholic fatty liver disease (Nafld). Pharmacol Res. (2023) 192:106786. doi: 10.1016/j.phrs.2023.106786
146. Francque S, Szabo G, Abdelmalek MF, Byrne CD, Cusi K, Dufour JF, et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat Rev Gastroenterol Hepatol. (2021) 18:24–39. doi: 10.1038/s41575-020-00366-5
147. Puengel T, Liu H, Guillot A, Heymann F, Tacke F, Peiseler M. Nuclear receptors linking metabolism, inflammation, and fibrosis in nonalcoholic fatty liver disease. Int J Mol Sci. (2022) 23:2668. doi: 10.3390/ijms23052668
148. Zarei M, Barroso E, Palomer X, Dai J, Rada P, Quesada-López T, et al. Hepatic regulation of vldl receptor by pparβ/δ and fgf21 modulates non-alcoholic fatty liver disease. Mol Metab. (2018) 8:117–31. doi: 10.1016/j.molmet.2017.12.008
149. Lan Q, Ren Z, Chen Y, Cui G, Choi IC, Ung COL, et al. Hepatoprotective effect of qushihuayu formula on non-alcoholic steatohepatitis induced by mcd diet in rat. Chin Med. (2021) 16:27. doi: 10.1186/s13020-021-00434-1
150. Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, gft505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. (2013) 58:1941–52. doi: 10.1002/hep.26461
151. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-ppar agonist lanifibranor in nash. N Engl J Med. (2021) 385:1547–58. doi: 10.1056/NEJMoa2036205
152. Murray A, Banota T, Guo GL, Smith LC, Meshanni JA, Lee J, et al. Farnesoid X receptor regulates lung macrophage activation and injury following nitrogen mustard exposure. Toxicol Appl Pharmacol. (2022) 454:116208. doi: 10.1016/j.taap.2022.116208
153. Li S, Ghoshal S, Sojoodi M, Arora G, Masia R, Erstad DJ, et al. The farnesoid X receptor agonist edp-305 reduces interstitial renal fibrosis in a mouse model of unilateral ureteral obstruction. FASEB J. (2019) 33:7103–12. doi: 10.1096/fj.201801699R
154. Shi T, Malik A, Yang Vom Hofe A, Matuschek L, Mullen M, Lages CS, et al. Farnesoid X receptor antagonizes macrophage-dependent licensing of effector T lymphocytes and progression of sclerosing cholangitis. Sci Transl Med. (2022) 14:eabi4354. doi: 10.1126/scitranslmed.abi4354
155. Kodama T, Takehara T, Hikita H, Shimizu S, Li W, Miyagi T, et al. Thrombocytopenia exacerbates cholestasis-induced liver fibrosis in mice. Gastroenterology. (2010) 138:2487–98, 98.e1-7. doi: 10.1053/j.gastro.2010.02.054
156. Watanabe M, Murata S, Hashimoto I, Nakano Y, Ikeda O, Aoyagi Y, et al. Platelets contribute to the reduction of liver fibrosis in mice. J Gastroenterol Hepatol. (2009) 24:78–89. doi: 10.1111/j.1440-1746.2008.05497.x
157. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. (2011) 53:2003–15. doi: 10.1002/hep.24315
Keywords: liver fibrosis, ECM, macrophage, heterogeneity, therapeutic strategies
Citation: Ma X, Qiu J, Zou S, Tan L and Miao T (2024) The role of macrophages in liver fibrosis: composition, heterogeneity, and therapeutic strategies. Front. Immunol. 15:1494250. doi: 10.3389/fimmu.2024.1494250
Received: 10 September 2024; Accepted: 31 October 2024;
Published: 20 November 2024.
Edited by:
Paula Vieira Teixeira Vidigal, Federal University of Minas Gerais, BrazilReviewed by:
Sujit K. Mohanty, United States Department of Agriculture (USDA), United StatesChao Yang, The First Affiliated Hospital of Xi’an Jiaotong University, China
Andrey Elchaninov, Avtsyn Research Institute of Human Morphology of FSBI “Petrovsky National Research Centre of Surgery”, Russia
Copyright © 2024 Ma, Qiu, Zou, Tan and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingting Miao, VGluZ3RpbmdtaWFvODg4QDE2My5jb20=
†These authors have contributed equally to this work