 Wu-xiao Wei
Wu-xiao Wei Ming-li Chen*†
Ming-li Chen*†- Guangxi University of Science and Technology First Affiliated Hospital, Liuzhou, China
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is a rare autoimmune disease, which is characterized by the immune system attacking astrocytes in the central nervous system, resulting in inflammation and damage to the nervous system. We reported a 41-year-old female patient with only drowsiness for 3 months, who was, otherwise, healthy with no other signs of meningoencephalitis or myelitis. There were no obvious abnormalities in her neurological and ophthalmic tests. Brain magnetic resonance imaging (MRI) plain scan + enhancement with the gadolinium contrast agent revealed patchy hypointensity on T1-weighted imaging, hyperintensity on T2-weighted imaging, hyperintensity on T2-weighted fluid-attenuated inversion recovery in the left basal ganglia, corona radiata, and local septum pellucida, with no enhancement in the enhanced lesions. Cerebrospinal fluid (CSF) revealed white blood cell count of 5.00 × 106/L, CSF protein of 828.53 mg/L, and glucose of 2.83 mmol/L. Aquaporin-4 (AQP4) antibody, N-methyl-D-aspartate receptor (NMDAR) antibody and GFAP antibody were all positive, whereas the remaining autoimmune encephalitis antibody tests were negative. Oncology screening [including head, chest, and whole-abdomen (involving the pelvic cavity) CT and tumor markers] did not reveal any obvious evidence of tumor presence. The patient received systemic treatment with high-dose intravenous injection of steroids combined with immunosuppressive agents, and the clinical and imaging features of the patients improved. To the best of our knowledge, reports on overlapping positivity of AQP4 antibody and NMDAR antibody in patients with GFAP astrocytopathy were still very rare. We hope to supplement the existing literature on this topic, review the relevant literature, and strive to increase the understanding toward GFAP astrocytopathy with overlapping autoimmune syndrome so as to enable early diagnosis and early treatment and to improve the clinical outcome of patients.
Introduction
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is a novel inflammatory autoimmune disease of the central nervous system (1, 3). The clinical manifestation of GFAP astrocytopathy is complex; moreover, it can coexist with a variety of antibodies, including aquaporin-4 (AQP4) antibody and N-methyl-D-aspartate receptor (NMDAR) antibody and is, therefore, called overlap syndrome (2). In this study, we presented a case of a patient with GFAP astrocytopathy with overlapping autoimmune syndrome, who was admitted and treated in our hospital. The patient received a long course of steroid combined with immunosuppressive agent treatment and achieved remarkable therapeutic effect.
In this case study, emphasizing the rarity of GFAP astrocytopathy combined with overlap syndromes aims to enhance clinical awareness among healthcare professionals. By providing detailed descriptions of the case, etiology, clinical manifestations, and diagnostic methods, it helps improve physicians’ ability to recognize such rare conditions, facilitating timely and accurate diagnosis and treatment. Such reports play a crucial role in expanding physicians’ knowledge base and increasing alertness to rare diseases.
Case report
A 41-year-old female patient residing in Liuzhou City, Guangxi Zhuang Autonomous Region, visited our hospital and was admitted as a result of “drowsiness for more than 3 months.” Her family complained that the patient experienced excessive daytime drowsiness without an obvious cause 3 months ago, with daytime sleep lasting more than 12 h. The symptoms persist without significant improvement. She denied a history of respiratory and digestive tract infections in the past 3 months; a history of chronic diseases such as “heart disease, hypertension, and kidney disease”; a history of infectious diseases including “tuberculosis, typhoid fever, dysentery, and viral hepatitis”; and a history of food and drug allergies. There were no obvious abnormalities in her neurological examinations, including time and place orientation, language assessment, cranial nerves, ophthalmic testing, movement, sensation, coordination, and gait. At the same time, her physical examination was normal.
The blood test showed a white blood cell count (WBC) of 15.16 × 109/L. Cerebrospinal fluid (CSF) analysis suggested a WBC of 5.00 × 106/L, CSF protein of 828.53 mg/L, and glucose of 2.83 mmol/L. CSF samples were sent to Guangxi Golden Field Medical Laboratory for testing of the twenty CSF autoimmune encephalitis antibodies through the cell-based assay. Its principle is to transfect the target antigen gene into mammalian cells to specifically express the corresponding antigen in mammalian cells. In addition, the green fluorescent protein was co-expressed during transfection as an internal reference for detection. Thereafter, the transfected cells were fixed onto the 96-well microplates and prepared into the antigen sheets. The specific antibodies in human serum, plasma, or CSF samples were detected semi-quantitatively by an indirect immunofluorescence method. The results were positive for anti-AQP4 antibody (+; 1:10), positive for anti–glutamate receptor (NMDA-type) Immunoglobulin G (IgG) antibody (+; 1:30), and positive for anti-GFAP antibody (+; 1:32) (Figure 1). The remaining autoimmune encephalitis antibodies including anti–glutamate receptor (AMPA1) antibody Immunoglobulin G (IgG), anti–glutamate receptor (AMPA2-type) antibody IgG, anti–leucine-rich glioma inactivated protein 1 (LGI 1) antibody IgG, anti–contact protein-associated protein 2 (CASPR2) antibody IgG, anti–gamma-aminobutyric acid–type B receptor (GABAB) antibody IgG, anti–Iglon family protein 5 (IgLON5) antibody IgG, anti–dipeptide similar protein 6 (DPPX) antibody IgG, anti–glycine receptor 1 (GlyR1) antibody IgG, anti–dopamine receptor 2 (DRD2) antibody IgG, anti–glutamate decarboxylase 2 (GAD65) antibody IgG, anti–metabolic glutamate receptor 5 (mGluR5) antibody IgG, anti–metabolic glutamate receptor 1 (mGluR1) antibody IgG, anti–synaptic protein-3α (Neurexin-3α) antibody IgG, anti–gamma-aminobutyric acid–type A receptor (GABAA) antibody, anti–Kelch-like protein 11 (KLHL11) antibody, anti–ganglionic acetylcholine receptor (ganglionic AChR) antibody, and anti–myelin oligodendrocyte glycoprotein antibody (MOG) were negative. Infectious disease screening by enzyme-linked immunosorbent assay suggested no abnormalities in hepatitis B surface antigen assay, hepatitis C antibody assay, human immunodeficiency virus antibody assay, treponema pallidum specific antibody assay, and syphilis non-specific antibody assay. Other laboratory tests, including antinuclear antibody assay, vasculitis test, folic acid, and vitamin B12, were normal. All other laboratory parameters were within normal limits.
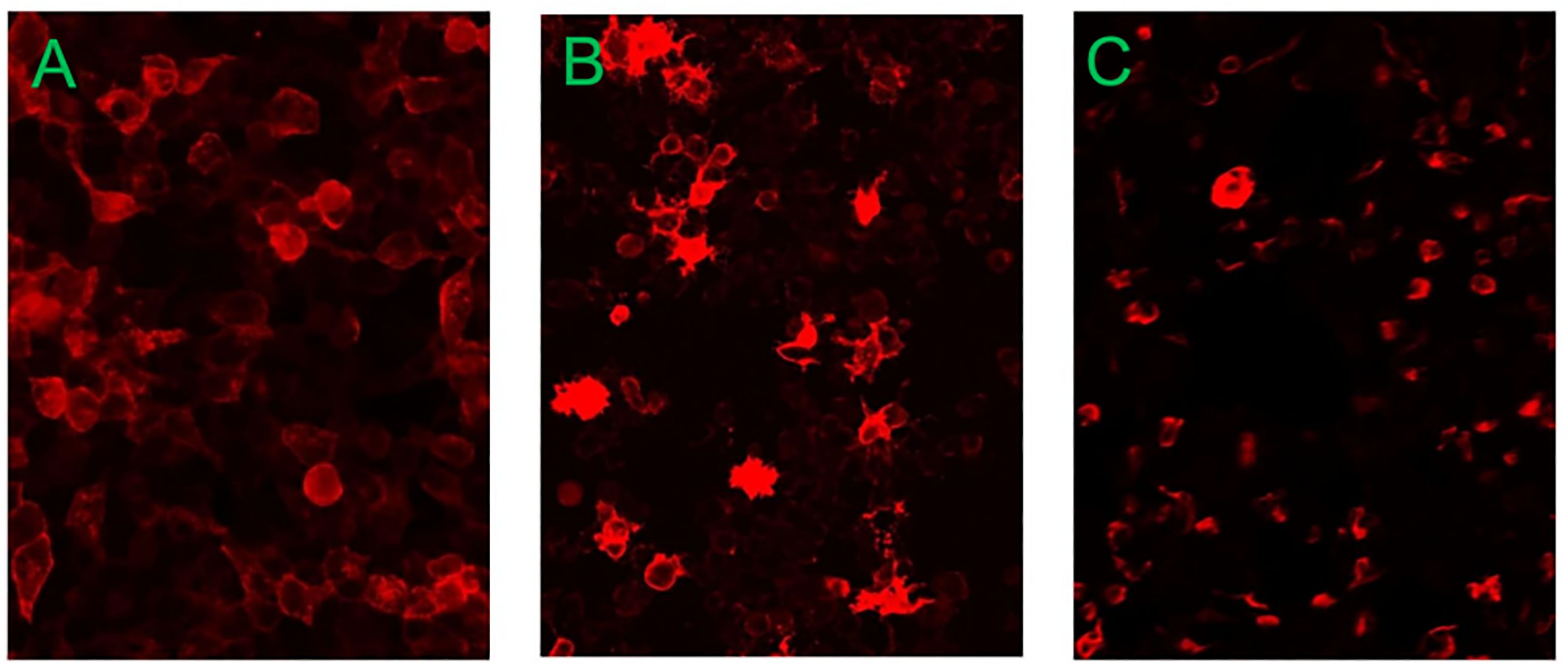
Figure 1. The results of CSF autoimmune encephalitis antibodies detected in Guangxi Golden Field Medical Laboratory were positive for anti–aquaporin-4 (AQP4) antibody (+; 1:10), positive for anti–glutamate receptor (NMDA-type) IgG antibody (+; 1:30), and positive for anti-GFAP antibody (+; 1:32). (A) Anti–aquaporin-4 antibody. (B) Anti–NMDA receptor antibody. (C) Anti-GFAP antibody.
The electroencephalogram (EEG) imaging indicates that, during awake and quiet closed-eye state, bilateral occipital regions exhibit predominant alpha rhythm activity at 9.0–10.0 Hz and 20–50 μV, with a basic symmetry between the left and right sides. Each lead shows minimal low-amplitude beta and theta waves, with widespread increase in alpha activity (Figure 2).
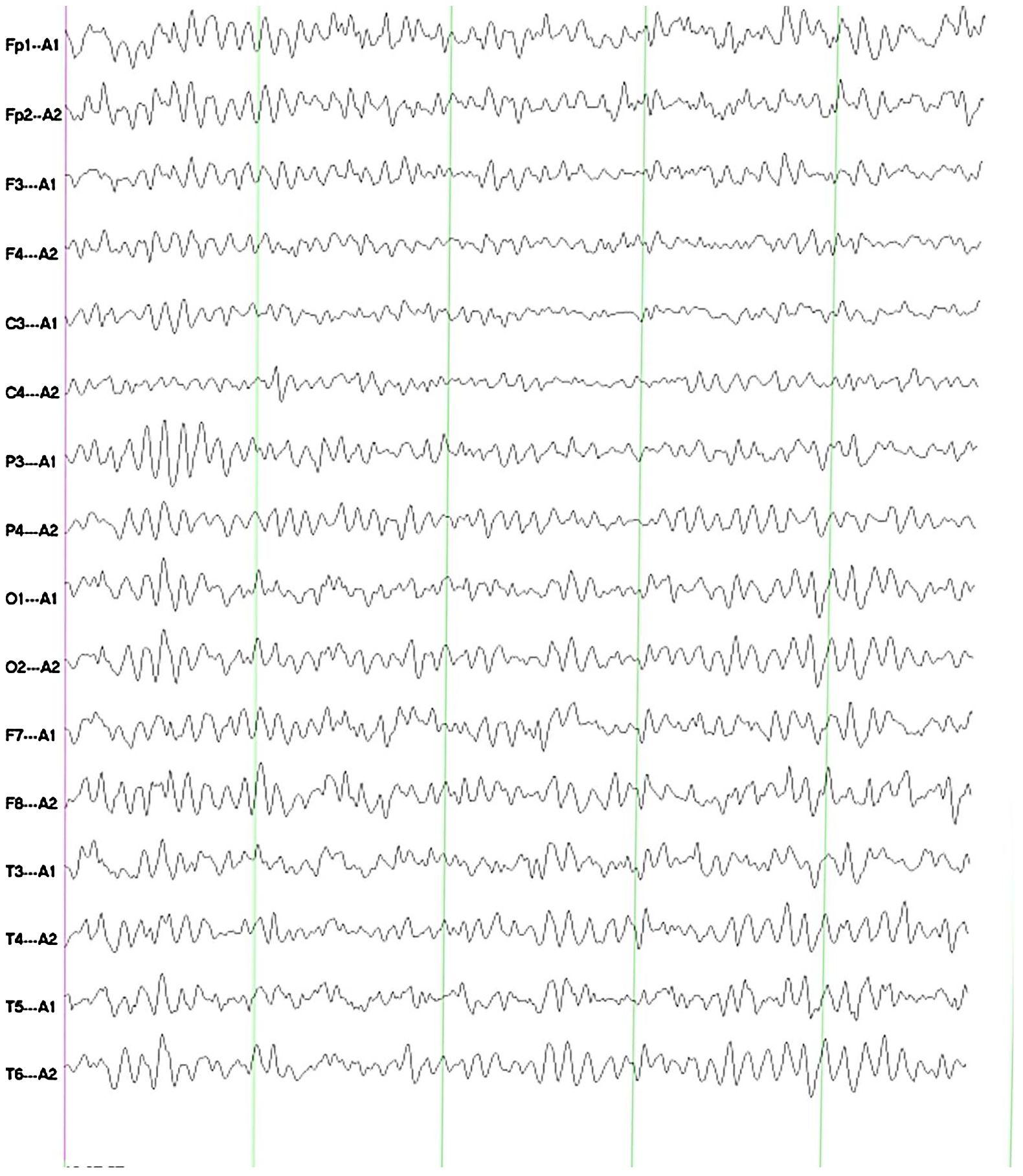
Figure 2. The EEG imaging indicates that, during awake and quiet closed-eye state, bilateral occipital regions exhibit predominant alpha rhythm activity at 9.0–10.0 Hz and 20–50 μV, with a basic symmetry between the left and right sides. Each lead shows minimal low-amplitude beta and theta waves, with widespread increase in alpha activity.
Brain magnetic resonance imaging (MRI) plain scan + enhancement with the gadolinium contrast agent revealed patchy hypointensity on T1WI, hyperintensity on T2WI, and hyperintensity on T2-FLAIR in the left basal ganglia, corona radiata, and local septum pellucida, with no enhancement in the enhanced lesions (Figures 3A–D). Spinal MRI showed no abnormality. A follow-up MRI was performed 6 months later, which demonstrated the nearly total regression of the previous findings (Figures 3E–H).
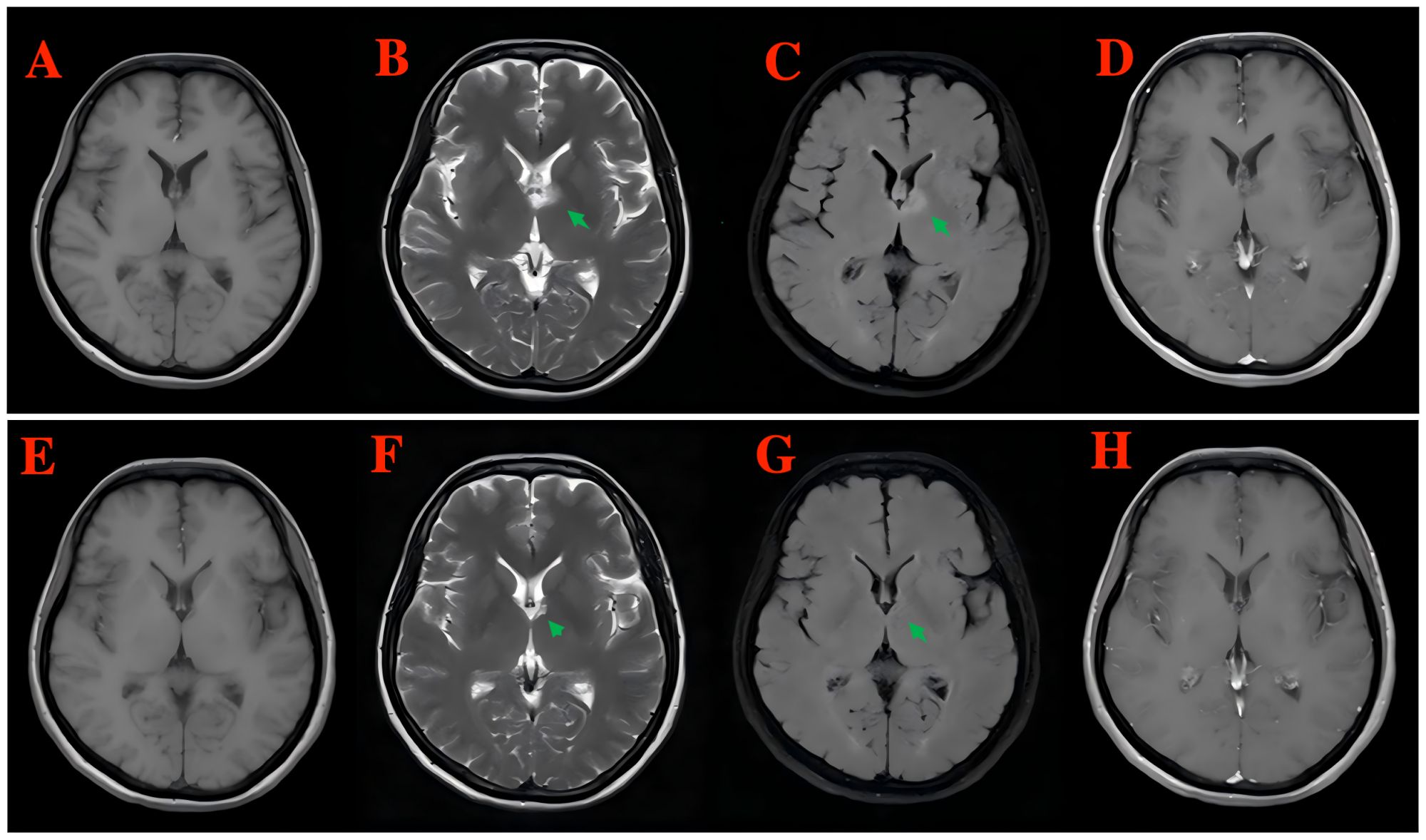
Figure 3. Brain MRI plain scan + enhancement with the gadolinium contrast agent revealed patchy hypointensity on T1WI, hyperintensity on T2WI, and hyperintensity on T2-FLAIR in the left basal ganglia, corona radiata, and local septum pellucida, with no enhancement in the enhanced lesions [(A) T1WI, (B) T2WI, (C) T2-FLAIR, and (D) enhanced T1-weighted MRI]. A follow-up MRI was performed 6 months later, which demonstrated the nearly total regression of the previous findings [(E) T1WI, (F) T2WI, (G) T2-FLAIR, and (H) enhanced T1-weighted MRI].
The patient received intravenous injection of methylprednisolone (500 mg/day for 5 days, consecutively), followed by a gradual dose reduction combined with mycophenolate mofetil (1 g, once daily) based on the hospital guidelines, and significant clinical and radiological improvements were observed. At present, the patient is still under follow-up, remains asymptomatic, and has no recurrence.
Discussion
GFAP astrocytopathy is an inflammatory central nervous system disorder first reported and named by the Lennon team at the Mayo Clinic in the USA in 2016 (1). Currently, the epidemiology of GFAP astrocytopathy remains unclear, but, in a population-based comparative study on the incidence and prevalence of autoimmune and infectious encephalitis in Olmsted County, USA, the prevalence of GFAP astrocytopathy was found to be 0.0006% (0.6/100,000). In 2017, Flanagan et al. identified GFAP antibody targeting astrocytes as the specific biomarker for the disease (4). GFAP astrocytopathy has an acute or subacute onset, and some patients have predromal fever and a history of infection, with the main manifestations of symptoms of meningitis (headache and neck pain), encephalitis (delirium, tremors, seizures, or psychiatric symptoms), and myelitis (paresthesia and myasthenia), and optic neuritis (impaired loss) (5, 6). In this case, drowsiness was the only clinical manifestation of GFAP astrocytopathy, and this condition is rarely reported (7).
GFAP, as a type III intermediate filament protein, is expressed in cells and serves as a key component of the intermediates of mature astrocytes. GFAP not only functions as a biomarker for astrocytes but also participates in multiple biological processes, including maintenance of astrocyte morphology, blood-brain barrier, synaptic plasticity, regulation of cell proliferation, and transport of vesicles and lysosomes in astrocytes (8, 9). The pathogenesis of GFAP astrocytopathy has not been well understood yet, which may be related to the abnormal activation of the tumor necrosis factor pathway or GFAP peptide–specific CD8+ T lymphocytes, resulting in damage to the astrocytes (10). As shown in some studies, CSF examination in patients with GFAP astrocytopathy is dominated by inflammation, and most patients can experience elevated WBC and protein level (11). In some research, approximately 40% of patients with GFAP astrocytopathy have one or more co-existing neuronal autoantibodies, and multiple co-existing neuronal autoantibodies have been reported in some patients with GFAP astrocytopathy, with NMDAR antibody being the most common co-existing antibody, followed by AQP4 antibody (1, 4, 12). According to relevant literature report, patients first diagnosed with neuromyelitis optica spectrum disease positive for AQP4 develop GFAP antibody in their second attack and show typical clinical features of GFAP astrocytopathy (13). In this study, the patient did not have clinical symptoms, signs, or imaging evidence for a diagnosis of neuromyelitis optica spectrum disease and was positive for AQP4 antibody and NMDAR antibody, contributing to the diagnosis of this rare GFAP astrocytopathy positive for antibodies. GFAP antibody, AQP4 antibody, and NMDAR antibody are expressed in teratoma. When GFAP antibody combined with AQP4 antibody and NMDAR antibody is positive, the prediction rate is as high as 71% (14). However, we completed tumor marker testing and CT plain scan of the head, chest, and whole abdomen (including the pelvic cavity) and found no evidence of tumor. According to Flanagan et al., 35 out of 102 patients (34%) had tumors, and 66% of the tumors were detected within 2 years of symptom onset, including ovarian teratoma, adenocarcinoma, and glioma (4), which might be related to the short onset time of the disease and required long-term follow-up.
Currently, there are few research reports on the specific EEG changes associated with GFAP astrocytopathy. Some studies have reported that children with GFAP astrocytopathy may exhibit the EEG phenomenon known as extreme deltabrush (EDB) (15–18).Additionally, EDB has been identified as a specific EEG feature of NMDAR antibody encephalitis, although the underlying mechanisms remain unclear (19). Studies have indicated that patients with high antibody titers and the presence of EDB often present with more severe symptoms and may even experience conditions such as coma and status epilepticus, necessitating early intensive care (20). In this study, the patient’s EEG primarily showed minimal low-amplitude beta and theta waves in all leads, with widespread increase in alpha activity. EEG examination is convenient, non-invasive, and relatively cost-effective. The identification of abnormal EEG waveforms is of great significance for clinical diagnosis and disease assessment; thus, further research is warranted.
Head and spinal MRI is the preferred option of imaging examination for GFAP astrocytopathy. More than 50% of head and spinal MRI examinations for GFAP astrocytopathy show hypointensity on T1WI, whereas hyperintensity on T2WI and T2-FLAIR. The lesions are mainly located in the basal ganglia, followed by the thalamus, and most of them show bilateral symmetrical distribution. The typical head imaging feature is linear radial perivascular enhancement that is perpendicular to the ventricles (4, 20). In our case, although the lesions were located in the left basal ganglia, there was no typical imaging evidence of the disease.
For the time being, no unified diagnostic criteria have been developed for GFAP astrocytopathy, making the diagnosis particularly challenging. In our case, the presence of multiple antibodies, including AQP4, NMDAR, and GFAP, further complicates the diagnosis of primary GFAP astrocytopathy. Our case should be differentiated from non-convulsive status epilepticus (NCSE), viral encephalitis, and Wernicke’s encephalopathy (WE). For patients with persistent drowsiness, it is crucial to rule out NCSE, as it can present with altered consciousness without obvious convulsions. Long-term EEG monitoring is essential for its diagnosis (21–23). Additionally, central nervous system inflammation, such as viral encephalitis, particularly herpes simplex encephalitis (HSE), must also be excluded. HSE often presents with characteristic imaging findings, such as involvement of the temporal lobes and positive viral markers in CSF (24, 25).Furthermore, WE should be considered in the differential diagnosis. WE is a neurological disorder caused by thiamine (vitamin B1) deficiency and is typically characterized by a clinical triad: ocular motor dysfunction, ataxia, and mental status changes. Although the clinical presentation of WE can vary, the most common features include acute or subacute onset of altered mental status, ataxia, and nystagmus (26). Based on the patient’s clinical manifestations, physical examination findings, response to auxiliary investigations, and treatment outcomes, the final diagnosis was determined to be GFAP astrocytopathy with overlapping autoimmune syndrome. The patient responded well to the treatment regimen, showing significant clinical and radiological improvements following intravenous Methylprednisolone administration for 5 consecutive days, followed by a gradual dose reduction and mycophenolate mofetil therapy as per hospital guidelines.
The treatments for acute-phase GFAP astrocytopathy mainly include high-dose glucocorticoids (500–1,000 mg/day), intravenous injection of immunoglobulin, and plasma exchange therapy. Some patients require long-term oral administration of steroids and immunosuppressive agents such as mycophenolate mofetil, azathioprine, or rituximab to prevent disease recurrence (3, 5, 7, 12, 27). After active treatment, a majority of patients have a good prognosis, whereas some may develop disease recurrence, and a few may experience a variety of dysfunction or even death (5, 28).
Conclusion
Currently, research regarding the diagnosis of GFAP astrocytopathy is scarce, and most physicians have insufficient understanding of the diagnosis of the diagnosis, which may easily lead to missed diagnosis and misdiagnosis. This case study adds to the limited research available on the presence of AQP4 antibody and NMDAR antibody in patients with GFAP astrocytopathy with overlapping positivity. Early detection and timely treatment are essential for the treatment of GFAP astrocytopathy, the improvement of long-term prognosis, and the reduction of mortality, which can thus avoid missed diagnosis and misdiagnosis and improve the clinical outcome of patients.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants or patients/participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
WW: Writing – review & editing, Writing – original draft. MC: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. LM: Writing – original draft, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research reported in this publication was supported by The Project for Development and Implementation of Appropriate Medical and Health Technologies in Guangxi (S2021121). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
GFAP, autoimmune glial fibrillary acidic protein; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; CT, computed tomography; EEG, electroencephalogram; AQP4, aquaporin-4; NMDAR, methyl-D-aspartate receptor.
References
1. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
2. Yang X, Xu H, Ding M, Huang Q, Chen B, Yang H, et al. Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodies. Front Neurol. (2018) 9:251. doi: 10.3389/fneur.2018.00251
3. Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. (2018) 83:166–77. doi: 10.1002/ana.25131
4. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. (2017) 81:298–309. doi: 10.1002/ana.24881
5. Kunchok A, Zekeridou A, McKeon A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol. (2019) 32:452–8. doi: 10.1097/WCO.0000000000000676
6. Liu L, Fang B, Qiao Z, Di X, Ma Q, Zhang J, et al. Clinical manifestation, auxiliary examination features, and prognosis of GFAP autoimmunity: a Chinese cohort study. Brain Sci. (2022) 12:1662. doi: 10.3390/brainsci12121662
7. Tang M, Huang S, Guo W, Zhou J, Huang Z, Li W, et al. Case report: Excessive daytime sleepiness as a presenting manifestation of autoimmune glial fibrillary acidic protein astrocytopathy. Front Immunol. (2023) 14:1302514. doi: 10.3389/fimmu.2023.1302514
8. Alexopoulos H, Akrivou S, Mastroyanni S, Antonopoulou M, Dinopoulos A, Giorgi M, et al. Postherpes simplex encephalitis: a case series of viral-triggered autoimmunity, synaptic autoantibodies and response to therapy. Ther Adv Neurol Disord. (2018) 11:1–16. doi: 10.1177/1756286418768778
9. Shan F, Long Y, Qiu W. Autoimmune glial fibrillary acidic protein astrocytopathy: a review of the literature. Front Immunol. (2018) 9:2802. doi: 10.3389/fimmu.2018.02802
10. Yuan Z, Li H, Huang L, Fu C, Chen Y, Zhi C, et al. CD8+ T-cell predominance in autoimmune glial fibrillary acidic protein astrocytopathy. Eur J Neurol. (2021) 28:2121–5. doi: 10.1111/ene.14778
11. Zarkali A, Cousins O, Athauda D, Moses S, Moran N, Harikrishnan S. Glial fibrillary acidic protein antibody-positive meningoencephalomyelitis. Pract Neurol. (2018) 18:315–9. doi: 10.1136/practneurol-2017-001863
12. Long Y, Liang J, Xu H, Huang Q, Yang J, Gao C, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neurol. (2018) 25:477–83. doi: 10.1111/ene.13531
13. Lin H, Huang Y, Zeng H, Wang M, Guan S, Chen G, et al. Overlapping clinical syndromes in patients with glial fibrillary acidic protein IgG. Neuroimmunomodulation. (2020) 27:69–74. doi: 10.1159/000505730
14. Martinez-Hernandez E, Guasp M, Garcia-Serra A, Maudes E, Ariño H, Sepulveda M, et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology. (2020) 94:e2302–10. doi: 10.1212/WNL.0000000000009239
15. Theroux LM, Goodkin HP, Heinan KC, Quigg M, Brenton JN. Extreme delta brush and distinctive imaging in a pediatric patient with autoimmune GFAP astrocytopathy. Mult Scler Relat Disord. (2018) 26:121–3. doi: 10.1016/j.msard.2018.09.015
16. Moise AM, Karakis I, Herlopian A, Dhakar M, Hirsch LJ, Cotsonis G, et al. Continuous EEG findings in autoimmune encephalitis. J Clin Neurophysiol. (2021) 38:124–9. doi: 10.1097/WNP.0000000000000654
17. Fang H, Hu W, Jiang Z, Yang H, Liao H, Yang L, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in children: a retrospective analysis of 35 cases. Front Immunol. (2021) 12:761354. doi: 10.3389/fimmu.2021.761354
18. Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. (2012) 79:1094–100. doi: 10.1212/wnl.0b013e3182698cd8
19. Steriade C, Hantus S, Moosa AN, Rae-Grant AD. Extreme delta–With or without brushes: A potential surrogate marker of disease activity in anti-NMDA-receptor encephalitis. Clin Neurophysiol. (2018) 129:2197–204. doi: 10.1016/j.clinph.2018.02.130
20. Kimura A, Takekoshi A, Yoshikura N, Hayashi Y, Shimohata T. Clinical characteristics of autoimmune GFAP astrocytopathy. J Neuroimmunol. (2019) 332:91–8. doi: 10.1016/j.jneuroim.2019.04.004
21. Meierkord H, Holtkamp M. Non-convulsive status epilepticus in adults: clinical forms and treatment. Lancet Neurol. (2007) 6:329–39. doi: 10.1016/S1474-4422(07)70074-1
22. Sutter R, Stevens RD, Kaplan PW. Continuous electroencephalographic monitoring in critically ill patients: indications, limitations, and strategies. Crit Care Med. (2013) 41:1124–32. doi: 10.1097/CCM.0b013e318275882f
23. Leitinger M, Trinka E, Gardella E, Rohracher A, Kalss G, Qerama E, et al. Diagnostic accuracy of the Salzburg EEG criteria for non-convulsive status epilepticus: a retrospective study. Lancet Neurol. (2016) 15:1054–62. doi: 10.1016/S1474-4422(16)30137-5
24. Stahl JP, Mailles A. Herpes simplex virus encephalitis update. Curr Opin Infect Dis. (2019) 32:239–43. doi: 10.1097/QCO.0000000000000554
26. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neuro. (2007) 6:442–55. doi: 10.1016/S1474-4422(07)70104-7
27. Iorio R, Damato V, Evoli A, Gessi M, Gaudino S, Di Lazzaro V, et al. Clinical and immunological characteristics of the spectrum of GFAP autoimmunity: a case series of 22 patients. J Neurol Neurosurg Psychiatry. (2018) 89:138–46. doi: 10.1136/jnnp-2017-316583
Keywords: anti-GFAP antibody, aquaporin-4 antibody, NMDAR antibody, autoimmune glial fibrillary acidic protein astrocytopathy, central nervous system
Citation: Wei W-x, Chen M-l and Meng L (2024) Case report: Autoimmune glial fibrillary acidic protein astrocytopathy with overlapping autoimmune syndrome. Front. Immunol. 15:1485374. doi: 10.3389/fimmu.2024.1485374
Received: 23 August 2024; Accepted: 24 September 2024;
Published: 11 October 2024.
Edited by:
Eugenio Pucci, AST Fermo Marche Region Health System, ItalyCopyright © 2024 Wei, Chen and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-li Chen, MjYwMjM2NTU5QHFxLmNvbQ==
†These authors have contributed equally to this work