 Chin-King Looi1
Chin-King Looi1 Ee-Mun Loo2,3
Ee-Mun Loo2,3 Heng-Chee Lim2
Heng-Chee Lim2 Yik-Ling Chew2
Yik-Ling Chew2 Kok-Yong Chin4
Kok-Yong Chin4 Shiau-Chuen Cheah5
Shiau-Chuen Cheah5 Bey Hing Goh6,7,8
Bey Hing Goh6,7,8 Chun-Wai Mai2*
Chun-Wai Mai2*- 1School of Postgraduate Studies, International Medical University, Kuala Lumpur, Malaysia
- 2Faculty of Pharmaceutical Sciences, UCSI University, Kuala Lumpur, Malaysia
- 3Advanced Genomics Laboratory, AGTC Genomics, Kuala Lumpur, Malaysia
- 4Department of Pharmacology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
- 5Faculty of Medicine and Health Sciences, UCSI University, Port Dickson, Negeri Sembilan, Malaysia
- 6Sunway Biofunctional Molecules Discovery Centre, School of Medical and Life Sciences, Sunway University Malaysia, Bandar Sunway, Selangor Darul Ehsan, Malaysia
- 7Biofunctional Molecule Exploratory Research Group, School of Pharmacy, Monash University Malaysia, Bandar Sunway, Selangor Darul Ehsan, Malaysia
- 8College of Pharmaceutical Sciences, Zhejiang University, Zhejiang, China
Nasopharyngeal carcinoma (NPC) is a distinct malignancy of the nasopharynx and is consistently associated with the Epstein-Barr virus (EBV) infection. Its unique anatomical location and complex aetiology often result in advanced-stage disease at first diagnosis. While radiotherapy (RT) and chemotherapy have been the mainstays of treatment, they often fail to prevent tumour recurrence and metastasis, leading to high rates of treatment failure and mortality. Recent advancement in cell-based therapies, such as chimeric antigen receptor (CAR)-T cell therapy, have shown great promise in hematological malignancies and are now being investigated for NPC. However, challenges such as targeting specific tumour antigens, limited T cell persistence and proliferation, and managing treatment-related toxicities must be addressed. Extensive research is needed to enhance the effectiveness and safety of these therapies, paving the way for their integration into standard clinical practice for better management of NPC and a better quality of life for human health.
1 Introduction
Nasopharyngeal carcinoma (NPC) is an undifferentiated form of squamous cell carcinoma (SCC) formed from the uncontrolled growth of cells in the epithelium layer of the nasopharynx. While globally rare, it is prevalent in Southern China, Southeast Asia, North Africa and the Middle East. Among the Cantonese population in Southern China, the incidence is approximately 25-50 cases per 100,000, compared to less than 1 per 100,000 in other parts of the world (1).. This low incidence contributes to limited research and fewer diagnostic advancements compared to other cancer types, making it challenging to effectively identify and diagnose NPC. In 2020, the International Agency for Research on Cancer (IARC) reported 133,354 cases and 80,008 deaths of NPC worldwide, with 62,444 (46.8%) cases in China, and 36,747 (27.6%) cases in the rest of Southeast Asia (2). These findings highlight persistent disparities in NPC incidence and mortality globally, underscoring the urgent need for improved diagnostic tools and treatment strategies to address the NPC burden in affected regions and countries. Notably, NPC incidence may vary based on gender, age, Epstein-Barr virus (EBV) infection, and risk factors such as smoking, alcohol consumption, occupational exposures and dietary consumption of Cantonese-style salted fish (1, 3).
The conventional treatment modalities for NPC include chemotherapy, radiotherapy (RT) (4), and immunotherapeutic strategies (5). While emerging evidence highlights that surgery or nasopharyngectomy is effective in the salvage of recurrent tumours or metastasis after primary RT or when other treatments have failed (6), it has a significant disadvantages in treatment of primary NPC. The anatomical location of the nasopharynx limits the visual field, making surgical access difficult. Moreover, surgery is risky due to the proximity to critical structures such as the internal carotid artery, spinal cord, and cranial nerves, which can cause serious consequences if accidentally injured. Additionally, patient’s quality of life (QOL) has been shown to deteriorate significantly with surgical treatment. Therefore, these limitations have made non-surgical treatments the primary options for NPC (7). While RT has been the primary treatment since 1965, with a high 5-year overall survival rate of up to 90% with RT alone for Stage I NPC patients, treatment with RT has been associated with severe acute and late toxicities (4). Delivering a high radiation dose to targets while protecting radiosensitive organs such as the brain stem, spinal cord, temporal lobes, middle and inner ears, and parotid glands, which are anatomically surrounded by the nasopharynx, is particularly challenging (8). Additionally, chemotherapy can increase susceptibility to infection due to immunosuppression and may cause systemic adverse effects like vertigo and hair loss. Chemotherapy resistance is also a significant issue that often leads to therapeutic failure (9). Despite recent advancements in immunotherapies, especially immune checkpoint blockade therapy, treatment responses vary significantly among patients, and high rates of relapse with poor prognosis and treatment unresponsiveness remain significant challenges in improving patient’s outcomes (10).
In recent years, cell-based therapies, particularly those involving genetically modified T cells with chimeric antigen receptors (CAR), have shown promise in treating hematologic malignancies (11) and are being evaluated for NPC (12, 13). NPC is an epithelial malignancy closely associated with EBV infection and is characterised by intense infiltration of immune cells. Despite the abundance of tumour-infiltrating lymphocytes (TILs), many of these cells are highly activated and exhausted, expressing exhaustion markers such as lymphocyte activation gene (LAG-3), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and programmed cell death 1(PD-1) (14), while high programmed death ligand 1 (PD-L1) expression on NPC cells promotes immune evasion (15). In addition, EBV expresses a series of latent viral genes, including latent membrane proteins (LMP1 and LMP2), Epstein–Barr nuclear antigen (EBNA1), BamH1 A fragment rightward reading frame 1 (BARF1), non-polyadenylated and non-protein coding small EBV RNAs (EBER1 and EBER2), BamHI A rightward transcripts (BARTs) and BART microRNAs (miR-BARTs) (16). These genes support the growth of infected cells and serve as attractive targets for immunotherapy and cell-based therapies, such as CAR-T. LMPs are highly immunogenic and activate nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3) signalling pathways, leading to the expression of various downstream targets involved in chronic inflammatory responses. These include interleukins (IL-6, IL-10, TNF-α), chemokines (CCL4, CCL5, CXCL10), vascular endothelial growth factor (VEGF), and hypoxia-inducible factor (HIF)-1α, all of which are involved in immune evasion, cell growth and proliferation, invasion, metastasis, and epithelial-mesenchymal transition (EMT) (17). Targeting EBV-specific antigens with cell-based therapies may offer long-lasting effects and reduce the risk of recurrence, with fewer side effects compared to conventional treatments (18). Inspired by the success of adoptive EBV-targeted cytotoxic T lymphocyte (EBV-CTL) therapy in other EBV-associated diseases (19, 20), anti-EBV cell-based strategies are gaining attention as potential treatments to improve NPC prognosis (21–23).
In this review, we delve into the potential of cell-based therapies as the next frontier in the treatment of NPC. We critically discuss the general mechanism of action and breakthroughs in cellular therapies and provide examples of utilising engineered immune cells in targeting NPC. Additionally, we summarise findings from both completed and ongoing clinical trials related to adoptive cell therapies against NPC and other cancers. We also address the limitations associated with cellular therapies and suggest integrating these therapies with conventional treatments to improve safety and effectiveness, aiming to enhanced outcomes for advanced NPC patients.
Despite the promise of developing new agents that target essential cellular pathways in cancer progression, most advanced cancer patients have experienced relatively short-term benefits. Among the emerging biologic therapeutics, cell-based therapy, also known as cell therapy or cellular therapy, holds potential to treat many intractable human diseases, including cancer (24). The first practice of cell therapy was introduced in the late 1950s by E. Donnall Thomas, who pioneered the use of hematopoietic stem cell (HSC) transplantation for the treatment of leukemia (25). Since then, the field of cell therapy has continuously progressed, with ongoing investigations focused on clinical safety and efficacy. Notably, the Food and Drug Administration (FDA) has approved Provenge (sipuleucel-T; Dendreon), an autologous cellular immunotherapy for metastatic castration-resistant prostate cancer in 2010 (26). In 2017, the FDA approved two genetically modified autologous T cell immunotherapies, Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel), for acute lymphoblastic leukaemia (ALL) (27), and relapsed or refractory large B cell lymphoma (LBCL) (28), respectively. Additionally, BRG01, an engineered, allogeneic, EBV-targeting T cell therapy, has recently received both fast-track and orphan drug designation from the FDA for EBV-positive relapsed/metastatic NPC (29). As highlighted in the Cell Therapy Market Size, Share, & Trends Analysis Report, the global cell therapy market size valued at USD 4.77 billion in 2022, is expected to grow at a compound annual growth rate of 16.5% from 2023 to 2030. This underscores the increasing adoption and demand for cell therapy solutions worldwide, opening a new era of therapy for human diseases.
Cell therapy can be generally classified into two categories: (i) stem cell-based and (ii) non-stem cell-based. It typically involves the use of autologous or allogenic cells and may incorporate genetic engineering or manipulations in formulation to achieve therapeutic effect (24).
2 Stem cell-based therapies
When comes to cell therapy, it is logical to explore the therapeutic potential of stem cells. Stem cells can be found in both embryos and adult cells. They are undifferentiated cells capable of self-renewal and differentiation into specialised cell types based on their developmental potency. This potency gradually reduced from totipotency (in the zygote and early embryonic cells) to pluripotency (in embryonic stem cells, ESCs), multipotency (in hematopoietic stem cells, HSCs), and unipotency (in the dermatocytes) (30). In cancer treatment, stem cells used include adult stem cells (ASC), and pluripotent stem cells (PSC), Different stem cells exhibit varying capabilities in proliferation, migration, and differentiation, defining their suitability for antitumour therapy (31).
2.1 Adult stem cells
Adult stem cells (ASCs), also known as somatic or resident stem cells, are undifferentiated cells found in various tissues with limited self-renewal ability and differential potential (32). Examples of ASCs include HSCs, neural stem cells (NSCs), and mesenchymal stem cells (MSCs).
HSCs are predominantly found in bone marrow, peripheral blood (PB) and umbilical cord blood (UCB). They play a crucial role in regenerating blood cells and treating haematologic malignancies through HSC transplantation (HSCT), also known as bone marrow transplantation (33). This procedure, pioneered by E. Donnall Thomas in the late 1950s, involves infusing autologous or allogeneic stem cells to reconstruct a functional hematopoietic system (25). HSCT is now a standard treatment for various hematologic malignancies and have also been explored in solid tumours as immune cell progenitors, exhibiting synergistic effects with adoptive T cell immunotherapy and dendritic cell vaccines. Studies show that HSCs facilitate T cell trafficking, augment immunological rejection of invasive tumour cells, and can differentiate into immune-stimulating dendritic cells (DCs) within the TME (34). These findings highlight the multifaced role of HSCs not only as stem or progenitor cells for cell regeneration, but also as cells capable of synergistically enhancing the efficacy of immunotherapy against solid cancers. However, till today, there is limited evidence of HSCs in treating NPC patients. And thus, HSCs may have theoretical role against NPC, but no clinical study has confirmed its efficacy.
NSCs are self-renewable stem cells in the central nervous system, capable of differentiating into astrocytes, oligodendrocytes, and neural cells (35). Engineered NSCs have shown promise in treating brain cancers due to their ability to migrate to malignant brain masses (36). While the mechanism of NSC homing to tumour is not fully understood, hypoxia has been identified as a key factor. HIF-1α in the TME activates NSC chemoattractant production, such as cytokines, chemokines, and growth factors (37). This migratory behaviour positions NSCs as an effective tumour eradication therapy (38, 39). Similar to HSCs, currently no clinical study has confirmed NSC can eradicate NPC, and thus further study is warranted to investigate whether the NSC has its clinical value for NPC patients.
MSCs, derived from sources such as bone marrow, adipose tissues, PB, placenta, and umbilical cord, playing roles in tissue repair and regeneration due to its primary, non-specialised, non-haematopoietic, with rapid proliferation and differentiation potential. MSCs also exhibit immunomodulatory and anti-inflammatory properties, and are non-immunogenic, making them suitable for transplantation without immunosuppression. For example, bone marrow-derived MSCs inhibited CTL activation by downregulating natural killer group 2 member D protein (NKG2D) receptor expression and increasing the production of immunosuppressive factors (40). MSCs also promote Tregs differentiation to suppress allogeneic T cell response (41). MSCs also suppress tumour growth by modulating cell cycle signalling pathways and inducing apoptosis (42, 43). However, MSCs is a double edge sword in cancer therapy, in which MSCs can differentiate into cancer-associated fibroblasts (CAFs) in response to TGF-β released from tumour cells, leading to the promotion of tumour growth and progression (44, 45).
It is also worth noting that MSCs release exosomes that facilitate cell communication (46) and modulate disease progression (47, 48). In NPC, MSC-derived exosomes are associated with elevated expression of fibroblast growth factor (FGF)-19, enhancing tumour growth, migration, and metastasis through the activation of the FGF19-fibroblast growth factor receptor 4 (FGFR4)-dependent ERK signaling cascade and promotion of EMT (49). However, some reports suggest that these exosomes can inhibit tumour growth by reversing its therapy resistance. For instance, MSC-derived exosomes with overexpressed tumour suppressor microRNA-34c-5p (miR-34c) have shown potential therapeutic value in reversing radioresistance and inhibiting malignant behavior in NPC cells (50). Similarly, exosomes from engineered human MSCs with elevated expression of microRNA-125a-3p (miR-125a) demonstrated the ability to suppress migration and vasculogenic mimicry formation in NPC cells, suggesting potential applications in anti-angiogenic therapy for cancer treatment (51).
In summary, HSC and NSC are currently still under investigation whether its potential can be translated to bedside for NPC patients. The dual role of MSCs as tumours promoter or suppressor (Table 1), may depend on tumour types, treatment doses, and duration. The role of MSCs as antitumour agents or therapeutic targets for NPC remains debated, and current evidence is insufficient to support the clinical application of MSC in NPC treatment. Further studies are needed to understand the complex interactions between MSCs and cancer, particularly the role of MSC-derived exosomes in carcinogenesis and therapy resistance.
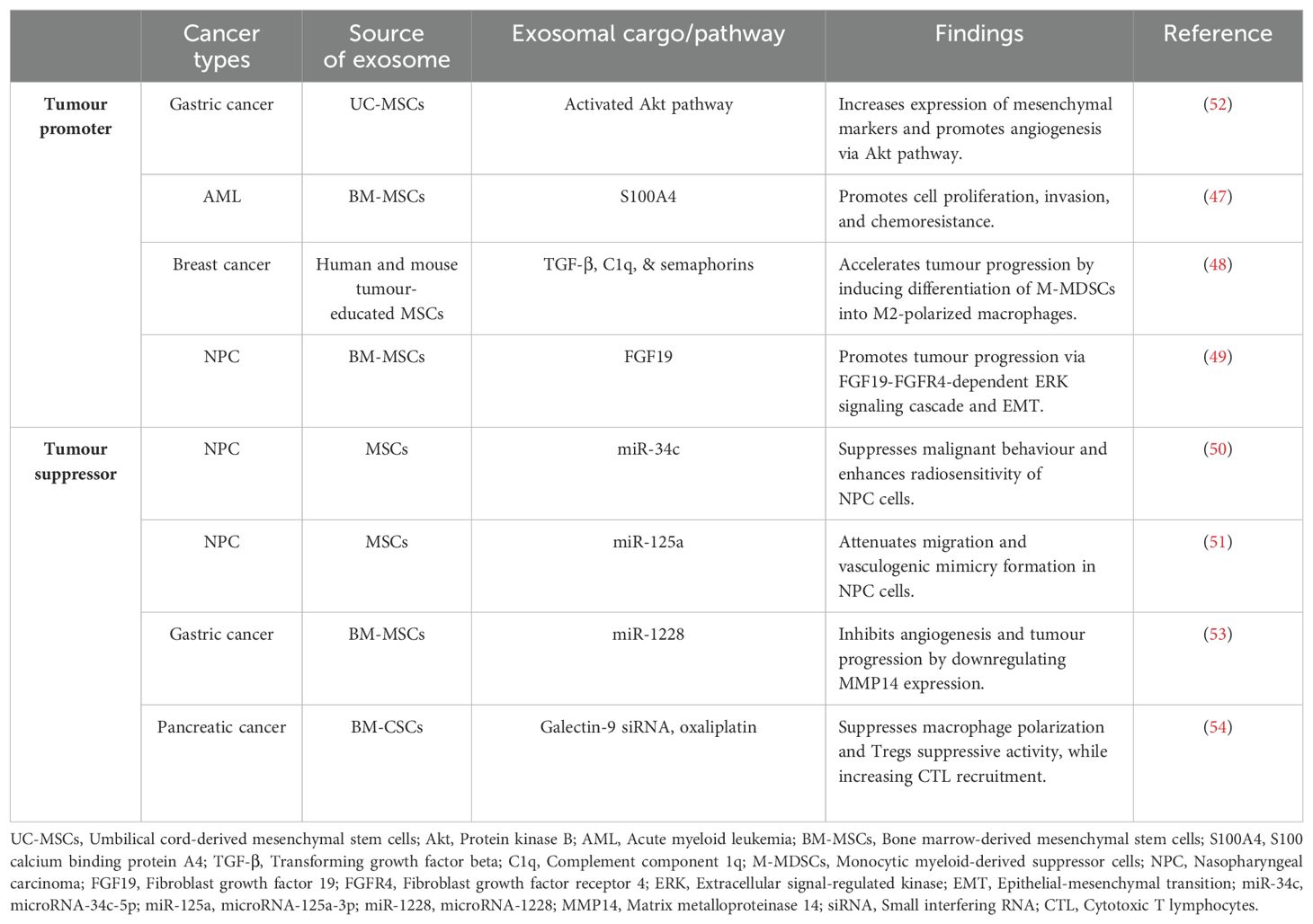
Table 1. Dual roles of MSC-derived exosomes in human cancers.
2.2 Pluripotent stem cells
Pluripotent stem cells (PSCs) can proliferate infinitely and differentiate into various specialised cell types. ESCs and induced pluripotent stem cells (iPSCs) are two types of human PSCs with significant implications for regenerative medicine and clinical research (55). The first human ESC line was derived by James Thomson and his team from in vitro-fertilised human embryos in 1998 (56). Due to ethical issues with ESCs (57), Takahashi and Yamanaka generated iPSCs from mouse embryonic and adult human somatic cells in 2006 and 2007, respectively (58, 59). These cells exhibit similar properties to ECSs, including pluripotent status, morphology, growth properties, surface antigen, gene expression, and telomerase activity, highlighting their potential in medical applications.
iPSCs can be genetically engineered into T cells or NK cells, which can effectively target and eradicate tumour cells. The generation of iPSC-derived T cells involves the induction of mesoderm specification and hematopoietic commitment, followed by T cell differentiation. Briefly, iPSCs collected from healthy donors are co-cultured on murine cell lines (C3H/10T1/2 or OP9) with morphogens, such as bone morphogenetic protein 4 (BMP4), VEGF, and fibroblast growth factor (FGF) to induce mesoderm specification. These cells are then co-cultured in a cocktail of cytokines to generate CD34+ HSCs (60). The CD34+ hematopoietic progenitors are transferred onto OP9 overexpressing the Notch ligand Delta-like 1 (DLL1) or DLL4 (OP9-DLL1 and OP9-DLL4) feeder cells to induce T cell development in the presence of cytokines (60). Similarly, iPSC-derived NK cells are generated from CD34+ hematopoietic progenitor cells using feeder-dependent culture method (61). However, murine-derived stroma feeder cells pose risks of cross-species contamination and complicate quality control due to the reliance on different serum and basal media. Therefore, feeder-free methods, such as using immobilized-DLL4 protein or DLL4-µbeads with lymphopoietic cytokines, have successfully induced T cell differentiation without feeder cells (62, 63). Researchers have also generated rejuvenated iPSC-derived antigen-specific T cells that retain the same antigen specificity and cytotoxicity as the original T cells, with higher proliferative capacity and reduced exhaustion markers (64, 65). iPSC-derived human papilloma virus (HPV)-specific CTLs have demonstrated more potent and sustained cytotoxic activity against cancer cells compared to original peripheral blood CTLs (66). Likewise, iPSC-derived EBV-specific CTLs persisted as central memory T cells in vivo for at least 6 months, continuously targeting EBV-associated lymphoma cells (67). Another study demonstrated that incorporating an inducible caspase-9 (iC9)-based suicide system into iPSC-derived EBV specific CTLs effectively suppressed tumour growth in vivo without compromising antigen-specific killing activity. This system also reduced cytokine release syndrome (CRS), enhancing the safety of T cell therapy (68).
NK cells derived from iPSCs exhibit greater cytotoxicity against a wide variety of cancers. By combining embryoid body (EB) formation with membrane-bound IL-21-expressing artificial APCs, functional NK cells can be efficiently produced from iPSCs. Briefly, iPSCs collected from healthy donors are grown under feeder-free conditions for a week, spun to aggregate into EBs, and cultured in feeder-free media with morphogens to induce the formation of HSC progenitors. The hematopoietic progenitor-containing EBs are then transferred to a feeder-free plate with NK differentiation media containing cytokines (namely IL-3, IL-15, IL-7, SCF, and Flt3L) to generate iPSC-derived NK cells (69). This method offers a safe and robust platform for producing clinically applicable immune cells. Preclinically, iPSC-derived NK cells have demonstrated effective cytotoxic responses against a variety of tumours in xenograft models. For example, iPSC-derived NK cells combined with anti-PD-1 therapy showed synergistic effects in ovarian cancer, delaying tumour progression and enhancing T cell recruitment and inflammatory cytokine production, transforming a “cold” TME into a “hot” TME (70). Although iPSC-NK cell therapy has shown promising results, it has yet to receive FDA approval. A recently completed phase I clinical trial (NCT03841110) involved evaluating the safety and efficacy of FT500, an off-the-shelf iPSC-NK cell product, in combination with one of three immune checkpoint inhibitors (nivolumab, pembrolizumab, or atezolizumab) in patients with solid tumours, including HNSCC (71). The administration of six doses of FT500 cells was well tolerated, with no serious adverse events, such as graft-versus-host disease (GvHD), cytokine release syndrome (CRS), neurotoxicity (NT), or host immune rejection. Nine out of 13 patients achieved stable disease, and one patient with Hodgkin lymphoma refractory to prior anti-PD-1 therapy experienced a 58% reduction in tumour size following the combination therapy (71). This trial provides clinical support for the high tolerability and potential antitumour efficacy of allogeneic iPSC-NK cell therapy.
Based on the evidence above, we advocate that iPSCs as a platform to provide an unlimited source of rejuvenated T cells or NK cells-based therapy. Such cell-based therapy holds promise as a safe and effective method for targeting NPC and other cancers. Further studies are warranted to investigate the possibility of introducing the CARs or transgenic T cell receptors (TCRs) into the iPSC-derived immune cells to further enhance their tumour-targeting capabilities while suicide gene-based safeguard system to ensure its safety.
3 Non-stem cell-based therapies
Non-stem cell-based therapies typically involve the use of somatic cells isolated from the human body and undergo processes such as propagation, expansion, and selection. These cells are subsequently administered to patients for various purposes, including curative, preventive, or diagnostic applications (72). Examples of non-stem cell-based therapies include chimeric antigen receptor (CAR)-T cells, engineered T cell receptor-T (TCR-T) cell therapy, tumour-infiltrating lymphocytes (TILs) therapy, NK cell therapy, CAR-NK cell therapy, aimed at enhancing their ability to recognise and eliminate malignant cells (72). Tables 2, 3 provide a summary of preclinical and clinical studies on cellular therapies for NPC.
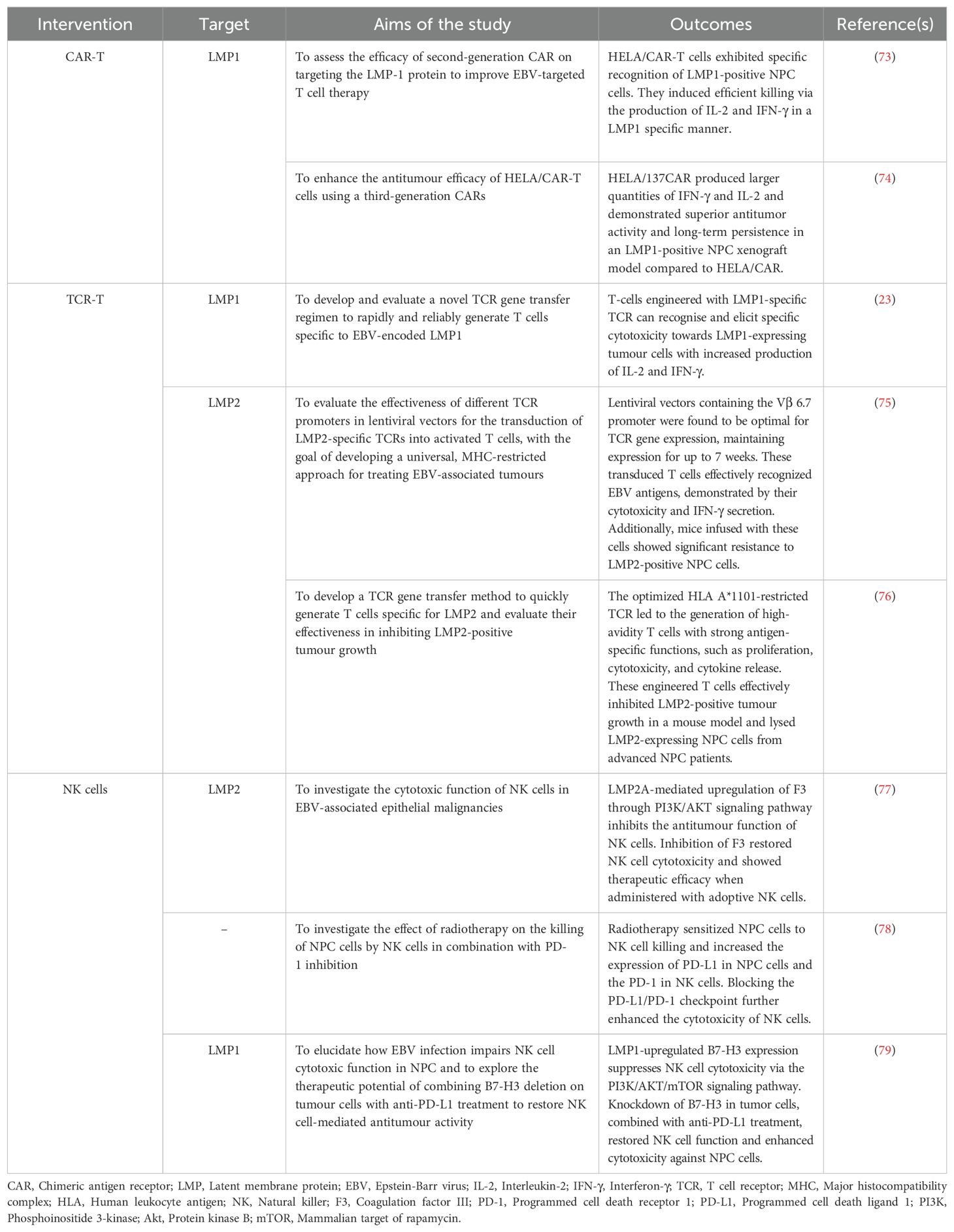
Table 2. Summary of preclinical studies on cellular therapies for NPC.
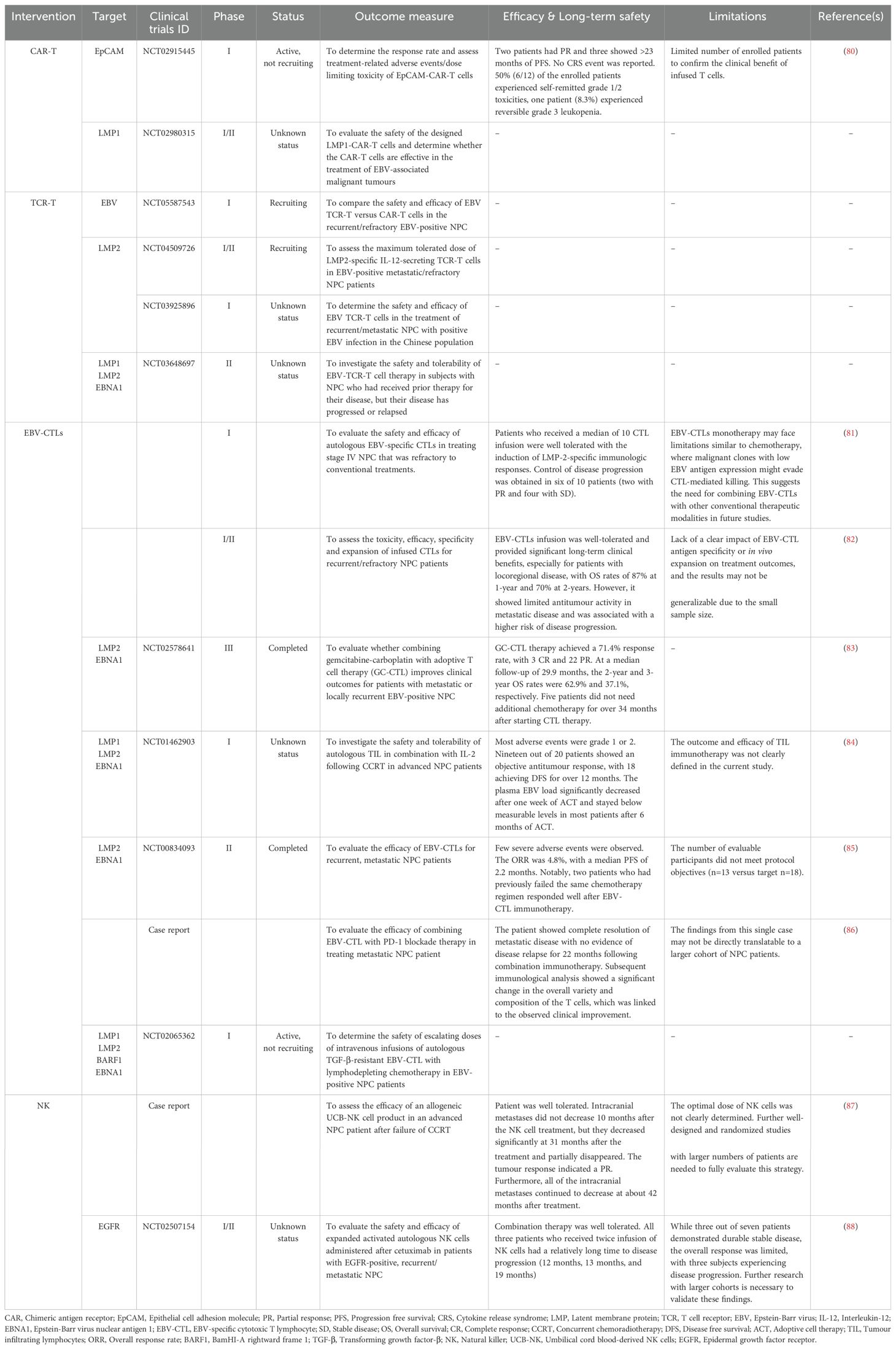
Table 3. Summary of clinical studies on cellular therapies for NPC.
3.1 Chimeric antigen receptors T cell
Chimeric antigen receptor (CAR)-T cells are a type of immunotherapy involving the genetic modification of a patient’s T cells to express a synthetic receptor known as CAR, which includes an antigen-recognition domain (often an antibody single-chain variable fragment, scFv), and an intracellular signalling domain (typically CD3ζ) (89). These CAR-T cells can selectively target tumour associated antigen (TAA) via scFv recognition domain, leading to tumour cell elimination through the production of inflammatory cytokines and cytolytic effectors, achieving a long-term potent antitumour activity (89).
CAR-T cell development has evolved over generations with improved efficacy (Figure 1). The first-generation CARs were developed in 1993 by Zelig Eshhar and consisted of a single signalling domain derived from the CD3ζ chain of the TCR (90). However, due to a low cytotoxicity, proliferation rate, and relatively short persistence in patients, the second-generation CARs were developed with an additional co-stimulatory domain (4-1BB/CD137 or CD28), resulting in enhanced T cell proliferation, cytotoxicity, and sustained response (91, 92). Soon, the third-generation CARs are further improved by including additional co-stimulatory domain, such as OX40/CD134 or CD137 (93, 94). CD28 and CD134 support the long-term T cell expansion and survival (95, 96), while CD137 to enhances T cell proliferation, IL-2 production, expression of anti-apoptotic genes to inhibit activation-induced cell death (AICD), and memory T cell development (97). Due to the undesirable clinical outcome, fourth-generation CARs, also known as T cell redirected for universal cytokine-mediated killing (TRUCKs) incorporated a nuclear factor of the activated T-cell (NFAT)-responsive promoter into the base of the second-generation CARs. This promoter can induce cytokine secretion, such as IL-7, IL-12, IL-15, and IL-18 upon CAR cell activation, augmenting T cell activation and killing of cancerous cells (98, 99). In the fifth-generation CARs, a truncated IL-2 receptor β (IL-2Rβ)-chain domain is introduced to provide a binding site for the transcription factor STAT3 and to activate JAK-STAT signalling pathway, further improving T cell activation, proliferation, and persistence (100).
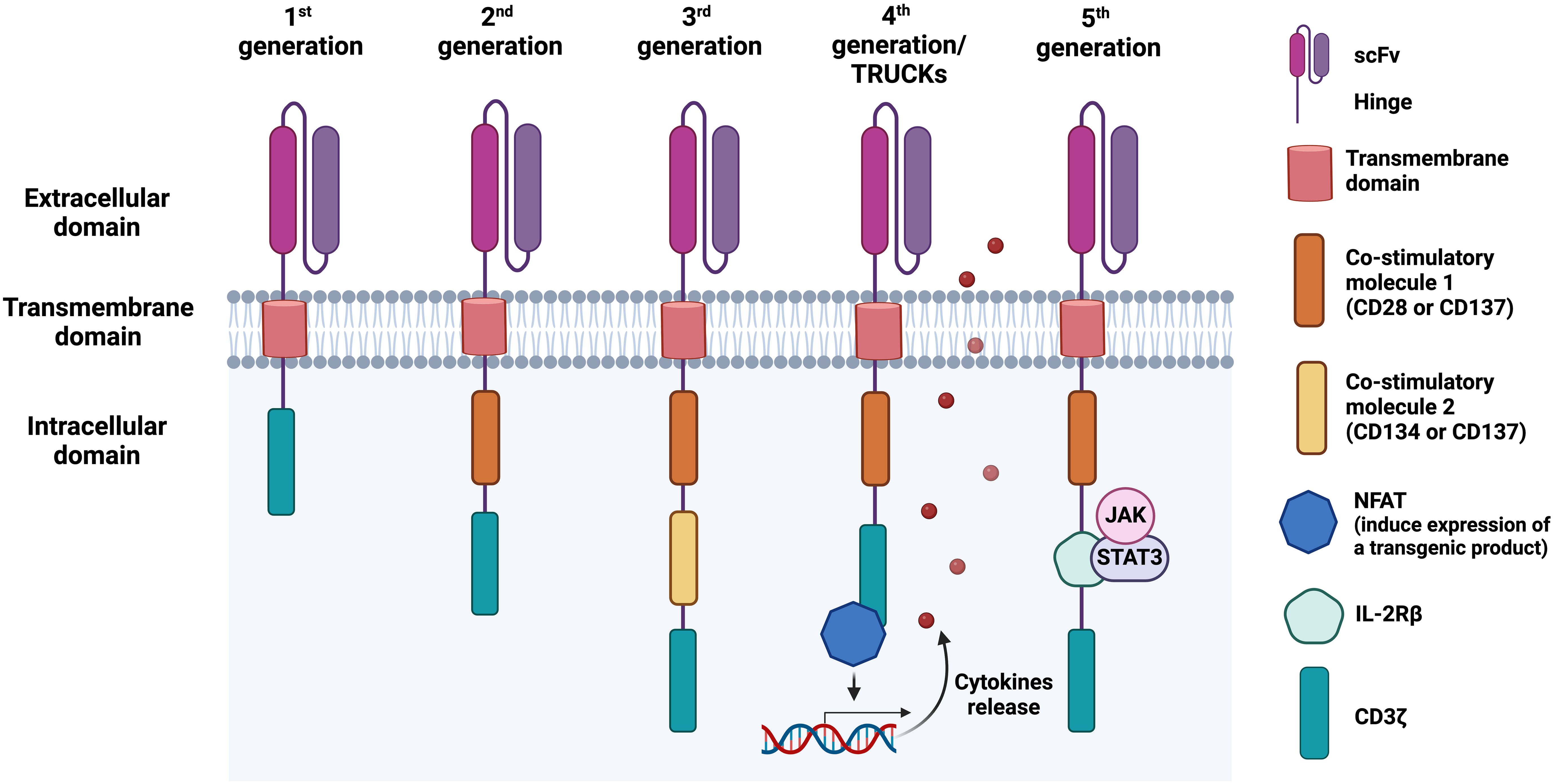
Figure 1. Chimeric antigen receptor (CAR) structure and its evolution from first to fifth generation. The first-generation of CARs consists of an antigen-binding domain, typically a single-chain variable fragment (scFv), followed by a hinge, a transmembrane domain, and an intracellular region, commonly the T cell receptor (TCR) signalling component CD3ζ. In the second and third CAR generations, one or two co-stimulatory domains are added, respectively, enhancing T cell activation and proliferation. Fourth-generation CARs, also known as T cell redirected for universal cytokine-mediated killing (TRUCKs), combine a second-generation CAR construct with additional functional elements, such as cytokine secretion modules. These modules enable CAR-engineered cells to secrete cytokines such as IL-12, which recruit immune cells to the tumour microenvironment (TME) upon antigen recognition, enhancing antitumour activity. The fifth-generation CAR-T cells, also referred to as the next generation, contain a truncated IL-2 receptor β (IL-2Rβ)-chain domain with a motif for binding transcription factors such as STAT3. This can lead to JAK/STAT activation and subsequent cytotoxic responses.
In recent years, CAR T-cell therapy has demonstrated remarkable success in treating relapsed or refractory haematological malignancies. As of January 2024, the FDA has approved six CAR-T cell products: Abecma and Carvykti for multiple myeloma, Breyanzi and Yescarta for large B-cell lymphoma, Kymriah for non-Hodgkin lymphomas, and Tecartus for B-cell acute lymphoblastic leukaemia. Four target CD19, while Abecma and Carvykti target B cell maturation antigen (BCMA) (11). Despite the success in haematological malignancies, CAR-T cell therapies have not received clinical approval for the treatment of solid tumours, including NPC, due to challenges such as, the lack of specific antigens, on-target off-tumour effects, and the complexity of the TME (11). Encouragingly, clinical trials are ongoing. A Phase 1 clinical trial (NCT02915445) targeting epithelial cell adhesion molecule (EpCAM) in solid tumours, including NPC, showed promising antitumour efficacy and an acceptable safety profile, with two patients showing a partial response and three experiencing more than 23 months of progression-free survival (PFS) (80). Engineered T cells targeting LMP1 in EBV-positive NPC cells have also demonstrated increased IFN-γ and IL-2 production, effectively killing LMP1-overexpressing NPC cells in vitro and reducing tumour growth in vivo (73, 74). The safety and efficacy of these CAR-T cells have been evaluated in a Phase I/II clinical trial (NCT02980315) for the treatment of EBV-associated NPC. Additionally, an ongoing early phase I study (NCT04657965) is assessing these engineered T cells in patients with LMP1-positive infectious diseases and haematological malignancies.
It is also worth noting that numerous challenges limit the therapeutic efficacy of CAR-T cells in both hematological malignancies and solid tumours (Figure 2). These challenges include antigen escape, poor tumour infiltration, low persistence, and the immunosuppressive nature of the TME (101). On-target off-tumour recognition is also a concern as many solid tumour antigens are expressed in normal tissues at varying levels. For example, HER2 CAR-T cells may induce severe side effects, including respiratory distress, cardiac arrest, and cytokine-release syndrome (CRS)-induced multi-organ failure. This is because HER2 is expressed not only by malignant cells, but also on normal epithelial cells in various tissues, making them off-targets by HER2 CAR-T cells (102). Therefore, selecting a suitable antigen for CAR-T cell therapy and enhancing the selective expression of CARs for targeting solid tumours are crucial considerations in current research. High rates of severe adverse events with fatal outcomes have prevented CAR-T cells from becoming first-line treatment. In a Phase I clinical trial (NCT01044069) for relapse B-cell ALL patients who received CD19-specific CAR T cells, potent antitumour efficacy of CAR-T cells was observed. However, 14 out of 53 patients experienced severe CRS, and tragically, one patient died from it (103). Neurotoxicity is also common after CAR-T cell therapy, with reports in 33 out of 53 patients, 22 of whom developed severe neurotoxicity within 28 days of CAR-T cell infusion (104). These challenges highlight the need for ongoing improvements and alternative approaches such as engineered TCR-T cells.
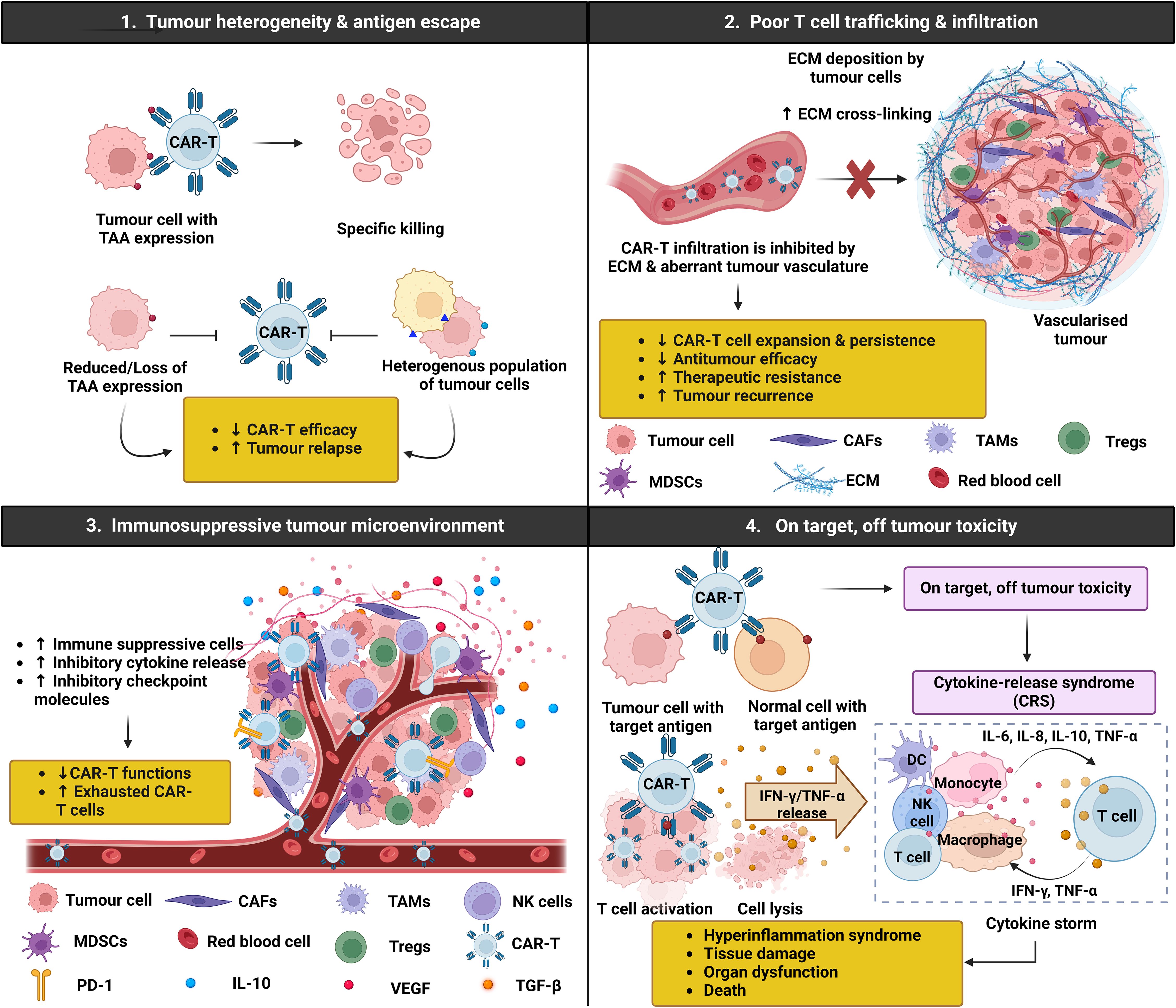
Figure 2. Chimeric antigen receptor (CAR)-T challenges in solid tumours. CAR-T cell immunotherapy has emerged as a promising therapeutic strategy for haematological malignancies, but its application in solid tumours presents several challenges and limitations that need to be addressed. Firstly, the heterogenous nature of tumour cells results in various genetic mutations and antigen expression patterns, making it difficult for CAR-T cells to effectively target all tumour cells. Additionally, tumour cells may undergo antigen escape, where they downregulate or lose the target antigen recognised by the CAR-T cells, allowing them to evade immune detection and destruction. Secondly, the clinical efficacy of CAR-T cell therapy is limited by the inability of CAR-T cells to traffic and infiltrate the tumour due to increased extracellular matrix (ECM) density and abnormal vasculature, ultimately hindering CAR-T cell diffusion and expansion. Thirdly, solid tumours often harbour a complex and highly immunosuppressive microenvironment, which can compromise the effectiveness of CAR-T cell therapy by dampening the immune response and promoting tumour growth. Moreover, higher surface expression of immune checkpoint molecules such as programmed death 1 (PD-1) on CAR-T cells can induce an exhausted phenotype, characterised by reduced cytokine production, proliferation, and cytotoxicity, limiting their capacity to control tumour growth. Lastly, CAR-T cells targeting tumour cells can induce the release of cytokines such as interferon (IFN)-γ and tumour necrosis factor (TNF)-α, activating other immune cells including dendritic cells (DCs), natural killer (NK) cells, monocytes, macrophages, and T cells. These cells further release pro-inflammatory cytokines, triggering a cascade reaction and ultimately contributing to the onset of cytokine release syndrome (CRS), a potentially severe and systemic inflammatory response.
3.2 Engineered T cell receptor-T cell therapy
The TCR is a crucial component on the T cell surface for recognising antigens on infected or tumour cells. It consists of two antigen-binding peptide chains and three CD3 subunits (ζζ homodimers and δϵ and γϵ heterodimers), forming an octamer complex. Most T cells have αβ TCRs, while a smaller subset has γδ TCRs. The TCR chains comprises an extracellular region for antigen recognition, a transmembrane region, and a shorter cytoplasmic tail linked to the CD3 complex for signal transduction (105). However, due to their lack of co-stimulatory functions, TCRs often require additional co-stimulatory signals for effective T cell activation.
Similar to CAR-T cell therapy, TCR-T cell therapy involves genetically engineering T cells with TCR genes to specifically target tumour-specific antigens (TSAs) or TAAs. Unlike CAR-T cells, which recognise antigens directly in an MHC-independent manner, TCR-T cells use naturally occurring TCRs that recognise peptides presented by MHC molecules on the cell surface (106). This involves the proteasomal degradation of cellular proteins, transport into the endoplasmic reticulum (ER), and complex formation with MHC molecules. The peptide-MHC complexes are then presented on the cell surface for T cell surveillance (107). It has been reported that TCR-T cells exhibit nearly 100- to 1000-fold greater sensitivity in recognising peptide-HLA complexes compared to CAR-T cells, enhancing immune recognition and the tumour elimination (108, 109). This high specificity leads to more selective and regulated T cell activation, reducing the risk of excessive activation and cytokine production, thus resulting in milder treatment-related toxicity compared to CAR-T cell therapy (110, 111). Therefore, TCR-T cells offer an enhanced therapeutic strategy for targeting a broader range of malignancies compared to CAR-T cell therapy.
Over the past decade, therapies involving the EBV-specific T cell infusion have shown remarkable clinical responses in treating post-transplant lymphoproliferative disease (PTLD), a life-threatening complication following organ transplantation (112). Subsequent studies have explored the use of TCRs specific to EBV viral antigens, such as EBNAs and LMPs, in treating EBV-associated malignancies, including NPC. For example, retroviral transduction of engineered EBV antigen-specific TCRs into primary human T cells have demonstrated increased IFN-γ production and efficient lysis of tumour cells (113). Cho and co-workers developed a TCR specific to LMP1 from HLA-A*0201 transgenic mice immunised with the minimal epitope LMP1166 (TLLVDLLWL). Infusion of these engineered T cells into immunocompromised mice revealed specific activation by low peptide concentrations and efficient recognition of LMP1-expressing tumour cells, demonstrating high avidity for antigen recognition in EBV-associated malignancies (23). Promising outcomes were also observed with LMP2-specific TCR, where transduced CTLs increased IFN-γ production and specific lysis of target cells. Infusion of these CTLs significantly attenuated the growth of LMP2-expressing tumours in vivo, improving survival rates in tumour-bearing mice (75). Additionally, transgenic T cells expressing LMP2-specific TCRs showed high avidity antigen-specific functions, including proliferation, cytotoxicity, and cytokine release (IFN-γ, TNF-α, and IL-2) against EBV-positive NPC cells. These engineered T cells effectively suppressed LMP2-positive tumour growth in an immunocompromised mouse model and specifically recognised and targeted LMP2-expressing NPC cells in advanced NPC patients (76). Taken together, these findings highlight the potential of TCR-T cell therapies in redirecting T cells to recognise EBV antigens, demonstrating efficacy in combating EBV-associated malignancies such as NPC.
At present, TCR-T cell therapies have not received FDA approval for clinical application in solid tumours other than melanoma. However, extensive efforts are ongoing to evaluate their safety and efficacy in other cancers, including NPC. TCR-T cells have also been developed to target cancer-associated viral antigens, such as EBV, HPV, and hepatitis B virus (HBV), and are being tested in multiple clinical trials. In a phase I/II clinical trial (NCT02280811) with HPV16-E6-specific TCR-T cells, two of 12 chemotherapy-refractory, metastatic HPV16-positive cancer patients experienced objective tumour responses without off-target toxicities or dose-limiting toxicities (114). In another phase I clinical trial (NCT03899415), HBV-targeted TCR T cells for HBV-related advanced HCC post-hepatectomy or radiofrequency ablation showed promising results. Among the eight patients, one achieved a partial response lasting 27.7 months, with only mild adverse events reported. Additionally, circulating HBV antigens and HBV DNA loads were reduced in all patients after TCR-T cell infusions, indicating effective targeting (115). For NPC, multiple clinical trials are focusing on TCR-T cells targeting EBV viral antigen such as LMP1 and LMP2. An ongoing phase I/II study (NCT05587543) is investigating LMP2-specific IL-12-secreting TCR-T cells in EBV-positive metastatic/refractory NPC patients. Another phase I trial (NCT03925896) is evaluating the safety and efficacy of LMP2-specific TCR T cells for HLA-A2, HLA-A11, and HLA-A24 recurrent and metastatic NPC patients. Additionally, a phase II study (NCT03648697) is assessing the safety and tolerability of LMP1, LMP2 and EBNA1-specific TCRs (YT-E001) in HLA-A02:01-/24:02-/11:01-positive recurrent or metastatic NPC patients. These high affinity TCRs targeting specific EBV antigens were screened from healthy donors and transduced into autologous T cells via lentiviral vectors.
Despite their promise, TCR-T cell therapies face challenges, including antigen selection, tumour heterogeneity, T cell exhaustion, and safety concerns. Ideal target antigen should be selectively and homogeneously expressed in tumours and presented via MHC class I to minimise off-target effects and treatment-related toxicity. However, solid tumours often exhibit antigenic variability, increasing the risk of antigen escape and the emergence of TCR-T cell-resistant tumours (116, 117). The immunosuppressive TME further impedes TCR-T cell infiltration and their efficacy in eliminating tumour cells (118). Additionally, prolonged TCR stimulation can induce T cell exhaustion, characterised by upregulated immune checkpoint proteins, such as PD-1, CTLA-4, T cell immunoglobulin and mucin domain-containing protein (TIM)-3, and LAG-3 (119). Tumours can also evade TCR-T cell recognition through mechanisms, such as downregulation of loss of MHC class I molecules through genetic alterations or epigenetic silencing (120). For example, non-responsive patients in a phase I/II clinical trial (NCT02858310) targeting HPV-16 E7 antigen showed tumour resistance due to defects in antigen presentation and interferon response pathways (121). Therefore, developing screening assays to identify genomic alterations associated with treatment resistance is important in order to improve TCR-T cell therapy efficacy.
3.3 Tumour-infiltrating lymphocytes therapy
Tumour-infiltrating lymphocytes (TILs) represent a subset of intratumour lymphocytes, and their adoption for treating advanced solid tumours has shown promising clinical outcomes. TIL therapy involves isolating these cells from the tumour, cultivating them in vitro, and reinfusing them into the patient with a high dose of IL-2 after lymphodepletion to enhance T cell survival and target tumour cells (122, 123). The efficacy of TIL therapy was first established by Steven Rosenberg in 1988 (124), and has now shown encouraging outcomes in NPC.
EBV antigens in NPC have led to the exploration of EBV-specific T cells as an alternative treatment. Studies showed that TILs isolated from NPC patients consists of a high frequency of CD4+ T cells that produce IFN-γ in response to EBNA1, aiding tumour regression (125). A number of clinical studies have delved into assessing the effectiveness of EBV-specific T cells in patients with NPC. These investigations have revealed promising trends, notably in the alteration of EBV DNA copy numbers and the augmentation of CTL levels, indicating a potential therapeutic benefit of EBV-specific T cell therapy in NPC patients. For instance, a phase I clinical study targeting EBV-positive NPC resistant to RT and chemotherapy with EBV-specific CTLs showed acceptable safety and modest objective responses. Among the 10 participants, two displayed partial responses and four maintained stable disease states (81). Building on this success, a subsequent phase II study in 23 recurrent or refractory NPC patients showed well-tolerated autologous EBV-CTL infusion, with 1-year and 2-year PFS rates of 65% and 52%, and overall survival rates of 87% and 70%, respectively (82). Furthermore, in a phase III clinical trial (NCT02578641), when EBV-CTLs were used to treat NPC patients after completing a first course of chemotherapy, the response rate increased to 71.4%, including three complete responses and 22 partial responses (83). A phase I study (NCT01462903) evaluating the safety and antitumour activity of TILs following CCRT in locoregionally advanced NPC (LANPC) patients showed promising results, including sustained antitumour activity, extended disease-free survival (> 12 months in 18 out of 20 patients), and enhanced EBV antigen-specific T cell responses (84). However, a completed phase II study (NCT00834093) evaluating EBV-specific CTLs in patients with recurrent, metastatic NPC demonstrated a poor overall response rate (ORR) and a median progression-free survival (PFS) of only 2.2 months. Of the 28 patients enrolled, 21 were treated, only one patient achieved a complete response, two experienced stable disease, and the remaining patients had disease progression. Interestingly, two patients who had previously failed the same chemotherapy regimen showed a renewed and robust response to chemotherapy after receiving EBV-CTL immunotherapy (85). These findings suggest that the efficacy of EBV-CTL immunotherapy may be enhanced when used in combination with other conventional therapies. Combination therapies, such as EBV-specific CTL with PD-1 blockade, have shown potential, as evidenced by a complete response with no evidence of disease relapse for 22 months in a metastatic EBV-positive NPC patient, suggesting potential for synergistic combination therapies (86). An ongoing phase I clinical trial (NCT02065362) takes a step further by examining the antitumour activity of TGF-β resistant, EBV-specific CTLs in patients with EBV-positive NPC. These genetically modified T cells incorporate a dominant negative receptor (DNR), conferring resistance to TGF-β and augmenting their efficacy in eliminating tumour cells. The ongoing or completed clinical approaches utilising EBV-specific TILs for treating NPC are summarised in Table 4.
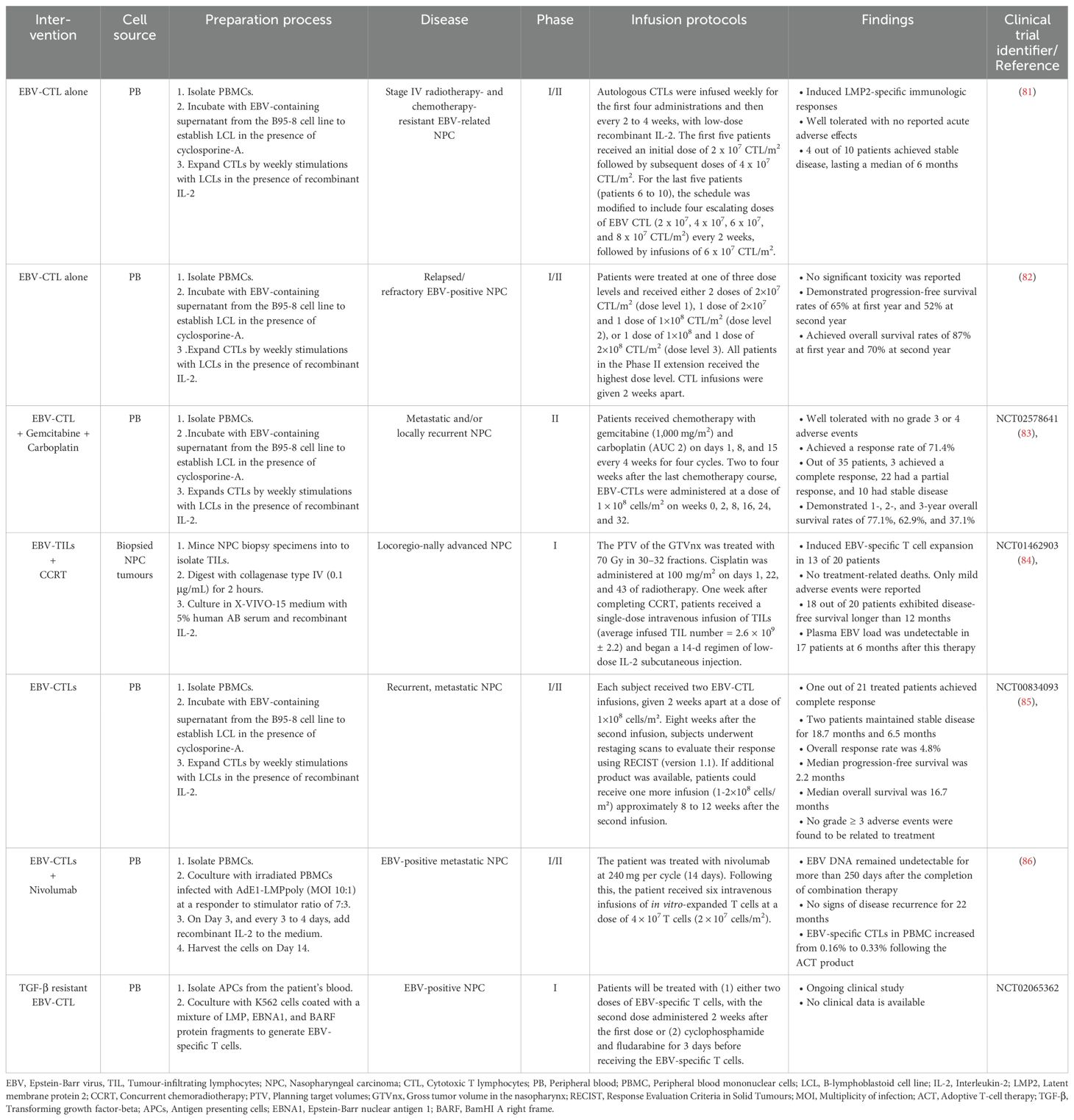
Table 4. Clinical trials of EBV-specific TIL therapy in NPC. .
Despite promising outcomes, TIL-based therapy encounters significant challenges. Initial steps involve invasive surgical resection for TIL isolation can be distressing and poses risks to patients (126). The heterogeneity of TILs in terms of antigen specificity and differentiation stages may affects their effectiveness against tumours, impacting the overall success of the therapy (127). Challenges in TIL expansion and preparation include the need for specialised facilities and technical expertise for TIL culture and expansion (128), and patient factors such as age and overall health may influence the success of ex vivo expansion (129). Furthermore, the lengthy process of isolation, expansion, and reintroduction into the patients’ body is around 3 weeks to 3 months, considered a prolonged duration that inevitably generates delays for patient intervention (130). Ongoing research aims to optimise expansion protocols and shorten production times, while exploring combination therapies involving TILs and conventional treatments that hold potential for synergistic efficacy in cancer therapy.
3.4 Natural killer cell therapy
Natural killer (NK) cells offer a promising alternative for cellular immunotherapy beyond T cells. As a key component of the innate immune system, NK cells play a vital role in cancer immune surveillance, eradicating tumour cells in an antigen-independent manner without requiring HLA matching (131). Their cytotoxic effects involve releasing perforin and granzymes, and activating killer activating receptors (KARs), leading to apoptosis via the expression of death ligands, such as TNF-α, FasL, and TRAIL. NK cells also modulate immunity by producing cytokines and chemokines, including IFN-γ, IL-10, CCL3, CCL4, and CCL5 (132). Several studies have confirmed the close relationship between NK cells and cancer development, with higher NK cell activity correlating with reducing susceptibility to oncogenic virus infection and improved survival (133, 134).
Adoptive transfer of autologous NK cells for cancer treatment is feasible due to easy sourcing and low risk of GVHD, but its limited tumour regression restricts clinical application (135). Researchers now focus on allogeneic NK cells, which offer advantages such as MHC-unrestricted immune recognition and lower GVHD incidence compared to CAR-T cell therapy. These cells, derived from PB, UCB, hESCs, iPSCs, or NK-92 cell lines, provide versatility for large-scale manufacturing and cryopreservation, allowing off-the-shelf availability (131). Among these, PB is the most accessible source but has limitations such as low cell counts (136), reduced proliferation and short lifespan (137), decreased cytotoxicity after cryopreservation (138), donor variability (139), and heterogenous NK cell populations, may potentially lead to variable NK cell function (140).
In contrast, UCB-derived NK cells offer several advantages over PB, including easier collection and long-term cryopreservation (141), better proliferation (142), enhanced bone marrow homing ability (143), and lower GVHD incidence (144). However, they have higher CD161 expression, limiting maturity and response to foreign antigens (143). The lower expression of adhesion molecules such as intercellular adhesion molecule (ICAM)-1 also limit their capacity to form conjugates with target cells (142, 145). They also exhibit higher expression of inhibitory molecules such as NKG2A/CD94, lower expression of CD16 (receptor that facilitates ADCC), CD57 (NK cell maturation marker), and KIR2DL4 (activating receptor), and reduced production of perforin, granzyme B, IFN-γ, and cell surface FasL and TRAIL, reducing cytotoxicity against tumour cells (146). However, cytokines such as IL-2, IL-12, IL-15, and IL-18 can restore UCB-NK cell cytotoxicity (147), and monoclonal antibodies such as monalizumab can enhance NK cell activity by inhibit the inhibitory function of NKG2A (148).
As discussed earlier, hESCs/iPSCs can differentiate into NK cells, offering an alternative source for allogeneic NK cells by providing homogenous cell populations that can be grown indefinitely. Their potential for genetic engineering with CARs makes them a promising strategy to generate standardised, off-the-shelf NK cells with enhanced expansion, in vivo persistence, and improved antitumour activity (149, 150). Immortalised NK cell lines such as NK-92, which can be expanded ex vivo, are also promising (151, 152). NK-92 shows high antitumour activity and are the only cell line approved for CAR-NK-92 clinical trials (153). It was established in 1992 from a non-Hodgkin lymphoma patient, showing characteristics of early NK cells with the expression of CD56, CD2, and CD7, but lack CD3 (154). The continuous growth of NK-92 is IL-2 dependent, and despite lacking CD16, they exhibit significant cytotoxicity due to the expression of activating receptors, including NKp30, NKp46, NKG2D, and CD28, with almost complete loss of inhibitory killer cell immunoglobulin-like receptors (KIRs) on the surface, except for KIR2DL4. NK-92 cells also express high levels of cytotoxic effector molecules, such as perforin, granzyme B, FasL, TRAIL, and TNF-α, consistently inducing high cytotoxic activity against tumour cells (155). In order to minimise the risk of developing lymphoma in recipients due to the origin of NK-92 cells, they require irradiation before infusion to prevent continuous growth while maintaining their cytotoxicity (156). Multiple phase I trials (NCT00990717, NCT00900809) have evaluated the safety and efficacy of irradiated NK-92 cells, showing favourable clinical outcomes, further reinforcing the potential of their therapeutic application (157, 158). However, irradiation may limit their expansion capacity, potentially diminishing overall antitumour efficacy (159).
There is growing research on NK cell-based therapy for NPC. LMP2 antigens are potential targets for NK cells, with targeted clearance of LMP2-containing cells showing robust antitumour effects and minimal toxicity to normal cells. During EBV infection, LMP2A induces F3 expression via the PI3K/Akt signalling pathway, negatively affecting NK cell activation (77). F3 promotes platelet aggregation, potentially aiding cancer metastasis and evading NK cell surveillance by downregulating NK2GD ligands and suppressing IFN-γ release (160). Additionally, platelet-derived TGF-β further enhances tumour metastasis by inhibiting the expression of CD226 and CD96 on NK cell surface, protecting tumour cells from being recognised by NK cells (161). Inhibition of F3 with the administration of NK-92MI (independent of exogenous IL-2) or UCB-derived NK cells has been shown to restore NK cell antitumour function in an NPC xenograft mouse model (77). UCB-derived NK cells also suppressed brain metastasis in a recurrent NPC patient post chemoradiotherapy, showing significant reduction over two years, with sustained efficacy (87). Combining RT with PD-1 inhibition synergistically increases NK cell-mediated killing of NPC cells in vitro and in vivo (78). Additionally, LMP1 induced B7-H3 expression via PI3K/AKT/mTOR pathway activation, leading to a reduction of NK cell cytotoxicity. Deleting B7-H3 in tumour cells and using anti-PD-L1 treatment restored NK cell function and enhanced cytotoxicity against NPC cells in xenograft models, suggesting the potential when combined with NK cell-based immunotherapy and immune checkpoint blockade against EBV-associated NPC (79). In a recent Phase I/II study (NCT02507154), Lim and co-workers assessed the safety and efficacy of combining cetuximab (anti-EGFR) with autologous expanded NK cells in EGFR-positive, recurrent and/or metastatic NPC patients who had failed at least two prior lines of chemotherapy. The treatment was well tolerated, with three out of seven patients achieved stable disease after the first NK cell infusion and experiencing a relatively long time to disease progression, lasting up to 19 months with two NK cell infusions (88).
3.5 CAR-NK cell therapy
NK-92 cells, known for their long-term cryopreservation capability and uniform population, are increasingly integrated with cancer-targeting CARs (CAR-NK-92). The first-generation CAR-NK cells feature a single signalling domain (CD3ζ), which is insufficient for potent killing. In contrast, the second- and third-generation CAR-NK cells incorporate additional co-stimulatory molecular motifs, such as CD28 or CD137 (4-1BB) for improved efficacy (162). For instance, anti-CD-19/CD22 bispecific CAR-NK-92 cells incorporating a CD3ζ/CD137 signalling domain exhibited increased cytotoxicity against B cell lymphoma cells compared to anti-CD19 CAR-NK-92 cells alone (163). Third-generation CAR-NK-92 cells targeting HER1 in HNSCC demonstrated enhanced antitumour immune responses characterised by increased INF-γ secretion and CD107a expression, a degranulation marker. Despite their enhanced killing activity, challenges such as PD-L1 upregulation and expansion of CD44v6-positive (putative CSC marker) on surviving HNSCC cells have been reported (164). This suggests that relying solely on CARs targeting a single TAA may not be effective as monotherapy, potentially leading to tumour relapse and treatment resistance. Therefore, combination therapy approaches are necessary to enhance CAR-NK cell efficacy. For instance, second-generation CAR-NK-92 cells co-expressing anti-HER2 and soluble PD-1 significantly increased NK cell and T cell infiltration and effector molecule release, enhancing immunological antitumour efficacy in PD-L1+Her2+ breast cancer cells (165). Similarly, a third-generation CAR targeting epidermal growth factor receptor (EGFR) in NK-92 cells, combined with the kinase inhibitor cabozantinib, effectively lysed EGFR-positive renal cell carcinoma (RCC) cells and improved tumour homing (166).
Currently, only a few registered clinical trials on the US Clinical Trials Registry (ClinicalTrials.gov) investigate CAR-NK-92 for hematological or solid tumours. For instance, a phase I clinical trial (NCT02944162) assessed the safety profile and clinical efficacy of third-generation CAR-NK-92 cells targeting CD33 in relapsed/refractory AML (167). Although no significant clinical efficacy was observed due to the decreased cytotoxic potential following irradiation, the therapy demonstrated safety with a high cell dose (five billion cells per patient) (167). Another ongoing phase I clinical trial (NCT03383978) is evaluating the safety and tolerability of a second-generation HER-2 specific CAR-NK-92 for recurrent glioblastoma. The trial has shown a favourable safety profile with local intracerebral injections of up to 1 x 108 HER2-CAR-NK-92 cells (168). New advancements include high-affinity Fc-receptor-expressing NK cells (haNKs), derived from NK-92 cells, which can be genetically engineered to express CARs, augmenting their targeting capabilities and cytotoxic potential (169). CAR-haNK cells targeting PD-L1 have demonstrated successful recognition and targeting of heterogeneous tumour cell populations, including both T cell-sensitive and T cell-resistant tumour cells, in an HNSCC mouse model, effectively overcoming immune resistance (170). A phase II clinical trial (NCT04847466) is evaluating the effectiveness of irradiated PD-L1 CAR-haNK cells in combination with the PD-1 inhibitor pembrolizumab plus N-803 (ALT-803, an IL-15 agonist) in patients with recurrent or metastatic gastric cancer and HNSCC. Preliminary studies (NCT04050709) have shown PD-L1 CAR-haNK cells at a dose of 2 x 109 cells twice weekly are well-tolerated, with no dose-limiting toxicities or CRS, supporting their advancement to phase II clinical trial (171). While studies have not yet evaluated CAR-NK products in NPC patients, the promising therapeutic efficacy observed in other solid tumours suggests that exploring genetic engineering of the ex vivo expanded NK cells could offer clinical benefits in NPC treatments.
Despite their promise, CAR-NK-92 cells require irradiation before infusion, which affects their viability and cytotoxicity more rapidly than non-irradiated cells (172). Therefore, the dosage and impact of irradiation on CAR-NK-92 cells needs to be carefully considered in future clinical trials. Given the short lifespan of irradiated CAR-NK-92 cells, shortening the interval between infusions may improve therapeutic efficacy. Similar to CAR-T cells, CAR-NK-92 cells exhibit target-dependent cytotoxicity and may induce on-target, off-tumour toxicity if target antigens are expressed in normal tissues (173). To address this, strategies like incorporating suicide genes into CAR constructs can serve as a safety switch, enabling control over engineered cell activity and facilitating elimination if necessary. This approach could improve safety and effectiveness of CAR therapies (174, 175).
4 Strategies to improve the efficacy of cell-based therapies in NPC
As discussed earlier, despite the encouraging results of cell-based therapies in hematological malignancies, their application in solid tumours is still in the clinical trial phase and faces numerous challenges. In this section, we will explore several strategies aimed at overcoming the insufficient infiltration of adoptively transferred cell types to the tumour site and improving the efficacy of the cellular therapies.
4.1 Alteration of chemokine expression profile
Chemokines are small cytokines that regulate immune cell migration and trafficking. The chemokine expression profile of solid tumours is complex and influenced immune cell recruitment to the TME and tumour growth (176). Strategies targeting chemokine and their receptors are increasingly used in immunotherapy to enhance the effectiveness of CAR-engineered immune cells. For instance, CAR-T cells engineered with IL-7 and CCR2b (7x2b CAR-T) showed improved survival and migration to the tumour site by boosting IFN-γ, IL-2, and granzyme production (177). Similarly, co-expressing CXCR1 in CAR-NK cells targeting NKG2D ligands exhibit better tumour trafficking and growth inhibition (178). Additionally, genetically modified oncolytic viruses (OVs) can alter the tumour chemokine profile, aiding in antigen presentation and boosting chemokine production to recruit immune cells into the TME (179). Studies show that intratumoral administration of a CXCL11-armed tumour selective vaccinia OV increases tumour-specific CTLs and granzyme B production, while reducing immunosuppressive cytokines in the TME of a syngeneic mouse mesothelioma model, leading to enhanced cytotoxic activities of CTLs (180). In another study, Moon and co-workers evaluated the synergistic effects of CXCL11 and mesothelin-redirected CAR-T cells. While CAR/CXCL11 showed limited T cell trafficking, VV.CXCL11, an oncolytic vaccinia virus producing CXCL11, effectively increased T cell infiltration and improved antitumour efficacy after adoptive T cell therapy (181). Thus, combining oncolytic virotherapy with adoptive T cell transfer holds promise for enhancing NPC therapy efficacy.
4.2 Targeting extracellular matrix and stromal cells
Solid tumours are enriched with extracellular matrix (ECM), stromal cells, and immunosuppressive cells, creating barriers that hinder immune cell penetration and infiltration (182). To tackle the problem, researchers have engineered CAR-T cells to target ECM components and cancer-associated fibroblasts (CAFs). For instance, CAR-T cells expressing heparanase (HPSE) can break down ECM components and reduce fibrosis, improving immune cell infiltration and antitumour effects (183). Fibroblast activation protein (FAP), a marker distinguishing CAFs from their normal counterparts, has been found in over 90% of epithelial cancers, including NPC, and is often correlated with poor prognosis (184). Preclinical studies shows that CAR-T cells targeting FAP can eliminate CAFs, suppress myeloid-derived suppressor cells (MDSCs) recruitment, and enhance CTL and CAR-T cell survival (185). At present, only two clinical trials have been conducted using anti-FAP CAR-T cells. A phase I clinical trial in malignant pleural mesothelioma (NCT01722149) reported that localized injection of these CAR-T cells was well tolerated with ongoing antitumour immune responses (186, 187). Another phase I clinical trial (NCT03932565) is evaluating the safety of fourth-generation CAR-T cells targeting Nectin4 and FAP, combined with IL-7, CCL19, or IL-12 for advanced Nectin4-positive solid tumours. Pre-clinical studies show that Nectin4-targeted CAR-T cells with IL-7 and CCL19 help prevent CAR-T cell exhaustion by reducing immune checkpoint expression, while FAP-targeted CAR-T cells with IL-12 enhance immune cells recruitment (188). These combination therapies show potential for improving treatment outcomes in NPC.
4.3 CAR-T cells secreting bispecific T-cell engagers
Over the past few decades, bispecific antibodies (BsAbs), especially bispecific T cell engagers (BiTEs), have proven effective in treating hematologic malignancies by binding two different antigens to direct immune cells to tumour (189). BiTEs facilitate direct interaction between T cells and tumour cells directly by binding CD3 on T cells and TAA on tumour cells, bypassing the need for antigen-presenting cells (APCs) (190). This approach helps overcome antigen loss and variability, as seen with EGFR variant III (EGFRvIII) CAR-T cells secreting BiTEs to target glioblastoma cells while activating bystander T cells, enhancing the antitumour response against heterogeneous tumours (191). A study by Yin and co-workers showed that BiTE-secreting T cells EGFR and interleukin-13 receptor alpha 2 (IL13Rα2) exhibited superior antitumour activity with higher sensitivity and specificity compared to their CAR-T counterparts in glioblastoma model (192). Overall, these findings suggest that BiTE-secreting CAR-T cells could be a promising approach to address challenges associated with antigen heterogeneity in solid tumours. While research on using BiTEs to enhance CAR-T efficacy in targeting NPC is limited, targeting LMPs expressed on EBV-infected cell surfaces, but not on normal cells, could be explored as a potential approach for BsAbs. This concept is supported by second-generation CAR-T cells targeting LMP1 in LMP1-positive NPC cells, demonstrating specific killing of NPC cells and inhibition of tumour growth in xenograft model (73).
5 Strategies to improve the safety of cell-based therapies in NPC
While cell-based therapies represent an innovative treatment in the oncology field, showing promising results in multiple clinical trials, they also carry the risk of potentially life-threatening or even fatal toxicities. In a recent study, Fusaroli and co-workers review post-approval adverse events associated with tisagenlecleucel and axicabtagene ciloleucel between October 2017 and September 2020 using the FDA Adverse Events Reporting System (FAERS) database. This database supports the FDA’s post-marketing safety surveillance program for drug and therapeutic biologic products. The study identified a total of 3225 reports (1793 for axicabtagene ciloleucel and 1433 for tisagenlecleucel), with CRS and neurotoxicity reported as the two major complications. Notably, 75% of these events occurred within the first 10 days of CAR-T therapy (193). Thus, enhancing the safety of cell-based therapies in NPC is critical for their efficacy and clinical application. This section explores strategies aimed at mitigating risks associated with these therapies, ensuring they are both effective and safe for patients.
5.1 Improving the safety of CAR-T by DNAX activation protein of 12 kDa (DAP12)
An increasing body of evidence suggests that CAR toxicity may be linked to the synthetic nature of the receptor design (194). To address this, researchers have constructed a natural multi-chain immunoreceptor CAR using DAP12 instead of CD3ζ. DAP12, a 12-kDa transmembrane adaptor protein with a single immunoreceptor tyrosine-based activation motif (ITAM), was originally found to activate NK cells and is involved in transmitting activating signals from various receptors (195). Studies show that DAP12-based CARs offer superior antitumour activity and safety than CD3ζ-based CARs, with improved antigen-specific cytotoxicity, TIL proliferation, reduced toxicity, and lower production of pro-inflammatory cytokines (196, 197), highlighting the potential of DAP12 in mitigating the risk of CRS. For instance, a phase I clinical trial (ChiCTR1800016584) of CD19-KIRS2/DAP12-BB CAR-T cells reported complete responses in all patients with low toxicity (198). Similarly, CAR-NK cells incorporating DAP12 have shown promising results in treating solid tumours. In a recent study, Xiao and co-workers constructed a CAR-NK cell by combining the NKG2D receptor with DAP12, which showed significant therapeutic effects and lower toxicity in mice with solid tumours. This approach also led to positive outcomes in three patients with metastatic colorectal cancer (199). Building on these results, a pilot clinical trial (NCT03415100) has been launched to evaluate the safety and feasibility of these CAR-NK cells for treating metastatic solid tumours.
5.2 Incorporation of suicide genes to address the challenge of toxicity
Another strategy to enhance the safety of CAR-based cell therapy is the engineering of suicide genes such as inducible caspase 9 (iCasp9) into the CAR construct. These suicide genes serve as a safety switch that can induce cell death upon activation by an external agent, such as drug or antibody (174, 175). For instance, the iCasp9 gene is often used in CAR-based cell therapy in conjunction with a small, bio-inert molecule AP1903 (Rimiducid), which acts as a chemical inducer of dimerization (CID) (200). When administered, AP1903 binds to the CID domain fused to iCasp9, leading to the formation of homodimers and subsequent activation of caspase 9. This activation triggers apoptotic cell death specifically in the CAR-engineered immune cells that express high levels of the transgene, allowing for the selective removal of inappropriately activated cells and thus providing a safety mechanism to manage potential toxicities or adverse events associated with CAR-T cell therapy (200). The efficacy of iCasp9 in eliminating CAR-engineered immune cells to counteract serious adverse event in CAR-based cell immunotherapy has been demonstrated in several preclinical studies (175, 201). Furthermore, several early phase clinical trials (NCT05239143, NCT03016377, NCT03696784 and NCT03721068) are ongoing to evaluate the safety and efficacy of this approach in patients with hematological malignancies or solid tumours, including NPC.
6 Efficient combinations of cellular therapies with conventional therapies
Conventional treatments such as RT and chemotherapy have long been standard for managing various malignancies. However, these approaches alone are often insufficient for eradicating large solid tumours or metastases, leading to recurrence or refractory disease. Additionally, the efficacy of immunotherapy may be restricted by an immunologically cold or immunosuppressive TME and its clinical success has primarily been limited to haematological malignancies (202, 203). Preclinical and clinical studies, however, suggested that combining conventional treatments with adoptive cell therapies can produce a synergistic anticancer effect, where RT or chemotherapy can relieve immune suppression, improve immune cell trafficking to the tumour sites, and enhance the antitumour activity of cytotoxic immune cells (204). For instance, a phase I clinical trial (NCT01462903) evaluating adoptive TIL immunotherapy following CCRT in locoregionally advanced NPC patients reported promising outcomes. Briefly, patients received RT (70 Gy) and cisplatin (100 mg/m2) on day 1, 22, and 43, followed by infusion of an average of 2.6 × 109 TILs (range 1.3 - 6.3 × 109) one week after CCRT. Of the 23 enrolled patients, 16 achieved a complete response by the end of CCRT, 19 maintained a complete response three months after adoptive cell transfer, and 18 experienced disease-free survival for over 12 months. This study demonstrated that CCRT prior to TIL infusion reduced tumour burden, decreased neutrophil and lymphocyte counts, and enhanced the expansion of EBV-antigen-specific T cells, leading to sustained antitumour activity and a robust anti-EBV immune response (84). Similarly, Chia and co-workers showed that metastatic and/or locally recurrent NPC patients who received a combination regimen of four cycles of gemcitabine and carboplatin, followed by up to six doses of EBV-specific CTLs, demonstrated a better response rate. Briefly, patients received 1000 mg/m2 of gemcitabine and AUC2 carboplatin on Day 1, 8, and 15 of each 28-day cycle, for a total of four cycles. This was followed by an autologous T cell infusion, with a median total CTL dose of 9.6 × 108 cells (range: 6.3–10.3 × 108 cells). Of the 38 patients enrolled, 35 received combination therapy. Among these patients, three achieved a complete response, 22 experienced a partial response, 10 had stable disease, and none developed progressive disease, resulting in a response rate of 71.4% compared to 42.9% during the CTL immunotherapy phase alone. Additionally, with a median follow-up of 29.9 months, the study reported a median progression-free survival of 7.6 months, surpassing the median PFS of 3.7 months only observed during the CTL immunotherapy phase alone (83).
In order to achieve this synergistic antitumour response, it is important to determine the order of administration, dosing, and volume of chemotherapy and RT when combined with cellular therapies to minimise toxicity while enhancing the efficacy of adoptive immune cells. For instance, RT can be administered prior to CAR-T cell therapy to reduce tumour burden and lessen the severity of CRS and neurotoxicity by decreasing the number of tumour cells for the CAR-T cells to target (205, 206). Poor MHC expression, low neoantigen load, and low density of infiltrating T lymphocytes are frequently associated with poor therapeutic response. Hence, in such conditions, it is necessary to upregulate the expression of neoantigens, converting the TME from cold to hot before receiving cellular therapies (207). To address this, lower doses of chemotherapy can be used to reduce immunosuppressive effects and toxicity, making it more compatible with cellular therapies. Shurin and co-workers demonstrated that low, non-cytotoxic concentrations of chemotherapeutic agents can upregulate the expression of antigen-presenting machinery components and co-stimulatory molecules on DCs, enhancing their ability to present antigens to antigen-specific T cells (208). Besides that, chemotherapy and RT have shown their ability to induce immunogenic cell death, characterised by the release of damage-associated molecular patterns (DAMPs) such as adenosine triphosphate (ATP), high mobility group box 1 protein (HMGB1), and calreticulin (CRT). These DAMPs are recognised by Toll-like receptor 4, which promotes the maturation and activation of DCs, thereby enhancing antigen presentation to CTLs and boosting the antitumour immune response (209, 210). McDonnell and co-workers also showed that systemic gemcitabine therapy can restore the capacity of suppressed or immature-like tumour-infiltrating DCs to cross-present antigens, thereby enhancing the DCs’ ability to present antigens to antigen-specific T cells and induce T cell activation (211). In the presence of high levels of immunosuppressive cells, it is recommended to deplete these cells or suppress their functions before administering cellular therapies. For instance, lymphodepleting chemotherapy with a combination of cyclophosphamide and fludarabine is usually given a few days before T cell infusion. These agents effectively eradicate Tregs and increase the production of homeostatic cytokine such as IL-15, which prolongs CAR-T cell expansion and persistence, thereby boosting their curative effects (212, 213). Chemotherapy can also sensitise tumour cells to cellular therapies. For example, chemotherapy-induced upregulation of mannose-6-phosphate receptors on the tumour cell surface enhances the penetration of T cells into the tumour site and increases the permeability of tumour cells to granzyme B in a perforin-independent manner. This increased permeability makes tumour cells, including bystander tumour cells that do not express tumour antigen, more susceptible to CTL-mediated cytotoxicity (214, 215). Similarly, Makowska and co-workers found that RT significantly increased the immunogenicity of NPC cells, leading to greater NK cell-induced killing compared to non-irradiated NPC cells. RT also upregulates the expression of PD-L1 on tumour cell surface, further enhancing the antitumour cytotoxicity of NK cells in combination with PD-L1/PD-1 blockade (78).
In the setting of NPC, it is well known that intensity-modulated radiation therapy (IMRT) alone or combined with chemotherapy has become the primary treatment for early or locally advanced patients. Hence, an effective approach to improving the homing and activation of infused immune cells, allowing their proper expansion without compromising overall immunity, would be to combine both high-dose and low-dose irradiation. Hypofractionated RT has been shown to be effective and well tolerated in patients with initial distant metastases in a phase II clinical trial (NCT03598218), compared to those who received IMRT (216). It is suggested that hypofractionated RT not only can directly kill tumour cells but also induces immunogenic cell death, releasing pro-inflammatory cytokines and DAMPs to enhance CTL-mediated cytotoxicity (217). Early preclinical studies have indeed shown that a hypofractionated regimen (8 Gy x 3) is superior to a single fraction of 20 Gy in promoting an antitumour immune response in combination with anti-CTLA-4 therapy, as evidenced by a significant increase in the number of CD4+ T cells and CTLs within the TME (218). Consistently, Vanpouille-Box and co-workers revealed that delivering 24 Gy in three fractions of 8 Gy promotes DC recruitment and CTL infiltration through IFN-β secretion and cGAS-STING pathway activation, enabling synergistic tumour rejection with CTLA-4 blockade therapy. However, when the single fraction dose exceeds 12 Gy, it induces the expression of DNA exonuclease Trex1, which attenuates tumour immunogenicity by degrading tumour DNA within the cytoplasm of tumour cells, leading to insufficient DC recruitment and activation of the CTL-mediated antitumour response (219). High-dose irradiation can also cause vascular damage, creating a hypoxic TME that limits CTL infiltration and leads to RT resistance (217). In contrast, low-dose RT (1 to 4 Gy) promotes immune cell infiltration without significant toxicity, reverses the suppressive function of immune cells, and inflames cold tumours, making it compatible with other anticancer treatments (220). As reported in a phase III clinical trial (NCT02456506), hyperfractionated RT significantly decreased the rate of severe adverse events and improved overall survival in patients with locally advanced recurrent NPC compared to IMRT (221). Therefore, combining a low-dose irradiation delivered in a large volume with a high dose delivered in a limited volume would improve the expansion, homing, and activation of infused T cells. Further studies are necessitated to determine optimal doses and fractionation schedules for activating the antitumour immune response while avoiding immune suppression to ensure the action of both endogenous and infused T cells.
Finally, a comprehensive evaluation and adjustment of immune cell infusion dosage are crucial for achieving optimal treatment effects. Previous studies have indicated that administering a single low dose of 2 x 105 CAR-T cells/kg was effective enough in inducing a complete response with no CRS observed in patients with high tumour burden, compared to those who received a higher dose of 2 x 107 CAR-T cells/kg (222). However, a high relapse rate was reported in this study, suggesting that a reduction in CAR-T cells may impair long-term efficacy. Therefore, dose fractionation or split dosing is recommended, wherein CAR-T cells are administered in multiple doses in the form of dose escalation. This approach aim to control T cell expansion and inflammatory cytokine secretion, striking a balance between long-term efficacy and safety of CAR-T cell therapy (223). Frey and co-workers compared three CAR-T cell infusion schemes in a pilot/phase I (NCT01029366) & and phase II (NCT02030847) study: high-dose single infusion (HDS, 5 x 108 CAR-T cells), low-dose single or fractionated infusion (LD, 5 x 107 CAR-T cells), and high-dose fractionated infusion (HDF, 5 x 108 CAR-T cells). In the fractionated infusion scheme, CAR T-cells were administered over three days (Day 1, 10%; Day 2, 30%; and Day 3, 60%). Among these groups, 20 patients in the HDF cohort achieved a complete response rate of 90% with manageable CRS, compared to the HDS cohort (n = 6), where only three patients achieved complete responses, and three patients died from CRS. Although the LD cohort (n = 9) experienced manageable CRS, only 33% patients achieved complete responses. The 2-year survival rate for the HDF cohort was 73%, compared to 22% and 17% in the LD and HDS cohorts, respectively (223). Similar fractionated dosing schemes were also evaluated in another clinical trial (NCT04309981). Administration of CAR-T cells in a fractionated manner with a booster dose induced sustained responses in patients. Of the 30 patients who received the fractionated CAR-T cells dosing, 80% experienced grade 1-2 CRS, and no neurotoxic events were reported (224). Collectively, these findings suggest that fractionated dosing of CAR-T cell infusion represents a promising strategy to ensure the safety of infused T cells without compromising their efficacy. However, further studies are needed to validate this approach across different cancer types and disease burdens, as well as to optimise the timing and dosing of infusion in order to achieve long-term favourable clinical outcomes.
In summary, combining cellular therapies with existing treatment modalities for NPC involves carefully designing synergistic combinations, sequencing, and dosing strategies. Integrating conventional treatments with cellular therapies holds promise for enhancing therapeutic efficacy and overcoming resistance. However, continuous monitoring and adaptive strategies are important for optimising patient outcomes and managing potential toxicities.
7 Conclusions and future perspective
Despite the promising outcomes demonstrated in a vast majority of preclinical studies on cell-based therapies, there have been relatively few clinical trials conducted in NPC. This indicates that sufficient clinical evidence is still lacking to fully support the implementation of these therapies into standard clinical practice. The limitations of the existing studies include small patient sample sizes, which can lead to false-positive results and reduced statistical power. Furthermore, many of these studies are non-randomised and lack control groups, which compromises the validity and generalizability of the findings (225). Additionally, long-term toxicity data are unavailable due to the short duration of observation and post-treatment follow up. This absence of long-term data makes it challenging to assess the sustained safety and efficacy of cell-based therapies. To address these issues, future research should focus on conducting larger, randomised controlled trials with extended follow-up periods to gather comprehensive data on both the benefits and potential risks of these therapies (226). This will provide a robust evidence base to support the clinical integration of cell-based therapies for NPC.
The evolution of cancer immunotherapy has revolutionised cancer treatment, offering an alternative approach to improve the survival and quality of life for NPC patients. Immunotherapy, by redirecting effector immune cells to selectively target tumour cells, offers a significant advantage over conventional treatments like RT and chemotherapy. This approach not only enhances the host’s antitumour response but also reduces treatment-related adverse events. Likewise, the integration of cellular therapies into the treatment regimen for NPC represents a transformative shift in cancer care. Traditional treatments such as RT and chemotherapy, while effective, often come with significant toxicities and limitations. Cellular therapies offer a targeted approach to overcoming these challenges. NPC, often associated with EBV, makes this cancer a suitable candidate for cellular therapies due to its expression of potentially targetable tumour-associated viral antigens. This suitability is enhanced by the capacity to genetically engineer both stem cells and non-stem cells for specific tumour cell recognition and stable expression of a variety of antitumour agents, which holds immense clinical potential. These precision therapies can potentially lead to more effective tumour control, sparing normal tissues and reducing the systemic toxicities associated with chemotherapy and RT. By priming the immune system, cellular therapies can reduce both primary and acquired resistance and offer long-lasting protection against cancer recurrence. Engineered T cells, for example, can persist in the body, providing continuous surveillance and the capability to respond to tumour relapse. This ongoing immune surveillance can significantly improve long-term patient outcomes.
However, the clinical application of cellular therapies in solid tumours, including NPC, encounters challenges arising from tumour heterogeneity and the immunosuppressive TME, potentially compromising the therapeutic efficacy. Safety concerns, including the development of GVHD, on-target, off-tumour cytotoxicity, and CRS, present additional hurdles that ongoing and future clinical trials must effectively address. To overcome these challenges and further enhance treatment outcomes, combinatorial approaches may prove pivotal. By combining cellular therapies with existing modalities, such as conventional treatments and immunotherapy, improved efficacy in targeting cancer cells and a reduction in cancer recurrence rates can be achieved. While the exploration of cell-based therapy in NPC lags behind its application in other cancers, promising findings from published and emerging research underscore its potential to significantly improve clinical outcomes for NPC patients. Not forgetting there is a need to integrate the recent cancer discoveries, ranging from cancer immunology (227), the role of epigenetic in cancer (3, 228), novel drug delivery system (229) to increase its clinical benefits and to reduce its side effects. More comprehensive studies are therefore required to further refine the efficacy and safety of cellular therapies, paving the way for their potential integration into mainstream clinical settings for the improved management of NPC.
Author contributions
C-KL: Data curation, Formal analysis, Visualization, Writing – original draft. E-ML: Data curation, Formal analysis, Visualization, Writing – original draft. H-CL: Data curation, Validation, Writing – review & editing. Y-LC: Data curation, Validation, Writing – review & editing. K-YC: Data curation, Validation, Writing – review & editing. S-CC: Supervision, Writing – review & editing. B-HG: Data curation, Validation, Writing – review & editing. C-WM: Conceptualization, Resources, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors would like to express their sincere gratitude to research grants provided by the Malaysia Ministry of Higher Education through the Fundamental Research Grant Scheme (FRGS/1/2023/SKK10/UCSI/02/1) and UCSI University Research Excellence & Innovation Grant (REIG-FPS-2023/038).
Acknowledgments
The authors would like to express their sincere gratitude to UCSI University, Sunway University Malaysia, and Universiti Kebangsaan Malaysia for their kind support and library resource for completing this review. All figures in this review paper were created by the authors using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang ET, Ye W, Zeng Y, Adami H. The evolving epidemiology of nasopharyngeal carcinoma. Cancer Epidemiology Biomarkers Prev. (2021) 30:1035–47. doi: 10.1158/1055-9965.EPI-20-1702
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
3. Looi CK, Foong LC, Chung FFL, Khoo ASB, Loo EM, Leong CO, et al. Targeting the crosstalk of epigenetic modifications and immune evasion in nasopharyngeal cancer. Cell Biol Toxicol. (2023) 39:2501–26. doi: 10.1007/s10565-023-09830-9
4. Sun X, Li X, Chen Q, Tang L, Mai H. Future of radiotherapy in nasopharyngeal carcinoma. Br J Radiol. (2019) 92:20190209. doi: 10.1259/bjr.20190209
5. Teow S, Yap H, Peh S. Epstein-barr virus as a promising immunotherapeutic target for nasopharyngeal carcinoma treatment. J Pathog. (2017) 2017:7349268. doi: 10.1155/2017/7349268
6. Thamboo A, Patel VS, Hwang PH. 5-year outcomes of salvage endoscopic nasopharyngectomy for recurrent nasopharyngeal carcinoma. J Otolaryngology-Head Neck Surg. (2021) 50:12. doi: 10.1186/s40463-020-00482-x
7. Ding X, Liu Y, Hua Y, Zou X, Wang Z, Xie Y, et al. Clinical advances in nasopharyngeal carcinoma surgery and a video demonstration. Visualized Cancer Med. (2021) 2:2. doi: 10.1051/vcm/2021001
8. Wei WI, Kwong DL. Current management strategy of nasopharyngeal carcinoma. Clin Exp otorhinolaryngology. (2010) 3:1–12. doi: 10.3342/ceo.2010.3.1.1
9. Kubeček O, Paterová P, Novosadová M. Risk factors for infections, antibiotic therapy, and its impact on cancer therapy outcomes for patients with solid tumors. Life. (2021) 11:1387. doi: 10.3390/life11121387
10. Dobosz P, Stępień M, Golke A, Dzieciątkowski T. Challenges of the immunotherapy: perspectives and limitations of the immune checkpoint inhibitor treatment. Int J Mol Sci. (2022) 23:2847. doi: 10.3390/ijms23052847
11. Chen Y, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers. (2023) 15:663. doi: 10.3390/cancers15030663
12. Prasad M, Ponnalagu S, Zeng Q, Luu K, Lang SM, Wong HY, et al. Epstein-Barr virus-induced ectopic CD137 expression helps nasopharyngeal carcinoma to escape immune surveillance and enables targeting by chimeric antigen receptors. Cancer Immunology Immunotherapy. (2022) 71:2583–96. doi: 10.1007/s00262-022-03183-8
13. Liu WN, So WY, Harden SL, Fong SY, Wong MXY, Tan WWS, et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci Adv. (2022) 8:eadd1187. doi: 10.1126/sciadv.add1187
14. Gong L, Kwong DL, Dai W, Wu P, Li S, Yan Q, et al. Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma. Nat Commun. (2021) 12:1–18. doi: 10.1038/s41467-021-21795-z
15. Liu X, Shen H, Zhang L, Huang W, Zhang S, Zhang B. Immunotherapy for recurrent or metastatic nasopharyngeal carcinoma. NPJ Precis Oncol. (2024) 8:101. doi: 10.1038/s41698-024-00601-1
16. Looi CK, Hii L, Chung FF, Mai C, Lim W, Leong C. Roles of inflammasomes in epstein–barr virus-associated nasopharyngeal cancer. Cancers. (2021) 13:1786. doi: 10.3390/cancers13081786
17. Lo AK, Dawson CW, Lung HL, Wong K, Young LS. The role of EBV-encoded LMP1 in the NPC tumor microenvironment: from function to therapy. Front Oncol. (2021) 11:640207. doi: 10.3389/fonc.2021.640207
18. Choi I, Wang Z, Ke Q, Hong M, Qian Y, Zhao X, et al. Signaling by the Epstein–Barr virus LMP1 protein induces potent cytotoxic CD4 and CD8 T cell responses. Proc Natl Acad Sci. (2018) 115:E686–95. doi: 10.1073/pnas.1713607115
19. Rooney CM, Ng C, Loftin S, Smith CA, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. (1995) 345:9–13. doi: 10.1016/S0140-6736(95)91150-2
20. Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, et al. Long–term restoration of immunity against Epstein–Barr virus infection by adoptive transfer of gene–modified virus–specific T lymphocytes. Nat Med. (1996) 2:551–5. doi: 10.1038/nm0596-551
21. Lin C, Lo W, Lee T, Ren Y, Hwang S, Cheng Y, et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8 T-cell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. (2002) 62:6952–8.
22. Comoli P, De Palma R, Siena S, Nocera A, Basso S, Del Galdo F, et al. Adoptive transfer of allogeneic Epstein–Barr virus (EBV)-specific cytotoxic T cells with in vitro antitumor activity boostsLMP2-specific immune response in a patient with EBV-related nasopharyngeal carcinoma. Ann Oncol. (2004) 15:113–7. doi: 10.1093/annonc/mdh027
23. Cho H, Kim U, Shin A, Won J, Lee H, Sohn H, et al. A novel Epstein–Barr virus-latent membrane protein-1-specific T-cell receptor for TCR gene therapy. Br J Cancer. (2018) 118:534–45. doi: 10.1038/bjc.2017.475
24. Kim I. A brief overview of cell therapy and its product. J Korean Assoc Oral Maxillofac Surgeons. (2013) 39:201–2. doi: 10.5125/jkaoms.2013.39.5.201
25. Thomas ED, Lochte HL Jr., Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. (1957) 257:491–6. doi: 10.1056/NEJM195709122571102
26. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. (2011) 17:3520–6. doi: 10.1158/1078-0432.CCR-10-3126
27. O'Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. (2019) 25:1142–6. doi: 10.1158/1078-0432.CCR-18-2035
28. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. (2019) 25:1702–8. doi: 10.1158/1078-0432.CCR-18-2743
29. Victoria J. T-Cell Therapy Racks Fast Track, Orphan Designations for EBV-Nasopharyngeal Carcinoma (2023). Available online at: https://www.cgtlive.com/view/t-cell-therapy-racks-fast-track-orphan-designations-for-ebv-nasopharyngeal-carcinoma (accessed December 27, 2023).
30. Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. (2019) 10:1–22. doi: 10.1186/s13287-019-1165-5
31. Chu D, Nguyen TT, Tien NLB, Tran D, Jeong J, Anh PG, et al. Recent progress of stem cell therapy in cancer treatment: molecular mechanisms and potential applications. Cells. (2020) 9:563. doi: 10.3390/cells9030563
32. Gurusamy N, Alsayari A, Rajasingh S, Rajasingh J. Adult stem cells for regenerative therapy. Prog Mol Biol Trans Sci. (2018) 160:1–22. doi: 10.1016/bs.pmbts.2018.07.009
33. Lee JY, Hong S. Hematopoietic stem cells and their roles in tissue regeneration. Int J Stem Cells. (2020) 13:1–12. doi: 10.15283/ijsc19127
34. Flores C, Pham C, Snyder D, Yang S, Sanchez-Perez L, Sayour E, et al. Novel role of hematopoietic stem cells in immunologic rejection of Malignant gliomas. Oncoimmunology. (2015) 4:e994374. doi: 10.4161/2162402X.2014.994374
35. Ma DK, Bonaguidi MA, Ming G, Song H. Adult neural stem cells in the mammalian central nervous system. Cell Res. (2009) 19:672–82. doi: 10.1038/cr.2009.56
36. Joo KM, Park IH, Shin JY, Jin J, Kang BG, Kim MH, et al. Human neural stem cells can target and deliver therapeutic genes to breast cancer brain metastases. Mol Ther. (2009) 17:570–5. doi: 10.1038/mt.2008.290
37. Kim SU. Neural stem cell-based gene therapy for brain tumors. Stem Cell Rev Rep. (2011) 7:130–40. doi: 10.1007/s12015-010-9154-1
38. Jiang W, Yang Y, Mercer-Smith AR, Valdivia A, Bago JR, Woodell AS, et al. Development of next-generation tumor-homing induced neural stem cells to enhance treatment of metastatic cancers. Sci Adv. (2021) 7:eabf1526. doi: 10.1126/sciadv.abf1526
39. Cordero A, Ramsey MD, Kanojia D, Fares J, Petrosyan E, Schwartz CW, et al. Combination of tucatinib and neural stem cells secreting anti-HER2 antibody prolongs survival of mice with metastatic brain cancer. Proc Natl Acad Sci. (2022) 119:e2112491119. doi: 10.1073/pnas.2112491119
40. Li M, Sun X, Kuang X, Liao Y, Li H, Luo D. Mesenchymal stem cells suppress CD8 T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2, 3-dioxygenase and transforming growth factor-β. Clin Exp Immunol. (2014) 178:516–24. doi: 10.1111/cei.12423
41. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4 CD25highFOXP3 regulatory T cells. Stem Cells. (2008) 26:212–22. doi: 10.1634/stemcells.2007-0554
42. Opo FDM, Moulay M, Alrefaei GI, Alsubhi NH, Alkarim S, Rahman MM. Effect of Co-culturing both placenta-derived mesenchymal stem cells and their condition medium in the cancer cell (HepG2) migration, damage through apoptosis and cell cycle arrest. Saudi J Biol Sci. (2023) 30:103519. doi: 10.1016/j.sjbs.2022.103519
43. Chen F, Zhong X, Dai Q, Li K, Zhang W, Wang J, et al. Human umbilical cord MSC delivered-soluble TRAIL inhibits the proliferation and promotes apoptosis of B-ALL cell in vitro and in vivo. Pharmaceuticals. (2022) 15:1391. doi: 10.3390/ph15111391
44. Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells. (2016) 34:2536–47. doi: 10.1002/stem.2412
45. Aoto K, Ito K, Aoki S. Complex formation between platelet-derived growth factor receptor β and transforming growth factor β receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget. (2018) 9:34090. doi: 10.18632/oncotarget.26124
46. Lin Z, Wu Y, Xu Y, Li G, Li Z, Liu T. Mesenchymal stem cell-derived exosomes in cancer therapy resistance: recent advances and therapeutic potential. Mol Cancer. (2022) 21:179. doi: 10.1186/s12943-022-01650-5
47. Lyu T, Wang Y, Li D, Yang H, Qin B, Zhang W, et al. Exosomes from BM-MSCs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance via upregulation of S100A4. Exp Hematol Oncol. (2021) 10:24. doi: 10.1186/s40164-021-00220-7
48. Biswas S, Mandal G, Roy Chowdhury S, Purohit S, Payne KK, Anadon C, et al. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J Immunol. (2019) 203:3447–60. doi: 10.4049/jimmunol.1900692
49. Shi S, Zhang Q, Xia Y, You B, Shan Y, Bao L, et al. Mesenchymal stem cell-derived exosomes facilitate nasopharyngeal carcinoma progression. Am J Cancer Res. (2016) 6:459.
50. Wan F, Chen K, Sun Y, Chen X, Liang R, Chen L, et al. Exosomes overexpressing miR-34c inhibit Malignant behavior and reverse the radioresistance of nasopharyngeal carcinoma. J Trans Med. (2020) 18:12. doi: 10.1186/s12967-019-02203-z
51. Wan F, Zhang H, Hu J, Chen L, Geng S, Kong L, et al. Mesenchymal stem cells inhibits migration and vasculogenic mimicry in nasopharyngeal carcinoma via exosomal MiR-125a. Front Oncol. (2022) 12:781979. doi: 10.3389/fonc.2022.781979
52. Gu H, Ji R, Zhang X, Wang M, Zhu W, Qian H, et al. Exosomes derived from human mesenchymal stem cells promote gastric cancer cell growth and migration via the activation of the Akt pathway. Mol Med Rep. (2016) 14:3452–8. doi: 10.3892/mmr.2016.5625
53. Chang L, Gao H, Wang L, Wang N, Zhang S, Zhou X, et al. Exosomes derived from miR-1228 overexpressing bone marrow-mesenchymal stem cells promote growth of gastric cancer cells. Aging (Albany NY). (2021) 13:11808–21. doi: 10.18632/aging.202878
54. Zhou W, Zhou Y, Chen X, Ning T, Chen H, Guo Q, et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials. (2021) 268:120546. doi: 10.1016/j.biomaterials.2020.120546
55. Yamanaka S. Pluripotent stem cell-based cell therapy—promise and challenges. Cell Stem Cell. (2020) 27:523–31. doi: 10.1016/j.stem.2020.09.014
56. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. (1998) 282:1145–7. doi: 10.1126/science.282.5391.1145
57. Cohen CB. Ethical issues in embryonic stem cell research. JAMA. (2001) 285:1439–40. doi: 10.1001/jama.285.11.1439
58. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
59. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
60. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. (2013) 31:928–33. doi: 10.1038/nbt.2678
61. Ni Z, Knorr DA, Clouser CL, Hexum MK, Southern P, Mansky LM, et al. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J Virol. (2011) 85:43–50. doi: 10.1128/JVI.01774-10
62. Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun. (2021) 12:430. doi: 10.1038/s41467-020-20658-3
63. Trotman-Grant AC, Mohtashami M, De Sousa Casal J, Martinez EC, Lee D, Teichman S, et al. DL4-μbeads induce T cell lineage differentiation from stem cells in a stromal cell-free system. Nat Commun. (2021) 12:5023. doi: 10.1038/s41467-021-25245-8
64. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8 T cells. Cell Stem Cell. (2013) 12:31–6. doi: 10.1016/j.stem.2012.12.006
65. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. (2013) 12:114–26. doi: 10.1016/j.stem.2012.11.002
66. Honda T, Ando M, Ando J, Ishii M, Sakiyama Y, Ohara K, et al. Sustainable tumor-suppressive effect of iPSC-derived rejuvenated T cells targeting cervical cancers. Mol Ther. (2020) 28:2394–405. doi: 10.1016/j.ymthe.2020.07.004
67. Ando M, Ando J, Yamazaki S, Ishii M, Sakiyama Y, Harada S, et al. Long-term eradication of extranodal natural killer/T-cell lymphoma, nasal type, by induced pluripotent stem cell-derived Epstein-Barr virus-specific rejuvenated T cells in vivo. Haematologica. (2020) 105:796–807. doi: 10.3324/haematol.2019.223511
68. Ando M, Nishimura T, Yamazaki S, Yamaguchi T, Kawana-Tachikawa A, Hayama T, et al. A safeguard system for induced pluripotent stem cell-derived rejuvenated T cell therapy. Stem Cell Rep. (2015) 5:597–608. doi: 10.1016/j.stemcr.2015.07.011
69. Bock AM, Knorr D, Kaufman DS. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs). JoVE (Journal Visualized Experiments). (2013) 74:e50337. doi: 10.3791/50337
70. Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci Trans Med. (2020) 12:eaaz5618. doi: 10.1126/scitranslmed.aaz5618
71. Hong D, Patel S, Patel M, Musni K, Anderson M, Cooley S, et al. 380 Preliminary results of an ongoing phase I trial of FT500, a first-in-class, off-the-shelf, induced pluripotent stem cell (iPSC) derived natural killer (NK) cell therapy in advanced solid tumors. J Immunother Cancer. (2020) 8:A231–2. doi: 10.1136/jitc-2020-SITC2020.0380
72. El-Kadiry AE, Rafei M, Shammaa R. Cell therapy: types, regulation, and clinical benefits. Front Med. (2021) 8:756029. doi: 10.3389/fmed.2021.756029
73. Tang X, Zhou Y, Li W, Tang Q, Chen R, Zhu J, et al. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J Biomed Res. (2014) 28:468. doi: 10.7555/JBR.28.20140066
74. Tang X, Tang Q, Mao Y, Huang X, Jia L, Zhu J, et al. CD137 co-stimulation improves the antitumor effect of LMP1-specific chimeric antigen receptor T cells in vitro and in vivo. OncoTargets Ther. (2019) 12:9341–50. doi: 10.2147/OTT.S221040
75. Yang D, Shao Q, Sun H, Mu X, Gao Y, Jiang R, et al. Evaluation of Epstein-Barr virus latent membrane protein 2 specific T-cell receptors driven by T-cell specific promoters using lentiviral vector. J Immunol Res. (2011) 2011:716926. doi: 10.1155/2011/716926
76. Zheng Y, Parsonage G, Zhuang X, MaChado LR, James CH, Salman A, et al. Human leukocyte antigen (HLA) A* 1101-restricted Epstein-Barr virus–specific T-cell receptor gene transfer to target nasopharyngeal carcinoma. Cancer Immunol Res. (2015) 3:1138–47. doi: 10.1158/2326-6066.CIR-14-0203-T
77. Duan X, Chen H, Zhou X, Liu P, Zhang X, Zhu Q, et al. EBV infection in epithelial Malignancies induces resistance to antitumor natural killer cells via F3-mediated platelet aggregation. Cancer Res. (2022) 82:1070–83. doi: 10.1158/0008-5472.CAN-21-2292
78. Makowska A, Lelabi N, Nothbaum C, Shen L, Busson P, Tran TTB, et al. Radiotherapy combined with PD-1 inhibition increases NK cell cytotoxicity towards nasopharyngeal carcinoma cells. Cells. (2021) 10:2458. doi: 10.3390/cells10092458
79. Chen H, Duan X, Deng X, Huang Y, Zhou X, Zhang S, et al. EBV-upregulated B7-H3 inhibits NK cell–mediated antitumor function and contributes to nasopharyngeal carcinoma progression. Cancer Immunol Res. (2023) 11:830–46. doi: 10.1158/2326-6066.CIR-22-0374
80. Li D, Guo X, Yang K, Yang Y, Zhou W, Huang Y, et al. EpCAM-targeting CAR-T cell immunotherapy is safe and efficacious for epithelial tumors. Sci Adv. (2023) 9:eadg9721. doi: 10.1126/sciadv.adg9721
81. Comoli P, Pedrazzoli P, Maccario R, Basso S, Carminati O, Labirio M, et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J Clin Oncol. (2005) 23:8942–9. doi: 10.1200/JCO.2005.02.6195
82. Louis CU, Straathof K, Bollard CM, Ennamuri S, Gerken C, Lopez TT, et al. Adoptive transfer of EBV-specific T cells results in sustained clinical responses in patients with locoregional nasopharyngeal carcinoma. J immunotherapy (Hagerstown Md.: 1997). (2010) 33:983–90. doi: 10.1097/CJI.0b013e3181f3cbf4
83. Chia W, Teo M, Wang W, Lee B, Ang S, Tai W, et al. Adoptive T-cell transfer and chemotherapy in the first-line treatment of metastatic and/or locally recurrent nasopharyngeal carcinoma. Mol Ther. (2014) 22:132–9. doi: 10.1038/mt.2013.242
84. Li J, Chen Q, He J, Li Z, Tang X, Chen S, et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology. (2015) 4:e976507. doi: 10.4161/23723556.2014.976507
85. Huang J, Fogg M, Wirth LJ, Daley H, Ritz J, Posner MR, et al. Epstein-Barr virus-specific adoptive immunotherapy for recurrent, metastatic nasopharyngeal carcinoma. Cancer. (2017) 123:2642–50. doi: 10.1002/cncr.v123.14
86. Smith C, McGrath M, Neller MA, Matthews KK, Crooks P, Le Texier L, et al. Complete response to PD-1 blockade following EBV-specific T-cell therapy in metastatic nasopharyngeal carcinoma. NPJ Precis Oncol. (2021) 5:24. doi: 10.1038/s41698-021-00162-7
87. Jin Y, Yang W, Zou S, Sun Z, Wu C, Yang Z. Chemoradiotherapy combined with NK cell transfer in a patient with recurrent and metastatic nasopharyngeal carcinoma inducing long-term tumor control: a case report. Medicine. (2020) 99:e22785. doi: 10.1097/MD.0000000000022785
88. Lim CM, Liou A, Poon M, Koh LP, Tan LK, Loh KS, et al. Phase I study of expanded natural killer cells in combination with cetuximab for recurrent/metastatic nasopharyngeal carcinoma. Cancer Immunology Immunotherapy. (2022) 71:2277–86. doi: 10.1007/s00262-022-03158-9
89. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol. (2016) 13:370–83. doi: 10.1038/nrclinonc.2016.36
90. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
91. Krause A, Guo H, Latouche J, Tan C, Cheung NV, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. (1998) 188:619–26. doi: 10.1084/jem.188.4.619
92. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
93. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. J Immunol. (2004) 172:104–13. doi: 10.4049/jimmunol.172.1.104
94. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
95. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. (2003) 3:939–51. doi: 10.1038/nri1248
96. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. (2009) 229:173–91. doi: 10.1111/j.1600-065X.2009.00766.x
97. Zhu Y, Chen L. CD137 in the regulation of T cell response to antigen. CD137 Pathway: Immunol Dis. (2007), 83–96. doi: 10.1007/0-387-32829-7_6
98. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
99. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol. (2020) 13:1–16. doi: 10.1186/s13045-020-00890-6
100. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang C, Saso K, et al. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
101. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
102. Liu X, Zhang N, Shi H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget. (2017) 8:62730. doi: 10.18632/oncotarget.17528
103. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
104. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discovery. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
105. Xu X, Li H, Xu C. Structural understanding of T cell receptor triggering. Cell Mol Immunol. (2020) 17:193–202. doi: 10.1038/s41423-020-0367-1
106. Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. (2023) 9:eadf3700. doi: 10.1126/sciadv.adf3700
107. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. (2019) 290:127–47. doi: 10.1111/imr.v290.1
108. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
109. Gudipati V, Rydzek J, Doel-Perez I, Gonçalves VDR, Scharf L, Königsberger S, et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol. (2020) 21:848–56. doi: 10.1038/s41590-020-0719-0
110. D'Angelo SP, Van Tine BA, Attia S, Blay J, Strauss SJ, Valverde Morales CM, et al. SPEARHEAD-1: A phase 2 trial of afamitresgene autoleucel (Formerly ADP-A2M4) in patients with advanced synovial sarcoma or myxoid/round cell liposarcoma. J Clin Oncol. (2021) 39:Suppl. 11504. doi: 10.1200/JCO.2021.39.15_suppl.11504
111. Ishihara M, Nishida Y, Kitano S, Kawai A, Muraoka D, Momose F, et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int J Cancer. (2023) 152:2554–66. doi: 10.1002/ijc.v152.12
112. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood J Am Soc Hematol. (2010) 115:925–35. doi: 10.1182/blood-2009-08-239186
113. Dudaniec K, Westendorf K, Nössner E, Uckert W. Generation of Epstein-Barr virus antigen-specific T cell receptors recognizing immunodominant epitopes of LMP1, LMP2A, and EBNA3C for immunotherapy. Hum Gene Ther. (2021) 32:919–35. doi: 10.1089/hum.2020.283
114. Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-cell receptor gene therapy for human papillomavirus–associated epithelial cancers: a first-in-human, phase I/II study. J Clin Oncol. (2019) 37:2759–68. doi: 10.1200/JCO.18.02424
115. Meng F, Zhao J, Tan AT, Hu W, Wang S, Jin J, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol Int. (2021) 15:1402–12. doi: 10.1007/s12072-021-10250-2
116. Liu Y, Yan X, Zhang F, Zhang X, Tang F, Han Z, et al. TCR-T immunotherapy: The challenges and solutions. Front Oncol. (2022) 11:794183. doi: 10.3389/fonc.2021.794183
117. Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell. (2019) 179:219–235. e21. doi: 10.1016/j.cell.2019.08.032
118. Xiao Z, Todd L, Huang L, Noguera-Ortega E, Lu Z, Huang L, et al. Desmoplastic stroma restricts T cell extravasation and mediates immune exclusion and immunosuppression in solid tumors. Nat Commun. (2023) 14:5110. doi: 10.1038/s41467-023-40850-5
119. Grywalska E, Pasiarski M, Góźdź S, Roliński J. Immune-checkpoint inhibitors for combating T-cell dysfunction in cancer. OncoTargets Ther. (2018) 11:6505. doi: 10.2147/OTT.S150817
120. Balasubramanian A, John T, Asselin-Labat M. Regulation of the antigen presentation machinery in cancer and its implication for immune surveillance. Biochem Soc Trans. (2022) 50:825–37. doi: 10.1042/BST20210961
121. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. (2021) 27:419–25. doi: 10.1038/s41591-020-01225-1
122. Zhao Y, Deng J, Rao S, Guo S, Shen J, Du F, et al. Tumor infiltrating lymphocyte (TIL) therapy for solid tumor treatment: progressions and challenges. Cancers. (2022) 14:4160. doi: 10.3390/cancers14174160
123. Tsimberidou A, Van Morris K, Vo HH, Eck S, Lin Y, Rivas JM, et al. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J Hematol Oncol. (2021) 14:1–22. doi: 10.1186/s13045-021-01115-0
124. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
125. Lin X, Gudgeon NH, Hui EP, Jia H, Qun X, Taylor GS, et al. CD4 and CD8 T cell responses to tumour-associated Epstein–Barr virus antigens in nasopharyngeal carcinoma patients. Cancer Immunology Immunotherapy. (2008) 57:963–75. doi: 10.1007/s00262-007-0427-8
126. Mullinax JE, Egger ME, McCarter M, Monk BJ, Toloza EM, Brousseau S, et al. Surgical considerations for tumor tissue procurement to obtain tumor-infiltrating lymphocytes for adoptive cell therapy. Cancer J. (2022) 28:285–93. doi: 10.1097/PPO.0000000000000608
127. Poschke IC, Hassel JC, Rodriguez-Ehrenfried A, Lindner KA, Heras-Murillo I, Appel LM, et al. The outcome of ex vivo TIL expansion is highly influenced by spatial heterogeneity of the tumor T-cell repertoire and differences in intrinsic in vitro growth capacity between T-cell clones. Clin Cancer Res. (2020) 26:4289–301. doi: 10.1158/1078-0432.CCR-19-3845
128. Morotti M, Albukhari A, Alsaadi A, Artibani M, Brenton JD, Curbishley SM, et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br J Cancer. (2021) 124:1759–76. doi: 10.1038/s41416-021-01353-6
129. Liem NT, Van Phong N, Kien NT, Anh BV, Huyen TL, Thao CT, et al. Phase I clinical trial using autologous ex vivo expanded NK cells and cytotoxic T lymphocytes for cancer treatment in Vietnam. Int J Mol Sci. (2019) 20:3166. doi: 10.3390/ijms20133166
130. Betof Warner A, Corrie PG, Hamid O. Tumor-infiltrating lymphocyte therapy in melanoma: facts to the future. Clin Cancer Res. (2023) 29:1835–54. doi: 10.1158/1078-0432.CCR-22-1922
131. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discovery. (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
132. Du N, Guo F, Wang Y, Cui J. NK cell therapy: A rising star in cancer treatment. Cancers. (2021) 13:4129. doi: 10.3390/cancers13164129
133. Garcia-Iglesias T, del Toro-Arreola A, Albarran-Somoza B, del Toro-Arreola S, Sanchez-Hernandez PE, Ramirez-Dueñas MG, et al. Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer. (2009) 9:186. doi: 10.1186/1471-2407-9-186
134. Nersesian S, Schwartz SL, Grantham SR, MacLean LK, Lee SN, Pugh-Toole M, et al. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Trans Oncol. (2021) 14:100930. doi: 10.1016/j.tranon.2020.100930
135. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
136. Theilgaard-Mönch K, Raaschou-Jensen K, Palm H, Schjødt K, Heilmann C, Vindeløv L, et al. Flow cytometric assessment of lymphocyte subsets, lymphoid progenitors, and hematopoietic stem cells in allogeneic stem cell grafts. Bone Marrow Transplant. (2001) 28:1073–82. doi: 10.1038/sj.bmt.1703270
137. Zhang Y, Wallace DL, De Lara CM, Ghattas H, Asquith B, Worth A, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. (2007) 121:258–65. doi: 10.1111/j.1365-2567.2007.02573.x
138. Mark C, Czerwinski T, Roessner S, Mainka A, Hörsch F, Heublein L, et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat Commun. (2020) 11:5224. doi: 10.1038/s41467-020-19094-0
139. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res. (2015) 62:341–56. doi: 10.1007/s12026-015-8664-y
140. Jonges LE, Albertsson P, Van Vlierberghe R, Ensink NG, Johansson BR, Van De Velde C, et al. The phenotypic heterogeneity of human natural killer cells: presence of at least 48 different subsets in the peripheral blood. Scand J Immunol. (2001) 53:103–10. doi: 10.1046/j.1365-3083.2001.00838.x
141. Nham T, Poznanski SM, Fan IY, Vahedi F, Shenouda MM, Lee AJ, et al. Ex vivo-expanded natural killer cells derived from long-term cryopreserved cord blood are cytotoxic against primary breast cancer cells. J Immunotherapy. (2018) 41:64–72. doi: 10.1097/CJI.0000000000000192
142. Dalle J, Menezes J, Wagner É, Blagdon M, Champagne J, Champagne MA, et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res. (2005) 57:649–55. doi: 10.1203/01.PDR.0000156501.55431.20
143. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. (2012) 73:248–57. doi: 10.1016/j.humimm.2011.12.015
144. Kim Y, Broxmeyer HE. Immune regulatory cells in umbilical cord blood and their potential roles in transplantation tolerance. Crit Rev Oncol. (2011) 79:112–26. doi: 10.1016/j.critrevonc.2010.07.009
145. Tanaka H, Kai S, Yamaguchi M, Misawa M, Fujimori Y, Yamamoto M, et al. Analysis of natural killer (NK) cell activity and adhesion molecules on NK cells from umbilical cord blood. Eur J Haematol. (2003) 71:29–38. doi: 10.1034/j.1600-0609.2003.00081.x
146. Wang Y, Xu H, Zheng X, Wei H, Sun R, Tian Z. High expression of NKG2A/CD94 and low expression of granzyme B are associated with reduced cord blood NK cell activity. Cell Mol Immunol. (2007) 4:377–82.
147. Alnabhan R, Madrigal A, Saudemont A. Differential activation of cord blood and peripheral blood natural killer cells by cytokines. Cytotherapy. (2015) 17:73–85. doi: 10.1016/j.jcyt.2014.08.003
148. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43. e13. doi: 10.1016/j.cell.2018.10.014
149. Zhu H, Kaufman DS. An improved method to produce clinical-scale natural killer cells from human pluripotent stem cells. In: Kaneko S, editor. In Vitro Differentiation of T-Cells. Methods in Molecular Biology, vol. 2048. New York, NY: Humana (2019). p. 107–19. doi: 10.1007/978-1-4939-9728-2_12
150. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–192. e5. doi: 10.1016/j.stem.2018.06.002
151. Tam YK, Martinson JA, Doligosa K, Klingemann HG. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy. (2003) 5:259–72. doi: 10.1080/14653240310001523
152. Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. (2012) 14:1131–43. doi: 10.3109/14653249.2012.700767
153. Bergman H, Sissala N, HÄgerstrand H, Lindqvist C. Human NK-92 cells function as target cells for human NK cells–implications for CAR NK-92 therapies. Anticancer Res. (2020) 40:5355–9. doi: 10.21873/anticanres.14543
154. Gong J, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. (1994) 8:652–8.
155. Maki G, Klingemann H, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. (2001) 10:369–83. doi: 10.1089/152581601750288975
156. Liu Q, Xu Y, Mou J, Tang K, Fu X, Li Y, et al. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19 Malignancy in a mouse model. Cytotherapy. (2020) 22:552–62. doi: 10.1016/j.jcyt.2020.06.003
157. Williams BA, Law AD, Routy B, Gupta V, Wang X, Chaboureau A, et al. A phase I trial of NK-92 cells for refractory hematological Malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. (2017) 8:89256. doi: 10.18632/oncotarget.19204
158. Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou J, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. (2017) 19:1225–32. doi: 10.1016/j.jcyt.2017.07.008
159. Navarrete-Galvan L, Guglielmo M, Cruz Amaya J, Smith-Gagen J, Lombardi VC, Merica R, et al. Optimizing NK-92 serial killers: gamma irradiation, CD95/Fas-ligation, and NK or LAK attack limit cytotoxic efficacy. J Trans Med. (2022) 20:1–13. doi: 10.1186/s12967-022-03350-6
160. Maurer S, Kropp KN, Klein G, Steinle A, Haen SP, Walz JS, et al. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology. (2018) 7:e1364827. doi: 10.1080/2162402X.2017.1364827
161. Cluxton CD, Spillane C, O'Toole SA, Sheils O, Gardiner CM, O'Leary JJ. Suppression of Natural Killer cell NKG2D and CD226 anti-tumour cascades by platelet cloaked cancer cells: Implications for the metastatic cascade. PLoS One. (2019) 14:e0211538. doi: 10.1371/journal.pone.0211538
162. Gong Y, Klein Wolterink RG, Wang J, Bos GM, Germeraad WT. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. (2021) 14:1–35. doi: 10.1186/s13045-021-01083-5
163. Kim H, Han M, Kim M, Kim H, Im HJ, Kim N, et al. CD19/CD22 bispecific chimeric antigen receptor-NK-92 cells are developed and evaluated. Oncol Lett. (2023) 25:1–9. doi: 10.3892/ol.2023.13822
164. Nowak J, Bentele M, Kutle I, Zimmermann K, Lühmann JL, Steinemann D, et al. CAR-NK cells targeting HER1 (EGFR) show efficient anti-tumor activity against head and neck squamous cell carcinoma (HNSCC). Cancers. (2023) 15:3169. doi: 10.3390/cancers15123169
165. Xia W, Chen J, Hou W, Chen J, Xiong Y, Li H, et al. Engineering a HER2-CAR-NK cell secreting soluble programmed cell death protein with superior antitumor efficacy. Int J Mol Sci. (2023) 24:6843. doi: 10.3390/ijms24076843
166. Zhang Q, Tian K, Xu J, Zhang H, Li L, Fu Q, et al. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. (2017) 2017:6915912. doi: 10.1155/2017/6915912
167. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. (2018) 8:1083–9.
168. Strassheimer F, Strecker MI, Zhang C, Mildenberger IC, Harter PN, Tonn T, et al. P12. 04 Synergistic effects of combination therapy of CAR-NK cells and anti-PD-1 antibody result in high efficacy against advanced stage orthotopic glioblastoma grafts in a syngeneic mouse model and induce protective anti-tumor immunity in vivo. Neuro-oncology. (2019) 21:iii60. doi: 10.1093/neuonc/noz126.215
169. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon Maurice Y, I I, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359. doi: 10.18632/oncotarget.13411
170. Lee MY, Robbins Y, Sievers C, Friedman J, Sater HA, Clavijo PE, et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J Immunother Cancer. (2021) 9:e002128. doi: 10.1136/jitc-2020-002128
171. Seery TE, Kistler M, Lee JH, Soon-Shiong P. Quilt-3.064: An open-label phase I study of PD-L1 t-haNK in subjects with locally advanced or metastatic solid cancers. J Clin Oncol. (2020) 38:Suppl. TPS3152. doi: 10.1200/JCO.2020.38.15_suppl.TPS3152
172. Zuo P, Li Y, He C, Wang T, Zheng X, Liu H, et al. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front Immunol. (2023) 14:1145706. doi: 10.3389/fimmu.2023.1145706
173. Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: From bench to bedside. biomark Res. (2022) 10:1–19. doi: 10.1186/s40364-022-00364-6
174. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol. (2014) 5:254. doi: 10.3389/fphar.2014.00254
175. Guercio M, Manni S, Boffa I, Caruso S, Di Cecca S, Sinibaldi M, et al. Inclusion of the inducible caspase 9 suicide gene in CAR construct increases safety of CAR. CD19 T cell therapy in B-cell Malignancies. Front Immunol. (2021) 12:755639. doi: 10.3389/fimmu.2021.755639
176. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
177. Li G, Zhang Q, Han Z, Zhu Y, Shen H, Liu Z, et al. IL-7 and CCR2b co-expression-mediated enhanced CAR-T survival and infiltration in solid tumors. Front Oncol. (2021) 11:734593. doi: 10.3389/fonc.2021.734593
178. Ng YY, Tay JC, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Therapy-Oncolytics. (2020) 16:75–85. doi: 10.1016/j.omto.2019.12.006
179. Eckert EC, Nace RA, Tonne JM, Evgin L, Vile RG, Russell SJ. Generation of a tumor-specific chemokine gradient using oncolytic vesicular stomatitis virus encoding CXCL9. Mol Therapy-Oncolytics. (2020) 16:63–74. doi: 10.1016/j.omto.2019.12.003
180. Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. (2016) 5:e1091554. doi: 10.1080/2162402X.2015.1091554
181. Moon EK, Wang LS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. (2018) 7:e1395997. doi: 10.1080/2162402X.2017.1395997
182. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. (2020) 6:160. doi: 10.3389/fmolb.2019.00160
183. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
184. Chen J, Yang P, Xiao Y, Zhang Y, Liu J, Xie D, et al. Overexpression of α-sma-positive fibroblasts (CAFs) in nasopharyngeal carcinoma predicts poor prognosis. J Cancer. (2017) 8:3897. doi: 10.7150/jca.20324
185. Liu Y, Sun Y, Wang P, Li S, Dong Y, Zhou M, et al. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18. 2-targeted CAR-T against pancreatic cancer. J Trans Med. (2023) 21:255. doi: 10.1186/s12967-023-04080-z
186. Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive Malignant pleural mesothelioma (FAPME-1). BMC Cancer. (2012) 12:615. doi: 10.1186/1471-2407-12-615
187. Curioni A, Britschgi C, Hiltbrunner S, Bankel L, Gulati P, Weder W, et al. A phase I clinical trial of Malignant pleural mesothelioma treated with locally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol. (2019) 30:v501. doi: 10.1093/annonc/mdz253.052
188. Li F, Zhao S, Wei C, Hu Y, Xu T, Xin X, et al. Development of Nectin4/FAP-targeted CAR-T cells secreting IL-7, CCL19, and IL-12 for Malignant solid tumors. Front Immunol. (2022) 13:958082. doi: 10.3389/fimmu.2022.958082
189. Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. biomark Res. (2021) 9:38. doi: 10.1186/s40364-021-00294-9
190. Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic Malignancies. J Hematol Oncol. (2021) 14:75. doi: 10.1186/s13045-021-01084-4
191. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
192. Yin Y, Rodriguez JL, Li N, Thokala R, Nasrallah MP, Hu L, et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol Ther. (2022) 30:2537–53. doi: 10.1016/j.ymthe.2022.05.011
193. Fusaroli M, Isgrò V, Cutroneo PM, Ferrajolo C, Cirillo V, Del Bufalo F, et al. Post-marketing surveillance of CAR-T-cell therapies: Analysis of the FDA adverse event reporting system (FAERS) database. Drug Saf. (2022) 45:891–908. doi: 10.1007/s40264-022-01194-z
194. Helsen CW, Hammill JA, Lau VW, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun. (2018) 9:3049. doi: 10.1038/s41467-018-05395-y
195. Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. (1998) 391:703–7. doi: 10.1038/35642
196. Wang E, Wang L, Tsai C, Bhoj V, Gershenson Z, Moon E, et al. Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res. (2015) 3:815–26. doi: 10.1158/2326-6066.CIR-15-0054
197. Ng Y, Tay JC, Li Z, Wang J, Zhu J, Wang S. T cells expressing NKG2D CAR with a DAP12 signaling domain stimulate lower cytokine production while effective in tumor eradication. Mol Ther. (2021) 29:75–85. doi: 10.1016/j.ymthe.2020.08.016
198. Sun M, Xu P, Wang E, Zhou M, Xu T, Wang J, et al. Novel two-chain structure utilizing KIRS2/DAP12 domain improves the safety and efficacy of CAR-T cells in adults with r/r B-ALL. Mol Therapy-Oncolytics. (2021) 23:96–106. doi: 10.1016/j.omto.2021.08.014
199. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
200. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:117198. doi: 10.3389/fphar.2014.00235
201. Ventin M, Cattaneo G, Jia J, Arya S, Maggs L, Sun Y, et al. B7-H3-targeted CAR T cell with an inducible caspase 9 suicide gene effectively eradicates uveal melanoma liver metastases. J Immunother Cancer. (2023) 11:A360. doi: 10.1136/jitc-2023-SITC2023.0317
202. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol. (2019) 10:168. doi: 10.3389/fimmu.2019.00168
203. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:1–16. doi: 10.1186/s13287-020-02128-1
204. Wang AX, Ong XJ, D’Souza C, Neeson PJ, Zhu JJ. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front Immunol. (2023) 14:1140541. doi: 10.3389/fimmu.2023.1140541
205. Yu Q, Zhang X, Wang N, Li C, Zhang Y, Zhou J, et al. Radiation prior to chimeric antigen receptor T-cell therapy is an optimizing bridging strategy in relapsed/refractory aggressive B-cell lymphoma. Radiotherapy Oncol. (2022) 177:53–60. doi: 10.1016/j.radonc.2022.10.018
206. Fan J, Adams A, Sieg N, Heger J, Gödel P, Kutsch N, et al. Potential synergy between radiotherapy and CAR T-cells-a multicentric analysis of the role of radiotherapy in the combination of CAR T cell therapy. Radiotherapy Oncol. (2023) 183:109580. doi: 10.1016/j.radonc.2023.109580
207. Kwon M, Jung H, Nam G, Kim I. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J Controlled Release. (2021) 331:321–34. doi: 10.1016/j.jconrel.2021.01.009
208. Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. (2009) 183:137–44. doi: 10.4049/jimmunol.0900734
209. Ashrafizadeh M, Farhood B, Musa AE, Taeb S, Najafi M. Damage-associated molecular patterns in tumor radiotherapy. Int Immunopharmacol. (2020) 86:106761. doi: 10.1016/j.intimp.2020.106761
210. Solari JIG, Filippi-Chiela E, Pilar ES, Nunes V, Gonzalez EA, Figueiró F, et al. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer. (2020) 20:1–14. doi: 10.1186/s12885-020-06964-5
211. McDonnell AM, Lesterhuis WJ, Khong A, Nowak AK, Lake RA, Currie AJ, et al. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur J Immunol. (2015) 45:49–59. doi: 10.1002/eji.201444722
212. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. (2005) 26:111–7. doi: 10.1016/j.it.2004.12.003
213. Bot A, Rossi JM, Jiang Y, Navale L, Shen Y, Sherman M, et al. Cyclophosphamide and fludarabine conditioning chemotherapy induces a key homeostatic cytokine profile in patients prior to CAR T cell therapy. Blood. (2015) 126:4426. doi: 10.1182/blood.V126.23.4426.4426
214. Ramakrishnan R, AsSudani D, Nagaraj S, Hunter T, Cho H, Antonia S, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. (2010) 120:1111–24. doi: 10.1172/JCI40269
215. Ramakrishnan R, Huang C, Cho H, Lloyd M, Johnson J, Ren X, et al. Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res. (2012) 72:5483–93. doi: 10.1158/0008-5472.CAN-12-2236
216. Liu J, Zhang B, Su Y, Qin G, Kong X, Mo Y, et al. Hypofractionated radiotherapy compared with conventionally fractionated radiotherapy to treat initial distant metastases in nasopharyngeal carcinoma: A multicenter, prospective, randomized, phase II trial. Radiotherapy Oncol. (2023) 187:109815. doi: 10.1016/j.radonc.2023.109815
217. Wang Y. Advances in hypofractionated irradiation-induced immunosuppression of tumor microenvironment. Front Immunol. (2021) 11:612072. doi: 10.3389/fimmu.2020.612072
218. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti–CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
219. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
220. Gao L, Zhang A. Low-dose radiotherapy effects the progression of anti-tumor response. Trans Oncol. (2023) 35:101710. doi: 10.1016/j.tranon.2023.101710
221. You R, Liu Y, Xie Y, Lin C, Duan C, Chen D, et al. Hyperfractionation compared with standard fractionation in intensity-modulated radiotherapy for patients with locally advanced recurrent nasopharyngeal carcinoma: a multicentre, randomised, open-label, phase 3 trial. Lancet. (2023) 401:917–27. doi: 10.1016/S0140-6736(23)00269-6
222. Turtle CJ, Hanafi L, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4: CD8 composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
223. Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol. (2020) 38:415–22. doi: 10.1200/JCO.19.01892
224. Oliver-Caldés A, González-Calle V, Cabañas V, Español-Rego M, Rodríguez-Otero P, Reguera JL, et al. Fractionated initial infusion and booster dose of ARI0002h, a humanised, BCMA-directed CAR T-cell therapy, for patients with relapsed or refractory multiple myeloma (CARTBCMA-HCB-01): a single-arm, multicentre, academic pilot study. Lancet Oncol. (2023) 24:913–24. doi: 10.1016/S1470-2045(23)00222-X
225. Secondino S, Zecca M, Licitra L, Gurrado A, Schiavetto I, Bossi P, et al. T-cell therapy for EBV-associated nasopharyngeal carcinoma: preparative lymphodepleting chemotherapy does not improve clinical results. Ann Oncol. (2012) 23:435–41. doi: 10.1093/annonc/mdr134
226. McGrath E, Chabannon C, Terwel S, Bonini C, Kuball J. Opportunities and challenges associated with the evaluation of chimeric antigen receptor T cells in real-life. Curr Opin Oncol. (2020) 32:427–33. doi: 10.1097/CCO.0000000000000665
227. Ng MSF, Kwok I, Tan L, Shi C, Cerezo-Wallis D, Tan Y, et al. Deterministic reprogramming of neutrophils within tumors. Science. (2024) 383:eadf6493. doi: 10.1126/science.adf6493
228. Mungly SB, Peter EP, Hii LW, Mai CW, Chung FFL. Epigenetic drug interventions in breast cancer: a narrative review of current research and future directions. Prog Microbes Mol Biol. (2024) 7:a00000448. doi: 10.36877/pmmb.a0000448
Keywords: nasopharyngeal carcinoma, stem cell, chimeric antigen receptor (CAR)-T cell, engineered T cell receptor-T (TCR-T) cell, tumour-infiltrating lymphocytes (TIL), natural killer (Nk) cell
Citation: Looi C-K, Loo E-M, Lim H-C, Chew Y-L, Chin K-Y, Cheah S-C, Goh BH and Mai C-W (2024) Revolutionizing the treatment for nasopharyngeal cancer: the impact, challenges and strategies of stem cell and genetically engineered cell therapies. Front. Immunol. 15:1484535. doi: 10.3389/fimmu.2024.1484535
Received: 22 August 2024; Accepted: 24 September 2024;
Published: 10 October 2024.
Edited by:
Laura Belver, Josep Carreras Leukaemia Research Institute (IJC), SpainReviewed by:
Zhenyu Dai, Stanford University, United StatesYingchun He, Hunan University of Chinese Medicine, China
Copyright © 2024 Looi, Loo, Lim, Chew, Chin, Cheah, Goh and Mai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Wai Mai, bWFpY3dAdWNzaXVuaXZlcnNpdHkuZWR1Lm15; bWFpLmNodW53YWlAZ21haWwuY29t