 Alexandre Aubert1,2,3†
Alexandre Aubert1,2,3† Jenna Goeres1,2,3†
Jenna Goeres1,2,3† Amy Liu1,2,3†Martin Kao1,2,3†Katlyn C. Richardson1,2,3†Karen Jung1,2,3†
Amy Liu1,2,3†Martin Kao1,2,3†Katlyn C. Richardson1,2,3†Karen Jung1,2,3† Boris Hinz4,5†Richard I. Crawford2,6†
Boris Hinz4,5†Richard I. Crawford2,6† David J. Granville1,2,3*†
David J. Granville1,2,3*†- 1International Collaboration on Repair Discoveries (ICORD) Centre, Vancouver Coastal Health Research Institute (VCHRI), University of British Columbia (UBC), Vancouver, BC, Canada
- 2Department of Pathology and Laboratory Medicine, University of British Columbia (UBC), Vancouver, BC, Canada
- 3British Columbia Professional Firefighters’ Burn and Wound Healing Group, Vancouver Coastal Health Research Institute (VCHRI), Vancouver, BC, Canada
- 4Laboratory of Tissue Repair and Regeneration, Keenan Research Institute for Biomedical Science of the St. Michael’s Hospital, Toronto, ON, Canada
- 5Faculty of Dentistry, University of Toronto, Toronto, ON, Canada
- 6Department of Dermatology and Skin Science, University of British Columbia (UBC), Vancouver, BC, Canada
Keloid scars (KS) and hypertrophic scars (HS) are fibroproliferative wound healing defects characterized by excessive accumulation of extracellular matrix (ECM) in the dermis of affected individuals. Although transforming growth factor (TGF)-β is known to be involved in the formation of KS and HS, the molecular mechanisms responsible for its activation remain unclear. In this study we investigated Granzyme B (GzmB), a serine protease with established roles in fibrosis and scarring through the cleavage of ECM proteins, as a potential new mediator of TGF-β activation in KS and HS. Increased GzmB-positive mast cells were identified in the dermis of KS and HS but not healthy skin controls. Elevated levels of substance P, a neuropeptide involved in mast cell degranulation, suggest that GzmB is released extracellularly, as confirmed by the significant reduction of the established extracellular GzmB substrate decorin in KS and HS. Similarly, presence of latent TGF-β binding protein 1 (LTBP1), a protein involved in the extracellular tethering of latent TGF-β, was disrupted proximal to the dermal-epidermal junction (DEJ) of GzmBhigh KS and HS lesions. Using LTBP1-enriched medium as well as purified LTBP1, its cleavage by GzmB was confirmed in vitro. Increased TGF-β/Smad signaling pathway was observed in keratinocytes treated with GzmB-digested LTBP1 and was abolished by the addition of a pan-TGF-β inhibitor, suggesting that GzmB cleavage of LTBP1 contributes to TGF-β activation. In dermal fibroblasts, GzmB also cleaved cell-derived LTBP1 and induced TGF-β activation through the cleavage of one or more unidentified fibroblast-secreted proteins. Altogether, the present results suggest that GzmB contributes to KS and HS through ECM remodeling and TGF-β activation.
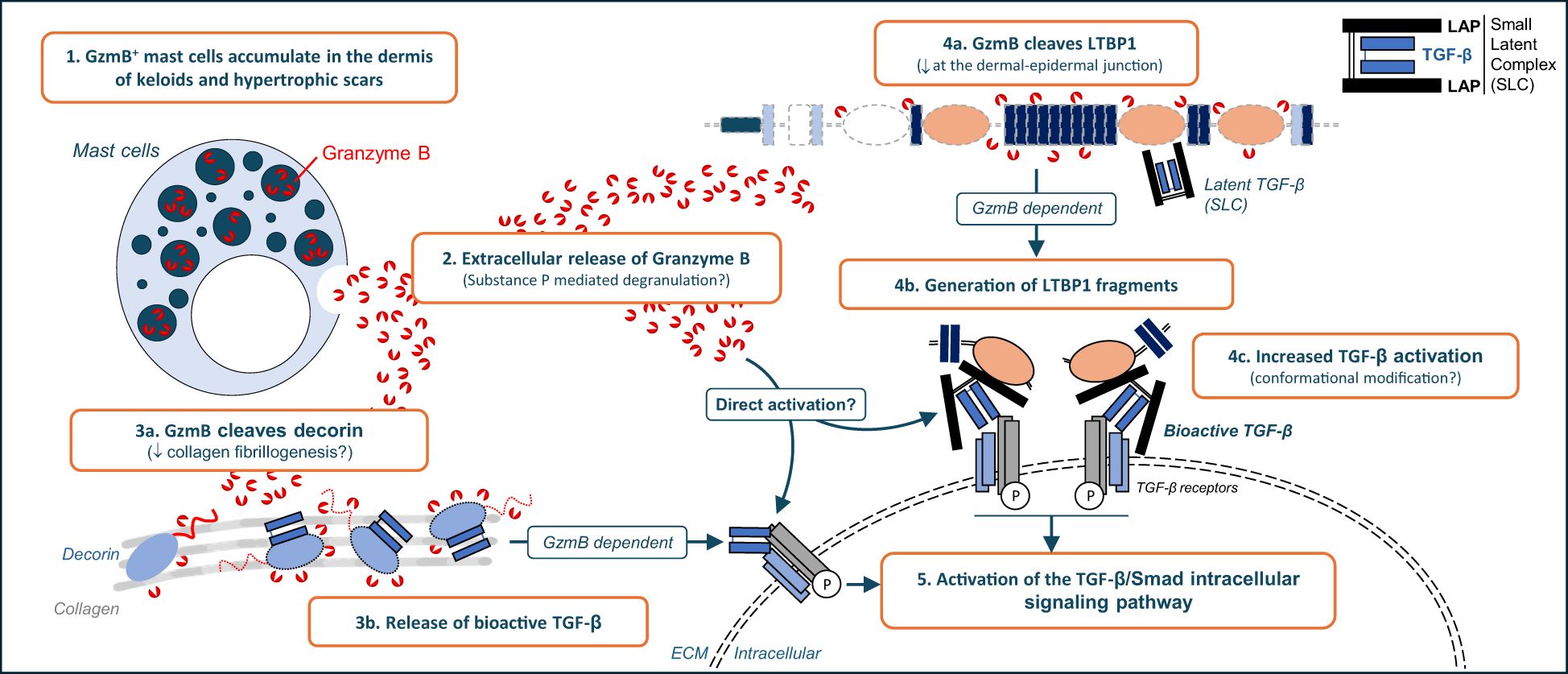
Graphical Abstract. Graphical Abstract.
Introduction
Keloid scars (KS) and hypertrophic scars (HS) represent two forms of pathologic scarring characterized by excessive extracellular matrix (ECM) accumulation, fibroblast proliferation, aberrant proteolysis, and secretion of pro-inflammatory mediators (1). Stemming from a connective tissue response to trauma, inflammation, surgery, or burns, KS and HS represent major aesthetic burdens for patients and are often associated with reduced quality of life due to mobility issues, pruritus, pain, and discomfort (1).
KS and HS share numerous hallmark features including chronic inflammation (2), infiltration of mast cells and macrophages (3, 4), as well as dermal fibrosis (5); however, they also exhibit distinct histological and underlying molecular attributes. KS are rounded protuberances that continuously grow beyond the confines of the wound margins and rarely regress over time, while HS are raised lesions that remain confined within the wound margins (1). In opposition to the increased type I to type III collagen ratio leading to the formation of abnormally thick collagen bundles in KS, HS are characterized by wavy type-III collagen rich bundles accumulating in the dermis (1). Clinical management options for KS and HS overlap and include surgery, occlusive dressing, compression therapies, and intralesional steroid administration (6). Nevertheless, efficacy of these approaches can vary between patients, with adverse side effects and potential recurrence, especially for KS. There is consequently an urgent need to better understand the pathobiology of KS and HS to develop more effective and safer therapies.
Another key molecular commonality between KS and HS is the up-regulated activity of transforming growth factor (TGF)-β compared to healthy skin (5). TGF-β exists in three isoforms in humans (β1, β2 and β3) and is produced by many cell types in an inactive form. Synthesized as a disulfide bonded dimeric precursor, TGF-β is proteolytically separated from its latency associated pro-peptide LAP by furin protein convertase during intracellular processing (7). Though cleaved, TGF-β and LAP remain non-covalently attached to form a small latent complex (SLC), where the LAP can act as a molecular “straitjacket” to prevent TGF-β from interacting with its cell surface receptors (8). While the SLC can be found in a soluble extracellular form, it is typically tethered to a presenting protein partner. In the ECM, the SLC is covalently bound to latent TGF-β binding protein-1 (LTBP1) to create a large latent complex (LLC) that is anchored to the fibronectin- and fibrillin-rich ECM (9). Bioactive TGF-β can also be sequestered into the skin dermis by interacting with decorin (10), a structural dermatan-sulfate proteoglycan involved in collagen fibrillogenesis (11).
Extracellular activation of TGF-β is a tightly regulated process that can be achieved through different mechanisms depending on the cell type involved. TGF-β can be activated independent of proteases through conformational modifications induced by mechanical stress (8, 12). In fact, active TGF-β is released by cell contractile forces applied to the SLC by integrins from the αv family, including αvβ6 expressed by epithelial cells (13), αvβ1 (14) and αvβ5 (15) expressed by fibroblastic cells, as well as αvβ8 predominantly expressed by immune cells (16). Likewise, TGF-β bioactive sites appear to be unmasked upon binding of the SLC to ECM molecules such as thrombospondin-1 (17), ADAMTS1 (18), and members of the tenascin family (19). Finally, soluble TGF-β can also be fully released from the SLC by proteolytic degradation of the LAP pro-peptide by extracellular proteases, including but not limited to plasmin (20), calpain (21), and matrix metalloproteinases (MMPs (22, 23)). Once activated, TGF-β can engage TGF-β receptors type I (TβRI, also known as ALK5) and type II (TβRII) at the cell surface to promote a canonical intracellular signaling pathway resulting in Smad2 and Smad3 phosphorylation (7). In cooperation with Smad4, phospho-Smad2/3 can translocate into the nucleus where they act as transcription factors to regulate the expression of TGF-β responsive genes involved in cytostasis and apoptosis (24), immuno-modulation (25), myofibroblast differentiation (26), as well as ECM protein synthesis (27).
TGF-β has been extensively investigated in the context of wound healing, fibrosis, and scarring (28) and is believed to play an important role in the pathogenesis of KS and HS (29). Recent characterization of the keloidal matrisome by mass spectrometry revealed significantly elevated levels of TGF-β1, as well as several of its extracellular activators, in the ECM of KS (30). A dysregulated TβRI/TβRII ratio (31), alongside increased Smad2 expression (32), is observed in fibroblasts derived from KS and HS which demonstrate a higher expression of αSMA, collagens and connective tissue growth factor (CTGF) after TGF-β stimulation compared to healthy fibroblasts (33–36). Alteration of the TGF-β signaling pathway is also possibly linked to the loss and/or reduction of decorin in KS (37, 38) and HS (39, 40). In vitro, the addition of decorin to HS fibroblasts inhibits their activation into scar-producing myofibroblasts by TGF-β (33), and decorin overexpression in keloidal fibroblasts reduces type I and type III collagen synthesis (37). Nevertheless, the molecular mechanisms involved in the regulation of TGF-β bioavailability in KS and HS remain to be fully elucidated.
Granzyme B (GzmB) is a serine protease with aspase-like activity extensively studied for its implications in immune cell-mediated apoptosis. Initially identified in the lytic granules of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, GzmB is delivered into the cytoplasm of targeted cells during immunological synapse formation (41). Once internalized aided by the pore forming protein perforin, GzmB can promote both caspase-dependent and -independent apoptosis through the cleavage of specific intracellular substrates (41). Nevertheless, either passively released by CTLs (42, 43) or produced by non-immune cells as well as other immune cells without cytotoxic potential [reviewed in (44)], GzmB can be secreted and accumulates in the extracellular space in conditions characterized by dysregulated or excessive inflammation. Due to the lack of an endogenous extracellular inhibitor in humans (45, 46), GzmB retains its proteolytic activity outside of cells and can contribute to disease progression through multiple extracellular pathways [previously reviewed in (44)].
In contrast to its absence in healthy individuals, GzmB accumulates extracellularly in a wide range of dermatological conditions including atopic dermatitis (47), psoriasis (48), and autoimmune blistering diseases (49, 50). GzmB is also elevated in human pressure injuries (51) as well as in several animal models of metabolically (52) or diabetically impaired (53, 54) wound healing. In these contexts, GzmB can directly contribute to both scarring and/or delayed wound closure through the cleavage of specific ECM proteins – fibronectin and vitronectin (55), type IV (56) and type VII (49) collagens, laminin (55), and biglycan (57). GzmB also directly cleaves decorin, leading to impaired collagen fibrillogenesis, associated with the release and subsequent activation of TGF-β (54, 57). To our knowledge, the role of GzmB has never been evaluated in KS and HS.
To investigate the implication of GzmB in KS and HS we immunostained human skin samples and detected GzmB+ mast cells exhibiting degranulation profiles accumulating in the dermis of KS and HS but not healthy skin controls. Levels of decorin, a well characterized extracellular GzmB substrate, were significantly reduced in the dermis of KS and HS. We also identified the loss of LTBP1 at the dermal epidermal junction of GzmBhigh, but not GzmBlow, lesions. In vitro, recombinant human LTBP1 was proteolytically cleaved by recombinant human GzmB. Increased activation of the TGF-β/Smad signaling pathway was also identified in cells treated with LTBP1 (conditioned medium or purified recombinant protein) pre-digested by GzmB. This GzmB/LTBP1-mediated Smad2 phosphorylation was abolished by a pan-TGF-β inhibitor, suggesting that GzmB cleavage of LTBP1 release bioactive TGF-β in vitro. Finally, we also identified the ability of GzmB to promote a direct TGF-β activation through the proteolysis of fibroblast-secreted proteins, independent of LAP degradation. Altogether, through the cleavage of specific ECM molecules including decorin and LTBP1, mast-cell derived GzmB may contribute to latent TGF-β activation and to the fibrotic response observed in KS and HS.
Results
GzmB accumulates in KS and HS
GzmB was assessed in the skin of 10 KS (2M/8F, mean age 32), 10 HS (6M/4F, mean age 48), and 6 healthy controls (1M/5F, mean age 45) by immunohistochemistry (patient information available in Table 1). Compared to healthy skin, the number of GzmB-positive (GzmB+) cells were significantly higher in the upper (Figures 1A, B) and lower (Figures 1C, D) dermis of HS. While the number of GzmB+ cells in KS appeared non-significant compared to healthy skin controls, a trend in the accumulation of GzmB+ cells was identified in the lower dermis (p=0.09, Figure 1D). No significant differences in the number of GzmB+ cells were observed between KS and HS.
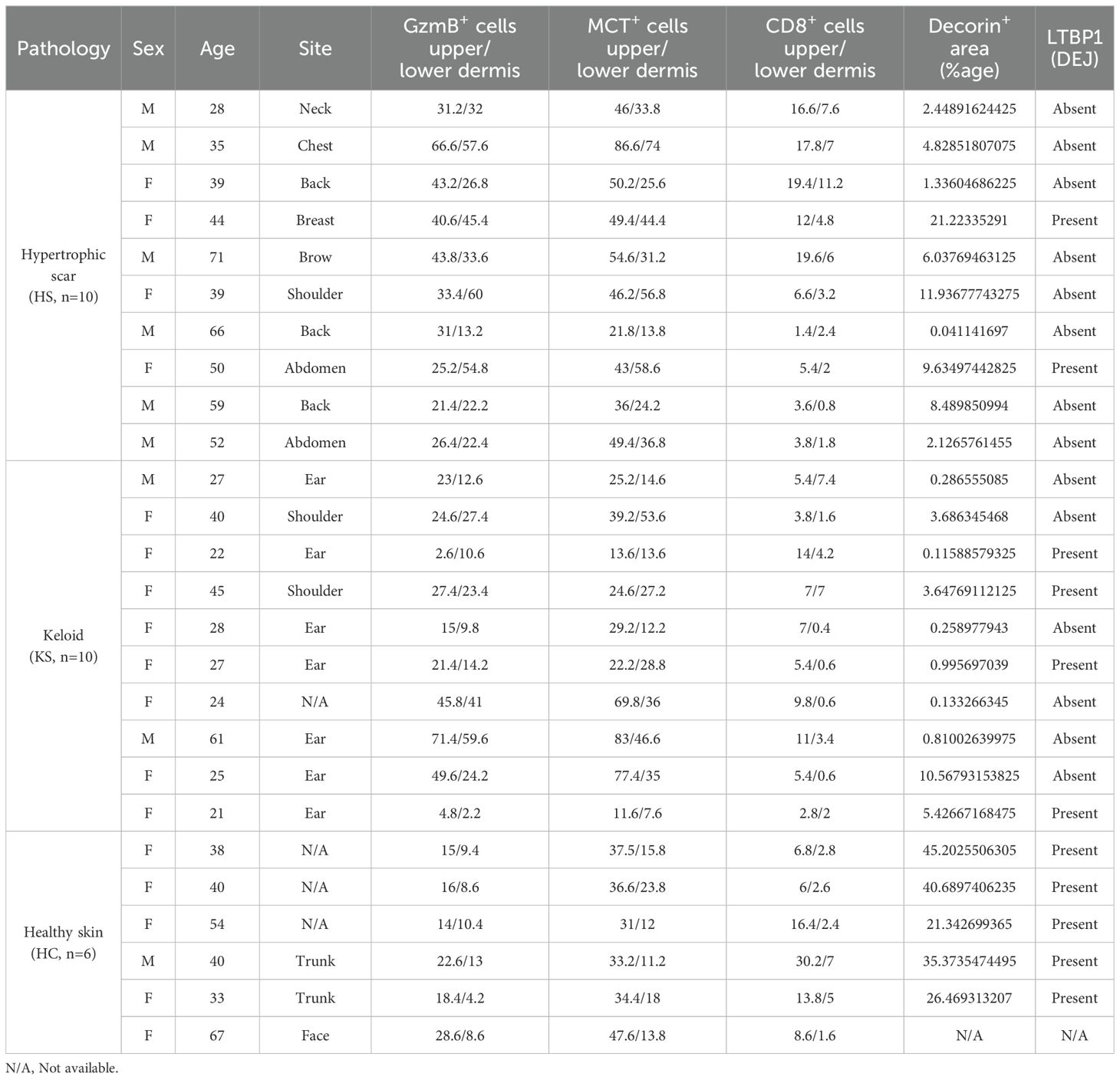
Table 1. Individual patient information.
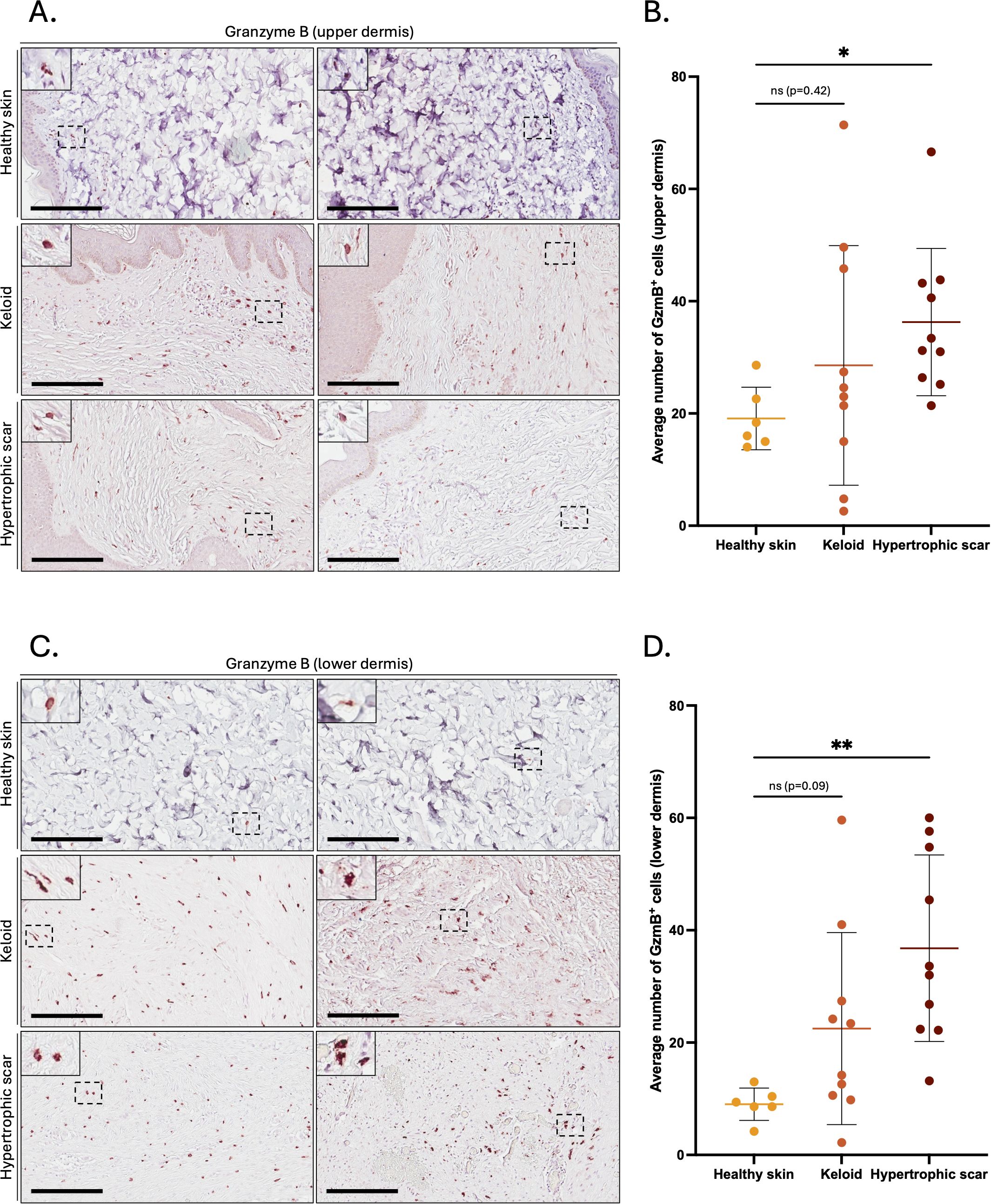
Figure 1. Granzyme B positive cells in the dermis of keloids and hypertrophic scars. (A, C) GzmB immunostaining performed on the upper (A) and lower (C) skin dermis of healthy controls and patients with keloids or hypertrophic scars. The inset shows high magnification of regions of interest. Bars, 200 μm. (B, D) Quantification of the average number of GzmB+ cells in the upper (B) and lower (D) dermis of healthy skin controls (n=6, orange), keloids (n=10, pink) and hypertrophic scars (n=10, brown) samples. Results are represented as mean ± SD. *p < 0.05, **p < 0.005. ns, not significant.
Mast cells are the primary source of GzmB in KS and HS
Based on the localization of GzmB in dermal areas characterized by immune cell infiltration, we next investigated the cell source of the protease by staining immune cell markers. Few CD8+ T-cells were detected in the upper and lower dermis of KS and HS (Supplementary Figure 1A), with no statistical differences compared to healthy skin controls (Supplementary Figures 1B, C). While a weak correlation was observed between the number of CD8+ and GzmB+ cells in the upper dermis of KS and HS (r=0.45, p=0.045, Supplementary Figure 1D), no correlation was observed in the lower dermis (r=0.17, p=0.49, Supplementary Figure 1E). This result indicates that CD8+ T-cells may not contribute to GzmB production in KS and HS.
In contrast, mast cells (defined by expression of mast cell tryptase, or MCT) significantly accumulated in the lower dermis of HS compared to healthy skin controls (Figures 2A–C). The numbers of MCT+ and GzmB+ cells in the upper (r=0.92, p>0.001, Figure 2D) and lower (r=0.88, p>0.001, Figure 2E) dermis of KS and HS (n=20) were significantly positively correlated, indicating mast cells as a major source of GzmB in these conditions. Co-immunofluorescence (Figure 3A) and tyramide signal amplification (TSA) (Supplementary Figure 2) assays revealed strong co-localization between MCT and GzmB, confirming mast cells as the main producer of the protease in KS and HS.
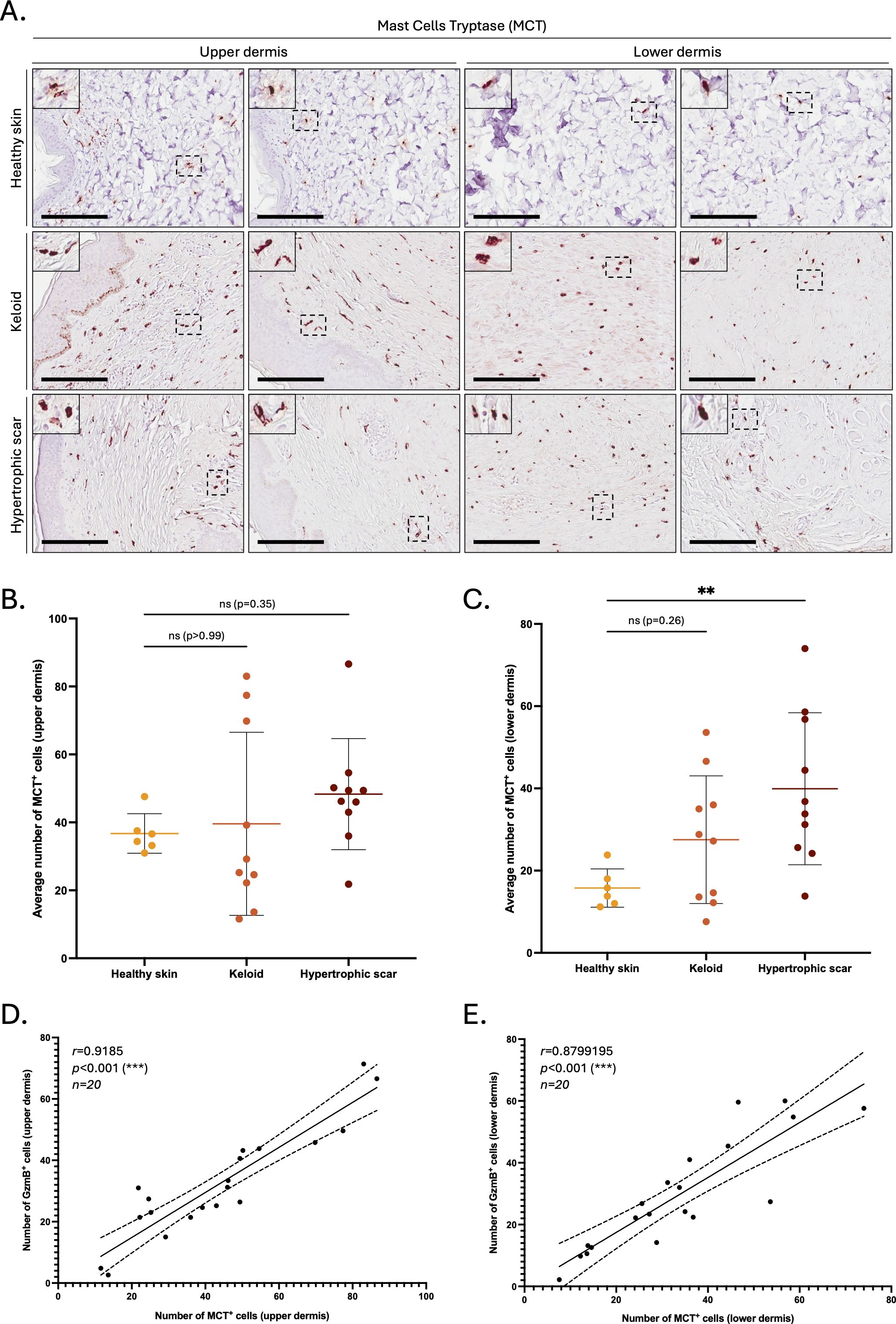
Figure 2. The number of MCT+ cells is positively correlated to the number of GzmB+ cells in the dermis of keloids and hypertrophic scars. (A) Mast Cell Tryptase (MCT) immunostaining performed on the upper and lower skin dermis of healthy controls and patients with keloids or hypertrophic scars. The inset shows high magnification of regions of interest. Bars, 200 μm. (B, C) Quantification of the average number of MCT+ cells in the upper (B) and lower (C) dermis of healthy skin controls (n=6, orange), keloids (n=10, pink) or hypertrophic scars (n=10, brown) samples. Results are represented as mean ± SD. (D, E) Correlation between the average number of GzmB+ and MCT+ cells in the upper (D) and lower (E) dermis of keloids or hypertrophic scars samples (n=20). **p < 0.01, ***p < 0.001. ns, not significant.
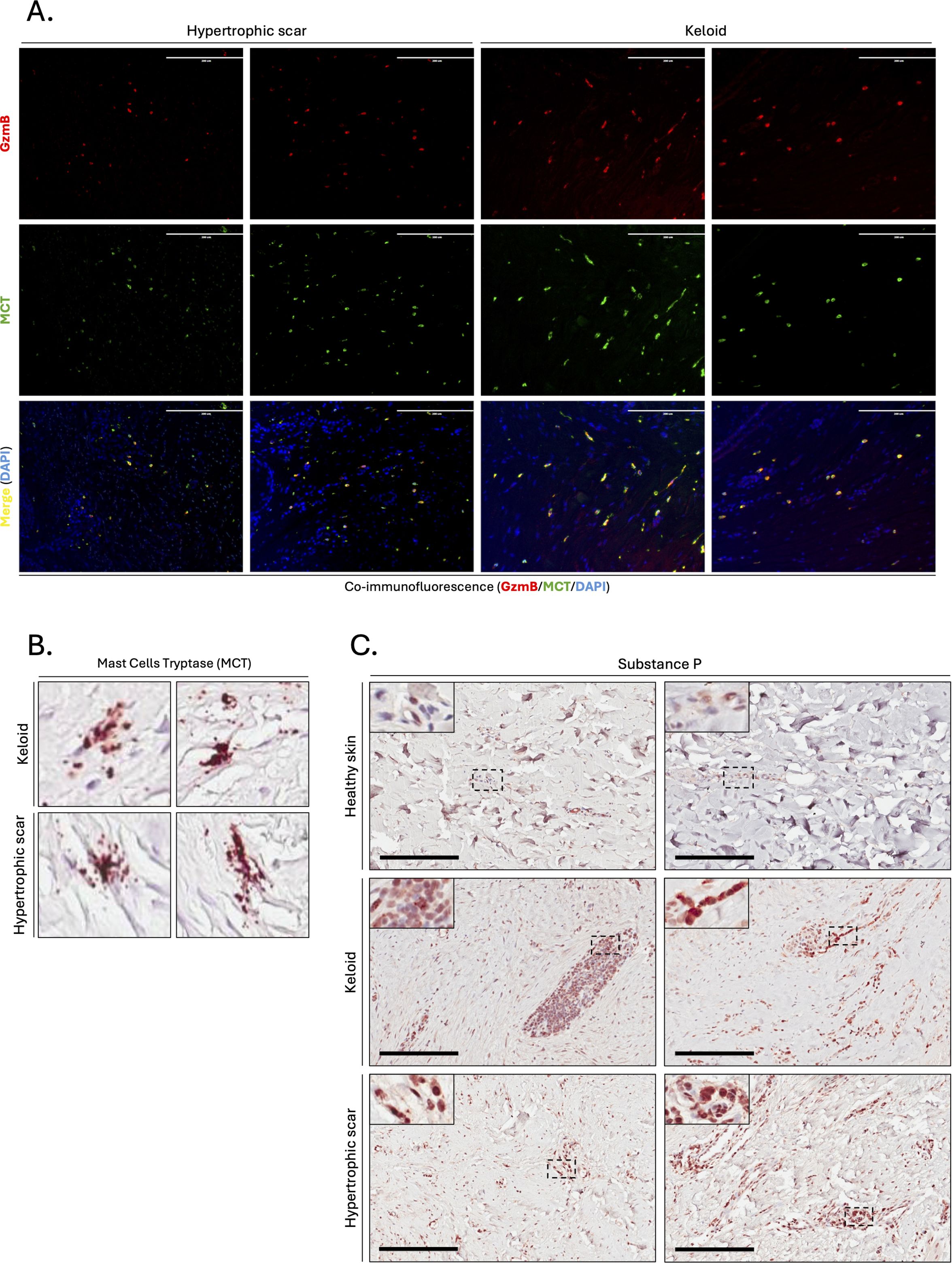
Figure 3. Mast cells are the primary source of GzmB in keloids and hypertrophic scars. (A) Co-immunofluorescence staining of GzmB (red) and MCT (green) in the skin dermis of keloids and hypertrophic scars. Merge shows overlap of the two channels and co-stained cells appear in yellow. Cell nuclei are stained with DAPI (blue). Scale, 200 μm. (B) MCT immunostaining demonstrating the presence of degranulating mast cells in the dermis of keloids and hypertrophic scars. (C) Substance P immunostaining performed on the skin dermis of healthy controls and patients with keloids or hypertrophic Scars. The inset shows high magnification of regions of interest. Bars, 200 μm.
Since some of the mast cells in KS and HS exhibit active granule releas (Figure 3B), we immunostained skin dermis for Substance P, a neuropeptide previously identified as mediator of human mast cell degranulation (58). Increased presence of Substance P+ cells was observed in the dermis of KS and HS compared to healthy skin controls (Figure 3C), suggesting that mast cells release GzmB into the ECM of KS and HS through Substance P-mediated degranulation.
Decorin and LTBP1 are reduced in KS and HS
Next, we investigated possible targets of mast-cell derived GzmB in KS and HS and first focused on decorin, an established extracellular GzmB substrate (57). In contrast to the abundance of decorin in healthy skin controls (Figure 4A), the intensity of decorin staining was significantly reduced in KS and HS (Figure 4B). While still partially detectable in the papillary dermis, decorin was almost completely absent from the reticular dermis of KS and HS (Figures 4A, B).
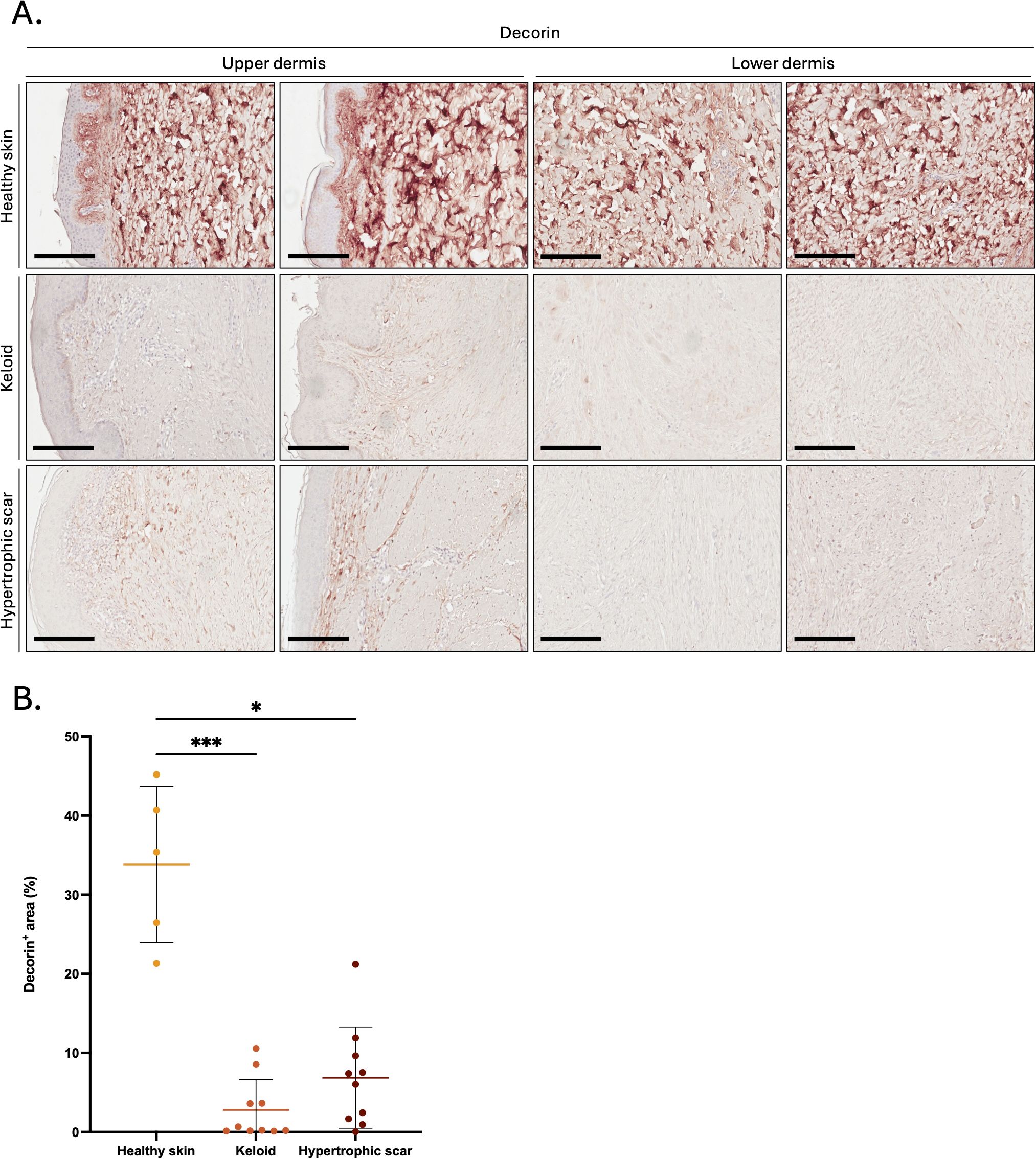
Figure 4. Decorin is reduced in the dermis of keloids and hypertrophic scars. (A) Decorin immunostaining performed on the upper and lower skin dermis of healthy controls and patients with keloids or hypertrophic scars. Bars, 200 μm. (B) Quantification of the decorin positive area (in %age of the total area) in the dermis of healthy skin controls (n=5, orange), keloids (n=10, pink) or hypertrophic scars (n=10, brown) samples. Results are represented as mean ± SD. *p < 0.05, ***p < 0.001.
As decorin content was particularly low around the dermal epidermal junction (DEJ) of KS and HS, we investigated the levels of LTBP1, a large ECM protein located at the DEJ (59) and involved in the extracellular anchoring of latent TGF-β (9). Compared to healthy skin controls, reduced LTBP1 was observed at the DEJ of KS and HS (Figure 5A), with a more pronounced reduction in HS (absent from 8 out of 10 stained samples, Supplementary Figure 3 and Table 1) than in KS (absent from 6 out of 10 stained samples, Supplementary Figure 4 and Table 1). When pathological skin sections were divided into two groups based on the presence of LTBP1 at the DEJ (LTBP1+ versus LTBP1-), the average number of GzmB+ (Figure 5B) and MCT+ (Figure 5C) cells was significantly higher in the upper dermis of LTBP1- skin lesions. Consequently, we hypothesized that GzmB may cleave LTBP1, resulting in its reduction at the DEJ of KS and HS.
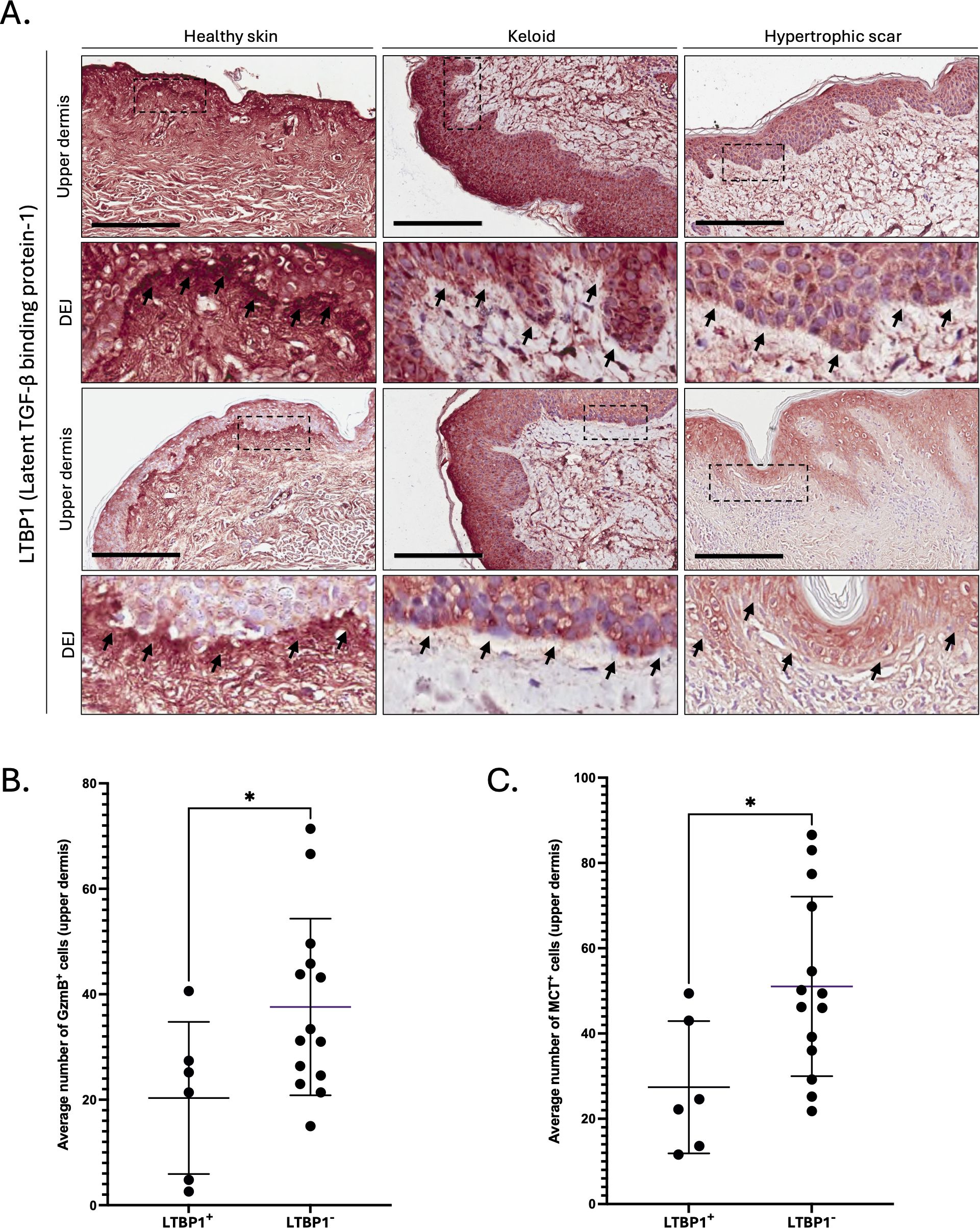
Figure 5. LTBP1 is reduced at the dermal epidermal junction of GzmB+ and MCT+ keloids and hypertrophic scars. (A) Latent TGF-β Binding Protein-1 (LTBP1) immunostaining performed on the upper dermis of healthy controls (n=5) and patients with keloids (n=10) or hypertrophic scars (n=10). The inset shows high magnification of the dermal epidermal junction (DEJ). Scale, 200 μm. (B, C) Average number of GzmB+ (B) and MCT+ (C) cells in the upper dermis of LTBP1+ (n=6) and LTBP1- (n=14) keloids and hypertrophic scars. Results are represented as mean ± SD. *p < 0.05.
GzmB cleaves LTBP1 in vitro
Using the GrabCas software, a bio-informatic tool used to predict the cleavage of protease with aspase-like activities (60), we identified 7 potential GzmB cleavage sites in the LTBP1 amino-acid sequence (Figure 6). Those cleavage sites, mostly located on the central Ca2+ binding Epidermal Growth Factor (EGF)-domains of LTBP1, are compatible with the release of a C-terminal fragment containing the SLC binding site (Figure 6, red arrowhead).
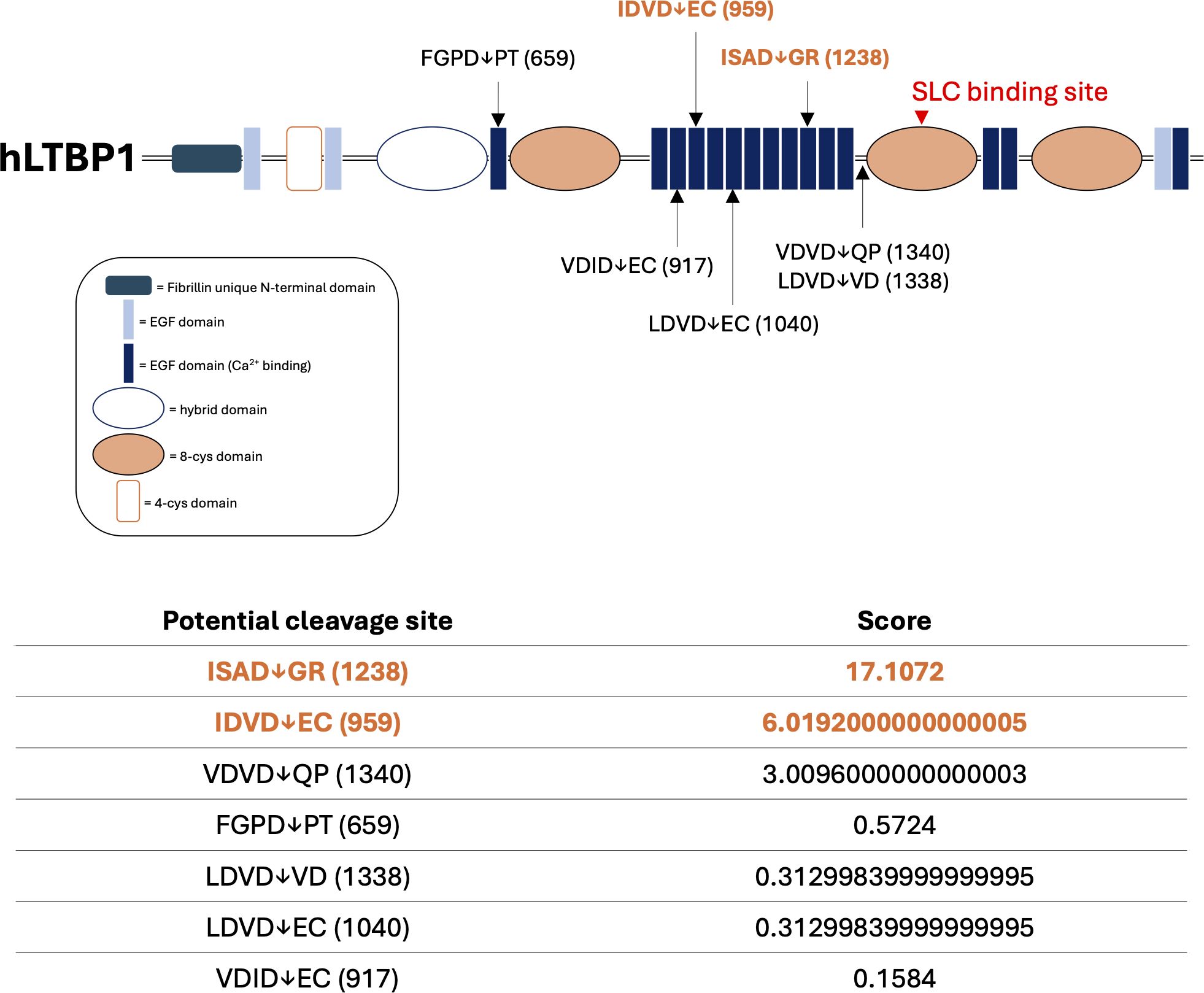
Figure 6. In silico prediction identifies LTBP1 as a potential new GzmB substrate. Schematic representation of predicted GzmB cleavage sites on human Latent TGF-β Binding Protein-1 (hLTBP1) amino-acid sequence. Letters represent the single amino acid code of the tetrapeptide preceding as well as the dipeptide following the expected cleavage site. Arrows represent the location of the expected cleavage site (after aspartic acid, D) and correspond to the amino acid location referenced between brackets. The score obtained using the GrabCas software is a readout of the cleavage susceptibility. The red arrowhead corresponds to the region responsible for the interaction between LTBP1 and the small latent complex (SLC). EGF, epidermal growth factor. Cys, cysteine.
Next, we investigated GzmB cleavage of LTBP1 in a cell free cleavage assay. Using CHO cells stably transfected with a plasmid encoding LTBP1 (CHO-LTBP1 (61)), we generated LTBP1-enriched conditioned media (LTBP1 CM). After protein precipitation, LTBP1 CM was incubated with GzmB for different time points followed by western blot analysis of the digestion products. Compared to non-digested controls, we identified a reduced LTBP1 molecular weight in the presence of GzmB (Figure 7A), indicating that GzmB can cleave LTBP1 from conditioned medium in vitro. A morphological switch towards fibroblastic-like features was also observed in CHO-LTBP1 cultured in presence of 100 nM GzmB (Supplementary Figure 5A, arrowheads). This partial epithelial to mesenchymal transition (EMT) phenotype was not observed in CHO-LTBP1 treated with PBS or with a lower GzmB concentration (50 nM). Consequently, we hypothesized that GzmB may modulate latent TGF-β bioavailability through the cleavage of LTBP1, thereby regulating the induction of EMT in epithelial cells (Supplementary Figure 5A).
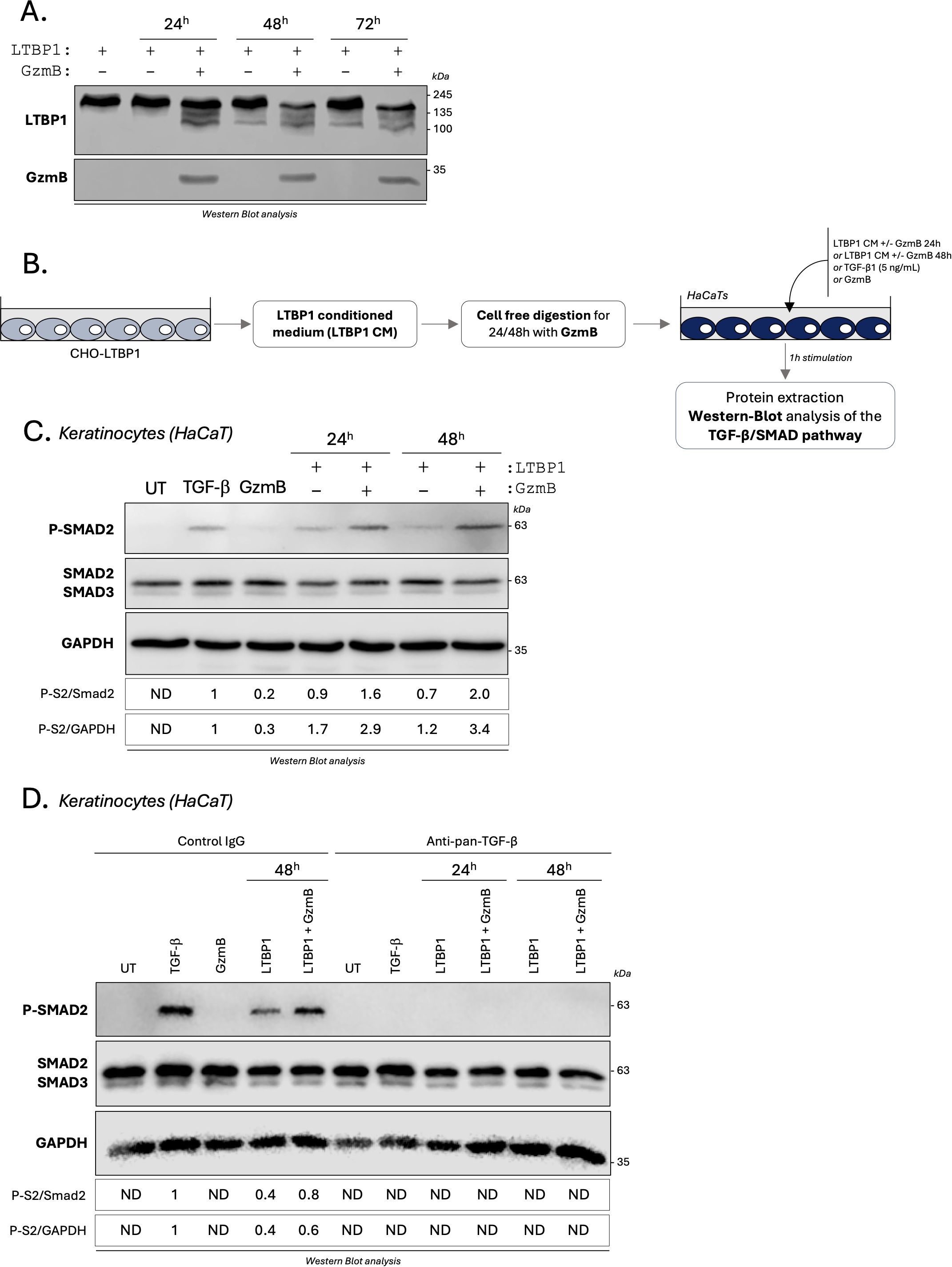
Figure 7. GzmB cleavage of LTBP1 in vitro activates TGF-β/Smad signaling pathway in epithelial cells. (A) LTBP1 conditioned media (from the same harvest) was precipitated, resuspended in GzmB digestion buffer, and incubated with (or without) 200 nM rhGzmB for several time point at 37°C and analyzed by western blot probing against LTBP1 (upper panel) and GzmB (lower panel). (B) Schematic representation of the experimental procedure used to investigate how GzmB digestion of LTBP1 modulates the TGF-β/Smad signaling pathway in keratinocytes (HaCaTs). (C) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in HaCaTs cultured for 1h as described in (B). (D) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in HaCaTs cultured for 1h as described in (B) in presence of an anti-pan-TGF-β inhibitor or an isotype-matched control IgG. UT, untreated; ND, non-detectable.
GzmB cleavage of LTBP1 activates TGF-β/Smad signaling pathway in epithelial cells
To characterize the functional consequences of GzmB-dependent LTBP1 cleavage on latent TGF-β activation, LTBP1 CM was digested with GzmB and subsequently used to treat human keratinocytes for 1 h (Figure 7B, optimized in Supplementary Figure 5B). Treatments with active TGF-β1 or GzmB alone were used as positive and negative controls respectively, and Smad2 phosphorylation was assessed in keratinocyte lysates as a readout for activated TGF-β1 signaling (Figure 7B). To ensure that the observed effect was dependent on LTBP1 cleavage and independent of decorin degradation, the absence of decorin from LTBP1 CM was confirmed by western blot analysis (Supplementary Figure 5C).
In contrast to the faint Smad2 phosphorylation observed in untreated or GzmB-treated keratinocytes (Figure 7C), treatment of HaCaT keratinocytes with GzmB-digested LTBP1 CM resulted in an increased Smad2 phosphorylation compared to the non-digested LTBP1 CM, with a 1.7-fold increase for 24h and 2.8-fold for 48h digestion (Figure 7C). To confirm the involvement of TGF-β in this process, we performed the same experiments in the presence of a pan-TGF-β inhibitor or an isotype match control antibody (Figure 7D). While Smad2 phosphorylation was unaffected by the control antibody, treatment with the pan-TGF-β inhibitor completely abolished Smad2 phosphorylation in all conditions (Figure 7D). This observation confirmed that GzmB can promote TGF-β dependent Smad2 phosphorylation through the generation of LTBP1 fragments. At the used concentration (200 nM), GzmB alone does not promote Smad2 phosphorylation in keratinocytes (Figures 7C, D).
To mitigate the effect of growth factors in the culture medium, we produced and purified recombinant human (rh)LTBP1 from conditioned medium of stably transfected CHO cells (Supplementary Figure 6A). Using cell free cleavage assay, we confirmed the ability of GzmB to cleave purified rhLTBP1 in vitro (Supplementary Figure 6A). Keratinocytes treated with rhLTBP1 for 1h demonstrated an increased and dose dependent Smad2-phosphorylation compared to non-treated controls (Supplementary Figure 6B). Pre-digestion of rhLTBP1 by GzmB resulted in a 2-fold increased Smad2 phosphorylation compared to the non-digested rhLTBP1 condition in keratinocytes (Supplementary Figure 6C), thus confirming the results obtained using CHO-derived LTBP1 CM (Figure 7).
As an additional indicator for TGF-β1 activity, p38 and p44/42 non-canonical TGF-β signaling pathways were also assessed in epithelial cells (62). Compared to the non-treated condition, LTBP1 CM promoted the phosphorylation of MAPK p38 (Supplementary Figure 7A) and p44/42 (Supplementary Figure 7B). Despite the ability of GzmB to directly activate both MAPK pathways, we observed no differences in p38 phosphorylation (Supplementary Figure 7A) as well as a reduction of p44/42 phosphorylation (Supplementary Figure 7B) for LTBP1 CM pre-digested with GzmB. Additionally, the LTBP1/GzmB-mediated MAPK activation was still detectable in the presence of the pan-TGF-β inhibitor (Supplementary Figures 7C, D), suggesting that this effect may be independent of TGF-β and directly mediated by LTBP1.
GzmB cleavage of LTBP1 activates TGF-β/Smad signaling pathway in dermal fibroblasts
Based on the fibroproliferative nature of KS and HS and because fibroblasts are the primary source of ECM molecules in fibrosis, the same experiments were performed using primary human dermal fibroblasts. While only a small increase in Smad2 phosphorylation was observed in primary dermal fibroblasts cultured with GzmB-digested LTBP1 CM compared with non-digested LTBP1 CM (Figure 8A), a 2-fold increase in Smad2 phosphorylation was observed when primary dermal fibroblasts were treated with GzmB-digested rhLTBP1 compared to the non-digested protein (Supplementary Figure 8). Together, these findings show that GzmB-dependent LTBP1 cleavage activates TGF-β signaling in dermal fibroblasts.
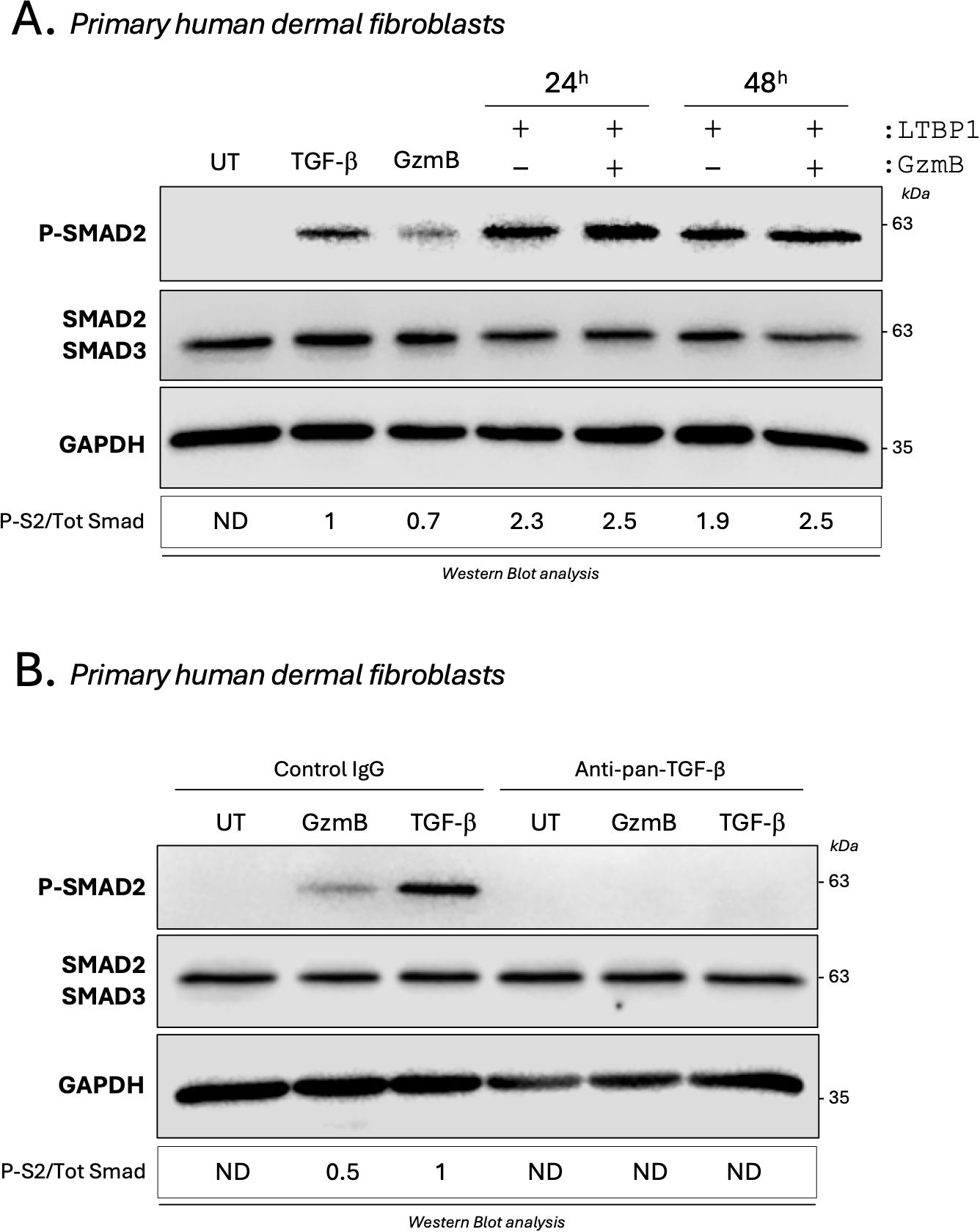
Figure 8. GzmB promotes a direct and TGF-β dependent Smad2 phosphorylation in dermal fibroblasts. (A) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in primary human dermal fibroblasts cultured for 1h as described in Figure 7B. (B) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in untreated (UT) primary human dermal fibroblasts or primary human dermal fibroblasts treated for 1h with TGF-β1 (5 ng/mL) or 200 nM GzmB in presence of an anti-pan-TGF-β inhibitor or an isotype-matched control IgG. UT, untreated; ND, non-detectable.
GzmB promotes TGF-β activation through the cleavage of one or more fibroblast secreted protein(s)
Unexpectedly, and in opposition to what was previously observed in keratinocytes (Figures 7C, D), a marked Smad2 phosphorylation was detectable in primary dermal fibroblasts treated with 200 nM GzmB alone (Figure 8A). This phosphorylation was completely abolished in the presence of the pan-TGF-β inhibitor (Figure 8B), indicating that GzmB itself can directly activate latent TGF-β secreted by dermal fibroblasts.
We next investigated whether this direct activation of the TGF-β/Smad signaling pathway was regulated by GzmB-dependent proteolysis of secreted proteins. To do so, primary human dermal fibroblasts were starved for 24h to generated fibroblast-derived CM enriched in secreted proteins. In a cell free system, fibroblast CM was incubated for 1h with GzmB to generate protein fragments. Then, and in order to mitigate the direct effect of GzmB, the protease was inhibited using its specific extracellular inhibitor VTI-1002 (Figure 9A). The digested fibroblast medium, thus containing protein fragments and inactivated GzmB, was used to treat dermal fibroblasts or keratinocytes for 1h followed by western blot analysis of the TGF-β/Smad signaling pathway (Figure 9A). A no inhibitor control, as well as a VTI-1002 alone control, were also included.
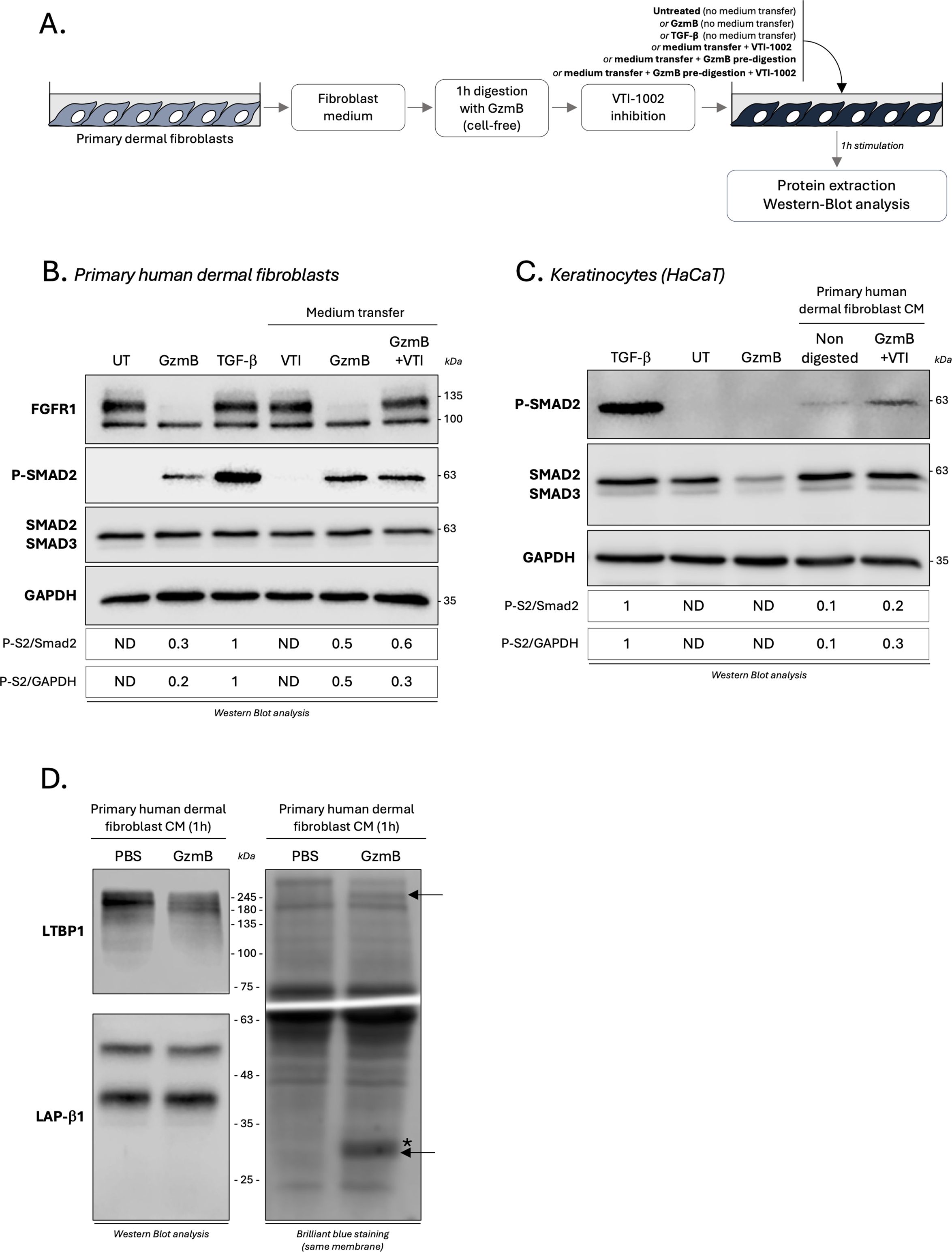
Figure 9. GzmB promotes TGF-β activation through the cleavage of one or more fibroblast secreted protein(s). (A) Schematic representation of the experimental procedure used to investigate how GzmB proteolysis of primary dermal fibroblast CM modulates the TGF-β/Smad signaling pathway. (B) Western blot analysis showing FGFR1, P-Smad2, total Smad2/3 and GAPDH levels in primary dermal human fibroblasts cultured for 1h as described in (A). (C) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in HaCaTs cultured for 1h as described in (A). (D) Western blot analysis demonstrating the ability of GzmB to cleave LTBP1 (top left panel) but not LAP-β1 (bottom left panel) from primary dermal fibroblast CM after 1h incubation. The right panel represent brilliant blue staining of the same membrane. De novo generated fragments are indicated by a black arrow and GzmB in indicated by a (*). UT, untreated; ND, non-detectable; CM, conditioned medium.
While VTI-1002 inhibited GzmB proteolytic activity as demonstrated by the nullification of fibroblast growth factor receptor 1 (FGFR1) cleavage, a strong Smad2 phosphorylation in primary dermal fibroblast was still detectable after GzmB inhibition (Figure 9B). Similar results were obtained using keratinocytes, with a 2- to 3-fold increased Smad2 phosphorylation when cultured in the presence of primary dermal fibroblast CM pre-digested with GzmB compared to non-digested controls (Figure 9C). As previously reported (Figures 7C, D), GzmB alone does not promote Smad2 phosphorylation in keratinocytes (Figure 9C).
Using dermal fibroblast CM digested for 1h with GzmB (cell free system), we confirmed the ability of GzmB to cleave fibroblast-derived LTBP1 by immunoblotting (Figure 9D, left panel). We also identified the presence of two newly generated degradation products in the CM of fibroblasts incubated with GzmB (Figure 9D, right panel, black arrows). As the intensity and molecular weight of the LAP-β1 pro-peptide (a component of the SLC) remained the same after 1h incubation with GzmB (Figure 9D, left panel), these results suggest a mechanism whereby GzmB promotes indirect latent TGF-β activation from fibroblast medium through the cleavage of one (or more) unidentified secreted protein(s).
Discussion
Tethered to the ECM as part of the SLC or the LLC, TGF-β is activated extracellularly in a tightly regulated process that involves ECM molecules, cell surface integrins, and/or extracellular proteases. While increased TGF-β signaling in fibrosis is well-established (28, 29), little is known about the molecular mechanisms involved in the regulation of its extracellular activation in KS and HS. Results from this study suggest that extracellular GzmB contributes to KS and HS pathology through the cleavage of ECM molecules involved in the regulation of latent TGF-β activation.
In the dermis of patients with HS, a significant accumulation of GzmB-expressing cells, especially mast cells, was observed. Compared to healthy skin controls, increased Substance P, a mediator of human mast cell degranulation, was also evident in both KS and HS (58). While the cell source of Substance P remains to be elucidated, these findings suggest that GzmB+ mast cells are a key source of extracellular GzmB in KS and HS. Since mast cells do not express perforin (63), it is possible that Substance P-mediated mast cell degranulation results in the accumulation of GzmB in the extracellular milieu. Consequently, as previously reported in other dermatological conditions (49–51, 54), GzmB may contribute to KS and HS through perforin-independent extracellular mechanisms. Nevertheless, extracellular release of GzmB after Substance P-mediated mast cell degranulation will have to be confirmed experimentally.
In addition to confirming the reduction of decorin in the dermis of KS (37, 38) and HS (39, 40), we reported the reduction of LTBP1 at the DEJ of KS and HS and confirmed its cleavage by GzmB in vitro. While we cannot exclude that LTBP1 reduction observed in KS and HS is due to its downregulation at a transcriptional level or to its degradation by other proteases (notably Bone Morphogenic Protein (BMP) 1 (64) and membrane-type 1 MMP (MT1-MMP) (23)), the abundance of GzmB+ mast cells in the upper dermis of LTBP1-negative lesions implicates GzmB as a potential contributor of its degradation in human samples.
In vitro, GzmB-dependent cleavage of LTBP1 promoted Smad2 phosphorylation in epithelial cells, most likely by generating LTBP1 fragments with increased ability to activate TGF-β. This mechanism, similar to the one previously identified for BMP-1 cleavage of thrombospondin-1 (65), might explain the partial EMT observed in CHO-LTBP1 cells treated with GzmB. A deeper analysis of the induced TGF-β/Smad signaling pathway, notably the downstream transcription of TGF-β targeted genes involved in EMT or ECM molecule synthesis, will help to decipher the precise roles of the GzmB/LTBP1 axis in latent TGF-β activation. It is also worth noting that fibrillin-1 and fibronectin, two LTBP1-interacting glycoproteins involved in the extracellular tethering of the LLC (9), are established GzmB substrates validated in vivo in different disease models (52, 66). Consequently, and in addition to the direct cleavage of LTBP1, GzmB may impair the extracellular anchoring of LTBP1 to the fibrillin-/fibronec,n-rich matrix, with potential consequences in biomechanical properties of KS and HS lesions. A similar mechanism has been notably reported in burn wounds, where GzmB dependent cleavage of decorin impaired wound remodeling through the modification of collagen bundle organization (54). This hypothesis can be evaluated by performing atomic force microscopy in KS and HS samples (67).
In the present study, epithelial cells and fibroblasts responded differently to GzmB stimulation. While no Smad2 phosphorylation was observed in keratinocytes treated with the protease, TGF-β-dependent Smad2 phosphorylation was observed in primary dermal fibroblasts treated with GzmB. In addition to confirming the cleavage of fibroblast-derived LTBP1, we also identified the presence of two protein fragments in the conditioned media of fibroblast digested with GzmB. Consequently, and keeping in mind that GzmB does not cleave the LAP pro-peptide, it is plausible that GzmB contributes to latent TGF-β activation through the cleavage of fibroblast-secreted ECM molecules that are not produced by keratinocytes. Such mechanism could involve several ECM molecules involved in TGF-β activation and already identified as GzmB substrates, such as LTBP1, decorin (57), thrombospondin-1 (68), fibrillin-1 (66), and/or tenascin-C (69). While this hypothesis will require in vitro and in vivo experimental validation, GzmB-mediated TGF-β activation also needs to be re-evaluated in primary cells extracted from KS and HS, as fibroblasts from these two conditions exhibit increased sensitivity to TGF-β stimulation compared to healthy dermal fibroblasts (33–36). Using patient-derived primary cells will also help us to decipher whether LTBP1 expression is altered at the transcriptional level in KS and HS.
Over the past 10 years, TGF-β-based monotherapies have only demonstrated mild successes in the context of KS and HS (5). This may be due to the fact that while TGF-β1 and β2 isoforms are pro-fibrotic, TGF-β3 is anti-fibrotic (70, 71). As GzmB is, to our knowledge, exclusively involved in TGF-β1 activation, targeting the protease in the context of wound healing might be a promising strategy to specifically tackle TGF-β pro-fibrotic effects. Of note, topical inhibition of GzmB reduced scarring and promoted the healing of burn wounds (54), and reduced ECM accumulation as well as myofibroblast differentiation in a model of cardiac fibrosis (72). While the burn model is relevant in the context of studying HS, a role for GzmB in KS has never been assessed in vivo. Such an approach is challenged by the difficulty of implementing a physiologically relevant KS model in mice, which usually requires the implantation of human keloid explants (or cells derived from human KS samples) in immunocompromised animals (73). Nevertheless, performing this model in a GzmB deficient background will be the next step in understanding the precise roles of the protease in the development of KS.
Altogether, this work provides a novel mechanism for GzmB-induced TGF-β activation in the context of scarring and fibrosis.
Materials and methods
Study approvals
Use of human biospecimens for this study was approved by the University of British Columbia (Clinical research ethics board no: H23-01301). Skin biopsies from healthy skins as well as keloid and hypertrophic scars were collected and provided by Dr. Richard Crawford (Department of Dermatology, University of British Columbia, Vancouver, BC, Canada). Patient information is available in Table 1.
Immunohistochemistry, tyramide signal amplification and co-immunofluorescence
Immunostaining was performed as previously established (47, 49). Briefly, after dewaxing and rehydration, antigen retrieval was performed in 10 mM sodium citrate buffer (Sigma) containing 0.05% Tween-20 (Sigma-Aldrich) pH 6 for 20 min in a steamer. Sections were then cooled down for 20 min at room temperature and endogen peroxidases were quenched by 3% H2O2 (v/v). Non-specific sites were then blocked for 30 min at RT followed by primary antibody incubation overnight at 4°C (diluted in blocking solution). The next day, after 30 min to 1h incubation with the secondary antibody (diluted in blocking solution), HRP was coupled with the biotinylated secondary antibody for 30 min using the VECTASTAIN Elite ABC-Peroxidase kit (Vector laboratories).
Immunohistochemistry
Revelation was performed using the Vector NovaRED Substrate Kit (Vector laboratories). Sections were counterstained with modified Harris Hematoxylin (Sigma-aldrich), blued in 0.5% Lithium Carbonate and mounted using the xylene-based Cytoseal™ 60 medium (Epredia). Slides were scanned using the APERIO CS2 slide scanner (Leica).
Tyramide signal amplification (TSA)
Revelation was performed according to the manufacturer recommendations. First revelation was performed using the Green Fluorescein Amplification Reagent (Akokabio) diluted at 1:50 in TBS-T for 5 min at RT. Avidin and Biotin free sites were then blocked for 15 min each at RT (Akokabio), followed by 30 min blocking and overnight incubation at 4°C with the second primary antibody (diluted in blocking solution). The next day, after secondary antibody incubation and HRP coupling, the second revelation was performed using the Red Fluorescein Amplification Reagent (Akokabio) diluted at 1:50 in TBS-T for 5 min at RT. Nuclei were then stained for 3 min with DAPI (1:1,000 in PBS) and sections were mounted using the water-based Hydromount medium (Electron Microscopy Sciences). Pictures were taken using a fluorescence inverted microscope (EVOS FL, Life Technology) and analyzed using the FIJI software (SciJava).
Co-Immunofluorescence
After de-waxing, rehydration and antigen retrieval, sections were permeabilized for 10 min with 0.5% tritton-X100 (FisherBiotech) and blocked in 5% FBS for 30 min prior co-incubation overnight at 4°C with both primary antibodies (diluted in blocking solution). The next day, after 1h of co-incubation with A593/A488-coupled secondary antibodies, nuclei were stained for 3 min with DAPI (1:1,000 in PBS) and sections were mounted using the water-based Hydromount medium (Electron Microscopy Sciences). Pictures were taken using a fluorescence inverted microscope (EVOS FL, Life Technology) and analyzed using the FIJI software (SciJava). Blocking conditions as well as primary and secondary antibodies dilution are detailed in Table 2.
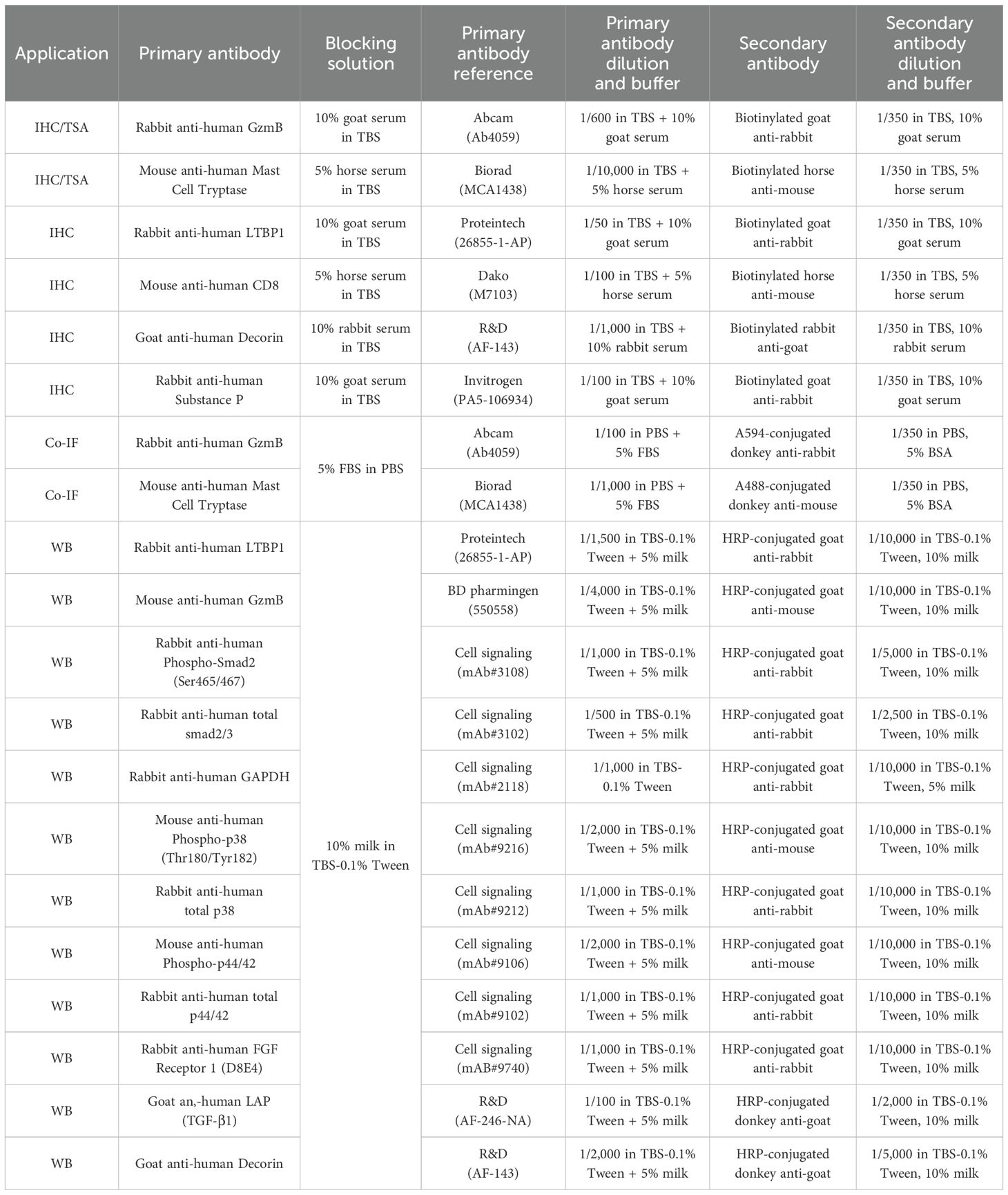
Table 2. List of primary and secondary antibodies used for immunohistochemistry (IHC), tyramide signal amplification (TSA), co-immunofluorescence (Co-IF) and Western blotting (WB).
Prediction of GzmB cleavage sites in human LTBP1
The protein sequence of human LTBP1 was obtained from Uniprot (assession no.: Q14766-1) and assessed for potential GzmB cleavage sites using GraBCas (v1.0) software with a cutoff of 0.1 (60). Cleavage sites were mapped to their respective regions in the full-length LTBP1 according to the domain organization available on Uniprot and displayed in (9).
Cell culture and generation of LTBP1 conditioned medium
Kera,nocytes (HaCaTs) and primary human dermal fibroblasts were maintained in complete Dulbecco modified Eagle’s medium (DMEM) containing 10% (v/v) Fetal Bovine Serum (FBS, Gibco) and 1% (v/v) Penicillin-Streptomycin (PS, Sigma). Chinese Hamster Ovary (CHO) cells stably transfected with the plasmid encoding LTBP1 (hereafter named CHO-LTBP1) was a gift from Boris Hinz (University of Toronto, Ontario, Canada) and were maintained in DMEM containing 10% (v/v) FBS, 1% (v/v) PS and selected with 1 mg/mL Geneticin (G418 sulfate, Agilent technology). To generate LTBP1-conditioned medium, CHO-LTBP1 were cultured to over-confluency for 3 weeks under serum deprivation and culture medium was collected 3 times a week. Conditioned media enriched in LTBP1 were cleared of cellular debris by centrifugation at 300 g for 10min at room temperature and stored at -20°C. Presence of LTBP1 in CM was assessed by immunoblotting.
Recombinant human LTBP1 production and purification
CHO cells transfected with the pSecTag/FRT/V5-His-TOPO vector encoding LTBP1-EGFP (61) were donated from Boris Hinz (University of Toronto, Ontario, Canada) and were maintained in complete DMEM containing 10% (v/v) Fetal Bovine Serum (FBS, Gibco), 1% (v/v) Penicillin-Streptomycin (PS, Sigma) and 1 mg/mL Geneticin (G418, Gibco). Stably transfected CHO cells were culture to overconfluence for 2 weeks under serum deprivation. Culture medium was collected 3 times a week, centrifuged at 300g for 5 minutes to remove cellular debris and stored at -20°C. Prior purification, LTBP1-EGFP enriched conditioned medium was dialyzed 3 times (twice for 2 hours, then overnight) at 4°C in PBS using 6-8 kDa dialysis membrane (SpectrumLabs). LTBP1-EGFP conditioned medium was then directly applied on HisTrap HP His Tag protein purification columns (5 mL, Cytiva Life Sciences) using the ÄKRA Start Chromatography System (Cytiva Life Sciences). Column was washed using PBS containing 20 mM imidazole and eluted using 300 mM imidazole diluted in PBS. Fractions enriched in LTBP1-EGFP were analyzed by Brilliant Blue Staining (MiliporeSigma) and immunoblotting, pulled together, and dialyzed 3 times overnight at 4°C in PBS, PBS containing 0.2% (v/v) chloroform, and PBS before storage at -80°C. LTBP1-EGFP concentration was determined using the Pierce BCA protein Assay Kit (Thermo Fisher Scientific).
In vitro cell free digestion assay
Conditioned medium
1 mL of LTBP1 CM from the same set of harvest was precipitated with 10% final Tri-chloroacetic acid (Sigma-aldrich) for 20 min on ice and proteins were pelleted by centrifugation at 14,324 g for 10 min at 4°C. After removal of the supernatant, protein pellets were washed twice with cold 100% ethanol (VWR) before being resuspended in 50 μL of GzmB digestion buffer (50 mM Tris in PBS, pH 7.4). 200 nM GzmB (Bon Opus) or an equivalent volume of PBS was then added to the samples and incubated at 37°C for 24, 48 or 72h. Reactions were stopped by adding 6X protein loading buffer (final concentration of 1X) followed by heat denaturation at 95°C for 10min and analysis by immunoblotting. For cell treatment experimenta,ons, 1 mL of nonprecipitated CHO-LTBP1 CM from the same set of harvest was incubated with 200 nM GzmB (or equivalent volume of PBS) at 37°C for 24 or 48h before being use as culture medium.
Purified recombinant human LTBP1
1 μg of rhLTBP1-EGFP was digested with rhGzmB (200 nM, Bon Opus) in 50 nM Tris (Fisher Bioreagents) diluted in PBS, pH 7.4, at 37°C for 24h (final volume of 50 μL). Reactions were stopped by adding 6X protein loading buffer (final concentration of 1X) followed by heat denaturation at 95°C for 10 min and analysis by immunoblotting.
Cell stimulation and protein extraction
1,000,000 HaCaTs or 500,000 primary dermal human fibroblasts were seeded in 6 well plates (Corning) and cultured for 24h in complete medium before being serum starved for 16 to 24h. For medium transfer experimentations, the supernatant was removed post starvation and cells were cultured in the presence of LTBP1 CM (from the same set of harvest) predigested with 200 nM GzmB for 24 or 48h at 37°C, or with primary human dermal fibroblast CM predigested with 200 nM GzmB for 1h at 37°C. TGF-β1 (from 5 to 10 ng/mL, PeproTech) or GzmB (280 ng or 200 nM, depending on the experimental design and the digestion volume) stimulations were used as both positive and negative controls. After 1h stimulation, cells were washed twice with cold PBS and total proteins were extracted using radioimmunoprecipitation assay (RIPA) extraction buffer containing EDTA-free protease inhibitor and phosphatase inhibitors (Roche) for 20min on ice. After centrifugation at 13,200g for 10min at 4°C, supernatant containing solubilized proteins was stored at -20°C prior to immunoblotting. Protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Sigma-Aldrich).
For TGF-β neutralization experiments, mouse monoclonal IgG anti-TGF-β1,2,3 (MAB1835, Clone 1D11) antibody was purchased from R&D Systems and isotype-matched mouse Kappa Globulin (clone p3.6.2.8.1) used as negative control was purchased from Invitrogen (14-4714-85). In both cases, cells (or LTBP1 CM) were pre-incubated for 10 min at 37°C in the presence of 2.5 μg/mL of either anti pan-TGF-β or isotype-matched antibody before stimulation. For GzmB inhibition experiments, VTI-1002 (viDA Therapeutics, Vancouver, Canada, BC) diluted in DMSO was used at a final concentration of 1 or 2 μM.
Phase contrast analysis
500,000 CHO-LTBP1 cells were seeded in a 6-well plates for 24h in complete medium before being serum starved overnight. The next day, medium was replaced with fresh FBS free DMEM and cells were treated with GzmB (50 or 100 nM) or an equivalent volume of PBS. After 48h treatment, media was replaced by fresh FBS free medium, and cells were treated another time with the same amount of GzmB or PBS. Cells were cultured for another 5 days in the presence of the protease (total duration of the treatment: 7 days) followed by morphological analysis. Phase contrast pictures were taken using a digital inverted microscope (EVOS FL, Life Technology) at 10X magnification.
Immunoblotting
20 μg of total proteins extracted from keratinocytes or primary human dermal fibroblasts were analyzed by SDS-PAGE in Tris-Glycine buffer (TG, composed of Tris-base from Fisher Bioreagents and Glycine from Fisher chemical) containing Sodium-Dodecyl-Sulfate (SDS, BIO-RAD) before being transferred onto a Polyvinylidene Fluoride membrane (PVDF 0.2 μm pore size, BIO-RAD) at 0.4 A for 2h in TG buffer containing 10% Ethanol (VWR). Membranes were blocked using 10% (w/v) non-fat dry milk diluted in Tris-buffered saline (TBS, Tris-base and NaCl from Fisher Bioreagents) containing 0.1% (v/v) Tween-20 at room temperature for 1h and incubated overnight at 4°C with primary antibody (orbital shaker). The next day, HRP-conjugated secondary antibody was incubated for 1h at RT and revelation was performed using the SuperSignal™ West Pico PLUS chemiluminescent Substrate (Thermo Scientific). Images were acquired on LI-COR Odyssey Fc system (LI-COR Bio-sciences, Lincoln, NE) and analyzed/quantified using the FIJI software (SciJava). Primary and secondary antibodies dilution are detailed in Table 2. For Supplementary Figure 3C, 200 ng of recombinant human decorin was used as control and purchased from Abnova.
Quantification and statistical analysis
Quantification of the number of GzmB+, MCT+ and CD8+ cells in the upper and lower dermis has been performed according to the following methods: each immunobiological section has been pictured at 10 different areas (20X zoom from the 20X magnification) and ranked from one to ten. Then, 5 pictures were randomly selected, and the number of positive cells was counted. The mean was then calculated as the average number of positive cells from these 5 pictures. For decorin quantification, images were deconvoluted on FIJI (SciJava) using the “deconvolution” plugin and measured using a 60/240 threshold.
The R software (and its graphical user interface R Commander, T Studio Team, PBC, Boston, MA) and Prism software (GraphPad Software, San Diego, CA, USA) were used for graphical representations of data and statistical analyses. Mean comparison between healthy skin, keloid scars and hypertrophic scars was performed using the Kruskal-Wallis test (non-parametric equivalent of ANOVA) followed by Dunn’s multiple comparison (vs. healthy skin). Correlation coefficients r between GzmB and MCT, as well as GzmB and CD8, were determined using the Spearman rank-order (non-parametric equivalent of the Pearson product-moment) correlation coefficient. Comparison of the average number of GzmB+/MCT+ cells in the upper dermis of LTBP1+ and LTBP1- lesions were performed using unpaired Student’s t-test (parametric) after normality (Shapiro-Wilk test) and homoscedasticity (Fisher’s exact test) validation. p values < 0.05 were considered as statistically significant and are indicated in the figure legends.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by University of British Columbia Clinical Research Ethics Board: H23-01301. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin because the biospecimens were de-identified with the exception of age and sex and the study team did not have access to any clinical data. The samples were collected for diagnostic purposes and the excess tissue sections would otherwise be discarded.
Author contributions
AA: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Resources, Visualization, Writing – original draft, Writing – review & editing. JG: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Writing – review & editing. AL: Investigation, Methodology, Writing – review & editing. MK: Investigation, Methodology, Writing – review & editing. KR: Investigation, Methodology, Writing – review & editing. KJ: Funding acquisition, Writing – review & editing. BH: Resources, Writing – review & editing. RC: Data curation, Resources, Writing – review & editing. DG: Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded in part by grants-in-Aid from the Canadian Institutes for Health Research (DJG - #479268), National Sciences and Engineering Research Council of Canada (DJG), the British Columbia Professional Firefighters’ Burn Fund (DJG), and the International Brotherhood of Electrical Workers (IBEW) Local 258 (DJG). AA is the recipient of the Arthritis Society Canada Training Postdoctoral Fellowship. BH’s research is supported by grant from the Canadian Institutes of Health Research (#375597 and #190081) and support from the John Evans Leadership funds (#36050 and #38861) and innovation funds (‘Fibrosis Network, #36349’) from the Canada Foundation for Innovation (CFI) and the Ontario Research Fund (ORF). KR is the recipient of Canada Graduate Scholarships -Doctoral Award (#172616).
Acknowledgments
We thank Simon Cheung for the preparation of keloid and hypertrophic scar sections, as well as Megan A. Pawluk for the extraction and isolation of primary human dermal fibroblasts.
Conflict of interest
DG is a co-founder and Chief Officer for viDA Therapeutics Vancouver, Canada, BC which developed VTI-1002, a granzyme B inhibitor used in this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1484462/full#supplementary-material
Supplementary Figure 1 | (A) CD8 immunostaining performed on the upper and lower skin dermis of healthy controls and patients with keloids or hypertrophic scars. Bars, 200 μm. (B, C) Quantification of the average number of CD8+ T-cells in the upper (B) and lower (C) dermis of healthy skin controls (n=6, orange), keloids (n=10, pink) or hypertrophic scars (n=10, brown) samples. Results are represented as mean ± SD. (D, E) Correlation between the average number of GzmB+ and CD8+ cells in the upper (D) and lower (E) dermis of keloids or hypertrophic scars samples (n=20). *p < 0.05. ns, not significant.
Supplementary Figure 2 | Multiplex staining with Tyramide Signal Amplification (TSA) of GzmB (green) and MCT (orange) in the skin dermis of keloids or hypertrophic scars. Scale, 20 μm. Merge showcases overlap of the two channels and co-stained cells appear in yellow. Cell nuclei are stained with DAPI (blue).
Supplementary Figure 3 | Immunostaining demonstrating the presence (left panel, 2 out of 10) or absence (right panel, 8 out of 10) of LTBP1 at the dermal epidermal junction of hypertrophic scars (n=10). Scale, 200 μm.
Supplementary Figure 4 | Immunostaining demonstrating the presence (left panel, 4 out of 10) or absence (right panel, 6 out of 10) of LTBP1 at the dermal epidermal junction of keloids (n=10). Scale, 200 μm.
Supplementary Figure 5 | (A) Phase-contrast microscopy performed on Chinese Hamster Ovary (CHO) cells stably transfected with a plasmid encoding LTBP1 and cultured for 7 days under serum deprivation and in presence of PBS or GzmB (50 nM and 100 nM). Arrowheads are pointing at cells undergoing partial EMT and displaying fibroblastic-like features. Bars, 400 μm. (B) Western blot analysis showing P-Smad2 and GAPDH levels in HaCaTs cultured in presence of LTBP1 CM for 30 min, 1h, 2h and 3h. (C) Western blot analysis showing the presence of LTBP1 (upper panel) and decorin (lower panel) in 1 mL of precipitated LTBP1 CM or 200 ng of recombinant human decorin.
Supplementary Figure 6 | (A) 1 μg of rhLTBP1 purified by affinity chromatography was incubated with (or without) 280 ng rhGzmB (corresponding to 200 nM in a final digestion volume of 50 μL) for 24h at 37°C and analyzed by western blot probing against LTBP1 (upper panel) and GzmB (lower panel). (B) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in HaCaTs cultured for 1h in presence of TGF-β (3.5 ng/mL) or increasing quantity of purified rhLTBP1. (C) Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in HaCaTs cultured for 1h in presence of TGF-β (10 ng/mL), GzmB (280 ng/mL), non-digested rhLTBP1 (1 μg) or rhLTBP1 (1 μg) pre-digested for 24h with 200 nM GzmB (280 ng) and pre-incubated for 10 min with 2 μM VTI-1002. Digestions were performed in a final volume of 50 μL. UT, untreated. ND, non-detectable.
Supplementary Figure 7 | (A, B) Western blot analysis showing P-p38, total p38 and GAPDH levels (A), as well as P-p44/42, total p44/42, and GAPDH (B) levels in HaCaTs cultured for 1h as described in (Figure 7B). (C, D) Western blot analysis showing P-p38, total p38 and GAPDH levels (C), as well as P-p44/42, total p44/42, and GAPDH (D) levels in HaCaTs cultured for 1h as described in (Figure 7B) in presence (or not) of an anti-pan-TGF-β inhibitor. UT, untreated. ND, non-detectable.
Supplementary Figure 8 | Western blot analysis showing P-Smad2, total Smad2/3 and GAPDH levels in primary dermal human fibroblasts cultured for 1h in presence of TGF-β (5 ng/mL), GzmB (280 ng/mL), non-digested rhLTBP1 (1 μg) or rhLTBP1 (1 μg) pre-digested for 24h with 200 nM GzmB (280 ng). Digestions were performed in a final volume of 50 μL. UT, untreated. ND, non-detectable.
References
1. Wolfram D, Tzankov A, Pülzl P, Piza-Katzer H. Hypertrophic scars and keloids—A review of their pathophysiology, risk factors, and therapeutic management. Dermatologic Surgery. (2009) 35:171–81. doi: 10.1111/j.1524-4725.2008.34406.x
2. Wang ZC, Zhao WY, Cao Y, Liu YQ, Sun Q, Shi P, et al. The roles of inflammation in keloid and hypertrophic scars. Front Immunol. (2020) 11:603187. doi: 10.3389/fimmu.2020.603187
3. Butzelaar L, Schooneman DPM, Soykan EA, Talhout W, Ulrich MMW, Van Den Broek LJ, et al. Inhibited early immunologic response is associated with hypertrophic scarring. Exp Dermatol. (2016) 25:797–804. doi: 10.1111/exd.2016.25.issue-10
4. Feng C, Shan M, Xia Y, Zheng Z, He K, Wei Y, et al. Single-cell RNA sequencing reveals distinct immunology profiles in human keloid. Front Immunol. (2022) 13:940645. doi: 10.3389/fimmu.2022.940645
5. Zhang T, Wang XF, Wang ZC, Lou D, Fang QQ, Hu YY, et al. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. Biomedicine Pharmacotherapy. (2020) 129:110287. doi: 10.1016/j.biopha.2020.110287
6. Berman B, Maderal A, Raphael B. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatol Surg. (2017) 43:S3–18. doi: 10.1097/DSS.0000000000000819
7. Massagué J, Sheppard D. TGF-β signaling in health and disease. Cell. (2023) 186:4007–37. doi: 10.1016/j.cell.2023.07.036
8. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature. (2011) 474:343–9. doi: 10.1038/nature10152
9. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol. (2015) 47:47:44–53. doi: 10.1016/j.matbio.2015.05.005
10. Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor- fJ by the proteoglycan decorin. Nature. (1990) 346:281–4. doi: 10.1038/346281a0
11. Kalamajski S, Aspberg A, Oldberg Å. The decorin sequence SYIRIADTNIT binds collagen type I. J Biol Chem. (2007) 282:16062–7. doi: 10.1074/jbc.M700073200
12. Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, et al. The single-molecule mechanics of the latent TGF-β1 complex. Curr Biol. (2011) 21:2046–54. doi: 10.1016/j.cub.2011.11.037
13. Munger JS, Huang X, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, et al. A mechanism for regulating pulmonary inflammation and fibrosis: the integrin αvβ6 binds and activates latent TGF β1. Cell. (1999) 96:319–28. doi: 10.1016/S0092-8674(00)80545-0
14. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol. (2007) 179:1311–23. doi: 10.1083/jcb.200704042
15. Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, et al. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc Res. (2014) 102:407–17. doi: 10.1093/cvr/cvu053
16. Worthington JJ, Kelly A, Smedley C, Bauché D, Campbell S, Marie JC, et al. Integrin αvβ8-mediated TGF-β Activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity. (2015) 42:903–15. doi: 10.1016/j.immuni.2015.04.012
17. Schultz-Cherry S. Thrombospondin causes activation of latent transforming growth factor- beta secreted by endothelial cells by a novel mechanism [published erratum appears in J Cell Biol 1993 Sep;122(5):following 1143. J Cell Biol. (1993) 122:923–32. doi: 10.1083/jcb.122.4.923
18. Bourd-Boittin K, Bonnier D, Leyme A, Mari B, Tuffery P, Samson M, et al. Protease profiling of liver fibrosis reveals the ADAM metallopeptidase with thrombospondin type 1 motif, 1 as a central activator of transforming growth factor beta. Hepatology. (2011) 54:2173–84. doi: 10.1002/hep.24598
19. Aubert A, Mercier-Gouy P, Aguero S, Berthier L, Liot S, Prigent L, et al. Latent TGF-β Activation is a hallmark of the tenascin family. Front Immunol. (2021) 12:613438. doi: 10.3389/fimmu.2021.613438
20. Pedrozo HA, Schwartz Z, Robinson M, Gomez R, Dean DD, Bonewald LF, et al. Potential mechanisms for the plasmin-mediated release and activation of latent transforming growth factor-β1 from the extracellular matrix of growth plate chondrocytes1. Endocrinology. (1999) 140:5806–16. doi: 10.1210/endo.140.12.7224
21. Abe M, Oda N, Sato Y. Cell-associated activation of latent transforming growth factor-β by calpain. J Cell Physiol. (1998) 174:186–93. doi: 10.1002/(SICI)1097-4652(199802)174:2<186::AID-JCP6>3.0.CO;2-K
22. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. (2000) 14:163–76. doi: 10.1101/gad.14.2.163
23. Tatti O, Vehvilainen P, Lehti K, Keskioja J. MT1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res. (2008) 314:2501–14. doi: 10.1016/j.yexcr.2008.05.018
24. Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. (2003) 3:807–20. doi: 10.1038/nrc1208
25. Lodyga M, Hinz B. TGF-β1 – A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol. (2020) 101:123–39. doi: 10.1016/j.semcdb.2019.12.010
26. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast. Am J Pathology. (2007) 170:1807–16. doi: 10.2353/ajpath.2007.070112
27. Branton MH, Kopp JB. TGF-β and fibrosis. Microbes Infection. (1999) 1:1349–65. doi: 10.1016/S1286-4579(99)00250-6
28. Penn JW, Grobbelaar AO, Rolfe KJ. The role of the TGF-β family in wound healing, burns and scarring: a review. Int J Burns Trauma. (2012) 2:18–28.
29. Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: The paradigm of skin fibrosis — Pathomechanisms and treatment. Matrix Biol. (2016) 51:37–46. doi: 10.1016/j.matbio.2016.01.013
30. Zhang S, Liu B, Wang W, Lv L, Gao D, Chai M, et al. The “Matrisome” reveals the characterization of skin keloid microenvironment. FASEB J. (2021) 35. doi: 10.1096/fj.202001660RR
31. Bock O, Yu H, Zitron S, Bayat A, Ferguson M, Mrowietz U. Studies of transforming growth factors beta 1-3 and their receptors I and II in fibroblast of keloids and hypertrophic scars. Acta Dermato-Venereologica. (2005) 1:1–1. doi: 10.1080/00015550410025453
32. Xie JL, Qi SH, Pan S, Xu YB, Li TZ, Liu XS, et al. Expression of smad protein by normal skin fibroblasts and hypertrophic scar fibroblasts in response to transforming growth factor 1. Dermatologic Surg. (2008) 34:1216–25. doi: 10.1111/j.1524-4725.2008.34261.x
33. Zhang Z, Garron TM, Li XJ, Liu Y, Zhang X, Li YY, et al. Recombinant human decorin inhibits TGF-β1-induced contraction of collagen lattice by hypertrophic scar fibroblasts. Burns. (2009) 35:527–37. doi: 10.1016/j.burns.2008.08.021
34. Bettinger DA, Yager DR, Diegelmann RF, Cohen KI. The effect of TGF-β on keloid fibroblast proliferation and collagen synthesis. Plastic and reconstructive surgery(1996). Available online at: https://journals.lww.com/plasreconsurg/fulltext/1996/10000/the_effect_of_tgf:_on_keloid_fibroblast.12.aspx (Accessed August 1, 2024).
35. Younai S, Nichter LS, Wellisz T, Reinisch J, Nimni ME, Tuan TL. Modulation of collagen synthesis by transforming growth factor-β in keloid and hypertrophic scar fibroblasts. Ann Plast Surg. (1994) 33:148–54. https://journals.lww.com/annalsplasticsurgery/fulltext/1994/08000/modulation_of_collagen_synthesis_by_transforming.5.aspx (Accessed August 1, 2024).
36. Colwell AS, Phan TT, Kong W, Longaker MT, Lorenz PH. Hypertrophic scar fibroblasts have increased connective tissue growth factor expression after transforming growth factor-β Stimulation Plast Reconstructive Surg. (2005) 116:1387–90. doi: 10.1097/01.prs.0000182343.99694.28
37. Lee WJ, Ahn HM, Roh H, Na Y, Choi I, Lee JH, et al. Decorin-expressing adenovirus decreases collagen synthesis and upregulates MMP expression in keloid fibroblasts and keloid spheroids. Exp Dermatol. (2015) 24:591–7. doi: 10.1111/exd.2015.24.issue-8
38. Meenakshi J, Vidyameenakshi S, Ananthram D, Ramakrishnan KM, Jayaraman V, Babu M. Low decorin expression along with inherent activation of ERK1,2 in ear lobe keloids. Burns. (2009) 35:519–26. doi: 10.1016/j.burns.2008.07.012
39. Sayani K, Dodd CM, Nedelec B, Shen YJ, Ghahary A, Tredget EE, et al. Delayed appearance of decorin in healing burn scars. Histopathology. (2000) 36:262–72. doi: 10.1046/j.1365-2559.2000.00824.x
40. Honardoust D, Varkey M, Hori K, Ding J, Shankowsky HA, Tredget EE. Small leucine-rich proteoglycans, decorin and fibromodulin, are reduced in postburn hypertrophic scar. Wound Repair Regeneration. (2011) 19:368–78. doi: 10.1111/j.1524-475X.2011.00677.x
41. Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest. (2009) 89:1195–220. doi: 10.1038/labinvest.2009.91
42. Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol. (1995) 25:1071–9. doi: 10.1002/eji.1830250432
43. Prakash MD, Bird CH, Bird PI. Active and zymogen forms of granzyme B are constitutively released from cytotoxic lymphocytes in the absence of target cell engagement. Immunol Cell Biol. (2009) 87:249–54. doi: 10.1038/icb.2008.98
44. Aubert A, Jung K, Hiroyasu S, Pardo J, Granville DJ. Granzyme serine proteases in inflammation and rheumatic diseases. Nat Rev Rheumatol. (2024) 20:361–76. doi: 10.1038/s41584-024-01109-5
45. Kurschus FC, Kleinschmidt M, Fellows E, Dornmair K, Rudolph R, Lilie H, et al. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. (2004) 562:87–92. doi: 10.1016/S0014-5793(04)00187-5
46. Tremblay GM, Wolbink AM, Cormier Y, Hack CE. Granzyme activity in the inflamed lung is not controlled by endogenous serine proteinase inhibitors. J Immunol. (2000) 165:3966–9. doi: 10.4049/jimmunol.165.7.3966
47. Turner CT, Zeglinski MR, Richardson KC, Santacruz S, Hiroyasu S, Wang C, et al. Granzyme B contributes to barrier dysfunction in oxazolone-induced skin inflammation through E-cadherin and FLG cleavage. J Invest Dermatol. (2021) 141:36–47. doi: 10.1016/j.jid.2020.05.095
48. Kamata Y, Kimura U, Matsuda H, Tengara S, Kamo A, Umehara Y, et al. Relationships among plasma granzyme B level, pruritus and dermatitis in patients with atopic dermatitis. J Dermatol Science. (2016) 84:266–71. doi: 10.1016/j.jdermsci.2016.09.009
49. Russo V, Klein T, Lim DJ, Solis N, MaChado Y, Hiroyasu S, et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Sci Rep. (2018) 8:9690. doi: 10.1038/s41598-018-28070-0
50. Hiroyasu S, Zeglinski MR, Zhao H, Pawluk MA, Turner CT, Kasprick A, et al. Granzyme B inhibition reduces disease severity in autoimmune blistering diseases. Nat Commun. (2021) 12:302. doi: 10.1038/s41467-020-20604-3
51. Turner CT, Bolsoni J, Zeglinski MR, Zhao H, Ponomarev T, Richardson K, et al. Granzyme B mediates impaired healing of pressure injuries in aged skin. NPJ Aging Mech Dis. (2021) 7:6. doi: 10.1038/s41514-021-00059-6
52. Hiebert PR, Wu D, Granville DJ. Granzyme B degrades extracellular matrix and contributes to delayed wound closure in apolipoprotein E knockout mice. Cell Death Differ. (2013) 20:1404–14. doi: 10.1038/cdd.2013.96
53. Hsu I, Parkinson LG, Shen Y, Toro A, Brown T, Zhao H, et al. Serpina3n accelerates tissue repair in a diabetic mouse model of delayed wound healing. Cell Death Dis. (2014) 5:e1458–8. doi: 10.1038/cddis.2014.423
54. Shen Y, Zeglinski MR, Turner CT, Raithatha SA, Wu Z, Russo V, et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp Mol Med. (2018) 50:1–11. doi: 10.1038/s12276-018-0095-0
55. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. (2005) 280:23549–58. doi: 10.1074/jbc.M412001200
56. Matsubara JA, Tian Y, Cui JZ, Zeglinski MR, Hiroyasu S, Turner CT, et al. Retinal distribution and extracellular activity of granzyme B: A serine protease that degrades retinal pigment epithelial tight junctions and extracellular matrix proteins. Front Immunol. (2020) 11:574. doi: 10.3389/fimmu.2020.00574
57. Boivin WA, Shackleford M, Vanden Hoek A, Zhao H, Hackett TL, Knight DA, et al. Granzyme B cleaves decorin, biglycan and soluble betaglycan, releasing active transforming growth factor-β1. PLoS One. (2012) 7:e33163. doi: 10.1371/journal.pone.0033163
58. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. (2008) 123:398–410. doi: 10.1111/j.1365-2567.2007.02705.x
59. Raghunath M, Unsöld C, Bruckner-Tuderman L, Kubitscheck U, Peters R, Meuli M. The cutaneous microfibrillar apparatus contains latent transforming growth factor-β Binding protein-1 (LTBP-1) and is a repository for latent TGF-β1. J Invest Dermatol. (1998) 111:559–64. doi: 10.1046/j.1523-1747.1998.00339.x
60. Backes C, Kuentzer J, Lenhof HP, Comtesse N, Meese E. GraBCas: a bioinformatics tool for score-based prediction of Caspase- and Granzyme B-cleavage sites in protein sequences. Nucleic Acids Res. (2005) 33:W208–13. doi: 10.1093/nar/gki433
61. Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, et al. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J Cell Biol. (2014) 207:283–97. doi: 10.1083/jcb.201402006
62. Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. (2009) 19:128–39. doi: 10.1038/cr.2008.328
63. Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. (2007) 14:1768–79. doi: 10.1038/sj.cdd.4402183
64. Ge G, Greenspan DS. BMP1 controls TGFβ1 activation via cleavage of latent TGFβ-binding protein. J Cell Biol. (2006) 175:111–20. doi: 10.1083/jcb.200606058
65. Anastasi C, Rousselle P, Talantikite M, Tessier A, Cluzel C, Bachmann A, et al. BMP-1 disrupts cell adhesion and enhances TGF-β activation through cleavage of the matricellular protein thrombospondin-1. Sci Signal. (2020) 13:eaba3880. doi: 10.1126/scisignal.aba3880
66. Chamberlain CM, Ang LS, Boivin WA, Cooper DM, Williams SJ, Zhao H, et al. Perforin-independent extracellular granzyme B activity contributes to abdominal aortic aneurysm. Am J Pathology. (2010) 76:1038–49. doi: 10.2353/ajpath.2010.090700
67. Prigent L, Mercier P, Bovio S, Aubert A, Liot S, Lambert E, et al. Analysis of biomechanical properties of mouse skin dermis through atomic force microscopy: Application to demonstrate a sexual dimorphism. Exp Dermatol. (2023) 32:1016–27. doi: 10.1111/exd.14807
68. Obasanmi G, Zeglinski MR, Hardie E, Wilhelm AC, Turner CT, Hiroyasu S, et al. Granzyme B contributes to choroidal neovascularization and age-related macular degeneration through proteolysis of thrombospondin-1. Lab Invest. (2023) 103:100123. doi: 10.1016/j.labinv.2023.100123
69. Aubert A, Liu A, Kao M, Goeres J, Richardson KC, Nierves L, et al. Granzyme B cleaves tenascin-C to release its C-terminal domain in rheumatoid arthritis. JCI Insight. (2024) 9:e181935. doi: 10.1172/jci.insight.181935
70. Xue K, Zhang J, Li C, Li J, Wang C, Zhang Q, et al. The role and mechanism of transforming growth factor beta 3 in human myocardial infarction-induced myocardial fibrosis. J Cell Mol Medi. (2019) 23:4229–43. doi: 10.1111/jcmm.2019.23.issue-6
71. Wu Y, Peng Y, Gao D, Feng C, Yuan X, Li H, et al. Mesenchymal stem cells suppress fibroblast proliferation and reduce skin fibrosis through a TGF-β3-dependent activation. Int J Lower Extremity Wounds. (2015) 14:50–62. doi: 10.1177/1534734614568373
72. Shen Y, Cheng F, Sharma M, Merkulova Y, Raithatha SA, Parkinson LG, et al. Granzyme B deficiency protects against angiotensin II–induced cardiac fibrosis. Am J Pathology. (2016) 186:87–100. doi: 10.1016/j.ajpath.2015.09.010
Keywords: keloids, hypertrophic scars, granzymes, inflammation, extracellular matrix remodeling, TGF-β activation
Citation: Aubert A, Goeres J, Liu A, Kao M, Richardson KC, Jung K, Hinz B, Crawford RI and Granville DJ (2025) Potential implications of granzyme B in keloids and hypertrophic scars through extracellular matrix remodeling and latent TGF-β activation. Front. Immunol. 15:1484462. doi: 10.3389/fimmu.2024.1484462
Received: 21 August 2024; Accepted: 20 December 2024;
Published: 16 January 2025.
Edited by:
Fehim Esen, Istanbul Medeniyet University, TürkiyeReviewed by:
Minghua Wu, University of Texas Health Science Center at Houston, United StatesXiaoxiao Pang, Stomatological Hospital of Chongqing Medical University, China
Copyright © 2025 Aubert, Goeres, Liu, Kao, Richardson, Jung, Hinz, Crawford and Granville. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David J. Granville, ZGdyYW52aWxsZUBpY29yZC5vcmc=
†ORCID: Alexandre Aubert, orcid.org/0000-0002-2646-2284
Jenna Goeres, orcid.org/0009-0004-8670-0346
Amy Liu, orcid.org/0009-0001-0928-5298
Martin Kao, orcid.org/0009-0003-1141-011X
Katlyn C. Richardson, orcid.org/0000-0001-9856-8558
Karen Jung, orcid.org/0009-0008-0349-3281
Boris Hinz, orcid.org/0000-0002-0526-393X
Richard I. Crawford, orcid.org/0000-0002-8669-1898
David J. Granville, orcid.org/0000-0001-6985-2816