 Sung-Ting Chuang
Sung-Ting Chuang Oscar Alcazar
Oscar Alcazar Brandon Watts
Brandon Watts Midhat H. Abdulreda
Midhat H. Abdulreda Peter Buchwald
Peter Buchwald- 1Diabetes Research Institute, Miller School of Medicine, University of Miami, Miami, FL, United States
- 2Department of Surgery, Miller School of Medicine, University of Miami, Miami, FL, United States
- 3Department of Microbiology and Immunology, Miller School of Medicine, University of Miami, Miami, FL, United States
- 4Department of Ophthalmology, Miller School of Medicine, University of Miami, Miami, FL, United States
- 5Department of Molecular and Cellular Pharmacology, Miller School of Medicine, University of Miami, Miami, FL, United States
As part of our work to develop small-molecule inhibitors (SMIs) of the CD40-CD40L(CD154) costimulatory protein-protein interaction, here, we describe the ability of two of our most promising SMIs, DRI-C21041 and DRI-C21095, to prolong the survival and function of islet allografts in two murine models of islet transplantation (under the kidney capsule and in the anterior chamber of the eye) and to prevent autoimmune type 1 diabetes (T1D) onset in NOD mice. In both transplant models, a significant portion of islet allografts (50%-80%) remained intact and functional long after terminating treatment, suggesting the possibility of inducing operational immune tolerance via inhibition of the CD40-CD40L axis. SMI-treated mice maintained the structural integrity and function of their islet allografts with concomitant reduction in immune cell infiltration as evidenced by direct longitudinal imaging in situ. Furthermore, in female NODs, three-month SMI treatment reduced the incidence of diabetes from 80% to 60% (DRI-C21041) and 25% (DRI-C21095). These results (i) demonstrate the susceptibility of this TNF superfamily protein-protein interaction to small-molecule inhibition, (ii) confirm the in vivo therapeutic potential of these SMIs of a critical immune checkpoint, and (iii) reaffirm the therapeutic promise of CD40-CD40L blockade in islet transplantation and T1D prevention. Thus, CD40L-targeting SMIs could ultimately lead to alternative immunomodulatory therapeutics for transplant recipients and prevention of autoimmune diseases that are safer, less immunogenic, more controllable (shorter half-lives), and more patient-friendly (i.e., suitable for oral administration, which makes them easier to administer) than corresponding antibody-based interventions.
1 Introduction
Immune checkpoints (ICPs), critical regulators of immune responses, involve cell surface protein-protein interactions (PPIs) that can either inhibit or stimulate T cell mediated immune reactions; therefore, ICP modulation has considerable therapeutic potential for treatment of cancer on one hand and for treatment of autoimmune diseases on the other (1–3). Coinhibitory or costimulatory PPIs belong to two main families: the immunoglobulin superfamily (IgSF), e.g., CD28–CD80/86, CTLA4(CD152)–CD80/86, ICOS(CD278)–ICOS-L(CD275), and PD-L1(CD274)–PD-1(CD279) and the TNFR–TNF superfamily (TNFSF), e.g., CD40–CD40L(CD154), OX40(CD134)–OX40L(CD252), and 4-1BB(CD137)–4-1BB-L. The development of ICP inhibitors that target coinhibitory interactions, such as CD80–CTLA4 and PD-1–PD-L1, to overcome immunological anergy and invigorate the T effector cells restoring their ability to destroy cancer cells was named “Breakthrough of the Year” in 2013 by Science magazine (4). The subsequent development of immuno-oncology with the introduction of antibodies against PD-1 (nivolumab, pembrolizumab), PD-L1 (atezolizumab, durvalumab, and avelumab), and CTLA4 (ipilimumab) has led to a paradigm shift in the treatment of cancer (5).
The therapeutic applicability of biologics such as antibodies is, however, often hindered by their immunogenicity, which is elicited by their protein nature (6), and exacerbated by their long elimination half-lives (typically two to three weeks (7)), which makes it difficult to rapidly abolish unwanted side effects when they occur (8). Not surprisingly, antibodies tend to have more post-market safety issues than traditional small-molecule drugs (9). Moreover, the use of immunomodulatory biologics is further complicated by the high likelihood of unwanted adverse reactions that include cytokine release syndrome, serious and protracted infections, malignancy, and anaphylaxis among others (10). Biologics-based cancer immunotherapies have been found to cause immune-related adverse events (irAEs) in a large fraction of treated patients, sometimes as high as 50% (11). In general, small molecules may represent viable, safer alternatives that can also be orally bioavailable. This is particularly important for prospective preventive therapies, for example in type 1 diabetes (T1D), because such therapies have to be patient-friendly (12) to encourage the adherence and compliance needed for such long-term treatments to achieve efficacy (13, 14).
However, the modulation of PPIs with small molecules can be challenging because the corresponding protein interfaces lack well-defined binding sites such as those typically needed for the strong binding of such small-molecule ligands. Nevertheless, during the last two decades, tens of PPI-targeting small-molecule inhibitors (SMIs) reached preclinical development (15–22), and three are already approved by the FDA for clinical use: • venetoclax (ABT-199; Venclexta®/Venclyxto®), an inhibitor targeting PPIs in the B cell lymphoma 2 (BCL-2) family approved in 2015 for the treatment of chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), and acute myeloid leukemia (AML) (23); • lifitegrast (SAR 1118; Xiidra®), an LFA-1–ICAM-1 inhibitor approved in 2016 for the treatment of dry eye (24); and • fostemsavir (BMS-663068; Rukobia®), a prodrug of temsavir that blocks gp120–CD4 binding approved in 2020 as an antiretroviral for adults living with HIV/AIDS (25). Such SMIs of PPIs (SMIPPIs) can lead to novel therapies that, compared to antibodies, are less immunogenic, more controllable (due to their shorter half-lives), easier to administer and thus more patient-friendly (by virtue of being suitable for oral administration), and more cell- and tissue-permeable (due to their better biodistribution properties). Notably, even in the field of TNF-targeting therapeutics, where the treatment of rheumatoid arthritis (RA) has been revolutionized by the use of anti-TNF biologics, such as etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab, which have been considered among the most transformative drugs of the 1985–2010 period (26), there is increasing interest in SMIs. One such SMI developed by Sanofi, SAR441566, is currently in Phase 2 clinical trials for RA treatment (e.g., NCT06073093) (27, 28).
Along these lines, we focused on identifying SMIs of the CD40(TNFRSF5) – CD40L(CD154, TNFSF5) costimulatory PPI (29) because blockade of this ICP has been consistently shown to be a highly effective immunomodulatory therapy in general (29–34) and for pancreatic islet transplantation in particular. For the latter, CD40–CD40L inhibition allowed the engraftment of allogeneic (and sometimes even xenogeneic) islets resulting in long-term insulin independence with the possibility for induced operational tolerance in various mouse (35–45) and nonhuman primate (NHP) allo-transplant models (46–52). Furthermore, CD40L inhibition has shown great therapeutic promise in the prevention of autoimmune diseases (29, 31, 34, 53) including T1D (54–61). It has been suggested that targeting the CD40–CD40L PPI may provide “the next major novel class of costimulatory inhibitors to treat autoimmune disease” (62). Notably, with the realization that the thromboembolic complications encountered with the first-generation of CD40L antibodies like ruplizumab (hu5c8) were driven by their Fc region and can be avoided (20, 63–70), there is a resurgence of interest in CD40–CD40L blockade as exemplified by the title of a recent review: “Phoenix from the flames: Rediscovering the role of the CD40-CD40L pathway…” (71). Indeed, second-generation Fc-silent anti-CD40L antibodies retain immunomodulatory activity but do not activate platelets (71–74). Examples include • letolizumab (BMS-986004; Bristol-Myers Squibb) for the treatment of immune thrombocytopenic purpura (ITP) and graft versus host disease (GVHD) (73); • dapirolizumab pegol (CDP7657; UCB Pharma) for systemic lupus erythematosus (SLE) – currently in Phase 3 clinical trials (75, 76); • frexalimab (SAR441344, INX-021; Sanofi) for multiple sclerosis (MS) – currently in Phase 2 trials (77); • tegoprubart (AT-1501; Eledon) for amyotrophic lateral sclerosis (ALS) – currently in Phase 2a trials (78); • TNX-1500 (Tonix), which has shown efficacy in NHP transplant models (79); and • dazodalibep (HZN-4920, VIB4920; Viela Bio, MedImmune, and Horizon Therapeutics) to prevent autoimmune RA – currently in Phase 2 studies (80, 81), as well as Sjögren’s disease – currently in Phase 2 with very promising results (82). Tegoprubart has also shown efficacy in kidney and islet transplants in NHPs (83) and early clinical trials. Iscalimab (CFZ533), a human Fc-silenced, non-depleting, IgG1 anti-CD40 monoclonal antibody was shown to be effective in Sjögren’s disease in a large Phase 2b study conducted at 71 sites in 23 countries (84). Thus, the CD40–CD40L interaction remains an attractive therapeutic target in islet transplant applications and for the prevention of T1D.
We have designed and developed SMIs for the CD40–CD40L PPI (85, 86), explored structure-activity relationships for more than 50 newly synthesized compounds, and identified lead SMIs that showed CD40L-inhibitory activity as well as selectivity in vitro and in vivo (87, 88). We now report further long-term in vivo studies showing that two of our most promising CD40–CD40L SMIPPIs, DRI-C21041 and DRI-C21095, were able to • significantly prolong the survival of islet allografts in two mouse models of islet transplantation in different sites and • prevent the onset of autoimmune diabetes in the non-obese diabetic (NOD) mouse model of T1D.
2 Materials and methods
2.1 Drugs
DRI-C21041 and DRI-C21095 were synthesized either in-house or by WuXi AppTec (Kowloon, Hong Kong) as described before in detail (87, 88). Structures were confirmed by high-resolution mass spectrometry (HRMS) and NMR; purities were confirmed to be ≥95% by HPLC. The anti-CD40L mouse antibody (clone MR-1) was purchased from Bio X Cell (Lebanon, NH, USA).
2.2 Animal care and treatment
Mice used for these studies were obtained from Jackson Laboratories (Bar Harbor, ME, USA). The following strains were used: NOD (NOD/ShiLtJ; strain # 001976), C57BL/6 (C57BL/6J; strain # 000664), and DBA/2 (DBA/2J; strain # 000671). All animal studies were reviewed and approved by the University of Miami Institutional Animal Care and Use Committee (IACUC). Procedures were conducted according to the guidelines of the Committee on Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources (National Research Council, Washington, DC, USA). Animals were housed in micro-isolated cages with free access to autoclaved food and water in Virus Antibody Free (VAF) rooms managed by the Department of Veterinary Resources of the University of Miami.
2.3 Prevention of diabetes onset in NOD mice
For the diabetes prevention studies, young female NOD mice (n = 5/group) were subcutaneously (s.c.) treated twice daily (b.i.d.) between the ages of 5 and 18 weeks with DRI-C21095 and DRI-C21041 at 20 and 12.5 mg/kg, respectively, administered in 20% hydroxypropyl-β-cyclodextrin (HPβCD) solution as vehicle. A corresponding control group was treated with vehicle only. Mice were monitored for diabetes onset by glycosuria once a week starting from week 10 until 40 weeks of age. Once hyperglycosuria was detected, monitoring was switched to daily glycemia measurements using portable glucometers (Contour Next; Bayer, Mishawaka, USA). Animals with three consecutive readings of blood glucose (BG) >300 mg/dL were considered diabetic and humanely euthanized for tissue analysis. Animals remaining normoglycemic by week 40 of age were euthanized shortly thereafter and blood as well as pancreas, spleen, lymph nodes, and other organs were collected for various analyses.
2.4 Immunophenotyping
Mouse splenocytes were collected by mashing spleens through a 70 μm cell strainer, subjected to ACK lysis to remove erythrocytes, and resuspended in Hank’s buffer. Cells were diluted in trypan blue and counted in a hemocytometer. One million viable cells were incubated with Live and Dead solution (Zombie UV, BioLegend, San Diego, CA, USA; cat. no. 423107) in PBS for 20 min at 4°C. Cells were washed and incubated with universal Fc blocker containing anti-mouse CD16/32 (clone 2.4G2) for 20 min at 4°C. Then, cells were incubated for 30 min at room temperature (RT) protected from light in a master mix for surface staining by the following fluorescence-labeled antibodies against NK-1.1 (BD Biosciences, Franklin Lakes, NJ, USA; cat. no. 741715); CD62L (BioLegend; cat. no. 104450); CD8a (BD Biosciences; cat. no. 553036); CD44 (BD Biosciences; cat. no. 562464); CD25 (BioLegend; cat. no. 102016); CD4 (BioLegend; cat. no. 100492); and CD3 (Invitrogen, Carlsbad, CA, USA; cat. no. 56-0032-82) at dilutions according to the recommendations of the suppliers. Cells were washed twice in PBS buffer supplemented with 1% BSA (VWR/Avantor, Radnor, PA, USA; cat. no. 0332), incubated with FIX/PERM (Thermo-Fisher Scientific, Waltham, MA, USA; kit cat. no. 00-5523-00) for 30 min at 4°C, and then washed twice with PERM buffer (Thermo-Fisher Scientific; kit cat. no. 00-5523-00);. Afterwards, cells were subjected to intracellular staining with anti-FoxP3 (Invitrogen; cat. on. 15-5773-82), in PERM buffer for 30 min at 4°C, then washed twice with wash buffer (1% BSA in PBS). After centrifugation, wash buffer was added to resuspend the cells before analysis. Single-color compensation samples for flow cytometry were prepared using the same antibodies with UltraComp eBeads (Thermo Fisher Scientific; cat. no. 01-3333-42). Data were acquired using a Cytek Aurora flow cytometer (Cytek, Fremont, CA, USA) and analyzed with Kaluza software (Beckman Coulter, Brea, CA, USA).
2.5 Islet isolation, transplantation in the kidney of C57BL/6 mice, and treatment
Pancreatic islets used for transplantation were obtained by enzymatic digestion of DBA/2 donor mouse pancreata, followed by purification on density gradients using protocols standardized at the Preclinical Cell Processing and Translational Models Core of our institute (Diabetes Research Institute, DRI) (37, 40). After overnight culture, isolated islets were implanted in fully anesthetized male and female recipient C57BL/6 mice under the kidney subcapsular space, as previously described (37, 40). Prior to transplantation, diabetes was induced with a single dose of streptozotocin (STZ) treatment (200 mg/kg, i.v.) and mice confirmed to be hyperglycemic (i.e., three consecutive non-fasting BG readings > 300 mg/dL) were transplanted with 500 islet equivalents (IEQ) under the kidney capsule. DRI-C21095 and DRI-C21041 treatments (10, 12.5, 15, and 20 mg/kg) were administered s.c. in 20% HPβCD vehicle twice daily (b.i.d.) starting the day before transplant (day -1) until day 30 post-transplant. Mice treated with anti-CD40L antibody (MR-1) received 250 μg administered once a day (s.c.) on days -1, 0, 3, and 7. Transplanted mice remaining normoglycemic at 60 days post-transplant underwent unilateral nephrectomy of the graft-bearing kidney to confirm function of the islet graft and were euthanized shortly thereafter.
2.6 In vivo imaging of islets transplanted in the ACE of C57BL/6 mice and treatment
In these studies, normoglycemic C57BL/6 mice were transplanted with allogeneic DBA/2 donor islets in the anterior chamber of the eye (ACE). Recipient mice were not pretreated with STZ since the ACE-islet grafts are monitored directly through the transparent cornea. The recipients also expressed green fluorescent protein (GFP) in either macrophages or CD8 T cells; these mice were generated by crossing FVB-Tg(Csf1r-icre)1Jwp/J (strain # 021024; Jackson Laboratories) or C57BL/6-Tg(Cd8a-cre)1Itan/J (strain # 008766; Jackson Laboratories), respectively, with B6.Ai38(RCL-GCaMP3)lox/lox (strain # 014538; Jackson Laboratories) and breeding for >10 generations to reach homozygosity. Homozygous mice received 50 IEQs in one eye (DBA/2 islets), as previously described in detail (89). Five to seven mice were used per treatment group. MR-1 treatment was as described above. Mice treated with DRI-C21095 (20 mg/kg) or DRI-C21041 (12.5 mg/kg) received s.c. injections twice daily (b.i.d.) in 20% HPβCD vehicle up to day 60 post-transplantation. The structural integrity of ACE-transplanted islets was longitudinally monitored by direct visualization and their survival was assessed by quantitative analysis of individual islets volume, as previously described in detail (52, 89–92). In brief, islets engrafted on top of the iris were mapped in digital images of the eye acquired during the first week after transplantation and revisited during the repeated imaging sessions in these longitudinal studies. The islets were imaged noninvasively through the cornea by confocal microscopy using the 633 nm laser backscatter (reflection) (90), and the islet-infiltrating immune cells (macrophages and T cells) were visualized based on the GFP expression. Three-dimensional (3D) confocal micrographs were acquired using a 20× water immersion objective in z-stacks spanning the full height of the ACE-transplanted islets, and analysis of the individual islet volume was performed in the 3D images. Survival of individual islets was defined when the individual islet maintained its volume above 70% relative to its corresponding baseline (acquired during the first week after transplantation). Immune cell infiltration into the islets was quantified based on the GFP volume inside individual islets and expressed as the percentage of the corresponding islet volume. Volumetric analysis in the images was performed using Volocity software version 6.3.1 (Quorum Technologies Inc.; Puslinch, ON, Canada).
2.7 Immunofluorescence
Harvested tissues were immediately placed in a 15 mL Falcon tube containing 4% paraformaldehyde (PFA), incubated at room temperature (RT) for 4 h, and washed three times with PBS for a total of 15 min. They were transferred into a tube containing 30% (w/v) sucrose solution and incubated at 4°C overnight. Eyes were then mounted in Tissue-Tek O.C.T. (Sakura Finetek, Torrance, CA, USA) and snap frozen by placing the tissue mold on dry ice. Tissue sections (14 μm thick) were collected on glass slides and kept at –80°C until analysis. For immunofluorescence staining, samples were subjected to antigen retrieval by standard protocol, washed three times with PBS, and permeabilized for 1 h at RT with 0.3% Triton X-100 in 10% Fc Block (SuperBlock Blocking Buffer, Pierce, Rockford, IL, USA) or normal goat serum. After blocking for at least 1 h, samples were incubated overnight at 4°C with anti-insulin (guinea pig, 1:500; Dako, Carpinteria, CA, USA) and anti-glucagon (rabbit, 1:500; Abcam, Cambridge, MA, USA) primary antibodies. Sections were washed 5 times with PBS plus 0.05% Tween 20 and then incubated with the secondary antibodies: goat anti-rabbit AlexaFluor 488 and goat anti-guinea pig AlexaFluor 647 (both at 1:200; Life Technologies, Carlsbad, CA, USA). Nuclear counterstain was performed by DAPI (1:2000; Life Technologies). Slides were washed another 5 times with PBS plus 0.05% Tween 20 and 2 times with PBS alone before applying mounting media (Clear-MOUNT with TRIS, Electron Microscopy Science, Hatfield, PA, USA) and cover slips. Immunofluorescence imaging was performed using a Leica Stellaris 5 confocal microscope (Leica, Deerfield, IL, USA).
2.8 Pharmacokinetic evaluation in mice
Pharmacokinetics (PK) in mice was evaluated by quantifying plasma concentration in blood samples (50 μL) drawn from the orbital sinus at predefined time points (0, 0.5, 1, 2, 4, 6, and 24 h) following administration of a single dose (30 mg/kg, s.c.) to C57BL/6 mice (n = 4; 10-week-old males and females). Plasma concentrations were obtained using LC/MS performed at the Mass Spectrometry Laboratory, Department of Chemistry, University of Florida (Gainesville, FL, USA) on a ThermoFinnigan, LTQ X mass spectrometer with a ThermoScientific Hypersil GOLD aQ (150 x 2.1 mm, 3 μm) column (Thermo Fisher Scientific, Waltham, MA, USA) using mobile phases containing 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Concentrations were calculated from the corresponding peak areas following a calibration curve in the 0.8 to 100 μg/mL range. Experimental concentration data obtained this way were fitted with a standard one-compartment, first-order absorption, first-order elimination PK model (87, 93) implemented as a custom equation in GraphPad Prism (GraphPad, La Jolla, CA, USA; version 10.1.2) to estimate the elimination rate constant k10 and the corresponding elimination half-life, t1/2 = ln(2)/k10.
2.9 Statistics and data fitting
The percent of functioning islet allografts in the transplantation models and the diabetes-free survival of NOD mice in the T1D prevention model were shown as Kaplan-Meier survival plots with asterisks denoting statistically significant differences versus the vehicle-treated group as determined by the log rank (Mantel-Cox) test using GraphPad Prism (GraphPad; version 10.1.2) (*p < 0.05, **p < 0.01).
3 Results
3.1 Prolongation of survival and function of islet allografts transplanted in the kidney subcapsular space
We evaluated the immunosuppressive efficacy of our SMIs targeting the CD40–CD40L PPI (DRI-C21041 and DRI-C21095; Figure 1) in an allogeneic islet transplantation model with full MHC-mismatch between donors and recipients. DBA/2 donor islets (MHC Class-I H-2Kd) were transplanted under the kidney capsule of STZ-induced diabetic C57BL/6 (MHC Class-I H-2Kb) mouse recipients that were then treated with various drug regimens. The ability of these recipients to maintain normoglycemia following the transplant was assessed via monitoring of blood glucose levels. DRI-C21095 and DRI-C21041 were administered at doses of 10, 12.5, 15, and 20 mg/kg (b.i.d., s.c.) from days -1 to 30 after transplant. Matched control mice treated with vehicle only (20% HPβCD) or the mouse anti-CD40L monoclonal antibody (MR-1; on days -1, 0, 3, 7 after transplant) were included as negative and positive controls, respectively (Figure 2). These studies revealed a noticeable dose-dependency: the two lowest doses of DRI-C21095 produced no effect, whereas the highest doses of both compounds produced significant prolongation of graft survival and function in 40%–50% of recipients even well after termination of treatment in a manner similar to the positive control MR-1 antibody. Unilateral nephrectomy of the graft-bearing kidney in DRI-C treated mice that remained non-diabetic (normoglycemic) as of day 60 post-transplant resulted in these mice becoming hyperglycemic (BG > 300 mg/dL), thereby confirming the sustained survival and function of the islet allografts in association with the DRI-C treatments.
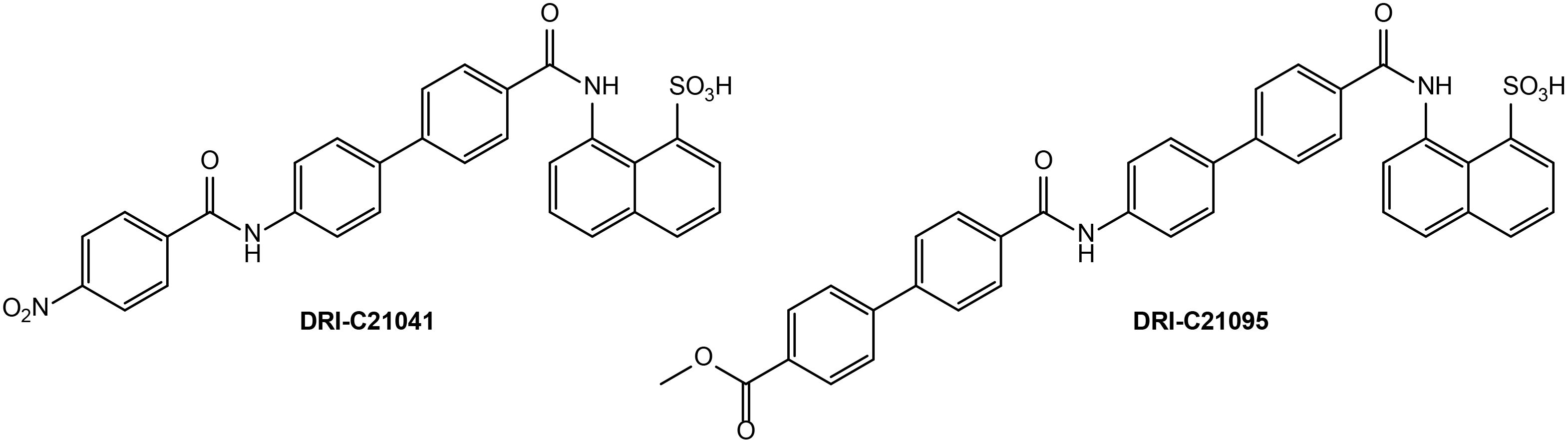
Figure 1. Chemical structures of DRI-C21041 and DRI-C21095, the SMIPPI compounds evaluated in the present study.
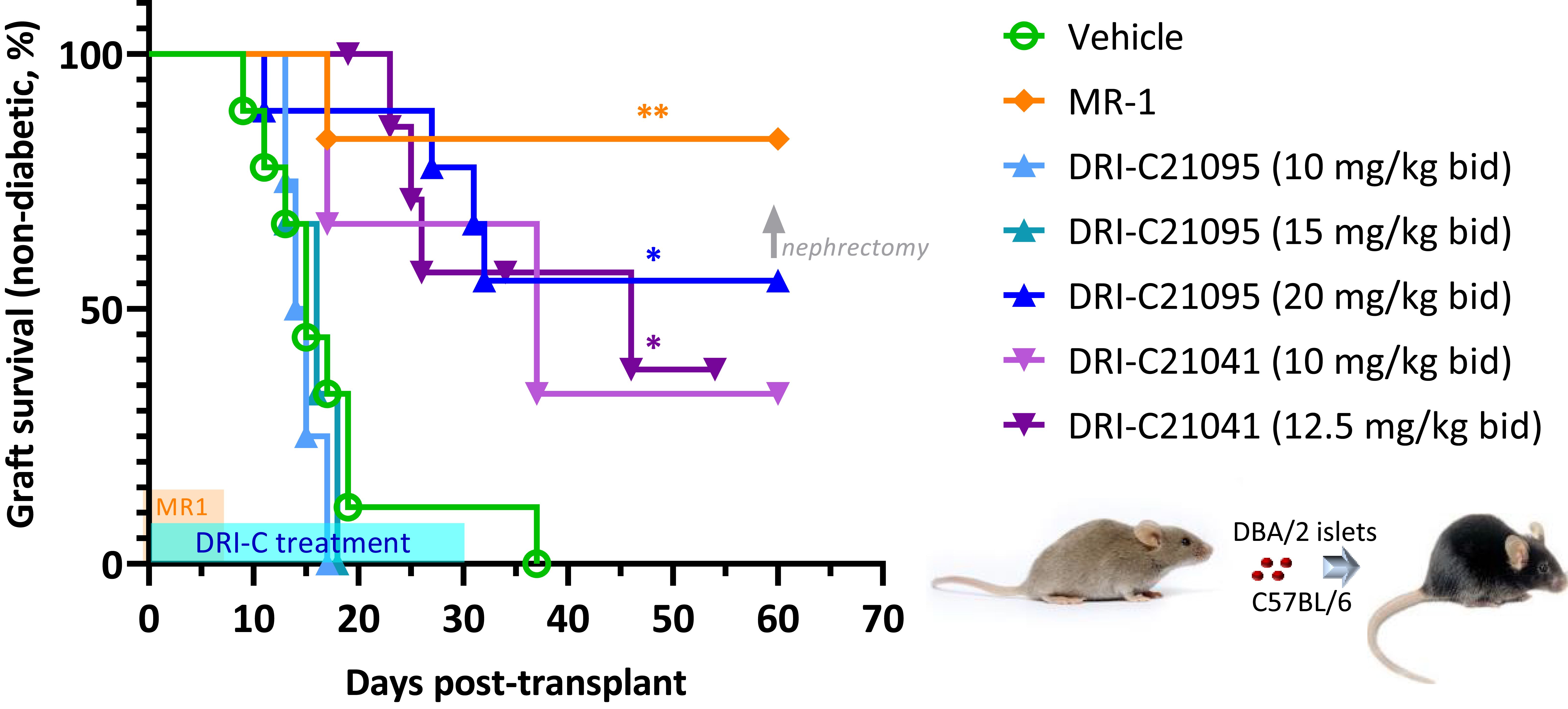
Figure 2. DRI-C21041 and DRI-C21095 prolong survival of islet allografts in the kidney subcapsular space of full MHC-mismatched recipients. Islets isolated from DBA/2 donors were transplanted under the kidney capsule of STZ-induced diabetic C57BL/6 mice (500 IEQ). DRI-C21095 and DRI-C21041 were administered s.c. at doses of 10–20 mg/kg (b.i.d.) in 20% HPβCD vehicle. DRI-C treatments in separate groups started on day -1 and continued until day 30, as shown in light blue on the time axis. Control mice treated with the vehicle received treatment during the same period. A positive control group was treated with anti-CD40L monoclonal antibody (mAb; clone MR-1) at a daily dose of 250 μg/mouse (~10 mg/kg) on days -1, 0, 3, and 7, as indicated by the orange-colored box along the time axis. Nephrectomy on day 60 in DRI-C treated mice maintaining normoglycemia (non-diabetic) confirmed the graft function upon return to hyperglycemia after removal of the islet graft-bearing kidney (BG > 300 mg/dL). Asterisks denote statistically significant differences versus the vehicle-treated group as determined by log rank (Mantel-Cox) test (*p < 0.05, **p < 0.01; n = 3–9 mice).
3.2 Prolongation of survival of islet allografts transplanted in the ACE
We also evaluated the immunosuppressive efficacy of our SMIPPIs in a different model of islet transplantation using the anterior chamber of the eye (ACE) as transplant site (52, 89–92). DRI-C21095 (20 mg/kg) and DRI-C21041 (12.5 mg/kg) were administered (b.i.d., s.c.) starting on the day of transplant (day 0) and maintained until day 60 post-transplant (Figure 3). As before, DBA/2 mice were used as islet donors and C57BL/6 mice as recipients to maintain full MHC-mismatch. In these studies, the recipients were normoglycemic and expressed GFP in either macrophages or T cells – this allowed direct in vivo visualization of these cells and the tracking of their infiltration into the ACE-transplanted islets. Survival of the islet allografts was monitored via longitudinal noninvasive imaging and quantitative volume analysis, taking advantage of the transparency of the transplant site (Figures 4A–C). In this model, DRI-C21041 was particularly effective: 80% of recipients maintained the structural integrity and function of their islet allografts up to the end of follow-up (115 days post-transplant), significantly longer than did the vehicle-treated and untreated control mice that rejected their grafts with a median of 28 days (Figure 3). Rejection in the controls was similar to that in other controls in our previous ACE studies (median survival of 21 days) (52). While the overall graft survival in the MR-1 treated mice in this study was for some reason less than what we previously observed in a similar model (52), MR-1 treatment still resulted in significant prolongation in the survival of the ACE-transplanted islet allografts compared to the control groups (Figure 3).
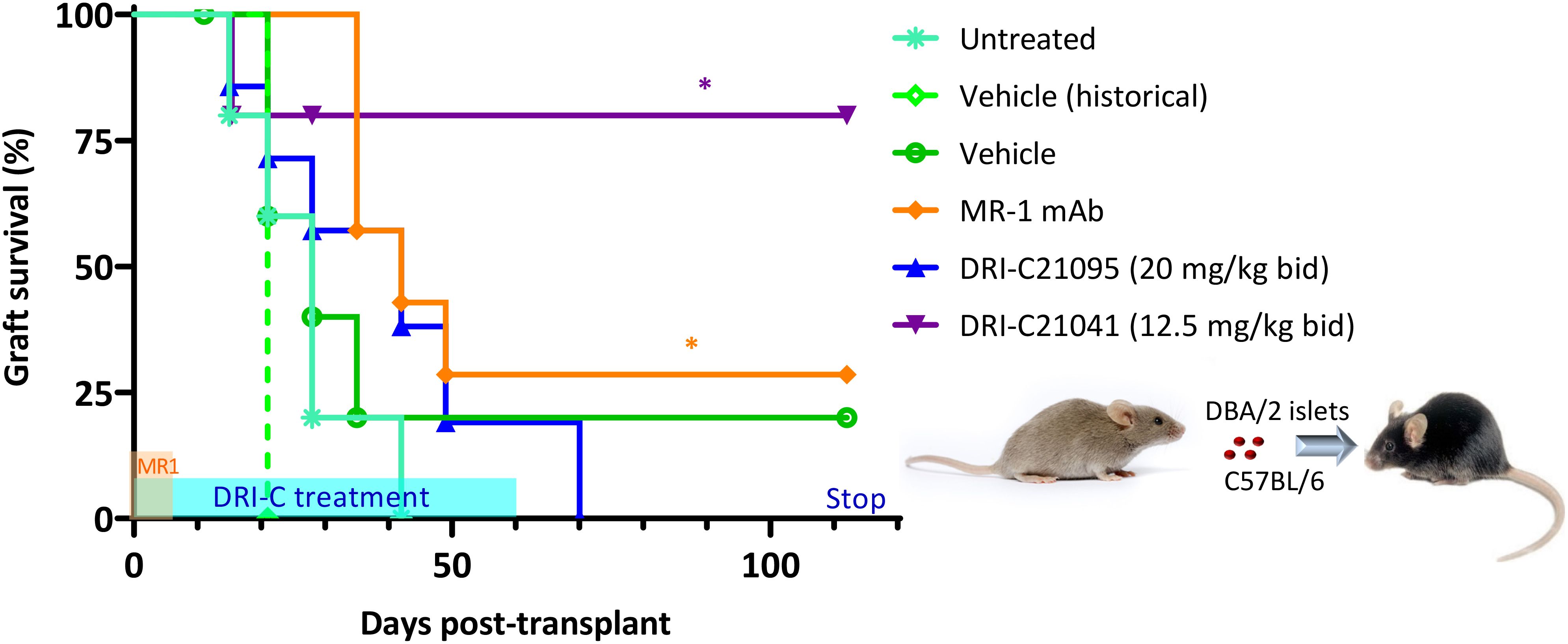
Figure 3. DRI-C21041 and DRI-C21095 prolong survival of islet allografts in the ACE model. Islets isolated from DBA/2 donors were transplanted into the anterior chamber of the eye (ACE) of C57BL/6 mice treated with DRI-C21095 (20 mg/kg, b.i.d.), DRI-C21041 (12.5 mg/kg, b.i.d.) – both s.c. in 20% HPβCD vehicle up to day 60, vehicle only, and MR-1 anti-CD40L mAb (250 μg/mouse, ~10 mg/kg, on days -1, 0, 3, 7 of transplant). Survival of the ACE-transplanted islet allografts was assessed by longitudinal volumetric analysis, as detailed in Methods. Most mice treated with DRI-C21041 did not reject their allografts long-term, even after treatment was stopped, until the end of follow-up (day 115); see Figures 4A–C for longitudinal islet images and volume analysis. Asterisks denote statistically significant differences versus the untreated group, log rank (Mantel-Cox) test (*p < 0.05; n = 5–7 mice).
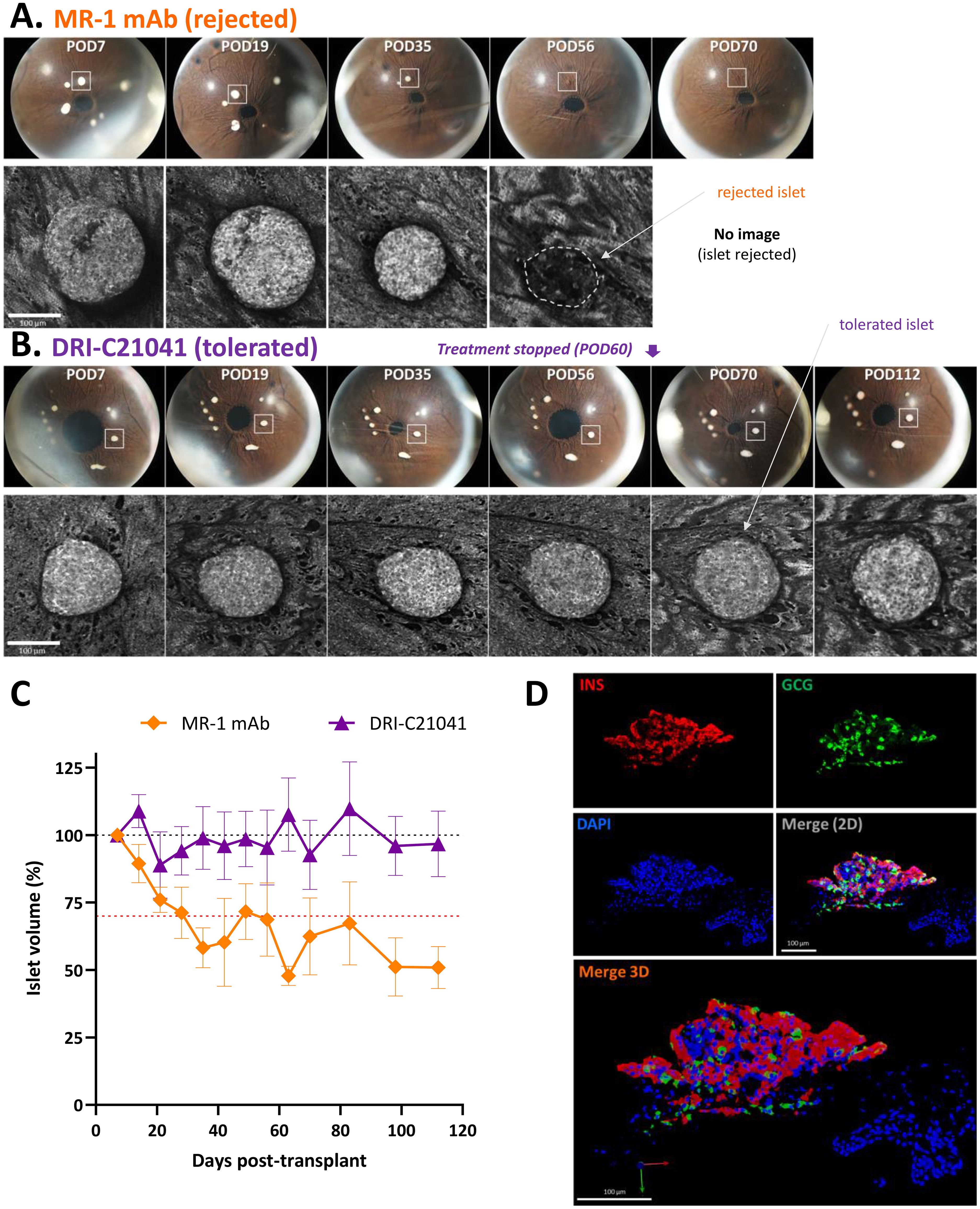
Figure 4. Imaging of ACE-transplanted islet grafts and quantification of their structural integrity over time. (A, B) Direct noninvasive and longitudinal imaging of ACE islets. Representative longitudinal images of ACE-transplanted DBA/2 islets in C57BL/6 mouse recipients treated either with MR-1 mAb (A) or DRI-C21041 (12.5 mg/kg b.i.d.) (B) from the study shown in Figure 3. Images showing the whole eye (top rows) with the islet highlighted in the longitudinal confocal micrographs (lower rows) marked with a white square. The shown MR-1–treated mouse rejected its islets by day 56 post-transplant (POD = post-operative day) and the DRI-C21041–treated mouse tolerated its islets until the end of the follow-up. The change in the structural integrity and volume of the rejected islet in the MR-1 treated mouse was evident, whereas islets in the DRI-C21041 treated mouse maintained their integrity and volume until the end of follow-up (last day of imaging, POD112). See C for overall longitudinal changes in the islet volume in MR-1– and DRI-C21041–treated mice. (C) Structural integrity of islet allografts is maintained in DRI-C21041 treated mice. Average islet volume per treatment group for DRI-C21041 and MR-1 mAb shown as determined by in vivo volumetric analysis in 3D confocal z-stacks of the individual islets and normalized to baseline (means ± SEM for n = 19–22 islets from 3–4 mice per group). (D) Fluorescence confocal micrographs confirming positive insulin and glucagon immunostaining in tolerated ACE-transplanted islet allografts in DRI-C21041 treated recipients. Representative immunofluorescence images of an ACE-transplanted DBA/2 islet in a section of the eye of a C57BL/6 recipient mouse that was treated with DRI-C21041 (12.5 mg/kg, b.i.d., s.c. up to day 60) showing positive immunostaining for insulin (red) and glucagon (green), thus, further confirming function of the islet allograft until the end of the follow-up on day 115 after the transplant. DAPI nuclear counterstain was used (blue), and slides were imaged at 40× magnification.
A distinctive advantage of the ACE-transplantation is that graft integrity can be monitored by direct noninvasive visualization, and longitudinal images of the same individual islets can be used to document the survival of the graft. This is illustrated in Figures 4A, B with two representative sets of longitudinal images showing the whole eye with pancreatic islets engrafted on top of the iris and the corresponding confocal micrographs of representative islets from an MR-1 treated mouse, which rejected its islets by day 56, and a DRI-C21041 treated mouse that did not reject its islet grafts up to the end of the follow-up on day 115 post-transplant. For the rejected islet shown, a reduction of its volume was already evident by day 35 (POD35), and the islet was no longer visible by day 56 post-transplant due to its complete immune destruction (Figure 4A). In contrast, the structural integrity of the islet in the tolerant DRI-C21041 treated mouse was maintained, and its volume remained unchanged until the end of the experiment (Figure 4B). In vivo volumetric analysis in confocal z-stacks (i.e., in 3D) of the individual islets in the DRI-C21041 and MR-1 treated mice highlighted the difference in the average change in islet volume between the two groups over time (Figure 4C). Further, ex vivo analysis in such islets (after necropsy) by immunostaining for glucagon and insulin also confirmed the sustained graft function in the DRI-C21041 treated recipients until the end of the studies on day 115 post-transplant (Figure 4D).
3.3 Suppression of immune cell infiltration
Because islets transplanted in the ACE can be followed via direct noninvasive imaging, we monitored in situ the graft infiltration by GFP-expressing macrophages and T cells (see Methods for details) and quantified the in vivo infiltration kinetics in mice that either rejected or tolerated their islet allografts. Regardless of treatment, macrophages initially infiltrated islets to similar degrees in recipients that ended up either rejecting or tolerating their allografts, but their relative abundance noticeably increased in rejected islets whose volume was progressively reduced (Figure 5). Additional studies in mice with GFP-expressing T cells further showed that blocking the CD40–CD40L interaction with either our SMIs or MR-1 decreased the T cell infiltration into the islet allografts. Interestingly, while the abundance of macrophages within tolerated islets remained relatively unchanged in the DRI-C21041 and MR-1 treated recipients until the end of follow-up on post-transplant day 112 (Figure 5), tolerant mice treated with our DRI-C compounds had markedly reduced initial T cell infiltration in the first 21 days after the transplant compared to the MR-1 treated counterparts, and this persisted until the end of the follow-up on day 112 post-transplant in DRI-C21041 treated mice (Figure 6).
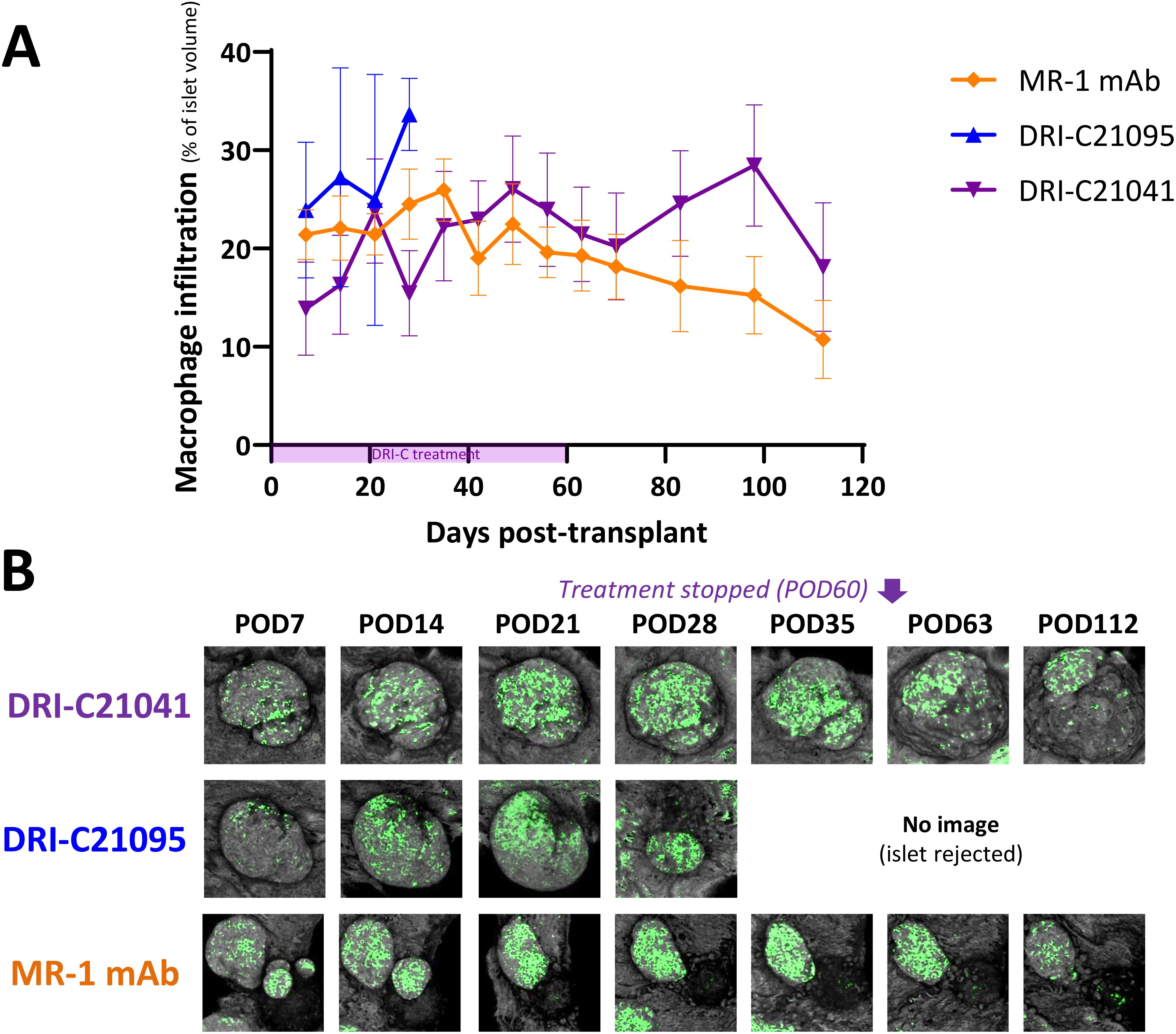
Figure 5. Time-course of macrophage infiltration into rejected versus tolerated islet allografts. (A) Longitudinal analysis of macrophage infiltration into rejected or tolerated islet allografts (n = 4–16) in selected representative recipient mice (n = 1–2) from the various treatment groups and (B) representative longitudinal images of rejected or tolerated single islets from selected recipients. Macrophages in these mice were visualized based on GFP expression and islets by backscatter (see Methods for details). The same islets were repeatedly imaged at the indicated time-points until rejection onset or stop of follow-up on day 112 post-transplant (POD = post-operative day). Images shown as max projection of z-stacks of confocal micrographs spanning the entire islets and where the infiltration analysis was performed in 3D (see Methods). Infiltration data shown as the means ± SEM of the individual islets in the imaged mice from the various treatment groups. Not all mice in each group were imaged, and data shown for DRI-C21095- and vehicle-treated as well as naïve controls correspond to mice that rejected their islet allografts.
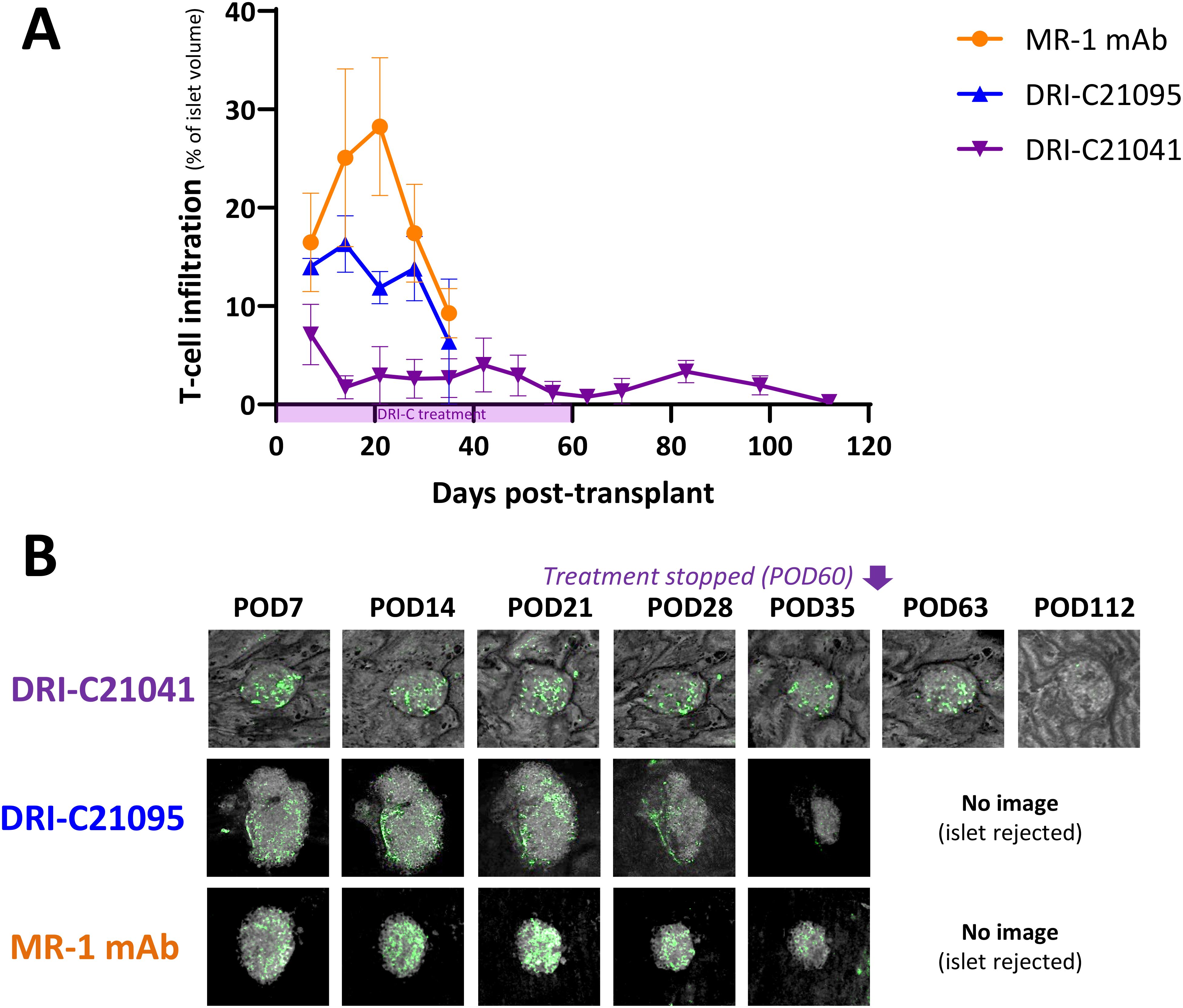
Figure 6. T cell infiltration kinetics into rejected versus tolerated islet allografts. (A) Time-course of T cell infiltration quantified in islets (n = 3–12) of selected representative recipient mice (n = 1–2) from the various treatment groups and (B) representative longitudinal images of single islets from selected recipients that either rejected or tolerated their ACE-transplanted islets. T cells in these mice were visualized based on GFP expression in the recipients and islets by backscatter (see Methods for details). Images (z-stack confocal micrographs shown as max projection) show the same islets imaged noninvasively and longitudinally at the indicated time-points until rejection onset or stop of follow-up. Infiltration data shown as the means ± SEM for each treatment group until imaging was stopped either due to islet rejection in the followed/imaged mice or at the end of follow-up of tolerant mice (POD112).
3.4 Prevention of diabetes onset
In addition to evaluating the immune suppressive effects of our SMIPPIs in allogeneic islet transplant models, we also assessed their ability to prevent the onset of autoimmune T1D in NOD mice in an exploratory study using female animals as customary due to their higher incidence rate of spontaneous diabetes. In light of previous publications studying CD40–CD40L inhibition in the same model (54, 60, 94), we administered treatments here from 5 to 18 weeks of age and monitored the diabetes incidence by regular glycemia measurements during an extended follow-up period until the mice reached 40 weeks of age. Results indicated that while hyperglycemia (i.e., diabetes onset) occurred in the typical 80% range in the control-treated animals, the incidence was reduced in the groups treated with our SMIs: it was reduced to 60% and 25% in the DRI-C21041- and DRI-C21095-treated mice, respectively (Figure 7). The median age of diabetes-free survival was 26 weeks in the vehicle-treated controls versus 31 weeks in those treated with DRI-C21041 and >40 weeks in the DRI-C21095 treated mice since they did not cross the 50% threshold (“undetermined”). We also performed exploratory immunophenotyping to identify indications of possible lasting treatment effects on immune cell subpopulations in the mice that did not become diabetic. Results can be considered as indicative only since they are from a small number of mice (one or two per group) and well after the termination of treatment (at week 40 vs 18 of age), nevertheless, they suggested possible persistent reduction in natural killer cells (NK; NK1.1+CD3–) and central memory T cells (CD4 and CD8 Tcm; CD44+CD62L+) and a concomitant increase in regulatory T cells (Treg; CD25+FoxP3+) (Supplementary Figure 1).
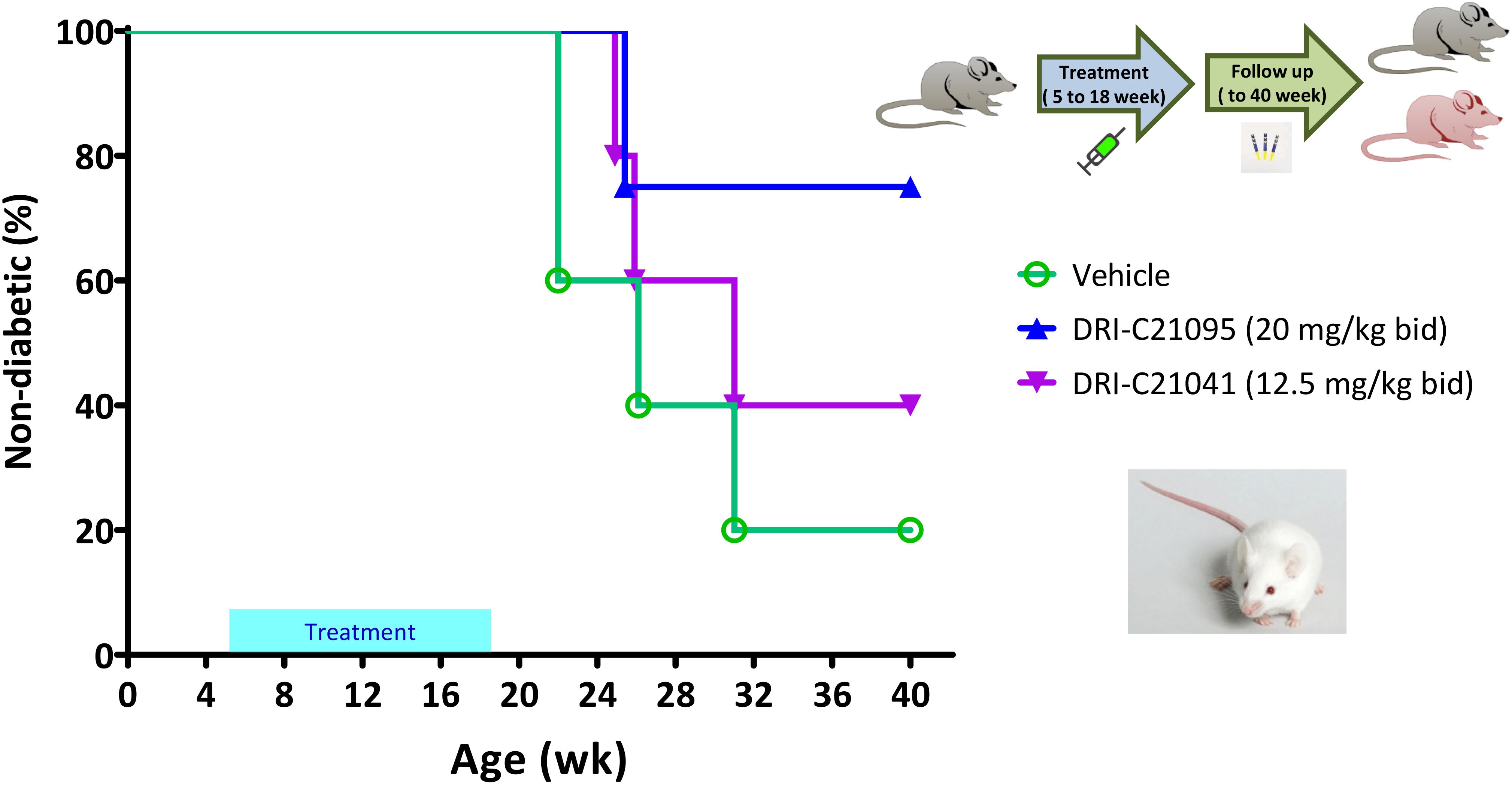
Figure 7. Prevention of diabetes in NOD mice by DRI-C21041 and DRI-C21095. NOD mice were treated from 5 to 18 weeks of age with DRI-C21095 and DRI-C21041 at doses of 20 and 12.5 mg/kg b.i.d., respectively. Control mice were treated with the vehicle alone during the same period. Mice were monitored for diabetes onset up to 40 weeks of age, and the percentage (%) of mice surviving diabetes-free (non-diabetic) in each treatment group is shown in a Kaplan-Meier survival plot (n = 4–5 mice per group). The median age of diabetes-free survival was 26 weeks in the vehicle-treated controls versus 31 weeks in those treated with DRI-C21041 and remained undetermined in the DRI-C21095–treated mice (did not cross the 50% threshold).
3.5 Pharmacokinetics
Because DRI-C21041 performed unexpectedly well in the ACE allo-transplant model compared to the ester-containing DRI-C21095 (Figure 1), which has shown higher potency in vitro (87, 88) and performed well in the kidney allo-transplant and diabetes prevention models, we carried out a brief pharmacokinetic (PK) evaluation in C57BL/6 mice. A single dose (30 mg/kg) was administered s.c. (same route as in the studies used for efficacy assessments), and plasma concentrations were quantified in samples collected at predefined time-points after the injection (0, 0.5, 1, 2, 4, 6, and 24 h). Results indicated that DRI-C21041 has an acceptably long half-life (t1/2 = 10.8 h) (Figure 8), considerably better than the ester-containing DRI-C21045 that we evaluated in an earlier study (t1/2 ≈ 2 h) (87). This is not surprising as ester-containing compounds are susceptible to quick metabolic degradation by abundantly present esterases, often resulting in shorter half-lives – an effect further exacerbated in rodents as they tend to metabolize ester-containing drugs (especially aliphatic esters) much faster than humans due to their known higher esterase activity (95).
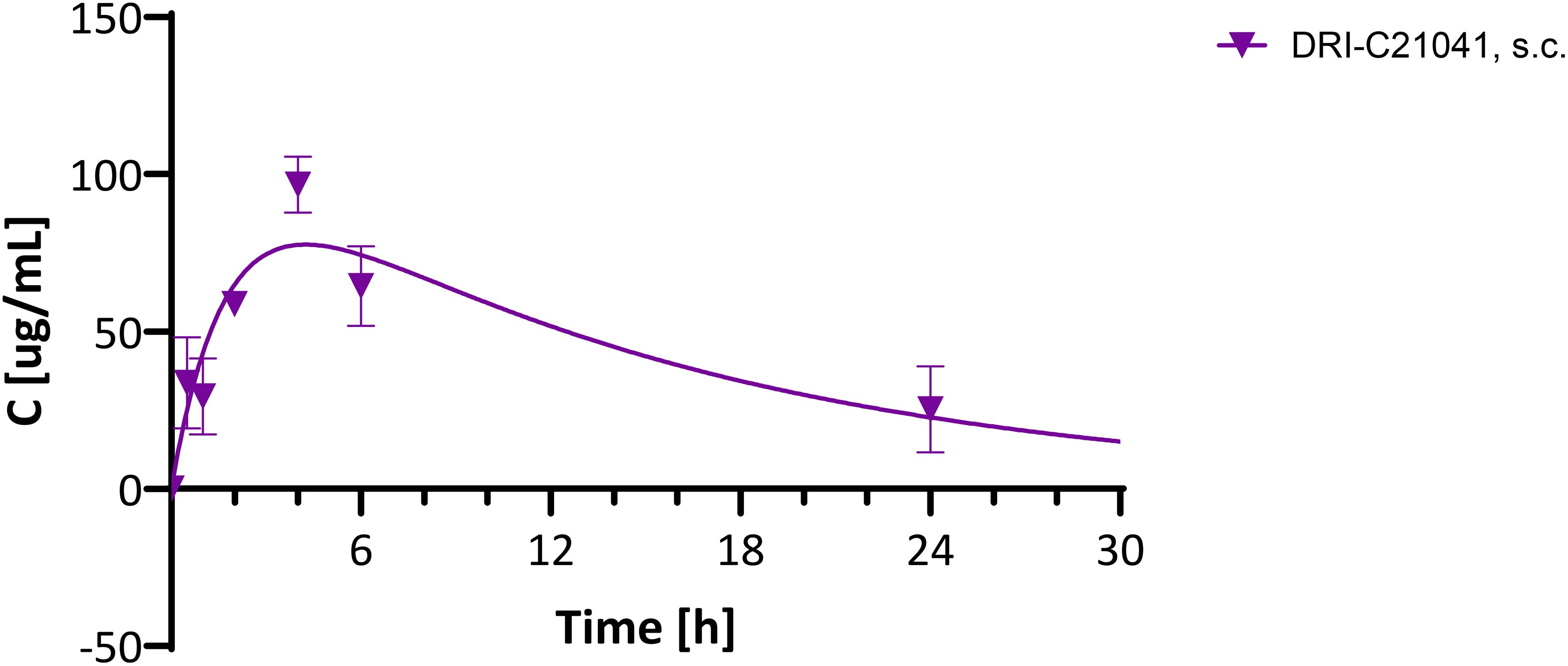
Figure 8. Pharmacokinetic evaluation of DRI-C21041 in mice. Concentration-time profile of DRI-C21041 in plasma following a single dose administration (30 mg/kg, s.c.) to male and female C57BL/6 mice. Experimental data (mean ± SD, n = 2–4 mice; purple symbols) shown fitted with a standard one compartment, first order absorption, first order elimination PK model (line).
4 Discussion
T1D, a disease in which the β-cells of the pancreatic islets are destroyed by an autoimmune process resulting in lifelong insulin deficiency (96–99), affects more than 8 million individuals worldwide and, as prevalence increases due to improved life-expectancy, it now affects about 1 in 200 adults and children older than 10 years of age in the United States (99–102). More than a century after the introduction of treatment with insulin, T1D still represents a disease with considerable therapeutic need, not only because of the burden of constant administration of exogenous insulin, but also because chronic and degenerative complications still occur in a sizable fraction of T1D patients despite considerable improvements in diabetes management and care (103–105). There is still excess mortality and loss of 10–20 life-years among those diagnosed with T1D even in high-income countries, and a much higher loss, >40 life-years, in low-income countries (101, 106, 107). Compared to the general population, all-cause mortality risk is about three-fold higher in people with T1D (108). Alarmingly, the incidence of T1D is increasing worldwide at a rate of 2–5% per year (97, 109–111), a phenomenon that seems to be occurring with other autoimmune diseases as well (112). Notably, despite many clinical trials of various immune interventions to prevent its onset, T1D remains the only autoimmune condition without a truly effective immunotherapy. Even the most successful immune interventions explored so far (e.g., cyclosporine, otelixizumab, rituximab, and abatacept) have achieved only a few months delay in the autoimmune-driven decline of insulin production (113–115). Teplizumab, which was recently approved by the FDA as the very first disease-modifying intervention for T1D (116), has shown a somewhat more promising delay in T1D onset (median of about 2 years) (117, 118), but it is restricted to select high-risk patients. Therefore, the need for effective immunotherapies for T1D remains critical, and controlling the associated autoimmune process will likely require novel immunomodulatory approaches (119). As ICP targets show particular promise (62), SMIPPIs targeting immune costimulation, such as those discussed here (i.e., CD40–CD40L), could ultimately lead to effective immunomodulatory therapeutics for the prevention of T1D and possibly other autoimmune diseases as well.
Furthermore, improved immunosuppressive therapies are also desperately needed for organ- and cell-transplant recipients to reduce or eliminate the serious side effects associated with current therapies that require chronic administration to avoid rejection and maintain the graft function. Costimulatory blockade is particularly promising in this aspect as well because it can be antigen/activation-specific and thus, less broadly immunosuppressive than current therapies and can lead to tolerance by allowing antigen recognition in the absence of costimulation (52, 63, 120–122). Because islets and especially β-cells are sensitive to commonly used immunosuppressive drugs (123, 124), there is an even more pronounced need for refined immunosuppressive strategies in β-cell replacement therapies, which can restore metabolic control more efficiently than exogeneous insulin therapy and prevent the serious long-term complications and co-morbidities associated with T1D (125–128). This is especially important now that (i) the FDA approved islet transplantation as a cell transplant therapy for the treatment of T1D (129) and (ii) stem cell therapies, which can provide renewable sources of insulin producing cells and promote the applicability of transplant therapy on a much wider scale, are beginning to show promise in T1D and have reached the clinical development phase (130–133).
In our previous work, we identified novel DRI-C compounds that inhibited the CD40–CD40L PPI with high nanomolar to low micromolar potency (IC50) and with >30-fold selectivity versus other TNF superfamily PPIs, namely OX40–OX40L, BAFFR–BAFF, and TNF-R1–TNFα (87, 88). Protein thermal shift analysis suggested that these SMIPPIs bind CD40L rather than CD40. The lead compounds used here, DRI-C21095 and DRI-C21041, inhibited CD40–CD40L binding in our cell-free ELISA assay with IC50 values of 19 and 87 nM, respectively (88). Their activities were also confirmed in cell-based assays. For example, DRI-C21041 inhibited the CD40L-induced activation of NF-κB biosensor cells with an IC50 of 10.3 μM and that of primary human B lymphocytes with an IC50 of 13.2 μM (87). DRI-C21095 had an IC50 of 6.0 μM in the NF-κB biosensor cell assay (88). In vivo activity was also confirmed previously in the short-term assay of alloantigen-induced T cell expansion in a draining lymph node (87, 88). Here, we confirmed the in vivo efficacy of our lead SMIs in longer-term transplantation- and autoimmune disease-relevant mouse models: two full MHC-mismatched allogeneic islet transplantation models (in the kidney subcapsular space and the anterior chamber of the eye - ACE, respectively) and a T1D prevention model in the diabetes-prone NOD mice.
The studies with islet allografts under the kidney capsule confirmed the immunosuppressive effects of both SMIs in a dose-dependent manner. At their highest doses tested here, both DRI-C21041 (12.5 mg/kg b.i.d.) and DRI-C21095 (20 mg/kg b.i.d.) significantly prolonged the survival and function of islet allografts in ~50% of recipients long after terminating the treatments (Figure 2) at doses that are roughly equivalent with that of the MR-1 antibody used as positive control (250 μg/mouse ≈ 10 mg/kg) considering the differences in molecular weights, potencies (IC50), and elimination half-lives (t1/2). Similarly, studies in the ACE-platform confirmed the long-term survival of islet allografts in recipients treated with either compound, and 80% of the DRI-C21041 treated recipients maintained structurally intact islets for >50 days after stopping treatment (Figure 3). We further exploited the ability of the ACE-platform that allows the detailed 3D spatial mapping of cellular level allo- and auto-immune reactions in and around the grafts and the longitudinal in situ tracking of infiltrating immune cells to derive kinetic profiles characteristic of specific cell subtypes. These studies revealed that treatment with the DRI-C compounds markedly reduced the infiltration of the allografts by T effector cells (Figure 6). Given the overwhelming evidence that allo- and auto-immune responses against ACE-transplanted islets mirror those in the pancreas and other transplant sites (92, 134–140), the current findings suggest that our SMIPPIs likely inhibited the activation and priming of T effector cells, which in turn reduced their infiltration into the islet allografts and consequently promoted the long-term survival and function of the islets in both transplant sites (i.e., kidney and ACE) and, possibly, the native pancreas of the NOD mice that never progressed to diabetes (Figure 7). Our exploratory immunophenotyping study in splenocytes also indicated a trend towards reduced central memory T cells among splenocytes and a concomitant increase in Tregs in association with DRI-C21041 and DRI-C21095 treatments in NOD mice (Supplementary Figure 1), further suggestive of peripheral immune regulation. These results are in overall agreement with effects expected from blockade of this important costimulatory interaction between CD40 and CD40L, which is mainly expressed on activated CD4+ T cells, known to play important roles in the promotion of germinal center formation and the production of class-switched antibodies as well as in the triggering of pro-inflammatory responses (34). Hence, the CD40–CD40L ICP represents an important therapeutic target in transplant therapy (33) and autoimmune diseases (34) as demonstrated by the large number of novel biologics being developed against it including those mentioned in the Introduction, such as letolizumab, dapirolizumab pegol, frexalimab, tegoprubart, TNX-1500, dazodalibep, or iscalimab.
The NOD mouse, an inbred strain developed from a line originally intended as a cataract-prone mouse strain (141), is by far the most commonly used animal model for autoimmune T1D (142–147). NOD mice develop diabetes spontaneously (typically, in about 60%–90% of females and 20%–40% of males between the age of 12 to 30 weeks) reproducing many aspects of the human autoimmune disease (e.g., presence of islet-specific autoantibodies, inflammation of pancreatic islets, and dependence on MHC alleles), but also with some important differences such as more severe insulitis and gender bias. An important role for the CD40–CD40L axis in T1D development has been already shown (55, 57): for example, diabetes did not develop in CD40L–/– NOD mice (56) and treatment with anti-CD40L mAb (54, 94) or CD40 peptide-inhibitors (60) abrogated the disease in regular NOD mice if treatment was initiated early enough. Our results in the NOD model are promising as they showed considerable reduction in the rate of onset (Figure 7); however, they must be treated with caution since, on one hand, they were obtained on a relatively small number of mice and, on the other, results in NODs in general do not translate well to humans. More than one hundred different successful preventive interventions in NOD mice have been reported already while their therapeutic success in humans has remained limited (143, 145). Nevertheless, the current findings showing the effect of our SMIPPIs in preventing T1D onset in NOD mice support their efficacy and further development for potential clinical application. DRI-C21095 seemed particularly effective in the NOD T1D onset prevention model (Figure 7) whereas DRI-C21041 in the ACE islet allotransplant model (Figure 3); future studies will explore the potential relevance of this for different clinical applications including in diabetic transplant recipients where both the allo- and autoimmune responses are present.
5 Conclusion
Results here confirm the in vivo therapeutic potential of DRI-C21041 and DRI-C21095, our new SMIs of the CD40–CD40L costimulatory PPI, as novel immune modulatory agents. Both agents showed considerable promise in two allogeneic islet transplant models, even suggesting the possibility of inducing operational immune tolerance, as well as in preventing autoimmune diabetes in the NOD mouse model. DRI-C21041 showed better in vivo efficacy than anticipated, especially in the ACE allotransplant model, likely due to its longer elimination half-life, which should allow for once daily administration in future studies. In summary, the current findings not only demonstrate the long-term in vivo efficacy of our new SMIs, but they also (i) provide further evidence that the CD40–CD40L PPI is susceptible to small-molecule inhibition and (ii) reinforce the potential for the induction of operational transplant immune tolerance and the prevention of T1D onset through the inhibition of this costimulatory ICP. Thus, further investigation of SMIs of the CD40–CD40L PPI is warranted, as they could ultimately lead to novel alternative immunomodulatory therapeutics that are safer, less immunogenic, more controllable (e.g., have shorter half-lives), and more patient-friendly (by being easier to administer – possibly through the oral route) than protein-based biologics.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by University of Miami Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
STC: Investigation, Methodology, Writing – review & editing. OA: Investigation, Methodology, Writing – review & editing. BW: Investigation, Methodology, Writing – review & editing. MHA: Data curation, Formal analysis, Funding acquisition, Methodology, Supervision, Visualization, Writing – review & editing. PB: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Financial support by the Diabetes Research Institute Foundation is gratefully acknowledged. Partial support for these studies was through grant R56AI130330 from the National Institute of Allergy and Infectious Diseases (NIAID). Funding for the Mass Spectrometry Research and Education Center at the University of Florida was provided by the National Institute of Health (NIH) S10 OD021758-01A1 and S10 OD030250-01A1.
Acknowledgments
The authors would like to thank Joel Szust as well as Yelena Gadea and Greycy Vega from the Preclinical Cell Processing and Translational Models Core of DRI for their excellent help with the islet isolations and animal care. The authors are grateful to Damir Bojadzic for his help with some of the experiments in the early stages of this work. They also thank Katie D. Heiden and Kari Basso from the Mass Spectrometry Laboratory at the University of Florida for their service.
Conflict of interest
The authors declare the following competing financial interest: The University of Miami has filed a patent on the DRI-C compounds with PB as inventor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VV declared a shared affiliation, with no collaboration, with the authors to the handling editor at the time of the review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1484425/full#supplementary-material
Abbreviations
ACE, anterior chamber of the eye; ALS, amyotrophic lateral sclerosis; BG, blood glucose; GFP, green fluorescent protein; HPβCD, hydroxypropyl-β-cyclodextrin; IEQ, islet equivalent; MS, multiple sclerosis; NHP, nonhuman primate; NOD, non-obese diabetic (mice); PK, pharmacokinetic; PPI, protein-protein interaction; POD, post-operative day; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SMI, small-molecule inhibitor; SMIPPI, small-molecule inhibitor of protein-protein interaction; STZ, streptozotocin; T1D, type 1 diabetes; TNF, tumor necrosis factor; TNFSF, TNF superfamily.
References
1. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
2. Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: Signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol. (2022) 86:137–50. doi: 10.1016/j.semcancer.2022.03.014
3. Huang C, Zhu HX, Yao Y, Bian ZH, Zheng YJ, Li L, et al. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J Autoimmun. (2019) 104:102333. doi: 10.1016/j.jaut.2019.102333
4. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. (2013) 342:1432–3. doi: 10.1126/science.342.6165.1432
5. Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discovery. (2016) 15:235–47. doi: 10.1038/nrd.2015.35
6. Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discovery. (2008) 7:21–39. doi: 10.1038/nrd2399
7. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. (2017) 6:576–88. doi: 10.1002/psp4.12224
8. Huck BR, Kötzner L, Urbahns K. Small molecules drive big improvements in immuno-oncology therapies. Angew Chem Int Ed Engl. (2018) 57:4412–28. doi: 10.1002/anie.201707816
9. Downing NS, Shah ND, Aminawung JA, Pease AM, Zeitoun JD, Krumholz HM, et al. Postmarket safety events among novel therapeutics approved by the US Food and Drug Administration between 2001 and 2010. J Am Med Assoc (JAMA). (2017) 317:1854–63. doi: 10.1001/jama.2017.5150
10. Sathish JG, Sethu S, Bielsky MC, de Haan L, French NS, Govindappa K, et al. Challenges and approaches for the development of safer immunomodulatory biologics. Nat Rev Drug Discovery. (2013) 12:306–24. doi: 10.1038/nrd3974
11. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. (2019) 16:563–80. doi: 10.1038/s41571-019-0218-0
12. Giannoukakis N, Phillips B, Trucco M. Toward a cure for type 1 diabetes mellitus: diabetes-suppressive dendritic cells and beyond. Pediatr Diabetes. (2008) 9:4–13. doi: 10.1111/j.1399-5448.2008.00401.x
13. Cochrane GM, Horne R, Chanez P. Compliance in asthma. Respir Med. (1999) 93:763–9. doi: 10.1016/s0954-6111(99)90260-3
14. Moia M, Mantovani LG, Carpenedo M, Scalone L, Monzini MS, Cesana G, et al. Patient preferences and willingness to pay for different options of anticoagulant therapy. Intern Emerg Med. (2013) 8:237–43. doi: 10.1007/s11739-012-0844-3
15. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discovery. (2004) 3:301–17. doi: 10.1038/nrd1343
16. Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. (2007) 450:1001–9. doi: 10.1038/nature06526
17. Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life. (2010) 62:724–31. doi: 10.1002/iub.383
18. Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein-protein interactions. Chem Rev. (2014) 114:4695–748. doi: 10.1021/cr400698c
19. Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discovery. (2016) 15:533–50. doi: 10.1038/nrd.2016.29
20. Bojadzic D, Buchwald P. Toward small-molecule inhibition of protein-protein interactions: General aspects and recent progress in targeting costimulatory and coinhibitory (immune checkpoint) interactions. Curr Top Med Chem. (2018) 18:674–99. doi: 10.2174/1568026618666180531092503
21. Buchwald P. Developing small-molecule inhibitors of protein-protein interactions involved in viral entry as potential antivirals for COVID-19. Front Drug Discovery. (2022) 2:2022.898035. doi: 10.3389/fddsv.2022.898035
22. Wu Y, Yang Z, Cheng K, Bi H, Chen J. Small molecule-based immunomodulators for cancer therapy. Acta Pharm Sin B. (2022) 12:4287–308. doi: 10.1016/j.apsb.2022.11.007
23. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
24. Gadek TR, Burdick DJ, McDowell RS, Stanley MS, Marsters JC Jr., Paris KJ, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. (2002) 295:1086–9. doi: 10.1126/science.295.5557.1086
25. Meanwell NA, Krystal MR, Nowicka-Sans B, Langley DR, Conlon DA, Eastgate MD, et al. Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J Med Chem. (2018) 61:62–80. doi: 10.1021/acs.jmedchem.7b01337
26. Kesselheim AS, Avorn J. The most transformative drugs of the past 25 years: a survey of physicians. Nat Rev Drug Discovery. (2013) 12:425–31. doi: 10.1038/nrd3977
27. Vugler A, O'Connell J, Nguyen MA, Weitz D, Leeuw T, Hickford E, et al. An orally available small molecule that targets soluble TNF to deliver anti-TNF biologic-like efficacy in rheumatoid arthritis. Front Pharmacol. (2022) 13:2022.1037983. doi: 10.3389/fphar.2022.1037983
28. Chédotal H, Narayanan D, Povlsen K, Gotfredsen CH, Brambilla R, Gajhede M, et al. Small-molecule modulators of tumor necrosis factor signaling. Drug Discovery Today. (2023) 28:103575. doi: 10.1016/j.drudis.2023.103575
29. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065x.2009.00782.x
30. Burkly LC. CD40 pathway blockade as an approach to immunotherapy. Adv Exp Med Biol. (2001) 489:135–52. doi: 10.1007/978-1-4615-1277-6_12
31. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. (2004) 22:307–28. doi: 10.1146/annurev.immunol.22.012703.104533
32. Daoussis D, Andonopoulos AP, Liossis SN. Targeting CD40L: a promising therapeutic approach. Clin Diagn Lab Immunol. (2004) 11:635–41. doi: 10.1128/CDLI.11.4.635-641.2004
33. Pinelli DF, Ford ML. Novel insights into anti-CD40/CD154 immunotherapy in transplant tolerance. Immunotherapy. (2015) 7:399–410. doi: 10.2217/imt.15.1
34. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv Drug Delivery Rev. (2019) 141:92–103. doi: 10.1016/j.addr.2018.12.005
35. Parker DC, Greiner DL, Phillips NE, Appel MC, Steele AW, Durie FH, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. (1995) 92:9560–4. doi: 10.1073/pnas.92.21.9560
36. Zheng XX, Markees TG, Hancock WW, Li Y, Greiner DL, Li XC, et al. CTLA4 signals are required to optimally induce allograft tolerance with combined donor-specific transfusion and anti-CD154 monoclonal antibody treatment. J Immunol. (1999) 162:4983–90. doi: 10.4049/jimmunol.162.8.4983
37. Molano RD, Berney T, Li H, Cattan P, Pileggi A, Vizzardelli C, et al. Prolonged islet graft survival in NOD mice by blockade of the CD40-CD154 pathway of T-cell costimulation. Diabetes. (2001) 50:270–6. doi: 10.2337/diabetes.50.2.270
38. Molano RD, Berney T, Pileggi A, Ricordi C, Burkly L, Rothstein D, et al. Prolonged survival of allogeneic islet grafts in NOD mice treated with a combination of anti-CD45RB and anti-CD154 antibodies. Transplant Proc. (2001) 33:248–9. doi: 10.1016/s0041-1345(00)01998-9
39. Berney T, Pileggi A, Molano RD, Poggioli R, Zahr E, Ricordi C, et al. The effect of simultaneous CD154 and LFA-1 blockade on the survival of allogeneic islet grafts in nonobese diabetic mice. Transplantation. (2003) 76:1669–74. doi: 10.1097/01.tp.0000092525.17025.d0
40. Molano RD, Pileggi A, Berney T, Poggioli R, Zahr E, Oliver R, et al. Prolonged islet allograft survival in diabetic NOD mice by targeting CD45RB and CD154. Diabetes. (2003) 52:957–64. doi: 10.2337/diabetes.52.4.957
41. Inverardi L, Linetsky E, Pileggi A, Molano RD, Serafini A, Paganelli G, et al. Targeted bone marrow radioablation with 153Samarium-lexidronam promotes allogeneic hematopoietic chimerism and donor-specific immunologic hyporesponsiveness. Transplantation. (2004) 77:647–55. doi: 10.1097/01.TP.0000112436.26473.A2
42. Masaki H, Appel MC, Leahy L, Leif J, Paquin L, Shultz LD, et al. Anti-mouse CD154 antibody treatment facilitates generation of mixed xenogeneic rat hematopoietic chimerism, prevents wasting disease and prolongs xenograft survival in mice. Xenotransplantation. (2006) 13:224–32. doi: 10.1111/j.1399-3089.2006.00290.x
43. Mai G, Bucher P, Morel P, Mei J, Bosco D, Andres A, et al. Anti-CD154 mAb treatment but not recipient CD154 deficiency leads to long-term survival of xenogeneic islet grafts. Am J Transplant. (2005) 5:1021–31. doi: 10.1111/j.1399-3089.2007.00402.x
44. Mai G, del Rio ML, Tian J, Ramirez P, Buhler L, Rodriguez-Barbosa JI. Blockade of the PD-1/PD-1L pathway reverses the protective effect of anti-CD40L therapy in a rat to mouse concordant islet xenotransplantation model. Xenotransplantation. (2007) 14:243–8. doi: 10.1111/j.1399-3089.2007.00402.x
45. Jung DY, Kim EY, Joo SY, Park JB, Moon C, Kim SH, et al. Prolonged survival of islet allografts in mice treated with rosmarinic acid and anti-CD154 antibody. Exp Mol Med. (2008) 40:1–10. doi: 10.3858/emm.2008.40.1.1
46. Kenyon NS, Fernandez LA, Lehmann R, Masetti M, Ranuncoli A, Chatzipetrou M, et al. Long-term survival and function of intrahepatic islet allografts in baboons treated with humanized anti-CD154. Diabetes. (1999) 48:1473–81. doi: 10.2337/diabetes.48.7.1473
47. Kenyon NS, Chatzipetrou M, Masetti M, Ranuncoli A, Oliveira M, Wagner JL, et al. Long-term survival and function of intrahepatic islet allografts in rhesus monkeys treated with humanized anti-CD154. Proc Natl Acad Sci USA. (1999) 96:8132–7. doi: 10.1073/pnas.96.14.8132
48. Koulmanda M, Smith RN, Qipo A, Weir G, Auchincloss H, Strom TB. Prolonged survival of allogeneic islets in cynomolgus monkeys after short-term anti-CD154-based therapy: nonimmunologic graft failure? Am J Transplant. (2006) 6:687–96. doi: 10.1111/j.1600-6143.2006.01235.x
49. Cardona K, Korbutt GS, Milas Z, Lyon J, Cano J, Jiang W, et al. Long-term survival of neonatal porcine islets in nonhuman primates by targeting costimulation pathways. Nat Med. (2006) 12:304–6. doi: 10.1038/nm1375
50. van der Windt DJ, Bottino R, Casu A, Campanile N, Smetanka C, He J, et al. Long-term controlled normoglycemia in diabetic non-human primates after transplantation with hCD46 transgenic porcine islets. Am J Transplant. (2009) 9:2716–26. doi: 10.1111/j.1600-6143.2009.02850.x
51. Bottino R, Knoll MF, Graeme-Wilson J, Klein EC, Ayares D, Trucco M, et al. Safe use of anti-CD154 monoclonal antibody in pig islet xenotransplantation in monkeys. Xenotransplantation. (2017) 24:e12283. doi: 10.1111/xen.12283
52. Abdulreda MH, Berman DM, Shishido A, Martin C, Hossameldin M, Tschiggfrie A, et al. Operational immune tolerance towards transplanted allogeneic pancreatic islets in mice and a non-human primate. Diabetologia. (2019) 62:811–21. doi: 10.1007/s00125-019-4814-4
53. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: The dark side of a great activator. Semin Immunol. (2009) 21:293–300. doi: 10.1016/j.smim.2009.05.012
54. Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, et al. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol. (1997) 159:4620–7. doi: 10.4049/jimmunol.159.9.4620
55. Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, et al. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity. (2002) 16:403–15. doi: 10.1016/s1074-7613(02)00290-x
56. Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. (2004) 114:979–87. doi: 10.1172/JCI200420483
57. Baker RL, Wagner DH Jr., Haskins K. CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J Autoimmun. (2008) 31:385–92. doi: 10.1016/j.jaut.2008.09.001
58. Wagner DH Jr., Vaitaitis G, Sanderson R, Poulin M, Dobbs C, Haskins K. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc Natl Acad Sci USA. (2002) 99:3782–7. doi: 10.1073/pnas.052247099
59. Baker RL, Mallevaey T, Gapin L, Haskins K. T cells interact with T cells via CD40-CD154 to promote autoimmunity in type 1 diabetes. Eur J Immunol. (2012) 42:672–80. doi: 10.1002/eji.201142071
60. Vaitaitis GM, Olmstead MH, Waid DM, Carter JR. Wagner DH, Jr. A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia. (2014) 57:2366–73. doi: 10.1007/s00125-014-3342-5
61. Vaitaitis GM, Waid DM, Yussman MG, Wagner DH Jr. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology. (2017) 152:243–54. doi: 10.1111/imm.12761
62. Spicer P, Runkel L. Costimulatory pathway targets for autoimmune and inflammatory conditions: clinical successes, failures, and hope for the future. Expert Opin Investig Drugs. (2019) 28:99–106. doi: 10.1080/13543784.2019.1557146
63. Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discovery. (2013) 12:147–68. doi: 10.1038/nrd3930
64. Song Y, Buchwald P. TNF superfamily protein-protein interactions: feasibility of small-molecule modulation. Curr Drug Targets. (2015) 16:393–408. doi: 10.2174/1389450116666150223115628
65. Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. (2000) 6:114. doi: 10.1038/72162
66. Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. (2003) 48:719–27. doi: 10.1002/art.10856
67. Koyama I, Kawai T, Andrews D, Boskovic S, Nadazdin O, Wee SL, et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation. (2004) 77:460–2. doi: 10.1097/01.tp.0000110291.29370.c0
68. Roth GA, Zuckermann A, Klepetko W, Wolner E, Ankersmit HJ, Moser B, et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation. (2004) 78:1238–9. doi: 10.1097/01.tp.0000135457.69220.5b
69. Couzin J. Drug discovery. Magnificent obsession. Sci. (2005) 307:1712–5. doi: 10.1126/science.307.5716.1712
70. Mirabet M, Barrabes JA, Quiroga A, Garcia-Dorado D. Platelet pro-aggregatory effects of CD40L monoclonal antibody. Mol Immunol. (2008) 45:937–44. doi: 10.1016/j.molimm.2007.08.006
71. Ramanujam M, Steffgen J, Visvanathan S, Mohan C, Fine JS, Putterman C. Phoenix from the flames: Rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun Rev. (2020) 19:102668. doi: 10.1016/j.autrev.2020.102668
72. Xie JH, Yamniuk AP, Borowski V, Kuhn R, Susulic V, Rex-Rabe S, et al. Engineering of a novel anti-CD40L domain antibody for treatment of autoimmune diseases. J Immunol. (2014) 192:4083–92. doi: 10.4049/jimmunol.1303239
73. Kim SC, Wakwe W, Higginbotham LB, Mathews DV, Breeden CP, Stephenson AC, et al. Fc-Silent anti-CD154 domain antibody effectively prevents nonhuman primate renal allograft rejection. Am J Transplant. (2017) 17:1182–92. doi: 10.1111/ajt.14197
74. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther. (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
75. Tocoian A, Buchan P, Kirby H, Soranson J, Zamacona M, Walley R, et al. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus. (2015) 24:1045–56. doi: 10.1177/0961203315574558
76. Furie RA, Bruce IN, Dorner T, Leon MG, Leszczynski P, Urowitz M, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology. (2021) 60:5397–407. doi: 10.1093/rheumatology/keab381
77. Vermersch P, Granziera C, Mao-Draayer Y, Cutter G, Kalbus O, Staikov I, et al. Inhibition of CD40L with frexalimab in multiple sclerosis. N Engl J Med. (2024) 390:589–600. doi: 10.1056/nejmoa2309439
78. Perrin S, Gill A, Gill C, Vieira F, Thompson K, Lincecum J, et al. The development and characterization of AT1501, an anti CD40L antibody lacking Fc effector function. Am J Transplant. (2021) 21:Abstr. 512. https://atcmeetingabstracts.com/abstract/the-development-and-characterization-of-at1501-an-anti-cd40l-antibody-lacking-fc-effector-function/.
79. Miura S, Abady Z, Pollok F, Ma M, Kinoshita K, Fogarty S, et al. TNX-1500, an Fc-modified anti-CD154 antibody, prolongs nonhuman primate cardiac allograft survival. J Heart Lung Transplant. (2022) 41:S148. doi: 10.1016/j.healun.2022.01.348
80. Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Trans Med. (2019) 11:eaar6584. doi: 10.1126/scitranslmed.aar6584
81. Singh AK, Goerlich CE, Zhang T, Lewis BGT, Hershfeld A, Mohiuddin MM. CD40-CD40L blockade: update on novel investigational therapeutics for transplantation. Transplantation. (2023) 107:1472–81. doi: 10.1097/tp.0000000000004469
82. St Clair EW, Baer AN, Ng WF, Noaiseh G, Baldini C, Tarrant TK, et al. CD40 ligand antagonist dazodalibep in Sjögren's disease: a randomized, double-blinded, placebo-controlled, phase 2 trial. Nat Med. (2024) 30:1583–92. doi: 10.1038/s41591-024-03009-3
83. Anwar IJ, Berman DM, DeLaura I, Gao Q, Willman MA, Miller A, et al. The anti-CD40L monoclonal antibody AT-1501 promotes islet and kidney allograft survival and function in nonhuman primates. Sci Trans Med. (2023) 15:eadf6376. doi: 10.1126/scitranslmed.adf6376
84. Fisher BA, Mariette X, Papas A, Grader-Beck T, Bootsma H, Ng WF, et al. Safety and efficacy of subcutaneous iscalimab (CFZ533) in two distinct populations of patients with Sjögren's disease (TWINSS): week 24 results of a randomised, double-blind, placebo-controlled, phase 2b dose-ranging study. Lancet. (2024) 404:540–53. doi: 10.1016/s0140-6736(24)01211-x
85. Margolles-Clark E, Jacques-Silva MC, Ganesan L, Umland O, Kenyon NS, Ricordi C, et al. Suramin inhibits the CD40–CD154 costimulatory interaction: a possible mechanism for immunosuppressive effects. Biochem Pharmacol. (2009) 77:1236–45. doi: 10.1016/j.bcp.2009.01.001
86. Margolles-Clark E, Umland O, Kenyon NS, Ricordi C, Buchwald P. Small molecule costimulatory blockade: organic dye inhibitors of the CD40-CD154 interaction. J Mol Med. (2009) 87:1133–43. doi: 10.1007/s00109-009-0519-3
87. Chen J, Song Y, Bojadzic D, Tamayo-Garcia A, Landin AM, Blomberg BB, et al. Small-molecule inhibitors of the CD40-CD40L costimulatory protein-protein interaction. J Med Chem. (2017) 60:8906–22. doi: 10.1021/acs.jmedchem.7b01154
88. Bojadzic D, Chen J, Alcazar O, Buchwald P. Design, synthesis, and evaluation of novel immunomodulatory small molecules targeting the CD40-CD154 costimulatory protein-protein interaction. Molecules. (2018) 23:1153. doi: 10.3390/molecules23051153
89. Abdulreda MH, Caicedo A, Berggren P-O. Transplantation into the anterior chamber of the eye for longitudinal, non-invasive in vivo imaging with single-cell resolution in real-time. J Vis Exp. (2013) 73):e50466. doi: 10.3791/50466
90. Abdulreda MH, Faleo G, Molano RD, Lopez-Cabezas M, Molina J, Tan Y, et al. High-resolution, noninvasive longitudinal live imaging of immune responses. Proc Natl Acad Sci USA. (2011) 108:12863–8. doi: 10.1073/pnas.1105002108
91. Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A, et al. Real-time immune cell interactions in target tissue during autoimmune-induced damage and graft tolerance. J Exp Med. (2014) 211:441–56. doi: 10.1084/jem.20130785
92. Abdulreda MH, Molano RD, Faleo G, Lopez-Cabezas M, Shishido A, Ulissi U, et al. In vivo imaging of type 1 diabetes immunopathology using eye-transplanted islets in NOD mice. Diabetologia. (2019) 62:1237–50. doi: 10.1007/s00125-019-4879-0
93. Buchwald P, Juhász A, Bell C, Pátfalusi M, Howes J. Bodor N. A pharmacogenetics-based unified parent-metabolite pharmacokinetic model incorporating acetylation polymorphism for talampanel in humans. J Pharmacokinet Pharmacodyn. (2005) 32:377–400. doi: 10.1007/s10928-005-0001-y
94. Nanji SA, Hancock WW, Luo B, Schur CD, Pawlick RL, Zhu LF, et al. Costimulation blockade of both inducible costimulator and CD40 ligand induces dominant tolerance to islet allografts and prevents spontaneous autoimmune diabetes in the NOD mouse. Diabetes. (2006) 55:27–33. doi: 10.2337/diabetes.55.01.06.db04-1154
95. Buchwald P. Structure-metabolism relationships: steric effects and the enzymatic hydrolysis of carboxylic esters. Mini-Rev Med Chem. (2001) 1:101–11. doi: 10.2174/1389557013407403
96. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
97. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/s0140-6736(13)60591-7
98. Lemos JRN, Hirani K, von Herrath M. Immunological and virological triggers of type 1 diabetes: insights and implications. Front Immunol. (2023) 14:2023.1326711. doi: 10.3389/fimmu.2023.1326711
99. Quattrin T, Mastrandrea LD, Walker LSK. Type 1 diabetes. Lancet. (2023) 401:2149–62. doi: 10.1016/s0140-6736(23)00223-4
100. Green A, Hede SM, Patterson CC, Wild SH, Imperatore G, Roglic G, et al. Type 1 diabetes in 2017: global estimates of incident and prevalent cases in children and adults. Diabetologia. (2021) 64:2741–50. doi: 10.1007/s00125-021-05571-8
101. Gregory GA, Robinson TIG, Linklater SE, Wang F, Colagiuri S, de Beaufort C, et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. (2022) 10:741–60. doi: 10.1016/s2213-8587(22)00218-2
102. Fang M, Wang D, Selvin E. Prevalence of type 1 diabetes among US children and adults by age, sex, race, and ethnicity. J Am Med Assoc (JAMA). (2024) 331:1411–3. doi: 10.1001/jama.2024.2103
103. Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. (2006) 55:1463–9. doi: 10.2337/db05-1423
104. Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, et al. Changes in diabetes-related complications in the United States, 1990-2010. N Engl J Med. (2014) 370:1514–23. doi: 10.1056/NEJMoa1310799
105. Zheng Y, Ma Q, Qi X, Zhu Z, Wu B. Prevalence and incidence of mild cognitive impairment in adults with diabetes in the United States. Diabetes Res Clin Pract. (2023) 205:110976. doi: 10.1016/j.diabres.2023.110976
106. Livingstone SJ, Levin D, Looker HC, Lindsay RS, Wild SH, Joss N, et al. Estimated life expectancy in a Scottish cohort with type 1 diabetes, 2008-2010. J Am Med Assoc (JAMA). (2015) 313:37–44. doi: 10.1001/jama.2014.16425
107. Rawshani A, Sattar N, Franzen S, Rawshani A, Hattersley AT, Svensson AM, et al. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study. Lancet. (2018) 392:477–86. doi: 10.1016/s0140-6736(18)31506-x
108. von Scholten BJ, Kreiner FF, Gough SCL, von Herrath M. Current and future therapies for type 1 diabetes. Diabetologia. (2021) 64:1037–48. doi: 10.1007/s00125-021-05398-3
109. Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. J Am Med Assoc (JAMA). (2014) 311:1778–86. doi: 10.1001/jama.2014.3201
110. Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T, et al. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989-2013: a multicentre prospective registration study. Diabetologia. (2019) 62:408–17. doi: 10.1007/s00125-018-4763-3
111. Wagenknecht LE, Lawrence JM, Isom S, Jensen ET, Dabelea D, Liese AD, et al. Trends in incidence of youth-onset type 1 and type 2 diabetes in the USA, 2002-18: results from the population-based SEARCH for Diabetes in Youth study. Lancet Diabetes Endocrinol. (2023) 11:242–50. doi: 10.1016/s2213-8587(23)00025-6
112. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis. (2015) 3:151–5. doi: 10.12691/ijcd-3-4-8
113. Skyler JS, Ricordi C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes. (2011) 60:1–8. doi: 10.2337/db10-1114
114. Skyler JS. Prevention and reversal of type 1 diabetes - past challenges and future opportunities. Diabetes Care. (2015) 38:997–1007. doi: 10.2337/dc15-0349
115. Jacobsen LM, Schatz D. Immunotherapy-based strategies for treatment of type 1 diabetes. Horm Res Paediatr. (2024) 97:ePub. doi: 10.1159/000542002
116. Hirsch JS. FDA approves teplizumab: a milestone in type 1 diabetes. Lancet Diabetes Endocrinol. (2023) 11:18. doi: 10.1016/s2213-8587(22)00351-5
117. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. (2019) 381:603–13. doi: 10.1056/nejmoa1902226
118. Sims EK, Bundy BN, Stier K, Serti E, Lim N, Long SA, et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci Trans Med. (2021) 13:eabc8980. doi: 10.1126/scitranslmed.abc8980
119. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: building a bridge to a cure for type 1 diabetes. Science. (2021) 373:510–6. doi: 10.1126/science.abh1654
120. Vincenti F, Luggen M. T cell costimulation: a rational target in the therapeutic armamentarium for autoimmune diseases and transplantation. Annu Rev Med. (2007) 58:347–58. doi: 10.1146/annurev.med.58.080205.154004
121. Sharpe AH. Mechanisms of costimulation. Immunol Rev. (2009) 229:5–11. doi: 10.1111/j.1600-065x.2009.00784.x
122. Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunol Rev. (2009) 229:271–93. doi: 10.1111/j.1600-065X.2009.00781.x
123. Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by β cell regeneration. J Clin Invest. (2007) 117:2553–61. doi: 10.1172/jci32959
124. Zahr E, Molano RD, Pileggi A, Ichii H, Jose SS, Bocca N, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation. (2007) 84:1576–83. doi: 10.1097/01.tp.0000296035.48728.28
125. Shapiro AM, Pokrywczynska M, Ricordi C. Clinical pancreatic islet transplantation. Nat Rev Endocrinol. (2017) 13:268–77. doi: 10.1038/nrendo.2016.178
126. Lablanche S, Vantyghem MC, Kessler L, Wojtusciszyn A, Borot S, Thivolet C, et al. Islet transplantation versus insulin therapy in patients with type 1 diabetes with severe hypoglycaemia or poorly controlled glycaemia after kidney transplantation (TRIMECO): a multicentre, randomised controlled trial. Lancet Diabetes Endocrinol. (2018) 6:527–37. doi: 10.1016/S2213-8587(18)30078-0
127. Vantyghem MC, Chetboun M, Gmyr V, Jannin A, Espiard S, Le Mapihan K, et al. Ten-year outcome of islet alone or islet after kidney transplantation in type 1 diabetes: A prospective parallel-arm cohort study. Diabetes Care. (2019) 42:2042–9. doi: 10.2337/dc19-0401
128. Markmann JF, Rickels MR, Eggerman TL, Bridges ND, Lafontant DE, Qidwai J, et al. Phase 3 trial of human islet-after-kidney transplantation in type 1 diabetes. Am J Transplant. (2021) 21:1477–92. doi: 10.1111/ajt.16174
129. Stabler CL, Russ HA. Regulatory approval of islet transplantation for treatment of type 1 diabetes: Implications and what is on the horizon. Mol Ther. (2023) 31:3107–8. doi: 10.1016/j.ymthe.2023.10.011
130. Johnson JD. The quest to make fully functional human pancreatic beta cells from embryonic stem cells: climbing a mountain in the clouds. Diabetologia. (2016) 59:2047–57. doi: 10.1007/s00125-016-4059-4
131. Siehler J, Blochinger AK, Meier M, Lickert H. Engineering islets from stem cells for advanced therapies of diabetes. Nat Rev Drug Discovery. (2021) 20:920–40. doi: 10.1038/s41573-021-00262-w
132. de Koning EJP, Carlotti F. Stem cell-based islet replacement therapy in diabetes: A road trip that reached the clinic. Cell Stem Cell. (2021) 28:2044–6. doi: 10.1016/j.stem.2021.11.008
133. The Lancet Diabetes and Endocrinology. Stem-cell therapy for diabetes: the hope continues. Lancet Diabetes Endocrinol. (2024) 12:775. doi: 10.1016/S2213-8587(24)00314-0
134. Mojibian M, Harder B, Hurlburt A, Bruin JE, Asadi A, Kieffer TJ. Implanted islets in the anterior chamber of the eye are prone to autoimmune attack in a mouse model of diabetes. Diabetologia. (2013) 56:2213–21. doi: 10.1007/s00125-013-3004-z
135. Chmelova H, Cohrs CM, Chouinard JA, Petzold C, Kuhn M, Chen C, et al. Distinct roles of beta-cell mass and function during type 1 diabetes onset and remission. Diabetes. (2015) 64:2148–60. doi: 10.2337/db14-1055
136. Schmidt-Christensen A, Hansen L, Ilegems E, Fransen-Pettersson N, Dahl U, Gupta S, et al. Imaging dynamics of CD11c(+) cells and Foxp3(+) cells in progressive autoimmune insulitis in the NOD mouse model of type 1 diabetes. Diabetologia. (2013) 56:2669–78. doi: 10.1007/s00125-013-3024-8
137. Berclaz C, Schmidt-Christensen A, Szlag D, Extermann J, Hansen L, Bouwens A, et al. Longitudinal three-dimensional visualisation of autoimmune diabetes by functional optical coherence imaging. Diabetologia. (2016) 59:550–9. doi: 10.1007/s00125-015-3819-x
138. Benson RA, Garcon F, Recino A, Ferdinand JR, Clatworthy MR, Waldmann H, et al. Non-invasive multiphoton imaging of islets transplanted into the pinna of the NOD mouse ear reveals the immediate effect of anti-CD3 treatment in autoimmune diabetes. Front Immunol. (2018) 9:2018.01006. doi: 10.3389/fimmu.2018.01006
139. Ilegems E, Dicker A, Speier S, Sharma A, Bahow A, Edlund PK, et al. Reporter islets in the eye reveal the plasticity of the endocrine pancreas. Proc Natl Acad Sci USA. (2013) 110:20581–6. doi: 10.1073/pnas.1313696110
140. Ellis CE, Mojibian M, Ida S, Fung VCW, Skovso S, McIver E, et al. Human A2-CAR T cells reject HLA-A2 + human islets transplanted into mice without inducing graft-versus-host disease. Transplantation. (2023) 107:e222–e33. doi: 10.1097/tp.0000000000004709
141. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu (Exp Anim). (1980) 29:1–13. doi: 10.1538/expanim1978.29.1_1
142. Leiter EH. The NOD mouse: a model for insulin-dependent diabetes mellitus. Curr Protoc Immunol. (2001) 15:9.1–.9.23. doi: 10.1002/0471142735.im1509s24
143. Roep BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nat Rev Immunol. (2004) 4:989–97. doi: 10.1038/nri1502
144. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
145. Shoda LK, Young DL, Ramanujan S, Whiting CC, Atkinson MA, Bluestone JA, et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity. (2005) 23:115–26. doi: 10.1016/j.immuni.2005.08.002
146. Leiter EH, SChile A. Genetic and pharmacologic models for type 1 diabetes. Curr Protoc Mouse Biol. (2013) 3:9–19. doi: 10.1002/9780470942390.mo120154
Keywords: CD154, costimulatory inhibition, immune checkpoint, immunosuppression, islet transplantation, protein-protein interaction, small-molecule drug, type 1 diabetes (T1D)
Citation: Chuang S-T, Alcazar O, Watts B, Abdulreda MH and Buchwald P (2024) Small-molecule inhibitors of the CD40–CD40L costimulatory interaction are effective in pancreatic islet transplantation and prevention of type 1 diabetes models. Front. Immunol. 15:1484425. doi: 10.3389/fimmu.2024.1484425
Received: 21 August 2024; Accepted: 28 October 2024;
Published: 13 November 2024.
Edited by:
Sina Naserian, Hôpital Paul Brousse, FranceReviewed by:
Pradeep Shrestha, University of Texas MD Anderson Cancer Center, United StatesVighnesh Vetrivel Venkatasamy, University of Miami, United States
Takayuki Anazawa, Kyoto University, Japan
Copyright © 2024 Chuang, Alcazar, Watts, Abdulreda and Buchwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Buchwald, cGJ1Y2h3YWxkQG1lZC5taWFtaS5lZHU=; Midhat H. Abdulreda, bWFiZHVscmVkYUBtaWFtaS5lZHU=