 Maria Francesca Viola
Maria Francesca Viola Eliana Franco Taveras
Eliana Franco Taveras Elvira Mass
Elvira Mass- Developmental Biology of the Immune System, Life and Medical Sciences (LIMES) Institute, University of Bonn, Bonn, Germany
Macrophages are integral components of the innate immune system that colonize organs early in development and persist into adulthood through self-renewal. Their fate, whether they are replaced by monocytes or retain their embryonic origin, depends on tissue type and integrity. Macrophages are influenced by their environment, a phenomenon referred to as developmental programming. This influence extends beyond the local tissue microenvironment and includes soluble factors that can reach the macrophage niche. These factors include metabolites, antibodies, growth factors, and cytokines, which may originate from maternal diet, lifestyle, infections, or other developmental triggers and perturbations. These influences can alter macrophage transcriptional, epigenetic, and metabolic profiles, affecting cell-cell communication and tissue integrity. In addition to their crucial role in tissue immunity, macrophages play vital roles in tissue development and homeostasis. Consequently, developmental programming of these long-lived cells can modulate tissue physiology and pathology throughout life. In this review, we discuss the ontogeny of macrophages, the necessity of developmental programming by the niche for macrophage identity and function, and how developmental perturbations can affect the programming of macrophages and their subtissular niches, thereby influencing disease onset and progression in adulthood. Understanding these effects can inform targeted interventions or preventive strategies against diseases. Finally, understanding the consequences of developmental programming will shed light on how maternal health and disease may impact the well-being of future generations.
1 Introduction
Tissue-resident macrophages are integral components of the innate immune system, ubiquitously present across all organs. Initially described by Nobel laureate Ilya Metchnikov as professional phagocytes, macrophages play a crucial role in the clearance of invading pathogens, apoptotic corpses, immune complexes and other cellular debris, thereby maintaining tissue homeostasis. Over the years, our understanding of macrophages has evolved beyond their role as mere ‘garbage cleaners’ to recognizing their vital, tissue-specific functions (1). These specialized roles are directed by signals from the surrounding cells within the tissue, collectively known as the ‘macrophage niche,’ which provides cues that tailor macrophage activity to meet local requirements. Additionally, macrophage function is likely influenced by their ontogeny and the factors and stimuli encountered during early life (Figure 1). Here, we review the current knowledge on the developmental programming of tissue-resident macrophages, and how this imprinting may affect tissue development and homeostasis.
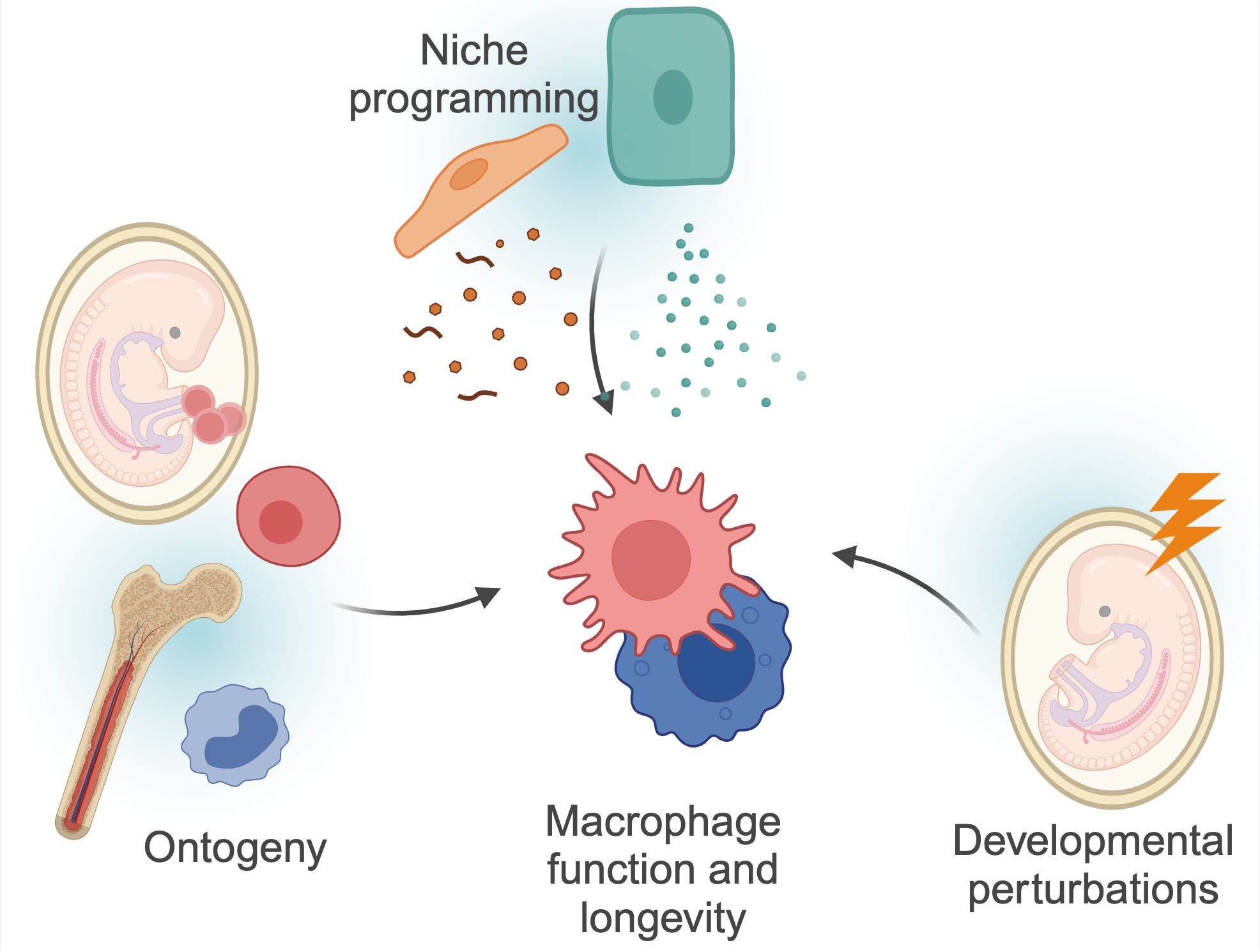
Figure 1. Factors determining macrophage functions. Macrophage function and longevity is a product of numerous, factors. Macrophages can derive from progenitors seeded within the embryo, or from differentiation of circulating monocytes. These cells are then further instructed by local cues and instructions from surrounding cells, the so-called macrophage ‘niche’. Finally, as discussed in more detail in this review, macrophages can be programmed by triggers during development, and retain this information throughout their lifespan. These factors are interconnected; for example, niche perturbations can impact monocyte recruitment, and developmental programming may also impact niche programming itself. Created using BioRender.
2 Macrophage ontogeny and longevity
Tissue-resident macrophages develop within the embryo and the adult mouse in a series of sequential developmental waves (Figure 2). At embryonic developmental day (E)8.5, erythro-myeloid progenitors (EMPs) in the yolk sac give rise to pre-macrophages (pMacs), which colonize the embryonic tissue (2, 3). These pMacs are present within the tissues during early development and differentiate into tissue-resident macrophages in parallel to organogenesis, thereby establishing cellular cross-talks with neighboring cells already early in life (4–6). In addition, at E10.5, hematopoietic stem cells (HSCs) emerge within the aorta-gonado-mesonephros (AGM) region from where they migrate to the fetal liver at E11.5 (7–10). There they expand and produce different lineages of leukocytes, and then finally migrate to the fetal bone marrow shortly before birth. At postnatal stages, bone marrow HSCs give rise to circulating monocytes, which can migrate into tissues and differentiate into monocyte-derived macrophages (MdMs) (4, 10). Albeit a fetal liver monocyte origin has been proposed for macrophages, there is to date no direct experimental evidence in mice showing that HSC-derived monocytes differentiate into macrophages during embryogenesis. Indeed, the fate-mapping mouse model Ms4a3Cre; Rosa26tdTomato specific to granulocyte-monocyte progenitors (GMP) has demonstrated that no tissue-resident macrophages are GMP-derived at birth (11).
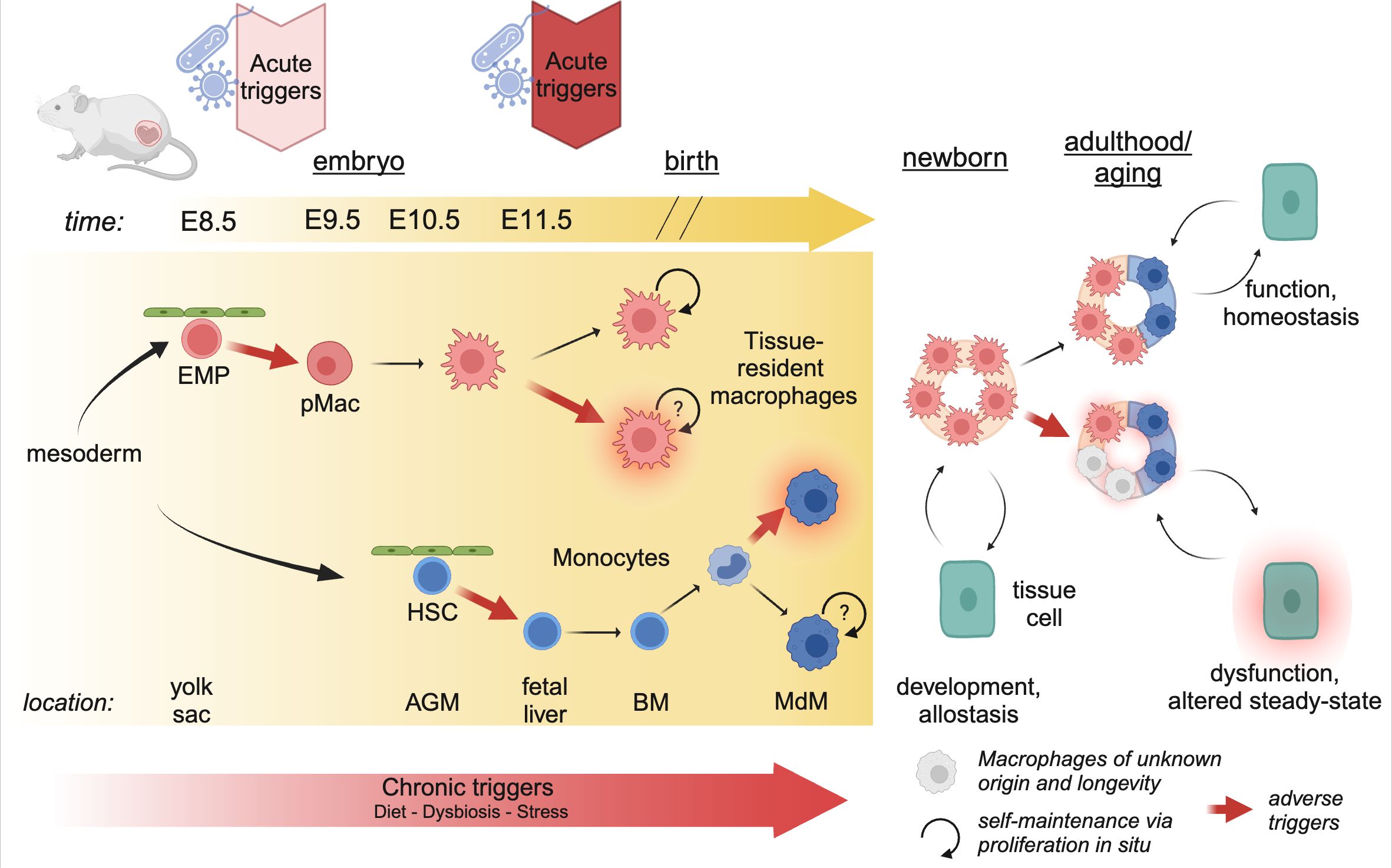
Figure 2. Developmental perturbations during hematopoiesis. The progenitors of tissue-resident macrophages develop in a series of waves during embryogenesis. As early as E8.5, erythro-myeloid progenitors (EMP) in the yolk sac give rise to pre-Macrophages (pMacs), which colonize the developing tissues and give rise to long-lived, proliferating tissue-resident macrophages. At E10.5, hematopoietic stem cells (HSCs) leave the aorta-gonado-mesonephros (AGM) region and migrate to the fetal liver. Here they expand, before migrating to the bone-marrow (BM) just before birth. In postnatal mice, bone-marrow derived monocytes can enter the tissue and differentiate into macrophages (MdM), possibly with different life cycles. The staggered timing of these developmental waves underlies the differential impact triggers can have. Acute triggers, depending on their timing, may program EMP/pMacs and/or HSCs, while chronic triggers may affect all lineages giving rise to macrophages. Created using BioRender.
A plethora of work has demonstrated that yolk sac-derived tissue-resident macrophages can proliferate and self-maintain within the tissue for months or even years (1). Using diverse tamoxifen-inducible fate-mapping models, it has been suggested that, with age, yolk sac-derived cells cease to proliferate and can be replaced by MdMs (12). The rate of replenishment by MdMs varies greatly between tissues; the tissue-resident macrophages of the brain parenchyma, microglia, self-maintain throughout life with little to no replenishment by MdMs; on the other end, macrophages in the gut lamina propria display a high turnover and are rapidly replaced by MdMs (reviewed excellently elsewhere) (1, 13). However, we would like to highlight that it has not yet been established whether MdMs that are starting to seed developing tissues after birth can become long-lived and proliferative as their yolk-sac derived counterparts (Figure 2). In any case, every tissue contains a mix of yolk sac-derived and monocyte-derived macrophages which dynamically changes over time, or as a consequence of inflammation or infection.
The distinct longevity of yolk sac-derived macrophages and MdMs highlights an intriguing aspect of developmental programming (Figure 2). Indeed, the first long-lived macrophages, such as microglia and Kupffer cells, are present as early as E9.5-E10.5 (3). Consequently, perturbations occurring during these early developmental stages can have long-lasting effects, as these cells proliferate in situ and give rise to adult macrophage clones that retain the information experienced by the progenitor macrophages. Conversely, developmental programming can also affect HSCs, which, in adulthood, give rise to MdMs. The impact of these perturbations depends on their duration and nature. Consequently, developmental disturbances can lead to varied outcomes based on the type of perturbation and the specific stage of development at which it occurs.
3 Niche programming vs. developmental programming
Once macrophages have colonized their tissue of residence, local cues determine macrophage longevity, identity and function. For instance, the expression of colony-stimulating factor 1 (CSF1) and IL-34 is essential for the self-maintenance of different macrophage populations (14). Further, niche programming constitutes factors such as IL-13 and colony-stimulating factor 2 (CSF2) for alveolar macrophages, transforming growth factor beta (TGFb), bone morphogenetic protein 9 (BMP9) and desmosterol for tissue-resident macrophages of the liver, Kupffer cells (15, 16). These niche programming events have been mostly studied during adulthood using macrophage depletion models. However, during development the intercellular interactions of a macrophage may be completely different than what we observe during steady-state adulthood. One such example is Kupffer cells, which act as primary macrophages for erythroblasts during embryogenesis, facilitating their maturation (17, 18). Additionally, these cells interact with fetal liver hematopoietic stem cells, thereby regulating their differentiation potential (19, 20). At this early time point, there are no hepatocytes present that could imprint Kupffer cell identity and drive gene expression of core Kupffer cell factors, such as Id3, which can already be detected as early as at E10.5 (21). Only from E14.5 onwards hepatocytes differentiate and directly interact with Kupffer cells (21). Finally, during the first weeks of life, Kupffer cells migrate to the lumen of sinusoids where they start to actively sample blood components and circulating cells (22). Another example is that of microglia, the tissue-resident macrophages of the brain. Indeed, it has been well-characterized that microglia participate in neuronal development early in life, and then switch to a neuro-supportive function in adulthood (23–25). It is probable that all long-lived macrophages undergo similar dynamic alterations in their cellular interaction partners during embryogenesis. We hypothesize that these interactions are essential for beneficial developmental programming of yolk sac-derived macrophages, enabling them to fulfill their tissue-specific functions necessary for maintaining tissue homeostasis later in life.
Beyond direct interactions, environmentally encoded developmental triggers, such as the maternal microbiota and food intake, may be essential for proper macrophage function. These factors, which may be interlinked, can lead to a state of chronic low-grade inflammation that can impact the developing fetus. However, also acute stressors during gestation may play a role, as has been demonstrated for stress and immune activation via, for example, bacterial or viral infections. The timing of these perturbations may also play a role in determining the outcome; indeed, while chronic stress, such as obesity, impacts all stages of development, acute stress may have a more restricted impact depending on when it occurs. Altogether, the core functions of adult macrophages are likely influenced by prenatal and early-life perturbations, which can have lasting impacts on macrophage precursors, including EMPs and pMacs, as well as on maturing macrophages, potentially through epigenetic mechanisms. However, developmental programming may have beneficial or detrimental outcomes, depending on the stimuli. From an evolutionary perspective, early-life or maternal priming of immune cells confer advantages for the growth and survival of offspring. For instance, measle-containing vaccines are associated with lower mortality in the first years after birth. Maternal priming has also demonstrated its importance in this context, as children with maternal antibodies from vaccination against measles have shown lower mortality rates (26). However, not all stimuli result beneficial outcomes (see below). Given the crucial role of macrophages in tissue maturation, their developmental programming can profoundly impact tissue function and homeostasis. This concept aligns with Barker’s theory of the “Developmental Origin of Health and Disease,” which suggests that environmental exposures during development can determine health and disease risk in adulthood. Of note, in addition to programming macrophage progenitors, developmental perturbations may also reprogram niche cells, which in turn, could impact macrophage function. Conversely, developmental reprogramming of macrophages may be masked by niche programming, which displays remarkable resilience and plasticity to external perturbations (1). The precise target of developmental programming may not always be clear throughout studies. However, as the topic of macrophage programming by the niche has been excellently covered elsewhere (27), and direct studies investigating the combined influence of developmental and niche programming are still lacking, we will here focus on how developmental perturbations may impact macrophages directly.
The nature of developmental perturbations affecting offspring immunity can be considered a regional issue. Indeed, in countries of the Global South, bacterial and viral infections, and accompanying antibiotic treatment, during gestation are far more common than those of the Global North (28–30). However, the exponential increase in obesity and metabolic disorders in all regions of the world poses a novel and unprecedented challenge, as these disorders have shown to have wide-ranging consequences on offspring immune function (31). Understanding the consequences of harmful developmental programming is of fundamental importance to consider its corresponding socioeconomic impact and to develop targeted interventions to safeguard the health of future generations.
Finally, while most studies focus on the maternal contribution to offspring immunity, increasing evidence suggests that developmental programming can also be transmitted from the paternal side via the sperm epigenome (32–34). However, in the following review we will focus on the maternal impact on offspring macrophage programming, highlighting current knowledge about maternal immune activation (MIA) associated with stress and infections, diet, as well as the role of maternal microbiome. An overview of models available to study the impact of developmental programming on the offspring is shown in Table 1.
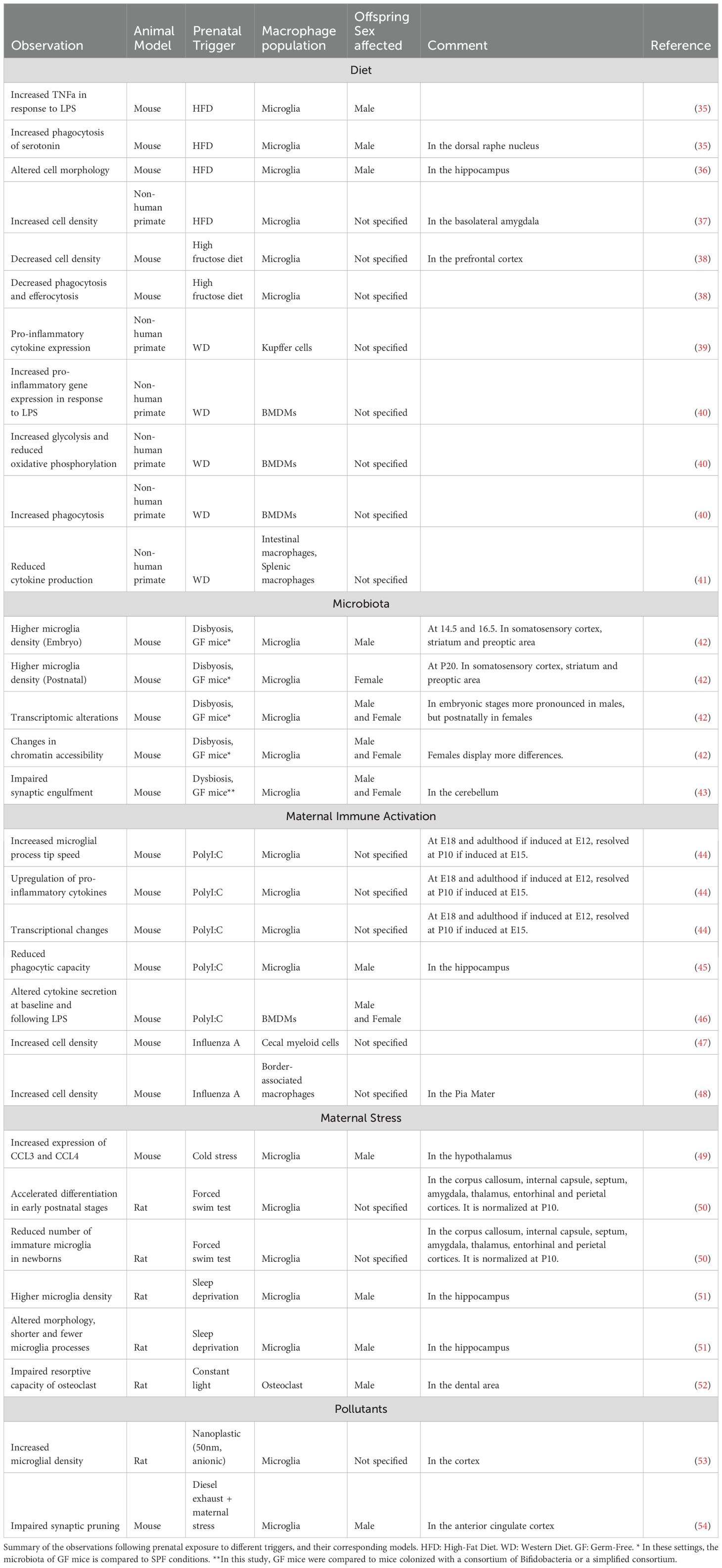
Table 1. Observed macrophage phenotypes and functions after a trigger.
4 Potential triggers of developmental programming
4.1 Diet
Research on maternal diet has focused mainly on maternal high-fat diet (HFD) or maternal ‘Western-style’ diet (WD). While both diets are rich in fat, WD is also high in sucrose, whereas HFD has a sucrose content comparable to a regular chow diet. However, drawing parallels across different studies is hampered by the specific composition of the maternal diet, e.g., the level of fat content and the source of dietary fat (corn oil, peanut oil, soybean oil, sunflower oil, or lard), which can lead to microbiota perturbations and subsequent downstream effects (55). Despite these challenges, it is widely accepted that maternal obesity, regardless of the specific diet, leads to alterations in the offspring’s immune system, particularly affecting macrophage responses to inflammatory stimuli. Microglia and Kupffer cells may represent key targets of developmental programming through the maternal diet. Indeed, both microglia and Kupffer cells are seeded early during embryonic development, and receive very little input from circulating cells throughout life. As such, information programmed in these cells during early development can have long-lasting effects on tissue function. Nevertheless, while a large volume of studies has focused on the impact of maternal diet on offspring microglia, much less is known about resident macrophages in other tissues.
There is compelling evidence suggesting that maternal obesity has a profound impact on neurodevelopment through alterations in microglia. These effects are notably sex and region-specific. For instance, microglia from male embryos of mothers fed a HFD showed increased TNFa production in response to LPS (35). Moreover, maternal HFD leads to increased CD68 immunoreactivity and increased phagocytosis of serotonin by microglia in the dorsal raphe nucleus of male offspring. This results in decreased serotonin availability in both fetal and adult brains, contributing to sex-specific behavioral outcomes. These changes were absent in a Cx3cr1CreERT2; Tlr4fl/fl conditional knockout model, implicating TLR4 signaling in these processes. This study further found increased endotoxin levels in placenta and fetal tissue of HFD-exposed offspring compared to offspring born to control diet (CD)-fed mice, suggesting that the adverse effects of maternal HFD on neuronal function are partly mediated by increased endotoxin and TLR4 activation (56). This evidence supports the hypothesis that the inflammatory environment induced by maternal obesity may contribute to the higher incidence of neuropsychiatric disorders, such as ADHD, in male offspring (57).
In mice, the dorsal hippocampus of juvenile offspring from HFD-fed mothers displayed altered microglial morphology, while overall microglial count remained unchanged (36, 58). In non-human primates, juvenile offspring of HFD-fed mothers displayed increased microglial numbers in the basolateral amygdala (37). While the functional consequences of increased microglial density remain to be elucidated, these findings suggest that maternal obesity can significantly disrupt microglia homeostasis in offspring, potentially affecting neurodevelopment.
While the consequences of maternal obesity on neurodevelopment have been well-characterized, emerging evidence suggests that other maternal dietary preferences may also have a profound effect on offspring brain development. A recent preprint highlights how maternal high-fructose diet led to decreased microglial density, decreased synaptic pruning, and deficient efferocytosis, which was reflected by an increase in uncleared apoptotic cells. Consequently, changes in microglia core functions lead to cognitive defects and anxiety-like behavior in the offspring. This was mediated via the high affinity fructose transporter SLC2A5 (GLUT5), as its deletion in neonates completely reversed microglia dysfunction and cognitive defects (38). Similarly, a recent study evidenced that maternal high-salt diet could affect synaptic plasticity and memory in the offspring, however it is unclear if these effects were mediated by microglia (59). Taken together, microglia clearly harbor the capacity to memorize cues from the maternal diet during gestation, which can then have profound consequences on the offspring neurodevelopment.
Kupffer cells also represent an interesting candidate for developmental programming by maternal diet. Indeed, Kupffer cells are predominantly fetal-derived and are only replaced very slowly by monocyte-derived macrophages during steady-state conditions (6). In line, in a non-human primate model it has been shown that a maternal WD leads to increased liver macrophage pro-inflammatory cytokine expression (Il-1b, TNFa, Il-6) in the offspring (39). It is intriguing to speculate that this developmental programming may contribute to the increased prevalence of fatty liver disease observed in the offspring of overweight mice, and potentially, humans (60–62).
Finally, while alterations in the long-lived resident macrophage populations can lead to developmental changes during tissue development, maternal obesity also impacts the progenitors of circulating monocytes in the bone marrow, which increasingly colonize tissue-resident macrophage niches as the organism ages. These changes have been observed to occur already in progenitors present in the fetal liver, which can lead to permanent changes in the bone marrow that persist throughout life. In mice, maternal WD leads to remodeling of fetal HSCs, skewing toward myeloid and B cell differentiation over HSC self-renewal (63). Similarly, in non-human primates, maternal WD induces transcriptional reprogramming of HSCs in the fetal liver, with upregulation of pathways involved in TNF signaling, oxidative phosphorylation and antigen processing and presenting (64). Of note, these transcriptional changes in HSCs from maternal obese offspring were epigenetically imprinted (40). In line with these findings, bone marrow-derived macrophages (BMDMs) differentiated from adult offspring of WD-fed non-human primates presented heightened pro-inflammatory gene expression (Il-1b, TNF) following LPS stimulation. This was accompanied by increased glycolysis, reduced oxidative phosphorylation, and increased phagocytosis of fluorescent bioparticles (40). These changes can impact tissue homeostasis as this cellular reprogramming may also occur in vivo in MdMs that increasingly colonize tissues. However, the biological consequences of such reprogramming remain unclear and may affect specific organs differently. An organ of particular interest regarding the functional consequences of developmental HSC programming is the intestine, where macrophages undergo high turnover by MdMs. Intriguingly, in a non-human primate model of maternal WD, offspring macrophages displayed a reduced production of TNFa and CCL20 and decreased capacity to respond to bacterial stimulation (41). Similarly, splenic macrophages, which also display turnover by MdM, also displayed reduced secretion of TNFa and IFNa in offspring from maternal obese mothers (41).
Taken together, maternal diet can have a significant impact on offspring immunity. The functional consequences of such developmental programming differentially impact fetal-derived macrophages and MdM, which can thus impact tissue homeostasis on different timescales and contexts. Further studies are warranted to fully elucidate the mechanisms involved.
4.2 Microbiota
Microbiota is a collective term to refer to the consortia of microorganisms that reside within an individual. The microbiota profits from the host’s environment, and in return, provides the host with advantages, such as protection against pathogens, digestion of otherwise undigestible dietary components and the production of dietary metabolites (65). Additionally, the microbiota is known to modulate the host’s immune system. Remarkably, commensal-derived metabolites contribute to fetal development and can directly shape the immune system of both mother and offspring. It is well-known that, beyond the maternal gut microbiota in prenatal stages, exposure to the vaginal and skin microbiota during birth or lactation is crucial for the correct priming of the immune system. In human babies born from vaginal delivery (VD), the infant’s microbiome is rich in Bifidobacteria, Lactobacillus, and Bacteriodotes and reduced in Enterococci and Klebsiella, in comparison to caesarean section-born infants. Microbiome in mouth, meconium and nose of C-Section (CS) babies resembled the mother’s skin, while VD babies harbored commensals that resembled more the mother’s vaginal microbiota (66). Interestingly, CS infants need about one year to achieve the microbiota baseline of their VD counterparts (67, 68). CS has long-term impacts on health since it has been linked to immune-system dysregulation and immaturity and higher risk of non-communicative diseases like type-1-diabetes, allergies, and obesity (69–72). Despite extensive descriptions of the impact of vaginal microbiota exposure and long-term health of offspring, the concrete influence on macrophages remains unknown.
Typical environments for studying the influence of microbiota in animal models include the use of germ-free (GF) mice, which are devoid of microorganisms. When mice are inoculated with specific, known bacterial strains they are termed gnotobiotic. The gnotobiotic setup allows researchers to study the roles of individual bacterial strains (monocolonization) or defined consortia of microorganisms (73). Specific-pathogen-free (SPF) mice, which have more heterogeneous microbiota, can vary between facilities and among individuals or communities within them (e.g. cages). Microbiota manipulation can also involve the usage of antibiotics or cohousing with animals harboring different commensals, such as pet shop mice, wild mice or wildlings (74). Taken together, these tools can be leveraged to assess the impact of the microbiota on individual tissues or specific cell types. One limitation to be noted in the context of early-life exposure is that GF mice develop and are born under GF condition, which makes it difficult to elucidate if observations result from prenatal or postnatal exposures, or from both. It should also be taken into consideration that these mice are often maintained throughout generations in GF facilities, so it is unclear whether observed phenotypes relate to gestation, or to the cumulative effect of several generations in the absence of microbiota.
In the brain, studies comparing GF and SPF embryos revealed an impaired microglial density at E14.5, E16.5 and postnatally in both the somatosensory cortex and the striatum. These observations were sex- and age-specific; males exhibited higher microglial density during embryonic stages while females showed higher density postnatally (42). Further analysis using ATAC-seq on GF and SPF embryos and adults revealed a different chromatin accessibility. Overall, SPF embryos displayed increased accessibility in regions related to core microglial functions and LPS responses (42, 75). The differentially accessible regions were mainly related to microglial functions and LPS responses.
A study published by Luck et al. focusing on post-natal mice during the first to third week of life identified a crucial link between early-life commensals and microglia in neurodevelopmental processes (43). The study involved mice born under GF conditions that were subsequently either maintained as GF or exposed to colonization with either a Bifidobacterium species or a conventional microbiota. The absence of a microbiome resulted in impaired synaptic engulfment of cerebellar Purkinje cells by microglia, a process crucial for proper neuronal maturation and function. This caused an impaired firing rate of the Purkinje cells, as determined by electrophysiology. Additionally, microglia from GF conditions were fewer in number and displayed lower expression levels of CD68 and CD36 in the postnatal stages. CD68 is a glycoprotein associated with phagocytosis, and CD36, although primarily known as a fatty acid transporter, plays a role in the phagocytosis of apoptotic cells and modulation of inflammatory responses (76). Altogether, these observations indicate that exposure to microbiota in this early period of development significantly influences microglial function.
The microbiome can synthesize essential vitamins de novo and produce short-chain fatty acids (SCFA) through fermentation of dietary fibers (77, 78). Among SCFA, acetate is defined as the most important in modulating the maturation, morphology, and function of microglia (79, 80). Mice deficient for SCFA receptors displayed microglia phenotypes of GF mice (80). Furthermore, administration of these metabolites in GF mice restored microglia density, maturity, and morphology (80). Studies show that maternal acetate and other SCFA can cross the placenta and influence the acetate levels of the offspring but it is not clear through which mechanisms they can influence developing microglia or other tissue-resident macrophages (81, 82). A leading hypothesis suggests that, as microglia fail to express Gpr43 and Gpr410 (G protein-coupled receptors sensitive to SCFAs), cytokine signals from peripheral cells harboring those GPCRs influence the epigenetic regulation of microglia (83). Interestingly, a recently published article showed the influence of the SCFA butyrate in the process of efferocytosis (84). In the study, mice raised in GF conditions or treated with antibiotics exhibit a decreased clearance of apoptotic cells compared to SPF mice. Although this study was performed in adult peritoneal macrophages, and the phenotype of antibiotic-treated animals was rescued by fecal transplants, it remains crucial to elucidate the consequences of impaired efferocytosis or other core macrophage functions specifically during development.
Taken together, it is clear that the microbiota can influence immunity in the offspring. However, it should be noted that diet can profoundly influence the microbiota, suggesting that these two developmental triggers are interlinked. Indeed, WD has been shown to reduce microbial complexity, thereby reducing production of SCFAs including acetate (85). Furthermore, while microglia serve as perfect candidates to study the microbiota’s effects on long-term developmental programming, studies have also highlighted changes occurring in other tissue-resident macrophage subsets, including the gut, the spleen and the liver (86–88). However, most of these changes are normalized following commensal recolonization, suggesting they reflect a direct response to the presence or absence of commensals, rather than developmental programming of macrophages themselves. These findings further highlight the resilience and plasticity of the niche, which is able to restore macrophage function upon cessation of the perturbation. Further studies should focus on teasing apart these mechanisms.
4.3 Maternal immune activation
Maternal immune activation (MIA), an inflammatory event occurring during gestation, has been linked to neurodevelopmental disorders and increased prevalence of psychiatric disorders later in life (reviewed in (89)). As microglia develop exclusively from YS progenitors early in development, and persist throughout life via self-maintenance, these cells are of particular interest in the context of developmental programming, in particular due to their role in supporting neuronal development and connectivity early in life (reviewed elsewhere recently (90)). It is now widely accepted that MIA leads to microglial activation, transcriptional changes, and functional consequences in the offspring, however, the lack of standardization across studies hampers study interpretation. In particular, gestational timing of MIA induction, the gender of the offspring and the severity of the inflammation have been shown to determine study outcomes (reviewed in (91)). In addition, the method of MIA induction also varies across studies; most commonly used are LPS, to mimic a bacterial infection, and PolyI:C a synthetic analog of double-stranded RNA (dsRNA), which mimics a viral infection. However, several authors have directly employed administration of pro-inflammatory cytokines during gestation to mimic the consequences of MIA. Finally, bacterial or viral infections can also be employed to induce MIA. With regards to the latter, evidence is accumulating that MIA may also impact other tissue-resident macrophage populations in the body, as detailed below.
4.3.1 PolyI:C
MIA has been shown to induce changes in microglial gene expression, function and motility in the offspring. Indeed, MIA induced at E12 or E15 results in increased microglial process tip speed in the offspring at E18, as well as in response to LPS challenge in adulthood (44). Process motility is a physiological function that allows microglia to constantly sample the environment and respond to potential stimuli, therefore these changes may reflect a reduced capacity of microglia to respond to neuronal needs and potential damage. These changes were accompanied by upregulation of pro-inflammatory cytokines, in particular IL-6, in fetal microglia following MIA. With regards to microglial motility and behavioral outcomes, MIA at E12 induces deficits that persist into adulthood, while MIA at later gestational stages results in transient alterations, most of which resolve by postnatal day 10 (44). However, MIA at E15 can still induce transcriptional changes in hippocampal microglia, which persist into adulthood, in particular upregulation of genes involved in inflammatory response, cell migration, and phagocytosis. In line, adult microglia from E15 MIA offspring display reduced phagocytic capacity (45). Of interest, the authors demonstrated that the transcriptional changes, which were accompanied by behavioral deficits, could be partially restored by postnatal treatment with minocycline, suggesting that developmental programming of microglia may be reversible and could be an interesting therapeutic target in psychiatric disorders (45).
Of particular importance in the effects of MIA on microglia is IL-6, as IL-6 knockout mice or the administration of an IL-6 neutralizing antibody show ameliorated microglial outcome following MIA (92). However, IL-6 blockade does not rescue behavioral defects in adult MIA offspring, suggesting that other mechanisms may also be involved (93). In line, Yu et al. recently demonstrated that MIA induces downregulation of Gpr56 in microglia, and that microglia-specific Gpr56 abrogation phenocopied the neurodevelopmental defects and autism-like behaviors observed in MIA offspring. Conversely, microglia-specific upregulation of Gpr56 was able to ameliorate behavioral deficits in MIA offspring (94). The critical role of microglia in mediating the neurodevelopmental effects of MIA in the offspring is further highlighted by a study by Ikezu et al., that demonstrated that behavioral deficits and synaptic abnormalities were abrogated by depleting microglia postnatally in the offspring (95).
The consequences of MIA on microglia have been well-documented, however whether other tissue-resident macrophage populations are affected is hitherto unknown. Of interest, MIA can also alter the immune response of bone-marrow derived macrophages in the offspring. In a murine model of maternal polyI:C injection, BMDMs obtained from PolyI:C exposed offspring had altered cytokine secretion at baseline and following LPS stimulation (46). Further studies are warranted to fully elucidate the consequences of MIA on peripheral tissue-resident macrophages and HSCs of the offspring.
4.3.2 Influenza A
Influenza A virus (IAV) infection during pregnancy has been shown to increase the risks of preterm birth, low birth weight, and even infant mortality, suggesting that gestational viral infections can have profound consequences on the offspring (96). However, in humans, it is not clear if these adverse outcomes are a direct result of altered offspring immunity. In mice, offspring born to mothers infected with IAV during gestation were more susceptible to heterologous infections compared to offspring from control or polyI:C-treated mothers. These effects were partly mediated by the imprinting of alveolar macrophages; transfer of alveolar macrophages from offspring of control mothers improved disease recovery and clearance of viral titers, despite an increased frequency of alveolar macrophage in maternal IAV offspring. Mechanistically, IAV infection during gestation altered hematopoiesis in the bone marrow of the offspring, leading to increased numbers of HSCs, c-Kit+ progenitors, and common myeloid progenitors (CMPs) in maternal IAV offspring compared to control or polyI:C-treated offspring (97). These alterations in hematopoiesis might also explain the increase in myeloid cells in the caecal patch, a large lymphoid aggregate in the intestine, in offspring from IAV-infected mothers compared to controls (47). However, the biological relevance of the increased myeloid cells in the cecal patch remains unknown. Finally, it has been shown that maternal IAV infection can lead to increased numbers of border-associated macrophages (BAMs) in the pia mater of the offspring, while microglial numbers remained unchanged (48). While the functional consequences of this expansion are unknown, it is intriguing to speculate that these cells, which lie at the interface between brain and periphery, may expand to protect the developing brain from peripheral danger (98).
4.4 Prenatal stress
The term ‘prenatal stress’ refers to psychological or physiological stressors experienced during pregnancy that can affect the developing fetus. In response to such challenges, the body activates its stress response systems, which help adjust to demands by mobilizing energy, modulating the immune system, and promoting behavior and cognition to improve survival (99–101). The stress response systems include the sympatho-adrenomedullary and the hypothalamic pituitary adrenal gland (HPA) axis (102, 103). While the sympatho-adrenomedullary system is responsible for rapid, acute responses, we will focus on the HPA axis, which is decisive for understanding the effects of chronic stress.
One of the most common prenatal stressors is related to disordered maternal nutrition, either through food restriction or excess, which was described in more detail in the diet chapter (102, 104–106). Other known stressors include the disruption of the maternal circadian rhythm and the usage of artificial glucocorticoids (107–109). Additionally, traumatic life experiences that are related to depression and anxiety can also impact the offspring (110). Population studies have reported that infants exposed to maternal psychological stress during pregnancy are at higher risk for infection-related hospitalizations, with this risk being higher in boys (110, 111).
Stress arises from the inability to adapt to stressors, leading to chronic activation of the HPA axis and prolonged exposure to glucocorticoids, which can be detrimental to offspring (101, 112). Glucocorticoids (corticosterone in rodents, fish, and amphibians, and cortisol in humans and guinea pigs) are essential hormones for the stress response and various physiological processes (113). They play a crucial role in the development of prenatal and postnatal organ maturity (114) and are instrumental in the maturation of the cardiovascular and immune systems (103, 115, 116). As birth approaches, glucocorticoid levels gradually increase to support the metabolic demands of the mother (117). However, imbalanced and prolonged glucocorticoid exposure can adversely affect offspring prenatally by altering cardiovascular functions and the immune system, thereby increasing the risk of metabolic and psychiatric diseases (102, 112, 118, 119).
The placenta and the fetus are equipped with mechanisms that protect from maternal glucocorticoids, primarily through the expression of the enzyme 11ß-hydroxysteroid dehydrogenase type 2 (11ß-HSD2), which breaks down active glucocorticoids (120). In humans, this enzyme is expressed from the 5th week of pregnancy in the placenta and it is effective in neutralizing most maternal glucocorticoids (121). Nevertheless, this mechanism cannot cope with prolonged, high levels of the hormone, or with synthetic glucocorticoids, which could be metabolized less efficiently than endogenous glucocorticoids.
Prenatal stress in mice is modeled using various methods. Different stressors applied to pregnant dams include light stress, sleep deprivation, restraint, exposure to foot shock, cold stress, and the forced swimming test (49–51, 122). These different stress paradigms share the common feature of inducing measurable maternal hormonal changes that can be transferred to the fetus. As discussed in previous sections, microglia are ideal candidates for developmental programming due to their long lifespan. Consequently, most studies have focused on these cells and the impact of their reprogramming on neurodevelopment. Microglia can sense stress through receptors sensitive to glucocorticoids, such as NR3C1 and NR2C2, whose transcripts have been detected in embryonal stages (123, 124). One proposed mechanism, described in postnatal microglia, indicates that these cells exhibit pro-inflammatory responses upon exposure to glucocorticoids (124, 125). Although glucocorticoids are typically described as immunosuppressive, they can also exert pro-inflammatory effects through the so-called “permissive effect” when administered in basal doses (125, 126).
In a model of prenatal stress using a forced swim test, pregnant rats subjected to forced swimming exhibited a reduction of immature microglia and accelerated differentiation in the newborn brains of their offspring, as indicated by an increased number of ramified microglia. This effect normalized after 10 days of life (50). Similarly, prenatal stress induced by sleep deprivation and other stressors has been shown to alter microglial density and induce region-specific morphological changes in microglia (51, 127–129). In the hippocampus, maternal sleep deprivation resulted in decreased and shorter microglial processes, along with reduced neurogenesis. Interestingly, the administration of pioglitazone in adolescent mice mitigated cognitive deficits by enhancing PPARγ-dependent microglia-mediated neurogenesis, suggesting that the developmental reprogramming of microglia by prenatal stress may be partially reversible (51). Furthermore, in the hypothalamus, Rosin et al. demonstrated that ventricular-residing microglia respond prenatally to stress in a model of maternal cold stress (49). Maternal cold stress affected neuronal specification and glial fate in a sex-dependent manner, impacting male but not female brains. This effect was reversed by the removal of microglia with the PLX5622 diet, highlighting the critical role of microglia as intergenerational messengers. Interestingly, a recent study in adult mice showed that a foamy-like macrophage within the adrenal gland arises from chronic and acute stress (130). This macrophage subtype can modulate glucocorticoid production by restricting steroidogenesis via Trem2 and TGF-β. Giving the long-lived nature of macrophages and their capacity to react timely to exposures, it would be intriguing to investigate whether developmental programming may impact adrenal macrophages, which thereby can directly modulate systemic stress responses.
Epigenetic changes represent another possible mechanism of action in response to prenatal stress. A meta-analysis of independent human longitudinal studies associating prenatal stress with newborn DNA methylation revealed that abuse during pregnancy was linked to methylation of CpG regions, such as those on chromosome 7 annotating the Myeloid Differentiation Primary Response 88 (MYD88) gene (51). This gene, related to immune system function, is also crucial for the development of the central nervous system. Nevertheless, further studies are required to elucidate these effects and determine whether they have a direct impact on macrophage development and function.
Finally, one study focused on osteoclasts, which are highly specialized bone-resident macrophages (52). Osteoclasts, derived from EMPs at birth, are responsible for bone resorption by dissolving bone minerals (131). This process generates the bone marrow cavity and facilitates tooth eruption (131). Fontanetti et al. induced stress in pregnant rats by exposing them to constant light during the last 10 days of gestation. This exposure resulted in impaired bone remodeling and reduced osteoclast activity in the dental area and impaired tooth eruption in the offspring born to stressed mothers (52). In conclusion, while prenatal stress significantly impacts fetal development by altering immune responses, microglial function, and bone remodeling, future studies are needed to elucidate the precise mechanisms and long-term effects of these changes, particularly regarding the role of epigenetic modifications.
4.5 Pollutants
Environmental pollutants also play a significant role in shaping the developing immune system. Multiple studies have demonstrated that a large number of foodstuffs destined for human consumption are contaminated with plastic particles smaller than 5mm, known as micro- and nanoplastics (132–136). While the impact of micro- and nanoplastic exposure on human health is just beginning to be elucidated, the effects of these contaminants on macrophage programming during development remain unclear. Of note, recent studies found microplastic contamination in all human placental samples analyzed, and in 75% of all breast milk samples, suggesting that these contaminants may pose a hitherto unknown risk to offspring health and immunity (53, 137). Although rodent studies of gestational microplastic exposure are limited due to significant differences in placental structure between humans and rodents, they may offer insights into how microplastics affect offspring immunity and development indirectly by causing immune activation in the placenta. For instance, a recent study in rats showed increased microglial density in the offspring of microplastic-exposed mothers compared to controls, accompanied by behavioral changes (138). Given the size-dependent capacity of plastic particles to pass the human placental barrier, it is imperative that future studies clarify the impact of micro- and nanoplastics on offspring immunity, especially in models that more accurately mimic the human fetal-placental interface (139).
Similarly, murine models of gestational air pollution exposure have shown region- and sex-specific alterations in microglial density (54, 140). A recent study combining diesel exhaust particle exposure and maternal stress demonstrated profound behavioral changes in male offspring only, accompanied by impaired microglial pruning during postnatal development in the anterior cingulate cortex (141). These findings suggest that individual environmental triggers can have a cumulative effect on offspring macrophage function and overall immune development.
5 Concluding remarks
Macrophages originating from progenitor cells that arise from the yolk sac, colonize the tissues during development and persist through self-maintenance. Both local and developmental cues shape their identity, longevity, and function. Macrophages play an important role in homeostasis, contributing to the active regulation of their environment, but also in allostasis, by preparing the individual for forthcoming challenges later in life. For this reason, developmental programming could be understood as either beneficial or detrimental depending on the stimuli, their intensity, and the time window in which they appear. Additionally, individual factors including offspring gender, duration of the stimuli, and nature of the stimuli significantly impact outcomes.
While substantial evidence indicates that early development is a critical moment for programming of cellular responses, more research is needed to identify developmentally sensitive periods. Furthermore, more sex-inclusive studies are essential to uncover underlying mechanisms, as sex plays a significant role in determining immunological responses, macrophage function, and macrophage replenishment by recruited monocytes (142, 143). The microbiota, which can vary meaningly across mouse facilities, and certainly among human individuals, also plays a crucial role in shaping the immune system. It is important to recognize that the microbiota of laboratory mice does not reflect the natural microbiota of wild mice. Using lab-strain embryos transferred into wild mice or co-housing laboratory mice with wild mice (wildlings) will provide more accurate models for natural immune phenotypes (74). Upcoming studies on wildlings will enhance our understanding of macrophage development in natural environments.
Finally, recent evidence indicates that also environmental pollutants, such as micro- and nano-plastics, or air pollutants, during gestation may also impact offspring macrophage function. The impact of such exposures may be massive, and may disproportionately affect specific global regions more than others, highlighting the need to rapidly increase our understanding of the impact of these pollutants on the future generations, in order to accurately inform policies and global guidelines.
Understanding the origin of macrophages and the impact of maternal-derived developmental triggers is crucial for assessing the consequences and potential interventions to mitigate adverse effects. Future research using fate-mapper models will clarify macrophage origin and determine further whether certain stressors or their timing influence macrophage longevity and monocyte replacement in certain tissues. Consequently, macrophages can be considered intergenerational messengers, storing information received early in life, including during gestation, and thereby influencing health in adulthood and beyond. This perspective underscores the importance of safeguarding the period before and during pregnancy to create a favorable environment for offspring development.
In conclusion, while the evidence strongly supports the role of developmental programming in influencing macrophage function, we recognize that the local tissue environment and niche programming may also play a part. Although direct studies investigating the combined influence of developmental and niche programming are still lacking, we are confident that further research in this area will provide greater clarity. This review underscores the need to explore how both developmental cues and tissue-specific factors shape macrophage identity over time.
Author contributions
MFV: Writing – original draft, Writing – review & editing. EFT: Writing – original draft, Writing – review & editing. EM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work of this review was supported by the Jürgen Manchot PhD-Fellowship (to EFT), and the EMBO Postdoctoral fellowship 873-2023 (to MFV), by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy-EXC2151-390873048 (to EM), SFB 1454 - Project ID 432325352 – (to EM), FOR5547 – Project-ID 503306912 (to EM). EM is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 851257). This work was supported by the Open Access Publication Fund of the University of Bonn.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. (2023) 9:563–79. doi: 10.1038/s41577-023-00848-y
2. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
3. Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353. doi: 10.1126/science.aaf4238
4. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis [Immunity 38 (2013) 79-91. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2013.05.008
5. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
6. Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/SCIENCE.1219179
7. Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. (2010) 464:112–5. doi: 10.1038/NATURE08761
8. Boisset JC, Van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. (2010) 464:116–20. doi: 10.1038/NATURE08764
9. Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. (2010) 464:108–11. doi: 10.1038/NATURE08738
10. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
11. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell. (2019) 178:1509–25.e19. doi: 10.1016/j.cell.2019.08.009
12. Lazarov T, Juarez-Carreño S, Cox N, Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature. (2023) 618:698–707. doi: 10.1038/s41586-023-06002-x
13. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
14. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. (2012) 13:753–60. doi: 10.1038/ni.2360
15. Cohen M, Giladi A, Gorki AD, Solodkin DG, Zada M, Hladik A, et al. Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell. (2018) 175:1031–44.e18. doi: 10.1016/J.CELL.2018.09.009
16. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the Kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity. (2019) 51:638–54.e9. doi: 10.1016/j.immuni.2019.08.017
17. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. (2014) 5:3. doi: 10.3389/FPHYS.2014.00003
18. Palis J. Interaction of the macrophage and primitive erythroid lineages in the mammalian embryo. Front Immunol. (2017) 7:669. doi: 10.3389/FIMMU.2016.00669
19. Kayvanjoo AH, Splichalova I, Bejarano DA, Huang H, Mauel K, Makdissi N, et al. Fetal liver macrophages contribute to the hematopoietic stem cell niche by controlling granulopoiesis. Elife. (2024) 13:e86493. doi: 10.7554/eLife.86493
20. Lewis K, Yoshimoto M, Takebe T. Fetal liver hematopoiesis: from development to delivery. Stem Cell Res Ther. (2021) 12:139. doi: 10.1186/S13287-021-02189-W
21. Yang L, Wang WH, Qiu WL, Guo Z, Bi E, Xu CR. A single-cell transcriptomic analysis reveals precise pathways and regulatory mechanisms underlying hepatoblast differentiation. Hepatology. (2017) 66:1387. doi: 10.1002/HEP.29353
22. Liang Y, Kaneko K, Xin B, Lee J, Sun X, Zhang K, et al. Temporal analyses of postnatal liver development and maturation by single-cell transcriptomics. Dev Cell. (2022) 57:398–414.e5. doi: 10.1016/J.DEVCEL.2022.01.004
23. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
24. Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall GP, et al. Microglia instruct subventricular zone neurogenesis. Glia. (2006) 54:815–25. doi: 10.1002/GLIA.20419
25. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
26. Baydemir I, Dulfer EA, Netea MG, Domínguez-Andrés J. Trained immunity-inducing vaccines: Harnessing innate memory for vaccine design and delivery. Clin Immunol. (2024) 261:109930. doi: 10.1016/J.CLIM.2024.109930
27. Guilliams M, Thierry GR, Bonnardel J, Bajenoff M. Establishment and maintenance of the macrophage niche. Immunity. (2020) 52:434–51. doi: 10.1016/J.IMMUNI.2020.02.015
28. Orwa SA, Gudnadottir U, Boven A, Pauwels I, Versporten A, Vlieghe E, et al. Global prevalence of antibiotic consumption during pregnancy: A systematic review and meta-analysis. J Infect. (2024) 89:106189. doi: 10.1016/J.JINF.2024.106189
29. Sheikh J, Lawson H, Allotey J, Yap M, Balaji R, Kew T, et al. Global variations in the burden of SARS-CoV-2 infection and its outcomes in pregnant women by geographical region and country’s income status: a meta-analysis. BMJ Glob Health. (2022) 7:e010060. doi: 10.1136/BMJGH-2022-010060
30. Velu PP, Gravett CA, Roberts TK, Wagner TA, Zhang JSF, Rubens CE, et al. Epidemiology and aetiology of maternal bacterial and viral infections in low- and middle-income countries. J Glob Health. (2011) 1:171.
31. Boutari C, Mantzoros CS. A 2022 update on the epidemiology of obesity and a call to action: as its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism. (2022) 133. doi: 10.1016/J.METABOL.2022.155217
32. Kleeman EA, Gubert C, Hannan AJ. Transgenerational epigenetic impacts of parental infection on offspring health and disease susceptibility. Trends Genet. (2022) 38:662. doi: 10.1016/J.TIG.2022.03.006
33. Zheng X, Li Z, Wang G, Wang H, Zhou Y, Zhao X, et al. Sperm epigenetic alterations contribute to inter- and transgenerational effects of paternal exposure to long-term psychological stress via evading offspring embryonic reprogramming. Cell Discovery. (2021) 7:101. doi: 10.1038/S41421-021-00343-5
34. Katzmarski N, Domínguez-Andrés J, Cirovic B, Renieris G, Ciarlo E, Le Roy D, et al. Transmission of trained immunity and heterologous resistance to infections across generations. Nat Immunol. (2021) 22:1382–90. doi: 10.1038/S41590-021-01052-7
35. Edlow AG, Glass RM, Smith CJ, Tran PK, James K, Bilbo S. Placental macrophages: A window into fetal microglial function in maternal obesity. Int J Dev Neurosci. (2019) 77:60–8. doi: 10.1016/J.IJDEVNEU.2018.11.004
36. Bordeleau M, Lacabanne C, Fernández De Cossío L, Vernoux N, Savage JC, González-Ibáñez F, et al. Microglial and peripheral immune priming is partially sexually dimorphic in adolescent mouse offspring exposed to maternal high-fat diet. J Neuroinflamm. (2020) 17:264. doi: 10.1186/S12974-020-01914-1
37. Dunn GA, Mitchell AJ, Selby M, Fair DA, Gustafsson HC, Sullivan EL. Maternal diet and obesity shape offspring central and peripheral inflammatory outcomes in juvenile non-human primates. Brain Behav Immun. (2022) 102:224–36. doi: 10.1016/J.BBI.2022.02.024
38. Wang Z, Lipshutz A, Liu Z-L, Trzeciak AJ, Miranda IC, Martínez de la Torre C, et al. Rojas WS, Saavedra PH V, et al. Early life high fructose exposure disrupts microglia function and impedes neurodevelopment. bioRxiv. (2023). doi: 10.1101/2023.08.14.553242
39. Thorn SR, Baquero KC, Newsom SA, El Kasmi KC, Bergman BC, Shulman GI, et al. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes. (2014) 63:2702–13. doi: 10.2337/DB14-0276
40. Nash MJ, Dobrinskikh E, Soderborg TK, Janssen RC, Takahashi DL, Dean TA, et al. Maternal diet alters long-term innate immune cell memory in fetal and juvenile hematopoietic stem and progenitor cells in nonhuman primate offspring. Cell Rep. (2023) 42:112393. doi: 10.1016/J.CELREP.2023.112393
41. Sureshchandra S, Doratt BM, Mendza N, Varlamov O, Rincon M, Marshall NE, et al. Maternal obesity blunts antimicrobial responses in fetal monocytes. Elife. (2023) 12:e81320. doi: 10.7554/eLife.81320
42. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. (2018) 172:500–16.e16. doi: 10.1016/J.CELL.2017.11.042
43. Luck B, Engevik MA, Ganesh BP, Lackey EP, Lin T, Balderas M, et al. Bifidobacteria shape host neural circuits during postnatal development by promoting synapse formation and microglial function. Sci Rep. (2020) 10:7737. doi: 10.1038/S41598-020-64173-3
44. Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep. (2020) 10:21378. doi: 10.1038/S41598-020-78294-2
45. Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. (2017) 7:e1120. doi: 10.1038/TP.2017.80
46. Onore CE, Schwartzer JJ, Careaga M, Berman RF, Ashwood P. Maternal immune activation leads to activated inflammatory macrophages in offspring. Brain Behav Immun. (2014) 38:220–6. doi: 10.1016/J.BBI.2014.02.007
47. Liong S, Miles MA, Mohsenipour M, Liong F, Hill-Yardin EL, Selemidis S. Influenza A virus infection during pregnancy causes immunological changes in gut-associated lymphoid tissues of offspring mice. Am J Physiol Gastrointest Liver Physiol. (2023) 325:G230–8. doi: 10.1152/AJPGI.00062.2023
48. Otero AM, Connolly MG, Gonzalez-Ricon RJ, Wang SS, Allen JM, Antonson AM. Influenza A virus during pregnancy disrupts maternal intestinal immunity and fetal cortical development in a dose- and time-dependent manner. Mol Psychiatry. (2023) 10.1038/s41380-024-02648-9. doi: 10.1101/2023.12.18.572222
49. Rosin JM, Sinha S, Biernaskie J, Kurrasch DM. A subpopulation of embryonic microglia respond to maternal stress and influence nearby neural progenitors. Dev Cell. (2021) 56:1326–45.e6. doi: 10.1016/J.DEVCEL.2021.03.018
50. Gómez-González B, Escobar A. Prenatal stress alters microglial development and distribution in postnatal rat brain. Acta Neuropathol. (2010) 119:303–15. doi: 10.1007/S00401-009-0590-4
51. Han Y, Wang J, Zhao Q, Xie X, Song R, Xiao Y, et al. Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav Immun. (2020) 87:568–78. doi: 10.1016/J.BBI.2020.02.002
52. Fontanetti PA, De Lucca RC, Mandalunis PM, Vermouth NT. Impairment of rat tooth eruption in pups born to mothers exposed to chronic stress during pregnancy. Arch Oral Biol. (2013) 58:1643–51. doi: 10.1016/J.ARCHORALBIO.2013.08.003
53. Ragusa A, Notarstefano V, Svelato A, Belloni A, Gioacchini G, Blondeel C, et al. Raman microspectroscopy detection and characterisation of microplastics in human breastmilk. Polymers (Basel). (2022) 14. doi: 10.3390/POLYM14132700
54. Zhang J, Yang Y, Al-Ahmady ZS, Du W, Duan J, Liao Z, et al. Maternal exposure to PM2.5 induces cognitive impairment in offspring via cerebellar neuroinflammation and oxidative stress. Ecotoxicol Environ Saf. (2023) 249:114425. doi: 10.1016/j.ecoenv.2022.114425
55. An J, Wang Q, Yi S, Liu X, Jin H, Xu J, et al. The source of the fat significantly affects the results of high-fat diet intervention. Sci Rep. (2022) 12:1–11. doi: 10.1038/s41598-022-08249-2
56. Ceasrine AM, Devlin BA, Bolton JL, Green LA, Jo YC, Huynh C, et al. Maternal diet disrupts the placenta-brain axis in a sex-specific manner. Nat Metab. (2022) 4:1732–45. doi: 10.1038/S42255-022-00693-8
57. Rodriguez A, Miettunen J, Henriksen TB, Olsen J, Obel C, Taanila A, et al. Maternal adiposity prior to pregnancy is associated with ADHD symptoms in offspring: evidence from three prospective pregnancy cohorts. Int J Obes (Lond). (2008) 32:550–7. doi: 10.1038/SJ.IJO.0803741
58. Musillo C, Creutzberg KC, Collacchi B, Ajmone-Cat MA, De Simone R, Lepre M, et al. Bdnf-Nrf-2 crosstalk and emotional behavior are disrupted in a sex-dependent fashion in adolescent mice exposed to maternal stress or maternal obesity. Transl Psychiatry. (2023) 13:399. doi: 10.1038/S41398-023-02701-1
59. Ge Q, Hu X, Ma N, Sun M, Zhang L, Cai Z, et al. Maternal high-salt diet during pregnancy impairs synaptic plasticity and memory in offspring. FASEB J. (2021) 35:e21244. doi: 10.1096/FJ.202001890R
60. Oben JA, Mouralidarane A, Samuelsson AM, Matthews PJ, Morgan ML, Mckee C, et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol. (2010) 52:913–20. doi: 10.1016/J.JHEP.2009.12.042
61. Hagström H, Simon TG, Roelstraete B, Stephansson O, Söderling J, Ludvigsson JF. Maternal obesity increases the risk and severity of NAFLD in offspring. J Hepatol. (2021) 75:1042–8. doi: 10.1016/J.JHEP.2021.06.045
62. Huang H, Splichalova I, Balzer NR, Seep L, Franco Taveras E, Radwaniak C, et al. Developmental programming of Kupffer cells by maternal obesity causes fatty liver disease in the offspring. Res Square Preprint. (2023). doi: 10.21203/rs.3.rs-3242837/v1
63. Kamimae-Lanning AN, Krasnow SM, Goloviznina NA, Zhu X, Roth-Carter QR, Levasseur PR, et al. Maternal high-fat diet and obesity compromise fetal hematopoiesis. Mol Metab. (2014) 4:25–38. doi: 10.1016/J.MOLMET.2014.11.001
64. Sureshchandra S, Chan CN, Robino JJ, Parmelee LK, Nash MJ, Wesolowski SR, et al. Maternal Western-style diet remodels the transcriptional landscape of fetal hematopoietic stem and progenitor cells in rhesus macaques. Stem Cell Rep. (2022) 17:2595–609. doi: 10.1016/J.STEMCR.2022.10.003
65. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/SCIENCE.1241165
66. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U.S.A. (2010) 107:11971–5. doi: 10.1073/PNAS.1002601107
67. Martin R, Makino H, Yavuz AC, Ben-Amor K, Roelofs M, Ishikawa E, et al. Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PloS One. (2016) 11:e0158498. doi: 10.1371/JOURNAL.PONE.0158498
68. Reyman M, van Houten MA, van Baarle D, Bosch AATM, Man WH, Chu MLJN, et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun. (2019) 10:1–12. doi: 10.1038/s41467-019-13014-7
69. Pei Z, Heinrich J, Fuertes E, Flexeder C, Hoffmann B, Lehmann I, et al. Cesarean delivery and risk of childhood obesity. J Pediatr. (2014) 164:1068–73.e2. doi: 10.1016/J.JPEDS.2013.12.044
70. Thavagnanam S, Fleming J, Bromley A, Shields MD, Cardwell CR. A meta-analysis of the association between Caesarean section and childhood asthma. Clin Exp Allergy. (2008) 38:629–33. doi: 10.1111/J.1365-2222.2007.02780.X
71. Bager P, Wohlfahrt J, Westergaard T. Caesarean delivery and risk of atopy and allergic disease: meta-analyses. Clin Exp Allergy. (2008) 38:634–42. doi: 10.1111/J.1365-2222.2008.02939.X
72. Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. (2008) 51:726–35. doi: 10.1007/S00125-008-0941-Z
73. Thomson CA, Morgan SC, Ohland C, McCoy KD. From germ-free to wild: modulating microbiome complexity to understand mucosal immunology. Mucosal Immunol. (2022) 15:1085–94. doi: 10.1038/S41385-022-00562-3
74. Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. (2019) 365. doi: 10.1126/SCIENCE.AAW4361
75. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. (2016) 353:aad8670. doi: 10.1126/science.aad8670
76. Ioghen O, Chițoiu L, Gherghiceanu M, Ceafalan LC, Hinescu ME. CD36 – A novel molecular target in the neurovascular unit. Eur J Neurosci. (2021) 53:2500–10. doi: 10.1111/ejn.15147
77. Magnúsdóttir S, Ravcheev D, De Crécy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. (2015) 6:148. doi: 10.3389/FGENE.2015.00148
78. LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. (2013) 24:160–8. doi: 10.1016/J.COPBIO.2012.08.005
79. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. (2014) 6:263ra158. doi: 10.1126/SCITRANSLMED.3009759
80. Erny D, Hrabě de Angelis AL, Prinz M. Communicating systems in the body: how microbiota and microglia cooperate. Immunology. (2017) 150:7–15. doi: 10.1111/IMM.12645
81. Hu M, Eviston D, Hsu P, Mariño E, Chidgey A, Santner-Nanan B, et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat Commun. (2019) 10:3031. doi: 10.1038/S41467-019-10703-1
82. Pronovost GN, Yu KB, Coley-O’Rourke EJL, Telang SS, Chen AS, Vuong HE, et al. The maternal microbiome promotes placental development in mice. Sci Adv. (2024) 9:eadk1887. doi: 10.1126/sciadv.adk1887
83. Zhou Y, Xie L, Schröder J, Schuster IS, Nakai M, Sun G, et al. Dietary fiber and microbiota metabolite receptors enhance cognition and alleviate disease in the 5xFAD mouse model of Alzheimer’s disease. J Neurosci. (2023) 43:6460–75. doi: 10.1523/JNEUROSCI.0724-23.2023
84. Saavedra PHV, Trzeciak A, Wang Z, Saitz Rojas W, Zago G, Docampo MD, et al. A microbiota-derived metabolite instructs peripheral efferocytosis. bioRxiv. (2022). doi: 10.1101/2022.08.17.504322
85. Agus A, Denizot J, Thévenot J, Martinez-Medina M, Massier S, Sauvanet P, et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep. (2016) 6:19032. doi: 10.1038/SREP19032
86. McDonald B, Zucoloto AZ, Yu IL, Burkhard R, Brown K, Geuking MB, et al. Programing of an Intravascular Immune Firewall by the Gut Microbiota Protects against Pathogen Dissemination during Infection. Cell Host Microbe. (2020) 28:660–8.e4. doi: 10.1016/J.CHOM.2020.07.014
87. Schmidt F, Dahlke K, Batra A, Keye J, Wu H, Friedrich M, et al. Microbial colonization in adulthood shapes the intestinal macrophage compartment. J Crohns Colitis. (2019) 13:1173–85. doi: 10.1093/ECCO-JCC/JJZ036
88. Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. (2014) 15:374–81. doi: 10.1016/J.CHOM.2014.02.006
89. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. (2016) 353:772. doi: 10.1126/SCIENCE.AAG3194
90. Thion MS, Garel S. Microglial ontogeny, diversity and neurodevelopmental functions. Curr Opin Genet Dev. (2020) 65:186–94. doi: 10.1016/J.GDE.2020.06.013
91. Woods RM, Lorusso JM, Potter HG, Neill JC, Glazier JD, Hager R. Maternal immune activation in rodent models: A systematic review of neurodevelopmental changes in gene expression and epigenetic modulation in the offspring brain. Neurosci Biobehav Rev. (2021) 129:389–421. doi: 10.1016/J.NEUBIOREV.2021.07.015
92. Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
93. Gumusoglu SB, Fine RS, Murray SJ, Bittle JL, Stevens HE. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav Immun. (2017) 65:274–83. doi: 10.1016/J.BBI.2017.05.015
94. Yu D, Li T, Delpech JC, Zhu B, Kishore P, Koshi T, et al. Microglial GPR56 is the molecular target of maternal immune activation-induced parvalbumin-positive interneuron deficits. Sci Adv. (2022) 8. doi: 10.1126/SCIADV.ABM2545
95. Ikezu S, Yeh H, Delpech JC, Woodbury ME, Van Enoo AA, Ruan Z, et al. Inhibition of colony stimulating factor 1 receptor corrects maternal inflammation-induced microglial and synaptic dysfunction and behavioral abnormalities. Mol Psychiatry. (2021) 26:1808–31. doi: 10.1038/S41380-020-0671-2
96. Newsome K, Alverson CJ, Williams J, McIntyre AF, Fine AD, Wasserman C, et al. Outcomes of infants born to women with influenza A(H1N1)pdm09. Birth Defects Res. (2019) 111:88–95. doi: 10.1002/BDR2.1445
97. Jacobsen H, Walendy-Gnirß K, Tekin-Bubenheim N, Kouassi NM, Ben-Batalla I, Berenbrok N, et al. Offspring born to influenza A virus infected pregnant mice have increased susceptibility to viral and bacterial infections in early life. Nat Commun. (2021) 12:4957. doi: 10.1038/S41467-021-25220-3
98. Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. (2020) 181:557–73.e18. doi: 10.1016/J.CELL.2020.03.021
99. Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. (2015) 22:20–32. doi: 10.1159/000362724
100. Exton JH. Regulation of gluconeogenesis by glucocorticoids. Monogr Endocrinol. (1979) 12:535–46. doi: 10.1007/978-3-642-81265-1_28
101. Chrousos GP, Gold PW. The concepts of stress and stress system disorders: overview of physical and behavioral homeostasis. JAMA. (1992) 267:1244–52. doi: 10.1001/jama.1992.03480090092034
102. Facchi JC, de Lima TAL, de Oliveira LR, Costermani H de O, Miranda GDS, de Oliveira JC. Perinatal programming of metabolic diseases: The role of glucocorticoids. Metabolism. (2020) 104:154047. doi: 10.1016/J.METABOL.2019.154047
103. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: Outcomes. Nat Rev Endocrinol. (2014) 10:391–402. doi: 10.1038/NRENDO.2014.73
104. Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R528–38. doi: 10.1152/AJPREGU.00316.2007
105. Armitage J, Poston L, Taylor P. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res. (2008) 36:73–84. doi: 10.1159/000115355
106. Smart JL, Dobbing J. Vulnerability of developing brain. II. Effects of early nutritional deprivation on reflex ontogeny and development of behaviour in the rat. Brain Res. (1971) 28:85–95. doi: 10.1016/0006-8993(71)90526-9
107. Arias A, Schander JA, Bariani MV, Correa F, Domínguez Rubio AP, Cella M, et al. Dexamethasone-induced intrauterine growth restriction modulates expression of placental vascular growth factors and fetal and placental growth. Mol Hum Reprod. (2021) 27. doi: 10.1093/MOLEHR/GAAB006
108. Cui Z, Xu H, Wu F, Chen J, Zhu L, Shen Z, et al. Maternal circadian rhythm disruption affects neonatal inflammation via metabolic reprograming of myeloid cells. Nat Metab. (2024) 6:899–913. doi: 10.1038/S42255-024-01021-Y
109. Xu T, Zhao M, Li H, Zhou X, Liu B, Sun M, et al. Antenatal Dexamethasone Exposure Impairs the High-Conductance Ca2+-Activated K+ Channels via Epigenetic Alteration at Gene Promoter in Male Offspring. Arterioscler Thromb Vasc Biol. (2020) 40:E284–95. doi: 10.1161/ATVBAHA.120.314905
110. Robinson M, Carter KW, Pennell CE, Jacoby P, Moore HC, Zubrick SR, et al. Maternal prenatal stress exposure and sex-specific risk of severe infection in offspring. PloS One. (2021) 16. doi: 10.1371/JOURNAL.PONE.0245747
111. Nielsen NM, Hansen AV, Simonsen J, Hviid A. Prenatal stress and risk of infectious diseases in offspring. Am J Epidemiol. (2011) 173:990–7. doi: 10.1093/AJE/KWQ492
112. Costa TJ, De Oliveira JC, Giachini FR, Lima VV, Tostes RC, Bomfim GF. Programming of vascular dysfunction by maternal stress: immune system implications. Front Physiol. (2022) 13:787617. doi: 10.3389/FPHYS.2022.787617
113. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol. (2019) 10:1545. doi: 10.3389/FIMMU.2019.01545
114. Wood CE, Walker CD. Fetal and neonatal HPA axis. Compr Physiol. (2015) 6:33–62. doi: 10.1002/CPHY.C150005
115. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: Mechanisms. Nat Rev Endocrinol. (2014) 10:403–11. doi: 10.1038/NRENDO.2014.74
116. Grier DG, Halliday HL. Effects of glucocorticoids on fetal and neonatal lung development. Treat Respir Med. (2004) 3:295–306. doi: 10.2165/00151829-200403050-00004
117. Jung C, Ho JT, Torpy DJ, Rogers A, Doogue M, Lewis JG, et al. A longitudinal study of plasma and urinary cortisol in pregnancy and postpartum. J Clin Endocrinol Metab. (2011) 96:1533–40. doi: 10.1210/JC.2010-2395
118. Rog-Zielinska EA, Richardson RV, Denvir MA, Chapman KE. Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J Mol Endocrinol. (2014) 52:R125–35. doi: 10.1530/JME-13-0204
119. Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab. (2007) 3:479–88. doi: 10.1038/NCPENDMET0515
120. Solano ME, Holmes MC, Mittelstadt PR, Chapman KE, Tolosa E. Antenatal endogenous and exogenous glucocorticoids and their impact on immune ontogeny and long-term immunity. Semin Immunopathol. (2016) 38:739–63. doi: 10.1007/s00281-016-0575-z
121. Alfaidy N, Gupta S, DeMarco C, Caniggia I, Challis JRG. Oxygen regulation of placental 11 beta-hydroxysteroid dehydrogenase 2: physiological and pathological implications. J Clin Endocrinol Metab. (2002) 87:4797–805. doi: 10.1210/JC.2002-020310
122. Mendez N, Abarzua-Catalan L, Vilches N, Galdames HA, Spichiger C, Richter HG, et al. Timed maternal melatonin treatment reverses circadian disruption of the fetal adrenal clock imposed by exposure to constant light. PloS One. (2012) 7:e42713–. doi: 10.1371/journal.pone.0042713
123. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U.S.A. (2016) 113:E1738–46. doi: 10.1073/PNAS.1525528113
124. Frank MG, Fonken LK, Watkins LR, Maier SF. Microglia: neuroimmune-sensors of stress. Semin Cell Dev Biol. (2019) 94:176–85. doi: 10.1016/J.SEMCDB.2019.01.001
125. Fonken LK, Frank MG, Gaudet AD, D’Angelo HM, Daut RA, Hampson EC, et al. Neuroinflammatory priming to stress is differentially regulated in male and female rats. Brain Behav Immun. (2018) 70:257–67. doi: 10.1016/J.BBI.2018.03.005
126. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. (1984) 5:25–44. doi: 10.1210/EDRV-5-1-25
127. Bittle J, Stevens HE. The role of glucocorticoid, interleukin-1β, and antioxidants in prenatal stress effects on embryonic microglia. J Neuroinflamm. (2018) 15:44. doi: 10.1186/S12974-018-1079-7
128. Carloni E, Ramos A, Hayes LN. Developmental stressors induce innate immune memory in microglia and contribute to disease risk. Int J Mol Sci. (2021) 22:13035. doi: 10.3390/IJMS222313035
129. Zhao Q, Peng C, Wu X, Chen Y, Wang C, You Z. Maternal sleep deprivation inhibits hippocampal neurogenesis associated with inflammatory response in young offspring rats. Neurobiol Dis. (2014) 68:57–65. doi: 10.1016/J.NBD.2014.04.008
130. Xu Y, Patterson MT, Dolfi B, Zhu A, Bertola A, Schrank PR, et al. Adrenal gland macrophages regulate glucocorticoid production through Trem2 and TGF-β. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.174746
131. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J, et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. (2019) 568:541–5. doi: 10.1038/S41586-019-1105-7
132. Kosuth M, Mason SA, Wattenberg EV. Anthropogenic contamination of tap water, beer, and sea salt. PLoS One. (2018) 13:e0194970. doi: 10.1371/JOURNAL.PONE.0194970
133. Liebezeit G, Liebezeit E. Non-pollen particulates in honey and sugar. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. (2013) 30:2136–40. doi: 10.1080/19440049.2013.843025
134. Liebezeit G, Liebezeit E. Synthetic particles as contaminants in German beers. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. (2014) 31:1574–8. doi: 10.1080/19440049.2014.945099
135. Oliveri Conti G, Ferrante M, Banni M, Favara C, Nicolosi I, Cristaldi A, et al. Micro- and nano-plastics in edible fruit and vegetables. The first diet risks assessment for the general population. Environ Res. (2020) 187:109677. doi: 10.1016/J.ENVRES.2020.109677
136. Schymanski D, Goldbeck C, Humpf H-U, Fürst P. Analysis of microplastics in water by micro-Raman spectroscopy: Release of plastic particles from different packaging into mineral water. Water Res. (2018) 129:154–62. doi: 10.1016/j.watres.2017.11.011
137. Garcia MA, Liu R, Nihart A, El Hayek E, Castillo E, Barrozo ER, et al. Quantitation and identification of microplastics accumulation in human placental specimens using pyrolysis gas chromatography mass spectrometry. Toxicol Sci. (2024) 199:81–8. doi: 10.1093/TOXSCI/KFAE021
138. Tian L, Zhang Y, Chen J, Liu X, Nie H, Li K, et al. Effects of nanoplastic exposure during pregnancy and lactation on neurodevelopment of rat offspring. J Hazard Mater. (2024) 474:134800. doi: 10.1016/j.jhazmat.2024.134800
139. Medley EA, Spratlen MJ, Yan B, Herbstman JB, Deyssenroth MA. A systematic review of the placental translocation of micro- and nanoplastics. Curr Environ Health Rep. (2023) 10:99–111. doi: 10.1007/s40572-023-00391-x
140. Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, et al. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. (2012) 26:4743–54. doi: 10.1096/fj.12-210989
141. Block CL, Eroglu O, Mague SD, Smith CJ, Ceasrine AM, Sriworarat C, et al. Prenatal environmental stressors impair postnatal microglia function and adult behavior in males. Cell Rep. (2022) 40:111161. doi: 10.1016/j.celrep.2022.111161
142. Bain CC, Gibson DA, Steers NJ, Boufea K, Louwe PA, Doherty C, et al. Rate of replenishment and microenvironment contribute to the sexually dimorphic phenotype and function of peritoneal macrophages. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.abc4466
Keywords: macrophage, niche, developmental programming, maternal immune activation (MIA), hematopoiesis
Citation: Viola MF, Franco Taveras E and Mass E (2024) Developmental programming of tissue-resident macrophages. Front. Immunol. 15:1475369. doi: 10.3389/fimmu.2024.1475369
Received: 03 August 2024; Accepted: 22 October 2024;
Published: 07 November 2024.
Edited by:
Florent Ginhoux, Singapore Immunology Network (A*STAR), SingaporeReviewed by:
Stoyan Ivanov, INSERM U1065 Centre Méditerranéen de Médecine Moléculaire, FranceRafael S. Czepielewski, Augusta University, United States
Copyright © 2024 Viola, Franco Taveras and Mass. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elvira Mass, ZWx2aXJhLm1hc3NAdW5pLWJvbm4uZGU=
†These authors have contributed equally to this work