 Lugang Wang1,2†
Lugang Wang1,2† Zhen Zhang
Zhen Zhang Peng Wu
Peng Wu Yi Zhang
Yi Zhang Xinfeng Chen
Xinfeng Chen- 1Biotherapy Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
- 2Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
- 3State Key Laboratory of Esophageal Cancer Prevention & Treatment, Zhengzhou, Henan, China
- 4School of Life Sciences, Zhengzhou University, Zhengzhou, Henan, China
- 5Engineering Key Laboratory for Cell Therapy of Henan Province, Zhengzhou, Henan, China
- 6Tianjian Laboratory of Advanced Biomedical Sciences, Academy of Medical Sciences, Zhengzhou University, Zhengzhou, Henan, China
The tumor microenvironment (TME) provides essential conditions for the occurrence, invasion, and spread of cancer cells. Initial research has uncovered immunosuppressive properties of the TME, which include low oxygen levels (hypoxia), acidic conditions (low pH), increased interstitial pressure, heightened permeability of tumor vasculature, and an inflammatory microenvironment. The presence of various immunosuppressive components leads to immune evasion and affects immunotherapy efficacy. This indicates the potential value of targeting the TME in cancer immunotherapy. Therefore, TME remodeling has become an effective method for enhancing host immune responses against tumors. In this study, we elaborate on the characteristics and composition of the TME and how it weakens immune surveillance and summarize targeted therapeutic strategies for regulating the TME.
1 Introduction
In the history of the struggle against cancer, from the initial belief that cancer is a tumor to its recognition as a complex systemic ailment, the use of the body’s immune system to specifically target and kill tumors has evolved (1). The TME, the latest research direction in cancer studies, has been confirmed by multiple studies as a key player in the development of tumors. The TME symbolizes the environment of cancer cells and embodies an intricate biome comprising vascular networks, extracellular matrices, immune cells, and various adjacent elements enveloping cancerous cells. Elements within the TME communicate and mutually regulate both cellular and noncellular components (2, 3). The “dysfunctional” state of tumor blood vessels fosters features such as a lack of oxygen and acidic conditions within the TME, creating an immunosuppressive microenvironment. Furthermore, the extracellular matrix, along with fibroblast-produced fibronectin and proteoglycans, provides crucial support for the proliferation and mobility of cancerous cells, influencing the invasive behavior and metastatic capabilities of tumors.
Recent years have seen notable advancements in cancer immunotherapy, specifically through the utilization of immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1 (PD-1) and its ligand (PD-L1), offering hope to many patients with advanced cancer (1). Enhancing the effectiveness of immunotherapy remains a challenge, given that the immunosuppressive features of TMEs enable tumors to evade immune detection, thereby rendering current treatment strategies ineffective (4). Hence, a thorough study of the composition and attributes of the TME, along with the targeted exploration of therapeutic approaches, is of great significance. This study focuses on exploring the intricate mechanisms by which the TME influences cancer progression, examining insights from foundational studies and practical clinical approaches, and devising potent tactics to improve immunotherapeutic outcomes.
2 TME overview and its characteristics
TME is a complex system made up of various cells and molecules that play a critical role in tumor progression and treatment resistance. Along with tumor cells, the TME includes endothelial cells, fibroblasts, and immune cells from both the innate and adaptive systems, whose early infiltration is vital for controlling tumors (5). Tumor cells interact with surrounding cells and structures within this environment to drive growth, invasion, and spread. Key features of the TME, such as hypoxia, low pH, high interstitial pressure, tumor vascular hyperpermeability, and inflammatory response, create conditions that support cancer cell survival while also making treatment more challenging (Figure 1). Furthermore, TME’s immunosuppressive nature, driven by cells like myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T-cells (Tregs), limits the effectiveness of immunotherapy (Figure 1). These cells promote tumor development by secreting immunosuppressive molecules, such as transforming growth factor beta (TGF-β), or by modulating immune checkpoint molecules (6, 7). Understanding these intricate interactions is crucial for developing new therapeutic strategies.
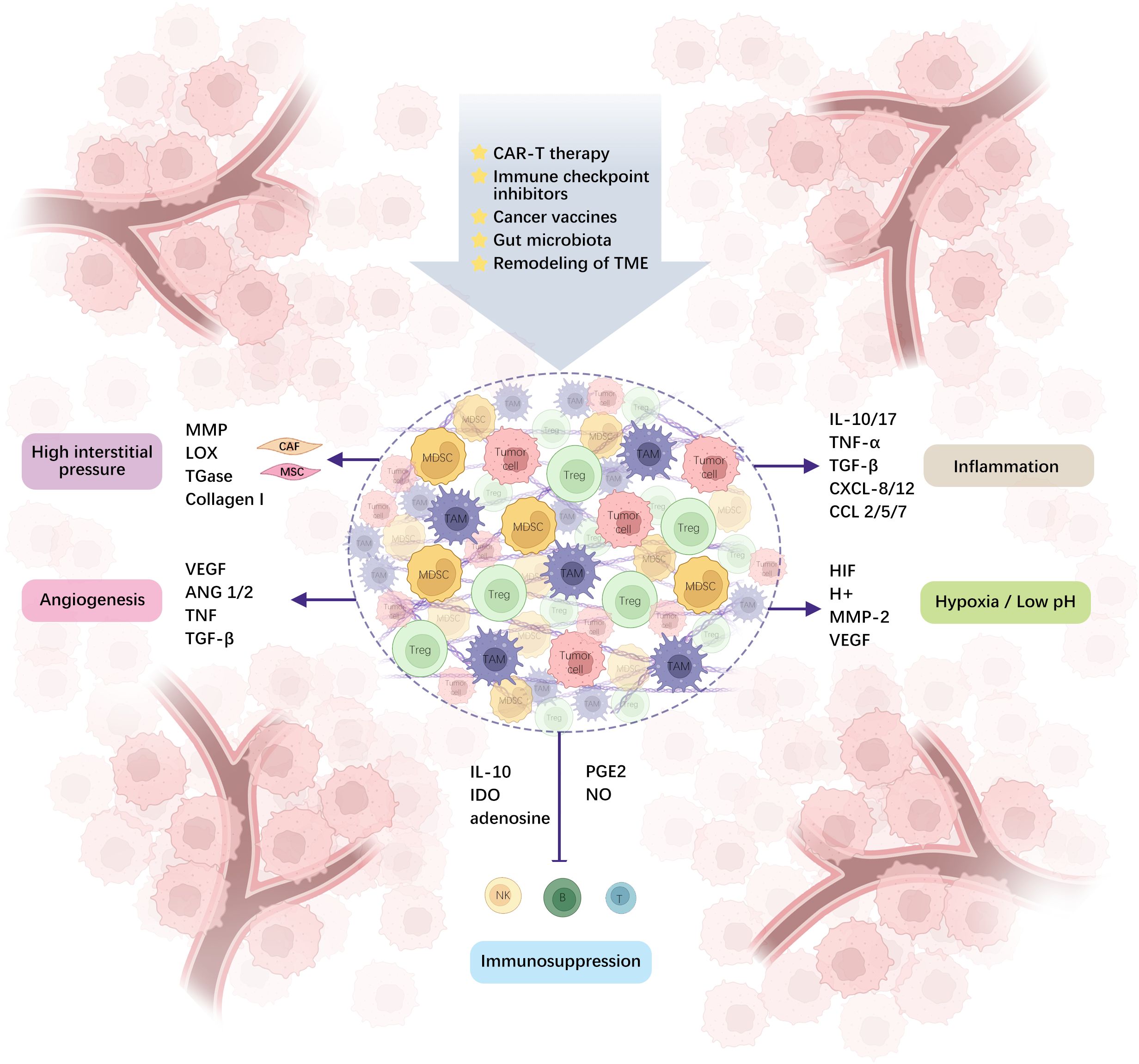
Figure 1. TME is comprised of different cells and molecules, and the interplay between immunosuppressive cells and tumor cells creates a microenvironment of hypoxia, acidity, high interstitial pressure, tumor-incapacitating angiogenesis, inflammation, and immunosuppression via diverse molecular pathways. (ANG1/2: angiopoietin-1 and 2, IDO: Indoleamine 2,3-dioxygenase, PGE2: prostaglandin E2, CAR-T: chimeric antigen receptor T-cell).
2.1 Hypoxia
Hypoxia is a common characteristic of TME in various cancers. The rapid growth and high metabolic activity of tumor tissues increase the demand for oxygen and other energy substrates, such as glucose, leading to hypoxia. The tumor vasculature and hypoxic microenvironment act as complementary factors, with hypoxia stimulating the heightened production of erythropoietin and vascular endothelial growth factor (VEGF), promoting the growth of vascular endothelial cells in tumors. However, a disordered and chaotic arrangement within these blood vessels leads to their “non-functionality,” which exacerbates hypoxic conditions in the TME.
Hypoxia activates several adaptive pathways, including those involving hypoxia-inducible factor (HIF) and nuclear factor κB (NF-κB) (8). Hypoxic signals interact with additional cellular pathways to modify the malignant behavior of cancer cells, weaken the capabilities of cytotoxic T-lymphocytes (CTLs), and recruit Tregs, thereby reducing tumor immunogenicity and ultimately affecting cancer treatment outcomes. Under hypoxic conditions, HIF triggers the production of a range of immune-related elements. For instance, HIF-1α suppresses the function of CTLs and reduces the infiltrative capacity of immune cells by enhancing levels of molecules such as VEGF, PD-L1, and TGF-β (9, 10). Research has demonstrated that HIF-1α triggers increased expression of PD-L1 in cancer and myeloid cells. Bailey et al. demonstrated that targeting HIF-1α can enhance immune tolerance function associated with PD-1/PD-L1 checkpoints within normal tissues and eliminate immune escape mechanisms (11). Tian et al. showed that targeting the HIF-1α/STAT3/NF-κB signaling cascade reduces PD-L1 expression, boosts the proportion of CTLs, decreases Tregs, and diminishes immune suppression factors (12).
2.2 Low PH
The pH of the TME often decreases, resulting in an acidic environment. This is primarily due to inadequate oxygen supply, which compels cancer cells to rely on anaerobic glycolysis to meet their energy needs, consequently leading to lactate accumulation. Simultaneously, ion exchange proteins located on tumor cell membranes consistently move H+ ions from the intracellular environment to the extracellular space to prevent self-acidification. To varying degrees, these cellular responses contribute to TME acidification, resulting in an overall acidic environment. This low pH environment can inhibit cytotoxic reactions mediated by CD8+ T-lymphocytes and suppress the generation of interferon (IFN)-γ and other related cytokines by TH1 cells (13). Acidic conditions may increase the spread of cancer cells, thereby affecting their function in the immune system. Estrella et al. demonstrated that the administration of oral sodium bicarbonate is effective in increasing acidity levels around tumors, ultimately hindering tumor progression and preventing local spreading. This finding supports the theory that tumor invasion is influenced by acidity levels (14).
Lactate has different effects on immune cells, regardless of its pH-lowering properties. Lactate accumulation within TME has been demonstrated to inhibit CD8+ T cell function through multiple mechanisms. First and foremost, alterations in metabolic pathways represent the most significant impact. Lactate can modulate the intracellular pH of CD8+ T cells, thereby disrupting their metabolic processes, including glycolysis, oxidative phosphorylation, and the tricarboxylic acid (TCA) cycle (15, 16). This metabolic dysregulation leads to reduced Adenosine-5’-triphosphate (ATP) production, diminished effector function, and increased susceptibility to exhaustion. Furthermore, lactate induces the expression of inhibitory receptors such as PD-1 and CTLA-4 on T lymphocytes, which strengthens their interaction with corresponding ligands on tumor cells and Tregs (17). This interaction suppresses CD8+ T cell activity and facilitates tumor immune evasion. Studies have shown that lactate can modulate the epigenetic landscape of CD8+ T cells by influencing histone deacetylase (HDAC) and DNA methyltransferase activity (18). These epigenetic alterations inhibit the expression of genes critical for T cell activation and survival, further impairing their antitumor function. Another study revealed an unexpected immunoprotective role of lactate in antitumor immunity. It demonstrated that lactate, by inhibiting HDAC activity, enhances a subset of CD8+ T cells expressing stem-like TCF-1 (19). Lactate also influences the crosstalk between CD8+ T cells and other immune cells within TME. For instance, lactate promotes the differentiation of MDSCs and Tregs, while enhancing the expression of inhibitory molecules on their surfaces (17). The complex interplay of these immunosuppressive factors further diminishes CD8+ T cell functionality. Additionally, lactate enhances the activity of fibroblasts and myofibroblasts, leading to the production of extracellular matrix components that create a physical barrier, impeding T cell infiltration and function (20, 21). Researchers have shown that lactate can shift macrophages towards tolerogenic M2 types and modify the metabolism of Treg cells to sustain their functionality under glucose-deficient conditions. Therefore, tumor cells can evade elimination by starving effector T-cells of necessary nutrients. Additionally, they can aid the regulatory cell population by utilizing metabolic pathways, which may promote immune suppression in the TME (22). The inhibition of enzymes related to the glycolytic pathway due to the acidic microenvironment, particularly phosphofructokinase-1, leads to bidirectional interactions between tumor metabolism and the microenvironment pH (16). This bidirectional interaction further exacerbates the immunosuppressive state, thereby complicating the effectiveness of immunotherapy.
2.3 High interstitial pressure
Tumor-stromal fibrosis refers to the presence of abundant collagen fibers and other extracellular matrix components around the tumor, forming a state of high pressure. The stroma, a critical component of tissues and organs, consists of a variety of specialized connective tissue cells, such as fibroblasts, stromal cells, osteoblasts, and chondrocytes. These cells collaborate to provide structural support and maintain tissue integrity. The extracellular matrix (ECM) plays a crucial role in supporting cell structure and function within the stroma. The tumor stroma contributes to the TME by fostering treatment resistance, making it a significant factor in cancer therapy outcomes. Fibroblasts are the main cell type that produce collagen fibers and regulate collagen synthesis and degradation in the tumor stroma. Fibrosis restricts the spread and movement of tumor cells, thereby affecting tumor angiogenesis and oxygen supply. Studies have shown that the progression of breast tumors usually involves elevated accumulation, linking, and disrupted cleavage of type I collagen (23). Collagen restructuring occurs primarily because of the increased activity and expression of a range of enzymes (including lysyl oxidase (LOX), transglutaminase (TGase), and matrix metalloproteinase (MMP)) within the dynamic tumor matrix. Enhanced crosslinking of collagen and subsequent stiffening of tissues enhance the biomechanical responses of breast tissue, promoting the progression of breast tumors (24).
2.4 Tumor vascular hyperpermeability
In tumor cells, different transcription factors, such as HIF, can induce the production of pro-angiogenic factors, such as VEGF and platelet-derived growth factor (PDGF), thereby stimulating the de novo generation of blood vessels within the tumor and leading to the formation of a unique tumor vasculature, also referred to as the tumor-associated vasculature system (25) (Figure 2). The vascular system associated with tumors differs from that of healthy tissues in its complex, leaky, and disordered structure, which leads to the establishment of a drug-resistant TME (26). Cytokines such as tumor necrosis factor (TNF) and TGF-β are released by tumor cells. These cytokines affect endothelial cell adhesion and permeability, resulting in increased vascular permeability. Increased permeability results in tumor vessel leakage, which can impair tumor blood circulation and increase “intratumoral fluid pressure”. Anomalous circulation hinders drug transport and fosters tumor invasiveness, spread, immune suppression, inflammation, fibrosis, and resistance to treatment owing to oxygen deprivation (27).
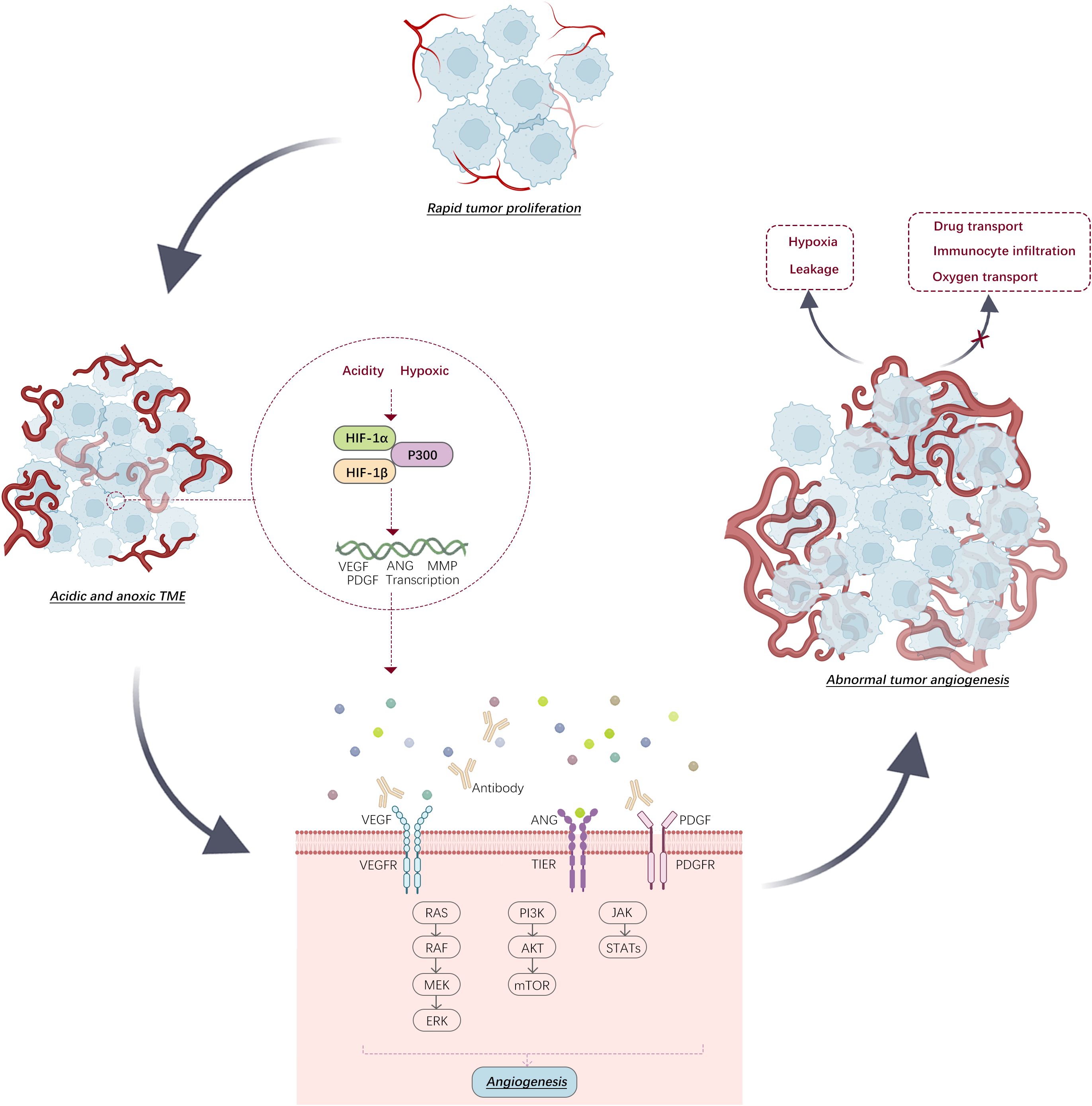
Figure 2. In environments that are both acidic and low in oxygen, transcription of angiogenic factors by HIF stimulates tumor angiogenesis. This progression can be hindered by specific antibodies. (VEGFR: vascular endothelial growth factor receptor, PDGFR: platelet-derived growth factor receptor).
2.5 Inflammatory response
During the development of tumors in hypoxic and acidic environments, both tumor tissues and adjacent tissue cells undergo extensive apoptosis, releasing cell fragments and chemotactic factors that attract inflammatory cells and trigger the release of inflammatory substances. Simultaneously, tumor growth induces an immune reaction, causing inflammatory cells to accumulate in the area and provoke a strong inflammatory reaction. Inflammation is a basic innate immune response that disrupts tissue homeostasis but can have a dual effect in promoting cancer (28). In the TME, inflammatory responses mainly occur via the activation of immune cells and the generation of inflammatory mediators. In the context of tumor initiation and progression, inflammatory mediators act more like a balance between anti-tumor and pro-tumor inflammatory mediators, dividing tumor immune editing into three distinct phases: elimination, balance, and evasion. Tumor-associated dendritic cells, MDSCs, tumor-associated macrophages (TAMs), Th17 cells, Tregs, and cancer cells induce immune suppression by secreting various immunosuppressive cytokines and molecules (2). Among these, TAMs, the primary type of inflammatory cells in the TME, sustain significant inflammation. TAMs induce a shift from M1 to M2 polarization by upregulating inflammatory molecules (e.g., IL-10, TGF-β) and secreting chemokine components (e.g., CCL2, CCL5, CCL7, CXCL8, CXCL12) to maintain an immune suppressive phenotype. In a study on colon cancer, it was reported that during the initial stages of inflammation-associated colon cancer, bacterial lipopolysaccharide (LPS) regulates the expression of CCL2 in colonic epithelial cells, which in turn regulates the accumulation of monocyte-like macrophages (MLMs). Furthermore, this stimulation increases the production of IL-1β by MLMs, inducing Th cells to produce IL-17 and promote inflammation, driving the onset of cancer (29).
Recent reports (30) support the association between chronic inflammation, IL-6 cytokine signaling, and resistance to immunotherapy. IL-6 and related cytokines are critical in the interplay between inflammation and cancer. Within TME, IL-6 and its family members promote tumor cell proliferation, survival, and angiogenesis by activating signaling pathways such as JAK/STAT, PI3K/AKT, and MAPK. IL-6 induces the expression of anti-apoptotic proteins, such as Bcl-2 and BCL-XL, in cancer cells, thereby enhancing their resistance to apoptosis. The IL-6 cytokine family also exerts profound effects on immune cells, promoting the differentiation of T cells into Th17 cells, which secrete pro-inflammatory cytokines that further support tumor growth (31). Hailemichael et al. revealed in cancer models receiving ICIs therapy that IL-6R inhibition alleviates ICI-related toxicity by reducing Th17 differentiation, pro-inflammatory cytokine secretion, and neutrophil chemokine production, while simultaneously enhancing tumor immunity by increasing Th1 and CD8+ T effector cells (32). Beyond IL-6, other members of the IL-6 cytokine family, such as IL-11, leukemia inhibitory factor (LIF), and oncostatin M (OSM), also play critical roles in the interplay between inflammation and cancer (31). For instance, IL-11 exhibits potent pro-tumorigenic effects in gastric and colorectal cancers. In a study on hepatocellular carcinoma (HCC), IL-11 was shown to promote tumor cell proliferation and resistance to apoptosis by activating the STAT3 signaling pathway, contributing to cancer recurrence in postoperative HCC patients (33). Given the multifaceted roles of the IL-6 cytokine family in cancer progression and immunosuppression, various strategies have been developed to target this cytokine family in cancer immunotherapy. One primary approach is the use of monoclonal antibodies (mAbs) that directly target IL-6 or its receptor components. Tocilizumab, an IL-6 receptor antagonist, and Siltuximab, an anti-IL-6 antibody, have been shown to inhibit IL-6 signaling; they were initially studied for treating cytokine release syndrome, and clinical and preclinical studies are now underway to evaluate their efficacy in a variety of cancers (34, 35). Another approach involves the development of small molecule inhibitors targeting IL-6-activated signaling pathways. For example, JAK inhibitors like Ruxolitinib and Baricitinib have been used to block the JAK/STAT pathway, a key conduit for IL-6 signal transduction. These inhibitors have demonstrated efficacy in treating certain hematologic malignancies, and their potential is currently being explored in solid tumors (Table 1).
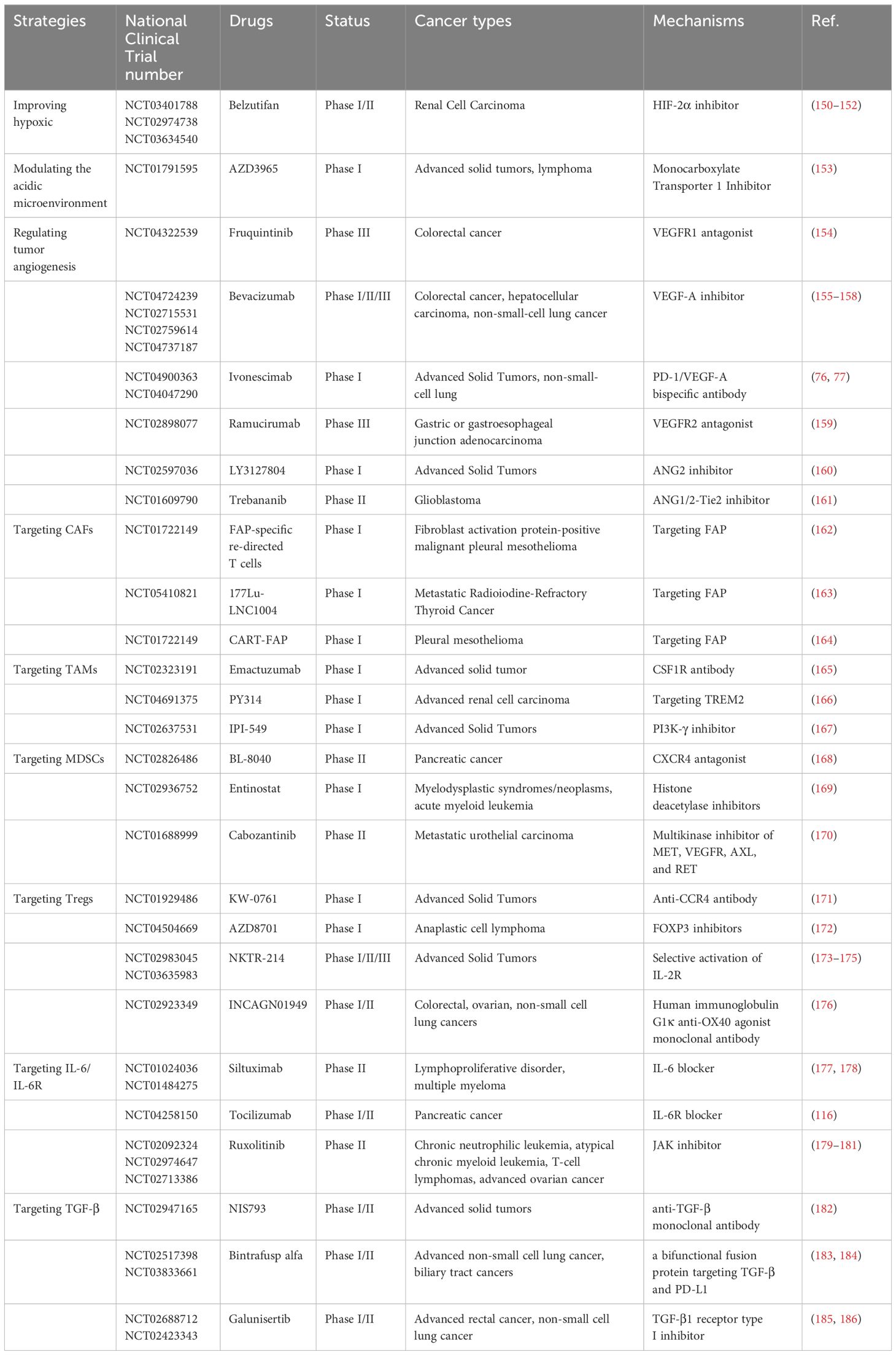
Table 1. Drugs targeting TME in clinical studies.
2.6 Immunosuppressive nature
Within the TME, interactions occur between cancer and stromal cells, which function together to enhance tumor growth and spread. The rise in immunosuppressive cells and molecules, inhibits antitumor immunocyte activity and creates conditions enabling immune evasion.
MDSCs are a collection of highly immunosuppressive cells whose main function is to inhibit immune reactions and shield tumor cells from attack (36). This cluster of highly immature bone marrow-derived immune cells primarily includes polymorphonuclear and monocytic MDSCs (37). MDSCs suppress immune cell activity via cell-to-cell contact mechanisms involving interactions between surface molecules and receptors. Additionally, MDSCs are capable of imparting immunosuppressive properties by generating extracellular factors such as nitric oxide (NO), opioid-like peptides, TGF-β, and IL-10. The surface receptor TIM-3 on MDSCs binds to T-lymphocytes and upregulates PD-L1 expression, thus inhibiting T-lymphocyte functionality. MDSCs promote angiogenesis and the development of pre-metastatic niches through mechanisms unrelated to immunity. They achieve this through the secretion of increased levels of VEGF and basic fibroblast growth factor (bFGF). These factors significantly affect the reshaping of the TME, thereby facilitating tumor progression (38, 39).
TAMs predominantly exhibit an M2 phenotype and are strongly correlated with unfavorable patient outcomes. Hypoxic and highly lactate-rich TMEs promote inflammation-suppressive polarization. A study focusing on tumor acidosis elucidated that TAMs sense the acidic environment, leading to high cAMP expression and subsequent induction of the transcription factor ICE, resulting in the polarization of TAMs towards a pro-tumor phenotype (40). High levels of AMP bind to adenosine receptors, such as A2AR and A2BR, on the surface of macrophages, promoting M2 polarization (41). Tumor-derived prostaglandin E2 (42), fatty acids (43), and α-ketoglutarate (44) are among the various agents that induce M2 polarization of macrophages, promoting a pro-tumor phenotype.
In oxygen-deprived TMEs, Tregs contribute to tumor progression while simultaneously hindering antitumor immunity. Treg cells engage with immune cells such as CD4+ Th and CD8+ CTLs, inhibiting their activity and function by binding to surface markers such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1. Tregs undergoing apoptosis release elevated amounts of ATP, which, in the presence of CD39 and CD73, degrades into adenosine and exerts immunosuppressive effects through A2A receptors on T-cells. Tregs release related factors such as TGF-β and IL-10/35, downregulating anti-tumor immunity and inhibiting antigen presentation by dendritic cells (DCs), further dampening immune responses by lymphocytes (45). In patients with cancer, a poor prognosis is often linked to the heightened presence of FoxP3+ Tregs within tumors. This correlation provides a solid foundation for exploring Treg targeting in cancer immunotherapy (46).
TGF-β, secreted by tumor cells, cancer-associated fibroblasts, and immunosuppressive cells, mediates tumorigenesis and progression through engagement with the TGF-β receptor (TGF-βR) family, activation of Smad transcription factors, and subsequent regulation of downstream gene expression (47). Among the TGF-β family members, the TGF-β1 isomerase exerts the most profound influence. Studies have elucidated that TGF-β1 participates in immunosuppression via multiple mechanisms, creating a favorable milieu for tumor growth and dissemination (48). Upon binding to the TGF-βR, TGF-β1 activates receptor-activated Smads (R-Smads), predominantly Smad2 and Smad3. These R-Smads form complexes with the common Smad (Co-Smad, Smad4), translocate to the nucleus, and modulate the expression of specific genes, including various factors pertinent to effector T cell functionality, such as IFN-γ and TNF-α. TGF-β1 downregulates the expression of these cytokines, diminishing the cytotoxicity and antitumor capabilities of effector T cells (49). TGF-β1 also suppresses the differentiation of Th1 and Th2 cells by downregulating the expression of T-bet and GATA3 through Smad-dependent pathways. Moreover, TGF-β1 modulates T cell activation and function through Smad-independent signaling pathways, including the MAPK, PI3K/Akt, and Rho GTPase pathways (50). Within TME, TGF-β1 potentiates the differentiation of immunosuppressive cells, thereby enhancing the immunosuppressive status of the microenvironment. It has been shown that TGF-β1 predominantly upregulates Foxp3 expression through the classical Smad-dependent pathway, promoting the differentiation of Tregs (50). TGF-β1 also facilitates the polarization of M2-type TAMs, further exacerbating immunosuppression (51). Recent evidence suggests that TGF-β1 activates the Smad2/3 complex to upregulate the expression of SOX18. Overexpression of SOX18, in turn, increases the transcription levels of PD-L1 and CXCL12, promoting the accumulation of immunosuppressive TAMs and Tregs within the microenvironment, thereby exacerbating the progression of hepatocellular carcinoma (52). In another study focusing on acute myeloid leukemia (AML), Wang et al. discovered that Glycoprotein A Repetitions Predominant (GARP), expressed on the surface of Tregs, can activate and release bioactive TGF-β1. This active TGF-β1 can induce dysfunction in natural killer (NK) cells of AML patients and facilitate early AML relapse (53). Additionally, TGF-β1 directly impairs the antigen-presenting ability of DCs by downregulating the expression of co-stimulatory molecules on DCs (such as CD80 and CD86) and by reducing the expression of major histocompatibility complex (MHC) class I molecules or influencing antigen processing (54). Increasing evidence indicates that TGF-β1 upregulates the expression of exhaustion markers such as PD-1, TIM-3, and LAG-3, driving effector T cells into a state of exhaustion (55, 56). These immunosuppressive effects synergize to afford TGF-β1 a formidable role in promoting tumorigenesis within the TME. Targeted interventions against the TGF-β1 signaling pathway are considered a pivotal strategy to augment antitumor immune responses, demonstrating certain promise in preclinical and clinical investigations (53, 57) (Table 1).
3 Immunotherapeutic strategies targeting the TME
The TME, as a crucial milieu for tumor growth and development, with hypoxic and acidic features, along with the presence of immunosuppressive cells and tumor vasculature, collectively creates a “pro-tumor, anti-immune” microenvironment. Therefore, targeting immunosuppressive components within the TME has emerged as a potent and effective strategy for reshaping the TME.
3.1 Improvement of hypoxic environment
Hypoxia is frequently observed in the TME, whereby a hypoxic environment can alter the metabolism of cancer cells and enhance their potential for invasion and metastasis while rendering tumors resistant to standard treatment modalities such as radiotherapy and chemotherapy. Strategies to increase oxygen delivery by pharmacological vasodilation to increase blood flow to the tumor have been proposed, but the non-functional nature of the tumor vasculature makes it challenging to improve blood flow to tumor tissues in practice (58). In recent years, researchers have explored methods for treating tumors in hypoxic environments.
Multiple studies have indicated that inhibition of HIF and its subsequent target genes is a successful method. Salman et al. demonstrated that the small molecule 32-134D successfully hindered gene activation controlled by HIF-1/2 in HCC in a mouse model. This action halts the growth of human and murine HCC tumors, increasing the elimination rate from 25% to 67% (59). Other research showed that honokiol (HNK) downregulates levels of HIF-1α and its associated factors (such as PDK1, HK2, and GLUT1), inhibiting HIF-1α-dependent glycolysis and inducing cancer cell death through apoptosis (60). Yan et al. demonstrated that HIF-2α is essential for enhancing chemical resistance in breast cancer cells exposed to low levels of oxygen. They identified the specific pathway through which HIF-2α influences this resistance, involving SOD2, mtROS, PDI/GRP78, and the unfolded protein response (UPR). This suggests that developing inhibitors targeting HIF-2α could represent a potentially effective strategy to combat chemoresistance in hypoxic breast cancer (61). Li et al. used RNA-seq for whole transcriptome analysis and intercellular space analysis to discover that P4HA2 stabilizes HIF-1α protein to activate genes downstream, thereby resulting in decreased levels of ROS in bladder cancer cells. This ultimately leads to resistance to erdafitinib. The discovery of P4HA2 provides a potential and valuable target for targeted therapy of acquired resistance in bladder cancer (62). CAR-T-cell therapy is a promising form of immunotherapy for combating cancer, given its limitations in solid tumors. Researchers have developed various combination therapies to enhance their efficacy. High levels of HIF-1α expression in individuals with cervical cancer are associated with unfavorable long-term outcomes. However, a recent study reported that PX-478, an inhibitor of HIF-1α, significantly degrades the cytotoxic activity of mesothelin-CAR-T-cells (meso-CAR-T-cells) targeting cervical cancer cells and induces T-cell exhaustion (63). Nevertheless, Luo et al. showed that when PX-478 is used in combination with ICIs, it promotes T-cell-induced apoptosis of tumor cells. Furthermore, in mice treated with both PX-478 and anti-PD-1 antibodies, researchers have observed a marked slowing of tumor development along with increased survival rates (64).
Tumor autophagy is a key biological process within the extensive regulatory network of HIFs. HIFs promote autophagy by regulating downstream target genes, including autophagy-related genes. Autophagy activation can further stabilize HIF proteins, creating a positive feedback circuit that contributes to tumor resistance (9). Research by Li et al. revealed that the N6-methyladenosine (m6A) reader protein induced by HIF-1α stimulates the expression of autophagy-related genes ATG2A and ATG14 in a manner sensitive to m6A, which is linked to hypoxia-induced autophagy and the advancement of HCC (65). In another study, under hypoxic conditions, HIF-1α was shown to induce upregulation of the tumor cell MALAT1, further promoting the growth and spread of triple-negative breast cancer (TNBC) cells by autophagy activation. Conversely, inhibition of autophagy in TNBC cells under anoxic conditions can be achieved by targeting MALAT1 with mirR141-3p. These findings indicate a significant interaction between HIF-1α, Malat1, and miR-141 in tumor progression (66). Several studies have shown that targeting HIF suppresses tumor growth (Table 1). However, the interactions of combination therapies within the complex TME are worth considering, and the mechanisms underlying the interactions between HIF inhibitors and various immunotherapies warrant further investigation.
In addition to targeted therapies to improve the hypoxic environment, preclinical models have been used to study physical approaches to alleviate hypoxia. In a lung cancer mouse model, exposure to additional oxygen (in breathing O2, levels rose from 21% to 60%) demonstrated that exogenous oxygen supplementation can reduce HIF-1α levels along with its downstream proteins, reversing the hypoxic, adenosine-rich TME, thereby promoting tumor regression (67). However, the risks involved in high-dose oxygen supply must be considered in clinical applications. Another similar method is hyperbaric oxygen (HBO) therapy, which is frequently employed as a supplementary treatment for a range of pathological conditions but has not yet been approved for cancer treatment. However, as an adjunctive therapy, HBO has demonstrated capabilities in restraining tumor progression. For example, research by Liu et al. indicated that HBO promotes the distribution of PD-1 antibodies and penetration of T-cells into tumor tissues by reducing the primary constituents of the ECM, disrupting hypoxia-induced immune suppression (68). Owing to the abnormal vascularity of tumor tissues, improving tumor tissue oxygen levels through physical means has inherent limitations. In recent years, researchers have focused on developing a new type of delivery system that accelerates the breakdown of internal H2O2 to produce O2 in tumor tissues. Yao et al. encapsulated catalase and a HIF-1 blocker within the aqueous compartments of liposomes to alleviate the hypoxic state in tumors. This approach enhances the sensitivity of iodine-125 brachytherapy and achieves the eradication of esophageal cancer (69).
3.2 Modulation of acidic TME
The acidic TME can drive cancer cells to migrate toward blood vessels while hindering the action of immune cells. However, restoring the acidic environment can reverse this immune suppression by enhancing the effectiveness of checkpoint inhibitor therapy targeting PD-1 antibodies (70). Lactic acid production, mediated by the enzyme lactate dehydrogenase (LDH), is a primary factor that leads to an acidic TME. Additionally, tumor vascular integrity, changes in the ECM, inflammatory responses, cell apoptosis, expression of ion-exchange proteins, and factors released by tumor cells collectively result in an acidic TME.
Currently, multiple studies are dedicated to overcoming the low pH characteristic of the TME to inhibit cancer cell growth and suppress immune-related cells. The most promising strategy is to therapeutically target major pathways involved in lactic acid production by inhibiting glycolysis. Zhang et al. used vesicular nanoparticles that contained cationic lipids to decrease the expression of LDH A (LDHA) in cancer cells. This causes a shift in acetate metabolism, a decrease in lactate levels, and the stabilization of the acidic TME. This therapy increases the migration of CD8+ T-lymphocytes and NK cells, reduces the quantity of immunosuppressive T-lymphocytes, and effectively suppresses cancer progression in immunologically active melanoma and breast tumor models (70). Another preclinical model of Ewing’s sarcoma demonstrated that inhibiting LDH to target glycolysis can suppress tumor growth (71). Another approach for targeting lactic acid is to inhibit monocarboxylate transport proteins (MCT1 and MCT4). Currently, MCT1 inhibitors (e.g., AZD3965) have successfully entered clinical trials (Table 1). Relevant ex vivo experiments have shown that simultaneously blocking MCT1 and MCT4 significantly prolongs overall survival in cancer models (72). Based on advancements in nanobiotechnology, Li et al. developed a stratified fibrous device for localized drug administration. The fibrous framework incorporates proton-pump inhibitors that are steadily released to impede the outflow of hydrogen ions from inside tumor cells. Experiments conducted inside living organisms revealed that this localized drug administration device renewed pH levels in the TME from 6.8 to 7.2, demonstrating remarkable immunotherapeutic effects (73). Although numerous strategies have been tested, whether they can be used in clinical settings remains unclear and requires further exploration.
3.3 Regulation of tumor angiogenesis
The “abnormalization” of tumor blood vessels is a crucial factor in leading to the low oxygen and acidic TME, making the normalization of tumor vasculature crucial for cancer therapy. Researchers have proposed various strategies to enhance the efficacy of immunotherapy targeting abnormal tumor vasculature. One strategy involves utilizing anti-angiogenic medications, such as VEGF blockers, to reduce tumor vasculature formation and increase vascular permeability (Figure 2). Bevacizumab, an anti-VEGF antibody, is a commonly used antiangiogenic medication. Combination therapy of bevacizumab with other immunotherapies has demonstrated clinically significant overall survival benefits in certain cancers, such as mesothelioma and advanced colorectal cancer (Table 1).
Another strategy involves combining anti-angiogenic agents with additional treatments, including ICIs and adoptive cell therapy (ACT). A clinical study targeting TNBC reported that combination therapy with low-dose VEGFR2 inhibition and anti-PD-1 therapy showed excellent tolerability and efficacy. Minimal inhibition of VEGFR2 leads to enhanced infiltration of immunocytes, promoting CD8+ T-lymphocytes to secrete osteopontin, which in turn induces tumor cells to produce TGF-β, upregulating PD-1 levels in immunocytes and thereby improving the effectiveness of ICIs (74). Abnormal tumor vasculature restricts the entry of immune cells into the tumor interior, thereby affecting the efficacy of immunotherapy. Shrimali et al. demonstrated that by disrupting the VEGF/VEGFR-2 axis with anti-VEGF antibodies to “normalize” the tumor vascular system and using it in conjunction with ACT, antibody therapy significantly promotes immune cell infiltration. Furthermore, this combination therapy significantly inhibited the growth of vascularized melanoma tumors (P = 0.009) and improved mouse survival rates (P = 0.003) (75). Ivonescimab, a recently authorized bi-specific antibody intended for the treatment of advanced solid malignancies, targets VEGF and PD-1. Data from phase I trials have demonstrated significant clinical advantages (76, 77). Currently, dozens of anti-angiogenic drugs that block the VEGF signaling pathway have been authorized for cancer therapy. Various antiangiogenic agents targeting ANG1, ANG2, and the tyrosine kinase receptor TIE2 pathway are currently undergoing clinical trials (Table 1). Furthermore, studies have demonstrated that anti-VEGF antibodies can prevent TAMs from differentiating into an M2-like phenotype and eliminate VEGF-induced DC maturation inhibition (78).
3.4 Targeting of substrates
Cancer-associated fibroblasts (CAFs) are the predominant stromal cells in tumor tissues. In certain types of cancer, the content of CAFs is inversely correlated with treatment outcomes in patients with cancer undergoing chemotherapy and immunotherapy (79). This is because anti-cancer drugs are easily restricted by stromal barriers; therefore, regulating CAFs could provide an effective auxiliary method to enhance sensitivity to anti-cancer drugs (80).
Fibroblast activation protein (FAP) is specifically expressed on proliferating fibroblasts, and its presence on the surface of CAFs can be detected in many types of cancer with poor prognoses. FAP is increasingly viewed as a promising target for therapy and molecular imaging applications (81). Considering the widely used CAR-T therapy, the integration of FAP expression with CAR-T-cells appears to offer a potentially effective treatment approach. Schuberth et al. were the first to show that ACT therapy targeting FAP effectively suppresses the proliferation of FAP-expressing cancer cells within the peritoneal cavity in murine models. This study underscores the promising capability of FAP CAR-T-cell therapy to specifically eliminate malignant cells that overexpress FAP (82). Because of the carcinogenic properties of FAP-positive CAFs, it can be reasonably assumed that employing CAR-T-cells directed at FAP may enhance patient prognosis. This was confirmed using an orthotopic TNBC mouse model. Das et al. used engineered FAP UCAR-T-cells to target CAFs, and their results showed significant efficacy in depleting CAFs and reducing fibrosis. Additionally, the co-administration of ACT therapy and anti-PD-1 checkpoint inhibitors resulted in a noteworthy reduction in tumor size and prolonged the overall survival of mice (83). In a study concerning pancreatic cancer, FAP CAR-T-cell therapy blocked the infiltration of MDSCs and increased the persistence of T-lymphocytes (84). Hence, targeting FAP could represent an approach for reshaping the TME and preventing immune escape. Related preclinical T-cell studies and clinical trials are ongoing (Table 1).
In comparison with the systemic targeting of FAP therapy, Zhen et al. described nanoparticle-based photoimmunotherapy (nano-PIT) (85). This strategy conjugates FAP single-chain fragment variable (scFv) to the surface of ferritin, and when exposed to light, nano-PIT can efficiently eradicate CAFs within tumors, further inhibiting the secretion of CXCL12 and ECM deposition. This significantly enhances T-cell infiltration and inhibits tumor growth. Other approaches that target FAP include antibodies that inhibit the proteolytic activity of FAP and FAP-targeted vaccines (86, 87).
During cancer development, normal fibroblasts are stimulated by various cytokines to differentiate into CAFs. The main cytokines involved in this process include TGF-β, bFGF, and IL-1β. Research indicates that this activation is largely driven by TGF-β (88). Therefore, by exploiting this mechanism, researchers have designed another approach to eliminating CAFs by targeting TGF-β. Yang et al. constructed a nanodrug delivery system (HA-LSL/siTGF-β) designed for the effective transport of siRNA aimed at TGF-β to simultaneously silence TGF-β in stromal and cancer cells, reducing stromal cell deposition and promoting nanoparticle penetration. The findings of that study verified that this strategy improved oxygenation and the effective delivery of ICIs. Additionally, a reduction in the percentage of immunosuppressive cells such as Tregs and MDSCs has been observed (89). Pei and colleagues introduced an innovative strategy for sequential targeting that initially targets stromal TGF-β signaling, reverses CAF activation, and weakens the dense stromal barrier to facilitate subsequent drug delivery. These findings indicate that, in contrast to the use of gemcitabine alone, this sequential targeting method significantly extended the survival of mice affected by pancreatic cancer (90).
3.5 Cell-based immunotherapy
Cellular therapy, an emerging modality within the realm of tumor immunotherapy, primarily aims to modulate and augment the patient’s intrinsic immune response against tumor cells, thereby achieving the goal of tumor eradication. In recent years, researchers have been diligently working on the development of various cellular therapeutic strategies, including CAR-T cells, CAR-NK cells, TCR-engineered T cells, and tumor-infiltrating lymphocytes (TILs), to target the intricate biological characteristics of TME.
3.5.1 CAR-T-cell therapy
CAR-T-cell therapy represents an innovative immunotherapeutic strategy that entails the genetic modification of patients’ lymphocytes to express CARs directed against tumor-associated antigens. CAR design integrates domains that bind to antigens with those that activate CTLs, enabling CAR-T-cells to identify and destroy cancerous cells (91). Using gene editing techniques, T-cells are endowed with a scFv, which is formed by connecting the light chain (VL) and heavy chain (VH) from a monoclonal antibody through peptide linkage, retaining antibody specificity and affinity for the cognate antigen. Upon identifying the tumor antigen, downstream co-stimulatory signaling molecules activate the T-cells, which further release cytokines such as IL-2 and IFN-γ, thereby inducing apoptosis of target cells (92). CAR-T-cells binding to target antigens through scFv offers species-specific recognition that is independent of MHC expression, avoiding tumor escape through MHC regulation and conferring the capability of CAR-T-cells to identify non-peptide antigens (93). By altering the receptor specificity of CAR-T-cells, the immunosuppressive components can be effectively identified, eliminating immune-suppressive components.
Studies have shown that targeting F4/80 (a macrophage marker) by CAR-T-cells can deplete TAMs and improve survival outcomes in animal models of ovarian and pancreatic cancers (94). Rodriguez-Garcia et al. demonstrated that TAM subsets expressing folate receptor β (FRβ) exhibit immune-suppressive M2-like characteristics. Meanwhile, eliminating FRβ-expressing TAMs within the TME by CAR-T-cells brings about the infiltration of CD8+ T-lymphocytes (95). Additionally, Li et al. conducted in vitro experiments showing that three types of innate T-lymphocytes (including mucosal-associated invariant T (MAIT) cells, invariant NK T (iNKT) cells, and gamma delta T (γδT) cells) possess significant TAM-killing capabilities and verified the heightened anticancer abilities of CAR-engineered innate T-lymphocytes (96). Because CCR4 is mainly expressed by Th2/17 cells and Tregs, Watanabe et al. developed anti-CCR4 CAR T-cells and demonstrated their capacity to selectively deplete Tregs. They successfully induced long-term remission and excellent anti-cancer outcomes in an animal model of human T-cell transplantation (97). Zannikou et al. demonstrated that MDSCs in the TME of glioblastomas express IL-15Rα. Utilizing CAR-T-cells engineered with IL-15 offers a two-pronged approach for targeting both cancer cells and the MDSCs found in glioblastomas. This approach enhances the recruitment of CD8+ T-lymphocytes, B-lymphocytes, and NK cells to the tumor site (98). Combination therapy has become a mainstream approach because of the complexity of the TME. Wu et al. analyzed the collective effects of GPC3-CAR T-cells and sorafenib in HCC models with normal immune function and immune deficiency. The results revealed that sorafenib boosts the anti-cancer effect of CAR-T-cells by stimulating IL-12 secretion by TAMs and triggering apoptosis of cancer cells (99). A similar conclusion was confirmed in another study in which lenvatinib upregulated the levels of CXCL10 and CCL8 in tumors, thereby boosting CAR-T-cell infiltration and decreasing immune escape (100).
Within the TME in multiple myeloma, high concentrations of TGF-β lead to an immunosuppressive environment. To address this issue, Alabanza et al. developed a unique B-cell maturation antigen (BCMA) CAR. This CAR includes a dominant-negative TGF-β receptor II (DNR II), which lacks the kinase structural domain. This DNR II confers T-cells with the ability to resist the suppressive characteristics of TGF-β, demonstrating improved anti-tumor cytotoxicity in a mouse tumor model overexpressing TGF-β (57). The same modification has demonstrated enhanced anti-cancer outcomes in prostate (101) and ovarian cancer (102) models. Recent research has demonstrated that SMAD7 (a TGF-β signaling inhibitor) can eliminate communication between TGF-β and NF-κB signaling pathways by downregulating TGF-βRI. Constructing a CAR structure on engineered T-cells that co-express SMAD7 and HER2 shows resistance to TGF-β-induced T-cell exhaustion (103). Separate research has indicated that utilizing CRISPR/Cas9 gene editing technology to eliminate endogenous TGF-βRII can decrease Treg conversion and inhibit the exhaustion of CAR-T-cells (104).
Recent findings suggest that immune-inhibitory cell-derived IL-10 reduces lymphocyte infiltration by upregulating the expression of PD-L1 in monocytes (105). This has been confirmed in glioblastoma (106), bladder cancer (107), and drug-resistant multiple myeloma (108), thus establishing a foundation for the surveillance and targeting of IL-10. Sullivan et al. disrupted IL-10 signaling using IL-10 receptor-blocking antibodies, which reversed immune suppression mediated by myeloid cells (109). Given the characteristic overexpression of IL-10R in most AML cells, Chen et al. developed a new ligand-driven CAR-T-cell model focused on IL-10R, which demonstrated excellent cell-killing abilities against AML cells in laboratory and animal models. However, this study revealed the potential risks involved in reshaping the suppressive TME by boosting IL-10 signaling (110). Contrary to the notion that IL-10 promotes immunosuppressive TMEs, it activates tumor-resident CD8+ T-lymphocytes (111). Guo et al. revealed that boosting oxidative phosphorylation by extending the half-life of an IL-10-Fc fusion protein could effectively strengthen the activity of terminally exhausted CD8+ tumor-infiltrating lymphocytes (112). Recent research has confirmed that modification of CAR-T-cells to release IL-10 preserves the intact mitochondrial structure and function of T-cells, boosting oxidative phosphorylation via reliance on mitochondrial pyruvate carriers. Therefore, the growth and effectiveness of CAR-T-cells can be enhanced (113).
3.5.2 Other cell-based therapies
Due to TCR-T cells have the ability to target a broader range of tumor-associated antigens, they have been developed to target intracellular tumor antigens presented by MHC molecules. Preclinical models have demonstrated that they can effectively infiltrate the TME and exert potent antitumor effects (114). The high-density ECM and abnormal vasculature in TME often impede T cell infiltration. To overcome this barrier, researchers have developed TCR-T cells expressing specific chemokine receptors, such as CXCR2 and CXCR4, to enhance their infiltration (115). Tumor cells within TME frequently evade immune detection through mechanisms such as antigen loss or downregulation of MHC expression. Multispecific TCR-T cells, including bispecific TCR-T cells, are engineered to recognize multiple tumor antigens, thereby reducing the risk of treatment failure due to antigen loss (116). In recent years, owing to the broad-spectrum antitumor activity of NK cells and their lower likelihood of developing immune tolerance, CAR-NK cell therapy has emerged as a novel immunotherapy, combining CAR technology with NK cells. CAR-NK cells avoid MHC restrictions and have minimal CRS risk. Additionally, because CAR-NK cells lack TCR expression and possess distinctive immunomodulatory characteristics, they do not induce graft-versus-host disease (GVHD) (117). Modified CAR-NK cells can better circumvent immune checkpoint mechanisms, thereby reducing the risk of tumor immune evasion (118). These features position CAR-NK cells as a next-generation cell therapy that is currently considered superior to CAR-T cells. Given that TGF-β secretion suppresses the effector functions of NK cells and reduces their recruitment, most NK cells within TME are immature and exhibit low cytotoxic capacity (119). A study in HCC demonstrated that NK cells expressing DNR II combined with CARs targeting GPC3 or AFP could effectively resist TGF-β-mediated inhibition and enhance the tumor-killing capability of CAR-NK cells (120). In a recent phase 1/2 trial for CD19-positive B cell malignancies, a CAR-NK cell expressing an anti-CD19 CAR and IL-15 (CAR19/IL-15) was designed. As anticipated, no significant CRS, neurotoxicity, or GVHD was observed. Moreover, the one-year overall survival rate reached 68% (121). Tumor cells evade immune surveillance through various mechanisms; however, TILs with their tumor-specific cytotoxic activity, can effectively recognize and kill tumor cells, particularly those that have escaped other immunotherapies. Given the functional exhaustion and limited proliferative capacity of T cells within the TME of solid tumors, Chen et al. developed TILs engineered to express CD28 and PD-1 molecules, which have shown long-term and stable efficacy in patients with renal and colorectal cancers (122). In conclusion, given the complexity of the TME, cell-based immunotherapy represents a promising opportunity in cancer immunotherapy.
3.6 ICIs
Cancer cells can hijack the immune system to maintain self-tolerance through inhibitory pathways that typically act as checkpoints to prevent cell death induced by T-cell activation, reduce T-cell cytotoxic effects, and minimize excessive inflammation that can damage healthy tissues (123). However, immune evasion is facilitated by immune checkpoints. Multiple immune checkpoint targets have been discovered; however, only ICIs targeting CTLA-4 and PD-1/PD-L1 have been approved and are widely available.
CTLA-4 is a negative-regulatory molecule that inhibits activation signals by competitively binding to members of the B7 family of molecules (such as CD80 and CD86) (124). This competitive binding inhibits cell proliferation and promotes cell cycle arrest. In addition to its intracellular functions that weaken T-cell activity, CTLA-4 modulates T-cell activation through several extracellular mechanisms. CTLA-4, which is found in Tregs, reduces the levels of CD80/CD86 co-stimulatory molecules in antigen-presenting cells (APCs). This reduction at the molecular level impairs APCs’ capacity to effectively activate T-lymphocytes (125). PD-L1, a PD-1 ligand, plays a crucial regulatory role in the immune system by binding to PD-1. Recent evidence suggests that PD-L1 activation of PD-1 preferentially dephosphorylates the costimulatory receptor CD28 rather than the T-cell receptor (TCR) by recruiting Src homology-2-containing protein tyrosine phosphatase 2 (SHP2), thereby inhibiting T-cell function by deactivating CD28 signaling (126). Suppression of T-cell activation and proliferation results in the impaired capability of T-lymphocytes to effectively target and attack tumor cells. Moreover, PD-L1 expression leads to immune evasion by hindering the function of immune cells (127) (Figure 3).
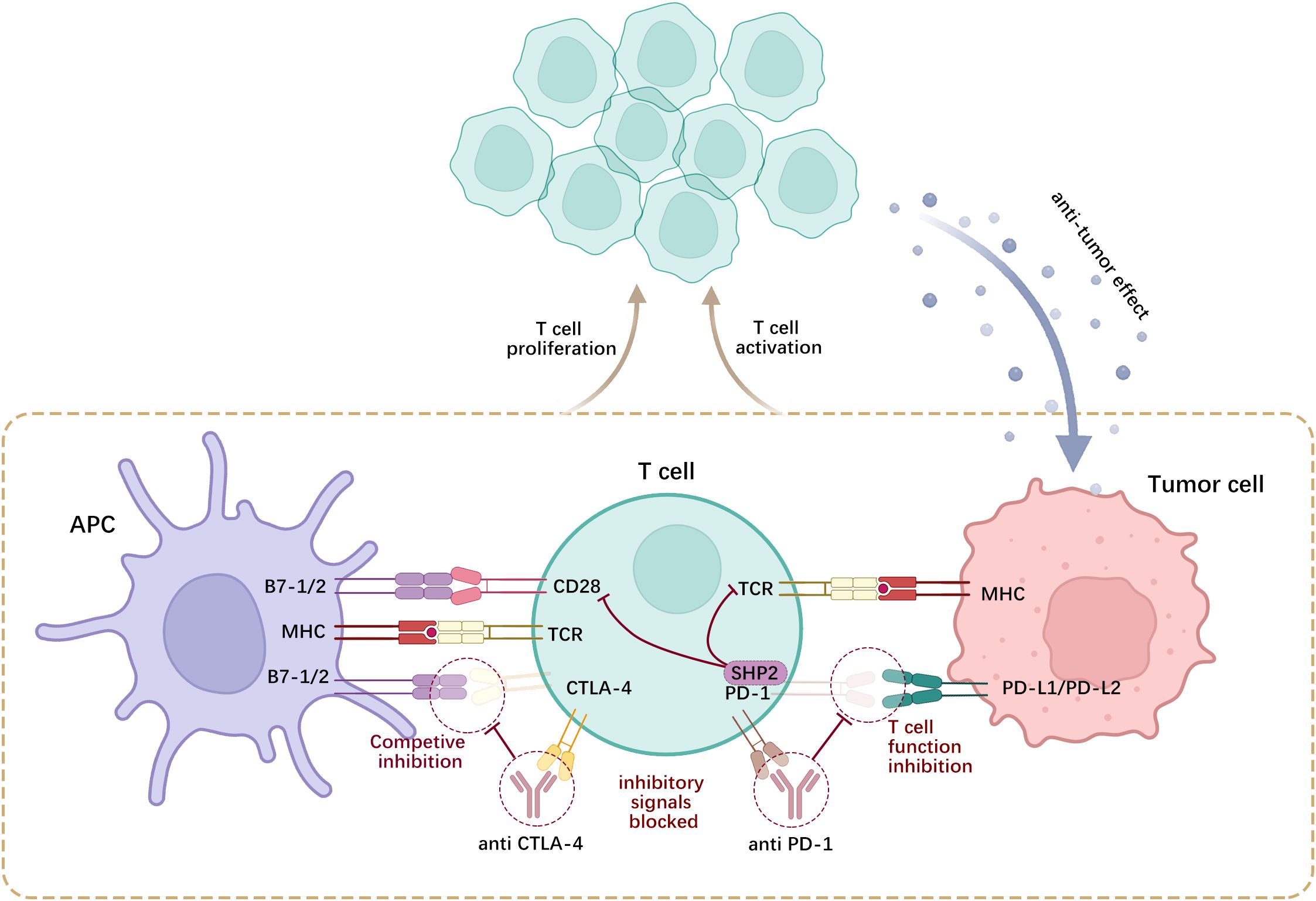
Figure 3. CTLA-4 and PD-1 are expressed on activated T-cells, wherein CTLA-4 competitively antagonizes CD28 and binds to B7-1/2 on the surface of APCs, while PD-1 engages with PD-L1/2 on tumor cells or APCs. Such interactions curtail the proliferation of self-reactive T-lymphocytes and the generation of cytokines.
Following the FDA’s endorsement of ipilimumab (anti-CTLA4) in 2011 for metastatic melanoma therapy, regulatory agencies worldwide have approved at least 11 different versions of this drug for > 90 additional indications. The first PD-1 immune checkpoint inhibitor, nivolumab, was approved in 2014, demarking a significant entry into ICI therapy. To date, the Food and Drug Administration (FDA) has approved a total of 10 monoclonal antibodies against PD-1/PD-L1. In comparison, there are currently only two approved CTLA-4 monoclonal antibodies: ipilimumab and tremelimumab. Numerous studies have demonstrated the efficacy of ICIs in treating a diverse array of cancers, including advanced melanoma (128) and non-small cell lung cancer (129).
Recent data indicate that the FDA-approved therapeutic agent tislelizumab has been used to treat adult individuals diagnosed with unresectable or metastatic esophageal squamous cell carcinoma (ESCC) who have undergone prior systemic chemotherapy. Data from a global phase 3 RATIONALE 302 trial showed that within the intention-to-treat (ITT) population, the median overall survival (OS) for patients treated with Tislelizumab was 8.6 months. In contrast, for patients receiving chemotherapy, the median OS was 6.3 months, demonstrating an extension in survival for individuals who had undergone prior systemic therapy (130). Current treatments involving ICIs face challenges, the major one being the influence of the TME. The TME hosts numerous immune inhibitory cells that can suppress T-lymphocyte function via diverse mechanisms, thereby affecting the effectiveness of ICIs (131). Therefore, researchers are developing new strategies to enhance the effectiveness of ICIs in treating cancer, including combination therapy with other anti-cancer agents such as immunotherapy, targeted therapy, and chemotherapy. Moreover, there has been an investigation into amalgamating ICIs with immune regulators to significantly enhance therapeutic results.
In a phase 3 clinical trial of patients with advanced renal cell carcinoma who had not received prior treatment, the co-administration of cabozantinib with nivolumab and ipilimumab led to a notably extended progression-free survival duration when compared with the use of nivolumab and ipilimumab as standalone treatments. The study’s experimental cohort demonstrated a 12-month progression-free survival (PFS) rate of 0.57, marginally surpassing the control group’s 0.49 rate (132). In a phase 3 study evaluating atezolizumab combined with cabozantinib versus cabozantinib alone in individuals with renal cell carcinoma who had received prior ICI therapy, researchers discovered that the median PFS did not significantly differ between the two cohorts. Joint management fails to improve clinical results and is associated with increased toxicity (133). These divergent results highlight that ICI and targeted therapy combination treatments are not necessarily suitable for all cancer patients and need to be personalized based on specific patient conditions and cancer characteristics. However, combination therapy with chemotherapy has demonstrated excellent OS rates. In a trial targeting terminal biliary tract cancer, pembrolizumab in combination with gemcitabine and cisplatin demonstrated a significant enhancement in OS compared with pembrolizumab alone. This improvement was statistically significant and clinically meaningful, suggesting that the addition of pembrolizumab can lead to superior patient outcomes. No new safety concerns have been identified regarding the use of pembrolizumab in co-administered therapy (134). Another evaluation of gastric/esophageal adenocarcinoma patients showed that the 12-month OS with ICIs combined with chemotherapy is superior to that with chemotherapy alone, supporting the co-administration of nivolumab alongside chemotherapy as a primary therapeutic option for terminal gastric or esophageal adenocarcinoma (135). Five-year follow-up results for the combination of pembrolizumab with two chemotherapeutic agents in nonsquamous cell lung cancer confirm this finding (136). Combination therapy targeting both the CTLA-4 and PD-1 pathways is more effective than monotherapy because these pathways operate via distinct mechanisms. Cadonilimab, a bispecific antibody targeting PD-1 and CTLA-4, has been tested in phase 1b/2 clinical trials in individuals with terminal cancers (137). Numerous studies have evaluated the efficacy and safety of ICIs.
3.7 Gut microbiota
ICIs have been used in clinical practice for a few decades and provide high survival rates for most patients with cancer. Nevertheless, this treatment does not benefit all patients because a substantial number exhibit either primary or acquired drug resistance (138). Emerging findings suggest that the gut microbiome is essential for immune therapy, as evidenced by clinical studies showing how the gut microbiota influences responses to such treatments (139). Research has indicated that the composition of the gut microbiota can influence the responses of patients with cancer to ICIs. Specific types of gut microbiota have been observed to boost the effectiveness of ICIs, thereby improving the survival rates of patients with cancer (140). Various mechanisms enable the gut microbiota to influence the effectiveness of immunotherapy, including the generation of metabolites, control of the development and function of lymphocytes, and adjustment of overall inflammatory responses.
Toll-like receptors (TLRs), a type of pattern recognition receptor, are broadly expressed in both intestinal epithelial cells (IECs) and resident immune cells in the lamina propria. Signal molecules provided by the gut microbiota, including LPS, flagellin, and bacterial peptidoglycan, interact with pattern recognition receptors to recognize and initiate downstream signaling cascades, ultimately influencing tumor immune responses (141) (Figure 4). Research conducted employing a mouse model of colon cancer indicated that metastasis-related protease cathepsin K (CTSK), which is secreted by tumors, can be activated by signals from the gut microbiota. This activation allows CTSK to interact with TLR4 and trigger the polarization of TAMs to the M2 phenotype through an mTOR-dependent pathway. This leads to the infiltration and metastasis of colorectal cancer (CRC) cells by activating the NF-κB pathway (142). Similarly, in an esophageal cancer study, it was identified that LPS exacerbates the invasive and migratory behaviors of esophageal cancer cells through the TLR4/NF-κB axis (143). Certain microbiota-producing toxins, such as enterotoxigenic Bacteroides fragilis (ETBF), promote the formation of colonic epithelial cell tumors through their toxins and IL-17 production, leading to MDSC differentiation, which upregulates arginase 1 (ARG1) and inducible nitric oxide synthase 2 (NOS2), produces NO, and inhibits T-lymphocyte proliferation, thus promoting tumorigenesis. In contrast, the gut microbiota can stimulate local or systemic immune cells to produce antitumor effects. For example, Bifidobacterium stimulates tumor-specific T-lymphocytes, promotes the accumulation of CD8+ T-lymphocytes in melanoma, and increases IFN production (144). When Helicobacter hepaticus (Hhep) was introduced into a CRC mouse model, there was a noticeable increase in infiltration by CTLs, resulting in suppression of tumor growth. Colonization by Hhep prompts the development of Hhep-specific T follicular helper (Tfh) cells, leading to an increase in the number of colonic Tfh cells. Additionally, the presence of Hhep supports the maturation of lymphoid structures responsible for draining tumors (145).
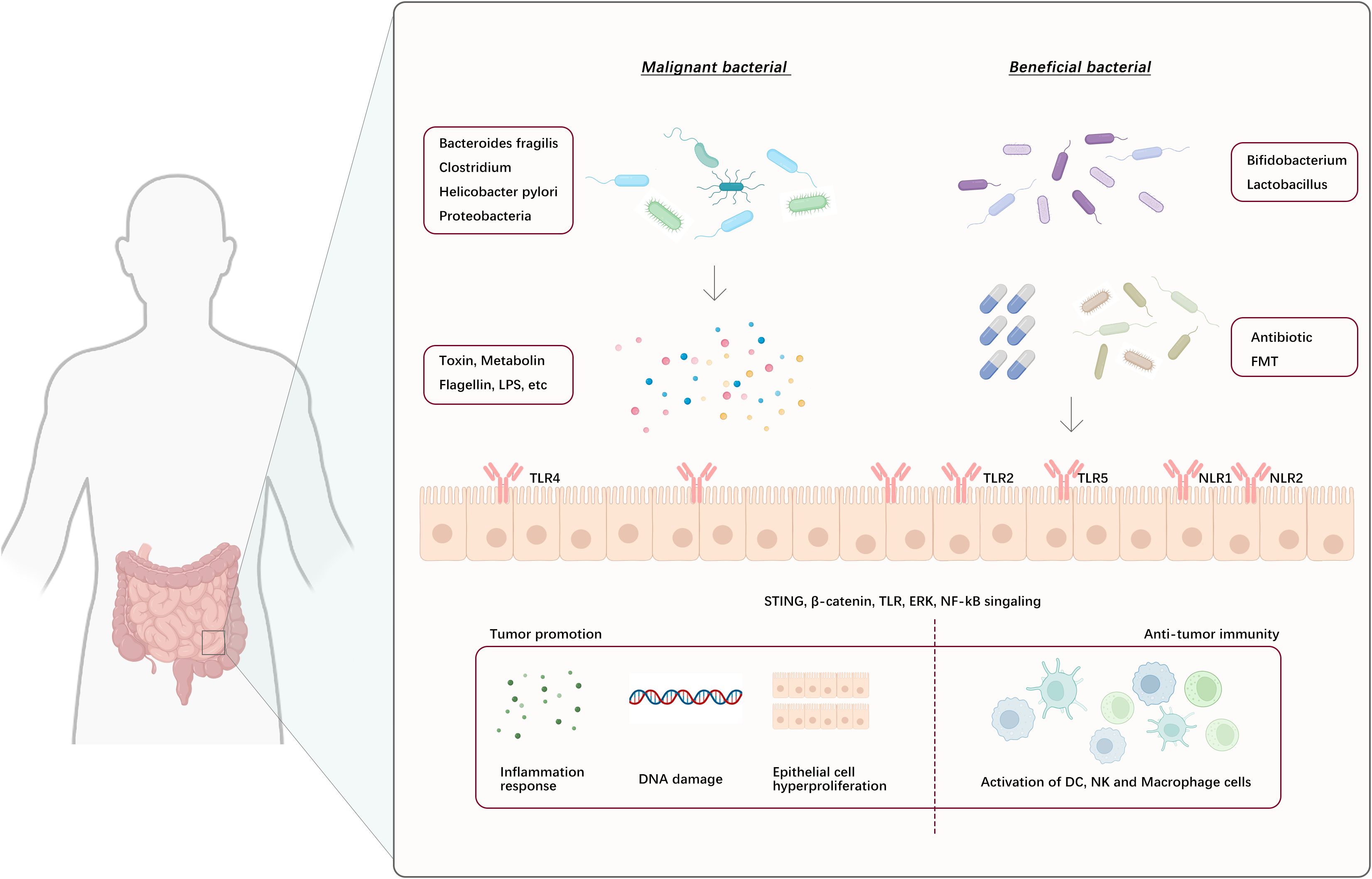
Figure 4. Gut microbes and their metabolites act on intestinal epithelial cells or bind to their surface pattern recognition receptors, eliciting downstream signaling cascade reflexes that lead to tumor-promoting and anti-tumor immune responses. (NLR1/2: NOD-like receptor 1 and 2, FMT: Fecal microbiota transplant).
Regulation of the gut microbiota to improve responses to immunotherapy can be achieved through various means, including the use of probiotics or microbial consortia to enhance beneficial bacteria, the use of antibiotics to reduce levels of harmful bacteria, and fecal microbiota transplantation to introduce beneficial microbiota (146–148) (Figure 4). These methods have shown promise for regulating the gut microbiome in both clinical and preclinical models. Moreover, using different gut microbiota characteristics to distinguish healthy individuals from patients with cancer can potentially serve as a method for predicting how patients with cancer will respond to ICI therapy (149). While still in their initial phases, these techniques provide great enthusiasm for optimizing immunotherapeutic outcomes.
4 Summary and prospects
The TME is an extremely complex system involving interactions between various cell types, cytokines, and signaling pathways. Within the realm of immunotherapy, the immunoregulatory role of the TME is particularly prominent and includes immune cell activity, activation of immune escape signaling pathways, and the influence of tumor stromal cells on immune cells. Due to the heterogeneity and complexity of the TME, the effectiveness of immunotherapy remains challenging. Hence, it is crucial to thoroughly explore the interplay between immunosuppressive TME and immunotherapy to improve therapeutic effectiveness. Emphasizing that in immunotherapy, regulation and intervention involving the TME are particularly important. Optimization of suppressive elements in the immune system and boosting factors that combat tumors is essential to enhance the effectiveness of immunocytes, improve their ability to identify cancer cells, and accelerate their removal.
Future research directions should include an in-depth analysis of the interplay between immunocytes, stromal cells, and cancer cells to improve the efficacy of cancer therapies, reduce drug toxicity, and explore novel immunotherapeutic strategies such as nanotechnology, gene editing, cell therapy, and manipulation of the gut microbiota. The emerging field of genomics has highlighted the significance of the gut microbiota in immune regulation and responses to immunotherapy. Harnessing the gut microbiota-cancer-immunity axis holds promise for opening new avenues in immunotherapy, the identification of novel immune-related markers, and the development of innovative treatment approaches. Advances in technologies such as single-cell sequencing and high-throughput screening have led to the discovery of new immune-related markers and potential therapeutic targets.
Author contributions
LW: Writing – original draft. LZ: Writing – original draft. ZZ: Writing – review & editing. PW: Writing – review & editing. YZ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. XC: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Healthy Talents Project of Henan Province 532(YXKC2020052 and YXKC2020051) and the Natural Science Foundation of China (No. 82370227, 82203548).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. (2020) 17:807–21. doi: 10.1038/s41423-020-0488-6
2. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
3. Gao Y, Liu S, Huang Y, Li F, Zhang Y. Regulation of anti-tumor immunity by metal ion in the tumor microenvironment. Front Immunol. (2024) 15:1379365. doi: 10.3389/fimmu.2024.1379365
4. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. (2020) 52:17–35. doi: 10.1016/j.immuni.2019.12.011
5. Gao FY, Li XT, Xu K, Wang RT, Guan XX. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun Signal. (2023) 21:28. doi: 10.1186/s12964-023-01043-1
6. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, et al. Dual inhibition of TGF-β and PD-L1: a novel approach to cancer treatment. Mol Oncol. (2022) 16:2117–34. doi: 10.1002/1878-0261.13146
7. Ping Y, Shan J, Qin H, Li F, Qu J, Guo R, et al. PD-1 signaling limits expression of phospholipid phosphatase 1 and promotes intratumoral CD8(+) T cell ferroptosis. Immunity. (2024) 57:2122–39.e9. doi: 10.1016/j.immuni.2024.08.003
8. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci. (2021) 22:5703. doi: 10.3390/ijms22115703
9. You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, et al. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev. (2021) 41:1622–43. doi: 10.1002/med.21771
10. Li L, Yang L, Fan Z, Xue W, Shen Z, Yuan Y, et al. Hypoxia-induced GBE1 expression promotes tumor progression through metabolic reprogramming in lung adenocarcinoma. Signal Transduct Target Ther. (2020) 5:54. doi: 10.1038/s41392-020-0152-8
11. Bailey CM, Liu Y, Liu M, Du X, Devenport M, Zheng P, et al. Targeting HIF-1α abrogates PD-L1-mediated immune evasion in tumor microenvironment but promotes tolerance in normal tissues. J Clin Invest. (2022) 132:e150846. doi: 10.1172/JCI150846
12. Tian X, Liu F, Wang Z, Zhang J, Liu Q, Zhang Y, et al. Modified Biejia Jianwan decoction restrains PD-L1-mediated immune evasion through the HIF-1α/STAT3/NF-κB signaling pathway. J Ethnopharmacol. (2024) 322:117577. doi: 10.1016/j.jep.2023.117577
13. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J, et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int J Mol Sci. (2020) 21:8363. doi: 10.3390/ijms21218363
14. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
15. Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science. (2022) 377:1519–29. doi: 10.1126/science.abj5104
16. Elia I, Rowe JH, Johnson S, Joshi S, Notarangelo G, Kurmi K, et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab. (2022) 34:1137–50.e6. doi: 10.1016/j.cmet.2022.06.008
17. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–18.e9. doi: 10.1016/j.ccell.2022.01.001
18. Zhang Q, Zhao Q, Li T, Lu L, Wang F, Zhang H, et al. Lactobacillus plantarum-derived indole-3-lactic acid ameliorates colorectal tumorigenesis via epigenetic regulation of CD8(+) T cell immunity. Cell Metab. (2023) 35:943–60.e9. doi: 10.1016/j.cmet.2023.04.015
19. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun. (2022) 13:4981. doi: 10.1038/s41467-022-32521-8
20. Linares JF, Cid-Diaz T, Duran A, Osrodek M, Martinez-Ordoñez A, Reina-Campos M, et al. The lactate-NAD(+) axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. (2022) 39:110792. doi: 10.1016/j.celrep.2022.110792
21. Andreucci E, Fioretto BS, Rosa I, Matucci-Cerinic M, Biagioni A, Romano E, et al. Extracellular lactic acidosis of the tumor microenvironment drives adipocyte-to-myofibroblast transition fueling the generation of cancer-associated fibroblasts. Cells. (2023) 12:939. doi: 10.3390/cells12060939
22. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
23. Preston SEJ, Bartish M, Richard VR, Aghigh A, Gonçalves C, Smith-Voudouris J, et al. Phosphorylation of eIF4E in the stroma drives the production and spatial organisation of collagen type I in the mammary gland. Matrix Biol. (2022) 111:264–88. doi: 10.1016/j.matbio.2022.07.003
24. Lian B, Yan S, Li J, Bai Z, Li J. HNRNPC promotes collagen fiber alignment and immune evasion in breast cancer via activation of the VIRMA-mediated TFAP2A/DDR1 axis. Mol Med. (2023) 29:103. doi: 10.1186/s10020-023-00696-5
25. Vimalraj S. A concise review of VEGF, PDGF, FGF, Notch, angiopoietin, and HGF signalling in tumor angiogenesis with a focus on alternative approaches and future directions. Int J Biol Macromol. (2022) 221:1428–38. doi: 10.1016/j.ijbiomac.2022.09.129
26. Braman N, Prasanna P, Bera K, Alilou M, Khorrami M, Leo P, et al. Novel radiomic measurements of tumor-associated vasculature morphology on clinical imaging as a biomarker of treatment response in multiple cancers. Clin Cancer Res. (2022) 28:4410–24. doi: 10.1158/1078-0432.CCR-21-4148
27. Tomita T, Kato M, Hiratsuka S. Regulation of vascular permeability in cancer metastasis. Cancer Sci. (2021) 112:2966–74. doi: 10.1111/cas.v112.8
28. Fernandes Q, Inchakalody VP, Bedhiafi T, Mestiri S, Taib N, Uddin S, et al. Chronic inflammation and cancer; the two sides of a coin. Life Sci. (2024) 338:122390. doi: 10.1016/j.lfs.2023.122390
29. Yang Y, Li L, Xu C, Wang Y, Wang Z, Chen M, et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut. (2020) 70:1495–506. doi: 10.1136/gutjnl-2020-320777
30. Naqash AR, McCallen JD, Mi E, Iivanainen S, Marie MA, Gramenitskaya D, et al. Increased interleukin-6/C-reactive protein levels are associated with the upregulation of the adenosine pathway and serve as potential markers of therapeutic resistance to immune checkpoint inhibitor-based therapies in non-small cell lung cancer. J Immunother Cancer. (2023) 11:e007310. doi: 10.1136/jitc-2023-007310
31. Soler MF, Abaurrea A, Azcoaga P, Araujo AM, Caffarel MM. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J Immunother Cancer. (2023) 11:e007530. doi: 10.1136/jitc-2023-007530
32. Hailemichael Y, Johnson DH, Abdel-Wahab N, Foo WC, Bentebibel SE, Daher M, et al. Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell. (2022) 40:509–23.e6. doi: 10.1016/j.ccell.2022.04.004
33. Wang D, Zheng X, Fu B, Nian Z, Qian Y, Sun R, et al. Hepatectomy promotes recurrence of liver cancer by enhancing IL-11-STAT3 signaling. EBioMedicine. (2019) 46:119–32. doi: 10.1016/j.ebiom.2019.07.058
34. Chen IM, Donia M, Chamberlain CA, Jensen AWP, Draghi A, Theile S, et al. Phase 2 study of ipilimumab, nivolumab, and tocilizumab combined with stereotactic body radiotherapy in patients with refractory pancreatic cancer (TRIPLE-R). Eur J Cancer. (2023) 180:125–33. doi: 10.1016/j.ejca.2022.11.035
35. Nguyen MLT, Bui KC, Scholta T, Xing J, Bhuria V, Sipos B, et al. Targeting interleukin 6 signaling by monoclonal antibody siltuximab on cholangiocarcinoma. J Gastroenterol Hepatol. (2021) 36:1334–45. doi: 10.1111/jgh.15307
36. Liu J, Liu J, Qin G, Li J, Fu Z, Li J, et al. MDSCs-derived GPR84 induces CD8(+) T-cell senescence via p53 activation to suppress the antitumor response. J Immunother Cancer. (2023) 11:e007802. doi: 10.1136/jitc-2023-007802
37. Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. (2021) 54:875–84. doi: 10.1016/j.immuni.2021.04.004
38. Xia C, Bai W, Deng T, Li T, Zhang L, Lu Z, et al. Sponge-like nano-system suppresses tumor recurrence and metastasis by restraining myeloid-derived suppressor cells-mediated immunosuppression and formation of pre-metastatic niche. Acta Biomater. (2023) 158:708–24. doi: 10.1016/j.actbio.2023.01.009
39. Cole K, Pravoverov K, Talmadge JE. Role of myeloid-derived suppressor cells in metastasis. Cancer Metastasis Rev. (2021) 40:391–411. doi: 10.1007/s10555-020-09947-x
40. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
41. Sun C, Wang B, Hao S. Adenosine-A2A receptor pathway in cancer immunotherapy. Front Immunol. (2022) 13:837230. doi: 10.3389/fimmu.2022.837230
42. Saha J, Sarkar D, Pramanik A, Mahanti K, Adhikary A, Bhattacharyya S. PGE2-HIF1α reciprocal induction regulates migration, phenotypic alteration and immunosuppressive capacity of macrophages in tumor microenvironment. Life Sci. (2020) 253:117731. doi: 10.1016/j.lfs.2020.117731
43. Li Z, Chen S, He X, Gong S, Sun L, Weng L. SLC3A2 promotes tumor-associated macrophage polarization through metabolic reprogramming in lung cancer. Cancer Sci. (2023) 114:2306–17. doi: 10.1111/cas.v114.6
44. Cai Z, Li W, Hager S, Wilson JL, Afjehi-Sadat L, Heiss EH, et al. Targeting PHGDH reverses the immunosuppressive phenotype of tumor-associated macrophages through α-ketoglutarate and mTORC1 signaling. Cell Mol Immunol. (2024) 21:448–65. doi: 10.1038/s41423-024-01134-0
45. Goldmann O, Nwofor OV, Chen Q, Medina E. Mechanisms underlying immunosuppression by regulatory cells. Front Immunol. (2024) 15:1328193. doi: 10.3389/fimmu.2024.1328193
46. Wang J, Gong R, Zhao C, Lei K, Sun X, Ren H. Human FOXP3 and tumour microenvironment. Immunology. (2023) 168:248–55. doi: 10.1111/imm.v168.2
47. Shi X, Yang J, Deng S, Xu H, Wu D, Zeng Q, et al. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J Hematol Oncol. (2022) 15:135. doi: 10.1186/s13045-022-01349-6
48. Wang J, Xiang H, Lu Y, Wu T. Role and clinical significance of TGF−β1 and TGF−βR1 in Malignant tumors (Review). Int J Mol Med. (2021) 47:55. doi: 10.3892/ijmm.2021.4888
49. Chen W. TGF-β Regulation of T cells. Annu Rev Immunol. (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
50. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. (2022) 7:eabi4613. doi: 10.1126/sciimmunol.abi4613
51. Li D, Zhang Q, Li L, Chen K, Yang J, Dixit D, et al. [amp]]beta;2-microglobulin maintains glioblastoma stem cells and induces M2-like polarization of tumor-associated macrophages. Cancer Res. (2022) 82:3321–34. doi: 10.1158/0008-5472.CAN-22-0507
52. Chen J, Feng W, Sun M, Huang W, Wang G, Chen X, et al. TGF-β1-induced SOX18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology. (2024) 167:264–80. doi: 10.1053/j.gastro.2024.02.025
53. Wang D, Sun Z, Zhu X, Zheng X, Zhou Y, Lu Y, et al. GARP-mediated active TGF-β1 induces bone marrow NK cell dysfunction in AML patients with early relapse post-allo-HSCT. Blood. (2022) 140:2788–804. doi: 10.1182/blood.2022015474
54. Laudisi F, Stolfi C, Monteleone I, Monteleone G. TGF-β1 signaling and Smad7 control T-cell responses in health and immune-mediated disorders. Eur J Immunol. (2023) 53:e2350460. doi: 10.1002/eji.202350460
55. Bao S, Jiang X, Jin S, Tu P, Lu J. TGF-β1 induces immune escape by enhancing PD-1 and CTLA-4 expression on T lymphocytes in hepatocellular carcinoma. Front Oncol. (2021) 11:694145. doi: 10.3389/fonc.2021.694145
56. Liang R, Zhu X, Lan T, Ding D, Zheng Z, Chen T, et al. TIGIT promotes CD8(+)T cells exhaustion and predicts poor prognosis of colorectal cancer. Cancer Immunol Immunother. (2021) 70:2781–93. doi: 10.1007/s00262-021-02886-8
57. Alabanza LM, Xiong Y, Vu B, Webster B, Wu D, Hu P, et al. Armored BCMA CAR T cells eliminate multiple myeloma and are resistant to the suppressive effects of TGF-β. Front Immunol. (2022) 13:832645. doi: 10.3389/fimmu.2022.832645
58. Jirtle RL. Chemical modification of tumour blood flow. Int J Hyperthermia. (1988) 4:355–71. doi: 10.3109/02656738809016490
59. Salman S, Meyers DJ, Wicks EE, Lee SN, Datan E, Thomas AM, et al. HIF inhibitor 32-134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J Clin Invest. (2022) 132:e156774. doi: 10.1172/JCI156774
60. Yi X, Qi M, Huang M, Zhou S, Xiong J. Honokiol inhibits HIF-1α-mediated glycolysis to halt breast cancer growth. Front Pharmacol. (2022) 13:796763. doi: 10.3389/fphar.2022.796763
61. Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. (2022) 29:1769–89. doi: 10.1038/s41418-022-00963-8
62. Li X, Li Y, Liu B, Chen L, Lyu F, Zhang P, et al. P4HA2-mediated HIF-1α stabilization promotes erdafitinib-resistance in FGFR3-alteration bladder cancer. FASEB J. (2023) 37:e22840. doi: 10.1096/fj.202201247R
63. Panahi Meymandi AR, Akbari B, Soltantoyeh T, Shahosseini Z, Hosseini M, Hadjati J, et al. PX-478, an HIF-1α inhibitor, impairs mesoCAR T cell antitumor function in cervical cancer. Front Oncol. (2024) 14:1357801. doi: 10.3389/fonc.2024.1357801
64. Luo F, Lu FT, Cao JX, Ma WJ, Xia ZF, Zhan JH, et al. HIF-1α inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non-small cell lung cancer. Cancer Lett. (2022) 531:39–56. doi: 10.1016/j.canlet.2022.01.027
65. Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang H, et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and Malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. (2021) 6:76. doi: 10.1038/s41392-020-00453-8
66. Xu F, Hu Y, Gao J, Wang J, Xie Y, Sun F, et al. HIF-1α/Malat1/miR-141 axis activates autophagy to increase proliferation, migration, and invasion in triple-negative breast cancer. Curr Cancer Drug Targets. (2023) 23:363–78. doi: 10.2174/1568009623666221228104833
67. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl). (2014) 92:1283–92. doi: 10.1007/s00109-014-1189-3
68. Liu X, Ye N, Liu S, Guan J, Deng Q, Zhang Z, et al. Hyperbaric oxygen boosts PD-1 antibody delivery and T cell infiltration for augmented immune responses against solid tumors. Adv Sci (Weinh). (2021) 8:e2100233. doi: 10.1002/advs.202100233
69. Yao X, Lu S, Feng C, Suo R, Li H, Zhang Y, et al. Tumor oxygenation nanoliposome synergistic hypoxia-inducible-factor-1 inhibitor enhanced Iodine-125 seed brachytherapy for esophageal cancer. Biomaterials. (2022) 289:121801. doi: 10.1016/j.biomaterials.2022.121801
70. Zhang YX, Zhao YY, Shen J, Sun X, Liu Y, Liu H, et al. Nanoenabled modulation of acidic tumor microenvironment reverses anergy of infiltrating T cells and potentiates anti-PD-1 therapy. Nano Lett. (2019) 19:2774–83. doi: 10.1021/acs.nanolett.8b04296
71. Yeung C, Gibson AE, Issaq SH, Oshima N, Baumgart JT, Edessa LD, et al. Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth in preclinical models of ewing sarcoma. Cancer Res. (2019) 79:5060–73. doi: 10.1158/0008-5472.CAN-19-0217
72. Khammanivong A, Saha J, Spartz AK, Sorenson BS, Bush AG, Korpela DM, et al. A novel MCT1 and MCT4 dual inhibitor reduces mitochondrial metabolism and inhibits tumour growth of feline oral squamous cell carcinoma. Vet Comp Oncol. (2020) 18:324–41. doi: 10.1111/vco.12551
73. Li Y, Yuan R, Luo Y, Guo X, Yang G, Li X, et al. A hierarchical structured fiber device remodeling the acidic tumor microenvironment for enhanced cancer immunotherapy. Adv Mater. (2023) 35:e2300216. doi: 10.1002/adma.202300216
74. Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin Cancer Res. (2020) 26:1712–24. doi: 10.1158/1078-0432.CCR-19-2179
75. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
76. Wang L, Luo Y, Ren S, Zhang Z, Xiong A, Su C, et al. A phase 1b study of ivonescimab, a programmed cell death protein-1 and vascular endothelial growth factor bispecific antibody, as first- or second-line therapy for advanced or metastatic immunotherapy-naive NSCLC. J Thorac Oncol. (2024) 19:465–75. doi: 10.1016/j.jtho.2023.10.014
77. Frentzas S, Austria Mislang AR, Lemech C, Nagrial A, Underhill C, Wang W, et al. Phase 1a dose escalation study of ivonescimab (AK112/SMT112), an anti-PD-1/VEGF-A bispecific antibody, in patients with advanced solid tumors. J Immunother Cancer. (2024) 12:e008037. doi: 10.1136/jitc-2023-008037
78. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
79. Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. (2021) 518:35–48. doi: 10.1016/j.canlet.2021.06.009
80. Xiao Z, Todd L, Huang L, Noguera-Ortega E, Lu Z, Huang L, et al. Desmoplastic stroma restricts T cell extravasation and mediates immune exclusion and immunosuppression in solid tumors. Nat Commun. (2023) 14:5110. doi: 10.1038/s41467-023-40850-5
81. Watabe T, Liu Y, Kaneda-Nakashima K, Shirakami Y, Lindner T, Ooe K, et al. Theranostics targeting fibroblast activation protein in the tumor stroma: (64)Cu- and (225)Ac-labeled FAPI-04 in pancreatic cancer xenograft mouse models. J Nucl Med. (2020) 61:563–9. doi: 10.2967/jnumed.119.233122
82. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of Malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. (2013) 11:187. doi: 10.1186/1479-5876-11-187
83. Das S, Valton J, Duchateau P, Poirot L. Stromal depletion by TALEN-edited universal hypoimmunogenic FAP-CAR T cells enables infiltration and anti-tumor cytotoxicity of tumor antigen-targeted CAR-T immunotherapy. Front Immunol. (2023) 14:1172681. doi: 10.3389/fimmu.2023.1172681
84. Liu Y, Sun Y, Wang P, Li S, Dong Y, Zhou M, et al. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J Transl Med. (2023) 21:255. doi: 10.1186/s12967-023-04080-z
85. Zhen Z, Tang W, Wang M, Zhou S, Wang H, Wu Z, et al. Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic T cell infiltration and tumor control. Nano Lett. (2017) 17:862–9. doi: 10.1021/acs.nanolett.6b04150
86. Zhang FF, Qiao Y, Xie Y, Liu C, Wu H, Wu JX, et al. Epitope-based minigene vaccine targeting fibroblast activation protein α induces specific immune responses and anti-tumor effects in 4 T1 murine breast cancer model. Int Immunopharmacol. (2022) 112:109237. doi: 10.1016/j.intimp.2022.109237
87. Xu M, Chen J, Zhang P, Cai J, Song H, Li Z, et al. An antibody-radionuclide conjugate targets fibroblast activation protein for cancer therapy. Eur J Nucl Med Mol Imaging. (2023) 50:3214–24. doi: 10.1007/s00259-023-06300-6
88. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. (2022) 21:104. doi: 10.1186/s12943-022-01569-x
89. Yang M, Qin C, Tao L, Cheng G, Li J, Lv F, et al. Synchronous targeted delivery of TGF-β siRNA to stromal and tumor cells elicits robust antitumor immunity against triple-negative breast cancer by comprehensively remodeling the tumor microenvironment. Biomaterials. (2023) 301:122253. doi: 10.1016/j.biomaterials.2023.122253
90. Pei Y, Chen L, Huang Y, Wang J, Feng J, Xu M, et al. Sequential targeting TGF-β Signaling and KRAS mutation increases therapeutic efficacy in pancreatic cancer. Small. (2019) 15:e1900631. doi: 10.1002/smll.201900631
91. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
92. Boulch M, Cazaux M, Cuffel A, Guerin MV, Garcia Z, Alonso R, et al. Tumor-intrinsic sensitivity to the pro-apoptotic effects of IFN-γ is a major determinant of CD4(+) CAR T-cell antitumor activity. Nat Cancer. (2023) 4:968–83. doi: 10.1038/s43018-023-00570-7
93. Hamieh M, Mansilla-Soto J, Rivière I, Sadelain M. Programming CAR T cell tumor recognition: tuned antigen sensing and logic gating. Cancer Discovery. (2023) 13:829–43. doi: 10.1158/2159-8290.CD-23-0101
94. Sánchez-Paulete AR, Mateus-Tique J, Mollaoglu G, Nielsen SR, Marks A, Lakshmi A, et al. Targeting macrophages with CAR T cells delays solid tumor progression and enhances antitumor immunity. Cancer Immunol Res. (2022) 10:1354–69. doi: 10.1158/2326-6066.CIR-21-1075
95. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. (2021) 12:877. doi: 10.1038/s41467-021-20893-2
96. Li YR, Brown J, Yu Y, Lee D, Zhou K, Dunn ZS, et al. Targeting immunosuppressive tumor-associated macrophages using innate T cells for enhanced antitumor reactivity. Cancers (Basel). (2022) 14:2749. doi: 10.3390/cancers14112749
97. Watanabe K, Gomez AM, Kuramitsu S, Siurala M, Da T, Agarwal S, et al. Identifying highly active anti-CCR4 CAR T cells for the treatment of T-cell lymphoma. Blood Adv. (2023) 7:3416–30. doi: 10.1182/bloodadvances.2022008327
98. Zannikou M, Duffy JT, Levine RN, Seblani M, Liu Q, Presser A, et al. IL15 modification enables CAR T cells to act as a dual targeting agent against tumor cells and myeloid-derived suppressor cells in GBM. J Immunother Cancer. (2023) 11:e006239. doi: 10.1136/jitc-2022-006239
99. Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined antitumor effects of sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol Ther. (2019) 27:1483–94. doi: 10.1016/j.ymthe.2019.04.020
100. Lu M, Zhang X, Gao X, Sun S, Wei X, Hu X, et al. Lenvatinib enhances T cell immunity and the efficacy of adoptive chimeric antigen receptor-modified T cells by decreasing myeloid-derived suppressor cells in cancer. Pharmacol Res. (2021) 174:105829. doi: 10.1016/j.phrs.2021.105829
101. Zanvit P, van Dyk D, Fazenbaker C, McGlinchey K, Luo W, Pezold JM, et al. Antitumor activity of AZD0754, a dnTGFβRII-armored, STEAP2-targeted CAR-T cell therapy, in prostate cancer. J Clin Invest. (2023) 133:e169655. doi: 10.1172/JCI169655
102. Li K, Xu J, Wang J, Lu C, Dai Y, Dai Q, et al. Dominant-negative transforming growth factor-β receptor-armoured mesothelin-targeted chimeric antigen receptor T cells slow tumour growth in a mouse model of ovarian cancer. Cancer Immunol Immunother. (2023) 72:917–28. doi: 10.1007/s00262-022-03290-6
103. Liang S, Zheng R, Zuo B, Li J, Wang Y, Han Y, et al. SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors. Cell Mol Immunol. (2024) 21:213–26. doi: 10.1038/s41423-023-01120-y
104. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5:e133977. doi: 10.1172/jci.insight.133977
105. Shiri AM, Zhang T, Bedke T, Zazara DE, Zhao L, Lücke J, et al. IL-10 dampens antitumor immunity and promotes liver metastasis via PD-L1 induction. J Hepatol. (2024) 80:634–44. doi: 10.1016/j.jhep.2023.12.015
106. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kückelhaus J, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun. (2022) 13:925. doi: 10.1038/s41467-022-28523-1
107. Xu Y, Zeng H, Jin K, Liu Z, Zhu Y, Xu L, et al. Immunosuppressive tumor-associated macrophages expressing interlukin-10 conferred poor prognosis and therapeutic vulnerability in patients with muscle-invasive bladder cancer. J Immunother Cancer. (2022) 10:e003416. doi: 10.1136/jitc-2021-003416
108. Zhang M, Chen J, Zhang H, Dong H, Yue Y, Wang S. Interleukin-10 increases macrophage-mediated chemotherapy resistance via FABP5 signaling in multiple myeloma. Int Immunopharmacol. (2023) 124:110859. doi: 10.1016/j.intimp.2023.110859
109. Sullivan KM, Jiang X, Guha P, Lausted C, Carter JA, Hsu C, et al. Blockade of interleukin 10 potentiates antitumour immune function in human colorectal cancer liver metastases. Gut. (2023) 72:325–37. doi: 10.1136/gutjnl-2021-325808
110. Chen N, Xu Y, Mou J, Rao Q, Xing H, Tian Z, et al. Targeting of IL-10R on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood Cancer J. (2021) 11:144. doi: 10.1038/s41408-021-00536-x
111. Salkeni MA, Naing A. Interleukin-10 in cancer immunotherapy: from bench to bedside. Trends Cancer. (2023) 9:716–25. doi: 10.1016/j.trecan.2023.05.003
112. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol. (2021) 22:746–56. doi: 10.1038/s41590-021-00940-2
113. Zhao Y, Chen J, Andreatta M, Feng B, Xie YQ, Wenes M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. (2024). doi: 10.1038/s41587-023-02060-8
114. Pang Z, Lu MM, Zhang Y, Gao Y, Bai JJ, Gu JY, et al. Neoantigen-targeted TCR-engineered T cell immunotherapy: current advances and challenges. biomark Res. (2023) 11:104. doi: 10.1186/s40364-023-00534-0
115. Idorn M, Skadborg SK, Kellermann L, Halldórsdóttir HR, Holmen Olofsson G, Met Ö, et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology. (2018) 7:e1450715. doi: 10.1080/2162402X.2018.1450715
116. Dao T, Xiong G, Mun SS, Meyerberg J, Korontsvit T, Xiang J, et al. A dual-receptor T-cell platform with Ab-TCR and costimulatory receptor achieves specificity and potency against AML. Blood. (2024) 143:507–21. doi: 10.1182/blood.2023021054
117. Karadimitris A. Cord blood CAR-NK cells: favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell. (2020) 37:426–7. doi: 10.1016/j.ccell.2020.03.018
118. Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. (2021) 14:73. doi: 10.1186/s13045-021-01083-5
119. Liu B, Zhu X, Kong L, Wang M, Spanoudis C, Chaturvedi P, et al. Bifunctional TGF-β trap/IL-15 protein complex elicits potent NK cell and CD8(+) T cell immunity against solid tumors. Mol Ther. (2021) 29:2949–62. doi: 10.1016/j.ymthe.2021.06.001
120. Thangaraj JL, Coffey M, Lopez E, Kaufman DS. Disruption of TGF-β signaling pathway is required to mediate effective killing of hepatocellular carcinoma by human iPSC-derived NK cells. Cell Stem Cell. (2024) 31:1327–1343.e5. doi: 10.1016/j.stem.2024.06.009
121. Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat Med. (2024) 30:772–84. doi: 10.1038/s41591-023-02785-8
122. Chen X, Zhao X, Mou X, Zhao J, Zhang Z, Zhang X, et al. PD-1-CD28-enhanced receptor and CD19 CAR-modified tumor-infiltrating T lymphocytes produce potential anti-tumor ability in solid tumors. BioMed Pharmacother. (2024) 175:116800. doi: 10.1016/j.biopha.2024.116800
123. Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discovery. (2021) 20:899–919. doi: 10.1038/s41573-021-00155-y
124. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
125. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
126. Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. (2022) 15:24. doi: 10.1186/s13045-022-01242-2
127. Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer. (2020) 1:681–91. doi: 10.1038/s43018-020-0075-x
128. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol. (2022) 40:127–37. doi: 10.1200/JCO.21.02229
129. Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. (2021) 22:198–211. doi: 10.1016/S1470-2045(20)30641-0
130. Shen L, Kato K, Kim SB, Ajani JA, Zhao K, He Z, et al. Tislelizumab versus chemotherapy as second-line treatment for advanced or metastatic esophageal squamous cell carcinoma (RATIONALE-302): A randomized phase III study. J Clin Oncol. (2022) 40:3065–76. doi: 10.1200/JCO.21.01926
131. Jenkins L, Jungwirth U, Avgustinova A, Iravani M, Mills A, Haider S, et al. Cancer-associated fibroblasts suppress CD8+ T-cell infiltration and confer resistance to immune-checkpoint blockade. Cancer Res. (2022) 82:2904–17. doi: 10.1158/0008-5472.CAN-21-4141
132. Choueiri TK, Powles T, Albiges L, Burotto M, Szczylik C, Zurawski B, et al. Cabozantinib plus nivolumab and ipilimumab in renal-cell carcinoma. N Engl J Med. (2023) 388:1767–78. doi: 10.1056/NEJMoa2212851
133. Pal SK, Albiges L, Tomczak P, Suárez C, Voss MH, de Velasco G, et al. Atezolizumab plus cabozantinib versus cabozantinib monotherapy for patients with renal cell carcinoma after progression with previous immune checkpoint inhibitor treatment (CONTACT-03): a multicentre, randomised, open-label, phase 3 trial. Lancet. (2023) 402:185–95. doi: 10.1016/S0140-6736(23)00922-4
134. Kelley RK, Ueno M, Yoo C, Finn RS, Furuse J, Ren Z, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2023) 401:1853–65. doi: 10.1016/S0140-6736(23)00727-4
135. Shitara K, Ajani JA, Moehler M, Garrido M, Gallardo C, Shen L, et al. Nivolumab plus chemotherapy or ipilimumab in gastro-oesophageal cancer. Nature. (2022) 603:942–8. doi: 10.1038/s41586-022-04508-4
136. Garassino MC, Gadgeel S, Speranza G, Felip E, Esteban E, Dómine M, et al. Pembrolizumab plus pemetrexed and platinum in nonsquamous non-small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 study. J Clin Oncol. (2023) 41:1992–8. doi: 10.1200/JCO.22.01989
137. Gao X, Xu N, Li Z, Shen L, Ji K, Zheng Z, et al. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): a multicentre, open-label, phase 1b/2 trial. Lancet Oncol. (2023) 24:1134–46. doi: 10.1016/S1470-2045(23)00411-4
138. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
139. Yang L, Li A, Wang Y, Zhang Y. Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy. Signal Transduct Target Ther. (2023) 8:35. doi: 10.1038/s41392-022-01304-4
140. Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. (2021) 374:1632–40. doi: 10.1126/science.aaz7015
141. Qu R, Zhang Y, Ma Y, Zhou X, Sun L, Jiang C, et al. Role of the gut microbiota and its metabolites in tumorigenesis or development of colorectal cancer. Adv Sci (Weinh). (2023) 10:e2205563. doi: 10.1002/advs.202205563
142. Li R, Zhou R, Wang H, Li W, Pan M, Yao X, et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. (2019) 26:2447–63. doi: 10.1038/s41418-019-0312-y
143. Peng Z, Wan P, Deng Y, Shen W, Liu R. Lipopolysaccharide exacerbates to the migration, invasion, and epithelial-mesenchymal transition of esophageal cancer cells by TLR4/NF-κB axis. Environ Toxicol. (2023) 38:1090–9. doi: 10.1002/tox.23750
144. Tripodi L, Feola S, Granata I, Whalley T, Passariello M, Capasso C, et al. Bifidobacterium affects antitumor efficacy of oncolytic adenovirus in a mouse model of melanoma. iScience. (2023) 26:107668. doi: 10.1016/j.isci.2023.107668
145. Overacre-Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A, et al. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity. (2021) 54:2812–24.e4. doi: 10.1016/j.immuni.2021.11.003
146. Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene. (2020) 39:4925–43. doi: 10.1038/s41388-020-1341-1
147. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. (2021) 371:602–9. doi: 10.1126/science.abb5920
148. Yadav MK, Kumari I, Singh B, Sharma KK, Tiwari SK. Probiotics, prebiotics and synbiotics: Safe options for next-generation therapeutics. Appl Microbiol Biotechnol. (2022) 106:505–21. doi: 10.1007/s00253-021-11646-8
149. Yonekura S, Terrisse S, Alves Costa Silva C, Lafarge A, Iebba V, Ferrere G, et al. Cancer induces a stress ileopathy depending on β-adrenergic receptors and promoting dysbiosis that contributes to carcinogenesis. Cancer Discovery. (2022) 12:1128–51. doi: 10.1158/2159-8290.CD-21-0999
150. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von Hippel-Lindau disease. N Engl J Med. (2021) 385:2036–46. doi: 10.1056/NEJMoa2103425
151. Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med. (2021) 27:802–5. doi: 10.1038/s41591-021-01324-7
152. Choueiri TK, McDermott DF, Merchan J, Bauer TM, Figlin R, Heath EI, et al. Belzutifan plus cabozantinib for patients with advanced clear cell renal cell carcinoma previously treated with immunotherapy: an open-label, single-arm, phase 2 study. Lancet Oncol. (2023) 24:553–62. doi: 10.1016/S1470-2045(23)00097-9
153. Halford S, Veal GJ, Wedge SR, Payne GS, Bacon CM, Sloan P, et al. A phase I dose-escalation study of AZD3965, an oral monocarboxylate transporter 1 inhibitor, in patients with advanced cancer. Clin Cancer Res. (2023) 29:1429–39. doi: 10.1158/1078-0432.CCR-22-2263
154. Dasari A, Lonardi S, Garcia-Carbonero R, Elez E, Yoshino T, Sobrero A, et al. Fruquintinib versus placebo in patients with refractory metastatic colorectal cancer (FRESCO-2): an international, multicentre, randomised, double-blind, phase 3 study. Lancet. (2023) 402:41–53. doi: 10.1016/S0140-6736(23)00772-9
155. Wang F, Jin Y, Wang M, Luo HY, Fang WJ, Wang YN, et al. Combined anti-PD-1, HDAC inhibitor and anti-VEGF for MSS/pMMR colorectal cancer: a randomized phase 2 trial. Nat Med. (2024) 30:1035–43. doi: 10.1038/s41591-024-02813-1
156. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. (2020) 21:808–20. doi: 10.1016/S1470-2045(20)30156-X
157. Zhou Q, Xu CR, Cheng Y, Liu YP, Chen GY, Cui JW, et al. Bevacizumab plus erlotinib in Chinese patients with untreated, EGFR-mutated, advanced NSCLC (ARTEMIS-CTONG1509): A multicenter phase 3 study. Cancer Cell. (2021) 39:1279–91.e3. doi: 10.1016/j.ccell.2021.07.005
158. Prager GW, Taieb J, Fakih M, Ciardiello F, Van Cutsem E, Elez E, et al. Trifluridine-tipiracil and bevacizumab in refractory metastatic colorectal cancer. N Engl J Med. (2023) 388:1657–67. doi: 10.1056/NEJMoa2214963
159. Xu RH, Zhang Y, Pan H, Feng J, Zhang T, Liu T, et al. Efficacy and safety of weekly paclitaxel with or without ramucirumab as second-line therapy for the treatment of advanced gastric or gastroesophageal junction adenocarcinoma (RAINBOW-Asia): a randomised, multicentre, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol. (2021) 6:1015–24. doi: 10.1016/S2468-1253(21)00313-7
160. Martin-Liberal J, Hollebecque A, Aftimos P, Jungels C, Martin-Romano P, Rodon J, et al. First-in-human, dose-escalation, phase 1 study of anti-angiopoietin-2 LY3127804 as monotherapy and in combination with ramucirumab in patients with advanced solid tumours. Br J Cancer. (2020) 123:1235–43. doi: 10.1038/s41416-020-1011-7
161. Lee EQ, Zhang P, Wen PY, Gerstner ER, Reardon DA, Aldape KD, et al. NRG/RTOG 1122: A phase 2, double-blinded, placebo-controlled study of bevacizumab with and without trebananib in patients with recurrent glioblastoma or gliosarcoma. Cancer. (2020) 126:2821–8. doi: 10.1002/cncr.32811
162. Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive Malignant pleural mesothelioma (FAPME-1). BMC Cancer. (2012) 12:615. doi: 10.1186/1471-2407-12-615
163. Fu H, Huang J, Zhao T, Wang H, Chen Y, Xu W, et al. Fibroblast activation protein-targeted radioligand therapy with 177Lu-EB-FAPI for metastatic radioiodine-refractory thyroid cancer: first-in-human, dose-escalation study. Clin Cancer Res. (2023) 29:4740–50. doi: 10.1158/1078-0432.CCR-23-1983
164. Hiltbrunner S, Britschgi C, Schuberth P, Bankel L, Nguyen-Kim TDL, Gulati P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol. (2021) 32:120–1. doi: 10.1016/j.annonc.2020.10.474
165. Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Perez Gracia JL, Stephen Hodi F, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J Immunother Cancer. (2022) 10:e004076. doi: 10.1136/jitc-2021-004076
166. Beckermann KE, Patnaik A, Winer I, Tan W, Bashir B, Kyriakopoulos CE, et al. A phase 1b open-label study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of py314 in combination with pembrolizumab in patients with advanced renal cell carcinoma. Invest New Drugs. (2024) 42:179–84. doi: 10.1007/s10637-024-01419-1
167. Hong DS, Postow M, Chmielowski B, Sullivan R, Patnaik A, Cohen EEW, et al. Eganelisib, a first-in-class PI3Kγ Inhibitor, in patients with advanced solid tumors: results of the phase 1/1b MARIO-1 trial. Clin Cancer Res. (2023) 29:2210–9. doi: 10.1158/1078-0432.CCR-22-3313
168. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. (2020) 26:878–85. doi: 10.1038/s41591-020-0880-x
169. Bewersdorf JP, Shallis RM, Sharon E, Park S, Ramaswamy R, Roe CE, et al. A multicenter phase Ib trial of the histone deacetylase inhibitor entinostat in combination with pembrolizumab in patients with myelodysplastic syndromes/neoplasms or acute myeloid leukemia refractory to hypomethylating agents. Ann Hematol. (2024) 103:105–16. doi: 10.1007/s00277-023-05552-4
170. Apolo AB, Nadal R, Tomita Y, Davarpanah NN, Cordes LM, Steinberg SM, et al. Cabozantinib in patients with platinum-refractory metastatic urothelial carcinoma: an open-label, single-centre, phase 2 trial. Lancet Oncol. (2020) 21:1099–109. doi: 10.1016/S1470-2045(20)30202-3
171. Fujikawa K, Saito T, Kurose K, Kojima T, Funakoshi T, Sato E, et al. Integrated analysis of phase 1a and 1b randomized controlled trials; Treg-targeted cancer immunotherapy with the humanized anti-CCR4 antibody, KW-0761, for advanced solid tumors. PloS One. (2023) 18:e0291772. doi: 10.1371/journal.pone.0291772
172. Revenko A, Carnevalli LS, Sinclair C, Johnson B, Peter A, Taylor M, et al. Direct targeting of FOXP3 in Tregs with AZD8701, a novel antisense oligonucleotide to relieve immunosuppression in cancer. J Immunother Cancer. (2022) 10:e003892. doi: 10.1136/jitc-2021-003892
173. Sharma M, Khong H, Fa’ak F, Bentebibel SE, Janssen LME, Chesson BC, et al. Bempegaldesleukin selectively depletes intratumoral Tregs and potentiates T cell-mediated cancer therapy. Nat Commun. (2020) 11:661. doi: 10.1038/s41467-020-14471-1
174. Diab A, Gogas H, Sandhu S, Long GV, Ascierto PA, Larkin J, et al. Bempegaldesleukin plus nivolumab in untreated advanced melanoma: the open-label, phase III PIVOT IO 001 trial results. J Clin Oncol. (2023) 41:4756–67. doi: 10.1200/JCO.23.00172
175. Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discovery. (2020) 10:1158–73. doi: 10.1158/2159-8290.CD-19-1510
176. Davis EJ, Martin-Liberal J, Kristeleit R, Cho DC, Blagden SP, Berthold D, et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. J Immunother Cancer. (2022) 10:e004235. doi: 10.1136/jitc-2021-004235
177. van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fosså A, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. (2014) 15:966–74. doi: 10.1016/S1470-2045(14)70319-5
178. Brighton TA, Khot A, Harrison SJ, Ghez D, Weiss BM, Kirsch A, et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin Cancer Res. (2019) 25:3772–5. doi: 10.1158/1078-0432.CCR-18-3470
179. Moskowitz AJ, Ghione P, Jacobsen E, Ruan J, Schatz JH, Noor S, et al. A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of JAK/STAT targeting in T-cell lymphomas. Blood. (2021) 138:2828–37. doi: 10.1182/blood.2021013379
180. Dao KT, Gotlib J, Deininger MMN, Oh ST, Cortes JE, Collins RH Jr., et al. Efficacy of ruxolitinib in patients with chronic neutrophilic leukemia and atypical chronic myeloid leukemia. J Clin Oncol. (2020) 38:1006–18. doi: 10.1200/JCO.19.00895
181. Landen CN, Buckanovich RJ, Sill MW, Mannel RS, Walker JL, DiSilvestro PA, et al. Phase I and randomized phase II study of ruxolitinib with frontline neoadjuvant therapy in advanced ovarian cancer: an NRG oncology group study. J Clin Oncol. (2024) 42:2537–45. doi: 10.1200/JCO.23.02076
182. Bauer TM, Santoro A, Lin CC, Garrido-Laguna I, Joerger M, Greil R, et al. Phase I/Ib, open-label, multicenter, dose-escalation study of the anti-TGF-β monoclonal antibody, NIS793, in combination with spartalizumab in adult patients with advanced tumors. J Immunother Cancer. (2023) 11:e007353. doi: 10.1136/jitc-2023-007353
183. Rajan A, Abdul Sater H, Rahma O, Agajanian R, Lassoued W, Marté JL, et al. Efficacy, safety, and biomarker analyses of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with advanced non-small cell lung cancer. J Immunother Cancer. (2024) 12:e008480. doi: 10.1136/jitc-2023-008480
184. Yoo C, Javle MM, Verdaguer Mata H, de Braud F, Trojan J, Raoul JL, et al. Phase 2 trial of bintrafusp alfa as second-line therapy for patients with locally advanced/metastatic biliary tract cancers. Hepatology. (2023) 78:758–70. doi: 10.1097/HEP.0000000000000365
185. Yamazaki T, Gunderson AJ, Gilchrist M, Whiteford M, Kiely MX, Hayman A, et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single-arm, phase 2 trial. Lancet Oncol. (2022) 23:1189–200. doi: 10.1016/S1470-2045(22)00446-6
Keywords: tumor microenvironment, immunosuppression, targeted, immunotherapy, cancer
Citation: Wang L, Zhang L, Zhang Z, Wu P, Zhang Y and Chen X (2024) Advances in targeting tumor microenvironment for immunotherapy. Front. Immunol. 15:1472772. doi: 10.3389/fimmu.2024.1472772
Received: 30 July 2024; Accepted: 16 September 2024;
Published: 03 October 2024.
Edited by:
Wenxue Ma, University of California, San Diego, United StatesReviewed by:
Anjali Barnwal, Duke University, United StatesDongyao Wang, University of Science and Technology of China, China
Copyright © 2024 Wang, Zhang, Zhang, Wu, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinfeng Chen, ZmVuZ3hpbmNoZW4xOTg1QDE2My5jb20=; Yi Zhang, eWl6aGFuZ0B6enUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship