 Chunxiao Niu
Chunxiao Niu Jiyan Zhang
Jiyan Zhang- Department of Molecular Immunology, Beijing Institute of Basic Medical Sciences, Beijing, China
Erythroid cells are the most abundant cells in the human body. In addition to their established function in gas-transportation, erythroid cells at various stages of differentiation have recently been shown to have immunomodulatory roles. Red blood cells may serve as modulators of innate and adaptive immunity, while their immature counterparts, CD71+ erythroid cells (CECs) have important immunomodulatory functions in various contexts. CECs are abundant in human cord blood and placenta, where they contribute to fetomaternal tolerance. CECs also accumulate in patients with infections, tumors, and anemia, and effectively suppress T cells by producing high levels of arginase, reactive oxygen species, programmed death-ligand 1, transforming growth factor β, and/or interleukin-10. Here, we systematically summarize the immunomodulatory functions of erythroid cells and propose some potential therapeutic applications based on their characteristics.
1 Introduction
In vertebrates, red blood cells (RBCs) are abundant in the circulation and are the main medium for oxygen transportation in the blood. In recent years, several studies have demonstrated that erythroid cells have additional functions beyond oxygen transport. Given their high level of production, vast numbers, and whole-body distribution, understanding of the immunomodulatory effects of erythroid cells has potential to provide novel targets for future immunotherapy approaches.
The immunoregulatory effects of erythroid cells were first discovered over 70 years ago, in 1953, when Nelson RA Jr discovered the phenomenon of immune-adherence between microorganisms and erythrocytes, which caused an immunologically specific reaction and enhanced phagocytosis (1). Subsequently, in 1979, the immunosuppression mediated by splenic nucleated erythrocytes was first revealed (2), followed by the work of Conway de Macario in 1980, linking immunosuppression with erythropoiesis in irradiated spleen-cell-transferred C57BL/6J mice (3). These studies revealed that nucleated erythrocytes can suppress primary and secondary antibody-mediated responses in vivo (4). A few years later, nucleated erythrocytes, which inhibit B-cell proliferation in humoral immune responses, were named erythroid immunosuppressor cells (5). Recent studies have demonstrated that erythroid cells modulate both innate and adaptive immune responses (6). The aims of this review were to introduce the basic features of erythropoiesis and to summarize the immunomodulatory functions of RBCs and CD71+ erythroid cells (CECs).
2 Erythropoiesis
Erythropoiesis is a constant, multi-stage process, which takes approximately 14 days in adult humans, who produce almost 200 billion RBCs every day, while mice generate more than 7000 erythrocytes per second (7, 8). During adulthood, steady state RBC generation occurs in the bone marrow, while damaged and/or senescent RBCs are recognized, internalized, and digested by splenic red pulp macrophages and Kupffer cells in the liver. This cycle of production and clearance creates steady-state RBC life spans of approximately 120 and 60 days in humans and mice, respectively (9).
2.1 Developmental stages of erythropoiesis
2.1.1 Embryonic hematopoiesis
In human embryos, erythropoiesis first occurs in the yolk sac, then transfers to the fetal liver and spleen, and finally becomes established in the bone marrow (10). Blood islands form from the mesoderm layer in the yolk sac, where primitive erythroid progenitor cells differentiate into primitive erythroblasts (PEs), which produce embryonic hemoglobin (α2ϵ2) (11). During weeks 6–8 of gestation, erythro-myeloid progenitors (EMPs) from the yolk sac begin to transfer to the fetal liver and spleen. The liver becomes the primary site of erythropoiesis during weeks 10–28 of gestation, while the spleen is the primary producer of RBCs during the second trimester (12, 13). At the end of the second trimester, erythropoiesis transfers to the bone marrow, which becomes the primary site of erythropoiesis until birth; fetal hemoglobin is produced to facilitate oxygen transport across the placenta during this stage (14). After birth, fetal hemoglobin output gradually decreases and is replaced by the adult form of hemoglobin. (Figure 1).
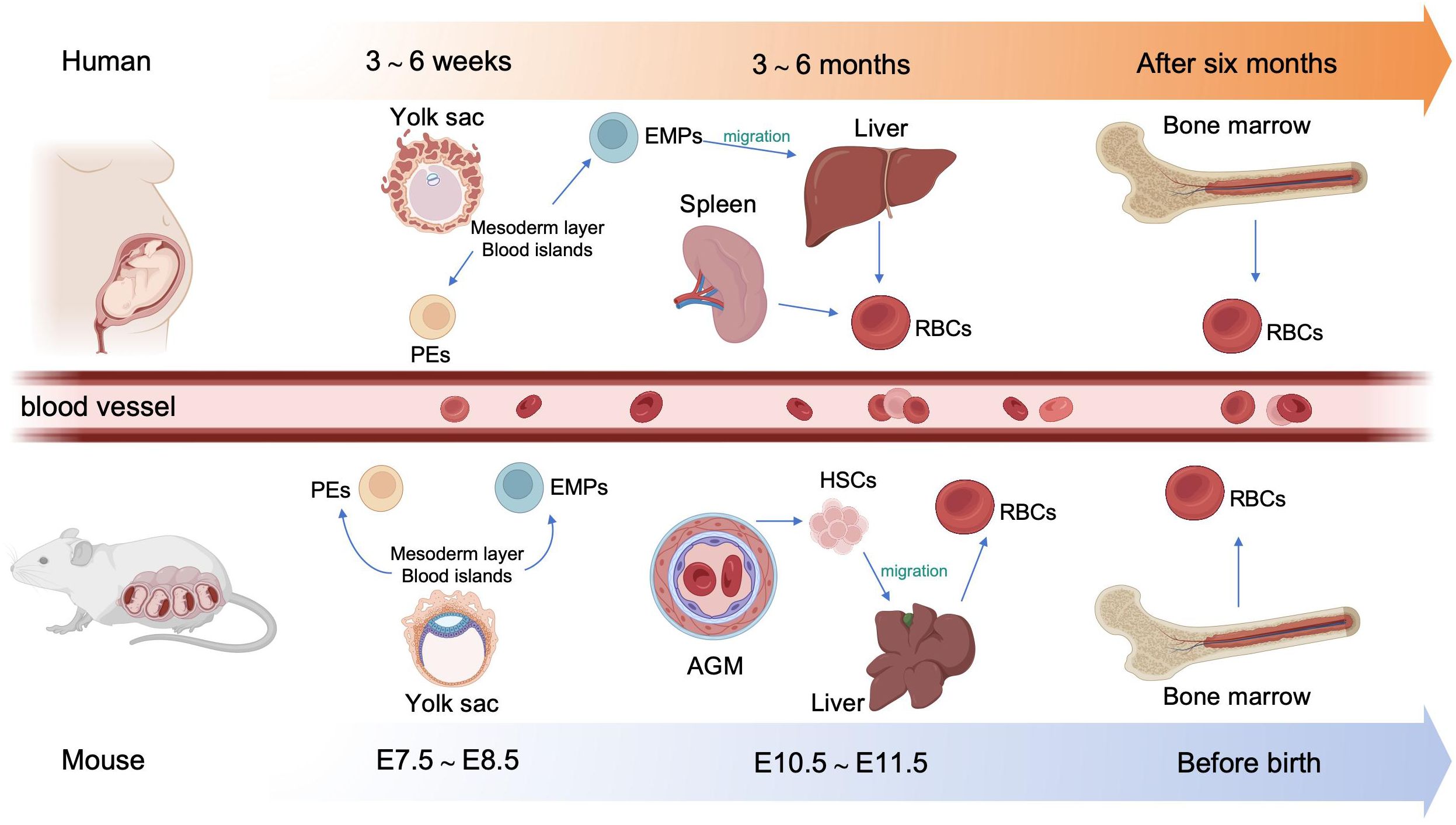
Figure 1. Overview of embryonic hematopoiesis in human and mouse. In human, erythropoiesis first occurs in the blood islands from the mesoderm layer of the yolk sac, generating primitive erythroblasts (PEs). Subsequently, erythromyeloid progenitors (EMPs) from the yolk sac migrate to the fetal liver and spleen. Finally erythropoiesis occurs in bone marrow. In mouse, primary erythropoiesis develops in the yolk sac. The yolk sac then atrophies and hematopoietic stem cells (HSCs) appear in aorta-gonad-mesonephros (AGM) region and transfer to the liver. Finally, erythropoiesis transfers to the bone marrow before birth.
In mouse embryos, hematopoiesis first emerges in the yolk sac at embryonic day 7.5 (E 7.5), and is characterized by the production of PEs, with diploid platelet progenitor cells and macrophages (15). Subsequently, EMPs emerge in the yolk sac at approximately E 8.25, which can generate erythroid colonies similar to those derived from adult bone marrow and have the capacity to produce multiple other myeloid lineages (16, 17). Soon afterwards, at around E10.5, hematopoietic stem cells (HSCs) appear in the dorsal aorta of the aorta-gonad-mesonephros region. Meanwhile, HSCs may also emerge from other hemogenic endothelial cells (ECs) within arteries in the umbilical cord, yolk sac, vitelline, cranial, and placental regions. These HSCs then migrate to the fetal liver, where they undergo a period of expansion, until they transfer to the bone marrow before birth (18) (Figure 1).
2.1.2 Stages of erythroid development
The development of erythroid cells during erythropoiesis can be divided into five stages. During the first stage, HSCs differentiate into megakaryocyte-erythroid progenitors (MEPs). The second stage is initiated by the differentiation of erythroid progenitor cells, followed by the appearance of burst-forming unit-erythroid (BFU-E) progenitors, and ends with the differentiation of colony-forming unit-erythroid (CFU-E) progenitors (19). The third stage begins with the development of pro-erythroblasts, followed sequentially by basophilic erythroblasts (Baso-E), polychromatic erythroblasts (Poly-E), and orthochromatic erythrocytes (Ortho-E). The fourth stage comprises reticulocytes, with mature erythroid cells formed in the fifth and final stage. Reticulocytes mature in the bone marrow, where they begin to eliminate mitochondria and other organelles, and subsequently enter the circulation to undergo further maturation into erythrocytes (20, 21). Erythroid cells gradually reduce their overall and nucleus size, while simultaneously increasing their hemoglobin content (10). Markers of erythropoiesis are listed in Table 1.
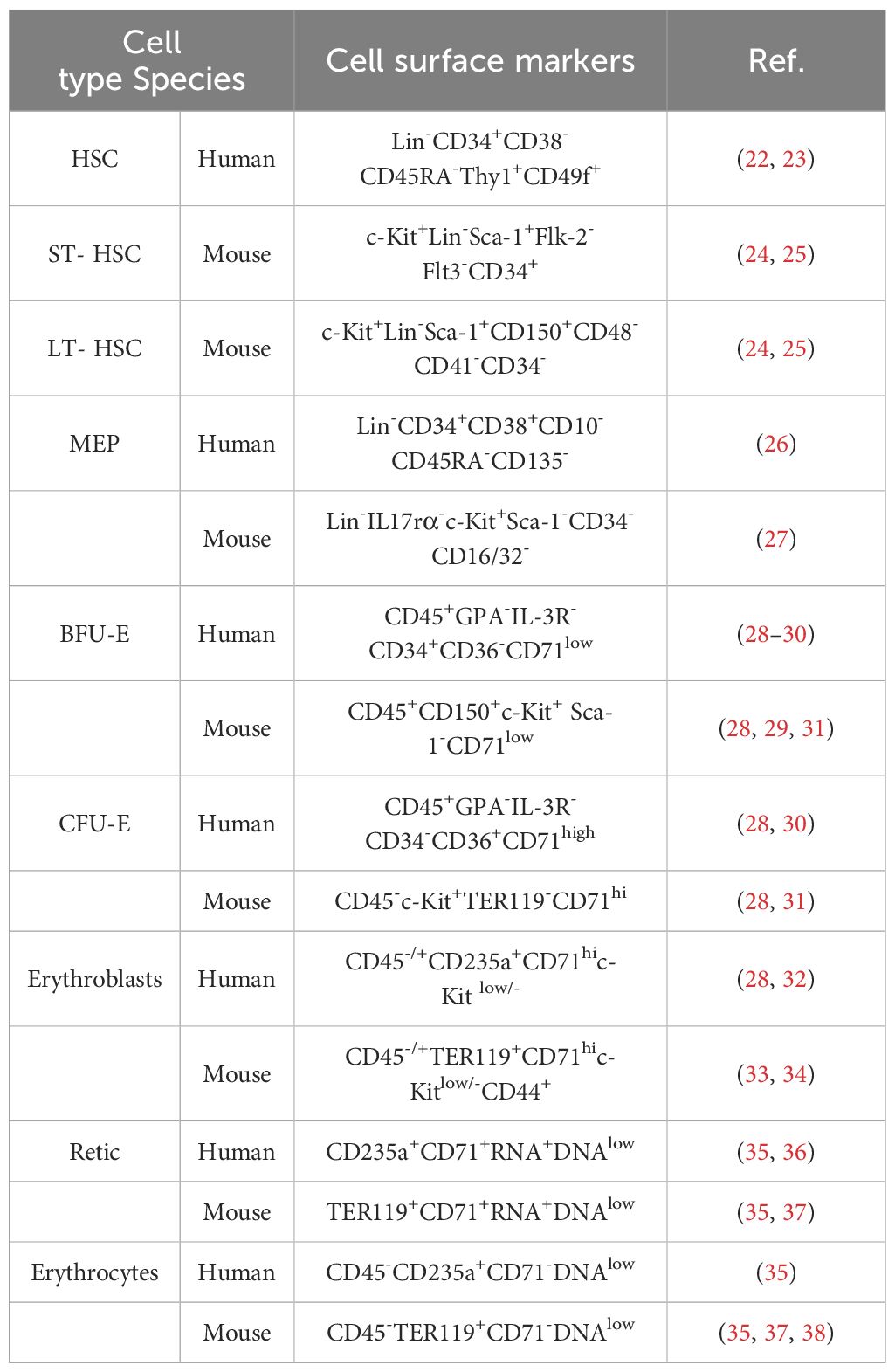
Table 1. Cell markers of the erythropoiesis.
2.2 Molecular regulation of erythropoiesis
The differentiation of HSCs to erythroid cells is regulated by various cytokines and growth factors (Table 2). The first stage of erythropoiesis is regulated by hematopoietic cytokines, such as stem cell factor (SCF; also known as c-Kit ligand), granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), thrombopoietin, IL-11, and transforming growth factor β (TGF-β). Further erythropoiesis is mainly regulated by erythropoietin (EPO), and iron metabolism is essential for hemoglobin synthesis. GATA1, GATA2, KLF1, and TAL1 are key transcription factors involved in erythropoiesis, while the transcription factors, FOG1, and BCL11A, regulate the expression of genes encoding enzymes associated with heme biosynthesis and hemoglobin production (62). Factors involved in the erythropoiesis are listed in Table 2.
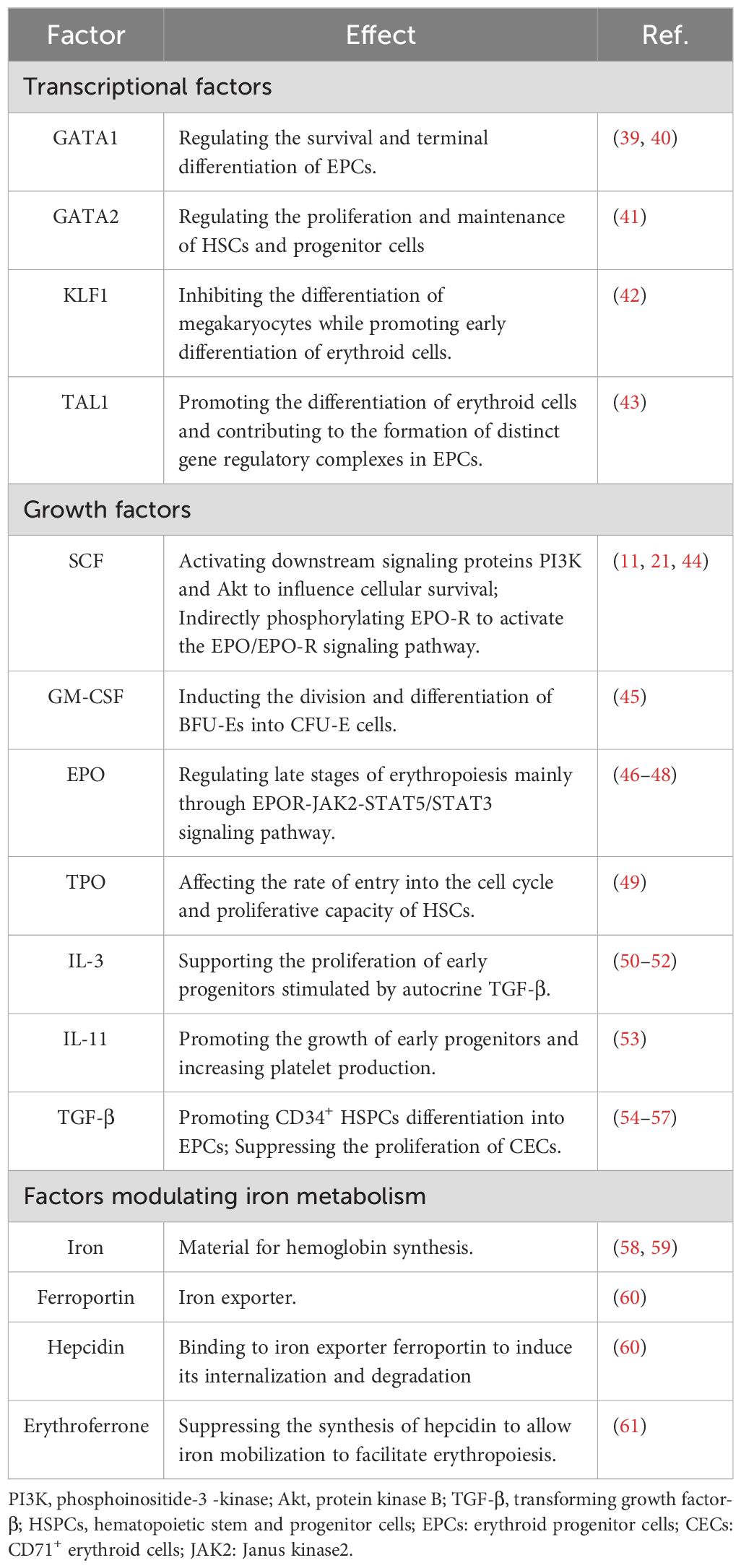
Table 2. Factors involved in the erythropoiesis.
2.3 Macrophages in erythropoiesis and erythrophagocytosis
2.3.1 Erythroblast islands
Erythroblastic islands (EBI), first discovered by Marcel Bessis in 1958, provide a specialized microenvironment for erythropoiesis (63). EBIs contain a central macrophage surrounded by maturing erythroblasts, and act as the erythroid precursor niche, which supports the bone marrow in producing RBCs at a rate of 2.5 million/second at homeostasis in adult humans (7). Terminal erythroid differentiation occurs within EBIs, where late CFU-Es mature into reticulocytes (64). Macrophages in the EBI secrete growth factors to support erythropoiesis, provide iron for hemoglobin synthesis, phagocytose extruded nuclei, and prevent toxic effects of free DNA (65, 66). Both mouse and human EBI macrophages express EPO-R, while EPO in the niche acts on erythroid cells and EBI macrophages simultaneously, to promote erythropoiesis. Under stress conditions (see section 2.4), RBCs are mainly produced through splenic erythropoiesis, which is distinct from bone marrow steady-state erythropoiesis (67). Impaired EPO-R signaling in splenic niche macrophages significantly inhibits the differentiation of stress erythroid progenitors (68). Further, EBI macrophage dysfunction can lead to specific erythroid hematological disorders (69).
2.3.2 Erythrophagocytosis
RBCs have a life span of around 120 days in the circulation. Macrophages have important roles in phagocytosis of aged or injured RBCs and contribute to iron recycling (70). RBC clearance is regulated by so called “eat me” and “don’t eat me” signals. Interaction of CD47 with SIRPα provides the “don’t eat me” signal (71). When RBCs undergo aging, “eat me” signals, such as phosphatidylserine (PS) and band 3, accumulate on their membranes in a process termed eryptosis (72). PS binds to Tim-1, Tim-4, Stabilin-2, or CD300 on macrophages, generating a pro-phagocytic signal, while band 3 interacts with CR-1 and Fc receptors to facilitate phagocytosis (73). PS also binds to platelets and ECs, which triggers pro-thrombotic risk and compromises the microcirculation (72). Enhanced eryptosis is observed in several clinical conditions, including malignancies (72). Tumor cells can directly interact with RBCs via galectin-4, leading to RBC aggregation (74). Together, RBC aggregation and augmented RBC adherence to the vascular wall due to enhanced eryptosis enable circulating tumor cells to stably roll along the vessel wall at a lower flow rate (75).
2.4 Stress erythropoiesis
Stress erythropoiesis is a stem cell-based tissue regeneration response that occurs in the spleen and fetal liver (76). Anemia or hypoxia accompanied by inflammation, which occur frequently during cancer development (77, 78), chronic infection (79), severe trauma (80), and chronic psychological stress (81, 82), disrupt the homeostasis between erythroid cell production through steady-state erythropoiesis and clearance of senescent or damaged erythroid cells by phagocytes, inducing stress erythropoiesis (79, 83); this process is regulated by bone morphogenetic protein 4 (BMP4), SCF, Hedgehog, EPO, growth-differentiation factor 15 (Gdf15), and glucocorticoids (GCs) (Figure 2) (84). Under homeostatic conditions, low EPO levels support terminal differentiation of only the most EPO-sensitive progenitors, while other erythroid progenitors undergo apoptosis; however, during stress erythropoiesis, increased EPO levels induce massive and rapid terminal differentiation of all erythroid progenitors (56). In addition, BMP4 and Hedgehog signals restrict the transition of short-term-HSCs to EPO-sensitive stress erythroid progenitors. Immature stress-induced erythroid progenitors maintain stem cell properties, including self-renewal, and can be serially transplanted (84–87). Further, BMP4 and SCF are required for the expansion of stress BFU-E spleen cells under hypoxic conditions (88), while Gdf15 regulates murine stress erythroid progenitor proliferation and controls stress erythropoiesis niche development (89). GCs are also essential for immature erythroid cell expansion during stress erythropoiesis, and act by binding and modulating the transcriptional activity of their cognate nuclear receptor, glucocorticoid receptor (GR) (90).
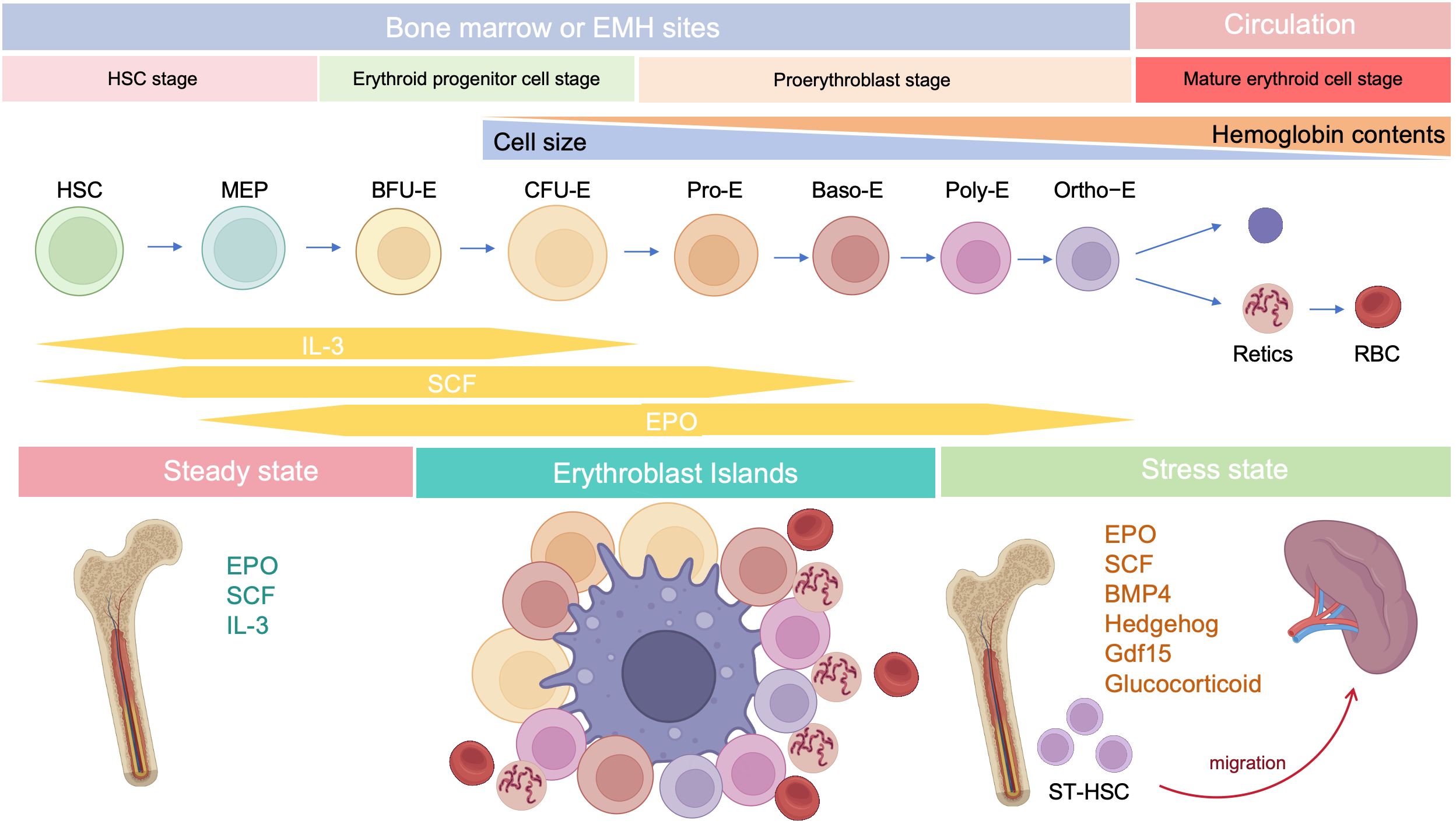
Figure 2. Developmental stages of erythropoiesis after birth. Under steady state, erythropoiesis occurs in the bone marrow, while stress erythropoiesis occurs mainly in the spleen. Erythropoiesis occurs in erythroblastic islands, which contains a central macrophage surrounded by developing erythroid cells.
Although biomarkers of BFU-E erythroid progenitors (Lin-cKit+CD71LowCD150+CD9+Sca-1-) responsive to stress erythropoiesis in the murine spleen are the same as those detected during steady state (91), whole genome transcriptional analysis demonstrated that mouse stress-BFU-E gene signatures are more BMP4-responsive and associated with erythropoiesis and proliferation, relative to those detected in the steady-state (92). Single-cell RNA-seq analysis of human stress-induced erythroid progenitors also revealed a distinct sub-population to that observed under steady-state erythropoiesis (93). Furthermore, splenic BFU-E exhibit different growth properties to their bone marrow counterparts; splenic BFU-E require only EPO to form colonies, while bone marrow BFU-E require EPO and a second factor (94).
Factors upstream of stress erythropoiesis have fundamental immunomodulatory effects. EPO is the principal cytokine regulating erythropoiesis through EPOR; however, EPOR is expressed not only on erythroid cells, but also on immune cells, such as macrophages, dendritic cells (DCs), mast cells, and lymphocytes (40). EPO can bind to EPOR and tissue-protective receptor (TPR, an EPOR/CD131 heterodimer), which are important in tissue protection and immune regulation (95). EPO inhibits the induction of genes encoding proinflammatory factors, such as TNF-α and inducible nitric oxide (NO) synthase (iNOS), in activated macrophages by decreasing NF-κB p65 activation (96). In addition, EPO suppresses DC maturation through the Jak2/STAT-3/SOCS1 pathway (97). Furthermore, EPO directly promotes regulatory T cell (Treg) proliferation, while inhibiting the expansion of conventional T cells via molecular crosstalk with the IL-2 pathway (98). GCs are required for regulation of stress erythroid progenitor expansion (90); however, GR signaling also has potent anti-inflammatory effects (99). Stress erythropoiesis produces more RBCs and CECs, and both populations possess considerable immunomodulatory functions under various conditions (see below for further details).
3 Immunomodulatory effects of RBCs
The link between RBCs and immune function was reported as early as 1953, when Nelson RA Jr. discovered the phenomenon of immune-adherence between erythrocytes and microorganisms, which augments phagocytosis (1). In 1991, RBCs were reported to bind to IL-8 and prevent its release into the blood, thereby limiting leukocyte stimulation (100). Data reported in 1993 demonstrated that RBCs can bind to several chemokine superfamily inflammatory peptides, indicating that RBCs may act as regulators of inflammatory processes (101). Unlike healthy RBCs, RBCs carrying mitochondria (Mito+ RBCs) augment inflammation. Furthermore, oxidative stress and RBC senescence generate a forward feedback cycle, resulting in the release of pro-inflammatory microparticles (MPs), Hb, heme, and iron, and the breakdown products generated by hemolysis have remarkable effects on immunological functions (Figure 3).
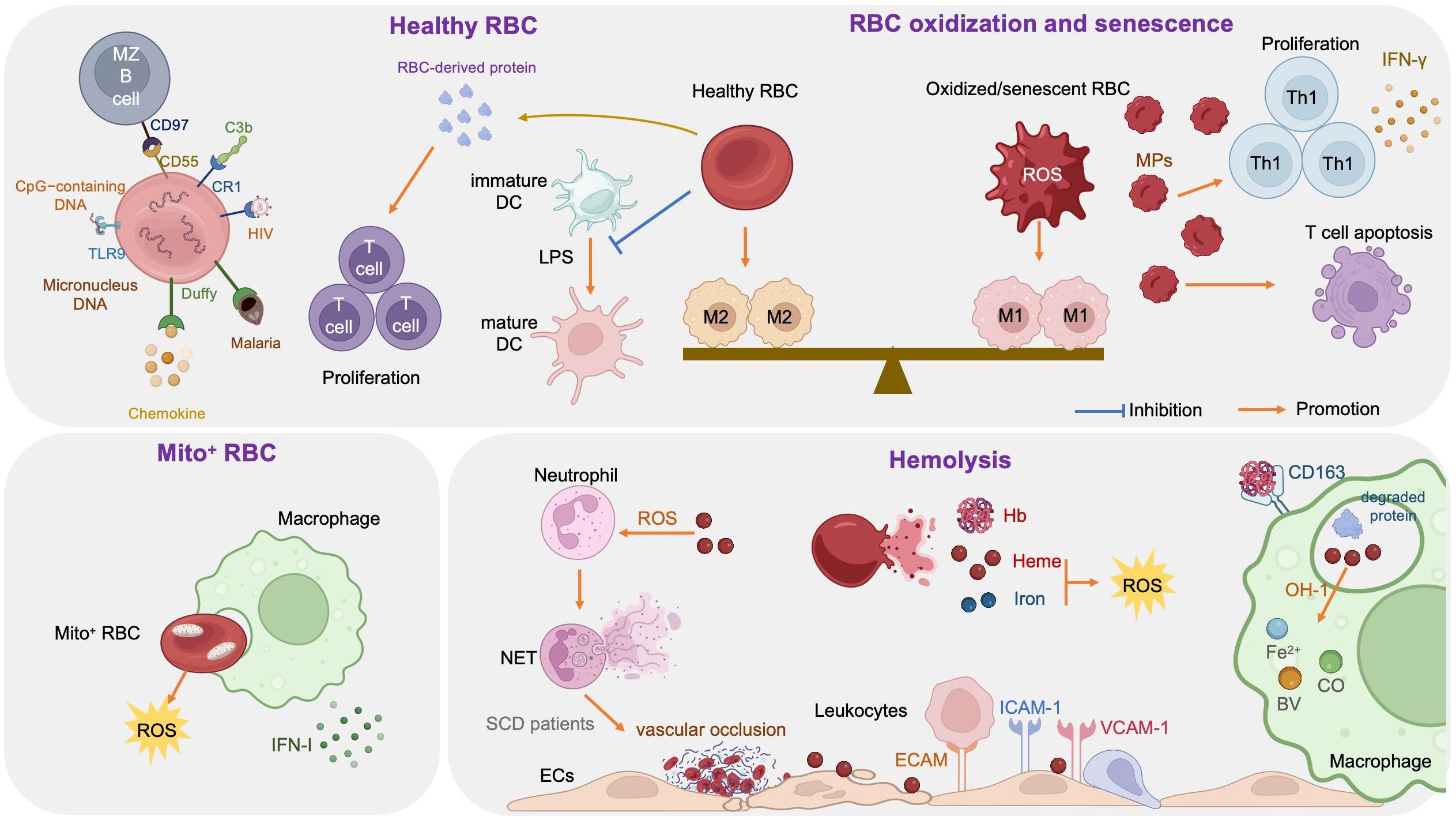
Figure 3. Immune regulation effects of red blood cells (RBCs).
3.1 Healthy RBCs and immune regulation
RBCs modulate innate and adaptive immunity mainly through their surface molecules (proteins, lipids, and carbohydrates) and potent antioxidant capacity (102); they express large amounts of the key complement regulators, CD55, CD59, and complement receptor type 1 (CR1, also referred to as CD35), where CD55 inactivates C3 convertases generated by all three complement activation pathways, CD59 prevents membrane attack complex formation by preventing C9 incorporation, and CR1 recognizes collagen-like regions of C1q, mannose-binding lectin, C3b, and C4b, to remove complement-tagged inflammatory particles. For example, in patients with HIV, the virus binds to RBCs via C1q-CR1 interaction (103). Further, CR1 is decreased on RBCs in patients with coronavirus disease 2019 (COVID-19), resulting in consistent inflammatory responses and tissue damage (104, 105).
Toll-like receptor 9 (TLR9), a nucleic acid sensing receptor, is expressed on the surface of mammalian RBCs. Under basal conditions, RBCs bind cell-free mitochondrial DNA (mtDNA) through TLR9 and mediate DNA scavenging to prevent unnecessary inflammation (106). Further, in the context of inflammation, TLR9 binds to CpG-containing DNA derived from bacteria, plasmodia, and mitochondria, which drives innate immune activation and red cell clearance (107). Erythroid-specific TLR9 deletion blocks erythrophagocytosis and decreases local and systemic cytokine production (107). During viral pneumonia and sepsis secondary to COVID-19, RBCs also exhibit protein oxidation, together with decreased antioxidant capacity, increased glycolysis, an altered membrane lipidome, and elevated mtDNA binding, which may contribute to anemia and disease severity (102, 108).
Moreover, RBCs can induce DCs toward an immature/tolerogenic phenotype in response to lipopolysaccharide (LPS), through a CD47-dependent mechanism (109). Mechanosensing by RBCs also ensures exposure of splenic type-2 conventional DCs to blood flow, allowing them to capture circulating antigens, while retaining them in the spleen through CD55-CD97 signaling (110); the same mechanism is also important for marginal B cell retention and function (111). In addition, Duffy blood antigen, primarily expressed on the surface of RBCs, can potently bind to multiple chemokines (112, 113).
Transfusion of fresh RBCs under noninflammatory conditions will reduce RBC clearance and therefore lessen macrophage loading with heme, as well as up-regulating heme oxygenase (HO), shifting macrophages toward the anti-inflammatory M2 state (112). Protein factors released from RBCs, such as Hb and peroxiredoxin II, can sustain normal and leukemic T cell growth and survival (113). RBCs can also synergize with TCR/CD3-mediated activation signals and enhance T cell survival and proliferation through a calcineurin-dependent mechanism (111).
3.2 Mito+ RBCs and immune regulation
Programmed mitochondrial removal occurs during normal erythropoiesis (114). A hypoxia-inducible factor-mediated metabolic switch and consequent activation of the ubiquitin-proteasome system precede, and are necessary for, autophagic mitochondria removal, and disruption of this pathway leads to accumulation of RBCs carrying mitochondria (Mito+ RBCs) (115). This process is defective in patients with systemic lupus erythematosus (SLE) (115) and sickle cell disease (SCD) (116), as well as in aged mtDNA mutator mice (117). In patients with SLE, Mito+ RBC levels are correlated with disease activity, and antibody-mediated Mito+ RBC internalization by macrophages induces type I interferon (IFN-I) production through cGAS/STING activation (115), while Mito+ RBCs may contribute to SCD pathophysiology via high reactive oxygen species (ROS) production (118).
3.3 Oxidized or senescent RBCs and immune regulation
RBCs are frequently exposed to various stressful conditions during their lifespan, including oxidative stress, osmotic shock, and mechanical squeezing (119), and consequently accumulate damage which influences their functions. Senescent RBCs show pathologic properties, including decreased deformability (120), MP release (121), increased hemin-carrying Hb (122), and surface antigen modification (123). RBC senescence occurs alongside oxidative stress and in turn becomes a source of ROS, which serves as an important signal of RBC senescence (124). Accumulation of oxidized lipids, such as 4-hydroxynonenal, may induce vascular inflammation (125, 126). At the molecular level, the major features of senescent RBCs are Band 3 clustering or breakdown (127), PS externalization (128), loss of glycophorin A, and reduction of CD47 expression (124). Consequently, senescent RBCs lose the ability to control LPS-induced DC maturation (129).
Oxidized or senescent RBCs or RBC-derived MPs are potential modifiers of T cell responses, which enhance mitogen-driven T cell proliferation and apoptosis through an antigen presenting cell- and cell contact-dependent mechanism, and regulate IFN-γ production from T helper 1 cells (124). Moreover, oxidized RBCs release Hb, heme, and iron which are both sources of radicals and able to activate ECs (130) and innate immune cells, such as monocytes (131), in a proinflammatory manner, as detailed below (see section 3.4). Stored RBCs display senescence-related changes, such as reduced structural integrity, MP release, and iron overload, and the transfusion of stored RBCs exacerbates existing lung inflammation and promotes lung injury, due to loss of Duffy antigen expression and their chemokine scavenging function during storage (132). Further, rapid clearance of transfused stored RBCs by macrophages polarizes the macrophages toward the classical M1 phenotype, with a huge Hb iron load (112, 133). In addition, packed RBCs suppress T cell proliferation via cell-cell contact and inhibit T cell activation via ROS-dependent signaling (134).
3.4 Hemolysis and immune regulation
RBCs are highly differentiated cells with an elegant structure that allows them to survive under continuous shear stress when transiting, making them ideal messengers between distant organs. The erythrocyte membrane skeleton is a polygonal 2D lattice structure, consisting of lipids, proteins, and carbohydrates (135, 136). The skeleton attaches to the cell membrane through the spectrin-actin junctional complex (adducin, dematin, and P4.1 interact with band 3, GPC/D, and Glut1) and the ankyrin complex (137). Disorders of the RBC cytoskeleton or dehydration cause hemolytic anemia, which is associated with altered immune regulation, as hemolysis breakdown products, including hemoglobin, heme, and iron, have remarkable effects on immunological functions (138).
3.4.1 Hemoglobin
Hemoglobin (Hb) is an iron-containing protein in RBCs formed from globin and heme (Fe2+ protoporphyrin-IX). When large amounts of Hb are released into the plasma from damaged RBCs, the scavenger protein haptoglobin (Hp) can rapidly bind with cell-free Hb, to generate a Hb-Hp complex, which neutralizes the pro-oxidative effects of Hb (139). When Hp binding capacity is saturated, heme in free Hb is easily oxidized to hemin (Fe3+ protoporphyrin-IX) in the circulation. Free Hb triggers vascular and organ dysfunction through extravascular translocation, NO inactivation, oxidative reactions, hemin release, and activation of downstream signaling pathways (see section 3.4.2) (139). Hp-Hb complexes bind to the CD163 receptor expressed on macrophages and hepatocytes and are subsequently digested, releasing heme into the cytoplasm (140). Free Hb enhances platelet activation by binding to ADP, as well as by abrogating the inhibitory effect of NO (141).
3.4.2 Heme
Heme is an important iron-containing porphyrin molecule and with crucial roles in cell protection, apoptosis, inflammation, and immune disorders (142). Hydrophobic hemin intercalates into cell membranes. Hydrogen peroxide from various sources splits the heme ring and releases free redox-active iron, which catalytically amplifies ROS production. Consequently, heme regulates inflammation mainly by acting as a pro-oxidant in macrophages, neutrophils, and ECs (143). Furthermore, heme can selectively bind to receptors, transcription factors, and enzymes (89).
Heme stimulates monocyte differentiation into splenic red pulp macrophages and bone marrow macrophages by promoting degradation of the transcriptional repressor, BACH1, and consequent expression of the transcription factor, SPI-C (144). Heme can also act as a pro-inflammatory second hit in macrophages by aggravating LPS-induced TLR4 signaling, or induce an anti-inflammatory response (M2 macrophages) via induction of SPI-C and HO-1, an inducible isoform of HO (145). Moreover, heme impairs phagocytosis by inhibiting cytoskeleton dynamics through the DOCT8/Cdc42 signaling pathway (146). Heme can also induce Treg expansion in purified T cell-monocyte cocultures by upregulating HO-1 in nonclassical monocytes (138).
Heme promotes neutrophil migration by stimulating macrophage-derived leukotriene B4 (147) and activating protein kinase C and G-protein-coupled receptors in neutrophils (148, 149), which induce chemokine expression and ROS production. During neutrophil development in patients with SCD, heme regulates neutrophil differentiation and can cause defective oxidative burst through HO-1 induction (150). Heme can also inhibit neutrophil apoptosis in vitro through the phosphoinositide 3-kinase (PI3K) and NF-κB pathways (151). Further, heme can induce neutrophil extracellular trap (NET) formation through ROS signaling, to protect the host against infections (152); however, in patients with SCD, NETs can enhance the adherence of erythrocytes and platelets to the endothelium and induce vascular occlusion or lung injury (153).
Free heme interacts with ECs and stimulates the expression of adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1), endothelial cell adhesion molecule (ECAM), vascular cell adhesion molecule 1 (VCAM-1), P-selectin, and others, through heme-mediated ROS and NF-κB signaling pathways (154). Leukocytes attach tightly to endothelium through adhesion molecules and migrate to tissue parenchyma, which promotes vascular occlusion and subsequent tissue ischemia (154, 155). In addition, cell-free heme and heme-loaded microvesicles activate the complement system via the alternative pathway in both serum and on the surface of ECs. Heme also upregulates P selectin, C3aR, and C5aR expression, and downregulates that of CD46, on ECs, which contributes to endothelial damage and vascular occlusion in patients with SCD (156).
3.4.3 Iron
Free heme is catabolized by HO into three products: biliverdin, carbon monoxide (CO), and Fe2+ (142), where biliverdin is converted to bilirubin, and both CO and bilirubin have potent anti-inflammatory and antioxidant properties, whereas Fe2+ enhances oxidative stress, thereby promoting ferroptosis (157). Fe2+ binds to the iron storage protein, ferritin, which has cytoprotective and anti-oxidative effects, as well as a role in iron storage. Ferritin was first discovered as a suppressor of granulocyte and macrophage production in 1981 (158). Further studies demonstrated that ferritin comprises two functionally distinct subunits: ferritin H and L (159). H-ferritin can suppress T cell proliferation in response to mitogens and impairs B cell maturation (159), as well as helping to mediate the protective effect of HO-1 against oxidative stress (160). Moreover, H-ferritin is a negative regulator of CXC chemokine receptor 4 in receptor-mediated cell migration (161). L-ferritin overexpression in LPS-induced Raw264.7 cells can significantly decrease the production of pro-inflammatory cytokines (TNF-α, IL-1β) and NO and inhibit MAPK and NF-κB activation (162).
4 Immunomodulatory effects of CECs
The term CECs refers to immature erythroid cells, including erythroblasts and reticulocytes, which are physiologically enriched in the spleen and cord blood of neonates, but rare in adult bone marrow (45). CECs are characterized by expression of CD71 and glycoprotein A (CD235a)/glycoprotein A-related protein (Ter119), as CD71+TER119+ cells in mice and CD71+CD235a+ cells in humans. CD71 is also known as transferrin receptor 1 (TfR-1), a type II transmembrane protein important in cellular iron uptake and iron metabolism (163). CD71 is a surface marker for erythroid cells from BFU-E to reticulocytes, which first appears in BFU-E, reaches its highest expression levels in Baso-E and Poly-E cells, then declines in Ortho-E cells, and finally disappears in mature erythrocytes (33).
There are three general CEC subtypes: early-stage CECs, EDMCs, and late-state CECs, each with differing immunosuppressive abilities. Erythropoietic tracking showed that CD45+CD71+TER119+ cells are enriched with stage I–III precursors, while CD45-CD71+TER119+ cells contain more terminally differentiated stage III–V erythroid cells (164). Recent studies have indicated that CECs at the earliest stages are more potent immune response suppressors (164–166). The various surface markers and functional properties of the three CEC subtypes are summarized in Table 3, the immunomodulatory effects of the CECs are summarized in Table 4 and Figure 4, and the immunomodulatory effects of the CECs in diseases are summarized in Table 5.
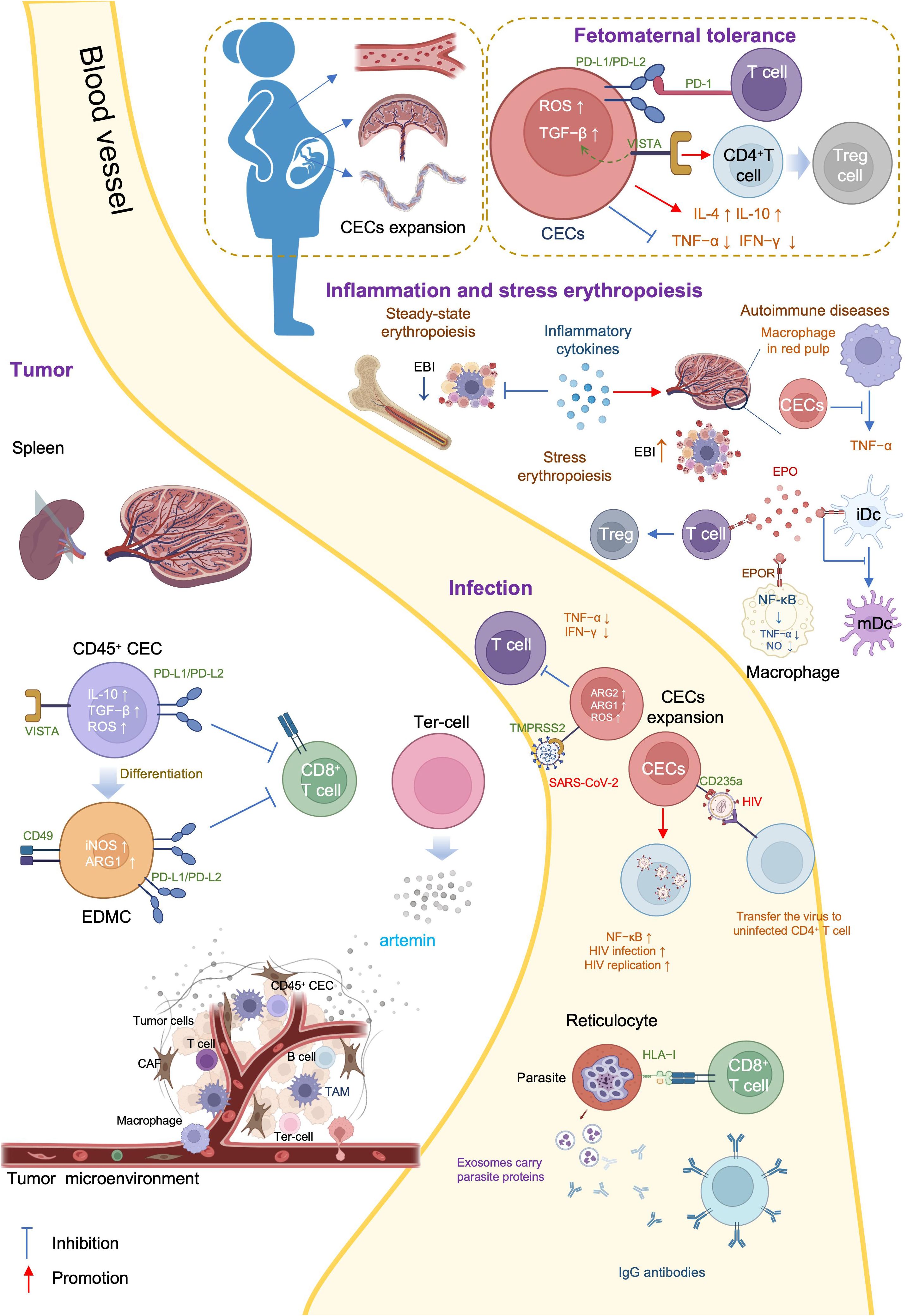
Figure 4. Mechanisms and immunoregulation effects of CECs.
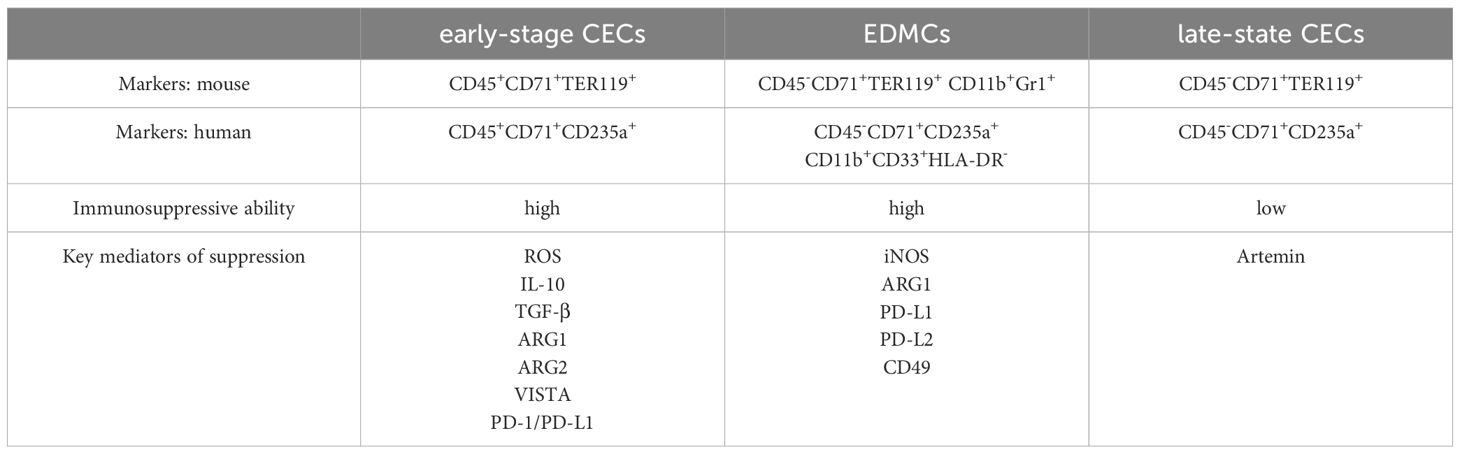
Table 3. Phenotypes of the CECs.
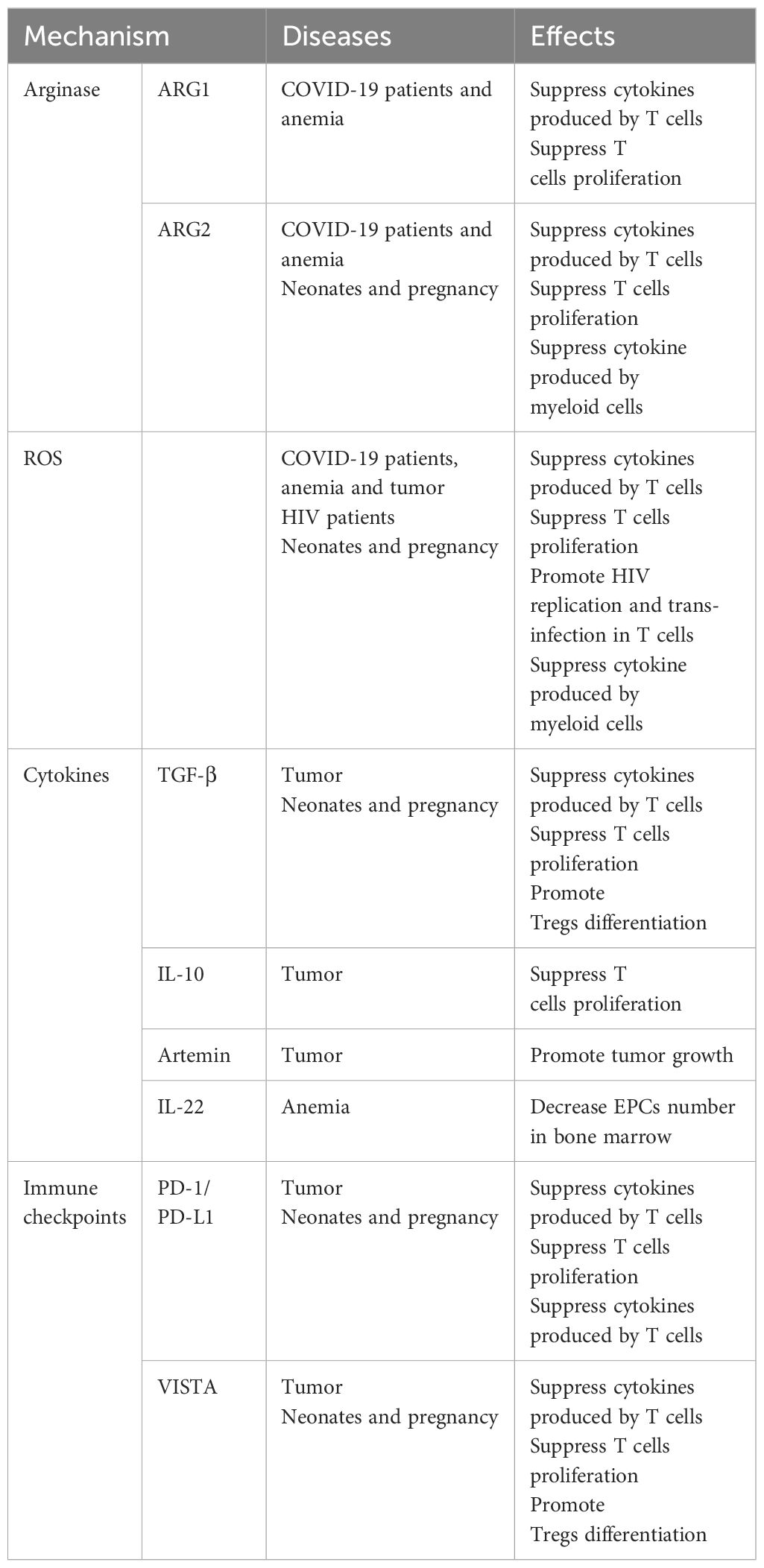
Table 4. Mechanisms of the CECs in immunoregulation.
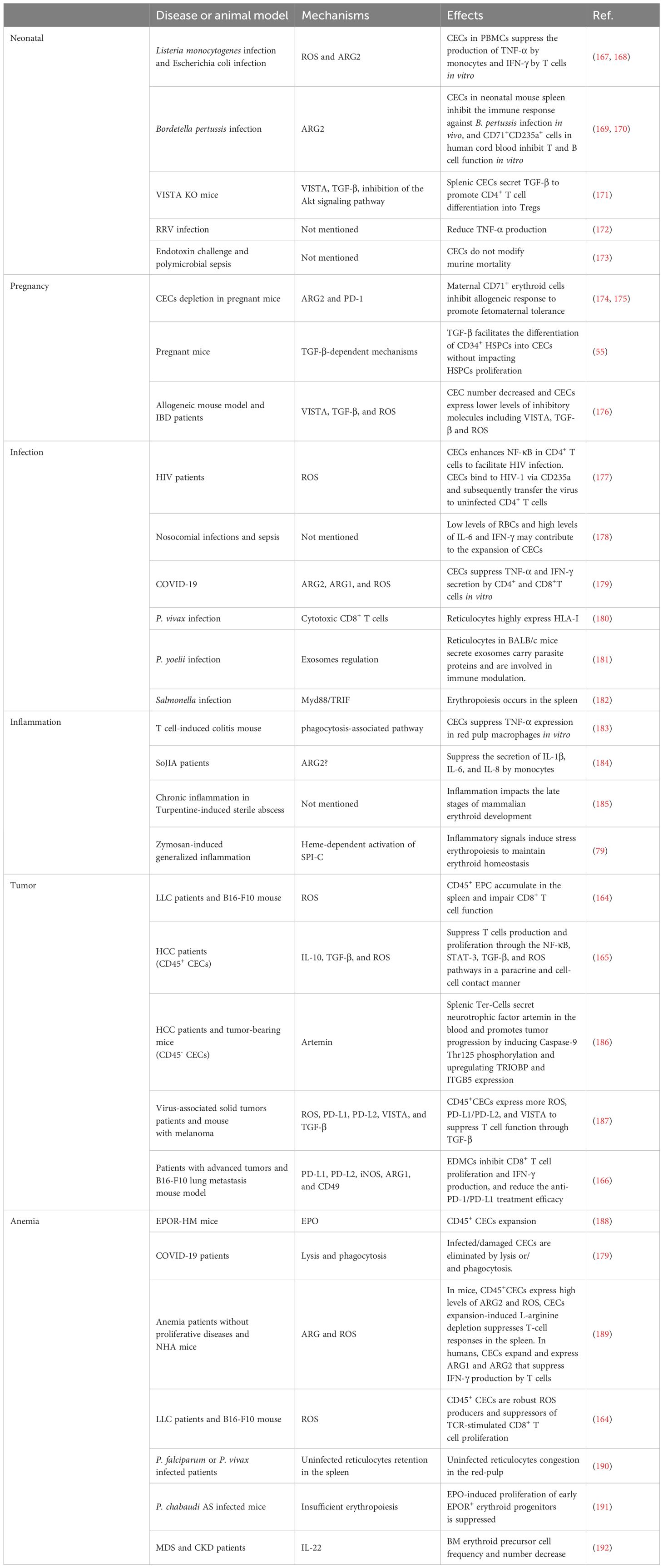
Table 5. Immunomodulatory effects of the CECs in diseases.
4.1 CECs in neonatal and pregnancy
Erythroid cells play a crucial role in immunological regulation during the neonatal period and in maternal-fetal tolerance. Mouse placental erythroid cells are mainly CD45+ and secrete the chemokines, CCL2, CCL3, CCL4, and CXCL1 (193). Further, CECs are abundant in mouse neonatal spleen and human cord blood, and possess unique immunosuppressive properties (167). CECs are abundant in the liver of children with biliary atresia (BA), and suppress the activation of hepatic mononuclear cells (172). Further, CECs are numerous in the peripheral blood of human newborns, but decline rapidly by 4 weeks of age (168).
CECs influence neonatal infections through various mechanisms. Bordetella pertussis is a common neonatal respiratory tract pathogen and CECs prevent the recruitment of immune cells to the mucosal infection site (167). CECs from human newborn peripheral blood mononuclear cells (PBMCs) suppress TNF-α production by CD14+ monocytes and IFN-γ production by T cells (168). Further, ablation of CECs enhances the innate immune response by increasing the production of protective cytokines, including IL-17, IFN-γ, TNF-α, and IL-12 in B. pertussis-infected lungs (169, 170).
L-arginine is essential for T cell proliferation and function (194). Arginase (ARG) depletes L-arginine, thereby inhibiting T cells, and is encoded by two recently-discovered genes, Arg1 and Arg2 (195). ARG1 is expressed in the cytosol, whereas ARG2 localizes to mitochondria. Neonatal and human cord blood CECs express ARG2 and ablation of CECs augments B. pertussis-specific T cell responses in the lung and spleen on re-infection or vaccination (170). In addition, ablation of CECs also induces enhanced systemic and mucosal B. pertussis-specific antibody responses (170). Accordingly, CECs in human cord blood can suppress T and B cell functions in vitro (170). Regarding innate immunity, CECs inhibit B. pertussis phagocytosis via ARG2 in vitro (169, 170). Such effects of CECs facilitate intestinal colonization with commensal microbes during the neonatal period (167). Depletion of CECs in neonatal mice renders them more resistant to infections by Listeria monocytogenes, Escherichia coli, and B. pertussis, indicating the protective effects against neonatal infectious diseases (167, 168); however, ablation of CD71+ cells failed to modify neonatal mortality in either a model of endotoxin challenge or a model of polymicrobial sepsis (173).
BA is a rare and progressive disease that develops in early infancy (196). Rhesus rotavirus (RRV) infection of neonatal mice induces an obstructive cholangiopathy, which is similar to BA (197). CECs expand in the liver of children with BA or RRV-infected mice and suppress the immune response by reducing TNF-α production. Preemptive depletion of hepatic CD71+ erythroid cells in neonatal mice augments the number of effector lymphocytes and delays RRV infection of the liver and extrahepatic bile duct, suppressing bile duct injury (172). Clearance of CECs before RRV infection renders mice resistant to RRV-induced BA, while repopulation of CD71+ erythroid cells after RRV inoculation promotes long-term survival (172).
CEC-mediated immunosuppression is crucial for fetomaternal tolerance. Both BALB/c and C57BL/6 female mice, and human women are enriched for CECs (198). Further, analysis of 155 umbilical cord blood samples showed that the proportion of CECs was 50-fold higher in cord blood than that in maternal blood (199). Erythropoiesis becomes active during pregnancy, and erythrocytes significantly expand in the peripheral blood (200). TGF-β has an important role in regulating the erythroid lineage differentiation potential of HSCs (201, 202). CECs in pregnant mice express more PD-L1/PD-L2 and suppress T cells expressing programmed cell death protein-1 (PD-1) at the fetomaternal interface (174). Maternal CECs inhibit IFN-γ and TNF-α production to protect the fetus against the allogeneic response. Further, fetal liver CECs also exhibit immunosuppressive activity. A recent transcriptional study demonstrated expression of high levels of galectin-9, galectin-1, and VISTA on the surface of neonatal splenic CECs. CD71+VISTA+ cells produce more TGF-β than CD71+VISTA− cells, and can promote CD4+ T cell differentiation into Tregs (171); however, CECs in human cord blood express negligible amounts of VISTA. Indeed, VISTA expression levels are significantly higher in placental CECs than those in cord blood (171). Thus, both maternal and fetal CECs are essential for fetomaternal tolerance (175). Accordingly, depletion of CECs in pregnant mice induces a proinflammatory immune response, by reducing IL-4 and IL-10 production, while increasing TNF-α and IL-6 levels in placental tissues, which in turn results in fetal resorption (175, 203); however, in pregnant women with inflammatory bowel disease (IBD), CECs are decreased in the peripheral blood, cord blood, and placenta tissue, and express lower levels of inhibitory molecules, including VISTA, TGF-β, and ROS. Accordingly, pregnant women with IBD have lower levels of Tregs and increased immune-activation. Patients with IBD are more likely to have a pro-inflammatory environment in the gastrointestinal tract, which leads to impairment of CECs during pregnancy (176).
4.2 CECs in infection
CECs not only function during neonatal infections, they participate in various infections throughout life.
Acquired immune deficiency syndrome is a systemic disease caused by human immunodeficiency virus (HIV), the genome of which comprises two copies of a 9749 nucleotide sequence packaged in each virion (204). CECs are expanded in the peripheral blood of patients with HIV and there is a positive correlation between CEC frequency and plasma viral load. When cocultured with CD4+ T cells, CECs exacerbate HIV-1 infection/replication, by enhancing NF-κB activation in CD4+ T cells to facilitate HIV infection (177). Meanwhile, CECs bind to HIV-1 via CD235a and subsequently transfer the virus to uninfected CD4+ T cells. Moreover, in the presence of antiretroviral therapy, CECs from HIV-infected individuals contain infective viral particles, which mediate HIV-1 trans-infection of CD4+ T cells (177). CECs are also significantly expanded and possess immunosuppressive properties in the blood of patients with COVID-19. With high levels of ARG2, ARG1, and ROS, CECs mediate immunosuppression by inhibiting CD4+ and CD8+ T cell production of TNF-α and IFN-γ in vitro (179). Furthermore, CD45+ CECs express ACE2, TMPRSS2, CD147, and CD26 and can be infected with SARS-CoV-2 (179).
CECs are also expanded in adult patients with sepsis and serve as predictors of 30-day mortality as well as nosocomial infection development. Low levels of RBCs and high levels of IL-6 and IFN-γ may contribute to the expansion of CECs in sepsis (178). During Salmonella infection, accumulation of CECs in the spleen and increased EPO production are dependent on Myd88/TRIF signaling (182); EPO neutralization reduces the population of CECs in the spleen and slightly improves the host immune response (182).
Malaria is an insect-borne infection caused by the bite of Anopheles mosquitoes, and a major global health problem, with approximately 247 million cases worldwide in 2021 and many more residents of endemic areas having asymptomatic parasitemia (chronic malaria) (205). Different species of malaria parasites exhibit distinct tropism (206). Plasmodium falciparum can invade all stages of erythrocytes while Plasmodium vivax and Plasmodium cynomolgi invade only reticulocytes (207, 208). P. vivax is the most widely distributed human malaria parasite and exhibits a strong preference for immature reticulocytes, with CD71 acting as an anchor receptor (209, 210). Reticulocytes have a more complex and enriched metabolic profile than mature erythrocytes, providing metabolic reservoirs for malaria parasites (206). P. vivax-infected reticulocytes express high levels of human leukocyte antigen class I (HLA-I), which can be specifically detected by cytotoxic CD8+ T cells to protect against intracellular parasites (180). In BALB/c mice, reticulocytes can secrete exosomes when infected by the reticulocyte-tropic non-lethal Plasmodium yoelii 17X strain (211). These reticulocyte-derived exosomes carry parasite proteins and are involved in antigen presentation. Mice immunized using purified exosomes produce IgG antibodies that can recognize P. yoelii-infected RBCs and show increased survival time and altered reticulocyte cell tropism of the parasite (181). Furthermore, during P. vivax infection, parasites invariably affect bone marrow CD71+ cells, inducing dyserythropoiesis and ineffective erythropoiesis (212). Identification and characterization of the reticulocyte receptor, metabolism, and the underlying mechanisms involved in malaria may provide insights to inform the development of novel antimalarial drugs and vaccines.
4.3 CECs in inflammation
Inflammation is the automatic defense response to tissue injury, and can be classified as acute and chronic, according to its duration. Inflammation modifies bone marrow hematopoiesis towards innate immune effector cells at the expense of lymphoid cells and erythrocytes (79). Inflammatory cytokines, such as TNF-α, limit steady-state erythropoiesis and promote granulopoiesis. Further, mature granulocytes contact the central macrophage of EBIs and alter EBI structures, leading to increased numbers of maturing granulocytes and fewer erythroid precursors (213). In chronic inflammation resulting from sterile abscesses, erythropoiesis is impaired at Ter119+ stages of erythroid development (185). Although inflammation inhibits erythropoiesis in the bone marrow, inflammatory signals induce stress erythropoiesis in the spleen, to maintain erythroid homeostasis. Inflammatory signaling through TLRs enhances erythrophagocytosis by splenic macrophages and augments expression of the transcription factor, SPI-C. In turn, SPI-C couples with TLR signaling to promote the expression of Gdf15 and Bmp4, which encode ligands that initiate the expansion of stress erythroid progenitors in the spleen (79). The spleen is the largest secondary lymphoid organ, and has a wide range of immunologic functions alongside its roles in erythropoiesis, and splenic erythropoiesis alters the histological structure of spleen to become rich in granulomatous lesions and devoid of clear separation between red and white pulp (214).
Autoimmune diseases comprise a range of disorders in which the immune response to self-antigens results in tissue damage or dysfunction (215). In patients with autoimmune diseases, CECs can inhibit inflammatory responses to prevent excessive inflammation and injury. Experimental autoimmune encephalomyelitis (EAE) is an autoimmune disease mainly mediated by specific sensitized CD4+ T cells, which serves as the best experimental model reflecting the autoimmune pathogenesis of human multiple sclerosis (216), and iron-deficient mice fail to develop EAE (217). Management using EPO or its non-erythropoietic derivatives consistently decreases EAE-associated TNF-α, IL-1β, and IL-1Ra production in the spinal cord, and IFN-γ by peripheral lymphocytes, which ameliorates chronic murine EAE (218). IBD inflammation spreads systemically and can cause complications, such as arthritis, cachexia, and anemia. In a CD45-deficient Rag1-deficient mouse model of T cell-induced colitis, an increased number of erythroid progenitors are found in the spleen. These CECs can suppress TNF-α expression in red pulp macrophages in a phagocytosis-dependent manner (183). Further, erythropoiesis-related genes are upregulated in PBMCs of patients with systemic-onset juvenile idiopathic arthritis (SoJIA) (184), while active SoJIA-driven CECs co-cultured with healthy donor monocytes suppress IL-1β, IL-6, and IL-8 secretion. Although ARG2 is the top upregulated gene in SoJIA-driven CECs, cytokine production from monocytes remains suppressed when they are treated using an arginase inhibitor (184).
4.4 CECs in tumor
Tumors are complex ecosystems, comprising tumor cells and various non-neoplastic cells (219), where non-neoplastic cells in the tumor microenvironment play critical roles in cancer development. Targeting the tumor microenvironment is considered a promising approach for cancer intervention (220). CECs are abundant in both the tumor microenvironment and the circulation and their levels can be used to predict tumor recurrence (165).
Tumor-associated myeloid cells include myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and neutrophils (221), which are important immune cell populations in the tumor microenvironment that are crucial for immune checkpoint blockade efficacy (222). MDSCs can be divided into at least two major subsets: mononuclear MDSCs (M-MDSCs, CD11b+Ly6G-Ly6Chigh) and polymorphonuclear MDSCs (PMN-MDSCs, CD11b+Ly6G+Ly6Clow) (223), where M-MDSCs exert more robust immunosuppression than PMN-MDSCs. Further, erythroid cells can differentiate into myeloid cells in tumors and mediate immunosuppression. Lineage tracking in patients with cancer and tumor-bearing mice revealed that > 30% of erythroid progenitor cells lose their developmental potential and switch to the myeloid lineage, and that these erythroid differentiated myeloid cells (EDMCs) are similar to their myeloid-originated counterparts at the transcription level (166). The phenotypes of EDMCs are CD45+CD235a+CD71+CD11b+CD33+HLA-DR- in patients with cancer and CD45+Ter119+CD71+CD11b+Gr1+ in tumor-bearing mice. EDMCs express more immune inhibitory molecules, including PD-L1, PD-L2, iNOS, ARG1, and CD49, than MDSCs, which may endow EDMCs with the ability to inhibit CD8+ T cell proliferation and IFN-γ production. Accordingly, EDMCs reduce the efficacy of anti-PD-1/PD-L1 treatment (166).
In tumors, CD45+ CECs exert a strong immune suppressive function, mainly by regulating T cells. In Lewis lung cancer, CD45+ CECs are induced by tumor growth-associated extramedullary hematopoiesis (EMH) in the spleen and their transcriptome closely resembles that of MDSCs. As robust immunosuppressors, CD45+ CECs hinder both CD8+ T cell priming in the spleen and effector function in peripheral tissues (164). In hepatocellular carcinoma (HCC) tissues, CD45+ CEC numbers are higher than those of CD45- CECs. Further, CD45+ CECs from patients with HCC inhibit CD4+ T cell proliferation and differentiation and suppress CD8+ T cell proliferation and cytotoxicity by generating factors including ROS, IL-10, and TGF-β (165). In patients with virus-associated solid tumors, substantially greater expansion of CECs occurs in the blood compared with that in healthy controls. CD45+ CECs have more immunosuppressive properties than their CD45- counterparts, mediated by higher levels of ROS, PD-L1/PD-L2, and VISTA. Further, the abundance of CECs in the circulation may be associated with anemia (187). Moreover, CECs in mice with melanoma secrete artemin, while this is not the case for VISTA+ CECs in patients with virus-associated solid tumors (187).
CD45- CECs have lower immunosuppression abilities than their CD45+ counterparts; however, they also play a crucial role in promoting tumor progression. One population of tumor-induced erythroblast-like cells (CD45-Ter119+CD71+, Ter-cells) derived from MEPs (186, 224), accumulate in the spleen of patients with terminal cancer and secret artemin, where artemin is a neurotrophic factor with an important role in cancer progression through its induction of Caspase-9 Thr125 phosphorylation, to maintain cell survival, and upregulation of TRIOBP and ITGB5 expression, to promote invasion. Blocking artemin, or its receptor, GFRα3, signaling inhibits HCC growth in vivo (186). In this context, the phenotype of Ter-cells is CD45-Ter119+ CD71+CD41+CD44+, and they mainly exist in the spleen of advanced-tumor bearing hosts; however, a few can also be found in the tumor. TGF-β and Smad3 activation contribute to Ter-cell generation. Moreover, serum artemin levels in patients with HCC are correlated with poor prognosis (186).
4.5 CECs in anemia
Anemia is a common blood disorder characterized by a decreased number of RBCs in the peripheral blood, which is defined as a hemoglobin level less than the 5th percentile for age (225). Anemia is the main cause of EPC expansion by increasing EPO concentration in response to oxygen deficit (226). In mice with anemia, CD45+ CECs expand in the spleen and express high levels of ARG2 and ROS. CEC expansion-induced L-arginine depletion suppresses T cell responses. In patients with anemia, CECs expand in the peripheral blood and express ARG1 and ARG2, which suppress IFN-γ production by T cells (189). Furthermore, human erythroleukemia-derived erythroid cell lines, including K562, HEL92.1.7, and TF-1, which express multiple erythroid-lineage markers, such as CD71 and CD235a, suppress T cells in an ARG- and ROS-dependent manner (189). Serum levels of IL-22 are increased in patients with anemia secondary to chronic kidney disease and myelodysplastic syndromes, and the IL-22 receptor, IL-22RA1, is present on erythroid precursors, with blockade of IL-22 signaling alleviating anemia in mice (192).
Anemia is also a common feature of sepsis (227). In patients with sepsis, RBC levels are negatively associated with CD45+ CEC frequency, suggesting that anemia may lead to CEC expansion through the EPO pathway (178). EPO can induce the expansion of CD45+ CECs, while EPO neutralization prevents infection-related CEC accumulation (188). In patients with COVID19, SARS-CoV-2 infection is associated with lower blood oxygen levels and the numbers of CECs in the blood are negatively correlated with hemoglobin levels; this may be due to the elimination of infected/damaged CECs by lysis and/or phagocytosis. Dexamethasone enhances the maturation of CECs to RBCs by downregulating ACE2 and TMPRSS in a dose-dependent manner (179).
In addition, anemia is very common among patients with cancer and tumor bearing animal models; approximately 30%–90% of patients with cancer have varying degrees of anemia, depending on the type of cancer (228). Immunosuppressive CECs can be detected in patients with cancer and anemia. Further, hematocrit, HGB levels, and mature RBC counts are decreased in the blood of mice after prolonged tumor-bearing, and HGB is negatively correlated with numbers of splenic CECs. Tumor-initiated anemia and EMH may act synergistically to cause splenic CEC accumulation (164): anemia induces EMH, whereas terminal differentiation is blocked in the presence of tumors. RNA sequencing of CD45+ and CD45- CECs generated by anemia induced in different ways revealed that CD45+ CECs differ significantly from their CD45- counterparts, particularly regarding signature genes defining the erythrocyte lineage and immunosuppression. Notably, ROS and NOX-2 are highly expressed in CD45+ CECs, particularly those from tumor-bearing individuals (164). EPO has been widely used to overcome hypoxia in patients with cancer. Recombinant human EPO and erythropoiesis-stimulating agents can promote EPC differentiation and maturation to RBCs, and thereby effectively treat anemia; however, these agents do not prolong the survival of patients with cancer (229–231). Immune checkpoint inhibitors (ICIs) targeting co-inhibitory molecules, including PD-1, PD-L1, and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), have been widely applied in the therapy of various tumors (232); however, EPO treatment in patients receiving anti-PD-L1 therapy reduce the therapeutic effects of this monoclonal antibody (187); the underlying mechanism involves EPO induction of continual differentiation of CECs into EDMCs, which mediate systemic immunosuppression against immune surveillance (166).
5 Future applications
5.1 Manipulation of CECs
Erythroid cells participate in several immune conditions and have important roles in regulating immune responses. Further, CECs may have beneficial effects in fetomaternal tolerance and autoimmune diseases; however, in contexts including infection, tumor, and anemia, CECs appear to exert detrimental effects (Table 6). Thus, further understanding of the immune regulatory mechanisms used by erythroid cells can provide new insights into pathogenic mechanisms, and CECs may serve as a novel target in immunological therapies (Figure 5). CECs can have opposite effects in different diseases, and different measures could be selected to manipulate CECs, according to context; for example, promotion/inhibition of CEC expansion, inhibition/promotion of CEC differentiation, and inhibition/promotion of CEC immunosuppressive properties.
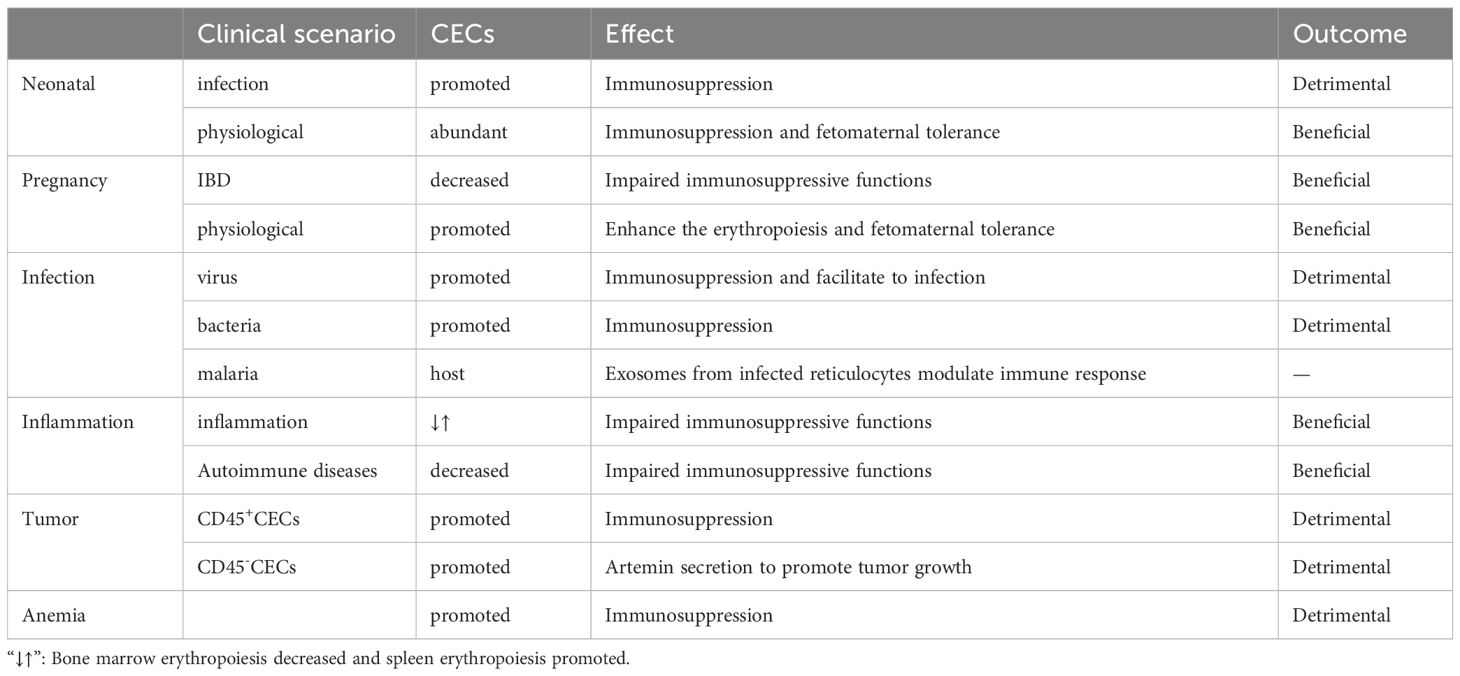
Table 6. Immunomodulatory effects of the CECs.
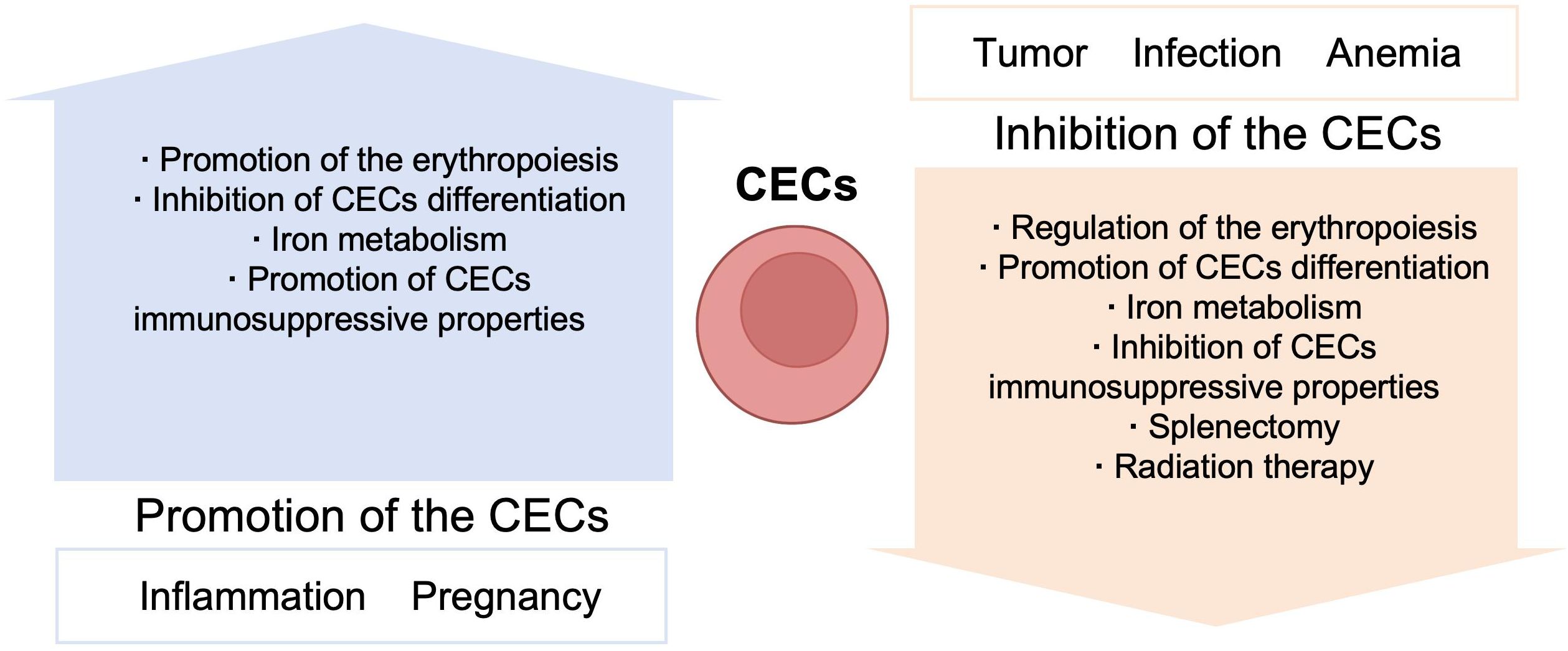
Figure 5. Manipulation of the CECs.
Under normal conditions, CECs suppress the immune response to protect tissue from immunologic injury; however, CECs may be impaired in autoimmune diseases and chronic inflammation. Thus, promoting CEC expansion may be beneficial in inflammatory disorders. In contrast, in patients with tumors, infection, or anemia, CECs expand and arrest at an early stage, to inhibit immune activity; therefore, preventing CEC expansion and promoting CEC differentiation may be promising therapies to attenuate immune evasion and enhance immune responses in these contexts.
5.1.1 Targeting CEC expansion signals
EPO is crucial in regulating the late stages of erythropoiesis, and EPO and EPO derivatives are widely used to treat different types of anemia (233–235). Clinical studies have demonstrated that EPO can significantly improve the management of anemia in patients with chronic renal insufficiency (236). Further, EPO administration for patients with cancer significantly improves hematological responses and decreases the need for RBC transfusion (237); however, caution is required in patients with cancer and anemia as, while EPO may influence the curative effect of ICI therapy by restoring Ter-cell numbers and serum artemin concentration (166), EPOR are present on various types of tumor cell and tumor cell lines, and EPO/EPOR may contribute to tumor progression and metastatic progression (238). EPO can also strongly suppress immune system activation and protect injured tissues from apoptosis, suggesting that it may be a promising therapeutic target in autoimmune diseases, allergy, and organ transplantation. As EPOR is expressed on various immune and tumor cells, the interactions between EPO and these cell types requires further study.
Other factors involved in stress erythropoiesis can also be targeted to modulate CEC expansion. GCs are established immunosuppressive steroid molecules secreted by the adrenal gland and regulated by the hypothalamic-pituitary-adrenal axis, and are widely used in the treatment of various immune disorders (239, 240). GCs can also be used to treat anemia by modeling human stress erythropoiesis, as they can both induce monocyte differentiation to EBI macrophages (241) and directly target EBI macrophages to promote erythroid expansion (242). Further, GCs can aid in the treatment of EPO-resistant anemia by stimulating progenitor self-renewal (243), while, in healthy humans, GC injection also accelerates erythropoiesis and increases total hemoglobin mass, which may help to prevent altitude sickness (244). Although GC application in immune disorders has been widely studied, whether GCs can be used to modulate CECs in these diseases awaits further in-depth investigations.
5.1.2 Promoting CEC differentiation
CECs expand and arrest in the early stages of maturation in patients with cancer and suppress immune responses. Therefore, promoting CEC differentiation is a novel therapeutic strategy to diminish the tumor-promoting effects of CECs.
TGF-β triggers differentiation arrest and promotes CEC expansion, as well as functioning as the main effector cytokine of CECs that regulate immunosuppression (245). Further, TGF-β and downstream Smad3 activation are important in splenic Ter-cell generation (186). Inhibitors targeting TGF-β and Smad2/3 signaling can stimulate CEC differentiation and promote their maturation, thereby neutralizing their suppressive effects (202, 246), and have been proven effective in mouse models of cancer. In addition, TGF-β-promoted immune escape of carcinoma cells can be flexibly treated using ionizing radiation combined with hyperthermia and ICIs. Numerous anti-cancer pharmacological interventions targeting TGF-β have undergone pre-clinical and clinical stage studies; however, although several anti-TGF-β-based immunotherapies were effective in preclinical trails (247–249), the results of subsequent clinical trials were disappointing, due to low efficacy and safety issues (250). Thus, further research to explore optimal combinations with other chemotherapies and improve specificity is needed.
p38 MAPK signaling is important in regulating erythropoiesis, and restrains EPC differentiation by regulating active GATA-1 degradation (251). Further, p38 MAPK signaling contributes to several biological functions, including inflammation and tumorigenesis, as well as cell proliferation, differentiation, apoptosis, and senescence (252–254). Therefore, p38 signaling inhibitors may be beneficial in patients with cancer as they have anti-tumor effects and can promote CEC maturation; however, similar to TGF-β inhibitors, although some p38 MAPK inhibitors have completed phase I and II trials, the results of clinical trials have been unsatisfactory due to high levels of systemic toxicity (254). Thus, further research is warranted to facilitate more comprehensive understanding of p38 MAPK signaling.
mTOR belongs to the PI3K-related kinase family of serine/threonine protein kinases and acts with Forkhead-box-class-O3 (FoxO3) to regulate erythropoiesis (255). FoxO3 inhibits mTOR and promotes CEC differentiation by inducing cell cycle exit of early-stage EPCs during ineffective erythropoiesis (255). mTOR inhibitors or FoxO3 inducers may be used to reduce ineffective erythropoiesis by promoting CEC maturation and inhibiting CEC proliferation (256); however, first-generation mTOR inhibitors showed limited sensitivity (257). Additional research is needed to enhance this type of therapy and overcome resistance.
Caspases are negative regulators of erythropoiesis through caspase-mediated degradation of the transcription factor, GATA-1 (258). In chronic inflammation, inflammasomes activate caspase-1 and skew the differentiation of HSPCs toward myeloid cells, resulting in neutrophilia and anemia. Caspase-1 inhibition rapidly upregulates GATA1 and promotes HSPC differentiation into erythroid cells (259). Caspase-1 is also involved in inflammatory processes and autoinflammation; therefore, it is of great interest to evaluate the effects of caspase inhibitors, such as colchicine (260), VRT-18858, VRT-043198, and sulfasalazine (261), on CEC expansion and differentiation in inflammation-associated anemia and autoimmune diseases.
Iron, a necessary component of hemoglobin and myoglobin, is essential in hemoglobin synthesis and erythroid cell proliferation (60), and iron deficiency leads to anemia. Thus, targeting iron and its metabolism is an effective way to ameliorate ineffective erythropoiesis and reduce accumulation of early-stage CECs. Several new agents to modulate iron metabolism, such as anti-hepcidin antibody (LY2787106) (262), anti-ferroportin antibody (LY2928057) (263), and anti-matriptase-2 antibody (RAP-536L and RLYB331), have been investigated (264), and are all beneficial in the treatment of anemia; hence, drug combinations incorporating these agents represent a potential superior option. RLYB331 prevents iron overload, ameliorates ineffective erythropoiesis, and limits the formation of toxic α-chain, while RAP-536L efficiently corrects anemia in β-thalassemic model mice. Combination treatment with RLYB331 and RAP-536L integrates their advantages, including hepcidin upregulation, alleviation of iron overload, and amelioration of ineffective erythropoiesis (264). Moreover, inflammation-inducible cytokines can directly suppress CEC differentiation as well as blocking intestinal iron absorption and causing iron-restricted erythropoiesis (265). Thus, therapies targeting pro-inflammatory cytokines can also promote CEC differentiation by increasing iron availability.
5.1.3 Modulation of CEC immunosuppressive properties
CECs modulate immune responses through multiple mechanisms, including L-arginine depletion by ARG, ROS, cytokines, and immune checkpoints. ARG inhibitors, such as boronic acid derivatives or L-arginine supplementation, may diminish the inhibitory effects of CECs on immune responses (189, 266). Similar to ARG inhibitors, targeting of ROS-generating proteins, including NOX enzymes, or use of a ROS inhibitor, such as N-acetylcysteine, may be helpful therapeutic strategies for autoimmune conditions or cancer (267). Further, targeting cytokines secreted by CECs is a promising strategy to attenuate CEC-induced immune evasion. TGF-β superfamily inhibitors can both ameliorate CEC suppressive effects and cooperate with EPO to promote RBC production and alleviate anemia (268); neutralization of TGF-β also reduces CEC expansion. Thus, targeting TGF-β signaling is a potentially promising strategy; however, as discussed above, a number of challenges are yet to be overcome. Late-stage CECs secrete artemin to promote tumor growth and invasiveness, and anti-artemin neutralizing antibody can inhibit tumor growth and increase the survival of tumor-bearing mice. Thus, the clinical utility of targeting artemin or related signaling pathways warrants exploration. Targeting immune checkpoints has revolutionized clinical oncology and antibodies targeting the PD-1/PD-L1 axis have proven effective in cancer therapy. CECs mediate immune response suppression by the PD-L1/PD-1 axis, and ICIs may, at least partially, suppress the tumor-promoting effects of CECs.
Importantly, mechanisms by which CECs induce immunosuppression also overlap with those used by many other immunomodulatory cells, including Tregs, MDSCs, and TAMs, among others. Therefore, therapeutic strategies targeting these mechanisms to modulate the properties of CECs may also influence other immunomodulatory cells, leading to unexpected effects.
5.1.4 Splenectomy and radiation
Splenomegaly occurs in patients with anemia or advanced cancer, where the spleen becomes a central organ of EMH, which generates suppressive cells, including CECs and myeloid cells (269). Splenectomy is associated with longer hospital stay and longer time to chemotherapy in patients with cancer but has no impact on overall or disease-free survival (270); however, for patients with advanced epithelial ovarian cancer or with splenic involvement, spleen resection is associated with longer survival (271, 272). Moreover, splenectomy leads to the depletion of MDSCs and promotes the activation of anti-tumor immunity (269), hence it may be beneficial for some patients with advanced cancer. Although there have been several clinical studies of splenectomy, more preclinical and clinical investigations are required.
Radiation is often used as an adjuvant therapy for tumors, and exhibits substantial versatility and efficacy in cancer treatment (273). Local irradiation can significantly decrease tumor-induced Ter-cell accumulation in the murine spleen (224). Further, patients with cancer who received local tumor ionizing radiation (IR) alongside PD-1 therapy exhibited IR-mediated reduction of Ter-cells, artemin, and GFRα3 (an artemin signaling partner associated with tumor regression) (224). Hence, radiation is an effective cancer treatment, and understanding the interactions when immunotherapies are combined with radiotherapy warrants further study.
5.2 Future applications of RBCs
Targeting RBCs is an underdeveloped therapeutic strategy; however, with their strong ability to contribute to material exchange and high immunocompatibility, RBCs have potential as drug delivery carriers (274, 275). RBC membrane-coated polymeric nanoparticles can effectively deliver doxorubicin in a mouse model of lymphoma (275). Further, RBC extracellular vesicles (RBCEVs) are taken up by leukemia cells with high efficiency, and may serve as a valid vehicle to deliver antisense oligonucleotides to leukemia cells (274). RBCEVs are also used as a vehicle for osteoclast-targeted delivery of anti-miR-214 oligonucleotides. TBP-CP05 is a functional peptide which binds to both CD63 on RBCEVs and receptors on osteoclasts, and TBP-CP05 binds with RBCEVs through CP05 and endows them with osteoclast-targeting ability. Intravenous injection of osteoclast-targeting RBCEVs significantly inhibits osteoclast activity, elevates osteoblast activity, and improves bone density in osteoporotic mice (276). Hence, RBCs have huge potential in cancer and clinical therapy as a novel type of nanoparticle-based RNA drug vehicle.
RBCs also have potential applications in disease diagnosis, prognosis, and monitoring (277). A considerable fraction of cell-free mtDNA, which is associated with trauma, autoimmune disease, sepsis, malignancy, cellular injury, and organ dysfunction, binds to the outer surface of RBCs and can serve as a biomarker (278–280). Furthermore, long DNA fragments which cover most nuclear and mitochondrial genome regions can be detected in RBCs from patients with cancer (281).
6 Perspectives on erythroid cell research
Erythroid cells possess complex immunoregulation functions at different stages of their development. Overall, the available evidence demonstrates the broad range of immunological properties possessed by these most abundant, but less appreciated, cells. Further studies should clarify the roles of erythroid cells at different stages of development and in various diseases and their underlying mechanisms, which could inform the development of new therapeutic strategies. Furthermore, recent studies have revealed erythropoiesis in the skull and dura (282, 283), and tissue-specific functions in these contexts are also of interest.
Author contributions
CN: Writing – original draft, Writing – review & editing. JZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Beijing Natural Science Foundation (Grant number: 5234033).
Acknowledgments
Figures were created by Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nelson RA Jr. The immune-adherence phenomenon; an immunologically specific reaction between microorganisms and erythrocytes leading to enhanced phagocytosis. Science. (1953) 118:733–7. doi: 10.1126/science.118.3077.733
2. Pavia CS, Stites DP. Immunosuppressive activity of murine newborn spleen cells. I. Selective inhibition of in vitro lymphocyte activation. Cell Immunol. (1979) 42:48–60. doi: 10.1016/0008-8749(79)90220-X
3. Conway de Macario E, Macario AJ. Immunosuppression associated with erythropoiesis in genetic low responder mice. Ann Immunol (Paris). (1980) 131C:397–404.
4. Elahi S. New insight into an old concept: role of immature erythroid cells in immune pathogenesis of neonatal infection. Front Immunol. (2014) 5:376. doi: 10.3389/fimmu.2014.00376
5. Tsyrlova IG. Erythroid immunosuppressor cells (Er suppressors) and their role in the regulation of immunity. Vestn Akad Med Nauk SSSR. (1991) 12:34–9.
6. Grzywa TM, Nowis D, Golab J. The role of CD71(+) erythroid cells in the regulation of the immune response. Pharmacol Ther. (2021) 228:107927. doi: 10.1016/j.pharmthera.2021.107927
7. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. (2014) 5:3. doi: 10.3389/fphys.2014.00003
8. Seu KG, Papoin J, Fessler R, Hom J, Huang G, Mohandas N, et al. Unraveling macrophage heterogeneity in erythroblastic islands. Front Immunol. (2017) 8:1140. doi: 10.3389/fimmu.2017.01140
9. Canny SP, Orozco SL, Thulin NK, Hamerman JA. Immune mechanisms in inflammatory anemia. Annu Rev Immunol. (2023) 41:405–29. doi: 10.1146/annurev-immunol-101320-125839
10. Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol. (2016) 173:206–18. doi: 10.1111/bjh.2016.173.issue-2
11. Caulier A, Sankaran VG. Molecular and cellular mechanisms that regulate human erythropoiesis. Blood. (2022) 139:2450–9. doi: 10.1182/blood.2021011044
12. Popescu DM, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver haematopoiesis. Nature. (2019) 574:365–71. doi: 10.1038/s41586-019-1652-y
13. Baron MH, Vacaru A, Nieves J. Erythroid development in the mammalian embryo. Blood Cells Mol Dis. (2013) 51:213–9. doi: 10.1016/j.bcmd.2013.07.006
14. Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. (2020) 136:2392–400. doi: 10.1182/blood.2020007645
15. Potts KS, Sargeant TJ, Markham JF, Shi W, Biben C, Josefsson EC, et al. A lineage of diploid platelet-forming cells precedes polyploid megakaryocyte formation in the mouse embryo. Blood. (2014) 124:2725–9. doi: 10.1182/blood-2014-02-559468
16. McGrath KE, Frame JM, Fegan KH, Bowen JR, Conway SJ, Catherman SC, et al. Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep. (2015) 11:1892–904. doi: 10.1016/j.celrep.2015.05.036
17. Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. (1999) 126:5073–84. doi: 10.1242/dev.126.22.5073
18. Chen MJ, Li Y, De Obaldia ME, Yang Q, Yzaguirre AD, Yamada-Inagawa T, et al. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. (2011) 9:541–52. doi: 10.1016/j.stem.2011.10.003
19. Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. (2013) 3:a011601. doi: 10.1101/cshperspect.a011601
20. Le Goff S, Boussaid I, Floquet C, Raimbault A, Hatin I, Andrieu-Soler C, et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood. (2021) 137:89–102. doi: 10.1182/blood.2019003439
21. Zhu L, He C, Guo Y, Liu H, Zhang S. Molecular regulatory mechanisms of erythropoiesis and related diseases. Eur J Haematol. (2023) 111:337–44. doi: 10.1111/ejh.v111.3
22. Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. (2017) 17:573–90. doi: 10.1038/nri.2017.53
23. Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. (2011) 333:218–21. doi: 10.1126/science.1201219
24. Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. (2009) 75:14–24. doi: 10.1002/cyto.a.v75a:1
25. Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. (2013) 13:102–16. doi: 10.1016/j.stem.2013.05.014
26. Kwon N, Thompson EN, Mayday MY, Scanlon V, Lu YC, Krause DS. Current understanding of human megakaryocytic-erythroid progenitors and their fate determinants. Curr Opin Hematol. (2021) 28:28–35. doi: 10.1097/MOH.0000000000000625
27. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. (2000) 404:193–7. doi: 10.1038/35004599
28. An X, Chen L. Flow cytometry (FCM) analysis and fluorescence-activated cell sorting (FACS) of erythroid cells. Methods Mol Biol. (2018) 1698:153–74. doi: 10.1007/978-1-4939-7428-3_9
29. Flygare J, Rayon Estrada V, Shin C, Gupta S, Lodish HF. HIF1alpha synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood. (2011) 117:3435–44. doi: 10.1182/blood-2010-07-295550
30. Li J, Hale J, Bhagia P, Xue F, Chen L, Jaffray J, et al. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood. (2014) 124:3636–45. doi: 10.1182/blood-2014-07-588806
31. Liu J, Zhang J, Ginzburg Y, Li H, Xue F, De Franceschi L, et al. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood. (2013) 121:e43–9. doi: 10.1182/blood-2012-09-456079
32. Hu J, Liu J, Xue F, Halverson G, Reid M, Guo A, et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood. (2013) 121:3246–53. doi: 10.1182/blood-2013-01-476390
33. Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci USA. (2009) 106:17413–8. doi: 10.1073/pnas.0909296106
34. Koulnis M, Pop R, Porpiglia E, Shearstone JR, Hidalgo D, Socolovsky M. Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flow-cytometric assay. J Vis Exp. (2011) 54:2809. doi: 10.3791/2809
35. An X, Chen L. Flow cytometric analysis of erythroblast enucleation. Methods Mol Biol. (2018) 1698:193–203. doi: 10.1007/978-1-4939-7428-3_11
36. Ovchynnikova E, Aglialoro F, Bentlage AEH, Vidarsson G, Salinas ND, von Lindern M, et al. DARC extracellular domain remodeling in maturating reticulocytes explains Plasmodium vivax tropism. Blood. (2017) 130:1441–4. doi: 10.1182/blood-2017-03-774364
37. Kalfa T, McGrath KE. Analysis of erythropoiesis using imaging flow cytometry. Methods Mol Biol. (2018) 1698:175–92. doi: 10.1007/978-1-4939-7428-3_10
38. Li W, Acker JP. Development and validation of a sensitive flow cytometric method for determining CECs in RBC products. Clin Chim Acta. (2022) 530:119–25. doi: 10.1016/j.cca.2022.03.029
39. Broxmeyer HE. Erythropoietin: multiple targets, actions, and modifying influences for biological and clinical consideration. J Exp Med. (2013) 210:205–8. doi: 10.1084/jem.20122760
40. Eggold JT, Rankin EB. Erythropoiesis, EPO, macrophages, and bone. Bone. (2019) 119:36–41. doi: 10.1016/j.bone.2018.03.014
41. Moriguchi T, Yamamoto M. A regulatory network governing Gata1 and Gata2 gene transcription orchestrates erythroid lineage differentiation. Int J Hematol. (2014) 100(5):417–24. doi: 10.1007/s12185-014-1568-0
42. Kim MY, Yan B, Huang S, Qiu Y. Regulating the regulators: the role of histone deacetylase 1 (HDAC1) in erythropoiesis. Int J Mol Sci. (2020) 21:417–24. doi: 10.3390/ijms21228460
43. Fechner J, Lausen J. Transcription factor TAL1 in erythropoiesis. Adv Exp Med Biol. (2024) 1459:243–58. doi: 10.1007/978-3-031-62731-6_11
44. Zheng ZF, Gou FL, Cheng T, Cheng H. PDGFRalpha(+) cell-derived SCF regulates hematopoiesis of adult mice. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2020) 28:1349–56. doi: 10.19746/j.cnki.issn.1009-2137.2020.04.046
45. Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. (2011) 118:6258–68. doi: 10.1182/blood-2011-07-356006
46. Bhoopalan SV, Huang LJ, Weiss MJ. Erythropoietin regulation of red blood cell production: from bench to bedside and back. F1000Res. (2020) 9:F1000 Faculty Rev-1153. doi: 10.12688/f1000research
47. Oikonomidou PR, Rivella S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. (2018) 32:130–43. doi: 10.1016/j.blre.2017.10.001
48. Tang P, Wang H. Regulation of erythropoiesis: emerging concepts and therapeutic implications. Hematology. (2023) 28:2250645. doi: 10.1080/16078454.2023.2250645
49. Kaushansky K, Broudy VC, Sitnicka E, Lofton-Day C, Grossmann A, Sprugel K. Do the preclinical effects of thrombopoietin correlate with its in vitro properties? Stem Cells. (1996) 14 Suppl 1:108–11. doi: 10.1002/stem.5530140713
50. Bohmer RM. IL-3-dependent early erythropoiesis is stimulated by autocrine transforming growth factor beta. Stem Cells. (2004) 22:216–24. doi: 10.1634/stemcells.22-2-216
51. Lewis JL, Marley SB, Blackett NM, Szydlo R, Goldman JM, Gordon MY. Interleukin 3 (IL-3), but not stem cell factor (SCF) increases self-renewal by human erythroid burst-forming units (BFU-E) in vitro. Cytokine. (1998) 10:49–54. doi: 10.1006/cyto.1997.0256
52. Umemura T, Papayannopoulou T, Stamatoyannopoulos G. The mechanism of expansion of late erythroid progenitors during erythroid regeneration: target cells and effects of erythropoietin and interleukin-3. Blood. (1989) 73:1993–8. doi: 10.1182/blood.V73.7.1993.1993
53. Teramura M, Kobayashi S, Yoshinaga K, Iwabe K, Mizoguchi H. Effect of interleukin 11 on normal and pathological thrombopoiesis. Cancer Chemother Pharmacol. (1996) 38 Suppl:S99–102. doi: 10.1007/s002800051048
54. Nakamura K, Smyth MJ. Aberrant erythropoiesis fuels tumor growth. Cell Res. (2018) 28:611–2. doi: 10.1038/s41422-018-0047-1
55. Fu Y, Li Z, Lin W, Yao J, Jiang X, Shu Q, et al. Extramedullary hematopoiesis contributes to enhanced erythropoiesis during pregnancy via TGF-beta signaling. Front Immunol. (2023) 14:1295717. doi: 10.3389/fimmu.2023.1295717
56. Villeval JL, Vainchenker W. Megakaryocytes tame erythropoiesis with TGFbeta1. Blood. (2020) 136:1016–7. doi: 10.1182/blood.2020006906
57. Di Giandomenico S, Kermani P, Molle N, Yabut MM, Abu-Zeinah G, Stephens T, et al. Megakaryocyte TGFbeta1 partitions erythropoiesis into immature progenitor/stem cells and maturing precursors. Blood. (2020) 136:1044–54. doi: 10.1182/blood.2019003276
58. Koury MJ, Ponka P. New insights into erythropoiesis: the roles of folate, vitamin B12, and iron. Annu Rev Nutr. (2004) 24:105–31. doi: 10.1146/annurev.nutr.24.012003.132306
59. Silvestri L, Nai A. Iron and erythropoiesis: A mutual alliance. Semin Hematol. (2021) 58:145–52. doi: 10.1053/j.seminhematol.2021.05.002
60. Correnti M, Gammella E, Cairo G, Recalcati S. Iron mining for erythropoiesis. Int J Mol Sci. (2022) 23(10):5341. doi: 10.3390/ijms23105341
61. Cai Y, Gao Y, Lv Y, Chen Z, Zhong L, Chen J, et al. Multicomponent comprehensive confirms that erythroferrone is a molecular biomarker of pan-cancer. Heliyon. (2024) 10:e26990. doi: 10.1016/j.heliyon.2024.e26990
62. Tsiftsoglou AS. Erythropoietin (EPO) as a key regulator of erythropoiesis, bone remodeling and endothelial transdifferentiation of multipotent mesenchymal stem cells (MSCs): implications in regenerative medicine. Cells. (2021) 10(8):2140. doi: 10.3390/cells10082140
64. Manwani D, Bieker JJ. The erythroblastic island. Curr Top Dev Biol. (2008) 82:23–53. doi: 10.1016/S0070-2153(07)00002-6
65. Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. (2008) 112:470–8. doi: 10.1182/blood-2008-03-077883
66. Costa-Junior DA, Souza Valente TN, Belisario AR, Carvalho GQ, Madeira M, Velloso-Rodrigues C. Association of ZBTB38 gene polymorphism (rs724016) with height and fetal hemoglobin in individuals with sickle cell anemia. Mol Genet Metab Rep. (2024) 39:101086. doi: 10.1016/j.ymgmr.2024.101086
67. Liao C, Prabhu KS, Paulson RF. Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood. (2018) 132:2580–93. doi: 10.1182/blood-2018-06-856831
68. Koury MJ, Bondurant MC. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. (1990) 248:378–81. doi: 10.1126/science.2326648
69. Li W, Guo R, Song Y, Jiang Z. Erythroblastic island macrophages shape normal erythropoiesis and drive associated disorders in erythroid hematopoietic diseases. Front Cell Dev Biol. (2020) 8:613885. doi: 10.3389/fcell.2020.613885
70. Borges MD, Sesti-Costa R. Macrophages: key players in erythrocyte turnover. Hematol Transfus Cell Ther. (2022) 44:574–81. doi: 10.1016/j.htct.2022.07.002
71. Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med. (2001) 193:855–62. doi: 10.1084/jem.193.7.855
72. Lang E, Bissinger R, Qadri SM, Lang F. Suicidal death of erythrocytes in cancer and its chemotherapy: A potential target in the treatment of tumor-associated anemia. Int J Cancer. (2017) 141:1522–8. doi: 10.1002/ijc.v141.8
73. Klei TR, Meinderts SM, van den Berg TK, van Bruggen R. From the cradle to the grave: the role of macrophages in erythropoiesis and erythrophagocytosis. Front Immunol. (2017) 8:73. doi: 10.3389/fimmu.2017.00073
74. Helwa R, Heller A, Knappskog S, Bauer AS. Tumor cells interact with red blood cells via galectin-4 - a short report. Cell Oncol (Dordr). (2017) 40:401–9. doi: 10.1007/s13402-017-0317-9
75. Xiao LL, Liu Y, Chen S, Fu BM. Effects of flowing RBCs on adhesion of a circulating tumor cell in microvessels. Biomech Model Mechanobiol. (2017) 16:597–610. doi: 10.1007/s10237-016-0839-5
76. Porayette P, Paulson RF. BMP4/Smad5 dependent stress erythropoiesis is required for the expansion of erythroid progenitors during fetal development. Dev Biol. (2008) 317:24–35. doi: 10.1016/j.ydbio.2008.01.047
77. Liu M, Jin X, He X, Pan L, Zhang X, Zhao Y. Macrophages support splenic erythropoiesis in 4T1 tumor-bearing mice. PloS One. (2015) 10:e0121921. doi: 10.1371/journal.pone.0121921
78. Sano Y, Yoshida T, Choo MK, Jiménez-Andrade Y, Hill KR, Georgopoulos K, et al. Multiorgan signaling mobilizes tumor-associated erythroid cells expressing immune checkpoint molecules. Mol Cancer Res. (2021) 19:507–15. doi: 10.1158/1541-7786.MCR-20-0746
79. Bennett LF, Liao C, Quickel MD, Yeoh BS, Vijay-Kumar M, Hankey-Giblin P, et al. Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-C. Sci Signal. (2019) 12(598):eaap7336. doi: 10.1126/scisignal.aap7336
80. Alamo IG, Kannan KB, Loftus TJ, Ramos H, Efron PA, Mohr AM. Severe trauma and chronic stress activates extramedullary erythropoiesis. J Trauma Acute Care Surg. (2017) 83:144–50. doi: 10.1097/TA.0000000000001537
81. Vignjević S, Budeč M, Marković D, Dikić D, Mitrović O, Mojsilović S, et al. Chronic psychological stress activates BMP4-dependent extramedullary erythropoiesis. J Cell Mol Med. (2014) 18:91–103. doi: 10.1111/jcmm.12167
82. McKim DB, Yin W, Wang Y, Cole SW, Godbout JP, Sheridan JF. Social stress mobilizes hematopoietic stem cells to establish persistent splenic myelopoiesis. Cell Rep. (2018) 25:2552–62.e3. doi: 10.1016/j.celrep.2018.10.102
83. Bennett LF, Liao C, Paulson RF. Stress erythropoiesis model systems. Methods Mol Biol. (2018) 1698:91–102. doi: 10.1007/978-1-4939-7428-3_5
84. Paulson RF, Shi L, Wu DC. Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol. (2011) 18:139–45. doi: 10.1097/MOH.0b013e32834521c8
85. Paulson RF, Hariharan S, Little JA. Stress erythropoiesis: definitions and models for its study. Exp Hematol. (2020) 89:43–54 e2. doi: 10.1016/j.exphem.2020.07.011
86. Harandi OF, Hedge S, Wu DC, McKeone D, Paulson RF. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest. (2010) 120:4507–19. doi: 10.1172/JCI41291
87. Xiang J, Wu DC, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood. (2015) 125:1803–12. doi: 10.1182/blood-2014-07-591453
88. Perry JM, Harandi OF, Paulson RF. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood. (2007) 109:4494–502. doi: 10.1182/blood-2006-04-016154
89. Hao S, Xiang J, Wu DC, Fraser JW, Ruan B, Cai J, et al. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv. (2019) 3:2205–17. doi: 10.1182/bloodadvances.2019000375
90. Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. (1999) 13:2996–3002. doi: 10.1101/gad.13.22.2996
91. Ji P. Finding erythroid stress progenitors: cell surface markers revealed. Haematologica. (2020) 105:2499–501. doi: 10.3324/haematol.2020.262493
92. Singbrant S, Mattebo A, Sigvardsson M, Strid T, Flygare J. Prospective isolation of radiation induced erythroid stress progenitors reveals unique transcriptomic and epigenetic signatures enabling increased erythroid output. Haematologica. (2020) 105:2561–71. doi: 10.3324/haematol.2019.234542
93. Fomukong HA, Kalu M, Aimola IA, Sallau AB, Bello-Manga H, Gouegni FE, et al. Single-cell RNA seq analysis of erythroid cells reveals a specific sub-population of stress erythroid progenitors. Hematology. (2023) 28:2261802. doi: 10.1080/16078454.2023.2261802
94. Lenox LE, Perry JM, Paulson RF. BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood. (2005) 105:2741–8. doi: 10.1182/blood-2004-02-0703
95. Peng B, Kong G, Yang C, Ming Y. Erythropoietin and its derivatives: from tissue protection to immune regulation. Cell Death Dis. (2020) 11:79. doi: 10.1038/s41419-020-2276-8
96. Nairz M, Schroll A, Moschen AR, Sonnweber T, Theurl M, Theurl I, et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-κB-inducible immune pathways. Immunity. (2011) 34:61–74. doi: 10.1016/j.immuni.2011.01.002
97. Yang C, Zhang Y, Wang J, Li L, Wang L, Hu M, et al. A novel cyclic helix B peptide inhibits dendritic cell maturation during amelioration of acute kidney graft rejection through Jak-2/STAT3/SOCS1. Cell Death Dis. (2015) 6:e1993. doi: 10.1038/cddis.2015.338
98. Cravedi P, Manrique J, Hanlon KE, Reid-Adam J, Brody J, Prathuangsuk P, et al. Immunosuppressive effects of erythropoietin on human alloreactive T cells. J Am Soc Nephrol. (2014) 25:2003–15. doi: 10.1681/ASN.2013090945
99. Escoter-Torres L, Caratti G, Mechtidou A, Tuckermann J, Uhlenhaut NH, Vettorazzi S. Fighting the fire: mechanisms of inflammatory gene regulation by the glucocorticoid receptor. Front Immunol. (2019) 10:1859. doi: 10.3389/fimmu.2019.01859
100. Darbonne WC, Rice GC, Mohler MA, Apple T, Hebert CA, Valente AJ, et al. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest. (1991) 88:1362–9. doi: 10.1172/JCI115442
101. Neote K, Darbonne W, Ogez J, Horuk R, Schall TJ. Identification of a promiscuous inflammatory peptide receptor on the surface of red blood cells. J Biol Chem. (1993) 268:12247–9. doi: 10.1016/S0021-9258(18)31379-6
102. Papadopoulos C, Spourita E, Tentes I, Steiropoulos P, Anagnostopoulos K. Red blood cell malfunction in COVID-19: molecular mechanisms and therapeutic targets. Viral Immunol. (2022) 35:649–52. doi: 10.1089/vim.2021.0212
103. Horakova E, Gasser O, Sadallah S, Inal JM, Bourgeois G, Ziekau I, et al. Complement mediates the binding of HIV to erythrocytes. J Immunol. (2004) 173:4236–41. doi: 10.4049/jimmunol.173.6.4236
104. Kisserli A, Schneider N, Audonnet S, Tabary T, Goury A, Cousson J, et al. Acquired decrease of the C3b/C4b receptor (CR1, CD35) and increased C4d deposits on erythrocytes from ICU COVID-19 patients. Immunobiology. (2021) 226:152093. doi: 10.1016/j.imbio.2021.152093
105. Lam LKM, Reilly JP, Rux AH, Murphy SJ, Kuri-Cervantes L, Weisman AR, et al. Erythrocytes identify complement activation in patients with COVID-19. Am J Physiol Lung Cell Mol Physiol. (2021) 321:L485–l9. doi: 10.1152/ajplung.00231.2021
106. Hotz MJ, Qing D, Shashaty MGS, Zhang P, Faust H, Sondheimer N, et al. Red blood cells homeostatically bind mitochondrial DNA through TLR9 to maintain quiescence and to prevent lung injury. Am J Respir Crit Care Med. (2018) 197:470–80. doi: 10.1164/rccm.201706-1161OC
107. Dobkin J, Mangalmurti NS. Immunomodulatory roles of red blood cells. Curr Opin Hematol. (2022) 29:306–9. doi: 10.1097/MOH.0000000000000734
108. Lam LKM, Murphy S, Kokkinaki D, Venosa A, Sherrill-Mix S, Casu C, et al. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Sci Transl Med. (2021) 13:eabj1008. doi: 10.1126/scitranslmed.abj1008
109. Schäkel K, von Kietzell M, Hänsel A, Ebling A, Schulze L, Haase M, et al. Human 6-sulfo LacNAc-expressing dendritic cells are principal producers of early interleukin-12 and are controlled by erythrocytes. Immunity. (2006) 24:767–77. doi: 10.1016/j.immuni.2006.03.020
110. Liu D, Duan L, Rodda LB, Lu E, Xu Y, An J, et al. CD97 promotes spleen dendritic cell homeostasis through the mechanosensing of red blood cells. Science. (2022) 375:eabi5965. doi: 10.1126/science.abi5965
111. Antunes RF, Brandão C, Carvalho G, Girão C, Arosa FA. Red blood cells carry out T cell growth and survival bioactivities that are sensitive to cyclosporine A. Cell Mol Life Sci. (2009) 66:3387–98. doi: 10.1007/s00018-009-0119-y
112. Yazdanbakhsh K, Bao W, Zhong H. Immunoregulatory effects of stored red blood cells. Hematol Am Soc Hematol Educ Program. (2011) 2011:466–9. doi: 10.1182/asheducation-2011.1.466
113. Antunes RF, Brandão C, Maia M, Arosa FA. Red blood cells release factors with growth and survival bioactivities for normal and leukemic T cells. Immunol Cell Biol. (2011) 89:111–21. doi: 10.1038/icb.2010.60
114. Zhang J, Ney PA. Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model. Autophagy. (2010) 6:405–8. doi: 10.4161/auto.6.3.11245
115. Caielli S, Cardenas J, de Jesus AA, Baisch J, Walters L, Blanck JP, et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell. (2021) 184:4464–79 e19. doi: 10.1016/j.cell.2021.07.021
116. Martino S, Arlet JB, Odièvre MH, Jullien V, Moras M, Hattab C, et al. Deficient mitophagy pathways in sickle cell disease. Br J Haematol. (2021) 193:988–93. doi: 10.1111/bjh.v193.5
117. Li-Harms X, Milasta S, Lynch J, Wright C, Joshi A, Iyengar R, et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood. (2015) 125:162–74. doi: 10.1182/blood-2014-07-586396
118. Jagadeeswaran R, Vazquez BA, Thiruppathi M, Ganesh BB, Ibanez V, Cui S, et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp Hematol. (2017) 50:46–52. doi: 10.1016/j.exphem.2017.02.003
119. Allison AC. Turnovers of erythrocytes and plasma proteins in mammals. Nature. (1960) 188:37–40. doi: 10.1038/188037a0
120. Diez-Silva M, Dao M, Han J, Lim CT, Suresh S. Shape and biomechanical characteristics of human red blood cells in health and disease. MRS Bull. (2010) 35:382–8. doi: 10.1557/mrs2010.571
121. Bosman GJ, Lasonder E, Groenen-Dopp YA, Willekens FL, Werre JM. The proteome of erythrocyte-derived microparticles from plasma: new clues for erythrocyte aging and vesiculation. J Proteomics. (2012) 76:203–10. doi: 10.1016/j.jprot.2012.05.031
122. You S, Park B, Lee MS. Accelerated RBC senescence as a novel pathologic mechanism of blood stasis syndrome in traditional East Asian medicine. Am J Transl Res. (2015) 7:422–9. doi: 10.1016/j.imr.2015.04.200
123. Fibach E. The redox balance and membrane shedding in RBC production, maturation, and senescence. Front Physiol. (2021) 12:604738. doi: 10.3389/fphys.2021.604738
124. Buttari B, Profumo E, Riganò R. Crosstalk between red blood cells and the immune system and its impact on atherosclerosis. BioMed Res Int. (2015) 2015:616834. doi: 10.1155/2015/616834
125. Leitinger N. Oxidized phospholipids as triggers of inflammation in atherosclerosis. Mol Nutr Food Res. (2005) 49:1063–71. doi: 10.1002/(ISSN)1613-4133
126. Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. (2005) 49:1044–9. doi: 10.1002/(ISSN)1613-4133
127. Kay MM, Bosman GJ, Johnson GJ, Beth AH. Band-3 polymers and aggregates, and hemoglobin precipitates in red cell aging. Blood Cells. (1988) 14:275–95.
128. Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci USA. (1998) 95:3077–81. doi: 10.1073/pnas.95.6.3077
129. Buttari B, Profumo E, Cuccu B, Straface E, Gambardella L, Malorni W, et al. Erythrocytes from patients with carotid atherosclerosis fail to control dendritic cell maturation. Int J Cardiol. (2012) 155:484–6. doi: 10.1016/j.ijcard.2011.12.068
130. Buttari B, Profumo E, Businaro R, Saso L, Capoano R, Salvati B, et al. Oxidized haemoglobin-driven endothelial dysfunction and immune cell activation: novel therapeutic targets for atherosclerosis. Curr Med Chem. (2013) 20:4806–14. doi: 10.2174/09298673113209990162
131. Buttari B, Profumo E, Di Cristofano C, Pietraforte D, Lionetti V, Capoano R, et al. Haemoglobin triggers chemotaxis of human monocyte-derived dendritic cells: possible role in atherosclerotic lesion instability. Atherosclerosis. (2011) 215:316–22. doi: 10.1016/j.atherosclerosis.2010.12.032
132. Mangalmurti NS, Xiong Z, Hulver M, Ranganathan M, Liu XH, Oriss T, et al. Loss of red cell chemokine scavenging promotes transfusion-related lung inflammation. Blood. (2009) 113:1158–66. doi: 10.1182/blood-2008-07-166264
133. Hod EA, Zhang N, Sokol SA, Wojczyk BS, Francis RO, Ansaldi D, et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood. (2010) 115:4284–92. doi: 10.1182/blood-2009-10-245001
134. Gerner MC, Bileck A, Janker L, Ziegler LS, Öhlinger T, Raeven P, et al. Packed red blood cells inhibit T-cell activation via ROS-dependent signaling pathways. J Biol Chem. (2021) 296:100487. doi: 10.1016/j.jbc.2021.100487
135. Reid ME, Mohandas N. Red blood cell blood group antigens: structure and function. Semin Hematol. (2004) 41:93–117. doi: 10.1053/j.seminhematol.2004.01.001
136. Li N, Chen S, Xu K, He MT, Dong MQ, Zhang QC, et al. Structural basis of membrane skeleton organization in red blood cells. Cell. (2023) 186:1912–29 e18. doi: 10.1016/j.cell.2023.03.017
137. Risinger M, Kalfa TA. Red cell membrane disorders: structure meets function. Blood. (2020) 136:1250–61. doi: 10.1182/blood.2019000946
138. Zhong H, Yazdanbakhsh K. Hemolysis and immune regulation. Curr Opin Hematol. (2018) 25:177–82. doi: 10.1097/MOH.0000000000000423
139. Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. (2013) 121:1276–84. doi: 10.1182/blood-2012-11-451229
140. Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. (2013) 18:2352–63. doi: 10.1089/ars.2012.4834
141. Helms CC, Marvel M, Zhao W, Stahle M, Vest R, Kato GJ, et al. Mechanisms of hemolysis-associated platelet activation. J Thromb Haemost. (2013) 11:2148–54. doi: 10.1111/jth.12422
142. Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, et al. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. (2003) 55:551–71. doi: 10.1124/pr.55.3.5
143. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. (2014) 5:115. doi: 10.3389/fphar.2014.00115
144. Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseño CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell. (2014) 156:1223–34. doi: 10.1016/j.cell.2014.01.069
145. Pradhan P, Vijayan V, Gueler F, Immenschuh S. Interplay of heme with macrophages in homeostasis and inflammation. Int J Mol Sci. (2020) 21(3):740. doi: 10.3390/ijms21030740
146. Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. (2016) 17:1361–72. doi: 10.1038/ni.3590
147. Monteiro AP, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, Porto BN, et al. Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol. (2011) 186:6562–7. doi: 10.4049/jimmunol.1002400
148. Graça-Souza AV, Arruda MA, de Freitas MS, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood. (2002) 99:4160–5. doi: 10.1182/blood.V99.11.4160
149. Porto BN, Alves LS, Fernández PL, Dutra TP, Figueiredo RT, Graça-Souza AV, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem. (2007) 282:24430–6. doi: 10.1074/jbc.M703570200
150. Evans C, Orf K, Horvath E, Levin M, de la Fuente J, Chakravorty S, et al. Impairment of neutrophil oxidative burst in children with sickle cell disease is associated with heme oxygenase-1. Haematologica. (2015) 100:1508–16. doi: 10.3324/haematol.2015.128777
151. Arruda MA, Rossi AG, de Freitas MS, Barja-Fidalgo C, Graça-Souza AV. Heme inhibits human neutrophil apoptosis: involvement of phosphoinositide 3-kinase, MAPK, and NF-kappaB. J Immunol. (2004) 173:2023–30. doi: 10.4049/jimmunol.173.3.2023
152. Kono M, Saigo K, Takagi Y, Takahashi T, Kawauchi S, Wada A, et al. Heme-related molecules induce rapid production of neutrophil extracellular traps. Transfusion. (2014) 54:2811–9. doi: 10.1111/trf.2014.54.issue-11
153. Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. (2013) 122:3892–8. doi: 10.1182/blood-2013-05-498311
154. Haines DD, Tosaki A. Heme degradation in pathophysiology of and countermeasures to inflammation-associated disease. Int J Mol Sci. (2020) 21(24):9698. doi: 10.3390/ijms21249698
155. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
156. Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres ML, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight. (2018) 3(12):e96910. doi: 10.1172/jci.insight.96910
157. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
158. Morikawa K, Oseko F, Morikawa S. A role for ferritin in hematopoiesis and the immune system. Leuk Lymphoma. (1995) 18:429–33. doi: 10.3109/10428199509059641
159. Zandman-Goddard G, Shoenfeld Y. Ferritin in autoimmune diseases. Autoimmun Rev. (2007) 6:457–63. doi: 10.1016/j.autrev.2007.01.016
160. Cheng HT, Yen CJ, Chang CC, Huang KT, Chen KH, Zhang RY, et al. Ferritin heavy chain mediates the protective effect of heme oxygenase-1 against oxidative stress. Biochim Biophys Acta. (2015) 1850:2506–17. doi: 10.1016/j.bbagen.2015.09.018
161. Li R, Luo C, Mines M, Zhang J, Fan GH. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. (2006) 281:37616–27. doi: 10.1074/jbc.M607266200
162. Fan Y, Zhang J, Cai L, Wang S, Liu C, Zhang Y, et al. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim Biophys Acta. (2014) 1843:2775–83. doi: 10.1016/j.bbamcr.2014.06.015
163. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. (2010) 142:24–38. doi: 10.1016/j.cell.2010.06.028
164. Zhao L, He R, Long H, Guo B, Jia Q, Qin D, et al. Late-stage tumors induce anemia and immunosuppressive extramedullary erythroid progenitor cells. Nat Med. (2018) 24:1536–44. doi: 10.1038/s41591-018-0205-5
165. Chen J, Qiao YD, Li X, Xu JL, Ye QJ, Jiang N, et al. Intratumoral CD45(+)CD71(+) erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. (2021) 499:85–98. doi: 10.1016/j.canlet.2020.12.003
166. Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell. (2022) 40:674–93 e7. doi: 10.1016/j.ccell.2022.04.018
167. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. (2013) 504:158–62. doi: 10.1038/nature12675
168. Elahi S, Vega-Lopez MA, Herman-Miguel V, Ramirez-Estudillo C, Mancilla-Ramirez J, Motyka B, et al. CD71(+) erythroid cells in human neonates exhibit immunosuppressive properties and compromise immune response against systemic infection in neonatal mice. Front Immunol. (2020) 11:597433. doi: 10.3389/fimmu.2020.597433
169. Dunsmore G, Bozorgmehr N, Delyea C, Koleva P, Namdar A, Elahi S. Erythroid Suppressor Cells Compromise Neonatal Immune Response against Bordetella pertussis. J Immunol. (2017) 199:2081–95. doi: 10.4049/jimmunol.1700742
170. Namdar A, Koleva P, Shahbaz S, Strom S, Gerdts V, Elahi S. CD71(+) erythroid suppressor cells impair adaptive immunity against Bordetella pertussis. Sci Rep. (2017) 7:7728. doi: 10.1038/s41598-017-07938-7
171. Shahbaz S, Bozorgmehr N, Koleva P, Namdar A, Jovel J, Fava RA, et al. CD71+VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-beta. PloS Biol. (2018) 16:e2006649. doi: 10.1371/journal.pbio.2006649
172. Yang L, Shivakumar P, Kinder J, Way SS, Donnelly B, Mourya R, et al. Regulation of bile duct epithelial injury by hepatic CD71+ erythroid cells. JCI Insight. (2020) 5(11):e135751. doi: 10.1172/jci.insight.135751
173. Wynn JL, Scumpia PO, Stocks BT, Romano-Keeler J, Alrifai MW, Liu JH, et al. Neonatal CD71+ Erythroid cells do not modify murine sepsis mortality. J Immunol. (2015) 195:1064–70. doi: 10.4049/jimmunol.1500771
174. Elahi S, Mashhouri S. Immunological consequences of extramedullary erythropoiesis: immunoregulatory functions of CD71(+) erythroid cells. Haematologica. (2020) 105:1478–83. doi: 10.3324/haematol.2019.243063
175. Delyea C, Bozorgmehr N, Koleva P, Dunsmore G, Shahbaz S, Huang V, et al. CD71(+) erythroid suppressor cells promote fetomaternal tolerance through arginase-2 and PDL-1. J Immunol. (2018) 200:4044–58. doi: 10.4049/jimmunol.1800113
176. Dunsmore G, Koleva P, Ghobakhloo N, Sutton R, Ambrosio L, Meng X, et al. Lower abundance and impaired function of CD71+ Erythroid cells in inflammatory bowel disease patients during pregnancy. J Crohns Colitis. (2019) 13:230–44. doi: 10.1093/ecco-jcc/jjy147
177. Namdar A, Dunsmore G, Shahbaz S, Koleva P, Xu L, Jovel J, et al. CD71(+) erythroid cells exacerbate HIV-1 susceptibility, mediate trans-infection, and harbor infective viral particles. mBio. (2019) 10(6):e02767-19. doi: 10.1128/mBio.02767-19
178. Zhao GJ, Jiang DW, Cai WC, Chen XY, Dong W, Chen LW, et al. CD71(+) erythroid cell expansion in adult sepsis: potential causes and role in prognosis and nosocomial infection prediction. Front Immunol. (2022) 13:830025. doi: 10.3389/fimmu.2022.830025
179. Shahbaz S, Xu L, Osman M, Sligl W, Shields J, Joyce M, et al. Erythroid precursors and progenitors suppress adaptive immunity and get invaded by SARS-CoV-2. Stem Cell Rep. (2021) 16:1165–81. doi: 10.1016/j.stemcr.2021.04.001
180. Junqueira C, Barbosa CRR, Costa PAC, Teixeira-Carvalho A, Castro G, Sen Santara S, et al. Cytotoxic CD8(+) T cells recognize and kill Plasmodium vivax-infected reticulocytes. Nat Med. (2018) 24:1330–6. doi: 10.1038/s41591-018-0117-4
181. Martin-Jaular L, Nakayasu ES, Ferrer M, Almeida IC, Del Portillo HA. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PloS One. (2011) 6:e26588. doi: 10.1371/journal.pone.0026588
182. Jackson A, Nanton MR, O’Donnell H, Akue AD, McSorley SJ. Innate immune activation during Salmonella infection initiates extramedullary erythropoiesis and splenomegaly. J Immunol. (2010) 185:6198–204. doi: 10.4049/jimmunol.1001198
183. Shim YA, Weliwitigoda A, Campbell T, Dosanjh M, Johnson P. Splenic erythroid progenitors decrease TNF-alpha production by macrophages and reduce systemic inflammation in a mouse model of T cell-induced colitis. Eur J Immunol. (2021) 51:567–79. doi: 10.1002/eji.202048687
184. Kanemasa H, Ishimura M, Eguchi K, Tanaka T, Nanishi E, Shiraishi A, et al. The immunoregulatory function of peripheral blood CD71(+) erythroid cells in systemic-onset juvenile idiopathic arthritis. Sci Rep. (2021) 11:14396. doi: 10.1038/s41598-021-93831-3
185. Prince OD, Langdon JM, Layman AJ, Prince IC, Sabogal M, Mak HH, et al. Late stage erythroid precursor production is impaired in mice with chronic inflammation. Haematologica. (2012) 97:1648–56. doi: 10.3324/haematol.2011.053397
186. Han Y, Liu Q, Hou J, Gu Y, Zhang Y, Chen Z, et al. Tumor-induced generation of splenic erythroblast-like ter-cells promotes tumor progression. Cell. (2018) 173:634–48 e12. doi: 10.1016/j.cell.2018.02.061
187. Bozorgmehr N, Okoye I, Mashhouri S, Lu J, Koleva P, Walker J, et al. CD71(+) erythroid cells suppress T-cell effector functions and predict immunotherapy outcomes in patients with virus-associated solid tumors. J Immunother Cancer. (2023) 11(5):e006595. doi: 10.1136/jitc-2022-006595
188. Dev A, Fang J, Sathyanarayana P, Pradeep A, Emerson C, Wojchowski DM. During EPO or anemia challenge, erythroid progenitor cells transit through a selectively expandable proerythroblast pool. Blood. (2010) 116:5334–46. doi: 10.1182/blood-2009-12-258947
189. Grzywa TM, Sosnowska A, Rydzynska Z, Lazniewski M, Plewczynski D, Klicka K, et al. Potent but transient immunosuppression of T-cells is a general feature of CD71(+) erythroid cells. Commun Biol. (2021) 4:1384. doi: 10.1038/s42003-021-02914-4
190. Kho S, Siregar NC, Qotrunnada L, Fricot A, Sissoko A, Shanti PAI, et al. Retention of uninfected red blood cells causing congestive splenomegaly is the major mechanism of anemia in malaria. Am J Hematol. (2024) 99:223–35. doi: 10.1002/ajh.27152
191. Chang KH, Stevenson MM. Malarial anaemia: mechanisms and implications of insufficient erythropoiesis during blood-stage malaria. Int J Parasitol. (2004) 34:1501–16. doi: 10.1016/j.ijpara.2004.10.008
192. Raundhal M, Ghosh S, Myers SA, Cuoco MS, Singer M, Carr SA, et al. Blockade of IL-22 signaling reverses erythroid dysfunction in stress-induced anemias. Nat Immunol. (2021) 22:520–9. doi: 10.1038/s41590-021-00895-4
193. Nazarov K, Perik-Zavodskii R, Perik-Zavodskaia O, Alrhmoun S, Volynets M, Shevchenko J, et al. Murine placental erythroid cells are mainly represented by CD45(+) immunosuppressive erythroid cells and secrete CXCL1, CCL2, CCL3 and CCL4 chemokines. Int J Mol Sci. (2023) 24(9):8130. doi: 10.3390/ijms24098130
194. Martí ILAA, Reith W. Arginine-dependent immune responses. Cell Mol Life Sci. (2021) 78:5303–24. doi: 10.1007/s00018-021-03828-4
195. Waddington SN. Arginase in glomerulonephritis. Kidney Int. (2002) 61:876–81. doi: 10.1046/j.1523-1755.2002.00236.x
196. Miyazaki N, Takami S, Uemura M, Oiki H, Takahashi M, Kawashima H, et al. Impact of gallbladder hypoplasia on hilar hepatic ducts in biliary atresia. Commun Med (Lond). (2024) 4:111. doi: 10.1038/s43856-024-00544-5
197. Xiao MH, Wu S, Liang P, Ma D, Zhang J, Chen H, et al. Mucosal-associated invariant T cells promote ductular reaction through amphiregulin in biliary atresia. EBioMedicine. (2024) 103:105138. doi: 10.1016/j.ebiom.2024.105138
198. Mashhouri S, Koleva P, Huynh M, Okoye I, Shahbaz S, Elahi S. Sex matters: physiological abundance of immuno-regulatory CD71+ Erythroid cells impair immunity in females. Front Immunol. (2021) 12:705197. doi: 10.3389/fimmu.2021.705197
199. Gomez-Lopez N, Romero R, Xu Y, Miller D, Unkel R, CM T, et al. Umbilical cord CD71+ erythroid cells are reduced in neonates born to women in spontaneous preterm labor. Am J Reprod Immunol. (2016) 76:280–4. doi: 10.1111/aji.12556
200. Tan EK, Tan EL. Alterations in physiology and anatomy during pregnancy. Best Pract Res Clin Obstet Gynaecol. (2013) 27:791–802. doi: 10.1016/j.bpobgyn.2013.08.001
201. Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. (2010) 6:265–78. doi: 10.1016/j.stem.2010.02.002
202. Gao X, Lee HY, da Rocha EL, Zhang C, Lu YF, Li D, et al. TGF-beta inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood. (2016) 128:2637–41. doi: 10.1182/blood-2016-05-718320
203. Elahi S. Neglected cells: immunomodulatory roles of CD71(+) erythroid cells. Trends Immunol. (2019) 40:181–5. doi: 10.1016/j.it.2019.01.003
204. Goodsell DS. Illustrations of the HIV life cycle. Curr Top Microbiol Immunol. (2015) 389:243–52. doi: 10.1007/82_2015_437
205. Poespoprodjo JR, Douglas NM, Ansong D, Kho S, Anstey NM. Malaria. Lancet. (2023) 402:2328–45. doi: 10.1016/S0140-6736(23)01249-7
206. Srivastava A, Creek DJ, Evans KJ, De Souza D, Schofield L, Muller S, et al. Host reticulocytes provide metabolic reservoirs that can be exploited by malaria parasites. PloS Pathog. (2015) 11:e1004882. doi: 10.1371/journal.ppat.1004882
207. Malleret B, Li A, Zhang R, Tan KS, Suwanarusk R, Claser C, et al. Plasmodium vivax: restricted tropism and rapid remodeling of CD71-positive reticulocytes. Blood. (2015) 125:1314–24. doi: 10.1182/blood-2014-08-596015
208. Kosaisavee V, Suwanarusk R, Chua ACY, Kyle DE, Malleret B, Zhang R, et al. Strict tropism for CD71(+)/CD234(+) human reticulocytes limits the zoonotic potential of Plasmodium cynomolgi. Blood. (2017) 130:1357–63. doi: 10.1182/blood-2017-02-764787
209. Venkatesan P. The 2023 WHO World malaria report. Lancet Microbe. (2024) 5:e214. doi: 10.1016/S2666-5247(24)00016-8
210. Martin-Jaular L, Elizalde-Torrent A, Thomson-Luque R, Ferrer M, Segovia JC, Herreros-Aviles E, et al. Reticulocyte-prone malaria parasites predominantly invade CD71hi immature cells: implications for the development of an in vitro culture for Plasmodium vivax. Malar J. (2013) 12:434. doi: 10.1186/1475-2875-12-434
211. Pattaradilokrat S, Cheesman SJ, Carter R. Congenicity and genetic polymorphism in cloned lines derived from a single isolate of a rodent malaria parasite. Mol Biochem Parasitol. (2008) 157:244–7. doi: 10.1016/j.molbiopara.2007.10.011
212. Brito MAM, Baro B, Raiol TC, Ayllon-Hermida A, Safe IP, Deroost K, et al. Morphological and transcriptional changes in human bone marrow during natural plasmodium vivax malaria infections. J Infect Dis. (2022) 225:1274–83. doi: 10.1093/infdis/jiaa177
213. Josselsohn R, Barnes BJ, Kalfa TA, Blanc L. Navigating the marrow sea towards erythromyeloblastic islands under normal and inflammatory conditions. Curr Opin Hematol. (2023) 30:80–5. doi: 10.1097/MOH.0000000000000756
214. Gomes AC, Moreira AC, Silva T, Neves JV, Mesquita G, Almeida AA, et al. IFN-γ-dependent reduction of erythrocyte life span leads to anemia during mycobacterial infection. J Immunol. (2019) 203:2485–96. doi: 10.4049/jimmunol.1900382
215. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. (2014) 35:347–69. doi: 10.1016/j.yfrne.2014.04.004
216. Munoz-Jurado A, Escribano BM, Tunez I. Animal model of multiple sclerosis: Experimental autoimmune encephalomyelitis. Methods Cell Biol. (2024) 188:35–60. doi: 10.1016/bs.mcb.2024.03.013
217. Grant SM, Wiesinger JA, Beard JL, Cantorna MT. Iron-deficient mice fail to develop autoimmune encephalomyelitis. J Nutr. (2003) 133:2635–8. doi: 10.1093/jn/133.8.2635
218. Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, et al. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. (2006) 172:27–37. doi: 10.1016/j.jneuroim.2005.10.016
219. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
220. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature. (2015) 521:94–8. doi: 10.1038/nature14395
221. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
222. Cheng X, Wang H, Wang Z, Zhu B, Long H. Tumor-associated myeloid cells in cancer immunotherapy. J Hematol Oncol. (2023) 16:71. doi: 10.1186/s13045-023-01473-x
223. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
224. Hou Y, Liang HL, Yu X, Liu Z, Cao X, Rao E, et al. Radiotherapy and immunotherapy converge on elimination of tumor-promoting erythroid progenitor cells through adaptive immunity. Sci Transl Med. (2021) 13(582):eabb0130. doi: 10.1126/scitranslmed.abb0130
226. Grzywa TM, Justyniarska M, Nowis D, Golab J. Tumor immune evasion induced by dysregulation of erythroid progenitor cells development. Cancers (Basel). (2021) 13(4):870. doi: 10.3390/cancers13040870
227. Elshinawy M, Kamal M, Nazir H, Khater D, Hassan R, Elkinany H, et al. Sepsis-related anemia in a pediatric intensive care unit: transfusion-associated outcomes. Transfusion. (2020) 60 Suppl 1:S4–9. doi: 10.1111/trf.v60.S1
228. Lefebvre P, Vekeman F, Sarokhan B, Enny C, Provenzano R, Cremieux PY. Relationship between hemoglobin level and quality of life in anemic patients with chronic kidney disease receiving epoetin alfa. Curr Med Res Opin. (2006) 22:1929–37. doi: 10.1185/030079906X132541
229. Arcasoy MO. Erythropoiesis-stimulating agent use in cancer: preclinical and clinical perspectives. Clin Cancer Res. (2008) 14:4685–90. doi: 10.1158/1078-0432.CCR-08-0264
230. Hedley BD, Allan AL, Xenocostas A. The role of erythropoietin and erythropoiesis-stimulating agents in tumor progression. Clin Cancer Res. (2011) 17:6373–80. doi: 10.1158/1078-0432.CCR-10-2577
231. Henke M, Laszig R, Rube C, Schafer U, Haase KD, Schilcher B, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. (2003) 362:1255–60. doi: 10.1016/S0140-6736(03)14567-9
232. Liu C, Ruan Y, Huang R, Fang L, Wu T, Lv Y, et al. Efficacy and safety of immune checkpoint inhibitors in solid tumor patients combined with chronic coronary syndromes or its risk factor: a nationwide multicenter cohort study. Cancer Immunol Immunother. (2024) 73:159. doi: 10.1007/s00262-024-03747-w
233. Dulmovits BM, Tang Y, Papoin J, He M, Li J, Yang H, et al. HMGB1-mediated restriction of EPO signaling contributes to anemia of inflammation. Blood. (2022) 139:3181–93. doi: 10.1182/blood.2021012048
234. Rainville N, Jachimowicz E, Wojchowski DM. Targeting EPO and EPO receptor pathways in anemia and dysregulated erythropoiesis. Expert Opin Ther Targets. (2016) 20:287–301. doi: 10.1517/14728222.2016.1090975
235. Del Prete S, Cinieri S, Lorusso V, Maiorino L, Pizza C, Pisano A, et al. Impact of anemia management with EPO on psychologic distress in cancer patients: results of a multicenter patient survey. Future Oncol. (2014) 10:69–78. doi: 10.2217/fon.13.161
236. Winearls CG, Oliver DO, Pippard MJ, Reid C, Downing MR, Cotes PM. Effect of human erythropoietin derived from recombinant DNA on the anaemia of patients maintained by chronic haemodialysis. Lancet. (1986) 2:1175–8. doi: 10.1016/S0140-6736(86)92192-6
237. Bohlius J, Wilson J, Seidenfeld J, Piper M, Schwarzer G, Sandercock J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst. (2006) 98:708–14. doi: 10.1093/jnci/djj189
238. Milano M, Schneider M. EPO in cancer anemia: benefits and potential risks. Crit Rev Oncol Hematol. (2007) 62:119–25. doi: 10.1016/j.critrevonc.2006.11.011
239. Ora J, Calzetta L, Matera MG, Cazzola M, Rogliani P. Advances with glucocorticoids in the treatment of asthma: state of the art. Expert Opin Pharmacother. (2020) 21:2305–16. doi: 10.1080/14656566.2020.1807514
240. Mitre-Aguilar IB, Moreno-Mitre D, Melendez-Zajgla J, Maldonado V, Jacobo-Herrera NJ, Ramirez-Gonzalez V, et al. The role of glucocorticoids in breast cancer therapy. Curr Oncol. (2022) 30:298–314. doi: 10.3390/curroncol30010024
241. Heideveld E, Hampton-O’Neil LA, Cross SJ, van Alphen FPJ, van den Biggelaar M, Toye AM, et al. Glucocorticoids induce differentiation of monocytes towards macrophages that share functional and phenotypical aspects with erythroblastic island macrophages. Haematologica. (2018) 103:395–405. doi: 10.3324/haematol.2017.179341
242. Falchi M, Varricchio L, Martelli F, Masiello F, Federici G, Zingariello M, et al. Dexamethasone targeted directly to macrophages induces macrophage niches that promote erythroid expansion. Haematologica. (2015) 100:178–87. doi: 10.3324/haematol.2014.114405
243. Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, et al. PPAR-α and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. (2015) 522:474–7. doi: 10.1038/nature14326
244. Nordsborg NB, Bonne TC, Breenfeldt Andersen A, Sørensen H, Bejder J. Glucocorticoids accelerate erythropoiesis in healthy humans-should the use in sports be reevaluated? Med Sci Sports Exerc. (2023) 55:1334–41. doi: 10.1249/mss.0000000000003156
245. Gao X, Lee HY, da Rocha EL, Zhang C, Lu YF, Li D, et al. TGF-β inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood. (2016) 128:2637–41. doi: 10.1182/blood-2016-05-718320
246. Teixeira AF, Ten Dijke P, Zhu HJ. On-target anti-TGF-beta therapies are not succeeding in clinical cancer treatments: what are remaining challenges? Front Cell Dev Biol. (2020) 8:605. doi: 10.3389/fcell.2020.00605
247. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
248. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
249. Niu M, Yi M, Wu Y, Lyu L, He Q, Yang R, et al. Synergistic efficacy of simultaneous anti-TGF-β/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J Hematol Oncol. (2023) 16:94. doi: 10.1186/s13045-023-01487-5
250. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. (2022) 21:104. doi: 10.1186/s12943-022-01569-x
251. Hernandez-Hernandez A, Ray P, Litos G, Ciro M, Ottolenghi S, Beug H, et al. Acetylation and MAPK phosphorylation cooperate to regulate the degradation of active GATA-1. EMBO J. (2006) 25:3264–74. doi: 10.1038/sj.emboj.7601228
252. Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. (2020) 21(6):1913. doi: 10.3390/ijms21061913
253. Zhang J, Shen B, Lin A. Novel strategies for inhibition of the p38 MAPK pathway. Trends Pharmacol Sci. (2007) 28:286–95. doi: 10.1016/j.tips.2007.04.008
254. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. (2009) 15:369–79. doi: 10.1016/j.molmed.2009.06.005
255. Zhang X, Campreciós G, Rimmelé P, Liang R, Yalcin S, Mungamuri SK, et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am J Hematol. (2014) 89:954–63. doi: 10.1002/ajh.v89.10
256. Franco SS, De Falco L, Ghaffari S, Brugnara C, Sinclair DA, Matte A, et al. Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica. (2014) 99:267–75. doi: 10.3324/haematol.2013.090076
257. Ayuk SM, Abrahamse H. mTOR signaling pathway in cancer targets photodynamic therapy in vitro. Cells. (2019) 8(5):431. doi: 10.3390/cells8050431
258. De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, et al. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. (1999) 401:489–93. doi: 10.1038/46809
259. Tyrkalska SD, Perez-Oliva AB, Rodriguez-Ruiz L, Martinez-Morcillo FJ, Alcaraz-Perez F, Martinez-Navarro FJ, et al. Inflammasome regulates hematopoiesis through cleavage of the master erythroid transcription factor GATA1. Immunity. (2019) 51:50–63 e5. doi: 10.1016/j.immuni.2019.05.005
260. Caruso F, Pedersen JZ, Incerpi S, Kaur S, Belli S, Florea RM, et al. Mechanism of caspase-1 inhibition by four anti-inflammatory drugs used in COVID-19 treatment. Int J Mol Sci. (2022) 23(3):1849. doi: 10.3390/ijms23031849
261. Speck-Planche A, Kleandrova VV, Scotti MT. In silico drug repurposing for anti-inflammatory therapy: virtual search for dual inhibitors of caspase-1 and TNF-alpha. Biomolecules. (2021) 11(12):1832. doi: 10.3390/biom11121832
262. Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL Jr., et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol. (2017) 10:73. doi: 10.1186/s13045-017-0427-x
263. Sheetz M, Barrington P, Callies S, Berg PH, McColm J, Marbury T, et al. Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br J Clin Pharmacol. (2019) 85:935–48. doi: 10.1111/bcp.13877
264. Wake M, Palin A, Belot A, Berger M, Lorgouilloux M, Bichon M, et al. A human anti-matriptase-2 antibody limits iron overload, alpha-globin aggregates, and splenomegaly in beta-thalassemic mice. Blood Adv. (2024) 8:1898–907. doi: 10.1182/bloodadvances.2023012010
265. Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. (2019) 133:40–50. doi: 10.1182/blood-2018-06-856500
266. Borek B, Gajda T, Golebiowski A, Blaszczyk R. Boronic acid-based arginase inhibitors in cancer immunotherapy. Bioorg Med Chem. (2020) 28:115658. doi: 10.1016/j.bmc.2020.115658
267. Cifuentes-Pagano ME, Meijles DN, Pagano PJ. Nox inhibitors & Therapies: rational design of peptidic and small molecule inhibitors. Curr Pharm Des. (2015) 21:6023–35. doi: 10.2174/1381612821666151029112013
268. Mies A, Platzbecker U. Increasing the effectiveness of hematopoiesis in myelodysplastic syndromes: erythropoiesis-stimulating agents and transforming growth factor-beta superfamily inhibitors. Semin Hematol. (2017) 54:141–6. doi: 10.1053/j.seminhematol.2017.06.004
269. Steenbrugge J, De Jaeghere EA, Meyer E, Denys H, De Wever O. Splenic hematopoietic and stromal cells in cancer progression. Cancer Res. (2021) 81:27–34. doi: 10.1158/0008-5472.CAN-20-2339
270. El Hajj H, Ferraioli D, Meus P, Beurrier F, Tredan O, Ray-Coquard I, et al. Splenectomy in epithelial ovarian cancer surgery. Int J Gynecol Cancer. (2023) 33:944–50. doi: 10.1136/ijgc-2022-003928
271. Pergialiotis V, Zachariou E, Lygizos V, Vlachos DE, Stamatakis E, Angelou K, et al. Splenectomy as part of maximal-effort cytoreductive surgery in advanced epithelial ovarian cancer. Cancers (Basel). (2024) 16(4):790. doi: 10.3390/cancers16040790
272. Sun H, Bi X, Cao D, Yang J, Wu M, Pan L, et al. Splenectomy during cytoreductive surgery in epithelial ovarian cancer. Cancer Manag Res. (2018) 10:3473–82. doi: 10.2147/CMAR.S172687
273. Yu J, Wang K, Tang Y, Zheng D. Applications and perspectives of tumor organoids in radiobiology (Review). Oncol Rep. (2024) 52(2):100. doi: 10.3892/or.2024.8759
274. Usman WM, Pham TC, Kwok YY, Vu LT, Ma V, Peng B, et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat Commun. (2018) 9:2359. doi: 10.1038/s41467-018-04791-8
275. Luk BT, Fang RH, Hu CM, Copp JA, Thamphiwatana S, Dehaini D, et al. Safe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumors. Theranostics. (2016) 6:1004–11. doi: 10.7150/thno.14471
276. Xu L, Xu X, Liang Y, Wen C, Ouyang K, Huang J, et al. Osteoclast-targeted delivery of anti-miRNA oligonucleotides by red blood cell extracellular vesicles. J Control Release. (2023) 358:259–72. doi: 10.1016/j.jconrel.2023.04.043
277. Dertinger SD, Chen Y, Miller RK, Brewer KJ, Smudzin T, Torous DK, et al. Micronucleated CD71-positive reticulocytes: a blood-based endpoint of cytogenetic damage in humans. Mutat Res. (2003) 542:77–87. doi: 10.1016/j.mrgentox.2003.08.004
278. Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PloS Med. (2013) 10:e1001577. doi: 10.1371/journal.pmed.1001577
279. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. (2011) 11:426–37. doi: 10.1038/nrc3066
280. Tamkovich S, Laktionov P. Cell-surface-bound circulating DNA in the blood: Biology and clinical application. IUBMB Life. (2019) 71:1201–10. doi: 10.1002/iub.v71.9
281. Liang N, Jiao Z, Zhang C, Wu Y, Wang T, Li S, et al. Mature red blood cells contain long DNA fragments and could acquire DNA from lung cancer tissue. Adv Sci (Weinh). (2023) 10:e2206361. doi: 10.1002/advs.202206361
282. Pérez-Jacoiste Asín MA, Ruiz Robles G. Skull erythropoiesis in a patient with congenital dyserythropoietic anaemia. Lancet. (2016) 387:787. doi: 10.1016/S0140-6736(15)00054-9
Keywords: immunoregulation, erythropoiesis, CD71 + erythroid cells, ROS, PD-L1, TGF-β
Citation: Niu C and Zhang J (2024) Immunoregulation role of the erythroid cells. Front. Immunol. 15:1466669. doi: 10.3389/fimmu.2024.1466669
Received: 18 July 2024; Accepted: 30 September 2024;
Published: 15 October 2024.
Edited by:
Qizhen Shi, Medical College of Wisconsin, United StatesReviewed by:
Tirthadipa Pradhan-Sundd, Versiti Blood Research Institute, United StatesNilam Mangalmurti, University of Pennsylvania, United States
Copyright © 2024 Niu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiyan Zhang, emhhbmdqeUBibWkuYWMuY24=