 Lei Zhang
Lei Zhang Futian Xu
Futian Xu Liyan Hou1*
Liyan Hou1*- 1Pharmacy Department, Weihai Central Hospital Affiliated to Qingdao University, Weihai, China
- 2Logistics Management Department, Weihai Central Hospital Affiliated to Qingdao University, Weihai, China
Diabetic kidney disease (DKD) is a severe microvascular complication of diabetes associated with high mortality and disability rates. Inflammation has emerged as a key pathological mechanism in DKD, prompting interest in novel therapeutic approaches targeting inflammatory pathways. Interleukin-6 (IL-6), a well-established inflammatory cytokine known for mediating various inflammatory responses, has attracted great attention in the DKD field. Although multiple in vivo and in vitro studies highlight the potential of targeting IL-6 in DKD treatment, its exact roles in the disease remains unclear. This review presents the roles of IL-6 in the pathogenesis of DKD, including immunoinflammation, metabolism, hemodynamics, and ferroptosis. In addition, we summarize the current status of IL-6 inhibitors in DKD-related clinical trials and discuss the potential of targeting IL-6 for treating DKD in the clinic.
1 Introduction
Diabetic kidney disease (DKD) is the leading cause of end-stage kidney disease (ESKD) worldwide and is the major contributor to diabetes mortality rate. Despite recent progress in the development of new drugs for DKD, the residual risk still remains (1). Identifying potential novel targets and therapeutic drugs for DKD is of great clinical significance. Numerous studies have highlighted inflammation as a crucial factor in the occurrence and progression of DKD (2, 3). DKD now is also acknowledged as an inflammatory disorder (4). Rayego-Mateos et al. propose prioritizing anti-inflammatory interventions in the management of DKD by 2030 (5).
Several important reviews highlight the critical role of inflammatory factors, especially interleukin-6 (IL-6), in the development of type 2 diabetes mellitus (T2DM) and its associated kidney diseases (2, 6). Research has demonstrated significantly elevated IL-6 levels in T2DM patients, which closely correlate with declining renal function (7). Several large cohort studies have confirmed the direct relationship between high circulating IL-6 levels and reduced kidney function in patients with chronic kidney disease (CKD) (8, 9). Compared to other cytokines such as interleukin-1 (IL-1) and tumor necrosis factor (TNF), anti-IL-6 therapy is more effective in improving serum lipoprotein levels in patients with rheumatoid arthritis (RA) (10). Additionally, IL-6 is more relevant to the 5-year all-cause and cardiovascular mortality risks in patients with CKD and ESKD than TNF and fibrosis markers (11). Among patients with ESKD, plasma IL-6 levels are more effective in predicting mortality risk than IL-1, interleukin-18 (IL-18), and TNF (12). Based on these findings, therapeutic strategies targeting IL-6 show significant potential in the management of DKD. For instance, studies using the IL-6 receptor blocker tocilizumab (TCZ) in mouse models have demonstrated its ability to ameliorate the pathological changes associated with DKD (13), further supporting the feasibility of targeting IL-6. The fact that TCZ has been successfully utilized in clinical settings for treating inflammatory diseases such as RA suggests that targeting IL-6 could represent a promising strategy for DKD. However, the exact physiological and pathological roles of IL-6 in DKD remain unresolved. In this article, we summarize the current understanding of IL-6 signaling in DKD to facilitate in-depth studies of potential therapeutic strategies for targeting IL-6 in the treatment of DKD in the future.
2 IL-6 and its signaling pathways
IL-6 plays a key role in multiple biological processes, including immune responses, vascular diseases, developmental processes, metabolic regulation. This pleiotropic activity has made it a focal point of research, particularly in inflammatory pathological states. When exploring the mechanisms of IL-6, it is important to understand its receptor system. The IL-6 receptor system includes both membrane-bound interleukin-6 receptor alpha chain (also known as mIL-6R) and soluble forms of IL-6Rα (also known as sIL-6R), with glycoprotein 130 (gp130, the IL-6Rβ subunit) playing a central role in signaling. IL-6Rα binds to IL-6, while gp130 is crucial for transmitting the signal inside the cell.
IL-6 actions are mediated by three distinct intracellular signaling pathways, as illustrated in Figure 1 (drawn by Figdraw). The first signaling pathway is the classical signaling pathway mediated through mIL-6R. IL-6 binds to mIL-6R and induces the homodimerization of the gp130 receptor chain, which recruits Janus kinases (JAKs) that activate each other and trigger the downstream signaling pathways, including the SHP-2/ERK MAPK pathway and the JAK/STAT pathway (14). This classical signaling pathway of IL-6 mainly occurs in a few types of cells, primarily found in immune cells (neutrophils, macrophages and CD4+ T cells) and resident cells (hepatocytes, pancreatic cells and podocytes) (15–17).
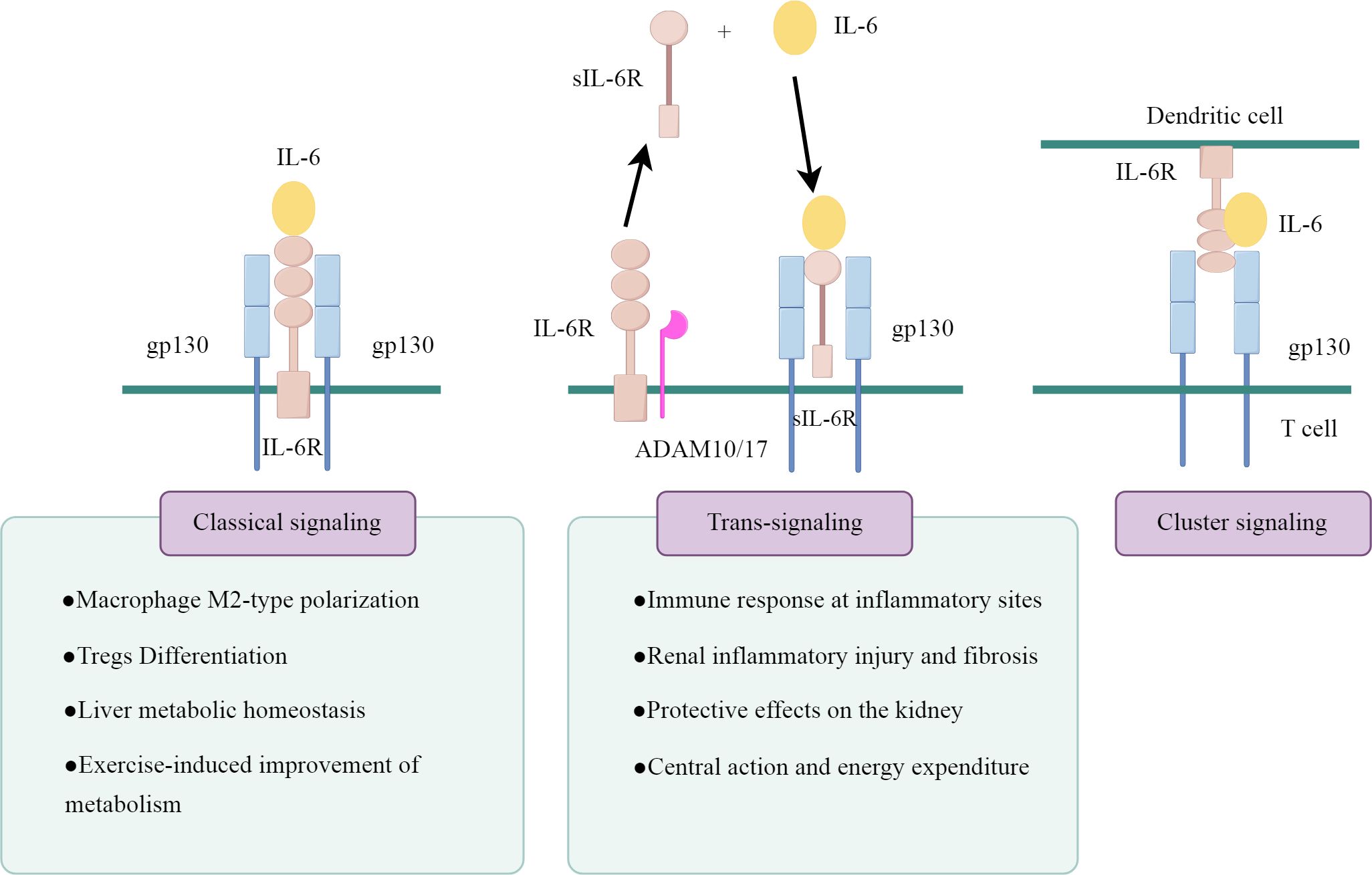
Figure 1. The signaling patterns of IL-6.
The second is the sIL-6R-mediated signaling system, known as IL-6 trans-signaling. sIL-6R is produced either by the proteolytic shedding of mIL-6R mediated by metalloenzymes such as a disintegrin and metalloprotease 10 (ADAM10) and a disintegrin and metalloprotease 17 (ADAM17), or by the selective splicing of IL-6R mRNA (18). sIL-6R binds to IL-6 to form a complex, which further activates gp130 and transmits the signal. gp130 is a signal transducer shared by all IL-6 family members, and theoretically, the IL-6/sIL-6R complex is capable of activating all cells in the body because of the widespread expression of gp130. However, this trans-signaling is highly regulated by naturally occurring soluble gp130 (sgp130), which binds to the IL-6/sIL-6R complex with high affinity and specifically blocks the IL-6 trans-signaling pathway, thus constituting a physiological buffer for the IL-6 trans-pathway (19).
In cells lacking mIL-6R, the trans-signaling pathway of IL-6 is their only signal transduction pathway. However, in cells expressing mIL-6R, the situation becomes more complex. Typically, these cells undergo parallel activation of both the classical and trans-signaling pathways. sIL-6R can competitively bind to IL-6 with mIL-6R, and the ratio of cell surface IL-6Rα/gp130 expression determines the strength of trans-signaling versus classical signal transduction (20). Moreover, when the molar concentration of sIL-6R exceeds that of IL-6, sgp130 can inhibit classical signal transduction (21).
The third pathway is cluster signaling pathway, primarily occurring between dendritic cells (DCs) and T cells in experimental mice (22). IL-6 binds to IL-6Rα on the surface of DCs, forming a complex that subsequently interacts with gp130 on the T cell membrane. This interaction induces gp130 dimerization, activating IL-6 signaling and initiating the Th17 response. However, its role in humans remains unconfirmed.
In summary, the IL-6 signaling system transmits information between cells through different receptor forms and regulatory mechanisms, thereby influencing disease progression. In recent years, researchers have also explored the pleiotropic roles of IL-6 at molecular genetic level. For instance, an IL-6 meta-analysis of genome-wide association studies (GWAS) identifies potential IL-6-modulating genes, including the interleukin-1 receptor antagonist gene (IL1RN), human leukocyte antigen (HLA), and IL-6R, which have roles in immunity and inflammation (23). Another genetic study links the IL6 single nucleotide polymorphism (SNP), inflammation and its association with ESKD (24). Future research on these novel aspects of IL-6 at the genetic level may help broaden the understanding of new potential signaling pathways, thereby elucidating the complex physiological functions of IL-6. In this context, the exploration of the relationship between IL-6 and DKD is of particular importance, as IL-6 plays a pivotal role in diabetes-related inflammatory responses and may significantly affect kidney structure and function.
3 The relationship between IL-6 and DKD
First, genetic studies focusing on immune responses, as well as systematic reviews and meta-analyses of GWAS, have revealed that IL-6 gene variants are associated with an increased risk of DKD (25–27). Second, IL-6 is associated with kidney injury in DKD. IL-6 expression is significantly upregulated in the kidneys of rat models with DKD (28, 29), and their kidney weight and proteinuria levels are correlated with renal IL-6 levels (28). Several human studies have indicated a marked increase in serum IL-6 levels among individuals with DKD (30–32). Furthermore, there exists a direct correlation between high glycated hemoglobin A1c (HbA1c) levels (33) and early glomerular structural abnormalities (31) in patients with T2DM in relation to serum IL-6 levels. Moreover, circulating IL-6 levels in patients with DKD are elevated compared to those in diabetic individuals without kidney disease (30, 32). IL-6 expression is found to be increased in the kidneys of patients with CKD or DKD (34, 35), particularly in areas of mesangial expansion, tubular infiltration and atrophy (35). Additionally, infiltrating immune cells in the damaged kidneys, including macrophages, T cells, and basophils, exhibit elevated levels of IL-6 expression (36–38), which further drives the progression of the inflammatory response.
More importantly, IL-6 emerges as a promising biomarker for predicting the progression of DKD. Clinical studies have shown that plasma IL-6 is an independent predictor of mortality in patients with advanced CKD (39), and plasma IL-6 levels in CKD patients increase with the CKD stage. Elevated plasma IL-6 levels in patients with T2DM are associated with deterioration in kidney function, independent of baseline kidney function or proteinuria (7). Several large cohort studies have confirmed a direct correlation between IL-6 levels and kidney function, with high IL-6 levels significantly associated with decreased kidney function in individuals with or without CKD (8, 9).
The abnormal expression of IL-6 in animal models and human studies of T2DM and DKD reveals its close relationship with these diseases, highlighting the necessity of focusing on the key roles of the inflammatory cytokine IL-6. Therefore, the potential mechanisms by which IL-6 affects DKD are worthy of further investigation.
4 Potential mechanisms of IL-6 Involvement in the regulation of DKD
The regulatory mechanisms of IL-6 in DKD are complex. Here, we explore the effects of IL-6 in relation to immunoinflammation, kidney cell function, glucolipid metabolism, renin-angiotensin-aldosterone system (RAAS) activation, and iron homeostasis, aiming to comprehensively elucidate the central role of IL-6 in the development of DKD.
4.1 Inflammatory immune cells: IL-6 regulates the initial and resolution phases of inflammation
From a pathological perspective, inflammation in DKD begins with an immune response initiated by the immune system, which is essential for the clearance of infectious factors. Once the threat signals have been eliminated, it is vital to restrain this response to prevent excessive tissue damage and reduce chronic inflammation (40). CD45+ immune cells play an important role in this process, so it is crucial to study the regulatory mechanisms of IL-6 on renal CD45+ immune cells.
In normal kidneys, a small number of CD45+ immune cells are detected; however, in the kidneys of DKD patients, these cells increase in the early stages (41) and may decrease in the late stages, leading to a dampened immune response and exacerbated tissue damage (42). The types of infiltrating immune cells vary across different renal regions: in diabetic glomeruli, macrophages predominate (43), whereas in crescentic glomerulonephritis (CGN), monocytes are prevalent within the glomeruli, and T cells are primarily found in the surrounding areas (41, 44). Monocytes (especially macrophages) and T cells play pivotal roles in renal injury and fibrosis (41, 44–46). These infiltrating immune cells, along with kidney cells, release various cytokines, including IL-6, which regulates immune cell activation and proliferation, modulates local inflammation, and participates in tissue repair.
Key events in the transition to the resolution phase include the apoptosis and clearance of infiltrating neutrophils, the activation of anti-inflammatory M2-type macrophages, and the differentiation of regulatory T cells (Tregs) (40). Interestingly, IL-6 plays an important role in both the initiation and resolution phases of inflammation. In the initiation of inflammation, neutrophils are the first to accumulate in tissues, followed by the release of sIL-6R, which initiates trans-signaling in the intrinsic renal cells, promoting the expression of inflammatory chemokines and intercellular adhesion molecules (47). IL-6 transactivation plays an important role in the regulation of neutrophil transport and clearance (48), driving the transition of neutrophil recruitment to monocyte recruitment, and inducing the shift from innate immunity to adaptive immunity (49).
Enhanced renal macrophage infiltration is evident throughout all stages of DKD. Macrophages exhibit two activation phenotypes: pro-inflammatory M1 and the anti-inflammatory M2. It has been demonstrated that M1 and M2 macrophages primarily infiltrate the glomeruli and interstitium of diabetic kidneys (41). In the early stages of DKD, M1-type macrophages are first recruited and then proliferate in the kidneys (50). Subsequently, IL-6 activates the IL-6Rα on the surface of M1 macrophages, inhibiting M1 proliferation while promoting the polarization of M2 macrophages. This process alleviates renal inflammation and facilitates tissue repair (51). The classical IL-6 signaling pathway may a potential therapeutic strategy to improve DKD, as its activation enhances the repair of inflammatory renal injuries (51, 52). However, in the late stages of DKD, M2 macrophages may transform into fibroblasts, contributing to fibrosis in chronic kidney disease, including DKD (53, 54). Thus, a deeper investigation into the phenotypic changes of macrophages and their roles at various stages of DKD is essential for optimizing treatment strategies.
DCs are essential antigen-presenting cells in the adaptive immune response, with IL-6 playing multiple roles in their regulation. IL-6 inhibits DC development (55) and maintains them in an immature state (56), thereby limiting their capacity for antigen presentation and T cell activation. However, under certain conditions, IL-6 can promote DC maturation and further enhance T cell responses (57). Additionally, IL-6 secreted by DCs induces the production of Th17 cells, promoting adaptive immune responses (58, 59).
T cells play a crucial role in the progression of DKD, particularly CD4+ T helper cells (Th). Effector CD4+ T helper cells can be classified into four primary subsets: Th1, Th2, Th17, and Treg. Studies have shown that the decreased Treg/Th17 ratio is an essential facilitator of worsening kidney function in diabetic patients (60, 61). IL-6 plays a significant role in regulating the Th17/Treg balance. Early studies suggest that IL-6 induces pathogenic Th17 responses in synergy with TGF-β while also suppressing Treg differentiation, ultimately resulting in tissue inflammation and damage (62). However, recent research indicates the duality of IL-6 action in vivo. The expression of mIL-6R is predominantly found in peripheral naive CD4+ or central memory T cells, whereas at inflammatory sites, IL-6 trans-signaling becomes essential for activated and memory T cells due to the downregulation of mIL-6R expression (63). At the initiation stage of inflammation, activation of the classical IL-6 pathway in naive CD4+ T cells leads to the development and functional maintenance of Th17 cells (64). At inflammatory sites, T cells rely on IL-6 trans-signaling to facilitate Th17 differentiation and recruitment (63, 65, 66) while simultaneously inhibiting apoptosis, which leads to persistent infiltration (67, 68). In the resolution phase of inflammation, the classical activation of IL-6 promotes Treg differentiation and their trafficking to the kidneys, aiding the repair of damaged kidneys (69). Several studies have emphasized that interference with the classic IL-6 pathway reduces M2 macrophages and enhances Th17 responses, worsening renal injury (51, 52). Further research has indicated that M2 macrophages regulate adaptive immune responses through various mechanisms, including promoting Treg differentiation, enhancing their ability to suppress Th17 cells, and facilitating the migration of Tregs to sites of renal inflammation (70–72), which indirectly inhibits Th17 cell activity. Based on these findings, selective interference with the IL-6 trans-signaling may exert better therapeutic effects on chronic inflammatory injury, but effects on initial immune responses must be carefully considered.
Global inhibition of IL-6 signaling enhances pro-inflammatory Th1 and Th17 responses in mouse kidneys, exacerbating tissue and functional damage (51). This is consistent with the dual mechanisms of IL-6, indicating that IL-6 may have a protective role under certain circumstances. In CGN models, elevated circulating sIL-6R levels are closely associated with the aggravation of renal injury (52). During advanced disease, inhibitors of the IL-6 pathway, especially those targeting IL-6 trans-signaling, significantly inhibit the recruitment of M2 macrophages, CD4+ T cells in mouse kidneys (73, 74), thereby alleviating renal fibrosis. These studies confirm the effectiveness of selective inhibition of IL-6 trans-signaling in controlling kidney inflammation.
IL-6 plays a complex and multifaceted role in the inflammatory process of DKD, participating in both the initiation and resolution of inflammation. In these processes, IL-6 exerts both pro-inflammatory and anti-inflammatory effects, with its specific actions related to the signaling patterns involved. Further studies should be conducted to elucidate the specific mechanisms of IL-6 in various types of cells and stages to develop more precise and effective therapeutic strategies. Moreover, attention should also be paid to the potential impacts of the dual roles of IL-6 for effective inflammation control.
4.2 Effects of IL-6 on renal cells: good or bad?
The kidney consists of renal parenchyma and interstitium, with the renal parenchyma serving as the primary functional component, encompassing the glomeruli, renal tubules, and renal vasculature. The expression levels of IL-6R mRNA and protein in the renal parenchyma are very low, thus impeding the transduction of IL-6 classical signaling in the kidneys (75). However, podocytes have been demonstrated to express membrane-bound IL-6R, allowing for the transduction of both classical and trans-signaling pathways of IL-6, while other renal cells predominantly transduce IL-6 trans-signaling (76). The analysis of differences in the effects of IL-6 signaling pathways among key kidney cell types, including podocytes, renal endothelial cells (RECs), mesangial cells (MCs), renal tubular epithelial cells (RTECs), and renal fibroblasts, will enhance the understanding of the “good or bad” effects of IL-6 on these cells, thereby uncovering the intrinsic relationship between IL-6 and DKD. The IL-6 signaling pathway in various kidney cells is depicted in Figure 2.
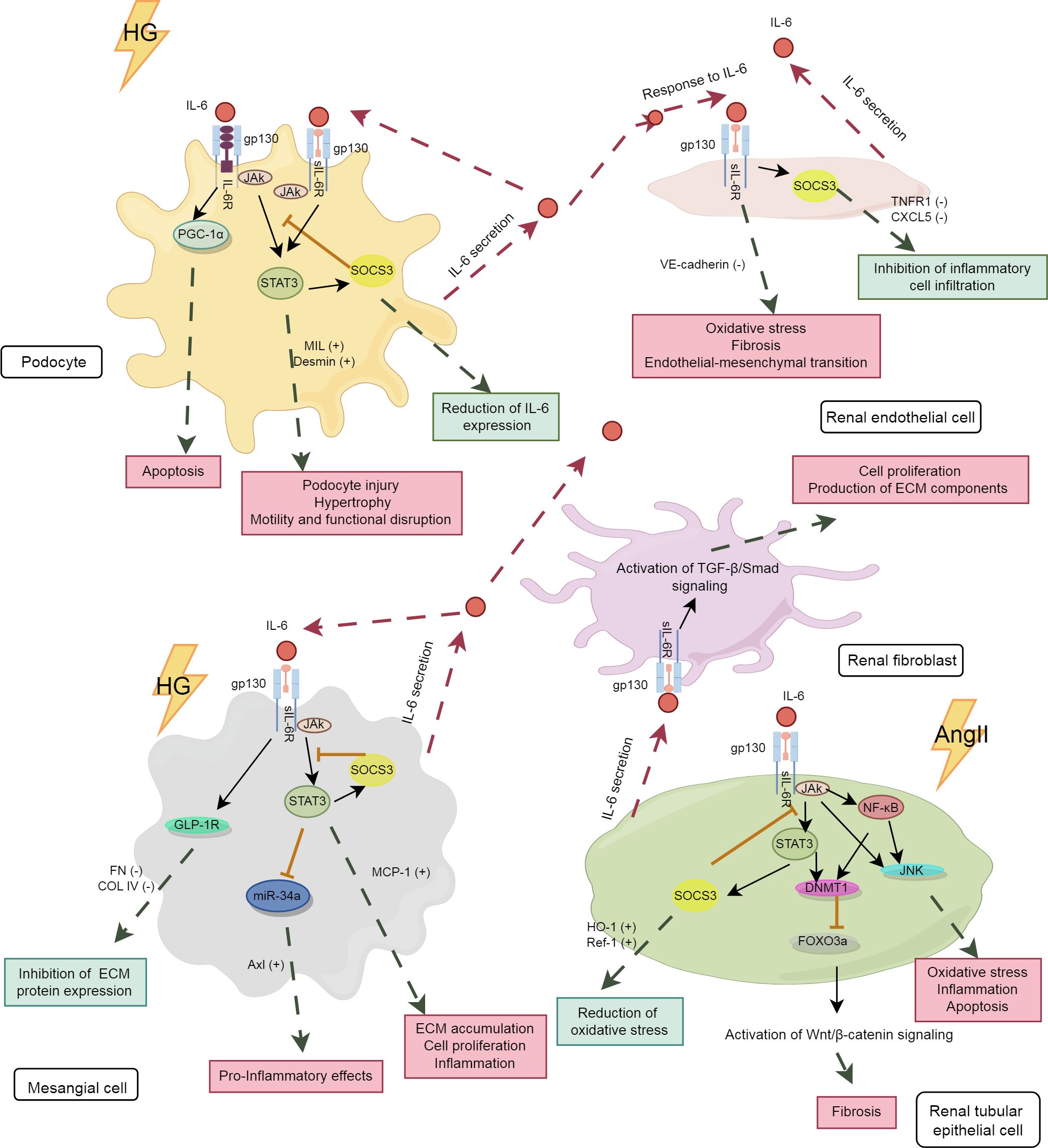
Figure 2. The IL-6 signaling pathway in kidney cells. The pink boxes in the panel denote the damaging effects, whereas the blue-green boxes represent the protective effects. HG, high glucose; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; TNFR1, tumor necrosis factor receptor 1; CXCL5, C-X-C motif chemokine ligand 5; GLP-1R,glucagon-like peptide-1 receptor; miR-34, microRNA-34; AngII, angiotensin II; MCP-1, monocyte chemoattractant protein-1; DNMT1, DNA methyltransferase 1; FOXO3a, forkhead box O 3a; HO-1, heme oxygenase-1; Ref-1, redox effector factor 1; VE-cadherin,vascular endothelial-cadherin; MLC, myosin light chain; COL IV, collagen type IV.
4.2.1 Podocytes and RECs
Podocytes and RECs are essential components of the glomerular filtration barrier. IL-6 plays a significant role in regulating the structure and function of both podocytes and RECs. As shown in Table 1, under diabetic conditions, high glucose induced podocytes to secrete IL-6, which activated the IL-6/gp130/PGC-1α and IL-6/gp130/JAK2/STAT3 signaling pathways, potentially leading to podocyte apoptosis (77), hypertrophy (78), and cytoskeletal disruption (79). Baseline IL-6 may exert a protective effect on podocytes, as IL-6-deficient mice develop acute kidney injury (AKI) with podocyte dysfunction (80). Under certain conditions, IL-6 secreted by podocytes promoted the expression of suppressor of cytokine signaling 3 (SOCS3) in RECs, thereby inhibiting IL-6 signaling and reducing neutrophil recruitment (81). However, these effects were not observed in co-cultures with human umbilical vein endothelial cells. This indicates the tissue-specific actions of IL-6. As the only renal cells expressing IL-6R, podocytes are studied to explore their specific signaling patterns. It has been shown that both classical and trans-signaling pathways of IL-6 are harmful to podocytes under high glucose conditions (76). More studies are needed to further elucidate the underlying mechanisms involved.
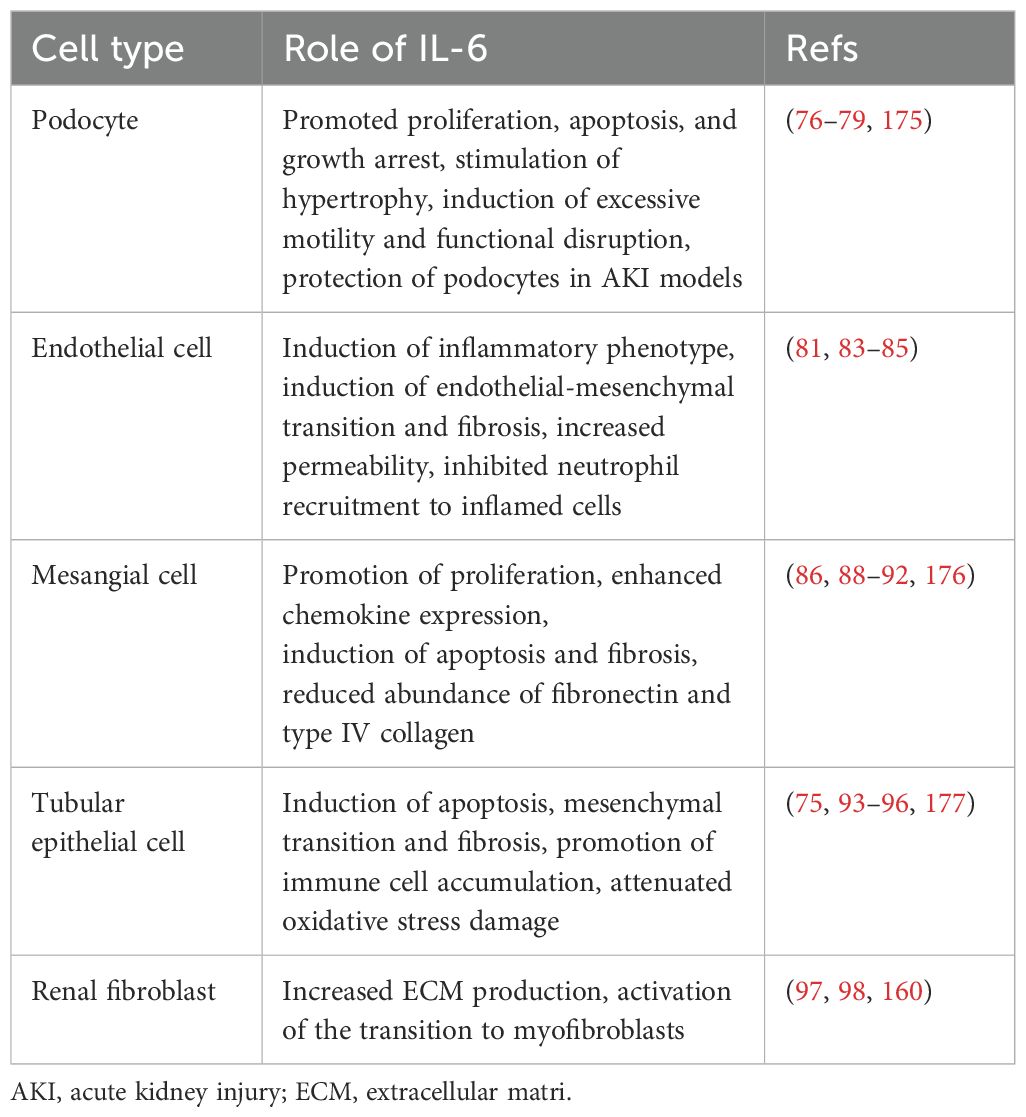
Table 1. Effects of IL-6 on kidney cells.
In addition, high glucose stimulates the secretion of IL-6 by RECs (82), inducing an inflammatory phenotype in RECs with increased levels of oxidative stress and fibrosis markers (83). Long-time exposure to IL-6 results in the loss of typical markers of RECs, contributing to renal fibrosis (84). In immunoglobulin A nephropathy, IL-6 suppressed the expression of vascular endothelial cadherin through trans-signaling pathway, causing increased permeability of RECs (85).
4.2.2 MCs
The persistent activation of the IL-6 pathway in MCs is an important mechanism of the early glomerulopathy in DKD. As an autocrine signal, IL-6 induces the expression of chemokines such as monocyte chemoattractant protein-1 (MCP-1) in MCs and influences immune cells through paracrine effects, thereby exacerbating immune-inflammatory damage in the glomeruli (86). Furthermore, studies have shown that the activation of Axl receptor tyrosine kinase (Axl) is closely associated with early glomerular hypertrophy in DKD (87), with the IL6/STAT3/miRNA-34a pathway playing a significant role in this process (88). In a hyperglycemic environment, the elevation of IL-6 in MCs led to cell apoptosis (89) and promoted cell proliferation, inflammation and fibrotic changes through the IL-6/JAK2/STAT3 pathway, thereby hastening the progression of DKD (90).
IL-6 trans-signaling plays an important role in regulating the inflammatory response of MCs. Its activation markedly induced MCP-1 synthesis in MCs, leading to glomerular inflammation (91). However, IL-6 may exhibit tissue-specific anti-inflammatory and pro-inflammatory effects under different conditions. For example, in RTECs, IL-6 induced increased expression of extracellular matrix (ECM) proteins, whereas it inhibited ECM protein expression in MCs (92). The pro-inflammatory fibrotic effect observed in RTECs cannot be simply defined as detrimental, as initial fibrosis can promote wound healing. More studies are needed to clarify the physiological significance of the differential effects of the IL-6 trans-signaling pathway in various tissues of the same organ.
4.2.3 RTECs and renal fibroblasts
Activation of renal fibroblasts and phenotypic conversion of RTECs are key factors in tubulointerstitial fibrosis (TIF). The activation of the IL-6 trans-pathway is closely associated with TIF. High glucose activated the IL-6/JAK2/STAT3 signaling pathway in RTECs, leading to inflammatory damage and cell apoptosis (93). Increased IL-6 induced epithelial-mesenchymal transition of RTECs (94) through the downstream STAT3 and DNMT1/FOXO3a/Wnt/β-catenin axis, promoting renal fibrotic lesions in AKI (95, 96). Furthermore, IL-6 released by RTECs promotes the proliferation of adjacent fibroblasts and the excessive production of fibrotic matrix (97, 98), accelerating renal fibrosis progression. Although IL-6 promotes renal injury in AKI (95, 99, 100), its trans-signaling also limits oxidative stress and promotes kidney repair (75), underscoring the dual role of IL-6 in tissue injury. This dual role may protect local tissues and organs from damage caused by acute inflammatory stimuli.
Animal studies further confirm the role of IL-6 in renal fibrosis and diabetic kidney injury. IL-6 knockdown markedly reduces renal fibrosis in type 1 diabetic mice (101). In type 2 diabetic nephropathy models, IL-6 neutralization inhibits nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome and downstream JAK2/STAT3 signaling, thereby alleviating oxidative stress and fibrotic injury (13, 90). Additionally, IL-6 elevates serum fibroblast growth factor 23 (FGF23) levels via sIL-6R (99), promoting the development of acute and chronic renal failure. In models of unilateral ureteral obstruction and ischemia reperfusion, specific blockade of IL-6 trans-signaling attenuates inflammation and fibrosis in renal tissues (74, 102). Consequently, the up-regulation of IL-6 trans-signal is closely associated with the progression of DKD.
4.3 Complexity of IL-6 regulation of glycolipid metabolism
Although IL-6 theoretically has the potential to regulate metabolism, adverse effects such as dyslipidemia have been observed with IL-6 inhibitors (e.g., TCZ) in clinical trials (103). This prompts us to revisit the complex roles of IL-6 in metabolic regulation. Therefore, we review the literature related to glycolipid metabolism, focusing on energy metabolism, T2DM models, and clinical trials, to explore the specific mechanisms by which IL-6 regulates glycolipid metabolism.
4.3.1 IL-6 regulates metabolism in a tissue-specific manner
IL-6 exhibits inconsistent effects among different peripheral target tissues of insulin, primarily including the liver, adipose tissue, and muscle, indicating the tissue-specific effects of IL-6 on metabolic regulation. The IL-6R on hepatocytes allows the intracellular classical signaling pathway that is essential for preventing metabolic changes and inflammation (104). Administration of IL-6-neutralizing antibodies to mice aggravates methionine-choline-deficient diet-induced hepatic steatosis (105), while hepatocyte-specific IL-6Rα deficiency leads to liver inflammation and reduced insulin sensitivity (106). In chronic liver disease, the downregulation of mIL-6R expression in hepatocytes favors the transduction of the IL-6 trans-signaling pathway, ultimately resulting in liver cirrhosis (107, 108). Increased plasma concentrations of IL-6 trans-signaling components show a clear correlation with HbA1c levels in patients with diabetes concomitant chronic liver disease (107). These studies suggest that the classical pathway mediated by hepatic IL-6R may maintain metabolic homeostasis in the early stages of the disease, but as the disease progresses, IL-6 trans-signaling predominates, leading to metabolic disturbances and inflammation.
IL-6 is strongly associated with lipid metabolic changes in both obese individuals and those with T2DM (109, 110), and adipose tissue is the primary source of IL-6 in vivo. Adipocytes predominantly transmit IL-6 trans-signaling (111), although there are also studies indicating the presence of IL-6R on adipocytes and suggesting its role in mediating liraglutide-induced hypoglycemic effects (112). Acute increases in IL-6 in adipose tissue have been shown to improve metabolism and blood glucose levels in obese mice (113), whereas prolonged elevations of IL-6 may contribute to increased lipolysis and leptin production, as well as insulin resistance (114). Moreover, IL-6/sIL-6R signaling pathways activated by high-fat diets have been found to exacerbate inflammation in adipose tissue of mice (115). Inhibition of IL-6 signaling in adipose tissue has been associated with reduced SOCS3 expression in hepatocytes and improved insulin sensitivity in mice. Selective inhibition of IL-6 in adipose tissue decreased SOCS3 expression in hepatocytes, thereby improving insulin sensitivity in mice (116). Thus, the chronic elevation of IL-6 in adipose tissue of obese patients is linked to metabolic deterioration.
Recent studies have indicated a connection between the classical IL-6 pathway and the metabolic benefits brought about by exercise. During exercise, IL-6, released by muscle tissue, stimulates the uptake and breakdown of glucose and fatty acids in muscle fibers through an osteocalcin-dependent mechanism by activating the IL-6 classical pathway in osteoblasts to optimize motor function (117). Moreover, IL-6 produced by muscles acts through IL-6R to support the retention and functional enhancement of Tregs in muscles, thereby improving muscle mass (118).
In addition to peripheral tissues (liver, adipose, and muscle), IL-6 is also involved in central metabolic regulation. Several studies have confirmed that central IL-6 activation can prevent high-fat-diet-induced obesity and insulin resistance in mice (119–121). Central IL-6 is closely related to body energy expenditure (120, 122). IL-6 trans-signaling plays a dominant role in its central effects. The trans-signaling of IL-6 in the paraventricular nucleus of the hypothalamus is essential for inhibiting feeding behavior and enhancing glucose homeostasis in mice (121).
4.3.2 IL-6 in models of T2DM
Patients with T2DM or DKD are usually accompanied by elevated circulating IL-6 levels. This chronic elevation of IL-6 similar to those observed in chronic inflammation, tends to disrupt the organismal metabolic homeostasis. TCZ treatment notably improved circulating glucagon, glucose and HbA1C levels in rhesus monkeys with spontaneous obesity or T2DM (123).
In contrast to chronic IL-6 actions, it is interesting to note that acute IL-6 infusions (at physiological concentrations) may improve the metabolism of patients with T2DM. In experimental designs, acute or intermittent IL-6 injections are often used to mimic the acute elevation of IL-6 post-exercise. Acutely elevated IL-6 has been shown to improve glucose tolerance in rodents and postprandial glucose in healthy volunteers (113, 124). In T2DM patients, acute infusions of IL-6 (up to the concentration after acute exercise), while not altering glucose turnover or postprandial glucose in patients, both notably reduced insulin levels in blood, implying an enhancement of insulin sensitivity (110, 124). This insulin-sparing effect may help to retain the function of pancreatic β-cells in diabetic patients. A single IL-6 injection improved insulin sensitivity in obese and type 2 diabetic mice, but failed to increase insulin secretion after pancreatic β-cell destruction in mice (113). It appears that the improvement of insulin sensitivity by IL-6 in T2DM patients is dependent on the preservation of pancreatic β-cell function. Furthermore, acute infusions of IL-6 can enhance fatty acid metabolism and systemic energy metabolism in elderly participants with T2DM (125), which emphasizes the lipolytic effects of IL-6 and may contribute to weight loss in them.
The pathological microenvironment induces changes in cell phenotypes, and the beneficial effects of IL-6 on metabolism observed in healthy volunteers cannot be simply extrapolated to diabetic patients. In healthy volunteers, acute IL-6 infusions delay gastric emptying and lower postprandial glucose levels. However, in patients with T2DM, IL-6 infusions delay gastric emptying but do not considerably reduce postprandial blood glucose levels (124). The underlying mechanisms of these differences remain unknown. It has been clearly demonstrated that individuals with T2DM show reduced responsiveness of myotubes to IL-6. While skeletal muscle cells from healthy volunteers can increase glucose uptake in response to IL-6 stimulation, myotubes from individuals with T2DM only respond to insulin in vitro, not to IL-6 (126).
Activation of the classical pathway of IL-6 may be important for improving metabolic disorders in patients with T2DM. Individuals with elevated serum levels of sIL-6R and/or sgp130 are at an increased risk of metabolic syndrome (127, 128). Prolonged exercise training can reduce plasma sIL-6R levels (129), thereby shifting the IL-6 signaling axis toward the classical IL-6 signaling mode. In addition, the researchers construct a novel compound, IC7Fc, which preserves the metabolic benefits of IL-6R. This compound improves hyperglycemia and glucose tolerance levels, while also reducing body weight and liver cirrhosis as well as promoting muscle and bone mass (130). Although blocking the trans-signaling pathway of IL-6 improves ATM accumulation induced by a high-fat diet, it does not alleviate systemic insulin resistance (131). Therefore, modulation of the IL-6 signaling pathway toward the classical model may be more effective in correcting metabolic disorders than simply blocking the trans-signaling pathway.
In the development of IL-6 agents, attention should also be paid to the potential impact of IL-6 blockade on the beneficial outcomes of exercise. A small-scale, double-blind clinical trial demonstrated that IL-6 blockade led to decreases in acute post-exercise and postprandial active GLP-1 concentrations among obese and type 2 diabetic individuals, albeit with a transient rather than sustained effect (132). Apart from that, another study showed that TCZ abolished exercise-induced reduction in visceral fat and increases circulating cholesterol levels in obese subjects (133).
4.3.3 Metabolic effects of IL-6 inhibitors observed in clinical trials
RA and diabetes share common pathogenic mechanisms, which involve chronic inflammation, autoimmunity, and insulin resistance and beyond. In recent years, research on RA has been shifting focus towards its complications of diabetes. Currently, the number of clinical trials investigating IL-6 inhibitors for diabetes treatment is limited. Therefore, large-scale clinical trials of TCZ in RA patients aid in the assessment of the metabolic effects of IL-6.
As shown in Table 2, beneficial effects on glucose metabolism and muscle mass are observed with TCZ therapy. In non-diabetic RA individuals, TCZ treatment notably improves insulin sensitivity and enhances muscle mass (134–136). In RA patients with T2DM, TCZ demonstrates a notable reduction in HbA1c levels and a decrease in daily prednisolone doses (137). Despite a case report also exists where RA patients developed type 1 diabetes after 17 months of TCZ treatment (137), the precise underlying mechanism remains unclear. Considering the genetic background of strong susceptibility genes in the patient, it is also possible that TCZ may delay the onset of type 1 diabetes.
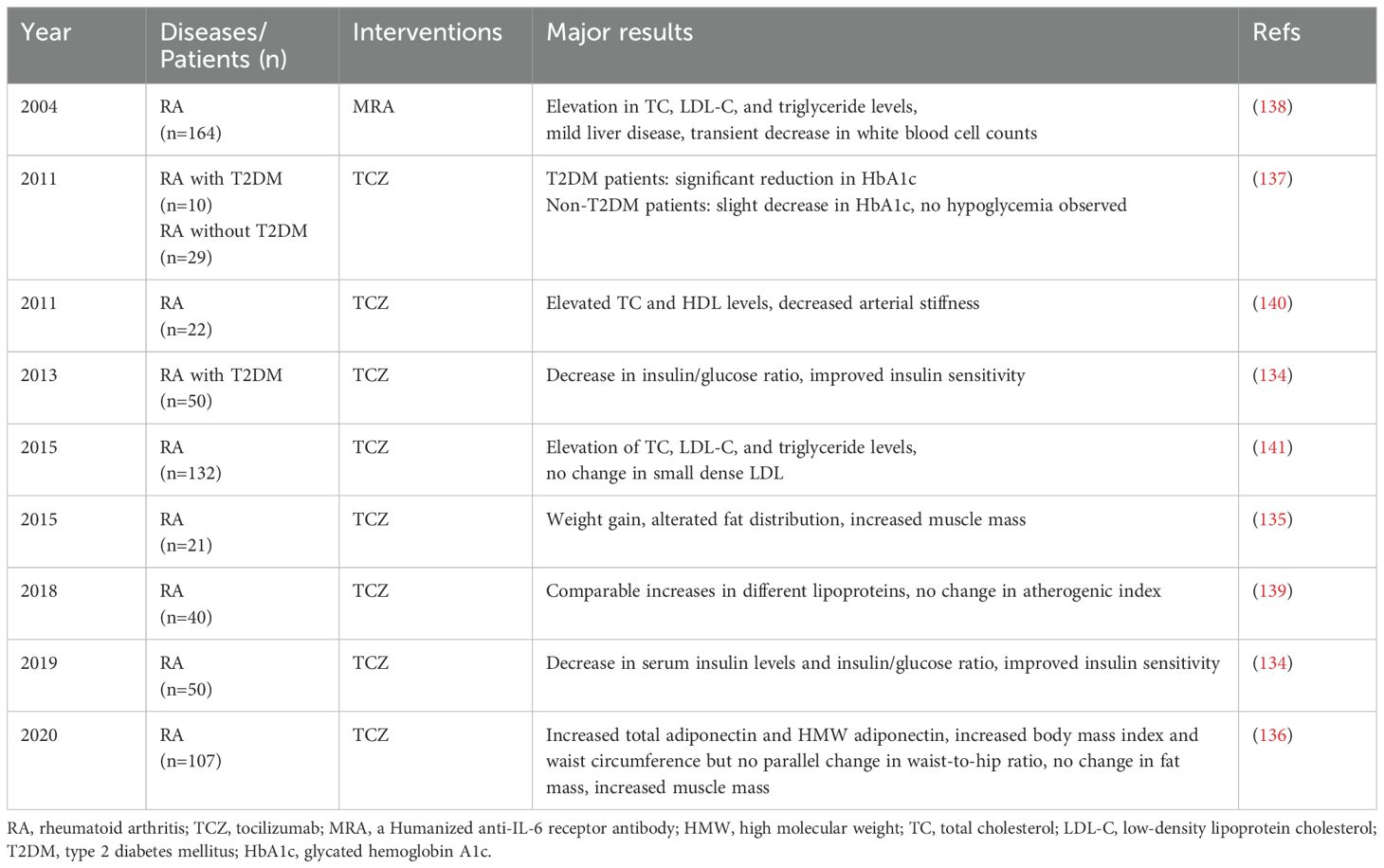
Table 2. Metabolic effects of IL-6 inhibitors observed in RA clinical trials.
During treatment with anti-IL-6 therapy, elevations in total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and triglyceride levels have been observed during TCZ treatment for RA (138). This phenomenon has triggered a deeper investigation into the metabolic effects of TCZ. Although elevated lipid levels are usually considered indicators of increased risk for metabolic diseases, the situation seems to be different in the context of TCZ treatment. The raised lipid levels induced by TCZ do not seem to result in increased cardiometabolic risks, but instead may lower the risk of cardiovascular events. This paradox can be explained by multiple factors.
First, the anti-inflammatory effects of TCZ may partially mitigate the potential risks associated with elevated lipid levels. TCZ leads to changes in different lipoproteins with a comparable gain, thus not affecting the atherogenic index (139), and potentially even alleviating arterial stiffness in patients (140). Importantly, the key pathogenic factor of atherosclerosis (small dense LDL) is not influenced by TCZ (141). Also, the increase in the levels of high-molecular-weight (HMW) adiponectin during TCZ treatment serves as a positive indicator for the improvement in metabolic diseases (136), suggesting that TCZ may play an important role in metabolic regulation.
Furthermore, studies in obese patients and those intolerant to statins have shown that anti-inflammatory therapy with IL-6 inhibitors plays a positive role in improving metabolism and disease progression in these patients. In a cohort study involving 1,153 obese participants, serum lipoprotein(a) (Lp(a)), a severe independent risk factor for cardiovascular diseases, was significantly associated with IL-6 levels. After TCZ treatment, Lp(a) levels were considerably reduced in patients (10), a change attributed specifically to the inhibition of IL-6 rather than to the suppression of inflammatory immune responses. The latest follow-up study of 13,970 statin-intolerant high-risk cardiovascular patients demonstrates that inflammation has a greater predictive value than LDL-C for future cardiovascular events and mortality (142), highlighting the significance of anti-inflammatory therapies in ameliorating metabolic disorders and reducing cardiovascular risk.
In conclusion, the clinical trials discussed above suggest that the improvements in insulin sensitivity and inflammation levels resulting from IL-6 blockade may lead to clinical benefits that outweigh the risks associated with elevated lipid levels.
4.4 IL-6 regulates the RAAS
The overactivation of the RAAS leads to progressive renal damage. Angiotensin II (AngII) is the primary effector of RAAS, generated from angiotensin I under the action of angiotensin-converting enzyme. Studies have shown that IL-6 is necessary for the renal damage caused by aberrant activation of RAAS.
IL-6 plays a key role in AngII-induced renal damage and fibrosis. Studies have shown that AngII stimulates IL-6 secretion from renal tubular cells (97) and elevates IL-6 expression in rat kidneys (143). In the absence of IL-6, AngII was unable to induce phosphorylation of JAK2/STAT3 in RTECs (144) and in murine kidneys (145). And IL-6 blockade inhibited mesangial cell proliferation induced by AngII (146). In IL-6 knockout mice, AngII-induced hypertension and urinary albumin excretion were lessened (145, 147), accompanied by a notable reduction in vascular endothelial inflammatory injury and oxidative stress (148). Moreover, L-6 deficiency decreased AngII-induced expression of renal fibrotic genes in mice (34, 149), thereby mitigating chronic kidney lesions. Multiple studies have shown that IL-6 further amplifies intrarenal AngII action by elevating the expression of renal angiotensinogen (AGT) (144, 150, 151), creating a vicious cycle that ultimately results in progressive renal function decline.
The mechanism by which IL-6 mediates the effects of AngII has attracted considerable interest. Multiple studies confirm the critical role of IL-6-mediated JAK2/STAT3 signaling in AngII-induced kidney injury (144, 145, 152). Furthermore, JAK-STAT signaling also underlies the increase in AGT levels induced by IL-6 (144, 153). JAK2/STAT3 activation in the kidney has been identified as a facilitator of AngII-induced kidney injury in diabetic patients (154). Thus, it is possible that the IL-6/JAK/STAT signaling pathway is a key downstream signal in RAAS-induced kidney injury.
4.5 IL-6 regulates renal iron homeostasis
Ferroptosis, a form of iron-dependent cell death characterized by lipid peroxidation, has been increasingly recognized for its close association with the onset and progression of DKD (155). Ferroportin (FPN) and hepcidin play key roles in mammalian iron homeostasis. The activation of the IL-6/Ca axis has been shown to enhance the presence of FPN on the plasma membrane of human embryonic kidney cells, which promotes iron efflux, and removes Fpn from the membrane via upregulation of hepcidin to restore iron homeostasis (156).
IL-6 is recognized as an inducer of ferroptosis involved in intervertebral disc degeneration and mastitis diseases (157, 158). Epitope-mimicking peptides are believed to be effective in mimicking antibody binding sites, inducing active immunity (159). In kidney diseases, the specific epitope mimics of TCZ were found to increase ferritin level, decrease lipid oxidation levels and renal cell apoptosis, attenuating kidney fibrosis (73). Besides, IL-6 mediates signaling crosstalk between ferroptotic kidney cells and surrounding fibroblasts, accelerating renal fibrosis (160). In autoimmune kidney diseases, IL-6 helps immune cells evade ferroptosis, promoting excessive activation of immune cells and worsening kidney disease. Blockade of IL-6 promotes ferroptosis in B cells and shows therapeutic effects in lupus nephritis (161). These findings highlight the intricate involvement of IL-6 in the regulation of ferroptosis and suggest that the impacts of IL-6 on ferroptosis are context-dependent.
5 The current status of IL-6 inhibitors in clinical trials related to DKD
The intrinsic relationship between IL-6 and DKD has been substantiated through various aspects in the aforementioned laboratory studies. In the process of translating targeted IL-6 therapy for DKD into clinical application, relevant clinical trials are being gradually conducted. Current IL-6 inhibitors in clinical trials for kidney disease mainly consist of TCZ, clazakizumab (an anti-IL-6R antibody), ziltivekimab (an IL-6 ligand inhibitor), and baricitinib (a JAK inhibitor). As shown in Table 3, the majority of studies evaluate the safety and efficacy of anti-IL-6 therapy in pre- and post-kidney transplant patients (NCT01594424, NCT03444103, NCT00106639, NCT00658359, NCT04561986, NCT03859388, NCT04779957, NCT03867617, NCT03380962, NCT03380377). According to the available data, IL-6 inhibitors effectively reduce immune rejection in post-kidney transplant patients with good tolerability (162–166).
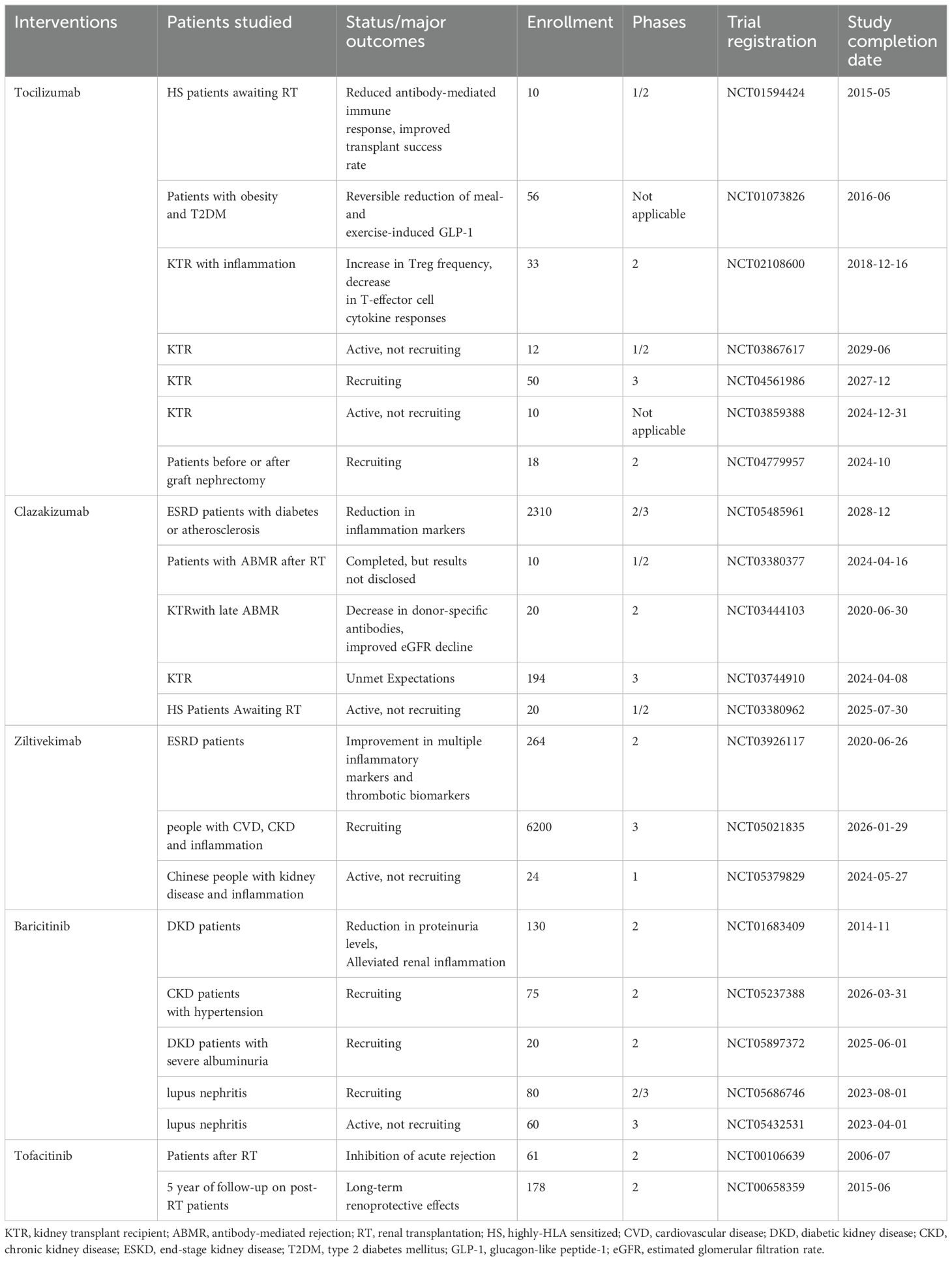
Table 3. IL-6 inhibitors in DKD-related clinical trials.
In addition, anti-IL-6 therapy also effectively improved inflammatory markers and proteinuria levels in patients with kidney disease, leading to a reduction of kidney inflammation. Previous results from a phase 2b clinical trial indicate that clazakizumab reduces serum inflammatory markers related to cardiovascular events in participants with ESKD complicated by diabetes or atherosclerosis (NCT05485961) (167). A Phase 3 trial is currently ongoing.
Furthermore, ziltivekimab shows improvements in multiple inflammatory markers and thrombotic biomarkers in ESKD patients with high cardiovascular risk (NCT03926117) (168). Trials are currently recruiting to assess the impact of ziltivekimab on cardiovascular risk in patients with cardiovascular disease, chronic kidney disease, and systemic inflammation (NCT05021835). The anti-inflammatory effects of TCZ have been evaluated in patients with post-kidney transplant inflammatory responses (164) (NCT02108600). Baricitinib has shown efficacy in reducing proteinuria levels and alleviating renal inflammation in T2DM patients with kidney diseases (169) (NCT01683409). Furthermore, studies are planned to evaluate the effect of baricitinib on urine protein in CKD (NCT05237388) patients and its impact on hard kidney endpoints in DKD patients with severe albuminuria (NCT05897372). Several studies (NCT05686746, NCT05432531) are planned to evaluate the effectiveness of baricitinib for lupus nephritis.
Overall, the results of these clinical trials provide robust evidence supporting the strategy of targeting IL-6 for treating DKD.
6 Conclusion
The current indications for IL-6 inhibitors have expanded from the initial rheumatoid arthritis, to more recent giant cell arteritis, and systemic sclerosis-associated interstitial lung disease (170, 171). Targeting IL-6 has shown significant potential for the development of treatments for immune-mediated inflammatory diseases. With the new concept of immunometabolism and the growing understanding of the crosstalk among immunity, iron metabolism and hemodynamics in preclinical studies, immunotherapy is highly likely to be a new breakthrough in treating DKD. The latest clinical trials are assessing the efficacy of IL-6 inhibitors on inflammatory markers relevant to cardiovascular and renal diseases.
In the investigation of targeted therapy strategies with anti-IL-6 agents for DKD, there are two points of concern. Firstly, further investigation is needed into the molecular mechanisms underlying the functions of the classical and trans-IL-6 signaling pathways. Both pathways transmit signals through the shared mediator gp130, but they can elicit distinct or even opposing effects. Thus, additional research at the molecular levels is necessary to elucidate the key mechanisms of this precise control and differential action, which would significantly advance the development of precision medicine for DKD. Some recent studies have focused on this. For example, Xu et al. have revealed the crosstalk between energy metabolism and IL-6 signaling, elucidating the molecular mechanisms underlying the distinct actions associated with reprogramming of glucose metabolism (172). Another study has demonstrated that changes in the binding parameters of the IL-6-gp130 receptor complex can lead to biased signaling (173). Specifically, modified cytokine-receptor binding dynamics can attenuate the pleiotropic effects of IL-6, thus mitigating potential side effects associated with IL-6-targeted treatments.
Secondly, in the development of IL-6-targeted therapy for DKD treatment, how to modulate the balance between the classical and trans-signaling pathways of IL-6 is also a challenging task. IL-6 trans-signaling-specific inhibitor olamkicept has been developed for treating ulcerative colitis with promising results in phase II clinical trials (174). An increasing number of studies are being redirected to the development of drugs that selectively inhibit IL-6 trans-signaling without affecting classical signaling. However, due to the diverse pathological manifestations in different diseases, targeting only the IL-6 trans-signaling pathway in DKD may not be the most effective approach, as demonstrated by the inefficacy of sgp130Fc in ameliorating insulin resistance induced by a high-fat diet in mice (131). In contrast, the recombinant IC7Fc, which is constructed with consideration of tuning of the balance between the two primary IL-6 pathways, has exhibited multiple beneficial effects in the context of type 2 diabetes. Therefore, in comparison to global inhibition of IL-6 or simply inhibition of the IL-6 trans-pathway, developing drugs that modulate the balance between IL-6 classical and trans-signaling pathways may be a more rational therapeutic strategy for DKD.
Author contributions
LZ: Conceptualization, Software, Writing – original draft, Writing – review & editing. FX: Conceptualization, Software, Writing – review & editing. LH: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Giugliano D, Maiorino MI, Bellastella G, Esposito K. The residual cardiorenal risk in type 2 diabetes. Cardiovasc diabetol. (2021) 20:36. doi: 10.1186/s12933-021-01229-2
2. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. (2011) 7:327–40. doi: 10.1038/nrneph.2011.51
3. Navarro-González JF, Mora-Fernández C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol: JASN. (2008) 19:433–42. doi: 10.1681/ASN.2007091048
4. Shikata K, Makino H. Microinflammation in the pathogenesis of diabetic nephropathy. J Diabetes Invest. (2013) 4:142–9. doi: 10.1111/jdi.2013.4.issue-2
5. Rayego-Mateos S, Rodrigues-Diez RR, Fernandez-Fernandez B, Mora-Fernández C, Marchant V, Donate-Correa J, et al. Targeting inflammation to treat diabetic kidney disease: the road to 2030. Kidney Int. (2023) 103:282–96. doi: 10.1016/j.kint.2022.10.030
6. Akbari M, Hassan-Zadeh V. IL-6 signalling pathways and the development of type 2 diabetes. Inflammopharmacology. (2018) 26:685–98. doi: 10.1007/s10787-018-0458-0
7. Sanchez-Alamo B, Shabaka A, Cachofeiro V, Cases-Corona C, Fernandez-Juarez G. Serum interleukin-6 levels predict kidney disease progression in diabetic nephropathy. Clin nephrol. (2022) 97:1–9. doi: 10.5414/CN110223
8. Hiramoto JS, Katz R, Peralta CA, Ix JH, Fried L, Cushman M, et al. Inflammation and coagulation markers and kidney function decline: the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. (2012) 60:225–32. doi: 10.1053/j.ajkd.2012.02.335
9. Amdur RL, Feldman HI, Gupta J, Yang W, Kanetsky P, Shlipak M, et al. Inflammation and progression of CKD: the CRIC study. Clin J Am Soc Nephrol: CJASN. (2016) 11:1546–56. doi: 10.2215/CJN.13121215
10. Müller N, Schulte DM, Türk K, Freitag-Wolf S, Hampe J, Zeuner R, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. (2015) 56:1034–42. doi: 10.1194/jlr.P052209
11. Kamińska J, Stopiński M, Mucha K, Jędrzejczak A, Gołębiowski M, Niewczas MA, et al. IL 6 but not TNF is linked to coronary artery calcification in patients with chronic kidney disease. Cytokine. (2019) 120:9–14. doi: 10.1016/j.cyto.2019.04.002
12. Zoccali C, Tripepi G, Mallamaci F. Dissecting inflammation in ESRD: do cytokines and C-reactive protein have a complementary prognostic value for mortality in dialysis patients? J Am Soc Nephrol: JASN. (2006) 17:S169–73. doi: 10.1681/ASN.2006080910
13. Wu R, Liu X, Yin J, Wu H, Cai X, Wang N, et al. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism: Clin experimental. (2018) 83:18–24. doi: 10.1016/j.metabol.2018.01.002
14. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (London England: 1979). (2012) 122:143–59. doi: 10.1042/CS20110340
15. Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A. (2008) 105:13163–8. doi: 10.1073/pnas.0801059105
16. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. (2012) 8:1237–47. doi: 10.7150/ijbs.4989
17. Schaper F, Rose-John S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth factor Rev. (2015) 26:475–87. doi: 10.1016/j.cytogfr.2015.07.004
18. Riethmueller S, Ehlers JC, Lokau J, Düsterhöft S, Knittler K, Dombrowsky G, et al. Cleavage site localization differentially controls interleukin-6 receptor proteolysis by ADAM10 and ADAM17. Sci Rep. (2016) 6:25550. doi: 10.1038/srep25550
19. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harbor Perspect Biol. (2018) 10(2):a028415. doi: 10.1101/cshperspect.a028415
20. Reeh H, Rudolph N, Billing U, Christen H, Streif S, Bullinger E, et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: fusing experimental insights and dynamic modelling. Cell Commun Signal. (2019) 17(1):46. doi: 10.1186/s12964-019-0356-0
21. Garbers C, Thaiss W, Jones GW, Waetzig GH, Lorenzen I, Guilhot F, et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J Biol Chem. (2011) 286:42959–70. doi: 10.1074/jbc.M111.295758
22. Heink S, Yogev N, Garbers C. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat Immunol. (2017) 18(1):74–85. doi: 10.1038/ni.3632
23. Ahluwalia TS, Prins BP, Abdollahi M, Armstrong NJ, Aslibekyan S, Bain L, et al. Genome-wide association study of circulating interleukin 6 levels identifies novel loci. Hum Mol Genet. (2021) 30(5):393–409. doi: 10.1093/hmg/ddab023
24. Rocha S, Valente MJ, Coimbra S, Catarino C, Rocha-Pereira P, Oliveira JG, et al. Interleukin 6 (rs1800795) and pentraxin 3 (rs2305619) polymorphisms-association with inflammation and all-cause mortality in end-stage-renal disease patients on dialysis. Sci Rep. (2021) 11:14768. doi: 10.1038/s41598-021-94075-x
25. Tziastoudi M, Stefanidis I, Hadjigeorgiou GM, Stravodimos K, Zintzaras E. A systematic review and meta-analysis of genetic association studies for the role of inflammation and the immune system in diabetic nephropathy. Clin Kidney J. (2017) 10:293–300. doi: 10.1093/ckj/sfx008
26. Chen B, Wu M, Zang C, Li Y, Xu Z. Association between IL-6 polymorphisms and diabetic nephropathy risk: A meta-analysis. Am J Med Sci. (2019) 358:363–73. doi: 10.1016/j.amjms.2019.07.011
27. Cui ZH, Lu XT, Xiao KL, Chen Y, Li HQ. Association of interleukin-6 -174G/C polymorphism with the risk of diabetic nephropathy in type 2 diabetes: A meta-analysis. Curr Med science. (2019) 39:250–8. doi: 10.1007/s11596-019-2027-1
28. Navarro JF, Milena FJ, Mora C, León C, García J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J nephrol. (2006) 26:562–70. doi: 10.1159/000098004
29. Sha J, Sui B, Su X, Meng Q, Zhang C. Alteration of oxidative stress and inflammatory cytokines induces apoptosis in diabetic nephropathy. Mol Med Rep. (2017) 16:7715–23. doi: 10.3892/mmr.2017.7522
30. Senthilkumar GP, Anithalekshmi MS, Yasir M, Parameswaran S, Packirisamy RM, Bobby Z. Role of omentin 1 and IL-6 in type 2 diabetes mellitus patients with diabetic nephropathy. Diabetes Metab syndrome. (2018) 12:23–6. doi: 10.1016/j.dsx.2017.08.005
31. Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol: JASN. (2005) 16 Suppl 1:S78–82. doi: 10.1681/ASN.2004110961
32. Sekizuka K, Tomino Y, Sei C, Kurusu A, Tashiro K, Yamaguchi Y, et al. Detection of serum IL-6 in patients with diabetic nephropathy. Nephron. (1994) 68:284–5. doi: 10.1159/000188281
33. Kado S, Nagase T, Nagata N. Circulating levels of interleukin-6, its soluble receptor and interleukin-6/interleukin-6 receptor complexes in patients with type 2 diabetes mellitus. Acta diabetologica. (1999) 36:67–72. doi: 10.1007/s005920050147
34. Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension (Dallas Tex: 1979). (2012) 59:136–44. doi: 10.1161/HYPERTENSIONAHA.111.173328
35. Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, et al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. (1995) 44:1233–8. doi: 10.2337/diab.44.10.1233
36. Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, et al. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol: JASN. (2005) 16:3315–25. doi: 10.1681/ASN.2003090757
37. Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in Dahl salt-sensitive rats. Am J Physiol Renal Physiol. (2016) 311:F555–61. doi: 10.1152/ajprenal.00594.2015
38. Doke T, Abedini A, Aldridge DL, Yang YW, Park J, Hernandez CM. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat Immunol. (2022) 23(6):947–59. doi: 10.1038/s41590-022-01200-7
39. Barreto DV, Barreto FC, Liabeuf S, Temmar M, Lemke HD, Tribouilloy C, et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. (2010) 77:550–6. doi: 10.1038/ki.2009.503
40. Schett G, Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun. (2018) 9:3261. doi: 10.1038/s41467-018-05800-6
41. Fu J, Sun Z, Wang X, Zhang T, Yuan W, Salem F, et al. The single-cell landscape of kidney immune cells reveals transcriptional heterogeneity in early diabetic kidney disease. Kidney Int. (2022) 102:1291–304. doi: 10.1016/j.kint.2022.08.026
42. Cucak H, Nielsen Fink L, Højgaard Pedersen M, Rosendahl A. Enalapril treatment increases T cell number and promotes polarization towards M1-like macrophages locally in diabetic nephropathy. Int immunopharmacol. (2015) 25:30–42. doi: 10.1016/j.intimp.2015.01.003
43. Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, et al. Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol: JASN. (2019) 30:533–45. doi: 10.1681/ASN.2018090896
44. Hooke DH, Gee DC, Atkins RC. Leukocyte analysis using monoclonal antibodies in human glomerulonephritis. Kidney Int. (1987) 31:964–72. doi: 10.1038/ki.1987.93
45. Kondo A, McGrady M, Nallapothula D, Ali H, Trevino AE, Lam A, et al. Spatial proteomics of human diabetic kidney disease, from health to class III. Diabetologia. (2024) 67(9):1962–79. doi: 10.1101/2023.04.12.534028
46. do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight. (2020) 5(3):e130651. doi: 10.1172/jci.insight.130651
47. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) 16:448–57. doi: 10.1038/ni.3153
48. Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol (Baltimore Md: 1950). (2008) 181:2189–95. doi: 10.4049/jimmunol.181.3.2189
49. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol (Baltimore Md: 1950). (2005) 175:3463–8. doi: 10.4049/jimmunol.175.6.3463
50. Yan J, Li X, Liu N, He JC. Relationship between macrophages and tissue microenvironments in diabetic kidneys. Biomedicines. (2023) 11(7):1889. doi: 10.3390/biomedicines11071889
51. Luig M, Kluger MA, Goerke B, Meyer M, Nosko A, Yan I, et al. Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J Am Soc Nephrol: JASN. (2015) 26:1597–607. doi: 10.1681/ASN.2014060620
52. Braun GS, Nagayama Y, Maruta Y, Heymann F, van Roeyen CR, Klinkhammer BM, et al. IL-6 trans-signaling drives murine crescentic GN. J Am Soc Nephrol: JASN. (2016) 27:132–42. doi: 10.1681/ASN.2014111147
53. Lin DW, Yang TM. Targeting macrophages: therapeutic approaches in diabetic kidney disease. Int J Mol Sci. (2024) 25(8):4350. doi: 10.3390/ijms25084350
54. Nakagawa M, Karim MR. Immunophenotypical characterization of M1/M2 macrophages and lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells. (2021) 10(2):257. doi: 10.3390/cells10020257
55. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. (2000) 1:510–4. doi: 10.1038/82763
56. Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol (Baltimore Md: 1950). (2004) 173:3844–54. doi: 10.4049/jimmunol.173.6.3844
57. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann rheumatic diseases. (2014) 73:2192–8. doi: 10.1136/annrheumdis-2013-203756
58. Liu J, Han C, Xie B, Wu Y, Liu S, Chen K, et al. Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat Immunol. (2014) 15:612–22. doi: 10.1038/ni.2898
59. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. (2007) 8:942–9. doi: 10.1038/ni1496
60. Zhang C, Xiao C, Wang P, Xu W, Zhang A, Li Q, et al. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Hum Immunol. (2014) 75:289–96. doi: 10.1016/j.humimm.2014.02.007
61. Ryba-Stanisławowska M, Skrzypkowska M, Myśliwiec M, Myśliwska J. Loss of the balance between CD4(+)Foxp3(+) regulatory T cells and CD4(+)IL17A(+) Th17 cells in patients with type 1 diabetes. Hum Immunol. (2013) 74:701–7. doi: 10.1016/j.humimm.2013.01.024
62. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
63. Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R, et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol (Baltimore Md: 1950). (2010) 184:2130–9. doi: 10.4049/jimmunol.0901528
64. Harbour SN, DiToro DF, Witte SJ. T(H)17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci Immunol. (2020) 5(49):eaaw2262. doi: 10.1126/sciimmunol.aaw2262
65. McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A. (2005) 102:9589–94. doi: 10.1073/pnas.0501794102
66. Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol (Baltimore Md: 1950). (2009) 182:613–22. doi: 10.4049/jimmunol.182.1.613
67. Curnow SJ, Scheel-Toellner D, Jenkinson W, Raza K, Durrani OM, Faint JM, et al. Inhibition of T cell apoptosis in the aqueous humor of patients with uveitis by IL-6/soluble IL-6 receptor trans-signaling. J Immunol (Baltimore Md: 1950). (2004) 173:5290–7. doi: 10.4049/jimmunol.173.8.5290
68. Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. (2000) 6:583–8. doi: 10.1038/75068
69. Hagenstein J, Melderis S, Nosko A, Warkotsch MT, Richter JV, Ramcke T, et al. A novel role for IL-6 receptor classic signaling: induction of RORγt(+)Foxp3(+) tregs with enhanced suppressive capacity. J Am Soc Nephrol: JASN. (2019) 30:1439–53. doi: 10.1681/ASN.2019020118
70. Shao X, Xu P, Ji L, Wu B, Zhan Y, Zhuang X, et al. Low-dose decitabine promotes M2 macrophage polarization in patients with primary immune thrombocytopenia via enhancing KLF4 binding to PPARγ promoter. Clin Transl Med. (2023) 13(7):e1344. doi: 10.1002/ctm2.v13.7
71. Sakai R, Ito M, Yoshimoto K, Chikuma S, Kurasawa T, Kondo T, et al. Tocilizumab monotherapy uncovered the role of the CCL22/17-CCR4(+) Treg axis during remission of crescentic glomerulonephritis. Clin Trans Immunol. (2020) 9:e1203. doi: 10.1002/cti2.v9.11
72. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34:566–78. doi: 10.1016/j.immuni.2011.03.018
73. Yang L, Guo J, Yu N, Liu Y, Song H, Niu J, et al. Tocilizumab mimotope alleviates kidney injury and fibrosis by inhibiting IL-6 signaling and ferroptosis in UUO model. Life Sci. (2020) 261:118487. doi: 10.1016/j.lfs.2020.118487
74. Chen W, Yuan H, Cao W, Wang T, Chen W, Yu H, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. (2019) 9:3980–91. doi: 10.7150/thno.32352
75. Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol: JASN. (2008) 19:1106–15. doi: 10.1681/ASN.2007070744
76. Lei CT, Su H, Ye C, Tang H, Gao P, Wan C, et al. The classic signalling and trans-signalling of interleukin-6 are both injurious in podocyte under high glucose exposure. J Cell Mol Med. (2018) 22(1):251–60. doi: 10.1111/jcmm.2018.22.issue-1
77. Kim DI, Park SH. Sequential signaling cascade of IL-6 and PGC-1α is involved in high glucose-induced podocyte loss and growth arrest. Biochem Biophys Res Commun. (2013) 435:702–7. doi: 10.1016/j.bbrc.2013.05.046
78. Jo HA, Kim JY, Yang SH, Han SS, Joo KW, Kim YS, et al. The role of local IL6/JAK2/STAT3 signaling in high glucose-induced podocyte hypertrophy. Kidney Res Clin practice. (2016) 35:212–8. doi: 10.1016/j.krcp.2016.09.003
79. He FF, Bao D, Su H, Wang YM, Lei CT, Zhang CY, et al. IL-6 increases podocyte motility via MLC-mediated focal adhesion impairment and cytoskeleton disassembly. J Cell Physiol. (2018) 233(9):7173–81. doi: 10.1002/jcp.v233.9
80. Lee SJ, Borsting E, Declèves AE, Singh P, Cunard R. Podocytes express IL-6 and lipocalin 2/ neutrophil gelatinase-associated lipocalin in lipopolysaccharide-induced acute glomerular injury. Nephron Exp Nephrol. (2012) 121:e86–96. doi: 10.1159/000345151
81. Kuravi SJ, McGettrick HM, Satchell SC, Saleem MA, Harper L, Williams JM, et al. Podocytes regulate neutrophil recruitment by glomerular endothelial cells via IL-6-mediated crosstalk. J Immunol (Baltimore Md: 1950). (2014) 193:234–43. doi: 10.4049/jimmunol.1300229
82. Tang LX, Wei B, Jiang LY, Ying YY, Li K, Chen TX, et al. Intercellular mitochondrial transfer as a means of revitalizing injured glomerular endothelial cells. World J Stem Cells. (2022) 14:729–43. doi: 10.4252/wjsc.v14.i9.729
83. Medica D, Franzin R, Stasi A. Extracellular vesicles derived from endothelial progenitor cells protect human glomerular endothelial cells and podocytes from complement- and cytokine-mediated injury. Cells. (2021) 10(7):1675. doi: 10.3390/cells10071675
84. Gui Z, Suo C, Wang Z, Zheng M, Fei S, Chen H, et al. Impaired ATG16L-dependent autophagy promotes renal interstitial fibrosis in chronic renal graft dysfunction through inducing EndMT by NF-κB signal pathway. Front Immunol. (2021) 12:650424. doi: 10.3389/fimmu.2021.650424
85. Yang YC, Fu H, Zhang B, Wu YB. Interleukin-6 downregulates the expression of vascular endothelial-cadherin and increases permeability in renal glomerular endothelial cells via the trans-signaling pathway. Inflammation. (2022) 45(6):2544–58. doi: 10.1007/s10753-022-01711-3
86. Hartner A, Goppelt-Struebe M, Hocke GM, Sterzel RB. Differential regulation of chemokines by leukemia inhibitory factor, interleukin-6 and oncostatin M. Kidney Int. (1997) 51:1754–60. doi: 10.1038/ki.1997.241
87. Nagai K, Arai H, Yanagita M, Matsubara T, Kanamori H, Nakano T, et al. Growth arrest-specific gene 6 is involved in glomerular hypertrophy in the early stage of diabetic nephropathy. J Biol Chem. (2003) 278:18229–34. doi: 10.1074/jbc.M213266200
88. Adams DE, Zhen Y, Qi X. Axl expression in renal mesangial cells is regulated by Sp1, Ap1, MZF1, and Ep300, and the IL-6/miR-34a pathway. Cells. (2022) 11(12):1869. doi: 10.3390/cells11121869
89. Zhu Y, Ruan CX, Wang J, Jiang FF, Xiong LS, Sheng X, et al. High glucose inhibits the survival of HRMCs and its mechanism. Eur Rev Med Pharmacol Sci. (2022) 26:5683–8. doi: 10.26355/eurrev_202208_29502
90. Zhang N, Zheng Q, Wang Y, Lin J, Wang H, Liu R, et al. Renoprotective effect of the recombinant anti-IL-6R fusion proteins by inhibiting JAK2/STAT3 signaling pathway in diabetic nephropathy. Front Pharmacol. (2021) 12:681424. doi: 10.3389/fphar.2021.681424
91. Coletta I, Soldo L, Polentarutti N, Mancini F, Guglielmotti A, Pinza M, et al. Selective induction of MCP-1 in human mesangial cells by the IL-6/sIL-6R complex. Exp nephrol. (2000) 8:37–43. doi: 10.1159/000059327
92. Chaudhari S, Yazdizadeh Shotorbani P, Tao Y, Davis ME, Mallet RT, Ma R. Inhibition of interleukin-6 on matrix protein production by glomerular mesangial cells and the pathway involved. Am J Physiol Renal Physiol. (2020) 318:F1478–f88. doi: 10.1152/ajprenal.00043.2020
93. Lv N, Li C, Liu X, Qi C, Wang Z. miR-34b alleviates high glucose-induced inflammation and apoptosis in human HK-2 cells via IL-6R/JAK2/STAT3 signaling pathway. Med Sci monitor: Int Med J Exp Clin Res. (2019) 25:8142–51. doi: 10.12659/MSM.917128
94. Li Y, Hu Q, Li C, Liang K, Xiang Y, Hsiao H, et al. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J Clin Invest. (2019) 129:1129–51. doi: 10.1172/JCI121987
95. Guo X, Zhu Y, Sun Y, Li X. IL-6 accelerates renal fibrosis after acute kidney injury via DNMT1-dependent FOXO3a methylation and activation of Wnt/β-catenin pathway. Int immunopharmacol. (2022) 109:108746. doi: 10.1016/j.intimp.2022.108746
96. Xiao Y, Yang N, Zhang Q, Wang Y, Yang S, Liu Z. Pentraxin 3 inhibits acute renal injury-induced interstitial fibrosis through suppression of IL-6/Stat3 pathway. Inflammation. (2014) 37:1895–901. doi: 10.1007/s10753-014-9921-2
97. Zhou Z, Ni J, Li J, Huo C, Miao N, Yin F, et al. RIG-I aggravates interstitial fibrosis via c-Myc-mediated fibroblast activation in UUO mice. J Mol Med (Berlin Germany). (2020) 98:527–40. doi: 10.1007/s00109-020-01879-x
98. Zhang Y, Qin X, Yang Y, Li J, Li X, Zou X, et al. Ginkgo biloba extract attenuates cisplatin-induced renal interstitial fibrosis by inhibiting the activation of renal fibroblasts through down-regulating the HIF-1α/STAT3/IL-6 pathway in renal tubular epithelial cells. Phytomedicine: Int J phytotherapy phytopharmacol. (2023) 115:154809. doi: 10.1016/j.phymed.2023.154809
99. Durlacher-Betzer K, Hassan A, Levi R, Axelrod J, Silver J, Naveh-Many T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. (2018) 94:315–25. doi: 10.1016/j.kint.2018.02.026
100. Ibrahim YF, Moussa RA, Bayoumi AMA, Ahmed AF. Tocilizumab attenuates acute lung and kidney injuries and improves survival in a rat model of sepsis via down-regulation of NF-κB/JNK: a possible role of P-glycoprotein. Inflammopharmacology. (2020) 28(1):215–30. doi: 10.1007/s10787-019-00628-y
101. Mohamed R, Jayakumar C, Ramesh G. Chronic administration of EP4-selective agonist exacerbates albuminuria and fibrosis of the kidney in streptozotocin-induced diabetic mice through IL-6. Lab investigation; J Tech Methods pathol. (2013) 93:933–45. doi: 10.1038/labinvest.2013.85
102. Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight. (2016) 1(13):e87023. doi: 10.1172/jci.insight.87023
103. Tanaka T, Narazaki M, Kishimoto T. Therapeutic targeting of the interleukin-6 receptor. Annu Rev Pharmacol toxicol. (2012) 52:199–219. doi: 10.1146/annurev-pharmtox-010611-134715
104. Kroy DC, Beraza N, Tschaharganeh DF, Sander LE, Erschfeld S, Giebeler A, et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatol (Baltimore Md). (2010) 51:463–73. doi: 10.1002/hep.23322
105. Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, et al. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab investigation; J Tech Methods pathol. (2010) 90:1169–78. doi: 10.1038/labinvest.2010.75
106. Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. (2010) 12:237–49. doi: 10.1016/j.cmet.2010.06.011
107. Gunes A, Schmitt C, Bilodeau L, Huet C, Belblidia A, Baldwin C, et al. IL-6 Trans-signaling is increased in diabetes, impacted by glucolipotoxicity, and associated with liver stiffness and fibrosis in fatty liver disease. Diabetes. (2023) 72(12):1820–34. doi: 10.2337/db23-0171
108. Lemmers A, Gustot T, Durnez A, Evrard S, Moreno C, Quertinmont E, et al. An inhibitor of interleukin-6 trans-signalling, sgp130, contributes to impaired acute phase response in human chronic liver disease. Clin Exp Immunol. (2009) 156:518–27. doi: 10.1111/j.1365-2249.2009.03916.x
109. Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, et al. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. (2002) 87:2084–9. doi: 10.1210/jcem.87.5.8450
110. Harder-Lauridsen NM, Krogh-Madsen R, Holst JJ, Plomgaard P, Leick L, Pedersen BK, et al. Effect of IL-6 on the insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. (2014) 306:E769–78. doi: 10.1152/ajpendo.00571.2013
111. Wueest S, Konrad D. The controversial role of IL-6 in adipose tissue on obesity-induced dysregulation of glucose metabolism. Am J Physiol Endocrinol Metab. (2020) 319(3):E607–e13. doi: 10.1152/ajpendo.00306.2020
112. Gutierrez AD, Gao Z, Hamidi V, Zhu L, Saint Andre KB, Riggs K, et al. Anti-diabetic effects of GLP1 analogs are mediated by thermogenic interleukin-6 signaling in adipocytes. Cell Rep Med. (2022) 3:100813. doi: 10.1016/j.xcrm.2022.100813
113. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. (2011) 17:1481–9. doi: 10.1038/nm.2513
114. Trujillo ME, Sullivan S, Harten I, Schneider SH, Greenberg AS, Fried SK. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J Clin Endocrinol Metab. (2004) 89:5577–82. doi: 10.1210/jc.2004-0603
115. Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci USA. (2020) 117(6):2751–60. doi: 10.1073/pnas.1920004117
116. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Sci (New York NY). (2008) 322:1539–43. doi: 10.1126/science.1160794
117. Chowdhury S, Schulz L, Palmisano B, Singh P, Berger JM, Yadav VK, et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J Clin Invest. (2020) 130:2888–902. doi: 10.1172/JCI133572
118. Becker M, Joseph SS, Garcia-Carrizo F, Tom RZ, Opaleva D, Serr I, et al. Regulatory T cells require IL6 receptor alpha signaling to control skeletal muscle function and regeneration. Cell Metab. (2023) 35:1736–51.e7. doi: 10.1016/j.cmet.2023.08.010
119. Sadagurski M, Norquay L, Farhang J, D'Aquino K, Copps K, White MF. Human IL6 enhances leptin action in mice. Diabetologia. (2010) 53:525–35. doi: 10.1007/s00125-009-1580-8
120. Mishra D, Richard JE, Maric I, Porteiro B, Häring M, Kooijman S, et al. Parabrachial interleukin-6 reduces body weight and food intake and increases thermogenesis to regulate energy metabolism. Cell Rep. (2019) 26:3011–26.e5. doi: 10.1016/j.celrep.2019.02.044
121. Timper K, Denson JL, Steculorum SM, Heilinger C, Engström-Ruud L, Wunderlich CM, et al. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 trans-Signaling. Cell Rep. (2017) 19:267–80. doi: 10.1016/j.celrep.2017.03.043
122. Katashima CK, de Oliveira Micheletti T. Evidence for a neuromuscular circuit involving hypothalamic interleukin-6 in the control of skeletal muscle metabolism. Sci Adv. (2022) 8(30):eabm7355. doi: 10.1126/sciadv.abm7355
123. Chen W, Cui W, Wu J, Zheng W, Sun X, Zhang J, et al. Blocking IL-6 signaling improves glucose tolerance via SLC39A5-mediated suppression of glucagon secretion. Metabolism: Clin experimental. (2023) 146:155641. doi: 10.1016/j.metabol.2023.155641
124. Lang Lehrskov L, Lyngbaek MP, Soederlund L, Legaard GE, Ehses JA, Heywood SE, et al. Interleukin-6 delays gastric emptying in humans with direct effects on glycemic control. Cell Metab. (2018) 27:1201–11.e3. doi: 10.1016/j.cmet.2018.04.008
125. Petersen EW, Carey AL, Sacchetti M, Steinberg GR, Macaulay SL, Febbraio MA, et al. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am J Physiol Endocrinol Metab. (2005) 288:E155–62. doi: 10.1152/ajpendo.00257.2004
126. Jiang LQ, Duque-Guimaraes DE, MaChado UF, Zierath JR, Krook A. Altered response of skeletal muscle to IL-6 in type 2 diabetic patients. Diabetes. (2013) 62:355–61. doi: 10.2337/db11-1790
127. Zuliani G, Galvani M, Maggio M, Volpato S, Bandinelli S, Corsi AM, et al. Plasma soluble gp130 levels are increased in older subjects with metabolic syndrome. role insulin resistance Atherosclerosis. (2010) 213:319–24. doi: 10.1016/j.atherosclerosis.2010.08.074
128. Weiss TW, Arnesen H, Seljeflot I. Components of the interleukin-6 transsignalling system are associated with the metabolic syndrome, endothelial dysfunction and arterial stiffness. Metabolism: Clin experimental. (2013) 62:1008–13. doi: 10.1016/j.metabol.2013.01.019
129. Nash D, Hughes MG, Butcher L, Aicheler R, Smith P, Cullen T. IL-6 signaling in acute exercise and chronic training: Potential consequences for health and athletic performance. Scand J Med Sci Sports. (2023) 33(1):4–19. doi: 10.1111/sms.14241
130. Findeisen M, Allen TL, Henstridge DC, Kammoun H, Brandon AE, Baggio LL, et al. Treatment of type 2 diabetes with the designer cytokine IC7Fc. Nature. (2019) 574:63–8. doi: 10.1038/s41586-019-1601-9
131. Kraakman MJ, Kammoun HL, Allen TL, Deswaerte V, Henstridge DC, Estevez E, et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab. (2015) 21:403–16. doi: 10.1016/j.cmet.2015.02.006
132. Ellingsgaard H, Seelig E, Timper K, Coslovsky M, Soederlund L, Lyngbaek MP, et al. GLP-1 secretion is regulated by IL-6 signalling: a randomised, placebo-controlled study. Diabetologia. (2020) 63:362–73. doi: 10.1007/s00125-019-05045-y
133. Wedell-Neergaard AS, Lang Lehrskov L, Christensen RH, Legaard GE, Dorph E, Larsen MK, et al. Exercise-induced changes in visceral adipose tissue mass are regulated by IL-6 signaling: A randomized controlled trial. Cell Metab. (2019) 29:844–55.e3. doi: 10.1016/j.cmet.2018.12.007
134. Castañeda S, Remuzgo-Martínez S, López-Mejías R, Genre F, Calvo-Alén J, Llorente I, et al. Rapid beneficial effect of the IL-6 receptor blockade on insulin resistance and insulin sensitivity in non-diabetic patients with rheumatoid arthritis. Clin Exp Rheumatol. (2019) 37:465–73.
135. Tournadre A, Pereira B, Dutheil F, Giraud C, Courteix D, Sapin V, et al. Changes in body composition and metabolic profile during interleukin 6 inhibition in rheumatoid arthritis. J cachexia sarcopenia muscle. (2017) 8:639–46. doi: 10.1002/jcsm.12189
136. Toussirot E, Marotte H, Mulleman D, Cormier G, Coury F, Gaudin P, et al. Increased high molecular weight adiponectin and lean mass during tocilizumab treatment in patients with rheumatoid arthritis: a 12-month multicentre study. Arthritis Res Ther. (2020) 22:224. doi: 10.1186/s13075-020-02297-7
137. Ogata A, Morishima A, Hirano T, Hishitani Y, Hagihara K, Shima Y, et al. Improvement of HbA1c during treatment with humanised anti-interleukin 6 receptor antibody, tocilizumab. Ann rheumatic diseases. (2011) 70:1164–5. doi: 10.1136/ard.2010.132845
138. Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis rheumatism. (2004) 50:1761–9. doi: 10.1002/art.20303
139. Cacciapaglia F, Anelli MG, Rinaldi A, Fornaro M, Lopalco G. Lipids and atherogenic indices fluctuation in rheumatoid arthritis patients on long-term tocilizumab treatment. Mediators Inflamm. (2018) 2018:2453265. doi: 10.1155/2018/2453265
140. Kume K, Amano K, Yamada S, Hatta K, Ohta H, Kuwaba N. Tocilizumab monotherapy reduces arterial stiffness as effectively as etanercept or adalimumab monotherapy in rheumatoid arthritis: an open-label randomized controlled trial. J Rheumatol. (2011) 38:2169–71. doi: 10.3899/jrheum.110340
141. McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann rheumatic diseases. (2015) 74:694–702. doi: 10.1136/annrheumdis-2013-204345
142. Ridker PM, Lei L, Louie MJ, Haddad T, Nicholls SJ, Lincoff AM. Inflammation and cholesterol as predictors of cardiovascular events among 13 970 contemporary high-risk patients with statin intolerance. Circulation. (2024) 149(1):28–35. doi: 10.1161/CIRCULATIONAHA.123.066213
143. Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, et al. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Supplement. (2002) 82:S12–22. doi: 10.1046/j.1523-1755.62.s82.4.x
144. Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Katsurada A, Navar LG, et al. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am J Physiol Renal Physiol. (2008) 295:F283–9. doi: 10.1152/ajprenal.00047.2008
145. Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension (Dallas Tex: 1979). (2010) 56:879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071
146. Moriyama T, Fujibayashi M, Fujiwara Y, Kaneko T, Xia C, Imai E, et al. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J Am Soc Nephrol: JASN. (1995) 6:95–101. doi: 10.1681/ASN.V6195
147. Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension (Dallas Tex: 1979). (2006) 48:1050–7. doi: 10.1161/01.HYP.0000248135.97380.76
148. Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arteriosclerosis thrombosis Vasc Biol. (2007) 27:2576–81. doi: 10.1161/ATVBAHA.107.153080
149. Manhiani MM, Quigley JE, Socha MJ, Motamed K, Imig JD. IL6 suppression provides renal protection independent of blood pressure in a murine model of salt-sensitive hypertension. Kidney Blood Pressure Res. (2007) 30:195–202. doi: 10.1159/000104094
150. Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol: JASN. (2001) 12:431–9. doi: 10.1681/ASN.V123431
151. O'Leary R, Penrose H, Miyata K, Satou R. Macrophage-derived IL-6 contributes to ANG II-mediated angiotensinogen stimulation in renal proximal tubular cells. Am J Physiol Renal Physiol. (2016) 310(10):F1000–7. doi: 10.1152/ajprenal.00482.2015
152. Recinos A 3rd, LeJeune WS, Sun H, Lee CY, Tieu BC, Lu M, et al. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis. (2007) 194:125–33. doi: 10.1016/j.atherosclerosis.2006.10.013
153. Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Urushihara M, Acres OW, et al. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol Cell Endocrinol. (2009) 311:24–31. doi: 10.1016/j.mce.2009.06.013
154. Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, et al. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol. (2004) 286:F653–9. doi: 10.1152/ajprenal.00163.2003
155. Wang H, Liu D, Zheng B, Yang Y, Qiao Y, Li S, et al. Emerging role of ferroptosis in diabetic kidney disease: molecular mechanisms and therapeutic opportunities. Int J Biol Sci. (2023) 19:2678–94. doi: 10.7150/ijbs.81892
156. Rosenblum SL, Bailey DK, Kosman DJ. Calcium and IL-6 regulate the anterograde trafficking and plasma membrane residence of the iron exporter ferroportin to modulate iron efflux. J Biol Chem. (2024) 300:107348. doi: 10.1016/j.jbc.2024.107348
157. Bin S, Xin L, Lin Z, Jinhua Z, Rui G, Xiang Z. Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced cartilage cell ferroptosis. Exp Mol pathol. (2021) 118:104570. doi: 10.1016/j.yexmp.2020.104570
158. Zhu G, Sui S, Shi F, Wang Q. Inhibition of USP14 suppresses ferroptosis and inflammation in LPS-induced goat mammary epithelial cells through ubiquitylating the IL-6 protein. Hereditas. (2022) 159:21. doi: 10.1186/s41065-022-00235-y
159. Yang L, Xing R, Li C, Liu Y, Sun L, Liu X, et al. Active immunization with Tocilizumab mimotopes induces specific immune responses. BMC Biotechnol. (2015) 15:46. doi: 10.1186/s12896-015-0161-9
160. Zhang Y, Zhang J, Feng D, Zhou H, Gui Z, Zheng M, et al. IRF1/ZNF350/GPX4-mediated ferroptosis of renal tubular epithelial cells promote chronic renal allograft interstitial fibrosis. Free Radical Biol Med. (2022) 193:579–94. doi: 10.1016/j.freeradbiomed.2022.11.002
161. Wang Z, Shen J. Neutrophil-derived IL-6 potentially drives ferroptosis resistance in B cells in lupus kidney. Mediators Inflamm. (2023) 2023:9810733. doi: 10.1155/2023/9810733
162. Busque S, Leventhal J, Brennan DC, Steinberg S, Klintmalm G, Shah T, et al. Calcineurin-inhibitor-free immunosuppression based on the JAK inhibitor CP-690,550: a pilot study in de novo kidney allograft recipients. Am J Transplant. (2009) 9:1936–45. doi: 10.1111/j.1600-6143.2009.02720.x
163. Busque S, Vincenti FG, Tedesco Silva H, O'Connell PJ, Yoshida A, Friedewald JJ, et al. Efficacy and safety of a tofacitinib-based immunosuppressive regimen after kidney transplantation: results from a long-term extension trial. Transplant direct. (2018) 4:e380. doi: 10.1097/TXD.0000000000000819
164. Chandran S, Leung J, Hu C, Laszik ZG, Tang Q. Interleukin-6 blockade with tocilizumab increases Tregs and reduces T effector cytokines in renal graft inflammation: A randomized controlled trial. Am J Transplant. (2021) 21(7):2543–54. doi: 10.1111/ajt.16459
165. Doberer K, Duerr M, Halloran PF, Eskandary F, Budde K, Regele H, et al. A randomized clinical trial of anti-IL-6 antibody clazakizumab in late antibody-mediated kidney transplant rejection. J Am Soc Nephrol. (2021) 32(3):708–22. doi: 10.1681/ASN.2020071106
166. Vo AA, Choi J, Kim I, Louie S, Cisneros K, Kahwaji J, et al. A phase I/II trial of the interleukin-6 receptor-specific humanized monoclonal (Tocilizumab) + Intravenous immunoglobulin in difficult to desensitize patients. Transplantation. (2015) 99:2356–63. doi: 10.1097/TP.0000000000000741
167. Chertow GM, Chang AM, Felker GM, Heise M. IL-6 inhibition with clazakizumab in patients receiving maintenance dialysis: a randomized phase 2b trial. Nat Med. (2024) 30(8):2328–36. doi: 10.1038/s41591-024-03043-1
168. Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet (London England). (2021) 397:2060–9. doi: 10.1016/S0140-6736(21)00520-1
169. Brosius FC, Tuttle KR, Kretzler M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia. (2016) 59:1624–7. doi: 10.1007/s00125-016-4021-5
170. Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. (2020) 8:963–74. doi: 10.1016/S2213-2600(20)30318-0
171. Ebata S, Yoshizaki-Ogawa A, Sato S, Yoshizaki A. New era in systemic sclerosis treatment: recently approved therapeutics. J Clin Med. (2022) 11(15):4631. doi: 10.3390/jcm11154631
172. Xu S, Deng KQ, Lu C, Fu X, Zhu Q, Wan S, et al. Interleukin-6 classic and trans-signaling utilize glucose metabolism reprogramming to achieve anti- or pro-inflammatory effects. Metabolism: Clin experimental. (2024) 155:155832. doi: 10.1016/j.metabol.2024.155832
173. Martinez-Fabregas J, Wilmes S. Kinetics of cytokine receptor trafficking determine signaling and functional selectivity. eLife. (2019) 8:e49314. doi: 10.7554/eLife.49314
174. Zhang S, Chen B, Wang B, Chen H, Li Y, Cao Q, et al. Effect of induction therapy with olamkicept vs placebo on clinical response in patients with active ulcerative colitis: A randomized clinical trial. JAMA. (2023) 329:725–34. doi: 10.1001/jama.2023.1084
175. Moutabarrik A, Nakanishi I, Ishibashi M. Interleukin-6 and interleukin-6 receptor are expressed by cultured glomerular epithelial cells. Scandinavian J Immunol. (1994) 40:181–6. doi: 10.1111/j.1365-3083.1994.tb03448.x
176. Ruef C, Budde K, Lacy J, Northemann W, Baumann M, Sterzel RB, et al. Interleukin 6 is an autocrine growth factor for mesangial cells. Kidney Int. (1990) 38:249–57. doi: 10.1038/ki.1990.193
177. Ranganathan P, Jayakumar C, Ramesh G. Proximal tubule-specific overexpression of netrin-1 suppresses acute kidney injury-induced interstitial fibrosis and glomerulosclerosis through suppression of IL-6/STAT3 signaling. Am J Physiol Renal Physiol. (2013) 304:F1054–65. doi: 10.1152/ajprenal.00650.2012
Keywords: IL-6, inflammation, diabetic kidney disease, IL-6 classical signaling, IL-6 trans-signaling pathway
Citation: Zhang L, Xu F and Hou L (2024) IL-6 and diabetic kidney disease. Front. Immunol. 15:1465625. doi: 10.3389/fimmu.2024.1465625
Received: 16 July 2024; Accepted: 03 December 2024;
Published: 19 December 2024.
Edited by:
Tarunveer Singh Ahluwalia, Steno Diabetes Center Copenhagen (SDCC), DenmarkReviewed by:
Latha P. Ganesan, The Ohio State University, United StatesIndumathi Manoharan, Augusta University, United States
Copyright © 2024 Zhang, Xu and Hou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyan Hou, aG91bGl5YW41Njc4OUBzaW5hLmNvbQ==