 Camillo Carrara
Camillo Carrara Blerina Mataj
Blerina Mataj Sara Gastoldi
Sara Gastoldi Piero Ruggenenti
Piero Ruggenenti Savino Sciascia
Savino Sciascia Dario Roccatello
Dario Roccatello- 1Unit of Nephrology, Azienda Socio-Sanitaria Territoriale Papa Giovanni XXIII, Bergamo, Italy
- 2Istituto di Ricerche Farmacologiche Mario Negri Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Bergamo, Italy
- 3University Center of Excellence on Nephrologic, Rheumatologic and Rare Diseases (ERK-Net, ERN-Reconnect and RITA-ERN Member) with Nephrology and Dialysis Unit and Center of Immuno-Rheumatology and Rare Diseases (CMID), Coordinating Center of the Interregional Network for Rare Diseases of Piedmont and Aosta Valley, San Giovanni Bosco Hub Hospital ASL Città di Torino and Department of Clinical and Biological Sciences, Turin, Italy
Catastrophic antiphospholipid syndrome (CAPS) is a life-threatening condition of small-vessel thrombosis with acute multiple-organ involvement and visceral damage. In this report, we present a case of a patient with CAPS who is refractory to conventional therapy. For the first time in a patient with CAPS, marked C5b-9 formation was demonstrated on microvascular endothelial cells, suggesting the usefulness of therapeutic complement inhibition in this setting. Eculizumab, a C5-blocking monoclonal antibody, is remarkably effective in the treatment of different forms of thrombotic microangiopathy by controlling complement system hyperactivation. It halted the “thrombotic storm” and promptly achieved full recovery of thrombocytopenia. However, kidney function did not recover, possibly because eculizumab was administered too late. Conceivably, the timing of treatment is crucial to achieving disease remission before irreversible structural damage occurs in target organs, thereby preventing their complete functional recovery.
Introduction
The antiphospholipid syndrome (APS) is the most common cause of acquired immune-mediated thrombophilia (1). APS may present with a variety of clinical phenotypes, including thrombosis in the veins and arteries, as well as obstetrical morbidity (2). The pathogenesis of APS is not yet completely elucidated; however, pathways involving several cells (including platelets, leucocytes, and endothelial cells), as well as activation of the coagulation and complement systems, are all known to be involved in the development of thrombosis (3). About 1% of APS patients (4) develop the highly thrombotic phenotype referred to as catastrophic APS (CAPS). CAPS is a life-threatening variant of APS, with a mortality rate of approximately 30%, that is characterized by multiple organ involvement in a short period of time, mainly due to small vessel occlusions, often showing refractoriness to standard treatment with anticoagulants, glucocorticoids, and plasma exchanges (PEX) (5). Evidence shows that uncontrolled complement activation plays a central role in the pathogenesis of APS, particularly in CAPS (6). The hypothesis of a crucial involvement of activated complement in the pathogenesis of the disease is corroborated by evidence that in affected patients, as observed for patients with atypical hemolytic uremic syndrome (aHUS), there is a high prevalence of complement regulatory gene mutations (7). Based on the observation that C5 complement inhibition radically improved outcomes in patients with aHUS, eculizumab has been successfully administered as a prophylactic treatment in a CAPS patient undergoing renal transplantation (8–10). Following this preliminary experience, there have been an increasing number of clinical reports describing the benefits of eculizumab therapy in patients with CAPS that have been recently collected in the “CAPS registry” (11, 12).
However, despite effective complement inhibition (13) and resolution of thrombocytopenia, as well as the life-threatening “thrombotic storm” in patients refractory to conventional therapy, eculizumab may occasionally fail to achieve functional recovery in target organs, including the kidney. The clinical outcome of the patient we are describing in the present report suggests that optimal timing of eculizumab treatment is crucial to achieving disease remission before irreversible structural damage occurs in target organs, potentially preventing their complete functional recovery.
Case report
We report the case of a 45-year-old man with a past medical history of triple-positive primary APS, characterized by a previous episode of pulmonary thromboembolism (PTE) and lower limb ulcers, who was on Warfarin and had a caval filter placed. He was also previously diagnosed with heparin-induced thrombocytopenia.
On 4 October 2022, he presented to our Emergency Department with intense asthenia and dyspnea. The arterial blood pressure was within the normal limits. His height and weight were, respectively, 176 cm and 76 kg. Laboratory tests showed severe pancytopenia (white blood cells: 2,760/μL, hemoglobin: 4.6 g/dL, platelets: 15,000/μL) along with B12 and folate deficiency. The computed tomography scan (CT) excluded a PTE and revealed numerous density alterations in the spleen parenchyma consistent with ischemic lesions. The patient received transfusion, and vitamin-K antagonist was discontinued, with the indication to start fondaparinux when the platelet count becomes higher than 20,000–30,000/μL.
After discharge from the emergency department, further laboratory examinations highlighted a polyglandular autoimmune syndrome, characterized by atrophic gastritis with anti-gastric parietal cells antibodies and autoimmune hypothyroidism. Pancytopenia at that time was considered to be based on vitamin deficiency, so a supplementing treatment with vitamin B12 and folate was started. Due to the improvement of thrombocytopenia (platelets up to 40,000/μL), fondaparinux was soon initiated. After 10 days, he was admitted again to our hospital, this time with the main complaint of abdominal pain and fever. The CT scans revealed acute acalculous cholecystitis and an extension of the splenic infarctions. Initial laboratory tests (Table 1) revealed thrombocytopenia and increased C-reactive protein (CRP) level. Empiric antibiotic therapy with piperacillin/tazobactam was started, along with hydration and parenteral nutrition, which resulted in mild clinical improvement and an initial CRP reduction. During hospitalization, he started complaining of severe headaches. Brain CT scans together with magnetic resonance imaging, were performed, which showed small ischemic lesions in different cortical areas and in the white matter. Considering the association of APS with severe thrombocytopenia and multiple organ infarcts, a diagnosis of CAPS was made. Fondaparinux dosage was increased, and corticosteroid treatment (3 days of intravenous methylprednisolone at the doses of 1 g + 0.5 g + 0.5 g, continuing thereafter with prednisone at 1 mg/kg/day, with subsequent tapering) was started along with hydroxychloroquine. Workup for autoimmune disorders ruled out systemic lupus erythematosus (SLE) (Table 1), and ADAMTS13 activity was found to be normal, with no ADAMTS13 inhibitor autoantibodies identified. Schistocytes were negative throughout the entire disease course. Notably, there was a marked C4 hypocomplementemia, together with low C3, while haptoglobin was consumed in only one determination. Due to persistent thrombocytopenia, on 2 November, he received a 5-day course of intravenous immunoglobulins (2 g/kg total dose), with no clinical or biochemical improvement (Figure 1). Therefore, rituximab therapy was added (two doses of 375 mg/m2 administered a week apart). Simultaneously, he developed a rapidly progressive renal failure with an active urinary sediment (microhematuria and proteinuria of 0.8 g/24 h) and arterial hypertension. Renal biopsy was considered too risky due to ongoing anticoagulation and severe thrombocytopenia, especially since the expected histological finding, given the prolonged anuria and tissue ischemia, would likely have been diffuse cortical necrosis. On 18 November, the patient was started on pulsed methylprednisolone (1 g + 0.5 g + 0.5 g), with subsequent tapering and three sessions of PEX. Unfortunately, kidney failure progressed, and hemodialysis treatment was started on 28 November. The patient did not show clinical improvement and continued to experience abdominal pain and thrombocytopenia, despite PEX and steroids. Marked C5b-9 formation was found on the microvascular endothelial cells (208% on resting and 263% on activated endothelial cells, normal values < 150%) (Figure 2), which completely normalized after exposure to eculizumab in vitro (13). The assessment of complement regulatory genes through NGS revealed a polymorphic heterozygous deletion of the CFHR3/CFHR1 genes.
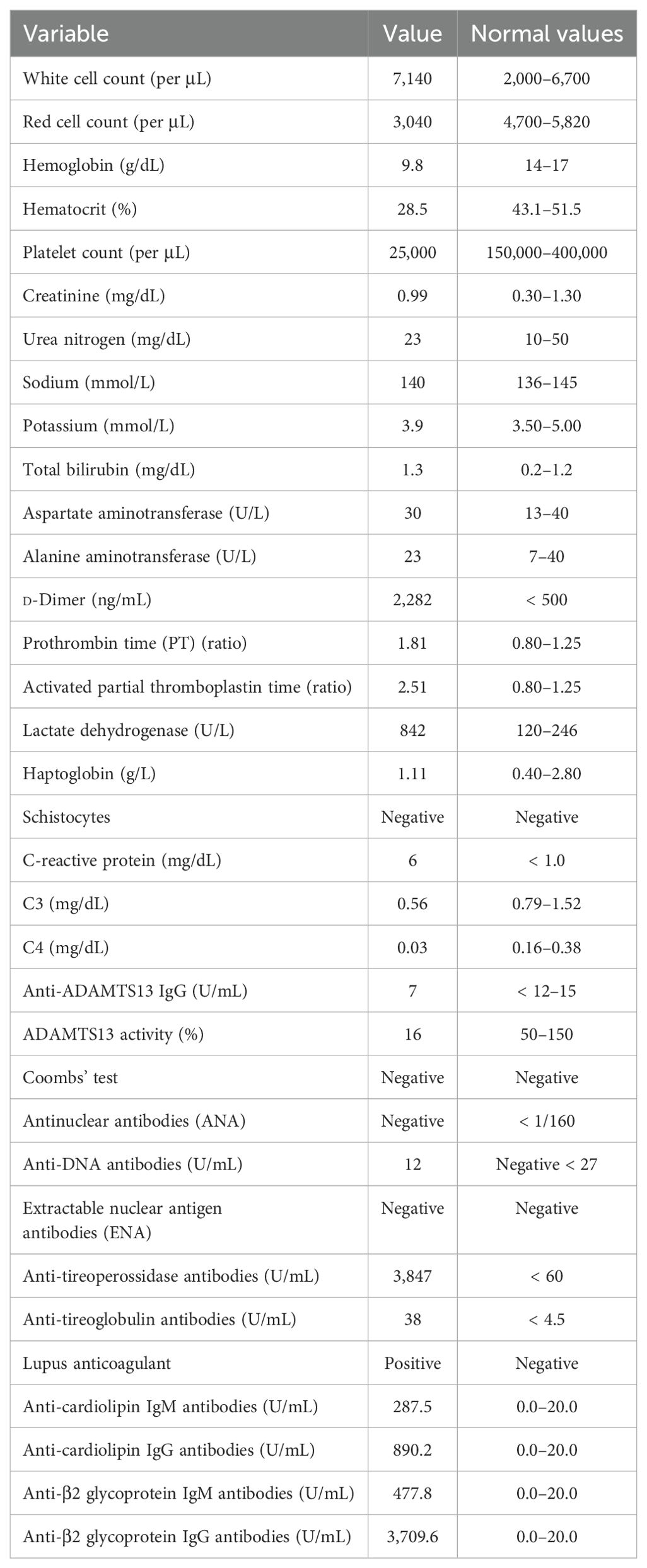
Table 1. Laboratory values of the patient at baseline.

Figure 1. Patient treatment and outcome from admission until hospital discharge and follow-up.
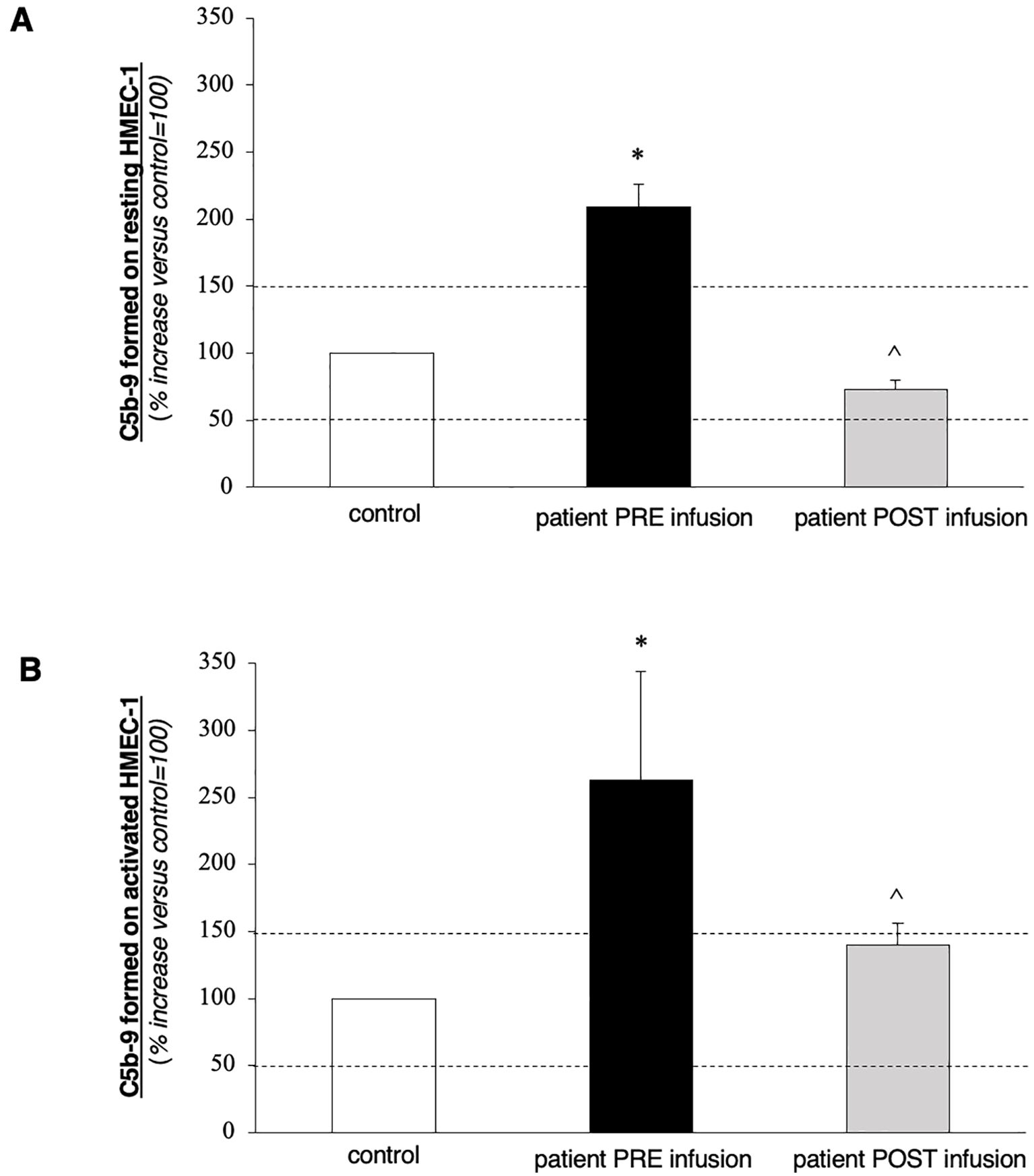
Figure 2. Ex vivo C5b-9 deposition on cultured human microvascular endothelial cell (HMEC-1) line induced by serum of the patient collected before (PRE-infusion) and 10 days after (POST-infusion) eculizumab treatment. (A, B) HMEC-1 resting (A) or activated with ADP (B) were incubated for 2 h with serum (diluted 1:2 with test medium, Hank’s Balanced Salt Solution (HBSS) with 0.5% Bovine Serum Albumin (BSA)) from the patient or with a control serum pool. At the end of incubation, cells were washed, fixed, and stained with rabbit antihuman complement C5b-9 complex antibody followed by Fluorescein Isothiocyanate (FITC)-conjugated secondary antibody. Fluorescence microscopy was used to view the fluorescent staining on the endothelial cell surface, and the HMEC-1 area covered by C5b-9 staining was calculated by automatic edge detection (Image J software) in 15 high-power fields. For each sample, the highest and lowest values were discarded, and the mean of the other 13 fields was calculated. Values were expressed as the percentage of C5b-9 deposits induced by a pool of sera from 10 healthy controls run in parallel (reference, 100%). Dashed lines indicate the upper and lower limits of the normal range. Data are reported ± SE. *p < 0.0001 versus control serum pool; °p < 0.0001 versus patient PRE-infusion. Statistical analysis: ANOVA.
On 16 December, eculizumab was started (900 mg for 4 weeks, followed by 1,200 mg at week 5), and then continued every 2 weeks thereafter. The patient received the vaccinations recommended for meningococcal and pneumococcal diseases. In addition, due to concerns about profound immunosuppression, he also received oral cotrimoxazole for the prevention of Pneumocystis carinii pneumonia. On 9 January 2023, C5b-9 formation on the microvascular endothelial cells was found to be within the normal range (72% on resting and 140% on activated endothelial cells). The patient was discharged with improving thrombocytopenia (platelet count of 78 × 109/L) but was still on hemodialysis, receiving eculizumab every 2 weeks. Anticoagulation therapy was shifted back to a vitamin-K antagonist. At 2 months follow-up, his platelet count kept increasing (127,000/μL), but he remained anuric.
Discussion
Here, we present the case of a patient affected by APS who developed multiorgan thrombosis, severe thrombocytopenia, and dialysis-dependent kidney failure in the context of CAPS. The patient was treated with anticoagulants, steroids, intravenous immunoglobulins, PEX, and rituximab without benefit. Eventually, eculizumab administration led to the rapid normalization of platelet count and halted the “thrombotic storm”. However, kidney function did not recover, and the patient remained dialysis-dependent. During the active phase of the disease, we were able to demonstrate higher-than-normal C5b-9 deposit formation on the microvascular endothelial cells, which completely normalized after treatment with the C5-blocker. Genetic analysis revealed a polymorphic heterozygous deletion of CFHR3/CFHR1 genes. Thus, we found a minor genetic abnormality in the complement system that could have predisposed to the development of CAPS upon exposure to a non-identified potential trigger.
CAPS is the most severe and rare form of APS, characterized by acute multiple-organ involvement, small-vessel thrombosis, and visceral damage (4). Complement has been identified as a critical pathway for the development of CAPS (14). Current knowledge supports the combination of high-dose glucocorticoids and the anticoagulant, heparin, as first-line treatment, with the addition of PEX and/or intravenous immunoglobulins for those with this life-threatening situation (5). Given the rarity and complexity of CAPS, as well as the absence of a clear consensus on the optimal timing of PEX initiation, it is challenging to determine whether earlier intervention would have significantly altered the final outcome in this case. Despite this standard treatment, CAPS is still associated with significant mortality. Several case reports suggest the efficacy of eculizumab in the management of CAPS (15–21). Furthermore, data from the “CAPS registry”, which includes 584 patients, of whom 6.7% were treated with eculizumab, suggest that complement inhibition should be considered in some patients with CAPS refractory to previous therapies, particularly if they present with features of aHUS (12). In the CAPS episode, 74.4% of patients treated with eculizumab recovered from the CAPS episode without recurrence of thrombosis during a median follow-up of 10.7 months. Among them, 64.1% presented complete remission and 10.3% had partial remission (improvement of hematologic features, but persisting signs of organ damage). All patients with thrombocytopenia showed an improvement in platelet count.
In a recent case series of patients with APS and thrombotic microangiopathy treated with eculizumab, an improvement in laboratory parameters was reported as early as the first week after the initiation of eculizumab. This suggests that complement inhibition might be considered earlier in the course of the disease (22). Timing in the administration of the anticomplement drug seems to be important in determining patient outcomes, especially the evolution of kidney function, as suggested in the “French cohort” (23). A total of 11 patients with severe CAPS and triple APS positivity were included in this study. Despite full doses of anticoagulants, corticosteroids, and PEX, the patients’ conditions continued to worsen. Overall, five patients had significant improvement in the few days after the first dose of eculizumab. Compared with nonresponders, responders had a less severe history of APS and a higher frequency of SLE. There was a higher number of patients who required hemodialysis before eculizumab in nonresponders. Among the four responders with renal failure, renal function recovered in three who were not on hemodialysis before eculizumab; the last patient continued to have dialysis-dependent renal failure. The authors concluded that patients with refractory CAPS respond inconsistently to eculizumab. Similar to our case, also in the French cohort, thrombocytopenia improved more rapidly and frequently after eculizumab, which may be of importance in sustaining optimal anticoagulation. The delay between the onset of CAPS and the administration of eculizumab raises the question of the benefit of earlier administration to avoid the occurrence of irreversible lesions, such as renal cortical necrosis that usually impairs renal function (24).
In conclusion, our case report demonstrates that inhibition of the complement system using eculizumab is a valuable strategy in the setting of CAPS. C5b-9 formation on cultured microvascular endothelial cells, a sophisticated test that we performed for the first time in a patient with CAPS, could represent a reliable marker of complement activation in the solid phase, and its inhibition is a reliable marker of the complement-inhibitory effect of eculizumab not only in aHUS but also in this setting. The timely administration of drugs (as early as possible) seems to be a crucial factor in preventing irreversible kidney structural damage and achieving full kidney function recovery.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CC: Writing – original draft, Writing – review & editing. BM: Writing – original draft, Writing – review & editing. SG: Investigation, Resources, Writing – original draft. PR: Writing – original draft, Writing – review & editing. SS: Writing – original draft, Writing – review & editing. DR: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
This case report was conceived and finalized as part of the master’s degree in Practical Rheumatology at the University of Turin. We thank Marina Noris and Miriam Galbusera, who supervised the complement tests. We thank the medical staff and the nurses of the Emergency Department of Azienda Socio-Sanitaria Territoriale Papa Giovanni XXIII for taking care of the patient with great dedication. We thank the nurses of the Nephrology Unit of Azienda Socio-Sanitaria Territoriale Papa Giovanni XXIII for taking care of the patient and performing the dialysis sessions. We thank Dr. Valentina Portalupi and Dr. Alessia Gennarini, two nephrologists at the Azienda Socio-Sanitaria Territoriale Papa Giovanni XXIII, who prescribed eculizumab infusions. Finally, we thank the European Reference Network for Rare Kidney Diseases (ERKNet), of which we are proud members.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. (2010) 376:1498–509. doi: 10.1016/S0140-6736(10)60709-X
2. Schreiber K, Sciascia S, De Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. (2018) 4:17103. doi: 10.1038/nrdp.2017.103
3. Sciascia S, Amigo M-C, Roccatello D, Khamashta M. Diagnosing antiphospholipid syndrome: “extra-criteria” manifestations and technical advances. Nat Rev Rheumatol. (2017) 13:548–60. doi: 10.1038/nrrheum.2017.124
4. Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, De Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. (2015) 74:1011–8. doi: 10.1136/annrheumdis-2013-204838
5. Asherson RA, Cervera R, De Groot PG, Erkan D, Boffa M-C, Piette J-C, et al. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. (2003) 12:530–4. doi: 10.1191/0961203303lu394oa
6. Oku K, Nakamura H, Kono M, Ohmura K, Kato M, Bohgaki T, et al. Complement and thrombosis in the antiphospholipid syndrome. Autoimmun Rev. (2016) 15:1001–4. doi: 10.1016/j.autrev.2016.07.020
7. Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. (2020) 135:239–51. doi: 10.1182/blood.2019003863
8. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 360:544–6. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19179329.
9. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. (2012) 8:622–33. doi: 10.1038/nrneph.2012.195
10. Legendre CM, Licht C, Loirat C. Eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med. (2013) 369:1377–80. doi: 10.1056/NEJMc1308826
11. Lonze BE, Singer AL, Montgomery RA. Eculizumab and renal transplantation in a patient with CAPS. N Engl J Med. (2010) 362:1744–5. doi: 10.1056/NEJMc0910965
12. López-Benjume B, Rodríguez-Pintó I, Amigo MC, Erkan D, Shoenfeld Y, Cervera R, et al. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry. Autoimmun Rev. (2022) 21:103055. doi: 10.1016/j.autrev.2022.103055
13. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. (2014) 124:1715–26. doi: 10.1182/blood-2014-02-558296
14. Chaturvedi S, Brodsky RA, McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol. (2019) 10:449. doi: 10.3389/fimmu.2019.00449
15. Kronbichler A, Frank R, Kirschfink M, Szilágyi Á, Csuka D, Prohászka Z, et al. Efficacy of eculizumab in a patient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: a case report. Medicine. (2014) 93:e143. doi: 10.1097/MD.0000000000000143
16. Strakhan M, Hurtado-Sbordoni M, Galeas N, Bakirhan K, Alexis K, Elrafei T. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. (2014) 2014:1–7. doi: 10.1155/2014/704371
17. Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. JCR: J Clin Rheumatol. (2015) 21:311–3. doi: 10.1097/RHU.0000000000000290
18. Barratt-Due A, Fløisand Y, Orrem HL, Kvam AK, Holme PA, Bergseth G, et al. Complement activation is a crucial pathogenic factor in catastrophic antiphospholipid syndrome. Rheumatology. (2016) 55:1337–9. doi: 10.1093/rheumatology/kew040
19. Guillot M, Rafat C, Buob D, Coppo P, Jamme M, Rondeau E, et al. Eculizumab for catastrophic antiphospholipid syndrome—a case report and literature review. Rheumatology. (2018) 57:2055–7. doi: 10.1093/rheumatology/key228
20. Ruffatti A, Tarzia V, Fedrigo M, Calligaro A, Favaro M, Macor P, et al. Evidence of complement activation in the thrombotic small vessels of a patient with catastrophic antiphospholipid syndrome treated with eculizumab. Autoimmun Rev. (2019) 18:561–3. doi: 10.1016/j.autrev.2019.03.015
21. Skoczynska M, Crowther MA, Chowaniec M, Ponikowska M, Chaturvedi S, Legault K. Thrombotic microangiopathy in the course of catastrophic antiphospholipid syndrome successfully treated with eculizumab: case report and systematic review of the literature. Lupus. (2020) 29:631–9. doi: 10.1177/0961203320917460
22. Kello N, Khoury LE, Marder G, Furie R, Zapantis E, Horowitz DL. Secondary thrombotic microangiopathy in systemic lupus erythematosus and antiphospholipid syndrome, the role of complement and use of eculizumab: Case series and review of literature. Semin Arthritis Rheumatism. (2019) 49:74–83. doi: 10.1016/j.semarthrit.2018.11.005
23. Yelnik CM, Miranda S, Mékinian A, Lazaro E, Quéméneur T, Provot F, et al. Patients with refractory catastrophic antiphospholipid syndrome respond inconsistently to eculizumab. Blood. (2020) 136:2473–7. doi: 10.1182/blood.2020007499
Keywords: antiphospholipid syndrome, complement system, eculizumab, kidney failure, timing, catastrophic antiphospholipid antibody syndrome (CAPS), C5b-9
Citation: Carrara C, Mataj B, Gastoldi S, Ruggenenti P, Sciascia S and Roccatello D (2024) Case report: Timing of eculizumab treatment in catastrophic antiphospholipid syndrome. Front. Immunol. 15:1460317. doi: 10.3389/fimmu.2024.1460317
Received: 05 July 2024; Accepted: 20 August 2024;
Published: 10 September 2024.
Edited by:
Maria Giovanna Danieli, Università Politecnica delle Marche, ItalyReviewed by:
Carlo Perricone, University of Perugia, ItalyLaura Andreoli, University of Brescia, Italy
Copyright © 2024 Carrara, Mataj, Gastoldi, Ruggenenti, Sciascia and Roccatello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Blerina Mataj, YmxlcmluYS5tYXRhakB1bmltaS5pdA==