 Li-Jun Ling1,2†Qiong Zhou3†Fan Zhang4,5Wen-Jia Lei4,5Meng-Die Li4,5
Li-Jun Ling1,2†Qiong Zhou3†Fan Zhang4,5Wen-Jia Lei4,5Meng-Die Li4,5 Jiang-Wen Lu4,5
Jiang-Wen Lu4,5 Wang-Sheng Wang4,5*
Wang-Sheng Wang4,5* Kang Sun4,5*
Kang Sun4,5* Hao Ying1,2*
Hao Ying1,2*- 1Department of Obstetrics, Shanghai First Maternity and Infant Hospital, School of Medicine, Tongji University, Shanghai, China
- 2Shanghai Key Laboratory of Maternal Fetal Medicine, Shanghai Institute of Maternal-Fetal Medicine and Gynecologic Oncology, Shanghai, China
- 3Department of Obstetrics and Gynecology, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 4Center for Reproductive Medicine, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 5Shanghai Key Laboratory for Assisted Reproduction and Reproductive Genetics, Shanghai, China
Introduction: Fetal membrane inflammation is an integral event of parturition. However, excessive pro-inflammatory cytokines can impose threats to the fetus. Coincidentally, the fetal membranes express abundant 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1), which generates biologically active cortisol to promote labor through induction of prostaglandin synthesis. Given the well-recognized anti-inflammatory actions of glucocorticoids, we hypothesized that cortisol regenerated in the fetal membranes might be engaged in restraining fetus-hazardous pro-inflammatory cytokine production for the safety of the fetus, while reserving pro-labor effect on prostaglandin synthesis to ensure safe delivery of the fetus.
Methods: The hypothesis was examined in human amnion tissue and cultured primary human amnion fibroblasts as well as a mouse model.
Results: 11β-HSD1 was significantly increased in the human amnion in infection-induced preterm birth. Studies in human amnion fibroblasts showed that lipopolysaccharide (LPS) induced 11β-HSD1 expression synergistically with cortisol. Cortisol completely blocked NF-κB-mediated pro-inflammatory cytokine expression by LPS, but STAT3-mediated cyclooxygenase 2 expression, a crucial prostaglandin synthetic enzyme, remained. Further studies in pregnant mice showed that corticosterone did not delay LPS-induced preterm birth, but alleviated LPS-induced fetal organ damages, along with increased 11β-HSD1, cyclooxygenase 2, and decreased pro-inflammatory cytokine in the fetal membranes.
Discussion: There is a feed-forward cortisol regeneration in the fetal membranes in infection, and cortisol regenerated restrains pro-inflammatory cytokine expression, while reserves pro-labor effect on prostaglandin synthesis. This dual role of cortisol regeneration can prevent excessive pro-inflammatory cytokine production, while ensure in-time delivery for the safety of the fetus.
Introduction
Inflammation of the fetal membranes plays a crucial role in labor onset in both term and preterm by inducing a wealth of labor-inducing factors including prostaglandins, matrix metalloproteases and pro-inflammatory cytokines etc. (1–3). Based on the presence or absence of infection, inflammation of the fetal membranes can be classified into infectious and sterile inflammation. Although sterile inflammation of the fetal membranes is an integral event of normal parturition, uncontrolled inflammation, particularly in infection-induced chorioamnionitis, can lead to preterm birth by producing excessive pro-inflammatory cytokines (4, 5). In addition to induction of labor, excessive pro-inflammatory cytokines can also pose fatal threats to the fetus by developing fetal inflammatory response syndrome (FIRS), a condition of fetal systemic inflammation with rapid elevation of pro-inflammatory cytokines (such as IL-1β and IL-6) and leucocytes in the fetal circulation (6, 7). FIRS can lead to multiple fetal organ damages, neonatal sepsis, and even fetal death (8–11). Therefore, it is of utmost importance to balance inflammation reactions of the fetal membranes precisely to allow the baby to be delivered safely. Here we examined whether there exists such a mechanism to balance the inflammatory reactions of the fetal membranes in normal and infection-induced parturition.
Glucocorticoids (GCs) are a class of steroid hormones which are recognized to have powerful anti-inflammatory actions with inhibition of nuclear factor-κB (NF-κB) as one of the central mechanisms (12). The adrenal cortex is known to be the primary site of de novo synthesis of glucocorticoids (13). In addition to the de novo synthesis in adrenal glands, there also exists an alternative glucocorticoid-regenerating pathway in virtually all glucocorticoid target tissues (14–17). In these tissues, biologically active glucocorticoids can be regenerated from their biologically inactive keto counterparts by 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) within the endoplasmic reticulum (18, 19). Specifically, 11β-HSD1 converts biologically inactive cortisone into active cortisol in humans, while converting inert 11-dehydrocorticoserone into active corticosterone in rodents (19). This intracellular glucocorticoid regenerating machinery is believed to be a supplemental gating mechanism to the circulating glucocorticoids to ensure the presence of adequate glucocorticoids for activation of the low affinity glucocorticoid receptor (GR) (14–16).
Notably, the human fetal membranes express abundant 11β-HSD1 and boast the largest glucocorticoid regenerating capacity among fetal tissues (20–22), which increases with gestational age, and further increases at parturition (23, 24). As a result, cortisol concentrations increase in the fetal membranes and amniotic fluid towards the end of gestation, reaching peak levels at parturition (fetal membranes: 6.58 ± 1.61 μM (25, 26), amniotic fluid: 97.66 ± 7.17 nM (27)). Of interest, 11β-HSD1 expression is under the synergistic feedforward induction by glucocorticoids and pro-inflammatory cytokines in the fetal membranes (28–30), indicating that glucocorticoid regeneration capacity may be greatly enhanced by inflammation of the fetal membranes. We and others have demonstrated that one of the prominent roles of glucocorticoid regenerated by 11β-HSD1 is to promote parturition by inducing prostaglandin E2 and F2α synthesis, and remodeling extracellular matrix (ECM) in the amnion layer of the fetal membranes (31–36), the events crucial for labor onset and membrane rupture (24, 26, 37–39). Of interest, a NF-κB-independent pathway has been revealed to mediate the prostaglandin inducing, and ECM remodeling effects of glucocorticoids in the fetal membranes with the involvement of GR interacting with other transcription factors such as STAT3, CREB and C/EBPδ (24, 26, 33).
Given the central role of NF-κB in inflammatory reactions (40), the powerful repressive effect of glucocorticoids on NF-κB (41) and the NF-κB-independent labor-inducing effects of glucocorticoids in the fetal membranes (24, 26, 33), we postulated that glucocorticoids regenerated in the fetal membranes might play a dual role in parturition. On the one hand, glucocorticoids might initiate parturition through NF-κB-independent actions, while restraining inflammatory reactions through NF-κB-dependent actions on the other hand. As such, fetal damage by excessive pro-inflammatory cytokines at parturition could be possibly avoided or ameliorated, particularly in infection-induced chorioamnionitis. As such, boosting glucocorticoid regeneration in the fetal membranes should be considered to increase the chance of fetal survival in infection-induced chorioamnionitis. In this study, we examined the hypothesis by using both cultured human primary amnion fibroblasts, a major cell type synthesizing both pro-labor and pro-inflammatory factors in the fetal membranes, and a mouse model of infection-induced preterm labor, with the ultimate goal to determine whether it is beneficial to enhance glucocorticoid regeneration in the fetal membranes when the fetus is threatened by intrauterine infection.
Materials and methods
Collection of the human amnion
The human amnion was collected from either elective cesarean section at term (designated as term none labor, TNL), or spontaneous labor at term without infection (designated as term labor, TL), or preterm labor with infection (designated as preterm labor with infection, PL) with written informed consent under a protocol approved by the ethics committee of Shanghai First Maternal and Infant Hospital, Tongji University School of Medicine. The amnion from the TNL group was used for cell culture. The amnion from the TL and PL groups was snap-frozen in liquid nitrogen for protein extraction. Criteria for determining intrauterine infection in preterm birth included elevated body temperature and white blood cell count, and detection of pathogens in culture of uterine secretion or placental swab. Bacterial culture, antimicrobial susceptibility testing and Gram staining were used for identification of the pathogen.
Preparation of human amnion fibroblasts
The amnion from TNL was used for isolation of amnion fibroblasts. After delivery, the inner amnion layer was identified at the rupture site of the fetal membranes, and carefully peeled off the chorion layer by hands. The amnion was rapidly transferred to the laboratory in sterile PBS at 4°C within 30 minutes. After thoroughly washing the surface blood of the amnion with sterile PBS, the amnion was digested twice with 0.125% trypsin (#27250-018, Life Technologies Inc., Grand Island, NY) to remove epithelial cells, and further digested with 0.1% Type I collagenase (#C0130, Sigma, St. Louis, MO) to isolate fibroblasts. Isolated fibroblasts were harvested by centrifugation and 1 × 106 cells were plated in a 6-well plate for culture in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% Fetal Bovine Serum (FBS) and antibiotics (Life Technologies Inc) at 37°C in 5% CO2-95% air. This method of cell isolation yields high purity of amnion fibroblast (>95%), which have been verified previously by staining with mesenchymal (vimentin and fibroblast-specific protein 1) and epithelial (cytokeratin-7) markers (35, 42, 43).
Treatment of human amnion fibroblasts
In this study, primary human amnion mesenchymal cells without any passage were used. Three days after plating, the primary amnion fibroblasts were treated with the following reagents in FBS-free medium. Fibroblasts were treated with cortisol (1 μM; Sigma), lipopolysaccharide (LPS, from E. coli serotype 0111:B4, 10 ng/mL; #L2630, Sigma), or combination of cortisol (1 μM) and LPS (10 ng/mL) for 24 hours to investigate the effect on 11β-HSD1, cyclooxygenase-2 (COX-2, the rate-limiting enzyme in prostaglandin synthesis), IκB-α (the NF-κB inhibitory protein) and pro-inflammatory cytokines (IL-1β and IL-6), and for 3 hours to study the effect on the phosphorylation of STAT3 and p65 (a subunit of NF-κB). Cells were treated with cortisone (10 μM; Sigma) with or without 11β-HSD1 inhibitor 10-J (1 μM; Millipore, Billerica, MA) for 24 hours to examine the involvement of 11β-HSD1 in the effect of cortisone. The cells were treated with cortisol and LPS with or without STAT3 antagonist S3I-201 (50 μM; Selleck, Houston, TX) or toll-like receptor-4 (TLR-4) antagonist TAK-242 (10 μM; R&D systems, Minneapolis, MN) or small interference RNA (siRNA)-mediated knockdown of p65 for 24 hours to examine their role in the induction of COX-2 by cortisol and LPS. Cells were transfected with siRNA (50 nM) against RELA (encoding p65) (5′-CCACUUUGGUGUUUCAUAAtt-3′) (GenePharma Co., Shanghai, China) or randomly scrambled siRNA with an electroporator (175 V, 5 ms) (Nepa Gene, Chiba, Japan), and were treated with reagents after recovery for 3 days. The knockdown efficiency was 91.5 + 2.6% (Supplementary Figure 1). After treatment, cells were harvested for RNA and protein extraction, and the conditioned culture medium was collected for cytokine measurement with a multiplex kit (Bio-Rad, Austin, TX). The treatment regimen of all reagents was based on previous studies (26, 44–48).
Measurement of cortisol regeneration in human amnion fibroblasts
To measure cortisol regeneration activity of 11β-HSD1 under the induction by cortisol and LPS, amnion fibroblasts were pretreated with cortisol (1 μM) in the presence or absence of LPS (10 ng/mL) for 24 hours. After removal of the treatments by washing, the cells were incubated with cortisone (10 μM), the substrate of 11β-HSD1, for 6 hours. The conditioned culture medium was then collected for cortisol measurement with a cortisol ELISA kit (#500360, Cayman chemical, Ann Arbor, MI).
Quantitative real-time polymerase chain reaction and western blotting
The abundance of HSD11B1 (encoding 11β-HSD1), PTGS2 (encoding COX-2), IL1B (encoding IL-1β), IL6 (encoding IL-6), NFKBIA (encoding IκB-α) and GAPDH was determined with qRT-PCR. The relative mRNA abundance was quantitated using the 2-△△Ct method. The primer sequences used for qRT-PCR were illustrated in Supplementary Table 1. The abundance of 11β-HSD1, IκB-α, COX-2, total p65, phosphorylated p65 at Ser536 (p-p65), total STAT3, phosphorylated STAT3 at Tyr705 (p-STAT3) and GAPDH was determined with Western blotting. The antibody information was detailed in Supplementary Table 2. The bands were visualized using a G-BOX iChemi Chemiluminescence Image Capture system (Syngene, Cambridge, U.K.). The ratio of band intensities of 11β-HSD1, IκB-α, COX-2 or total p65, over that of GAPDH was used to indicate the target protein abundance. The ratio of phosphorylated protein over total protein was used to indicate the level of protein phosphorylation. Detailed methods of qRT-PCR and Western blotting have been described previously (49).
Animal study
Animal study was conducted following a standard protocol for animal care which was approved by Department of Laboratory Animal Science, Tongji University. C57BL/6 mice (Ziyuan, Hangzhou, China) aging from 10 to 13 weeks were mated overnight. The presence of a vaginal plug next morning was counted as gestational day 0.5 (GD 0.5). Pregnant mice were given LPS (from E. coli serotype 0111:B4, 20 μg per dam (50)) with or without corticosterone (5 μM) intraperitoneally on GD 16.5. An equal volume of vehicle was given as control. Some mice were allowed to delivery spontaneously and euthanized after observing the pregnancy outcomes. Some mice were sacrificed for collection of amniotic fluid, fetal membranes and fetal organs (lungs and brains). All mice were euthanized by dislocation of cervical vertebrae after administration of anesthetics (Zoletil 50, 50mg/kg, Virbac, Carros, France). The mice were housed and experimented at the Department of Laboratory Animal Science, Tongji University. The fetal lung was stained with hematoxylin-Eosin (HE), and the fetal brain was stained with immunohistochemistry for ionized calcium binding adaptor molecule 1 (Iba1) for examination of injury. The number of alveoli in three visual fields was counted under a 20x objective and averaged, which was used to evaluate the degree of lung injury. Iba1 expression in frozen brain sections was examined by immunohistochemistry as described previously (24). Fetal organs including fetal brains and fetal membranes, and amniotic fluid were frozen in liquid nitrogen for protein extraction for measurements of pro-inflammatory cytokines (IL-1β and IL-6) and Iba1 with enzyme immunoassay kits (Abclonal, Wuhan, China) and Western blotting respectively.
Statistical analysis
All data are reported as means ± SEM. The number of experiments for human amnion fibroblasts represents cell preparations from different subjects. After normality examination, paired Student’s t-test or one-way ANOVA followed by the Newman-Keuls multiple comparison test was performed where appropriate. Significance was set at P < 0.05.
Results
Increased 11β-HSD1 expression in the human amnion in chorioamnionitis
Previously, we have demonstrated that 11β-HSD1 expression is significantly increased in the human amnion in TL without infection (24, 33). Here, we compared 11β-HSD1 abundance in the amnion tissue between the PL group with infection and the TL group without infection. The demographic and clinical features of recruited subjects for this study were illustrated in Table 1. The results showed that 11β-HSD1 abundance was further increased in the human amnion in preterm birth with either Gram-negative bacteria or other pathogen infection when compared with that in normal spontaneous labor at term without infection (Figure 1). Further analysis showed that the increase was more pronounced in infection with Gram-negative bacteria than infection with other microorganisms (Figure 1). Detailed information on the pathogen detected in preterm birth was shown in Supplementary Table 3. Since the expression of 11β-HSD1 increases with gestational age (23), the higher expression of 11β-HSD1 in the amnion in the PL group with infection than the TL group without infection indicates that infection may be a further inducer of 11β-HSD1 expression in addition to the labor process per se.
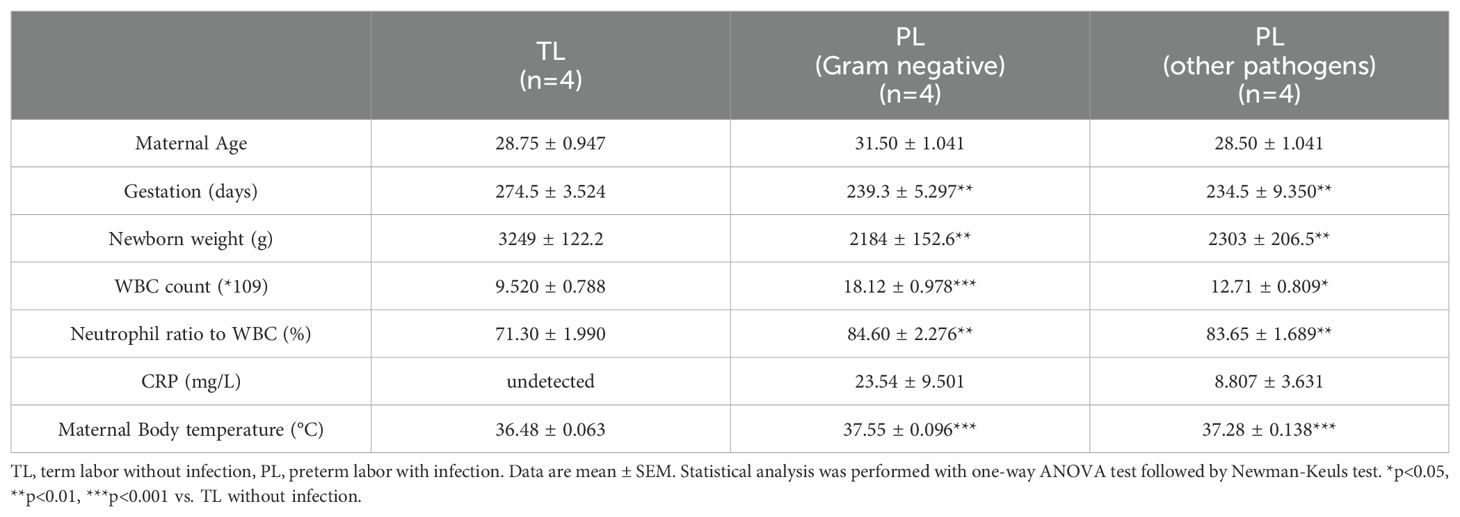
Table 1. Demographic and clinical characteristics of recruited pregnant women.
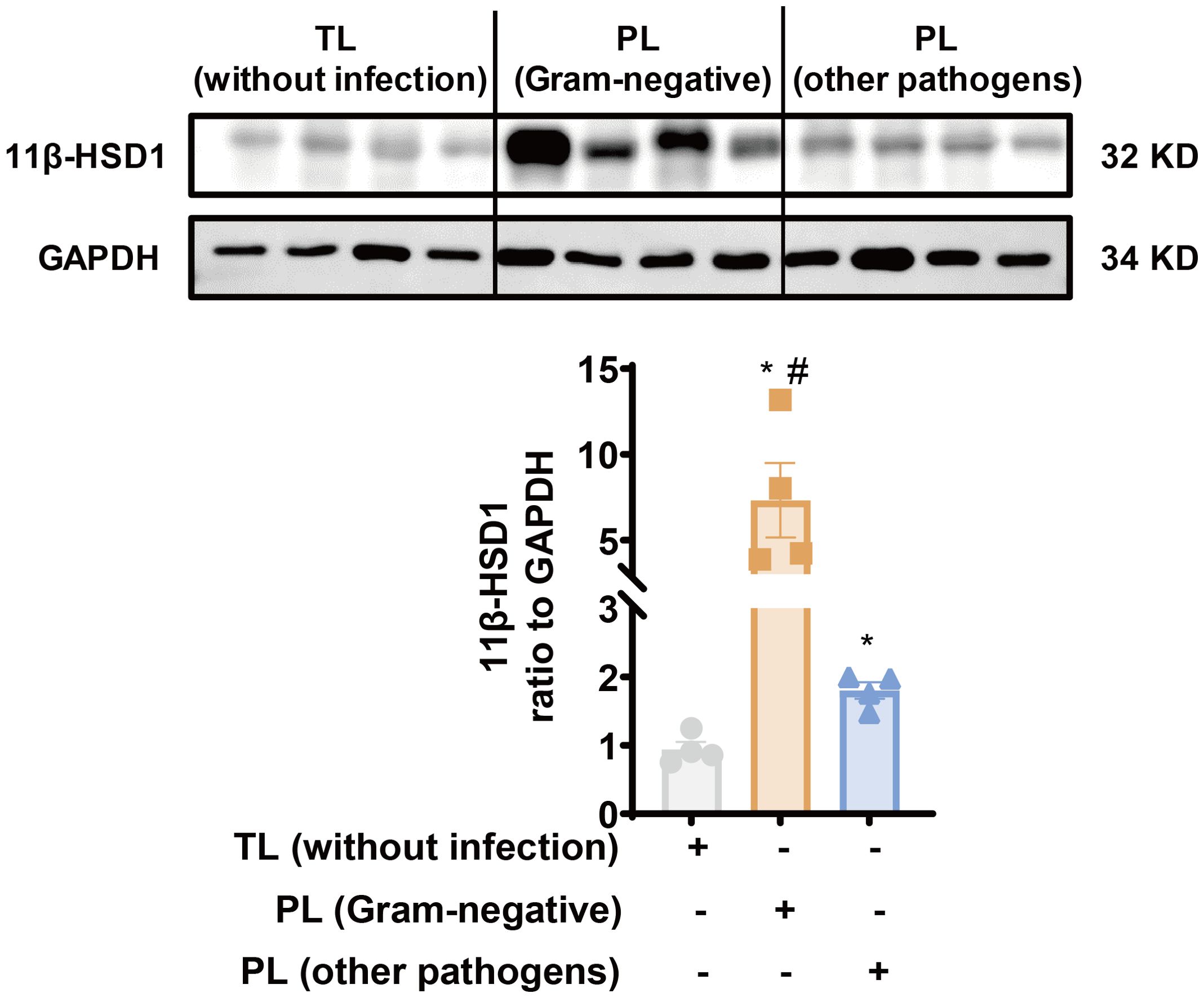
Figure 1. Increased 11β-HSD1 expression in the human amnion in infection-induced chorioamnionitis. Western blotting analysis showing increased 11β-HSD1 levels in the human amnion in either PL with Gram-negative bacterial infection (n=4) or PL with other pathogen infection (n=4) as compared to TL without infection (n=4). A more pronounced increase was observed in PL with Gram-negative bacterial infection. Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test. *p<0.05 vs. TL without infection, #p<0.05 vs. PL (other pathogens).
Synergistic induction of 11β-HSD1 expression by LPS and glucocorticoids in human amnion fibroblasts
Next, we examined whether LPS of the gram-negative bacteria could induce 11β-HSD1 expression synergistically with glucocorticoids. We found that both cortisol (1 μM) and LPS (10 ng/mL) induced 11β-HSD1 mRNA, protein and cortisol regeneration in human amnion fibroblasts (Figures 2A, B). Moreover, the induction was synergistic when cortisol and LPS were combined (Figures 2A, B). Although cortisone (10 μM) had no effect on 11β-HSD1 mRNA and protein on its own, combination of cortisone and LPS also induced 11β-HSD1 mRNA and protein synergistically, which was significantly attenuated by an 11β-HSD1 inhibitor 10-J (1 μM) (Figure 2C), suggesting that the enhancement required the conversion of cortisone to cortisol by 11β-HSD1. These results indicate that further enhancement of cortisol regeneration in preterm birth with infection is ascribed at least in part to the synergistic induction of 11β-HSD1 by LPS and glucocorticoids.
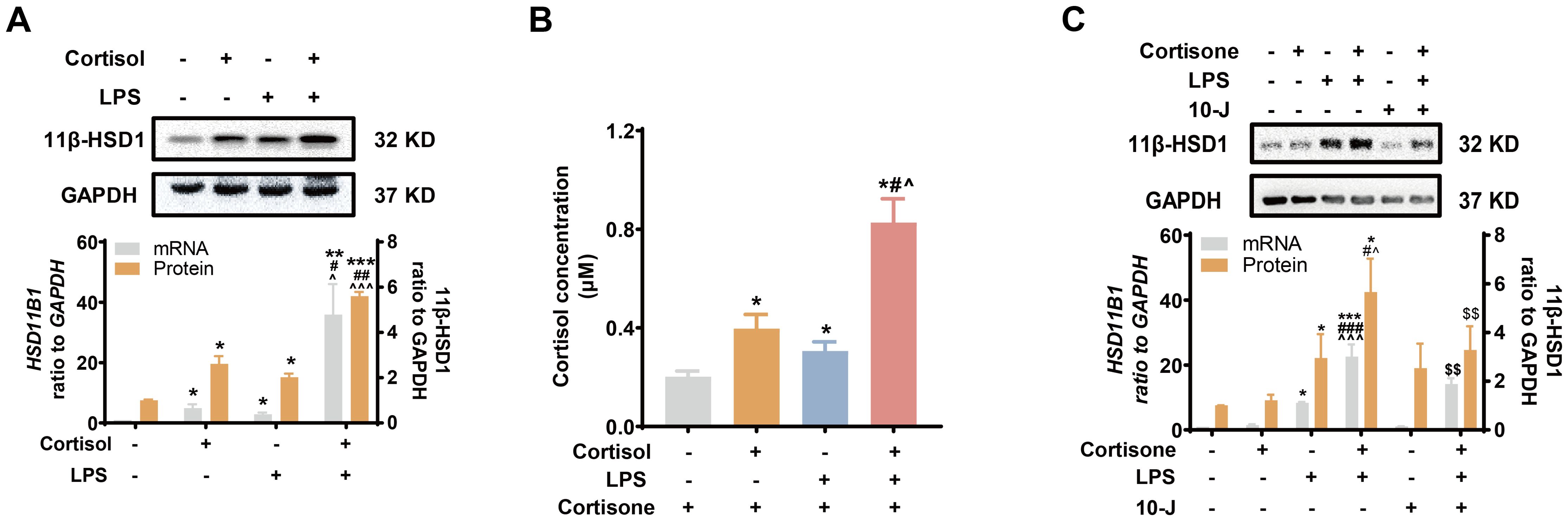
Figure 2. Synergistic induction of 11β-HSD1 expression by LPS and glucocorticoids in human amnion fibroblasts. (A, B) Effect of cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours) on 11β-HSD1 expression and cortisol regeneration (n=4). For the study of cortisol regeneration, the cells were pretreated with cortisol (1 μM) in the presence or absence of LPS (10 ng/mL) for 24 hours. After removal of the treatments by washing, the cells were incubated with cortisone (10 μM), the substrate of 11β-HSD1, for 24 hours. The conditioned culture medium was then collected for cortisol measurement. (C) Effect of 11β-HSD1 inhibitor 10-J (1 μM; 24 hours) on the synergistic induction of 11β-HSD1 mRNA (n=4) and protein (n=5) by cortisone (10 μM; 24 hours) and LPS (10 ng/mL; 24 hours). Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test. *p<0.05, **p<0.01, ***p<0.001 vs. control (without treatment), #p<0.05, ##p<0.01, ###p<0.001 vs. cortisol or cortisone, ^p<0.05, ^^^p<0.001 vs. LPS, $$p<0.01 vs. cortisol or cortisone plus LPS.
Effect of glucocorticoids on the induction of COX-2 and pro-inflammatory cytokines by LPS in human amnion fibroblasts
LPS (10 ng/mL) induced both pro-inflammatory cytokines (IL-1β and IL-6) and COX-2 drastically in amnion fibroblasts (Figures 3A–E), both of which were inhibited by cortisol (1 μM) (Figures 3A, B, D) or cortisone (10 μM) (Figures 3C, E). The inhibition by cortisone could be blocked by 10-J (1 μM) (Figures 3C, E), indicating again that the effect required the conversion of cortisone to cortisol by 11β-HSD1. Cortisol (1 μM) or cortisone (10 μM) alone significantly decreased pro-inflammatory cytokines (IL-1β and IL-6) but increased COX-2 levels (Figures 3A–E) though the effect of cortisone was not as great as that of cortisol (Figures 3C, E). Notably, although combination of LPS with cortisol or cortisone significantly suppressed the induction of COX-2 by LPS, the suppression was incomplete, and the remaining increase was about the same level as the induction by cortisol or cortisone alone (Figures 3D, E). However, the same concentration of cortisol or cortisone completely blocked the induction of pro-inflammatory cytokines by LPS (Figures 3A–C). These results suggest that a differential mechanism may exist in the induction of COX-2 by LPS and cortisol in human amnion fibroblasts.
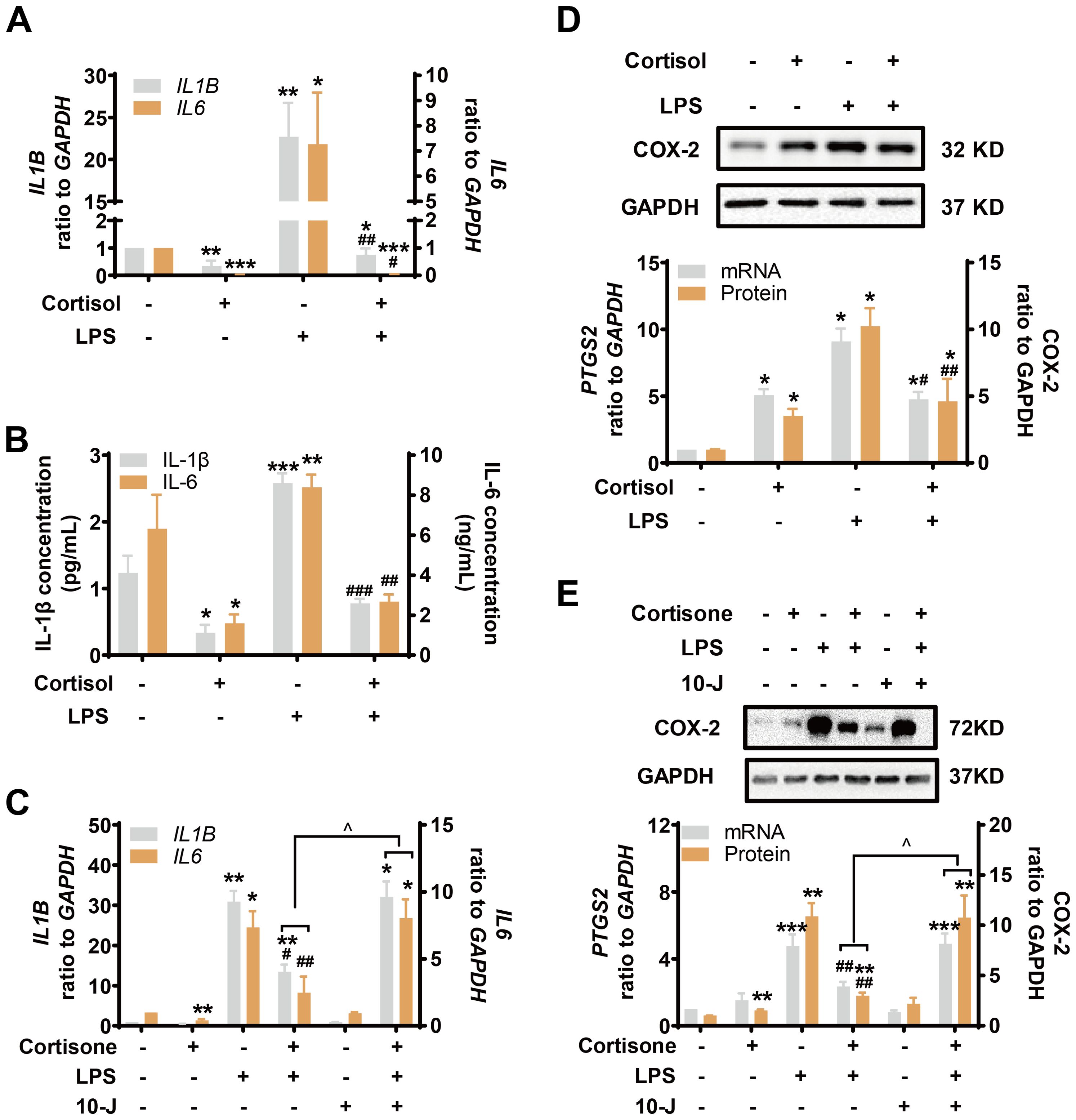
Figure 3. Simultaneous induction of COX-2 and inhibition of pro-inflammatory cytokines by glucocorticoids in the presence and absence of LPS in human amnion fibroblasts. (A, B) Effect of cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours) on IL-1β and IL-6 mRNA expression (A) and secretion (B) (n=3). (C) Effect of cortisone (10 μM; 24 hours), LPS (10 ng/mL; 24 hours) and 11β-HSD1 inhibitor 10-J (1 μM; 24 hours) on IL-1β and IL-6 mRNA (n=4). (D) Effect of cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours) on COX-2 mRNA and protein (n=4). (E) Effect of cortisone (10 μM; 24 hours), LPS (10 ng/mL; 24 hours) and 11β-HSD1 inhibitor, 10-J (1 μM; 24 hours) on COX-2 mRNA (n=4) and protein (n=5). Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test. *p<0.05, **p<0.01, ***p<0.001 vs. control (without treatment), #p<0.05, ##p<0.01, ###p<0.001 vs. LPS, ^p<0.05 vs cortisone plus LPS.
Role of the TLR-4/NF-κB and STAT3 pathways in the effects of LPS and glucocorticoids in human amnion fibroblasts
Inhibition of TLR-4, the receptor mediating the effect of LPS, with TAK-242 (10 μM) blocked the induction of COX-2 and pro-inflammatory cytokines by LPS (10 ng/mL), but not the induction of COX-2 by cortisol alone (1 μM) or the remaining induction of COX-2 by the combination of LPS and cortisol in human amnion fibroblasts (Figure 4A). NF-κB, which is normally sequestered by IκB protein in the cytoplasm, plays a central role in the mediation of infection-induced inflammation upon liberation from IκB (40). Small interference RNA-mediated knock-down of the p65 subunit of NF-κB significantly inhibited the induction of pro-inflammatory cytokines (Figure 4C) and COX-2 by LPS (10 ng/mL), but not the induction of COX-2 by cortisol (1 μM) (Figure 4B). Of interest, both cortisol (1 μM) and LPS (10 ng/mL) increased NFKBIA (encoding IκB-α) mRNA in human amnion fibroblasts (Figure 4D). However, cortisol (1 μM) increased while LPS (10 ng/mL) decreased IκB-α protein levels, and the inhibition by LPS could be blocked by cortisol (1 μM) (Figure 4E). Moreover, the basal and LPS (10 ng/mL)-induced p65 phosphorylation, an indicator of NF-κB activation, were also inhibited by cortisol (1 μM) (Figure 4F). By contrast, inhibition of STAT3 with S3I-201 (50 μM) blocked not only the induction of COX-2 by cortisol alone (1 μM), but also the remaining induction of COX-2 expression with the combination of LPS and cortisol (Figure 5A), but not the induction by LPS (10 ng/mL) alone (Figure 5A). Consistently, cortisol (1 μM) increased STAT3 phosphorylation (Figure 5B). These results indicate that the TLR-4/NF-κB pathway mediates the induction of COX-2 and pro-inflammatory cytokines by LPS, and STAT3 pathway mediates the induction of COX-2 by cortisol alone. The latter may account for the remaining induction of COX-2 with the combination of cortisol and LPS.
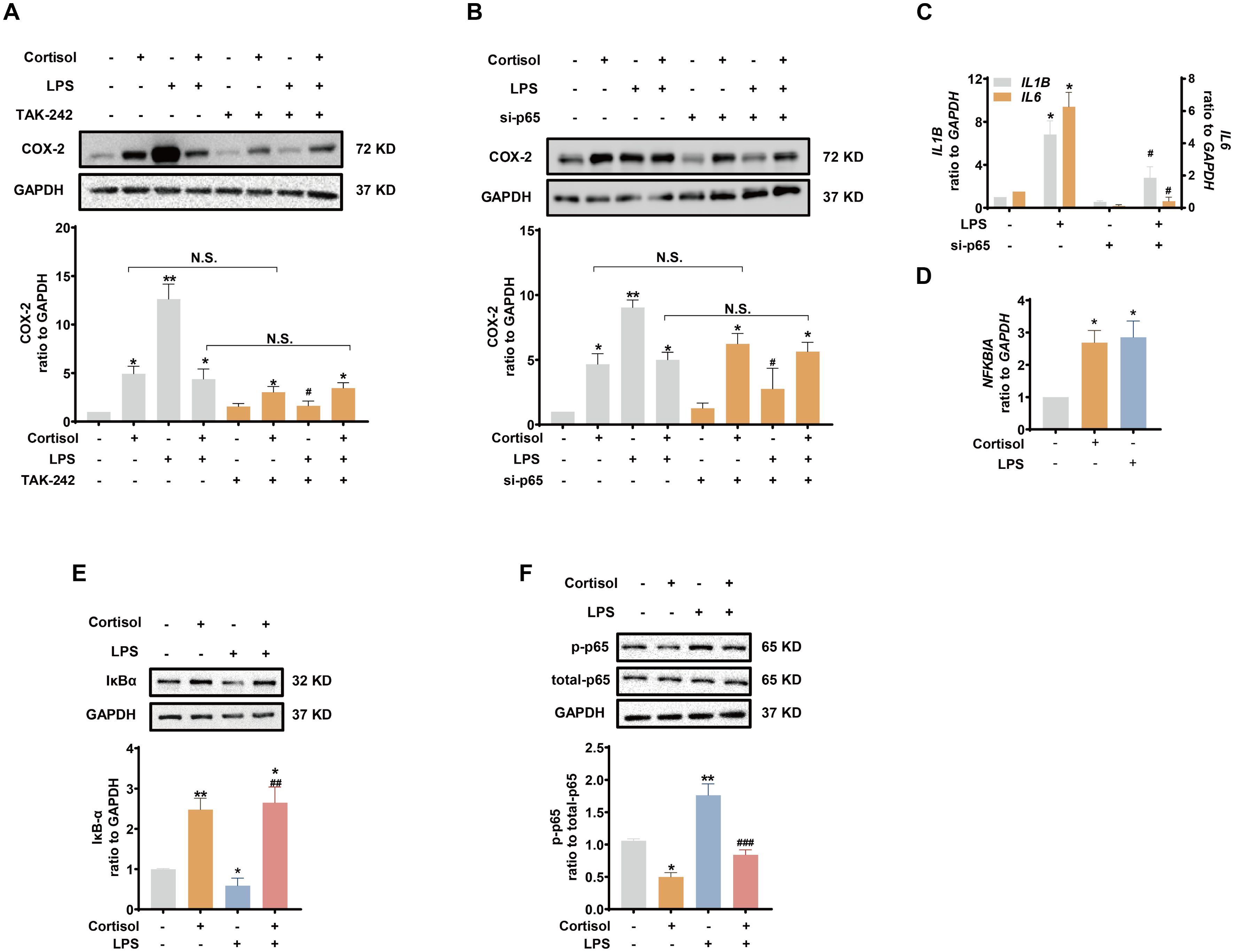
Figure 4. Involvement of NF-κB in the attenuation of LPS-induced pro-inflammatory cytokines and COX-2 expression by glucocorticoids in human amnion fibroblasts. (A, B) Effect of TLR-4 inhibitor TAK-242 (10 μM; 24 hours) (A, n=4) and siRNA-mediated knockdown of p65 (si-p65) (B, n=3) on the induction of COX-2 by cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours). (C) Effect of si-p65 on the induction of IL1B and IL6 mRNA by LPS (10 ng/mL; 24 hours) (n=4). (D, E) Effect of cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours) on NFKBIA mRNA (D) and IκB-α protein (E) abundance (n=3). (F) Effect of cortisol (1 μM; 3 hours) and LPS (10 ng/mL; 3 hours) on p65 phosphorylation at ser536 (p-p65) (n=3). Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls s test. *p<0.05, **p<0.01 vs. control (without treatment), #p<0.05, ##p<0.01, ###p<0.001 vs. LPS. N.S., none significance.
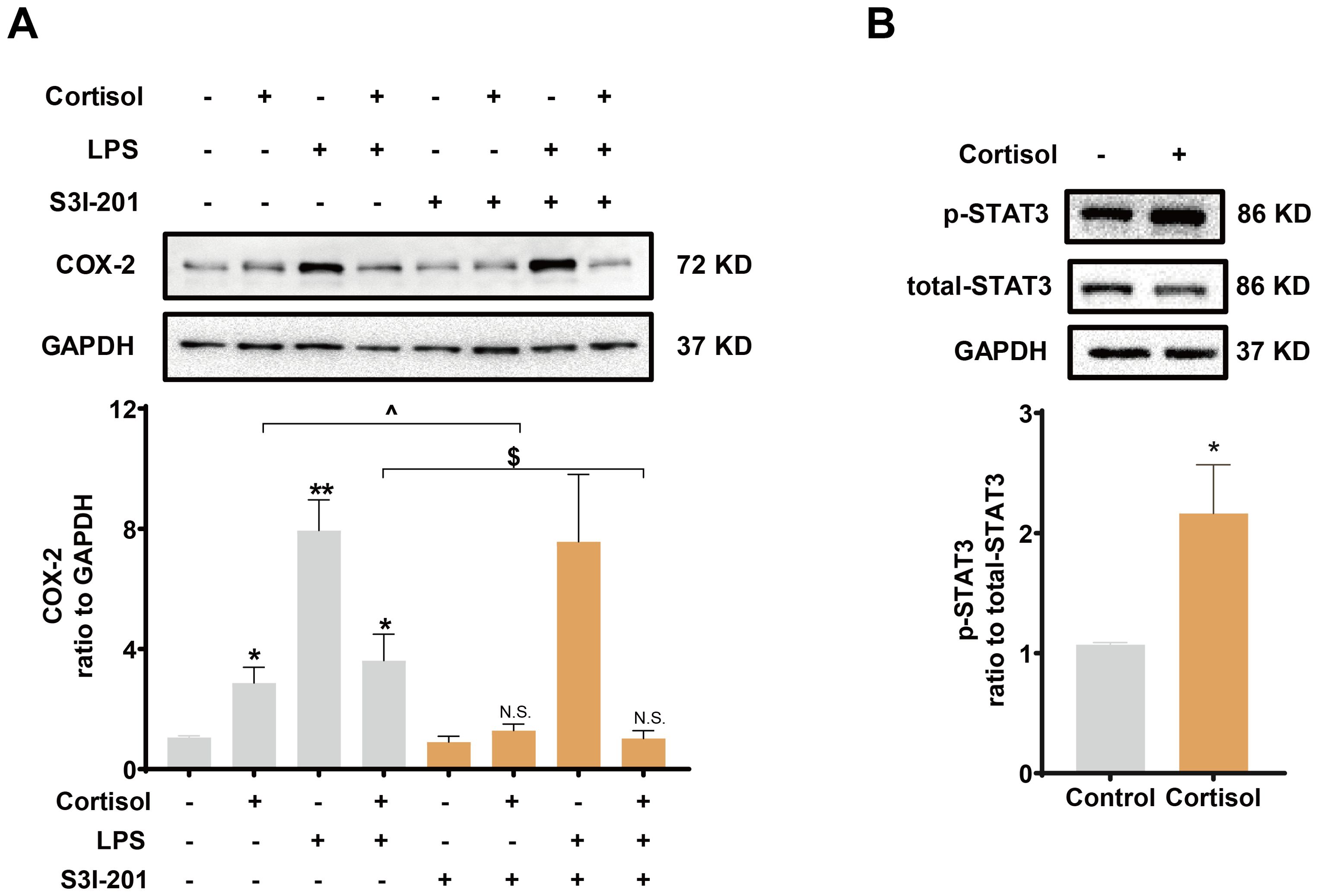
Figure 5. Role of STAT3 in the induction of COX-2 by cortisol in the presence or absence of LPS in human amnion fibroblasts. (A) Effect of STAT3 inhibitor S3I-201 (50 μM; 24 hours; n=5) on the induction of COX-2 by cortisol (1 μM; 24 hours) and LPS (10 ng/mL; 24 hours). (B) Effect of cortisol (1 μM; 3 hours) on STAT3 phosphorylation at Tyr705 (p-STAT3) (n=4). Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test (A) or paired Student’s t-test (B). *p<0.05, vs. control (without treatment), ^p<0.05, $p<0.05 between lined groups, N.S., none significance vs. control.
Corticosterone ameliorates fetal organ injury induced by LPS in pregnant mice
LPS (20 μg per dam) with or without corticosterone (1 μg per dam) was administered intraperitoneally to pregnant mice on GD 16.5 to mimic intrauterine Gram-negative bacteria infection with or without glucocorticoid treatment (Figure 6A). Preterm delivery occurred in all mice about 20 hours after LPS injection (Figure 6B). Corticosterone co-treatment failed to change the course of preterm birth induced by LPS (Figure 6B), but alleviated LPS-induced fetal organ damages (Figures 6C–F). Stereomicroscopic examination of the fetal lung and brain showed that LPS caused bleeding and edema in both organs (Figure 6C). Microscopic examination of HE-stained fetal lung section showed a reduction in alveoli along with collapsed bronchial wall with LPS administration (Figures 6D, E). Immunohistochemical staining and Western blotting showed increased Iba-1 abundance, a marker of brain response to injury (51–53), in the fetal brain with LPS administration (Figures 6D, F). Corticosterone co-treatment alleviated those lesions and the increase in Iba-1 caused by LPS (Figures 6C–F).
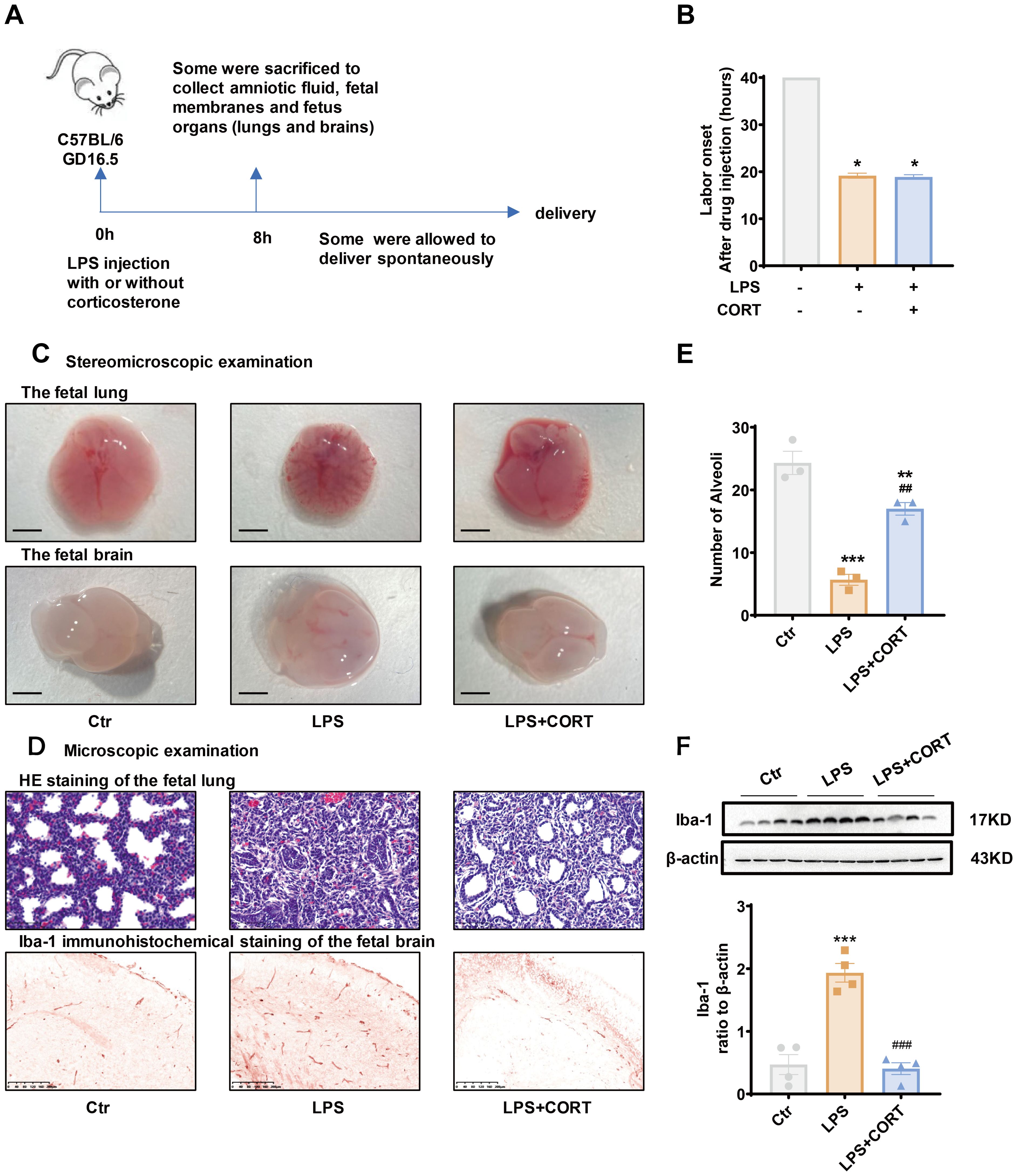
Figure 6. Effect of corticosterone on LPS-induced preterm birth and fetal organ damage in pregnant mice. (A) Diagram illustrating experiment procedure in pregnant mice. (B) Effect of corticosterone (1 μg per dam) and LPS (20 μg per dam) on the onset of labor. (C) Stereomicroscopic examination of the fetal lung and brain following injection of LPS (20 μg per dam) and corticosterone (1 μg per dam). Scale bars, 3mm. (D) Microscopic examination of the fetal lung with hematoxylin-eosin (HE) staining and the fetal brain with Iba-1 immunohistochemical staining following injection of LPS (20 μg per dam) and corticosterone (1 μg per dam). Scale bars, 200μm. (E) Number of alveoli per field of view in the fetal lung with HE staining (n =3). (F) Western blotting showing the effect of LPS (20 μg per dam) and corticosterone (1 μg per dam) on Iba-1 abundance in the fetal brain (n =4). GD, gestational days. CORT, corticosterone. Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test. *p<0.05, **p<0.01, ***P<0.001 vs. control (without treatment), ##p<0.01, ###p<0.001 vs. LPS.
ELISA showed that increases in IL-1β and IL-6 in the amniotic fluid and fetal membranes induced by LPS were blocked by corticosterone co-treatment (Figure 7A). By contrast, the increase in 11β-HSD1 induced by LPS in the fetal membranes was enhanced by corticosterone co-treatment (Figures 7B, C). Although corticosterone co-treatment also suppressed the induction of COX-2 by LPS in the fetal membranes, only partial suppression was observed (Figures 7B, D). The remaining COX-2 induction with LPS and corticosterone co-treatment was still significant compared to the control group, which may be attributed to the induction by corticosterone per se. In line with the notion, the induction of p65 phosphorylation in the fetal membranes that mediates the induction of pro-inflammatory cytokine and COX-2 expression by LPS, was completely blocked by corticosterone co-treatment, while STAT3 phosphorylation, a major mediator of COX-2 expression by glucocorticoids, remained in the LPS and corticosteroid co-treatment group (Figures 7B, E, F).
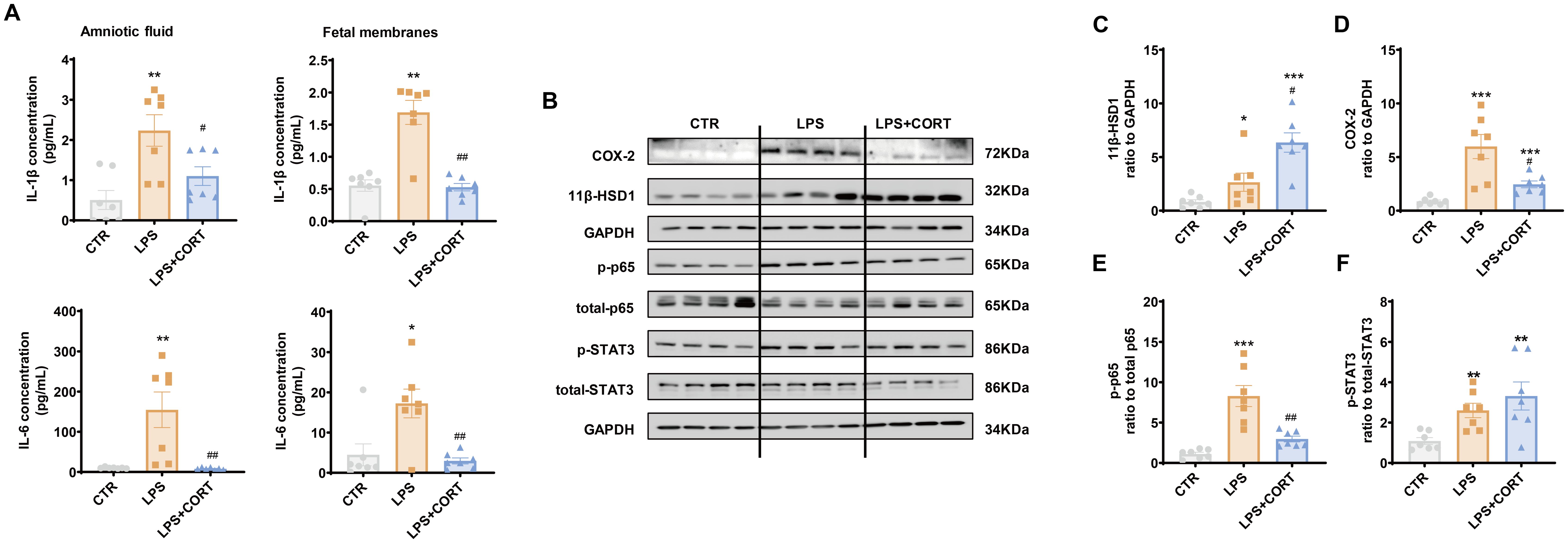
Figure 7. Effect of LPS and corticosterone injection on the abundance of pro-inflammatory cytokines, COX-2, 11β-HSD1, p-p65 and p-STAT3 in mouse intrauterine tissues. (A) Effect of LPS (20 μg per dam) and corticosterone (1 μg per dam) injection on IL-1β and IL-6 abundance in the amnion fluid and fetal membranes (n = 7 each group). (B–F) Effect of LPS (20 μg per dam) and corticosterone (1 μg per dam) injection on 11β-HSD1, COX-2, p65 phosphorylation at Ser536 (p-p65) and STAT3 phosphorylation at Tyr705 (p-STAT3) in the fetal membranes (n=7 each group). Data are mean ± SEM. Statistical analysis was performed with one-way ANOVA test followed by Newman-Keuls test. *p<0.05, **p<0.01, ***P<0.001 vs. control (without treatment), #p<0.05, ##p<0.01 vs. LPS.
Discussion
The inner amnion layer of the fetal membranes, being the most resilient layer, plays a vital role in maintaining the integrity of the fetal membranes and sterile intrauterine environment. Beyond its physical barrier function, the amnion is also a source of a variety of bioactive substances that contribute to parturition and immunomodulation (20, 54, 55). In this study, we demonstrated that increased glucocorticoid regeneration by 11β-HSD1 in the amnion layer of the fetal membranes might play a dual role in parturition. On the one hand, glucocorticoid accumulation may promote parturition through stimulation of prostaglandin synthesis via induction of COX-2 expression by a NF-κB-independent pathway such as STAT3 as demonstrated in this study as well as in previous studies (24, 26, 37–39). On the other hand, glucocorticoids may also restrain the production of pro-inflammatory cytokines through a NF-κB-dependent pathway to avoid excessive pro-inflammatory cytokine production at parturition. Of interest, we found that 11β-HSD1 expression could be further enhanced when infection was present, particularly in Gram-negative bacteria infection. LPS of the Gram-negative bacteria could induce 11β-HSD1 expression synergistically with glucocorticoids. These results indicate that the pro-parturition and anti-inflammatory actions of glucocorticoids in the fetal membranes may be further strengthened when infection is present. We believe that this synergistic enhancement of glucocorticoid regeneration is a self-protective measure of the fetus threatened by in-utero infection so that brakes can be applied in time to prevent excessive pro-inflammatory cytokine production while not affecting the timing of labor for the safety of the fetus.
In addition to infection-induced chorioamnionitis, sterile inflammation also exists in the fetal membranes in normal parturition with increased expression of pro-inflammatory cytokines including IL-1β and TNF-α (56). Our previous studies have demonstrated that these pro-inflammatory cytokines can synergize with cortisol to induce 11β-HSD1 expression in the fetal membranes as well (28, 30), indicating that cortisol regeneration may also play a dual role in in normal parturition.
Our previous studies have shown that the induction of COX-2 by glucocorticoids in human amnion fibroblasts involves the interaction of GR with a myriad of transcription factors including STAT3, CREB, and C/EBPδ (24, 26, 57). In this study, we demonstrated again that STAT3 was involved in the induction of COX-2 by glucocorticoids in human amnion fibroblasts, and inhibition of STAT3 blocked the induction of COX-2 by glucocorticoids either on its own or in combination with LPS. Because inhibition of STAT3 was unable to block the induction of COX-2 by LPS, we believe that the remaining induction of COX-2 with the combination of cortisol and LPS is due to the induction by cortisol per se through a NF-κB-independent pathway rather than due to the incomplete blockade of LPS-induced COX-2 by cortisol. These NF-κB-independent labor-inducing actions of glucocorticoids may remain to help the fetus deliver in time so that detrimental effects of excessive pro-inflammatory cytokines on the fetus can be avoided at parturition. Together with the well-described effects of glucocorticoids on fetal organ maturation (58–60), these actions of glucocorticoids would increase the likelihood of fetal survival when infection-induced preterm birth is inevitable.
In addition to COX-2 induction, glucocorticoid regeneration may also promote membrane rupture at parturition by inhibiting collagen cross-linking, inducing collagen cleavage via an NF-κB-independent pathway in human fetal membranes (32–34). Moreover, glucocorticoids may also promote parturition by stimulating estrogen synthesis and progesterone withdrawal via induction of aromatase and 20α-hydroxysteroid dehydrogenase (20α-HSD) expression in human placenta and fetal membranes respectively (25, 61, 62). These pro-parturition effects of glucocorticoids may offset the parturition-delaying effect brought on by the anti-inflammatory effect of glucocorticoids, which may explain why preterm birth still occurred in the dam that received co-administration of LPS and corticosterone in this study. However, the LPS-induced damage on the fetal organs was greatly alleviated by corticosterone treatment, which is more likely to be a benefit of the anti-inflammatory action of glucocorticoids either regenerated endogenously in intrauterine tissues or from glucocorticoid treatment.
As we demonstrated previously, cortisol not only induced prostaglandin synthesis but also ECM remodeling and progesterone withdrawal in the fetal membranes (25, 32–34, 37–39). Although cortisol regeneration in the fetal membranes has been shown to be implicated in these pivotal events of parturition, it is intriguing that systemic administration of glucocorticoids, particularly dexamethasone, often fails to affect gestational age, which may be attributed to the potent negative feedback effects of systemic administration of glucocorticoids on maternal and fetal hypothalamus-pituitary-adrenal axes. These negative feedback effects may obscure the pro-laboring effects of endogenous steroids synthesized by the maternal and fetal adrenals. However, these complicated effects of systemic administration of glucocorticoids do not necessarily mean that cortisol regenerated locally in the fetal membranes plays no role in parturition. As a matter of fact, clinical observations showed that intra-amniotic injection of glucocorticoids indeed shortened gestational age (63–66).
In addition to glucocorticoids, there are other factors such as galectin-1, prolactin, progesterone, and IL-10 have been reported to inhibit infection-induced inflammatory responses in the intrauterine tissues (67–70). It would be of interest to investigate the interaction of cortisol with these factors in the fetal membranes. In patients with nasal polyps, glucocorticoid treatment has been shown to enhance galectin-1 expression in their surgical resection (71). We are not sure whether this interaction also exists in the fetal membranes. In the case of progesterone, the situation is more complicated as progesterone also carries other pregnancy-maintaining actions in addition to its anti-inflammatory actions. In the fetal membranes, cortisol has been shown to induce progesterone functional withdrawal by upregulating its degradation enzyme 20α-HSD (25).
In summary, our study illustrates that glucocorticoid regeneration is implicated in both pro-parturition and anti-inflammatory actions in the fetal membranes, which may minimize fetus-hazardous effects imposed by pro-inflammatory cytokines at parturition (Figure 8), and help the fetus escape from the in-utero environment affluent in pro-inflammatory cytokines at parturition. This dual role of glucocorticoid regeneration in the fetal membranes may be of particular importance for the survival of the fetus in infection-induced chorioamnionitis. However, it should be kept in mind that over inhibition of the immune responses by glucocorticoids may also exacerbate bacterial infection. For the safety and survival of the preterm baby under conditions of chorioamnionitis, it is important to deliver the baby timely with functional lungs. In consideration of the pro-laboring and lung maturing effects of glucocorticoids, it is may be beneficial to give glucocorticoids along with adequate antibiotics when the fetus is endangered by intrauterine infection. Therefore, it presents a tricky task to balance these effects of glucocorticoids in infection-induced chorioamnionitis in the future.
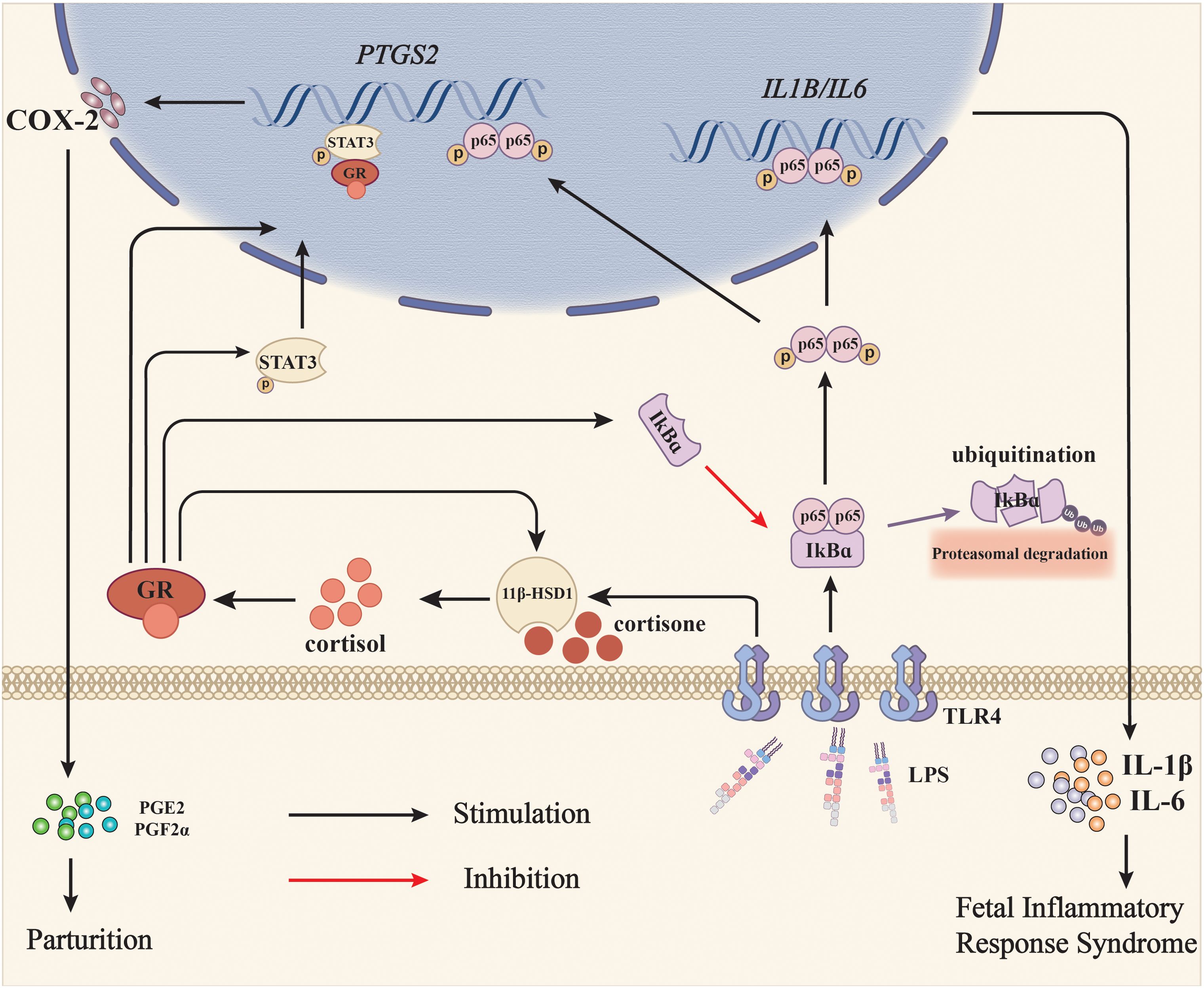
Figure 8. Diagram illustrating the role of glucocorticoid regeneration of the fetal membranes in parturition with infection-induced chorioamnionitis. When intrauterine infection is present in pregnancy, LPS from Gram-negative bacteria activates NF-κB via the TLR4 receptor, which subsequently induces the expression of pro-inflammatory cytokines such as IL-1β and IL-6, and COX-2, the rate-limiting enzyme in prostaglandin synthesis. Although prostaglandins synthesized by COX-2 are pro-parturition, excessive pro-inflammatory cytokines may harm the fetus and lead to fetal inflammatory response syndrome (FIRS). Meanwhile, LPS also induces 11β-HSD1 expression either by itself or in synergy with glucocorticoids, resulting in increased conversion of biologically inactive cortisone to bioactive cortisol. Cortisol not only suppresses LPS-induced pro-inflammatory cytokine and COX-2 expression via inhibition of NF-κB, but also induces COX-2 expression on its own through NF-κB-independent pathways such as activation of STAT3, thereby minimizing the fetus-hazardous effects of pro-inflammatory cytokines at parturition and helping the fetus escape from the in-utero environment affluent in pro-inflammatory cytokines at the same time. Black arrows indicate stimulation and the red arrow indicates inhibition.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the ethics committee of Shanghai First Maternal and Infant Hospital, Tongji University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by Department of Laboratory Animal Science, Tongji University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
L-JL: Writing – original draft, Investigation, Methodology, Validation. QZ: Writing – original draft, Resources. FZ: Investigation, Methodology, Writing – original draft. W-JL: Methodology, Writing – original draft. M-DL: Investigation, Writing – original draft. J-WL: Investigation, Writing – original draft. W-SW: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. KS: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. HY: Funding acquisition, Project administration, Resources, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by National Key R & D Program of China (2022YFC2704602); National Natural Science Foundation of China (81830042, 82271717 and 82271719); Shanghai Municipal Science and Technology Commission Research Fund (21140903800); Program of Shanghai Academic/Technology Research Leader (23XD1402700); Innovative Research Team of High-Level Local Universities in Shanghai (SHSMU-ZLCX20210201); Three-Year Action Plan for Strengthening the Construction of the Public Health System in Shanghai (GWVI-11.1-36); and Shanghai’s Top Priority Research Center Construction Project (2023ZZ02002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1459489/full#supplementary-material
Abbreviations
11β-HSD1, 11β-hydroxysteroid dehydrogenase 1; COX-2, cyclooxygenase 2; DMEM, Dulbecco’s modified Eagle’s medium; ECM, extracellular matrix; FBS, Fetal Bovine Serum; FIRS, fetal inflammatory response syndrome; GCs, Glucocorticoids; GD, gestational day; GR, glucocorticoid receptor; HE, hematoxylin-Eosin; Iba1, ionized calcium binding adaptor molecule 1; IL-1β, interleukin-1β; IL-6, interleukin-6; IκB-α, NFκB inhibitor alpha; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; TNL, term none labor without infection; TL, term labor without infection; PL, preterm labor with infection; qRT-PCR, quantitative real-time polymerase chain reaction; TLR4, toll-like receptor 4; 20α-hydroxysteroid dehydrogenase, 20α-HSD.
References
1. Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel LA, Nien JK. Inflammation in preterm and term labour and delivery. Semin fetal neonatal Med. (2006) 11:317–26. doi: 10.1016/j.siny.2006.05.001
2. Menon R. Fetal inflammatory response at the fetomaternal interface: A requirement for labor at term and preterm. Immunol Rev. (2022) 308:149–67. doi: 10.1111/imr.13075
3. Menon R, Behnia F, Polettini J, Richardson LS. Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes. Semin immunopathology. (2020) 42:431–50. doi: 10.1007/s00281-020-00808-x
4. Romero R, Espinoza J, Kusanovic JP, Gotsch F, Hassan S, Erez O, et al. The preterm parturition syndrome. BJOG: an Int J obstetrics gynaecology. (2006) 113 Suppl 3:17–42. doi: 10.1111/j.1471-0528.2006.01120.x
5. Galinsky R, Polglase GR, Hooper SB, Black MJ, Moss TJ. The consequences of chorioamnionitis: preterm birth and effects on development. J pregnancy. (2013) 2013:412831. doi: 10.1155/2013/412831
6. Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am J obstetrics gynecology. (2015) 213:S29–52. doi: 10.1016/j.ajog.2015.08.040
7. Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, et al. The fetal inflammatory response syndrome. Clin obstetrics gynecology. (2007) 50:652–83. doi: 10.1097/GRF.0b013e31811ebef6
8. Romero R, Savasan ZA, Chaiworapongsa T, Berry SM, Kusanovic JP, Hassan SS, et al. Hematologic profile of the fetus with systemic inflammatory response syndrome. J perinatal Med. (2011) 40:19–32. doi: 10.1515/JPM.2011.100
9. Korzeniewski SJ, Romero R, Cortez J, Pappas A, Schwartz AG, Kim CJ, et al. A "multi-hit" model of neonatal white matter injury: cumulative contributions of chronic placental inflammation, acute fetal inflammation and postnatal inflammatory events. J perinatal Med. (2014) 42:731–43. doi: 10.1515/jpm-2014-0250
10. Lu HY, Zhang Q, Wang QX, Lu JY. Contribution of histologic chorioamnionitis and fetal inflammatory response syndrome to increased risk of brain injury in infants with preterm premature rupture of membranes. Pediatr neurology. (2016) 61:94–8.e1. doi: 10.1016/j.pediatrneurol.2016.05.001
11. Jung E, Romero R, Yeo L, Diaz-Primera R, Marin-Concha J, Para R, et al. The fetal inflammatory response syndrome: the origins of a concept, pathophysiology, diagnosis, and obstetrical implications. Semin fetal neonatal Med. (2020) 25:101146. doi: 10.1016/j.siny.2020.101146
12. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. (2005) 353:1711–23. doi: 10.1056/NEJMra050541
13. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. (2000) 21:55–89. doi: 10.1210/edrv.21.1.0389
14. Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. (2004) 25:831–66. doi: 10.1210/er.2003-0031
15. Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. (2013) 93:1139–206. doi: 10.1152/physrev.00020.2012
16. Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology. (2001) 142:1371–6. doi: 10.1210/endo.142.4.8114
17. Wang W, Chen ZJ, Myatt L, Sun K. 11beta-HSD1 in human fetal membranes as a potential therapeutic target for preterm birth. Endocr Rev. (2018) 39:241–60. doi: 10.1210/er.2017-00188
18. Tomlinson JW, Stewart PM. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract Res Clin Endocrinol Metab. (2001) 15:61–78. doi: 10.1053/beem.2000.0119
19. Chapman KE, Kotelevtsev YV, Jamieson PM, Williams LJ, Mullins JJ, Seckl JR. Tissue-specific modulation of glucocorticoid action by the 11 beta-hydroxysteroid dehydrogenases. Biochem Soc Trans. (1997) 25:583–7. doi: 10.1042/bst0250583
20. Murphy BE. Chorionic membrane as an extra-adrenal source of foetal cortisol in human amniotic fluid. Nature. (1977) 266:179–81. doi: 10.1038/266179a0
21. Murphy BE. Ontogeny of cortisol-cortisone interconversion in human tissues: a role for cortisone in human fetal development. J Steroid Biochem. (1981) 14:811–7. doi: 10.1016/0022-4731(81)90226-0
22. Sun K, Yang K, Challis JR. Differential expression of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes. J Clin Endocrinol Metab. (1997) 82:300–5. doi: 10.1210/jcem.82.1.3681
23. Alfaidy N, Li W, MacIntosh T, Yang K, Challis J. Late gestation increase in 11beta-hydroxysteroid dehydrogenase 1 expression in human fetal membranes: a novel intrauterine source of cortisol. J Clin Endocrinol Metab. (2003) 88:5033–8. doi: 10.1210/jc.2002-021915
24. Lu JW, Wang WS, Zhou Q, Ling LJ, Ying H, Sun Y, et al. C/EBPdelta drives key endocrine signals in the human amnion at parturition. Clin Trans Med. (2021) 11:e416. doi: 10.1002/ctm2.416
25. Lu J-W, Lei W-J, Ling L-J, Wang L-Y, Lin Y-K, Zhang F, et al. Cortisol stimulates local progesterone withdrawal through induction of AKR1C1 in human amnion fibroblasts at parturition. Endocrinology. (2022) 163:bqac148. doi: 10.1210/endocr/bqac148
26. Wang W, Guo C, Zhu P, Lu J, Li W, Liu C, et al. Phosphorylation of STAT3 mediates the induction of cyclooxygenase-2 by cortisol in the human amnion at parturition. Sci Signaling. (2015) 8:ra106. doi: 10.1126/scisignal.aac6151
27. Ohrlander S, Gennser G, Eneroth P. Plasma cortisol levels in human fetus during parturition. Obstet Gynecol. (1976) 48:381–7.
28. Sun K, Myatt L. Enhancement of glucocorticoid-induced 11beta-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology. (2003) 144:5568–77. doi: 10.1210/en.2003-0780
29. Yang Z, Guo C, Zhu P, Li W, Myatt L, Sun K. Role of glucocorticoid receptor and CCAAT/enhancer-binding protein alpha in the feed-forward induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J endocrinology. (2007) 195:241–53. doi: 10.1677/JOE-07-0303
30. Li W, Gao L, Wang Y, Duan T, Myatt L, Sun K. Enhancement of cortisol-induced 11beta-hydroxysteroid dehydrogenase type 1 expression by interleukin 1beta in cultured human chorionic trophoblast cells. Endocrinology. (2006) 147:2490–5. doi: 10.1210/en.2005-1626
31. Guller S, Kong L, Wozniak R, Lockwood CJ. Reduction of extracellular matrix protein expression in human amnion epithelial cells by glucocorticoids: a potential role in preterm rupture of the fetal membranes. J Clin Endocrinol Metab. (1995) 80:2244–50. doi: 10.1210/jcem.80.7.7608287
32. Wang LY, Wang WS, Wang YW, Lu JW, Lu Y, Zhang CY, et al. Drastic induction of MMP-7 by cortisol in the human amnion: implications for membrane rupture at parturition. FASEB J. (2019) 33:2770–81. doi: 10.1096/fj.201801216R
33. Liu C, Guo C, Wang W, Zhu P, Li W, Mi Y, et al. Inhibition of lysyl oxidase by cortisol regeneration in human amnion: implications for rupture of fetal membranes. Endocrinology. (2016) 157:4055–65. doi: 10.1210/en.2016-1406
34. Mi Y, Wang W, Zhang C, Liu C, Lu J, Li W, et al. Autophagic degradation of collagen 1A1 by cortisol in human amnion fibroblasts. Endocrinology. (2017) 158:1005–14. doi: 10.1210/en.2016-1829
35. Mi Y, Wang W, Lu J, Zhang C, Wang Y, Ying H, et al. Proteasome-mediated degradation of collagen III by cortisol in amnion fibroblasts. J Mol Endocrinol. (2018) 60:45–54. doi: 10.1530/JME-17-0215
36. Wang W, Liu C, Sun K. Induction of Amnion Epithelial Apoptosis by Cortisol via tPA/Plasmin System. Endocrinology. (2016) 157:4487–98. doi: 10.1210/en.2016-1464
37. Casey ML, MacDonald PC, Mitchell MD. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis. A paradox? J Clin Invest. (1985) 75:1852–7. doi: 10.1172/JCI111899
38. Zakar T, Hirst JJ, Mijovic JE, Olson DM. Glucocorticoids stimulate the expression of prostaglandin endoperoxide H synthase-2 in amnion cells. Endocrinology. (1995) 136:1610–9. doi: 10.1210/endo.136.4.7895671
39. Guo C, Wang W, Liu C, Myatt L, Sun K. Induction of PGF2alpha synthesis by cortisol through GR dependent induction of CBR1 in human amnion fibroblasts. Endocrinology. (2014) 155:3017–24. doi: 10.1210/en.2013-1848
40. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal transduction targeted Ther. (2017) 2:17023–. doi: 10.1038/sigtrans.2017.23
41. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. (1995) 270:286–90. doi: 10.1126/science.270.5234.286
42. Zhang C, Wang W, Liu C, Lu J, Sun K. Role of NF-kappaB/GATA3 in the inhibition of lysyl oxidase by IL-1beta in human amnion fibroblasts. Immunol Cell Biol. (2017) 95:943–52. doi: 10.1038/icb.2017.73
43. Sun K, Ma R, Cui X, Campos B, Webster R, Brockman D, et al. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin H synthase type 2 but not microsomal prostaglandin E synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J Clin Endocrinol Metab. (2003) 88:5564–71. doi: 10.1210/jc.2003-030875
44. Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. (2010) 78:257–68. doi: 10.1038/ki.2010.154
45. Huang CJ, Acevedo EO, Mari DC, Randazzo C, Shibata Y. Glucocorticoid inhibition of leptin- and lipopolysaccharide-induced interleukin-6 production in obesity. Brain Behav Immun. (2014) 35:163–8. doi: 10.1016/j.bbi.2013.10.004
46. Mantov N, Zrounba M, Brollo M, Grassin-Delyle S, Glorion M, David M, et al. Ruxolitinib inhibits cytokine production by human lung macrophages without impairing phagocytic ability. Front Pharmacol. (2022) 13:896167. doi: 10.3389/fphar.2022.896167
47. Lei WJ, Zhang F, Lin YK, Li MD, Pan F, Sun K, et al. IL-33/ST2 axis of human amnion fibroblasts participates in inflammatory reactions at parturition. Mol Med. (2023) 29:88. doi: 10.1186/s10020-023-00668-9
48. Zhang F, Lu JW, Lei WJ, Li MD, Pan F, Lin YK, et al. Paradoxical induction of ALOX15/15B by cortisol in human amnion fibroblasts: implications for inflammatory responses of the fetal membranes at parturition. Int J Mol Sci. (2023) 24:10881. doi: 10.3390/ijms241310881
49. Zhang F, Sun K, Wang WS. Identification of a feed-forward loop between 15(S)-HETE and PGE2 in human amnion at parturition. J Lipid Res. (2022) 63:100294. doi: 10.1016/j.jlr.2022.100294
50. Gan XW, Wang WS, Lu JW, Ling LJ, Zhou Q, Zhang HJ, et al. De novo synthesis of SAA1 in the placenta participates in parturition. Front Immunol. (2020) 11:1038. doi: 10.3389/fimmu.2020.01038
51. Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, et al. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer's disease. Glia. (2016) 64:1562–72. doi: 10.1002/glia.23024
52. Estrada SM, Thagard AS, Dehart MJ, Damicis JR, Dornisch EM, Ippolito DL, et al. The orphan nuclear receptor Nr4a1 mediates perinatal neuroinflammation in a murine model of preterm labor. Cell Death disease. (2020) 11:11. doi: 10.1038/s41419-019-2196-7
53. Mallard C, Tremblay ME, Vexler ZS. Microglia and neonatal brain injury. Neuroscience. (2019) 405:68–76. doi: 10.1016/j.neuroscience.2018.01.023
54. Menon R, Richardson LS, Lappas M. Fetal membrane architecture, aging and inflammation in pregnancy and parturition. Placenta. (2019) 79:40–5. doi: 10.1016/j.placenta.2018.11.003
55. Li WJ, Lu JW, Zhang CY, Wang WS, Ying H, Myatt L, et al. PGE2 vs PGF2alpha in human parturition. Placenta. (2021) 104:208–19. doi: 10.1016/j.placenta.2020.12.012
56. Behnia F, Sheller S, Menon R. Mechanistic differences leading to infectious and sterile inflammation. Am J Reprod Immunol. (2016) 75:505–18. doi: 10.1111/aji.12496
57. Zhu XO, Yang Z, Guo CM, Ni XT, Li JN, Ge YC, et al. Paradoxical stimulation of cyclooxygenase-2 expression by glucocorticoids via a cyclic AMP response element in human amnion fibroblasts. Mol endocrinology. (2009) 23:1839–49. doi: 10.1210/me.2009-0201
58. Agnew EJ, Ivy JR, Stock SJ, Chapman KE. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J Mol Endocrinol. (2018) 61:R61–73. doi: 10.1530/JME-18-0077
59. Bolt RJ, van Weissenbruch MM, Lafeber HN, Delemarre-van de Waal HA. Glucocorticoids and lung development in the fetus and preterm infant. Pediatr pulmonology. (2001) 32:76–91. doi: 10.1002/ppul.1092
60. Grier DG, Halliday HL. Effects of glucocorticoids on fetal and neonatal lung development. Treatments Respir Med. (2004) 3:295–306. doi: 10.2165/00151829-200403050-00004
61. Wang WS, Liu C, Li WJ, Zhu P, Li JN, Sun K. Involvement of CRH and hCG in the induction of aromatase by cortisol in human placental syncytiotrophoblasts. Placenta. (2014) 35:30–6. doi: 10.1016/j.placenta.2013.10.018
62. Wang W, Li J, Ge Y, Li W, Shu Q, Guan H, et al. Cortisol induces aromatase expression in human placental syncytiotrophoblasts through the cAMP/Sp1 pathway. Endocrinology. (2012) 153:2012–22. doi: 10.1210/en.2011-1626
63. Craft I, Brummer V, Horwell D, Morgan H. Betamethazone induction of labour. Proc R Soc Med. (1976) 69:827–8. doi: 10.1177/003591577606901119
64. Mati JK. Corticosteroids and the initiation of parturition in the human. East Afr Med J. (1973) 50:462–5.
65. Mati JK, Horrobin DF, Bramley PS. Induction of labour in sheep and in humans by single doses of corticosteroids. Br Med J. (1973) 2:149–51. doi: 10.1136/bmj.2.5859.149
66. Nwosu UC, Wallach EE, Bolognese RJ. Initiation of labor by intraamniotic cortisol instillation in prolonged human pregnancy. Obstet Gynecol. (1976) 47:137–42.
67. Kammala AK, Mosebarger A, Radnaa E, Rowlinson E, Vora N, Fortunato SJ, et al. Extracellular Vesicles-mediated recombinant IL-10 protects against ascending infection-associated preterm birth by reducing fetal inflammatory response. Front Immunol. (2023) 14:1196453. doi: 10.3389/fimmu.2023.1196453
68. Gomez-Chavez F, Castro-Leyva V, Espejel-Nunez A, Zamora-Mendoza RG, Rosas-Vargas H, Cancino-Diaz JC, et al. Galectin-1 reduced the effect of LPS on the IL-6 production in decidual cells by inhibiting LPS on the stimulation of IkappaBzeta. J Reprod Immunol. (2015) 112:46–52. doi: 10.1016/j.jri.2015.07.002
69. Flores-Espinosa P, Vega-Sanchez R, Mancilla-Herrera I, Bermejo-Martinez L, Preciado-Martinez E, Olmos-Ortiz A, et al. Prolactin selectively inhibits the LPS-induced chemokine secretion of human foetal membranes. J maternal-fetal neonatal medicine: Off J Eur Assoc Perinatal Medicine Fed Asia Oceania Perinatal Societies Int Soc Perinatal Obstet. (2020) 33:4083–9. doi: 10.1080/14767058.2019.1596255
70. Pineda-Torres M, Flores-Espinosa P, Espejel-Nunez A, Estrada-Gutierrez G, Flores-Pliego A, Maida-Claros R, et al. Evidence of an immunosuppressive effect of progesterone upon in vitro secretion of proinflammatory and prodegradative factors in a model of choriodecidual infection. BJOG: an Int J obstetrics gynaecology. (2015) 122:1798–807. doi: 10.1111/1471-0528.13113
Keywords: glucocorticoid, fetal membranes, chorioamnionitis, 11β-HSD1, preterm birth
Citation: Ling L-J, Zhou Q, Zhang F, Lei W-J, Li M-D, Lu J-W, Wang W-S, Sun K and Ying H (2024) The dual role of glucocorticoid regeneration in inflammation at parturition. Front. Immunol. 15:1459489. doi: 10.3389/fimmu.2024.1459489
Received: 04 July 2024; Accepted: 19 August 2024;
Published: 03 September 2024.
Edited by:
Tamas Zakar, The University of Newcastle, AustraliaReviewed by:
Lauren Stafford Richardson, University of Texas Medical Branch at Galveston, United StatesVeronica Zaga-Clavellina, Instituto Nacional de Perinatología (INPER), Mexico
Copyright © 2024 Ling, Zhou, Zhang, Lei, Li, Lu, Wang, Sun and Ying. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kang Sun, c3VuZ2FuZ3JlbmppQHNqdHUuZWR1LmNu; Wang-Sheng Wang, d2FuZ3NoZW5nX3dhbmdAc2p0dS5lZHUuY24=; Hao Ying, c3RlcGhlbnlpbmdfMjAxMUAxNjMuY29t
†These authors have contributed equally to this work