 Benedetta Rambaldi
Benedetta Rambaldi Giuliana Rizzuto1,2
Giuliana Rizzuto1,2 Martino Introna
Martino Introna- 1Dipartimento di Oncologia ed Ematologia, Ospedale Papa Giovanni XXIII, Bergamo, Italy
- 2Molecular and Translational Medicine Doctoral Program (DIMET), University of Milano-Bicocca, Monza, Italy
- 3Department of Oncology and Hematology, Università degli Studi di Milano, Milan, Italy
Although allogeneic hematopoietic cell transplantation (HCT) represents a curative approach for many patients with hematological diseases, post-transplantation relapse occurs in 20-50% of cases, representing the primary cause of treatment failure and mortality. Alloreactive donor T cells are responsible for the graft versus leukemia (GvL) effect, which represents the key mechanism for the long-term curative effect of HCT. However, the downside is represented by graft versus host disease (GvHD), largely contributing to transplant-related mortality (TRM). Multiple factors play a role in regulating the delicate balance between GvL and GvHD, such as the optimization of the donor HLA and KIR match, the type of graft source, and the adaptive use of post-transplant cellular therapy. In addition to the standard donor lymphocyte infusion (DLI), several attempts were made to favor the GvL effect without increasing the GvHD risk. Selected DLI, NK DLI, activated DLI and more sophisticated genetically engineered cells can be employed. In this scenario, cytokine-induced killer (CIK) cells represent a suitable tool to boost GvL while minimizing GvHD. CIK cells are T lymphocytes activated in culture in the presence of monoclonal antibodies against CD3 (OKT3), interferon-gamma (IFN-g), and interleukin-2 (IL-2), characterized by the expression of markers typical of NK cells and T cells (CD3+, CD56+, with a prevalent CD8+ phenotype). CIK cells can mediate cytotoxicity through both MHC and non-MHC restricted recognition, which is the so‐called “dual‐functional capability” and display minimum alloreactivity. Allogeneic CIK cells showed a favorable rate of response, especially in the setting of minimal residual disease, with a rate of GvHD not exceeding 25%. Finally, the CIK cell platform can be adapted for chimeric antigen receptor (CAR) cell strategy, showing promising results in both preclinical and clinical settings. In this review, we describe the main immunological basis for the development of the GvL and the possible cellular therapy approaches used to boost it, with a particular focus on the use of CIK cells.
1 Introduction
Allogeneic hematopoietic cell transplantation (HCT) remains a curative approach for many patients with malignant and non-malignant hematological diseases (1). Acute myeloid leukemia (AML) represents the most frequent indication for HCT, accounting for 38% of transplants in Europe (2). In medically fit patients, HCT represents the most used post-remission therapy, reducing relapse incidence by eliminating residual leukemic cells (3). However, post-transplantation relapse occurs in 20-50% of patients, still representing the primary cause of treatment failure and mortality (4–6).
The anti-leukemic effect of transplant is due to the conditioning regimen, consisting of high-dose chemotherapy with or without total body irradiation (TBI), and to the donor immune surveillance. The conditioning regimen aims not only to kill residual leukemic cells but also to reduce recipient bone marrow hematopoietic and immune cells. The intensity of the conditioning used depends on the fitness and age of the patient (7, 8). Although myeloablative conditioning (MAC) showed better activity in controlling the disease recurrence in AML and in myelodysplastic syndrome (MDS) patients, compared to reduced intensity conditioning (RIC) (9), the use of RIC regimens increased over time, allowing the treatment of elderly or unfit patients too.
Alloreactive donor T cells are responsible for the graft versus leukemia (GvL) effect, which represent the key mechanism for the long-term curative effect of HCT, especially after RIC HCT (10). However, donor T cells are also responsible for alloreactivity against normal tissues, leading to graft versus host disease (GvHD), largely contributing to the transplant-related mortality (TRM) (6). The first description of the GvL effect goes back to the experimental mouse model, in which the animals injected with leukemic cells were treated with TBI followed by the administration of isologous or homologous myeloid tissue. Both graft sources could restore the hematopoietic compartment, but only the latter one displayed the ability to protect from leukemia. However, it was also associated to lethal toxicity (diarrhea) (11). These preliminary experiments showed for the first time the tight connection between GvL and GvHD. In 1994, Gale and colleagues reproduced these data in patients, showing that HCT from sibling donors can potentially cure patients with AML, while a minor effect was seen using a syngeneic donor (12, 13).
Post-transplant relapse is often driven by the ability of the tumor to escape from the immunological surveillance, hampering the GvL activity (14). Principal mechanisms of immune escape after HCT are loss of mismatched human leukocyte antigen (HLA) haplotype (15–17) HLA downregulation (18–20), and inhibition of allogeneic T cells through overexpression of inhibitory receptors or the perturbation of anti- and pro-inflammatory cytokines (19). Moreover, different strategies are used to modulate the donor T cell alloreactivity and prevent GvHD, such as the optimization of the donor match, the T cell depletion (both in vivo and ex vivo), and several immunosuppressive drugs (21). However, the same approaches could hamper the desired GvL activity, increasing the risk of disease relapse. For these reasons, multiple strategies were tested to boost the GvL activity after transplant, including different cellular therapy approaches, such as donor lymphocyte infusion (DLI), selected DLI, activated DLI, cytokine-induced memory-like (CIML) NK cells, and cytokine-induced killer (CIK) cells. Finally, genetically modified lymphocytes using chimeric antigen receptor (CAR), represent a potent tool to boost the GvL in specific disease setting, where a target antigen is available.
This review aims to describe the main immunological basis for the development of the GvL after HCT and the possible cellular therapy approaches used to boost it, with a particular focus on the use of CIK cells.
2 Immunological basis of GvL: donor selection through HLA, KIR matching and graft source
The GvL activity can be predicted and modulated during the selection of the suitable transplant donor, in terms of HLA matching, killer Ig-like receptor (KIR) alloreactivity, and type of graft source (Figure 1).
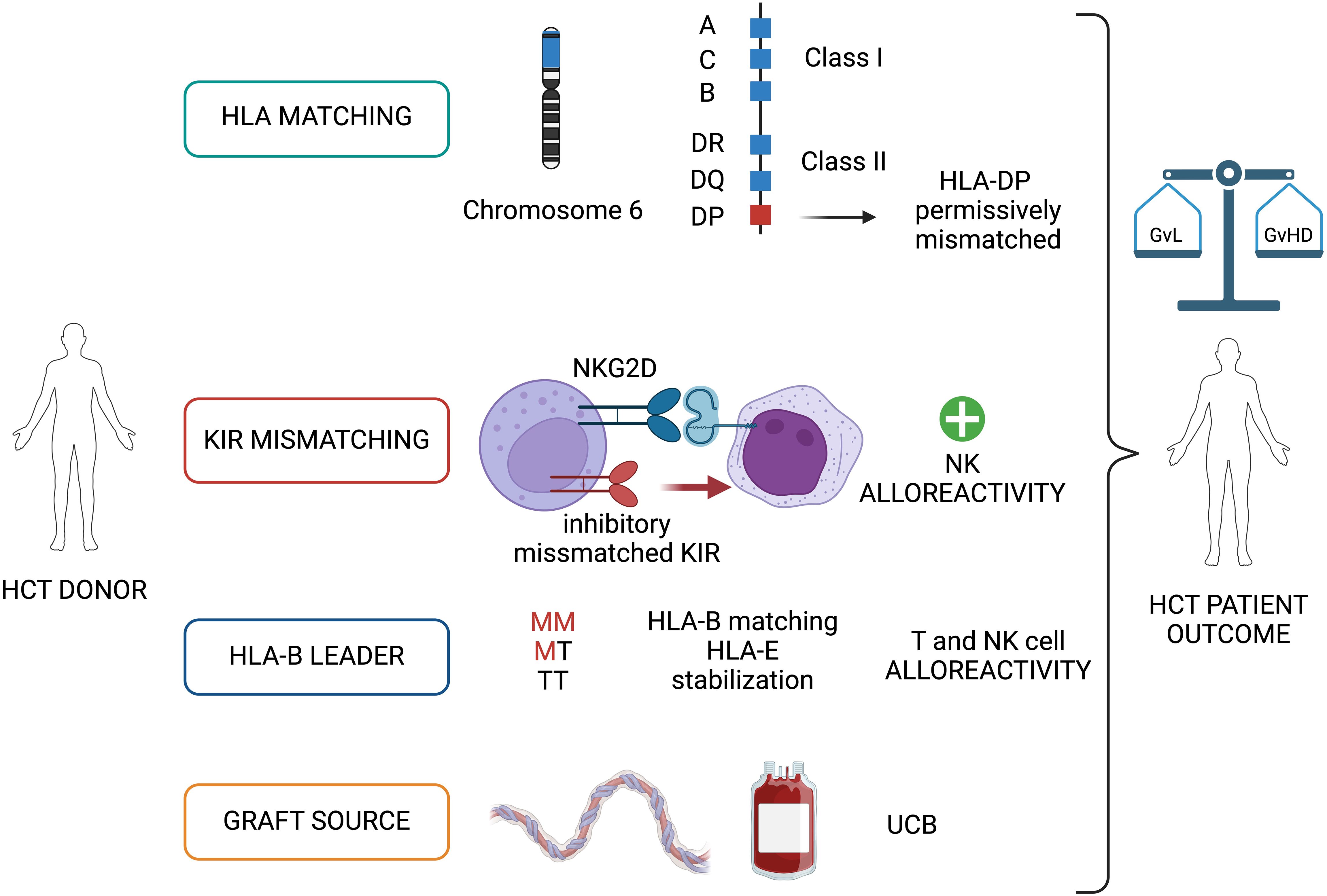
Figure 1. Graft versus Leukemia (GvL) immunological basis: donor selection through HLA, KIR matching and graft source. The donor-recipient matching for HLA and KIR, and the type of graft source (UCB) are detrimental factors during the selection of the HCT donor and contribute to the final balance between a GvL and GvHD. HCT, allogeneic hematopoietic cell transplantation; KIR, killer Ig-like receptor; UCB, umbilical cord blood; M, methionine; T, threonine; GvHD, graft versus host disease. Image created with BioRender.com.
2.1 HLA matching
The HLA genes, which are located on the short arm of chromosome 6 (6p21.3), encode for the molecules responsible for presenting antigens to T lymphocytes, making these genes the key players of the alloreactivity. The HLA loci are divided into two subclasses: HLA class I and HLA class II. Generally, HLA class I alleles -A, -B, and -C are expressed on all nucleated cells and display antigen to CD8+ T cells, while HLA class II alleles -DR, -DQ, -DP are expressed on antigen-presenting cells (APCs) and initiate a response by CD4+ T cells, playing an important role in recognizing non-self, infected or malignant cells. In the thymus, T cells with various T cell receptors (TCRs) undergo positive selection and negative selection, among which those that do not respond to the HLA-antigen peptide complex or have a strong response to self-antigens are eliminated (22). In addition to classic HLA class I and HLA class II alleles, there are also fewer polymorphic genes in these regions, including HLA-E, HLA-F, and HLA-G in HLA class I regions and HLA-DO and HLA-DM in HLA class II regions. These less polymorphic genes are also important regulators of the immune system. To date, more than 30,000 HLA alleles have been identified (23).
Considering the role of HLA proteins in the discrimination between the self and non-self, the search for an HLA-matched donor is crucial in the setting of tissue and organ transplantation, especially for HCT (24). HLA-matched related donors (MRD) generally share both alleles of HLA-A, -B, -C, -DR, -DQ, and -DP (12 of 12) because they have inherited the same parental copies of chromosome 6 encompassing the major histocompatibility complex (MHC). In MRD HCT the targets of T-cell alloreactivity are almost exclusively minor histocompatibility antigens (mHAgs), mediated by donor naïve T cells. This explains the lower rate of GvHD but also a less potent GvL effect in MRD HCT compared to transplants performed with alternative donors (25). In HLA-matched unrelated donor (MUD) HCT, patients and their HLA-matched donors carry the same 8 of 8 alleles at HLA-A, -B, -C, and -DRB1, without sharing their respective genetic backgrounds. Genetic disparity between unrelated individuals frequently includes the HLA-DRB3/4/5, -DQ, and -DP loci which are targets of direct T-cell alloreactivity. In this case, both naïve and memory donor T cells can recognize different recipient HLA-restricted peptides regardless of their derivation from novel or recall antigens. Due to strong linkage disequilibrium between HLA-DRB1 and -DRB3/4/5 and -DQ, mismatches for these genes are relatively rare in MUD. In contrast, HLA-DP mismatches are present in over 80% of MUD HCT, representing a key aspect of both GvL and GvHD in this transplant setting (26, 27). Moreover, HLA-DR+ leukemias and lymphomas generally co-express HLA-DP, making it an attractive GvL target (28). Of note, homozygosity for HLA-DP genes represents a frequent event, often resulting in unidirectional (only graft-versus-host [GvH] or host-versus-graft [HvG] direction) HLA-DP mismatches in MUD-HCT. Indeed, HLA-DPB1 mismatches are associated with increased risks of acute GvHD after 8 of 8 HLA-matched MUD HCT, but also with a significantly reduced risk of leukemia relapse, resulting in no net impact on overall survival (OS) (25). The notion of permissive mismatches is based on accumulating evidence suggesting that limited alloreactivity is sufficient for GvL, whereas aggressive alloreactivity can lead to clinically uncontrollable GvHD (29). In this view, permissive mismatches are those eliciting limited alloreactivity, shifting the balance from GvHD to GvL (30, 31). Current guidelines for patients transplanted for malignant disease recommend selection of an HLA-DPB1 allele-matched or permissively mismatched MUD, available with a probability of at least 70% (32). Also, Rutten and colleagues, in ex vivo analysis, first demonstrated the importance of CD4+ T cells alloreactive to HLA-DP in patients responding to DLI after MUD HCT (33). Moreover, to enhance the GvL effect, some groups propose to use in vitro expanded cytolytic T-lymphocytes (CTL) selected from the CD4+ naïve T cells alloreactive against HLA-DP specificities (34).
In the absence of an HLA-matched donor, the use of an alternative donor, such as an HLA-mismatched unrelated donor (MMUD, less than 8/8 alleles at HLA-A, -B, -C, and -DRB1), umbilical cord blood (UCB) or haploidentical related donor, has increased the number of patients that can access to the transplant procedure. However, in the MMUD setting, even a single allele mismatch negatively impacts patient outcomes after transplant (35). Specific strategies are needed to achieve the attenuation of the donor T cell alloreactivity in haploidentical HCT. The use of high-dose post-transplant cyclophosphamide (PTCy) has become the most frequently used due to its relatively easy applicability (36). Other strategies are represented by the use of rabbit-derived anti-thymoglobulin polyclonal antibodies (REFS) granulocyte-colony stimulating factor (G-CSF) mobilized bone marrow grafts with extensive immunosuppression (37) or ex vivo graft manipulation, such as CD34 megadose (38, 39) or a/b T cell depletion (40). Finally, the Perugia group proposed a novel approach using a T-cell-depleted graft and a subsequent infusion of regulatory and conventional T cells, without any post-transplant immune suppression (41).
Altogether, the GvL effect can benefit from a careful choice of the HLA-compatible donor, particularly in the setting of permissive HLA-DP mismatched MUD (Figure 1).
2.2 NK cell alloreactivity and KIR matching
A special emphasis is being placed on using natural killer (NK) cells to harness both innate and adaptive immunity after HCT (42). NK cells are uniquely regulated by activating and inhibitory receptors and can mediate a critical GvL effect, also referred to as NK cell alloreactivity, without mediating GvHD (43–46). NK cell alloreactivity can potentially provide a better antitumor effect, as documented by lower relapse rates and better survival in patients with higher NK cell numbers early post-transplant (47, 48). Cytotoxic activity of NK cells is mediated primarily by a balance between inhibitory and activating receptors expressed on the cell surface, the former being mainly accounted KIRs that recognize HLA class I molecules on the surface of target cells (49). When NK cells encounter the matching HLA-class I ligand for their inhibitory KIR, they are considered “educated” or “licensed” and refrain from an attack on healthy tissues under a steady state. When NK cells are accustomed to this inhibitory signal and subsequently encounter a cell that does not express the appropriate KIR-ligand (“missing ligand”), they can mount an effector response, if the target also expresses stress-ligands that trigger activating NK-cell receptors (such as natural killer group 2 member D, NKG2D, Figure 1). Due to the lack of exposure to their corresponding ligand, unlicensed NK cells are “un-educated” and hyporesponsive at a steady state rather than being triggered by self-tissues lacking the ligand (50). Several models of donor-recipient NK cell alloreactivity have been proposed. The KIR ligand incompatibility (ligand–ligand) model, in which NK cells will react and kill host cells that lack the HLA class I ligand(s) for inhibitory KIR, was first proposed by the Perugia group (51). Alloreactive NK cells in the GvH direction helped promote engraftment and GvL, resulting in a reduced risk of leukemia relapse and better survival in adults with AML without increasing the rate of GvHD (52). An alternative model called the receptor-ligand or missing-ligand model, proposed that NK cells will react if at least one KIR gene expressed in the donor’s NK cell repertoire does not recognize any of the HLA molecules in the recipient’s ligand repertoire. This model showed a better ability to predict NK alloreactivity and lower the risk of leukemia relapse in a pediatric study of patients with high-risk leukemia given CD34+ selected haploidentical graft (53). The educational/missing licensing proof model required that the NK cells were educated against a specific antigen in order to become alloreactive when encounter a recipient cell that lack its cognate ligand (54). Other groups have used the KIR-haplotype model which takes into consideration the presence or absence of a B-KIR haplotype in the donor, as a measure of enrichment for activating versus inhibitory KIRs. The use of this model demonstrated a reduced risk of leukemia relapse when patients were transplanted from donors with centromeric B-haplotypes (55, 56). Finally, the KIR genotyping model, analyzes KIR genes between donor and recipient to observe a correlation between KIR match/mismatch and transplant outcomes. This model has shown discrepant results in studies with different HCT platforms (57).
Although a definitive and easy-to-apply model for the evaluation of donor-recipient KIR alloreactivity is not currently available in clinical practice, the presence of a KIR mismatched have shown to positive impact the prognosis of leukemia patients after transplant, augmenting the NK-mediated GvL.
2.3 T and NK cells alloreactivity through the HLA-B leader
The last factor to take into consideration for modulating T and NK cell alloreactivity is the HLA-B leader (Figure 1). The leader sequence refers to the peptide encoded by exon 1 of the 7 exons that comprise the HLA-B gene. It has been shown that in HLA-B, the leader sequence encodes methionine (M) or threonine (T) at position -21 and can give rise to TT, MT, or MM genotypes. In the setting of MMUD, there is an increased risk of severe acute GvHD when the patient has an M in the leader sequence and when the leader sequence is mismatched (58). Moreover, the leader sequence of HLA class-I molecules is presented by the non-classical class I molecule, HLA-E, and stabilizes HLA-E expression on the cell surface, enhancing binding to receptors on NK cells. It is hypothesized that HLA-B presentation by HLA-E and subsequent recognition by NK cells may contribute to improvements in non-relapse mortality (NRM) that have been demonstrated in several HCT settings (MMUD, haploidentical, and UCB HCT) (59–61).
The HLA-B leader status contributes regulating the GvL/GvHD balance and may inform relapse and NRM risk after HCT.
2.4 Graft source and alloreactivity: the case of UCB transplant
Multiple data are now favoring the use of UCB HCT for patients with high-risk leukemia, suggesting the role of the graft source in determining the GvL effect (Figure 1). In the retrospective analysis performed by Milano and colleagues, 582 consecutive patients with acute leukemia or myelodysplastic syndrome received a first myeloablative hematopoietic-cell transplant from an UCB (140 patients), an MUD, or an MMUD (344 and 98 patients, respectively). Authors observed that among patients with pre-transplant minimal residual disease, the probability of OS after receipt of a transplant from a UCB was at least as favorable as that after receipt of a transplant from an MUD and was significantly higher than the probability after receipt of a transplant from an MMUD. Furthermore, the probability of relapse was lower in the UCB group than in either of the other groups, suggesting a higher GvL activity in the UCB group (62). In addition, the pediatric study by Verneris and colleagues revealed an enhanced GvL effect in acute leukemia patients after transplantation with 2 partially HLA-matched UCB units compared to 1 single unit UCB (63). In another prospective, multicenter, pediatric study recently published, 367 patients affected by AML/MDS undergoing T-cell replete HCT were analyzed. One-hundred and twelve patients underwent a UCB HCT, the remaining 255 received other cell sources. Although a higher incidence of poor prognosis features in the UCB group, these patients showed an unexpectedly favorable EFS (64.1%). In a multivariable analysis, the UCB cohort had significantly improved EFS, time to relapse, and reduced chronic GvHD, with some evidence of improved OS. The effect appeared similar regardless of the minimal residual disease (MRD) status, suggesting that UCB HCT without serotherapy may be the optimal transplant option for children with myeloid malignancy (64). Finally, so far, there are no reports of HLA loss, as a strategy of leukemia immune escape, in the setting of UCB transplantation (Prof Luca Vago, personal communication).
UCB represents a suitable graft source to boost GvL after HCT and should be considered in the setting of high-risk disease.
2.5 Graft manipulation and alloreactivity
Different approaches were used to modify the graft in order to reduce the incidence of GvHD while maintaining the GvL effect (Table 1). The first strategy used the CD34+ megadose in the setting of haploidentical transplantation, without the addition of any other immunosuppression (38). This strategy showed low incidence of relapse and remarkably low incidence of GvHD. Another approach includes the depletion of possible alloreactive T cells, defined as α/β TCR positive cells, in association with ATG and rituximab. This strategy was mostly developed in the pediatric haploidentical setting by Locatelli and colleagues with promising results (40). Again, the group of Perugia further expanded the concept of CD34+ megadose, with the Treg/Tcon graft strategy, where a known dose of haploidentical regulatory (Treg) and conventional (Tcon) T cells were administrated to the patient. This strategy showed remarkably low incidence of both acute and chronic GvHD with a favorable relapse incidence (65, 66).
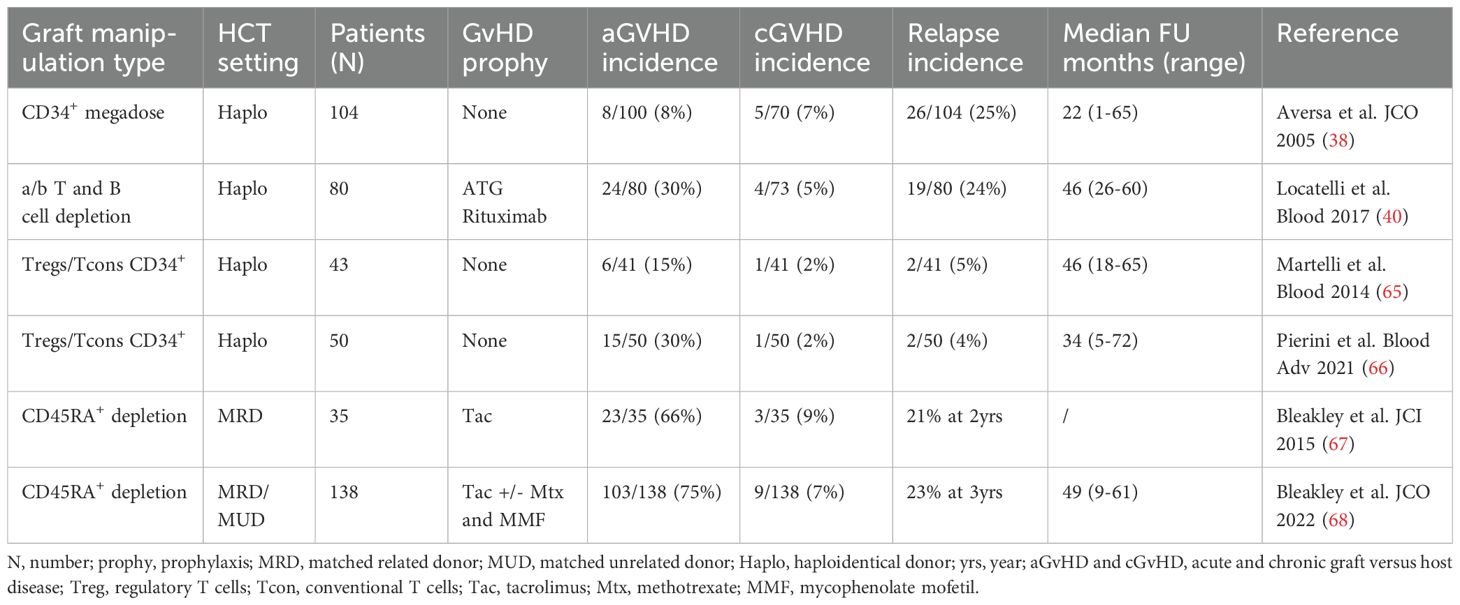
Table 1. Selection of main clinical trials on prophylactic graft manipulation strategies to prevent GvHD and enhance GvL.
Finally, Bleakley and colleagues, elegantly tested the effect of a naïve T cell depleted graft in the setting of MRD or MUD, by eliminating the CD45RA+ cells from the graft. These trials showed a non-detrimental impact on relapse incidence with low cGvHD, but an aGvHD still above 50% (67, 68).
3 Cellular therapies to boost GvL effect
Adaptive and innate immunity are both crucial in maintaining the delicate balance between GvHD and GvL effect. The major players in this scenario are the T and NK cells lymphocytes. Different strategies were tested to use unmodified or modified T and NK cells in the setting of HCT to prevent or treat leukemia recurrence (Figure 2). Here we describe the major utilized cellular therapy approaches, highlining the ability of CIK cells to combine both T and NK cell properties.
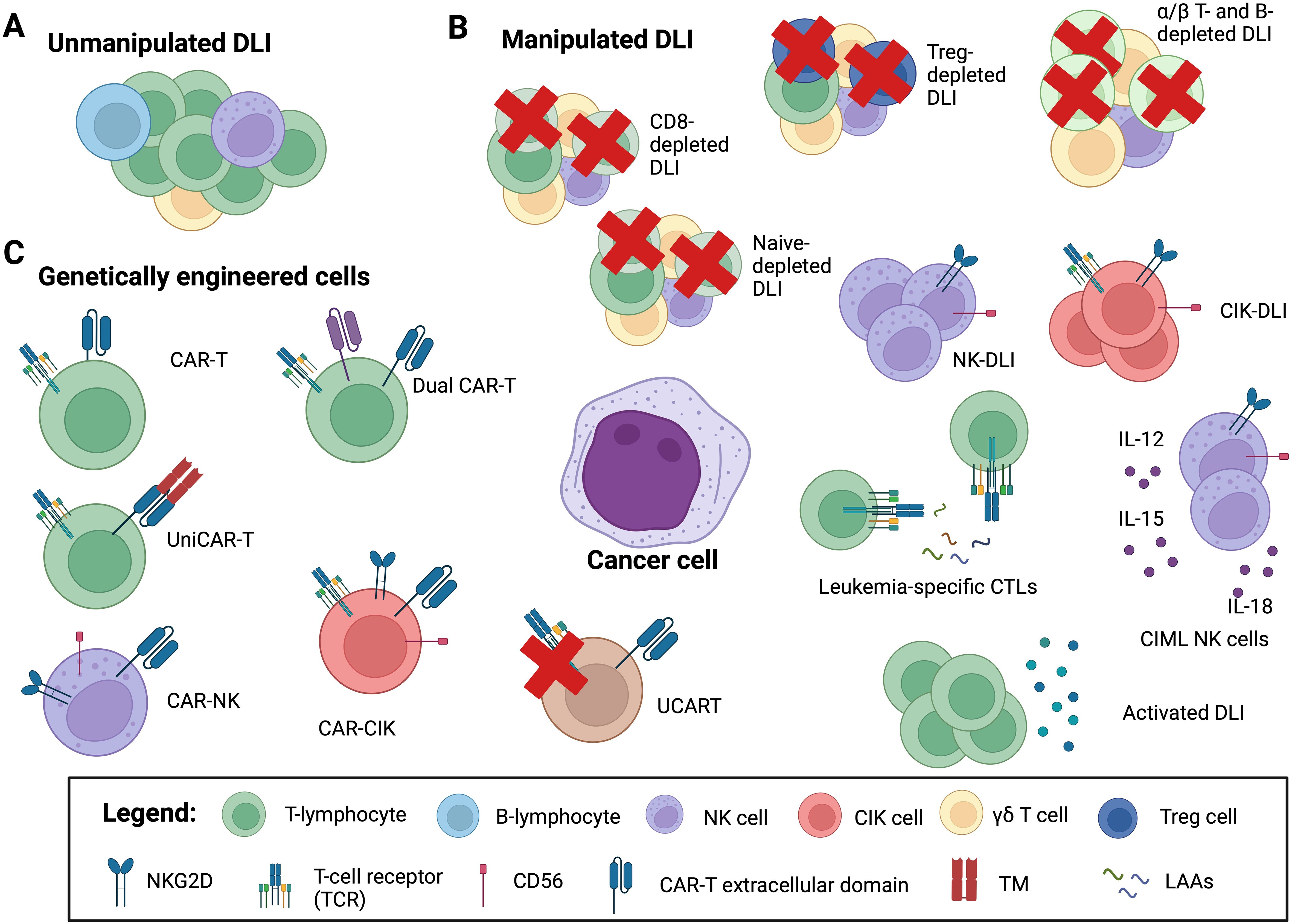
Figure 2. Adoptive cellular therapy in the post-HCT setting. Adoptive cellular therapy strategies include: (A) unmanipulated donor lymphocyte infusions (DLI); (B) manipulated DLI: CD8+ T cell depleted DLI, α/β T and B cell depleted DLI and CD45RA+ naïve T cell depleted DLI to mitigate the risk of graft versus host disease (GvHD); regulatory T cell (Treg) depleted DLI, to enhance graft versus leukemia (GvL) activity; natural killer (NK) cell and cytokine-induced killer cell (CIK) DLI to both minimize GvHD and increase GvL; cytokine-induced memory-like (CIML) NK cell DLI for enhancing NK killing capacity and persistence; activated DLI, either with cytokine (IL-2, IL-15, IL-21) or with dendritic cells or G-CSF primed DLI to augment graft versus leukemia (GvL) activity; leukemia-specific CTLs (cytotoxic T lymphocytes) and leukemia-associated antigens (LAAs) to redirect specificity; (C) genetically engineer cells: chimeric antigen receptor (CAR) T cells, genetically engineered to recognize and kill targets: allogeneic CAR-T; “universal” CAR (UniCAR) cells are inactive under physiologic conditions, but can recognize a soluble adaptor, called targeting module (TM), directed against the specific target; CAR-NK and CAR-CIK cells, to minimize the risk of GvHD; dual-CAR-T target multiple antigens simultaneously to circumvent immune escape arising from antigen-negative disease; UCART where the endogenous TCR can be disrupted in mature T cells to abrogate GvHD potential. Image created with BioRender.com.
3.1 Secondary HCT
Although outcomes for AML/MDS patients relapsed after HCT are quite disappointing, a second HCT may offer a chance of survival in selected patients, representing a valid cellular therapy option. However, a second HCT is generally limited to medically fit patients due a high rate of TRM, reaching up to 73.5% in the first series of studies (69). A more encouraging TRM was reported with the introduction of RIC regimens, ranging from 30% to zero, mostly associated with a short interval between the two transplants and the disease status at the time of the second HCT (70). A recent retrospective single-center study analyzed data from 407 consecutive patients with relapsed AML/MDS after the first HCT. Sixty-two patients had a second HCT (15%) and 345 did not. The 2-year cumulative incidence rates of NRM and relapse following the second HCT were 26% and 50%, respectively. Patients who underwent a second HCT had a better outcome, compared to patients who did not, with 5-year OS rates of 25% and 7%, respectively. In this study, the use of a second HCT was associated with longer survival especially in patients with longer remission after the first HCT (71). Since the loss of mismatch, HLA is one of the most relevant and understood mechanisms of immune escape after HCT (15, 16, 72, 73), a careful choice of the second HCT donor should be mandatory (74). In addition, emerging data on the use of easily accessible, alternative donor sources, such as UCB or haploidentical donors, may promote the practice of switching donors for second allotransplants. Indeed, in patients who relapsed with an HLA loss, a second HCT using a donor with a different HLA haplotype provided promising results (16). Different retrospective studies have tried to determine which is the best donor for the second HCT, with different results. In one single center experience including 40 patients undergone a second HCT, authors showed that an allograft with a new mismatched haplotype may improve outcomes following a second HCT (75). However, in a retrospective German registry study on 179 patients undergone a second HCT, changing the stem cell donor for the second transplant did not provide a significant advantage for disease recurrence or long-term survival, compared with the choice of the original donor for the second transplant (76). Of note, no haploidentical or UCB transplants were included in this analysis. Moreover, in a retrospective multicenter EBMT study, including 556 patients who underwent a second HCT, similar outcomes were observed in patients after a second HCT with the same or different matched donor. In this analysis, patients were divided into three groups based on the type of the second HCT donor: same donor, different matched donor, or haploidentical donor. The two-year leukemia-free survival (LFS) rate was 23.5%, 23.7%, and 21.8%, respectively. In addition, a non-T-cell depleted haploidentical transplant was associated with a similar relapse rate, but higher NRM (77). A different study from the EBMT conducted in acute lymphoblastic leukemia (ALL) patients demonstrated that T-cell replete haploidentical donors in second HCT had a 1.5-fold higher 2-year OS (49% vs. 31%) albeit non statically difference in OS when choosing either a MUD or haploidentical donor (78). Finally, some groups showed the feasibility of a UCB for a second HCT. In a retrospective Japanese analysis of 263 consecutive patients with different hematological malignancies, OS, NRM, and relapse at 2-years were 16.7%, 46.9%, and 40.6%, respectively. In multivariate analysis, younger age (<50), good PS (0-1), and long interval between HCTs (≧1 year) showed a superior OS (79). Similar positive results were collected in a pediatric series treated with a second/third UCB HCT, showing 3-year OS and EFS of 69.2% and 64.9%, respectively. The cumulative incidence of TRM at 3 years was 19.2%. Notably, no toxicity-related deaths were seen in the 16 patients who received UCB after 2015, suggesting an improvement in transplant care in this setting. The 3-year cumulative incidence of relapse was 15.9%, remarkably low for these very high-risk patients (80). In our center, we retrospectively collected data from 61 patients undergone a second HCT. Forty-five patients received the second HCT for disease relapse, while 16 for graft failure. Seventy percent of the patients were conditioned using a reduced-intensity regimen. In 54% of the patients a different donor was chosen for the second HCT and in 6 patients a UCB was used. Concerning disease status at the second HCT, 36% were in CR and 64% had active disease. At a median follow-up of 8.36 months (range: 0.2 – 276.3), 18 patients were alive. Twenty-seven patients died due to disease recurrence or progression. The 5-year cumulative incidence of NRM was 22%. The 5-year OS and PFS rates were 29% and 28%, respectively. Patients undergoing second HCT due to graft failure exhibited a 56% survival rate, higher compared to ones transplanted for disease recurrence (20%) (81).
Overall, recent data have shown an acceptable NRM and reasonable long-term survival in selected patients who underwent a second HCT.
3.2 Donor lymphocyte infusion
DLI can induce a GvL effect, defined as the ability of donor T lymphocytes to recognize and kill leukemia cells. The efficacy and toxicity of a DLI are hard to predict and depend on the wide range of different cell populations present in a leukapheresis product. In the DLI product, the majority of lymphocytes consists of CD3+ T cells (75-80%) (α/β T cells, regulatory T cells, and gd T cells), followed by NK cells (5-20%) and B cells (5%) (82, 83). ab T cells represent the largest and best-studied effector population, mediating alloreactivity by recognizing minor histocompatibility antigens (MiHA). Unmodified DLIs are the simplest way to increase GvL reactivity after HCT (Figure 2). However, studies have given different results, due to multiple variables, such as the type of conditioning regimen, in vivo/ex vivo T-cell depletion, post-transplant immunosuppression, the source of DLI, (unstimulated leukapheresis or G-CSF-stimulated products), fresh or cryopreserved products, the interval between the HCT and the first DLI, cell doses, and the number of infusions (82, 84). From the beginning, DLI was successfully employed in chronic myeloid leukemia (CML) (85), while less impressive results were observed in patients affected by AML (86, 87).
DLI therapy can be classified based on treatment intention into:
1. therapeutic, when used for the treatment of hematological relapse, often performed in conjunction with another anti-leukemic treatment;
2. preemptive, when used for the treatment of minimal residual disease or mixed chimerism;
3. prophylactic, when used in patients in disease remission but with a high risk of relapse (82, 83).
DLI was also tested in combination with other anti-leukemic drugs for both relapse treatment or maintenance after HCT. Schroreder and colleagues analyzed 154 patients with AML and MDS who relapsed after HCT and were treated with 5-azacytidine (AZA). All patients received a median number of 4 courses of AZA of those, 105 patients received concomitant DLI. Complete and partial remission rates were observed in 27% and 6%, respectively. The OS at 2 years was 29%, in MDS patients the 2 years OS was 66% and correlated with disease burden. Therefore, the authors conclude that AZA and DLI is an effective and well-tolerated treatment option in R/R patients after HCT, particularly for those with low disease burden (88). Later, in 2021, Guillaume retrospectively analyzed a cohort of 77 AML and MDS patients considered at high risk based on either their genomic or clinical status at transplantation. Following allogeneic transplantation, they received at least 1 cycle of prophylactic or preemptive low-dose AZA with or without escalating doses of DLI. Almost one-half of the patients were able to receive the full 12 cycles of AZA, and a majority (79%) received at least one DLI. Among the whole cohort, 19 patients relapsed, with a cumulative incidence of relapse (CIR) was 22% at 24 months. OS and progression-free survival (PFS) at 24 months were 70.8% and 68.3%, respectively. Grade II-IV acute GvHD and chronic GvHD were 27.4% and 45%, respectively (89).
Finally, some studies compared the efficacy of DLI in treating disease relapse with a second HCT. In one retrospective study 89 patients with a relapse disease or a graft failure, were treated with a second HCT (56%) or DLI (44%). The same HCT donor of the first transplant was used in 50% of the patients. The median number of DLI administered was 2 (range 1-11). This study showed that a second HCT may improve outcomes when performed for relapse if patients achieve a CR at the time of the second HCT, while DLI may be reserved for patients with active disease (90). In a retrospective EBMT study involving 418 adults with relapsed AML who received a second HCT (N=137) or DLI (N=281) there was no difference in OS whether a second HCT or DLI was prescribed, with a 5-year OS of 19% and 15% for second HCT and DLI, respectively. The best outcomes seem to be achieved in patients relapsing after 6 months from the first HCT or those in complete remission at the time of either second HCT or DLI (91).
The most feared adverse event following DLI therapy is the onset of GvHD, mediated by the infusion of donor lymphocytes, not controlled by immunosuppressive therapy (92). Depending from the DLI source and from the study the incidence of aGvHD and cGvHD varies from 20-60% and 20-40%, respectively (82). For this reason, efforts have been made to develop more targeted cellular therapy strategies to promote the GvL effect without increasing the GvHD effect. Table 2 outlines the major studies using modified or manipulated DLI divided according to the strategy used into different groups: selected T cell DLI, NK cell DLI, antigen-specific DLI and activated DLI. The first three groups include products of specific lymphocyte subset, such as Treg-depleted DLI, CD8+ αβ-depleted T cells, ab T cells against specific leukemic antigens (WT1, NPM1) and NK DLI, while the latter group includes products activated using different cytokine cocktails or drugs, such as CIK cells or cytokine-induced memory like (CIML) NK cells (Figure 2).
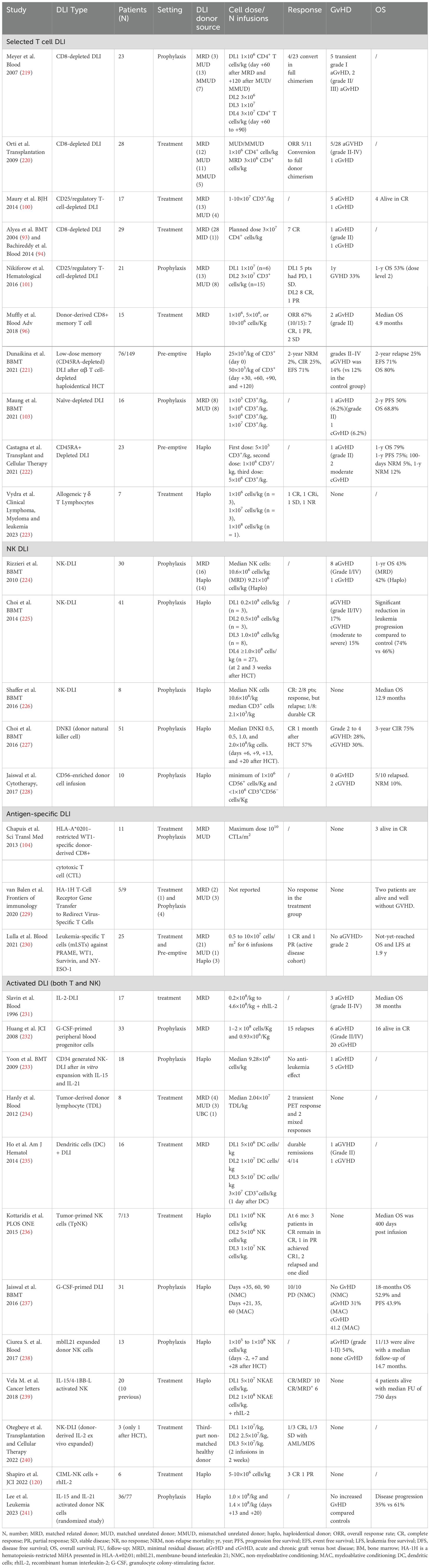
Table 2. Clinical trials on modified donor lymphocyte infusion (DLI): selected T cell, NK cell, antigen-specific, and activated DLI.
DLI has been shown to boost the GvL activity and to improve the patient outcome, especially in the setting of MRD and pre-emptive settings. However, the risk of subsequent GvHD is considerable and may increase the NRM, thus reducing the OS benefit of this cellular therapy.
3.3 Selected DLI
In order to reduce the GvHD risk and maintain a GvL effect, the DLI product can be manipulated. Back to 2004, Alyea and colleagues published a study using CD8+ depleted DLI for the treatment of disease recurrence after HCT. Patients were targeted to receive 3x107 CD4+ T cells/kg. Nine patients were enrolled, 3 CML, 3 myeloma, 2 CLL, and 1 NHL. CD8 depletion was highly specific, with a median recovery of CD4+ cells of 75%. All CML patients achieved a complete molecular remission. A CLL patient demonstrated a complete response. One patient developed grade II acute GVHD and subsequently chronic GVHD (93). A subsequent study demonstrated that response to DLI was associated with quantity of preexisting marrow CD8+ T cells and local reversal of T-cell exhaustion (94).
Murine models showed that CD8+CD44hi memory T (TM) cells could eradicate malignant cells without inducing GvHD (95). Muffly and colleagues evaluated the feasibility and safety of infusing freshly isolated and purified donor-derived CD8+ TM cells into patients relapsed after HCT. Fifteen patients received CD8+ TM cells at escalating doses, 9 had active disease and 13 received cytoreduction before cell infusion. No adverse infusion events or dose-limiting toxicities occurred. GvHD developed in 1 patient (grade 2 liver) and 10 patients (67%) maintained or achieved a response. Median EFS and OS were 4.9 months (1-19.3 months) and 19.6 months (5.6 months to not reached), respectively (96).
Regulatory T cells (Treg) are CD4+ T lymphocytes that express CD25 and FoxP3 and are negative for the CD127 marker. These cells play a crucial role in modulating the immune response and may reduce the GvL activity (97–99). Some groups have tried to eliminate these cells from the DLI product to boost the anti-tumor activity. Maury and colleagues treated 17 patients with relapses of various hematological malignancies after HCT. All patients had failed to respond to standard DLI and had no history of GvHD. Treg-depleted DLI were prepared by depleting CD25+ cells from donor leukapheresis using anti-CD25 magnetic microbeads. Compared with unmanipulated DLI, Treg-depleted DLI showed a better GvL effect, however with an increased GvHD incidence (6 out of 17 patients). With a mean follow-up of 6-years, Treg-depleted DLI treatment was associated with improved survival among the whole trial cohort (100). Colleagues from Dana-Farber Cancer Institute explored the use of Treg-depleted DLI in a phase I study treating 21 patients with different hematologic malignancies who had relapsed after transplantation. Seven subjects (33%) developed GvHD by 1 year, including one patient who died. The 1-year survival rate was 53% among patients treated with dose level 2. A shorter period between relapse and infusion was associated with response at dose level 2. Compared to unmodified DLI in 14 contemporaneous patients meeting study eligibility, Treg-depleted DLI treatment was associated with a better response rate and improved EFS (101).
The DLI product can also be manipulated to reduce the risk of GvHD, especially in the setting of mismatched donors. In a Phase II multicenter study of 23 patients with acute leukemia, a haploidentical, naïve T cells-enriched product, depleted of recipient-alloreactive T cells, called ATIR101 was infused in a single dose at 1 month after a T-cell depleted haploidentical HCT. Patients received a MAC conditioning with anti-thymocyte globulin (ATG) as a sole GvHD prophylaxis. NRM at 1-year was inferior for patients receiving ATIR101 concerning recipients of conventional haploidentical HCT, as well as improved GRFS (102). In another Phase I study, CD45RA+ naïve T-cells depleted DLI was given at 2 months following HLA-identical non-myeloablative HCT for hematologic malignancies. One patient developed grade 2 acute GvHD of skin and GI, and one moderate chronic GvHD of the lungs following the DLI. After a median follow-up of 2.8 years, 2-year PFS and OS were 50% and 68.8%, respectively (103). An ongoing phase I study (NCT03939585) is testing the safety of prophylactic TCR-αβ T and B cells depleted DLI infused on day 28 following HCT from MRD or haploidentical donor. This study hypothesizes that this DLI enriched in NK and TCR-γδ T cells displays a better GvL effect without increasing the risk of GvHD.
Finally, manufacturing T cells that can selectively target leukemia associated antigens (LAAs) represents an opportunity to promote antileukemic activity without inducing GvHD. Wilms tumor antigen 1 (WT1) is a transcription factor overexpressed in some leukemias. HLA-A*0201–restricted WT1-specific donor-derived CD8+cytotoxic T cell (CTL) clones were safely administered after HCT in 11 patients. Transferred cells exhibited direct evidence of antileukemic activity in two patients: a transient response in one patient with advanced progressive disease and the induction of prolonged remission in a patient with MRD. Additionally, 3 treated patients at high risk for relapse after HCT survived without leukemia relapse, GVHD, or additional anti-leukemic treatment. In this study, the exposure of CTLs to interleukin-21 (IL-21) increased in vivo CTL survival (104). Another strategy is to edit the TCR to be specific for a tumor antigen (105). However, TCR gene transfer results in competition for surface expression and inappropriate pairing between the exogenous and endogenous TCR chains, resulting in suboptimal activity and potentially harmful unpredicted specificities. To overcome this limitation, Bonini and colleagues designed zinc-finger nucleases (ZFNs) promoting the disruption of endogenous TCR β and α chain genes. ZFN-treated lymphocytes lacked CD3/TCR surface expression. These cells were subsequently infected with a lentiviral to express a WT1-specific TCR. these TCR-edited cells did not mediate off-target reactivity while maintaining anti-tumor activity in vivo (106).
Selected DLI have shown to reduce the risk of GvHD, while maintain the desirable GvL activity. However, the best approach to apply has not been defined yet.
3.4 Natural killer cell strategy: NK DLI and cytokine-induced memory-like NK cells
Several studies have shown a correlation between transplant outcomes and an impaired NK cell recovery, especially after haploidentical transplant with PTCy, suggesting the need to boost NK cells recovery early after transplant (47, 107–109). NK cells have long been utilized in the field of cellular therapy, leveraging their ability to recognize and kill tumor cells in a non-MHC-mediated manner (49). Moreover, they exhibit a better toxicity profile compared to T cells, reducing the risk of GvHD and the need for autologous sources, and opening the possibility of off-the-shelf, ready-to-use products. An important limitation of NK cell therapy is associated with their limited ability to persist in vivo and maintain immunologic memory (42, 110–112).
Various forms of NK cells have been tested in clinical settings, including haploidentical NK cells, cord blood-derived NK cells, stem cell-derived NK cells, NK-CAR cells, and CIML NK cells (113). Devillier and colleagues tested in a phase I study the prophylactic infusion of IL-2 activated NK cells after HCT from MRD with favorable results (114). Colleagues from MD Anderson employed an NK expansion method using K562 feeder cells expressing membrane-bound IL21 and 4-1BBL (FC21) to prepare NK DLI. In this phase I/II study, 25 patients received 3 prophylactic doses of donor NK cells administered on days −2, +7, and +28 after haploidentical HCT. After a median follow-up of 24 months, the 2-year relapse incidence and the LFS were improved compared to contemporaneous 160 case-match controls from the CIBMTR dataset (4% vs. 38%, and 66% vs. 44%, respectively). Only one relapse occurred in the study group, in a patient with a high level of donor-specific anti-HLA antibodies (DSA) presented before transplantation. Acute GvHD occurred in 10 patients and no patient developed chronic GvHD (115). Other trials testing the prophylactic NK cell infusions following HCT are ongoing (NCT03300492 and NCT02452697).
Interleukin 15 (IL-15) is a cytokine that stimulates both CD8+ T cell and NK cell antitumor responses. Romee and colleagues first tested this hypothesis in patients with myeloid malignancies relapsed after HCT using the IL-15 superagonist complex ALT-803. Thirty-three patients were treated with ALT-803 without observing any no dose-limiting toxicities or GvHD. Responses were observed in 19% of evaluable patients, including 1 complete remission lasting 7 months (116). The same group explored the use of recombinant human IL-15 (rhIL-15) after lymphodepleting chemotherapy and haploidentical NK cells. Of 26 treated patients, 36% had robust in vivo NK-cell expansion at day 14, and 32% achieved CR. The use of rhIL-15 was associated with cytokine release syndrome (CRS) in 56% of the cases and some cases of neurological toxicity (117). This study included some patients relapsed after a previous HCT.
CIML NK cells are generated ex vivo after cultivation with IL-12, IL-15, and IL-18, conferring a typical memory-like phenotype, an increased ability to secrete cytokines and exert cytotoxicity, and enhanced in vivo persistence (weeks/months) (118). Furthermore, they displayed excellent anti-leukemic activity both in vitro and in vivo. The first phase I human study (NCT01898793) showed the safety and initial efficacy of allogeneic CIML NK cells, along with concurrent administration of IL-2, in relapsed/refractory (R/R) AML patients (objective response rate - ORR 5/9 patients, including 4 complete responses) (119). These results were confirmed by a subsequent phase I study, demonstrating the safety and initial efficacy of CIML NK cells even in the context of relapse after HCT (120). This study reported the results of the first 6 treated patients, showing a rapid expansion of NK cells, lasting for several months. Fever and cytopenia were reported as the most common adverse events.
NK cell-based DLI have shown promising results, breaking down the GvHD incidence and enhancing the GvL effect. However, the poor persistence of the NK cells remains a major limitation of this approach, leading to an unsatisfactory duration of response and limited benefit in terms of patients OS.
3.5 Cytokine-induced killer cells
CIK cells represent a rare T cell subpopulation, recapitulating both T and NK cell features. CIK cells are obtained starting from peripheral blood mononuclear cells activated in culture in the presence of monoclonal antibodies against CD3 (OKT3), interferon-gamma (IFN-g), and interleukin-2 (IL-2) (121). IL‐2 and fresh culture medium need to be supplemented regularly until after 14-21 days of culture. At this point, an enrichment of CIK cells is observed, characterized by the expression of markers typical of NK cells and T cells, especially defined as CD3+, CD56+, with a prevalent CD8+ phenotype (122). Of note, CIK cells are a cluster of heterogeneous cells comprising CD3+CD56− T cells, CD3−CD56+ NK cells, and CD3+CD56+ NKT cells (121, 123). CIK cells have the unique ability to recognize and kill malignant or infected cells, both through TCR-mediated cytotoxicity typical of T cells and through non-MHC restricted recognition, similar to NK cells, which is the so‐called “dual‐functional capability” (124–126). Indeed, as T cells, CIK cells express the polyclonal TCR repertoire. However, similarly to NK cells, they show an increased expression of cytotoxic receptors including NKG2D, DNAX accessory molecule-1 (DNAM-1), TRAIL, FasL, LFA-1, low density of NKp30 and CD16, and inducing the secretion of perforin and granzyme, but lack expression of NKp44, NKp46, KIR2DL1, KIR2DL2, KIR3DL1, NKG2A, CD94 (122, 127). Thanks to these characteristics, CIK cells have the unique ability to induce a GvL effect with a low risk of exacerbating GvHD (128–131).
In 2010 the International Registry on CIK cells (IRCC) was established, to collect and evaluate clinical trials using CIK cells for the treatment of different types of cancer. In the last update from the IRCC, a total of 106 clinical trials including 10,225 patients were collected, of which 4,889 patients in over 30 distinct tumor entities were treated with CIK cells alone or in combination with conventional or novel therapies. Of note, only 8 and 5 studies used allogeneic CIK cells and UCB-derived CIK cells, respectively. CIK dose varies between studies, with a wide range of 7.9 × 108 to 7.9 × 1010. The number of infusions also varied according to the treatment efficiency or adverse effects. Ten studies reported that more cycles of CIK cell infusion were significantly related to the prolonged OS and PFS (132). In this meta-analysis including mostly solid tumor trials, the ORR was 38% with 9 studies reporting a significantly increased 5‐year survival rate. Moreover, mild adverse effects were observed and the rate of acute and chronic GvHD was modest and in most cases was controlled by the administration of immunosuppressive corticosteroids (133, 134). Initial studies using autologous CIK cells showed feasibility and no adverse events, but limited efficacy (135–137). Subsequent clinical trials tested allogeneic CIK cells in the setting of post-HCT relapse (138–148) Results of these trials are summarized in Table 3. Globally, these data confirmed the safe profile of CIK cells and the low rate of GvHD (not exciding 25% for aGvHD).
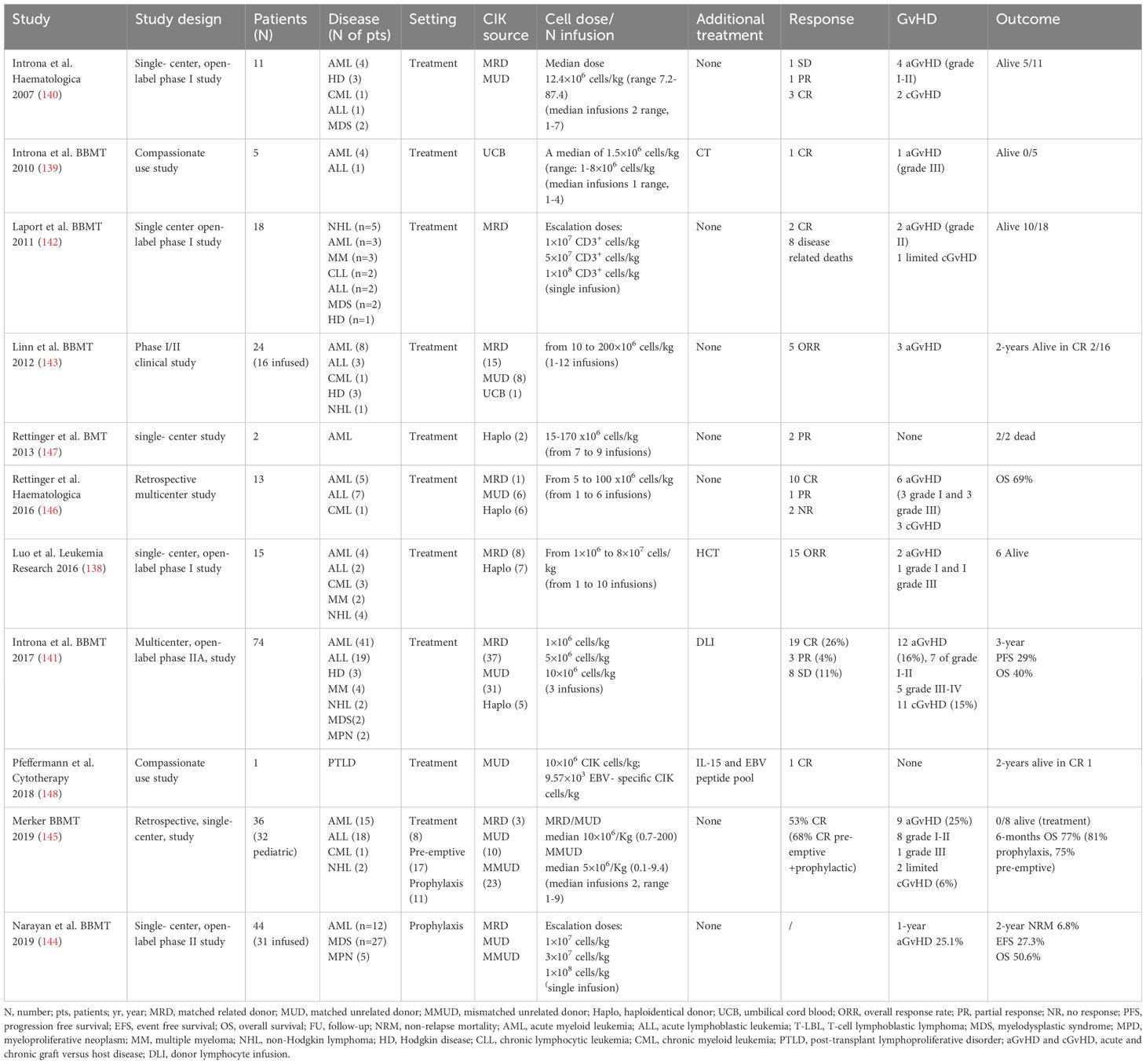
Table 3. Clinical trials on cytokine-induced killer (CIK) cell after HCT.
Our group showed promising results generated by phase I studies demonstrating the feasibility and safety of clinical-grade allogeneic CIK cell production (139, 140). Moreover, we conducted a phase II study on the sequential use of DLI and CIK in 74 patients relapsed post-transplant, demonstrating a low incidence of GvHD and adequate disease control. Acute GvHD was observed in 16% of patients, of these, 7 presented with grade I-II and in 5 with grade III-IV. Chronic GvHD manifested in 15% of cases. A CR was observed in 26%, partial response (PR) in 4%, stable disease (SD) in 11%, and progression in 56% of cases. PFS at 1 and 3 years was 31% and 29%, respectively (141). Other groups have confirmed the low rates of acute and chronic GvHD after CIK infusion as a treatment for post-HCT relapse (142, 144, 145). In addition, a recent retrospective analysis from the Frankfurt group, comparing CIK cell infusions with DLI in a pediatric enriched cohort of HCT patients showed a low rate of acute and chronic GvHD and exciting outcomes when used in a prophylactic/pre-emptive setting (145). Of note, in this study, 63% of patients had a MMUD donor and CIK cells were produced by adding IL-15 and harvested after a shorter period (10-12 days of culture). In view of this low risk of alloreactivity, a phase I/II trial using haploidentical donor derived CIK cells for post-HCT relapse is currently ongoing at our institution (NCT03821519).
Finally, CIK cell therapy provides a suitable platform for combination strategies with monoclonal antibody (anti-CD20) (149), bispecific drugs, such as blinatumomab (anti-CD3 and anti-CD19) (150), immune checkpoint inhibitors including PD‐1, PD‐L1, KIR, LAG‐3, or TIM‐3 (151) or gene modification through the insertion of Chimeric Antigen Receptor (CAR) (152). The latter strategy has also been tested for AML (153–155). Also, some studies demonstrated that CIK cells stimulated by or armored with IL‐6, IL‐7, IL‐12, IL‐15, IL‐21 or thymoglobulin manifested phenotype alteration, proliferation improvement, and cytotoxicity enhancement (156–160).
Data from different trials conducted in various institutions worldwide have shown the ability of CIK cell infusions to boost the GvL activity, with high rate of response especially in the setting of MRD and pre-emptive therapy and with a minimum impact on GvHD onset, leading to an OS benefit.
4 Genetically engineered cell therapy strategies
4.1 Chimeric antigen receptor T cells
CAR-T cell therapy is an emerging novel therapeutic strategy in the context of relapsed/refractory lymphomas and leukemias, including AML (161). CARs have revolutionized the concept of immunotherapy by combining the specificity of monoclonal antibodies with the cytotoxic activity mediated by T-cells against tumor-associated antigens (TAA). The CAR consists of an extracellular antigen-binding domain, derived from the fusion of a variable portion of the light and heavy chain of immunoglobulins (scFv), and an intracellular portion characterized by one or more activating domains for T-cells, particularly the zeta chain of CD3 belonging to the T-cell receptor (TCR) complex (162). In the beginning, the approach for CAR-T cell development involved the use of viral vectors (adenovirus or lentivirus). However, the high production costs and complex manufacturing processes limited their clinical use. The introduction of non-viral transfections, using transposons through the Sleeping Beauty (SB) system, has proven to be an efficient and safe alternative (163, 164). Besides the choice of CAR construct design and the method of transgene insertion into the cell genome (viral or non-viral), the selection of the effector cell can have an impact on the treatment outcome.
Autologous CAR-T cell therapy has shown excellent results in the treatment of B-cell malignancies, such as B-cell acute lymphoblastic leukemia (B-ALL), B-cell non-Hodgkin lymphomas (B-NHL), chronic lymphocytic leukemia (CLL), and multiple myeloma (MM) (165–173). Since 2017, six different CAR-T cell therapies have been approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), including 4 anti-CD19 CAR-Ts and 2 anti-BCMA CAR-Ts for the treatment of B-ALL, B-NHL, and MM (174).
Differently, the use of allogeneic T cells poses unique challenges owing to potential alloreactivity. Allogeneic CAR-T cells can be collected either from an allogeneic donor (donor-derived) or from patients who relapsed after HCT (patient-derived). Table 4 reported studies using either autologous or allogeneic CAR source as long as administrated after an HCT. Patient-derived donor T cells may be expected to carry a lesser risk of acute and chronic GVHD if the CAR T cells are generated from tolerized cells. Of note, data on patient-derived allogeneic CAR cells were collected from trials using mostly autologous CAR-T outside the setting of HCT. In these trials, the rate of GVHD was often not reported, but some of these studies show low rate or absence of GvHD after CAR-T cell therapy (166, 175–182). However, more recent studies did report some incidence of both aGvHD and cGvHD (183, 184). Unfortunately, the value of donor chimerism at the time of lymphocyte apheresis was nearly always not available.
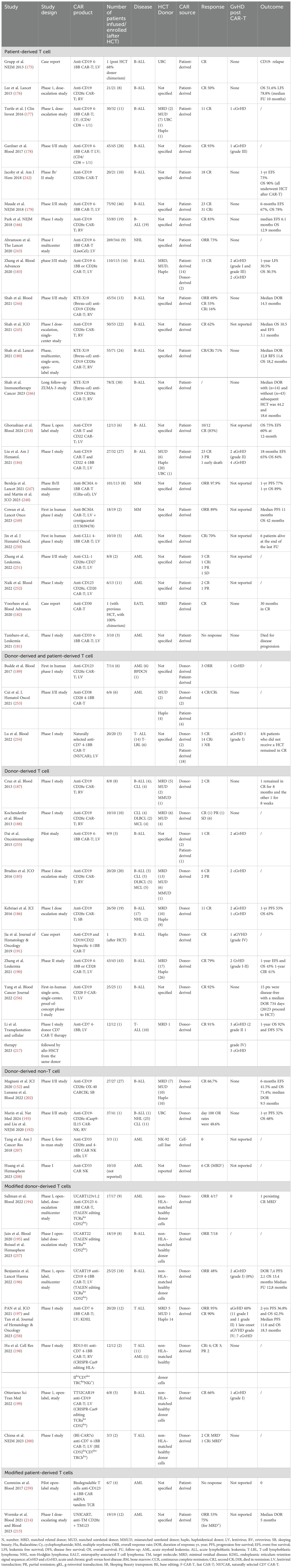
Table 4. Clinical trials on chimeric antigen receptor (CAR) cell therapy after HCT.
Surprisingly, initial reports using donor-derived allogeneic CAR-T cells showed a low incidence of GvHD. These studies, however, did not include lymphodepleting regimens and showed limited efficacy (185–188) More recent trials have tested allogeneic CAR-T cells with lymphodepletion in the setting of disease recurrence after HCT (189, 190) and even haploidentical HCT (184, 191) with positive results.
However, several strategies are under investigation to mitigate the risk of causing or aggravating GvHD. One possibility is to choose different effector cells other than T cells, such as CIK cells (152) or NK cells (192, 193). Another possibility is to perform gene editing to delete the endogenous TCR (194–200). Interestingly, with the exemption of two studies (196, 197), no GvHD was reported in this series of patients. Finally, also the CAR-T cell production method plays a role in improving treatment efficacy. The goal is to favor protocols that promote the differentiation of T cells into stem memory cells (SCM) or central memory T cells (CM), which have shown better anti-tumor activity compared to effector memory T cells (EM) (201).
CAR cell therapy has shown unprecedented results in different hematologic diseases, however data on the use of allogeneic products is still limited.
4.2 Alternative effector cell sources: genetically modified CIK and NK cells
CIK cells can be employed as alternative effectors for CAR cell therapy. Anti-CD19 CAR-CIK cells have shown an excellent safety profile and good response rates in patients with post-HCT R/R B-ALL (152, 202). In this study, no GVHD following CAR-CIK was reported. Preclinical studies using CAR-CIK cells against CD33 and CD123 have shown promising results also in the setting of AML (153–155). CAR-CIK cells were also tested in vitro against CD44v6 with encouraging results (203). In addition, one case report form China, showed a transient response in an AML patient treated with autologous CAR-CIK cells direct against CD33 (204). Finally, preclinical data showed that CAR cell activity can be enhanced by increasing BM homing through the expression of specific chemokines (CXCR4) (205) or by armoring them to constitutively secrete cytotoxic cytokine (IL-18) (206).
CAR-NK cells can be generated from either NK tumor lines (NK92), induced pluripotent stem cells (iPSCs) differentiated into NK cells, or isolated NK cells from peripheral or UCB (113). UCB-derived CAR-NK cells demonstrated impressive clinical efficacy in CD19-positive diseases (192, 193). In this study, only one patient presented a post-HCT relapse/refractory disease and no case of GvHD was reported. NK92-derived CAR-NK cells, targeting CD33 and constitutively secreting IL-2, were used to treat 3 patients with relapsed/refractory AML. This study observed no toxicity but also no efficacy (207). NK cells derived from NK92 do not express some activating/cytotoxic receptors such as CD16 and NKp44, representing a limitation of this approach. Huang and colleagues presented preliminary data from a phase I study on multiple infusions of allogeneic CAR-NK cells targeting CD33 in 10 patients with relapsed/refractory AML, reporting no toxicity, and 6 out of 10 patients achieved MRD-negative complete remission by day 28 (208). Allogeneic CAR-NK cells targeting CD123, derived from the peripheral blood of healthy donors, demonstrating anti-leukemic activity in vitro and in vivo without toxicity to the normal healthy myeloid compartment (209). Finally, Dong and colleagues proposed an innovative approach, where NK cells were induced to differentiate into CIML-like NK cells and then transduced with a CAR recognizing the neopeptide derived from cytosolic oncogenic nucleophosmin-1 (NPM1c), presented by HLA-A2 (210). Currently, several clinical trials are recruiting patients with relapsed/refractory AML for treatment with CAR-NK cells targeting different epitopes: CD123, CD33, CCL1, and NKG2DL (211).
Both allogeneic donor-derived CAR-CIK and CAR-NK cell strategies have shown to exert a potent GvL activity with a minimum GvL toxicity. Long-term survival analysis and data on CAR cell persistence are needed to select the best strategy.
4.3 Novel strategy to improve CAR-T cell therapy
One goal for the improvement of cellular therapy with CAR-T cells is the use of fit, ready to use donor T cells, without exacerbating the risk of GvHD. Gene editing can be used to manipulate the T cell, by deleting the endogenous TCR, allowing the use of allogeneic non matched or third part T cells. Most studies on T-cell editing initially began by deleting a TCR gene, then introducing a CAR using a retroviral or lentiviral vector (212).
UCART are gene-edited CAR-T cells engineered using a TALEN editing strategy, to knock down the TCR and the CD52 receptor. TALENs are chimeric proteins that contain two functional domains: a DNA-recognition transcription activator-like effector (TALE) and a nuclease domain. This technology allows the use of third-party T cells, tearing down the risk of GvHD and the use of alemtuzumab within the lymphodepleting regimen without the risk of CAR-T depletion. Indeed in 3 different studies only 2 cases of grade I aGvHD were reported (194–196).
In the setting of anti-CD7 CAR T cells, different editing strategies were used to block the expression of CD7, the T cell receptor (TCR), and human leukocyte antigen (HLA) class II, to avoid CAR T cell fratricide, GvHD, and rejection, respectively (197–200). Furthermore, in the study of Hu and colleagues, the expression of the NK inhibitor (NKi) and the common cytokine receptor gamma chain (gc) was induced to enhance CAR-T cell cytotoxic activity (198).
However, an interesting mouse study from the Memorial Sloan Kettering Cancer Center showed that allogeneic second generation CD28 CAR-T cells have less GvHD capacity while maintaining an anti-lymphoma capability compared to first generation or second generation 4-1BB CAR-T cells (213). These data suggested a natural tolerogenic profile of CD28 CAR-T cells, while maintaining their endogenous TCR.
Another approach to reduced CAR-T cell toxicity involves the “universal” CAR-T cells (UniCAR). This second-generation autologous CAR construct using a CD28 costimulatory domain is designed to bind a soluble target module (TM) able to bind the tumor antigen (CD123). In this phase I study, 19 patients were treated, of which 12 after a relapse post-HCT. The ORR was 53% and no case of GvHD was reposted (214, 215). Interestingly, treatment-related toxicities quickly resolved with the suspension of the target module administration.
Finally, to overcome the selection of the therapeutic target considering the shared expression of antigens by both myeloid precursors and malignant cells, valid alternatives include post-CAR-T cell HCT (216, 217), targeting AML specific antigens (such as NPM1) (210), and the use of dual-targeting CAR-T cells capable of recognizing more than one target and fully activated only when both antigens are expressed on the target cell (155). In addition, by targeting multiple antigens, the dual-CAR construct can potentially overcome the tumor immune escape arising from antigen-negative disease, such as in the case of CD19 loss (218).
Although the real risk of GvHD after donor-derived CAR-T cells is still not well defined yet, novel gene editing strategies can be effectively employed to mitigate the T cell alloreactivity in the context of allogeneic or third-part CAR-T cell therapy.
5 Conclusions
Despite the successful rate of cure after HCT, a considerable fraction of patients still experiences disease recurrence or persistence. CIK cell-based therapy represents a suitable approach to boost the GvL activity without increasing GvHD. Indeed, CIK cells offer an allogeneic cell platform with a tolerogenic profile and can be considered also for a third-part ready to use cellular therapy. In addition, novel strategy using CAR-CIK cells showed efficacy with limited toxicity and no report of GvHD in a setting of heavily pretreated patients. Major limitations are represented by the limited efficacy in the setting of overt hematological relapse and lack of suitable targets and severe toxicities in the myeloid setting. Moreover, high-level evidence coming from phase III randomized controlled trials (RCTs) is needed in this field. Future perspective includes the use of prophylactic CIK cell infusion, the use of novel modified CIK cells, such as armored-CIK cells or CAR-CIK cells for the treatment of disease recurrence after HCT.
Author contributions
BR: Writing – review & editing, Writing – original draft, Visualization, Project administration, Methodology, Data curation, Conceptualization. GR: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. AR: Writing – review & editing, Writing – original draft, Visualization, Conceptualization. MI: Writing – review & editing, Writing – original draft, Visualization, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Associazione Italiana per la Ricerca sul Cancro, Grant/Award Number: 22791; 5x1000 ISM, Grant/Award Number: 21147.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Copelan EA. Hematopoietic Stem-Cell Transplantation. N Engl J Med. (2006) 354:1813–26. doi: 10.1056/NEJMra052638
2. Passweg JR, Baldomero H, Chabannon C, Basak GW, de la Cámara R, Corbacioglu S, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transplant. (2021) 56:1651–64. doi: 10.1038/s41409-021-01227-8
3. Dholaria B, Savani BN, Hamilton BK, Oran B, Liu HD, Tallman MS, et al. Hematopoietic Cell Transplantation in the Treatment of Newly Diagnosed Adult Acute Myeloid Leukemia: An Evidence-Based Review from the American Society of Transplantation and Cellular Therapy. Transplant Cell Ther. (2021) 27:6–20. doi: 10.1016/j.bbmt.2020.09.020
4. Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. (2016) 127:62–70. doi: 10.1182/blood-2015-07-604546
5. Keating AK, Langenhorst J, Wagner JE, Page KM, Veys P, Wynn RF, et al. The influence of stem cell source on transplant outcomes for pediatric patients with acute myeloid leukemia. Blood Adv. (2019) 3:1118–28. doi: 10.1182/bloodadvances.2018025908
6. Bolon YT AR, Allbee-Johnson M, Estrada-Merly N LS. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR summary slide. (2023).
7. Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the Intensity of Conditioning Regimens: Working Definitions. Biol Blood Marrow Transplant. (2009) 15:1628–33. doi: 10.1016/j.bbmt.2009.07.004
8. Spyridonidis A, Labopin M, Savani BN, Niittyvuopio R, Blaise D, Craddock C, et al. Redefining and measuring transplant conditioning intensity in current era: a study in acute myeloid leukemia patients. Bone Marrow Transplant. (2020) 55:1114–25. doi: 10.1038/s41409-020-0803-y
9. Scott BL, Pasquini MC, Fei M, Fraser R, Wu J, Devine SM, et al. Myeloablative versus Reduced-Intensity Conditioning for Hematopoietic Cell Transplantation in Acute Myelogenous Leukemia and Myelodysplastic Syndromes—Long-Term Follow-Up of the BMT CTN 0901 Clinical Trial. Transplant Cell Ther. (2021) 27:483.e1–6. doi: 10.1016/j.jtct.2021.02.031
10. Zilberberg J, Feinman R, Korngold R. Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol Blood Marrow Transplant. (2015) 21:1000–7. doi: 10.1016/j.bbmt.2014.11.001
11. Barnes D, Corp M, Loutit J, Neal F. Treatment of murine leukaemia with X rays and homologous bone marrow; preliminary communication. Br Med J. (1956) 2:626–7. doi: 10.1136/bmj.2.4993.626
12. Gale RP, Horowitz MM, Ash RC, Champlin RE, Goldman JM, Rimm AA, et al. Identical-twin bone marrow transplants for leukemia. Ann Intern Med. (1994) 120:646–52. doi: 10.7326/0003-4819-120-8-199404150-00004
13. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb H-J, et al. Graft-Versus-Leukemia Reactions After Bone Marrow Transplantation. Blood. (1990) 75:555–62. doi: 10.1182/blood.V75.3.555.555
14. Zeiser R, Vago L. Mechanisms of immune escape after allogeneic hematopoietic cell transplantation. Blood. (2019) 133:1290–7. doi: 10.1182/blood-2018-10-846824
15. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MTL, et al. Loss of Mismatched HLA in Leukemia after Stem-Cell Transplantation. N Engl J Med. (2009) 361:478–88. doi: 10.1056/NEJMoa0811036
16. Crucitti L, Crocchiolo R, Toffalori C, Mazzi B, Greco R, Signori A, et al. Incidence, risk factors and clinical outcome of leukemia relapses with loss of the mismatched HLA after partially incompatible hematopoietic stem cell transplantation. Leukemia. (2015) 29:1143–52. doi: 10.1038/leu.2014.314
17. Ahci M, Toffalori C, Bouwmans E, Crivello P, Brambati C, Pultrone C, et al. A new tool for rapid and reliable diagnosis of HLA loss relapses after HSCT. Blood. (2017) 130:1270–3. doi: 10.1182/blood-2017-05-784306
18. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
19. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. (2019) 25:603–11. doi: 10.1038/s41591-019-0400-z
20. Adachi Y, Sakai T, Terakura S, Shiina T, Suzuki S, Hamana H, et al. Downregulation of HLA class II is associated with relapse after allogeneic stem cell transplantation and alters recognition by antigen-specific T cells. Int J Hematol. (2022) 115:371–81. doi: 10.1007/s12185-021-03273-w
21. Penack O, Marchetti M, Ruutu T, Aljurf M, Bacigalupo A, Bonifazi F, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol. (2020) 7:e157–67. doi: 10.1016/S2352-3026(19)30256-X
22. von Boehmer H. Positive and negative selection of the αβ T-cell repertoire. vivo. Curr Opin Immunol. (1991) 3:210–5. doi: 10.1016/0952-7915(91)90052-3
23. Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res. (2020) 48:D948–55. doi: 10.1093/nar/gkz950
24. Anasetti C, Amos D, Beatty PG, Appelbaum FR, Bensinger W, Buckner CD, et al. Effect of HLA Compatibility on Engraftment of Bone Marrow Transplants in Patients with Leukemia or Lymphoma. N Engl J Med. (1989) 320:197–204. doi: 10.1056/NEJM198901263200401
25. Fleischhauer K, Shaw BE. HLA-DP in unrelated hematopoietic cell transplantation revisited: challenges and opportunities. Blood. (2017) 130:1089–96. doi: 10.1182/blood-2017-03-742346
26. Shaw BE, Gooley TA, Malkki M, Madrigal JA, Begovich AB, Horowitz MM, et al. The importance of HLA-DPB1 in unrelated donor hematopoietic cell transplantation. Blood. (2007) 110:4560–6. doi: 10.1182/blood-2007-06-095265
27. Fleischhauer K, Shaw BE, Gooley T, Malkki M, Bardy P, Bignon J-D, et al. Effect of T-cell-epitope matching at HLA-DPB1 in recipients of unrelated-donor haemopoietic-cell transplantation: a retrospective study. Lancet Oncol. (2012) 13:366–74. doi: 10.1016/S1470-2045(12)70004-9
28. Ibisch C, Gallot G, Vivien R, Diez E, Jotereau F, Garand R, et al. Recognition of leukemic blasts by HLA-DPB1-specific cytotoxic T cell clones: a perspective for adjuvant immunotherapy post-bone marrow transplantation. Bone Marrow Transplant. (1999) 23:1153–9. doi: 10.1038/sj.bmt.1701768
29. Petersdorf W, Malkki M, O’hUigin C, Carrington M, Gooley T, Haagenson M, et al. High HLA-DP Expression and Graft-versus-Host Disease. N Engl J Med. (2024) 373:599–609. doi: 10.1056/NEJMoa1500140
30. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. (2003) 9:1144–50. doi: 10.1038/nm915
31. van Bergen CAM, van Luxemburg-Heijs SAP, de Wreede LC, Eefting M, von dem Borne PA, van Balen P, et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J Clin Invest. (2017) 127:517–29. doi: 10.1172/JCI86175
32. Tram K, Stritesky G, Wadsworth K, Ng J, Anasetti C, Dehn J. Identification of DPB1 Permissive Unrelated Donors Is Highly Likely. Biol Blood Marrow Transplant. (2017) 23:81–6. doi: 10.1016/j.bbmt.2016.10.021
33. Rutten CE, van Luxemburg-Heijs SAP, Halkes CJM, van Bergen CAM, Marijt EWA, Oudshoorn M, et al. Patient HLA-DP&x2013;Specific CD4+ T Cells from HLA-DPB1&x2013;Mismatched Donor Lymphocyte Infusion Can Induce Graft-versus-Leukemia Reactivity in the Presence or Absence of Graft-versus-Host Disease. Biol Blood Marrow Transplant. (2013) 19:40–8. doi: 10.1016/j.bbmt.2012.07.020
34. Herr W, Eichinger Y, Beshay J, Bloetz A, Vatter S, Mirbeth C, et al. HLA-DPB1 mismatch alleles represent powerful leukemia rejection antigens in CD4 T-cell immunotherapy after allogeneic stem-cell transplantation. Leukemia. (2017) 31:434–45. doi: 10.1038/leu.2016.210
35. Morishima Y, Kashiwase K, Matsuo K, Azuma F, Morishima S, Onizuka M, et al. Biological significance of HLA locus matching in unrelated donor bone marrow transplantation. Blood. (2015) 125:1189–97. doi: 10.1182/blood-2014-10-604785
36. Luznik L, O’Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. (2008) 14:641–50. doi: 10.1016/j.bbmt.2008.03.005
37. Huang X, Liu DH, Liu KY, Xu LP, Chen H, Han W, et al. Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological malignancies. Bone Marrow Transplant. (2006) 38:291–7. doi: 10.1038/sj.bmt.1705445
38. Aversa F, Terenzi A, Tabilio A, Falzetti F, Carotti A, Ballanti S, et al. Full Haplotype-Mismatched Hematopoietic Stem-Cell Transplantation: A Phase II Study in Patients With Acute Leukemia at High Risk of Relapse. J Clin Oncol. (2005) 23:3447–54. doi: 10.1200/JCO.2005.09.117
39. Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of High-Risk Acute Leukemia with T-Cell–Depleted Stem Cells from Related Donors with One Fully Mismatched HLA Haplotype. N Engl J Med. (1998) 339:1186–93. doi: 10.1056/NEJM199810223391702
40. Locatelli F, Merli P, Pagliara D, Li Pira G, Falco M, Pende D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood. (2017) 130:677–85. doi: 10.1182/blood-2017-04-779769
41. Velardi A, Mancusi A, Ruggeri L, Pierini A. How adoptive transfer of components of the donor immune system boosts GvL and prevents GvHD in HLA-haploidentical hematopoietic transplantation for acute leukemia. Bone Marrow Transplant. (2024) 59:301–5. doi: 10.1038/s41409-024-02199-1
42. Vivier E, Rebuffet L, Narni-Mancinelli E, Cornen S, Igarashi RY, Fantin VR. Natural killer cell therapies. Nature. (2024) 626:727–36. doi: 10.1038/s41586-023-06945-1
43. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. NK cell education after allogeneic transplantation: dissociation between recovery of cytokine-producing and cytotoxic functions. Blood. (2011) 118:2784–92. doi: 10.1182/blood-2011-04-347070
44. Vacca P, Montaldo E, Croxatto D, Moretta F, Bertaina A, Vitale C, et al. NK Cells and Other Innate Lymphoid Cells in Hematopoietic Stem Cell Transplantation. Front Immunol. (2016) 7:188. doi: 10.3389/fimmu.2016.00188
45. Simonetta F, Alvarez M, Negrin RS. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front Immunol. (2017) 8:465. doi: 10.3389/fimmu.2017.00465
46. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
47. Russo A, Oliveira G, Berglund S, Greco R, Gambacorta V, Cieri N, et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: dynamics and clinical implications. Blood. (2018) 131:247–62. doi: 10.1182/blood-2017-05-780668
48. Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, Yong ASM, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia. (2007) 21:2145–52. doi: 10.1038/sj.leu.2404892
49. Caligiuri MA. Human natural killer cells. Blood. (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
50. Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. (2011) 32:364–72. doi: 10.1016/j.it.2011.06.001
51. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of Natural Killer Cell Alloreactivity in HLA-Mismatched Hematopoietic Stem Cell Transplantation. Blood. (1999) 94:333–9. doi: 10.1182/blood.V94.1.333.413a31_333_339
52. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Sci (80- ). (2002) 295:2097–100. doi: 10.1126/science.1068440
53. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of Antileukemia Effects of Allogeneic NK Cells 1. J Immunol. (2004) 172:644–50. doi: 10.4049/jimmunol.172.1.644
54. Nowak J, Kościńska K, Mika-Witkowska R, Rogatko-Koroś M, Mizia S, Jaskuła E, et al. Donor NK cell licensing in control of malignancy in hematopoietic stem cell transplant recipients. Am J Hematol. (2014) 89:E176–83. doi: 10.1002/ajh.23802
55. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood. (2009) 113:726–32. doi: 10.1182/blood-2008-07-171926
56. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. (2010) 116:2411–9. doi: 10.1182/blood-2010-05-283051
57. Downing J, D’Orsogna L. High-resolution human KIR genotyping. Immunogenetics. (2022) 74:369–79. doi: 10.1007/s00251-021-01247-0
58. Petersdorf EW, Stevenson P, Bengtsson M, De Santis D, Dubois V, Gooley T, et al. HLA-B leader and survivorship after HLA-mismatched unrelated donor transplantation. Blood. (2020) 136:362–9. doi: 10.1182/blood.2020005743
59. Sajulga R, Bolon Y-T, Maiers MJ, Petersdorf EW. Assessment of HLA-B genetic variation with an HLA-B leader tool and implications in clinical transplantation. Blood Adv. (2022) 6:270–80. doi: 10.1182/bloodadvances.2021004561
60. Solomon SR, Aubrey MT, Zhang X, Jackson KC, Roark CL, Freed BM, et al. Lineage-Specific Relapse Prediction After Haploidentical Transplantation With Post-Transplant Cyclophosphamide Based on Recipient HLA-B-Leader Genotype and HLA-C-Group KIR Ligand. Transplant Cell Ther. (2022) 28:601.e1–8. doi: 10.1016/j.jtct.2022.06.023
61. Petersdorf EW, Gooley T, Volt F, Kenzey C, Madrigal A, McKallor C, et al. Use of the HLA-B leader to optimize cord blood transplantation. Haematologica. (2020) 106:3107–14. doi: 10.3324/haematol.2020.264424
62. Milano F, Gooley T, Wood B, Woolfrey A, Flowers ME, Doney K, et al. Cord-Blood Transplantation in Patients with Minimal Residual Disease. N Engl J Med. (2016) 375:944–53. doi: 10.1056/NEJMoa1602074
63. Verneris MR, Brunstein CG, Barker J, MacMillan ML, DeFor T, McKenna DH, et al. Relapse risk after umbilical cord blood transplantation: enhanced graft-versus-leukemia effect in recipients of 2 units. Blood. (2009) 114:4293–9. doi: 10.1182/blood-2009-05-220525
64. Horgan C, Mullanfiroze K, Rauthan A, Patrick K, Butt NA, Mirci-Danicar O, et al. T-cell replete cord transplants give superior outcomes in high-risk and relapsed/refractory pediatric myeloid malignancy. Blood Adv. (2023) 7:2155–65. doi: 10.1182/bloodadvances.2022009253
65. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. (2014) 124:638–44. doi: 10.1182/blood-2014-03-564401
66. Pierini A, Ruggeri L, Carotti A, Falzetti F, Saldi S, Terenzi A, et al. Haploidentical age-adapted myeloablative transplant and regulatory and effector T cells for acute myeloid leukemia. Blood Adv. (2021) 5:1199–208. doi: 10.1182/bloodadvances.2020003739
67. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell–depleted stem cell grafts. J Clin Invest. (2015) 125:2677–89. doi: 10.1172/JCI81229
68. Bleakley M, Sehgal A, Seropian S, Biernacki MA, Krakow EF, Dahlberg A, et al. Naive T-Cell Depletion to Prevent Chronic Graft-Versus-Host Disease. J Clin Oncol. (2022) 40:1174–85. doi: 10.1200/JCO.21.01755
69. Sanders JE, Buckner CD, Clift RA, Fefer A, McGuffin R, Storb R, et al. Second marrow transplants in patients with leukemia who relapse after allogeneic marrow transplantation. Bone Marrow Transplant. (1988) 3:11–9.
70. Göksoy HS, Arat M. The use of second allogeneic hematopoietic stem cell transplantation for hematologic malignancies relapsed after the first: Does it worth to do? Transfus Apher Sci. (2016) 54:91–8. doi: 10.1016/j.transci.2016.01.020
71. Yerushalmi Y, Shem-Tov N, Danylesko I, Canaani J, Avigdor A, Yerushalmi R, et al. Second hematopoietic stem cell transplantation as salvage therapy for relapsed acute myeloid leukemia/myelodysplastic syndromes after a first transplantation. Haematologica. (2023) 108:1782–92. doi: 10.3324/haematol.2022.281877
72. Vago L, Toffalori C, Ahci M, Lange V, Lang K, Todaro S, et al. Incidence of HLA Loss in a Global Multicentric Cohort of Post-Transplantation Relapses: Results from the Hlaloss Collaborative Study. Blood. (2018) 132:818. doi: 10.1182/blood-2018-99-112142
73. Rovatti PE, Gambacorta V, Lorentino F, Ciceri F, Vago L. Mechanisms of Leukemia Immune Evasion and Their Role in Relapse After Haploidentical Hematopoietic Cell Transplantation. Front Immunol. (2020) 11:147. doi: 10.3389/fimmu.2020.00147
74. Vago L, Ciceri F. Choosing the Alternative. Biol Blood Marrow Transplant. (2017) 23:1813–4. doi: 10.1016/j.bbmt.2017.09.009
75. Imus PH, Blackford AL, Bettinotti M, Iglehart B, Dietrich A, Tucker N, et al. Major Histocompatibility Mismatch and Donor Choice for Second Allogeneic Bone Marrow Transplantation. Biol Blood Marrow Transplant. (2017) 23:1887–94. doi: 10.1016/j.bbmt.2017.07.014
76. Christopeit M, Kuss O, Finke J, Bacher U, Beelen DW, Bornhäuser M, et al. Second allograft for hematologic relapse of acute leukemia after first allogeneic stem-cell transplantation from related and unrelated donors: The role of donor change. J Clin Oncol. (2013) 31:3259–71. doi: 10.1200/JCO.2012.44.7961
77. Shimoni A, Labopin M, Finke J, Ciceri F, Deconinck E, Kröger N, et al. Donor selection for a second allogeneic stem cell transplantation in AML patients relapsing after a first transplant: a study of the Acute Leukemia Working Party of EBMT. Blood Cancer J. (2019) 9:88. doi: 10.1038/s41408-019-0251-3
78. Kharfan-Dabaja MA, Labopin M, Bazarbachi A, Ciceri F, Finke J, Bruno B, et al. Comparing outcomes of a second allogeneic hematopoietic cell transplant using HLA-matched unrelated versus T-cell replete haploidentical donors in relapsed acute lymphoblastic leukemia: a study of the Acute Leukemia Working Party of EBMT. Bone Marrow Transplant. (2021) 56:2194–202. doi: 10.1038/s41409-021-01317-7
79. Watanabe O, Yamamoto H, Yamaguchi K, Kageyama K, Kaji D, Taya Y, et al. Single CBT As 2nd Allo-SCT Is Promising for Selected Patients with Relapsed Hematopoietic Malignancies. Blood. (2023) 142:7096. doi: 10.1182/blood-2023-188946
80. Troullioud Lucas AG, Boelens JJ, Prockop SE, Curran KJ, Bresters D, Kollen W, et al. Excellent leukemia control after second hematopoietic cell transplants with unrelated cord blood grafts for post-transplant relapse in pediatric patients. Front Oncol. (2023) 13:1221782. doi: 10.3389/fonc.2023.1221782
81. Castelli M, Grassi A, Algarotti A, Micò MC, Lussana F, Finazzi MC, et al. Outcomes of second allogeneic hematopoietic stem cell transplantation for hematologic diseases: a single-center real-world experience. EBMT. (2024).
82. Schmid C, Kuball J, Bug G. Defining the Role of Donor Lymphocyte Infusion in High-Risk Hematologic Malignancies. J Clin Oncol. (2021) 39:397–418. doi: 10.1200/JCO.20.01719
83. Pagliuca S, Schmid C, Santoro N, Simonetta F, Battipaglia G, Guillaume T, et al. Donor lymphocyte infusion after allogeneic haematopoietic cell transplantation for haematological malignancies: basic considerations and best practice recommendations from the EBMT. Lancet Haematol. (2024) 11:e448–58. doi: 10.1016/S2352-3026(24)00098-X
84. Maurer K, Antin JH. The graft versus leukemia effect: donor lymphocyte infusions and cellular therapy. Front Immunol. (2024) 15:1328858. doi: 10.3389/fimmu.2024.1328858
85. Kolb HJ, Mittermuller J, Clemm C, Holler E, Ledderose G, Brehm G, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. (1990) 76:2462–5. doi: 10.1182/blood.v76.12.2462.2462
86. Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. (2004) 103:767–76. doi: 10.1182/blood-2003-02-0342
87. Schmid C, Labopin M, Nagler A, Bornhäuser M, Finke J, Fassas A, et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: A retrospective risk factors analysis and comparison with other strategies by the EBMT acute leukem. J Clin Oncol. (2007) 25:4938–45. doi: 10.1200/JCO.2007.11.6053
88. Schroeder T, Rachlis E, Bug G, Stelljes M, Klein S, Steckel NK, et al. Treatment of Acute Myeloid Leukemia or Myelodysplastic Syndrome Relapse after Allogeneic Stem Cell Transplantation with Azacitidine and Donor Lymphocyte Infusions&x2014;A Retrospective Multicenter Analysis from the German Cooperative Transplant Study Gro. Biol Blood Marrow Transplant. (2015) 21:653–60. doi: 10.1016/j.bbmt.2014.12.016
89. Guillaume T, Thépot S, Peterlin P, Ceballos P, Bourgeois AL, Garnier A, et al. Prophylactic or Preemptive Low-Dose Azacitidine and Donor Lymphocyte Infusion to Prevent Disease Relapse following Allogeneic Transplantation in Patients with High-Risk Acute Myelogenous Leukemia or Myelodysplastic Syndrome. Transplant Cell Ther. (2021) 27:839.e1–6. doi: 10.1016/j.jtct.2021.06.029
90. Al-Shaibani E, Bautista R, Lipton JH, Kim DD, Viswabandya A, Kumar R, et al. Comparison of Outcomes After Second Allogeneic Hematopoietic Cell Transplantation Versus Donor Lymphocyte Infusion in Allogeneic Hematopoietic Cell Transplant Patients. Clin Lymphoma Myeloma Leuk. (2022) 22:e327–34. doi: 10.1016/j.clml.2021.11.004
91. Kharfan-Dabaja MA, Labopin M, Polge E, Nishihori T, Bazarbachi A, Finke J, et al. Association of Second Allogeneic Hematopoietic Cell Transplant vs Donor Lymphocyte Infusion With Overall Survival in Patients With Acute Myeloid Leukemia Relapse. JAMA Oncol. (2018) 4:1245–53. doi: 10.1001/jamaoncol.2018.2091
92. Scarisbrick JJ, Dignan FL, Tulpule S, Gupta ED, Kolade S, Shaw B, et al. A multicentre UK study of GVHD following DLI: Rates of GVHD are high but mortality from GVHD is infrequent. Bone Marrow Transplant. (2015) 50:62–7. doi: 10.1038/bmt.2014.227
93. Alyea EP, Canning C, Neuberg D, Daley H, Houde H, Giralt S, et al. CD8+ cell depletion of donor lymphocyte infusions using cd8 monoclonal antibody-coated high-density microparticles (CD8-HDM) after allogeneic hematopoietic stem cell transplantation: a pilot study. Bone Marrow Transplant. (2004) 34:123–8. doi: 10.1038/sj.bmt.1704536
94. Bachireddy P, Hainz U, Rooney M, Pozdnyakova O, Aldridge J, Zhang W, et al. Reversal of in situ T-cell exhaustion during effective human antileukemia responses to donor lymphocyte infusion. Blood. (2014) 123:1412–21. doi: 10.1182/blood-2013-08-523001
95. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. (2003) 112:101–8. doi: 10.1172/JCI17601
96. Muffly L, Sheehan K, Armstrong R, Jensen K, Tate K, Rezvani AR, et al. Infusion of donor-derived CD8+ memory T cells for relapse following allogeneic hematopoietic cell transplantation. Blood Adv. (2018) 2:681–90. doi: 10.1182/bloodadvances.2017012104
97. Thornton AM, Shevach EM. CD4+CD25+ Immunoregulatory T Cells Suppress Polyclonal T Cell Activation In Vitro by Inhibiting Interleukin 2 Production. J Exp Med. (1998) 188:287–96. doi: 10.1084/jem.188.2.287
98. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
99. Pacini CP, Soares MVD, Lacerda JF. The impact of regulatory T cells on the graft-versus-leukemia effect. Front Immunol. (2024) 15:1339318. doi: 10.3389/fimmu.2024.1339318
100. Maury S, Redjoul R, Cabanne L, Vigouroux S, Legros L, Cohen JL. Regulatory T-cell depletion in donor lymphocyte infusions for haematological malignancies: long-term outcomes from a prospective study. Br J Haematol. (2014) 166:452–5. doi: 10.1111/bjh.12856
101. Nikiforow S, Kim HT, Daley H, Reynolds C, Jones KT, Armand P, et al. A phase I study of CD25/regulatory T-cell-depleted donor lymphocyte infusion for relapse after allogeneic stem cell transplantation. Haematologica. (2016) 101:1251–9. doi: 10.3324/haematol.2015.141176
102. Roy DC, Walker I, Maertens J, Lewalle P, Olavarria E, Selleslag D, et al. ATIR101 administered after T-cell-depleted haploidentical HSCT reduces NRM and improves overall survival in acute leukemia. Leukemia. (2020) 34:1907–23. doi: 10.1038/s41375-020-0733-0
103. Maung KK, Chen BJ, Barak I, Li Z, Rizzieri DA, Gasparetto C, et al. Phase I dose escalation study of naive T-cell depleted donor lymphocyte infusion following allogeneic stem cell transplantation. Bone Marrow Transplant. (2021) 56:137–43. doi: 10.1038/s41409-020-0991-5
104. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-Reactive CD8+ T Cells Can Mediate Antileukemic Activity and Persist in Post-Transplant Patients. Sci Transl Med. (2013) 5:174ra27–174ra27. doi: 10.1126/scitranslmed.3004916
105. Casucci M, Bondanza A, Falcone L, Provasi E, Magnani Z, Bonini C. Genetic engineering of T cells for the immunotherapy of haematological malignancies. Tissue Antigens. (2012) 79:4–14. doi: 10.1111/j.1399-0039.2011.01799.x
106. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. (2012) 18:807–15. doi: 10.1038/nm.2700
107. Rambaldi B, Kim HT, Reynolds C, Rai SC, Arihara Y, Kubo T, et al. Impaired T- And NK-cell reconstitution after haploidentical HCT with posttransplant cyclophosphamide. Blood Adv. (2021) 5:352–64. doi: 10.1182/bloodadvances.2020003005
108. Vago L, Forno B, Sormani MP, Crocchiolo R, Zino E, Di Terlizzi S, et al. Temporal, quantitative, and functional characteristics of single-KIR–positive alloreactive natural killer cell recovery account for impaired graft-versus-leukemia activity after haploidentical hematopoietic stem cell transplantation. Blood. (2008) 112:3488–99. doi: 10.1182/blood-2007-07-103325
109. McCurdy SR, Radojcic V, Tsai H-L, Vulic A, Thompson E, Ivcevic S, et al. Signatures of GVHD and relapse after posttransplant cyclophosphamide revealed by immune profiling and machine learning. Blood. (2022) 139:608–23. doi: 10.1182/blood.2021013054
110. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
111. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. (2010) 28:955–9. doi: 10.1200/JCO.2009.24.4590
112. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
113. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. (2021) 18:85–100. doi: 10.1038/s41571-020-0426-7
114. Devillier R, Calmels B, Guia S, Taha M, Fauriat C, Mfarrej B, et al. Phase I Trial of Prophylactic Donor-Derived IL-2-Activated NK Cell Infusion after Allogeneic Hematopoietic Stem Cell Transplantation from a Matched Sibling Donor. Cancers (Basel). (2021) 13:2673. doi: 10.3390/cancers13112673
115. Ciurea SO, Kongtim P, Soebbing D, Trikha P, Behbehani G, Rondon G, et al. Decrease post-transplant relapse using donor-derived expanded NK-cells. Leukemia. (2022) 36:155–64. doi: 10.1038/s41375-021-01349-4
116. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. (2018) 131:2515–27. doi: 10.1182/blood-2017-12-823757
117. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. (2019) 3:1970–80. doi: 10.1182/bloodadvances.2018028332
118. Berrien-Elliott MM, Wagner JA, Fehniger TA. Human Cytokine-Induced Memory-Like Natural Killer Cells. J Innate Immun. (2015) 7:563–71. doi: 10.1159/000382019
119. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123 LP–357ra123. doi: 10.1126/scitranslmed.aaf2341
120. Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J Clin Invest. (2022) 132:e154334. doi: 10.1172/JCI154334
121. Schmidt-Wolf IGH, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. (1991) 174:139–49. doi: 10.1084/jem.174.1.139
122. Franceschetti M, Pievani A, Borleri G, Vago L, Fleischhauer K, Golay J, et al. Cytokine-induced killer cells are terminallydifferentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp Hematol. (2009) 37:616–628.e2. doi: 10.1016/j.exphem.2009.01.010
123. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front Immunol. (2017) 8:774. doi: 10.3389/fimmu.2017.00774
124. Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol. (1993) 21:1673–9.
125. Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. (2011) 118:3301–10. doi: 10.1182/blood-2011-02-336321
126. Valgardsdottir R, Capitanio C, Texido G, Pende D, Cantoni C, Pesenti E, et al. Direct involvement of CD56 in cytokine-induced killer&x2013;mediated lysis of CD56+ hematopoietic target cells. Exp Hematol. (2014) 42:1013–1021.e1. doi: 10.1016/j.exphem.2014.08.005
127. Verneris MR, Karami M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. (2015) 125:2583. doi: 10.1182/blood-2015-03-634667
128. Verneris MR, Baker J, Edinger M, Negrin RS. Studies of ex vivo activated and expanded CD8+ NK-T cells in humans and mice. J Clin Immunol. (2002) 22:131–6. doi: 10.1023/a:1015415928521
129. Rambaldi A, Biagi E, Bonini C, Biondi A, Introna M. Cell-based strategies to manage leukemia relapse: efficacy and feasibility of immunotherapy approaches. Leukemia. (2015) 29:1–10. doi: 10.1038/leu.2014.189
130. Introna M, Correnti F. Innovative Clinical Perspectives for CIK Cells in Cancer Patients. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19020358
131. Baker J, Verneris MR, Ito M, Shizuru JA, Negrin RS. Expansion of cytolytic CD8+ natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon γ production. Blood. (2001) 97:2923–31. doi: 10.1182/blood.V97.10.2923
132. Gu Y, Lv H, Zhao J, Li Q, Mu G, Li J, et al. Influence of the number and interval of treatment cycles on cytokine-induced killer cells and their adjuvant therapeutic effects in advanced non-small-cell lung cancer (NSCLC). Int Immunopharmacol. (2017) 50:263–9. doi: 10.1016/j.intimp.2017.07.006
133. Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IGH. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol. (2015) 141:839–49. doi: 10.1007/s00432-014-1864-3
134. Zhang Y, Schmidt-Wolf IGH. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J Cell Physiol. (2020) 235:9291–303. doi: 10.1002/jcp.29827
135. Leemhuis T, Wells S, Scheffold C, Edinger M, Negrin RS. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. Biol Blood Marrow Transplant. (2005) 11:181–7. doi: 10.1016/j.bbmt.2004.11.019
136. Olioso P, Giancola R, Di Riti M, Contento A, Accorsi P, Iacone A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematol Oncol. (2009) 27:130–9. doi: 10.1002/hon.886
137. Wang Y, Bo J, Dai H, Lu X, Lv H, Yang B, et al. CIK cells from recurrent or refractory AML patients can be efficiently expanded in vitro and used for reduction of leukemic blasts in vivo. Exp Hematol. (2013) 41:241–252.e3. doi: 10.1016/j.exphem.2012.10.014
138. Luo Y, Zeng H-Q, Shen Y, Zhang P, Lou S-F, Chen L, et al. Allogeneic hematopoietic stem cell transplantation following donor CIK cell infusion: A phase I study in patients with relapsed/refractory hematologic malignancies. Leuk Res. (2016) 48:6–10. doi: 10.1016/j.leukres.2016.06.006
139. Introna M, Pievani A, Borleri G, Capelli C, Algarotti A, Micò C, et al. Feasibility and Safety of Adoptive Immunotherapy with CIK Cells after Cord Blood Transplantation. Biol Blood Marrow Transplant. (2010) 16:1603–7. doi: 10.1016/j.bbmt.2010.05.015
140. Introna M, Borleri G, Conti E, Franceschetti M, Barbui AM, Broady R, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: A phase I study. Haematologica. (2007) 92:952–9. doi: 10.3324/haematol.11132
141. Introna M, Lussana F, Algarotti A, Gotti E, Valgardsdottir R, Micò C, et al. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. (2017) 23:2070–8. doi: 10.1016/j.bbmt.2017.07.005
142. Laport GG, Sheehan K, Baker J, Armstrong R, Wong RM, Lowsky R, et al. Adoptive immunotherapy with cytokine-induced killer cells for patients with relapsed hematologic malignancies after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2011) 17:1679–87. doi: 10.1016/j.bbmt.2011.05.012
143. Linn Y-C, Niam M, Chu S, Choong A, Yong H-X, Heng K-K, et al. The anti-tumour activity of allogeneic cytokine-induced killer cells in patients who relapse after allogeneic transplant for haematological malignancies. Bone Marrow Transplant. (2012) 47:957–66. doi: 10.1038/bmt.2011.202
144. Narayan R, Benjamin JE, Shah O, Tian L, Tate K, Armstrong R, et al. Donor-Derived Cytokine-Induced Killer Cell Infusion as Consolidation after Nonmyeloablative Allogeneic Transplantation for Myeloid Neoplasms. Biol Blood Marrow Transplant. (2019) 25:1293–303. doi: 10.1016/j.bbmt.2019.03.027
145. Merker M, Salzmann-Manrique E, Katzki V, Huenecke S, Bremm M, Bakhtiar S, et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biol Blood Marrow Transplant. (2019) 25:1281–92. doi: 10.1016/j.bbmt.2019.03.004
146. Rettinger E, Huenecke S, Bonig H, Merker M, Jarisch A, Soerensen J, et al. Interleukin-15-activated cytokine-induced killer cells may sustain remission in leukemia patients after allogeneic stem cell transplantation: Feasibility, safety and first insights on efficacy. Haematologica. (2016) 101:e153–6. doi: 10.3324/haematol.2015.138016
147. Rettinger E, Bonig H, Wehner S, Lucchini G, Willasch A, Jarisch A, et al. Feasibility of IL-15-activated cytokine-induced killer cell infusions after haploidentical stem cell transplantation. Bone Marrow Transplant. (2013) 48:1141–3. doi: 10.1038/bmt.2013.19
148. Pfeffermann L-M, Pfirrmann V, Huenecke S, Bremm M, Bonig H, Kvasnicka H-M, et al. Epstein-Barr virus–specific cytokine-induced killer cells for treatment of Epstein-Barr virus–related malignant lymphoma. Cytotherapy. (2018) 20:839–50. doi: 10.1016/j.jcyt.2018.04.005
149. Pievani A, Belussi C, Klein C, Rambaldi A, Golay J, Introna M. Enhanced killing of human B-cell lymphoma targets by combined use of cytokine-induced killer cell (CIK) cultures and anti-CD20 antibodies. Blood. (2011) 117:510–8. doi: 10.1182/blood-2010-06-290858
150. Golay J, Martinelli S, Alzani R, Criboli S, Albanese C, Gotti E, et al. Cord blood–derived cytokine-induced killer cells combined with blinatumomab as a therapeutic strategy for CD19+ tumors. Cytotherapy. (2018) 20:1077–88. doi: 10.1016/j.jcyt.2018.06.003
151. Poh SL, Linn YC. Immune checkpoint inhibitors enhance cytotoxicity of cytokine-induced killer cells against human myeloid leukaemic blasts. Cancer Immunol Immunother. (2016) 65:525–36. doi: 10.1007/s00262-016-1815-8
152. Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping Beauty–engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. (2020) 130:6021–33. doi: 10.1172/JCI138473
153. Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GMP, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. (2013) 161:389–401. doi: 10.1111/bjh.12282
154. Rotiroti MC, Buracchi C, Arcangeli S, Galimberti S, Valsecchi MG, Perriello VM, et al. Targeting CD33 in Chemoresistant AML Patient-Derived Xenografts by CAR-CIK Cells Modified with an Improved SB Transposon System. Mol Ther. (2020) 28:1974–86. doi: 10.1016/j.ymthe.2020.05.021
155. Perriello VM, Rotiroti MC, Pisani I, Galimberti S, Alberti G, Pianigiani G, et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv. (2023) 7:2855–71. doi: 10.1182/bloodadvances.2022008762
156. Bonanno G, Iudicone P, Mariotti A, Procoli A, Pandolfi A, Fioravanti D, et al. Thymoglobulin, interferon-γ and interleukin-2 efficiently expand cytokine-induced killer (CIK) cells in clinical-grade cultures. J Transl Med. (2010) 8:129. doi: 10.1186/1479-5876-8-129
157. Lin G, Wang J, Lao X, Wang J, Li L, Li S, et al. Interleukin-6 Inhibits Regulatory T Cells and Improves the Proliferation and Cytotoxic Activity of Cytokine-induced Killer Cells. J Immunother. (2012) 35:337–43. https://journals.lww.com/immunotherapy-journal/fulltext/2012/05000/interleukin_6_inhibits_regulatory_t_cells_and.5.aspx.
158. Rettinger E, KuçI S, Naumann I, Becker P, Kreyenberg H, Anzaghe M, et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy. (2012) 14:91–103. doi: 10.3109/14653249.2011.613931
159. Iudicone P, Fioravanti D, Cicchetti E, Bonanno G, Pandolfi A, Scocchera R, et al. Cytotoxic potential of interleukin-15 stimulated cytokine induced killer (CIK) against epithelial cancer cell lines. Cytotherapy. (2014) 16:S23. doi: 10.1016/j.jcyt.2014.01.070
160. Zoll B, Lefterova P, Ebert O, Huhn D, von Ruecker A, Schmidt-Wolf IGH. Modulation of cell surface markers on nk-like t lymphocytes by using IL-2, IL-7 or IL-12 in vitro stimulation. Cytokine. (2000) 12:1385–90. doi: 10.1006/cyto.2000.0733
161. Vanhooren J, Dobbelaere R, Derpoorter C, Deneweth L, Van Camp L, Uyttebroeck A, et al. CAR-T in the Treatment of Acute Myeloid Leukemia: Barriers and How to Overcome Them. HemaSphere. (2023) 7:e937–7. doi: 10.1097/HS9.0000000000000937
162. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U.S.A. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
163. Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, et al. Sleeping beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. (2008) 16:580–9. doi: 10.1038/sj.mt.6300404
164. Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the sleeping beauty system. Cancer Res. (2008) 68:2961–71. doi: 10.1158/0008-5472.CAN-07-5600
165. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
166. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/nejmoa1709919
167. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/nejmoa1707447
168. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/nejmoa1804980
169. Locke FL, Miklos DB, Jacobson CA, Perales M-A, Kersten M-J, Oluwole OO, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. (2022) 386:640–54. doi: 10.1056/nejmoa2116133
170. Bishop MR, Dickinson M, Purtill D, Barba P, Santoro A, Hamad N, et al. Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. N Engl J Med. (2022) 386:629–39. doi: 10.1056/nejmoa2116596
171. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/nejmoa1103849
172. Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-Specific chimeric antigen Receptor-modified T cells after failure of ibrutinib. J Clin Oncol. (2017) 35:3010–20. doi: 10.1200/JCO.2017.72.8519
173. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/nejmoa2024850
174. Chen L, Xie T, Wei B, Di DL. Current progress in CAR-T cell therapy for tumor treatment (Review). Oncol Lett. (2022) 24:1–12. doi: 10.3892/ol.2022.13478
175. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
176. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
177. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
178. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
179. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/nejmoa1709866
180. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. (2021) 398:491–502. doi: 10.1016/S0140-6736(21)01222-8
181. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. (2021) 35:3282–6. doi: 10.1038/s41375-021-01232-2
182. Voorhees TJ, Ghosh N, Grover N, Block J, Cheng C, Morrison K, et al. Long-term remission in multiply relapsed enteropathy-associated T-cell lymphoma following CD30 CAR T-cell therapy. Blood Adv. (2020) 4:5925–8. doi: 10.1182/bloodadvances.2020003218
183. Zhang X, Lu XA, Yang J, Zhang G, Li J, Song L, et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. (2020) 4:2325–38. doi: 10.1182/bloodadvances.2020001466
184. Liu S, Deng B, Yin Z, Lin Y, An L, Liu D, et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. (2021) 96:671–9. doi: 10.1002/ajh.26160
185. Brudno JN, Somerville RPT, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol. (2016) 34:1112–21. doi: 10.1200/JCO.2015.64.5929
186. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. (2016) 126:3363–76. doi: 10.1172/JCI86721
187. Cruz CRY, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. (2013) 122:2965–73. doi: 10.1182/blood-2013-06-506741
188. Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. (2013) 122:4129–39. doi: 10.1182/blood-2013-08-519413
189. Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood. (2017) 130:811. doi: 10.1182/blood.V130.Suppl_1.811.811
190. Zhang C, Wang X-Q, Zhang R-L, Liu F, Wang Y, Yan Z-L, et al. Donor-derived CD19 CAR-T cell therapy of relapse of CD19-positive B-ALL post allotransplant. Leukemia. (2021) 35:1563–70. doi: 10.1038/s41375-020-01056-6
191. Jia H, Wang Z, Wang Y, Liu Y, Dai H, Tong C, et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J Hematol Oncol. (2019) 12:57. doi: 10.1186/s13045-019-0741-6
192. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
193. Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B cell tumors: a phase 1/2 trial. Nat Med. (2024) 30:772–84. doi: 10.1038/s41591-023-02785-8
194. Sallman DA, DeAngelo DJ, Pemmaraju N, Dinner S, Gill S, Olin RL, et al. Ameli-01: A Phase I Trial of UCART123v1.2, an Anti-CD123 Allogeneic CAR-T Cell Product, in Adult Patients with Relapsed or Refractory (R/R) CD123+ Acute Myeloid Leukemia (AML). Blood. (2022) 140:2371–3. doi: 10.1182/blood-2022-169928
195. Jain N, Roboz GJ, Konopleva M, Liu H, Jabbour E, Poirot C, et al. Preliminary Results of Balli-01: A Phase I Study of UCART22 (allogeneic engineered T-cells expressing anti-CD22 Chimeric Antigen Receptor) in Adult Patients with Relapsed or Refractory (R/R) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood. (2020) 136:7–8. doi: 10.1182/blood-2020-138594
196. Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol. (2022) 9:e833–43. doi: 10.1016/S2352-3026(22)00245-9
197. Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J Clin Oncol. (2021) 39:3340–51. doi: 10.1200/JCO.21.00389
198. Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge W, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res. (2022) 32:995–1007. doi: 10.1038/s41422-022-00721-y
199. Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. (2022) 14:eabq3010. doi: 10.1126/scitranslmed.abq3010
200. Chiesa R, Georgiadis C, Syed F, Zhan H, Etuk A, Gkazi SA, et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N Engl J Med. (2023) 389:899–910. doi: 10.1056/nejmoa2300709
201. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. (2008) 118:294–305. doi: 10.1172/JCI32103
202. Lussana F, Magnani CF, Gaipa G, Galimberti S, Gritti G, Belotti D, et al. Final Results of Phase I/II Study of Donor-Derived CAR T Cells Engineered with Sleeping Beauty in Pediatric and Adult Patients with B-Cell Acute Lymphoblastic Leukemia Relapsed Post-HSCT. Blood. (2022) 140:4568–9. doi: 10.1182/blood-2022-165448
203. Circosta P, Donini C, Gallo S, Giraudo L, Gammaitoni L, Rotolo R, et al. Full chimaeric CAR.CIK from patients engrafted after allogeneic haematopoietic cell transplant: Feasibility, anti-leukaemic potential and alloreactivity across major human leukocyte antigen barriers. Br J Haematol. (2023) 200:64–9. doi: 10.1111/bjh.18469
204. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
205. Biondi M, Tettamanti S, Galimberti S, Cerina B, Tomasoni C, Piazza R, et al. Selective homing of CAR-CIK cells to the bone marrow niche enhances control of the acute myeloid leukemia burden. Blood. (2023) 141:2587–98. doi: 10.1182/blood.2022018330
206. Debenedette M, Purdon TJ, McCloud JR, Plachco A, Norris M, Gamble A, et al. CD19-CAR Cytokine Induced Killer Cells Armored with IL-18 Control Tumor Burden, Prolong Mouse Survival and Result in In Vivo Persistence of CAR-CIK Cells in a Model of B-Cell Acute Lymphoblastic Leukemia. Blood. (2023) 142:6826. doi: 10.1182/blood-2023-178793
207. Tang X, Yang L, Li Z, Nalin A, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. (2018) 8:1083–9. http://www.ajcr.us/files/ajcr0078782.pdf%0Ahttp://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emexa&NEWS=N&AN=622906093.
208. Huang R, Wen Q, Wang X, Yan H, MA Y, Wang M, et al. P522: off-the-shelf CD33 Car-NK cell therapy for relapse/refractory aml: first-in-human, phase i trial. HemaSphere. (2023) 7. doi: 10.1097/01.HS9.0000968996.69938.df
209. Caruso S, De Angelis B, Del Bufalo F, Ciccone R, Donsante S, Volpe G, et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J Hematol Oncol. (2022) 15:163. doi: 10.1186/s13045-022-01376-3
210. Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci. (2022) 119:e2122379119. doi: 10.1073/pnas.2122379119
211. Christodoulou I, Solomou EE. A Panorama of Immune Fighters Armored with CARs in Acute Myeloid Leukemia. Cancers (Basel). (2023) 15:1–17. doi: 10.3390/cancers15113054
212. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
213. Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. (2017) 23:242–9. doi: 10.1038/nm.4258
214. Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler M-E, Middeke JM, et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood. (2021) 137:3145–8. doi: 10.1182/blood.2020009759
215. Wermke M, Metzelder S, Kraus S, Sala E, Vucinic V, Fiedler W, et al. Updated Results from a Phase I Dose Escalation Study of the Rapidly-Switchable Universal CAR-T Therapy UniCAR-T-CD123 in Relapsed/Refractory AML. Blood. (2023) 142:3465. doi: 10.1182/blood-2023-177867
216. Yongxian H, Mingming Z, Tingting Y, Zhuomao M, Guoqing W, Ruirui J, et al. Sequential CD7 CAR T-Cell Therapy and Allogeneic HSCT without GVHD Prophylaxis. N Engl J Med. (2024) 390:1467–80. doi: 10.1056/NEJMoa2313812
217. Li Z, An N, Yang K, Meng F, Xu T, Peng X, et al. Donor CD7 Chimeric Antigen Receptor T Cell Bridging to Allogeneic Hematopoietic Stem Cell Transplantation for T Cell Hematologic Malignancy. Transplant Cell Ther. (2023) 29:167–73. doi: 10.1016/j.jtct.2022.11.013
218. Ghorashian S, Lucchini G, Richardson R, Nguyen K, Terris C, Guvenel A, et al. CD19/CD22 targeting with cotransduced CAR T cells to prevent antigen-negative relapse after CAR T-cell therapy for B-cell ALL. Blood. (2024) 143:118–23. doi: 10.1182/blood.2023020621
219. Meyer RG, Britten CM, Wehler D, Bender K, Hess G, Konur A, et al. Prophylactic transfer of CD8-depleted donor lymphocytes after T-cell–depleted reduced-intensity transplantation. Blood. (2007) 109:374–82. doi: 10.1182/blood-2006-03-005769
220. Orti G, Lowdell M, Fielding A, Samuel E, Pang K, Kottaridis P, et al. Phase I Study of High-Stringency CD8 Depletion of Donor Leukocyte Infusions After Allogeneic Hematopoietic Stem Cell Transplantation. Transplantation. (2009) 88:1312–8. https://journals.lww.com/transplantjournal/fulltext/2009/12150/phase_i_study_of_high_stringency_cd8_depletion_of.12.aspx.
221. Dunaikina M, Zhekhovtsova Z, Shelikhova L, Glushkova S, Nikolaev R, Blagov S, et al. Safety and efficacy of the low-dose memory (CD45RA-depleted) donor lymphocyte infusion in recipients of αβ T cell-depleted haploidentical grafts: results of a prospective randomized trial in high-risk childhood leukemia. Bone Marrow Transplant. (2021) 56:1614–24. doi: 10.1038/s41409-021-01232-x
222. Castagna L, Valli V, Timofeeva I, Capizzuto R, Bramanti S, Mariotti J, et al. Feasibility and Efficacy of CD45RA+ Depleted Donor Lymphocytes Infusion After Haploidentical Transplantation With Post-Transplantation Cyclophosphamide in Patients With Hematological Malignancies. Transplant Cell Ther. (2021) 27:478.e1–5. doi: 10.1016/j.jtct.2021.03.010
223. Vydra J, Cosimo E, Lesný P, Wanless RS, Anderson J, Clark AG, et al. A Phase I Trial of Allogeneic γδ T Lymphocytes From Haploidentical Donors in Patients With Refractory or Relapsed Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk. (2023) 23:e232–9. doi: 10.1016/j.clml.2023.02.003
224. Rizzieri DA, Storms R, Chen D-F, Long G, Yang Y, Nikcevich DA, et al. Natural Killer Cell-Enriched Donor Lymphocyte Infusions from A 3-6/6 HLA Matched Family Member following Nonmyeloablative Allogeneic Stem Cell Transplantation. Biol Blood Marrow Transplant. (2010) 16:1107–14. doi: 10.1016/j.bbmt.2010.02.018
225. Choi I, Yoon SR, Park S-Y, Kim H, Jung S-J, Jang YJ, et al. Donor-Derived Natural Killer Cells Infused after Human Leukocyte Antigen–Haploidentical Hematopoietic Cell Transplantation: A Dose-Escalation Study. Biol Blood Marrow Transplant. (2014) 20:696–704. doi: 10.1016/j.bbmt.2014.01.031
226. Shaffer BC, Le Luduec J-B, Forlenza C, Jakubowski AA, Perales M-A, Young JW, et al. Phase II Study of Haploidentical Natural Killer Cell Infusion for Treatment of Relapsed or Persistent Myeloid Malignancies Following Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant. (2016) 22:705–9. doi: 10.1016/j.bbmt.2015.12.028
227. Choi I, Yoon SR, Park S-Y, Kim H, Jung S-J, Kang Y-L, et al. Donor-Derived Natural Killer Cell Infusion after Human Leukocyte Antigen–Haploidentical Hematopoietic Cell Transplantation in Patients with Refractory Acute Leukemia. Biol Blood Marrow Transplant. (2016) 22:2065–76. doi: 10.1016/j.bbmt.2016.08.008
228. Jaiswal SR, Zaman S, Nedunchezhian M, Chakrabarti A, Bhakuni P, Ahmed M, et al. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced i. Cytotherapy. (2017) 19:531–42. doi: 10.1016/j.jcyt.2016.12.006
229. van Balen P, Jedema I, van Loenen MM, de Boer R, van Egmond HM, Hagedoorn RS, et al. HA-1H T-Cell Receptor Gene Transfer to Redirect Virus-Specific T Cells for Treatment of Hematological Malignancies After Allogeneic Stem Cell Transplantation: A Phase 1 Clinical Study. Front Immunol. (2020) 11:1804. doi: 10.3389/fimmu.2020.01804
230. Lulla PD, Naik S, Vasileiou S, Tzannou I, Watanabe A, Kuvalekar M, et al. Clinical effects of administering leukemia-specific donor T cells to patients with AML/MDS after allogeneic transplant. Blood. (2021) 137:2585–97. doi: 10.1182/blood.2020009471
231. Slavin S, Naparstek E, Nagler A, Ackerstein A, Samuel S, Kapelushnik J, et al. Allogeneic cell therapy with donor peripheral blood cells and recombinant human interleukin-2 to treat leukemia relapse after allogeneic bone marrow transplantation. Blood. (1996) 87:2195–204. doi: 10.1182/blood.V87.6.2195.bloodjournal8762195
232. Huang X-J, Wang Y, Liu D-H, Xu L-P, Chen H, Chen Y-H, et al. Modified Donor Lymphocyte Infusion (DLI) for the Prophylaxis of Leukemia Relapse after Hematopoietic Stem Cell Transplantation in Patients with Advanced Leukemia—Feasibility and Safety Study. J Clin Immunol. (2008) 28:390–7. doi: 10.1007/s10875-008-9193-4
233. Yoon SR, Lee YS, Yang SH, Ahn KH, Lee J-H, Lee J-H, et al. Generation of donor natural killer cells from CD34+ progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. (2010) 45:1038–46. doi: 10.1038/bmt.2009.304
234. Hardy NM, Fellowes V, Rose JJ, Odom J, Pittaluga S, Steinberg SM, et al. Costimulated tumor-infiltrating lymphocytes are a feasible and safe alternative donor cell therapy for relapse after allogeneic stem cell transplantation. Blood. (2012) 119:2956–9. doi: 10.1182/blood-2011-09-378398
235. Ho VT, Kim HT, Kao G, Cutler C, Levine J, Rosenblatt J, et al. Sequential infusion of donor-derived dendritic cells with donor lymphocyte infusion for relapsed hematologic cancers after allogeneic hematopoietic stem cell transplantation. Am J Hematol. (2014) 89:1092–6. doi: 10.1002/ajh.23825
236. Kottaridis PD, North J, Tsirogianni M, Marden C, Samuel ER, Jide-Banwo S, et al. Two-Stage Priming of Allogeneic Natural Killer Cells for the Treatment of Patients with Acute Myeloid Leukemia: A Phase I Trial. PLoS One. (2015) 10:e0123416. doi: 10.1371/journal.pone.0123416
237. Jaiswal SR, Zaman S, Chakrabarti A, Sen S, Mukherjee S, Bhargava S, et al. Improved Outcome of Refractory/Relapsed Acute Myeloid Leukemia after Post-Transplantation Cyclophosphamide-Based Haploidentical Transplantation with Myeloablative Conditioning and Early Prophylactic Granulocyte Colony-Stimulating Factor&x2013;Mobilized D. Biol Blood Marrow Transplant. (2016) 22:1867–73. doi: 10.1016/j.bbmt.2016.07.016
238. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood. (2017) 130:1857–68. doi: 10.1182/blood-2017-05-785659
239. Vela M, Corral D, Carrasco P, Fernández L, Valentín J, González B, et al. Haploidentical IL-15/41BBL activated and expanded natural killer cell infusion therapy after salvage chemotherapy in children with relapsed and refractory leukemia. Cancer Lett. (2018) 422:107–17. doi: 10.1016/j.canlet.2018.02.033
240. Otegbeye F, Cooper B, Caimi P, Zamborsky K, Reese-Koc J, Hillian A, et al. A Phase I Study to Determine the Maximum Tolerated Dose of ex Vivo Expanded Natural Killer Cells Derived from Unrelated, HLA-Disparate Adult Donors. Transplant Cell Ther. (2022) 28:250.e1–8. doi: 10.1016/j.jtct.2022.02.008
241. Lee K-H, Yoon SR, Gong J-R, Choi E-J, Kim HS, Park C-J, et al. The infusion of ex vivo, interleukin-15 and -21-activated donor NK cells after haploidentical HCT in high-risk AML and MDS patients—a randomized trial. Leukemia. (2023) 37:807–19. doi: 10.1038/s41375-023-01849-5
242. Jacoby E, Bielorai B, Avigdor A, Itzhaki O, Hutt D, Nussboim V, et al. Locally produced CD19 CAR T cells leading to clinical remissions in medullary and extramedullary relapsed acute lymphoblastic leukemia. Am J Hematol. (2018) 93:1485–92. doi: 10.1002/ajh.25274
243. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
244. Shah BD, Bishop MR, Oluwole OO, Logan AC, Baer MR, Donnellan WB, et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood. (2021) 138:11–22. doi: 10.1182/blood.2020009098
245. Shah NN, Lee DW, Yates B, Yuan CM, Shalabi H, Martin S, et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J Clin Oncol. (2021) 39:1650–9. doi: 10.1200/JCO.20.02262
246. Shah BD, Cassaday RD, Park JH, Houot R, Oluwole OO, Logan AC, et al. Impact of prior therapies and subsequent transplantation on outcomes in adult patients with relapsed or refractory B-cell acute lymphoblastic leukemia treated with brexucabtagene autoleucel in ZUMA-3. J Immunother Cancer. (2023) 11:e007118. doi: 10.1136/jitc-2023-007118
247. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (London England). (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
248. Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene Autoleucel, an Anti–B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J Clin Oncol. (2022) 41:1265–74. doi: 10.1200/JCO.22.00842
249. Cowan AJ, Pont MJ, Sather BD, Turtle CJ, Till BG, Libby EN 3rd, et al. Wood B, Gooley T, et al. &x3b3;-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: a phase 1, first-in-human trial. Lancet Oncol. (2023) 24:811–22. doi: 10.1016/S1470-2045(23)00246-2
250. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol. (2022) 15:88. doi: 10.1186/s13045-022-01308-1
251. Zhang H, Bu C, Peng Z, Li G, Zhou Z, Ding W, et al. Characteristics of anti-CLL1 based CAR-T therapy for children with relapsed or refractory acute myeloid leukemia: the multi-center efficacy and safety interim analysis. Leukemia. (2022) 36:2596–604. doi: 10.1038/s41375-022-01703-0
252. Naik S, Madden RM, Lipsitt A, Lockey T, Bran J, Rubnitz JE, et al. Safety and Anti-Leukemic Activity of CD123-CAR T Cells in Pediatric Patients with AML: Preliminary Results from a Phase 1 Trial. Blood. (2022) 140:4584–5. doi: 10.1182/blood-2022-170201
253. Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, et al. CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. (2021) 14:4–8. doi: 10.1186/s13045-021-01092-4
254. Lu P, Liu Y, Yang J, Zhang X, Yang X, Wang H, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. (2022) 140:321–34. doi: 10.1182/blood.2021014498
255. Dai H, Zhang W, Li X, Han Q, Guo Y, Zhang Y, et al. Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology. (2015) 4:e1027469. doi: 10.1080/2162402X.2015.1027469
256. Yang J, He J, Zhang X, Li J, Wang Z, Zhang Y, et al. Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J. (2022) 12:104. doi: 10.1038/s41408-022-00694-6
257. Boissel N, Chevallier P, Curran K, Schiller G, Liu H, Larson R, et al. P1408: Updated results of the phase I balli-01 trial of UCART22, an anti-CD22 allogeneic car-T cell product, in patients with relapsed or refractory (R/R) CD22+ B-cell acute lymphoblastic leukemia (B-ALL). HemaSphere. (2023) 7:146. https://journals.lww.com/hemasphere/fulltext/2023/08003/p1408:updated_results_of_the_phase_i_balli_01.1304.aspx.
258. Tan Y, Shan L, Zhao L, Deng B, Ling Z, Zhang Y, et al. Long-term follow-up of donor-derived CD7 CAR T-cell therapy in patients with T-cell acute lymphoblastic leukemia. J Hematol Oncol. (2023) 16:34. doi: 10.1186/s13045-023-01427-3
Keywords: hematopoietic cell transplant, donor lymphocyte infusion (DLI), cytokine-induced killer (CIK) cells, leukemia, graft versus leukemia (GVL), chimeric antigen receptor (CAR)
Citation: Rambaldi B, Rizzuto G, Rambaldi A and Introna M (2024) Genetically modified and unmodified cellular approaches to enhance graft versus leukemia effect, without increasing graft versus host disease: the use of allogeneic cytokine-induced killer cells. Front. Immunol. 15:1459175. doi: 10.3389/fimmu.2024.1459175
Received: 03 July 2024; Accepted: 30 September 2024;
Published: 24 October 2024.
Edited by:
Mahasweta Gooptu, Dana–Farber Cancer Institute, United StatesReviewed by:
Mauro Di Ianni, University of Studies G. d’Annunzio Chieti and Pescara, ItalyAntonio Rosato, University of Padua, Italy
Copyright © 2024 Rambaldi, Rizzuto, Rambaldi and Introna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benedetta Rambaldi, ZGV0dGEucmFtYmFsZGlAaG90bWFpbC5pdA==; YnJhbWJhbGRpQGFzc3QtcGcyMy5pdA==