 Luhong Cao
Luhong Cao Guixiang Huang2†
Guixiang Huang2† Jiangang Fan
Jiangang Fan Zhiyue Ma
Zhiyue Ma- 1Department of Otolaryngology, Head and Neck Surgery, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Emergency Surgery, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
- 3Department of Pulmonary and Critical Care Medicine, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
N6-methyladenosine (m6A) is considered the most prevalent methylation modification in messenger RNA (mRNA) that critically impacts head and neck cancer (HNC) pathogenesis and development. Alterations of m6A methylation related proteins are closely related to the progression, therapeutic effect, and prognosis of HNC. The human innate immune system activates immune pathways through pattern recognition receptors, which can not only resist pathogen infection, but also play a vital role in tumor immunity. Emerging evidence has confirmed that m6A methylation affects the activation of innate immune pathways such as TLR, cGAS-STING, and NLR by regulating RNA metabolism, revealing its potential mechanisms in the innate immune response of tumor cells. However, the relevant research is still in its infancy. This review elaborates the biological significance of RNA m6A methylation in HNC and discusses its potential regulatory relationship with TLR, cGAS-STING, and NLR pathways, providing a new perspective for in-depth understanding of the role of RNA methylation in the innate immune mechanism and therapeutic application of HNC.
1 Introduction
Head and neck cancer (HNC) typically refers to a group of neoplasms originating from the oral cavity, pharynx, larynx, nasal cavity, paranasal sinus, and salivary gland, including oral cavity cancer, nasopharyngeal carcinoma, laryngeal cancer, hypopharyngeal cancer, paranasal sinus cancer, salivary gland cancer, and thyroid cancer. HNC ranks the seventh most incident cancer, with an estimated 890,000 new cases in 2018 and 450,000 of these predicted to be fatal (1). In the United States, HNC accounts for 3% of all cancer cases (51,540 new cases) and results in 1.5% of all cancer deaths (10,030 deaths) (2). Head and neck squamous cell carcinoma (HNSCC) is the most common pathological type of head and neck neoplasms (3). Approximately 1.08 million new HNSCC cases are expected per year by 2030 (4). In general, the incidence of HNC shows a rising trend year by year. Early detection and comprehensive treatment can significantly improve the survival and quality of life of patients. Therefore, it is particularly important to strengthen the understanding of the relevant molecular mechanisms regulating the occurrence and development of HNC.
Innate immunity serves as the first line of defense against various external threats, consisting of a variety of complex mechanisms to rapidly and widely respond to pathogen invasion (5). Innate immunity also plays a vital role in tumorigenesis and progression by affecting the formation, growth, and metastasis of tumors, as well as the effect of immunotherapy (6). On the one hand, the innate immune system can repress the development of tumors by recognizing and clearing abnormal cells. Innate immune cells such as natural killer (NK) cells and macrophages can recognize and kill tumor cells that have undergone abnormalities, thereby preventing their constant growth and spread (7). On the other hand, the innate immune system is critical for the treatment of tumors. For example, enhancing the ability of innate immunity to attack tumors by increasing the activity of NK cells or inhibiting immunosuppressive pathways has become one of the important therapeutic strategies for tumors (8, 9).
RNA modifications usually affect RNA splicing, stability, localization, translation, and RNA-RNA/RNA-protein interactions. N6-methyladenosine (m6A) methylation is one of the most prevalent post-transcriptional modifications, which is closely related to the occurrence and prognosis of human malignancies (10–12). m6A is a modified base that exists in all RNA types, including ribosomal RNA (rRNA), transfer RNA (tRNA), and small nuclear RNA (snRNA), as well as mRNA of cellular and viral origin. Recent studies have demonstrated the important role of m6A RNA methylation in tumor innate immune response. For example, m6A methylation guarantees the homeostasis of NK cells and tumor immune surveillance. Loss of m6A methyltransferase METTL3 alters NK cell homeostasis and inhibits NK cell infiltration and function in the tumor microenvironment (13). Meanwhile, YTHDF2 deficiency impairs the antitumor and antiviral activities of NK cells (14). In addition to the regulation on NK cells, m6A methylation mediated by METTL3 can promote the activation of dendritic cells (DCs) (15). METTL3 also drives the polarization of M1 macrophages by directly methylating STAT1 mRNA (16).
Therefore, understanding the mechanism of m6A RNA methylation in innate immune response during HNC is of great significance for developing new immunotherapeutic strategies and improving the therapeutic effect of patients. Here, we comprehensively review and explore the involvement of m6A methylation in the pathogenesis and development of HNC, as well as its different mechanisms in the signal transduction mediated by different nucleic acid sensor receptors (TLR, cGAS-STING, NOD) in the innate immune response of HNC, thereby providing a new theoretical framework for the treatment of HNC.
2 Molecular mechanisms of m6A methylation
m6A is a dynamic and reversible methylation modification at the N6-position of adenosine. As the most prevalent internal modification on eukaryotic mRNA, m6A mainly appears in the RRACH sequences (R = A/G, H = A/C/U) near the stop codon and 3’-untranslated region (3’-UTR). m6A has been demonstrated to critically regulate various processes of RNA metabolism, such as stability, splicing, and translation (17). Typically, m6A modification is installed by m6A methyltransferases, or writers, such as METTL3/14, removed by demethylases, or erasers, including FTO and ALKBH5, and recognized by m6A-binding proteins such as YTHDF1, termed readers (18).
1. m6A writers are methyltransferases responsible for installing methyl groups on target RNAs, including METTL3, METTL14, WTAP, RBM15/15B, ZC3H13, and KIAA1429 (VIRMA) (Figure 1). The core component of m6A writers is the methyltransferase complex (MTC) mainly composed of METTL3, METTL4, and WTAP. METTL3 is the core subunit with catalytic activity; METTL14 has substrate recognition function, and WTAP is responsible for recruiting METTL3 and METTL14. Only when METTL3 and METTL14 form a heterodimer as the core of the MTC, the activity of METTL3 is enhanced significantly, thus activating its spatial structure) (19, 20). m6A methylation is sequence specific. m6A methylation mediated by METTL3/14 complex is preferentially enriched in RRACH (R = A or G; H = A, C, or U) motifs, which are widely used to recognize m6A methylation (21).
2. m6A erasers realize a dynamic and reversible process by removing internal m6A from RNA, in which demethylases FTO and ALKBH5 play important roles (Figure 1). FTO is the first demethylase discovered. Jia et al. have revealed the potent m6A demethylase activity of FTO (22). Mechanistically, FTO catalyzes the demethylation of m6A by utilizing cofactors 2-oxoglutarate and Fe2+, gradually generating N6-hydroxymethyladenosine and N6-formyladenosine (23). Rubio et al. have found that FTO preferentially demethylates m6Am rather than m6A, reducing the stability of m6Am mRNAs (24). Wei et al. have shown that FTO mediates internal m6A and cap m6Am demethylation of polyadenylated RNA with differential substrate preferences in nucleus versus cytoplasm, in which the internal m6A demethylation correlates with changes in transcript level (25). Therefore, the subcellular localization of FTO affects its ability to perform different RNA modifications. Another demethylase, ALKBH5, is found to be highly expressed in testis. The demonstration of its demethylase activity provides the first evidence of reversible post-transcriptional modification in mRNA. ALKBH5 functions as a unique m6A regulator that specifically regulates m6A modifications without affecting other types of RNA modifications. It demethylates specific transcripts at the 3’ UTR and accelerates mRNA export from the nucleus to the cytoplasm (26).
3. m6A readers are RNA binding proteins that affect RNA splicing, nuclear transport, stability, translation, and decay by specifically recognizing and anchoring RNA with m6A modification (27). Reader proteins include YTH family, IGF2BP, HNRNPC, HNRNPA2B1, and eIF3 (Figure 1). YTH family proteins, including YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3, have RNA binding domains and are the first group of m6A binding proteins discovered. YTHDC1 is associated with mRNA splicing, epigenetic silencing mediated by non-coding RNA XIST, and nuclear export of mRNA (28, 29). YTHDC2, the largest member of the YTH family, also preferentially binds m6A within the consensus motif, which can improve translation efficiency while reducing the aggregation rate of its target mRNA (30). YTHDF protein exerts different functions at each end. Its C-terminal YTH domain helps it bind m6A-modified mRNA, while the N-terminal region can freely bind various cofactors to achieve expression diversity) (31). YTHDF1 was initially confirmed to bind to methylated mRNA transcripts near stop codons, and its overall distribution pattern was similar to that of m6A sites on mRNA. Mechanistic studies have revealed that YTHDF1 interacts with the translation initiation mechanism to enhance the translation efficiency of its target RNA (32). Of note, it has been unraveled that the three members of the DF protein family function together to mediate primarily the degradation of m6A-modified mRNAs, rather than to play a role in facilitating translation (33).
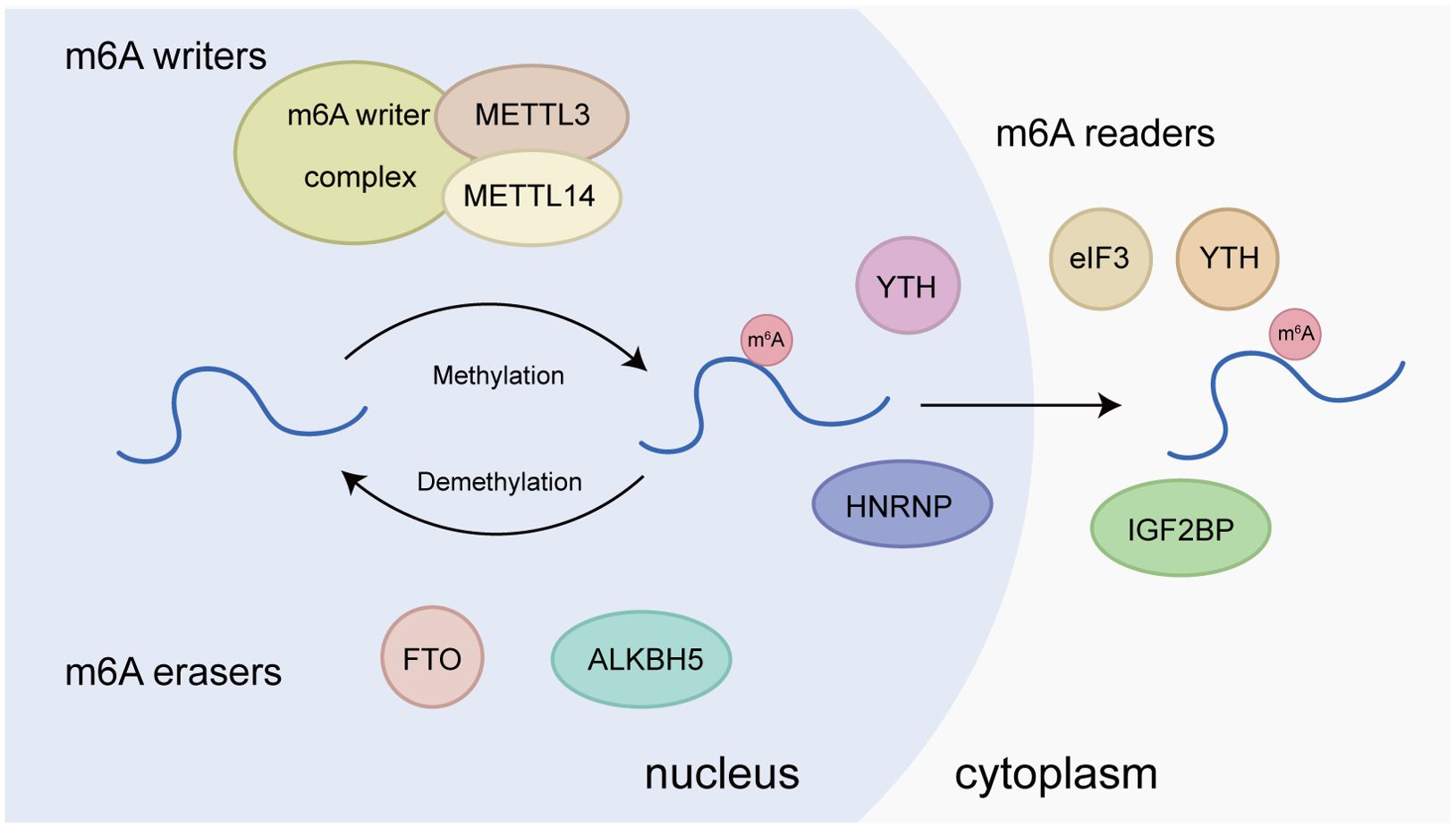
Figure 1. Molecular basis of m6A RNA methylation. m6A “writers”, including METTL3, METTL14, WTAP, RBM15/15B, ZC3H13, and KIAA1429, are the methyltransferases responsible for installing methyl groups on target RNAs. m6A “erasers”, including FTO and ALKBH5, are m6A demethylases. m6A “readers”, such as YTHDF1/2/3, YTHDC1, HNRNPA2B1, HNRNPC, eIF3, FMR1, and LRPPRC, are proteins that recognize m6A modification and exhibit various functions.
In conclusion, changes in the expression of m6A methylation regulators directly affect the RNA modification status of tumor cells, further modulating key biological processes such as gene expression, cell proliferation, and immune escape. In-depth studies of the way that m6A methylation regulates these biological processes can better unravel molecular mechanisms underlying the development and progression of tumors. In the clinical setting, understanding the role of m6A methylation is helpful in developing new biomarkers for tumor diagnosis, prognostic assessment, and treatment response prediction. In addition, drugs targeting m6A methylation regulators, such as METTL3 inhibitors and FTO inhibitors, are expected to become new anticancer drugs and increase the efficacy of existing treatments.
3 Expression and influence of m6A in HNC
RNA m6A methylation and its mechanisms are altered in a variety of HNCs, such as nasopharyngeal carcinoma (NPC), oral squamous cell carcinoma (OSCC), laryngeal squamous cell carcinoma (LSCC), and thyroid cancer (TC), leading to tumorigenesis, progression, and metastasis through dysregulation of coding and non-coding transcripts and their associated functional pathways (Figure 2, Table 1).
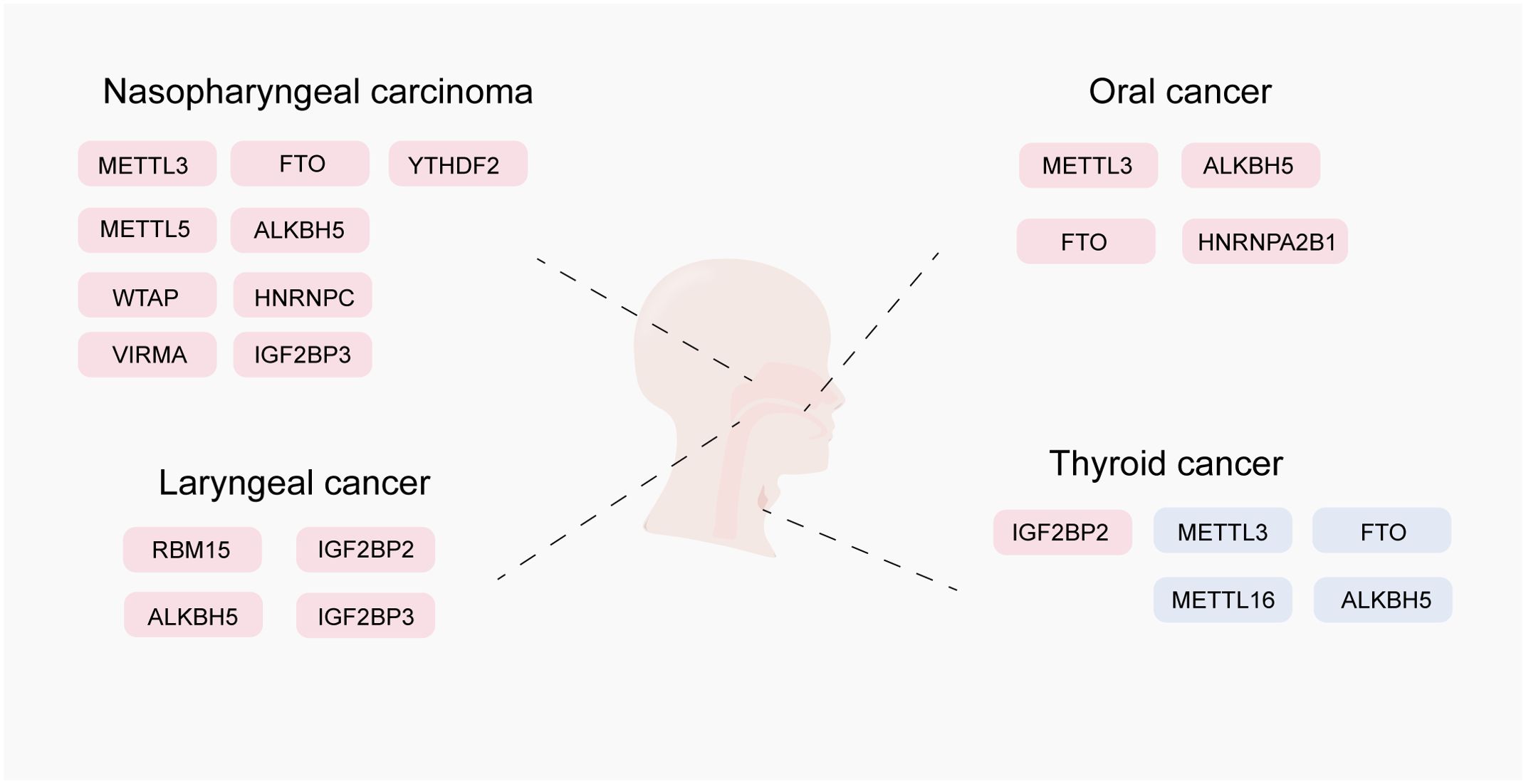
Figure 2. Expression and role of m6A RNA modification in head and neck cancer. This figure shows the oncogenic and anti-oncogenic effects of N6-methyladenosine (m6A) modifiers, including m6A writers (METTL3, METTL3, METTL16, VIRMA, and WTAP), m6A erasers (FTO and ALKBH5), and m6A readers (HNRNPC, IGF2BP3, IGF2BP3, YTHDF1, YTHDF2, and HNRNPA2B1). Positively regulated (upregulated) targets of m6A modifiers are labeled in red, whereas negatively regulated (downregulated) targets are labeled in blue.
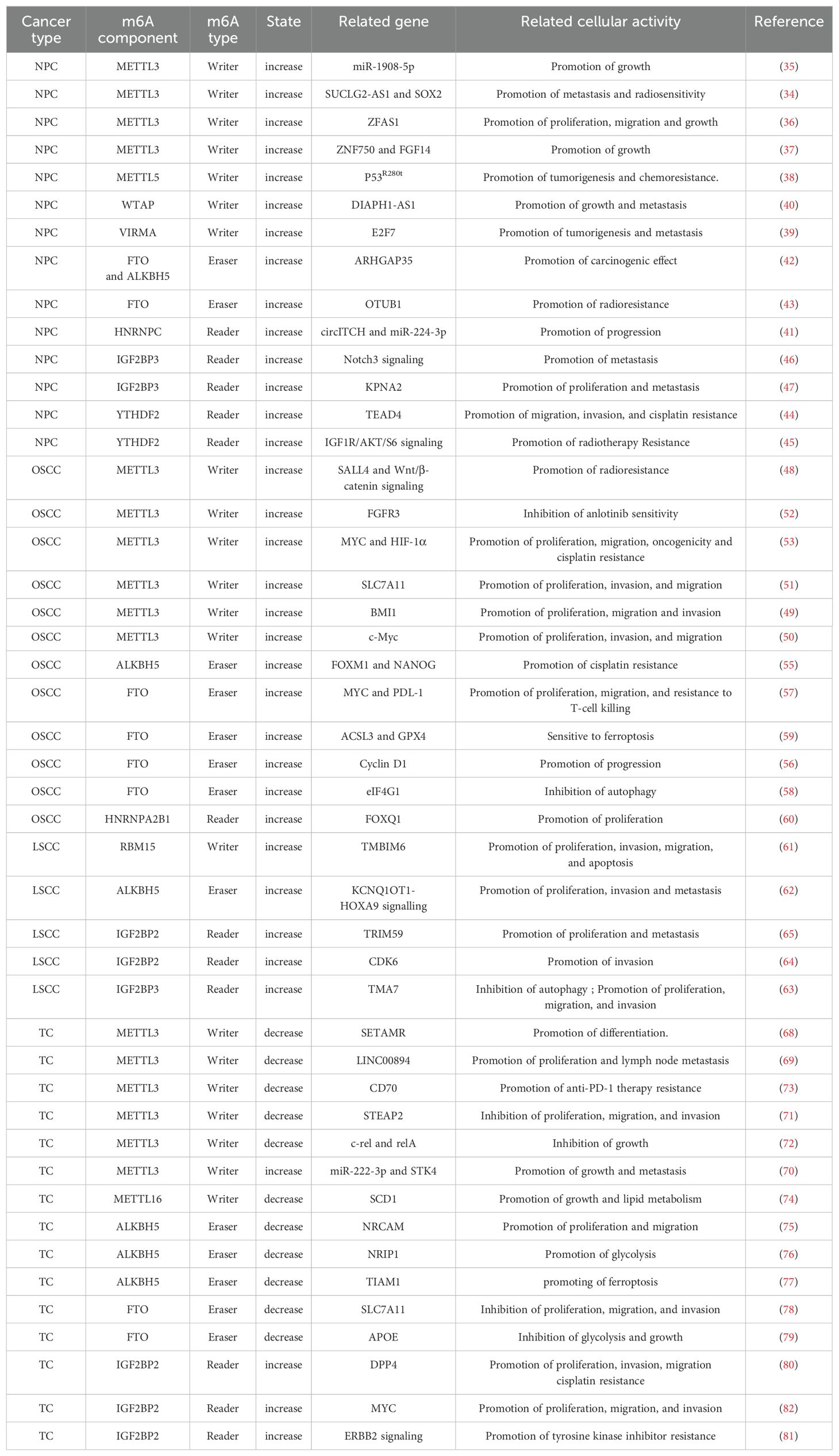
Table 1. Role of RNA m6A modification and its target genes in head and neck tumors.
m6A binding proteins have complex regulatory mechanisms in NPC. The m6A writer METTL3 mediates m6A modification in the SUCLG2-AS1 transcript, which subsequently recognizes and stabilizes IGF2BP3. SUCLG2-AS1 regulates the expression of SOX2 by forming long-range chromatin loop, thereby regulating the metastasis and radiosensitivity of NPC (34). Moreover, METTL3 can mediate the m6A modification of miR-1908-5p (35) and lncRNA ZFAS1 (36) to affect NPC cells. METTL3 also mediates the m6A methylation of ZNF750 to decrease its expression in NPC, thereby promoting NPC progression (37). The m6A writer METTL5 enhances the translation of HSF4b to activate the transcription of HSP90B1, which binds to the gain of function p53R280T protein and prevents its ubiquitination-dependent degradation, thereby promoting NPC tumorigenesis and chemoresistance (38). VIRMA is highly expressed in NPC, which elevates the expression of E2F7 in an m6A-dependent manner to promote the proliferation and metastasis of NPC cells in vitro and in vivo (39). WTAP expression is significantly upregulated in NPC, and lncRNA DIAPH1-AS1 is identified as the m6A target of WTAP. DIAPH1-AS1 acts as a molecular linker to promote the formation of MTDH-LASP1 complex and elevate LASP1 expression, ultimately promoting NPC growth and metastasis (40). m6A reader HNRNPC mediates the downregulation of circITCH to prevent miR-224-3p chelation and promote NPC tumorigenesis (41). A higher expression of FTO and ALKBH5 in tissues of patients with advanced NPC predicts a poorer prognosis. ARHGAP35 acts as a downstream target of FTO and ALKBH5 to induce NPC tumorigenesis) (42). In addition, FTO confers radioresistance to NPC cells by promoting OTUB1-mediated anti-ferroptosis (43). YTHDF2 recognizes WTAP-mediated TEAD4 m6A methylation to promote its stability and lead to abnormal upregulation of TEAD4, thereby promoting NPC migration, invasion, and cisplatin resistance (44). YTHDC2 also promotes radiotherapy resistance of NPC cells by activating the IGF1R/ATK/S6 signaling axis (45). IGF2BP3 expression is significantly increased in metastatic NPC. IGF2BP3 maintains NOTCH3 mRNA stability by inhibiting CCR4-NOT complex-mediated deadenylation (in an m6A dependent manner), thereby maintaining the activation of NOTCH3 signal and increasing the transcription of downstream genes related to stem cells, and ultimately promoting tumor metastasis (46). IGF2BP3 can also promote the proliferation and metastasis of NPC cells by affecting the stability of m6A-modified KPNA2 (47).
m6A methylation modification plays a vital role in the occurrence and development of OSCC. The m6A writer protein METTL3 exerts multiple functions in OSCC. METTL3/SALL4 axis promotes cancer stem cell (CSC) phenotype and radiotherapy resistance in OSCC via the Wnt/β-catenin signaling pathway (48). METTL3 can recognize m6A residues on BMI1 3’-UTR and bind to m6A reading protein IGF2BP1 to promote BMI1 translation, thereby inducing OSCC cell proliferation and metastasis (49). METTL3 enhances c-Myc stability through YTHDF1-mediated m6A modification to promote tumorigenesis in OSCC (50). METTL3 can also strengthen the mRNA stability of SLC7A11 through m6A-mediated IGF2BP2 binding, thereby promoting OSCC progression (51). Furthermore, m6A is implicated in the drug resistance of OSCC. METTL3-mediated FGFR3 m6A modification plays a key role in the sensitivity of anlotinib in OSCC (52). METTL3-HIF-1α-MYC establishes a positive feedback loop to promote OSCC progression, which accelerates the proliferation, migration, carcinogenicity, and cisplatin resistance of OSCC cells (53). DDX3 is a human DEAD-box RNA helicase involved in RNA metabolism and translation) (54). DDX3 directly regulates m6A through the demethylase ALKBH5, thus reducing the transcription of cancer stem cell transcription factors FOXM1 and NANOG, and eventually leading to chemotherapy resistance (55). m6A demethylase FTO can regulate the cell cycle of oral cancer and promote the cancer progression by regulating the expression of cyclin D1 (56). Arecoline-induced FTO, a high-risk factor of OSCC, elevates the stability and expression of PD-L1 transcript by mediating m6A modification and MYC activity, respectively. PD-L1 upregulation enables OSCC cells to have better proliferation, migration, and resistance to T cell killing (57). FTO can also regulate autophagy and affect tumorigenesis by targeting the eIF4G1 gene encoding in OSCC (58). In addition, high-expression of FTO in OSCC cells promotes ferroptosis by regulating ACSL3 and GPX4 (59). m6A reader protein HNRNPA2B1 is highly expressed in OSCC and enhances the malignant phenotype of OSCC by stabilizing m6A-modified FOXQ1 mRNA, which ultimately aggravates the malignancy and tumorigenicity of OSCC (60).
LSCC is a malignant tumor originated from laryngeal squamous cell lesions. The overall mRNA m6A methylation level is significantly increased in LSCC patients. RBM15, as the “writer” of methyltransferase, is notably upregulated in LSCC and associated with poor prognosis. Knockdown of RBM15 reduces the proliferation, invasion, migration, and apoptosis of LSCC in vitro and in vivo (61). ALKBH5-mediated m6A modification of KCNQ1OT1 regulates LSCC cell proliferation, migration, and invasion through the upregulation of HOXA9 (62). IGF2BP3 manipulates TMA7-mediated autophagy and cisplatin resistance in LSCC via m6A RNA methylation (63). IGF2BP2 overexpression in LSCC promotes the CDK6 mRNA stability and protein expression, thus increasing the expression of CDK6 and promoting LSCC cell proliferation and invasion in vitro and tumor growth in xenograft tumor models (64). IGF2BP2 can also stabilize circMMP9 in an m6A-dependent manner, and then circMMP9 recruits ETS1 to control TRIM59 transcription, thereby promoting LSCC progression via the PI3K/AKT signaling pathway (65).
Increasing evidences have revealed the close correlation between aberrant m6A modification and the occurrence and development of TC. Xu et al. have found differential expression patterns of multiple m6A-related genes between TC patients and normal population in The Cancer Genome Atlas (TCGA) dataset, including METTL3, YTHDC2, WTAP, YTHDF1, ALKBH5, METTL14, YTHDC1, FTO, ZC3H13, YTHDF2, and RBM15. By verifying the gene signatures of three Gene Expression Omnibus (GEO) datasets (GSE33630, GSE35570, and GSE60542), they conclude that m6A modification affects the prognosis of TC (66). Through bioinformatics analysis, Wang et al. have revealed that the expressions of METTL3, YTHDC1, FTO, METTL14, RBM15, YTHDF3, WTAP, ALKBH5, METTL16, YTHDC, and YTHDF1 in TC tissues are significantly lower than those in normal thyroid tissues (67). Since there are multiple subtypes of TC with different clinical and pathological characteristics, the function of m6A modification proteins in different subtypes of TC needs further study and verification. METTL3 expression in TC cells, especially in anaplastic thyroid cancer (ATC) cells, is significantly lower than that in normal thyroid cells. METTL3-mediated m6A modification controls SETMAR expression, and SETMAR significantly regulates TC cell proliferation, epithelial mesenchymal transition (EMT), thyroid differentiation related gene expression, radioiodine uptake, and sensitivity to MAPK inhibitor based redifferentiation therapy (68). METTL3 accelerates papillary thyroid cancer (PTC) progression by enhancing LINC00894 mRNA stability and upregulating LINC00894 expression via m6A-YTHDC2-dependent pathway (69). In addition, METTL3 induces miR-222-3p expression, thereby inhibiting STK4 expression and promoting the malignancy and metastasis of TC (70). Some studies have also reported that METTL3 acts as a thyroid tumor suppressor. METTL3-mediated STEAP2 m6A modification plays a tumor suppressive role in PTC by blocking Hedgehog signaling pathway and EMT (71). METTL3 cooperates with YTHDF2 through the c-Rel and NF-κB pathways to change Tan invasion and repress tumor growth, thereby inhibiting the occurrence and development of PTC (72). Overexpression of METTL3 can enhance the efficacy of anti-PD-1 therapy in PTC and ATC. Low expression of METTL3 in TC cells leads to demethylation and stabilization of CD70 mRNA in a YTHDF2-dependent manner. Subsequent upregulation of CD70 protein increases the abundance of immunosuppressive Tregs and terminally exhausted T cells, thereby inducing resistance to anti-PD-1 therapy (73). METTL16 is lowly expressed in PTC tissues, resulting in elevated SCD1 expression and enhanced lipid metabolism in PTC cells, which promote PTC progression and lead to poor clinical prognoses (74). circNRCAM is highly expressed in PTC cells, which enhances PTC cell proliferation, invasion, and migration by interacting with ALKBH5 and inhibiting miR-506-3p) (75). ALKBH5 modifies circNRIP1 to regulate PKM2 expression by competing for the sponge effect of miR-541-5p and miR-3064-5p, thereby affecting the glycolytic function of PTC cells (76). ALKBH5 overexpression inhibits TC cell proliferation, increases Fe2+ and reactive oxygen species levels, decreases GPX4 and SLC7A11 proteins, and eventually retards TC progression (77). FTO is downregulated in PTC and acts as a tumor suppressor gene. FTO reduces the expression of SLC7A11 through ferroptosis to suppress the development of PTC) (78). Meanwhile, FTO inhibits the expression of APOE via IGF2BP2-mediated m6A modification and regulates IL-6/JAK2/STAT3 signaling pathway to inhibit the glycolytic metabolism of PTC, thereby repressing tumor growth (79). IGF2BP2 is upregulated in PTC and interacts with DPP4 in an m6A-dependent manner to activate the NF-KB pathway and promote lymph node metastasis. Knockdown of IGF2BP2 increases the sensitivity of PTC cells to cisplatin to a certain extent (80). Moreover, IGF2BP2-dependent activation of ERBB2 signaling is involved in acquired resistance to tyrosine kinase inhibitors (81). By competitively binding to miR-204, lncRNA MALAT1 upregulates IGF2BP2 via m6A modification and enhances MYC expression, thereby stimulating the proliferation, migration, and invasion of TC cells, accompanied by the attenuation of tumor growth and apoptosis (82).
To sum up, aberrant m6A methylation affects the occurrence and development of HNC at all levels. Notably, the expression of some m6A writers and erasers are both upregulated in HNC, and their functions may be dependent on m6A modification. Nevertheless, the specific mechanisms and targets of these proteins may differ. Research on the specific roles and interactions of these proteins can reveal the complex regulatory mechanisms of m6A modification in diseases, thereby offering important clues for understanding the molecular mechanism of HNC and also providing guidance for future research and treatment of HNC.
4 Potential regulatory role of m6A methylation in innate immune pathways in HNC
Modern immunology believes that the recognition of tumor by innate immunity can be divided into antibacterial immune recognition and emergency immune recognition, but attempts to identify the molecular pathways shared by all tumors have been unsuccessful, and there is no evidence that the common determinants or patterns expressed by tumor cells can be recognized by the immune system. In contrast, immune cells can sense several types of modifications associated with various tumors, but not specific to these tumors. Nucleic acid detection is a typical example of using infection detection pathways to promote the recognition of growing tumors and trigger tumor specific immune responses, thereby coordinating downstream immune responses (83, 84). In the immune recognition of HNC, nucleic acid sensing involves toll like receptors (TLRs) (85) and cytosolic nucleic acid sensors such as stimulator of interferon genes (STING) (86) and NOR like receptors (NLRs) (87). All these immune recognition and immune response processes can be regulated by m6A methylation.
4.1 TLR pathway
TLRs are a class of pattern recognition receptors (PRRs) that can recognize pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs), thereby initiating immune responses (88). When ligands appear and bind to TLRs, dimers are formed and cascade signals are generated, which subsequently initiate the MyD88 pathway or TIR domain-containing adaptor inducing IFN-beta (TRIF) pathway (89). The activation of TLR signal transduction originates from the domain of cytosolic Toll/IL-1 receptor (TIR), which interacts with MyD88, an adaptor protein contained in the TIR domain, to initiate interleukin 1R related kinase (IRAK). Tumor necrosis factor receptor-associated factor (TRAF) is a molecule that assists in sending signals from IRAK to the nucleus, which further signals to the nucleus and leads to the activation of mitogen-activated protein kinases (MAPKs) and nuclear factor-kappaB (NF-κB) (90). In the TRIF pathway, TRIF interacts with TRAF6 and TRAF3. TRAF6 recruits the receptor-interacting protein 1 (RIP1), which in turn activates the transforming growth factor beta-activated kinase 1 (TAK1), leading to the activation of NF-κB and MAPKs and the induction of inflammatory cytokines (91). In contrast, TRAF3 recruits IKK related kinases TANK-binding kinase 1 (TBK1) and inducible IkappaB kinase inhibitor (IKKi) as well as nuclear factor (NF)-kappaB essential modulator (NEMO) for interferon regulatory factor 3 (IRF3) phosphorylation. Subsequently, IRF3 forms dimers and translocates from the cytoplasm into the nucleus, thereby inducing the expression of type I interferon (IFN) (92). Previous studies have shown the crucial implication of TLRs in the occurrence and proliferation of malignant tumors. On the one hand, cancer-related TLR regulation can elevate the susceptibility to infection. For example, TLR1 SNP associated with lymphoma can indicate enhanced susceptibility to Helicobacter pylori infection in lymphoma patients (93). TLR1 polymorphism provides an additional step for Helicobacter pylori infection to induce further oncogenic transformation (93). These infections can participate in the process of tumorigenesis through the further regulation of TLRs. For example, Helicobacter pylori lipopolysaccharide has the ability to induce TLR4 expression, and this regulation leads to gastric epithelial cell proliferation via the MEK1/2-ERK1/2 MAP kinase pathway (94). Therefore, TLR activation induced by various ways facilitates the carcinogenesis process by releasing pro-inflammatory cytokines and anti-apoptotic factors, recruiting immune cells, and promoting the proliferation of cells in the tumor microenvironment (TME), thereby creating a tumor friendly environment.
TLRs are critically participate in the occurrence and development of HNC. TLR2, TLR4, and TLR9 are expressed in primary tumors, neck metastatic tumors, and recurrent tumors of oral tongue squamous cell carcinoma (OTSCC) ranging from the tumor surface to the invasive front, which may be one of the important factors to promote the invasion of OTSCC (95, 96). TLR2 is expressed on keratinocytes of OTSCC and specifically regulates the growth and survival of tumor cells by promoting immune escape and inhibiting apoptosis (97). TLR3 is overexpressed in both HNC and squamous cell carcinoma, and the TLR3 rs3775291 polymorphism mutation genotype is associated with poor survival in patients with oral cancer (98). TLR4 is also a key mediator in the tumorigenesis of HNC. TLR4 expression is upregulated in oral cancer, and elevated TLR4 expression drives the transition of epithelial cells to mesenchymal cells, thereby promoting metastasis, differentiation, and proliferation, and resulting in poor survival and exacerbating disease severity (99). TLR9 promotes the migration of oral cancer cells HB in vitro through the activation of its cognate ligand CpG ODN (100). In contrast, OSCC patients with higher levels of TLR9 but not TLR2 or TLR4 present much worse 10-year overall survival (101). In addition, various types of TLRs are also highly expressed in NPC, LSCC, and TC (101–104).
Tong et al. have demonstrated that METTL3 is a positive regulator of the innate response of macrophages. Mechanistically, the transcript of Irakm gene, which encodes a negative regulator of TLR4 signaling, is highly m6A modified. METTL3 deletion abates m6A modification on Irakm mRNA and slows down its degradation, resulting in the increase of Irakm expression, which ultimately inhibits TLR signal-mediated macrophage activation and promotes tumor growth (105). Knockdown of the methyltransferase METTL3 regulates the alternative splicing of MyD88, a participant in the TLR-mediated antiviral immune pathway, resulting in the generation of the MyD88 splice variant, which is known to inhibit the production of inflammatory cytokines (106). Depletion of METTL3 reduces the accumulation of inflammatory cytokines and inhibits the activation of NF-κB and MAPK signaling pathways (Figure 3A). By comparing m6A-modified RNA and unmodified RNA, it is found that the m6A-modified RNA does not induce TLR3 transformed cells to produce detectable levels of IL-8, but also inhibits the signal transduction activity by TLR7 and TLR8 (107), while unmodified RNA can activate all these human TLRs. The inhibition of RNA-mediated immunostimulation is directly proportional to the number of modified nucleosides present in RNA. Moreover, the presence of m6A modification reduces RNA-mediated activation of monocyte-derived dendritic cells (DCs) more effectively than m5C or Ψ modification (106, 107). Briefly, m6A methylation may have complex and critical effects on TLRs and HNC pathogenesis, providing important insights into the immune mechanism of HNC and the development of therapeutic strategies for HNC.
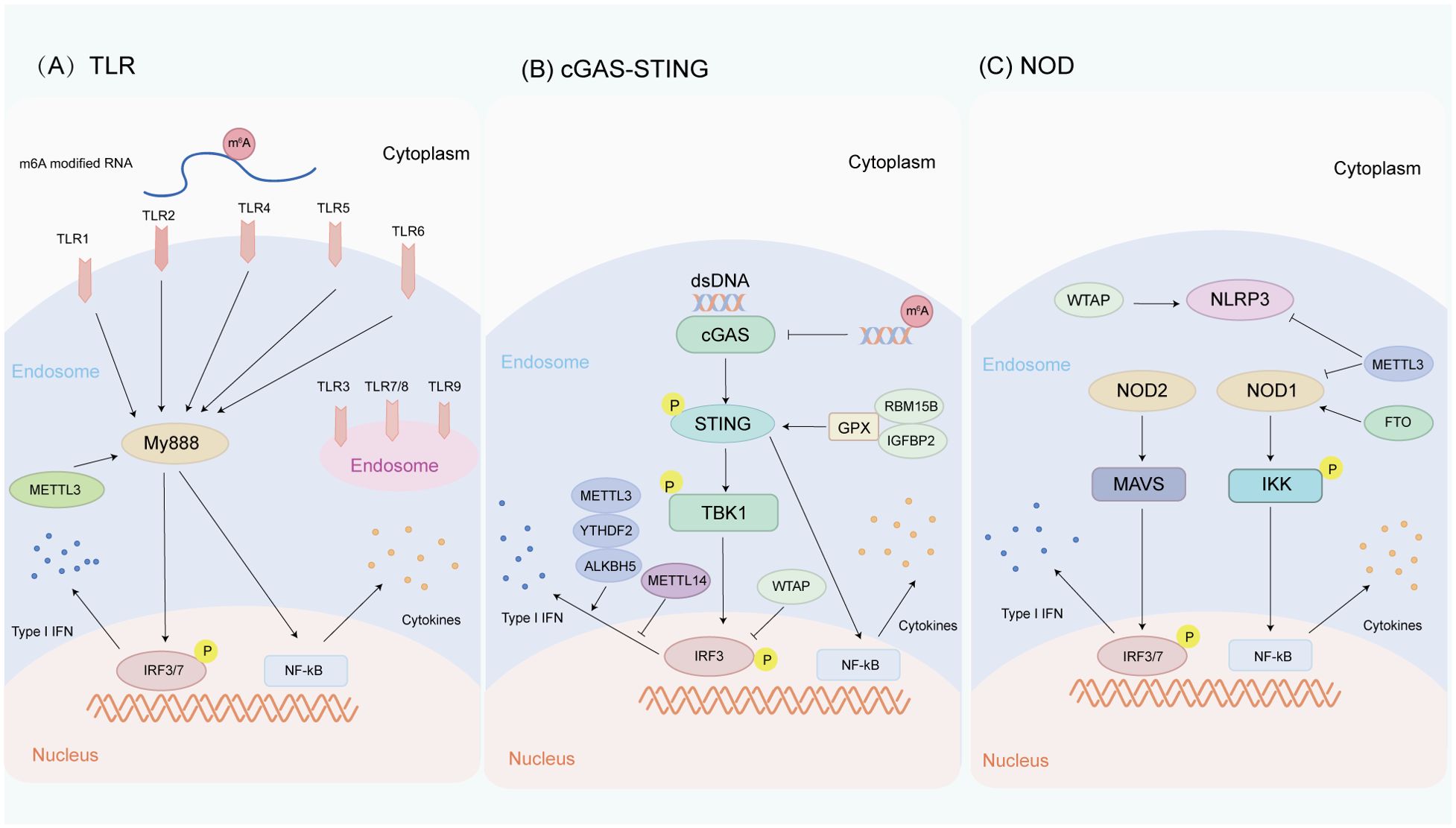
Figure 3. Innate immune pathways are regulated by m6A methylation in HNC. (A) Relationship between m6A methylation and TLR pathway. TLR1, TLR2, TLR4, TLR5, and TLR6 are located on the cell surface, while TLR3, TLR7, TLR8, and TLR9 are located in the endosomal/lysosomal part. METTL3 regulates the alternative splicing of MyD88, a participant in the TLR-mediated antiviral immune pathway. In addition, the difference between m6A-modified RNA and unmodified RNA can cause different TLR signaling activities. (B) Relationship between m6A methylation and cGAS-STING pathway. cGAS is an intracellular DNA sensor that activates the IFN pathway by recognizing dsDNA in the cytoplasm. RBM15B and IGFBP2 activate the cGAS-STING pathway by methylating GPX4 mRNA, thereby increasing the activity of the cGAS-STING pathway. METTL3, YTHDF2, AKLBH5, METTL14, and WTAP regulate the production of type I IFNs triggered by dsDNA. (C) Relationship between m6A methylation and NLR pathway. NLRs are a class of intracellular pattern recognition receptors. METTL3 and FTO regulate NOD1-mediated NF-κB signal transduction WTAP promotes NLRP3 mRNA m6A methylation, thereby upregulating NLRP3 inflammasome activation, while knockdown of METTL14 inhibits NLRP3 inflammasome activation.
4.2 cGAS-STING pathway
The cGAS-STING pathway is a sensor for cytosolic double stranded DNA (dsDNA) in central cells that allows innate immunity to respond to infection, inflammation, and cancer (108, 109). cGAS recognizes a variety of cytosolic dsDNA, including DNA with viral, apoptotic, exosomal, mitochondrial, micronucleus, and retroelement sources. cGAS binds directly to dsDNA and subsequently catalyzes the production of cyclic GMP-AMP (cGAMP) (110). After the cGAMP of stimulation, STING dimers translocate from the endoplasmic reticulum to perinuclear microsomes through the Golgi apparatus to recruit and activate TBK1, further phosphorylating IRF3 and upregulating the expression of type I IFN (111). In addition, STING can activate the NF-κB pathway in activated B cells by binding to IκB kinase (IKK) and NF-κB inducible kinase (NIK) (112). The activated NF-κB pathway cooperates with the TBK1-IRF3 pathway to induce the expression of type I IFNs (113). Type I IFNs have a variety of immunostimulatory functions, which can promote the maturation, migration, and activation of immune cells such as DCs, T cells, and NK cells (114). The possibility of DNA exposure to cytosolic DNA sensors increases in tumor cells, which induces the production of IFN and wakes up the host immune response mediated by the infiltration of immune cells (such as T cells and NK cells), thys inhibiting the progression of tumor cells (115). The cGAS-STING sting pathway downregulates the expression of anti-apoptotic protein BCL2 and upregulates the abundance of pro-apoptotic protein BAX (116). BAX-mediated mitochondrial outer membrane permeabilization and simultaneous caspase-9-driven caspase-3 activation contribute to apoptosis (117). Therefore, the intact cGAS-STING is an important regulator of cancer cell growth and immune surveillance. Due to selective pressure, surviving cancer cells often have defects in the cGAS-STING pathway. It has been detected that the activation of cGAS-STING is often impaired by epigenetic hypermethylation in a variety of cancers (118). All these findings suggest that understanding the regulatory mechanism of cGAS-STING pathway and its alternations in different cancer types is crucial for developing new immunotherapeutic strategies.
Human papillomavirus (HPV) is closely associated with HNSCC, and HPV16 is the primary high-risk HPV type (119). In HPV16-positive HNSCC cells, the cGAS-STING pathway is blocked due to the highly conserved LXCXE motif of its 16E7 oncogene, which in turn leads to the blockade of the relevant immune response (120, 121). In HNSCC, STING expression is also associated with enhanced immune infiltration in the tumor microenvironment and hence is considered as a favorable prognostic factor (122). By analyzing the STING expression of HNSCC cohort in TCGA, it is found that low STING mRNA is indicative of poor overall survival (123). HNSCC patients often receive DNA damage therapy (i.e., radiotherapy and cisplatin), and data show that low STING expression is associated with treatment resistance (123). In TCGA, the prognosis of OSCC patients with high expression of cGAS-STING is better (124). In multiple solid tumor types, impaired cGAS-STING signaling is associated with poor prognosis and low immune cell infiltration. However, some studies have found the specific increase of STING expression in intrinsic tumor cells with the progression of LSCC, although no correlation between STING and CGAs has been observed at the protein level (125). The low expression of MYC in HNSCC induces DNA damage related cGAS-STING pathway, which increases the recruitment of chemokines by CD8 T cells, promotes tumor immune response, and inhibits the proliferation and migration of HNSCC (126). In NPC patients, TRIM21 promotes the degradation of VDAC2 via K48-linked ubiquitination, which inhibits pore formation by VDAC2 oligomers for mitochondrial DNA (mtDNA) release, thereby inhibiting type I IFN responses following radiation exposure (127). Therefore, understanding the activation of cGAS-STING pathway in HNC is conducive to formulating suitable immunotherapy plans.
HPV infection (oropharyngeal carcinoma) and EB virus infection (nasopharyngeal carcinoma) are the high-risk factors of HNC. m6A modification has been reported to regulate cGAS-STING signal transduction. Rubio et al. observed that the m6A methyltransferase METTL14 and demethylase ALKBH5 controlled the production of type I IFNs triggered by dsDNA (128) (Figure 3B). Roni et al. have revealed that m6A acts as a negative regulator of IFN response by determining the rapid turnover of IFN mRNA and thus promoting viral transmission (129). Ge et al. have found that WTAP, as one of the “writers” of m6A, participates in the intracellular WTAP-IRF3/IFNAR1 axis as a negative feedback pathway to fine tune the activation of type I IFN signaling (130). Melania et al. have unveiled that the cytoplasmic recognition of m6A methylated dsDNA enhances the activation of macrophages and dendritic cells, which may be due to the fact that m6A methylation changes the immunogenicity of cytoplasmic DNA, affects the secondary structure of DNA, and causes DNA bending, thus affecting the binding affinity between dsDNA and cGAS and promoting cGAS dimerization or its enzymatic activity (131). Chen et al. have found that RBM15B and IGFBP2 mediate m6A-modified GPX4 to promote cGAS-STING signaling activation by maintaining redox homeostasis in colorectal adenocarcinoma, thereby promoting its anticancer immune response (132). The newly discovered nuclear DNA sensor hnRNPA2B1 is also an m6A reader that can partially promote DNA-induced innate immunity by promoting mRNA m6A modification of cGAS, IFI16, and STING (133). TRIM29 is also an important innate immunomodulator, which upregulates TRIM29 translation in cisplatin-resistant ovarian cancer cells through YTHDF1-mediated m6A modification (134). Prior studies showed that TRIM29 could promote DNA and RNA viral infections by modulating DNA and RNA sensor-mediated type I interferon pathways (135, 136). A recent study also revealed that TRIM29 enhanced PERK-mediated immune responses to endoplasmic reticulum stress to facilitate viral myocarditis caused by cardiotropic viruses (137). Given the critical role of TRIM29 in regulating innate immunity and cancer, it is a worthwhile direction for in-depth research to explore whether m6A modification can regulate TRIM29 expression to influence innate immune responses in HNC. These findings highlight the important impact of m6A methylation on the cGAS-STING pathway in HNC and indicate its potential as a hotspot for future HNC research.
4.3 NOD pathway
NLRs are a class of intracellular pattern recognition receptors (PRRs) that recognize PAMPs or DAMPs in the cytosol and activate NF-κB complexes, initiate the assembly of a variety of protein complexes including inflammasomes, and lead to the secretion of pro-inflammatory and chemotactic cytokines (138, 139). NLRs usually consist of three domains, including an N-terminal regulatory domain, a nucleotide oligomerization binding domain (NOD), and a C-terminal leucine-rich repeat domain (LRR) (140). The combination of five different functional elements in the N-terminal domain, namely the acidic transactivation domain (AD), pyrin domain (PYD), caspase recruitment domain (CARD), death effector domain (DED), and baculovirus inhibitor repeat domain (BIR), divides NLRs into five subfamily members: NLRA, NLRB, NLRC, NLRP, and NLRX (141). The NLRP receptor subfamily consists of 14 members characterized by the presence of an N-terminal pyrin (PYD) effector domain (142), which has conserved sequence motifs found in more than 20 human proteins. NLRP1, NLRP3, and NLRC4 can assemble a multi-molecular complex called “inflammasome”. The formation of inflammasomes leads to caspase-1 activation and IL-1 family cytokine maturation, thereby transmitting apoptotic and inflammatory signals. Deletion of NLR family members including NOD1, NOD2, NLRP3, NLRC4, and NLRP12 also accelerates carcinogenicity (143–145). However, the overexpression of NLR pathway components mediates the occurrence of inflammation and also drives cancer. Some studies have directly shown that NLRs can participate in immune monitoring (146, 147), and NLRs are also related to antitumor immunity through the link between IL-18 and increased NK cell antitumor activity (148, 149). Therefore, NLRs mediate the balance between inflammation and repair to maintain the homeostasis of each tissue, and malignant tumors may occur if tilted in either direction.
Biopsy results of nasal cancer and HNC have shown that cancer cells first show consistent and strong expression of NOD1 and NAIP, while normal epithelial cells show a broader NLR spectrum with the presence of NOD1, NOD2, NLRP1, NLRP3, and NAIP (150). NOD1 stimulation induces an immune response in tumor cells different from that in normal epithelial cells (150). NOD1 and NOD2 genes are expressed in human OSCC cell line YD-10B, which may trigger immune responses via the MAPK pathway. Stimulation with the NOD2 agonist MDP can inhibit cell growth by inducing apoptosis (151). NLRP3 inflammasome acts as a negative regulator of tumorigenesis in HNSCC (152, 153), and blocking NLRP3 inflammasome can also delay the tumor-bearing speed in HNSCC mice (153). Therefore, the regulation of NOD signal transduction possesses significant potential in the treatment of HNC.
m6A RNA methylation plays a pivotal role in the regulation of NOD1 signaling pathway. Cai et al. have found that the NLP signaling pathway may be a target of METTL3. During LPS-induced inflammatory response, METTL3 deletion upregulates the NOD1 pathway in macrophages, but does not affect the NOD2 pathway. METTL3 knockdown increases the mRNA stability of NOD1 and RIPK2 in a YTHDF1- and YTHDF2-dependent manner, thereby activating the NOD1 pathway (154). Xu et al. have noted that knockdown of METTL3 in HepG2 cells significantly increases the mRNA expressions of NOD1, RIPK2, NFKBIA, and NF-κB p65 mRNA, while FTO overexpression significantly increases the mRNA expressions of NOD1 and NF-κB p65 and decreases the mRNA expression of NFKBIA (155) (Figure 3C). Cao et al. have verified that NLRP3 is a downstream target of METTL14 by m6A RNA immunoprecipitation, and knockdown of METTL14 inhibits the activation of NLRP3 inflammasome and alleviates acute lung injury in vivo and in vitro (156). Lan has found that WTAP promotes NLRP3 mRNA m6A methylation, thereby activating NLRP3 inflammasome and further inducing pyroptosis and inflammatory responses of HK-2 cells (157). Overall, these studies clarify the role of m6A in regulating NLR-mediated signal transduction in different diseases, while its specific role in NLR-mediated transduction in HNC needs further exploration.
5 Challenges and prospects
Despite considerable progress in understanding the regulation of m6A methylation on innate immune pathways, the role and mechanism of m6A methylation in different tumors remain elusive due to the high heterogeneity of tumor cells. By virtue of constantly developing technologies, such as high-throughput sequencing and single-cell analysis, we are expected to have a deeper understanding of the distribution of m6A methylation in a variety of HNCs and its impact on innate immune pathways. Further research on the mechanism of m6A methylation in tumors, particularly its regulatory effect on the innate immune pathway, can reveal the mechanism of immune escape in tumors and provide theoretical support for the development of novel immunotherapies in the clinic. For example, targeting m6A methylation regulators can enhance the recognition and clearance of tumor cells by the immune system, thereby increasing the efficacy of immunotherapy. In addition, further research also provides a basis for the discovery of new biomarkers, which can contribute to better tumor diagnosis and prognostic assessment. Nowadays, a growing number of small molecules are emerging as potential drugs targeting m6A regulators for cancer treatment. For example, corresponding competitive inhibitors, such as the METTL3 inhibitor UZH1a, were designed with virtual screening based on adenosine, a substrate analog of m6A regulators (158). Components with an m6A inhibitory activity, including the FTO inhibitor emodin (159) and the METTL3 inhibitor quercetin (160), were identified from natural products. Based on their old use, some clinical drugs, metformin, have also been found to have an m6A inhibitory activity (161). These small molecule modulators of m6A regulators target the epigenetic level and precisely intervene in RNA modification, effectively disrupting the survival mechanism of tumor cells and providing a new path for cancer therapy.
In conclusion, it is crucial for the development of new therapeutic strategies to investigate the mechanism of m6A methylation in tumor innate immune pathways. At present, research on the role of m6A methylation in innate immune pathways during HNC is still in its preliminary stage. Accordingly, it is necessary to further investigate the mechanism by which m6A methylation affects tumor recognition by innate immunity, which is crucial for promoting the understanding of tumor-immune interactions and developing new therapeutic strategies.
Author contributions
LC: Writing – original draft. GH: Writing – original draft. JF: Supervision, Writing – original draft. XL: Conceptualization, Software, Writing – review & editing. ZM: Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
3. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. (2018) 18:269–82. doi: 10.1038/nrc.2018.11
4. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. (2019) 144:1941–53. doi: 10.1002/ijc.v144.8
5. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
6. Paludan SR, Pradeu T, Masters SL, Mogensen TH. Constitutive immune mechanisms: mediators of host defence and immune regulation. Nat Rev Immunol. (2021) 21:137–50. doi: 10.1038/s41577-020-0391-5
7. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
8. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
9. Laskowski TJ, Biederstadt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. (2022) 22:557–75. doi: 10.1038/s41568-022-00491-0
10. Huang H, Weng H, Chen J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. (2020) 37:270–88. doi: 10.1016/j.ccell.2020.02.004
11. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. (2018) 28:507–17. doi: 10.1038/s41422-018-0034-6
12. Lin H, Wang Y, Wang P, Long F, Wang T. Mutual regulation between N6-methyladenosine (m6A) modification and circular RNAs in cancer: impacts on therapeutic resistance. Mol Cancer. (2022) 21:148. doi: 10.1186/s12943-022-01620-x
13. Song H, Song J, Cheng M, Zheng M, Wang T, Tian S, et al. METTL3-mediated m(6)A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun. (2021) 12:5522. doi: 10.1038/s41467-021-25803-0
14. Ma S, Yan J, Barr T, Zhang J, Chen Z, Wang LS, et al. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J Exp Med. (2021) 218:e20210279. doi: 10.1084/jem.20210279
15. Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. (2019) 10:1898. doi: 10.1038/s41467-019-09903-6
16. Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am J Physiol Cell Physiol. (2019) 317:C762–75. doi: 10.1152/ajpcell.00212.2019
17. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
18. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. (2019) 18:176. doi: 10.1186/s12943-019-1109-9
19. Huang J, Yin P. Structural insights into N(6)-methyladenosine (m(6)A) modification in the transcriptome. Genomics Proteomics Bioinf. (2018) 16:85–98. doi: 10.1016/j.gpb.2018.03.001
20. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
21. Wei J, He C. Site-specific m(6)A editing. Nat Chem Biol. (2019) 15:848–9. doi: 10.1038/s41589-019-0349-8
22. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
23. Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. (2013) 4:1798. doi: 10.1038/ncomms2822
24. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)A(m) in the 5’ cap controls mRNA stability. Nature. (2017) 541:371–5. doi: 10.1038/nature21022
25. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell. (2018) 71:973–985 e5. doi: 10.1016/j.molcel.2018.08.011
26. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
27. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. (2018) 28:616–24. doi: 10.1038/s41422-018-0040-8
28. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. (2016) 537:369–73. doi: 10.1038/nature19342
29. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. (2017) 6:e31311. doi: 10.7554/eLife.31311
30. Bailey AS, Batista PJ, Gold RS, Chen YG, de Rooij DG, Chang HY, et al. The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. Elife. (2017) 6:e26116. doi: 10.7554/eLife.26116
31. Li F, Zhao D, Wu J, Shi Y. Structure of the YTH domain of human YTHDF2 in complex with an m(6)A mononucleotide reveals an aromatic cage for m(6)A recognition. Cell Res. (2014) 24:1490–2. doi: 10.1038/cr.2014.153
32. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
33. Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. (2020) 181:1582–1595.e18. doi: 10.1016/j.cell.2020.05.012
34. Hu X, Wu J, Feng Y, Ma H, Zhang E, Zhang C, et al. METTL3-stabilized super enhancers-lncRNA SUCLG2-AS1 mediates the formation of a long-range chromatin loop between enhancers and promoters of SOX2 in metastasis and radiosensitivity of nasopharyngeal carcinoma. Clin Transl Med. (2023) 13:e1361. doi: 10.1002/ctm2.v13.9
35. Zhou Y, Li W. Methyltransferase-like 3-mediated m6A modification of miR-1908-5p contributes to nasopharyngeal carcinoma progression by targeting homeodomain-only protein homeobox. Environ Toxicol. (2024) 39:1631–40. doi: 10.1002/tox.24032
36. Peng J, Zheng H, Liu F, Wu Q, Liu S. The m6A methyltransferase METTL3 affects autophagy and progression of nasopharyngeal carcinoma by regulating the stability of lncRNA ZFAS1. Infect Agent Cancer. (2022) 17:1. doi: 10.1186/s13027-021-00411-1
37. Zhang P, He Q, Lei Y, Li Y, Wen X, Hong M, et al. m(6)A-mediated ZNF750 repression facilitates nasopharyngeal carcinoma progression. Cell Death Dis. (2018) 9:1169. doi: 10.1038/s41419-018-1224-3
38. Chen B, Huang Y, He S, Yu P, Wu L, Peng H. N(6)-methyladenosine modification in 18S rRNA promotes tumorigenesis and chemoresistance via HSF4b/HSP90B1/mutant p53 axis. Cell Chem Biol. (2023) 30:144–158.e10. doi: 10.1016/j.chembiol.2023.01.006
39. Zheng ZQ, Huang ZH, Liang YL, Zheng WH, Xu C, Li ZX, et al. VIRMA promotes nasopharyngeal carcinoma, tumorigenesis, and metastasis by upregulation of E2F7 in an m6A-dependent manner. J Biol Chem. (2023) 299:104677. doi: 10.1016/j.jbc.2023.104677
40. Li ZX, Zheng ZQ, Yang PY, Lin L, Zhou GQ, Lv JW, et al. WTAP-mediated m(6)A modification of lncRNA DIAPH1-AS1 enhances its stability to facilitate nasopharyngeal carcinoma growth and metastasis. Cell Death Differ. (2022) 29:1137–51. doi: 10.1038/s41418-021-00905-w
41. Zhou Q, Song W, Li X, Lin J, Zhu C, Cao L, et al. N6-Methyladenosine reader HNRNPC-mediated downregulation of circITCH prevents miR-224-3p sequestering and contributes to tumorigenesis in nasopharyngeal carcinoma. Environ Toxicol. (2024) 39:2893–907. doi: 10.1002/tox.24139
42. Yang Z, Zhang S, Xiong J, Xia T, Zhu R, Miao M, et al. The m(6)A demethylases FTO and ALKBH5 aggravate the Malignant progression of nasopharyngeal carcinoma by coregulating ARHGAP35. Cell Death Discovery. (2024) 10:43. doi: 10.1038/s41420-024-01810-0
43. Huang WM, Li ZX, Wu YH, Shi ZL, Mi JL, Hu K, et al. m6A demethylase FTO renders radioresistance of nasopharyngeal carcinoma via promoting OTUB1-mediated anti-ferroptosis. Transl Oncol. (2023) 27:101576. doi: 10.1016/j.tranon.2022.101576
44. Wang YQ, Wu DH, Wei D, Shen JY, Huang ZW, Liang XY, et al. TEAD4 is a master regulator of high-risk nasopharyngeal carcinoma. Sci Adv. (2023) 9:eadd0960. doi: 10.1126/sciadv.add0960
45. He JJ, Li Z, Rong ZX, Gao J, Mu Y, Guan YD, et al. m(6)A reader YTHDC2 promotes radiotherapy resistance of nasopharyngeal carcinoma via activating IGF1R/AKT/S6 signaling axis. Front Oncol. (2020) 10:1166. doi: 10.3389/fonc.2020.01166
46. Chen B, Huang R, Xia T, Wang C, Xiao X, Lu S, et al. The m6A reader IGF2BP3 preserves NOTCH3 mRNA stability to sustain Notch3 signaling and promote tumor metastasis in nasopharyngeal carcinoma. Oncogene. (2023) 42:3564–74. doi: 10.1038/s41388-023-02865-6
47. Du M, Peng Y, Li Y, Sun W, Zhu H, Wu J, et al. MYC-activated RNA N6-methyladenosine reader IGF2BP3 promotes cell proliferation and metastasis in nasopharyngeal carcinoma. Cell Death Discovery. (2022) 8:53. doi: 10.1038/s41420-022-00844-6
48. Huang J, Li H, Yang Z, Liu R, Li Y, Hu Y, et al. SALL4 promotes cancer stem-like cell phenotype and radioresistance in oral squamous cell carcinomas via methyltransferase-like 3-mediated m6A modification. Cell Death Dis. (2024) 15:139. doi: 10.1038/s41419-024-06533-9
49. Liu L, Wu Y, Li Q, Liang J, He Q, Zhao L, et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m(6)A Methylation in Oral Squamous Cell Carcinoma. Mol Ther. (2020) 28:2177–90. doi: 10.1016/j.ymthe.2020.06.024
50. Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, et al. METTL3 Facilitates Oral Squamous Cell Carcinoma Tumorigenesis by Enhancing c-Myc Stability via YTHDF1-Mediated m(6)A Modification. Mol Ther Nucleic Acids. (2020) 20:1–12. doi: 10.1016/j.omtn.2020.01.033
51. Xu L, Li Q, Wang Y, Wang L, Guo Y, Yang R, et al. m(6)A methyltransferase METTL3 promotes oral squamous cell carcinoma progression through enhancement of IGF2BP2-mediated SLC7A11 mRNA stability. Am J Cancer Res. (2021) 11:5282–98.
52. Chen J, Li S, Huang Z, Cao C, Wang A, He Q. METTL3 suppresses anlotinib sensitivity by regulating m(6)A modification of FGFR3 in oral squamous cell carcinoma. Cancer Cell Int. (2022) 22:295. doi: 10.1186/s12935-022-02715-7
53. Wang C, Kadigamuwa C, Wu S, Gao Y, Chen W, Gu Y, et al. RNA N6-methyladenosine (m6A) methyltransferase-like 3 facilitates tumorigenesis and cisplatin resistance of arecoline-exposed oral carcinoma. Cells. (2022) 11(22):3605. doi: 10.3390/cells11223605
54. Geissler R, Golbik RP, Behrens SE. The DEAD-box helicase DDX3 supports the assembly of functional 80S ribosomes. Nucleic Acids Res. (2012) 40:4998–5011. doi: 10.1093/nar/gks070
55. Shriwas O, Priyadarshini M, Samal SK, Rath R, Panda S, Das MSK, et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m(6)A-demethylation of FOXM1 and NANOG. Apoptosis. (2020) 25:233–46. doi: 10.1007/s10495-020-01591-8
56. Hirayama M, Wei FY, Chujo T, Oki S, Yakita M, Kobayashi D, et al. FTO demethylates cyclin D1 mRNA and controls cell-cycle progression. Cell Rep. (2020) 31:107464. doi: 10.1016/j.celrep.2020.03.028
57. Li X, Chen W, Gao Y, Song J, Gu Y, Zhang J, et al. Fat mass and obesity-associated protein regulates arecoline-exposed oral cancer immune response through programmed cell death-ligand 1. Cancer Sci. (2022) 113:2962–73. doi: 10.1111/cas.v113.9
58. Wang F, Liao Y, Zhang M, Zhu Y, Wang W, Cai H, et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in Malignant development of oral squamous cell carcinoma. Oncogene. (2021) 40:3885–98. doi: 10.1038/s41388-021-01820-7
59. Wang Z, Li H, Cai H, Liang J, Jiang Y, Song F, et al. FTO sensitizes oral squamous cell carcinoma to ferroptosis via suppressing ACSL3 and GPX4. Int J Mol Sci. (2023) 24(22):16339. doi: 10.3390/ijms242216339
60. Wang X, Zhi M, Zhao W, Deng J. HNRNPA2B1 promotes oral squamous cell carcinogenesis via m(6) A-dependent stabilization of FOXQ1 mRNA stability. IUBMB Life. (2024) 76(7):437–50. doi: 10.1002/iub.v76.7
61. Wang X, Tian L, Li Y, Wang J, Yan B, Yang L, et al. RBM15 facilitates laryngeal squamous cell carcinoma progression by regulating TMBIM6 stability through IGF2BP3 dependent. J Exp Clin Cancer Res. (2021) 40:80. doi: 10.1186/s13046-021-01871-4
62. Li Y, Yan B, Wang X, Li Q, Kan X, Wang J, et al. ALKBH5-mediated m6A modification of lncRNA KCNQ1OT1 triggers the development of LSCC via upregulation of HOXA9. J Cell Mol Med. (2022) 26:385–98. doi: 10.1111/jcmm.17091
63. Yang L, Yan B, Qu L, Ren J, Li Q, Wang J, et al. IGF2BP3 Regulates TMA7-mediated Autophagy and Cisplatin Resistance in Laryngeal Cancer via m6A RNA Methylation. Int J Biol Sci. (2023) 19:1382–400. doi: 10.7150/ijbs.80921
64. Tang X, Tang Q, Li S, Li M, Yang T. IGF2BP2 acts as a m(6)A modification regulator in laryngeal squamous cell carcinoma through facilitating CDK6 mRNA stabilization. Cell Death Discovery. (2023) 9:371. doi: 10.1038/s41420-023-01669-7
65. Li J, Cao H, Yang J, Wang B. IGF2BP2-m6A-circMMP9 axis recruits ETS1 to promote TRIM59 transcription in laryngeal squamous cell carcinoma. Sci Rep. (2024) 14:3014. doi: 10.1038/s41598-024-53422-4
66. Xu N, Chen J, He G, Gao L, Zhang D. Prognostic values of m6A RNA methylation regulators in differentiated Thyroid Carcinoma. J Cancer. (2020) 11:5187–97. doi: 10.7150/jca.41193
67. Wang X, Fu X, Zhang J, Xiong C, Zhang S, Lv Y. Identification and validation of m(6)A RNA methylation regulators with clinical prognostic value in Papillary thyroid cancer. Cancer Cell Int. (2020) 20:203. doi: 10.1186/s12935-020-01283-y
68. Zhang W, Ruan X, Huang Y, Zhang W, Xu G, Zhao J, et al. SETMAR facilitates the differentiation of thyroid cancer by regulating SMARCA2-mediated chromatin remodeling. Adv Sci (Weinh). (2024) 11(32):e2401712. doi: 10.1002/advs.202401712
69. Zhou X, Chang L, Liang Q, Zhao R, Xiao Y, Xu Z, et al. The m6A methyltransferase METTL3 drives thyroid cancer progression and lymph node metastasis by targeting LINC00894. Cancer Cell Int. (2024) 24:47. doi: 10.1186/s12935-024-03240-5
70. Lin S, Zhu Y, Ji C, Yu W, Zhang C, Tan L, et al. METTL3-induced miR-222-3p upregulation inhibits STK4 and promotes the Malignant behaviors of thyroid carcinoma cells. J Clin Endocrinol Metab. (2022) 107:474–90. doi: 10.1210/clinem/dgab480
71. Zhu Y, Peng X, Zhou Q, Tan L, Zhang C, Lin S, et al. METTL3-mediated m6A modification of STEAP2 mRNA inhibits papillary thyroid cancer progress by blocking the Hedgehog signaling pathway and epithelial-to-mesenchymal transition. Cell Death Dis. (2022) 13:358. doi: 10.1038/s41419-022-04817-6
72. He J, Zhou M, Yin J, Wan J, Chu J, Jia J, et al. METTL3 restrains papillary thyroid cancer progression via m(6)A/c-Rel/IL-8-mediated neutrophil infiltration. Mol Ther. (2021) 29:1821–37. doi: 10.1016/j.ymthe.2021.01.019
73. Ning J, Hou X, Hao J, Zhang W, Shi Y, Huang Y, et al. METTL3 inhibition induced by M2 macrophage-derived extracellular vesicles drives anti-PD-1 therapy resistance via M6A-CD70-mediated immune suppression in thyroid cancer. Cell Death Differ. (2023) 30:2265–79. doi: 10.1038/s41418-023-01217-x
74. Li Q, Wang Y, Meng X, Wang W, Duan F, Chen S, et al. METTL16 inhibits papillary thyroid cancer tumorigenicity through m(6)A/YTHDC2/SCD1-regulated lipid metabolism. Cell Mol Life Sci. (2024) 81:81. doi: 10.1007/s00018-024-05146-x
75. Wang C, Wang Z, Fu L, Du J, Ji F, Qiu X. CircNRCAM up-regulates NRCAM to promote papillary thyroid carcinoma progression. J Endocrinol Invest. (2024) 47:1215–26. doi: 10.1007/s40618-023-02241-x
76. Ji X, Lv C, Huang J, Dong W, Sun W, Zhang H. ALKBH5-induced circular RNA NRIP1 promotes glycolysis in thyroid cancer cells by targeting PKM2. Cancer Sci. (2023) 114:2318–34. doi: 10.1111/cas.v114.6
77. Li W, Huang G, Wei J, Cao H, Jiang G. ALKBH5 inhibits thyroid cancer progression by promoting ferroptosis through TIAM1-Nrf2/HO-1 axis. Mol Cell Biochem. (2023) 478:729–41. doi: 10.1007/s11010-022-04541-x
78. Ji FH, Fu XH, Li GQ, He Q, Qiu XG. FTO prevents thyroid cancer progression by SLC7A11 m6A methylation in a ferroptosis-dependent manner. Front Endocrinol (Lausanne). (2022) 13:857765. doi: 10.3389/fendo.2022.857765
79. Huang J, Sun W, Wang Z, Lv C, Zhang T, Zhang D, et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J Exp Clin Cancer Res. (2022) 41:42. doi: 10.1186/s13046-022-02254-z
80. Wang W, Ding Y, Zhao Y, Li X. m6A reader IGF2BP2 promotes lymphatic metastasis by stabilizing DPP4 in papillary thyroid carcinoma. Cancer Gene Ther. (2024) 31:285–99. doi: 10.1038/s41417-023-00702-2
81. Sa R, Liang R, Qiu X, He Z, Liu Z, Chen L. IGF2BP2-dependent activation of ERBB2 signaling contributes to acquired resistance to tyrosine kinase inhibitor in differentiation therapy of radioiodine-refractory papillary thyroid cancer. Cancer Lett. (2022) 527:10–23. doi: 10.1016/j.canlet.2021.12.005
82. Ye M, Dong S, Hou H, Zhang T, Shen M. Oncogenic Role of Long Noncoding RNAMALAT1 in Thyroid Cancer Progression through Regulation of the miR-204/IGF2BP2/m6A-MYC Signaling. Mol Ther Nucleic Acids. (2021) 23:1–12. doi: 10.1016/j.omtn.2020.09.023
83. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
84. Li X, Dai H, Wang H, Han W. Exploring innate immunity in cancer immunotherapy: opportunities and challenges. Cell Mol Immunol. (2021) 18:1607–9. doi: 10.1038/s41423-021-00679-8
85. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. (2009) 9:57–63. doi: 10.1038/nrc2541
86. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
87. Kent A, Blander JM. Nod-like receptors: key molecular switches in the conundrum of cancer. Front Immunol. (2014) 5:185. doi: 10.3389/fimmu.2014.00185
88. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
89. Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
90. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
91. Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. (2016) 100:27–45. doi: 10.1189/jlb.2RI1115-531R
92. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
93. Forman D, Burley VJ. Gastric cancer: global pattern of the disease and an overview of environmental risk factors. Best Pract Res Clin Gastroenterol. (2006) 20:633–49. doi: 10.1016/j.bpg.2006.04.008
94. Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun. (2010) 78:468–76. doi: 10.1128/IAI.00903-09
95. Makinen LK, Atula T, Hayry V, Jouhi L, Datta N, Lehtonen S, et al. Predictive role of Toll-like receptors 2, 4, and 9 in oral tongue squamous cell carcinoma. Oral Oncol. (2015) 51:96–102. doi: 10.1016/j.oraloncology.2014.08.017
96. Makinen LK, Ahmed A, Hagstrom J, Lehtonen S, Makitie AA, Salo T, et al. Toll-like receptors 2, 4, and 9 in primary, metastasized, and recurrent oral tongue squamous cell carcinomas. J Oral Pathol Med. (2016) 45:338–45. doi: 10.1111/jop.12373
97. Ng LK, Rich AM, Hussaini HM, Thomson WM, Fisher AL, Horne LS, et al. Toll-like receptor 2 is present in the microenvironment of oral squamous cell carcinoma. Br J Cancer. (2011) 104:460–3. doi: 10.1038/sj.bjc.6606057
98. Zeljic K, Supic G, Jovic N, Kozomara R, Brankovic-Magic M, Obrenovic M, et al. Association of TLR2, TLR3, TLR4 and CD14 genes polymorphisms with oral cancer risk and survival. Oral Dis. (2014) 20:416–24. doi: 10.1111/odi.2014.20.issue-4
99. Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. (2009) 69:3105–13. doi: 10.1158/0008-5472.CAN-08-3838
100. Chuang HC, Chou MH, Chien CY, Chuang JH, Liu YL. Triggering TLR3 pathway promotes tumor growth and cisplatin resistance in head and neck cancer cells. Oral Oncol. (2018) 86:141–9. doi: 10.1016/j.oraloncology.2018.09.015
101. Han N, Zhang Z, Jv H, Hu J, Ruan M, Zhang C. Culture supernatants of oral cancer cells induce impaired IFN-alpha production of pDCs partly through the down-regulation of TLR-9 expression. Arch Oral Biol. (2018) 93:141–8. doi: 10.1016/j.archoralbio.2018.06.006
102. Ruuskanen M, Leivo I, Minn H, Vahlberg T, Haglund C, Hagstrom J, et al. Expression of toll-like receptors in non-endemic nasopharyngeal carcinoma. BMC Cancer. (2019) 19:624. doi: 10.1186/s12885-019-5816-9
103. Sikora J, Frydrychowicz M, Kaczmarek M, Brzezicha B, Mozer-Lisewska I, Szczepanski M, et al. TLR receptors in laryngeal carcinoma - immunophenotypic, molecular and functional studies. Folia Histochem Cytobiol. (2010) 48:624–31. doi: 10.2478/v10042-010-0077-0
104. Hagstrom J, Heikkila A, Siironen P, Louhimo J, Heiskanen I, Maenpaa H, et al. TLR-4 expression and decrease in chronic inflammation: indicators of aggressive follicular thyroid carcinoma. J Clin Pathol. (2012) 65:333–8. doi: 10.1136/jclinpath-2011-200402
105. Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m(6)A as a positive regulator of macrophage activation. Sci Adv. (2021) 7(18):eabd4742. doi: 10.1126/sciadv.abd4742
106. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J Cell Mol Med. (2018) 22:2558–68. doi: 10.1111/jcmm.2018.22.issue-5
107. Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. (2005) 23:165–75. doi: 10.1016/j.immuni.2005.06.008
108. Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. (2013) 14:19–26. doi: 10.1038/ni.2491
109. Samson N, Ablasser A. The cGAS-STING pathway and cancer. Nat Cancer. (2022) 3:1452–63. doi: 10.1038/s43018-022-00468-w
110. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
111. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347(6227):aaa2630. doi: 10.1126/science.aaa2630
112. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
113. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
114. de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. (2007) 282:20053–7. doi: 10.1074/jbc.R700006200
115. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. (2018) 49:754–763.e4. doi: 10.1016/j.immuni.2018.09.016
116. Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell. (2018) 34:361–78. doi: 10.1016/j.ccell.2018.05.013
117. Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. (2010) 29:1762–73. doi: 10.1038/emboj.2010.50
118. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. (2016) 14:282–97. doi: 10.1016/j.celrep.2015.12.029
119. Bratman SV, Bruce JP, O’Sullivan B, Pugh TJ, Xu W, Yip KW, et al. Human papillomavirus genotype association with survival in head and neck squamous cell carcinoma. JAMA Oncol. (2016) 2:823–6. doi: 10.1001/jamaoncol.2015.6587
120. Shaikh MH, Bortnik V, McMillan NA, Idris A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb Pathog. (2019) 132:162–5. doi: 10.1016/j.micpath.2019.05.004
121. Bortnik V, Wu M, Julcher B, Salinas A, Nikolic I, Simpson KJ, et al. Loss of HPV type 16 E7 restores cGAS-STING responses in human papilloma virus-positive oropharyngeal squamous cell carcinomas cells. J Microbiol Immunol Infect. (2021) 54:733–9. doi: 10.1016/j.jmii.2020.07.010
122. Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J Clin Invest. (2020) 130:1635–52. doi: 10.1172/JCI129497
123. Hayman TJ, Baro M, MacNeil T, Phoomak C, Aung TN, Cui W, et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat Commun. (2021) 12:2327. doi: 10.1038/s41467-021-22572-8
124. Zhu C, Li J, Yao M, Fang C. Potential for treatment benefit of STING agonists plus immune checkpoint inhibitors in oral squamous cell carcinoma. BMC Oral Health. (2021) 21:506. doi: 10.1186/s12903-021-01813-8
125. Viculin J, Degoricija M, Vilovic K, Gabela I, Frankovic L, Vrdoljak E, et al. Elevated tumor cell-intrinsic STING expression in advanced laryngeal cancer. Cancers (Basel). (2023) 15(13):3510. doi: 10.3390/cancers15133510
126. Liu S, Qin Z, Mao Y, Zhang W, Wang Y, Jia L, et al. Therapeutic targeting of MYC in head and neck squamous cell carcinoma. Oncoimmunology. (2022) 11:2130583. doi: 10.1080/2162402X.2022.2130583
127. Li JY, Zhao Y, Gong S, Wang MM, Liu X, He QM, et al. TRIM21 inhibits irradiation-induced mitochondrial DNA release and impairs antitumour immunity in nasopharyngeal carcinoma tumour models. Nat Commun. (2023) 14:865. doi: 10.1038/s41467-023-36523-y
128. Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. (2018) 32:1472–84. doi: 10.1101/gad.319475.118
129. Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol. (2019) 20:173–82. doi: 10.1038/s41590-018-0275-z
130. Ge Y, Ling T, Wang Y, Jia X, Xie X, Chen R, et al. Degradation of WTAP blocks antiviral responses by reducing the m(6) A levels of IRF3 and IFNAR1 mRNA. EMBO Rep. (2021) 22:e52101. doi: 10.15252/embr.202052101
131. Balzarolo M, Engels S, de Jong AJ, Franke K, van den Berg TK, Gulen MF, et al. m6A methylation potentiates cytosolic dsDNA recognition in a sequence-specific manner. Open Biol. (2021) 11:210030. doi: 10.1098/rsob.210030
132. Chen B, Hong Y, Zhai X, Deng Y, Hu H, Tian S, et al. m6A and m5C modification of GPX4 facilitates anticancer immunity via STING activation. Cell Death Dis. (2023) 14:809. doi: 10.1038/s41419-023-06241-w
133. Wang L, Wen M, Cao X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. (2019) 365(6454):eaav0758. doi: 10.1126/science.aav0758
134. Hao L, Wang JM, Liu BQ, Yan J, Li C, Jiang JY, et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118878. doi: 10.1016/j.bbamcr.2020.118878
135. Li W, Song Y, Du Y, Huang Z, Zhang M, Chen Z, et al. Duck TRIM29 negatively regulates type I IFN production by targeting MAVS. Front Immunol. (2022) 13:1016214. doi: 10.3389/fimmu.2022.1016214
136. Li Q, Lin L, Tong Y, Liu Y, Mou J, Wang X, et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discovery. (2018) 4:13. doi: 10.1038/s41421-018-0010-9
137. Wang J, Lu W, Zhang J, Du Y, Fang M, Zhang A, et al. Loss of TRIM29 mitigates viral myocarditis by attenuating PERK-driven ER stress response in male mice. Nat Commun. (2024) 15:3481. doi: 10.1038/s41467-024-44745-x
138. Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev. (2015) 95:149–78. doi: 10.1152/physrev.00009.2014
139. Sundaram B, Tweedell RE, Prasanth KS, Kanneganti TD. The NLR family of innate immune and cell death sensors. Immunity. (2024) 57:674–99. doi: 10.1016/j.immuni.2024.03.012
140. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. (2007) 27:549–59. doi: 10.1016/j.immuni.2007.10.002
141. Zhou Y, Yu S, Zhang W. NOD-like receptor signaling pathway in gastrointestinal inflammatory diseases and cancers. Int J Mol Sci. (2023) 24(19):14511. doi: 10.3390/ijms241914511
142. Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. (2009) 119:3502–11. doi: 10.1172/JCI40599
143. Werts C, Rubino S, Ling A, Girardin SE, Philpott DJ. Nod-like receptors in intestinal homeostasis, inflammation, and cancer. J Leukoc Biol. (2011) 90:471–82. doi: 10.1189/jlb.0411183
144. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. (2011) 20:649–60. doi: 10.1016/j.ccr.2011.10.022
145. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. (2012) 36:742–54. doi: 10.1016/j.immuni.2012.03.012
146. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
147. Huang TT, Ojcius DM, Young JD, Wu YH, Ko YF, Wong TY, et al. The anti-tumorigenic mushroom Agaricus blazei Murill enhances IL-1beta production and activates the NLRP3 inflammasome in human macrophages. PloS One. (2012) 7:e41383. doi: 10.1371/journal.pone.0041383
148. Nishio S, Yamada N, Ohyama H, Yamanegi K, Nakasho K, Hata M, et al. Enhanced suppression of pulmonary metastasis of Malignant melanoma cells by combined administration of alpha-galactosylceramide and interleukin-18. Cancer Sci. (2008) 99:113–20. doi: 10.1111/j.1349-7006.2007.00636.x
149. Zheng JN, Pei DS, Mao LJ, Liu XY, Sun FH, Zhang BF, et al. Oncolytic adenovirus expressing interleukin-18 induces significant antitumor effects against melanoma in mice through inhibition of angiogenesis. Cancer Gene Ther. (2010) 17:28–36. doi: 10.1038/cgt.2009.38
150. Millrud CR, Kvarnhammar AM, Tajti J, Munck-Wikland E, Uddman R, Cardell LO. Nod-like receptors in head and neck squamous cell carcinoma. Acta Otolaryngol. (2013) 133:1333–44. doi: 10.3109/00016489.2013.831476
151. Yoon HE, Ahn MY, Kwon SM, Kim DJ, Lee J, Yoon JH. Nucleotide-binding oligomerization domain 2 (NOD2) activation induces apoptosis of human oral squamous cell carcinoma cells. J Oral Pathol Med. (2016) 45:262–7. doi: 10.1111/jop.2016.45.issue-4
152. Wang H, Luo Q, Feng X, Zhang R, Li J, Chen F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer. (2018) 18:500. doi: 10.1186/s12885-018-4403-9
153. Huang CF, Chen L, Li YC, Wu L, Yu GT, Zhang WF, et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. (2017) 36:116. doi: 10.1186/s13046-017-0589-y
154. Cai Y, Yu R, Kong Y, Feng Z, Xu Q. METTL3 regulates LPS-induced inflammatory response via the NOD1 signaling pathway. Cell Signal. (2022) 93:110283. doi: 10.1016/j.cellsig.2022.110283
155. Luo J, Wang F, Sun F, Yue T, Zhou Q, Yang C, et al. Targeted inhibition of FTO demethylase protects mice against LPS-induced septic shock by suppressing NLRP3 inflammasome. Front Immunol. (2021) 12:663295. doi: 10.3389/fimmu.2021.663295
156. Cao F, Chen G, Xu Y, Wang X, Tang X, Zhang W, et al. METTL14 contributes to acute lung injury by stabilizing NLRP3 expression in an IGF2BP2-dependent manner. Cell Death Dis. (2024) 15:43. doi: 10.1038/s41419-023-06407-6
157. Lan J, Xu B, Shi X, Pan Q, Tao Q. WTAP-mediated N(6)-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett. (2022) 27:51. doi: 10.1186/s11658-022-00350-8
158. Moroz-Omori EV, Huang D, Kumar BR, Cheriyamkunnel SJ, Bochenkova E, Dolbois A, et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem. (2021) 16:3035–43. doi: 10.1002/cmdc.202100291
159. Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. (2012) 134:17963–71. doi: 10.1021/ja3064149
160. Du Y, Yuan Y, Xu L, Zhao F, Wang W, Xu Y, et al. Discovery of METTL3 small molecule inhibitors by virtual screening of natural products. Front Pharmacol. (2022) 13:878135. doi: 10.3389/fphar.2022.878135
Keywords: m6A methylation, TLR, cGAS-STING, NLR, innate immune, head and neck cancer
Citation: Cao L, Huang G, Fan J, Liu X and Ma Z (2024) Role of N6-methyladenosine methylation in head and neck cancer and its regulation of innate immune pathways. Front. Immunol. 15:1458884. doi: 10.3389/fimmu.2024.1458884
Received: 04 July 2024; Accepted: 09 September 2024;
Published: 30 September 2024.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Zhenhua Chen, City of Hope, United StatesGuangchuan Wang, Jinzhou Medical University, China
Copyright © 2024 Cao, Huang, Fan, Liu and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangang Fan, ZW50c2NmamdAMTYzLmNvbQ==; Xingren Liu, bGl1eGluZ3JlbjgyNzBAMTYzLmNvbQ==; Zhiyue Ma, bWF6aGl5dWUyMTQyQDE2My5jb20=
†These authors share first authorship