 Seo Young Choi1†
Seo Young Choi1† Parkyong Song2†Ji Sun Hwang3You Kyeong Lee1Mi Song Shin1Hong-Joo Son1
Parkyong Song2†Ji Sun Hwang3You Kyeong Lee1Mi Song Shin1Hong-Joo Son1 Yu-Jin Kim1
Yu-Jin Kim1 Wanil Kim4*
Wanil Kim4* Kwang Min Lee1*
Kwang Min Lee1*- 1Department of Life Science and Environmental Biochemistry, Pusan National University, Miryang, Republic of Korea
- 2Department of Convergence Medicine, Pusan National University School of Medicine, Yangsan, Republic of Korea
- 3New Drug Development Center, Daegu–Gyeongbuk Medical Innovation Foundation, Daegu, Republic of Korea
- 4Department of Biochemistry, Department of Convergence Medical Science, and Institute of Medical Science, School of Medicine, Gyeongsang National University, Jinju, Republic of Korea
The liver is vulnerable to various hepatotoxins, including carbon tetrachloride (CCl4), which induces oxidative stress and apoptosis by producing reactive oxygen species (ROS) and activating the mitogen-activated protein kinase (MAPK) pathway. Cereblon (CRBN), a multifunctional protein implicated in various cellular processes, functions in the pathogenesis of various diseases; however, its function in liver injury remains unknown. We established a CRBN-knockout (KO) HepG2 cell line and examined its effect on CCl4-induced hepatocellular damage. CRBN-KO cells exhibited reduced sensitivity to CCl4-induced cytotoxicity, as evidenced by decreased levels of apoptosis markers, such as cleaved caspase-3, and aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities. CRBN deficiency enhanced antioxidant defense, with increased superoxide dismutase activity and glutathione ratios (GSH/GSSG), as well as reduced pro-inflammatory cytokine expression. Mechanistically, the protective effects of CRBN deficiency appeared to involve the attenuation of the MAPK-mediated pathways, particularly through decreased phosphorylation of JNK and ERK. Overall, these results suggest the crucial role of CRBN in mediating the hepatocellular response to oxidative stress and inflammation triggered by CCl4 exposure, offering potential clinical implications for liver injury in a wide range of liver diseases.
1 Introduction
The human liver plays a crucial role in the detoxification and metabolic conversion of chemicals, drugs, and harmful substances (1). Detoxification generates reactive oxygen species (ROS) that contribute to oxidative damage, cellular disturbance, and hepatotoxicity. Excessive ROS levels pose a significant risk of cellular toxicity, resulting in oxidative stress (2).
Oxidative stress exhibits several detrimental effects on cells. Oxidized proteins become vulnerable to proteasomal degradation, and reactive oxygen species (ROS) disrupt mitochondrial integrity, altering permeability and releasing pro-apoptotic factors, such as cytochrome C (3). Increased mitochondrial permeability activates caspase-3, leading to cell death (4). Oxidative stress, resulting from an imbalance between ROS production and antioxidant defense mechanisms, is implicated in various diseases, including cancer, arteriosclerosis, diabetes, neurodegeneration, and acute and chronic liver conditions (5–7).
Carbon tetrachloride (CCl4) is a hepatotoxin frequently used to induce liver damage in experimental models. In the liver, CCl4 induces free radical generation via metabolic processes. CCl4 undergoes metabolic activation through a cytochrome P-450-dependent process, further leading to the production of free radicals that initiate inflammation (8). CCl4 also causes the peroxidation of cellular and organelle membrane lipids, inhibits protein synthesis, and disrupts cell morphology, ultimately culminating in apoptosis and necrosis of hepatocytes (9, 10). Thus, understanding the mechanisms of cellular antioxidant defenses is crucial for mitigating the liver damage caused by oxidative stress.
Cereblon (CRBN) is a multifunctional protein that plays diverse roles in cellular physiology and disease pathogenesis. Initially identified as a gene associated with mild forms of autosomal recessive non-syndromic mental retardation (ARNSMR) (11), CRBN has been implicated in various cellular processes (12–14). CRBN is a target for thalidomide-induced birth defects and a substrate receptor within the Cullin-RING E3 ubiquitin ligase (CRL) complex (15). Through direct interaction with the alpha subunit of AMP-activated protein kinase (AMPK, a pivotal metabolic sensor), CRBN inhibits the enzymatic activity of AMPK, suggesting its potential role in metabolic disorders (16, 17). The dysregulation of CRBN function has been implicated in various diseases, including cancer, neurodegenerative disorders, and developmental abnormalities (18–20). However, the physiological role(s) of CRBN in liver injury and its underlying molecular mechanisms have not been investigated yet.
In this study, we investigated the potential hepatoprotective role of CRBN in hepatocellular injury. We specifically explored the effect of CRBN knockout on CCl4-induced liver damage in HepG2 cells and the underlying molecular pathways through which CRBN mitigates hepatocellular injury. Our findings provide insights into novel therapeutic approaches for liver diseases in multiple pathological contexts.
2 Materials and methods
2.1 Cell culture and CCl4 treatment
HepG2 human hepatoblastoma cell line was purchased from the Korean Cell Line Bank (Seoul, South Korea) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Waltham, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone), 100 units/mL penicillin, and 100 µg/mL streptomycin sulfate. α-Mouse liver 12 (AML12; immortalized mouse hepatocytes) cell line was a kind gift of Prof. Hueng-Sik Choi (School of Biological Sciences and Technology, Chonnam National University, Republic of Korea) and cultured in DMEM/F-12 medium (Gibco, Eggenstein, Germany) supplemented with 10% heat-inactivated FBS, 0.1% insulin-transferrin-selenium (ITS; Sigma, Burligton, USA), 40 ng/ml dexamethasone (Sigma, USA), 100 units/mL penicillin, and 100 µg/mL streptomycin sulfate. The cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. For the following experiments, the cells were seeded in a 12-well plate and allowed to reach 90% confluency. Subsequently, the cells were treated with 1 mL of the indicated concentrations of CCl4 dissolved in DMEM containing 0.25% DMSO (21). The control treatment included only DMEM containing 0.25% DMSO. After 1 h of CCl4 treatment, the cells and culture medium were collected separately. The cells were lysed with RIPA buffer for western blot analysis, homogenized according to the manufacturer’s protocol for measuring SOD activity and GSH/GSSG ratio, and total RNA was extracted using TRIzol reagent. The culture medium was used to measure the AST and ALT activities.
2.2 Generation of CRBN knockout cell line using CRISPR-Cas9-mediated gene editing
The CRISPR/Cas9 Gene Knockout Kit specific for the human CRBN gene (sc-418922) or control (sc-412142) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Plasmids containing the CRISPR/Cas9 system were transfected into HepG2 cells using the Lipofectamine transfection reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. Two days after transfection, the cells were treated with 2 μg/mL puromycin (Sigma, St. Louis, MO, USA) for 3 days to select for transfected cells. Subsequently, serial dilutions were performed in 96-well plates to obtain one cell per well for clonal selection. The expression of CRBN was analyzed using western blotting.
2.3 AST and ALT activity
AST and ALT activities in the supernatant after exposure of cells to 0.1% CCl4 were determined using the colorimetric assay kit (AST; #K753-100, ALT; #K752- 100, Biovision Co., CA, USA), according to manufacturer’s instructions.
2.4 Antioxidant parameters
SOD activity and the GSH/GSSG ratio were determined using commercially available assay kits (SOD; #S311, GSH/GSSG; #G257, Dojindo Molecular Technologies, Kumamoto, Japan), according to the manufacturer’s instructions.
2.5 Western blotting
Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. After blocking with 3% BSA in TBS-T (137 mM NaCl, 20 mM Tris-Cl, pH 7.6, 0.1% Tween 20), the membranes were incubated with various primary antibodies, including anti-cleaved caspase-3 (cell signaling 9664), anti-CRBN (cell Signaling 71810), anti (MAPK; Erk1/2) (R&D systems MAB1576), anti-phospho-p44/42 MAPK (Erk1/2) (cell signaling 4377), anti-p38 MAPK (cell signaling 8690), anti-phospho-p38 MAPK (cell signaling 4511), anti-JNK (cell signaling 9252), anti-phospho-JNK (cell signaling 4668), and anti-tubulin (Sigma-Aldrich T6199). The blots were then incubated with a secondary antibody [anti-rabbit horseradish peroxidase-conjugated or anti-mouse horseradish peroxidase conjugate; Santa Cruz Biotechnology)], and the protein bands were visualized using an enhanced chemiluminescence detection system.
2.6 Cell viability assay
Cell viability was measured using the water-soluble tetrazolium salt 1 assay (WST-1; EZ-CyTox, Dogen, Korea), according to the manufacturer’s instructions. The optical density of the control cells was considered 100% viable.
2.7 Quantitative realtime PCR
IL-1β, IL-6, TNF-α, and COX-2 gene expression was quantified using quantitative real-time polymerase chain reaction (qRT-PCR). Briefly, the total RNA of HepG2 cells was extracted using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Total RNA (2 µg) was used as a template for synthesizing the first-strand of cDNA using the reverse transcriptase reaction mixture (Takara, Shiga, Japan). For qRT-PCR, the PCR mix (FastStart Universal SYBR Green Master; Roche, Switzerland) used in the reaction system and thermocycler conditions were set according to the manufacturer’s instructions. Relative expression data were normalized to GAPDH and are presented as a ratio relative to the control. The primer sequences used for the PCR analyses are listed in Table 1.

Table 1 Oligonucleotide sequences used in RT-PCR analysis.
2.8 Statistical analysis
Significant differences between groups were determined using two-tailed unpaired Student’s t-tests, and multiple comparisons were performed using one-way or two-way repeated-measures ANOVA. Data are expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical significance was set at P < 0.05, as indicated in the figure legends.
3 Results
3.1 Dose-dependent cytotoxicity of carbon tetrachloride in HepG2 cells
To establish a baseline for understanding the effect of CCl4 on hepatocytes, we initially assessed the cytotoxicity of various concentrations of CCl4 in HepG2 cells, a human liver cancer cell line that is used to model hepatocyte function in vitro (22, 23). This was crucial for determining the effective dose that induced observable cellular damage without immediate lethality, thereby facilitating subsequent investigations into the protective role of CRBN. Treatment with 0.05–0.08% CCl4 did not significantly affect cell viability compared to the untreated controls. However, 0.1% CCl4 exhibited a significant reduction in cell viability, establishing this as the optimal concentration for inducing a measurable and consistent toxic effect (Figure 1A). We confirmed these effects by measuring the activity of caspase-3, an indicator of apoptosis, using western blotting (24). Increased cleaved caspase-3 levels were evident at 0.1% CCl4 (Figures 1B, C).
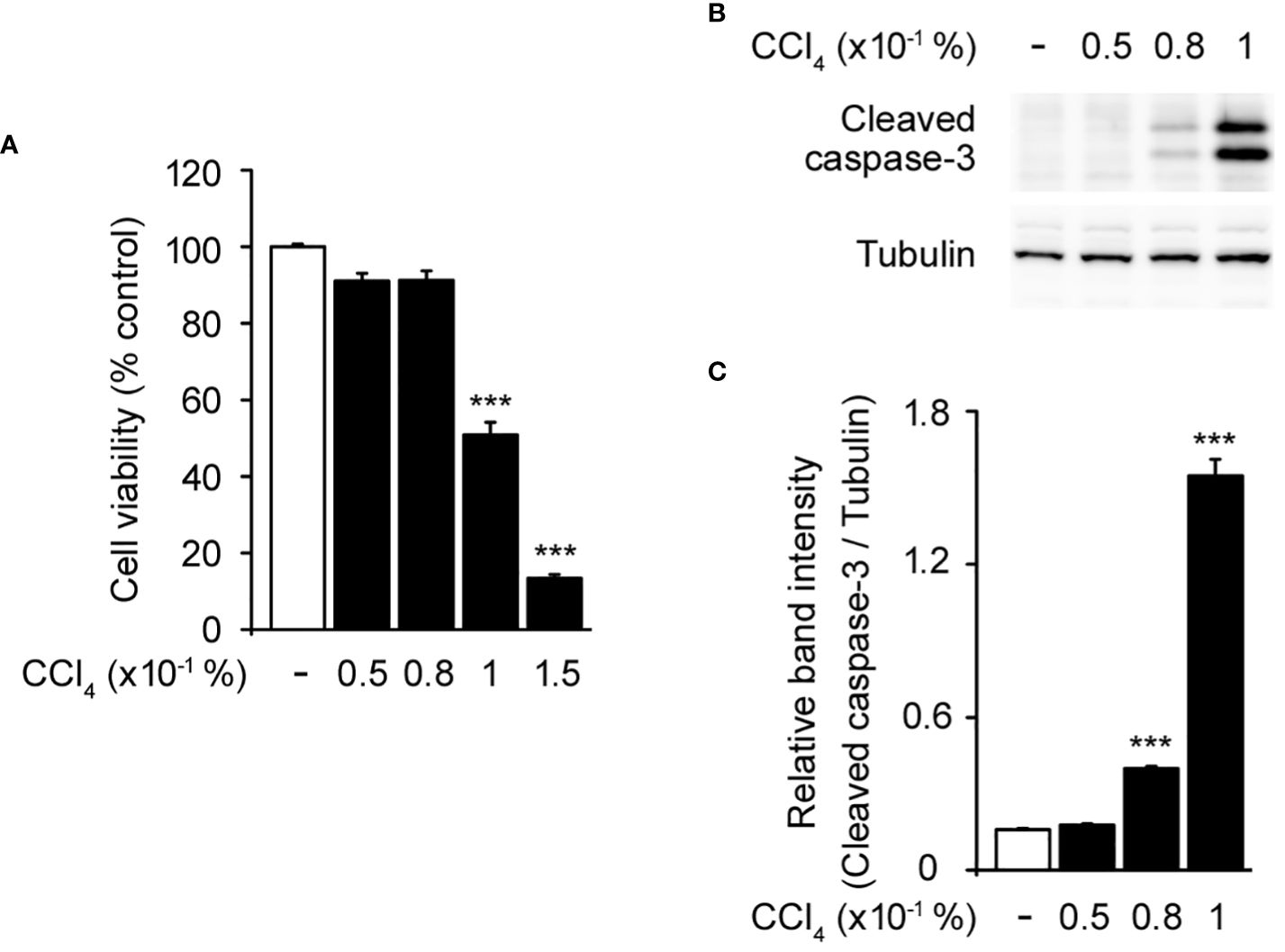
Figure 1 Toxic effects of carbon tetrachloride (CCl4) on HepG2 cells. (A) Cytotoxicity of CCl4 on HepG2 cells. (B) Western blot of the cleaved caspase-3 in HepG2 cells. (C) Relative band intensity of cleaved caspase-3 quantified using densitometric analysis and then normalized to that of tubulin. HepG2 cells were incubated with different concentrations of CCl4 (0.05, 0.08, 0.1, and 0.15%) for 1 h. Values in the bar graphs are the mean ± SEM of at least three independent experiments. ***P < 0.005, significantly different from the control group.
3.2 CRBN deficiency prevented CCl4-stimulated hepatotoxicity in vitro
To investigate the functional involvement of CRBN in liver injury, we used CRISPR-Cas9 technology to develop CRBN knockout (CRBN KO) and control (Ctrl) HepG2 cells. The cells were treated with CCl4 and its morphological changes were evaluated with inverted microscopy and quantitative analysis of cell viability using the WST-1 assay. CCl4 treatment significantly inhibited cell confluence and caused cell death in control cells, which is in agreement with previous studies (1, 25, 26). However, CCl4-induced morphological alterations were prevented in the CRBN KO HepG2 cells (Figure 2A). A comparison of cell viability between Ctrl and CRBN KO cells in the presence of CCl4 revealed that CRBN KO cells exhibited improved viability against CCl4-induced cytotoxicity (Figure 2B).
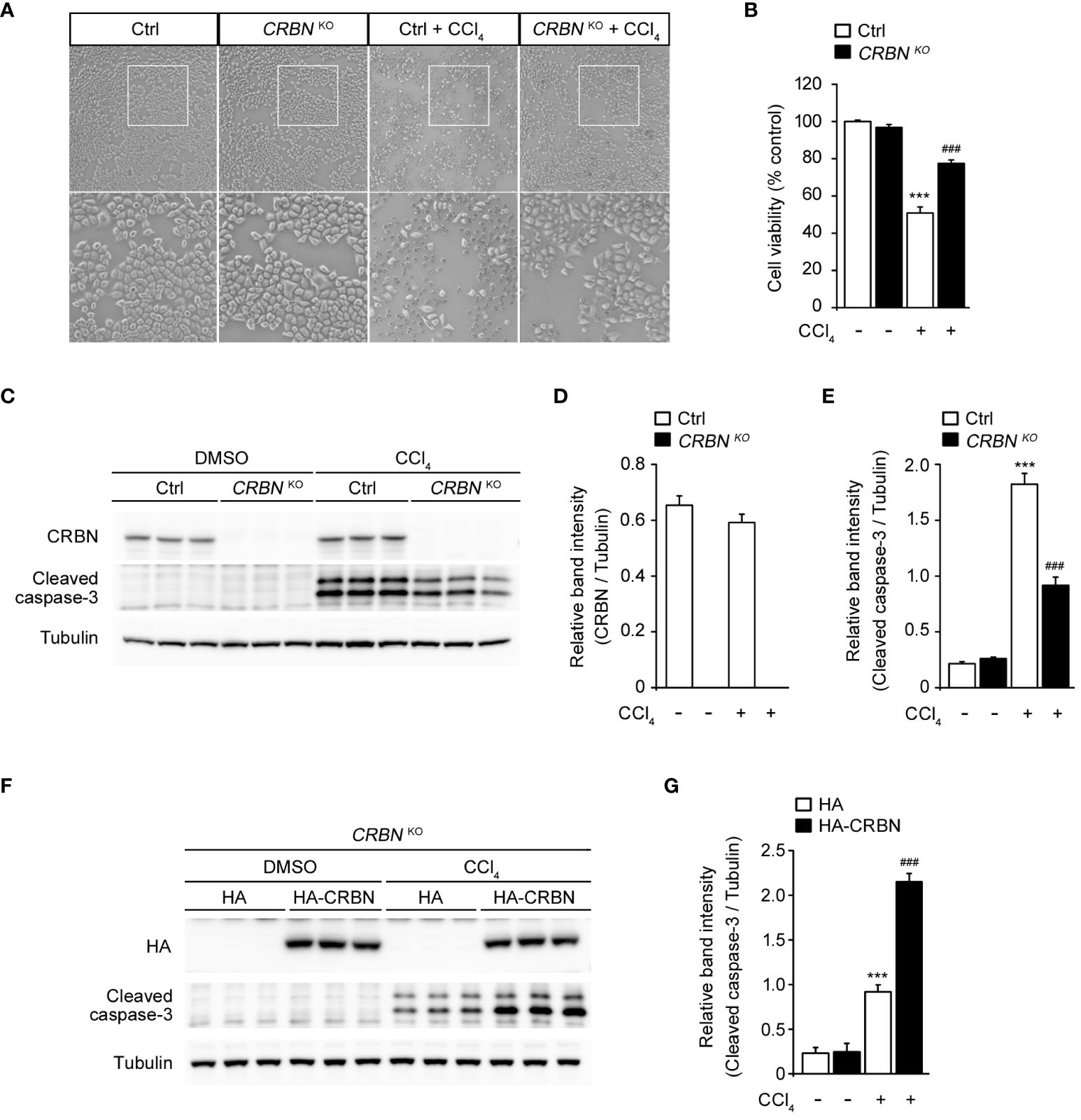
Figure 2 Effect of CRBN knockout (KO) on CCl4-induced damage in HepG2 cells. (A) Representative microscopic images of HepG2 cells (Ctrl and CRBN KO) treated with DMSO or CCl4, respectively. The regions outlined in white in the upper images are shown in the bottom panels. Original magnification, ×100. (B) Relative cell viability presented using the WST-1 of the cells from (A). (C) Representative western blotting analysis of CRBN, cleaved caspase-3, and tubulin in HepG2 cells (Ctrl and CRBN KO) treated with either DMSO or CCl4. Tubulin was used as a loading control. (D) Relative band intensity of CRBN quantified using densitometric analysis and then normalized to that of tubulin. (E) Relative band intensity of cleaved caspase-3 quantified using densitometric analysis and then normalized to that of tubulin. (F) Representative Western blotting analysis of HA, cleaved caspase-3, and tubulin in CRBN KO treated with either DMSO or CCl4. Tubulin was used as a loading control. (G) Relative band intensity of cleaved caspase-3 was quantified using densitometric analysis and then normalized to that of tubulin. Ctrl-and CRBN KO HepG2 cells were treated with either DMSO or CCl4 (0.1%) for 1 h. The values in the bar graphs represent the mean ± SEM of at least three independent experiments. *Statistical differences (*P < 0.05, **P < 0.01, ***P < 0.005) compared to the control HepG2 cells not treated with CCl4. #Statistical differences (#P < 0.05, ##P < 0.01, ###P < 0.005) compared to the control HepG2 cells treated with CCl4.
Thus, we next performed western blotting for the protein expression of cleaved caspase-3 to confirm the resistance to cell death conferred by CRBN KO (Figure 2C). CCl4-treated CRBN KO cells showed a significantly decreased cleaved caspase-3 level compared with CCl4-treated ctrl cells (Figures 2C, E). Notably, endogenous CRBN expression was not affected by CCl4 treatment (Figures 2C, D). These results suggested that CRBN is an essential component of apoptotic signaling in CCl4-induced hepatocellular injury in vitro.
To further validate the functional role of CRBN in CCl4-stimulated hepatotoxicity, we attempted to rescue the phenotype of the CRBN deficiency by exogenously expressing HA-tagged CRBN in CRBN KO HepG2 cells followed by CCl4 treatment (Figure 2F). Western blotting revealed that overexpression of CRBN markedly increased the levels of cleaved caspase-3 in CRBN KO cells compared to mock-transfected CRBN KO cells treated with CCl4 (Figure 2G), further confirming the critical role of CRBN in mediating CCl4-induced hepatocellular injury and indicating that the observed protective effects are attributable to CRBN deficiency.
3.3 CRBN deletion ameliorated CCl4-induced oxidative stress in HepG2 cell lines
To clarify the protective effect of CRBN KO against hepatocellular injury, we examined aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity in HepG2 cells after exposure to CCl4. AST and ALT are enzymes primarily found in hepatocytes and are released into the bloodstream upon liver injury. Therefore, measuring AST and ALT activity serves as a sensitive and reliable method for assessing hepatocellular injury (8). CCl4-treated ctrl cells exhibited markedly elevated AST and ALT activities compared with CCl4 untreated ctrl cells (Figures 3A, B). This observation is consistent with previous studies that demonstrated the hepatotoxic effects of CCl4 and the release of AST and ALT into the culture media as markers of hepatocellular injury (8, 27). In contrast, CRBN KO HepG2 cells treated with CCl4 exhibited a notable reduction in AST and ALT activities compared with control HepG2 cells treated with CCl4.
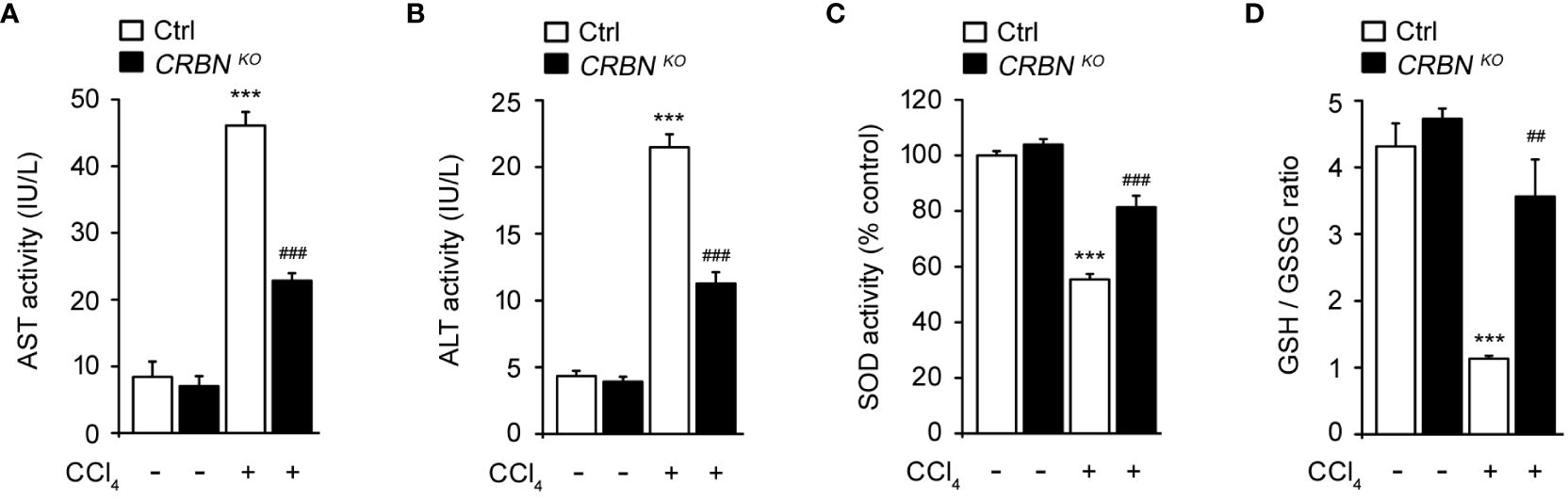
Figure 3 Effect of CRBN loss on the biochemical parameters and antioxidant enzyme activity of CCl4-treated HepG2 cells. (A) AST activity of HepG2 cells. (B) ALT activity of HepG2 cells. (C) SOD activity. (D) GSH/GSSG ratio. Ctrl-and CRBN KO HepG2 cells were treated with either DMSO or CCl4 (0.1%) for 1 h. The values in the bar graphs represent the mean ± SEM of at least three independent experiments. *Statistical differences (*P < 0.05, **P < 0.01, ***P < 0.005) compared to the control HepG2 cells not treated with CCl4. #Statistical differences (#P < 0.05, ##P < 0.01, ###P < 0.005) compared to the control HepG2 cells treated with CCl4.
Oxidative stress is essential in the progression of liver injury. Intracellular antioxidants, such as superoxide dismutase (SOD) and glutathione (GSH), play crucial roles in protecting the liver from oxidative stress-induced damage by neutralizing harmful reactive oxygen species (ROS) (28). Thus, SOD activity and the GSH/GSSG ratio were also examined. CCl4-treated ctrl cell showed a notable decrease in SOD activity and the GSH/GSSG ratio compared with CCl4 untreated ctrl cells (Figures 3C, D). These reductions in SOD activity and the GSH/GSSG ratio indicate an imbalance between ROS production and antioxidant defense mechanisms, resulting in oxidative stress and cellular damage caused by CCl4 treatment. In contrast, CRBN deficiency group exposed to CCl4 exhibited the preservation of endogenous SOD activity and the GSH/GSSG ratio compared with the control group exposed to CCl4.
These findings further support the protective role of CRBN KO against hepatocellular damage, implying a potential mechanism by which CRBN modulates oxidative stress and protects against liver injury.
3.4 CRBN deficiency resulted in the defective expression of the genes involved in CCl4-induced inflammation
Inflammation plays a critical role in hepatic injury progression. To evaluate the effect of CRBN deficiency on CCl4-induced inflammation, we assessed the mRNA levels of proinflammatory genes, including IL-1b, IL-6, TNF-a, and COX-2, in cells following CCl4 exposure. CCl4-treated ctrl cells showed a significant upregulation in IL-1b, IL-6, TNF-a, and COX-2 expression compared with CCl4 untreated ctrl cells (Figure 4). This demonstrated that CCl4-induced pro-inflammation, indicating an inflammatory response triggered by hepatocellular injury. Conversely, CRBN KO HepG2 cells treated with CCl4 exhibited a notable decrease in IL-1b, IL-6, TNF-a, and COX-2 mRNA levels compared with control HepG2 cells treated with CCl4. These results demonstrated that CRBN deletion attenuated the upregulation of pro-inflammatory genes induced by CCl4 exposure in HepG2 cells, suggesting the importance of CRBN in modulating inflammatory responses in the liver.
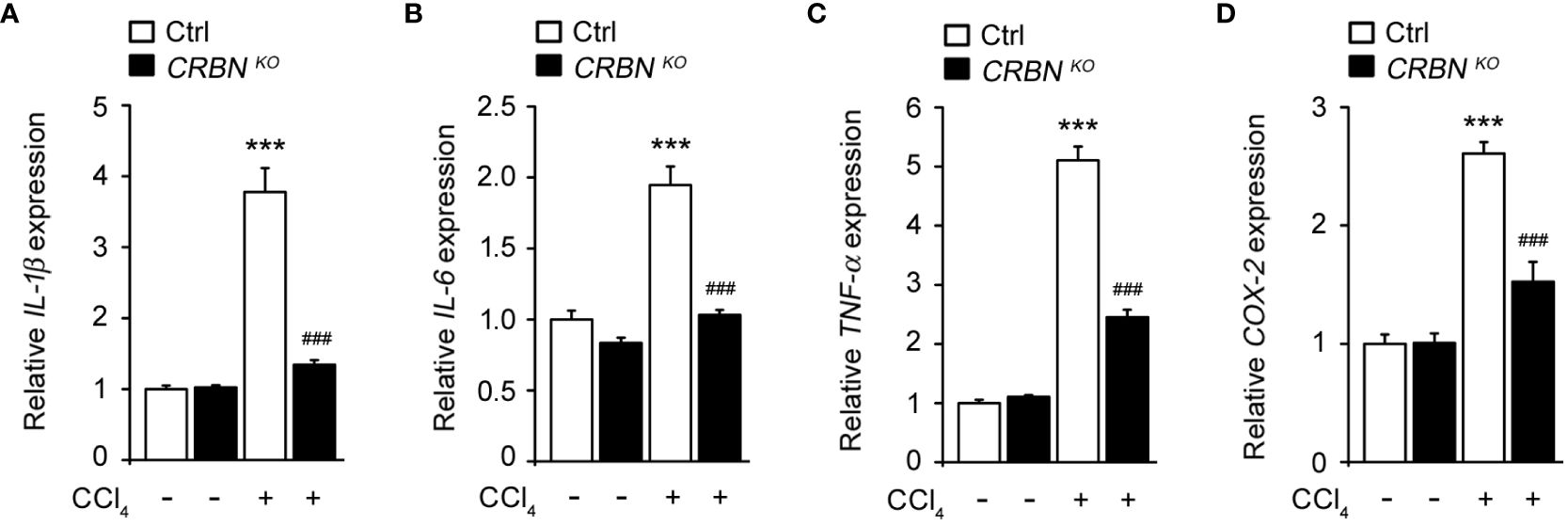
Figure 4 Effect of CRBN KO on pro-inflammatory cytokine mRNA expression in the HepG2 cells treated with CCl4. Total RNA was isolated from HepG2 cells (Ctrl and CRBN KO) treated with DMSO or CCl4, and subjected to quantitative real-time PCR analysis to determine the expression of (A) IL-1β, (B) IL-6, (C) TNF-α, and (D) COX-2. Expression levels were normalized to GAPDH mRNA levels. Fold changes in mRNA levels relative to that of the control HepG2 cells not treated with CCl4, which were set arbitrarily at 1.0, are shown. The values in the bar graphs represent the mean ± SEM of at least three independent experiments. *Statistical differences (*P < 0.05, **P < 0.01, ***P < 0.005) compared to the control HepG2 cells not treated with CCl4. #Statistical differences (#P < 0.05, ##P < 0.01, ###P < 0.005) compared to the control HepG2 cells treated with CCl4.
3.5 CRBN KO attenuated CCl4-induced mitogen-activated protein kinase (MAPK) pathway
Mitogen-activated protein kinases (MAPKs) are pivotal regulators of cellular processes and play critical roles in proliferation, apoptosis, and immune defense (29). MAPKs form three distinct pathways in mammalian cells, the ERK, JNK kinase, and p38 pathways. Each pathway responds to specific extracellular signals, and regulates various cellular processes (30). CCl4-induced oxidative stress initiates apoptosis via the activation of the MAPK family (31–33). Therefore, we investigated the function of the MAPK pathway in the molecular mechanisms underlying the protective effects of CRBN knockout against CCl4-induced cell damage.
CCl4 treatment of control cells significantly increased the levels of phospho-JNK, phospho-ERK, and phospho-p38 MAPK proteins compared with CCl4-untreated control cells (Figure 5), consistent with previous reports (34, 35). However, the absence of CRBN decreased the CCl4-induced phosphorylation of JNK and ERK, whereas the phosphorylation of p38 MAPK remained unaffected. (Figure 5). Notably, the total protein levels of the three MAPKs did not change, regardless of CCl4 treatment or CRBN knockout. These observations indicate that CRBN knockout attenuates the activation of JNK and ERK in response to CCl4-induced oxidative stress, but not that of p38 MAPK, suggesting a molecular mechanism underlying the protective effects of CRBN knockout against CCl4-induced cell damage and the potential involvement of the MAPK pathway in mediating these effects.
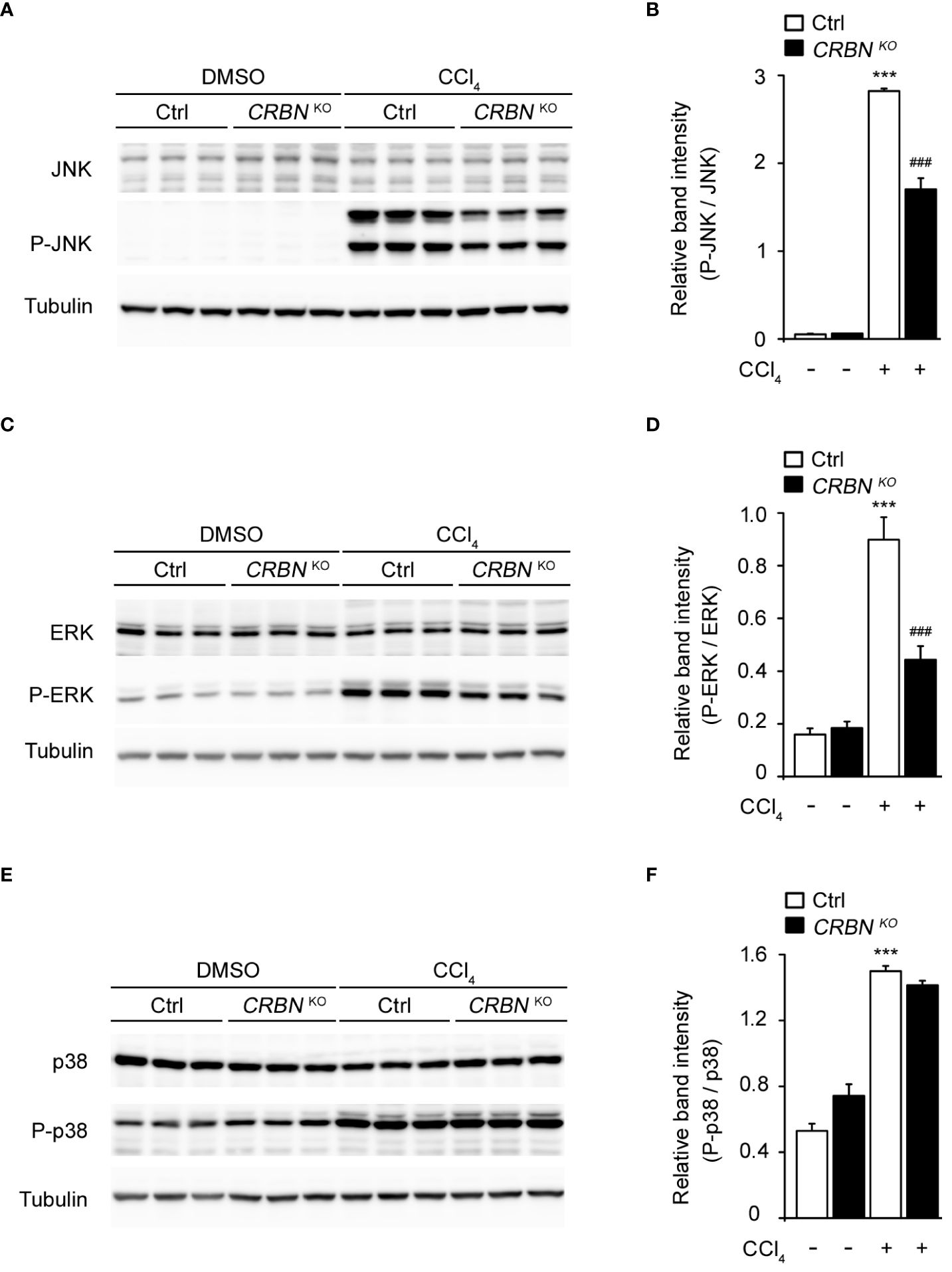
Figure 5 Effect of CRBN deletion on ERK 1/2, JNK 1/2, and p38 expression in the HepG2 cells treated with CCl4. (A) Representative western blotting analysis of JNK and P-JNK. Tubulin was used as the loading control. (B) Relative band intensity of P-JNK quantified using densitometric analysis and then normalized to that of JNK. (C) Representative western blotting analysis of ERK and P-ERK. (D) Relative band intensity of P-ERK to ERK. (E) Representative western blotting analysis of p38 and P-p38. (F) Relative band intensity of P-p38 to p38. Ctrl-and CRBN KO HepG2 cells were treated with DMSO or CCl4. The values in the bar graphs represent the mean ± SEM of at least three independent experiments. *Statistical differences (*P < 0.05, **P < 0.01, ***P < 0.005) compared to the control HepG2 cells not treated with CCl4. #Statistical differences (#P < 0.05, ##P < 0.01, ###P < 0.005) compared to the control HepG2 cells treated with CCl4.
3.6 CRBN knockdown mitigated CCl4-induced hepatic toxicity in AML12 cells
To further validate the protective role of CRBN deficiency against CCl4-induced hepatocellular injury, we extended our study to another cell line using TD-165, a well characterized CRBN degrader (18, 36) in AML-12 cells, which are derived from mouse hepatocytes.
We first confirmed the effectiveness of the CRBN degrader by assessing CRBN expression levels. Treatment with TD-165 significantly reduced CRBN expression in AML12 cells, demonstrating successful knockdown (Figures 6A, B).

Figure 6 Effects of CRBN knockdown (KD) against CCl4-induced damage in AML12 cells. AML12 cells were treated with or without TD-165 (1μM) for 24 h and subsequently exposed to CCl4 (0.1%) for an additional 1 h. Cell lysates were analyzed by Western blot. (A) Representative Western blotting analysis of CRBN, cleaved caspase-3, and tubulin. Tubulin was used as a loading control. (B) Relative band intensity of CRBN was quantified using densitometric analysis and then normalized to that of tubulin. (C) Relative band intensity of cleaved caspase-3 was quantified using densitometric analysis and then normalized to that of tubulin. *Statistical differences (*P < 0.05, **P < 0.01, ***P < 0.005) compared to the DMSO control group without TD-165. #Statistical differences (#P < 0.05, ##P < 0.01, ###P < 0.005) compared to the corresponding treatment group without TD-165.
To examine the impact of CRBN knockdown against CCl4-induced hepatic toxicity, we measured the levels of cleaved caspase-3. Western blot analysis revealed that CCl4-treated CRBN knockdown AML12 cells showed a significantly decreased cleaved caspase-3 level compared with CCl4-treated ctrl AML12 cells (Figure 6C), consistent with our findings in HepG2 cells. These results suggest that the protective role of CRBN deficiency against CCl4-induced hepatic toxicity is not limited to HepG2 cells and can be replicated in AML12 cells, further supporting the protective role of CRBN deficiency against hepatocellular injury.
4 Discussion
The liver is a prime target for reactive oxygen species (ROS), and it possesses an intricate antioxidant system that preserves redox balance within its tissues. Elevated ROS levels can disrupt this balance, leading to oxidative stress, which is heavily implicated in liver diseases and various chronic and degenerative conditions (9, 37). In a CCl4-induced liver injury model, the triggered oxidative stress leads to lipid peroxidation that damages the membranes of hepatocytes. This damage results in a significant release of pro-inflammatory chemokines and cytokines, ultimately exacerbating liver damage (38). Given its ability to mimic chronic liver disease, CCl4 has been commonly used as a hepatotoxin in numerous experimental models (39–41).
CRBN performs multifunctional roles in cellular physiology and in a wide array of human diseases (42–44). However, the hepatoprotective role of CRBN against CCl4-induced hepatic injury remains unknown. Our study demonstrated that CRBN KO may confer protection against CCl4-induced acute liver injury, potentially through mechanisms involving the inhibition of inflammatory responses and attenuation of oxidative stress.
CRBN KO cells exhibited improved viability and reduced apoptosis in response to CCl4 treatment compared with control cells. The observed decrease in caspase-3 activation, a hallmark of apoptosis, along with lower AST and ALT activities, suggests a substantial reduction in hepatocellular damage. These findings not only underline the potential hepatoprotective attributes of CRBN modulation, but also advance our understanding of the molecular interactions involved in liver toxicity.
The reduced pro-inflammatory cytokine expression in CRBN-KO cells following CCl4 treatment also highlights the important anti-inflammatory effects of CRBN deletion. Given the role of inflammation in the exacerbation of liver damage, the ability to suppress inflammatory mediators can be highly beneficial in clinical settings where inflammation significantly contributes to disease progression (45). Additionally, we explored antioxidant defense mechanisms by examining SOD activity and the GSH/GSSG ratios. The preservation of these antioxidants in the CRBN-KO cells further supports the role of CRBN in oxidative stress, which is a key player in the pathogenesis of liver injury. By maintaining a balance between ROS production and antioxidant defense, CRBN-KO cells are better equipped to reduce oxidative stress and the likelihood of cellular damage and death. Our study also revealed that the protective mechanism likely involved the modulation of MAPK signaling pathways. Specifically, the reduced phosphorylation of JNK and ERK in CRBN KO cells treated with CCl4 compared with that in ctrl cells treated with CCl4 indicates a targeted disruption of these pathways, which mediate cellular responses to stress and damage. Interestingly, p38 MAPK phosphorylation remained unchanged, suggesting the selective involvement of certain MAPK pathways in the protective effects conferred by CRBN deletion. This selectivity may be pivotal for therapeutic strategies aimed at minimizing hepatocellular injury without interfering with other cellular functions regulated by p38 MAPK.
The rescue experiment, where re-expression of CRBN in CRBN KO HepG2 cells reversed the protective effects, further confirms the critical role of CRBN in mediating CCl4-induced hepatocellular injury. Additionally, our observations in AML12 cells suggest that the protective role of CRBN deficiency extends across different hepatocyte models, indicating the broader applicability of CRBN’s protective effects against CCl4-induced hepatotoxicity.
Our study conclusively demonstrates the hepatoprotective capabilities of CRBN in CCl4-induced hepatocellular injury. Specifically, we found that CRBN plays a critical role in mitigating damage, at least in part, through mechanisms that reduce oxidative stress and inflammation, and through the inhibition of MAPK pathway-induced apoptosis. Although further animal studies or clinical trials are needed to validate our findings and enhance the translational potential of our results, the present study provides deeper insights into the molecular mechanisms of liver disease and opens new avenues for targeted therapies to prevent or mitigate liver injury in a range of pathological contexts.
In conclusion, the present study not only highlights the protective role of CRBN deletion against CCl4-induced hepatocellular injury, but also provides a foundation for further investigations into gene-targeted therapies for liver injury across a spectrum of liver diseases.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
SC: Writing – original draft, Validation, Investigation, Formal analysis, Data curation. PS: Writing – review & editing, Validation, Investigation, Formal analysis, Data curation. JH: Writing – review & editing, Validation, Formal analysis, Data curation. YL: Writing – review & editing, Resources, Formal analysis. MS: Writing – review & editing, Resources, Data curation. H-JS: Writing – review & editing, Resources. Y-JK: Writing – review & editing, Resources. WK: Writing – review & editing, Writing – original draft, Supervision, Data curation, Conceptualization. KL: Writing – review & editing, Writing – original draft, Validation, Supervision, Investigation, Data curation, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Research Foundation of Korea (NRF), funded by the Korea government (MSIT), awarded to KL (2019R1C1C1007045), WK (No. 2023-00219399), and PS (2020R1C1C1005500).
Acknowledgments
The authors acknowledge the resources and support of the Mosaic Core Lab facilities at the College of Natural Resources and Life Science, Pusan National University. We also thank Dr. Jong Yeon Hwang (Korea Research Institute of Chemical Technology) for kindly providing us with the TD-165.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang SN, Kim SH, Dey DK, Park SM, Nasif O, Bajpai VK, et al. 5-O-demethylnobiletin alleviates ccl(4)-induced acute liver injury by equilibrating ros-mediated apoptosis and autophagy induction. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22031083
2. Shields HJ, Traa A, Van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front Cell Dev Biol. (2021) 9:628157. doi: 10.3389/fcell.2021.628157
3. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. (2014) 20:8082–91. doi: 10.3748/wjg.v20.i25.8082
4. Stefanis L. Caspase-dependent and -independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. (2005) 11:50–62. doi: 10.1177/1073858404271087
5. Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. (1996) 19:257–67. doi: 10.2337/diacare.19.3.257
6. Reddy VP, Zhu X, Perry G, Smith MA. Oxidative stress in diabetes and alzheimer's disease. J Alzheimers Dis. (2009) 16:763–74. doi: 10.3233/JAD-2009-1013
7. Allameh A, Niayesh-Mehr R, Aliarab A, Sebastiani G, Pantopoulos K. Oxidative stress in liver pathophysiology and disease. Antioxidants (Basel). (2023) 12. doi: 10.3390/antiox12091653
8. Gonzalez LT, Minsky NW, Espinosa LE, Aranda RS, Meseguer JP, Perez PC. In vitro assessment of hepatoprotective agents against damage induced by acetaminophen and ccl(4). BMC Complement Altern Med. (2017) 17:39. doi: 10.1186/s12906-016-1506-1
9. Boll M, Weber LW, Becker E, Stampfl A. Mechanism of carbon tetrachloride-induced hepatotoxicity. Hepatocellular damage by reactive carbon tetrachloride metabolites. Z Naturforsch C J Biosci. (2001) 56:649–59. doi: 10.1515/znc-2001-7-826
10. Wang K, Yu D, Bai Y, Cao H, Guo J, Su Z. Isolation and purification of chitosan oligosaccharides (Mw </= 1000) and their protective effect on acute liver injury caused by ccl(4). Mar Drugs. (2024) 22. doi: 10.3390/md22030128
11. Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel atp-dependent lon protease gene in a kindred with mild mental retardation. Neurology. (2004) 63:1927–31. doi: 10.1212/01.WNL.0000146196.01316.A2
12. Jo S, Lee KH, Song S, Jung YK, Park CS. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J Neurochem. (2005) 94:1212–24. doi: 10.1111/j.1471-4159.2005.03344.x
13. Lee KM, Yang SJ, Park S, Choi YD, Shin HK, Pak JH, et al. Depletion of the cereblon gene activates the unfolded protein response and protects cells from er stress-induced cell death. Biochem Biophys Res Commun. (2015) 458:34–9. doi: 10.1016/j.bbrc.2015.01.054
14. Shi Q, Chen L. Cereblon: A protein crucial to the multiple functions of immunomodulatory drugs as well as cell metabolism and disease generation. J Immunol Res. (2017) 2017:9130608. doi: 10.1155/2017/9130608
15. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. (2010) 327:1345–50. doi: 10.1126/science.1177319
16. Lee KM, Jo S, Kim H, Lee J, Park CS. Functional modulation of amp-activated protein kinase by cereblon. Biochim Biophys Acta. (2011) 1813:448–55. doi: 10.1016/j.bbamcr.2011.01.005
17. Lee KM, Yang SJ, Kim YD, Choi YD, Nam JH, Choi CS, et al. Disruption of the cereblon gene enhances hepatic ampk activity and prevents high-fat diet-induced obesity and insulin resistance in mice. Diabetes. (2013) 62:1855–64. doi: 10.2337/db12-1030
18. Yang SJ, Jeon SJ, Van Nguyen T, Deshaies RJ, Park CS, Lee KM. Ubiquitin-dependent proteasomal degradation of ampk gamma subunit by cereblon inhibits ampk activity. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118729. doi: 10.1016/j.bbamcr.2020.118729
19. Zhou L, Xu G. Cereblon attenuates DNA damage-induced apoptosis by regulating the transcription-independent function of P53. Cell Death Dis. (2019) 10:69. doi: 10.1038/s41419-019-1317-7
20. Jeon S, Yoon YS, Kim HK, Han J, Lee KM, Seol JE, et al. Ablation of crbn induces loss of type I collagen and sch in mouse skin by fibroblast senescence via the P38 mapk pathway. Aging (Albany NY). (2021) 13:6406–19. doi: 10.18632/aging.202744
21. Ozerkan D, Ozsoy N, Yilmaz E. Vitamin D and melatonin protect the cell's viability and ameliorate the ccl4 induced cytotoxicity in hepg2 and hep3b hepatoma cell lines. Cytotechnology. (2015) 67:995–1002. doi: 10.1007/s10616-014-9738-8
22. Donato MT, Tolosa L, Gomez-Lechon MJ. Culture and functional characterization of human hepatoma hepg2 cells. Methods Mol Biol. (2015) 1250:77–93. doi: 10.1007/978-1-4939-2074-7_5
23. Bouma ME, Rogier E, Verthier N, Labarre C, Feldmann G. Further cellular investigation of the human hepatoblastoma-derived cell line hepg2: morphology and immunocytochemical studies of hepatic-secreted proteins. In Vitro Cell Dev Biol. (1989) 25:267–75. doi: 10.1007/BF02628465
24. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. (1999) 6:99–104. doi: 10.1038/sj.cdd.4400476
25. Pu X, Fu Y, Yang Y, Xu G. Ginkgo biloba extract alleviates ccl(4)-induced acute liver injury by regulating pi3k/akt signaling pathway. Heliyon. (2024) 10:e26093. doi: 10.1016/j.heliyon.2024.e26093
26. Qian A, Zhou L, Shi D, Pang Z, Lu B. Portulaca oleracea alleviates ccl(4)-induced acute liver injury by regulating hepatic S100a8 and S100a9. Chin Herb Med. (2023) 15:110–6. doi: 10.1016/j.chmed.2022.05.004
27. Pareek A, Godavarthi A, Issarani R, Nagori BP. Antioxidant and hepatoprotective activity of fagonia schweinfurthii (Hadidi) hadidi extract in carbon tetrachloride induced hepatotoxicity in hepg2 cell line and rats. J Ethnopharmacol. (2013) 150:973–81. doi: 10.1016/j.jep.2013.09.048
28. Sanchez-Valle V, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr Med Chem. (2012) 19:4850–60. doi: 10.2174/092986712803341520
29. Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2o2. Role in cell survival following oxidant injury. J Biol Chem. (1996) 271:4138–42. doi: 10.1074/jbc.271.8.4138
30. Cargnello M, Roux PP. Activation and function of the mapks and their substrates, the mapk-activated protein kinases. Microbiol Mol Biol Rev. (2011) 75:50–83. doi: 10.1128/MMBR.00031-10
31. Shu M, Huang DD, Hung ZA, Hu XR, Zhang S. Inhibition of mapk and nf-kappab signaling pathways alleviate carbon tetrachloride (Ccl4)-induced liver fibrosis in toll-like receptor 5 (Tlr5) deficiency mice. Biochem Biophys Res Commun. (2016) 471:233–9. doi: 10.1016/j.bbrc.2016.01.119
32. Ko IG, Jin JJ, Hwang L, Kim SH, Kim CJ, Han JH, et al. Polydeoxyribonucleotide Exerts Protective Effect against Ccl(4)-Induced Acute Liver Injury through Inactivation of Nf-Kappab/Mapk Signaling Pathway in Mice. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21217894
33. Li L, Lan Y, Wang F, Gao T. Linarin protects against ccl(4)-induced acute liver injury via activating autophagy and inhibiting the inflammatory response: involving the tlr4/mapk/nrf2 pathway. Drug Des Devel Ther. (2023) 17:3589–604. doi: 10.2147/DDDT.S433591
34. Bak J, Je NK, Chung HY, Yokozawa T, Yoon S, Moon JO. Oligonol ameliorates ccl(4)-induced liver injury in rats via the nf-kappa B and mapk signaling pathways. Oxid Med Cell Longev. (2016) 2016:3935841. doi: 10.1155/2016/3935841
35. Iida C, Fujii K, Kishioka T, Nagae R, Onishi Y, Ichi I, et al. Activation of mitogen activated protein kinase (Mapk) during carbon tetrachloride intoxication in the rat liver. Arch Toxicol. (2007) 81:489–93. doi: 10.1007/s00204-007-0181-x
36. Kim K, Lee DH, Park S, Jo SH, Ku B, Park SG, et al. Disordered region of cereblon is required for efficient degradation by proteolysis-targeting chimera. Sci Rep. (2019) 9:19654. doi: 10.1038/s41598-019-56177-5
37. Elkhamesy A, Refaat M, Gouida MSO, Alrdahe SS, Youssef MM. Diminished ccl(4) -induced hepatocellular carcinoma, oxidative stress, and apoptosis by co-administration of curcumin or selenium in mice. J Food Biochem. (2022) 46:e13845. doi: 10.1111/jfbc.13845
38. Thiesen LC, da Silva LM, Santin JR, Bresolin TMB, de Andrade SF, Amorim CM, et al. Hepatoprotective effect of maytenus robusta reiss extract on ccl(4)-induced hepatotoxicity in mice and hepg2 cells. Regul Toxicol Pharmacol. (2017) 86:93–100. doi: 10.1016/j.yrtph.2017.02.023
39. Bellassoued K, Ben Hsouna A, Athmouni K, van Pelt J, Makni Ayadi F, Rebai T, et al. Protective effects of mentha piperita L. Leaf essential oil against ccl(4) induced hepatic oxidative damage and renal failure in rats. Lipids Health Dis. (2018) 17:9. doi: 10.1186/s12944-017-0645-9
40. Wang R, Shen H, Zhang J, Li X, Guo Y, Zhao Z, et al. Dimethyl bisphenolate ameliorates carbon tetrachloride-induced liver injury by regulating oxidative stress-related genes. Molecules. (2023) 28. doi: 10.3390/molecules28247989
41. Bashandy SAE, Ebaid H, Al-Tamimi J, Hassan I, Omara EA, Elbaset MA, et al. Protective effect of daidzein against diethylnitrosamine/carbon tetrachloride-induced hepatocellular carcinoma in male rats. Biol (Basel). (2023) 12. doi: 10.3390/biology12091184
42. Kim HK, Seol JE, Ahn SW, Jeon S, Park CS, Han J. Cereblon: promise and challenges for combating human diseases. Pflugers Arch. (2021) 473:1695–711. doi: 10.1007/s00424-021-02624-0
43. Lee KM, Yang SJ, Choi JH, Park CS. Functional effects of a pathogenic mutation in cereblon (Crbn) on the regulation of protein synthesis via the ampk-mtor cascade. J Biol Chem. (2014) 289:23343–52. doi: 10.1074/jbc.M113.523423
44. Lee KJ, Lee KM, Jo S, Kang KW, Park CS. Induction of cereblon by nf-E2-related factor 2 in neuroblastoma cells exposed to hypoxia-reoxygenation. Biochem Biophys Res Commun. (2010) 399:711–5. doi: 10.1016/j.bbrc.2010.08.005
Keywords: inflammation, carbon tetrachloride, pro-inflammatory cytokines, hepatotoxicity, cereblon, MAP kinase, oxidative stress
Citation: Choi SY, Song P, Hwang JS, Lee YK, Shin MS, Son H-J, Kim Y-J, Kim W and Lee KM (2024) Cereblon deficiency ameliorates carbon tetrachloride-induced acute hepatotoxicity in HepG2 cells by suppressing MAPK-mediated apoptosis. Front. Immunol. 15:1457636. doi: 10.3389/fimmu.2024.1457636
Received: 01 July 2024; Accepted: 16 July 2024;
Published: 30 July 2024.
Edited by:
Siyoung Yang, Sungkyunkwan University, Republic of KoreaReviewed by:
Tae In Kam, Johns Hopkins University, United StatesHyejin Park, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
Copyright © 2024 Choi, Song, Hwang, Lee, Shin, Son, Kim, Kim and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wanil Kim, d2tpbUBnbnUuYWMua3I=; Kwang Min Lee, bGVla21AcHVzYW4uYWMua3I=
†These authors have contributed equally to this work