 Junwei Ge
Junwei Ge Xuan Yin
Xuan Yin Lujun Chen
Lujun Chen- 1Department of Tumor Biological Treatment, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China
- 2Jiangsu Engineering Research Center for Tumor Immunotherapy, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China
- 3Institute of Cell Therapy, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China
Regulatory T cells (Tregs), a subset of CD4+T cells marked by the expression of the transcription factor forkhead box protein 3 (Foxp3), are pivotal in maintaining immune equilibrium and preventing autoimmunity. In our review, we addressed the functional distinctions between Foxp3+Tregs and other T cells, highlighting their roles in autoimmune diseases and cancer. We uncovered the dual nature of Tregs: they prevented autoimmune diseases by maintaining self-tolerance while contributing to tumor evasion by suppressing anti-tumor immunity. This study underscored the potential for targeted therapeutic strategies, such as enhancing Treg activity to restore balance in autoimmune diseases or depleting Foxp3+Tregs to augment anti-tumor immune responses in cancer. These insights laid the groundwork for future research and clinical applications, emphasizing the critical role of Foxp3+Tregs in immune regulation and the advancement of next-generation immunotherapies.
1 Introduction
Regulatory T cells (Tregs) are a unique group of lymphocytes within the immune system, playing vital roles in maintaining immune equilibrium, preventing autoimmunity, and reducing inflammation. Beyond their immune functions, research has shown that Tregs also aid in skeletal muscle repair via IL-33 and support scar healing by expressing Sparc (1). Tregs are primarily identified by the surface markers CD4 and the intracellular transcription factor forkhead box protein 3 (Foxp3), the latter being essential for their regulatory function (2). Additionally, they express specific molecules like PD-1 (programmed cell death protein 1) and CTLA-4 (cytotoxic T-lymphocyte antigen-4), which are key to their regulatory roles (3). Tregs regulate the immune response through both contact-dependent mechanisms (direct interaction with other immune cells) and contact-independent mechanisms, such as secreting inhibitory cytokines like IL-10, IL-35, and TGF-β (4).
Tregs protect the body from excessive inflammation and autoimmune damage by controlling the activation of other immune cells and moderating the intensity of immune responses. They can suppress self-reactive T cells and B cells, reducing attacks on self-tissues (5). Furthermore, the immunosuppressive factors they secrete inhibit inflammatory and autoimmune responses, and Tregs modulate the activity and function of other immune cells by inhibiting stimulatory molecules. In the tumor microenvironment (TME), Tregs play a multifaceted role. They mitigate excessive inflammation and tissue damage, potentially providing protective effects during tumor development. However, they also facilitate tumor cells in evading immune attacks, contributing to cancer progression (6). By suppressing immune cells such as CD4+T cells, CD8+T cells, natural killer (NK) cells, and dendritic cells (DCs), Tregs promote tumor growth and progression (7).
Our present review provides a detailed outlook of Treg characteristics, focusing on their high expression of molecular markers. It delves into the interactions between Tregs and other cells, highlighting the differences in Treg function and phenotype. Additionally, this review delves into the latest advancements in immunotherapy targeting Tregs, encompassing the development of targeted treatment strategies, innovative drug development, and clinical applications related to Treg signaling pathways.
2 Characterization and subtypes of Tregs
Tregs were first identified as CD4+T cells characterized by their elevated expression of CD25 (interleukin-2 receptor α-chain, IL-2Rα). This marker binds strongly to IL-2, enhancing Treg immune tolerance via the CD25/STAT5 signaling pathway (8). Additionally, studies have demonstrated that CD25-Tregs exhibit a similar memory/effector phenotype compared to CD25+Tregs, yet they maintain their suppressive capacity. Specifically, CD25-Tregs effectively inhibit the proliferation of effector T cells (Teff) and the production of cytokines in vitro, particularly in the context of HIV-Tuberculosis co-infection (9). The specific expression of Foxp3 in Tregs is a critical factor for their development, regulating inhibitory functions and defining Treg lineage identity (10). Mutations that inactivate Foxp3 can result in severe autoimmune disorders, such as X-linked immune dysregulation polyendocrinopathy enteropathy syndrome (IPEX), multiple endocrine neoplasia, and enteropathy (11). Interestingly, in human CD4+CD25−cells, TCR stimulation in the presence of TGF-β leads to a transient induction of Foxp3 without conferring a suppressive phenotype while similar cells in mice exhibit strong suppressive functions in vitro under the same conditions (12).
CD25+Foxp3+Tregs, which develop and mature in the thymus, are known as thymic Tregs (tTregs) (13). Their development involves both T-cell receptor (TCR)-dependent and TCR-independent mechanisms. In the TCR-dependent pathway, thymic cells expressing TCR with intermediate affinity differentiate into CD4+CD25+Foxp3-Treg precursor cells. Meanwhile, the TCR-independent process relies on cytokines such as IL-2, IL-7, and IL-15 to regulate Foxp3 expression (14). Both precursor tTregs and mature Tregs display elevated expression of OX40, GITR, and TNFRII. When these molecules bind with their respective ligands, they drive the differentiation of thymic cells into tTregs (15). Furthermore, the Kcnk18 gene encodes K2P18.1, which has been shown to promote Foxp3 expression via the NF-κB and NFAT pathways, thereby supporting the development and maturation of tTregs (16) (Figure 1).
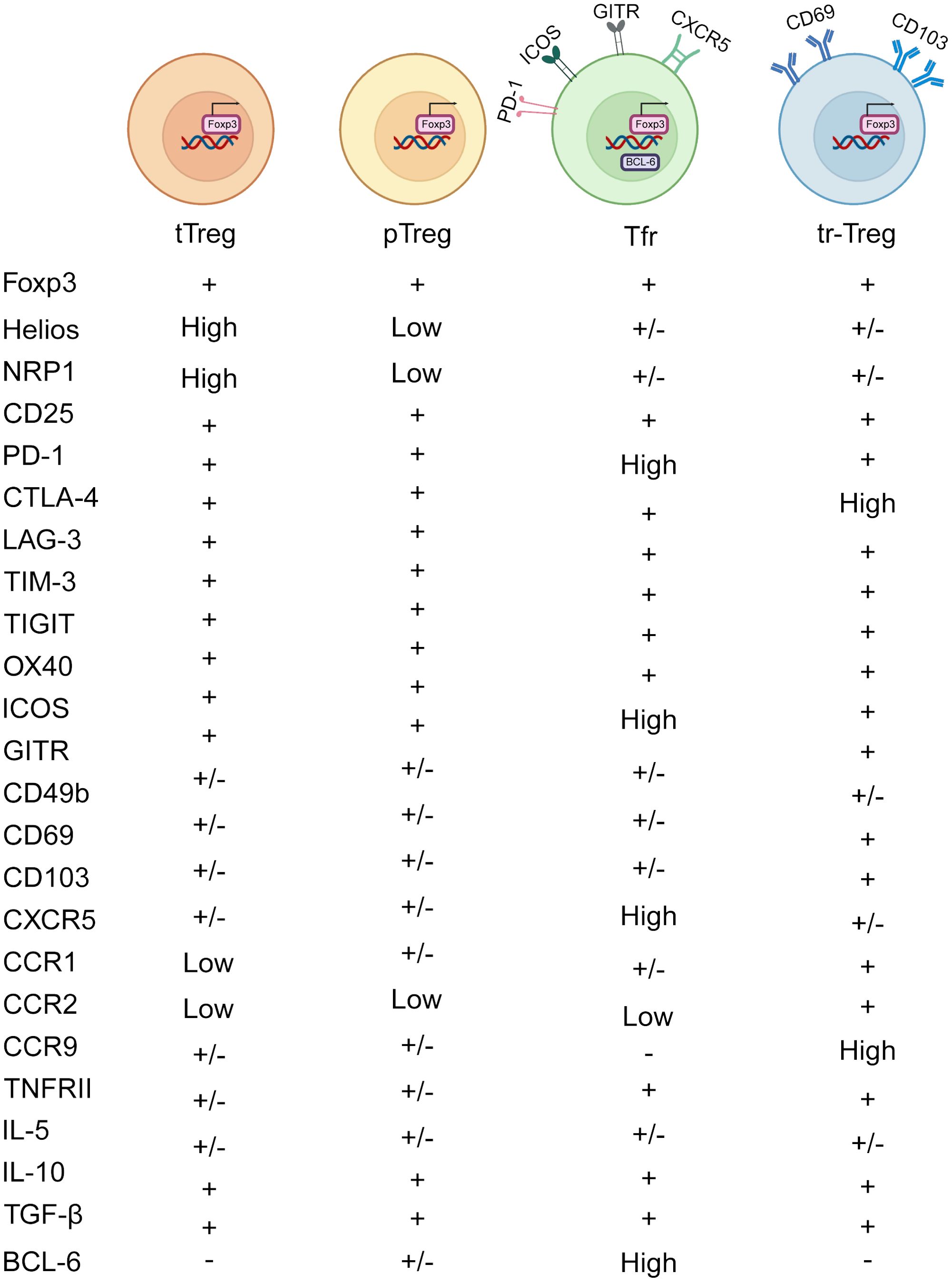
Figure 1. Heterogeneity of Treg subtypes. Treg cells comprise several subtypes based on their origin and function. tTregs are produced in the thymus, where they help maintain self-tolerance by inhibiting self-reactive T cells and facilitating central tolerance to self-antigens. pTregs are induced in peripheral lymphoid organs, mediating peripheral tolerance and regulating the intensity and scope of immune reactions. Tfr cells are enriched in lymph node follicles and splenic follicles, where they regulate B cell activation and antibody production within follicles, thus contributing to the regulation of humoral immune responses. Tr1 cells are distributed throughout peripheral tissues and lymphoid organs, producing abundant IL-10 and TGF-β to restrain immune cell activation and inflammation. tr-Tregs include subpopulations residing in tumor tissues, inflammatory tissues, and organ-specific tissues, regulating local immune responses and impacting tumor development, inflammation progression, and organ immune balance. CD103 and CD69 are classical markers of tr-Tregs, which may be upregulated in TI-Tregs.
Naïve CD4+T cells, when encountering antigen stimulation in peripheral tissues, can become peripheral Tregs (pTregs), especially under the influence of factors like IL-2, TGF-β, and retinoic acid (17). These pTregs primarily inhibit reactions against exogenous antigens, orchestrating tolerance at the periphery and maintaining tissue equilibrium. While neuropilin-1 (Nrp1) and Helios have been suggested as markers to distinguish tTregs from pTregs, it’s important to recognize that pTregs and inducible Tregs (iTregs) can also express Nrp1. This is because Nrp1 expression is positively regulated by TGF-β, leading TGF-β-induced iTregs to express Nrp1 as well (18). Furthermore, decreased expression of Nrp1 can be induced by IFN-γ, thereby impairing the activity and stability of Treg (19). Foxp3+Tregs generated by stimulating conventional T cells (Tconv) in the presence of TGF-β are referred to as iTregs. Compared to the more stable pTregs, iTregs may represent a less stable cell population with potential therapeutic applications in preventing autoimmune diseases and transplant rejection, primarily by inhibiting T cell activation (20). However, detailed studies on the mechanisms of iTreg action remain limited (Figure 1).
Follicular regulatory T cells (Tfr) are another subset within the Treg family, characterized by high levels of PD-1, GITR, ICOS, CXCR5, and BCL6 (21). Tfr cells play a crucial role in suppressing the differentiation of B cells into plasma cells, with neuritin enhancing BCL6 expression in Tfr cells to prevent this transition (22) (Figure 1).
Tissue-resident Tregs (tr-Tregs) are distributed across various non-lymphoid organs, such as the skin, lungs, liver, adipose tissue, skeletal muscle, small intestine, and large intestine (23). These tr-Tregs are pivotal in tissue repair and maintaining homeostasis, characterized by CD103 and CD69 (24). CD103, known as αE integrin facilitates the retention of lymphocytes within epithelial tissues and act as a receptor for E-cadherin. In different tissues, tr-Tregs perform indispensable functions. For instance, skin-resident Tregs are integral to wound healing and the regeneration of hair follicle stem cells (HFSCs) (25). Meanwhile, skeletal muscle-resident Tregs secrete amphiregulin (Areg), which promotes satellite cell differentiation and enhances muscle repair by inhibiting Tconv (26). Tr-Tregs are vital in the TME for promoting angiogenesis by releasing vascular endothelial growth factor (VEGF) and secreting higher levels of VEGF-A under hypoxic conditions, thereby supporting tumor growth and metastasis (27). Tr-Tregs exhibit increased expression of chemokine receptors such as CCR1, CCR2, and CCR9, along with immune checkpoint molecules like TIM-3, GITR, and CD39 (Figure 1).
3 Interaction between Tregs and other cells
3.1 Interaction between Tregs and DCs
DCs, vital immune sentinels found in nearly all peripheral tissues, play crucial roles in immune regulation and are primarily classified into three types: conventional DCs (cDCs), plasmacytoid DCs (pDCs), and monocyte-derived DCs (MoDCs), with cDCs subdivided into type 1 (cDC1) and type 2 (cDC2) (28). In immune responses, cDC1 cells primarily stimulate CD8+T cells and induce Th1 cell reactions by cross-presenting exogenous antigens, playing key roles in antiviral and antitumor immunity. In contrast, cDC2 cells activate CD4+T cells, contributing to inflammatory responses and immune regulation (29).
DCs activate naïve Tregs by providing three essential signals: presenting exogenous antigens via major histocompatibility complex (MHC) molecules, delivering co-stimulatory molecules (such as CD80 and CD86), and releasing cytokines (including TGF-β) to facilitate Treg development. Furthermore, DCs induce tolerance by expressing the immunoregulatory enzyme indoleamine 2,3-dioxygenase 1 (IDO1), maintaining self-antigen tolerance (30). Additionally, epithelial cells from the intestinal lining contribute to the generation and function of tolerogenic CD103+DCs, which could help regulate Tregs.
Tregs critically influence DCs by producing IL-10 and TGF-β to regulate DC initiation and function, and by limiting the recruitment of CCR5-dependent DCs in lymph nodes through the inhibition of CCR5 ligand production (31). Additionally, Tregs block IL-15Rα exposure on DCs, affecting DC-mediated NK cell proliferation, a regulatory mechanism particularly significant in autoimmune diseases as it controls DC/T cell autoreactivity triggered by NK cells (32). The interplay between Tregs and DCs is also crucial in anti-tumor immunity. Tumor-infiltrating Tregs (TI-Tregs) receive TCR signals from cDCs and downregulate CD80/CD86 expression on cDCs via CTLA-4, thereby inhibiting T cell activation and expansion (33). Furthermore, Tregs expressing CXCR3 co-localize with DCs producing CXCL9 in tumors, which affects the presentation of tumor-derived antigens and dampens the activity of anti-tumor CD8+T cells (Figure 2).
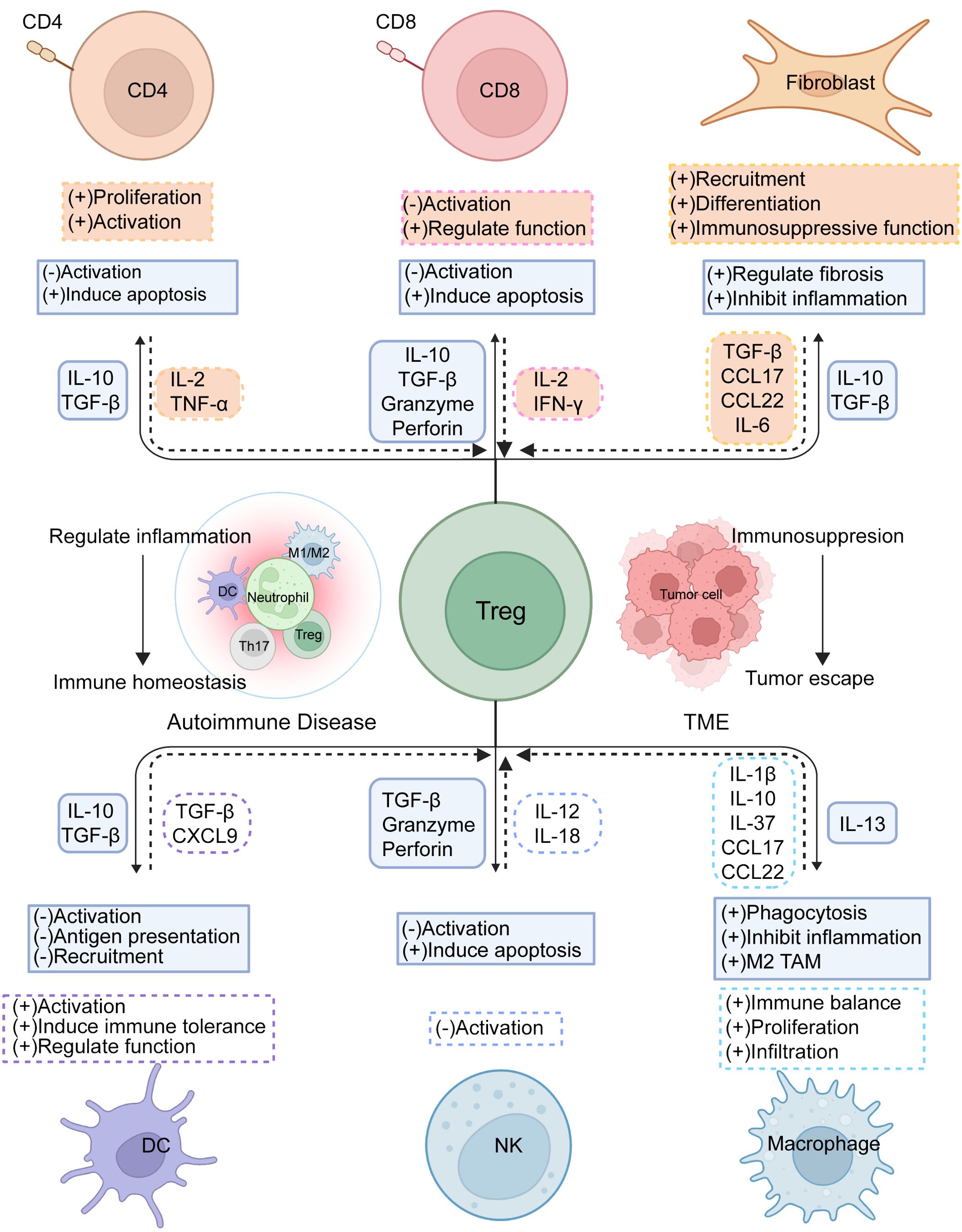
Figure 2. Tregs immunoregulation. Treg cells interact with diverse cells in the immune microenvironment to regulate immune stability and foster suppression within the TME. By expressing suppressive receptors and releasing inhibitory cytokines, Treg cells modulate the activation and differentiation of Teff cells, helping to restrain excessive immune reactions and prevent autoimmune conditions, while also governing immune cell-driven tumor control. Concurrently, other immune cells support Treg activation, proliferation, and functionality, collectively maintaining immune equilibrium.
3.2 Interplay between Tregs and NK cells
NK cells utilize a variety of pathways to eliminate pathogens and tumor cells, including granule enzyme and perforin pathways, as well as death receptor-mediated pathways. They also secrete chemokines and cytokines, playing a vital role in immune regulation. However, these functions can be partially suppressed by Tregs. Tregs modulate NK cell activity through several mechanisms. For instance, they produce TGF-β, which suppresses NKG2D expression and weakens NK cell responses (34). Additionally, Tregs can trigger NK cell apoptosis via a mechanism dependent on granzyme B and perforin, thus hindering tumor clearance (35). Another pathway involves inhibiting IL-2-induced NK cell activation by depriving NK cells of IL-2 signaling. Furthermore, IL-37 produced by Tregs can interact with the inhibitory receptor IL-1R8 on NK cells, causing phenotypic changes and reduced functionality (36).
Notably, NK cells also exert a significant influence on Treg activity (36). For instance, in type 1 diabetes, the upregulation of IL-12/IL-18 allows NK cells to enhance CD25 expression, directly competing with Tregs for IL-2, which reduces Treg suppression capability (37). In-depth research into the interactions between NK cells and Tregs can offer valuable insights into the regulatory mechanisms of the immune system. Understanding these dynamics could lead to the development of new therapeutic strategies to enhance immune responses against pathogens and tumors (Figure 2).
3.3 Interaction between Tregs and T lymphocytes
Tregs regulate antigen-presenting cells (APCs), primarily DCs, through various mechanisms, directly or indirectly suppressing Teff. These regulatory mechanisms involve the release of inhibitory cytokines and the expression of inhibitory receptors. While much research has focused on how Tregs inhibit CD4+T cells, they also modulate CD8+T cells, which are crucial effector cells in antitumor immune responses. CD8+T cells become activated by recognizing antigen peptides presented on MHC-I molecules and mature into cytotoxic effector T cells, leading to the death of target cells (38).
Tregs can release granzyme B and perforin to trigger apoptosis in CD8+T cells in the TME (35). In maintaining immune homeostasis, Tregs curb the expansion of CD8+T cells and prevent the spontaneous transition of memory CD8+T cells into effector cells (39). Additionally, Tregs mitigate the response of CD8+T cells to antigens. They employ multiple mechanisms to suppress CD8+T cell activity. For instance, the CTLA-4 molecule on Tregs binds to the B7 molecule on APCs, reducing CD8+T cell activation. Moreover, Tregs secrete inhibitory cytokines like IL-10 and TGF-β, which dampen CD8+T cell activity and function. These cytokines can impede APC activity or directly affect CD8+T cells, diminishing their antigen sensitivity, inhibiting cytokine production, and inducing apoptosis.
Furthermore, Tregs restrict the availability of IL-2 to suppress CD8+T cell activity and prevent APCs from producing chemokines CCL3, CCL4, and CCL5, thereby curtailing CD8+T cell migration and activation (40) (Figure 2). Gaining deeper insights into how Tregs suppress CD8+T cells can enhance our comprehension of immune regulation in both immune tolerance and diseases. This knowledge lays a crucial theoretical foundation for developing treatments for related disorders.
3.4 Interaction between Tregs and macrophages
Macrophages, originating from bone marrow, are widely distributed in various tissues, including the liver, lungs, skin, and lymph nodes. These immune cells have the capacity to detect, ingest, and decompose pathogens, playing a significant role in immune responses by demonstrating both pro-inflammatory and anti-inflammatory properties (41). Moreover, macrophages are involved in antigen presentation and contribute to the eradication of tumor cells. They are categorized into M1 macrophages, which are pro-inflammatory and secrete cytokines such as IL-1β, IL-6, IL-12, IL-23, and TNF-α, and M2 macrophages, which possess anti-inflammatory and immunoregulatory functions, producing cytokines like IL-10 and TGF-β (42).
An intricate network of interactions exists between macrophages and Tregs, playing essential roles in immune regulation. Tregs release IL-13, which induces macrophages to produce IL-10, enhancing the phagocytosis of apoptotic cells via a Vav1-Rac1-mediated mechanism (43). Additionally, LAG-3 on Tregs inhibits the CD40 and NF-κB signaling pathways in CX3CR1+macrophages, reducing the secretion of IL-1β and IL-23. This reduction indirectly affects innate lymphoid cells (ILC3) production, thereby regulating intestinal inflammation and maintaining intestinal homeostasis (44). Conversely, IL-1β produced by intestinal macrophages acts on ILC3s within the intestine, which subsequently generate granulocyte-macrophage colony-stimulating factor (GM-CSF) that induces myeloid cells, including DCs and macrophages, to produce retinoic acid and IL-10, thereby supporting the transformation and proliferation of Tregs and maintaining immune balance in the intestine (45). Additionally, the transcriptional co-repressors TLE3 and TLE4 in intestinal macrophages negatively regulate MMP9 transcription, inhibiting potential TGF-β activation and thus limiting the expansion of Tregs and Th17 cells (46).
The interplay between macrophages and Tregs within the TME is highly complex. Tregs influence macrophage function and polarization, affecting tumor progression. For example, Tregs indirectly enhance M2-type tumor-associated macrophages (TAMs) by preventing the release of IFN-γ from CD8+T cells, increasing the population of TAMs and supporting their activity (47). Moreover, the expression of LAG-3 and TIM-3 on Tregs prompts macrophages to produce IL-10, which lowers the levels of MHCII, CD80, CD86, and TNF-α, thereby modulating the pro-inflammatory activation of macrophages. Conversely, macrophages primarily support Treg cell proliferation and function, promoting their infiltration and activity within the TME. For instance, macrophages can stimulate Treg cell proliferation and induce IL-10 production by releasing inhibitory factors such as IL-37, which dampens the activation of cytotoxic T cells and NK cells, reducing their attacks on tumor cells (48). Chemokines secreted by macrophages, like CCL17 and CCL22, attract Tregs to the tumor area, enhancing Treg infiltration within the TME and further sustaining the tumor’s ability to evade immune detection (49). TAM-derived CXCL1 promotes the conversion of naive CD4+T cells into Tregs by activating the NF-κB/Foxp3 signaling pathway at the transcriptional level, fostering an immunosuppressive environment (50). Further exploration of these regulatory mechanisms and interaction modes can aid in developing more effective tumor immunotherapy strategies, leading to better treatment outcomes for cancer patients (Figure 2).
3.5 Interaction between Tregs and fibroblasts
Fibroblasts, essential mature connective tissue cells derived from mesenchymal origins, play significant roles in wound healing, extracellular matrix (ECM) remodeling, inflammation, and cancer progression (51). In rheumatoid arthritis (RA), fibroblasts activated by tumor necrosis factor (TNF) produce high levels of chemokines such as IL-6, CXCL12, and CCL2, which enhance the recruitment and attraction of monocytes. Similarly, in inflammatory bowel disease (IBD), fibroblasts contribute to inflammation through the secretion of factors like IL-11, IL-24, and IL-13RA2. These observations highlight the critical role of fibroblasts in both inflammation and fibrosis (52).
In the TME, normal fibroblasts act as barriers to tumor development, while cancer-associated fibroblasts (CAFs) enhance tumor characteristics, such as cancer cell growth and invasion, angiogenesis, and ECM remodeling. CAFs are classified into three types: myCAF (resembling myofibroblasts), iCAF (with inflammatory properties), and apCAF (with antigen-presenting features) (53). CAFs interact with Tregs through various mechanisms, collectively promoting tumor immune tolerance and tumorigenesis. They affect Treg activity and function by releasing different signaling molecules. Notably, TGF-β from CAFs has a dual role: it promotes CXCL13 secretion from CD4+T and CD8+T cells while decreasing IL-2 levels, leading to an increase in Treg populations, and additionally encourages the formation of the myCAF phenotype by downregulating IL1R1 expression (54). Chemokines CCL17 and CCL22 released by CAFs attract Tregs, further intensifying immune suppression. Additionally, CAFs trigger signal transducer and activator of transcription 3 (STAT-3) activation through IL-6 production, which recruits immunosuppressive IDO+DCs and inhibits T-cell proliferation while promoting Treg production (55). It has been observed that IL-6 release by CAFs induces the differentiation of CD73+γδ Tregs, which secrete more adenosine, thus enhancing immune suppression (56). Furthermore, INHBA/recombinant activin A in CAFs induces PD-L1 expression via an autocrine mechanism involving SMAD2-dependent signaling, promoting Treg differentiation (57). Activation by the transcription factor Snail1 also enhances interactions between CAFs and macrophages, increasing Treg activity (58). Our recent work has also indicated that Tregs promote MHC-II expression on CAFs by expressing IL1R2, thus facilitating Treg accumulation and manifesting Treg function (59) (Figure 2).
4 Tregs in autoimmune diseases/cancer
4.1 Tregs in autoimmune diseases
In autoimmune diseases, Tregs are crucial for regulating the inflammatory process. Their dysregulation is associated with several autoimmune conditions, including multiple sclerosis (MS), psoriasis, systemic lupus erythematosus (SLE), IBD such as Crohn’s disease and ulcerative colitis, and RA. Thus, maintaining the appropriate numbers and activity of Tregs is vital for sustaining normal host immunity and overall health. Key examples of Treg imbalance in patients will be explored in various autoimmune manifestations as below (Figure 3).
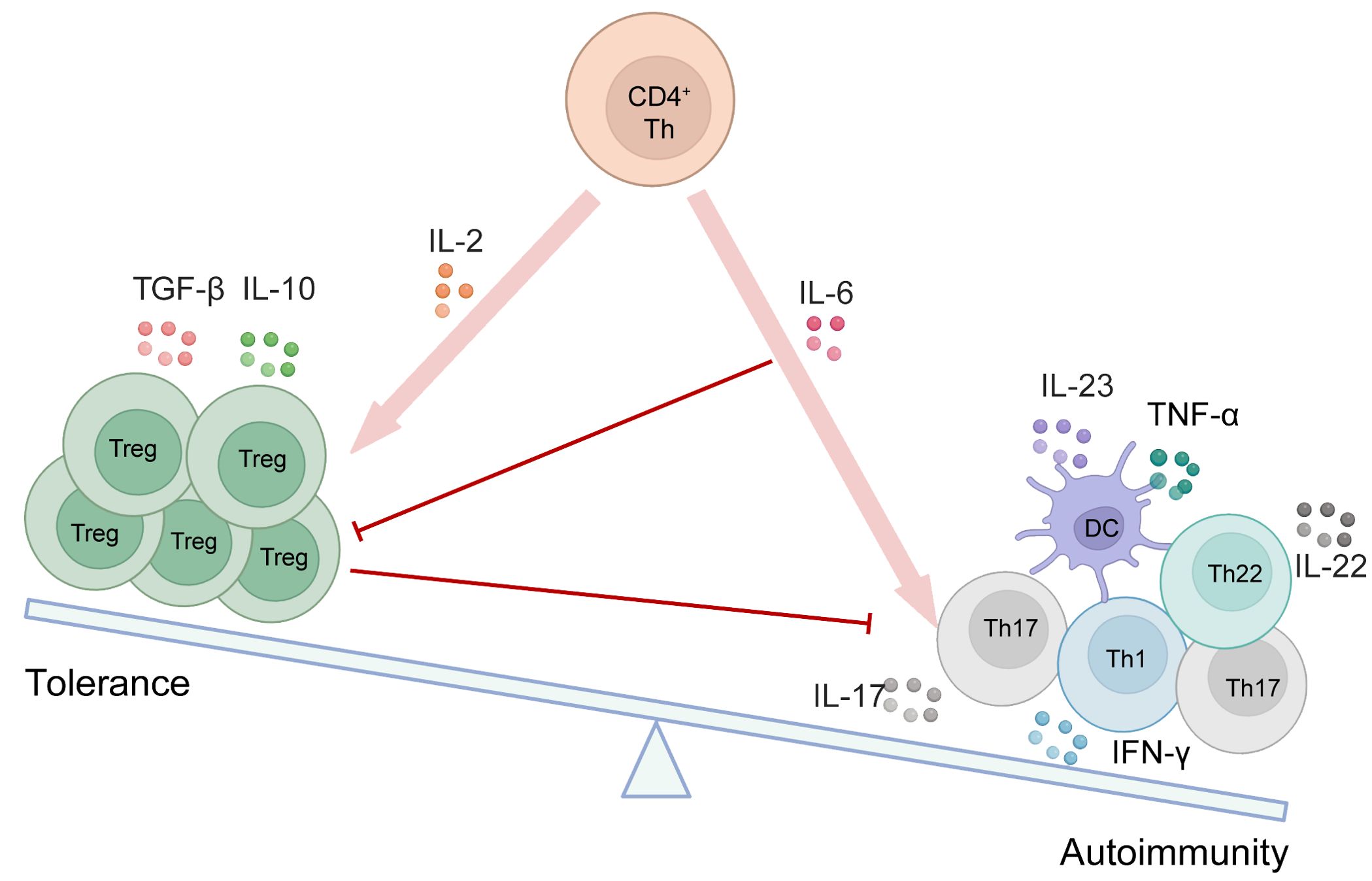
Figure 3. The equilibrium between Tregs and other immune cells is disrupted in autoimmune disorders. As depicted in the schematic, various cytokines contribute to the initiation and generation of different cell types. Red arrows indicate promotion, while red lines represent inhibition.
4.1.1 MS
MS is an autoimmune disease characterized by T cell-mediated demyelination and neurodegeneration within the central nervous system (CNS). Experimental autoimmune encephalomyelitis (EAE) in mice is the primary animal model used to study MS (60). Although the exact etiology of MS remains elusive, Th17 cells, which secrete IL-17 and IL-22, and Tregs, which produce IL-10 and TGF-β, are critical in the disease pathogenesis and in the EAE model. In MS patients, both the number and function of Tregs in the cerebrospinal fluid are diminished (61). IL-17 and IL-22 contribute to the disruption of the blood-brain barrier and facilitate the recruitment of CD4+T cells and neutrophils to the CNS, while IFN-γ primarily enhances macrophage recruitment (62). Tregs counteract Th17 cells throughout the course of MS by inhibiting self-reactive Th1 and Th17 cells. Furthermore, during the remission phase in EAE mice, Tregs accumulate in the CNS, with IL-10 playing a pivotal role in this process (63).
CD39, also known as nucleoside triphosphate diphosphohydrolase 1 (NTPDase1), is primarily responsible for breaking down extracellular ATP and ADP into AMP, which is subsequently converted into adenosine by CD73 (64). Adenosine has anti-inflammatory and immunomodulatory effects, making CD39 a crucial factor in regulating the immune response. Studies have shown that CD39’s regulatory function enhances the migratory behavior of both CD4+T cells and Tregs (including CCR5+Tregs and CCR6+Tregs) in the CNS and lymphatic drainage sites of EAE mice and facilitates their migration in vitro. Lack of CD39 eliminates the accumulation of Tregs in EAE and correlates with heightened Th1/Th17 signaling in the CNS and gut-associated lymphoid tissues (65).
IL-6 is a pivotal cytokine in promoting Th17 differentiation, and the IL-6 signaling pathway, along with STAT-3, plays a crucial role in inhibiting the development of iTregs. Consequently, abnormal activation of the IL-6/STAT-3 pathway can lead to reduced Treg numbers and function, disrupting the balance between Teff cells and Tregs. This imbalance exacerbates the onset and progression of autoimmune diseases (66).
4.1.2 Psoriasis
Psoriasis is a chronic autoimmune disorder characterized by the increased proliferation of skin keratinocytes, resulting in epidermal thickening and scaling (67). Clinically, psoriasis often manifests as erythematous plaques, scales, and skin inflammation, primarily caused by the accumulation of inflammatory cells, including CD4+T cells, CD8+T cells, NK cells, and DCs. The development of psoriasis is closely linked to the abnormal activation of the immune system, particularly involving the dysfunction of various T-cell subpopulations (68). This dysregulated activation leads to inflammatory responses and skin lesions, with pro-inflammatory cytokines such as IFN-γ, TNF-α, and IL-17 being secreted by cells like Th1, Th17, and Th22. DCs also play a crucial role in psoriasis by excessively releasing inflammatory mediators like IL-23 and TNF-α. The IL-23/IL-17 axis is recognized as a major immune pathway involved in the development of psoriasis (69).
Tregs play a crucial role in suppressing the activation of other immune cells and preventing autoimmune reactions, thereby maintaining immune homeostasis. However, research indicates that both the number and functionality of Tregs are often impaired in patients with psoriasis (70). This impairment can lead to a loss of the immune system’s regulatory function against self-tissues, triggering excessive inflammatory and immune responses, which further promote the development of psoriasis. Additionally, studies have shown a notable increase in γδT cell numbers in psoriatic lesions, which release pro-inflammatory cytokines like IL-17, potentially contributing to the formation and progression of skin lesions (71).
Izumo1R is a folate receptor present on CD4+T cells, particularly on Tregs. Although the precise role of Tregs in psoriasis requires further exploration, studies using a mouse model with Treg-specific Izumo1r deficiency have shown no significant impact on Treg development and homeostasis. However, this deficiency results in immune dysregulation in the skin, accompanied by the dysregulation of γδT cells, underscoring the importance of Izumo1R expression on Tregs in the development of psoriasis (72).
4.1.3 Other autoimmune diseases
Tregs also play a crucial role in various autoimmune diseases such as SLE, IBD, and RA. As immune regulatory cells, Tregs help maintain the balance of inflammatory responses through their unique immunomodulatory effects. In SLE, excessive expression of type I interferons (IFN-I) is a hallmark feature. IFN-I signaling affects the interplay between activated Tregs and Teff, compromising the regulatory role of Tregs in viral infections and within the TME. Research has identified an exhausted phenotype of Tregs in SLE patients, specifically CCR7loCD74hi Tregs. IFN-I signaling significantly contributes to the development of Treg cell exhaustion and functional impairments. Similarly, CD4+T-cell data from ulcerative colitis patients show analogous characteristics of Treg dysfunction (73). In IBD patients, increased levels of HLA-DR+CD38+T cells, including IL17A+Tregs that produce inflammatory cytokines, are observed in colon mucosal samples. Patients with Crohn’s disease exhibit elevated levels of IL1B+Tregs in peripheral blood mononuclear cells (74). Regarding RA, a diminished expression of EZH2 in CD4+T cells of affected individuals leads to the down-regulation of RUNX1 and the up-regulation of SMAD7, which in turn inhibits Foxp3 transcription and suppresses the differentiation of Tregs (75). In summary, Tregs are critically important across various autoimmune diseases, and their dysfunction can lead to immune imbalance and disease progression.
4.2 Immunotherapeutic strategy targeting Tregs in autoimmune diseases
Enhancing the immune system’s regulatory function is crucial for treating autoimmune conditions. Current strategies primarily focus on boosting Treg activity, which is vital for suppressing harmful immune responses (76). One promising approach involves targeting IL-2, a molecule essential for the growth and function of various immune cells, including Tregs. In a Phase I/II trial, researchers have identified the optimal dose of IL-2 for safely expanding Tregs in children with Type 1 Diabetes (T1D) (NCT01862120) (77). Another study has revealed that administering a low dose of IL-2 per day selectively activates and expands Tregs in patients with systemic sclerosis (SSc) without activating Teffs, paving the groundwork for a Phase II efficacy trial (NCT01988506) (78). Additionally, rapamycin has been shown to selectively expand Tregs while maintaining their suppressive phenotype and function, as demonstrated in a mouse model of T1D (79). Furthermore, the high expression of TNFR2 on Tregs can selectively eliminate autoreactive CD8+T cells, suggesting its potential benefits in treating autoimmune diseases (80). Despite its potential, administering low doses of IL-2 carries risks, including off-target effects due to effector cell activation and a short lifespan in the body. IL-2 muteins are engineered to overcome the limitations of conventional IL-2 therapies. By decreasing IL-2’s affinity for specific receptor subunits, such as CD122 or CD25, these modified proteins alter IL-2 signaling properties, thereby reducing Teff activation and enhancing Tregs selectivity (81). A novel IL-2 mutein has demonstrated significant efficacy in preclinical models by increasing Treg numbers and enhancing their suppressive activity, which extends the survival of allogeneic grafts and promotes antigen-specific tolerance (82). Additionally, compared to wild-type IL-2, these muteins have shown greater selectivity and efficacy in disease control within non-obese diabetic (NOD) models (81).
PolyTreg therapy which involves the isolation, in vitro expansion, and reinfusion of a patient’s own Tregs, designed to enhance both the number and function of Tregs. This technique is currently being tested in clinical trials for T1D (83). Additionally, combining polyTregs with low-dose IL-2 may further augment Treg numbers and functionality. A Phase I study indicated that this combination therapy could expand Tregs and cytotoxic T lymphocytes (CTLs) in T1D patients, with a favorable safety profile (NCT01210664, NCT02772679) (84). These findings suggest that the combination of Tregs with other therapies holds significant potential for treating autoimmune diseases, though further investigation and validation are necessary (Table 1).
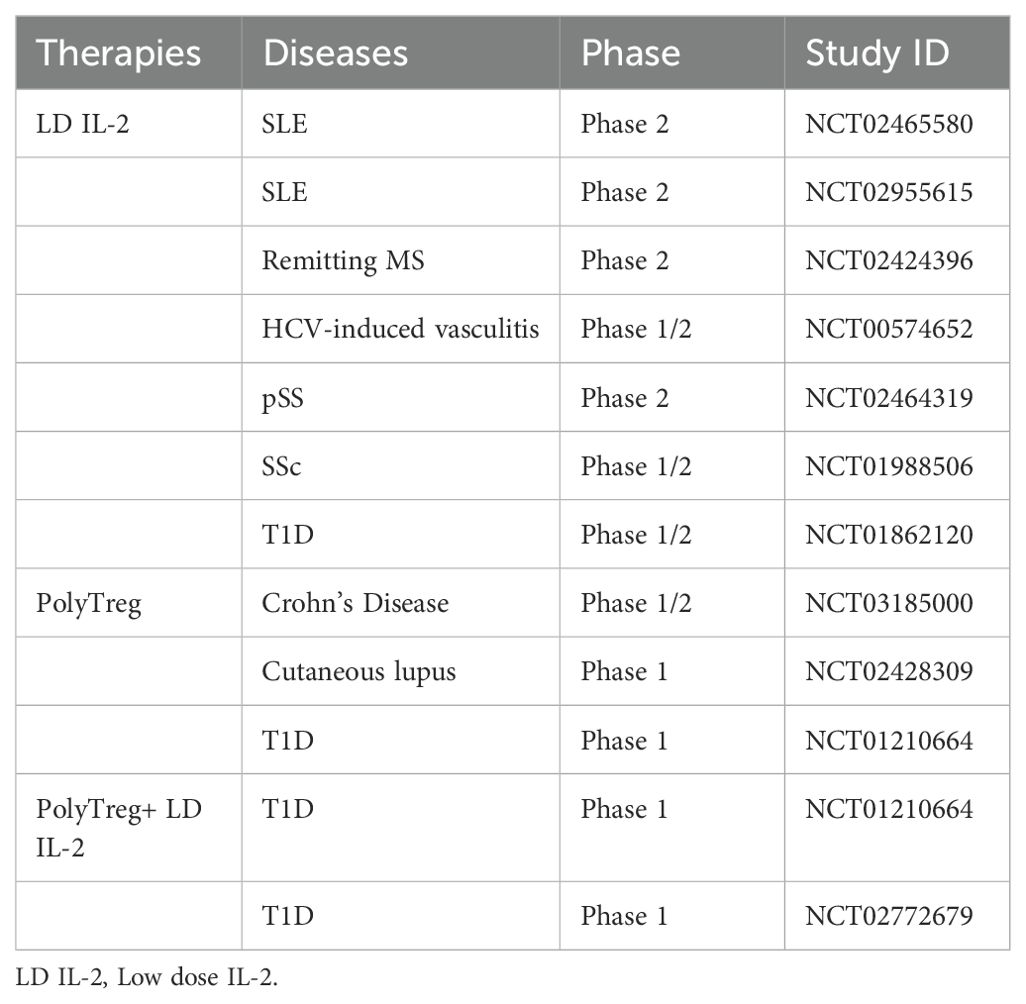
Table 1. Clinical trials to target Tregs to treat autoimmune diseases.
Another approach involves Treg-cell therapy based on genetic engineering to enhance Treg-cell function and redirect their targeting specificity, such as transgenic TCR therapy and chimeric antigen receptor (CAR) therapy (85). Since Tregs and self-reactive T cells possess a similar TCR repertoire, introducing a transgenic TCR into Tregs can efficiently direct them toward specific antigens of interest (86). However, the safety and efficacy of Tregs engineered with transgenic TCRs are still under investigation. Compared to TCR Tregs, CAR Tregs offer several advantages, including maintaining the phenotype and function of Tregs, retaining the ability to expand to therapeutic levels, and minimizing cytotoxicity to target cells. Additionally, CAR Tregs operate independently of the MHC and have a lower dependence on IL-2.
Autoimmune diseases are characterized by abnormalities of immune responses and inflammation, yet effective cures remain elusive. While Treg therapy shows promise in modulating the immune system, its efficacy is limited, necessitating further in-depth research. Currently, Treg therapy faces several challenges in treating autoimmune diseases, including unstable Treg cell function and inconsistent treatment outcomes. To overcome these challenges, it is essential to bolster foundational research, explore innovative treatment strategies, and conduct additional clinical trials to identify more effective therapies.
4.3 Tregs in cancer
TI-Tregs represent a critical mechanism for cancer evasion, posing a significant challenge to tumor-immune defenses and immunotherapy. In various cancers, such as liver cancer (87), gastric cancer (88), and bladder cancer (89), the presence of TI-Tregs often correlates with poor prognoses. The ratio of Tregs to CD8+T cells within tumors serves as a crucial prognostic factor, typically indicating unfavorable outcomes when skewed towards a higher proportion of Tregs (89). However, the role of Tregs in colorectal cancer (CRC) remains controversial, partly due to the diverse functions and phenotypes of Tregs (90).
Tregs can include both suppressive and non-inhibitory subsets. For example, peripheral blood Tregs can be divided into three main subsets based on the expression levels of Foxp3 and CD45RA (91), namely (1) Fr.I: Foxp3loCD45RA+, known as naive Tregs (nTregs); (2) Fr.II: Foxp3hiCD45RA-, known as effector Tregs (eTregs), characterized by high CTLA-4 expression and strong immunosuppressive functions; (3) Fr.III: Foxp3loCD45RA-, which primarily release inflammatory cytokines like IL-2 and IFN-γ.
The proportions of these different subsets in tumors can have varying impacts on prognosis. In most cancers, highly CTLA-4-expressing Fr.II eTregs are the predominant tumor-infiltrating Foxp3+Tregs. Saito et al. have further classified CRC into two types based on the frequency of Fr.III cells: Type A and Type B. Compared to Type B CRC, Type A CRC has fewer eTregs overall but higher in vitro suppressive activity, leading to a poor prognosis (92). Type B CRC has a higher frequency of Fr.III cells secreting IL-17 and IFN-γ, possibly originating from non-Tregs stimulated by IL-12 and TGF-β (93). These findings underscore the importance of assessing Treg subset distribution in tumor tissues (Table 2).
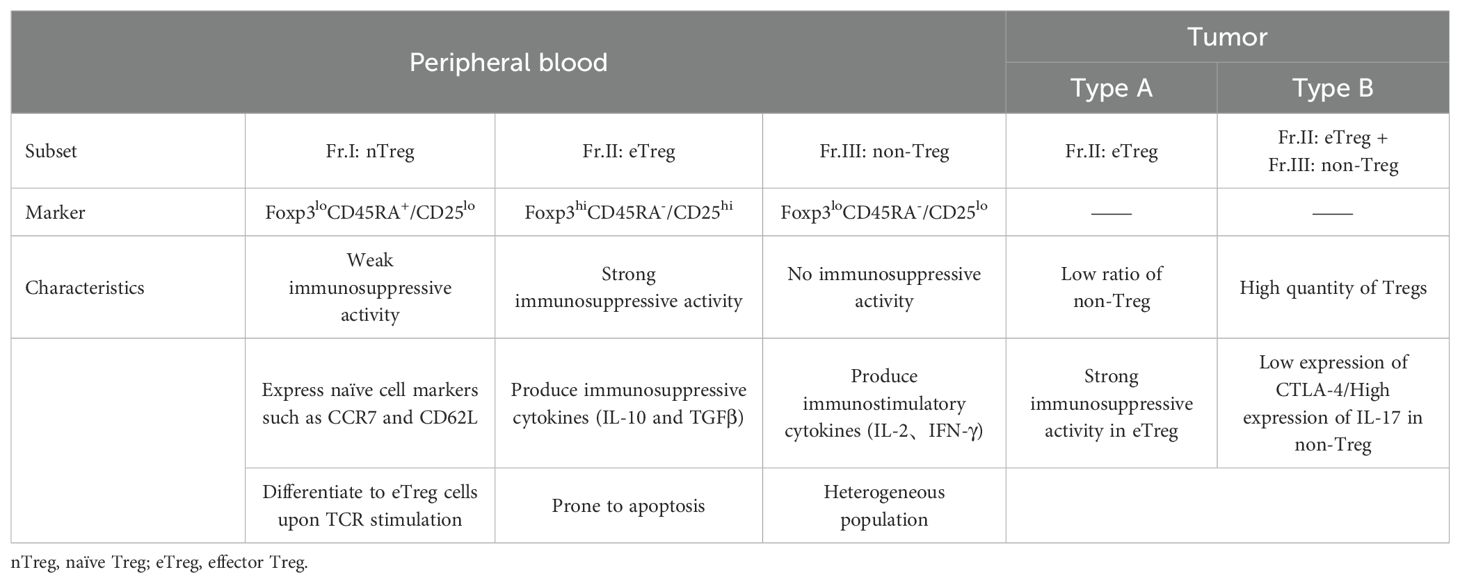
Table 2. Functional classification of human Tregs in the blood and tumors.
4.4 Immunotherapy for targeting Tregs in cancer
Research has shown that tr-Tregs exhibit diverse gene expression patterns similar to TI-Tregs. However, the differences between tr-Tregs and Tregs in normal tissue reflect the unique physiological characteristics of the TME, suggesting that tr-Tregs may have distinct functional and survival requirements (27). The increase and heightened activity of Tregs inhibit the immune system’s ability to attack tumors, thereby providing favorable conditions for tumor escape. Furthermore, by suppressing the function of T cells specific to tumor antigens, Tregs exacerbate immune tolerance, hindering the effectiveness of immunotherapy. Consequently, targeting Tregs has emerged as a crucial strategy to rejuvenate the immune system’s capacity to recognize and eliminate tumors (Table 3).
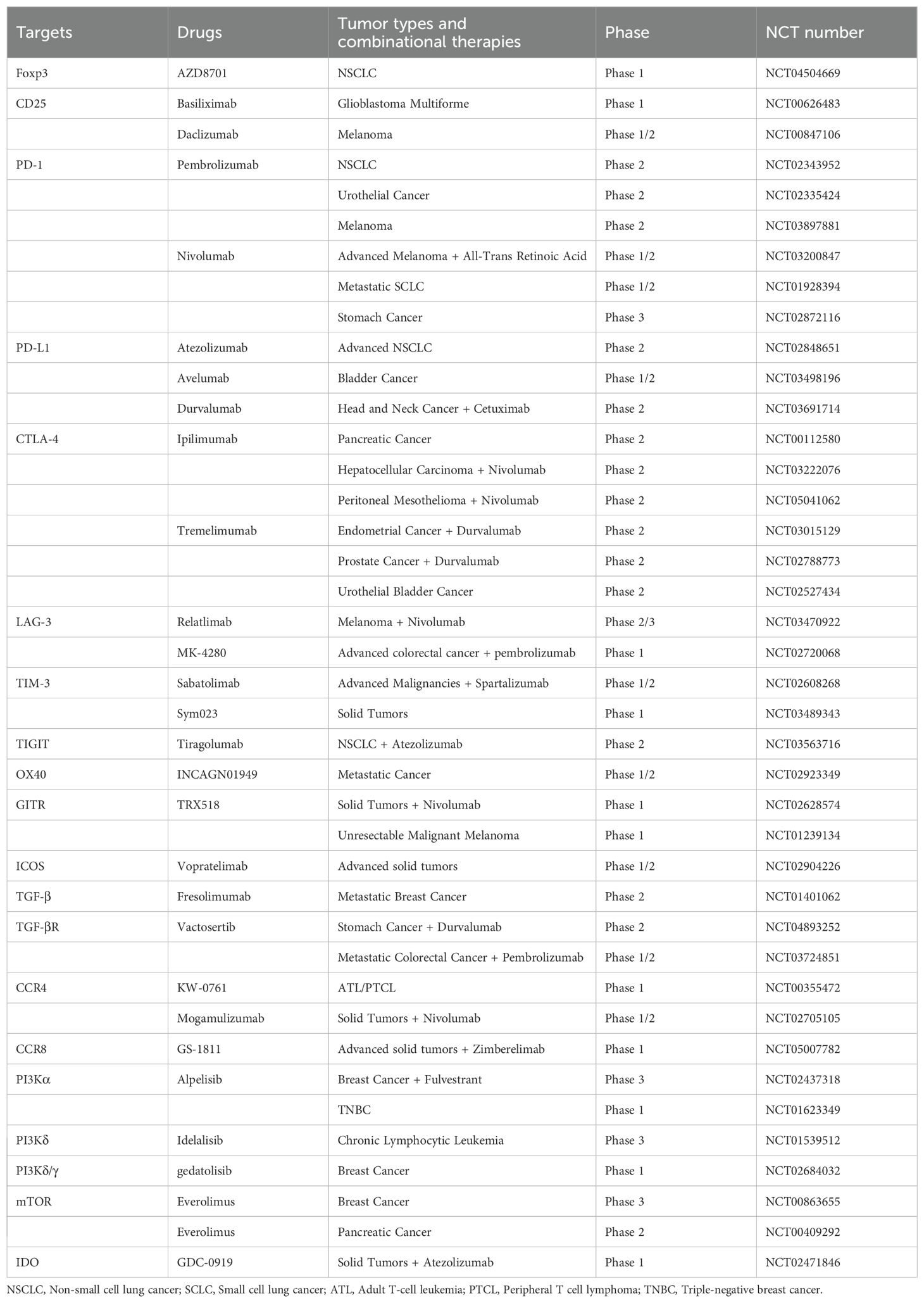
Table 3. Clinical trials to target Tregs in cancer patients.
4.4.1 Depleting Tregs
4.4.1.1 Targeting Foxp3
The expression of Foxp3 is critical for the development and function of Tregs. However, its overexpression can mediate the activation of Wnt/β-catenin signaling, which promotes tumor growth, metastasis, and epithelial-mesenchymal transition (EMT) (94). Studies have demonstrated that transient genetic depletion of Foxp3 in Tregs can enhance the efficacy of therapeutic vaccines against established melanoma tumors in preclinical models. This indicates that Foxp3 could be a highly attractive target in cancer immunotherapy (95). AZD8701, a first-in-class human clinical candidate antisense oligonucleotide (ASO) inhibitor targeting Foxp3, is currently being evaluated in Phase 1a/b clinical trials (NCT04504669) for its potential use in cancer treatment (96).
4.4.1.2 Targeting IL-2/CD25 signaling
CD25 is predominantly found on Tregs and has a high affinity for IL-2, facilitating the formation of the IL-2/IL-2 receptor complex, which collectively enhances Treg proliferation, survival, and functional integrity. Consequently, CD25 has emerged as a specific target for depleting Tregs in both mouse and human cancers. By altering the IL-2 signaling pathway, CD25 can influence Treg activity. For instance, Daclizumab inhibits the binding of IL-2 to CD25, adversely affecting Tregs (97). Nevertheless, this method has a limited therapeutic range since CD25 is upregulated not only on Tregs but also on Teff cells following activation. On the other hand, the chimeric anti-CD25 antibody basiliximab can prevent T-lymphocyte activation by blocking CD25 subunits on the IL-2 receptor. It has been documented that basiliximab targets CD4+CD25high cells while sparing CD4+CD25low cells (98).
4.4.2 Targeting immune checkpoints
4.4.2.1 Anti-PD-1/PD-L1
PD-1 is an immune checkpoint receptor extensively found on various immune cells, including T cells, B cells, and NK cells. In T cells, PD-1 predominantly appears on the surface of activated T cells. Its expression in Tregs is essential for maintaining their stability and function, promoting Foxp3 expression and Treg proliferation (99). Additionally, PD-1 suppresses TCR signal transduction in CD8+T cells by inhibiting the CD28 co-stimulatory signal, thus preventing overactivation of Teffs (100). In the TME, tumor cells and other immunosuppressive cells like macrophages and DCs often overexpress PD-L1. Tumor cells can also boost PD-L1 expression through IFN-γ secretion. The PD-L1/PD-1 interaction reduces T-cell production, cytokine release, and survival, aiding tumors in evading immune surveillance and destruction. Inhibitors targeting PD-1/PD-L1 have become critical therapies for various cancers, with PD-1 inhibitors such as Pembrolizumab showing favorable survival outcomes in patients with urothelial carcinoma (101), non-small cell lung cancer (NSCLC) (102), and melanoma (103). Nivolumab is used in clinical trials for refractory esophagogastric cancer and metastatic small cell lung cancer (SCLC) (104). Atezolizumab, which targets PD-L1, has been approved as a primary monotherapy for metastatic NSCLC patients with high PD-L1 levels and as an adjuvant treatment for resected stage II-IIIA NSCLC patients (105). However, some studies suggest that PD-1 blockade might lead to the expansion of highly suppressive PD-1+ eTregs, which can hinder anti-tumor immune responses, indicating limitations in monotherapy (106). Therefore, combining PD-1/PD-L1 inhibitors with other treatments like radiotherapy, chemotherapy, CTLA-4 inhibitors, or tumor vaccines may offer improved therapeutic outcomes.
4.4.2.2 Anti-CTLA-4
CTLA-4 plays a vital role in sustaining the immunosuppressive functions of Tregs through its constant expression on these cells. In mouse models, blocking CTLA-4 in vivo can trigger autoimmune diseases and disrupt the inhibitory effects mediated by Tregs in vitro (107). This underscores the essential role of CTLA-4-expressing Tregs in mediating immune suppression. Mechanistically, Tregs control the levels of CD80/CD86 on the surfaces of APCs via a CTLA-4-dependent pathway, preventing CD28 from binding to CD80/CD86 and thereby hindering the signaling necessary for T-cell proliferation. Besides interacting with CTLA-4 on T cells, CD80 also engages with PD-L1 on APCs. Studies have shown that reducing CD80/CD86 levels leads to an increase in PD-L1 on APCs, enabling Tregs to suppress not only naïve T cells but also Teff cells expressing PD-1 (108). Monoclonal antibodies (mAbs) targeting CTLA-4 have revolutionized cancer treatment by inducing lasting tumor regression, although their mechanisms of action are not fully understood. Initially, it was thought that anti-CTLA-4 antibodies worked by blocking CTLA-4, thereby enhancing the binding between CD28 and CD80/CD86 and reducing T-cell inhibition (109). However, recent findings suggest that these antibodies may also mediate antibody-dependent cellular cytotoxicity/phagocytosis (ADCC/P), leading to Treg depletion and enhancing CD8+T cell responses specific to tumor antigens (110). Additionally, CTLA-4 mAbs can activate myeloid cells and modify the TME through interactions with FcγR (111). These insights offer new perspectives on the mechanisms of therapies targeting CTLA-4. Clinically, combining PD-1 and CTLA-4 immune checkpoint inhibitors (ICIs) has significantly improved survival outcomes in patients with melanoma, highlighting the benefits of combination therapy (112).
4.4.2.3 Targeting other immune checkpoints
LAG-3, TIM-3, and TIGIT are inhibitory receptors found on Tregs, presenting new targets for immunotherapy. LAG-3 negatively regulates T cell function and mediates bidirectional signaling in APCs (113). During interactions between Tregs and DCs, the engagement of LAG-3 on Tregs enhances their activity, promoting immune tolerance and indirectly inhibiting DC function. Furthermore, the interaction of LAG-3 with its ligand, MHC class II, can downregulate cytokine secretion and proliferation of CD4+T cells (113). Current research is exploring anti-LAG-3 antibodies both alone and in combination with other immunotherapies for treating melanoma. Compared to LAG-3 blockade alone, the combination of LAG-3 and PD-1 inhibitors has shown a more promising response rate in patients with refractory melanoma (114). Relatlimab combined with Nivolumab as a first-line treatment for metastatic melanoma patients has demonstrated improved progression-free survival compared to PD-1 blockade alone (115). Favezelimab, an emerging mAb targeting LAG-3, has shown significant efficacy when combined with pembrolizumab in treating patients with advanced microsatellite-stable CRC (116).
Studies have demonstrated that TIM-3+Tregs express higher levels of cell markers such as FoxP3, LAG-3, and CTLA-4, indicating significantly enhanced suppressive functions. These Tregs specifically inhibit Th17 cells, highlighting the potential value of TIM-3 as a target for cancer therapy (117). Similarly, Phase I studies of the anti-TIM-3 antibody Sabatolimab, used either as a monotherapy or in various combinations (including with PD-1 inhibitors), are being conducted in patients with advanced solid tumors. Early results indicate that the combination of Sabatolimab and Spartalizumab is well-tolerated and exhibits anti-tumor activity (118).
Additionally, TIGIT is highly expressed on Tregs and suppresses Th1 and Th17 cells through the secretion of Fgl2. It may also contribute to the generation of tolerogenic DCs, thereby inhibiting the activation of Teff responses (119).Therapeutic strategies targeting TIGIT involve receptor-ligand blockade independent of Fc interactions, clearance of TIGIT-expressing Tregs through Fc-mediated mechanisms, and Fc-dependent modulation of myeloid cells (120). A clinical trial has revealed that the objective response rate (ORR) for NSCLC patients treated with both the anti-TIGIT antibody tiragolumab and atezolizumab is 31%, compared to 16% with atezolizumab monotherapy, highlighting the superior clinical benefits of combination therapy (120).
4.4.3 Targeting other co-stimulation molecules
Anti-tumor immunity can also be achieved by activating co-stimulatory receptors on T cells using agonistic antibodies. These co-stimulatory receptors, mainly from the tumor necrosis factor receptor superfamily (TNFRSF), include OX40, ICOS, and GITR. OX40 is a co-stimulatory molecule constantly expressed by Tregs and induced in activated Teffs. In an open-label phase I/II study, the anti-OX40 antibody, including the mAb INCAGN01949, is being assessed for safety and pharmacodynamics in treating advanced malignancies (NCT02923349) (121). Another novel anti-OX40 antibody, BAT6026, which has enhanced antibody-dependent cell cytotoxicity, facilitates intra-tumoral Treg depletion and is being utilized in cancer immunotherapy (122).
Therapeutic antibodies targeting GITR have shown therapeutic efficacy in preclinical tumor models, characterized by reduced and functionally altered intra-tumoral Tregs and enhanced anti-tumor CD8+T cell function (123). Currently, studies are ongoing to evaluate GITR agonists either alone or in combination therapies for patients with advanced solid tumors (NCT02583165 and NCT02628574) (124). The GITR agonist TRX518, both as a monotherapy and in combination with pembrolizumab or nivolumab, has demonstrated acceptable safety in phase I studies (125).
Vopratelimab, an ICOS agonist has shown good safety profiles in patients with advanced solid tumors, both as a monotherapy and in combination with nivolumab (NCT02904226). This therapy upregulates ICOS expression on CD4+T cells after T-cell priming, enhancing their proliferation and activation (126).
4.4.4 Targeting Treg cytokine secretion
The inhibition of the TGF-β pathway is garnering significant attention in cancer research. Currently, antibodies targeting TGF-β are categorized into two main types: those directly inhibiting TGF-β activity, such as neutralizing antibodies like Fresolimumab, SAR439459, and NIS793, and those targeting TGF-β receptors, including LY3200882, Vactosertib, and Galunisertib. These inhibitors can block TGF-β signaling pathways, with some already in clinical trials. For example, Fresolimumab has been assessed for its efficacy in patients with late-stage melanoma or renal cell carcinoma (127). Vactosertib in conjunction with Durvalumab is under examination for treating gastric cancer (NCT04893252), while its combination with Pembrolizumab is being tested for advanced CRC or gastric cancer (NCT03724851). Similarly, Galunisertib paired with Nivolumab is being assessed for NSCLC or hepatocellular carcinoma. Furthermore, therapies that concurrently target TGF-β and PD-L1 pathways have yielded promising results. Bintrafusp alfa, a bifunctional fusion protein that inhibits TGF-βRII and blocks PD-L1, has shown positive clinical outcomes in second-line therapy for NSCLC and first-line treatment for cervical cancer patients (128).
IL-10 and IL-35 play crucial roles in modulating the immune response to tumors. IL-10 supports Treg stability and phenotype by maintaining Foxp3 and TGF-β expression and influences Treg cell differentiation by inhibiting Akt phosphorylation and preserving Foxo1 function (129). IL-35 is overexpressed in various cancers, directly correlating with tumor progression and poor prognosis. Treg cell-derived IL-10 and IL-35 induce exhaustion in CD8+T cells by upregulating the expression of BLIMP-1 and genes associated with exhaustion, leading to CD8+T cell dysfunction (130). Neutralizing antibodies against IL-35 not only enhance gemcitabine chemotherapy but also significantly reduce microvascular density in mouse models of pancreatic cancer (131). Therefore, targeting IL-35 within the TME could provide a promising approach for tumor immunotherapy.
4.4.5 Inhibiting Treg migration
Another intriguing aspect of Tregs is their elevated expression of chemokine receptors CCR4 and CCR8, which presents exciting therapeutic opportunities. The CCR4 mAb KM2760 was originally employed to treat adult T-cell leukemia/lymphoma (ATLL). This antibody boosts ADCC by enhancing its affinity for FcγR on effector cells, aiming to reduce Treg numbers and initiate a potent anti-tumor immune response (132). Although Mogamulizumab, in combination with Nivolumab, does not show increased efficacy in a phase I/II study for advanced solid tumors, it does exhibit promising safety and tolerability profiles (133). Unlike Mogamulizumab, anti-CCR8 antibodies can selectively target and deplete Tregs, thereby enhancing immune responses against tumors (134). Additionally, research suggests that combining CCR8 targeting with anti-PD-1 therapy could effectively eliminate TI-Tregs (135). These findings open up new avenues for targeting Tregs via CCR4 and CCR8, positioning them as pivotal players in the future of tumor immunotherapy.
4.4.6 Targeting Tregs metabolism
Tumor metabolism can significantly impede the efficacy of immunotherapy, particularly through the actions of TI-Tregs. Therefore, targeting the metabolism of these Tregs holds considerable immunological and metabolic promise and could be a strategy to overcome metabolic barriers in immunotherapy. The Phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway not only regulates the expression of Foxp3 in Tregs but also plays a vital role in key metabolic processes like glycolysis (136).
Alpelisib, the pioneering PI3Kα inhibitor used for breast cancer treatment, has extended progression-free survival in patients with HR+/HER2- metastatic breast cancer carrying PIK3CA mutations when combined with Fulvestrant (NCT02437318) (137). Additionally, Alpelisib in combination with Olaparib has shown good tolerability in pre-treated triple-negative breast cancer (TNBC) patients (NCT01623349) (138). The PI3Kδ inhibitor Idelalisib is approved for treating B-cell malignancies (NCT01539512). Duvelisib, a PI3K-δ/γ inhibitor, has demonstrated clinical activity and safety in T-cell lymphomas (NCT01476657). Gedatolisib, which selectively targets all class I PI3K subtypes (p110α, p110β, p110γ, and p110δ) and mTOR, significantly avoids resistance development. By targeting these class I PI3K and mTOR, Gedatolisib has shown promising ORR in breast cancer treatment (NCT02684032) (139).
mTOR activity is often upregulated in human cancers. Combining Everolimus with hormone therapy has improved progression-free survival in patients with advanced HR+ breast cancer (NCT00863655), and it has also demonstrated good tolerability in treating metastatic pancreatic cancer patients (NCT00409292) (140). IDO, the primary enzyme in tryptophan (Trp) degradation metabolism, converts Trp into kynurenine (Kyn). Overexpression of IDO and the resulting accumulation of Kyn in tumors can inhibit Teff cells and activate Tregs by binding to the aryl hydrocarbon receptor (AHR) (141). Although several IDO1 inhibitors, including INCB024360, GDC-0919, and NLG802, are not yet approved by the US FDA for cancer therapy, some have advanced to clinical trials. For instance, the combination of Navoximod (GDC-0919) and the PD-L1 inhibitor Atezolizumab has shown acceptable safety and tolerability in patients with late-stage cancer (NCT02471846) (142). Further clinical investigation is necessary to target IDO as a blockade point, and exploring other targets related to amino acid metabolism remains to be an illustrated area.
5 Conclusion
Tregs, crucial regulators of immune homeostasis, are of significant interest in both autoimmune and anti-tumor immune clinical settings. Therapies targeting Tregs are currently being actively explored, either as standalone treatments or in combination with other therapeutic approaches such as immune checkpoint blockade, targeted vaccine therapy, radiotherapy, and chemotherapy, especially when combined with ICIs. Strategies to increase the number of Tregs can be applied in treating autoimmune conditions and other disorders requiring immunosuppression. Conversely, depleting Tregs holds great potential in cancer treatment, although it may lead to harmful autoimmune reactions. Therefore, targeting terminally differentiated eTregs rather than all Foxp3+T cells, is a crucial strategy. For instance, CTLA-4 inhibitors can either eliminate eTregs or diminish their suppressive function (143). Overall, despite some clinical success in targeting Tregs, a deeper understanding of their formation, maintenance, and function is crucial to improve precision-targeted immunotherapy against Tregs.
Author contributions
JG: Visualization, Writing – original draft, Writing – review & editing. XY: Writing – original draft. LC: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was supported by the National Natural Science Foundation of China (82172689, 82473269), the Key R&D Project of Jiangsu Province (BE2022721), Changzhou Medical Center of Nanjing Medical University (CZKYCMCC202202, CZKYCMCC202301).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. (2020) 142:1956–73. doi: 10.1161/CIRCULATIONAHA.120.046789
2. Lee W, Lee GR. Transcriptional regulation and development of regulatory T cells. Exp Mol Med. (2018) 50:e456–6. doi: 10.1038/emm.2017.313
3. Bayati F, Mohammadi M, Valadi M, Jamshidi S, Foma AM, Sharif-Paghaleh E. The therapeutic potential of regulatory T cells: challenges and opportunities. Front Immunol. (2020) 11:585819. doi: 10.3389/fimmu.2020.585819
4. Norton EG, Chapman NM, Chi H. Strengthening bonds via RyR2 inhibition helps immune suppression. J Clin Invest. (2023) 133:e172986. doi: 10.1172/JCI172986
5. Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. (2008) 124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x
6. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
7. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. (2022) 40:656–673.e657. doi: 10.1016/j.ccell.2022.04.011
8. Malek TR. The biology of interleukin-2. Annu Rev Immunol. (2008) 26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357
9. Angerami MT, Suarez GV, Vecchione MB, Laufer N, Ameri D, Ben G, et al. Expansion of CD25-Negative Forkhead Box P3-Positive T Cells during HIV and Mycobacterium tuberculosis Infection. Front Immunol. (2017) 8:528. doi: 10.3389/fimmu.2017.00528
10. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
11. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
12. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. (2007) 110:2983–90. doi: 10.1182/blood-2007-06-094656
13. Santamaria JC, Borelli A, Irla M. Regulatory T cell heterogeneity in the thymus: impact on their functional activities. Front Immunol. (2021) 12:643153. doi: 10.3389/fimmu.2021.643153
14. Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. (2008) 28:100–11. doi: 10.1016/j.immuni.2007.11.021
15. Kumar P, Marinelarena A, Raghunathan D, Ragothaman VK, Saini S, Bhattacharya P, et al. Critical role of OX40 signaling in the TCR-independent phase of human and murine thymic Treg generation. Cell Mol Immunol. (2019) 16:138–53. doi: 10.1038/cmi.2018.8
16. Ruck T, Bock S, Pfeuffer S, Schroeter CB, Cengiz D, Marciniak P, et al. K(2P)18.1 translates T cell receptor signals into thymic regulatory T cell development. Cell Res. (2022) 32:72–88. doi: 10.1038/s41422-021-00580-z
17. Clough JN, Omer OS, Tasker S, Lord GM, Irving PM. Regulatory T-cell therapy in Crohn's disease: challenges and advances. Gut. (2020) 69:942–52. doi: 10.1136/gutjnl-2019-319850
18. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. (2012) 209:1723–42. doi: 10.1084/jem.20120914
19. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Sander C et al: Interferon-γ Drives T(reg) Fragility to Promote Anti-tumor Immunity. Cell. (2017) 169:1130–1141.e1111. doi: 10.1016/j.cell.2017.05.005
20. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259:88–102. doi: 10.1111/imr.12160
21. Gonzalez-Figueroa P, Roco JA, Papa I, Núñez Villacís L, Stanley M, Linterman MA, et al. Follicular regulatory T cells produce neuritin to regulate B cells. Cell. (2021) 184:1775–1789.e1719. doi: 10.1016/j.cell.2021.02.027
22. Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol. (2017) 18:1249–60. doi: 10.1038/ni.3837
23. Lee J, Kim D, Min B. Tissue resident foxp3(+) regulatory T cells: sentinels and saboteurs in health and disease. Front Immunol. (2022) 13:865593. doi: 10.3389/fimmu.2022.865593
24. Sharma A, Rudra D. Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol. (2018) 9:883. doi: 10.3389/fimmu.2018.00883
25. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–1129.e1111. doi: 10.1016/j.cell.2017.05.002
26. Wu J, Ren B, Wang D, Lin H. Regulatory T cells in skeletal muscle repair and regeneration: recent insights. Cell Death Dis. (2022) 13:680. doi: 10.1038/s41419-022-05142-8
27. Glasner A, Plitas G. Tumor resident regulatory T cells. Semin Immunol. (2021) 52:101476. doi: 10.1016/j.smim.2021.101476
28. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
29. Müller AL, Casar C, Preti M, Krzikalla D, Gottwick C, Averhoff P, et al. Inflammatory type 2 conventional dendritic cells contribute to murine and human cholangitis. J Hepatol. (2022) 77:1532–44. doi: 10.1016/j.jhep.2022.06.025
30. Catani L, Sollazzo D, Trabanelli S, Curti A, Evangelisti C, Polverelli N, et al. Decreased expression of indoleamine 2,3-dioxygenase 1 in dendritic cells contributes to impaired regulatory T cell development in immune thrombocytopenia. Ann Hematol. (2013) 92:67–78. doi: 10.1007/s00277-012-1556-5
31. Kushwah R, Hu J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. (2011) 1:20. doi: 10.1186/2045-3701-1-20
32. Terme M, Chaput N, Combadiere B, Ma A, Ohteki T, Zitvogel L. Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+ self-reactive T cells. J Immunol. (2008) 180:4679–86. doi: 10.4049/jimmunol.180.7.4679
33. Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. (2021) 184:3998–4015.e3919. doi: 10.1016/j.cell.2021.05.027
34. Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. (2006) 176:1582–7. doi: 10.4049/jimmunol.176.3.1582
35. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
36. Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res. (2018) 6:766–75. doi: 10.1158/2326-6066.CIR-17-0498
37. Dean JW, Peters LD, Fuhrman CA, Seay HR, Posgai AL, Stimpson SE, et al. Innate inflammation drives NK cell activation to impair Treg activity. J Autoimmun. (2020) 108:102417. doi: 10.1016/j.jaut.2020.102417
38. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
39. Hosseinalizadeh H, Rabiee F, Eghbalifard N, Rajabi H, Klionsky DJ, Rezaee A. Regulating the regulatory T cells as cell therapies in autoimmunity and cancer. Front Med (Lausanne). (2023) 10:1244298. doi: 10.3389/fmed.2023.1244298
40. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
41. Sreejit G, Fleetwood AJ, Murphy AJ, Nagareddy PR. Origins and diversity of macrophages in health and disease. Clin Transl Immunol. (2020) 9:e1222. doi: 10.1002/cti2.1222
42. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
43. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–677.e666. doi: 10.1016/j.immuni.2018.07.015
44. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–352.e345. doi: 10.1016/j.immuni.2018.07.007
45. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1249288. doi: 10.1126/science.1249288
46. Li X, Zhang B, Zhang X, Yu S, Xue HH, Hu X. TLE3 and TLE4-coordinated colonic macrophage-CD4(+) T cell crosstalk maintains intestinal immune homeostasis. Mucosal Immunol. (2023) 16:50–60. doi: 10.1016/j.mucimm.2022.12.005
47. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the SREBP1-Dependent metabolic fitness of tumor-Promoting macrophages via repression of CD8(+) T cell-Derived interferon-γ. Immunity. (2019) 51:381–397.e386. doi: 10.1016/j.immuni.2019.06.017
48. La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. Targeting MARCO and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res. (2021) 81:956–67. doi: 10.1158/0008-5472.CAN-20-1885
49. Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 axis controls treg infiltration in the tumor microenvironment by inducing CCR4 expression in breast cancer. Front Immunol. (2022) 13:740588. doi: 10.3389/fimmu.2022.740588
50. Li J, Wang S, Wang N, Zheng Y, Yang B, Wang X, et al. Aiduqing formula inhibits breast cancer metastasis by suppressing TAM/CXCL1-induced Treg differentiation and infiltration. Cell Commun Signal. (2021) 19:89. doi: 10.1186/s12964-021-00775-2
51. Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. (2021) 184:3852–72. doi: 10.1016/j.cell.2021.06.024
52. Wei K, Nguyen HN, Brenner MB. Fibroblast pathology in inflammatory diseases. J Clin Invest. (2021) 131:e149538. doi: 10.1172/JCI149538
53. Broz MT, Ko EY, Ishaya K, Xiao J, De Simone M, Hoi XP, et al. Metabolic targeting of cancer associated fibroblasts overcomes T-cell exclusion and chemoresistance in soft-tissue sarcomas. Nat Commun. (2024) 15:2498. doi: 10.1038/s41467-024-46504-4
54. O'Connor RA, Martinez BR, Koppensteiner L, Mathieson L, Akram AR. Cancer-associated fibroblasts drive CXCL13 production in activated T cells via TGF-beta. Front Immunol. (2023) 14:1221532. doi: 10.3389/fimmu.2023.1221532
55. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. (2016) 5:e198. doi: 10.1038/oncsis.2016.7
56. Hu G, Cheng P, Pan J, Wang S, Ding Q, Jiang Z, et al. An IL6-adenosine positive feedback loop between CD73(+) γδTregs and CAFs promotes tumor progression in human breast cancer. Cancer Immunol Res. (2020) 8:1273–86. doi: 10.1158/2326-6066.CIR-19-0923
57. Hu Y, Recouvreux MS, Haro M, Taylan E, Taylor-Harding B, Walts AE, et al. INHBA(+) cancer-associated fibroblasts generate an immunosuppressive tumor microenvironment in ovarian cancer. NPJ Precis Oncol. (2024) 8:35. doi: 10.1038/s41698-024-00523-y
58. Bruch-Oms M, Olivera-Salguero R, Mazzolini R, Del Valle-Pérez B, Mayo-González P, Beteta Á, et al. Analyzing the role of cancer-associated fibroblast activation on macrophage polarization. Mol Oncol. (2023) 17:1492–513. doi: 10.1002/1878-0261.13454
59. Chen L, Huang H, Zheng X, Li Y, Chen J, Tan B, et al. IL1R2 increases regulatory T cell population in the tumor microenvironment by enhancing MHC-II expression on cancer-associated fibroblast. J Immunother Cancer. (2022) 10:e004585. doi: 10.1136/jitc-2022-004585
60. De Conto C, Oevermann A, Burgener IA, Doherr MG, Blum JW. Gastrointestinal tract mucosal histomorphometry and epithelial cell proliferation and apoptosis in neonatal and adult dogs. J Anim Sci. (2010) 88:2255–64. doi: 10.2527/jas.2009-2511
61. Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev. (2014) 259:231–44. doi: 10.1111/imr.12169
62. Álvarez-Sánchez N, Cruz-Chamorro I, López-González A, Utrilla JC, Fernández-Santos JM, Martínez-López A, et al. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav Immun. (2015) 50:101–14. doi: 10.1016/j.bbi.2015.06.021
63. McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. (2005) 175:3025–32. doi: 10.4049/jimmunol.175.5.3025
64. Caiazzo E, Bilancia R, Rossi A, Ialenti A, Cicala C. Ectonucleoside triphosphate diphosphohydrolase-1/CD39 affects the response to ADP of female rat platelets. Front Pharmacol. (2019) 10:1689. doi: 10.3389/fphar.2019.01689
65. Wang Y, Begum-Haque S, Telesford KM, Ochoa-Repáraz J, Christy M, Kasper EJ, et al. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes. (2014) 5:552–61. doi: 10.4161/gmic.29797
66. Aqel SI, Kraus EE, Jena N, Kumari V, Granitto MC, Mao L, et al. Novel small molecule IL-6 inhibitor suppresses autoreactive Th17 development and promotes T(reg) development. Clin Exp Immunol. (2019) 196:215–25. doi: 10.1111/cei.13258
67. Liu S, He M, Jiang J, Duan X, Chai B, Zhang J, et al. Triggers for the onset and recurrence of psoriasis: a review and update. Cell Commun Signal. (2024) 22:108. doi: 10.1186/s12964-023-01381-0
68. Li L, Lu J, Liu J, Wu J, Zhang X, Meng Y, et al. Immune cells in the epithelial immune microenvironment of psoriasis: emerging therapeutic targets. Front Immunol. (2023) 14:1340677. doi: 10.3389/fimmu.2023.1340677
69. Vičić M, Kaštelan M, Brajac I, Sotošek V, Massari LP. Current concepts of psoriasis immunopathogenesis. Int J Mol Sci. (2021) 22:11574. doi: 10.3390/ijms222111574
70. Yang L, Li B, Dang E, Jin L, Fan X, Wang G. Impaired function of regulatory T cells in patients with psoriasis is mediated by phosphorylation of STAT3. J Dermatol Sci. (2016) 81:85–92. doi: 10.1016/j.jdermsci.2015.11.007
71. Qi C, Wang Y, Li P, Zhao J. Gamma delta T cells and their pathogenic role in psoriasis. Front Immunol. (2021) 12:627139. doi: 10.3389/fimmu.2021.627139
72. Zarin P, Shwartz Y, Ortiz-Lopez A, Hanna BS, Sassone-Corsi M, Hsu YC, et al. Treg cells require Izumo1R to regulate γδT cell-driven inflammation in the skin. Proc Natl Acad Sci U.S.A. (2023) 120:e2221255120. doi: 10.1073/pnas.2221255120
73. Guo C, Liu Q, Zong D, Zhang W, Zuo Z, Yu Q, et al. Single-cell transcriptome profiling and chromatin accessibility reveal an exhausted regulatory CD4+ T cell subset in systemic lupus erythematosus. Cell Rep. (2022) 41:111606. doi: 10.1016/j.celrep.2022.111606
74. Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, et al. Single-Cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and crohn's disease. Gastroenterology. (2020) 159:591–608.e510. doi: 10.1053/j.gastro.2020.04.074
75. Xiao XY, Li YT, Jiang X, Ji X, Lu X, Yang B, et al. EZH2 deficiency attenuates Treg differentiation in rheumatoid arthritis. J Autoimmun. (2020) 108:102404. doi: 10.1016/j.jaut.2020.102404
76. Kishimoto TK, Fournier M, Michaud A, Rizzo G, Roy C, Capela T, et al. Rapamycin nanoparticles increase the therapeutic window of engineered interleukin-2 and drive expansion of antigen-specific regulatory T cells for protection against autoimmune disease. J Autoimmun. (2023) 140:103125. doi: 10.1016/j.jaut.2023.103125
77. Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: a Phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia. (2020) 63:1808–21. doi: 10.1007/s00125-020-05200-w
78. Barde F, Lorenzon R, Vicaut E, Rivière S, Cacoub P, Cacciatore C, et al. Induction of regulatory T cells and efficacy of low-dose interleukin-2 in systemic sclerosis: interventional open-label phase 1-phase 2a study. RMD Open. (2024) 10:e003500. doi: 10.1136/rmdopen-2023-003500
79. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
80. Ban L, Zhang J, Wang L, Kuhtreiber W, Burger D, Faustman DL. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc Natl Acad Sci U.S.A. (2008) 105:13644–9. doi: 10.1073/pnas.0803429105
81. Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci Immunol. (2020) 5:eaba5264. doi: 10.1126/sciimmunol.aba5264
82. Efe O, Gassen RB, Morena L, Ganchiku Y, Al Jurdi A, Lape IT, et al. A humanized IL-2 mutein expands Tregs and prolongs transplant survival in preclinical models. J Clin Invest. (2024) 134:e173107. doi: 10.1172/JCI173107
83. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
84. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight. (2021) 6:e147474. doi: 10.1172/jci.insight.147474
85. Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci. (2020) 21:7015. doi: 10.3390/ijms21197015
86. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduction Targeted Ther. (2023) 8:9. doi: 10.1038/s41392-022-01270-x
87. Zhou L, Wang J, Lyu SC, Pan LC, Shi XJ, Du GS, et al. PD-L1(+)NEUT, foxp3(+)Treg, and NLR as new prognostic marker with low survival benefits value in hepatocellular carcinoma. Technol Cancer Res Treat. (2021) 20:15330338211045820. doi: 10.1177/15330338211045820
88. Xu D, Liu X, Ke S, Guo Y, Zhu C, Cao H. CCL19/CCR7 drives regulatory T cell migration and indicates poor prognosis in gastric cancer. BMC Cancer. (2023) 23:464. doi: 10.1186/s12885-023-10882-7
89. Koll FJ, Banek S, Kluth L, Köllermann J, Bankov K, Chun FK, et al. Tumor-associated macrophages and Tregs influence and represent immune cell infiltration of muscle-invasive bladder cancer and predict prognosis. J Transl Med. (2023) 21:124. doi: 10.1186/s12967-023-03949-3
90. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. (2009) 27:186–92. doi: 10.1200/JCO.2008.18.7229
91. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
92. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res. (2022) 10:86. doi: 10.1186/s40364-022-00434-9
93. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. (2016) 22:679–84. doi: 10.1038/nm.4086
94. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. (2017) 16:124. doi: 10.1186/s12943-017-0700-1
95. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. (2010) 70:7788–99. doi: 10.1158/0008-5472.CAN-10-1736
96. Revenko A, Carnevalli LS, Sinclair C, Johnson B, Peter A, Taylor M, et al. Direct targeting of FOXP3 in Tregs with AZD8701, a novel antisense oligonucleotide to relieve immunosuppression in cancer. J Immunother Cancer. (2022) 10:e003892. doi: 10.1136/jitc-2021-003892
97. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr., et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. (2012) 4:134ra162. doi: 10.1126/scitranslmed.3003330
98. Okita R, Yamaguchi Y, Ohara M, Hironaka K, Okawaki M, Nagamine I, et al. Targeting of CD4+CD25high cells while preserving CD4+CD25low cells with low-dose chimeric anti-CD25 antibody in adoptive immunotherapy of cancer. Int J Oncol. (2009) 34:563–72.
99. Kim MJ, Kim K, Park HJ, Kim GR, Hong KH, Oh JH, et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat Immunol. (2023) 24:148–61. doi: 10.1038/s41590-022-01373-1
100. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. (2017) 355:1423–7. doi: 10.1126/science.aaf0683
101. Balar AV, Castellano D, O'Donnell PH, Grivas P, Vuky J, Powles T, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol. (2017) 18:1483–92. doi: 10.1016/S1470-2045(17)30616-2
102. Aguilar EJ, Ricciuti B, Gainor JF, Kehl KL, Kravets S, Dahlberg S, et al. Outcomes to first-line pembrolizumab in patients with non-small-cell lung cancer and very high PD-L1 expression. Ann Oncol. (2019) 30:1653–9. doi: 10.1093/annonc/mdz288
103. Weber JS, Carlino MS, Khattak A, Meniawy T, Ansstas G, Taylor MH, et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet. (2024) 403:632–44. doi: 10.1016/S0140-6736(23)02268-7
104. Ready NE, Ott PA, Hellmann MD, Zugazagoitia J, Hann CL, de Braud F, et al. Nivolumab monotherapy and nivolumab plus ipilimumab in recurrent small cell lung cancer: results from the checkMate 032 randomized cohort. J Thorac Oncol. (2020) 15:426–35. doi: 10.1016/j.jtho.2019.10.004
105. Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-Line treatment of PD-L1-Selected patients with NSCLC. N Engl J Med. (2020) 383:1328–39. doi: 10.1056/NEJMoa1917346
106. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U.S.A. (2019) 116:9999–10008. doi: 10.1073/pnas.1822001116
107. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U.S.A. (2016) 113:E2383–2392. doi: 10.1073/pnas.1603892113
108. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
109. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
110. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U.S.A. (2019) 116:609–18. doi: 10.1073/pnas.1812186116
111. Yofe I, Landsberger T, Yalin A, Solomon I, Costoya C, Demane DF, et al. Anti-CTLA-4 antibodies drive myeloid activation and reprogram the tumor microenvironment through FcγR engagement and type I interferon signaling. Nat Cancer. (2022) 3:1336–50. doi: 10.1038/s43018-022-00447-1
112. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell. (2019) 35:238–255.e236. doi: 10.1016/j.ccell.2019.01.003
113. Cai L, Li Y, Tan J, Xu L, Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. (2023) 16:101. doi: 10.1186/s13045-023-01499-1
114. Solinas C, Migliori E, De Silva P, Willard-Gallo K. LAG3: the biological processes that motivate targeting this immune checkpoint molecule in human cancer. Cancers (Basel). (2019) 11:1213. doi: 10.3390/cancers11081213
115. Tawbi HA, SChadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. (2022) 386:24–34. doi: 10.1056/NEJMoa2109970
116. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open. (2022) 7:100639. doi: 10.1016/j.esmoop.2022.100639
117. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. (2014) 44:2703–11. doi: 10.1002/eji.201344392
118. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
119. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
120. Guan X, Hu R, Choi Y, Srivats S, Nabet BY, Silva J, et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and T(reg) cells. Nature. (2024) 627:646–55. doi: 10.1038/s41586-024-07121-9
121. Davis EJ, Martin-Liberal J, Kristeleit R, Cho DC, Blagden SP, Berthold D, et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. J Immunother Cancer. (2022) 10:e004235. doi: 10.1136/jitc-2021-004235
122. Liang S, Zheng D, Liu X, Mei X, Zhou C, Xiao C, et al. BAT6026, a novel anti-OX40 antibody with enhanced antibody dependent cellular cytotoxicity effect for cancer immunotherapy. Front Oncol. (2023) 13:1211759. doi: 10.3389/fonc.2023.1211759
123. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med. (2019) 25:759–66. doi: 10.1038/s41591-019-0420-8
124. Balmanoukian AS, Infante JR, Aljumaily R, Naing A, Chintakuntlawar AV, Rizvi NA, et al. Safety and clinical activity of MEDI1873, a novel GITR agonist, in advanced solid tumors. Clin Cancer Res. (2020) 26:6196–203. doi: 10.1158/1078-0432.CCR-20-0452
125. Davar D, Zappasodi R, Wang H, Naik GS, Sato T, Bauer T, et al. Phase IB study of GITR agonist antibody TRX518 singly and in combination with gemcitabine, pembrolizumab, or nivolumab in patients with advanced solid tumors. Clin Cancer Res. (2022) 28:3990–4002. doi: 10.1158/1078-0432.CCR-22-0339
126. Yap TA, Gainor JF, Callahan MK, Falchook GS, Pachynski RK, LoRusso P, et al. First-in-Human phase I/II ICONIC trial of the ICOS agonist vopratelimab alone and with nivolumab: ICOS-High CD4 T-Cell populations and predictors of response. Clin Cancer Res. (2022) 28:3695–708. doi: 10.1158/1078-0432.CCR-21-4256
127. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, et al. Dual inhibition of TGF-β and PD-L1: a novel approach to cancer treatment. Mol Oncol. (2022) 16:2117–34. doi: 10.1002/1878-0261.13146
128. Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in second-Line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. (2020) 15:1210–22. doi: 10.1016/j.jtho.2020.03.003
129. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. (2009) 10:1178–84. doi: 10.1038/ni.1791
130. Damo M, Joshi NS. T(reg) cell IL-10 and IL-35 exhaust CD8(+) T cells in tumors. Nat Immunol. (2019) 20:674–5. doi: 10.1038/s41590-019-0389-y
131. Yi P, Yu W, Xiong Y, Dong Y, Huang Q, Lin Y, et al. IL-35: new target for immunotherapy targeting the tumor microenvironment. Mol Cancer Ther. (2024) 23:148–58. doi: 10.1158/1535-7163.MCT-23-0242
132. Ishida T, Iida S, Akatsuka Y, Ishii T, Miyazaki M, Komatsu H, et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T-Cell leukemia/lymphoma. Clin Cancer Res. (2004) 10:7529–39. doi: 10.1158/1078-0432.CCR-04-0983
133. Hong DS, Rixe O, Chiu VK, Forde PM, Dragovich T, Lou Y, et al. Mogamulizumab in combination with nivolumab in a phase I/II study of patients with locally advanced or metastatic solid tumors. Clin Cancer Res. (2022) 28:479–88. doi: 10.1158/1078-0432.CCR-21-2781
134. Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, et al. Fc-Optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res. (2021) 81:2983–94. doi: 10.1158/0008-5472.CAN-20-3585
135. Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer. (2021) 9:e001749. doi: 10.1136/jitc-2020-001749
136. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U.S.A. (2008) 105:7797–802. doi: 10.1073/pnas.0800928105
137. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, hormone receptor-Positive advanced breast cancer. N Engl J Med. (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
138. Batalini F, Xiong N, Tayob N, Polak M, Eismann J, Cantley LC, et al. Phase 1b clinical trial with alpelisib plus olaparib for patients with advanced triple-Negative breast cancer. Clin Cancer Res. (2022) 28:1493–9. doi: 10.1158/1078-0432.CCR-21-3045
139. Layman RM, Han HS, Rugo HS, Stringer-Reasor EM, Specht JM, Dees EC, et al. Gedatolisib in combination with palbociclib and endocrine therapy in women with hormone receptor-positive, HER2-negative advanced breast cancer: results from the dose expansion groups of an open-label, phase 1b study. Lancet Oncol. (2024) 25:474–87. doi: 10.1016/S1470-2045(24)00034-2
140. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. : Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
141. Guo Y, Liu Y, Wu W, Ling D, Zhang Q, Zhao P, et al. Indoleamine 2,3-dioxygenase (Ido) inhibitors and their nanomedicines for cancer immunotherapy. Biomaterials. (2021) 276:121018. doi: 10.1016/j.biomaterials.2021.121018
142. Jung KH, LoRusso P, Burris H, Gordon M, Bang YJ, Hellmann MD, et al. Phase I study of the indoleamine 2,3-Dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid tumors. Clin Cancer Res. (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
Keywords: Treg, autoimmune diseases, cancer, immunotherapy, checkpoint inhibitors
Citation: Ge J, Yin X and Chen L (2024) Regulatory T cells: masterminds of immune equilibrium and future therapeutic innovations. Front. Immunol. 15:1457189. doi: 10.3389/fimmu.2024.1457189
Received: 30 June 2024; Accepted: 19 August 2024;
Published: 03 September 2024.
Edited by:
Claude Lambert, Centre Hospitalier Universitaire (CHU) de Saint-Étienne, FranceReviewed by:
Iris Caramalho, Instituto Gulbenkian de Ciência, PortugalMichal Kuczma, Georgia State University, United States
Copyright © 2024 Ge, Yin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lujun Chen, Y2hlbmx1anVuQHN1ZGEuZWR1LmNu