
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 October 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1454747
 William H. Robinson1*
William H. Robinson1* David Fiorentino1Lorinda Chung1,2Larry W. Moreland3
David Fiorentino1Lorinda Chung1,2Larry W. Moreland3 Malavika Deodhar4Mary Beth Harler4Carrie Saulsbery4
Malavika Deodhar4Mary Beth Harler4Carrie Saulsbery4 Rebecca Kunder4
Rebecca Kunder4B-cell depletion therapy (BCDT) has been employed to treat autoimmune disease for ~20 years. Immunoglobulin G1 (IgG1) monoclonal antibodies targeting CD20 and utilizing effector function (eg, antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis) to eliminate B cells have historically been the predominant therapeutic approaches. More recently, diverse BCDT approaches targeting a variety of B-cell surface antigens have been developed for use in hematologic malignancies, including effector-function–enhanced monoclonal antibodies, chimeric antigen receptor T-cell (CAR-T) treatment, and bispecific T-cell engagers (TCEs). The latter category of antibodies employs CD3 engagement to augment the killing of target cells. Given the improvement in B-cell depletion observed with CAR-T and TCEs compared with conventional monospecific antibodies for treatment of hematologic malignancies and the recent case reports demonstrating therapeutic benefit of CAR-T in autoimmune disease, there is potential for these mechanisms to be effective for B-cell–mediated autoimmune disease. In this review, we discuss the various BCDTs that are being developed in autoimmune diseases, describing the molecule designs, depletion mechanisms, and potential advantages and disadvantages of each approach as they pertain to safety, efficacy, and patient experience. Additionally, recent advances and strategies with TCEs are presented to help broaden understanding of the potential for bispecific antibodies to safely and effectively engage T cells for deep B-cell depletion in autoimmune diseases.
The hallmark of autoimmune disease is recognition of self-antigens by the host immune system, leading to self-reactive T cells, autoantibodies, inflammation, and tissue damage (1). For decades, T cells were viewed as the major drivers of autoimmune disease, with B cells being only minor contributors. However, in the early 2000s anecdotal observations that anti-CD20 B-cell depletion therapy (BCDT) resulted in reduced disease measures in rheumatoid arthritis (RA) and other autoimmune diseases led to studies that transformed our appreciation of the role of B cells in multiple autoimmune diseases previously presumed to be mediated by T cells, including RA, multiple sclerosis (MS), systemic lupus erythematosus (SLE), and anti-neutrophil cytoplasmic antibodies associated vasculitis (1, 2). Because not all patients respond to treatment, and some BCDTs do not appear to fully deplete B-cell subsets that are important in autoimmune pathophysiology, including tissue-resident B cells or low-expressing CD20+ cells, it is now appreciated that deeper B-cell depletion from newer therapeutic approaches may provide increased efficacy in patients with autoimmune diseases (1). Approaches that offer 1) more extensive B-cell depletion beyond that achievable through conventional effector function mechanisms and 2) favorable safety and ease of administration appropriate for an outpatient setting are of particular interest for patients with chronic autoimmune diseases.
The array of immunotherapeutic BCDT approaches include monoclonal antibodies (mAbs) that target B-cell surface antigens, such as CD19 or CD20 (which are expressed on the surface of most cells of the B-cell lineage), B-cell specific chimeric antigen receptor T-cell (CAR-T) treatment, and, more recently, B-cell targeting bispecific T-cell engagers (TCEs) (1). Based on their efficacy and safety record in cancer, various BCDT approaches are now being repurposed for use in autoimmune diseases in which B cells play a central role (2).
In this review, we discuss the various BCDTs that are approved or are being developed for use in autoimmune disease. Whereas there is an abundance of recent literature describing cell therapy approaches for autoimmune diseases (3–5), the same is not true for TCEs. Therefore, additional focus on recent advances and strategies with TCEs is presented to help build a broader understanding of the potential for bispecific antibodies to safely and effectively engage T cells for deep B-cell depletion in autoimmune diseases.
Autoimmune diseases, which result from abnormalities of the adaptive immune system, can be distinguished from autoinflammatory diseases, which are caused by hyperactivation of the innate immune system (6). B cells play an important role in a variety of autoimmune diseases (1), including major rheumatic autoimmune afflictions, such as SLE, RA, and antineutrophil cytoplasmic antibodies–associated vasculitis, as well as other autoimmune diseases, such as MS, myasthenia gravis, Graves’ disease, immunoglobulin (Ig) A nephropathy, immune thrombocytopenia, systemic sclerosis, and idiopathic inflammatory myopathies.
Both B and T cells are central components of the adaptive immune system (2). B cells are continuously generated throughout a person’s life in the bone marrow. B cells are primarily known as antibody-producing cells (7); however, in addition to antibody secretion, B cells have important functions, including antigen presentation and cytokine secretion, which can impact T cells and ultimately contribute to autoimmune pathology (2). Given their pleiotropic roles in autoimmune pathophysiology, B cells are attractive therapeutic targets for a range of autoimmune diseases.
One of the earliest surface markers targeted to deplete B cells was CD20, which plays a role in B-cell receptor signaling, antigen response, and circulating memory B-cell development (8). Although more closely associated with B cells, CD20 expression has also been observed on T cells, possibly being acquired by T cells via trogocytosis from B cells (9). Like other surface markers, CD20 expression varies depending on the stage of B-cell development (Figure 1) (1). Although the majority of B cells express CD20, expression decreases during the plasmablast differentiation stage, and CD20 is not present on the surface of plasma cells or B-cell progenitors (10).
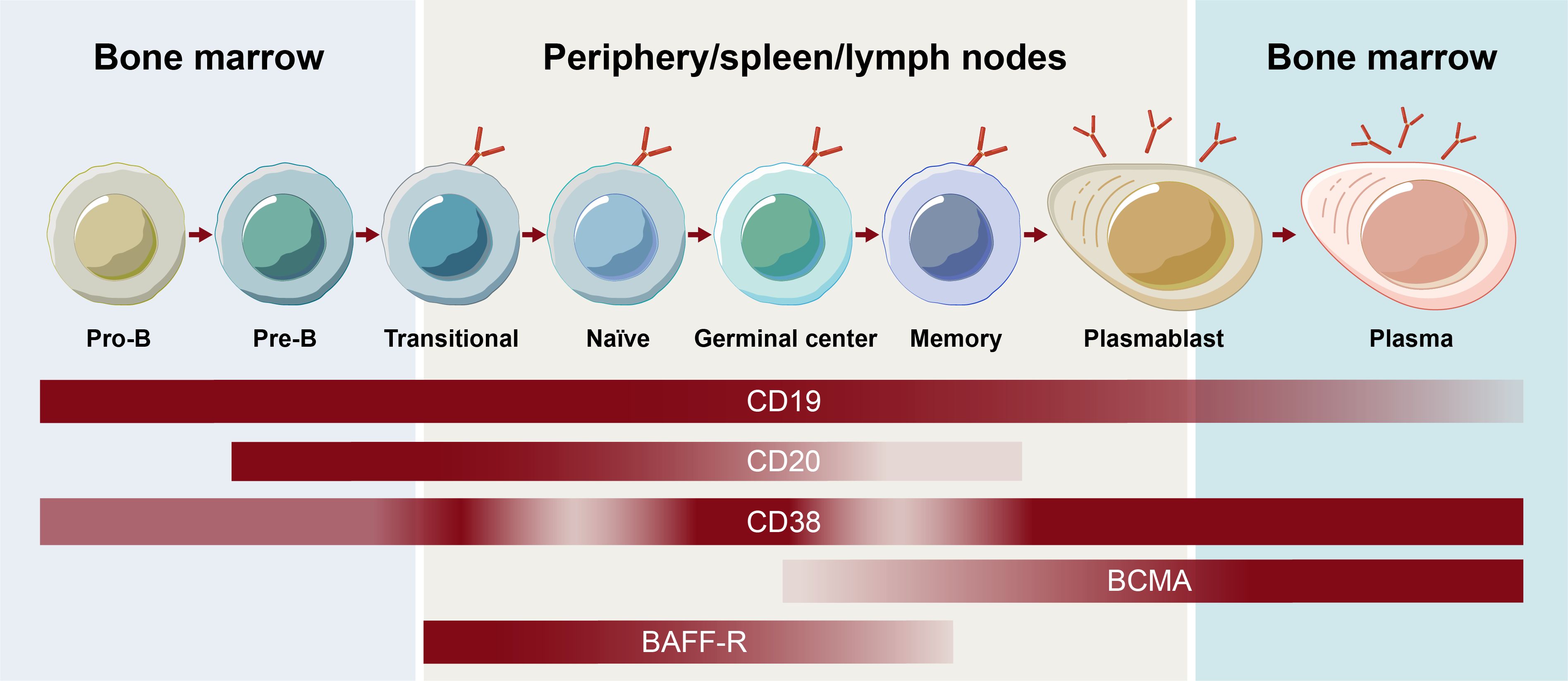
Figure 1. Receptor expression on B-cell subsets throughout B-cell development. BCMA, B-cell maturation antigen.
The contribution of the various subsets of B cells, plasmablasts, and plasma cells to autoimmune disease continues to be an active area of research. Memory B cells mature into short-lived plasmablast cells that, together with long-lived plasma cells, have been shown to be responsible for antibody secretion (2). Different surface antigens beyond CD20, including CD19, B-cell maturation antigen, CD38, and B-cell activating factor receptor, are expressed on unique subsets in the B cell lineage and play roles in the function and survival of B cells and/or plasma cells (Figure 1) (4). Assessing their effectiveness as BCDT targets continues to expand the understanding of the contribution of B-cell subsets in autoimmune disease and may ultimately allow for a more selective targeting of B-cell subsets to optimize the efficacy and safety of BCDT. Hence, an understanding of the breadth of current and emerging BCDT approaches is of high priority.
The era of BCDT began when the generation of mAbs with hybridomas became possible, leading to the creation of the first murine mAbs in 1975 (Figure 2) (10). The following decades of research demonstrated that these murine mAbs had activity against lymphoma and also later enabled the development of fully humanized antibodies. After approval of the first mAb, an anti-CD3 mAb (muromonab-CD3), for acute transplant rejection (1986), rituximab became the first anti-CD20 mAb, approved for relapsed/refractory low-grade or follicular non-Hodgkin’s lymphoma in 1997 and later for autoimmune diseases (initially for RA in 2006; Figure 2) (10).
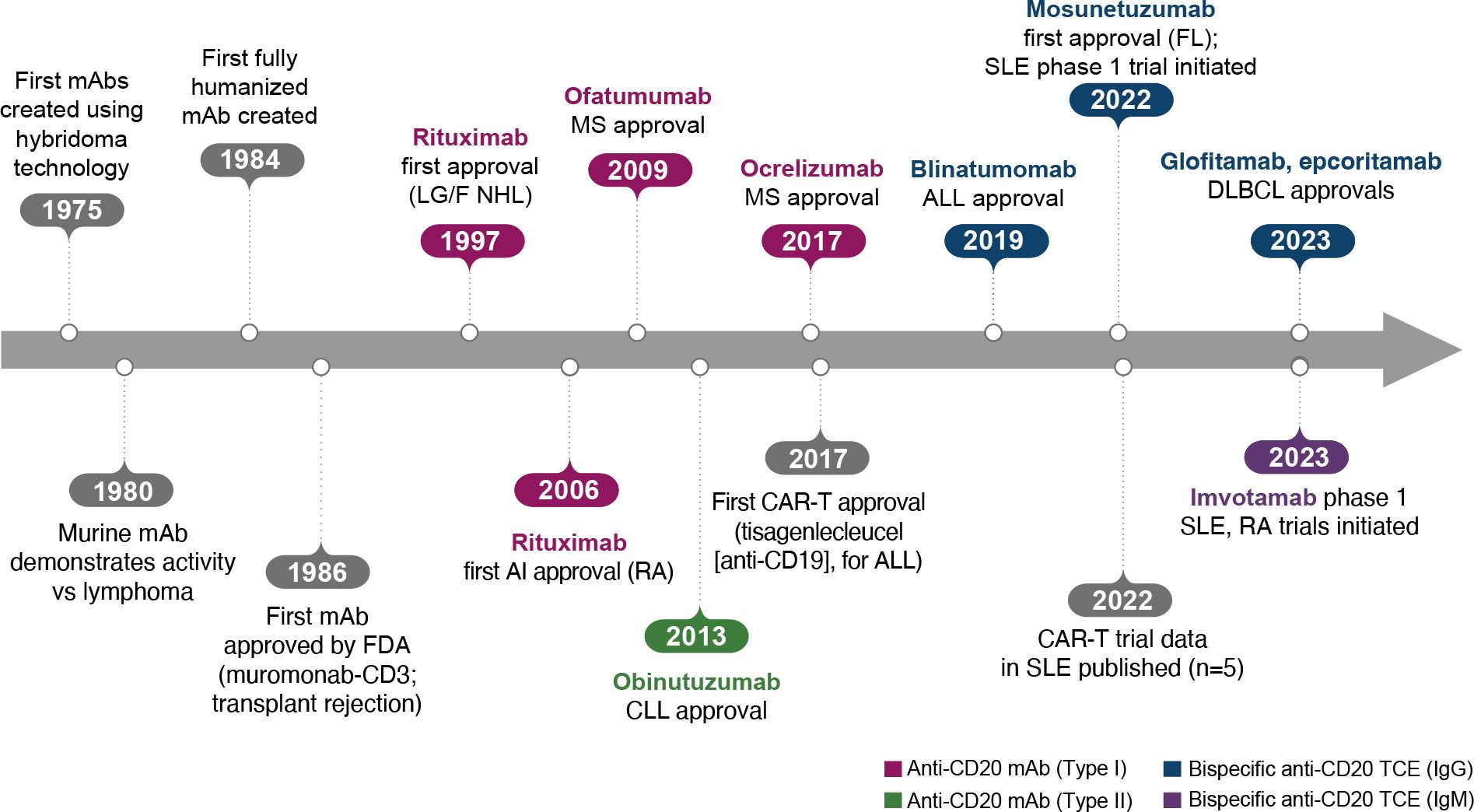
Figure 2. Key events in the BCDT timeline. ALL, acute lymphoblastic leukemia; BCDT, B-cell depletion therapy; CAR-T, chimeric antigen receptor T cell; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; FDA, US Food and Drug Administration; FL, follicular lymphoma; Ig, immunoglobulin; mAb, monoclonal antibody; LG/F NHL, relapsed/refractory low-grade or follicular non-Hodgkin’s lymphoma; MS, multiple sclerosis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; TCE, T-cell engager.
Anti-CD20 mAbs have been shown to be active across a broad range of autoimmune conditions, albeit with varying degrees of efficacy (Table 1). Existing anti-CD20 mAbs can be categorized into type I, capable of translocation/clustering of CD20 into lipid rafts, and type II, which does not have this ability (11–13). Type I mAbs can cause cell death via 2 main mechanisms: antibody-dependent cellular cytotoxicity (ADCC), via signaling through activating Fc gamma receptors (FcγRs) expressed on natural killer cells, and complement-dependent cytotoxicity (CDC), which is accentuated by lipid raft clustering and occurs via binding of C1q, initiating activation of the classical complement pathway. Type I mAbs can also induce antibody-dependent cellular phagocytosis (ADCP), which involves recognition of opsonized target cells by phagocytes and direct cell death (11, 12). Type II anti-CD20 mAbs, which do not induce clustering of CD20 into lipid rafts, are less effective at CDC, but were optimized to induce direct killing of cells through lysosome-mediated cell death. Antibody engineering to enhance effector function including ADCC and ADCP has been utilized to enhance the efficacy of anti-CD20 mAbs (11, 12, 14).
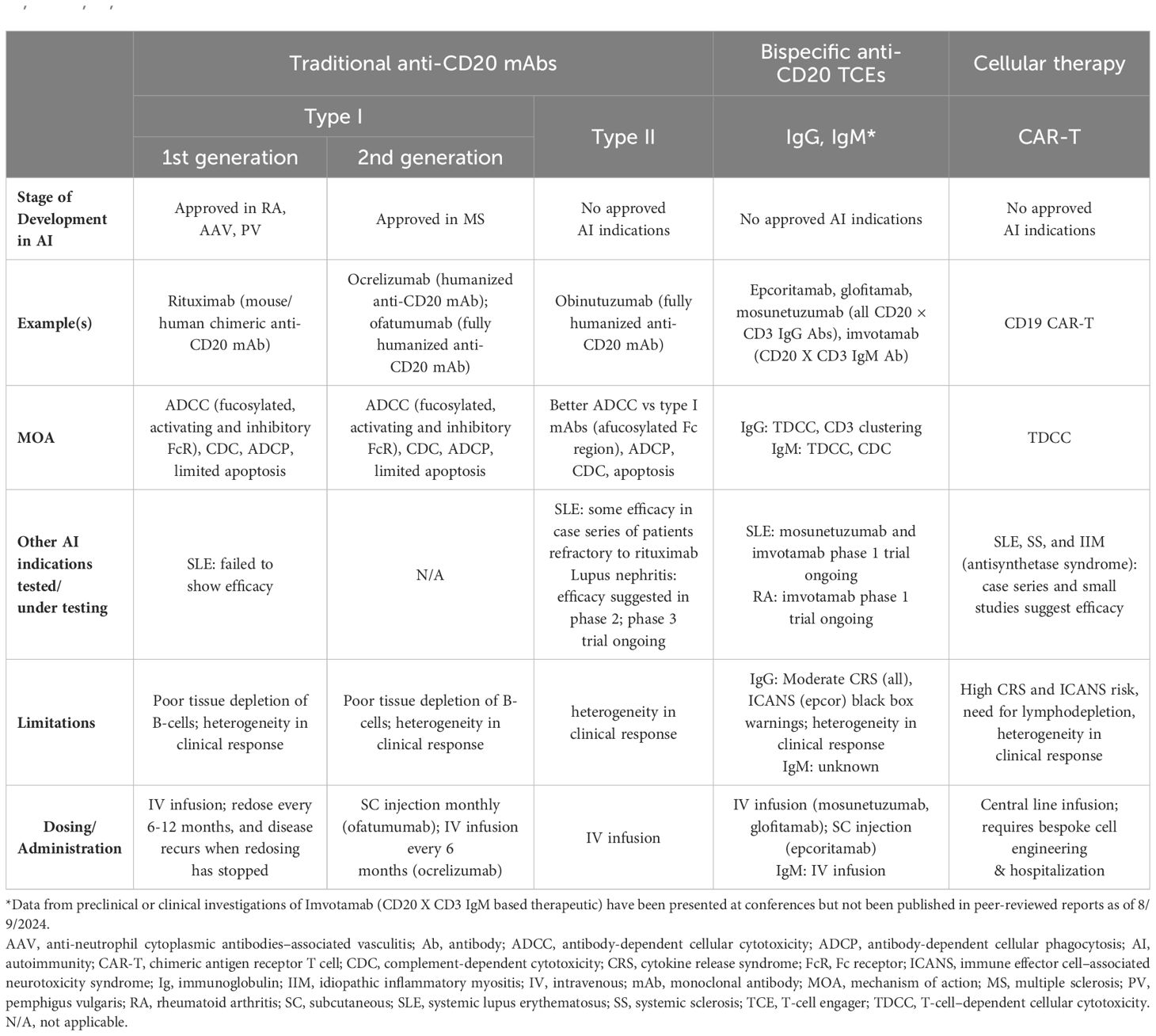
Table 1. Comparison of traditional mAbs, bispecific anti-CD20 TCEs, and CAR-T therapies (1, 2, 12, 13, 15–17, 30, 31, 33–37, 42–48, 50, 52–54, 56, 57, 60–62, 67, 68).
As a type I anti-CD20 mAb, the chimeric mouse/human rituximab relies on a combination of ADCC, CDC, ADCP, and direct cell death (10, 12, 13, 15) (Figure 3; Table 1). In addition to its use in lymphoma and leukemia, rituximab is approved for multiple autoimmune diseases (RA, granulomatosis with polyangiitis, microscopic polyangiitis, and pemphigus vulgaris) (16). However, rituximab was not found to be efficacious in patients with SLE (17) (Table 1). Even among diseases with demonstrated efficacy, not all patients respond to rituximab; anti-drug antibodies develop in approximately 30% of patients and low CD20-expressing B cells are not effectively depleted (18–21). Further, study of this type of anti-CD20 antibody in mice demonstrated the inability of effector function killing to deeply deplete B cells in the tissue (22). This may be due to insufficient effector cells and/or complement in the local tissue milieu, or the persistence of low expressing CD20+ cells, which cannot be effectively depleted through effector function due to the threshold expression requirement to activate these mechanisms. Intriguingly, in patients with RA, the lack of extensive depletion after rituximab was noted as a risk factor for relapse based on the level of circulating memory B cells (23–26). However, despite the lack of conclusive efficacy data, rituximab continues to be used in the management of SLE. It is also part of EULAR recommendations for SLE management (27), British Society of Rheumatology guideline for management of SLE (28), and American College of Rheumatology for treatment of lupus nephritis (29). To circumvent these and other limitations, next-generation type I anti-CD20 mAbs were developed, including ofatumumab (US Food and Drug Administration [FDA]–approved for MS in 2009) (30) and ocrelizumab (approved for MS in 2017; Figure 2) (31). Both ofatumumab and ocrelizumab are humanized mAbs, and as type I antibodies are capable of killing cells through the same mechanisms as rituximab, although ocrelizumab exhibits enhanced ADCC while ofatumumab is optimized for enhanced CDC (11, 12, 32). Ofatumumab is also administered subcutaneously, unlike rituximab and ocrelizumab, which are administered by intravenous infusion (16, 30, 31).
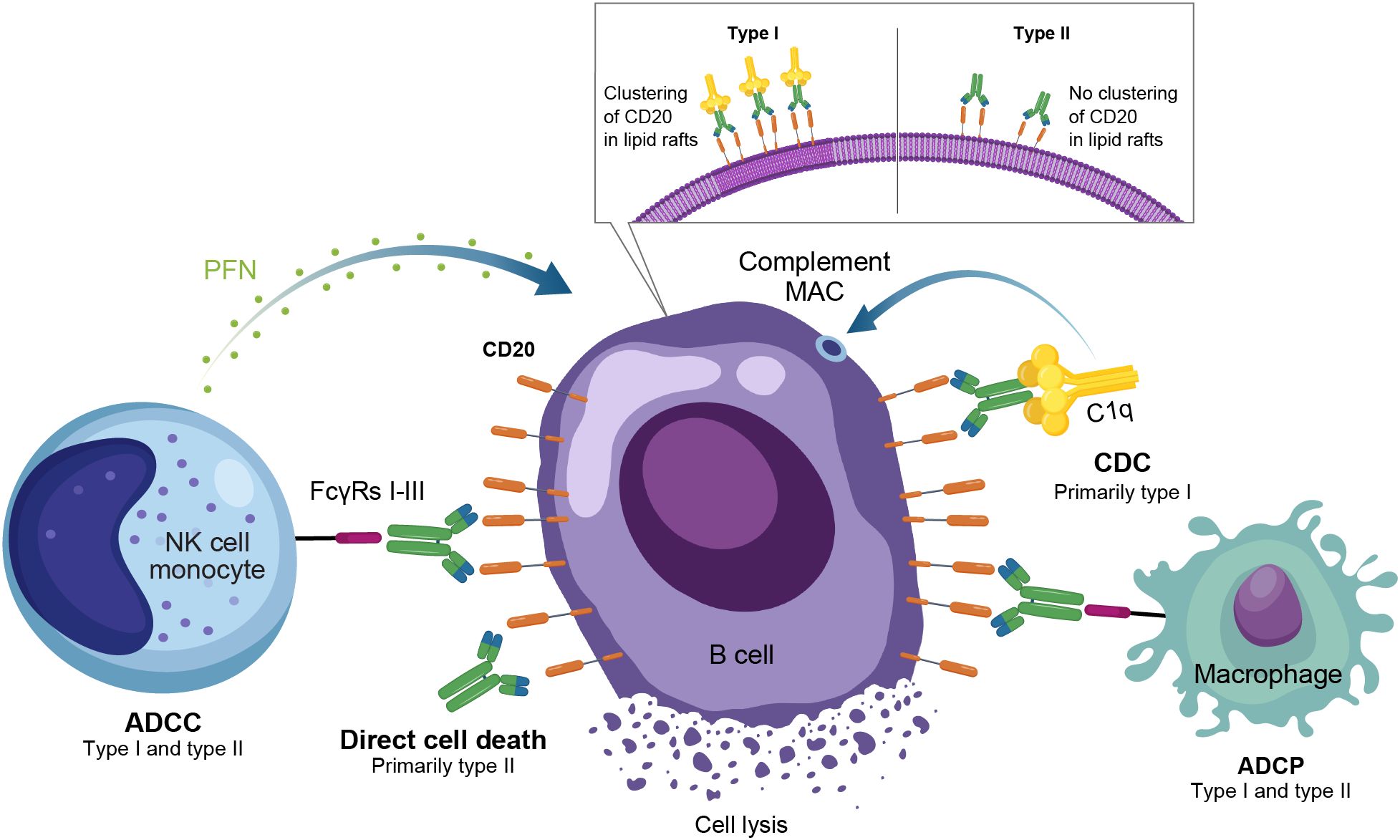
Figure 3. Overview of the MOA for anti-CD20 monoclonal antibodies in autoimmune disease. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; CDC, complement-dependent cytotoxicity; FcγR, Fc gamma receptor; MAC, membrane attack complex; MOA, mechanism of action; NK, natural killer; PFN, perforin.
To further enhance ADCC, the fully humanized type II anti-CD20 mAb obinutuzumab was glycoengineered to be afucosylated at asparagine 297 in the Fc region (Table 1) (12, 13, 33–35). Because fucosylation is known to reduce binding affinity of IgG for the FcγR, and thus ADCC, afucosylation has led to higher binding affinity for obinutuzumab to FcγRIIIa; consequently, obinutuzumab activates FcγRIII more potently and has increased ADCC compared with type I anti-CD20 mAbs (12, 13, 33–35). ADCC and ADCP are the main mechanisms through which Type II antibodies cause B-cell depletion. Obinutuzumab is indicated for patients with cancer, including chronic lymphocytic leukemia following FDA approval in 2013, and is being evaluated in autoimmune diseases (Figure 2) (36). Obinutuzumab has demonstrated efficacy in a case series of patients with SLE who were refractory to rituximab (37), and also showed improved renal responses compared with placebo and was well tolerated in a randomized, phase 2 trial in patients with lupus nephritis (38); obinutuzumab is currently being developed for both SLE and lupus nephritis. Emerging studies may help elucidate the extent to which improved anti-CD20 monoclonal IgGs can address the limitations of traditional mainstays of treatment such as rituximab.
Though CD20 antibodies remain the focus of B-cell depletion therapies, inebilizumab, an anti-CD19 approved for treatment of a subset of patients with NMSOD was also shown to cause long-lasting B-cell depletion that correlated significantly with disease outcomes in a post hoc analysis (39). It is currently also being developed in management of IgG4- related disease. Additionally, antibodies like ianalumumab which target anti-B cell activating factor (BAFF-R) are also being developed to target autoimmune diseases like SLE and Sjogrens syndrome and have shown promising safety profiles in preliminary reports (40).
Ublituximab is another CD20- targeting monoclonal antibody that was recently approved for treatment of relapsing forms of multiple sclerosis. B-cell depletion via ADCC is described as the primary mechanism of action for ubilituximab (41). Though ublituximab has been tested in hematological cancers and NMSOD, no other autoimmune conditions are currently under development.
A range of CAR-T therapies are currently being evaluated in autoimmune diseases, including both mono- and bispecific approaches, to deplete distinct populations in the B-cell lineage (Table 2). Recent advances using anti-CD19 CAR-T therapy offer early but compelling evidence for substantial therapeutic benefit through deep B-cell depletion, as reflected by treatment-free remission in a small number of patients with severe, treatment-refractory autoimmune diseases (42–48). Potential safety concerns, the need for preconditioning, hospitalization, and logistic considerations of CAR-T production and administration are areas that currently limit CAR-T from being a widely accessible therapy for broad use in autoimmunity (49).
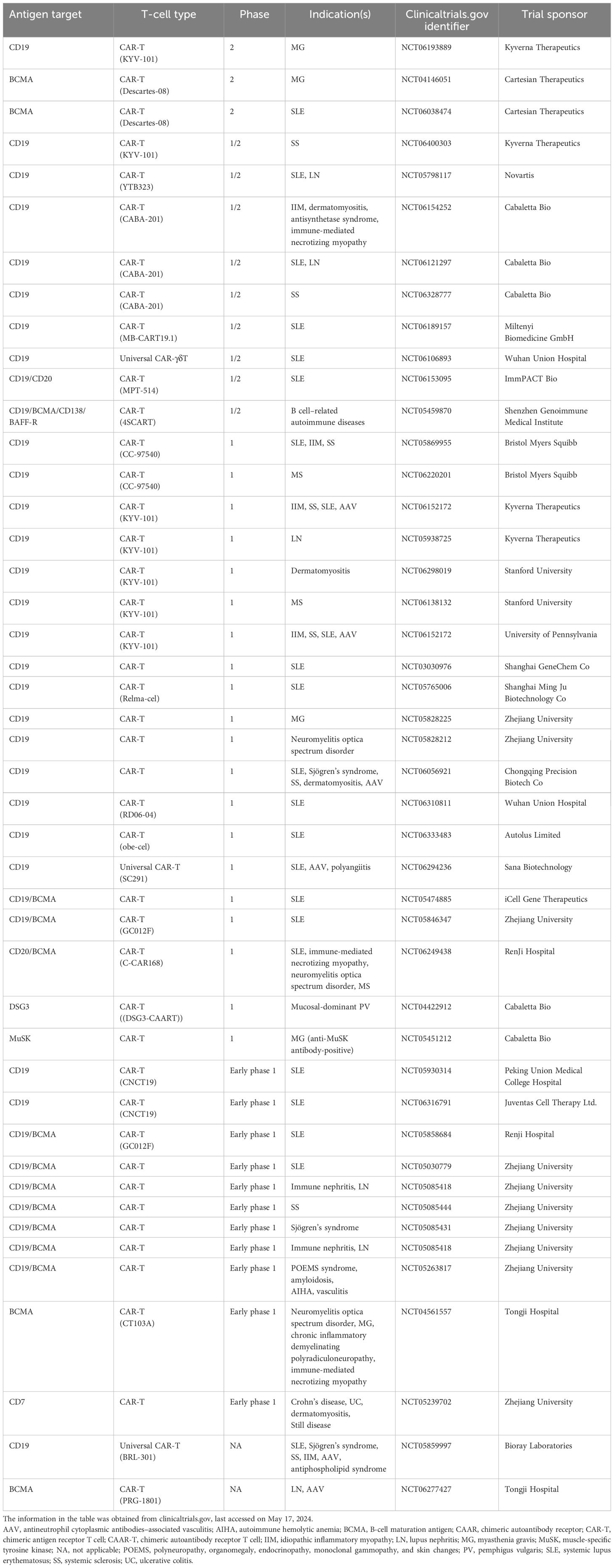
Table 2. Ongoing clinical trials with CAR-T in autoimmune diseases.
Autologous CAR-T therapy, initially developed for hematologic malignancies, requires genetic modification of an individual patient’s T cells to express specifically tailored synthetic receptors targeting cancer cells (50). Allogenic “off-the-shelf” CAR-T approaches, derived from healthy donors rather than individual patients, are also under development (51). CAR-Ts are activated through binding of the tumor-associated antigen to the chimeric antigen receptor (CAR). This results in T-cell secretion of cytokines and cytotoxins, such as granzyme and perforin, which ultimately kill the target tumor cells through T-cell dependent cellular cytotoxicity (Table 1, Figure 4) (52). The first CAR-T therapy approval in 2017 was for the CD19-targeted tisagenlecleucel in B cell precursor acute lymphoblastic leukemia (Figure 2) (53).
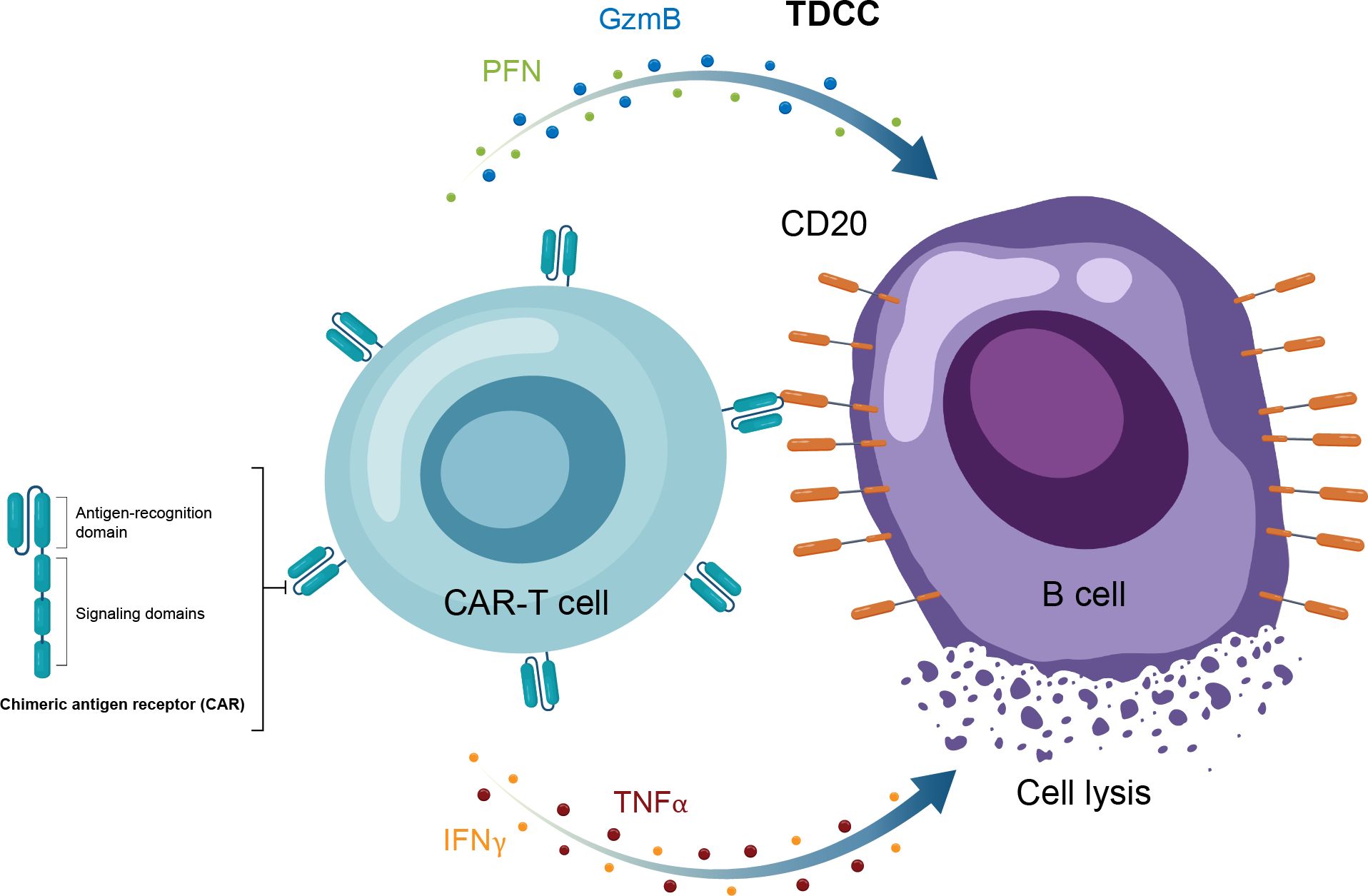
Figure 4. Overview of the MOA for CAR-T therapy in in autoimmune disease. CAR-T, chimeric antigen receptor T cell; GzmB, granzyme B; IFNγ, interferon gamma; MOA, mechanism of action; PFN, perforin; TNFα, tumor necrosis factor alpha.
Anti-CD19 CAR-T therapy in patients with various autoimmune conditions was first reported in 2021, including a small series of patients with treatment-refractory SLE, antisynthetase syndrome, and systemic sclerosis (42–48). Follow-up (median, 15 months) in these patients demonstrated impressive and durable clinical responses in the majority of patients treated with CAR-T therapy (54). These preliminary findings have resulted in initiation of a number of clinical trials focused on treatment of moderate-to-severe SLE and an array of other autoimmune diseases (Table 2).
Attendant to the promising efficacy with CAR-T therapy are several notable limitations, including a need for lymphodepletion and hospitalization, high costs of therapy, and serious safety risks. Two of the most important risks of administration are cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) (50, 53). These toxicities can often be managed with tocilizumab (with or without steroids) but can also be life-threatening. The long-term safety profile of CAR-T therapy, including the potential risk for secondary T-cell malignancy that is being investigated by the FDA (55), is yet to be fully characterized. In addition, the high financial burden of CAR-T therapies presents a significant limitation for many patients. Thus, despite compelling evidence of efficacy in a small number of patients, the safety, logistical, and cost drawbacks of CAR-T therapies have led to an unmet need for efficacious and well tolerated treatments for patients with autoimmune diseases who may require lifelong treatment. Advances in CAR-T from autologous to allogeneic cells may help to reduce some of the logistical concerns, as may other approaches, such as use of RNA instead of DNA-based CAR and the use of natural killer cells in CAR-based cell therapy.
A recent therapeutic advance in oncology, which is also being applied to autoimmune diseases, is use of bispecific immune cell engagers, which redirect effector cells to tumor cells (56, 57). T-cell engaging antibodies have binding specificity for target cells and effector T-cells, bridging the 2 by targeting specific antigens and an activating T-cell surface molecule resulting in cytotoxic cell death (58). In the context of oncology, the target cell is the tumor cell and the target antigen is a tumor-specific antigen, whereas in autoimmune disease, the target cell is a pathogenic cell expressing a target antigen or “address” that brings the T cell to that pathogenic cell to kill it. Structurally, these molecules have a binding arm directed to a tumor-associated antigen (eg, CD20) and at least 1 other arm targeting an activating receptor on an immune effector cell, with the most common approach to date being CD3 for T cells (58). For TCEs, the activated T cells then form an immune synapse with the target cells, with the TCE acting as the bridge. This leads to the release of cytokines, perforin and additional granzymes, causing target cell lysis but also carrying with it a risk of cytokine release syndrome and neurotoxicity (Figure 5) (57).
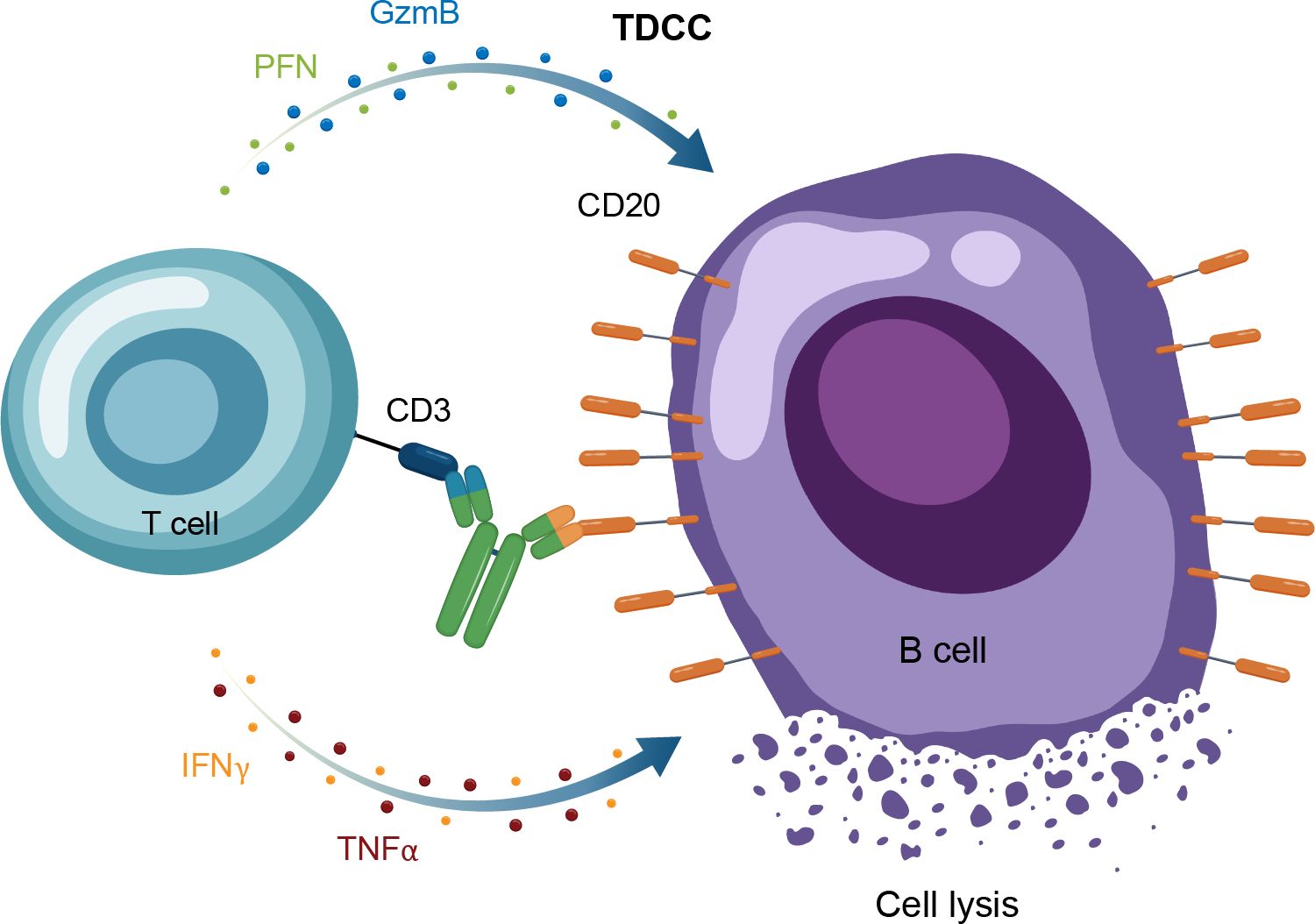
Figure 5. Overview of the MOA for IgG TCEs in autoimmune disease. GzmB, granzyme B; Ig, immunoglobulin; IFNγ, interferon gamma; MOA, mechanism of action; PFN, perforin; TDCC, T-cell dependent cellular cytotoxicity; TNFα, tumor necrosis factor alpha.
The CD19 × CD3–targeting agent blinatumomab was the first B-cell–depleting TCE approved by the FDA in 2014 (for B-cell precursor acute lymphoblastic leukemia; Figure 2) (59), and additional B-cell depleting TCEs, including those targeting CD20 × CD3, were approved thereafter (mosunetuzumab for follicular lymphoma in 2022; glofitamab, and epcoritamab for diffuse large B-cell lymphoma in 2023) (60–62). These bispecific CD20 × CD3 IgG molecules, which utilize CD3 clustering to generate strong T-cell activation and as a result T-cell dependent cellular cytotoxicity as the mechanism of action for cell killing (Table 1; Figure 5) (57), have black box warnings for CRS in their label (60–62), with instructions to utilize step-up dosing to reduce the incidence and severity of CRS. Epcoritamab also has a black box warning pertaining to ICANS risk, and is the only approved TCE that can be administered subcutaneously (61). Although these safety risks have seemingly limited the scope of use for TCEs outside of oncology, with no approved indications or available data in autoimmune diseases, a phase 1 trial with mosunetuzumab in patients with SLE was initiated in 2022, with primary data expected later this year (ClinicalTrials.gov Identifier: NCT05155345; Table 3). B-cell depletion and clinical improvement were recently reported using the CD19 × CD3 bispecific blinatumomab (administered as a continuous infusion for 5 days per cycle) to treat 6 patients with RA (2 cycles) (63) and a patient with rapidly progressive severe systemic sclerosis (4 cycles) (64).
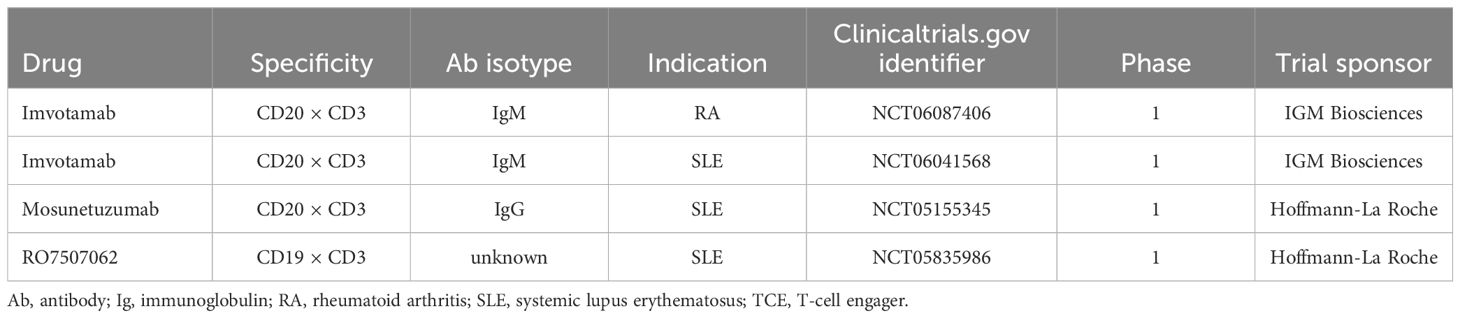
Table 3. Ongoing clinical trials with TCEs in autoimmune diseases.
Another TCE in development for use in autoimmune conditions is the CD20 × CD3 IgM molecule imvotamab (IGM-2323; Table 1). The structure of imvotamab consists of 10 high-affinity IgG CD20-binding domains attached to a pentameric IgM scaffold, with 1 anti-CD3 single-chain fragment variable attached to the J chain (65, 66). Although traditional mAbs, and approved TCEs, are typically built on an IgG framework, the IgM structure may be uniquely suited to use in a bispecific TCE due to the larger size and 10 high-affinity CD20 target binding sites in the IgM format.This results in IgM-based TCEs having high target avidity and potentially more controlled levels of T-cell activation as compared to corresponding IgG-based TCEs. Theoretically, this high avidity would enable imvotamab to cause depletion of low target-expressing cells, including activated memory B cells, using a combination of T-cell–dependent cellular cytotoxicity and CDC (Figure 6), with potentially less CD3 clustering that is exhibited by traditional IgG TCEs (Table 1). Preclinical data, as well as phase 1 data in patients with advanced B-cell malignancies, have suggested the potential for B-cell depletion in tissue and in low CD20+ expressors, without evidence of high levels of CRS and other adverse events (65, 66). Phase 1 clinical trials with imvotamab have been initiated in RA (ClinicalTrials.gov Identifier: NCT06087406) and SLE (ClinicalTrials.gov Identifier: NCT06041568), with data availability projected for 2025 for both trials (Table 3, Figure 2); an additional trial in myositis is planned. Based on the data obtained from these studies, the potential for an IgM-based TCE in patients with autoimmune diseases can be more thoroughly assessed.
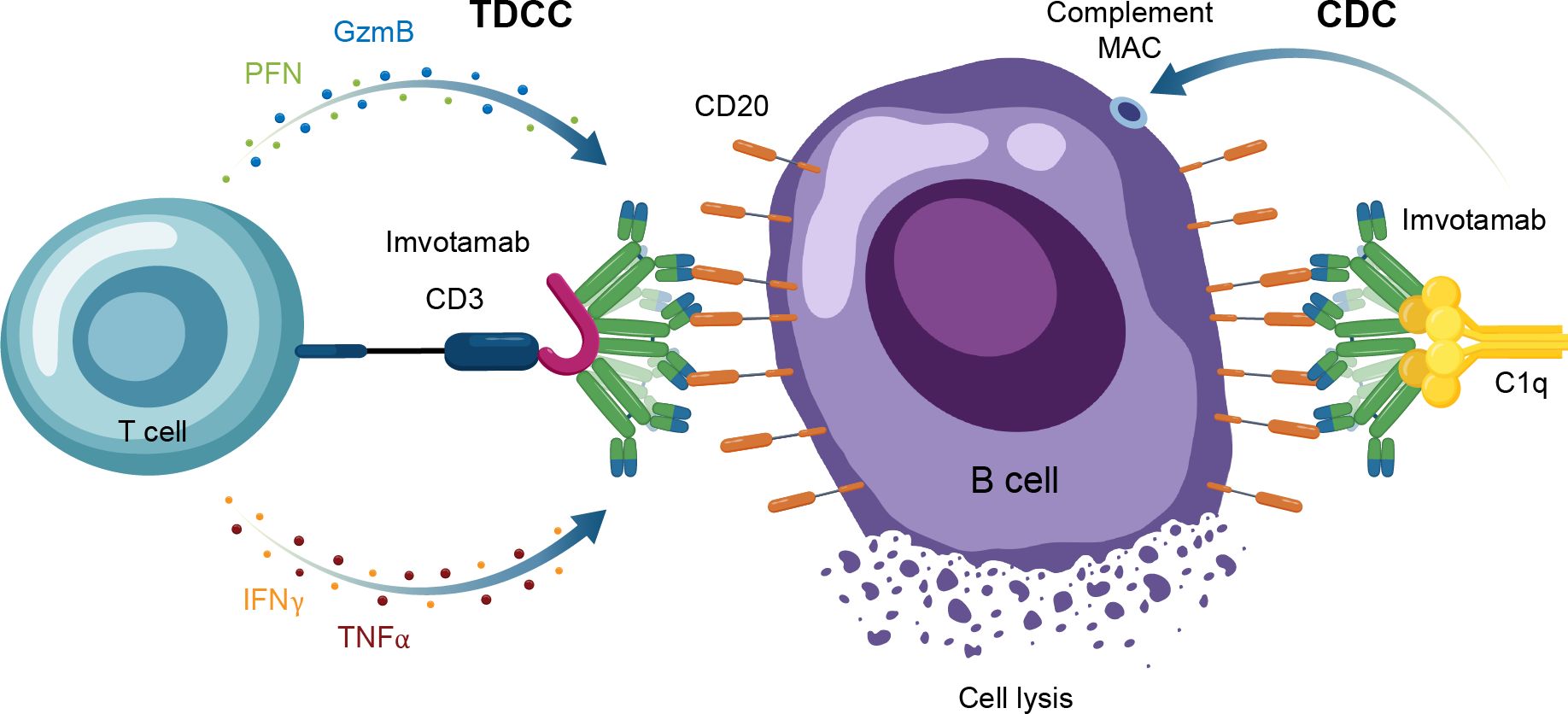
Figure 6. Overview of the MOA for IgM TCEs in autoimmune disease. CAR-T, chimeric antigen receptor T cell; CDC, complement-dependent cytotoxicity; GzmB, granzyme B; IFNγ, interferon gamma; MAC, membrane attack complex; MOA, mechanism of action; PFN, perforin; TDCC, T-cell dependent cellular cytotoxicity; TNFα, tumor necrosis factor alpha.
As the threads of the immune system are increasingly intertwining malignancy and autoimmune disease, leveraging oncology drug discovery and development for the treatment of autoimmunity is increasingly fruitful. The recent CAR-T data in patients with severe autoimmune diseases offer a view into the potential of deep BCDT. Off-the-shelf approaches, such as allogeneic cell therapy and TCEs, may have the potential to deliver better clinical outcomes than achievable with current therapies, but without many of the logistical, financial, and safety considerations associated with autologous CAR-T. Clinical studies are ongoing to characterize risks and benefits specifically in autoimmune conditions. Data from ongoing trials with TCEs in patients with autoimmune conditions (with mosunetuzuamb [SLE] and imvotamab [RA, SLE]), may shed more light on the early promise of these therapeutic agents in autoimmunity. Additionally, potential beyond repurposed TCEs from oncology exists to target other immune or nonimmune cell types (eg, fibroblasts), to utilize multispecific TCEs for tissue-specific delivery, and to engage alternative effector cells (eg, NK cells). The therapeutic landscape continues to evolve as we fully harness novel technologies to address specific cell types and pathways responsible for autoimmunity and to ultimately bring relief to those suffering from autoimmune diseases.
WR: Conceptualization, Writing – original draft, Writing – review & editing. DF: Conceptualization, Writing – review & editing. LC: Conceptualization, Writing – review & editing. LM: Conceptualization, Writing – review & editing. MD: Conceptualization, Visualization, Writing – review & editing. MH: Conceptualization, Writing – review & editing. CS: Conceptualization, Writing – review & editing. RK: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Preparation of this manuscript was funded by IGM Biosciences. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Medical writing support was provided by Chris Kirk, PhD, and Jeanne McKeon, PhD, of Lumanity Communications Inc., and was funded by IGM Biosciences.
WR has received consulting fees from IGM Biosciences. DF has received consulting fees from IGM Biosciences, Kyverna Therapeutics, Argenx, Priovant Therapeutics, Pfizer, Biogen, Bristol Meyers Squibb, and Serono. LC has received consulting fees from IGM Biosciences, Kyverna Therapeutics, Mitsubishi Tanabe, Lilly, Janssen, Mediar Therapeutics, and Genentech. LM serves as a member of independent data safety monitoring boards for Boehringer Ingelheim and Celltrion. MD, MH, CS, and RK are employees of IGM Biosciences.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AAV: antineutrophil cytoplasmic antibodies–associated vasculitis
ADCC: antibody-dependent cellular cytotoxicity
ADCP: antibody-dependent cellular phagocytosis
ANCA: anti-neutrophil cytoplasmic antibodies
AI: autoimmunity
AIHA: autoimmune hemolytic anemia
ALL: acute lymphoblastic leukemia
BCDT: B-cell depletion therapy
BCMA: B-cell maturation antigen
CAAR: chimeric autoantibody receptor
CAR: chimeric antigen receptor
CAR-T: chimeric antigen receptor
CLL: chronic lymphocytic leukemia
CDC: complement-dependent cytotoxicity
CRS: cytokine release syndrome
DLBCL: diffuse large B-cell lymphoma
FcγR: Fc gamma receptor
FDA: US Food and Drug Administration
FL: follicular lymphoma
GzmB: granzyme B
ICANS: immune effector cell–associated neurotoxicity syndrome
IFNγ: interferon gamma
Ig: immunoglobulin
IgG1: immunoglobulin G1
IIM: idiopathic inflammatory myopathy
IV: intravenous
LN: lupus nephritis
mAb: monoclonal antibody
MAC: membrane attack complex MG, myasthenia gravis
MOA: mechanism of action
MS: multiple sclerosis
MuSK: muscle-specific tyrosine kinase
NK: natural killer
NMOSD: neuromyelitis optica spectrum disorder
PFN: perforin
POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes
PV: pemphigus vulgaris
RA: rheumatoid arthritis
SC: subcutaneous
SLE: systemic lupus erythematosus
SS: systemic sclerosis
TCE: T-cell engager
TDCC: T-cell–dependent cellular cytotoxicity
TNFα: tumor necrosis factor alpha
UC: ulcerative colitis
1. Merino-Vico A, Frazzei G, van Hamburg JP, Tas SW. Targeting B cells and plasma cells in autoimmune diseases: From established treatments to novel therapeutic approaches. Eur J Immunol. (2023) 53:e2149675. doi: 10.1002/eji.202149675
2. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discovery. (2021) 20:179–99. doi: 10.1038/s41573-020-00092-2
3. Aghajanian H, Rurik JG, Epstein JA. CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab. (2022) 4:163–9. doi: 10.1038/s42255-022-00537-5
4. Zhang Z, Xu Q, Huang L. B cell depletion therapies in autoimmune diseases: monoclonal antibodies or chimeric antigen receptor-based therapy? Front Immunol. (2023) 14:1126421. doi: 10.3389/fimmu.2023.1126421
5. Blache U, Tretbar S, Koehl U, Mougiakakos D, Fricke S. CAR T cells for treating autoimmune diseases. RMD Open. (2023) 9:4. doi: 10.1136/rmdopen-2022-002907
6. Arakelyan A, Nersisyan L, Poghosyan D, Khondkaryan L, Hakobyan A, Loffler-Wirth H, et al. Autoimmunity and autoinflammation: A systems view on signaling pathway dysregulation profiles. PloS One. (2017) 12:e0187572. doi: 10.1371/journal.pone.0187572
7. Cano RLE, Lopera HDE. Introduction to T and B lymphocytes. In: Anaya JM, Shoenfeld Y, Rojas-Villarrag A, editors. Autoimmunity: from bench to bedside. El Rosario University Press, Bogota, Colombia (2013).
8. Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica. (2020) 105:1494–506. doi: 10.3324/haematol.2019.243543
9. Lee AYS. CD20(+) T cells: an emerging T cell subset in human pathology. Inflammation Res. (2022) 71:1181–9. doi: 10.1007/s00011-022-01622-x
10. Pierpont TM, Limper CB, Richards KL. Past, present, and future of rituximab-the world’s first oncology monoclonal antibody therapy. Front Oncol. (2018) 8:163. doi: 10.3389/fonc.2018.00163
11. Chan AC, Brunetta PG, Chin P. Ocrelizumab: a new generation of anti-CD20 mAb for treatment of multiple sclerosis. In: Fischer J, Klein C, Childers WE, editors. Successful drug discovery. Wiley-VCH Verlag GmbH & Co, Weinheim, Germany (2019).
12. Mariottini A, Muraro PA, Lunemann JD. Antibody-mediated cell depletion therapies in multiple sclerosis. Front Immunol. (2022) 13:953649. doi: 10.3389/fimmu.2022.953649
13. Herter S, Herting F, Mundigl O, Waldhauer I, Weinzierl T, Fauti T, et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol Cancer Ther. (2013) 12:2031–42. doi: 10.1158/1535-7163.MCT-12-1182
14. Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. (2018) 9:63–73. doi: 10.1007/s13238-017-0473-8
15. de Seze J, Maillart E, Gueguen A, Laplaud DA, Michel L, Thouvenot E, et al. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front Immunol. (2023) 14:1004795. doi: 10.3389/fimmu.2023.1004795
16. Rituxan® (rituximab) injection for intravenous use. South San Francisco, CA: Genentech, Inc (2021).
17. Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. (2010) 62:222–33. doi: 10.1002/art.27233
18. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol. (2021) 3:e489–e97. doi: 10.1016/S2665-9913(21)00107-7
19. Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. (2014) 66:740–9. doi: 10.1002/art.38270
20. Leclair V, Galindo-Feria AS, Dastmalchi M, Holmqvist M, Lundberg IE. Efficacy and safety of rituximab in anti-synthetase antibody positive and negative subjects with idiopathic inflammatory myopathy: a registry-based study. Rheumatol (Oxford). (2019) 58:1214–20. doi: 10.1093/rheumatology/key450
21. Bitoun S, Hassler S, Ternant D, Szely N, Gleizes A, Richez C, et al. Response to biologic drugs in patients with rheumatoid arthritis and antidrug antibodies. JAMA Netw Open. (2023) 6:e2323098. doi: 10.1001/jamanetworkopen.2023.23098
22. Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. (2005) 174:817–26. doi: 10.4049/jimmunol.174.2.817
23. Trouvin AP, Jacquot S, Grigioni S, Curis E, Dedreux I, Roucheux A, et al. Usefulness of monitoring of B cell depletion in rituximab-treated rheumatoid arthritis patients in order to predict clinical relapse: a prospective observational study. Clin Exp Immunol. (2015) 180:11–8. doi: 10.1111/cei.12481
24. Vos K, Thurlings RM, Wijbrandts CA, van Schaardenburg D, Gerlag DM, Tak PP. Early effects of rituximab on the synovial cell infiltrate in patients with rheumatoid arthritis. Arthritis Rheum. (2007) 56:772–8. doi: 10.1002/art.22400
25. Reddy VR, Pepper RJ, Shah K, Cambridge G, Henderson SR, Klein C, et al. Disparity in peripheral and renal B-cell depletion with rituximab in systemic lupus erythematosus: an opportunity for obinutuzumab? Rheumatol (Oxford). (2022) 61:2894–904. doi: 10.1093/rheumatology/keab827
26. Kamburova EG, Koenen HJ, Borgman KJ, ten Berge IJ, Joosten I, Hilbrands LB. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am J Transplant. (2013) 13:1503–11. doi: 10.1111/ajt.12220
27. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:736–45. doi: 10.1136/annrheumdis-2019-215089
28. Gordon C, Amissah-Arthur MB, Gayed M, Brown S, Bruce IN, D’Cruz D, et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatol (Oxford). (2018) 57:e1–e45. doi: 10.1093/rheumatology/kex286
29. Hahn BH, McMahon MA, Wilkinson A, Wallace WD, Daikh DI, Fitzgerald JD, et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). (2012) 64:797–808. doi: 10.1002/acr.21664
30. Kesimpta® (ofatumumab) injection for subcutaneous use. East Hanover, NJ: Novartis Pharmaceuticals Corporation (2024).
31. Ocrevus® (ocrelizumab) injection for intravenous use. South San Francisco, CA: Genentech, Inc (2024).
32. Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. (2004) 104:1793–800. doi: 10.1182/blood-2004-01-0039
33. Elias S, Kahlon S, Kotzur R, Kaynan N, Mandelboim O. Obinutuzumab activates FcgammaRI more potently than other anti-CD20 antibodies in chronic lymphocytic leukemia (CLL). Oncoimmunology. (2018) 7:e1428158. doi: 10.1080/2162402X.2018.1428158
34. Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. (2010) 115:4393–402. doi: 10.1182/blood-2009-06-225979
35. Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. (2018) 10:693–711. doi: 10.1080/19420862.2018.1466767
36. Gazyva® (obinutuzumab) injection for intravenous use. South San Francisco, CA: Genentech, Inc (2022).
37. Arnold J, Dass S, Twigg S, Jones CH, Rhodes B, Hewins P, et al. Efficacy and safety of obinutuzumab in systemic lupus erythematosus patients with secondary non-response to rituximab. Rheumatol (Oxford). (2022) 61:4905–9. doi: 10.1093/rheumatology/keac150
38. Furie RA, Aroca G, Cascino MD, Garg JP, Rovin BH, Alvarez A, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2022) 81:100–7. doi: 10.1136/annrheumdis-2021-220920
39. Bennett JL, Aktas O, Rees WA, Smith MA, Gunsior M, Yan L, et al. Association between B-cell depletion and attack risk in neuromyelitis optica spectrum disorder: An exploratory analysis from N-MOmentum, a double-blind, randomised, placebo-controlled, multicentre phase 2/3 trial. EBioMedicine. (2022) 86:104321. doi: 10.1016/j.ebiom.2022.104321
40. Cuker A, Al-Samkari H, Barcellini W, Cooper N, Ghanima W, Michel M, et al. Ianalumab, a novel anti-B-cell activating factor (BAFF) receptor (BAFF-R) monoclonal antibody (mAb) in development for immune thrombocytopenia (ITP) and warm autoimmune hemolytic anemia (wAIHA), has demonstrated a favorable safety profile in Sjögren’s syndrome (SjS), systemic lupus erythematosus (SLE) and chronic lymphocytic leukemia (CLL). Blood. (2023) 142:5427–8. doi: 10.1182/blood-2023-180055
42. Bergmann C, Muller F, Distler JHW, Gyorfi AH, Volkl S, Aigner M, et al. Treatment of a patient with severe systemic sclerosis (SSc) using CD19-targeted CAR T cells. Ann Rheum Dis. (2023) 82:1117–20. doi: 10.1136/ard-2023-223952
43. Mackensen A, Muller F, Mougiakakos D, Boltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
44. Merkt W, Freitag M, Claus M, Kolb P, Falcone V, Rohrich M, et al. Third-generation CD19.CAR-T cell-containing combination therapy in Scl70+ systemic sclerosis. Ann Rheum Dis. (2024). 12:4. doi: 10.1136/ard-2023-225174
45. Mougiakakos D, Kronke G, Volkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
46. Muller F, Boeltz S, Knitza J, Aigner M, Volkl S, Kharboutli S, et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. (2023) 401:815–8. doi: 10.1016/S0140-6736(23)00023-5
47. Pecher AC, Hensen L, Klein R, Schairer R, Lutz K, Atar D, et al. CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome. JAMA. (2023) 329:2154–62. doi: 10.1001/jama.2023.8753
48. Taubmann J, Knitza J, Muller F, Volkl S, Aigner M, Kleyer A, et al. Rescue therapy of antisynthetase syndrome with CD19-targeted CAR-T cells after failure of several B-cell depleting antibodies. Rheumatol (Oxford). (2024) 63:e12–e4. doi: 10.1093/rheumatology/kead330
49. Daamen AR, Lipsky PE. Potential and pitfalls of repurposing the CAR-T cell regimen for the treatment of autoimmune disease. Ann Rheum Dis. (2024) 83:696–9. doi: 10.1136/ard-2024-225638
50. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
51. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. [amp]]lsquo;Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
52. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen eceptor (CAR) T cells. Int J Mol Sci. (2019) 20:11. doi: 10.3390/ijms20061283
53. Kymriah® (tisagenlecleucel) suspension for intravenous infusion. East Hanover, NJ: Novartis Pharmaceuticals Corporation (2022).
54. Muller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Volkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - a case series with follow-up. N Engl J Med. (2024) 390:687–700. doi: 10.1056/NEJMoa2308917
55. US Food and Drug Administration. FDA investigating serious risk of T-cell Malignancy following BCMA-directed or CD19-directed autologous chimeric antigen receptor (CAR) T cell immunotherapies (2023). Available online at: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-serious-risk-t-cell-malignancy-following-bcma-directed-or-cd19-directed-autologous (Accessed March 21, 2024).
56. Tapia-Galisteo A, Alvarez-Vallina L, Sanz L. Bi- and trispecific immune cell engagers for immunotherapy of hematological Malignancies. J Hematol Oncol. (2023) 16:83. doi: 10.1186/s13045-023-01482-w
57. Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic Malignancies. J Hematol Oncol. (2021) 14:75. doi: 10.1186/s13045-021-01084-4
58. Fenis A, Demaria O, Gauthier L, Vivier E, Narni-Mancinelli E. New immune cell engagers for cancer immunotherapy. Nat Rev Immunol. (2024) 24:471–86. doi: 10.1038/s41577-023-00982-7
59. Blincyto® (blinatumomab) for injection for intravenous use. Thousand Oaks, CA: Amgen Inc (2023).
60. Columvi™ (glofitamab-gxbm) injection for intravenous use. South San Francisco, CA: Genentech, Inc (2023).
61. Epkinly™ (epcoritamab-bysp) injection for subcutaneous use. Plainsboro, NJ: Genmab US, Inc (2023).
62. Lunsumio™ (mosunetuzumab-axgb) injection for intravenous use. South San Francisco, CA: Genentech, Inc (2022).
63. Bucci L, Hagen M, Rothe T, Raimondo MG, Fagni F, Tur C, et al. Bispecific T cell engager therapy for refractory rheumatoid arthritis. Nat Med. (2024) 30:1593–601. doi: 10.1038/s41591-024-02964-1
64. Subklewe M, Magno G, Gebhardt C, Bucklein V, Szelinski F, Arevalo HJR, et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur J Cancer. (2024) 204:114071. doi: 10.1016/j.ejca.2024.114071
65. Domingo-Gonzalez R, Baribaud I, Oyasu M, Hart KC, Burns K, Pandey S, et al. Therapeutic potential of imvotamab, a CD20-targeted bispecific IgM T cell engager, for the treatment of refractory autoimmune disease patients (Poster #0583). San Diego, CA: ACR Convergence (2023).
66. Budde E, Gopal AK, Kim WS, Flinn I, Cheah CY, Nastoupil L, et al. A phase 1 dose-escalation study of IGM-2323, a novel anti-CD20 x anti-CD3 IgM T-cell engager in patients with advanced B-cell Malignancies. Blood. (2021) 138:132. doi: 10.1182/blood-2021-153355
67. Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U.S.A. (2017) 114:3485–90. doi: 10.1073/pnas.1702173114
Keywords: autoimmune disease, B-cell depletion therapy, effector function-enhanced monoclonal antibodies, imvotamab, T-cell engager
Citation: Robinson WH, Fiorentino D, Chung L, Moreland LW, Deodhar M, Harler MB, Saulsbery C and Kunder R (2024) Cutting-edge approaches to B-cell depletion in autoimmune diseases. Front. Immunol. 15:1454747. doi: 10.3389/fimmu.2024.1454747
Received: 25 June 2024; Accepted: 13 September 2024;
Published: 09 October 2024.
Edited by:
Marko Radic, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Gregg Joshua Silverman, New York University, United StatesCopyright © 2024 Robinson, Fiorentino, Chung, Moreland, Deodhar, Harler, Saulsbery and Kunder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William H. Robinson, d3JvYmluc0BzdGFuZm9yZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.