
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 02 October 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1453030
This article is part of the Research TopicCommunity Series in the Role of Complement in Health and Disease: Volume IIView all 16 articles
 Yingjin Kong1,2†
Yingjin Kong1,2† Naixin Wang1,2†
Naixin Wang1,2† Zhonghua Tong1,2
Zhonghua Tong1,2 Dongni Wang1,2
Dongni Wang1,2 Penghe Wang1,2
Penghe Wang1,2 Qiannan Yang1,2Xiangyu Yan1,2
Qiannan Yang1,2Xiangyu Yan1,2 Weijun Song1,2
Weijun Song1,2 Zexi Jin1,2
Zexi Jin1,2 Maomao Zhang1,2*
Maomao Zhang1,2*In the genesis and progression of cardiovascular and metabolic diseases (CVMDs), adipose tissue plays a pivotal and dual role. Complement factor D (CFD, also known as adipsin), which is mainly produced by adipocytes, is the rate-limiting enzyme of the alternative pathway. Abnormalities in CFD generation or function lead to aberrant immune responses and energy metabolism. A large number of studies have revealed that CFD is associated with CVMDs. Herein, we will review the current studies on the function and mechanism of CFD in CVMDs such as hypertension, coronary heart disease, ischemia/reperfusion injury, heart failure, arrhythmia, aortic aneurysm, obesity, insulin resistance, and diabetic cardiomyopathy.
Cardiovascular and metabolic diseases (CVMDs) constitute the principal causes of death globally. Cardiovascular diseases (CVDs) constitute more than two-thirds of the deaths associated with a high body mass index (BMI) (1). The pathogenesis of these diseases remains incompletely clarified. Although researchers have made great progress in the prevention and treatment of CVMDs, the morbidity, disability and mortality rates remain high.
The development and prognosis of CVMDS are closely related to adipose tissue. For each 5 kg/m2 increase in BMI, the risks of CVDs will increase varied from 10% to 49% (2). Adipose tissue is an energy storage organ as well as an active and inflammatory organ capable of releasing adipokines (3). Adipokines modulate lipid metabolism, glucose metabolism, inflammation, and blood pressure. CFD is a serine protease that is produced mainly by adipose tissue and macrophages. CFD is an essential rate-limiting enzyme in complement alternative pathway (AP). As an adipokine, CFD regulates metabolism by increasing adipocyte differentiation and lipid accumulation, protecting beta-cells, and promoting insulin secretion. As a complement factor, CFD amplifies complement cascade activation and protects cells from infection, while it induces low-grade inflammation (LGI) in CVDs.
Mounting evidence suggests that CFD plays a significant role in CVMDS by regulating immune system homeostasis and metabolic balance. CFD levels are implicated in the etiology of several CVMDs. In a large population-based cohort, participants with higher CFD have a greater adverse cardiovascular profiles (4). CFD is positively associated with CVDs incidence. For each 1 standard deviation increase in the plasma concentration of CFD, the incidence of CVDs is 15% greater (5). Increasing CFD levels are correlated with obesity, hypertension, LGI, and endothelial dysfunction (6). However, CFD often acts as a protective factor in diabetes and diabetic cardiomyopathy.
Given the controversial role of CFD in different contexts of cardiovascular system immunity and metabolism (Figure 1), summarizing the specific effects of CFD on CVMDs is crucial for further research. In this review, we summarize the progress of CFD in CVMDs in recent years.
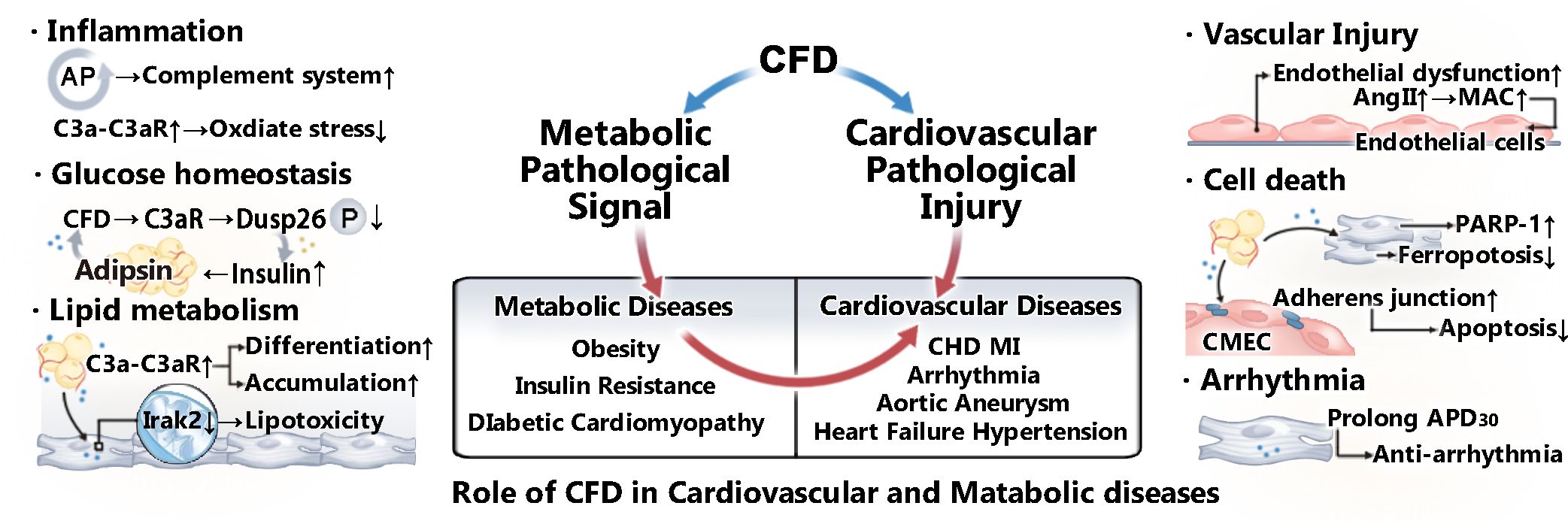
Figure 1. Role of CFD in CVMDs. CFD plays a role in various cardiovascular and metabolic diseases by regulating metabolic pathological signals and mediating cardiovascular pathological injury. CFD is closely related to the activation of the complement system. In addition, as an adipokine, it plays a vital role in lipid metabolism and glucose homeostasis. Moreover, studies have shown that CFD is associated with vascular injury, apoptosis, and arrhythmia.
CFD is a differentiation-dependent serine protease with a molecular weight of 24.4 kDa. Two variants of immature human CFD exist: variant 1, which is composed of 251 amino acids with an 18-amino acid leader sequence, and variant 2, which is composed of 260 amino acids with a 25-amino acid leader sequence (7). Apart from the pro-peptide, the two variants have the same polypeptide chain and form the same mature pro-CFD. As an inactive zymogen, pro-CFD consists of 235 amino acids and matures after the cleavage of a 6-amino acid peptide. Activated human CFD consists of a single serine protease domain of 228 amino acids with a catalytic triad (7). This process seems to occur during or immediately after secretion (8). Over 99% of plasma CFD exists in the form of CFD instead of the pro-CFD form (9). Human CFD is not glycosylated. In contrast, mouse CFD is highly glycosylated, possessing an molecular mass of approximately 42 and 45 kDa and being more stable in circulation (7, 10).
CFD circulates in a self-inhibited form and possesses relatively low proteolytic activity in blood (11). CFD has a narrow substrate specificity. CFD only binds to an open complement factor B (CFB) conformation in its Mg2+-dependent complex with C3b, triggering reversible conformational changes in the catalytic center of CFD (12). After the cleavage of CFB, CFD returns to an inactive resting-state conformation in circulation (13).
CFD is primarily and constitutively secreted by adipocytes, is also produced by macrophages and monocytes, and is synthesized by a small number of brain astrocytes (14). Among all the complement components, CFD has the lowest level in blood, ranging from 1 to 2 μg/mL, with no difference between healthy men and women (15). The CFD level in heart tissue depends on its serum level, whereas heart tissue expresses CFD mRNA at relatively low levels. Encircling the heart and blood vessels, epicardial adipose tissue (EAT) and pericardial adipose tissue (PVAT) synthesize CFD and deliver it to the heart via exosomes (16, 17). CFD is filtered through the glomerulus and then reabsorbed and decomposed by the renal tubules (7). It is estimated that the fractional catabolic rate of plasma CFD is 60% per hour, resulting in a lower serum concentration under nonpathological conditions (18). However, serum CFD levels can be influenced by renal dysfunction (19). CFD plasma levels increase approximately tenfold in patients with end-stage renal failure (20). However, CFD levels increase in the urine and decrease in the plasma of patients with Fanconi syndrome because of selective impairments in the renal tubular epithelium (21).
The role of CFD in protecting cells from infection has been well recognized. CFD deficiency causes insufficient terminal C5b-9 complement complex (TCC) generation, which ultimately increases the risk of meningococcal infection approximately 6000-fold (22). In families with CFD deficiency, family members show low AP activity and have meningitis and pneumonia (22, 23). CFD is also involved in other physiological processes. In previous studies, evidence indicating a central role of CFD in energy homeostasis and systemic metabolism has been put forward. CFD promotes the differentiation of adipocytes and the accumulation of lipids by activating the C3a-C3aR pathways in adipocytes and preadipocytes (24–26). In addition, CFD promotes the generation of acylation-stimulating protein (ASP) by catalyzing the conversion of C3 to C3a. As a C3 cleavage product of exopeptidase activity, ASP regulates lipid storage by increasing the activity of diacylglycerol O-acyltransferase 2 (27). CFD, which is upregulated by proliferator-activated receptor gamma (PPARγ) acetylation, inhibits Wnt/β-catenin signaling and thus primes bone marrow mesenchymal stem cell adipogenic differentiation in mice (28). As a diurnal factor, CFD plays an adipose-autonomous role in metabolic rhythms. In mouse adipose tissue, the expression of CFD exhibits a rhythm pattern, with a peak at zeitgeber time (zt) 12 and a trough at zt0 (29). No fluctuations in CFD are observed in the plasma (29). PPARγ acetylation upregulates CFD transcription, which destabilizes BMAL1 and mediates metabolic rhythms (29). CFD also plays a role in glucose metabolism. Several studies have indicated that CFD promotes insulin secretion and sustains β-cell function via the C3a-C3aR1 pathway (30).
CFD contributes to paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), geographic atrophy (GA), and various diseases (Table 1). Patients afflicted with PNH and aHUS exhibit excessive activation of the AP. CFD inhibitors decrease C3 fragment deposition on PNH erythrocytes and abate complement-mediated hemolysis (31). Complement dysregulation is a key component of the pathogenesis of systemic lupus erythematosus (SLE). MBL-associated serine protease (MASP)3 is likely a direct activator of pro-CFD in resting human blood, cleaving inactive pro-CFD into active CFD (32). Compared with healthy individuals, SLE patients exhibit increased MASP-1 and MASP-3 levels. In addition, CFD, CFB and MASP1/3 are deposited along the glomerulus in patients with lupus nephritis (LN), which demonstrates that AP is involved in the pathogenesis of LN (33).
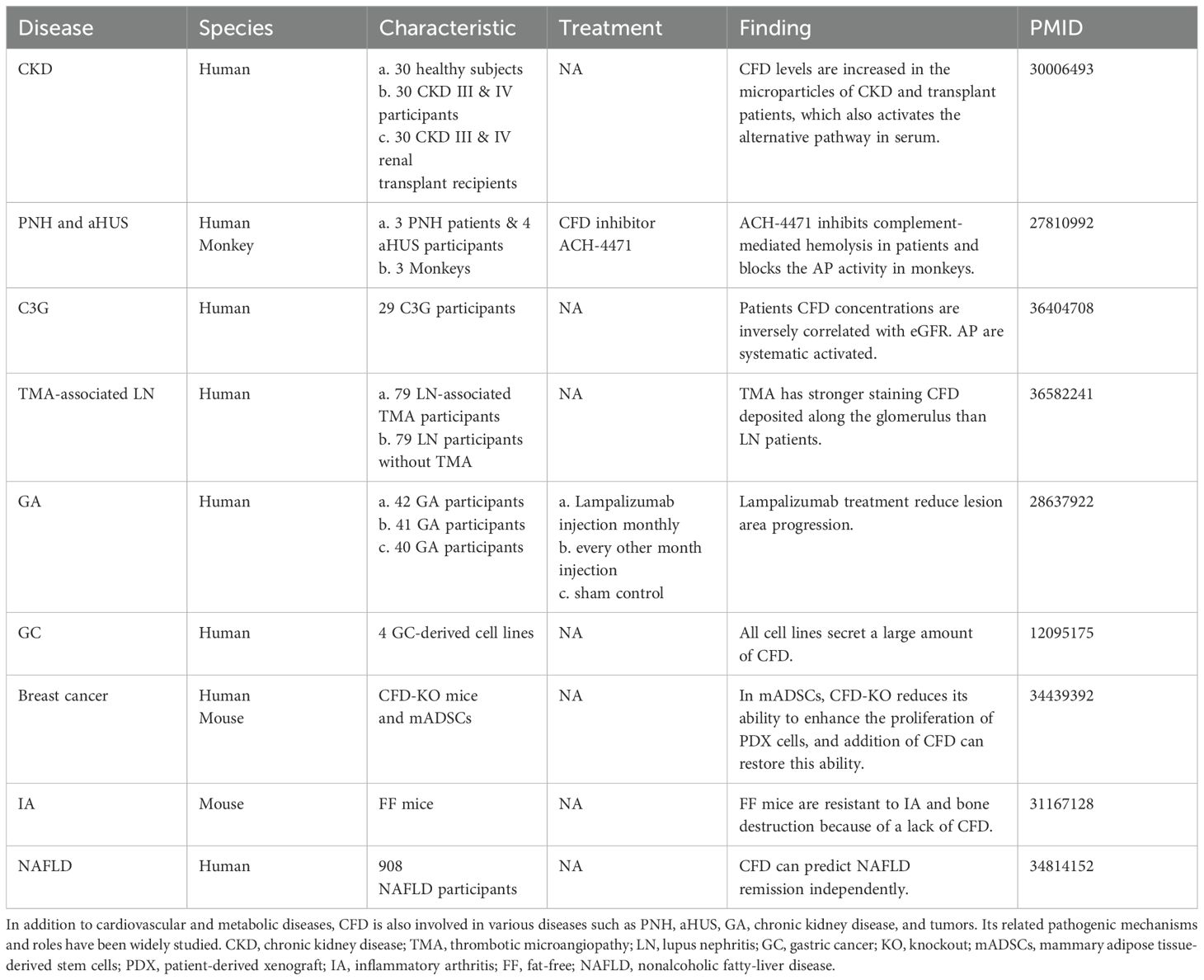
Table 1. Summary of the main studies investigating the effects of CFD in multiple diseases.
CFD, together with MASP-1, C3, and C5, participates in the hemostatic response. While playing a protective role in preventing bleeding, it also exacerbates thrombotic and inflammatory diseases. While research has shown that platelets secrete CFD and activate AP (34), research has shown that CFD deficiency in mice can prevent thrombosis, and the inhibition of CFD can reduce platelet activation during vascular injury (35). A decrease in CFD in heparinized human whole blood can inhibit platelet activation in a simulated extracorporeal circulation circuit (36).
CFD plays an important role in CVMDs through various pathological mechanisms (Figure 2), which we will discuss in detail later.
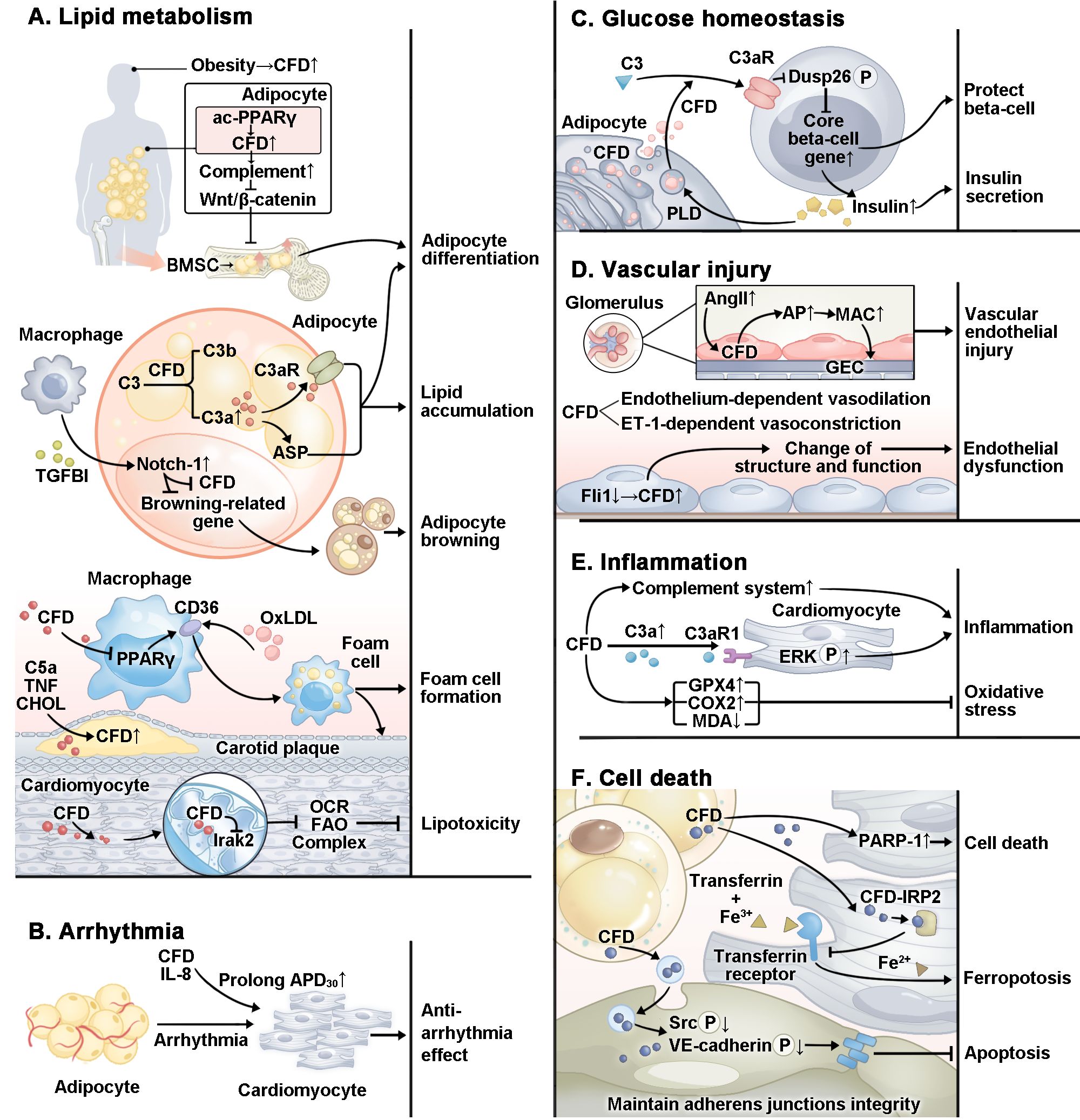
Figure 2. Specific mechanism by which CFD plays a role in CVMDs. In CVMDs, CFD plays a complex role. (A) CFD regulates adipocyte differentiation and lipid accumulation through multiple pathways. CFD promotes foam cell formation. However, in cardiomyocytes, CFD inhibits lipotoxicity. (B) CFD plays an antiarrhythmic role by prolonging APD30. (C) CFD protects beta-cells and promotes insulin secretion. (D) CFD is related to vascular endothelial injury and endothelial dysfunction. (E) CFD plays a pro-inflammatory role through the complement system and multiple pathways. In addition, studies have reported that CFD inhibits oxidative stress. (F) CFD protects cardiomyocyte against ferroptosis and maintains the integrity of cardiac microvascular endothelial cell adherens junctions. However, CFD activates PARP-1 and causes cardiomyocytes apoptosis. ac, acetylation; PPARγ, proliferator-activated receptor gamma; TGFBI, transforming growth factor-beta; OxLDL, oxidized low-density lipoprotein; TNF, tumor necrosis factor; CHOL, cholesterol; Irak2, interleukin-1 receptor-associated kinase 2; OCR, oxygen consumption rate; FAO, fatty acid oxidation; IL-8, interleukin 8; APD, action potential duration; Dusp26, dual specificity phosphatase 26; P, phosphorylation; PLD, phospholipase D; AngII, angiotensin II; GEC, glomerular endothelial cell; Fli1, friend leukemia virus integration 1; ERK, extracellular signal-regulated kinase; GPX4, glutathione peroxidase 4; COX2, cyclooxygenase-2; MDA, malondialdehyde; PARP-1, poly ADP-ribosepolymerase-1; IRP2, iron regulatory protein 2; Src, proto-oncogene tyrosine-protein kinase; VE, vascular endothelial.
Complement system activation involves three pathways: the classical pathway (CP), the lectin pathway (LP), and the AP (37). The activation of the CP is driven mainly by antigen-antibody complexes as well as C-reactive protein and other factors (38). LP is triggered by specific microbial polysaccharides. AP precedes CP and LP and is slowly self-activated by hydrolysis (38).
The three pathways mentioned above initiate complement activation, which converges on the common core component C3. C3 is cleaved into C3a and C3b by C3(H2O)Bb, which initiates signal amplification (39). CFD plays a promoting role in the generation of C3(H2O)Bb. In addition, initial pathways activation results in C5 convertase formation, which causes C5 to be cleaved into C5a and C5b. Moreover, the cleavage of C5 results in a labile conformation of C5b, which then binds successively to C6, C7, C8, and multiple copies of C9 to form the TCC, which is also called as the membrane attack complex (MAC) (37). The MAC can directly create transmembrane pores on cells and bacteria, leading to target swelling and eventual lysis (37).
Since complement cascade activation is a rapidly amplifying proinflammatory response, a variety of proteins that inhibit this reaction tightly regulate complement system activation in multiple ways, such as by modulating convertase activity and controlling the formation of the MAC. Complement factor I and complement factor H can limit the activity of C3 convertase and bind to C3b, preventing novel convertase formation (40).
AP activation starts with the slow spontaneous hydrolysis of C3 to form C3(H2O). MASP-3 is the exclusive activator of pro-CFD in resting human blood (41), promoting the proteolytic processing of pro-CFD to CFD (42). CFD cleaves C3(H2O) combined with CFB to the AP liquid phase C3 convertase C3(H2O)Bb. Through catalysis, C3 is cleaved into C3a and C3b. AP generates the majority of C3b (38). C3b later binds to a surface and recruits CFB to generate the C3bB complex. Subsequently, CFD, which binds to C3bB, becomes transiently active and cleaves CFB into Ba and Bb fragments, generating the AP cell surface C3 convertase C3bBb, which further promotes the generation of C3b. C3b binds to pathogens and acts as an opsonin to enhance phagocytosis. Additionally, when C3bBb accumulates and its local concentration reaches a certain level, C3b may bind to C3bBb, resulting in the generation of the C5 convertase C3bBb3b (43), which catalyzes downstream cascade reactions, resulting in the formation of MAC and lysis of the pathogen. Moreover, properdin combines with C3bBb to form C3bBbproperdin. This process stabilizes C3bBb, increasing its half-life by 5- to 10-fold (44).
In a variety of diseases, complement system activation leads to inflammatory damage, such as PNH and aHUS. Researchers have reported that it regulates the onset and development of ischemia/reperfusion (I/R) injury in recent years. Complement activation occurs early in acute myocardial infarction (AMI) (45). The elevation of C3d and sC5b9 occurs prior to the classical markers of myocardial necrosis in AMI patients (45).
AP has been regarded as a dual system (46). It both takes on a recognition function comparable to that of CP and LP and carries out an amplification function in the complement system. Since CP and LP activation generates C4bC2a, which forms C3b, triggering signal propagation via the amplification loop, AP amplification is a common feature of any kind of initial complement activation (46). AP amplification is responsible for approximately 80% of CP-induced complement terminal activation (47).
CFD is a pivotal rate-limiting enzyme in AP (Figure 3). The concentration of CFD is closely related to the activity and output of the AP and downstream amplification loop (7). The cleavage of CFB into Ba and Bb fragments by CFD is regarded as the rate-limiting stage for the generation of the C3 convertase C3(H2O)Bb. An increase in the CFD concentration enhances complement cascade activity. A lack of CFD has been shown to lead to inactive AP (22). More importantly, complement activation initiated by CP and LP can be amplified through the AP. C3b deposited on the cell surface by CP can serve as a site for AP C3 convertase formation (46). Under the catalysis of CFD, more C3bBb is produced and cleaves C3 to form more C3b deposited on the cell surface. Therefore, CFD amplifies the output of all three pathways through a positive feedback mechanism (48).
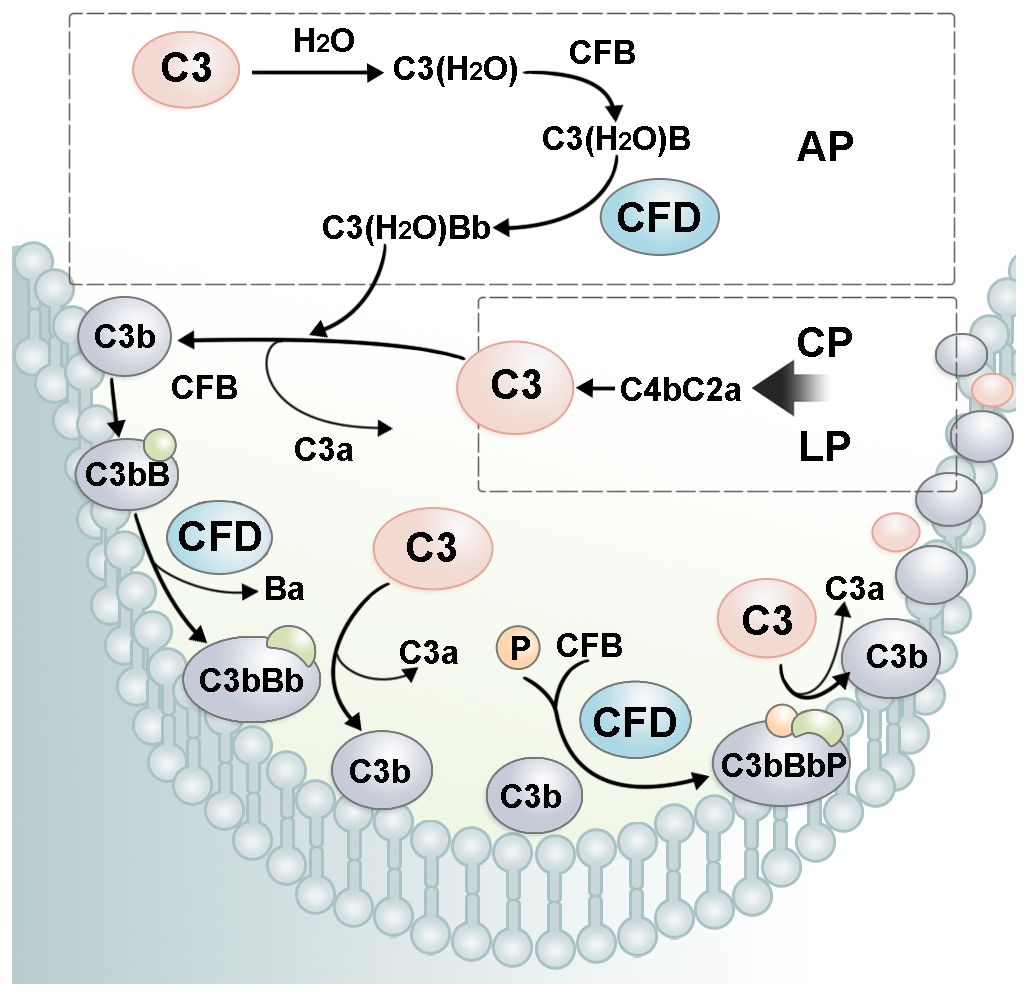
Figure 3. The role of CFD in the complement amplification loop. In AP, CFD cleaves C3(H2O)B into the alternative pathway fluid phase C3 convertase, C3(H2O)Bb. C3(H2O)Bb cleaves C3 into C3a and C3b. C3b recruits CFB on the cell surface. After cleavage by CFD, alternative pathway cell-surface C3 convertase, C3bBb, is formed. C3bBb cleaves C3 and forms more C3b, leading to a rapid amplification of complement cascade activity. P, properdin.
The findings summarized above emphasize the role of CFD in the complement system. We will briefly summarize the evidence that suggests possible roles of CFD (Figure 4) in each disease in the following text.
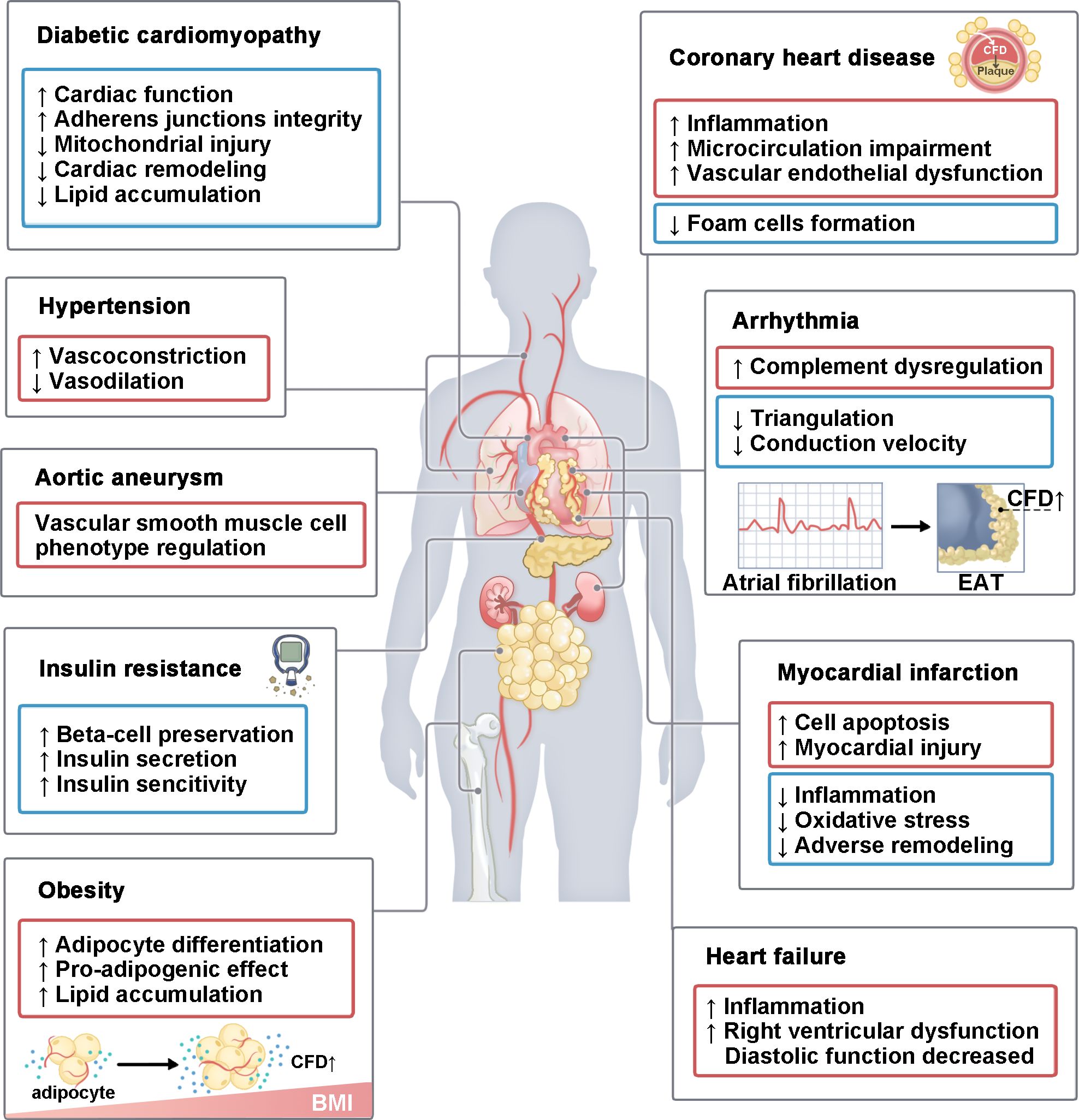
Figure 4. The role of CFD in various CVMDs. CFD plays a role in various CVMDs. It mainly includes diabetic cardiomyopathy, hypertension, aortic aneurysm, insulin resistance, obesity, coronary heart disease, arrhythmia, I/R injury and heart failure. This process involves multiple organs, including the heart, blood vessels, lungs, pancreas, kidneys, bones, and adipose tissue. In some diseases, CFD has both pathogenic and protective effects. BMI, body mass index; EAT, epicardial adipose tissue.
Many studies have verified the vital role of immune cytokines in hypertension and end-organ damage. Triggered by lifestyle factors, infections and autoimmunity, both chronic initiation and adaptive immune overactivation may result in high blood pressure and tissue damage (49). According to the Maastricht Study (4), participants with higher CFD are characterized by higher blood pressure. As an endogenous vascular elastase, CFD mediates the onset and development of pulmonary hypertension (PAH) (50). Moreover, CFD contributes to an obesity-related decrease in endothelium-dependent vasodilation and endothelin-1-dependent vasoconstriction (51).
As an autoimmune disease, systemic sclerosis (SSc) can lead to varying degrees of involvement in connective tissues, which can cause PAH in patients. The progression of PAH is related to elevated serine elastase activity (52). In patients with systemic sclerosis, CFD is increased in serum and is positively correlated with right ventricular systolic blood pressure and PAH. Patients with elevated CFD levels are at higher risk for PAH-related cardiac dysfunction (53). Importantly, high CFD levels are more specific to PAH than to brain natriuretic peptide levels. Mechanistically, progressive PAH is often associated with abnormalities in the structure and function of vascular and endothelial cells. In SSc, Friend leukemia virus integration 1 deficiency upregulates CFD in human dermal microvascular endothelial cells (19).
Innate immune response activation occurs when the embryo attaches to the uterus. Recent investigations have demonstrated that complement system dysregulation mediates the onset of preeclampsia, which is mainly characterized by gestational hypertension. CFD concentrations are reportedly increased in patients with preeclampsia during late gestation (54, 55). The concentration of complement terminal complexes in the placenta of preeclamptic women is higher than that in the placenta of normal women.
CFD is a risk factor for coronary heart disease (CHD) in postmenopausal women (56). According to a cross-sectional study among 288 women, serum CFD levels are elevated in patients with polycystic ovary syndrome and are influenced by BMI, homeostatic model assessment for insulin resistance and high-sensitivity C-reactive protein (57). Another study showed that CFD can serve as an independent predictor of carotid intima-media thickness (CIMT) (57). Furthermore, CFD may be correlated with the instability of atherosclerotic plaques in patients with CHD (58). A study conducted among 370 patients undergoing diagnostic coronary angiography demonstrated that serum CFD levels have the potential to predict future incidences of AMI and all-cause death in patients with coronary artery diseases (58). CFD is highly expressed in coronary atherosclerotic plaques and adventitial adipocyte tissue (58). This reflects the potential role CFD plays in the development of atherosclerosis.
CFD plays a pathogenic role in CHD through multiple pathways. First, EAT can release proinflammatory factors directly into the coronary lumen because it is close to myocardiocytes. Previous studies have demonstrated that circulating CFD is exposed to the vascular endothelial layer and is correlated with systemic AP activation, which is correlated with vascular endothelial dysfunction (59). Moreover, pericoronary adipose tissue is bidirectionally linked to endothelial dysfunction (60). In response to angiotensin II stimulation, CFD is overly expressed in glomerular endothelial cells and activates local complement, resulting in the formation and deposition of the MAC complex on endothelial cells and promoting vascular endothelial injury (61). Then, complement induces endothelial cells to release inflammatory cytokines, which may subsequently promote vascular dysfunction (23). An increasing level of CFD is related to low-grade inflammation and endothelial dysfunction (6). Coronary angiography assessments using optical coherence tomography have shown that serum CFD levels can partially predict thin-cap fibroatheromas, which are inflammatory plaques that are prone to cause acute coronary syndrome (62). In addition, atherosclerosis is usually associated with abnormal lipid metabolism, such as increased intracellular cholesterol uptake and decreased excretion. CFD is expressed in the aortic endothelium (63). The expression of CFD, C1q alpha and beta, C2, and C3 is increased in atherosclerotic carotid plaques from acute coronary syndrome patients primed with C5a and tumor necrosis factor followed by cholesterol crystals (64).
Overall, the majority of studies show a promoting effect of CFD on atherosclerosis development. However, some studies have suggested a negative correlation between CFD and arterial injury. In 483 obese adult subjects, circulating CFD concentrations are reduced in individuals with CIMT and asymptomatic carotid atherosclerosis (65). Moreover, the overexpression of CFD inhibits the development of atherosclerosis in mice (66). Mechanistically, CFD downregulates the PPARγ/CD36 pathway, suppresses oxidized low-density lipoprotein-induced lipid uptake by macrophages, and prevents the formation of foam cells (66). Therefore, more in-depth investigations are still required to determine the specific mechanism of CFD in the onset and progression of CHD.
I/R injury can occur during myocardial infarction (MI) or cardiovascular surgery. AMI can lead to the production and secretion of CFD, and the serum CFD level of patients with AMI is upregulated at admission (17). At the 1-month follow-up, this difference in CFD levels between AMI patients and the control group disappeared (17). Among MI rats, the CFD expression levels in the EAT are markedly greater than those in subcutaneous adipose tissue. Inhibiting CFD activity can alleviate myocardial injury after MI (67). CFD derived from EAT induces cell apoptosis through excessive activation of poly ADP-ribose polymerase-1 in H9c2 cells (67).
However, studies have also suggested that CFD has a cardioprotective effect during MI injury by reducing inflammation, alleviating oxidative stress, and improving adverse remodeling. Open heart surgery may induce I/R injury. In 32 patients who received elective on-pump coronary artery bypass grafting with cardiopulmonary bypass, plasma CFD declined after surgery and decreased to the lowest level (approximately 30% below the baseline values) at 24 hours after surgery (68). There is an inverse correlation between CFD and IL-6 levels in samples collected at the start of cardiopulmonary bypass and 1 minute after removal of the aortic cross-clamp (68). In one study, CFD overexpression significantly increased the survival rate, preserved the left ventricular ejection fraction, and alleviated myocardial pathological damage in mice after MI surgery (17). CFD alleviates lipid oxidative stress after MI by decreasing cyclooxygenase-2 expression and increasing glutathione peroxidase 4 levels (17). Further observations noted that the CFD treatment of neonatal mouse ventricular myocytes altered the expression of proteins related to ferroptosis and iron metabolism (17). Mechanistically, CFD interacts with IRP2 to regulate iron homeostasis by upregulating the iron storage-related protein ferritin heavy chain and downregulating the transferrin receptor, thereby protecting cardiomyocytes against ferroptosis and reducing cardiac remodeling after MI (17).
Experimental and clinical evidence points to inflammatory mediators as a major cause of the development of heart failure (HF), as they can lead to maladaptive processes within the myocardium (39). Uncontrolled complement activation can cause widespread inflammation and tissue damage in HF. C3 and CFD are highly abundant in the right ventricle (69). In HF patients, the plasma concentration of CFD is elevated, especially in those with New York Heart Association functional class III and IV. The concentrations of CFD and terminal complement complexes are associated with the N-terminal pro-B-type natriuretic peptide and C-reactive protein concentrations (70). Increased CFD levels are also correlated with measures of inflammatory response and deteriorated diastolic function (70). In line with this, increased CFD is closely associated with left ventricular dysfunction (p < 0.001), right ventricular dysfunction, and a higher frequency of diastolic dysfunction in patients with systemic sclerosis (53). In conclusion, the concentration of CFD is correlated with the disease severity of patients with HF.
Recently, research has demonstrated that the C3-CFD-C3aR axis plays a crucial role in right ventricular failure. C3a-C3aR1 activated extracellular signal-regulated kinase and upregulated HF- and inflammation-related pathways in rat cardiomyocytes. In mouse models of right ventricular failure, both C3 knockout and CFD knockout attenuate ventricular dysfunction (69).
EAT accumulates and expands in obese and insulin-resistant states. In addition to functioning as an energy reserve system, epicardial fat determines the local inflammatory milieu of the heart via the release of multiple inflammatory and anti-inflammatory adipokines. The expansion of EAT can be proarrhythmic, resulting in hypoxia, inflammatory cytokine release, and the infiltration of fat into the myocardium. Epicardial fat increases the occurrence of atrial arrhythmias (71). Gene expression analysis revealed that the CFD gene was highly abundant in the EAT of patients with atrial fibrillation (AF) (72).
However, another study noted that the expression of CFD is downregulated in patients with AF-related cardiogenic embolism stroke (AF-CE) (73). Although CFD is associated with neutrophils, it is negatively correlated with naive B cells, naive CD4+ T cells, and resting NK cells. Bioinformatic analysis and experiments further support that CFD can potentially serve as a diagnostic blood biomarker for AF-CE (73). Thus, the decrease in CFD may imply complement dysregulation, which predicts the risk of AF-CE.
In direct coculture, white fat-like adipocytes (hAdip) increase triangulation, prolong action potential duration, and slow the conduction velocity of human-induced pluripotent stem cells to ventricular cardiomyocytes (74). The presence of MCP-1, IL-6, IL-8, and CFD, which are correlated with the immune response and inflammation, increased in the hAdip-conditioned medium. However, CFD and IL-8 can decrease triangulation and shorten conduction velocity through increasing action potential duration 30 alone (74).
Abdominal aortic aneurysm is correlated with vascular smooth muscle cell apoptosis and inflammation, and chronic inflammatory reactions activate the AP and may lead to the progression of atherosclerotic aortic aneurysm aortic wall remodeling. CFD levels are elevated in patients with thoracic aortic aneurysms and abdominal aortic aneurysms (75, 76), suggesting that CFD may promote vascular smooth muscle cell phenotype regulation in the onset of aneurysms.
The expansion of adipose tissue is often accompanied by the accumulation of immune cells. Emerging evidence shows that obesity is correlated with chronic LGI. There is a positive correlation between C3 and various indicators of body fat (77). Studies have shown that the concentrations of CFD are correlated with the likelihood of obesity (78). In overweight/obese women with a BMI≥25 kg/m2, circulating CFD levels are higher than those in subjects with a BMI<25 kg/m2 (57). An increasing number of studies have shown that BMI is positively correlated with CFD (79). Placental macrophages Hofbauer cells secrete CFD. More CFD was released from the placenta of obese mothers than from that of lean mothers (27). Circulating fetal CFD is positively correlated with maternal BMI and maternal insulin resistance (27).
There is still controversy about the type of adiposity associated with CFD. On the one hand, a cross-sectional study suggested that CFD and leptin concentrations are positively associated with BMI and visceral fat mass in patients with cardiometabolic disorders (79). In adults with growth hormone deficiency (AGHD), serum CFD is correlated with total body fat, visceral fat, and insulin resistance. After growth hormone treatment and appropriate lipid-lowering therapy, CFD levels significantly decrease in visceral adipose tissue (80). In contrast, a study has shown that CFD is related to subcutaneous and intrathoracic adipose volumes yet shows no connection with visceral adiposity (78).
In contrast, some studies have found the opposite effect on CFD. Plasma CFD levels are significantly downregulated in high-fat diet (HFD)-fed mice (81) but are slightly increased in isocaloric HFD-fed mice (82). Intriguingly, it has also been reported that CFD levels rise slightly within 48 hours after HFD feeding in mice but then decrease below baseline levels after 6 weeks after initiation of a HFD (83). Plasma proteome analysis has shown that preventative exercise can only partially reverse the decrease in plasma CFD caused by HFD overconsumption (82). Exercise inhibits the CFD-secreted phosphoprotein 1 signaling pathway in diet-induced obese male mice (84). Promoting the browning of white adipose tissue is considered a potential strategy for protecting against obesity. Transforming growth factor-beta, which is derived from macrophages, suppresses CFD mRNA and browning-related gene expression in adipocytes by activating Notch-1 signaling (81).
CFD plays an important role in promoting insulin secretion, regulating blood glucose levels and improving insulin sensitivity. In plasma, the level of CFD is decreased in patients with type 2 diabetes mellitus (T2DM) (85). Compared to T2DM patients, T2DM patients with β-cell failure exhibit decreased levels of CFD mRNA in visceral and subcutaneous adipose tissues (30). Animals with genetically absent CFD have glucose intolerance because of insulinopenia (30). In diabetic mice, CFD preserves beta cells by catalyzing C3a formation and downregulating dual specificity phosphatase 26 expression in β cells (78). Furthermore, CFD activates C3a-C3aR1 to promote insulin secretion (30). In addition, insulin promotes CFD secretion from adipocytes by stimulating phospholipase D activity (86).
Diabetic cardiomyopathy causes fatty acid and lipid accumulation in cardiomyocytes, resulting in lipotoxicity and triggering progressive structural and functional remodeling of the heart. In diabetic cardiomyopathy mice, CFD levels in heart tissue, plasma, white adipose tissue, and brown adipose tissue are significantly decreased (87). Overexpression of CFD relieves cardiac remodeling and improves cardiac function both in mice and rats with diabetic cardiomyopathy (87, 88).
CFD alleviates diabetes-related coronary microvascular complications (87). Specifically, CFD maintains the integrity of cardiac microvascular endothelial cell adherens junctions by downregulating tyrosine-protein kinase Csk and inhibiting VE-cadherin phosphorylation, thereby protecting the microvasculature against hyperpermeability (87). The abnormal processing of mitochondrial calcium and decreased level of free matrix calcium lead to a decrease in cardiomyocyte function. Recent research has revealed that CFD binds to interleukin-1 receptor-associated kinase 2 (Irak2) and inhibits its mitochondrial translocation, downregulating the interaction of Irak2 with prohibition-optic atrophy protein 1 on mitochondria in cardiomyocytes of mice treated with DMC. Inhibition of the Irak2 signaling pathway improves fatty acid oxidation, protects mitochondrial structure and function, and alleviates lipid accumulation (88).
The central role of CFD in AP activation and the critical role of CFD in complement cascade amplification make this protein an effective pharmaceutical target for complement-related diseases. As an inhibitor targeting the proximal part of AP, CFD inhibitor selectively block AP activation, impede C3 cleavage, and limit signal amplification of the CP and LP without any effect on the initiation or terminal function of these two complement pathways (8). Since the CP and terminal pathway mediate the bacterial lysis, CFD inhibition potentially reduces the risk of infections compared with C5 blockade or even C3 blockade (31). Previous studies have shown that terminal complement deficiencies patients have increased risk of infection with Neisseria species compared to early complement pathway component deficiencies (89). Additionally, C5a participate in the activation of leukocytes to phagocytose pathogens (90). Moreover, inhibiting CFD does not block C3b-mediated opsonization or C3a-mediated inflammation. Thus, CFD inhibitor modulate complement activation while keep “protective” complement activation.
Although CFD inhibitor have the advantage of oral administration, its short half-life requires 2–3 administrations per day (91). In addition, since that low levels of CFD are sufficient for AP activation in serum, complete inhibition of CFD may be vital for therapeutic efficacy (8). Compared to C5 inhibitors targeting downstream pathways, CFD inhibitors cannot fully block complement activation (91). C3 activation by other plasma protease, bypassing factor D, has been described in specific clinical circumstances (89). As a part of the plasma contact activation system, kallikrein cleaves C3 to C3b and C3a (89). These fundings suggest that a combined treatment with C5 inhibitors and CFD inhibitors may achieve better clinical response.
At present, a number of CFD inhibitors have been designed and quite a few of them have entered clinical trials. Benefited by its distinct mechanism, CFD inhibitors may achieve more profound clinical benefits in the future.
Danicopan (ALXN2040) is an oral CFD inhibitor that is mainly used as an add-on therapy in patients with PNH (NCT04469465). Danicopan improved hemoglobin concentrations with no serious adverse events reported in the study (92). Additionally, Danicopan is currently in a phase II clinical trial for patients with GA (NCT05019521). Furthermore, BCX9930 is another oral CFD inhibitor used in phase II clinical trials for the treatment of PNH (NCT04702568) and C3 glomerulopathy (NCT05162066).
There are few anti-CFD antibodies currently in clinical trials. Lanpazumab is a humanized monoclonal antibody targeting the antigen-binding fragment of CFD (93). In a cardiopulmonary bypass simulation, anti-CFD monoclonal antibodies inhibited the formation of inflammatory factors and the activation of neutrophils and platelets (36). Anti-CFD antibodies significantly inhibited the formation of alternative, lectin, and soluble TCCs in SARS-CoV-2-infected monkeys (94). In addition, CFD antibody treatment reduced the levels of proinflammatory cytokines, chemokines, and the plasma endothelial lesion marker plasminogen activator inhibitor-1 and improved coagulation abnormalities (94).
This review has shown the clear function of CFD in a range of CVMDs and identifies the role of CFD as a biomarker in these diseases. Current research shows that it has multiple effects on the cardiovascular system. Although CFD promotes endothelial dysfunction and atherosclerosis development and mediates ventricular diastolic dysfunction, it alleviates oxidative stress and improves adverse remodeling. Furthermore, regarding metabolic diseases, CFD promotes adipose tissue accumulation, controls blood glucose levels, improves insulin sensitivity and relieves diabetic cardiomyopathy. Thus, CFD may mediate interweaving immune responses and energy metabolism.
A large number of studies have confirmed the association between CFD and CVMDs. Many articles have pointed out the predictive value and biomarker role of CFD in CVMDs. However, the role of CFD in each CVMDS remains elusive. More research is required to clarify the effect of CFD on CVMDs and to confirm the specific mechanism by which CFD regulates CVMDs. Understanding the specific mechanism of CFD in the onset and development of diseases is an important knowledge base for discoveries about the diagnostic value and clinical treatment of CFD in CVMDS. Furthermore, researchers may adopt different methods to assess the safety, toxicology profile, and therapeutic effect of CFD-targeting medicine on CVMDS in animal models and human patients.
Overall, CFD is a prospective biomarker and therapeutic target for CVMDs.
YK: Conceptualization, Investigation, Visualization, Writing – original draft. NW: Conceptualization, Investigation, Writing – review & editing. ZT: Investigation, Writing – review & editing. DW: Investigation, Writing – review & editing. PW: Investigation, Writing – review & editing. QY: Investigation, Writing – review & editing. XY: Investigation, Writing – review & editing. WS: Investigation, Writing – review & editing. ZJ: Investigation, Writing – review & editing. MZ: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from National Natural Science Foundation of China (82270525 to MZ) and Natural Science Foundation of Heilongjiang Province (GA23C006 to MZ).
We would like to express our appreciation to everyone involved in drafting and preparing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Collaborators GBDO, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
2. Kim MS, Kim WJ, Khera AV, Kim JY, Yon DK, Lee SW, et al. Association between adiposity and cardiovascular outcomes: an umbrella review and meta-analysis of observational and Mendelian randomization studies. Eur Heart J. (2021) 42:3388–403. doi: 10.1093/eurheartj/ehab454
3. Francisco V, Pino J, Gonzalez-Gay MA, Mera A, Lago F, Gomez R, et al. Adipokines and inflammation: is it a question of weight? Br J Pharmacol. (2018) 175:1569–79. doi: 10.1111/bph.14181
4. Hertle E, Arts IC, van der Kallen CJ, Feskens EJ, Schalkwijk CG, Stehouwer CD, et al. The alternative complement pathway is longitudinally associated with adverse cardiovascular outcomes. The CODAM study. Thromb Haemost. (2016) 115:446–57. doi: 10.1160/TH15-05-0439
5. Jin S, Eussen S, Schalkwijk CG, Stehouwer CDA, van Greevenbroek MMJ. Plasma factor D is cross-sectionally associated with low-grade inflammation, endothelial dysfunction and cardiovascular disease: The Maastricht study. Atherosclerosis. (2023) 377:60–7. doi: 10.1016/j.atherosclerosis.2023.06.079
6. Luc G, Empana JP, Morange P, Juhan-Vague I, Arveiler D, Ferrieres J, et al. Adipocytokines and the risk of coronary heart disease in healthy middle aged men: the PRIME Study. Int J Obes (Lond). (2010) 34:118–26. doi: 10.1038/ijo.2009.204
8. Barratt J, Weitz I. Complement factor D as a strategic target for regulating the alternative complement pathway. Front Immunol. (2021) 12:712572. doi: 10.3389/fimmu.2021.712572
9. Stanton CM, Yates JR, den Hollander AI, Seddon JM, Swaroop A, Stambolian D, et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2011) 52:8828–34. doi: 10.1167/iovs.11-7933
10. Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, et al. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. J Exp Med. (2010) 207:29–37. doi: 10.1084/jem.20090633
11. Forneris F, Burnley BT, Gros P. Ensemble refinement shows conformational flexibility in crystal structures of human complement factor D. Acta Crystallogr D Biol Crystallogr. (2014) 70:733–43. doi: 10.1107/S1399004713032549
12. Gavriilaki E, Papakonstantinou A, Agrios KA. Novel insights into factor D inhibition. Int J Mol Sci. (2022) 23:7216. doi: 10.3390/ijms23137216
13. Volanakis JE, Narayana SV. Complement factor D, a novel serine protease. Protein Sci. (1996) 5:553–64. doi: 10.1002/pro.5560050401
14. White RT, Damm D, Hancock N, Rosen BS, Lowell BB, Usher P, et al. Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J Biol Chem. (1992) 267:9210–3. doi: 10.1016/S0021-9258(19)50409-4
15. Milek M, Moulla Y, Kern M, Stroh C, Dietrich A, Schon MR, et al. Adipsin serum concentrations and adipose tissue expression in people with obesity and type 2 diabetes. Int J Mol Sci. (2022) 23:2222. doi: 10.3390/ijms23042222
16. Oikonomou EK, Antoniades C. The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol. (2019) 16:83–99. doi: 10.1038/s41569-018-0097-6
17. Man W, Song X, Xiong Z, Gu J, Lin J, Gu X, et al. Exosomes derived from pericardial adipose tissues attenuate cardiac remodeling following myocardial infarction by Adipsin-regulated iron homeostasis. Front Cardiovasc Med. (2022) 9:1003282. doi: 10.3389/fcvm.2022.1003282
18. Pascual M, Paccaud JP, Macon K, Volanakis JE, Schifferli JA. Complement activation by the alternative pathway is modified in renal failure: the role of factor D. Clin Nephrol. (1989) 32:185–93.
19. Miyagawa T, Taniguchi T, Saigusa R, Fukayama M, Takahashi T, Yamashita T, et al. Fli1 deficiency induces endothelial adipsin expression, contributing to the onset of pulmonary arterial hypertension in systemic sclerosis. Rheumatol (Oxford). (2020) 59:2005–15. doi: 10.1093/rheumatology/kez517
20. Pascual M, Steiger G, Estreicher J, Macon K, Volanakis JE, Schifferli JA. Metabolism of complement factor D in renal failure. Kidney Int. (1988) 34:529–36. doi: 10.1038/ki.1988.214
21. Volanakis JE, Barnum SR, Giddens M, Galla JH. Renal filtration and catabolism of complement protein D. N Engl J Med. (1985) 312:395–9. doi: 10.1056/NEJM198502143120702
22. Sprong T, Roos D, Weemaes C, Neeleman C, Geesing CL, Mollnes TE, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. (2006) 107:4865–70. doi: 10.1182/blood-2005-07-2820
23. Biesma DH, Hannema AJ, van Velzen-Blad H, Mulder L, van Zwieten R, Kluijt I, et al. A family with complement factor D deficiency. J Clin Invest. (2001) 108:233–40. doi: 10.1172/JCI12023
24. Barrère-Lemaire S, Vincent A, Jorgensen C, Piot C, Nargeot J, Djouad F. Mesenchymal stromal cells for improvement of cardiac function following acute myocardial infarction: a matter of timing. Physiol Rev. (2023) 104:659–725. doi: 10.1152/physrev.00009.2023
25. Song NJ, Kim S, Jang BH, Chang SH, Yun UJ, Park KM, et al. Small molecule-induced complement factor D (Adipsin) promotes lipid accumulation and adipocyte differentiation. PloS One. (2016) 11:e0162228. doi: 10.1371/journal.pone.0162228
26. Cianflone K, Roncari DA, Maslowska M, Baldo A, Forden J, Sniderman AD. Adipsin/acylation stimulating protein system in human adipocytes: regulation of triacylglycerol synthesis. Biochemistry. (1994) 33:9489–95. doi: 10.1021/bi00198a014
27. Sivakumar K, Bari MF, Adaikalakoteswari A, Guller S, Weickert MO, Randeva HS, et al. Elevated fetal adipsin/acylation-stimulating protein (ASP) in obese pregnancy: novel placental secretion via Hofbauer cells. J Clin Endocrinol Metab. (2013) 98:4113–22. doi: 10.1210/jc.2012-4293
28. Aaron N, Kraakman MJ, Zhou Q, Liu Q, Costa S, Yang J, et al. Adipsin promotes bone marrow adiposity by priming mesenchymal stem cells. Elife. (2021) 10:e69209. doi: 10.7554/eLife.69209
29. He Y, B’Nai Taub A, Yu L, Yao Y, Zhang R, Zahr T, et al. PPARgamma acetylation orchestrates adipose plasticity and metabolic rhythms. Adv Sci (Weinh). (2023) 10:e2204190. doi: 10.1002/advs.202204190
30. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell. (2014) 158:41–53. doi: 10.1016/j.cell.2014.06.005
31. Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. (2017) 102:466–75. doi: 10.3324/haematol.2016.153312
32. Oroszlan G, Kortvely E, Szakacs D, Kocsis A, Dammeier S, Zeck A, et al. MASP-1 and MASP-2 do not activate pro-factor D in resting human blood, whereas MASP-3 is a potential activator: kinetic analysis involving specific MASP-1 and MASP-2 inhibitors. J Immunol. (2016) 196:857–65. doi: 10.4049/jimmunol.1501717
33. Zhang B, Xing G. Thrombotic microangiopathy mediates poor prognosis among lupus nephritis via complement lectin and alternative pathway activation. Front Immunol. (2022) 13:1081942. doi: 10.3389/fimmu.2022.1081942
34. Noris M, Galbusera M. The complement alternative pathway and hemostasis. Immunol Rev. (2023) 313:139–61. doi: 10.1111/imr.13150
35. Ueda Y, Mohammed I, Song D, Gullipalli D, Zhou L, Sato S, et al. Murine systemic thrombophilia and hemolytic uremic syndrome from a factor H point mutation. Blood. (2017) 129:1184–96. doi: 10.1182/blood-2016-07-728253
36. Fung M, Loubser PG, Undar A, Mueller M, Sun C, Sun WN, et al. Inhibition of complement, neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J Thorac Cardiovasc Surg. (2001) 122:113–22. doi: 10.1067/mtc.2001.114777
37. Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol. (2017) 72:124–32. doi: 10.1016/j.semcdb.2017.06.009
38. Pangburn MK. Initiation of the alternative pathway of complement and the history of “tickover. Immunol Rev. (2023) 313:64–70. doi: 10.1111/imr.13130
39. Lappegard KT, Garred P, Jonasson L, Espevik T, Aukrust P, Yndestad A, et al. A vital role for complement in heart disease. Mol Immunol. (2014) 61:126–34. doi: 10.1016/j.molimm.2014.06.036
40. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
41. Dobo J, Szakacs D, Oroszlan G, Kortvely E, Kiss B, Boros E, et al. MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked. Sci Rep. (2016) 6:31877. doi: 10.1038/srep31877
42. Hayashi M, Machida T, Ishida Y, Ogata Y, Omori T, Takasumi M, et al. Cutting edge: role of MASP-3 in the physiological activation of factor D of the alternative complement pathway. J Immunol. (2019) 203:1411–16. doi: 10.4049/jimmunol.1900605
43. Schubart A, Flohr S, Junt T, Eder J. Low-molecular weight inhibitors of the alternative complement pathway. Immunol Rev. (2023) 313:339–57. doi: 10.1111/imr.13143
44. Blatt AZ, Pathan S, Ferreira VP. Properdin: a tightly regulated critical inflammatory modulator. Immunol Rev. (2016) 274:172–90. doi: 10.1111/imr.12466
45. Bavia L, Lidani KCF, Andrade FA, Sobrinho M, Nisihara RM, de Messias-Reason IJ. Complement activation in acute myocardial infarction: An early marker of inflammation and tissue injury? Immunol Lett. (2018) 200:18–25. doi: 10.1016/j.imlet.2018.06.006
46. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. (2008) 12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x
47. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. (2004) 138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x
48. Boyer DS, Schmidt-Erfurth U, van Lookeren Campagne M, Henry EC, Brittain C. The pathophysiology of geographic atrophy secondary to age-related macular degeneration and the complement pathway as a therapeutic target. Retina. (2017) 37:819–35. doi: 10.1097/IAE.0000000000001392
49. Madhur MS, Elijovich F, Alexander MR, Pitzer A, Ishimwe J, Van Beusecum JP, et al. Hypertension: do inflammation and immunity hold the key to solving this epidemic? Circ Res. (2021) 128:908–33. doi: 10.1161/CIRCRESAHA.121.318052
50. Zhu L, Wigle D, Hinek A, Kobayashi J, Ye C, Zuker M, et al. The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J Clin Invest. (1994) 94:1163–71. doi: 10.1172/JCI117432
51. Schinzari F, Tesauro M, Campia U, Cardillo C. Dysregulated adipokine secretory profile is associated with endothelial dysfunction in human obesity. Eur Heart J. (2020) 41. doi: 10.1093/ehjci/ehaa946.3032
52. Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. (2000) 6:698–702. doi: 10.1038/76282
53. Korman BD, Marangoni RG, Hinchcliff M, Shah SJ, Carns M, Hoffmann A, et al. Brief report: association of elevated adipsin levels with pulmonary arterial hypertension in systemic sclerosis. Arthritis Rheumatol. (2017) 69:2062–68. doi: 10.1002/art.40193
54. Poveda NE, Garces MF, Ruiz-Linares CE, Varon D, Valderrama S, Sanchez E, et al. Serum adipsin levels throughout normal pregnancy and preeclampsia. Sci Rep. (2016) 6:20073. doi: 10.1038/srep20073
55. Liu M, Luo X, Xu Q, Yu H, Gao L, Zhou R, et al. Adipsin of the alternative complement pathway is a potential predictor for preeclampsia in early pregnancy. Front Immunol. (2021) 12:702385. doi: 10.3389/fimmu.2021.702385
56. Prentice RL, Zhao S, Johnson M, Aragaki A, Hsia J, Jackson RD, et al. Proteomic risk markers for coronary heart disease and stroke: validation and mediation of randomized trial hormone therapy effects on these diseases. Genome Med. (2013) 5:112. doi: 10.1186/gm517
57. Gursoy Calan O, Calan M, Yesil Senses P, Unal Kocabas G, Ozden E, Sari KR, et al. Increased adipsin is associated with carotid intima media thickness and metabolic disturbances in polycystic ovary syndrome. Clin Endocrinol (Oxf). (2016) 85:910–17. doi: 10.1111/cen.13157
58. Ohtsuki T, Satoh K, Shimizu T, Ikeda S, Kikuchi N, Satoh T, et al. Identification of adipsin as a novel prognostic biomarker in patients with coronary artery disease. J Am Heart Assoc. (2019) 8:e013716. doi: 10.1161/JAHA.119.013716
59. Jalal D, Renner B, Laskowski J, Stites E, Cooper J, Valente K, et al. Endothelial microparticles and systemic complement activation in patients with chronic kidney disease. J Am Heart Assoc. (2018) 7:e007818. doi: 10.1161/JAHA.117.007818
60. Napoli G, Pergola V, Basile P, De Feo D, Bertrandino F, Baggiano A, et al. Epicardial and pericoronary adipose tissue, coronary inflammation, and acute coronary syndromes. J Clin Med. (2023) 12:7212. doi: 10.3390/jcm12237212
61. Wang Z, Zhang Z, Li Y, Zhang Y, Wei M, Li H, et al. Endothelial-derived complement factor D contributes to endothelial dysfunction in Malignant nephrosclerosis via local complement activation. Hypertens Res. (2023) 46:1759–70. doi: 10.1038/s41440-023-01300-3
62. Sun R, Qiao Y, Yan G, Wang D, Zuo W, Ji Z, et al. Association between serum adipsin and plaque vulnerability determined by optical coherence tomography in patients with coronary artery disease. J Thorac Dis. (2021) 13:2414–25. doi: 10.21037/jtd-21-259
63. Maresh JG, Shohet RV. In vivo endothelial gene regulation in diabetes. Cardiovasc Diabetol. (2008) 7:8. doi: 10.1186/1475-2840-7-8
64. Niyonzima N, Bakke SS, Gregersen I, Holm S, Sandanger O, Orrem HL, et al. Cholesterol crystals use complement to increase NLRP3 signaling pathways in coronary and carotid atherosclerosis. EBioMedicine. (2020) 60:102985. doi: 10.1016/j.ebiom.2020.102985
65. Zhang J, Teng F, Pan L, Guo D, Liu J, Li K, et al. Circulating adipsin is associated with asymptomatic carotid atherosclerosis in obese adults. BMC Cardiovasc Disord. (2021) 21:517. doi: 10.1186/s12872-021-02329-3
66. Duan Y, Zhang X, Zhang X, Lin J, Shu X, Man W, et al. Inhibition of macrophage-derived foam cells by Adipsin attenuates progression of atherosclerosis. Biochim Biophys Acta Mol Basis Dis. (2022) 1868:166533. doi: 10.1016/j.bbadis.2022.166533
67. Hao S, Zhang J, Pei Y, Guo L, Liang Z. Complement factor D derived from epicardial adipose tissue participates in cardiomyocyte apoptosis after myocardial infarction by mediating PARP-1 activity. Cell Signal. (2023) 101:110518. doi: 10.1016/j.cellsig.2022.110518
68. Laurikka A, Vuolteenaho K, Toikkanen V, Rinne T, Leppanen T, Tarkka M, et al. Adipocytokine resistin correlates with oxidative stress and myocardial injury in patients undergoing cardiac surgery. Eur J Cardiothorac Surg. (2014) 46:729–36. doi: 10.1093/ejcts/ezt634
69. Ito S, Hashimoto H, Yamakawa H, Kusumoto D, Akiba Y, Nakamura T, et al. The complement C3-complement factor D-C3a receptor signalling axis regulates cardiac remodelling in right ventricular failure. Nat Commun. (2022) 13:5409. doi: 10.1038/s41467-022-33152-9
70. Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, et al. The alternative complement pathway is dysregulated in patients with chronic heart failure. Sci Rep. (2017) 7:42532. doi: 10.1038/srep42532
71. Lin YK, Chen YC, Chang SL, Lin YJ, Chen JH, Yeh YH, et al. Heart failure epicardial fat increases atrial arrhythmogenesis. Int J Cardiol. (2013) 167:1979–83. doi: 10.1016/j.ijcard.2012.05.009
72. Zhao L, Ma Z, Guo Z, Zheng M, Li K, Yang X. Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. BioMed Pharmacother. (2020) 121:109634. doi: 10.1016/j.biopha.2019.109634
73. Ding Q, Xing J, Bai F, Shao W, Hou K, Zhang S, et al. C1QC, VSIG4, and CFD as potential peripheral blood biomarkers in atrial fibrillation-related cardioembolic stroke. Oxid Med Cell Longev. (2023) 2023:5199810. doi: 10.1155/2023/5199810
74. Morrissette-McAlmon J, Xu WR, Teuben R, Boheler KR, Tung L. Adipocyte-mediated electrophysiological remodeling of human stem cell - derived cardiomyocytes. J Mol Cell Cardiol. (2024) 189:52–65. doi: 10.1016/j.yjmcc.2024.02.002
75. Feridooni T, Zamzam A, Popkov M, Syed MH, Djahanpour N, Wheatcroft M, et al. Plasma complement component C2: a potential biomarker for predicting abdominal aortic aneurysm related complications. Sci Rep. (2022) 12:21252. doi: 10.1038/s41598-022-24698-1
76. Murakami Y, Nishigori M, Yagi H, Osaki T, Wakabayashi M, Shirai M, et al. Serum proteomic identification and validation of two novel atherosclerotic aortic aneurysm biomarkers, profilin 1 and complement factor D. Proteome Sci. (2023) 21:11. doi: 10.1186/s12953-023-00212-x
77. Al Haj Ahmad RM, Al-Domi HA. Complement 3 serum levels as a pro-inflammatory biomarker for insulin resistance in obesity. Diabetes Metab Syndr. (2017) 11 Suppl 1:S229–S32. doi: 10.1016/j.dsx.2016.12.036
78. Gomez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat Med. (2019) 25:1739–47. doi: 10.1038/s41591-019-0610-4
79. Schrover IM, van der Graaf Y, Spiering W, Visseren FL, Group SS. The relation between body fat distribution, plasma concentrations of adipokines and the metabolic syndrome in patients with clinically manifest vascular disease. Eur J Prev Cardiol. (2018) 25:1548–57. doi: 10.1177/2047487318790722
80. Wang Y, Zheng X, Xie X, Qian W, Ren Z, Chen Y, et al. Body fat distribution and circulating adipsin are related to metabolic risks in adult patients with newly diagnosed growth hormone deficiency and improve after treatment. BioMed Pharmacother. (2020) 132:110875. doi: 10.1016/j.biopha.2020.110875
81. Lee SG, Chae J, Woo SM, Seo SU, Kim HJ, Kim SY, et al. TGFBI remodels adipose metabolism by regulating the Notch-1 signaling pathway. Exp Mol Med. (2023) 55:520–31. doi: 10.1038/s12276-023-00947-9
82. Martinez-Huenchullan SF, Shipsey I, Hatchwell L, Min D, Twigg SM, Larance M. Blockade of high-fat diet proteomic phenotypes using exercise as prevention or treatment. Mol Cell Proteomics. (2021) 20:100027. doi: 10.1074/mcp.TIR120.002343
83. Asterholm IW, McDonald J, Blanchard PG, Sinha M, Xiao Q, Mistry J, et al. Lack of “immunological fitness” during fasting in metabolically challenged animals. J Lipid Res. (2012) 53:1254–67. doi: 10.1194/jlr.M021725
84. Shi Z, Wang L, Luan J, Yin L, Ji X, Zhang W, et al. Exercise promotes bone marrow microenvironment by inhibiting adipsin in diet-induced male obese mice. Nutrients. (2022) 15:19. doi: 10.3390/nu15010019
85. Tafere GG, Wondafrash DZ, Zewdie KA, Assefa BT, Ayza MA. Plasma adipsin as a biomarker and its implication in type 2 diabetes mellitus. Diabetes Metab Syndr Obes. (2020) 13:1855–61. doi: 10.2147/DMSO.S253967
86. Millar CA, Meerloo T, Martin S, Hickson GR, Shimwell NJ, Wakelam MJ, et al. Adipsin and the glucose transporter GLUT4 traffic to the cell surface via independent pathways in adipocytes. Traffic. (2000) 1:141–51. doi: 10.1034/j.1600-0854.2000.010206.x
87. Zhang X, Duan Y, Zhang X, Jiang M, Man W, Zhang Y, et al. Adipsin alleviates cardiac microvascular injury in diabetic cardiomyopathy through Csk-dependent signaling mechanism. BMC Med. (2023) 21:197. doi: 10.1186/s12916-023-02887-7
88. Jiang MY, Man WR, Zhang XB, Zhang XH, Duan Y, Lin J, et al. Adipsin inhibits Irak2 mitochondrial translocation and improves fatty acid beta-oxidation to alleviate diabetic cardiomyopathy. Mil Med Res. (2023) 10:63. doi: 10.1186/s40779-023-00493-5
89. Irmscher S, Doring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
90. Harris CL. Expanding horizons in complement drug discovery: challenges and emerging strategies. Semin Immunopathol. (2018) 40:125–40. doi: 10.1007/s00281-017-0655-8
91. Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. (2019) 10:1157. doi: 10.3389/fimmu.2019.01157
92. Lee JW, Griffin M, Kim JS, Lee Lee LW, Piatek C, Nishimura JI, et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): a double-blind, randomised, phase 3 trial. Lancet Haematol. (2023) 10:e955–e65. doi: 10.1016/S2352-3026(23)00315-0
93. Leung E, Landa G. Update on current and future novel therapies for dry age-related macular degeneration. Expert Rev Clin Pharmacol. (2013) 6:565–79. doi: 10.1586/17512433.2013.829645
Keywords: CFD, adipsin, inflammation, cardiovascular disease, metabolic diseases
Citation: Kong Y, Wang N, Tong Z, Wang D, Wang P, Yang Q, Yan X, Song W, Jin Z and Zhang M (2024) Role of complement factor D in cardiovascular and metabolic diseases. Front. Immunol. 15:1453030. doi: 10.3389/fimmu.2024.1453030
Received: 22 June 2024; Accepted: 16 September 2024;
Published: 02 October 2024.
Edited by:
Anna Swierzko, Polish Academy of Sciences, PolandReviewed by:
Hideharu Sekine, Fukushima Medical University, JapanCopyright © 2024 Kong, Wang, Tong, Wang, Wang, Yang, Yan, Song, Jin and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maomao Zhang, bWFvbWFvbHAxOTgzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.