 Jingjing Wei1†
Jingjing Wei1† Aolong Wang1†
Aolong Wang1† Bin Li1,2†Xingyuan Li1Rui Yu1Haitao Li1
Bin Li1,2†Xingyuan Li1Rui Yu1Haitao Li1 Xinlu Wang1*
Xinlu Wang1* Yongxia Wang1*
Yongxia Wang1* Mingjun Zhu1*
Mingjun Zhu1*- 1Heart Center, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, China
- 2Henan Evidence-based Medicine Center of Chinese Medicine, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, China
Systemic lupus erythematosus (SLE) is a prevalent autoimmune disease primarily characterized by the involvement of multiple systems and organs. Cardiovascular disease is the primary cause of mortality in patients with SLE, though the mechanisms underlying the increased cardiovascular risk in SLE patients remain unclear. Recent studies indicate that abnormal activation of programmed cell death (PCD) signaling and the crosstalk among various forms of cell death are critical in the immunopathogenesis of SLE. Furthermore, apoptosis, necroptosis, pyroptosis, NETosis, and ferroptosis are recognized as key cellular processes in the pathogenesis of SLE and are closely linked to cardiac involvement. This review uniquely explores the intricate crosstalk between apoptosis, necroptosis, and other cell death pathways, discussing their roles and interactions in the pathogenesis of cardiac involvement in SLE. Investigating the interplay between PCD signaling and cardiac involvement in SLE in understanding the disease’s underlying mechanisms and offers opportunities for new therapeutic interventions. The integration of precision medicine and innovative strategies targeting these complex pathways holds promise for enhancing the treatment prospects of SLE with cardiac involvement.
1 Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by systemic inflammation and abnormal production of pathogenic antibodies (1). The clinical manifestations of SLE are highly heterogeneous, potentially affecting multiple organs including the heart, skin, skeletal muscles, kidneys, lungs, and gastrointestinal tract (2). Over the past 20 years, advances in medical technology have decreased the mortality rate of SLE patients, raising the 5-year survival rate to over 90% (3). However, the standardized mortality rate of SLE patients remains 2.5 times higher than that of the general population (4). Cardiovascular disease (CVD) is the leading cause of premature death in SLE patients (5), with the risk of CVD-related mortality being 3 times higher in SLE patients than in the general population (6). To further reduce the mortality rate of SLE patients, it is urgent to investigate the pathological mechanisms underlying cardiac involvement in systemic lupus erythematosus.
Multiple forms of programmed cell death (PCD) have been described in SLE patients and are considered contributing factors to disease onset (7). These cell death mechanisms not only activate the immune system through the release of autoantigens and inflammatory mediators but also influence the course and severity of SLE by regulating immune cell functions and inflammatory responses, ultimately causing damage to multiple organs (8). Previous studies have shown enhanced apoptosis in SLE patients with cardiac involvement (9). Additionally, necroptosis, pyroptosis, NETosis, and ferroptosis are known to be closely associated with CVD (10–12). Recent research indicates that the crosstalk between different forms of cell death has a significant impact on the pathogenesis of SLE and cardiac involvement. For instance, the interaction between apoptosis and necroptosis can provoke a stronger immune response and inflammatory reaction, worsening cardiovascular damage (13). Additionally, emerging forms of cell death, such as pyroptosis and ferroptosis, are increasingly studied in SLE, and their roles in cardiac involvement are attracting growing attention (14, 15). Some clinical trials investigating cell death mechanisms have further elucidated potential therapeutic targets for cardiac involvement in SLE (Table 1). A comprehensive delineation of different pathways mediating different cell death mechanisms and their interactions will help elucidate the pathological basis in cardiac involvement of SLE patients, providing a theoretical foundation for developing new therapeutic strategies.

Table 1. Overview of clinical studies on cell death mechanisms.
2 Apoptosis and its contribution to cardiac involvement
Apoptosis is a form of programmed cell death that is activated via two pathways: the extrinsic pathway, which activates caspase-8 through specific receptor-ligand interactions, and the intrinsic pathway, which activates caspase-9 through the mitochondrial pathway, ultimately leading to apoptosis by activating caspase-3 (Figure 1A) (16). Additionally, the caspase-8 mediated apoptotic pathway can drive mitochondrial outer membrane permeabilization (MOMP) by cleaving BID, thereby activating caspase-9 and caspase-3 (17). The activation of caspase-3 leads to the production of apoptosis-derived membrane vesicles (AdMVs) containing double-stranded DNA (dsDNA). It is noteworthy that extracellular vesicles can be released during cellular death processes, including necroptosis, pyroptosis, and NETosis. The defective clearance of extracellular vesicles resulting from excessive cell death can trigger autoimmune responses, which is one of the pathological mechanisms of systemic lupus erythematosus (18, 19). The cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) signaling pathway is the primary effector by which cells sense and respond to the abnormal presence of dsDNA in the cytoplasm (20). In the serum of SLE patients, AdMVs induce type I interferon (IFN-1) production through the cGAS-STIN pathway (21). Notably, senescent monocytes in SLE can also promote IFN-α production via the STING pathway (22). IFN-1 is a key cytokine mediator in the pathogenesis of SLE, with elevated levels of interferon-stimulated genes in the blood of systemic lupus erythematosus patients (23). IFN-1 can mediate CD8+ T cell death in SLE patients by regulating nicotinamide adenine dinucleotide (NAD+) levels (24). Additionally, recent studies have confirmed that IFNα is associated with B cell activation and autoantibody production (25, 26). In summary, Apoptosis is closely related to SLE. Increased apoptosis and defective clearance of apoptotic cells lead to heightened autoantigen exposure, triggering autoimmune and inflammatory responses in SLE (7). This may be one of the reasons for the involvement of multiple organs.
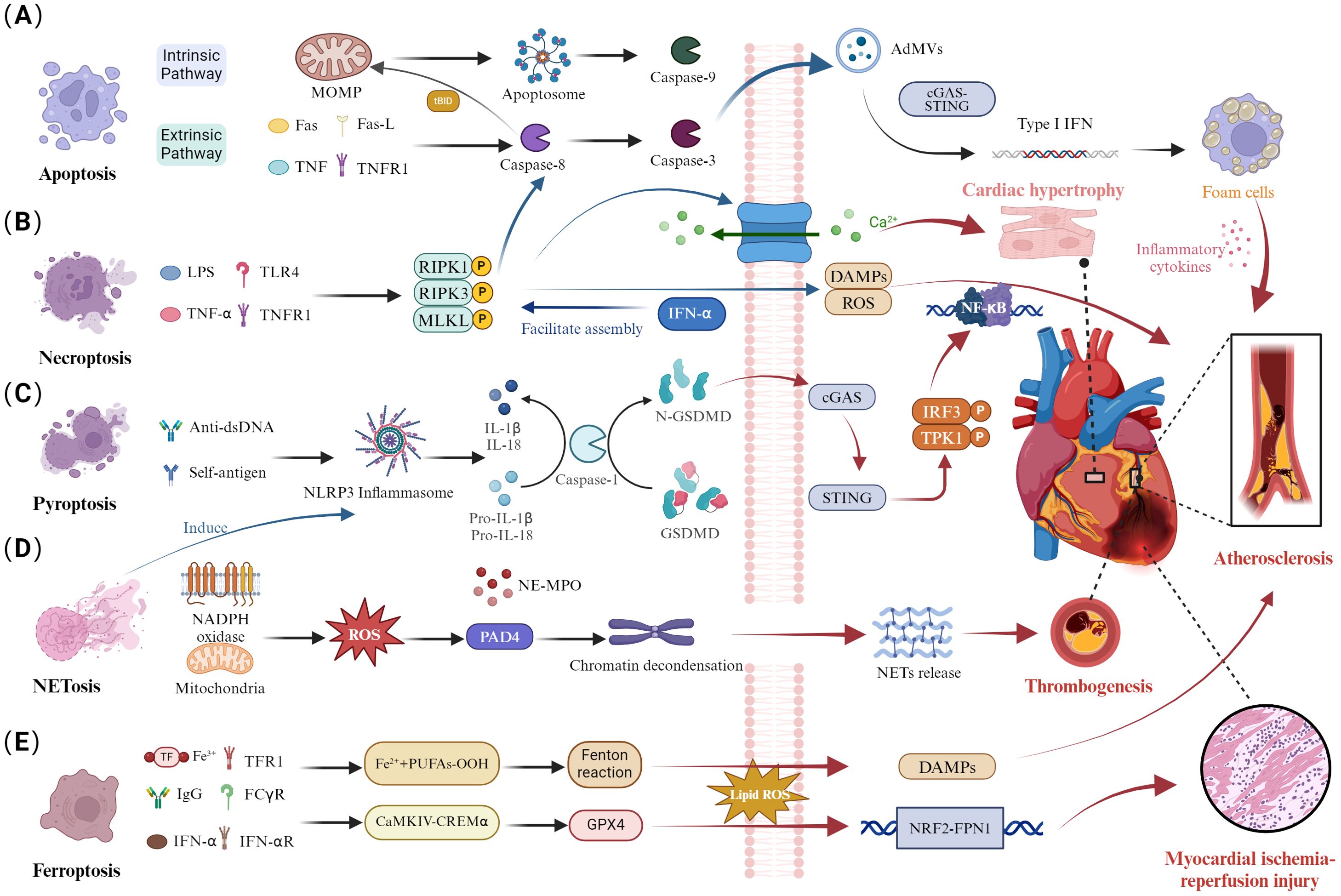
Figure 1. Dysregulation of cell death pathways and cardiac involvement in SLE. (A) Disruption of apoptosis. The extrinsic apoptotic pathway activates caspase-8 through specific receptor-ligand interactions, converting it to caspase-3. Additionally, caspase-8 cleaves Bid, inducing mitochondrial outer MOMP and activating caspase-9 within the apoptosome, triggering a caspase cascade. Activation of caspase-3 generates AdMVs, which induce IFN-1 production via the cGAS-STING pathway, influencing atherosclerotic plaque progression by enhancing foam cell formation. (B) Necroptosis disturbance. Activation of necroptosis through TNFR and TLR4 binding leads to caspase-8-mediated apoptosis. The necrosome is stabilized by RIPK1, RIPK3, and autophosphorylated MLKL, which promotes the binding of phosphorylated MLKL to the cell membrane. This process disrupts cell integrity, exposing DAMPs. The RIPK3/MLKL signaling pathway mediates calcium influx, promoting cardiomyocyte hypertrophy. (C) Pyroptotic imbalance. Anti-dsDNA antibodies and autoantigens activate the NLRP3 inflammasome, leading to caspase-1-mediated maturation and secretion of IL-1β and IL-18, along with GSDMD cleavage. Subsequently, GSDMD can mediate the cGAS/STING/TBK1/IRF3/NF-κB signaling pathway, contributing to the atherosclerosis process. (D) Abnormal NETosis. NADPH oxidase produces ROS and mitochondrial ROS, activating PAD4 and inducing chromatin decondensation. The nuclear translocation of NE and MPO leads to histone degradation and NETs release, contributing to arterial thrombosis formation. (E) Ferroptosis dysregulation. Autoantibodies and IFN-α synergistically induce the CaMKIV/CREMα axis, inhibiting GPX4 expression and increasing lipid-derived ROS, exacerbating ferroptosis. The NRF2/FPN1 signaling pathway is downregulated, leading to myocardial ischemia-reperfusion injury.
Previous studies indicated that patients with SLE have a significantly increased risk of CV (27, 28). Patients with both SLE and CVD exhibited higher levels of fas cell surface death receptor (FAS), tumor necrosis factor receptor 1(TNFR1), matrix metallopeptidase 1 (MMP-1), and MMP-7 compared to those without CVD (9). This suggested that FAS and TNFR1-mediated extrinsic apoptosis may be linked to the heightened cardiovascular risk in SLE patients. A clinical trial involving 80 SLE patients revealed that anti-dsDNA antibodies can promote monocyte apoptosis and activate the immune system, thereby driving the development of atherosclerosis (29). Additionally, studies have indicated that the mitochondrial deoxyribonucleic acid (mtDNA)-mediated cGAS-STING pathway is associated with endothelial-to-mesenchymal transition (30). Furthermore, apoptosis-mediated IFN-I may be a key factor in the cardiac involvement observed in SLE patients (31, 32). IFN-I induces apoptosis in endothelial progenitor cells and circulating angiogenic cells (33), promotes foam cell formation, activates endothelial and immune cells, and enhances the recruitment of pro-inflammatory leukocytes to the arteries (34, 35). This is a primary mechanism of atherosclerosis in SLE. A clinical trial indicated that Anifrolumab, an antibody blocking the IFN-I receptor, can reduce levels of neutrophil extracellular traps (NETs), TNF-α, and interleukin-10 (IL-10) while improving cholesterol efflux capacity (36). Overall, apoptosis and IFN-I generation may accelerate the progression of CVD in SLE. This mechanism could be a potential therapeutic target for reducing cardiovascular risk in SLE patients.
3 Necroptosis and its contribution to cardiac involvement
As we delve deeper into the specific mechanisms of apoptosis in SLE, it is essential to understand how this process interacts with other cell death pathways, such as necroptosis. Necroptosis is a form of cell death distinct from traditional apoptosis and necrosis pathways. This process is mediated by receptor-interacting protein 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like protein (MLKL) (Figure 1B) (37). The formation of the RIPK1/RIPK3 necrosome induces MLKL phosphorylation, which plays a role in the inflammation and immune response in SLE (38, 39). The crosstalk between apoptosis, necroptosis, and pyroptosis has emerged as a crucial pathological mechanism in SLE. The inhibition of caspase-8 activity in apoptosis can activate RIPK3 and MLKL phosphorylation, leading to necroptosis (16). Enhanced INF signaling in SLE patients can mediate the assembly of RIPK1 and RIPK3, increasing necroptosis levels (40). Interestingly, RIPK3 can mediate both apoptosis and necroptosis, depending on the levels of its molecular chaperones, heat shock protein 90 (Hsp90), and cell division cycle protein 37 (Cdc37). RIPK3 can aggregate with RIPK1, and caspase-8 to activate apoptosis (41). Additionally, the activation of RIPK3 and MLKL can promote the production of nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) and IL-1β, which are associated with pyroptosis (42). These findings indicate that necroptosis mediates the processes of apoptosis and pyroptosis.
MLKL, a critical factor in necroptosis, has increased mRNA levels in the peripheral blood mononuclear cells (PBMCs) of SLE patients (43). Meanwhile, the RIPK1/RIPK3/MLKL pathway-mediated necroptosis plays a significant role in various CVDs, including atherosclerosis, ischemia-reperfusion injury, myocardial infarction, and myocarditis (44, 45). We hypothesize that necroptosis might be a contributing factor to cardiac involvement in SLE. In patients with atherosclerosis, the expression levels of RIPK3 protein and MLKL are elevated. The necroptosis inhibitor Necrostatin-1 can reduce necrosome formation, thereby alleviating plaque instability and size (46). Additionally, studies indicate that RIPK1 may contribute to atherosclerosis by activating the NF-κB pathway and promoting inflammation (47). Interestingly, research also suggests that RIPK1 is involved in endothelial cell damage induced by oxidized low-density lipoprotein (ox-LDL), and the necroptosis inhibitor Necrostatin-1 can inhibit the NF-κB pathway (48). RIPK3 protein expression increases in models of cardiomyocyte hypertrophy, and the RIPK3/MLKL signaling pathway can mediate calcium influx, promoting cardiomyocyte hypertrophy (49). In mouse models, the RIPK3 inhibitor GSK872 can ameliorate diabetes-induced myocardial fibrosis (50). These studies elucidate the importance of RIPK1/RIPK3/MLKL pathway-mediated necroptosis in SLE and CVD. Despite the current lack of research on the necroptosis mechanisms in both conditions, targeting necroptosis might be a potential strategy for reducing CVD risk in SLE. In addition to apoptosis and necroptosis, pyroptosis is also an important factor in the pathological mechanism of cardiac involvement in SLE.
4 Pyroptosis and its contribution to cardiac involvement
Pyroptosis is a novel form of PCD characterized by membrane perforation, cell rupture, and release of inflammatory cytokines, including IL-1β and IL-18, mediated by the gasdermin D (GSDMD) protein family triggered by inflammasomes, promoting inflammatory responses and rapidly initiating the immune response process (Figure 1C) (51). NLRP3 can activate caspase-1, which cleaves IL-1β and IL-18 precursors into mature inflammatory factors IL-1β and IL-18, further exacerbating the occurrence of inflammatory responses and inducing pyroptosis (52). Moreover, GSDMD also plays a crucial role in SLE, where it is cleaved by caspase-1 into GSDMD-N, which translocates to the cell membrane and forms pores, facilitating the release of IL-1β and IL-18 (53). Importantly, abnormal aggregation or clearance defects of apoptotic cells in SLE patients can lead to exposure of self-antigens, accumulation of DNA and mtDNA, promoting the production of autoantibodies, and further enhancing the generation of IFN-1 (54, 55). Recent research using a SLE mouse model has confirmed that mtDNA promotes the oligomerization of GSDMD. The GSDMD inhibitor DSF can reduce the production of autoantibodies, suggesting GSDMD as a new therapeutic target for SLE (56). Additionally, DSF, as a GSDMD inhibitor, effectively suppresses the levels of IFN and pro-inflammatory cytokines in the PBMC of SLE patients.
The GSDMD, as a key mediator of cell pyroptosis, is closely associated not only with pathological process in SLE but also with the inflammatory response during acute myocardial infarction. Inhibition of GSDMD can reduce the area of myocardial infarction, exerting a cardioprotective function (57). Recent research has shown enhanced expression of GSDMD in atherosclerosis, where it can mediate the cGAS/STING/TBK1/IRF3/NF-κB signaling pathway, contributing to the process of atherosclerosis. GSDMD inhibitors can reduce the formation of atherosclerotic plaques by regulating the release of mtDNA and the STING pathway (58). Furthermore, PBMCs in SLE patients exhibit upregulated expression of NLRP3 and IL-1β (59). Previous studies have reported the involvement of NLRP3 inflammasomes and pro-inflammatory cytokines IL-1β and IL-18 in the development of various CVDs (10, 60), including acute myocardial infarction, atherosclerosis, and heart failure. Elevated levels of NLRP3 inflammasomes, caspase-1, and IL-1β are observed in the peripheral blood of patients with dilated cardiomyopathy, with NLRP3 levels serving as an independent risk factor for readmission within 6 months (61). The NLRP3 inhibitor MCC950 may represent a potential therapeutic avenue for ameliorating multi-organ involvement in patients with SLE (62). Recent research has confirmed that the MCC950 can alleviate oxidative stress and aging in myocardial cells, alleviate adrenaline-induced cardiac dysfunction in mice, and reduce the formation of atherosclerotic plaques and systemic inflammation (63, 64). Mounting evidence suggests that IL-1 and NLRP3 inflammasomes may serve as novel therapeutic targets for CVDs. Previous research has indicated that pyroptosis mediated by GSDMD and NLRP3 plays similar roles in both SLE and CVDs (65, 66). Moreover, GSDMD inhibitors exhibit significant efficacy in treating both SLE and CVDs, possibly offering a direction for future treatments of these conditions. In addition to the above-mentioned modes of cell death, NETosis also plays an important role in cardiac involvement in SLE.
5 NETosis and its contribution to cardiac involvement
NETs are web-like structures released by neutrophils into the extracellular space (67). The formation of NETs is accompanied by neutrophil death, termed NETosis, which is a unique cell death process distinct from apoptosis and necroptosis. It is known that the formation of NETosis is closely related to reactive oxygen species (ROS) produced by NADPH oxidase and mitochondria (Figure 1D). Activated peptidyl arginine deiminase 4 (PAD4) mediates histone citrullination and neutrophil elastase (NE)-mediated histone cleavage, facilitating chromatin decondensation, a critical process in the formation of Neutrophil Extracellular Traps (NETs) (68). Furthermore, the rupture of the nuclear and plasma membranes is essential for the extracellular release of chromatin and NET formation (69). The nuclear membrane acts as the primary physical barrier to chromatin release, while the nuclear lamina, composed of type A (A, C) and type B (B1, B2) lamin filaments, plays a critical role in maintaining its structural integrity (70). Recent research has revealed that the rupture of the nuclear membrane during NET formation is caused by kinase-mediated phosphorylation and disassembly of the nuclear lamina, rather than by proteolytic cleavage (71). For instance, cyclin-dependent kinases 4 and 6(CDK4/6), associated with nuclear envelope rupture, may regulate the phosphorylation of lamin A/C, although causality has not been experimentally confirmed (72). Notably, protein kinase C-α (PKC-α) plays a pivotal role in the phosphorylation of lamin B, facilitating the nuclear envelope rupture during NETosis (71). Recent studies have confirmed that NETs are an important source of mononucleosome circulating DNA, distinct from apoptotic cells (73). The DNA, granule proteins, and histones released during NETosis can become autoantigens in SLE (73). Furthermore, the formation of NETs can promote IFN secretion, accelerating inflammation and disease progression in SLE (74). Simultaneously, apoptotic cell microparticles in SLE patients drive the formation of NETosis (75, 76). Notably, GSDMD plays a significant role in pyroptosis in SLE. Recent studies have shown that GSDMD knockout inhibits NETs formation in SLE mouse models (56). Additionally, mitochondrial ROS participate in both NETs formation and IFN-1 production. Inhibition of mitochondrial ROS reduces IFN-1 production, alleviating disease progression in SLE (77, 78). These studies illustrate the crosstalk between pyroptosis, NETs, and apoptosis in SLE, ultimately leading to immune system dysregulation in SLE.
Recent studies have found elevated levels of NETs in the circulating blood of SLE patients (79). It is known that NETs are involved in processes closely related to CVDs, such as atherosclerosis (12), thrombosis (80), and adverse cardiac remodeling (81). Stanley Moore and colleagues conducted the first clinical study on the correlation between CVD and NETs in SLE patients. The results showed that NETs are related to CVDs such as arterial thrombosis and endothelial cell activation (82). The active components of NETs, such as DNA and histones, can regulate the levels of FXII and FXI (83, 84). The crosstalk between NETs and endothelial cell mechanisms jointly participates in thrombosis formation (85). Additionally, NETs can be elevated in patients with primary hypertension, but their levels decrease after ARB treatment. This phenomenon may be related to oxidative stress, autophagy, and PAD4-mediated histone citrullination induced by Ang II (86). A recent mouse study confirmed that Padi4-/- mice have a weakened hypertensive response to Ang II, reduced aortic inflammation, and improved endothelial cell-dependent vasorelaxation (87). Furthermore, PAD inhibition can alleviate atherosclerosis and arterial thrombosis formation (88). On the other hand, MPO contributes to CVD by promoting endothelial dysfunction, activating MMP, and causing LDL and HDL imbalances. MPO is associated with atherosclerotic plaques and adverse cardiovascular events (89). MPO inhibitors (AZM198) can stabilize plaques, and serum MPO-DNA complexes can serve as biomarkers for predicting adverse left ventricular remodeling post-PCI in myocardial infarction patients (90, 91). In summary, although MPO, PAD4, and NETs formation are related to CVD, extensive clinical trials and basic research are still ongoing. This suggests that NETs may become a common target for future treatment of SLE and CVD. As we consider the role of NETosis in SLE, it is crucial to also examine ferroptosis. The interaction between NETosis and ferroptosis adds another dimension to the complex network of cell death modalities in SLE.
6 Ferroptosis and its contribution to cardiac involvement
Ferroptosis is a form of non-apoptotic cell death characterized by abnormal iron metabolism, lipid metabolic disorders, and excessive accumulation of ROS (Figure 1E) (92). The glutathione (GSH)-glutathione peroxidase 4 (GPX4) axis and the accumulation of iron ions, which trigger the Fenton reaction, mediate lipid peroxidation, ultimately leading to ferroptosis (93). GPX4 is a key regulator in the ferroptosis process, capable of decomposing lipid peroxides (LPO), thereby inhibiting ferroptosis (94). Previous studies have shown that serum autoantibodies and IFN-α in SLE patients can regulate the CaMKIV/CREMα signaling pathway, inducing neutrophil ferroptosis by inhibiting GPX4 activity and leading to increased lipid ROS (95). Ferroptosis may be a key process in neutrophil death and reduction in SLE patients. Recent studies have confirmed that B cell ferroptosis is involved in the progression of SLE, potentially related to ferroptosis-mediated B cell differentiation and plasma cell composition. Ferroptosis inhibitors, such as Liproxstatin-1, inhibited the production of autoantibodies and malondialdehyde in SLE (96, 97). These studies highlight the significant role of ferroptosis in different cells during the progression of SLE.
Interestingly, ferroptosis is widely involved in the development of various CVDs, including atherosclerosis, myocardial infarction, ischemia-reperfusion injury, and heart failure (98). It is known that ox-LDL can induce endothelial dysfunction associated with atherosclerosis (99). Iron overload promotes macrophages to absorb ox-LDL and transform into foam cells, ultimately leading to atherosclerosis. In ox-LDL-treated mouse aortic endothelial cells, the ferroptosis inhibitor Ferrostatin-1 can regulate lipid peroxidation and ferroptosis. Ferrostatin-1 alleviates endothelial dysfunction and atherosclerosis by downregulating adhesion molecules and upregulating eNOS expression (100). Meanwhile, GPX4 plays a key role in ferroptosis. Previous studies have shown that the severity of atherosclerosis is negatively correlated with GPX4 expression (101). In I/R mouse models, GPX4 expression is significantly reduced, which may be related to the upregulation of ELAVL1 and the NRF2/FPN1 pathway (102, 103). Ferroptosis inhibitors, such as Liproxstatin-1, can protect the heart by regulating GPX4 expression (104). Although there is currently a lack of research on the shared mechanisms of ferroptosis in SLE and CVD, existing studies suggest that the ferroptosis process in SLE is closely related to CVD. This understanding may help us better explore the reasons for the higher risk of CVD in SLE patients.
7 Conclusion
Overall, increasing research provides compelling evidence for the involvement of various cell death modalities in cardiac complications associated with SLE, highlighting a higher risk of cardiovascular disease-related mortality in SLE patients. There is an urgent need to understand the common mechanisms between SLE and CVD to develop new therapeutic strategies. In this review, we systematically discuss the potential common mechanisms linking different types of cell death and immune response crosstalk in SLE patients and their contribution to cardiac involvement, revealing potential common targets. This may represent just the tip of the iceberg of SLE-related cardiac complications. Although numerous studies have shown that the crosstalk between different cell death modalities leads to autoimmune abnormalities in SLE, which may underlie the increased cardiovascular risk, more basic and clinical research is needed to confirm this. Understanding the crosstalk between cell death pathways not only elucidates the pathogenesis of cardiac involvement in SLE but also opens new avenues for therapeutic intervention. By comprehensively mapping these interactions, we can better identify novel targets for treatment, potentially improving outcomes for patients with SLE and reducing their cardiovascular risk. Future research should focus on elucidating these mechanisms further, with the aim of translating these findings into clinical applications.
Author contributions
JW: Writing – original draft, Writing – review & editing. AW: Writing – original draft, Writing – review & editing. BL: Writing – original draft, Writing – review & editing. XL: Supervision, Writing – review & editing. RY: Supervision, Writing – review & editing. HL: Supervision, Writing – review & editing. XW: Writing – review & editing, Conceptualization. YW: Writing – review & editing, Conceptualization. MZ: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (Grant No.82030120, No.82074226), Henan Province Traditional Chinese Medicine Scientific Research Special Project (Grant No. 2023ZY2030). Henan Province medical science and technology research Project (Grant No. LHGJ20220572).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
SLE, systemic lupus erythematosus; PCD, programmed cell death; CVD, cardiovascular disease; MOMP, membrane permeabilization; AdMVs, apoptosis-derived membrane vesicles; dsDNA, double-stranded deoxyribonucleic acid; cGAS-STING, cyclic GMP-AMP synthase-stimulator of interferon gene; IFN-1, type I interferon; NAD+, nicotinamide adenine dinucleotide; TNFR1, tumor necrosis factor receptor 1; MMP-1, matrix metallopeptidase 1; NETs, neutrophil extracellular traps; IL-10, interleukin-10; RIPK1, receptor-interacting protein 1; MLKL, mixed lineage kinase domain-like protein; NLRP3, nucleotide-binding oligomerization domain-like receptor protein 3; PBMCs, peripheral blood mononuclear cells; GSDMD, gasdermin D; ROS, reactive oxygen species; MPO, myeloperoxidase; NE, neutrophil elastase; PAD4, peptidyl arginine deiminase 4; GSH, glutathione; GPX4, glutathione peroxidase 4; LPO, lipid peroxides.
References
1. Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med. (2020) 172:ITC81–96. doi: 10.7326/AITC202006020
2. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. (2019) 393:2344–58. doi: 10.1016/S0140-6736(19)30546-X
3. Barber MRW, Drenkard C, Falasinnu T, Hoi A, Mak A, Kow NY, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. (2021) 17:515–32. doi: 10.1038/s41584-021-00668-1
4. Barber MRW, Falasinnu T, Ramsey-Goldman R, Clarke AE. The global epidemiology of SLE: narrowing the knowledge gaps. Rheumatol (Oxford). (2023) 62:i4–9. doi: 10.1093/rheumatology/keac610
5. Manadan AM, Kambhatla S, Gauto-Mariotti E, Okoli C, Block JA. Reasons for hospitalization and in-hospital mortality in adult systemic lupus erythematosus. ACR Open Rheumatol. (2020) 2:683–9. doi: 10.1002/acr2.11195
6. Taylor T, Anastasiou C, Ja C, Rush S, Trupin L, Dall'Era M, et al. Causes of death among individuals with systemic lupus erythematosus by race and ethnicity: A population-based study. Arthritis Care Res (Hoboken). (2023) 75:61–8. doi: 10.1002/acr.24988
7. Lou H, Ling GS, Cao X. Autoantibodies in systemic lupus erythematosus: From immunopathology to therapeutic target. J Autoimmun. (2022) 132:102861. doi: 10.1016/j.jaut.2022.102861
8. Xu Y, Li P, Li K, Li N, Liu H, Zhang X, et al. Pathological mechanisms and crosstalk among different forms of cell death in systemic lupus erythematosus. J Autoimmun. (2022) 132:102890. doi: 10.1016/j.jaut.2022.102890
9. Wigren M, Svenungsson E, Mattisson IY, Gustafsson JT, Gunnarsson I, Zickert A, et al. Cardiovascular disease in systemic lupus erythematosus is associated with increased levels of biomarkers reflecting receptor-activated apoptosis. Atherosclerosis. (2018) 270:1–7. doi: 10.1016/j.atherosclerosis.2018.01.022
10. Toldo S, Abbate A. The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat Rev Cardiol. (2024) 21:219–37. doi: 10.1038/s41569-023-00946-3
11. Zhang ZD, Shi CR, Li FX, Gan H, Wei Y, Zhang Q, et al. Disulfiram ameliorates STING/MITA-dependent inflammation and autoimmunity by targeting RNF115. Cell Mol Immunol. (2024) 21:275–91. doi: 10.1038/s41423-024-01131-3
12. Liu Y, Carmona-Rivera C, Moore E, Seto NL, Knight JS, Pryor M, et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front Immunol. (2018) 9:1680. doi: 10.3389/fimmu.2018.01680
13. Frostegård J. Systemic lupus erythematosus and cardiovascular disease. J Intern Med. (2023) 293:48–62. doi: 10.1111/joim.13557
14. Bello N, Meyers KJ, Workman J, Hartley L, McMahon M. Cardiovascular events and risk in patients with systemic lupus erythematosus: Systematic literature review and meta-analysis. Lupus. (2023) 32:325–41. doi: 10.1177/09612033221147471
15. Katz G, Smilowitz NR, Blazer A, Clancy R, Buyon JP, Berger JS. Systemic lupus erythematosus and increased prevalence of atherosclerotic cardiovascular disease in hospitalized patients. Mayo Clin Proc. (2019) 94:1436–43. doi: 10.1016/j.mayocp.2019.01.044
16. Ai Y, Meng Y, Yan B, Zhou Q, Wang X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol Cell. (2024) 84:170–9. doi: 10.1016/j.molcel.2023.11.040
17. Tan Q, Huang W, Zheng Y, Li M, Tao Y, Yu S. Unveiling the nexus: decoding interactions between regulated cell death and systemic lupus erythematosus pathogenesis for innovative therapeutic avenues. Rheumatol Autoimmun. (2024) 4:1–10. doi: 10.1002/rai2.12104
18. Zhao Y, Wei W, Liu ML. Extracellular vesicles and lupus nephritis - New insights into pathophysiology and clinical implications. J Autoimmun. (2020) 115:102540. doi: 10.1016/j.jaut.2020.102540
19. Liu ML, Williams KJ, Werth VP. Microvesicles in autoimmune diseases. Adv Clin Chem. (2016) 77:125–75. doi: 10.1016/bs.acc.2016.06.005
20. Chen C, Xu P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. (2023) 33:630–48. doi: 10.1016/j.tcb.2022.11.001
21. Kato Y, Park J, Takamatsu H, Aoki W, Aburaya S, Ueda M, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
22. Kuga T, Chiba A, Murayama G, Hosomi K, Nakagawa T, Yahagi Y, et al. Enhanced GATA4 expression in senescent systemic lupus erythematosus monocytes promotes high levels of IFNα production. Front Immunol. (2024) 15:1320444. doi: 10.3389/fimmu.2024.1320444
23. Caielli S, Wan Z, Pascual V. Systemic lupus erythematosus pathogenesis: interferon and beyond. Annu Rev Immunol. (2023) 41:533–60. doi: 10.1146/annurev-immunol-101921-042422
24. Buang N, Tapeng L, Gray V, Sardini A, Whilding C, Lightstone L, et al. Type I interferons affect the metabolic fitness of CD8+ T cells from patients with systemic lupus erythematosus. Nat Commun. (2021) 12:1980. doi: 10.1038/s41467-021-22312-y
25. Ferri DM, Nassar C, Manion KP, Kim M, Baglaenko Y, Muñoz-Grajales C, et al. Elevated levels of interferon-α Act directly on B cells to breach multiple tolerance mechanisms promoting autoantibody production. Arthritis Rheumatol. (2023) 75:1542–55. doi: 10.1002/art.42482
26. Baker T, Sharifian H, Newcombe PJ, Gavin PG, Lazarus MN, Ramaswamy M, et al. Type I interferon blockade with anifrolumab in patients with systemic lupus erythematosus modulates key immunopathological pathways in a gene expression and proteomic analysis of two phase 3 trials. Ann Rheum Dis. (2024) 83:1018–27. doi: 10.1136/ard-2023-225445
27. Lu X, Wang Y, Zhang J, Pu D, Hu N, Luo J, et al. Patients with systemic lupus erythematosus face a high risk of cardiovascular disease: A systematic review and Meta-analysis. Int Immunopharmacol. (2021) 94:107466. doi: 10.1016/j.intimp.2021.107466
28. Kain J, Owen KA, Marion MC, Langefeld CD, Grammer AC, Lipsky PE. Mendelian randomization and pathway analysis demonstrate shared genetic associations between lupus and coronary artery disease. Cell Rep Med. (2022) 3:100805. doi: 10.1016/j.xcrm.2022.100805
29. Patiño-Trives AM, Pérez-Sánchez C, Pérez-Sánchez L, Luque-Tévar M, Ábalos-Aguilera MC, Alcaide-Ruggiero L, et al. Anti-dsDNA antibodies increase the cardiovascular risk in systemic lupus erythematosus promoting a distinctive immune and vascular activation. Arterioscler Thromb Vasc Biol. (2021) 41:2417–30. doi: 10.1161/ATVBAHA.121.315928
30. Liu Q, Cheng Z, Huang B, Luo S, Guo Y. Palmitic acid promotes endothelial-to-mesenchymal transition via activation of the cytosolic DNA-sensing cGAS-STING pathway. Arch Biochem Biophys. (2022) 727:109321. doi: 10.1016/j.abb.2022.109321
31. Ambler WG, Kaplan MJ. Vascular damage in systemic lupus erythematosus. Nat Rev Nephrol. (2024) 20:251–65. doi: 10.1038/s41581-023-00797-8
32. Ding X, Xiang W, He X. IFN-I mediates dysfunction of endothelial progenitor cells in atherosclerosis of systemic lupus erythematosus. Front Immunol. (2020) 11:581385. doi: 10.3389/fimmu.2020.581385
33. Blachut D, Przywara-Chowaniec B, Tomasik A, Kukulski T, Morawiec B. Update of potential biomarkers in risk prediction and monitoring of atherosclerosis in systemic lupus erythematosus to prevent cardiovascular disease. Biomedicines. (2023) 11:2814. doi: 10.3390/biomedicines11102814
34. Yennemadi AS, Jordan N, Diong S, Keane J, Leisching G. The link between dysregulated immunometabolism and vascular damage: implications for the development of atherosclerosis in systemic lupus erythematosus and other rheumatic diseases. J Rheumatol. (2024) 51:234–41. doi: 10.3899/jrheum.2023-0833
35. Liu Y, Yu X, Zhang W, Zhang X, Wang M, Ji F. Mechanistic insight into premature atherosclerosis and cardiovascular complications in systemic lupus erythematosus. J Autoimmun. (2022) 132:102863. doi: 10.1016/j.jaut.2022.102863
36. Casey KA, Smith MA, Sinibaldi D, Seto NL, Playford MP, Wang X, et al. Modulation of cardiometabolic disease markers by type I interferon inhibition in systemic lupus erythematosus. Arthritis Rheumatol. (2021) 73:459–71. doi: 10.1002/art.41518
37. Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflamm. (2018) 15:199. doi: 10.1186/s12974-018-1235-0
38. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
39. Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H, et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J Autoimmun. (2019) 103:102286. doi: 10.1016/j.jaut.2019.05.014
40. Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. (2019) 26:332–47. doi: 10.1038/s41418-018-0122-7
41. Li D, Chen J, Guo J, Li L, Cai G, Chen S, et al. A phosphorylation of RIPK3 kinase initiates an intracellular apoptotic pathway that promotes prostaglandin2α-induced corpus luteum regression. Elife. (2021) 10:e67409. doi: 10.7554/eLife.67409
42. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA. (2017) 114:E961–9. doi: 10.1073/pnas.1613305114
43. Zhang M, Jie H, Wu Y, Han X, Li X, He Y, et al. Increased MLKL mRNA level in the PBMCs is correlated with autoantibody production, renal involvement, and SLE disease activity. Arthritis Res Ther. (2020) 22:239. doi: 10.1186/s13075-020-02332-7
44. Zhe-Wei S, Li-Sha G, Yue-Chun L. The role of necroptosis in cardiovascular disease. Front Pharmacol. (2018) 9:721. doi: 10.3389/fphar.2018.00721
45. Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. (2015) 6:e1975. doi: 10.1038/cddis.2015.316
46. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. (2016) 2:e1600224. doi: 10.1126/sciadv.1600224
47. Karunakaran D, Nguyen MA, Geoffrion M, Vreeken D, Lister Z, Cheng HS, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce NF-κB activation and atherogenesis in mice. Circulation. (2021) 143:163–77. doi: 10.1161/CIRCULATIONAHA.118.038379
48. An S, Qi Y, Zhang Z, Mo R, Hou L, Yao X, et al. Antagonism of receptor interacting protein 1 using necrostatin-1 in oxidized LDL- induced endothelial injury. BioMed Pharmacother. (2018) 108:1809–15. doi: 10.1016/j.biopha.2018.09.052
49. Xue H, Shi H, Zhang F, Li H, Li C, Han Q. RIP3 contributes to cardiac hypertrophy by influencing MLKL-mediated calcium influx. Oxid Med Cell Longev. (2022) 2022:5490553. doi: 10.1155/2022/5490553
50. Qiao S, Hong L, Zhu Y, Zha J, Wang A, Qiu J, et al. RIPK1-RIPK3 mediates myocardial fibrosis in type 2 diabetes mellitus by impairing autophagic flux of cardiac fibroblasts. Cell Death Dis. (2022) 13:147. doi: 10.1038/s41419-022-04587-1
51. You R, He X, Zeng Z, Zhan Y, Xiao Y, Xiao R. Pyroptosis and its role in autoimmune disease: A potential therapeutic target. Front Immunol. (2022) 13:841732. doi: 10.3389/fimmu.2022.841732
52. Ren W, Sun Y, Zhao L, Shi X. NLRP3 inflammasome and its role in autoimmune diseases: A promising therapeutic target. BioMed Pharmacother. (2024) 175:116679. doi: 10.1016/j.biopha.2024.116679
53. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
54. Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol. (2016) 28:159–73. doi: 10.1016/j.smim.2016.03.004
55. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. (2019) 366:1531–6. doi: 10.1126/science.aav4011
56. Miao N, Wang Z, Wang Q, Xie H, Yang N, Wang Y, et al. Oxidized mitochondrial DNA induces gasdermin D oligomerization in systemic lupus erythematosus. Nat Commun. (2023) 14:872. doi: 10.1038/s41467-023-36522-z
57. Jiang K, Tu Z, Chen K, Xu Y, Chen F, Xu S, et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J Clin Invest. (2022) 132:e151268. doi: 10.1172/JCI151268
58. Fan X, Han J, Zhong L, Zheng W, Shao R, Zhang Y, et al. Macrophage-derived GSDMD plays an essential role in atherosclerosis and cross talk between macrophages via the mitochondria-STING-IRF3/NF-κB axis. Arterioscler Thromb Vasc Biol. (2024) 44:1365–78. doi: 10.1161/ATVBAHA.123.320612
59. Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatol (Oxford). (2015) 54:324–31. doi: 10.1093/rheumatology/keu318
60. Vlachakis PK, Theofilis P, Kachrimanidis I, Giannakopoulos K, Drakopoulou M, Apostolos A, et al. The role of inflammasomes in heart failure. Int J Mol Sci. (2024) 25:5372. doi: 10.3390/ijms25105372
61. Luo B, Wang F, Li B, Dong Z, Liu X, Zhang C, et al. Association of nucleotide-binding oligomerization domain-like receptor 3 inflammasome and adverse clinical outcomes in patients with idiopathic dilated cardiomyopathy. Clin Chem Lab Med. (2013) 51:1521–8. doi: 10.1515/cclm-2012-0600
62. Wu X, Yang J, Wu J, Yang X. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome in systemic lupus erythematosus. BioMed Pharmacother. (2024) 172:116261. doi: 10.1016/j.biopha.2024.116261
63. Shi Y, Zhao L, Wang J, Liu S, Zhang Y, Qin Q. The selective NLRP3 inflammasome inhibitor MCC950 improves isoproterenol-induced cardiac dysfunction by inhibiting cardiomyocyte senescence. Eur J Pharmacol. (2022) 937:175364. doi: 10.1016/j.ejphar.2022.175364
64. Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. (2021) 11:19305. doi: 10.1038/s41598-021-98437-3
65. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. (2020) 126:1260–80. doi: 10.1161/CIRCRESAHA.120.315937
66. Shen S, Duan J, Hu J, Qi Y, Kang L, Wang K, et al. Colchicine alleviates inflammation and improves diastolic dysfunction in heart failure rats with preserved ejection fraction. Eur J Pharmacol. (2022) 929:175126. doi: 10.1016/j.ejphar.2022.175126
67. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev. (2017) 16:1160–73. doi: 10.1016/j.autrev.2017.09.012
68. Singh J, Boettcher M, Dölling M, Heuer A, Hohberger B, Leppkes M, et al. Moonlighting chromatin: when DNA escapes nuclear control. Cell Death Differ. (2023) 30:861–75. doi: 10.1038/s41418-023-01124-1
69. Liu ML, Lyu X, Werth VP. Recent progress in the mechanistic understanding of NET formation in neutrophils. FEBS J. (2022) 289:3954–66. doi: 10.1111/febs.16036
70. Goldberg MW, Huttenlauch I, Hutchison CJ, Stick R. Filaments made from A- and B-type lamins differ in structure and organization. J Cell Sci. (2008) 121:215–25. doi: 10.1242/jcs.022020
71. Li Y, Li M, Weigel B, Mall M, Werth VP, Liu ML, et al. Nuclear envelope rupture and NET formation is driven by PKCα-mediated lamin B disassembly. EMBO Rep. (2020) 21:e48779. doi: 10.15252/embr.201948779
72. Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, et al. Cell-cycle proteins control production of neutrophil extracellular traps. Dev Cell. (2017) 43:449–62.e5. doi: 10.1016/j.devcel.2017.10.013
73. Pisareva E, Mihalovičová L, Pastor B, Kudriavtsev A, Mirandola A, Mazard T, et al. Neutrophil extracellular traps have auto-catabolic activity and produce mononucleosome-associated circulating DNA. Genome Med. (2022) 14:135. doi: 10.1186/s13073-022-01125-8
74. Li M, Weng L, Yu D, Yang G, Hao J. Increased formation of neutrophil extracellular traps induced by autophagy and identification of autophagy-related biomarkers in systemic lupus erythematosus. Exp Dermatol. (2024) 33:e14881. doi: 10.1111/exd.14881
75. Dieker J, Tel J, Pieterse E, Thielen A, Rother N, Bakker M, et al. Circulating apoptotic microparticles in systemic lupus erythematosus patients drive the activation of dendritic cell subsets and prime neutrophils for NETosis. Arthritis Rheumatol. (2016) 68:462–72. doi: 10.1002/art.39417
76. Rother N, Pieterse E, Lubbers J, Hilbrands L, van der Vlag J. Acetylated histones in apoptotic microparticles drive the formation of neutrophil extracellular traps in active lupus nephritis. Front Immunol. (2017) 8:1136. doi: 10.3389/fimmu.2017.01136
77. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
78. Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci Med. (2020) 7:e000387. doi: 10.1136/lupus-2020-000387
79. Henning S, Reimers T, Abdulahad W, Fierro JJ, Doornbos-van der Meer B, Bootsma H, et al. Low density granulocytes and neutrophil extracellular trap formation are increased in incomplete systemic lupus erythematosus. Rheumatol (Oxford). (2024) keae300. doi: 10.1093/rheumatology/keae300
80. Pertiwi KR, van der Wal AC, Pabittei DR, Mackaaij C, van Leeuwen MB, Li X, et al. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb Haemost. (2018) 118:1078–87. doi: 10.1055/s-0038-1641749
81. Hofbauer TM, Mangold A, Scherz T, Seidl V, Panzenböck A, Ondracek AS, et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res Cardiol. (2019) 114:33. doi: 10.1007/s00395-019-0740-3
82. Moore S, Juo HH, Nielsen CT, Tyden H, Bengtsson AA, Lood C. Role of neutrophil extracellular traps regarding patients at risk of increased disease activity and cardiovascular comorbidity in systemic lupus erythematosus. J Rheumatol. (2020) 47:1652–60. doi: 10.3899/jrheum.190875
83. Nappi F, Bellomo F, Avtaar Singh SS. Worsening thrombotic complication of atherosclerotic plaques due to neutrophils extracellular traps: A systematic review. Biomedicines. (2023) 11:113. doi: 10.3390/biomedicines11010113
84. Ibrahim N, Eilenberg W, Neumayer C, Brostjan C. Neutrophil extracellular traps in cardiovascular and aortic disease: A narrative review on molecular mechanisms and therapeutic targeting. Int J Mol Sci. (2024) 25:3983. doi: 10.3390/ijms25073983
85. Yao M, Ma J, Wu D, Fang C, Wang Z, Guo T, et al. Neutrophil extracellular traps mediate deep vein thrombosis: from mechanism to therapy. Front Immunol. (2023) 14:1198952. doi: 10.3389/fimmu.2023.1198952
86. Chrysanthopoulou A, Gkaliagkousi E, Lazaridis A, Arelaki S, Pateinakis P, Ntinopoulou M, et al. Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension. JCI Insight. (2021) 6:e148668. doi: 10.1172/jci.insight.148668
87. Krishnan J, Hennen EM, Ao M, Kirabo A, Ahmad T, de la Visitación N, et al. NETosis drives blood pressure elevation and vascular dysfunction in hypertension. Circ Res. (2024) 134:1483–94. doi: 10.1161/CIRCRESAHA.123.323897
88. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. (2014) 114:947–56. doi: 10.1161/CIRCRESAHA.114.303312
89. Lin W, Chen H, Chen X, Guo C. The roles of neutrophil-derived myeloperoxidase (MPO) in diseases: the new progress. Antioxidants (Basel). (2024) 13:132. doi: 10.3390/antiox13010132
90. Chen W, Tumanov S, Kong SMY, Cheng D, Michaëlsson E, Bongers A, et al. Therapeutic inhibition of MPO stabilizes pre-existing high risk atherosclerotic plaque. Redox Biol. (2022) 58:102532. doi: 10.1016/j.redox.2022.102532
91. Yang K, Gao R, Chen H, Hu J, Zhang P, Wei X, et al. Myocardial reperfusion injury exacerbation due to ALDH2 deficiency is mediated by neutrophil extracellular traps and prevented by leukotriene C4 inhibition. Eur Heart J. (2024) 45:1662–80. doi: 10.1093/eurheartj/ehae205
92. Li S, Huang Y. Ferroptosis: an iron-dependent cell death form linking metabolism, diseases, immune cell and targeted therapy. Clin Transl Oncol. (2022) 24:1–12. doi: 10.1007/s12094-021-02669-8
93. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. (2021) 7:193. doi: 10.1038/s41420-021-00579-w
94. Zhang W, Liu Y, Liao Y, Zhu C, Zou Z. GPX4, ferroptosis, and diseases. Biomed Pharmacother. (2024) 174:116512. doi: 10.1016/j.biopha.2024.116512
95. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. (2021) 22:1107–17. doi: 10.1038/s41590-021-00993-3
96. Chen Q, Xiang M, Gao Z, Lvu F, Sun Z, Wang Y, et al. The role of B-cell ferroptosis in the pathogenesis of systemic lupus erythematosus. Clin Immunol. (2023) 256:109778. doi: 10.1016/j.clim.2023.109778
97. Yang B, Hou S, Huang S, Li H, Li Y. Ferroptosis inhibitor regulates the disease progression of systematic lupus erythematosus mice model through Th1/Th2 ratio. Curr Mol Med. (2023) 23:799–807. doi: 10.2174/1566524022666220525144630
98. Zhang Z, Yang Z, Wang S, Wang X, Mao J. Decoding ferroptosis: Revealing the hidden assassin behind cardiovascular diseases. BioMed Pharmacother. (2024) 176:116761. doi: 10.1016/j.biopha.2024.116761
99. Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE–mechanisms and management. Nat Rev Rheumatol. (2012) 8:214–23. doi: 10.1038/nrrheum.2012.14
100. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
101. Zhou Y, Zhou H, Hua L, Hou C, Jia Q, Chen J, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med. (2021) 171:55–68. doi: 10.1016/j.freeradbiomed.2021.05.009
102. Tian H, Xiong Y, Zhang Y, Leng Y, Tao J, Li L, et al. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones. (2021) 27:149–64. doi: 10.1007/s12192-022-01257-1
103. Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P, Zheng SY. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. (2021) 27:14. doi: 10.1186/s10020-021-00271-w
Keywords: programmed cell death, systemic lupus erythematosus, cardiac involvement, crosstalk, immune dysregulation
Citation: Wei J, Wang A, Li B, Li X, Yu R, Li H, Wang X, Wang Y and Zhu M (2024) Pathological mechanisms and crosstalk among various cell death pathways in cardiac involvement of systemic lupus erythematosus. Front. Immunol. 15:1452678. doi: 10.3389/fimmu.2024.1452678
Received: 21 June 2024; Accepted: 19 August 2024;
Published: 05 September 2024.
Edited by:
Jianan Zhao, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Dongyi He, Shanghai Guanghua Rheumatology Hospital, ChinaPing Jiang, Shanghai Jiao Tong University, China
Ming-Lin Liu, University of Pennsylvania, United States
Copyright © 2024 Wei, Wang, Li, Li, Yu, Li, Wang, Wang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinlu Wang, d2FuZ3hpbmx1MTEwQDEyNi5jb20=; Yongxia Wang, d3l4Y2h6aHFAMTYzLmNvbQ==; Mingjun Zhu, emh1bWluZ2p1bjMxN0AxNjMuY29t
†These authors have contributed equally to this work