
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 03 September 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1448724
 Jiazhi Yi1†Shuyun Wu2†Hongxia He3*
Jiazhi Yi1†Shuyun Wu2†Hongxia He3*Objectives: Previous observational epidemiological studies have identified a potential association between inflammatory bowel disease (IBD) and sarcoidosis. Nonetheless, the precise biological mechanisms underlying this association remain unclear. Therefore, we adopted a Mendelian randomization (MR) approach to investigate the causal relationship between IBD with genetic susceptibility to sarcoidosis, as well as to explore the potential mediating role.
Methods: The genetic associations were obtained from publicly available genome-wide association studies (GWASs) of European ancestry. The IBD dataset has 31,665 cases and 33,977 controls, consisting of 13,768 individuals with ulcerative colitis (UC) and 17,897 individuals with Crohn’s disease (CD). The genetic associations of sarcoidosis with 4,854 cases and 446,523 controls. A bidirectional causality between IBD and sarcoidosis was implemented to be determined by a two-sample MR approach. The inverse variance weighted (IVW) method was utilized as the main statistical method, and a series of sensitivity analyses were performed to detect heterogeneity and horizontal pleiotropy. A two-step MR approach was used to investigate whether the mediating pathway from IBD to sarcoidosis was mediated by PBC.
Results: The forward MR analysis indicated that genetic predisposition to IBD was significantly linked to an increased risk of sarcoidosis (OR = 1.088, 95% CI: 1.023–1.158, pIBD-sar = 7.498e-03). Similar causal associations were observed in CD (OR = 1.082, 95% CI: 1.028–1.138, pCD-sar = 2.397e-03) and UC (OR = 1.079, 95% CI: 1.006–1.158, pUC-sar = 0.034). Reverse MR analysis revealed that genetic susceptibility to sarcoidosis was correlated with an augmented risk of CD (OR = 1.306, 95% CI: 1.110–1.537, psar-CD = 1.290e-03) but not IBD or UC. The mediation analysis via two-step MR showed that the causal influence of IBD and CD on sarcoidosis effects was partly mediated by PBC, and the mediating effect was 0.018 (95% CI: 0.005–0.031, p = 7.596e-03) with a mediated proportion of 21.397% in IBD, and 0.014 (95% CI: 0.004–0.024, p = 7.800e-03) with a mediated proportion of 17.737% in CD.
Conclusions: The MR analysis provided evidence substantiating the causal effect of IBD (CD and UC) on an increased risk of sarcoidosis, with PBC playing a mediating role in IBD and CD. However, sarcoidosis only enhances the risk of developing CD, but not IBD or UC. These findings illuminate the etiology of sarcoidosis and contribute to the management of IBD patients.
Inflammatory bowel disease (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic and nonspecific disorder of the gastrointestinal tract (1–3). Despite significant progress in understanding IBD, the comprehension of etiological factors and the complex pathogenetic process remains incomplete (4). Sarcoidosis is a multisystem granulomatous disease that can affect any body organ at any age. This disease is highly heterogeneous and has an unpredictable clinical course (5–7). The etiology of sarcoidosis remains unknown; it is plausible that the disease is an immunological disorder par excellence, caused by one or several antigen exposures, where different genes induce an altered immune response after contact with specific antigenic stimuli that initiate and possibly perpetuate the granulomatous process (6).
The relationship between IBD and sarcoidosis has recently sparked people’s curiosity. Although both have traditionally been considered separate entities, IBD and sarcoidosis share similar clinical and histopathological manifestations (8). The potential pathological changes in each disease give rise to the formation of granulomas (9, 10) and result in clinical overlap of common organs (8, 11, 12). Mitchell et al. first demonstrated nearly 50% positivity of Kveim testing in CD patients, indicating a shared mechanism between CD and sarcoidosis (13). Other observational studies have shown that an elevated ratio of CD4/CD8 lymphocytes in bronchoalveolar lavage fluid was present in both CD and sarcoidosis (14). Additionally, similar responses to mycobacterial infection were observed in CD (15) and sarcoidosis tissues (16). The coexistence of UC and sarcoidosis has also been previously reported. Moreover, several studies have suggested their immunogenicity and pathogenicity associations (17, 18). Halling et al. confirmed a significantly increased risk of sarcoidosis in patients diagnosed with UC or (and) CD (19). However, apart from these observational studies, limited knowledge exists regarding the association between IBD and sarcoidosis.
Observational studies have indicated that the incidence of autoimmune liver disease primary biliary cholangitis (PBC) is higher in IBD patients than in healthy individuals (20). IBD and PBC share certain metabolites that are causally associated with both, such as isovalerylcarnitine, and may have analogous pathogenesis (21, 22). On the other hand, clinical features of overlap between PBC and morphologically proven sarcoidosis lesions have been demonstrated (23–28). However, the cause-and-effect relationship of IBD on PBC, and the causal link between PBC and sarcoidosis, are not widely known. Moreover, whether the causal effect of IBD on sarcoidosis is mediated by the role of PBC remains unclear.
Mendelian randomization (MR) is a method frequently employed for exploring causal links between risk factors and outcomes by utilizing single-nucleotide polymorphisms (SNPs) as instrumental variables (IVs) (29–32). Here, we employed a two-sample bidirectional MR analysis to assess the potential causal relationship of IBD, including CD and UC, with the risk for sarcoidosis, and whether PBC can act as a mediator in this connection.
This study was conducted following the reporting guideline of the Strengthening the Reporting of Observational Studies in Epidemiology (https://www.strobe-mr.org/).
We used a two-sample MR design: a genetic IV analysis based on summary-level data with SNPs as instruments for the risk factor. For causal estimates from MR studies to be valid, three assumptions must be adhered to (33): (1) the genetic variants are highly associated with the exposure, (2) the genetic variants are not associated with any potential confounder of the exposure–outcome association, and (3) the variants exclusively affect the outcome through the exposure. We first performed a two-sample bidirectional MR to investigate the causal relationship of IBD, UC, and CD with sarcoidosis risk. The IVs, for IBD encompassing CD and UC subtypes, were extracted from a GWAS: the International IBD Genetics Consortium (IIBDGC) by Liu et al., including 31,665 cases and 33,977 controls of European ancestry, with 13,768 UC and 17,897 CD cases (34). The genetic association data of sarcoidosis were obtained from a GWAS that consists of 4,854 cases and 446,523 controls (https://r11.finngen.fi/pheno/D3_SARCOIDOSIS) in the European population. We obtained genetic association data of PBC from a GWAS that consists of 8,021 European cases and 16,489 European controls (35) (Supplementary Table 1). If there are significant causal associations between IBD (including CD and UC) and PBC and both of them have causal relations on sarcoidosis, a two-step MR analysis was performed to investigate whether the mediating pathway from IBD to sarcoidosis was mediated by PBC. All GWAS summary data are publicly available and therefore no additional ethical approval or informed consent was required. All summary statistics used were GWAS analyses and no sample overlap was observed.
Genetic IVs were selected. Identified SNPs at each significance threshold (p < 5×10−8) were clumped for independence using PLINK clumping in the TwoSampleMR tool (36). Since there were too few SNPs with p-values less than 5×10−8 for sarcoidosis, we extended the threshold to 5×10−6 to select eligible genetic IVs, which had been applied to previous MR research (37). A strict cutoff of R2 < 0.001 and a window of 10,000 kb were used for clumping with the 1000 Genomes European data as the reference panel. The proportions of trait variance explained by the identified SNPs were calculated using the following formula: R2 = 2 × β2 × MAF × (1−MAF)/(2 × MAF × (1 − MAF) × β2 + 2 × MAF × (1 − MAF) × N × se(β)2) (38). In addition, we evaluated instrument strength using the F statistic, where F = (R2 × (N − k − 1))/k(1 − R2), to test the significant association of the genetic instruments with the exposure (39), and SNPs with F < 10 were excluded, as an F ≥ 10 indicates a relatively low risk of weak instrument bias in MR analysis (40). To avoid potential confounding, we investigated each instrument SNP in the PhenoScanner GWAS database (41) to assess any previous associations (p < 5×10−8) with plausible confounders (including obesity, celiac disease, psychological stress, physical activity, trunk fat percentage, waist circumference, alcohol intake frequency, BMI, hypothyroidism, psoriasis, rheumatoid arthritis, and education), which have been reported to affect IBD (42–52), PBC (53–56), and sarcoidosis (57–59). To meet the assumption that requires instruments to be associated with the outcome only through exposure, we excluded SNPs highly linked to the outcome. The effects of SNPs on exposure and outcome were then harmonized to ensure that the β values were signed to the same alleles. After data harmonization, we removed palindromic SNPs with intermediate allele frequencies (>0.42) and outlier pleiotropic SNPs via the heterogeneity test (modified Q statistics) using RadialMR (Version 1.0) with the p-value threshold of 0.05 (60). The remaining SNPs were used to perform MR analysis.
The inverse variance weighted (IVW) method was used as the main approach in this MR analysis, which provides accurate estimates in the absence of heterogeneity and directional pleiotropy between the exposure and outcome (61–63). The heterogeneity of the IVW model was assessed by Cochran’s Q test. If Cochran’s Q test suggested significant heterogeneity (p < 0.05), we turned from the fixed IVW model to the random-effects model. In addition, the MR Egger method was performed to estimate the causal effect, with the capability of identifying and accounting for any directional pleiotropy (64, 65). The MR Pleiotropy RESidual Sum and Outlier (MR-PRESSO) method was used to evaluate horizontal pleiotropy (66). If horizontal pleiotropy exists, horizontal pleiotropy was corrected by removing the outlier and determining whether there are substantial variations in the causal effects before and after the outlier removal. Moreover, the MR-Egger regression intercept term was used to assess the possible presence of horizontal pleiotropy, where deviation from zero (p < 0.05) indicates directional pleiotropy. All statistical analyses were performed in R 4.3.1 with the package TwoSampleMR (version 0.5.7) and MRPRESSO (Version 1.0).
The quantity and specific characteristics of SNPs selected for IBD, CD, UC phenotype, and sarcoidosis are shown in Supplementary Tables 2-6. Based on the aforementioned selection criteria, the linkage disequilibrium test was initially conducted to select SNPs associated with IBD (encompassing CD and UC) and sarcoidosis at first. After eliminating confounders and undergoing quality control (Supplementary Table 17), a total of 109, 102, and 64 SNPs related to sarcoidosis in association with IBD, CD, and UC, respectively, were obtained. Cochran’s Q statistics test identified statistical heterogeneity using IVW and MR-Egger (IVW IBD, CD, and UC for sarcoidosis, p = 6.295e-10, 4.064e-09, and 2.584e-06, respectively), as listed in Table 1; therefore, analyses were conducted using the IVW with the multiplicative random-effects model for IBD, CD, and UC (67). We observed a significantly causal association using the IVW method between IBD (CD and UC) and sarcoidosis (OR = 1.088, 95% CI: 1.023–1.158, pIBD-sar = 7.498e-03; OR = 1.082, 95% CI: 1.028-1.138, pCD-sar = 2.397e-03; OR = 1.079, 95% CI: 1.006-1.158, pUC-sar = 0.034) (Figure 1). The sensitivity analysis indicated no remarkable horizontal pleiotropy (Table 1). Additionally, the leave-one-out analysis showed that the genetically predicted causal associations between IBD (CD and UC) and sarcoidosis were not driven by a single SNP. Funnel plot, scatter plot, and forest plot of the MR are presented (Supplementary Figures 1-3). Thus, a positive link between genetic susceptibility of IBD (CD and UC) and risk of sarcoidosis was discovered in MR. Together, IBD with its subtypes and sarcoidosis are causally associated with MR estimation results, and all sensitivity analyses as qualitative control support the idea that the causal association between IBD (UC and CD) and sarcoidosis has weak bias.
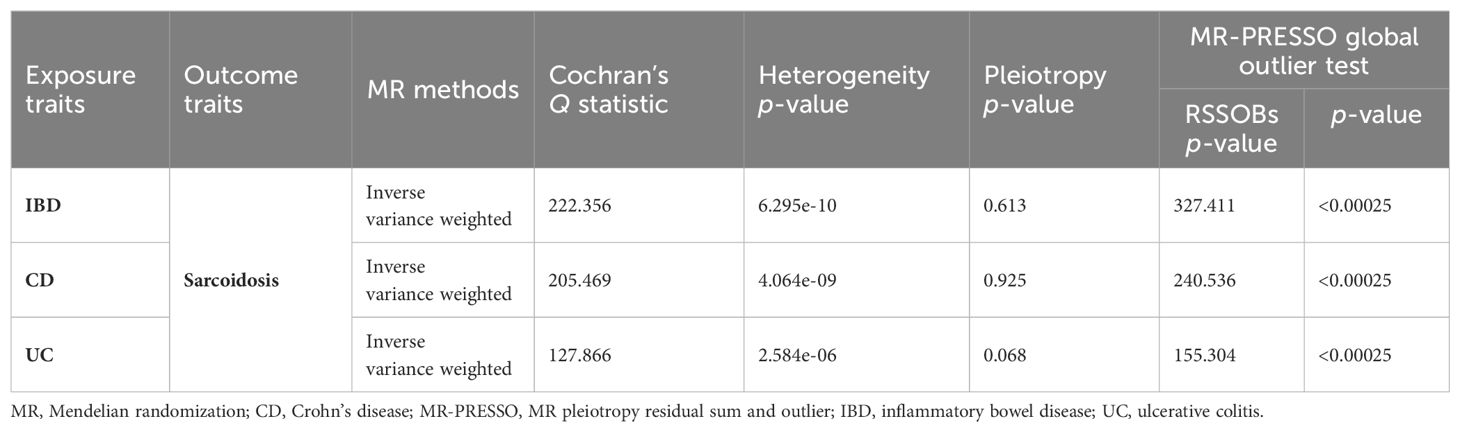
Table 1. Heterogeneity and pleiotropy analysis of IBD (CD and UC) and sarcoidosis using different analytical methods.
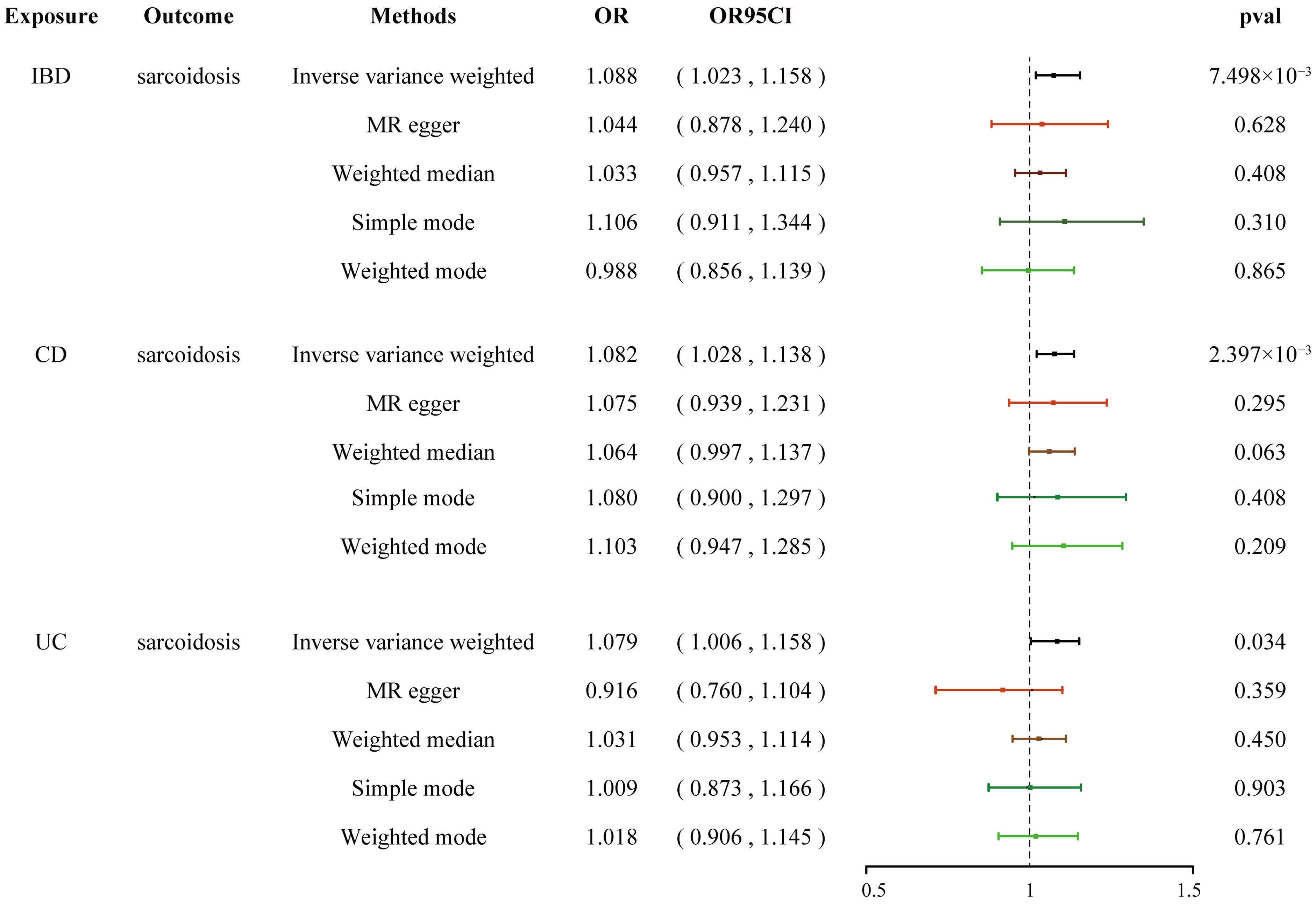
Figure 1. Forest plots shows the causal estimates given as OR and 95% confidence intervals for the effect of IBD, CD, and UC on sarcoidosis. IBD, inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis; OR, odds ratio.
We extended the threshold to 5×10−6 to select eligible IVs (37) linked to sarcoidosis and IBD (encompassing CD and UC). The F-statistics for the genetic instruments were consistent with an absence of weak instrument bias (Supplementary Table 7). After eliminating confounders and undergoing quality control (Supplementary Table 18), a total of two, two, and five SNPs were obtained related to IBD, CD, and UC in association with sarcoidosis (Supplementary Tables 8-10). Cochran’s Q test showed no evidence for heterogeneity of IBD (pIBD = 0.051), CD (pCD = 0.683), and UC (pUC = 0.083) (Supplementary Table 20); therefore, analyses were conducted using the IVW method with the multiplicative fixed-effects model. The results showed that sarcoidosis had a suggestive causal effect on CD (OR = 1.306, 95% CI: 1.110–1.537, psar-CD = 1.290e-03). However, we found no evidence of causal relationships of sarcoidosis with IBD (OR = 1.013, 95% CI: 0.796–1.289, psar-IBD = 0.919) and UC (OR = 1.104, 95% CI: 0.970–1.257, psar-CD = 0.132) (Supplementary Figure 4). The forest, funnel, and scatter plots of IBD and its sub-phenotypes are presented in Supplementary Figures 5-7. Thus, there is a positive link between genetic susceptibility of sarcoidosis on the risk of CD, but not IBD and UC.
We conducted a two-step MR analysis to investigate the mediating pathway from IBD to sarcoidosis via PBC. In the first step, genetic instruments for IBD (encompassing CD and UC) were used to estimate the causal effect of the exposure on PBC as the potential mediator. SNPs related to potential confounders and palindromic SNPs with intermediate allele frequencies (>0.42) were removed (Supplementary Table 19). Then, summary information on the PBC-related phenotype for the SNPs associated with IBD (CD and UC) is listed in Supplementary Tables 3 and 11–13. As listed in Table 2, the Cochran’s Q statistics test identified statistical heterogeneity using IVW and MR-Egger (IVW IBD, CD, and UC for PBC, p = 1.195e-13, 7.371e-11, and 7.160e-05, respectively); therefore, analyses were conducted using the IVW with the multiplicative random-effects model for IBD, CD, and UC. We observed that IBD and CD were causally associated with PBC, and the IVW method showed that genetically predicted IBD (OR = 1.204, 95% CI: 1.095–1.324, pIBD-PBC = 1.280e-04) and CD (OR = 1.154, 95% CI: 1.072–1.243, pCD-PBC = 1.420e-04) were significantly associated with an increased risk of PBC. Other methods did not reach statistical significance for the causal association between IBD and PBC. Similarly, excluding MR-Egger (OR = 1.037, 95% CI: 0.865–1.244, p = 0.695) and the Simple model (OR = 1.150, 95% CI: 0.988–1.339, p = 0.076), other methods supported the nominally significant causal association between CD and PBC (weighted median OR = 1.102, 95% CI: 1.020–1.192, p = 0.014; weighted model OR = 1.108, 95% CI: 1.026–1.196, p = 0.011) (Figure 2). However, MR analysis as a result of the IVW model implied that genetic susceptibility to UC had no causal association with PBC (OR = 1.066, 95% CI: 0.971–1.171, pUC-PBC = 0.178), and the weighted median, the weighted mode, the Simple model, and MR-Egger yielded similar patterns of effects (Figure 2). The leave-one-out analysis, funnel plot, scatter plot, and forest plot of the MR of IBD, CD, and UC on PBC risk are presented in Supplementary Figures 8-10. In the second step, we assessed the causal effect of the mediator on sarcoidosis risk using genetic instruments for the PBC-related phenotype, and to avoid potential confounding, any previous associations with plausible confounders (that is celiac disease) were removed (Supplementary Table 21). Since the Cochran’s Q statistics test identified a statistical heterogeneity (IVW PBC for sarcoidosis, p = 0.015) (Table 3), the Forest plot showed causal evidence for the effect of PBC on sarcoidosis by using the IVW method with the multiplicative random-effects model (OR = 1.102, 95% CI: 1.047–1.160, pPBC-Sar = 1.971e-04). However, other methods did not reach statistical significance for the causal association (Figure 3, Supplementary Figure 11). Then, given that significant causal effects of IBD and CD on PBC and sarcoidosis were found in the abovementioned analyses, the two-step MR analysis was performed to estimate the mediation effects of PBC between IBD/CD and sarcoidosis. We estimated the indirect effect of IBD and CD on sarcoidosis via PBC and obtained the mediation effect of PBC was 0.018 (95% CI: 0.005–0.031; p = 7.596e-03) in IBD and 0.014 (95% CI: 0.004–0.024; p = 7.800e-03) in CD, with a mediated proportion of 21.397% and 17.737%, respectively (Figure 4, Table 4).
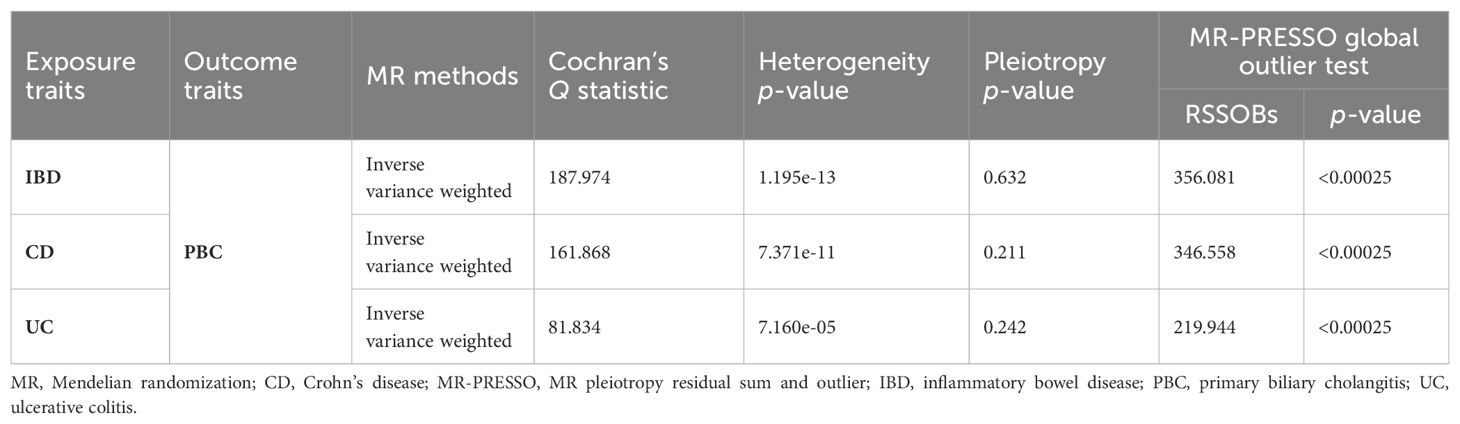
Table 2. Heterogeneity and pleiotropy analysis of IBD, CD, and UC with PBC using different analytical methods.
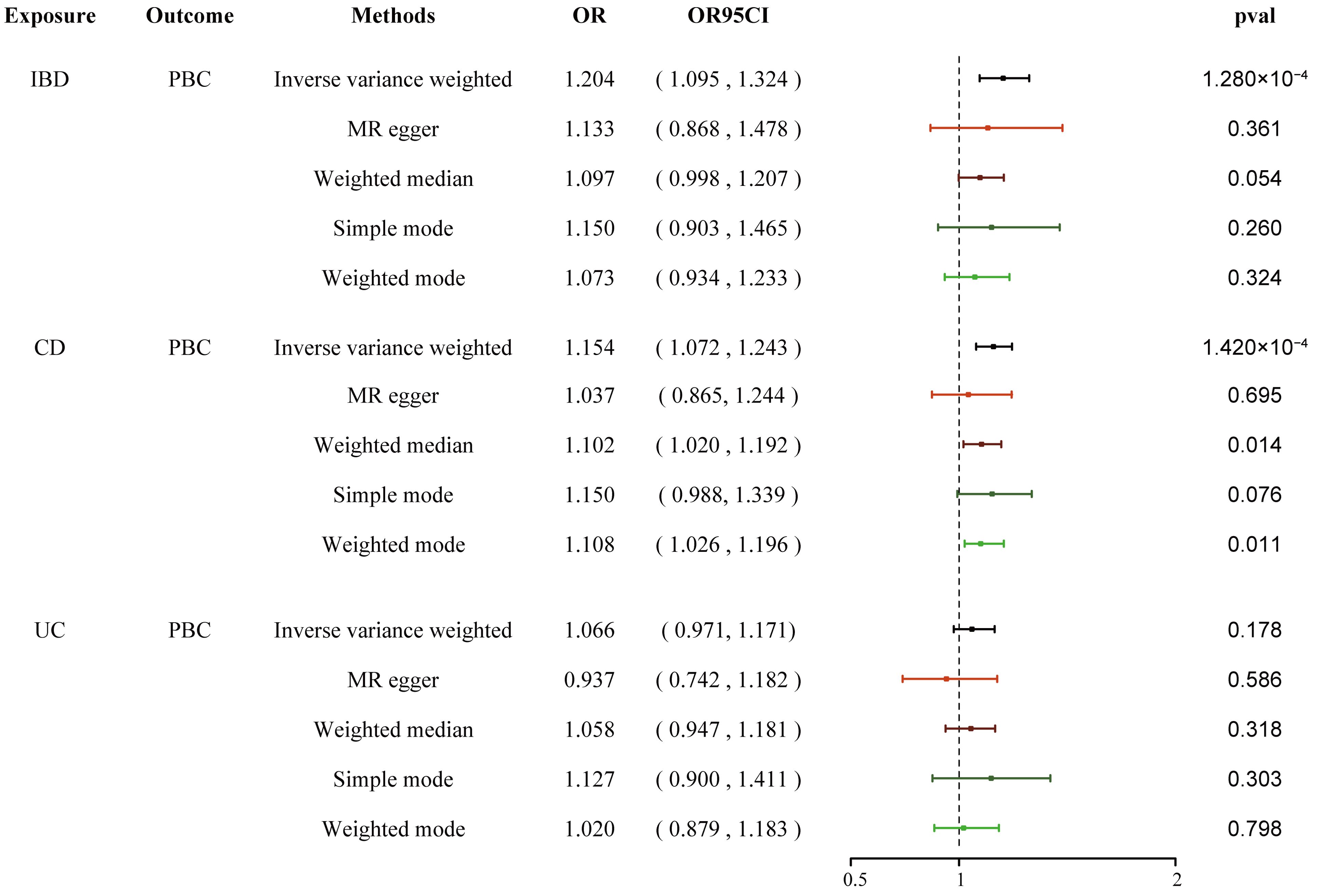
Figure 2. Causal estimates given as OR and 95% confidence intervals for the effect of IBD, CD, and UC on PBC. IBD, inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis; PBC, primary biliary cholangitis; OR, odds ratio.
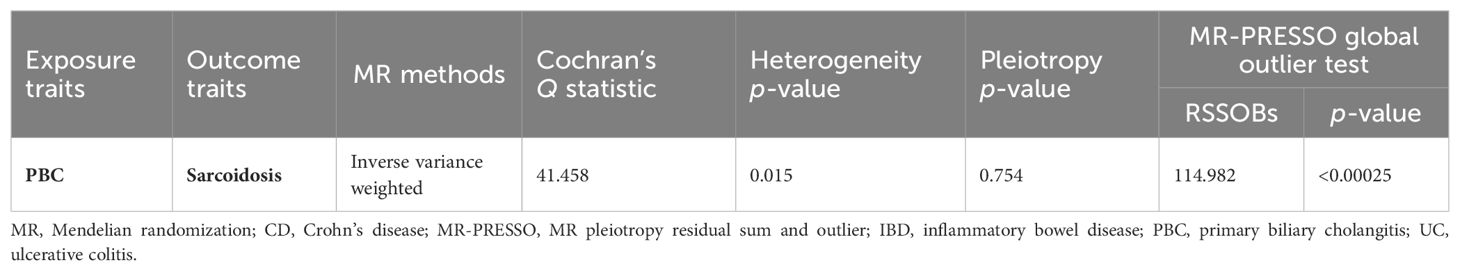
Table 3. Heterogeneity and pleiotropy analysis of PBC and sarcoidosis using different analytical methods.
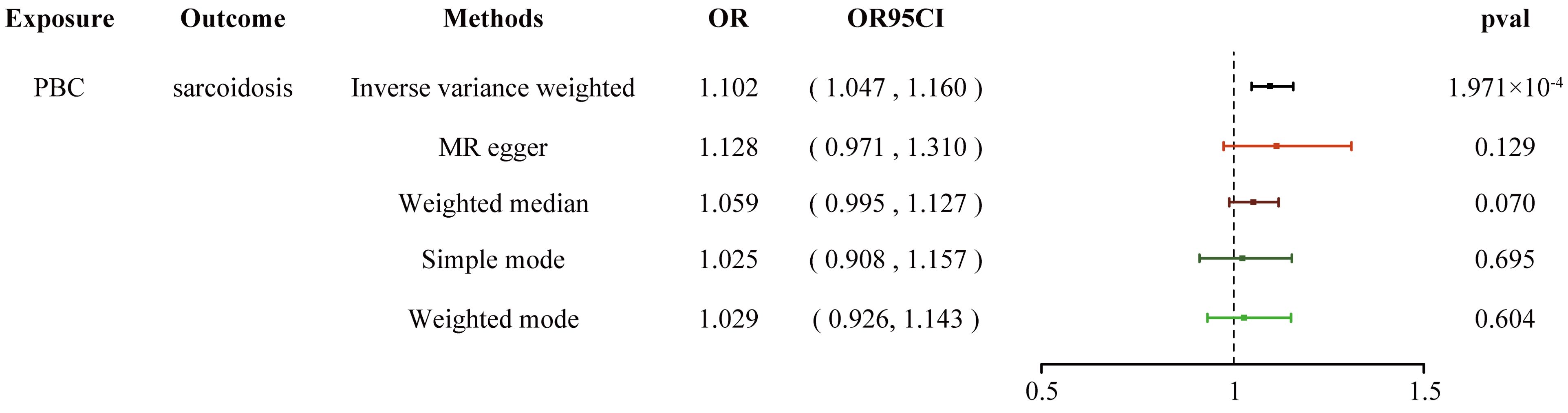
Figure 3. Forest plot shows the causal estimates given as OR and 95% confidence intervals for the effect of PBC on sarcoidosis. PBC, primary biliary cholangitis; OR, odds ratio.
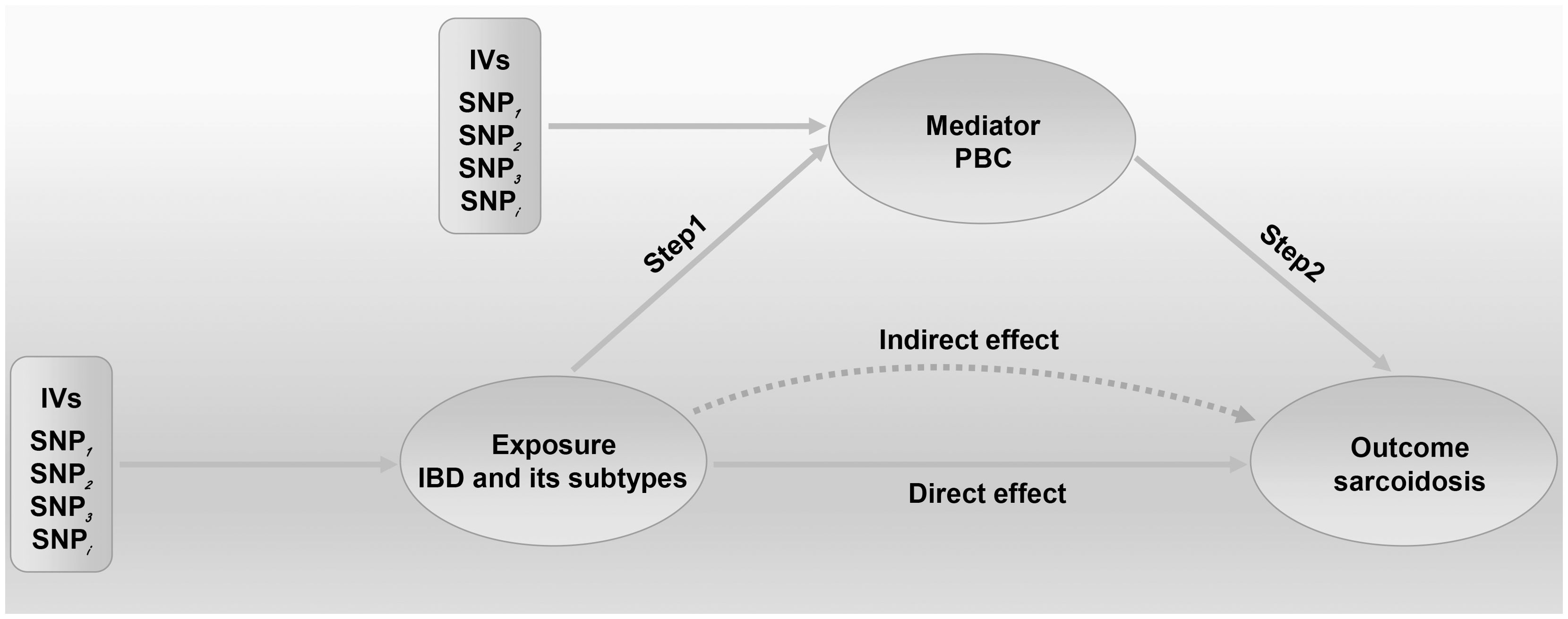
Figure 4. Two-step Mendelian randomization (MR) analysis framework shows the mediation analysis of the effect of IBD and CD on sarcoidosis via PBC. We extended the threshold to 5×10−8 or 5×10−6 to select eligible IVs in this MR study. Step 1 estimated the causal effect of the exposure on the potential mediator, and step 2 assessed the causal effect of the mediator on sarcoidosis risk. “Total effect” indicates the effect of IBD (CD) on sarcoidosis, “Direct effect A” indicates the effect of IBD (CD) on PBC, “Direct effect B” indicates the effect of PBC on sarcoidosis, and “mediation effect” indicates the effect of IBD (CD) on sarcoidosis through PBC. Total effect, Direct effect A, and Direct effect B were derived by IVW; IVs, instrumental variables; IVW, inverse variance weighted; IBD, inflammatory bowel disease; CD, Crohn’s disease; PBC, primary biliary cholangitis.
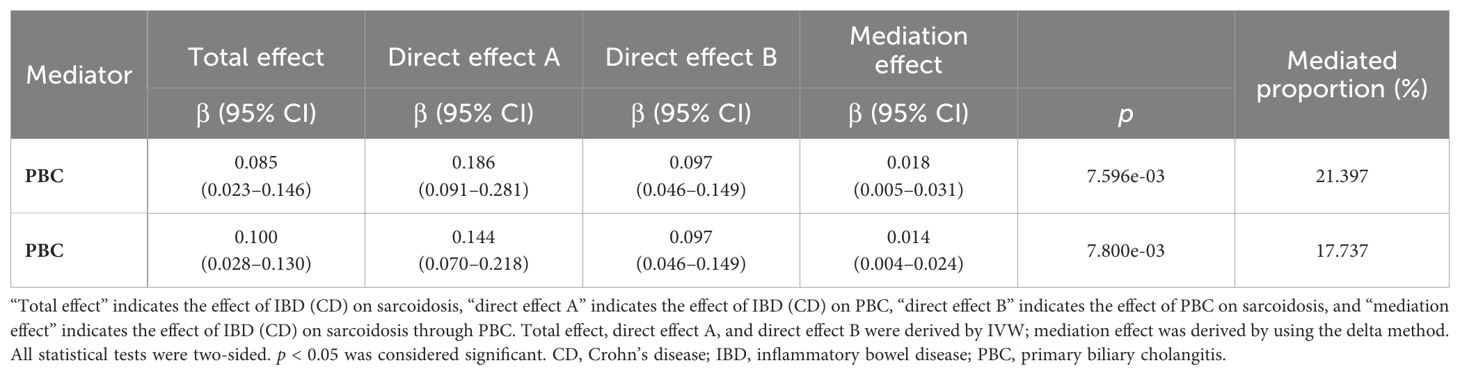
Table 4. The mediation effect of IBD and CD on Sarcoidosis via PBC.
In this MR analysis, we found a causal relationship between IBD and its two subtypes with sarcoidosis. Furthermore, we conducted a mediation analysis and demonstrated for the first time that the effect of IBD on sarcoidosis risk was partially mediated by PBC.
The etiology factors and the complex pathogenetic process of IBD are not yet fully understood. It has been suggested that IBD emerges in genetically susceptible individuals from a convergence of genetic risk, environmental factors, and gut microbiota (1–4). So far, several susceptible genes have been identified, many of which are clarified in other immune-mediated inflammatory diseases (IMIDs). However, apart from the extraintestinal manifestations, little is known about the association between IBD and other IMIDs. Because sarcoidosis is an IMID of unknown cause, clinicians approach it as a systemic disease with extra-pulmonary organ system manifestations including skin, lymph nodes, and the central nervous system (5–7). Evidence from observational case reports and retrospective study suggests that IBD and sarcoidosis often co-occur (8, 13, 14, 17–19), some of which indicated that IBD is linked to an increased risk of sarcoidosis (19). The GWAS has found that the HLA loci associated with IBD were also associated with sarcoidosis (68), which might affect the onset of sarcoidosis caused by IBD. However, given that there are only a few scattered reports rather than evidence from RCTs, the different study designs and the varying validities of diagnoses, population sizes, and confounders, for instance, ethnicity and economic and social status, all make the findings of these studies difficult to interpret. Moreover, it is difficult to distinguish whether it is the chronic inflammatory disease activity itself or the treatment regimen, including immunosuppressive drugs and biological agents, that increases a patient’s risk of sarcoidosis. Furthermore, it is difficult to distinguish whether it is the chronic inflammatory disease activity itself or the treatment regimen, including immunosuppressive drugs and biological agents, that increases a patient’s risk of sarcoidosis. We performed MR analysis to assess the evidence of causal association of IBD, CD, and UC with sarcoidosis. We found that the risks of sarcoidosis were elevated by 8.8%, 8.2%, and 7.9% for IBD, CD, and UC, respectively. Halling et al. (19) reported that IBD is linked to an increased risk of sarcoidosis, which is consistent with our estimate in terms of direction and magnitude. Moreover, there is a positive link between genetic susceptibility of sarcoidosis and the risk of CD; however, we found no evidence of causal relationships of sarcoidosis with IBD and UC.
Previous research indicates that the prevalence of any IMIDs is higher in IBD patients than in the general population (69). Most IMIDs are considered to be Th1 mediated, and the presence of Th17 cells in IBD and their ability to induce a Th1 response might explain this (70–73). Cumali Efe’s study has specifically analyzed the frequency of sarcoidosis in patients with PBC (74). Observational studies have shown that the frequency of PBC is higher in IBD patients (19); both share the same metabolites (21) and have an analogical pathogenesis causally associated with each other (22). The pathogenesis of PBC resembles those of CD and UC, which involves the pathogenicity and synergistic action of Th1 and Th17 cells (75–77). Moreover, some lines of evidence suggest that there is a shared genetic component between IBD and PBC (78, 79). We demonstrated that the promotion effect of IBD on sarcoidosis risk was partially mediated by PBC, which was partially in line with Zhang et al.’s recent study (80). These two simultaneous occurrences of uncommon disorders are all considered to be T cell-mediated diseases, suggesting that a common pathway might contribute to the granuloma formation in both diseases and influence each other.
Admittedly, there are limitations and restrictions in the current investigation. One of the constraints in this study is that we were compelled to rely solely on GWASs conducted in persons of European ancestry to estimate the causal effects due to the absence of extensive GWASs conducted in non-European ancestries. Thus, caution should be exercised when generalizing our findings to other ethnic groups. However, population stratification was not a potential bias in our study since European ancestry was predominant in all datasets. Then, regrettably, we must acknowledge the limitation linked to the use of the IEU GWAS database, which does not allow for subgroup analysis based on gender as conditions of IMIDs are more commonly found in female than in male patients. Therefore, we were unable to provide gender-adjusted ORs. Finally, the underlying mechanisms to determine the association of the causal pathway are needed.
In conclusion, this study demonstrated that IBD and its two subtypes CD and UC had an impact on sarcoidosis, which was partially mediated by PBC. If IBD truly increases the risk of sarcoidosis, promoting mucosal healing as a promising therapeutic strategy in IBD might help prevent PBC and sarcoidosis among individuals at elevated risk. This is of great significance in preventing their detrimental clinical process and restricting their psychological influence, and vice versa.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
JY: Conceptualization, Data curation, Software, Project administration, Investigation, Formal analysis, Writing – original draft. SW: Data curation, Project administration, Formal analysis, Methodology, Writing – original draft. HH: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The study reported here was conducted using the GWASs data from the IEU GWAS database and other prominent international consortiums, including the IIBDGC consortium. We would like to thank all participants and the abovementioned consortiums for their contribution.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1448724/full#supplementary-material
1. Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. (2008) 8:458–66. doi: 10.1038/nri2340
2. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Anderson CA et al: Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
3. Rudbaek JJ, Agrawal M, Torres J, Mehandru S, Colombel JF, Jess T. Deciphering the different phases of preclinical inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. (2024) 21:86–100. doi: 10.1038/s41575-023-00854-4
4. Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, Zaidi D, et al. Mason AL et al: Host immunoglobulin G selectively identifies pathobionts in pediatric inflammatory bowel diseases. Microbiome. (2019) 7:1. doi: 10.1186/s40168-018-0604-3
5. Drent M, Crouser ED, Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med. (2021) 385:1018–32. doi: 10.1056/NEJMra2101555
6. Rossides M, Darlington P, Kullberg S, Arkema EV. Sarcoidosis: Epidemiology and clinical insights. J Intern Med. (2023) 293:668–80. doi: 10.1111/joim.13629
7. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. (2007) 357:2153–65. doi: 10.1056/NEJMra071714
8. Jiang Y, Rim DS, Rodgers B, Ahlawat S. Sarcoidosis is associated with lower risks of penetrating disease and colectomy in hospitalized patients with inflammatory bowel disease. JGH Open. (2020) 4:1199–206. doi: 10.1002/jgh3.12423
9. Sostegni R, Daperno M, Pera A. Pulmonary manifestations of inflammatory bowel disease. Dig Liver Dis. (2007) 39:239–41. doi: 10.1016/j.dld.2006.11.007
10. Tunc B, Filik L, Bilgic F, Arda K, Ulker A. Pulmonary function tests, high-resolution computed tomography findings and inflammatory bowel disease. Acta Gastroenterol Belg. (2006) 69:255–60.
11. Orchard TR, Chua CN, Ahmad T, Cheng H, Welsh KI, Jewell DP. Uveitis and erythema nodosum in inflammatory bowel disease: clinical features and the role of HLA genes. Gastroenterology. (2002) 123:714–8. doi: 10.1053/gast.2002.35396
12. Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr., Bresnitz EA, et al. Johns CJ et al: Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. (2001) 164:1885–9. doi: 10.1164/ajrccm.164.10.2104046
14. Thao C, Lagstein A, Allen T, Dincer HE, Kim HJ. Crohn's disease-associated interstitial lung disease mimicking sarcoidosis: a case report and review of the literature. Sarcoidosis Vasc Diffuse Lung Dis. (2016) 33:288–91.
15. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
16. Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. (2005) 201:755–67. doi: 10.1084/jem.20040429
17. Jarrot PA, Dury S, Rakotomalala A, Vella-Boucaud J, Patey M, Deslee G, et al. Association of sarcoidosis and ulcerative colitis: a review of 20 cases. Sarcoidosis Vasc Diffuse Lung Dis. (2013) 30:212–6.
18. Fok KC, Ng WW, Henderson CJ, Connor SJ. Cutaneous sarcoidosis in a patient with ulcerative colitis on infliximab. J Crohns Colitis. (2012) 6:708–12. doi: 10.1016/j.crohns.2012.01.008
19. Halling ML, Kjeldsen J, Knudsen T, Nielsen J, Hansen LK. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. (2017) 23:6137–46. doi: 10.3748/wjg.v23.i33.6137
20. Liberal R, Gaspar R, Lopes S, Macedo G. Primary biliary cholangitis in patients with inflammatory bowel disease. Clin Res Hepatol Gastroenterol. (2020) 44:e5–9. doi: 10.1016/j.clinre.2019.05.002
21. Yu XH, Cao RR, Yang YQ, Lei SF. Identification of causal metabolites related to multiple autoimmune diseases. Hum Mol Genet. (2022) 31:604–13. doi: 10.1093/hmg/ddab273
22. Xiao WB, Liu YL. Primary biliary cirrhosis and ulcerative colitis: a case report and review of literature. World J Gastroenterol. (2003) 9:878–80. doi: 10.3748/wjg.v9.i4.878
23. Leff JA, Ready JB, Repetto C, Goff JS, Schwarz MI. Coexistence of primary biliary cirrhosis and sarcoidosis. West J Med. (1990) 153:439–41.
24. Sherman S, Nieland NS, Van Thiel DH. Sarcoidosis and primary biliary cirrhosis. Coexistence in a single patient. Dig Dis Sci. (1988) 33:368–74. doi: 10.1007/BF01535764
25. Ormann W. [Association of primary biliary liver cirrhosis with sarcoidosis–separate diseases or a new nosological entity]? Dtsch Med Wochenschr. (1987) 112:1082–5. doi: 10.1055/s-2008-1068198
26. Bories C, Certin M, Lavergne A, Galian A, Godeau P, Rambaud JC. [Primary biliary cirrhosis and sarcoidosis. Association or unique disease]? Gastroenterol Clin Biol. (1984) 8:851–5.
27. Maddrey WC. Sarcoidosis and primary biliary cirrhosis. Associated disorders? N Engl J Med. (1983) 308:588–90. doi: 10.1056/NEJM198303103081011
28. Kishor S, Turner ML, Borg BB, Kleiner DE, Cowen EW. Cutaneous sarcoidosis and primary biliary cirrhosis: A chance association or related diseases? J Am Acad Dermatol. (2008) 58:326–35. doi: 10.1016/j.jaad.2007.07.031
29. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. (2014) 23:R89–98. doi: 10.1093/hmg/ddu328
30. Gala H, Tomlinson I. The use of Mendelian randomisation to identify causal cancer risk factors: promise and limitations. J Pathol. (2020) 250:541–54. doi: 10.1002/path.5421
31. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. (2017) 318:1925–6. doi: 10.1001/jama.2017.17219
32. Boef AG, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. (2015) 44:496–511. doi: 10.1093/ije/dyv071
33. Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. (2007) 16:309–30. doi: 10.1177/0962280206077743
34. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
35. Cordell HJ, Fryett JJ, Ueno K, Darlay R, Aiba Y, Hitomi Y, et al. Gervais O et al: An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J Hepatol. (2021) 75:572–81. doi: 10.1016/j.jhep.2021.04.055
36. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. Daly MJ et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. (2007) 81:559–75. doi: 10.1086/519795
37. Kwok MK, Schooling CM. Herpes simplex virus and Alzheimer's disease: a Mendelian randomization study. Neurobiol Aging. (2021) 99:101 e111–101 e113. doi: 10.1016/j.neurobiolaging.2020.09.025
38. Zhang M, Tang J, Li W, Xue K, Wang Z, Chen Y, et al. Ma J et al: Schizophrenia mediating the effect of smoking phenotypes on antisocial behavior: A Mendelian randomization analysis. CNS Neurosci Ther. (2024) 30:e14430. doi: 10.1111/cns.14430
39. Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. (2011) 40:755–64. doi: 10.1093/ije/dyr036
40. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. (2012) 21:223–42. doi: 10.1177/0962280210394459
41. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. (2019) 35:4851–3. doi: 10.1093/bioinformatics/btz469
42. He Z, Fu T, Lu S, Sun Y, Zhang Y, Shi W, et al. Adiposity as a risk factor for inflammatory bowel disease and the mediating effect of metabolic and inflammatory status: A population-based cohort study. United Eur Gastroenterol J. (2023) 11:973–84. doi: 10.1002/ueg2.12468
43. Hsu TY, Shih HM, Wang YC, Lin LC, He GY, Chen CY, et al. Effect of alcoholic intoxication on the risk of inflammatory bowel disease: A nationwide retrospective cohort study. PloS One. (2016) 11:e0165411. doi: 10.1371/journal.pone.0165411
44. Je Y, Han K, Chun J, Kim Y, Kim JH, Hoon Youn Y, et al. Association of waist circumference with the risk of inflammatory bowel disease: a nationwide cohort study of 10 million individuals in Korea. J Crohns Colitis. (2023) 17:681–92. doi: 10.1093/ecco-jcc/jjac193
45. Rahmani J, Kord-Varkaneh H, Hekmatdoost A, Thompson J, Clark C, Salehisahlabadi A, et al. Body mass index and risk of inflammatory bowel disease: A systematic review and dose-response meta-analysis of cohort studies of over a million participants. Obes Rev. (2019) 20:1312–20. doi: 10.1111/obr.12875
46. Chu X, Chen X, Zhang H, Wang Y, Guo H, Chen Y, et al. Association of diet and outdoor time with inflammatory bowel disease: a multicenter casecontrol study using propensity matching analysis in China. Front Public Health. (2024) 12:1368401. doi: 10.3389/fpubh.2024.1368401
47. Bhagavathula AS, Clark CCT, Rahmani J, Chattu VK. Impact of body mass index on the development of inflammatory bowel disease: A systematic review and dose-response analysis of 15.6 million participants. Healthcare (Basel). (2021) 9:35. doi: 10.3390/healthcare9010035
48. Amarapurkar AD, Amarapurkar DN, Rathi P, Sawant P, Patel N, Kamani P, et al. Narawane N et al: Risk factors for inflammatory bowel disease: A prospective multi-center study. Indian J Gastroenterol. (2018) 37:189–95. doi: 10.1007/s12664-018-0850-0
49. Casella G, De Marco E, Antonelli E, Daperno M, Baldini V, Signorini S, et al. The prevalence of hyper- and hypothyroidism in patients with ulcerative colitis. J Crohns Colitis. (2008) 2:327–30. doi: 10.1016/j.crohns.2008.09.001
50. Kim MS, Song M, Kim S, Kim B, Kang W, Kim JY, et al. Khera AV et al: Causal effect of adiposity on the risk of 19 gastrointestinal diseases: a Mendelian randomization study. Obes (Silver Spring). (2023) 31:1436–44. doi: 10.1002/oby.23722
51. Alinaghi F, Tekin HG, Burisch J, Wu JJ, Thyssen JP, Egeberg A. Global prevalence and bidirectional association between psoriasis and inflammatory bowel disease-A systematic review and meta-analysis. J Crohns Colitis. (2020) 14:351–60. doi: 10.1093/ecco-jcc/jjz152
52. Wu S, Yi J, Wu B. Casual associations of thyroid function with inflammatory bowel disease and the mediating role of cytokines. Front Endocrinol (Lausanne). (2024) 15:1376139. doi: 10.3389/fendo.2024.1376139
53. French JA, Gow P, Simpson-Yap S, Collins K, Ng J, Angus PW, et al. Alcohol intake is associated with a decreased risk of developing primary biliary cholangitis. World J Hepatol. (2022) 14:1747–56. doi: 10.4254/wjh.v14.i9.1747
54. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. (2005) 42:1194–202. doi: 10.1002/(ISSN)1527-3350
55. Prince MI, Ducker SJ, James OF. Case-control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut. (2010) 59:508–12. doi: 10.1136/gut.2009.184218
56. Gao W, Peng C, Wang Z, Li Y, Liu M. Genetic association and causal relationship between multiple modifiable risk factors and autoimmune liver disease: a two-sample mendelian randomization study. J Transl Med. (2024) 22:425. doi: 10.1186/s12967-024-05247-y
57. Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. (2020) 26:527–34. doi: 10.1097/MCP.0000000000000715
58. Hena KM. Sarcoidosis epidemiology: race matters. Front Immunol. (2020) 11:537382. doi: 10.3389/fimmu.2020.537382
59. Fallahi P, Ferrari SM, Ruffilli I, Elia G, Biricotti M, Vita R, et al. The association of other autoimmune diseases in patients with autoimmune thyroiditis: Review of the literature and report of a large series of patients. Autoimmun Rev. (2016) 15:1125–8. doi: 10.1016/j.autrev.2016.09.009
60. Bowden J, Spiller W, Del Greco MF, Sheehan N, Thompson J, Minelli C, et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int J Epidemiol. (2018) 47:1264–78. doi: 10.1093/ije/dyy101
61. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. (2013) 37:658–65. doi: 10.1002/gepi.21758
62. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology. (2017) 28:30–42. doi: 10.1097/EDE.0000000000000559
63. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40:304–14. doi: 10.1002/gepi.21965
64. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
65. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. (2016) 45:1961–74. doi: 10.1093/ije/dyw220
66. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. (2018) 50:693–8. doi: 10.1038/s41588-018-0099-7
67. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. (2017) 36:1783–802. doi: 10.1002/sim.7221
68. Fischer A, Nothnagel M, Franke A, Jacobs G, Saadati HR, Gaede KI, et al. Schreiber S et al: Association of inflammatory bowel disease risk loci with sarcoidosis, and its acute and chronic subphenotypes. Eur Respir J. (2011) 37:610–6. doi: 10.1183/09031936.00049410
69. Bezzio C, Della Corte C, Vernero M, Di Luna I, Manes G, Saibeni S. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at the less frequent associations. Therap Adv Gastroenterol. (2022) 15:17562848221115312. doi: 10.1177/17562848221115312
70. Romagnani S. Th1/th2 cells. Inflammation Bowel Dis. (1999) 5:285–94. doi: 10.1097/00054725-199911000-00009
71. Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. (2009) 58:1152–67. doi: 10.1136/gut.2008.163667
72. Cao H, Diao J, Liu H, Liu S, Liu J, Yuan J, et al. The pathogenicity and synergistic action of th1 and th17 cells in inflammatory bowel diseases. Inflammation Bowel Dis. (2023) 29:818–29. doi: 10.1093/ibd/izac199
73. Lv L, Chen Z, Bai W, Hao J, Heng Z, Meng C, et al. Cao Y et al: Taurohyodeoxycholic acid alleviates trinitrobenzene sulfonic acid induced ulcerative colitis via regulating Th1/Th2 and Th17/Treg cells balance. Life Sci. (2023) 318:121501. doi: 10.1016/j.lfs.2023.121501
74. Efe C, Torgutalp M, Henriksson I, Alalkim F, Lytvyak E, Trivedi H, et al. Coppo C et al: Extrahepatic autoimmune diseases in primary biliary cholangitis: Prevalence and significance for clinical presentation and disease outcome. J Gastroenterol Hepatol. (2021) 36:936–42. doi: 10.1111/jgh.15214
75. Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 cytokine-induced downregulation of PPARgamma in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology. (2005) 41:1329–38. doi: 10.1002/(ISSN)1527-3350
76. Harada K, Van de Water J, Leung PS, Coppel RL, Ansari A, Nakanuma Y, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. (1997) 25:791–6. doi: 10.1002/(ISSN)1527-3350
77. Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. Ansari AA et al: IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. (2014) 59:1944–53. doi: 10.1002/hep.26979
78. Tanaka A, Leung PSC, Gershwin ME. The genetics and epigenetics of primary biliary cholangitis. Clin Liver Dis. (2018) 22:443–55. doi: 10.1016/j.cld.2018.03.002
79. Ostrowski J, Goryca K, Lazowska I, Rogowska A, Paziewska A, Dabrowska M, et al. Kluska A et al: Common functional alterations identified in blood transcriptome of autoimmune cholestatic liver and inflammatory bowel diseases. Sci Rep. (2019) 9:7190. doi: 10.1038/s41598-019-43699-1
Keywords: inflammatory bowel disease, sarcoidosis, mediating role, primary biliary cholangitis, Mendelian randomization
Citation: Yi J, Wu S and He H (2024) Causal association of inflammatory bowel disease with sarcoidosis and the mediating role of primary biliary cholangitis. Front. Immunol. 15:1448724. doi: 10.3389/fimmu.2024.1448724
Received: 13 June 2024; Accepted: 07 August 2024;
Published: 03 September 2024.
Edited by:
Jose Luis Fachi, Washington University in St. Louis, United StatesReviewed by:
Qida He, City University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2024 Yi, Wu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxia He, SG9odW5nSGFAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.