 Amy Armstrong
Amy Armstrong Yuting Tang
Yuting Tang Neelam Mukherjee
Neelam Mukherjee Nu Zhang
Nu Zhang Gang Huang
Gang Huang- 1Department of Cell Systems and Anatomy, Long School of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 2Department of Microbiology, Immunology, and Molecular Genetics, Long School of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 3Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 4Department of Urology, Long School of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 5Department of Pathology & Laboratory Medicine, Long School of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
There is a continuous cycle of activation and contraction in the immune response against pathogens and other threats to human health in life. This intrinsic yin-yang of the immune response ensures that inflammatory processes can be appropriately controlled once that threat has been resolved, preventing unnecessary tissue and organ damage. Various factors may contribute to a state of perpetual immune activation, leading to a failure to undergo immune contraction and development of cytokine storm syndromes. A literature review was performed to consider how the trajectory of the immune response in certain individuals leads to cytokine storm, hyperinflammation, and multiorgan damage seen in cytokine storm syndromes. The goal of this review is to evaluate how underlying factors contribute to cytokine storm syndromes, as well as the symptomatology, pathology, and long-term implications of these conditions. Although the recognition of cytokine storm syndromes allows for universal treatment with steroids, this therapy shows limitations for symptom resolution and survival. By identifying cytokine storm syndromes as a continuum of disease, this will allow for a thorough evaluation of disease pathogenesis, consideration of targeted therapies, and eventual restoration of the balance in the yin-yang immune response.
1 Introduction
The immune system must maintain a status quo through a balance of activation and contraction. While its main purpose is to mount a defense against external and internal threats to the host, it must also ensure that it does not cause excessive or unnecessary damage to the host in the process. A period of immune contraction is equally important to that of activation, allowing the immune response and body to return the baseline – status quo – state once the threat has been resolved (1–5). This complimentary and cyclic nature of the immune response is somewhat like that of the yin-yang concept in traditional Chinese philosophy (6) to ensure that there is a robust response to threats whilst still maintaining an equilibrium. As depicted in Figure 1, this philosophy consists of opposing yet interrelated forces. “Yin” is associated with contraction, passivity, and coldness, while “Yang” relates to expansion, activation, and warmth. Yin-yang is observed in different aspects of life such as night and day, cardinal directions of north and south, and traditional ideals of femininity and masculinity. The host demonstrates the yin-yang philosophy through activation and contraction in a normal immune response. The “yang” component is viewed through the initial response to microbial or internal threats by immune cells activation and inflammation, while the “yin” component is demonstrated through wound healing, immune cell death, and immunological memory after the threat is resolved. Both forces must be present and active to make certain a balanced immune response occurs.
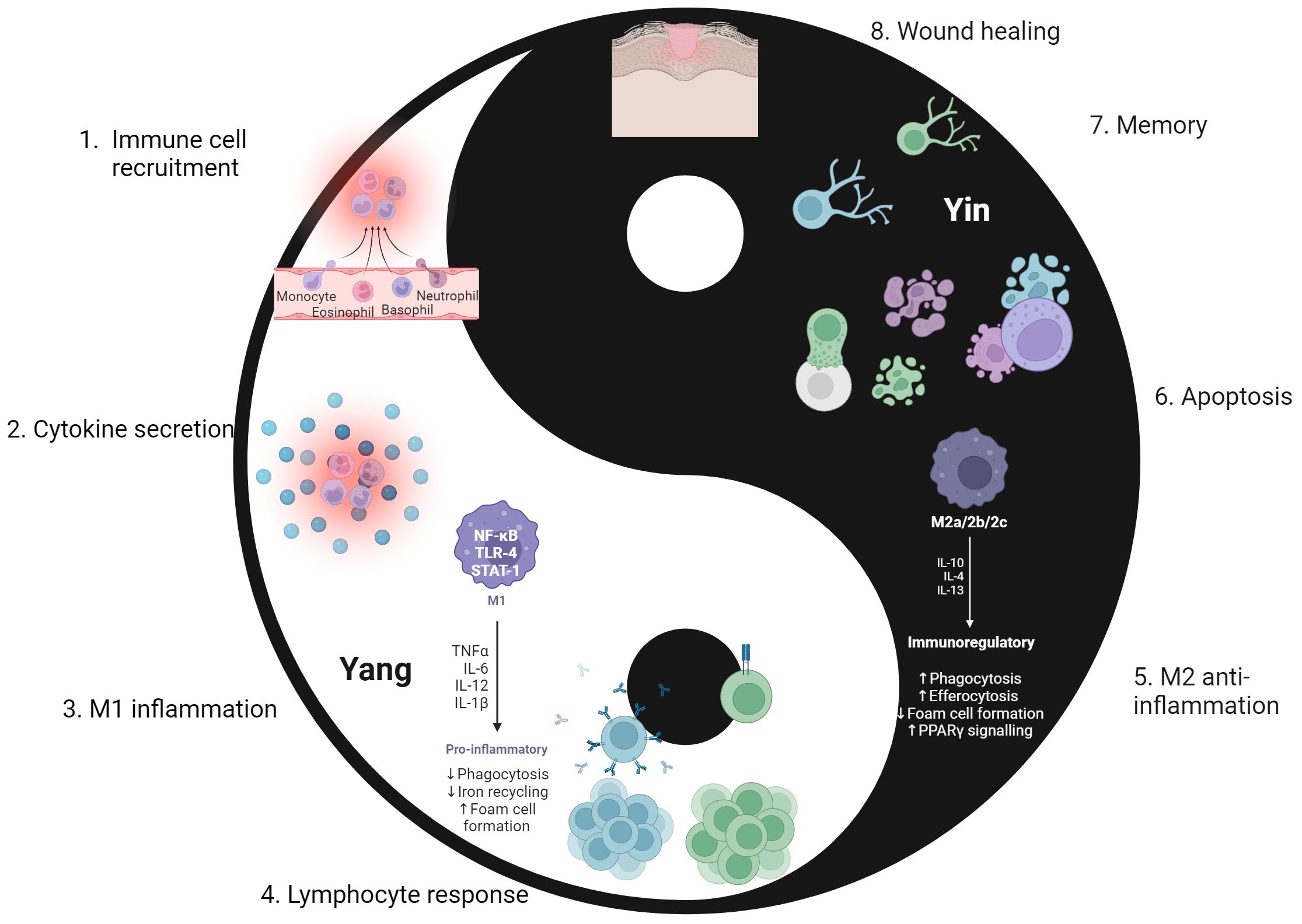
Figure 1. The cyclic “yin-yang” nature of the immune response. The immune response aligns with the yin-yang concept of complimentary forces. Yin represents a retractive or passive force, depicted as the immune contraction phase (5–8). Yang is the force that is active and expansive, as seen in the immune activation phase (1–4). Following repair of sites that were damaged by both pathogen and inflammatory processes, the immune system returns to a baseline and is poised to react to the next threat. 1. Upon encountering a pathogen, chemokine and cytokine gradients allow for recruitment of innate immune cells to the site of infection. 2. Innate immune cells secrete pro-inflammatory cytokines that cause further recruitment of immune cells, self and nearby immune cell activation, and the initiation of an inflammatory environment for further activation. 3. Stimulated from IFN-γ secretion from both self and other immune cells, macrophages differentiate into the pro-inflammatory M1 phenotype. This furthers the activation phase in the immune response. 4. Lymphocytes initiate the adaptive immune response, forming a more specific and targeted response to pathogens. This involves development, selection, activation, and differentiation into effector cells as lymphocytes enter the site of infection. 5. Successful elimination of the threat sets the stage for the contraction phase. Macrophages switch to the anti-inflammatory M2 phenotype, aiming to downregulate inflammatory processes from the activated phase. 6. Apoptosis of immune cells occurs, eliminating the excessive cells that were specifically recruited and activated for pathogen defense. This ensures the inflammatory and cytotoxic effector function of immune cells do not continue to persist. 7. Selected B and T lymphocytes undergo a complex process of differentiation into memory cells. Such memory lymphocytes residing in tissues or circulation for extended periods of time to serve as a record of past infection. This will allow for a faster and more robust response if the pathogen is encountered again. 8. Wound healing also occurs in steps 5-7, but its completion marks the end of the immune contraction phase. Following repair of sites that were damaged by both pathogen and inflammatory processes, the immune system returns to a baseline and is poised to react to the next threat. Created with BioRender.com.
When the balance is tipped in favor of activation for a prolonged period of time, the immune response becomes dysregulated and favors strong inflammatory responses. The exact mechanism driving a dysregulated immune response is not fully understood, but it is thought to be mediated by an interplay of different components that include genetics, immune cell dysfunction, and excessive secretion of pro-inflammatory cytokines (7). Although an immune response is present in most healthy individuals, it is important to emphasize that its specific response to perceived threats is not synonymous in all. Recent studies have postulated on how differences in responsiveness to vaccines and disease development or progression can be attributed to various factors such as age, genetics, and biological sex (8–11). This is representative of the diversity in the immune baseline of different individuals. The development of hyperinflammatory conditions can be helpful to understand how differences in the immune response can lead to varying outcomes.
Chemical signaling through chemokines and cytokines is responsible for the recruitment and maintenance of immune cells in the innate and adaptive immune responses. Innate immune cells sense microbial antigens by pattern recognition proteins, which initiates signaling pathways that activate NF-κB transcription factor for mass production of the cytokines IL-1, IL-6, IL-8, and TNF-α (12). These classical pro-inflammatory cytokines ensure that the inflammatory immune response is maintained through activation of endothelial cells, release of cytokines and chemokines, and recruitment of immune cells. With over fifty members, chemokines play an important role in the innate response through directing immune cells to sites of infections by gradients and assisting innate immune cells with communication, persistence, and activation of lymphocytes in the adaptive immune response (13). Lymphocytes in the adaptive immune response are similarly reliant on cytokines for activation, recruitment, and maintenance, such as IL-2 signaling needed for T lymphocyte survival (14). Chemical signaling is an integral part of the yang component in the immune response, but cytokine storm syndromes (CSS) can occur when it becomes overpowering.
A cytokine storm can occur when the activation phase of the immune response persists far beyond what is beneficial to the host. This is characterized by an unprecedented flood of pro-inflammatory mediators, resulting in a hyperinflammatory state. The term cytokine storm was initially documented in the context of a dangerous and uncontrolled system-wide immune response to T-cell engaging, antibody-mediated drugs such as with the OKT3 immunosuppressant to prevent organ rejection (15), and TGN1412 for treatment of B cell lymphoma and rheumatoid arthritis (16). The presence of a cytokine storm has also been discussed on the background of hyperinflammatory conditions such as septic shock (17), graft-versus-host disease (18), and hemophagocytic lymphohistiocytosis (19). More recently, the association of a cytokine storm in severe COVID-19 manifestations has reinvigorated interest in understanding the mechanism and pathology underlying this hyperinflammatory immune response (20–22). Fajgenbaum and June have provided an extensive review of a cytokine storm to consolidate the shared features of systemic cytokine release and multiorgan failure seen in cytokine storm syndromes (CSS), as well as guidance on therapeutic interventions (23). It is continuously necessary to consider how cytokine storm propagates a damaging and uncontrolled hyperinflammatory response in these individuals. Evaluating CSS allows for a deeper understanding of how various factors can culminate into a perpetual state of activation and disease pathology. A brief overview of major CSS in adult and pediatric patients has been summarized in Table 1. This current review aims to focus on seven of the most clinically relevant cytokine storm-mediated conditions: sepsis and septic shock, drug reaction with eosinophilia and systemic symptoms (DRESS), primary hemophagocytic lymphohistiocytosis (pHLH), secondary HLH and macrophage activation syndrome (sHLH/MAS), graft-versus-host disease (GVHD), immune-related adverse events (irAEs), and cytokine release syndrome with immune effector cell associated neurotoxicity syndrome (CRS/ICANS). The purpose is to explore how differences in the immune baseline of certain individuals disrupts the yin-yang of the immune response, leading to similar patterns of immune dysregulation and CSS. This immune baseline describes the state at which the host is not contending with pathogens or malignancies yet is still able to rapidly mount an immune response if a threat arises. For hyperinflammatory conditions, the activation phase is too strong and leads to pathological effects. Furthermore, long-term effects, sequalae, and promising therapeutic approaches for CSS are also reviewed.
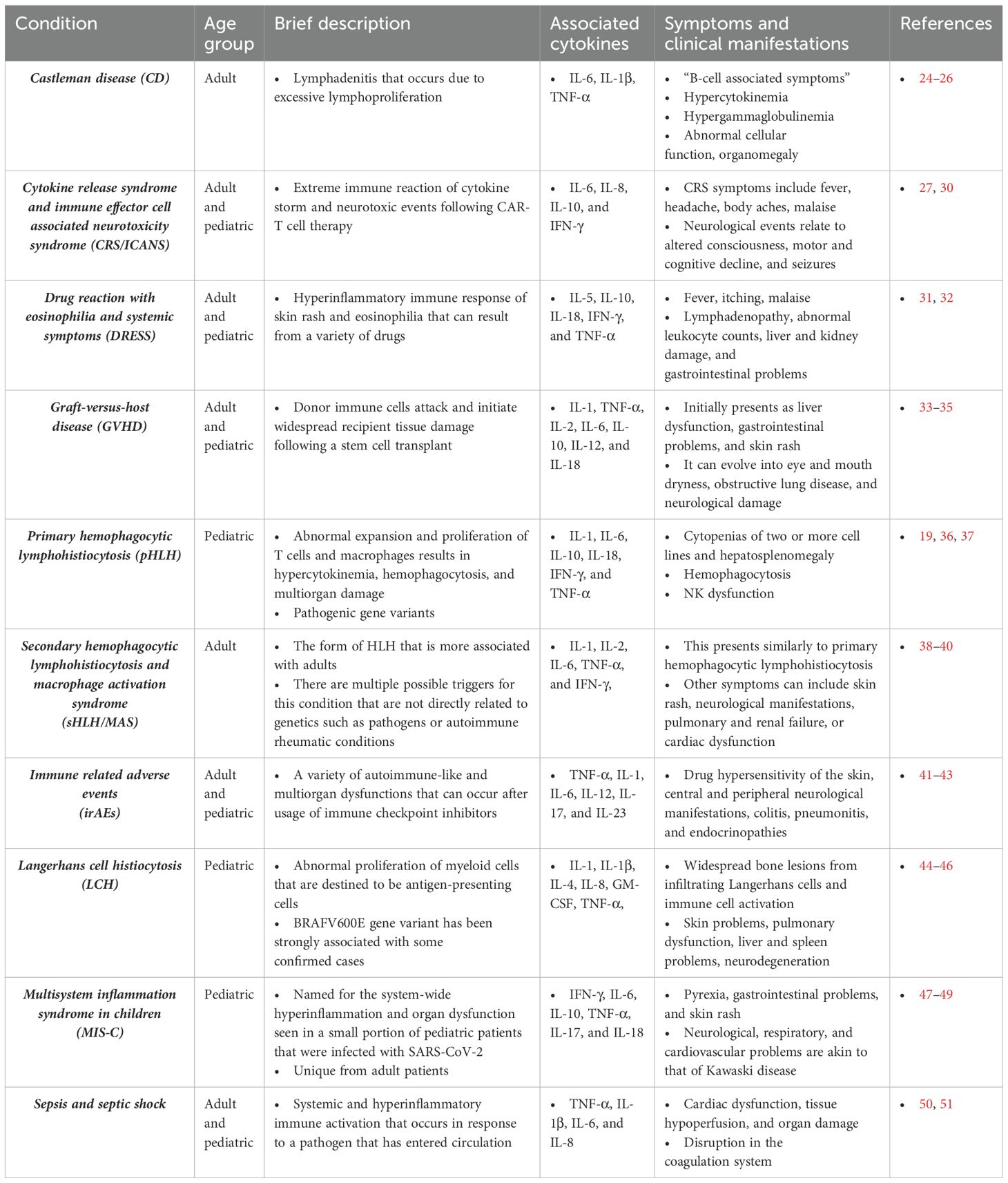
Table 1. Overview of major cytokine storm syndromes.
2 Sepsis and septic shock
By convention, sepsis and septic shock are described as an inappropriate system-wide response to a pathogen that has entered the bloodstream, involving excessive immune cell activation and subsequent secretion of pro-inflammatory mediators (52). As depicted in Figure 2, sepsis and septic shock begin as a normal immune response to a pathogen. When the pathogen enters the bloodstream, the immune response becomes overactive and may culminate into a septic condition. When the pathogen persists within the host, the secretion of classical pro-inflammatory cytokines also persists and leads to pathological consequences. The innate response is subject to widespread immune cell and complement activation for hyperinflammation (53). Persistence of microbial antigens in septic conditions also plays a role in the breakdown of the adaptive immune response. B and T lymphocytes present with an exhausted phenotype due to constant presentation with microbial antigens, leading to events of apoptosis and increased secretion of anti-inflammatory IL-10 (54). An overwhelming yang component of chemical signaling and immune cell activation leads to host tissue and organ damage in septic conditions.
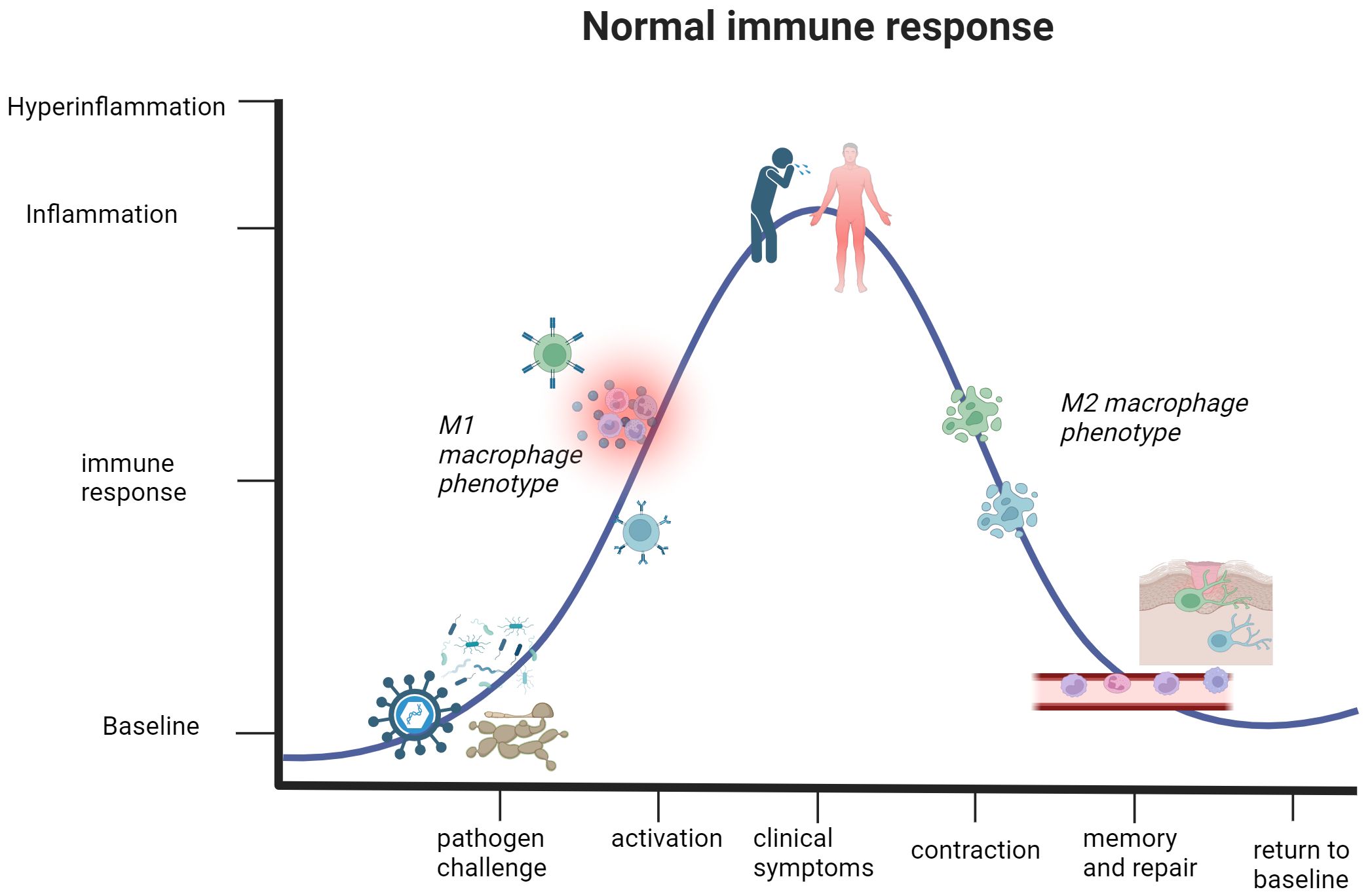
Figure 2. The immune response in healthy individuals undergoes a cycle of activation and contraction. This graph illustrates the trajectory of a normal immune response. Detection of a pathogen or threat to human health initiates immune activation and subsequent inflammation, partly thanks to M1 macrophages. The mounting immune response causes clinical presentation of symptoms in the host, but this activation phase will not reach a state of hyperinflammation that jeopardizes the safety and life of the host. Once the immune system eliminates the threat, the body enters the contraction phase defined by an anti-inflammatory environment and elimination of excess immune cells that are no longer necessary for active defense. Lastly, differentiation of quiescent memory cells and wound healing occurs. This represents a return to a baseline or steady state of being, which can respond to future threats. Created with BioRender.com.
A study of ICU patients diagnosed with infection-associated septic shock reported a 70% overall positivity rate for one or more active microbe, further specifying that there were 47%, 62%, and 19% positivity rates for gram-positive bacteria, gram-negative bacteria, and various fungal species, respectively (50). The systemic activation of the immune response results in compromise of the endothelial cell barrier, leading to hypotension and multiorgan failure that defines septic shock (51, 55). Other clinical manifestations include cellular dysfunctions of disseminated intravascular coagulation and thrombocytopenia, tissue hypoperfusion and systemic vasodilation from cardiac dysfunction, and organ failure of the lungs, kidneys, and liver (56–61). Common symptoms of septic shock include pyrexia, elevated respiratory rate, tachycardia, and malaise (62). It should be reiterated that sepsis and septic shock represent the most extreme immune response to a pathogen, not the norm. The active inflammatory response in sepsis and septic shock coincides with the yang component of the immune response. As the onset and progression of septic shock are only discussed in ICU patients (52), this alludes towards an underlying factor contributing to susceptibility of this condition. Various comorbidities, such as diabetes, cancer, lung disease, and kidney or liver failure, are associated with the onset and overall severity in septic shock (63, 64). Figures 2, 3 illustrate how the immune response to a pathogen can lead to either successful clearance and immunological memory or persistent hyperinflammation in septic shock and related conditions, respectively. In Table 1, we also provide a brief overview of this condition, as well as other cytokine storm-related conditions. The existence of preexisting conditions could create a disturbed immune baseline that lowers the threshold required for immune activation, leading to difficulty in entering a state of immune contraction once the threat has ceased (Figure 1).
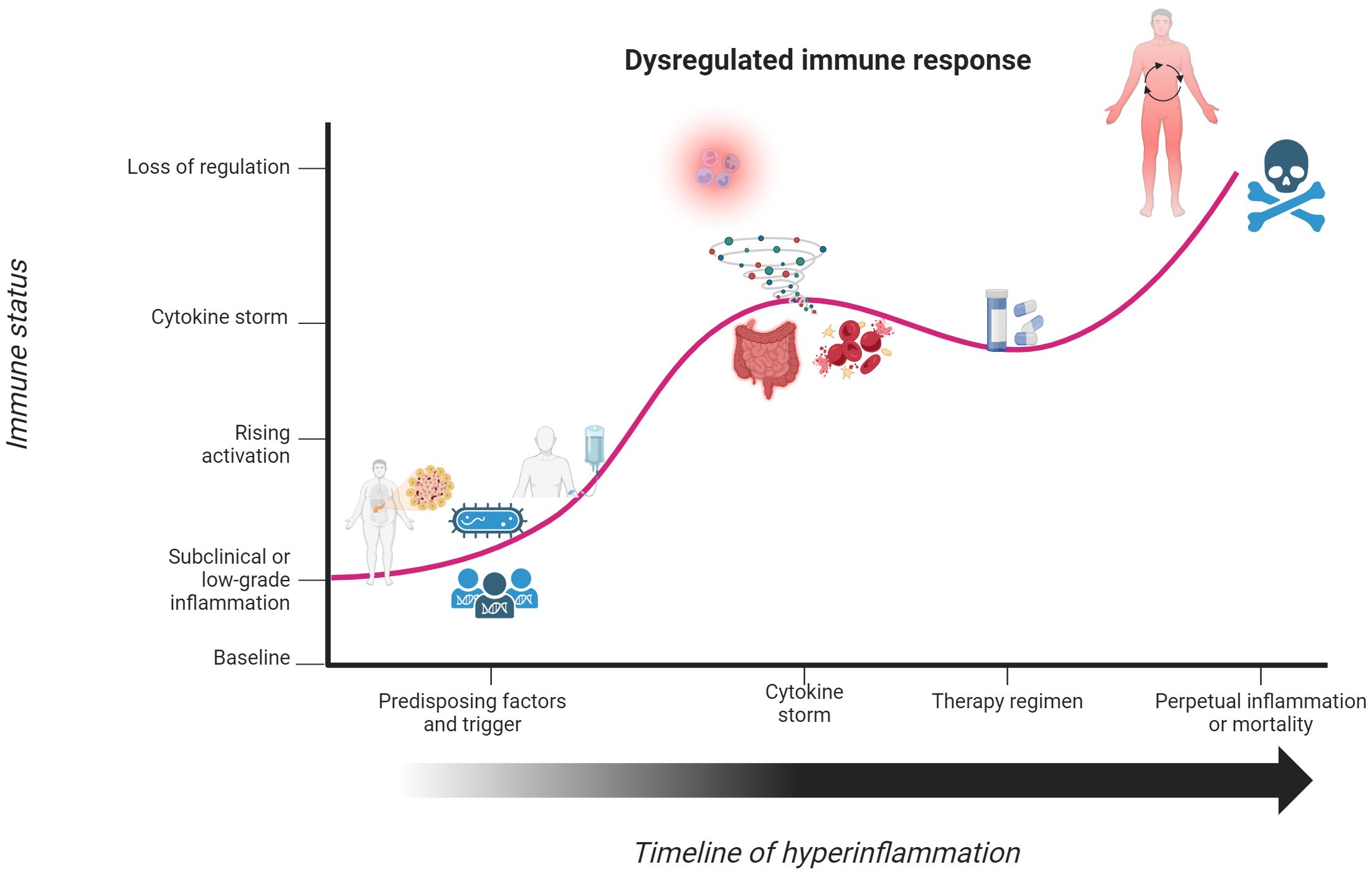
Figure 3. Underlying factors disrupt the yin-yang immune response and initiate a hyperinflammatory state. Individuals with underlying factors, such as chronic or pre-existing illnesses, cancer, or pathogenic gene variants, have an immune response that contrasts with that seen in Figure 2. In response to a perceived threat to the body or therapeutic trigger, individuals enter a state of immune activation to eliminate the threat. This immune response continues to mount until it culminates into a state of hyperinflammation or cytokine storm. Although steroids can temporarily stabilize the immune response, such effects do not fully correct the prolonged state of activation. This dysregulated activation phase of the immune response becomes so powerful and uncontrolled that patients enter a state of chronic hyperinflammation or meet their demise. Created with BioRender.com.
Given the potentially dire situation of sepsis and septic shock, there has been much interest in identifying biomarkers that can predict onset and severity in patients. C-reactive protein is elevated in this condition, but there is notable skepticism in using it as a biomarker because it is also elevated in many other conditions (58, 65). The rapid uptick of proinflammatory cytokines or cytokine storm has been more widely accepted and discussed as reliable biomarkers (66, 67). The cytokine profile of septic shock conditions was first detailed as elevations in TNF-α, IL-1β, IL-6, and IL-8 (68, 69). It has also been noted that elevations of IL-10 and CCL2 can be seen in certain patients with acute septic shock (70). A more in-depth discussion on the direct and indirect roles that various pro- and anti-inflammatory cytokines contribute towards septic shock progression has been described elsewhere (71). Although an elevation in proinflammatory cytokines is crucial for immune cell activation and response to a pathogen, septic shock patients are unable to control this activation phase. Corticosteroids are universally acknowledged as the standard of treatment for cytokine storm syndromes (CSS), but controversy remains on its true efficacy and sustainability in patient outcomes (72–75). This has been illustrated by the dismal reports of a 43.1% to 62% mortality rate within a 5-year follow-up period seen in three separate studies (76–78). Immunomodulatory drugs have been utilized as an alternative to steroids for sepsis and septic shock, but widespread approval and acceptance of this treatment regimen is far from complete (79, 80). For those that recover from septic shock, the dysregulated immune response is far from over. A strong yang component allows for hyperinflammation and organ damage seen in sepsis and sepsis shock, but some patients can develop a yin component that is equally detrimental. It is estimated that 33% of survivors may develop a chronic critical illness with a background of persistent inflammation, immunosuppression, and catabolism syndrome (PICS) (81). First proposed by physicians (82), PICS is characterized by recurrent infections due to a slew of opportunistic pathogens, wasting due to metabolic dysfunction, and low yet persistent levels of pro-inflammatory cytokines (83, 84). The damaging effects of septic shock can persist long after the illness has been treated, leaving the patient in an immunosuppressed state that directly opposes the hyperactive immune response from the cytokine storm. Yin and yang can be present in both the sequalae and main symptomatology of septic shock, but the bias towards either response is damaging. A combination of intrinsic factors and comorbid conditions position patients into a state of imbalance in the yin-yang immune response. From this point, a pathogenic trigger initiates a state of immune activation, leading to a cytokine storm and clinical manifestations of sepsis and septic shock (Figure 3).
3 Drug reaction with eosinophilia and systemic symptoms
The term “adverse drug reactions” was initially coined to describe individuals that developed skin rash and eosinophilia following usage of anticonvulsant medications (31). This condition is presently known as drug reaction with eosinophilia and systemic symptoms (DRESS), with reports expanding to include many commonly prescribed drugs and cancer immunotherapies (32, 85–88). The underlying immunopathology in DRESS is driven by components of both the innate and adaptive immune response. Helper T lymphocytes categorize drug antigens as foreign and launch a type 2 immune response to secrete IL-4, IL-5, and eotaxin (89, 90). These chemical signals then recruit eosinophils to sites of inflammation where they activate and release granules for further inflammation and tissue damage. It is also believed that latent viral reactivation in monocytes and macrophages are able to infect helper T cells and further contribute to the hyperinflammation seen in DRESS (90). The combined effects of drug hypersensitivity and viral reactivation lead to the cytokine storm and clinical manifestations seen in this condition.
Classical features of DRESS become apparent weeks to months after initial intake of the drug. These diverse symptoms include fever, skin eruptions, lymphadenopathy, and facial swelling (85). Histopathology of patients can reveal leukocytosis, mainly related to eosinophil infiltration into the skin (32). As described in Table 1, multiorgan manifestations are associated with this condition. Skin rash is one of the defining features of DRESS, characterized as having a maculopapular form and covering up to 50% of total body surface area (91, 92). Other clinical manifestations include pulmonary dysfunction, neurological problems such as speech and motor control deficiencies and altered levels of consciousness, liver failure, and acute kidney injury (89, 93–98). Organ manifestations in DRESS are reminiscent of those seen in other cytokine storm syndromes (CSS) (23). The low reports of this condition compared to the widespread and systemic usage of prescription drugs alludes to DRESS as an infrequent problem. While it is challenging to pinpoint the exact incidence of DRESS in the general population, it has been estimated to fall within the ranges of 1 in 1,000 to 1 in 100,000 (31). A study of electronic health records aligns with the lower range of incidence, reporting a prevalence of 2.18 out of 100,000 (98). The hyperinflammatory immune response to drugs and subsequent pathological consequences represent a strong bias towards the yang component of the immune response, directly contrasting with the balance illustrated in Figure 1. Elevated cytokines in DRESS further reveals its similarities to other CSS.
It has been postulated that reactivation of latent viruses, such as human herpesvirus 6 (HHV-6), Epstein-Barr virus (EBV), and cytomegalovirus (CMV), can overstimulate immune cells and lead to a cytokine storm, but is unclear whether viral reactivation is a mediator or consequence of DRESS (91, 99). A comparative analysis of patients diagnosed with COVID-19, COVID-19 with concurrent maculopapular rash, or DRESS found that there were similar elevations of CXCL9, CXCL10, CXCL11, IFN-γ, IL-10, TNF-α, and IL-18 across the three groups (100). There is a growing interest on how cytokine storm-like manifestations to different drugs can perpetuate the hyperinflammatory response in (drug reaction with eosinophilia and systemic symptoms (DRESS). It has also been shown that some DRESS patients have pathogenic variants in the CYP2C9*3 gene, resulting in diminished ability to metabolize drugs (101–103). The presence of latent viral reactivation and genetic abnormalities represent a disturbed yin-yang immune baseline, allowing for an immune response to drugs that is both hyperresponsive and damaging. To dampen this immune activation phase, topical skin steroids or systemic corticosteroids are used as a first-line therapeutic (104, 105). Alternative treatments, such as intravenous immunoglobulin and cyclosporin (106–109), can be used but are not as widely accepted nor implemented as steroids. The hyperinflammatory immune response seen in this condition can persist far beyond therapeutic intervention and resolution of symptomatology, as demonstrated in Figure 3.
Although not reported in all patients, there has been linkage of DRESS resolution and autoimmune sequalae (87, 91, 92, 94). In a retrospective study of 55 DRESS/drug-induced hypersensitivity (DIHS) patients, nine patients generated autoantibodies nearly a decade before the onset of an autoimmune condition (110). Furthermore, the development of autoimmune conditions occurred in a sequential order within 7.5 years of the initial observation. The disturbed yin-yang immune response in these individuals reaches far past the presentation of DRESS, prolonging the dysregulated immune baseline through the development of autoimmunity. For these individuals, the inability of the yin response to occur leaves them in a state of persistent activation. While usage of steroids as a first-line therapeutic provides immediate relief for symptomatology, the delayed presentation of autoimmunity indicates that it is unsuccessful in correcting the underlying immune dysregulation from DRESS.
4 Primary hemophagocytic lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a hematological malignancy defined by defective cytotoxic functioning of T and NK cells, leading to rapid expansion and activation of macrophages and lymphocytes for persistent inflammation through hypercytokinemia and hemophagocytosis (111, 112). Immunopathology of this condition demonstrates a complex interplay of immune cells and hyperinflammation. Overactivation of these cells is viewed as a compensatory mechanism for their inability to release perforin on and subsequently lyse infected or malignant target cells (113). The overabundance of cytokines such as IFN-γ leads to activation of macrophages, furthering the cytokine storm by the defective cytotoxic cells. Macrophages then become hyperactive and display the key feature of this condition through consumption of red blood cells (114). Dysfunction of multiple immune cell types is then seen in the widespread organ and tissue damage of HLH patients.
This condition is further characterized into having primary and secondary forms (115). A brief summary of pHLH has been provided in Table 1. Hypercytokinemia and rapid expansion of leukocytes in HLH aligns with the yang phase of the immune response. The primary HLH (pHLH) form is associated with pediatric patients, illustrated in two separate reports by which most clinical presentations and diagnoses occurred before the patient’s first birthday (36, 116). Commonly noted symptoms include fever, rash, jaundice, lethargy, persistent viral infections, and swollen lymph nodes. As discussed in the previous conditions, hyperinflammatory manifestations of pHLH are known to occur in a system-wide manner. Poor prognosis is associated with the 30-70% of patients that develop neurological manifestations related to altered levels of consciousness, focal issues, and seizures (117), reported by one retrospective study to be associated with a 39.1% mortality rate in pHLH patients (118). Additionally, another retrospective study found a strong linkage between liver failure and confirmed pHLH phenotype (119). The excessive immune cell activation and organ damage is characteristic of an overactive yang component of the immune response, as seen with septic shock and DRESS. One group reported a pHLH incidence of 1.2 cases per million children (120), signifying the rarity of this hyperinflammatory disorder. Similar to DRESS, the low incidence of this condition speaks to an intrinsic difference in the immune baseline of such children.
For pediatric patients, it is more relevant to focus on genetic or inborn errors as drivers of disease pathology. Compared to adults, pediatric cohorts have not experienced the passage of time that allows for exposure to environmental factors and culmination of genetic damage that drives adult-onset pathologies. The genes most commonly associated with primary hemophagocytic lymphohistiocytosis (pHLH) onset are divided into those that dysregulate cytotoxic functioning of T and NK cells (PRF1, UNC13D, and STX11), lead to the pigmentation disorders of Griscelli syndrome, Chediak-Higashi syndrome, and Hermansky-Pudlak syndrome (RAB27A, LYST and AP3B1), or interfere with inflammasome formation (XIAP, NLRC4, and CDC42) (121–123). Hypercytokinemia and pHLH are common end results of these gene defects, resulting from overactivation of CD8+ cells, NK cells, and macrophages that attempt to compensate for defective effector functions (37, 124, 125). As previously discussed, multiorgan damage and hyperinflammation ensue from these pathogenic gene variants. With regards to cytokine storm, both forms of HLH share the cytokine profile of elevated IL-1, IL-6, IL-10, IL-12, IL-18, IFN-γ, and TNF-α (125–127). Recent studies of HLH found that increased ratios of IL-10 to IFN-γ (128) or concurrent elevations in IL-10 and IL-13 (129) were associated with the pHLH form. Therapeutic options for pHLH patients also focus on controlling the cytokine storm. HLH-94 and HLH-2004 protocols provide guidance on controlling the system-wide inflammation, seen with first-line therapeutics of the corticosteroids prednisolone and methylprednisolone (130, 131).The chemotherapeutic etoposide is also described as a treatment option, but its usage is mainly concurrent with steroids (36, 132). A stem cell or bone marrow transplant could prove curative for pHLH, but it is not without its own set of risks. One retrospective study found that 39 out 61 pHLH patients that received a stem cell transplant died before the 5.5-year follow-up period, either due to graft failure or disease progression (133). Of 51 observable patients, some form of graft-versus-host disease (GVHD) occurred in 48% of them. The contraction or yin phase of the immune response forcibly induced by steroids only serves to temporarily placate the hyperinflammatory state of pHLH patients, leaving their stressed immune system open to attack by opportunistic pathogen. Even those cured of pHLH from a stem cell transplant are not free of a dysregulated yin-yang immune response as the threat of developing GVHD is ever-present.
5 Secondary hemophagocytic lymphohistiocytosis
Genetic abnormalities were initially credited with driving hemophagocytic lymphohistiocytosis (HLH) pathogenesis, but the later discovery of HLH symptoms in children diagnosed with juvenile rheumatoid arthritis provided a new perspective into disease onset (134). Patients experience the same yang component and immunopathology seen in pHLH, with overactivated macrophages and T cells driving a hyperactive immune response. Table 1 has provide an overview of this condition that will be further detailed in this section. Clinical features of fever, jaundice, skin rash, lymphadenopathy, and lethargy align with those described in pHLH. Termed as secondary HLH (sHLH), this form of HLH is characterized with having both an older age demographic and variety of triggers. A retrospective study of the US National Inpatient Sample database found a bimodal distribution of sHLH disease incidence in the age groups of 16-30 and 56-70 (135). This older age demographic is further illustrated by two separate studies by which the median age of sHLH diagnosis was reported to be over 50 years old (136, 137). Due to this older age demographic, non-genetic components are more implicated with disease when compared to pHLH. Rheumatic autoimmune conditions, such as systemic lupus erythematous (SLE), systemic idiopathic juvenile arthritis (sIJA), and adult-onset Still’s disease (AOSD), are commonly cited triggers for the macrophage activation syndrome (MAS) portion of sHLH (126, 138, 139). Other triggers of sHLH include pathogens like EBV and fungi, cancer, or treatment for hematological cancers, such as chimeric antigen receptor T (CAR T)-cell therapy (38, 39, 140). Underlying conditions or infections demonstrate an inclination towards an activated immune response, similar to that seen in the association of comorbidities with septic shock conditions. A simplified overview of different factors that contribute to cytokine storm syndromes (CSS) presentation in pediatric- or adult-associated conditions are summarized in Figure 4, as seen in pHLH and sHLH. Two separate genetic analyses of sHLH have also revealed a surprisingly large number of variants in pHLH-associated genes, including UNC13D, LYST, and PRF1 (141, 142). A variety of different factors, such as genetics and underlying conditions, can compound to initiate the cytokine storm and organ damage observed in sHLH.
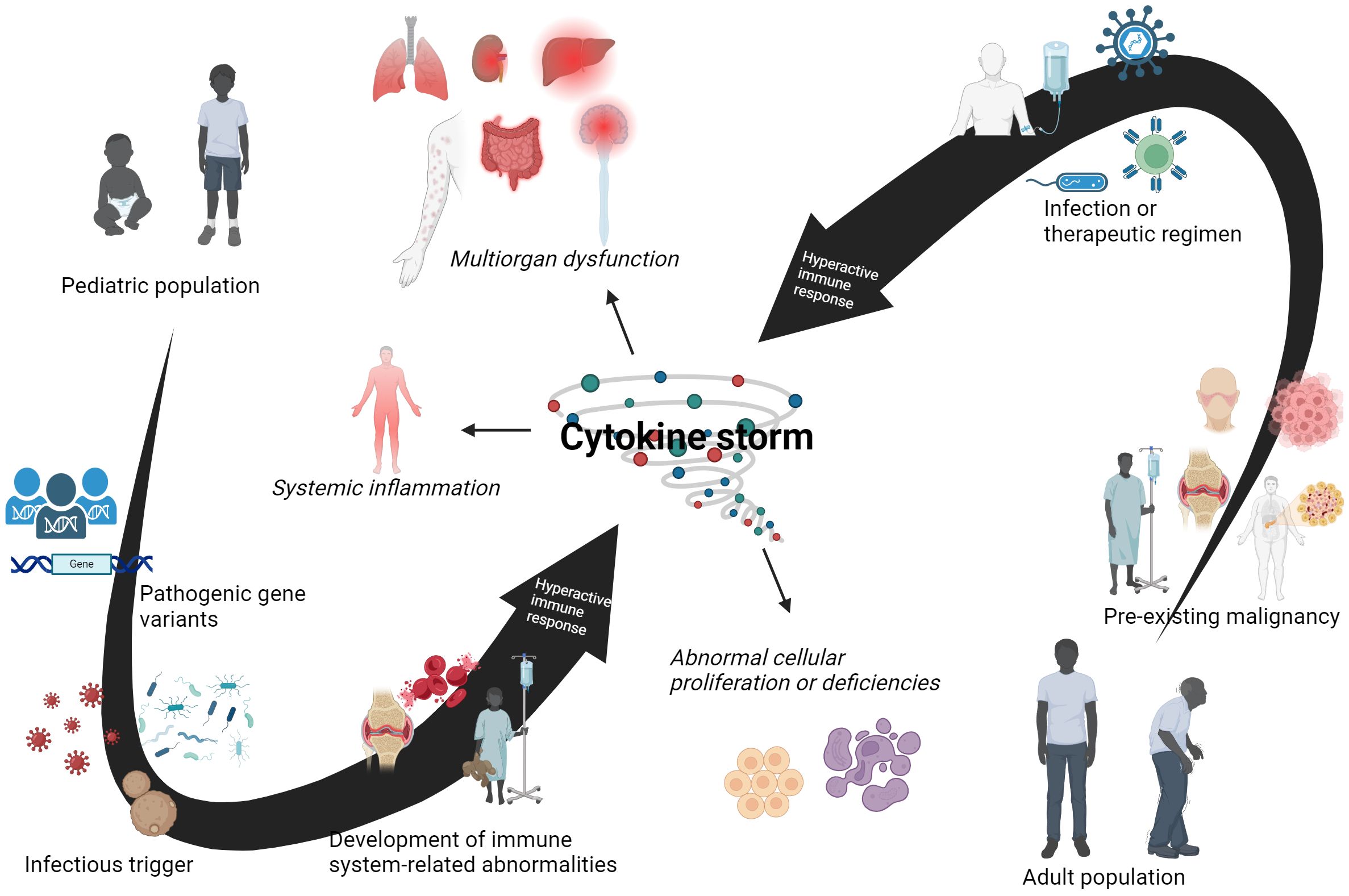
Figure 4. Different age-related factors contribute to cytokine storm syndromes (CSS). This is a schematic for intrinsic and extrinsic components in both pediatric and adult patients that can lead to the development of CSS. It is understood that different factors contribute to disease onset and progression in these two age groups. Pathogenic gene variants play a major role in a disturbed yin-yang immune baseline in pediatric patients, which is further exacerbated by subsequent encounters with infectious agents. Immune-related disorders can occur and culminate into CSS and related conditions in young patients. For adult populations, a pre-existing malignancy can be present before onset of CSS symptomatology. This imbalanced yin-yang immune response is hyperresponsive towards infectious or therapeutic triggers, leading to the development of a CSS in adult patients. However, there are exceptions to these observations. Pediatric patients can have a pre-existing illness that is not linked to genetic abnormalities. Adult patients with CSS have been identified to have pathogenic gene variants that may contribute to syndrome onset. An infectious trigger or therapeutic regimen may be enough for some adults to develop CSS. Nevertheless, both pediatric and adult patients are capable of developing CSS. Created with BioRender.com.
The cytokine storm of IL-2, IL-6, TNF-α, and IFN-γ observed in both forms of hemophagocytic lymphohistiocytosis (HLH) induces a hyperactivated state of both macrophages and cytotoxic T cells. This positive feedback loop of cytokine secretion, immune cell activation, and multiorgan damage is similar to the effects seen in sepsis and septic shock conditions (40). Furthermore, there is also discussion of elevated IL-1 and IL-18 in the cytokine storm of HLH (143). To delineate the two forms of HLH, there have been concerted efforts to determine cytokine profiles that are exclusively predictive for either form. When compared to pHLH, one study found that a cytokine profile with elevated IL-4 and IFN-γ was predictive for sHLH patients (144). Another study compared non-HLH vs. HLH adult patients, reporting an elevation of familiar cytokines such as IL-10, IL-18, IFN-γ, and TNF-α, while also noting some rarely discussed chemokines such as eotaxin, IL-7, and MCP-1 (145). Tang and colleagues also discuss a seminal study by which a novel mouse model of elevated IL-10 and IL-18 produced an sHLH phenotype (146). This variety of elevated pro-inflammatory cytokines underlies the complexity in elucidating the cytokine storm and perpetual immune activation that occurs. The disturbance of the yin-yang immune response seen in Figure 1 allows for an increased activation phase and cytokine storm development. Therapeutic guidelines follow that of pHLH by advising usage of etoposide and steroids as a broad attempt to quash the hyperinflammatory response (39, 117, 130–132). Rituximab for EBV-mediated sHLH and the anti-IL-1 cytokine therapy anakinra (147–150) have been explored as alternative options for treatment. These alternative therapeutic options aim to target specific mechanisms or components of the disease over induction of systemic immunosuppression through steroids. Although the field relies heavily on steroids to treat sHLH, statistics on sHLH survival rate do not speak to the efficacy of this treatment. When looking at ICU patients, one report found that 39% of sHLH patients died during the time of their stay (151) while a separate study estimated a 74% mortality rate at the 6-month follow-up (152). Given the high mortality rate of sHLH, this suggests that the yang immune response of patients is not complimented by the anti-inflammatory and retractive yin component. Patients still experience hyperinflammatory responses that persist throughout their lifetime, resulting in a complete breakdown of the yin-yang immune balance or mortality. The abysmal outcomes of sHLH indicate that novel therapeutic interventions are necessary to properly address the underlying hyperinflammation.
6 Graft-versus-host disease
In the mid-twentieth century, the term “secondary syndrome” was used to describe mice that died from multiorgan dysfunction following total-body irradiation and isologous transplantation of bone marrow cells from a donor mouse (153). This has since been renamed as graft-versus-host-disease (GVHD) and understood to occur due to donor T lymphocytes reacting to MHC molecules of the recipient, inducing a hyperinflammatory immune response and system-wide tissue damage (154, 155). The mismatch of human leukocyte antigen (HLA) is the main factor in driving GVHD onset. On a cellular level, donor T cells recognize both the self-peptides presented by recipient HLA class I and II molecules and polymorphic residues on HLA molecules themselves as foreign, thus initiating an inflammatory response (156). It is widely acknowledged that T lymphocytes in the adaptive immune response are responsible for initiating the cytokine storm and organ damage seen in GVHD. The yang or activation phase of the immune response is most prevalent in this condition. GVHD was originally categorized into acute (aGVHD) and chronic (cGVHD) forms depending on if main symptomatology occurred within 100 days of or post-100 days after a BMT (157–159). The field has since shifted towards a framework that uses the extent and severity of organ manifestations to typify this condition (160, 161). General features associated with GVHD onset include fever, rash and other skin eruptions, jaundice, diarrhea, nausea, and vomiting. Skin rash, gastrointestinal involvement, and liver dysfunction are considered to be some of the hallmarks of aGVHD onset (162–164), further expanding to reports of disturbed mucosal tissues of the eyes and mouth, dyspnea, and muscle aches in cGVHD (165–167). A concise summary of pathogenesis and clinical symptoms has been provided in Table 1. Acute and chronic forms of GVHD will not be considered separately due to the same common denominator of BMT as the initiator of pathology. As cytokine storm syndromes (CSS), GVHD and DRESS are similar in that onset is mediated by a hyperinflammatory reaction to therapeutic interventions intended to improve patients’ health. A bone marrow or stem cell transplant can be used to treat a variety of hematological disorders, autoimmune conditions, and inborn errors of metabolic function (168–170). Both malignancy-associated sHLH and GVHD have pre-existing conditions that serve as a predisposing factor for a hyperinflammatory immune response, although it is the treatment with a BMT that can specifically trigger GVHD. Figure 3 illustrates how multiple factors can contribute to GVHD onset, as well as other CSS.
As a known cytokine storm syndrome (CSS), there has been interest in the cytokine profile responsible for graft-versus-host disease (GVHD) onset and severity. Conditioning regimens are necessary to prepare the recipient for engraftment, but their very nature may serve to further exacerbate the yin-yang immune dysregulation in patients. Immunosuppression and tissue damage can create an inflammatory environment by which donor lymphocytes are stimulated to expand and rapidly secrete cytokines (171, 172). Some of the earliest work on this unique profile found IL-1 and TNF-α as some of the main contributors of GVHD (35). More recent reports of the cytokine profile in GVHD include elevated levels of IL-2, IL-6, IL-10, and IL-17 (173, 174). Cytokine storm can play a central role in GVHD pathogenesis, highlighted by the difference in T-cell response and associated cytokine secretion in the two forms of this condition. The initial presence of Th1 cells secreting IL-12 IL-18, IFN-γ, and TNF-α in the inflammatory phase of aGVHD can switch to Th2 and Th17 cells secreting IL-4, IL-17, and IL-2 with cGVHD (174, 175). This can be viewed as the immune system trying to complement the hyperinflammation with the yin component, but ultimately failing to, resulting in the occurrence of cGVHD. For treatment options, the National Comprehensive Cancer Network (NCCN) unanimously recommends the use of either topical steroids or systemic corticosteroids depending on the extent and severity of tissue and organ involvement (176). However, there is concern for patients that become unresponsive or steroid-refractory (sr-GVHD) (177–182). For these cases, other treatment options include the FDA-approved JAK 1/2 inhibitor for sr-GVHD, as well as cellular products or antithymocyte globulin (178, 182). One study on the prognosis of aGVHD patients noted that more than 50% of all patients died within 7 months of initial diagnosis, with 25.5% of all deaths within a year not related to the underlying malignancy or non-relapse mortality rate (NRM) (183). These findings are complimented by a study on cGVHD by which the NRM rate of 22% within a five-year follow-up period was attributed to progression of cGVHD infections and non-specified factors (184). While these statistics do not include malignancy-related deaths, it can be understood that the dysregulated immune baseline and heighted activation phase play roles in poor prognosis of GVHD. The presence of autoantibodies has also been documented in GVHD, but it is not known if it mediates or is resultant from this condition (185, 186). HLA mismatches, pre-existing malignancies, and conditioning regimens related to a BMT form a complex interplay that disturbs the immune baseline and culminates into GVHD. This yin-yang immune balance may be temporarily restored with steroids, but patient prognoses of further pathologies and poor survival rates underlie the need for more effective therapeutics.
7 Immune related adverse events
First approved by the FDA in 2011, immune checkpoint inhibitors (ICIs) represented a major stride in the mechanistic understanding of cancer immunology (187). Khan and Gerber nicely reviewed that ICIs function to target the immune checkpoint molecules of CTLA-4 and PD-1/PD-L1, which normally work to prevent T cell response to self-antigens and ensure peripheral tolerance is maintained (41). Cancer cells take advantage of these proteins to downregulate T-cell mediated responses against tumors. The therapeutic intervention of ICIs prevents T-cell engagement with checkpoint inhibitors and immunosuppressive signaling, allowing the cytotoxic T cells to continue to expand, secrete cytokines, and perform effector functions against cancer (42, 188). The blockage of checkpoint inhibitors can lead to a variety of damaging and autoimmune-like manifestations such as diabetes, myasthenia gravis, colitis, and rheumatic autoimmune conditions (189, 190). Favoring T-cell activation through ICIs interferes with the yin-yang balance of the immune system, allowing for the immunopathology of hyperinflammation and multiorgan and tissue damage. These manifestations represent a bias towards the yang component seen in Figure 1. Termed as immune-related adverse events (irAEs), these manifestations are an extreme side effect of ICIs. The ability of ICIs to prevent downregulation of T cell responses relates to its anticancer effects but is also directly responsible for irAEs. A detailed review of the various body systems associated with irAEs manifestations, as well as suggested therapeutic interventions, has been discussed elsewhere (43). As indicated in Table 1, the term irAEs is unique from previously described cytokine storm syndromes (CSS) because it is a catch-all for a slew of symptoms that can result from ICI usage rather than a specific syndrome with well-defined pathology (191–193). General symptoms of irAEs include fever, rash, fatigue, abdominal pain, and diarrhea. Like DRESS, malignancy-associated sHLH, and GVHD, this condition is also initiated by a therapeutic regimen intended to treat an underlying malignancy. One of the most definitive features of cancer is rapid and uncontrollable cell growth (194), representing an immune baseline that contrasts with the yin-yang cycle of cell growth and death that is normally maintained and depicted in Figure 1. Regarding immune checkpoint inhibitor (ICI) usage, multiple studies have depicted a bias towards an adult population. Two clinical studies on ICI usage found the median patient age to be over 50 years old (195, 196). Although other studies have enrolled patients as young as 18 to geriatric patients over 80 years old (197, 198), these studies demonstrate a bias towards an adult population. In comparison, the amount of literature detailing ICI usage and long-term outcomes in pediatric patients is sparse. Wong and colleagues suggest that the pronounced gap of ICIs in pediatric populations and subsequent development of irAEs may be due to conflicting reports on efficacy and differences in tumor biology (199). There is hesitation with ubiquitous application of ICIs in pediatric tumors due to low mutational burden that prevents neo-epitopes for T cells and absence of PD-L1 targeted by ICIs (200–203). As there is yet to exist a direct comparison of the frequency of irAEs in adult and pediatric populations, the possibility of an age-dependent presentation requires further elucidation. The combined effects of pre-existing malignancies and median therapeutic age may be associated with the hyperinflammatory response initiated by T cells, leading to the development of a cytokine storm and irAEs in adult patients.
Cytokines play a unique role in irAEs, demonstrated by their involvement with the specific cytokine release syndrome (CRS)-related immune related adverse events (irAEs) and a variety of other irAEs organ manifestations. In a query of 80,700 cases of irAEs from VigiBase, one report analyzed that 55 out of 58 confirmed cases of CRS-irAE lead to serious complications and the final outcome of 20 such cases were not available (204). Although reports of CRS-irAEs are relatively small in comparison to the totality of irAEs, its presentation can serve as a harbinger of both immediate and prolonged immune dysregulation. The shared cytokine profile amongst all irAEs is portrayed through the cytokine-targeted therapies that focus on elevated levels of TNF-α, IL-1, IL-6, IL-12, IL-17, and IL-23 (205). In a study of gastrointestinal cancer patients, it was observed that colitis-irAEs could be correlated with elevated serum levels of IL-6, IL-22, and SCF (206). Elevations of IL-6 and IL-17 have been discussed as well (207, 208) but there is no universal consensus for a cytokine profile for irAEs. The conflicting reports on cytokine elevations in irAEs could be the result of the diversity in manifestations and differences in measurements across different studies. Nevertheless, the field acknowledges that irAEs stem from the hyperactive immune response initiated by immune checkpoint inhibitors (ICIs). The first-line therapeutic for irAEs relies on broad application of corticosteroids (209, 210) resembling the regimen described in previous CSS. With more severe manifestations or presentation of a steroid refractory patient, anti-cytokine therapies such as the TNF-α inhibitor infliximab or IL-6 inhibitor tocilizumab have also been utilized (211, 212). Other irAEs-specific recommendations include hormone replacement therapy for thyroid dysfunction or rituximab to treat neurological and rheumatic manifestations (213–217). Although the presentation of low-grade irAEs has shown to be efficacious in improving anti-cancer responses and overall survival rate of patients (218, 219) the immune landscape may be altered for the worst. It has been estimated that approximately 40% of all ICI patients will develop a chronic irAE that persists at least three months after initiation of therapy, usually related to autoimmune endocrine disorders or rheumatic dysfunction (43, 190, 220, 221). For these patients, the yin phase of the immune response is unable to complement the activation response. Due to pre-existing cancer, the disturbed immune baseline of ICI patients is more responsive to immune-targeted therapeutic regimens. The prolonged activation phase in irAEs is a hyperinflammatory response that is not effectively addressed nor corrected with current therapeutic guidelines.
8 Cytokine release syndrome and immune effector cell associated neurotoxicity syndrome
Chimeric antigen receptor T (CAR T) cell therapy has ushered in a new age in the evolving landscape of cancer immunotherapy. This therapy was first approved to treat pediatric and young adult relapsed/refractory B-cell acute lymphoblastic leukemia (R/R B-cell ALL) (27) and has presently expanded to treat other hematological cancers. CAR T-cell therapy harnesses the community’s increased understanding and subsequent manipulation of cancer immunobiology to provide targeted and efficacious treatment options for more patients. The general process of CAR T-cell therapy involves reengineering the patient’s own T lymphocytes with a receptor that recognizes cell surface markers found on B cells and in their malignant transformation, such as CD19 and BCMA (28, 29). At least two prior lines of therapy must fail before becoming eligible for CAR T therapy, seen to be more rigid in multiple myeloma where four prior lines of therapy must be unsuccessful (30, 222). Pre-existing malignancy and multiple lines of failed therapy cause heightened stress in the body, tipping the yin-yang immune response towards hyperinflammation.
CAR T-cell therapy clinical trials of the past and present are wary of cytokine release syndrome (CRS) and accompanying neurotoxic manifestations or immune cell-associated neurotoxicity syndrome (ICANS) that can result (223–226) as has been illustrated in Figure 3. Manifestations are graded on a scale of 1 to 4 based upon increasing severity of presentations that can include fever, malaise, headache, dyspnea, and hypotension for CRS and aphasia, altered levels of consciousness, decline in motor control, tremors and seizures, and fatal cerebral edema for ICANS (227, 228). As with ICIs, targeted activation of T lymphocytes is the basis for immunopathology of CRS/ICANS. CAR T cells release cytokines upon engaging with the target antigen that cause both expansion of self and bystander activation of other immune cells, such as monocytes and macrophages (221). A positive feedback loop of cytokine secretion is initiated amongst the cells that leads to cytokine storm and endothelial cell damage. Neurotoxic effects are believed to arise from cytokine storm as well, with leakage of immune cells across a compromised blood-brain barrier directly or indirectly killing neurons (229). Cytokine release represents a double-edged sword in CAR T as it is necessary for their expansion but causes bystander immune cell activation and subsequent organ tissue.
Both the mild and severe clinical presentations of cytokine release syndrome/immune effector cell-associated neurotoxicity syndrome (CRS/ICANS) represent the yang component of the immune response. The main symptomatology of CRS/ICANS has also been touched upon in Table 1. Although other adverse events have been reported with CAR T-cell therapy usage (230), the amount of literature on CRS/ICANS and its direct relationship with a cytokine storm make these manifestations most appropriate for consideration in this review. A comparison of adult and pediatric clinical trials leaves room for consideration of age-based severity of ICANS manifestations. The JULIET trial for adult R/R B-cell ALL reported a 12% incidence of grades 3 and 4 ICANS, noting that glucocorticoids successfully resolved most cases (224). This data aligns with the recent UNIVERSAL trial for multiple myeloma in adult patients by which 12% of all patients had potential low grade neurological manifestations that were also resolved with steroid usage (226). In comparison, the ELIANA trial for pediatric and young adult R/R B-cell ALL found that 40% of all patients experienced some degree of neurological manifestations within 8 weeks of initiating therapy (231). While the 13% incidence of grade 3 ICANS mirrors that in the JULIET trial, this pediatric cohort is most noteworthy for the three cases of grade 3 ICANS events were still unresolved at time of therapy cessation or death. The persistence of neurological manifestations represents a disturbed immune baseline in certain pediatric patients that becomes hyperactive with CAR T-cell therapy. Age differences in the immune landscape may then contribute to a baseline that is prone to activation. It must be acknowledged that this perceived difference is not agreed upon by all, as Shalabi and colleagues assert that the incidence of ICANS was found to be similar in both adult and pediatric patients (232). As such, the possible disparity in ICANS incidence and severity deserves further research and consideration on how age can play a role in dysregulated immune responses. Similar to septic shock and HLH conditions, cytokine storm has central role in mediating inflammation in CRS/ICANS.
There is much interest in characterizing a cytokine profile that is predictive of both onset and severity of cytokine release syndrome/immune effector cell-associated neurotoxicity syndrome (CRS/ICANS). Elevated IL-6, IL-8, IL-10, and IFN-γ has been frequently discussed, with a majority of focus placed on IL-6 (28, 225, 226). A meta-analysis of CSF taken from ICANS patients also identified elevated IL-15, IL-10, GM-CSF, IL-2, IL-1RA, and CXCL10 (233). The identification of cytokines elevated in CRS/ICANS shows how a hyperinflammatory immune baseline in cancer patients allows for the development of a cytokine storm. Therapeutic regimens are not unique for this cytokine storm syndrome (CSS), concurrently using corticosteroids and tocilizumab as the first-line therapy (234, 235). Anakinra has also been discussed as both a prophylactic and reactive treatment (236–239). But as with previously mentioned cytokine storm syndromes (CSS), alterative treatment options have yet to replace steroids as the first-line therapeutic for CSS manifestations. The prevalence of steroid treatment is best understood through its mechanism of interfering with the transcription of pro-inflammatory genes, leading to a reduction in cytokine release and associated mediators that are needed for inflammatory processes to occur (240). Although broad enough to be applied to a variety of hyperinflammatory conditions, steroid usage has been linked to many adverse effects including gastrointestinal issues, hypertension, osteoporosis, and the development of cataracts and glaucoma (241, 242). Steroid-induced immunosuppression can dampen the hyperinflammation caused by a cytokine storm, but usage leaves patients vulnerable to both opportunistic pathogens and the risk of future clinical manifestations. There have even been case reports of delayed ICANS that occurred weeks to months after cessation of CAR T-cell therapy (243, 244). Once more, the yin-yang immune response of these individuals becomes fixed in a state of activation and unable to contract to the yin phase and return to a status quo. Usage of targeted therapies may be promising for improved outcomes, but there is yet to exist a therapeutic that is able to correct the inherent immune dysregulation that is present in different types of CSS.
9 Discussion and perspectives
The balance in the yin-yang immune response ensures that the powerful and potentially damaging effects of the immune system are constantly kept in check. This occurs through the cyclic activation and contraction phases that both allows for protective inflammatory processes to occur and subsequent downregulation once the threat has been resolved, ensuring the activation phase does not persist past what is beneficial to the host. Cytokine storm syndromes (CSS) represent a dysregulated yin-yang immune response that is predisposed towards an inflammatory or hyperactive immune baseline. The yang component of CSS patients is overactive and creates a hyperinflammatory environment that is not complemented by the contraction phase or yin component. Given the rarity and unique circumstances surrounding CSS, there is an interplay of factors that must be present for a hyperinflammatory response to occur. Age, genetics, hematological and cancerous malignancies, and pre-existing illnesses have varying degrees of involvement in nurturing a dysregulated immune response that is more receptive towards the activation phase. Once these individuals have a trigger, such as illness, pathogen, or therapeutic intervention, they become subject to cellular dysfunctions, multiorgan damage, and systemic inflammation that is representative of cytokine storm.
There has been limited success in counteracting this state of hyperinflammation to allow patients to return to a balanced yin-yang immune response. Steroids are universally applied as a first-line therapeutic for CSS but are wholly unsuccessful in long-term resolution of symptoms, instead causing a variety of undesirable side effects. Alternative treatment options have been used in place of steroids but have yet to be widely recommended or considered. Further development of immunosuppressive and autoimmune states suggests that the immune response is just as dysregulated as before CSS onset. Given the similarities in clinical manifestations, perfunctory yet inefficient use of steroids, and overall poor prognoses of CSS, it is evident that there is an unmet need for more targeted therapies. Given the similarities of the immunopathology and elevated pro-inflammatory cytokines in all conditions, the usage of cytokine-targeted therapies could be an alternative for a more focused treatment approach. By identifying and targeting the cytokine profile for individual conditions, this approach focuses on specific components of the disease and is less subject to unnecessary side effects from non-specific steroid usage. While cytokine-targeted therapies may not be the only solution, effective therapies focusing on the mechanism and immune background of disease onset should be considered. Recognizing the familiar patterns in CSS and affiliated conditions will be central to understanding the mechanisms underlying each disease pathology, trajectory of the immune response, and identifying targeted therapies that allow for restoration of a balance yin-yang immune response and improving the outcome.
Author contributions
AA: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. YT: Conceptualization, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. NM: Conceptualization, Formal analysis, Visualization, Writing – review & editing. NZ: Conceptualization, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. GH: Conceptualization, Formal analysis, Investigation, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Viganò S, Perreau M, Pantaleo G, Harari A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin Dev Immunol. (2012) 2012:485781. doi: 10.1155/2012/485781
2. Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. (2002) 3:619–26. doi: 10.1038/ni804
3. Robbins SH, Tessmer MS, Mikayama T, Brossay L. Expansion and contraction of the NK cell compartment in response to murine cytomegalovirus infection. J Immunol. (2004) 173:259–66. doi: 10.4049/jimmunol.173.1.259
4. Riera Romo M. Cell death as part of innate immunity: Cause or consequence? Immunology. (2021) 163:399–415. doi: 10.1111/imm.13325
5. Campisi L, Cummings RJ, Blander JM. Death-defining immune responses after apoptosis. Am J Transplant. (2014) 14:1488–98. doi: 10.1111/ajt.12736
6. Liu HH, Nichols C, Zhang H. Understanding yin-yang philosophic concept behind tai chi practice. Holist Nurs Pract. (2023) 37:E75–82. doi: 10.1097/HNP.0000000000000598
7. Long A, Kleiner A, Looney RJ. Immune dysregulation. J Allergy Clin Immunol. (2023) 151:70–80. doi: 10.1016/j.jaci.2022.11.001
8. Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. (2017) 17:21–9. doi: 10.1038/nri.2016.125
9. Liston A, Humblet-Baron S, Duffy D, Goris A. Human immune diversity: from evolution to modernity. Nat Immunol. (2021) 22:1479–89. doi: 10.1038/s41590-021-01058-1
10. Tsang JS, Dobaño C, VanDamme P, Moncunill G, Marchant A, Othman RB, et al. Improving vaccine-induced immunity: can baseline predict outcome? Trends Immunol. (2020) 4:457–65. doi: 10.1016/j.it.2020.04.001
11. Pulendran B. Systems vaccinology: probing humanity’s diverse immune systems with vaccines. Proc Natl Acad Sci U S A. (2014) 111:12300–6. doi: 10.1073/pnas.1400476111
12. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. (2021) 6:291. doi: 10.1038/s41392-021-00687-0
13. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. (2015) 7:a016303. doi: 10.1101/cshperspect.a016303
14. Chi H, Pepper M, Thomas PG. Principles and therapeutic applications of adaptive immunity. Cell. (2024) 187:2052–78. doi: 10.1016/j.cell.2024.03.037
15. Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected. N Engl J Med. (1989) 320:1420–1. doi: 10.1056/NEJM198905253202117. Erratum in: N Engl J Med 1989 Jul 6;321(1):63.
16. Attarwala H. TGN1412: from discovery to disaster. J Young Pharm. (2010) 2:332–6. doi: 10.4103/0975-1483.66810
17. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. (2017) 5:517–28. doi: 10.1007/s00281-017-0639-8
18. Ferrara JL. Cytokine dysregulation as a mechanism of graft versus host disease. Curr Opin Immunol. (1993) 5:794–9. doi: 10.1016/0952-7915(93)90139-j
19. Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am. (2012) 59:329–44. doi: 10.1016/j.pcl.2012.03.002
20. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
21. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. (2021) 93:250–6. doi: 10.1002/jmv.26232
22. García LF. Immune response, inflammation, and the clinical spectrum of COVID-19. Front Immunol. (2020) 11:1441. doi: 10.3389/fimmu.2020.01441
23. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
24. Ehsan N, Zahra F. Castleman disease. 2023 apr 27. In: StatPearls. StatPearls Publishing, Treasure Island (FL (2024).
25. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood. (2020) 135:1353–64. doi: 10.1182/blood.2019000931
26. Sumiyoshi R, Koga T, Kawakami A. Candidate biomarkers for idiopathic multicentric Castleman disease. J Clin Exp Hematop. (2022) 62:85–90. doi: 10.3960/jslrt.22010
27. Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy. (2020) 22:57–69. doi: 10.1016/j.jcyt.2019.12.004
28. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
29. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
30. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel). (2023) 15:663. doi: 10.3390/cancers15030663
31. De A, Rajagopalan M, Sarda A, Das S, Biswas P. Drug reaction with eosinophilia and systemic symptoms: an update and review of recent literature. Indian J Dermatol. (2018) 63:30–40. doi: 10.4103/ijd.IJD_582_17
32. Lu J, Thuraisingam T, Chergui M, Nguyen K. Nivolumab-associated DRESS syndrome: A case report. JAAD Case Rep. (2019) 5:216–8. doi: 10.1016/j.jdcr.2018.11.017
33. Justiz Vaillant AA, Modi P, Mohammadi O. Graft-versus-host disease. 2022 oct 10. In: StatPearls. StatPearls Publishing, Treasure Island (FL (2024).
34. Ratanatharathorn V, Ayash L, Lazarus HM, Fu J, Uberti JP. Chronic graft-versus-host disease: clinical manifestation and therapy. Bone Marrow Transplant. (2001) 28:121–9. doi: 10.1038/sj.bmt.1703111
35. Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood. (1992) 80:2964–8. doi: 10.1182/blood.V80.12.2964.2964
36. Nixon A, Roddick E, Moore K, Wild D. A qualitative investigation into the impact of hemophagocytic lymphohistiocytosis on children and their caregivers. Orphanet J Rare Dis. (2021) 16:205. doi: 10.1186/s13023-021-01832-2
37. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Program. (2013) 2013:605–11. doi: 10.1182/asheducation-2013.1.605
38. Nguyen TTT, Kim YT, Jeong G, Jin M. Immunopathology of and potential therapeutics for secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome: a translational perspective. Exp Mol Med. (2024) 56:559–69. doi: 10.1038/s12276-024-01182-6
39. Bauchmuller K, Manson JJ, Tattersall R, Brown M, McNamara C, Singer M, et al. Hemophagocytic lymphohistiocytosis in adult critical care. J Intensive Care Soc. (2020) 21:256–68. doi: 10.1177/1751143719893865
40. Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol. (2012) 10:726–32.
41. Khan S, Gerber DE. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin Cancer Biol. (2020) 64:93–101. doi: 10.1016/j.semcancer.2019.06.012
42. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. (2019) 16:563–80. doi: 10.1038/s41571-019-0218-0
43. Choi J, Lee SY. Clinical characteristics and treatment of immune-related adverse events of immune checkpoint inhibitors. Immune Netw. (2020) 20:e9. doi: 10.4110/in.2020.20.e9
44. Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. (2018) 379:856–68. doi: 10.1056/NEJMra1607548
45. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. (2020) 135:1319–31. doi: 10.1182/blood.2019000934
46. Kannourakis G, Abbas A. The role of cytokines in the pathogenesis of Langerhans cell histiocytosis. Br J Cancer Suppl. (1994) 23:S37–40.
47. Patel JM. Multisystem inflammatory syndrome in children (MIS-C). Curr Allergy Asthma Rep. (2022) 22:53–60. doi: 10.1007/s11882-022-01031-4
48. Esposito S, Principi N. Multisystem inflammatory syndrome in children related to SARS-coV-2. Pediatr Drugs. (2021) 23:119–29. doi: 10.1007/s40272-020-00435-x
49. Constantin T, Pék T, Horváth Z, Garan D, Szabó AJ. Multisystem inflammatory syndrome in children (MIS-C): Implications for long COVID. Inflammopharmacology. (2023) 31:2221–36. doi: 10.1007/s10787-023-01272-3
50. Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. (2009) 302:2323–9. doi: 10.1001/jama.2009.1754
51. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. (2016) 2:16045. doi: 10.1038/nrdp.2016.45
52. Vincent JL, Abraham E. The last 100 years of sepsis. Am J Respir Crit Care Med. (2006) 173:256–63. doi: 10.1164/rccm.200510-1604OE
53. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. (2017) 17:407–20. doi: 10.1038/nri.2017.36
54. Wiersinga WJ, van der Poll T. Immunopathophysiology of human sepsis. EBioMedicine. (2022) 86:104363. doi: 10.1016/j.ebiom.2022.104363
55. Gyawali B, Ramakrishna K, Dhamoon AS. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med. (2019) 7:2050312119835043. doi: 10.1177/2050312119835043
56. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. (2010) 2:e2010024. doi: 10.4084/MJHID.2010.024
57. Rello J, Valenzuela-Sánchez F, Ruiz-Rodriguez M, Moyano S. Sepsis: A review of advances in management. Adv Ther. (2017) 34:2393–411. doi: 10.1007/s12325-017-0622-8
58. Merx MW, Weber C. Sepsis and the heart. Circulation. (2007) 116:793–802. doi: 10.1161/CIRCULATIONAHA.106.678359
59. Hu Q, Hao C, Tang S. From sepsis to acute respiratory distress syndrome (ARDS): emerging preventive strategies based on molecular and genetic researches. Biosci Rep. (2020) 40:BSR20200830. doi: 10.1042/BSR20200830
60. Zarbock A, Gomez H, Kellum JA. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr Opin Crit Care. (2014) 20:588–95. doi: 10.1097/MCC.0000000000000153
61. Yan J, Li S, Li S. The role of the liver in sepsis. Int Rev Immunol. (2014) 33:498–510. doi: 10.3109/08830185.2014.889129
62. Basodan N, Al Mehmadi AE, Al Mehmadi AE, Aldawood SM, Hawsawi A, Fatini F, et al. Septic shock: management and outcomes. Cureus. (2022) 14:e32158. doi: 10.7759/cureus.32158
63. Sinapidis D, Kosmas V, Vittoros V, Koutelidakis IM, Pantazi A, Stefos A, et al. Progression into sepsis: an individualized process varying by the interaction of comorbidities with the underlying infection. BMC Infect Dis. (2018) 18:242. doi: 10.1186/s12879-018-3156-z
64. Esper AM, Moss M, Lewis CA, Nisbet R, Mannino DM, Martin GS. The role of infection and comorbidity: Factors that influence disparities in sepsis. Crit Care Med. (2006) 34:2576–82. doi: 10.1097/01.CCM.0000239114.50519.0E
65. Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. (2018) 9:754. doi: 10.3389/fimmu.2018.00754
66. Tang XD, Ji TT, Dong JR, Feng H, Chen FQ, Chen X, et al. Pathogenesis and treatment of cytokine storm induced by infectious diseases. Int J Mol Sci. (2021) 22:13009. doi: 10.3390/ijms222313009
67. Feuerecker M, Sudhoff L, Crucian B, Pagel JI, Sams C, Strewe C, et al. Early immune anergy towards recall antigens and mitogens in patients at onset of septic shock. Sci Rep. (2018) 8:1754. doi: 10.1038/s41598-018-19976-w
68. Thijs LG, Hack CE. Time course of cytokine levels in sepsis. Intensive Care Med. (1995) Suppl 2:S258–63. doi: 10.1007/BF01740764
69. Blackwell TS, Christman JW. Sepsis and cytokines: current status. Br J Anaesth. (1996) 77:110–7. doi: 10.1093/bja/77.1.110
70. Matsumoto H, Ogura H, Shimizu K, Ikeda M, Hirose T, Matsuura H, et al. The clinical importance of a cytokine network in the acute phase of sepsis. Sci Rep. (2018) 8:13995. doi: 10.1038/s41598-018-32275-8
71. Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, et al. Role of cytokines as a double-edged sword in sepsis. In Vivo. (2013) 27:669–84.
72. Marik PE. Steroids for sepsis: yes, no or maybe. J Thorac Dis. (2018) 10:S1070–3. doi: 10.21037/jtd.2018.04.35
73. Annane D. Corticosteroids for severe sepsis: an evidence-based guide for physicians. Ann Intensive Care. (2011) 1:7. doi: 10.1186/2110-5820-1-7
74. Liang H, Song H, Zhai R, Song G, Li H, Ding X, et al. Corticosteroids for treating sepsis in adult patients: A systematic review and meta-analysis. Front Immunol. (2021) 12:709155. doi: 10.3389/fimmu.2021.709155. Erratum in: Front Immunol. 2021 Nov 05;12:771779.
75. Gibbison B, López-López JA, Higgins JP, Miller T, Angelini GD, Lightman SL, et al. Corticosteroids in septic shock: a systematic review and network meta-analysis. Crit Care. (2017) 21:78. doi: 10.1186/s13054-017-1659-4
76. Rahmel T, Schmitz S, Nowak H, Schepanek K, Bergmann L, Halberstadt P, et al. Long-term mortality and outcome in hospital survivors of septic shock, sepsis, and severe infections: The importance of aftercare. PloS One. (2020) 15:e0228952. doi: 10.1371/journal.pone.0228952
77. Cuthbertson BH, Elders A, Hall S, Taylor J, MacLennan G, Mackirdy F, et al. Mortality and quality of life in the five years after severe sepsis. Crit Care. (2013) 17:R70. doi: 10.1186/cc12616
78. Wang HE, Szychowski JM, Griffin R, Safford MM, Shapiro NI, Howard G. Long-term mortality after community-acquired sepsis: a longitudinal population-based cohort study. BMJ Open. (2014) 4:e004283. doi: 10.1136/bmjopen-2013-004283
79. Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. (2014) 20:224–33. doi: 10.1016/j.molmed.2014.01.002
80. Steinhagen F, Schmidt SV, Schewe JC, Peukert K, Klinman DM, Bode C. Immunotherapy in sepsis - brake or accelerate? Pharmacol Ther. (2020) 208:107476. doi: 10.1016/j.pharmthera.2020.107476
81. Fenner BP, Darden DB, Kelly LS, Rincon J, Brakenridge SC, Larson SD, et al. Immunological endotyping of chronic critical illness after severe sepsis. Front Med (Lausanne). (2021) 7:616694. doi: 10.3389/fmed.2020.616694
82. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. (2012) 72:1491–501. doi: 10.1097/TA.0b013e318256e000
83. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin. (2017) 33:245–58. doi: 10.1016/j.ccc.2016.12.001
84. Chadda KR, Puthucheary Z. Persistent inflammation, immunosuppression, and catabolism syndrome (PICS): a review of definitions, potential therapies, and research priorities. Br J Anaesth. (2024) 132:507–18. doi: 10.1016/j.bja.2023.11.052
85. Choudhary S, McLeod M, Torchia D, Romanelli P. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome. J Clin Aesthet Dermatol. (2013) 6:31–7.
86. Behera SK, Das S, Xavier AS, Selvarajan S. DRESS syndrome: a detailed insight. Hosp Pract. (2018) 46:152–62. doi: 10.1080/21548331.2018.1451205
87. Calle AM, Aguirre N, Ardila JC, Cardona Villa R. DRESS syndrome: A literature review and treatment algorithm. World Allergy Organ J. (2023) 16:100673. doi: 10.1016/j.waojou.2022.100673
88. Ros J, Muñoz-Couselo E. DRESS syndrome due to vemurafenib treatment: switching BRAF inhibitor to solve a big problem. BMJ Case Rep. (2018) 2018, bcr2018224379. doi: 10.1136/bcr-2018-224379
89. Musette P, Janela B. New insights into drug reaction with eosinophilia and systemic symptoms pathophysiology. Front Med (Lausanne). (2017) 4:179. doi: 10.3389/fmed.2017.00179
90. Miyagawa F, Asada H. Current perspective regarding the immunopathogenesis of drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms (DIHS/DRESS). Int J Mol Sci. (2021) 22:2147. doi: 10.3390/ijms22042147
91. Stirton H, Shear NH, Dodiuk-Gad RP. Drug reaction with eosinophilia and systemic symptoms (DReSS)/drug-induced hypersensitivity syndrome (DiHS)-readdressing the DReSS. Biomedicines. (2022) 10:999. doi: 10.3390/biomedicines10050999
92. Cho YT, Yang CW, Chu CY. Drug reaction with eosinophilia and systemic symptoms (DRESS): an interplay among drugs, viruses, and immune system. Int J Mol Sci. (2017) 18:1243. doi: 10.3390/ijms18061243
93. Kardaun SH, Sekula P, Valeyrie-Allanore L, Liss Y, Chu CY, Creamer D, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. (2013) 169:1071–80. doi: 10.1111/bjd.12501
94. Ramirez GA, Ripa M, Burastero S, Benanti G, Bagnasco D, Nannipieri S, et al. Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS): Focus on the pathophysiological and diagnostic role of viruses. Microorganisms. (2023) 11:346. doi: 10.3390/microorganisms11020346
95. Lens S, Crespo G, Carrión JA, Miquel R, Navasa M. Severe acute hepatitis in the DRESS syndrome: Report of two cases. Ann Hepatol. (2010) 9:198–201. doi: 10.1016/S1665-2681(19)31663-1
96. Oliveira AM, Carvalho R, Martins A, Reis J. Acute hepatitis in the DRESS syndrome. GE Port J Gastroenterol. (2016) 23:304–8. doi: 10.1016/j.jpge.2016.06.001
97. Dagnon da Silva M, Domingues SM, Oluic S, Radovanovic M, Kodela P, Nordin T, et al. Renal manifestations of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome: A systematic review of 71 cases. J Clin Med. (2023) 12:4576. doi: 10.3390/jcm12144576
98. Wolfson AR, Zhou L, Li Y, Phadke NA, Chow OA, Blumenthal KG. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome identified in the electronic health record allergy module. J Allergy Clin Immunol Pract. (2019) 7:633–40. doi: 10.1016/j.jaip.2018.08.013
99. Chen YC, Chiu HC, Chu CY. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. (2010) 146:1373–9. doi: 10.1001/archdermatol.2010.198
100. Mitamura Y, Schulz D, Oro S, Li N, Kolm I, Lang C, et al. Cutaneous and systemic hyperinflammation drives maculopapular drug exanthema in severely ill COVID-19 patients. Allergy. (2022) 77:595–608. doi: 10.1111/all.14983
101. Nissen MS, Beier CP. DRESS after IV phenytoin associated with cytochrome P450 CYP2C9*3 homozygosity. Neurol Genet. (2018) 4:e272. doi: 10.1212/NXG.0000000000000272
102. Suvichapanich S, Jittikoon J, Wichukchinda N, Kamchaisatian W, Visudtibhan A, Benjapopitak S, et al. Association analysis of CYP2C9*3 and phenytoin-induced severe cutaneous adverse reactions (SCARs) in Thai epilepsy children. J Hum Genet. (2015) 60:413–7. doi: 10.1038/jhg.2015.47
103. Chung WH, Chang WC, Lee YS, Wu YY, Yang CH, Ho HC, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. (2014) 312:525–34. doi: 10.1001/jama.2014.7859
104. Schunkert EM, Divito SJ. Updates and insights in the diagnosis and management of DRESS syndrome. Curr Dermatol Rep. (2021) 10:192–204. doi: 10.1007/s13671-021-00348-z
105. Cabañas R, Ramírez E, Sendagorta E, Alamar R, Barranco R, Blanca-López N, et al. Spanish guidelines for diagnosis, management, treatment, and prevention of DRESS syndrome. J Investig Allergol Clin Immunol. (2020) 30:229–53. doi: 10.18176/jiaci.0480
106. Galvão VR, Aun MV, Kalil J, Castells M, Giavina-Bianchi P. Clinical and laboratory improvement after intravenous immunoglobulin in drug reaction with eosinophilia and systemic symptoms. J Allergy Clin Immunol Pract. (2014) 2:107–10. doi: 10.1016/j.jaip.2013.11.008
107. Joly P, Janela B, Tetart F, Rogez S, Picard D, D’Incan M, et al. Poor benefit/risk balance of intravenous immunoglobulins in DRESS. Arch Dermatol. (2012) 148:543–4. doi: 10.1001/archderm.148.4.dlt120002-c
108. Kirchhof MG, Wong A, Dutz JP. Cyclosporine treatment of drug-induced hypersensitivity syndrome. JAMA Dermatol. (2016) 152:1254–7. doi: 10.1001/jamadermatol.2016.2220
109. Zita S, Broussard L, Hugh J, Newman S. Cyclosporine in the treatment of drug reaction with eosinophilia and systemic symptoms syndrome: retrospective cohort study. JMIR Dermatol. (2023) 6:e41391. doi: 10.2196/41391
110. Mizukawa Y, Aoyama Y, Takahashi H, Takahashi R, Shiohara T. Risk of progression to autoimmune disease in severe drug eruption: risk factors and the factor-guided stratification. J Invest Dermatol. (2022) 142:960–68.e9. doi: 10.1016/j.jid.2021.11.008
111. Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. (2013) 139:713–27. doi: 10.1309/AJCP4ZDKJ4ICOUAT
112. Standage SW, Filipovich AH. Hemophagocytic lymphohistiocytosis syndromes. Pediatr Crit Care Med. (2014) 3:385–93. doi: 10.1007/978-1-4471-6416-6_26
113. Bseiso O, Zahdeh A, Isayed O, Mahagna S, Bseiso A. The role of immune mechanisms, inflammatory pathways, and macrophage activation syndrome in the pathogenesis of hemophagocytic lymphohistiocytosis. Cureus. (2022) 14:e33175. doi: 10.7759/cureus.33175
114. Sztajnbok F, Fonseca AR, Campos LR, Lino K, Rodrigues MCF, Silva RM, et al. Hemophagocytic lymphohistiocytosis and macrophage activation syndrome: two rare sides of the same devastating coin. Adv Rheumatol. (2024) 64:28. doi: 10.1186/s42358-024-00370-2
115. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. (1991) 18:29–33.
116. Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control. (2014) 21:301–12. doi: 10.1177/107327481402100406
117. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 16:1332–43. doi: 10.1182/blood.2019000936
118. Kim MM, Yum MS, Choi HW, Ko TS, Im HJ, Seo JJ, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol. (2012) 47:273–80. doi: 10.5045/kjh.2012.47.4.273
119. Hadžić N, Molnar E, Height S, Kovács G, Dhawan A, Andrikovics H, et al. High prevalence of hemophagocytic lymphohistiocytosis in acute liver failure of infancy. J Pediatr. (2022) 250:67–74.e1. doi: 10.1016/j.jpeds.2022.07.006
120. Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. (1991) 80:428–35. doi: 10.1111/j.1651-2227.1991.tb11878.x
121. Piva S, Bertoni M, Gitti N, Rasulo FA, Latronico N. Neurological complications of sepsis. Curr Opin Crit Care. (2023) 29:75–84. doi: 10.1097/MCC.0000000000001022
122. Bąbol-Pokora K, Wołowiec M, Popko K, Jaworowska A, Bryceson YT, Tesi B, et al. Molecular genetics diversity of primary hemophagocytic lymphohistiocytosis among polish pediatric patients. Arch Immunol Ther Exp (Warsz). (2021) 69:31. doi: 10.1007/s00005-021-00635-4
123. Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. (2016) 127:2672–81. doi: 10.1182/blood-2016-01-690636
124. Planas R, Felber M, Vavassori S, Pachlopnik Schmid J. The hyperinflammatory spectrum: from defects in cytotoxicity to cytokine control. Front Immunol. (2023) 14:1163316. doi: 10.3389/fimmu.2023.1163316
125. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. (2018) 13:27–49. doi: 10.1146/annurev-pathol-020117-043625
126. Soy M, Atagündüz P, Atagündüz I, Sucak GT. Hemophagocytic lymphohistiocytosis: a review inspired by the COVID-19 pandemic. Rheumatol Int. (2021) 41:7–18. doi: 10.1007/s00296-020-04636-y
127. Zhang HQ, Yang SW, Fu YC, Chen MC, Yang CH, Yang MH, et al. Cytokine storm and targeted therapy in hemophagocytic lymphohistiocytosis. Immunol Res. (2022) 70:566–77. doi: 10.1007/s12026-022-09285-w
128. Xu XJ, Luo ZB, Song H, Xu WQ, Henter JI, Zhao N, et al. Simple evaluation of clinical situation and subtypes of pediatric hemophagocytic lymphohistiocytosis by cytokine patterns. Front Immunol. (2022) 13:850443. doi: 10.3389/fimmu.2022.850443
129. Ou W, Zhao Y, Wei A, Ma H, Zhang Q, Zhang L, et al. Serum cytokine pattern in children with hemophagocytic lymphohistiocytosis. Ann Hematol. (2023) 102:729–39. doi: 10.1007/s00277-023-05132-6
130. Henter JI, Aricò M, Egeler RM, Elinder G, Favara BE, Filipovich AH, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. (1997) 28:342–7. doi: 10.1002/(sici)1096-911x(199705)28:5<342::aid-mpo3>3.0.co;2-h
131. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
132. Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
133. Cesaro S, Locatelli F, Lanino E, Porta F, Di Maio L, Messina C, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Hematologica. (2008) 93:1694–701. doi: 10.3324/haematol.13142
134. Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. (1985) 106:561–6. doi: 10.1016/s0022-3476(85)80072-x
135. Abdelhay A, Mahmoud AA, Al Ali O, Hashem A, Orakzai A, Jamshed S. Epidemiology, characteristics, and outcomes of adult hemophagocytic lymphohistiocytosis in the USA, 2006-19: a national, retrospective cohort study. EClinical Med. (2023) 62:102143. doi: 10.1016/j.eclinm.2023.102143
136. Karlsson T. Secondary hemophagocytic lymphohistiocytosis: Experience from the Uppsala University Hospital. Ups J Med Sci. (2015) 120:257–62. doi: 10.3109/03009734.2015.1064500
137. Zhou M, Li L, Zhang Q, Ma S, Sun J, Zhu L, et al. Clinical features and outcomes in secondary adult hemophagocytic lymphohistiocytosis. QJM. (2018) 111:23–31. doi: 10.1093/qjmed/hcx183
138. Carvelli J, Piperoglou C, Farnarier C, Vely F, Mazodier K, Audonnet S, et al. Functional and genetic testing in adults with HLH reveals an inflammatory profile rather than a cytotoxicity defect. Blood. (2020) 136:542–52. doi: 10.1182/blood.2019003664
139. Fadlallah MM, Salman SM, Fadlallah MM, Rahal H. Hemophagocytic syndrome and COVID-19: A comprehensive review. Cureus. (2023) 15:e36140. doi: 10.7759/cureus.36140
140. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
141. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–196.e4. doi: 10.1016/j.jaci.2015.06.048
142. Miao Y, Zhu HY, Qiao C, Xia Y, Kong Y, Zou YX, et al. Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol. (2019) 10:395. doi: 10.3389/fimmu.2019.00395
143. Zinter MS, Hermiston ML. Calming the storm in HLH. Blood. (2019) 134:103–4. doi: 10.1182/blood.2019001333
144. Chen Y, Wang Z, Luo Z, Zhao N, Yang S, Tang Y. Comparison of Th1/Th2 cytokine profiles between primary and secondary hemophagocytic lymphohistiocytosis. Ital J Pediatr. (2016) 42:50. doi: 10.1186/s13052-016-0262-7
145. Salunke B, Savarkar S, Patil VP. Hemophagocytic syndrome-an approach to the management. Indian J Crit Care Med. (2019) 23:S191–6. doi: 10.5005/jp-journals-10071-23251
146. Tang Y, Xu Q, Luo H, Yan X, Wang G, Hu L, et al. Excessive IL-10 and IL-18 trigger hemophagocytic lymphohistiocytosis-like hyperinflammation and enhanced myelopoiesis. J Allergy Clin Immunol. (2022) 150:1154–67. doi: 10.1016/j.jaci.2022.06.017
147. Schäfer EJ, Jung W, Korsten P. Combination immunosuppressive therapy including rituximab for epstein-barr virus-associated hemophagocytic lymphohistiocytosis in adult-onset still’s disease. Case Rep Rheumatol. (2016) 2016:8605274. doi: 10.1155/2016/8605274
148. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced hemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Hematol. (2013) 162:376–82. doi: 10.1111/bjh.12386
149. Baverez C, Grall M, Gerfaud-Valentin M, De Gail S, Belot A, Perpoint T, et al. Anakinra for the treatment of hemophagocytic lymphohistiocytosis: 21 cases. J Clin Med. (2022) 11:5799. doi: 10.3390/jcm11195799
150. Bami S, Vagrecha A, Soberman D, Badawi M, Cannone D, Lipton J, et al. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2020) 67:e28581. doi: 10.1002/pbc.28581
151. Jumic S, Nand S. Hemophagocytic lymphohistiocytosis in adults: associated diagnoses and outcomes, a ten-year experience at a single institution. J Hematol. (2019) 8:149–54. doi: 10.14740/jh592
152. Park HS, Kim DY, Lee JH, Lee JH, Kim SD, Park YH, et al. Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: an analysis of treatment outcome and prognostic factors. Ann Hematol. (2012) 91:897–904. doi: 10.1007/s00277-011-1380-3
153. Vriesendorp HM, Heidt PJ. History of graft-versus-host disease. Exp Hematol. (2016) 44:674–88. doi: 10.1016/j.exphem.2016.05.011
154. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. (2009) 373:1550–61. doi: 10.1016/S0140-6736(09)60237-3
155. Jacobsohn DA, Vogelsang GB. Acute graft versus host disease. Orphanet J Rare Dis. (2007) 2:35. doi: 10.1186/1750-1172-2-35
156. Petersdorf EW. Role of major histocompatibility complex variation in graft-versus-host disease after hematopoietic cell transplantation. F1000Res. (2017) 6:617. doi: 10.12688/f1000research.10990.1
157. Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. (2003) 9:215–33. doi: 10.1053/bbmt.2003.50026
158. Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. (1980) 69:204–17. doi: 10.1016/0002-9343(80)90380-0
159. Malard F, Holler E, Sandmaier BM, Huang H, Mohty M. Acute graft-versus-host disease. Nat Rev Dis Primers. (2023) 9:27. doi: 10.1038/s41572-023-00438-1
160. Lee SJ. Classification systems for chronic graft-versus-host disease. Blood. (2017) 129:30–7. doi: 10.1182/blood-2016-07-686642
161. Zeiser R, Blazar BR. Acute graft-versus-host disease - biologic process, prevention, and therapy. N Engl J Med. (2017) 377:2167–79. doi: 10.1056/NEJMra1609337
162. Flowers ME, Kansu E, Sullivan KM. Pathophysiology and treatment of graft-versus-host disease. Hematol Oncol Clin North Am. (1999) 13:1091–112. doi: 10.1016/s0889-8588(05)70111-8
163. Ramachandran V, Kolli SS, Strowd LC. Review of graft-versus-host disease. Dermatol Clin. (2019) 37:569–82. doi: 10.1016/j.det.2019.05.014
164. Yu J, Hamilton BK, Turnbull J, Stewart SK, Vernaya A, Bhatt V, et al. Patient-reported symptom burden and impact on daily activities in chronic graft-versus-host disease. Cancer Med. (2023) 12:3623–33. doi: 10.1002/cam4.5209
165. Filipovich AH. Diagnosis and manifestations of chronic graft-versus-host disease. Best Pract Res Clin Hematol. (2008) 21:251–7. doi: 10.1016/j.beha.2008.02.008
166. Pavletic SZ, Smith LM, Bishop MR, Lynch JC, Tarantolo SR, Vose JM, et al. Prognostic factors of chronic graft-versus-host disease after allogeneic blood stem-cell transplantation. Am J Hematol. (2005) 78:265–74. doi: 10.1002/ajh.20275
167. Bazinet A, Popradi G. A general practitioner’s guide to hematopoietic stem-cell transplantation. Curr Oncol. (2019) 26:187–91. doi: 10.3747/co.26.5033
168. Burns SO, Morris EC. How I use allogeneic HSCT for adults with inborn errors of immunity. Blood. (2021) 138:1666–76. doi: 10.1182/blood.2020008187
169. Tan EY, Boelens JJ, Jones SA, Wynn RF. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr. (2019) 7:433. doi: 10.3389/fped.2019.00433
170. Taylor M, Khan S, Stapleton M, Wang J, Chen J, Wynn R, et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: past, present, and future. Biol Blood Marrow Transplant. (2019) 25:e226–46. doi: 10.1016/j.bbmt.2019.02.012
171. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of gvHD and other HSCT-related major complications. Front Immunol. (2017) 8:79. doi: 10.3389/fimmu.2017.00079
172. MacDonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest. (2017) 127:2452–63. doi: 10.1172/JCI90593
173. Kumar S, Mohammadpour H, Cao X. Targeting cytokines in GVHD therapy. J Immunol Res Ther. (2017) 2:90–9.
174. Hill GR, Betts BC, Tkachev V, Kean LS, Blazar BR. Current concepts and advances in graft-versus-host disease immunology. Annu Rev Immunol. (2021) 39:19–49. doi: 10.1146/annurev-immunol-102119-073227
175. Jiang H, Fu D, Bidgoli A, Paczesny S. T cell subsets in graft versus host disease and graft versus tumor. Front Immunol. (2021) 12:761448. doi: 10.3389/fimmu.2021.761448
176. Saad A, de Lima M, Anand S, Bhatt VR, Bookout R, Chen G, et al. Hematopoietic cell transplantation, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2020) 18:599–634. doi: 10.6004/jnccn.2020.0021
177. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute graft versus host disease: A comprehensive review. Anticancer Res. (2017) 37:1547–55. doi: 10.21873/anticanres.11483
178. Garnett C, Apperley JF, Pavlů J. Treatment and management of graft-versus-host disease: improving response and survival. Ther Adv Hematol. (2013) 4:366–78. doi: 10.1177/2040620713489842
179. Wolff D, Fatobene G, Rocha V, Kröger N, Flowers ME. Steroid-refractory chronic graft-versus-host disease: treatment options and patient management. Bone Marrow Transplant. (2021) 56:2079–87. doi: 10.1038/s41409-021-01389-5
180. Malard F, Huang XJ, Sim JPY. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia. (2020) 34:1229–40. doi: 10.1038/s41375-020-0804-2
181. Toubai T, Magenau J. Immunopathology and biology-based treatment of steroid-refractory graft-versus-host disease. Blood. (2020) 136:429–40. doi: 10.1182/blood.2019000953
182. Murata M, Teshima T. Treatment of steroid-refractory acute graft-versus-host disease using commercial mesenchymal stem cell products. Front Immunol. (2021) 12:724380. doi: 10.3389/fimmu.2021.724380
183. Holtan SG, Yu J, Choe HK, Paranagama D, Tang J, Naim A, et al. Disease progression, treatments, hospitalization, and clinical outcomes in acute GVHD: a multicenter chart review. Bone Marrow Transplant. (2022) 57:1581–5. doi: 10.1038/s41409-022-01764-w
184. DeFilipp Z, Alousi AM, Pidala JA, Carpenter PA, Onstad LE, Arai S, et al. Nonrelapse mortality among patients diagnosed with chronic GVHD: an updated analysis from the Chronic GVHD Consortium. Blood Adv. (2021) 5:4278–84. doi: 10.1182/bloodadvances.2021004941
185. Hao B, Gao S, Sang YW, Wang L, Meng XQ, You JY. Potential value of autoantibodies as biomarkers of chronic graft-versus-host disease after allogeneic stem cell transplantation. J Zhejiang Univ Sci B. (2019) 20:849–60. doi: 10.1631/jzus.B1900205
186. Yang K, Chen Y, Qi H, Ye Y, Fan Z, Huang F, et al. Anti-ro52 autoantibodies are related to chronic graft-vs.-host disease after allogeneic hematopoietic stem cell transplantation. Front Immunol. (2020) 11:1505. doi: 10.3389/fimmu.2020.01505
187. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. (2020) 11:3801. doi: 10.1038/s41467-020-17670-y
188. Saibil SD, Ohashi PS. Targeting T cell activation in immuno-oncology. Curr Oncol. (2020) 27:S98–S105. doi: 10.3747/co.27.5285
189. Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. (2022) 19:254–67. doi: 10.1038/s41571-022-00600-w
190. Wang SJ, Dougan SK, Dougan M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer. (2023) 9:543–53. doi: 10.1016/j.trecan.2023.04.002
191. Chennamadhavuni A, Abushahin L, Jin N, Presley CJ, Manne A. Risk factors and biomarkers for immune-related adverse events: A practical guide to identifying high-risk patients and rechallenging immune checkpoint inhibitors. Front Immunol. (2022) 13:779691. doi: 10.3389/fimmu.2022.779691
192. Ibis B, Aliazis K, Cao C, Yenyuwadee S, Boussiotis VA. Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front Immunol. (2023) 14:1197364. doi: 10.3389/fimmu.2023.1197364
193. Conroy M, Naidoo J. Immune-related adverse events and the balancing act of immunotherapy. Nat Commun. (2022) 13:392. doi: 10.1038/s41467-022-27960-2
194. Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S, et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. (2013) 14:21087–113. doi: 10.3390/ijms141021087
195. Nardin C, Hennemann A, Diallo K, Funck-Brentano E, Puzenat E, Heidelberger V, et al. Efficacy of immune checkpoint inhibitor (ICI) rechallenge in advanced melanoma patients’ Responders to a first course of ICI: A multicenter national retrospective study of the french group of skin cancers (Groupe de cancérologie cutanée, GCC). Cancers (Basel). (2023) 15:3564. doi: 10.3390/cancers15143564
196. Reckamp KL, Redman MW, Dragnev KH, Minichiello K, Villaruz LC, Faller B, et al. Phase II randomized study of ramucirumab and pembrolizumab versus standard of care in advanced non-small-cell lung cancer previously treated with immunotherapy-lung-MAP S1800A. J Clin Oncol. (2022) 40:2295–306. doi: 10.1200/JCO.22.00912
197. Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. (2021) 384:1289–300. doi: 10.1056/NEJMoa2035716
198. Nebhan CA, Cortellini A, Ma W, Ganta T, Song H, Ye F, et al. Clinical outcomes and toxic effects of single-agent immune checkpoint inhibitors among patients aged 80 years or older with cancer: A multicenter international cohort study. JAMA Oncol. (2021) 7:1856–61. doi: 10.1001/jamaoncol.2021.4960
199. Wong SK, Nebhan CA, Johnson DB. Impact of patient age on clinical efficacy and toxicity of checkpoint inhibitor therapy. Front Immunol. (2021) 12:786046. doi: 10.3389/fimmu.2021.786046
200. Rahal Z, Abdulhai F, Kadara H, Saab R. Genomics of adult and pediatric solid tumors. Am J Cancer Res. (2018) 8:1356–86.
201. Nigro O, Ferrari A, Casanova M, Orbach D, Leruste A, Gatz SA, et al. Controversies on the possible role of immune checkpoint inhibitors in pediatric cancers: balancing irAEs and efficacy. Tumori. (2021) 107:276–81. doi: 10.1177/03008916211010214
202. Casey DL, Cheung NV. Immunotherapy of pediatric solid tumors: treatments at a crossroads, with an emphasis on antibodies. Cancer Immunol Res. (2020) 8:161–66. doi: 10.1158/2326-6066.CIR-19-0692
203. Aoki T, Hino M, Koh K, Kyushiki M, Kishimoto H, Arakawa Y, et al. Low frequency of programmed death ligand 1 expression in pediatric cancers. Pediatr Blood Cancer. (2016) 63:1461–64. doi: 10.1002/pbc.26018
204. Ceschi A, Noseda R, Palin K, Verhamme K. Immune checkpoint inhibitor-related cytokine release syndrome: analysis of WHO global pharmacovigilance database. Front Pharmacol. (2020) 11:557. doi: 10.3389/fphar.2020.00557
205. Les I, Martínez M, Pérez-Francisco I, Cabero M, Teijeira L, Arrazubi V, et al. Predictive biomarkers for checkpoint inhibitor immune-related adverse events. Cancers (Basel). (2023) 15:1629. doi: 10.3390/cancers15051629
206. Wang Y, Zou J, Li Y, Jiao X, Wang Y, Zhuo N, et al. Serological biomarkers predict immune-related adverse events and clinical benefit in patients with advanced gastrointestinal cancers. Front Immunol. (2022) 13:987568. doi: 10.3389/fimmu.2022.987568
207. Javaid A, Bennett C, Rao A, Spain L. Rare immune-related adverse events (irAEs): approach to diagnosis and management. Pharmaceut Med. (2024) 38:25–38. doi: 10.1007/s40290-023-00508-5
208. Jia XH, Geng LY, Jiang PP, Xu H, Nan KJ, Yao Y, et al. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J Exp Clin Cancer Res. (2020) 39:284. doi: 10.1186/s13046-020-01749-x
209. Trinh S, Le A, Gowani S, La-Beck NM. Management of immune-related adverse events associated with immune checkpoint inhibitor therapy: a minireview of current clinical guidelines. Asia Pac J Oncol Nurs. (2019) 6:154–60. doi: 10.4103/apjon.apjon_3_19
210. Schneider BJ, Naidoo J, Santomasso BD, Lacchetti C, Adkins S, Anadkat M, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J Clin Oncol. (2021) 39:4073–126. doi: 10.1200/JCO.21.01440
211. Yin Q, Wu L, Han L, Zheng X, Tong R, Li L, et al. Immune-related adverse events of immune checkpoint inhibitors: a review. Front Immunol. (2023) 14:1167975. doi: 10.3389/fimmu.2023.1167975
212. Verheijden RJ, van Eijs MJM, May AM, van Wijk F, Suijkerbuijk KPM. Immunosuppression for immune-related adverse events during checkpoint inhibition: an intricate balance. NPJ Precis Oncol. (2023) 7:41. doi: 10.1038/s41698-023-00380-1
213. Mosaferi T, Tsai K, Sovich S, Wilhalme H, Kathuria-Prakash N, Praw SS, et al. Optimal thyroid hormone replacement dose in immune checkpoint inhibitor-associated hypothyroidism is distinct from hashimoto’s thyroiditis. Thyroid. (2022) 32:496–504. doi: 10.1089/thy.2021.0685
214. Olsen TA, Zhuang TZ, Caulfield S, Martini DJ, Brown JT, Carthon BC, et al. Advances in knowledge and management of immune-related adverse events in cancer immunotherapy. Front Endocrinol (Lausanne). (2022), 13:779915. doi: 10.3389/fendo.2022.779915
215. Verma N, Jaffer M, Pina Y, Peguero E, Mokhtari S. Rituximab for immune checkpoint inhibitor myasthenia gravis. Cureus. (2021) 13:e16337. doi: 10.7759/cureus.16337
216. Lin JS, Wang DY, Mamlouk O, Glass WF, Abdelrahim M, Yee C, et al. Immune checkpoint inhibitor associated reactivation of primary membranous nephropathy responsive to rituximab. J Immunother Cancer. (2020) 8:e001287. doi: 10.1136/jitc-2020-001287
217. Iyer PC, Cabanillas ME, Waguespack SG, Hu MI, Thosani S, Lavis VR, et al. Immune-related thyroiditis with immune checkpoint inhibitors. Thyroid. (2018) 28:1243–51. doi: 10.1089/thy.2018.0116
218. Watson AS, Goutam S, Stukalin I, Ewanchuk BW, Sander M, Meyers DE, et al. Association of immune-related adverse events, hospitalization, and therapy resumption with survival among patients with metastatic melanoma receiving single-agent or combination immunotherapy. JAMA Netw Open. (2022) 5:e2245596. doi: 10.1001/jamanetworkopen.2022.45596
219. Romão R, Mendes AS, Ranchor R, Ramos MJ, Coelho J, Pichel RC, et al. Impact of immune-related adverse events on immune checkpoint inhibitors treated cancer patients’ Survival: single center experience and literature review. Cancers (Basel). (2023) 15:888. doi: 10.3390/cancers15030888
220. Patrinely JR, Johnson R, Lawless AR, Bhave P, Sawyers A, Dimitrova M, et al. Chronic immune-related adverse events following adjuvant anti-PD-1 therapy for high-risk resected melanoma. JAMA Oncol. (2021) 7:744–8. doi: 10.1001/jamaoncol.2021.0051
221. Goodman RS, Lawless A, Woodford R, Fa’ak F, Tipirneni A, Patrinely JR, et al. Extended follow-up of chronic immune-related adverse events following adjuvant anti-PD-1 therapy for high-risk resected melanoma. JAMA Netw Open. (2023) 6:e2327145. doi: 10.1001/jamanetworkopen.2023.27145
222. Wang JY, Wang L. CAR-T cell therapy: Where are we now, and where are we heading? Blood Sci. (2023) 5:237–48. doi: 10.1097/BS9.0000000000000173
223. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
224. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
225. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
226. Mailankody S, Matous JV, Chhabra S, Liedtke M, Sidana S, Oluwole OO, et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: phase 1 UNIVERSAL trial interim results. Nat Med. (2023) 29:422–29. doi: 10.1038/s41591-022-02182-7
227. Freyer CW, Porter DL. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic Malignancies. J Allergy Clin Immunol. (2020) 146:940–48. doi: 10.1016/j.jaci.2020.07.025
228. Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. (2020) 11:1973. doi: 10.3389/fimmu.2020.01973
229. Gu T, Hu K, Si X, Hu Y, Huang H. Mechanisms of immune effector cell-associated neurotoxicity syndrome after CAR-T treatment. WIREs Mech Dis. (2022) 14:e1576. doi: 10.1002/wsbm.1576
230. Chen X, Li P, Tian B, Kang X. Serious adverse events and coping strategies of CAR-T cells in the treatment of Malignant tumors. Front Immunol. (2022) 13:1079181. doi: 10.3389/fimmu.2022.1079181
231. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
232. Shalabi H, Nellan A, Shah NN, Gust J. Immunotherapy associated neurotoxicity in pediatric oncology. Front Oncol. (2022) 12:836452. doi: 10.3389/fonc.2022.836452
233. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
234. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
235. Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. (2021) 56:552–66. doi: 10.1038/s41409-020-01134-4
236. Messmer AS, Que YA, Schankin C, Banz Y, Bacher U, Novak U, et al. CAR T-cell therapy and critical care: A survival guide for medical emergency teams. Wien Klin Wochenschr. (2021) 133:1318–25. doi: 10.1007/s00508-021-01948-2
237. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
238. Yáñez L, Alarcón A, Sánchez-Escamilla M, Perales MA. How I treat adverse effects of CAR-T cell therapy. ESMO Open. (2020) 4:e000746. doi: 10.1136/esmoopen-2020-000746
239. Park JH, Nath K, Devlin SM, Sauter CS, Palomba ML, Shah G, et al. CD19 CAR T-cell therapy and prophylactic anakinra in relapsed or refractory lymphoma: phase 2 trial interim results. Nat Med. (2023) 29:1710–7. doi: 10.1038/s41591-023-02404-6
240. Barnes PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol. (2006) 148:245–54. doi: 10.1038/sj.bjp.0706736
241. Rice JB, White AG, Scarpati LM, Wan G, Nelson WW. Long-term systemic corticosteroid exposure: A systematic literature review. Clin Ther. (2017) 39:2216–29. doi: 10.1016/j.clinthera.2017.09.011
242. Langarizadeh MA, Ranjbar Tavakoli M, Abiri A, Ghasempour A, Rezaei M, Ameri A. A review on function and side effects of systemic corticosteroids used in high-grade COVID-19 to prevent cytokine storms. EXCLI J. (2021) 20:339–65. doi: 10.17179/excli2020-3196
243. Santomasso BD, Gust J, Perna F. How I treat unique and difficult-to-manage cases of CAR T-cell therapy-associated neurotoxicity. Blood. (2023) 141:2443–51. doi: 10.1182/blood.2022017604
Keywords: cytokine storm syndromes, cytokines, hyperinflammation, immune response, yin-yang
Citation: Armstrong A, Tang Y, Mukherjee N, Zhang N and Huang G (2024) Into the storm: the imbalance in the yin-yang immune response as the commonality of cytokine storm syndromes. Front. Immunol. 15:1448201. doi: 10.3389/fimmu.2024.1448201
Received: 12 June 2024; Accepted: 22 August 2024;
Published: 10 September 2024.
Edited by:
Sara Bobillo, Sant Joan de Déu Hospital, SpainReviewed by:
Carla Lima, Butantan Institute, BrazilManoussa Ethel Fanny, Xilio Therapeutics, Inc., United States
Copyright © 2024 Armstrong, Tang, Mukherjee, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Huang, aHVhbmdnMUB1dGhzY3NhLmVkdQ==