 Xuanyu Huang
Xuanyu Huang Ziwei Lan
Ziwei Lan Zhiping Hu
Zhiping Hu- Department of Neurology, The Second Xiangya Hospital of Central South University, Changsha, Hunan, China
Mast cells serve as crucial effector cells within the innate immune system and are predominantly localized in the skin, airways, gastrointestinal tract, urinary and reproductive tracts, as well as in the brain. Under physiological conditions, brain-resident mast cells secrete a diverse array of neuro-regulatory mediators to actively participate in neuroprotection. Meanwhile, as the primary source of molecules causing brain inflammation, mast cells also function as the “first responders” in brain injury. They interact with neuroglial cells and neurons to facilitate the release of numerous inflammatory mediators, proteases, and reactive oxygen species. This process initiates and amplifies immune-inflammatory responses in the brain, thereby contributing to the regulation of neuroinflammation and blood-brain barrier permeability. This article provides a comprehensive overview of the potential mechanisms through which mast cells in the brain may modulate neuroprotection and their pathological implications in various neurological disorders. It is our contention that the inhibition of mast cell activation in brain disorders could represent a novel avenue for therapeutic breakthroughs.
1 Introduction
Mast cells (MCs) are derived from hematopoietic progenitor cells and undergo maturation in vascular tissues, playing a role in both innate and adaptive immune responses (1). As sentinel cells of the immune system, MCs are predominantly distributed in anatomical regions that come into contact with the external environment, such as the skin, respiratory tract, gastrointestinal tract, and urinary tract (2). MCs possess the capability to degranulate and engage in cross-talk with diverse immune cells, thereby playing a pivotal role in safeguarding against pathogenic microorganisms and potential environmental threats (2, 3).
MCs are predominantly located within the vascular lumen of the brain membrane, entorhinal cortex, choroid plexus, olfactory bulb, midbrain, thalamus, and hypothalamus regions (4), where they engage in interactions with neurons, glial cells, and endothelial cells (5, 6). Furthermore, MCs are also found on the basal side of the blood-brain barrier (BBB) (6). MCs not only facilitate normal brain development and function, but also play a crucial role in the regulation of cognition and emotion (5, 7, 8). Additionally, MCs are considered the “first responders” of the brain, promptly detecting external stimuli and releasing inflammatory mediators and chemoattractants to recruit inflammatory cells in response to injury. They are pivotal in initiating, amplifying, and sustaining immune and neural responses (6). As one of the most prominent immune cell populations in the brain, MCs have emerged as a focal point of research in the context of brain disorders (9).
2 Mast cells and neuroinflammation
Neuroinflammation plays a crucial role in the pathogenesis and progression of various brain disorders, making it a prime target for therapeutic intervention (10, 11). Mounting evidence suggests that MCs, alongside traditional immune and inflammatory cells such as microglia and astrocytes, also contribute to the immunological and inflammatory processes within the brain (12, 13). MCs have the capacity to release cellular secretions containing immune and inflammatory mediators, such as histamine, β-tryptase, tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β), among others (14). It is noteworthy that MCs serve as a predominant source of histamine within the brain. Indeed, more than half of the total histamine content found in the central nervous system is attributed to the degranulation of MCs (12, 15). Additionally, MCs are the sole cells in the brain that preform and store TNF-α in advance (16). MCs also express receptors and ligands for various inflammation pathways, including the protease-activated receptor-2 (PAR2)-mitogen activated protein kinase (MAPK)-nuclear factor κb (NF-κB), histamine receptors 1 (H1R)/H4R-MAPK, and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)-NF-κB signaling cascades (17, 18).
In addition to their intrinsic functions, MCs have the potential for bidirectional signaling with microglia and astrocytes, leading to the induction and exacerbation of neuroinflammation (12) (Figure 1). Activated MCs can upregulate levels of cytokines and chemokines, thereby promoting the polarization of microglia towards pro-inflammatory phenotypes (19). Furthermore, MCs release proteolytic enzymes, which can activate PAR-2 receptors on microglia, triggering the release of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (17, 20, 21). Notably, MCs residing in the white matter have the capacity to instigate alterations in both the morphology and function of astrocytes. This is achieved through the action of multiple proteases that originate from mast cells. These proteases can trigger the release of IL-33 from astrocytes by engaging and activating specific intracellular signaling cascades. They activate the p38, ERK1/2 MAPKs, and NF-κB signaling pathways, thereby facilitating the astrocytic response to inflammation (22). Moreover, MC-derived histamine can modulate the activity of microglia and astrocytes by binding to multiple histamine receptors on these cells (5, 17, 23, 24). In turn, activated microglia and astrocytes can reciprocally influence MCs, leading to upregulation of PAR-2 and Toll-like receptors 2 (TLR2)/TLR4 expression in MCs and triggering the release of histamine, IL-6, and TNF-α (20, 25). IL-33 generated and secreted by activated astrocytes can also activate the suppression of tumorigenicity-2 (ST-2) receptor to stimulate microglia and MCs, thereby promoting the proliferation of the former and inducing the latter to produce IL-6, IL-8, and IL-13 (26–28). In conclusion, reciprocal activation of MCs, microglia, and astrocytes through diverse signaling pathways potentiates the inflammatory response, thereby exacerbating disease prognosis.
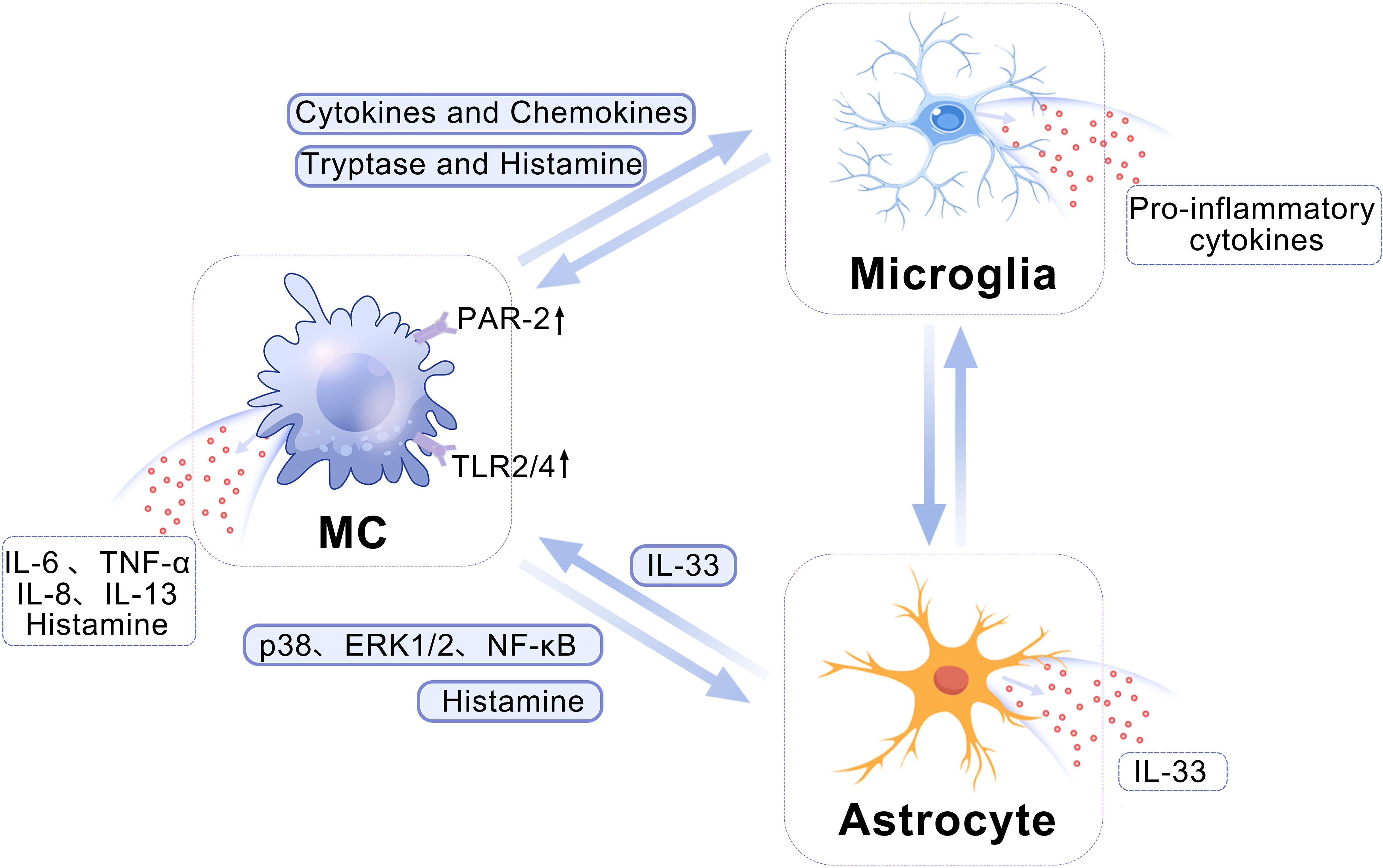
Figure 1. A schematic depicting the crosstalk among MCs, microglia, and astrocytes. MCs can modulate the pro-inflammatory phenotype of microglia by enhancing the expression of cytokines and chemokines. Additionally, MC-released tryptase and histamine can activate PAR-2 receptors on microglia, ultimately leading to the induction of a robust release of pro-inflammatory factors by microglia. The histamine released by MCs can stimulate the histamine receptors on astrocytes, thereby modulating their activation. Concurrently, MCs secrete diverse proteases that facilitate the release of IL-33 through activation of the p38, ERK1/2 MAPKs, and NF-κB signaling pathways in astrocytes. ERK, Extracellular-signal-regulated kinase; IL, interleukin; MAPK, mitogen-activated protein kinase; MC, mast cell; NF-κB, nuclear factor κB; PAR-2, protease-activated receptor 2.
3 Mast cells and neurons
MCs can engage with neurons via cell adhesion molecule-1 (CADM1), N-cadherin, and transgranulation (Figure 2). CADM1 plays a pivotal role in mediating the adhesion between MCs and neurons, facilitating information exchange between them, and is closely associated with neural immunity (29). The interaction between MCs and neurons is facilitated through synaptic-like structures, wherein N-cadherin assumes a crucial function (30). Research has demonstrated that N-cadherin participates in regulating pre- and post-synaptic structural modifications, promoting the establishment and maintenance of synaptic connections, as well as governing synaptic adhesion (30). Additionally, transgranulation enables MCs to release heparin into neurons, thereby disrupting calcium homeostasis and inhibiting neuronal responses (31). In turn, the activation of MCs can be induced by various neuropeptides such as substance P (SP), calcitonin gene-related peptide (CGRP), neurotensin (NT), and nerve growth factor (NGF) released by neurons, leading to the release of cytokines and chemokines like monocytic chemotactic protein 1 (MCP-1), IL-8, and C-C chemokine ligand 5 (CCL5) (32, 33).
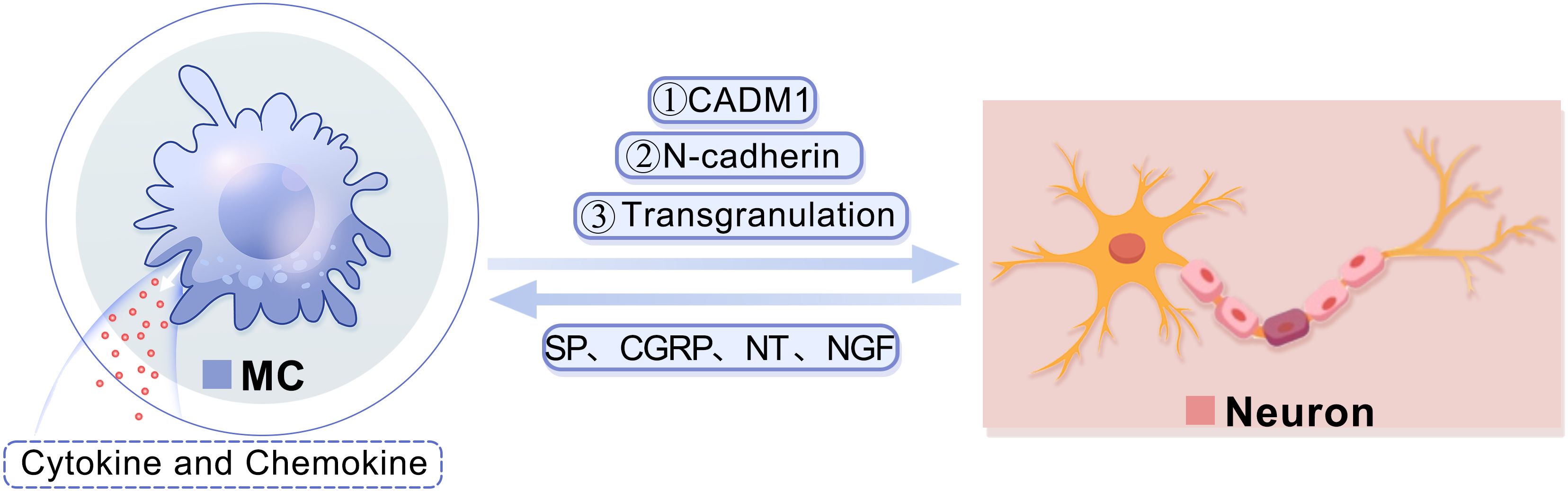
Figure 2. A diagram showing the interaction between MCs and neurons.MCs are capable of engaging in communication with neurons via CADM1. Furthermore, MCs have the ability to modulate the structural alterations of neuronal dendrites and synapses, facilitate the establishment and preservation of synaptic connections, and regulate synaptic adhesion by controlling N-cadherin. Additionally, MCs can release heparin into neurons through transgranulation to disrupt calcium homeostasis. Consequently, the release of various neuropeptides by neurons, such as SP, CGRP, NT, and NGF, can stimulate mast cells and prompt them to secrete cytokines and chemokines. CADM1, cell adhesion molecule-1; CGRP, calcitonin gene-related peptide; MC, mast cell; NGF, nerve growth factor; NT, neurotensin; SP, substance P.
Under normal physiological conditions, MCs contribute to neural protection and repair through the secretion of neuro-regulatory mediators (Table 1). Research has demonstrated that MCs secrete histamine, which can mitigate neuronal excitotoxicity induced by N-methyl-D-aspartatic acid (NMDA) and glutamate through the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway or by upregulating glutamate transporter currents and glutamine synthetase expression (34). Additionally, MCs are capable of promoting neuron growth by secreting NGF and inducing neuropeptide expression, lowering the firing threshold, and enhancing central nervous system transmission (35). Moreover, NGF binds to a specific receptor on the surface of microglia, enhancing membrane dynamics and endocytosis, while mediating the tyrosine kinase A pathway. This leads to an augmented phagocytic capacity for substances such as β-amyloid and others, thereby exerting neuroprotective effects (36). Under the stimulation of FcϵRI-mediated signaling, TLR ligands, and NGF, MCs synthesize and release angiogenin (37), which supports motor neuron survival and neurogenesis (38). Additionally, MCs in the hippocampus secrete serotonin, which plays a crucial role in regulating hippocampal-dependent behavior and enhancing neurogenesis (39).

Table 1. The principal neuroprotective mediators released by mast cells.
4 Mast cells and blood-brain barrier
The BBB serves as a highly selective and semipermeable interface between the brain parenchyma and the circulatory system, playing a pivotal role in upholding the normal function of the brain and the homeostasis of the internal environment (40). Research has revealed that MCs are frequently situated in close proximity to the active sites of matrix metalloproteinases (MMPs) (41). They influence the permeability of the BBB by relaeasing MMP. Initially, activated MMPs degrade a majority of the protein constituents within the extracellular matrix (ECM), such as collagen, elastin, fibronectin, and vitronectin (42, 43); subsequently, MMPs target cleavage sites of tight junctions (TJs) proteins, enabling brain microvascular endothelial cells (BMECs) to detach from the ECM (44); ultimately, key components of MMPs, namely MMP-2 and MMP-9, can directly break down microvascular basement membrane components, particularly type IV collagen (45, 46). Moreover, MCs secrete tryptase that are capable of activating PAR-2 receptors to enhance the expression of vascular cell adhesion molecule-1 (VCAM-1), TLR4, and TNF-α, while simultaneously reducing the expression of Occludin and Claudin-5, thereby inducing an increase in BBB permeability (47). Additionally, tryptase can directly compromise the integrity of the BBB by breaking down zonula occludens-1 (ZO-1), ZO-2, Occludin, and Claudin-5 proteins associated with TJs (48). Furthermore, MCs release substantial quantities of histamine which can bind to H1 and H2 receptors on endothelial cells leading to an upregulation in P-selectin expression and consequently elevating BBB permeability (49, 50).
The excessive increase in inflammatory factors following brain injury or disorder is the primary cause of BBB disruption (40). MCs represent a major source of these inflammatory factors, contributing to the disruption of the BBB in pathological conditions. For instance, the secretion of TNF-α by MCs can interact with the TNF-1 receptor or activate the NF-κB signaling pathway, thereby reducing the expression level of Claudin 5, leading to TJ disruption and subsequent BMEC death (51). Furthermore, IL-1β secreted by MCs disrupts the BBB through two mechanisms: firstly, it induces the expression of hypoxia-inducible factor-1 (HIF-1) and its gene target vascular endothelial growth factor-A (VEGF-A) in astrocytes, thus initiating BBB disruption (52). Secondly, IL-1β promotes the secretion of IL-6 and TNF-α, disrupting the paracellular pathway of BBB cells and increasing cell paracellular permeability (53).
5 Mast cells and brain disorders
5.1 Ischemic stroke
Ischemic stroke (IS) is the predominant form of stroke, accounting for approximately 87% of all stroke cases (54). The injuries associated with IS primarily encompass BBB disruption (55), oxidative stress (56), excitotoxicity (57), microvascular impairment (58), and neuroinflammation (59–62), ultimately leading to neuronal apoptosis (63). MCs play a crucial role in the pathogenic mechanisms of IS (Figure 3). Following IS, MCs perceive danger signals from the ischemic brain as early as 2-4 hours before microglia and astrocytes are activated (12, 64). They are among the first responders to be activated, contributing to disease progression and exacerbating neural damage by facilitating blood-brain barrier disruption and inflammatory infiltration (65). Relevant studies have demonstrated that rats with IS treated with MC activators exhibited a significant increase in brain edema, whereas those treated with MC stabilizers or MC-deficient rats showed reduced brain edema (65). Mast cells secrete TNF-α and tissue-type plasminogen activator, which can respectively augment the secretion of gelatinase by neighboring cells (66, 67) and activate MMP-9, thereby facilitating the disruption of the BBB (66, 68), hastening the progression of cerebral edema and increasing the risk of IS rat mortality (16). Furthermore, neutrophil infiltration commences several hours post Ischemia/Reperfusion (I/R) (69, 70), further exacerbating the accumulation of MMP-9 in microvessels (71), a process intricately linked to MCs (65). There are also reports in the literature indicating that following IS, meningeal MCs secrete IL-6, which can significantly exacerbate cerebral edema, enlarge the infarction area, and increase the number of granulocytes and activated macrophages in the brain (72). This finding is consistent with early clinical studies, where an elevated level of IL-6 in the cerebrospinal fluid of stroke patients was positively correlated with the size of the infarction area (73). Moreover, considering MCs’ strategic positioning near important blood vessels around the meninges and their interaction with cerebral circulation, they may function as potential “gatekeepers” to regulate immune cell infiltration into the brain during a stroke event (72).
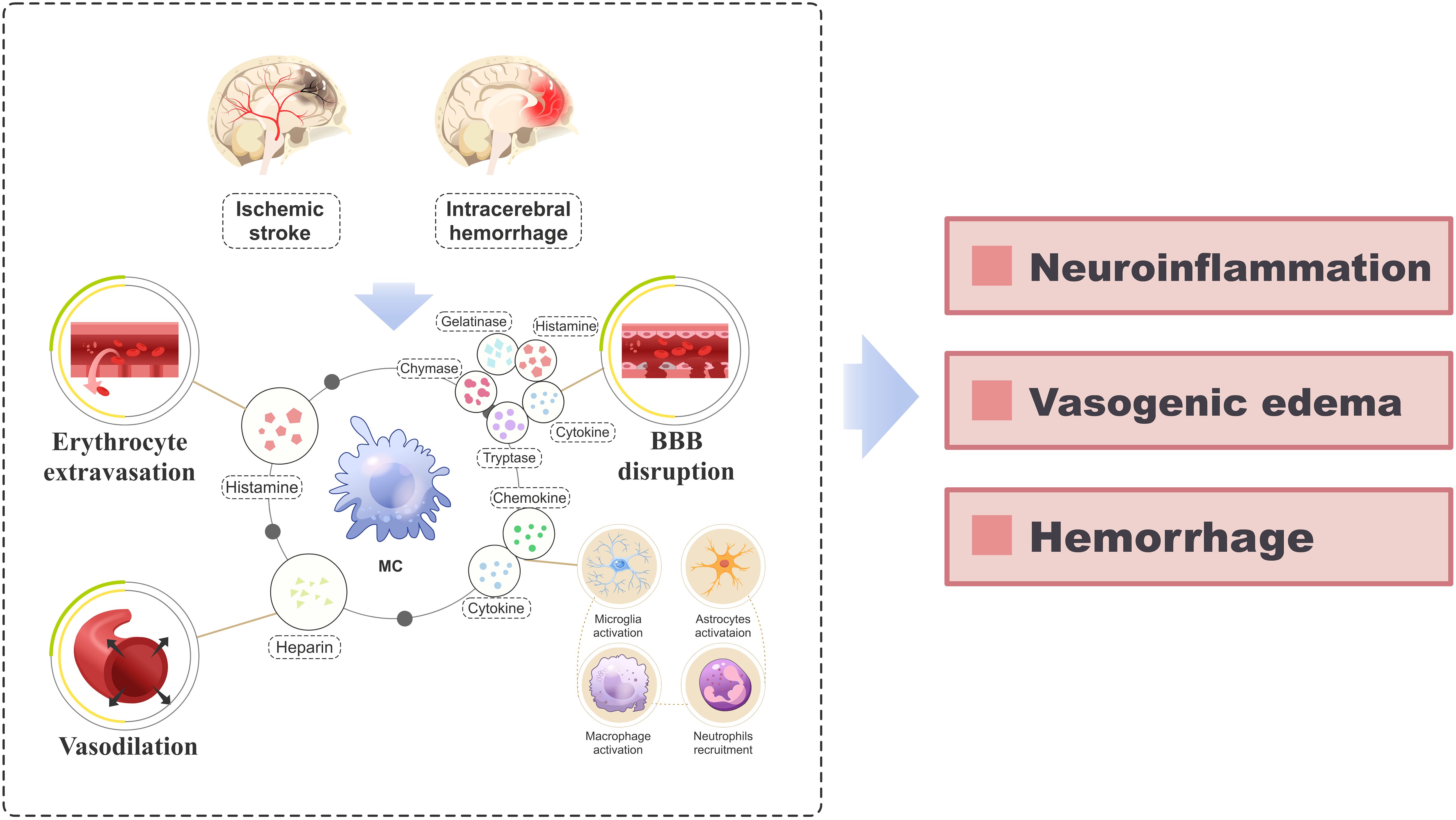
Figure 3. A schematic diagram elucidating the role of MCs in the pathogenesis of IS and ICH. After the occurrence of IS and ICH, activated MCs release a diverse array of vasoactive substances, cytokines, and proteolytic enzymes (including histamine, heparin, IL-6, TNF-α, IL-1β, tryptase, chymase, MMPs), which orchestrate erythrocyte extravasation, vasodilation and disruption of the BBB. Additionally, the release of various cytokines and chemokines from MCs triggers the recruitment and activation of neural immune cells (including microglia, astrocytes, macrophages, and neutrophils) from the periphery, perpetuating the inflammatory response. This pathological process ultimately culminates in the development of neuroinflammation, vasogenic edema, and hemorrhage. BBB, blood-brain barrier; IS, ischemic stroke; ICH, intracerebral hemorrhage; IL, interleukin; MC, mast cell; MMP, matrix metalloproteinases; TNF-α, tumor necrosis factor-alpha.
Recently conducted studies have provided novel insights into the targeted treatment of MCs in IS. Previous research has demonstrated the anti-inflammatory and neuroprotective properties of N-Palmitoylethanolamide-Oxazoline (PEA-OXA) (74, 75). Fusco et al. demonstrated that PEA-OXA significantly attenuates the activation and detachment of MCs, as well as the NF-κB pathway activation in IS rats, leading to a reduction in I/R-related lesion size, cytokine expression, and histological damage (76). In recent years, researchers have increasingly focused on the significant role of the brain-gut axis in stroke (77–79). Following IS, there is a rise in the number of MCs in the intestines of mice, accompanied by elevated levels of histamine and pro-inflammatory cytokines in the brain and circulating plasma (79), which are closely associated with neurological prognosis (80). Inhibiting MCs during IS can alleviate cerebral and intestinal MCs activation, neuroinflammation, peripheral inflammation, reduce neurological deficits, and improve prognosis (81). Furthermore, recent studies have identified high expression of the PTGS2 gene, linked to MCs activation specifically in IS. Inhibiting PTGS2 has been shown to decrease infarction volume and neurological deficits while improving neurological prognosis in IS rats. This indirectly confirms that MCs may play a pivotal role in IS pathogenesis, suggesting that targeting genes related to MCs activation could lead to new breakthroughs in IS treatment (82).
5.2 Intracerebral hemorrhage
Intracerebral hemorrhage (ICH) refers to non-traumatic bleeding occurring in the brain parenchyma (83). Brain injury following ICH encompasses primary injury caused by hematoma compression and stimulation, as well as secondary brain injury such as blood-brain barrier disruption, neuroinflammation, oxidative stress, cell autophagy and apoptosis (84–86), among which the severity of secondary brain injury is closely linked to prognosis (87). The role of MCs in the early stage of secondary brain injury after ICH and throughout the course of the disease has been demonstrated by numerous studies (88) (Figure 3). After ICH, the activation of MCs results in BBB damage, exacerbation of cerebral edema, worsening of neurological deficits, and increased mortality. However, inhibiting MC activation can alleviate these effects (89, 90). Furthermore, MCs release various immune and inflammatory mediators that directly contribute to neuroinflammation following ICH (88). It is worth noting that MCs also play a role in ICH rebleeding and hematoma expansion. Studies have demonstrated that MCs release endogenous anticoagulant heparin, which impairs coagulation function and leads to red cell extravasation, thereby contributing to hematoma expansion (91, 92). Stabilizing or inducing deficiency in MCs can reduce the size of hematomas in ICH rats (89).
Several studies have investigated the inhibition of MCs activation following ICH. Inhalation of hydrogen has been shown to exert neuroprotective effects in ICH by reducing Lyn kinase phosphorylation, thereby inhibiting MCs activation and degranulation (93). Intravenous immunoglobulin (IVIG) can inhibit MCs activation after ICH by activating the FcγRIIB/SHIP1/PIP3 pathway, thus reducing neuroinflammation and improving BBB permeability (94). Yang et al.’s study demonstrated that inhibition of the IRE1α/miR-125/Lyn signaling pathway can reduce MCs activation, degranulation, and neuroinflammation after ICH, leading to reduced cerebral edema, hematoma size, and improved neurological deficits (95). Recent studies have demonstrated that GW0742 can attenuate neuroinflammation induced by MCs in rats with GMH (Germinal matrix hemorrhage) through activation of the PPARβ/δ/CD300a/SHP1 pathway (96). In conclusion, MCs play a role in the complex pathological process of ICH, and targeting MCs may offer new prospects for ICH treatment. Nevertheless, the pathophysiological significance of MCs in ICH remains incompletely understood, necessitating further investigation.
5.3 Intracranial aneurysm and subarachnoid hemorrhage
Intracranial aneurysm (IA) is a complex condition characterized by pathological dilation of cerebral arteries (97, 98). Subarachnoid hemorrhage (SAH) represents a severe consequence of IA rupture, associated with high mortality and morbidity rates (99, 100). Ample evidence suggests that MCs play a significant role in the pathophysiology of both IA and SAH.
MCs contribute to the IA through diverse mechanisms (101, 102). The intact IA vessel wall harbors a substantial population of activated MCs, which orchestrate vascular inflammation by upregulating the expression of inflammation-related molecules (103–105). Additionally, MCs secrete tryptase and chymase, leading to the upregulation of MMP-2 and MMP-9 expression and activity, thereby contributing to the degeneration of the arterial wall extracellular matrix (106–108). This cascade further induces the expression of inducible nitric oxide synthase (iNOS), which in turn mediates smooth muscle cell apoptosis. Consequently, this process results in decreased arterial stiffness, medial thinning, and ultimately culminates in IA expansion (105). Moreover, MCs can induce the formation and remodeling of new blood vessels in IA by secreting a variety of angiogenic factors (109, 110). Additionally, the histamine and proteases released by MCs can respectively facilitate leakage (111) and rupture of newly formed blood vessels (109, 110, 112), ultimately resulting in microbleeding.
Analysis of human IAs revealed a significantly higher degree of MC infiltration in ruptured IAs compared to unruptured IAs (103). Activation of MCs markedly increased the rate of IA rupture, while stabilizing MCs or correcting MC defects significantly reduced the incidence of IA rupture in mice (113). Stromal Cell-Derived Factor-1 (SDF-1), a well-known chemokine for MCs, can induce pathological remodeling of the IA wall (114). Meanwhile, the MCs in the IA wall secrete tryptase, which can convert angiotensin I into angiotensin II, thus activating the renin-angiotensin system and promoting the rupture of the IA (115, 116). In addition, the TNF-α and Hepatocyte Growth Factor (HGF) released by MCs also play a crucial role in the rupture of the IA (117, 118).
IA rupture is highly prone to causing SAH. Inflammation plays a crucial role in the pathogenesis of SAH (119) and is also a pivotal determinant of prognosis (120). Following SAH, MCs not only directly contribute to inflammatory damage by releasing various inflammatory mediators but also activate microglia to exert a synergistic pro-inflammatory effect (47). Cerebral vasospasm is a common complication following SAH and significantly increases the risk of delayed ischemia, leading to poor prognosis (121). The adenosine A3 receptor (A3R), abundantly expressed on the membranes of MCs, is responsible for vascular constriction (122). Meanwhile, the activation of A3R can also exacerbate brain injury by inducing the release of tryptase, chymase, pro-inflammatory cytokines, and chemokines from MCs, resulting in the infiltration of inflammatory cells into the injured hemisphere (123).
In recent years, numerous scholars have conducted research on MC as a therapeutic target. Studies have revealed that intravenous injection of MSCs can activate the cyclooxygenase-2 (COX-2)-dependent pathway, suppress MC activation, and effectively prevent IA rupture (124). Simultaneously, the microvesicles from intravenous injection of MSCs can also enhance the levels of prostaglandin E2 (PGE2) and E-prostanoid 4 (EP4) receptor expression associated with MCs in the IA, thereby safeguarding the vascular wall and preventing IA rupture (125). After SAH, the inhibition of MCs has been shown to significantly reduce cerebral edema, neuroinflammation, and neurological deficits (47). Moreover, LJ529 has demonstrated the ability to inhibit MC degranulation through the A3R-PKCϵ-ALDH2 pathway, thereby mitigating MC-related inflammation in SAH (126). In addition to these preclinical studies, it is well-established that various cytokines and chemokines are implicated in the pathogenic mechanism of MCs in IA and SAH; however, the specific mechanism remains incompletely elucidated. Therefore, further research on MCs is necessary to explore novel therapeutic approaches for IA and SAH treatment.
5.4 Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative condition characterized by the prominent pathological features of extracellular amyloid-beta (Aβ) protein deposition, hyperphosphorylation of Tau protein, and synaptic loss leading to the development of neurofibrillary tangles (NFTs) and eventual neuronal death (127, 128). Research has demonstrated extensive MCs infiltration in the brains of AD patients, particularly in regions where Aβ protein accumulates (9, 129). This phenomenon is attributed to neuroglia cells within Aβ deposition sites releasing substantial amounts of chemoattractants for MC infiltration such as serum amyloid A. Furthermore, MCs themselves serve as early detectors of Aβ accumulation; Harcha et al. observed a rapid increase in MCs presence in the hippocampus and cortex prior to noticeable Aβ deposition (130).
MCs have been demonstrated to contribute to the development of AD by promoting Aβ formation and neuroinflammation (131) (Figure 4). Activation of corticotropin-releasing hormone (CRH) receptors on MCs by CRH, through the hypothalamic-pituitary-adrenal axis, stimulates MC activation and disrupts the BBB, facilitating the entry of peripheral inflammatory factors and cells into the brain, thereby activating neuroglia cells (132). This process also leads to Aβ formation and subsequent tau protein aggregation, NFT formation, ultimately contributing to AD development (133). Additionally, deposited Aβ can induce degranulation by activating Pannexin1 hemichannel (Panx1 HC) on MCs and recruit other immune cells to release substantial amounts of inflammatory factors, thus participating in AD-related neuroinflammation (130). Recent studies have revealed that various forms of Aβ including Aβ1-42, Aβ1-40, and Aβ-35 can stimulate MCs to secrete significant levels of pro-inflammatory cytokines; among these forms, Aβ1-42 exhibits the most potent effect (134).
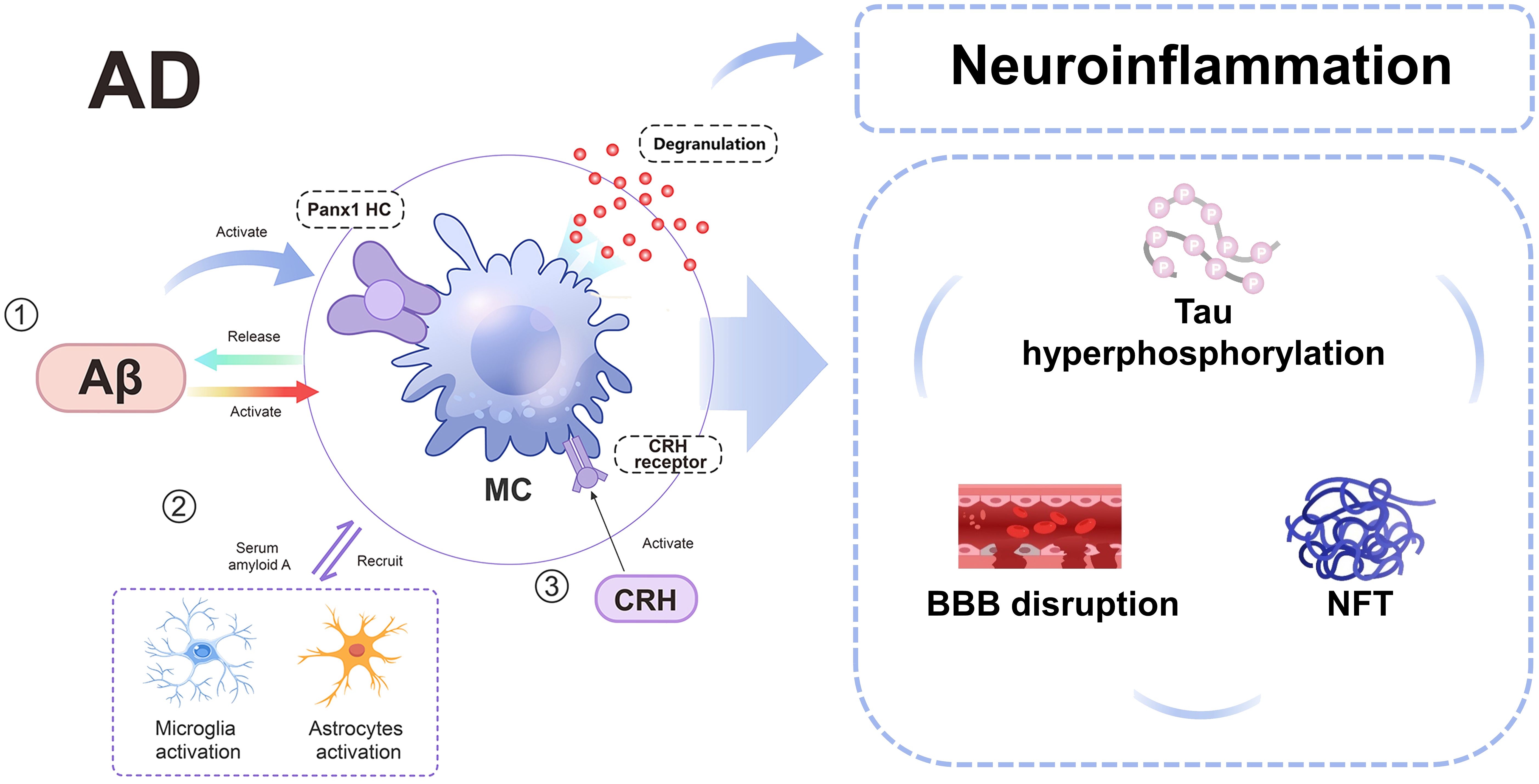
Figure 4. A schematic diagram illustrating the involvement of MCs in AD. In AD, MCs can be activated by Aβ, Panx1 HC, and CRH receptor, subsequently interacting with microglia and astrocytes. This interaction leads to disruption of the BBB, hyperphosphorylation of tau protein, and neurofibrillary tangles. AD, alzheimer’s disease; Aβ: amyloid-beta; BBB, blood-brain barrier; CRH, Corticotropin-releasing hormone; MC, mast cell; MMP, matrix metalloproteinases; NFT, neurofibrillary tangles; Panx1 HC, Pannexin1 hemichannel.
Recently, scholars have investigated the potential of MCs as a therapeutic target for AD. Lin et al. demonstrated that MC depletion in 5XFAD mice upregulated transcriptomic features of neuroprotective DAM, downregulated markers of reactive astrocytes, and improved hippocampal-dependent cognitive function (135). Furthermore, they discovered that dura mater MCs can survey cerebrospinal fluid to receive signals from the intracranial environment and respond by expressing immunomodulatory mediators that impact cognitive and neuroglial function (135). The tyrosine kinase inhibitor MC stabilizer masitinib has been demonstrated to exert neuroprotective effects by inhibiting the upregulation of BBB permeability by MCs and suppressing neuroinflammation at the onset of AD (136). These findings offer a preliminary foundation for exploring novel therapeutic targets for AD, but further research is necessary to elucidate and validate them.
5.5 Parkinson’s disease
Parkinson’s disease (PD) is an irreversible neurodegenerative condition characterized by the degeneration and loss of dopamine neurons in the substantia nigra pars compacta of the midbrain, along with the accumulation of misfolded α-synuclein (α-Syn), which forms eosinophilic inclusion bodies known as Lewy bodies (LB). These pathological changes are accompanied by neuroinflammation (137, 138).
MCs play a pivotal role in the pathogenesis of PD through diverse mechanisms (Figure 5). In the context of PD, activated MCs are capable of concomitantly releasing other pro-inflammatory mediators and substantial amounts of reactive oxygen species (ROS), thereby eliciting oxidative stress (6, 139, 140). This oxidative stress represents a fundamental contributor to neurodegeneration in PD (141). Histamine significantly contributes to PD pathogenesis. Elevated levels along with increased presence within substantia nigra have been observed both in PD patients as well as animal models (142–145). Local administration or systemic delivery has been shown to result in dopaminergic neuron death alongside worsening dyskinesia symptoms (21). The induction mechanism involves activation through H2R and H4R receptors leading to dopaminergic degeneration (146), while inhibiting H2R activation suppresses JNK/P38 phosphorylation thus reducing CASP3 activity which mitigates cellular apoptosis (146). Furthermore, blocking H4R hinders MCs activation within brain tissue resulting in reduced TNF-α release thereby ameliorating neurodegeneration including LB-like neuropathological changes (147, 148). Notably, MCs serve as a primary source accounting for over 50% total brain-histamine secretion (14), suggesting potential therapeutic promise lies within regulating their secretory function for future PD treatments.
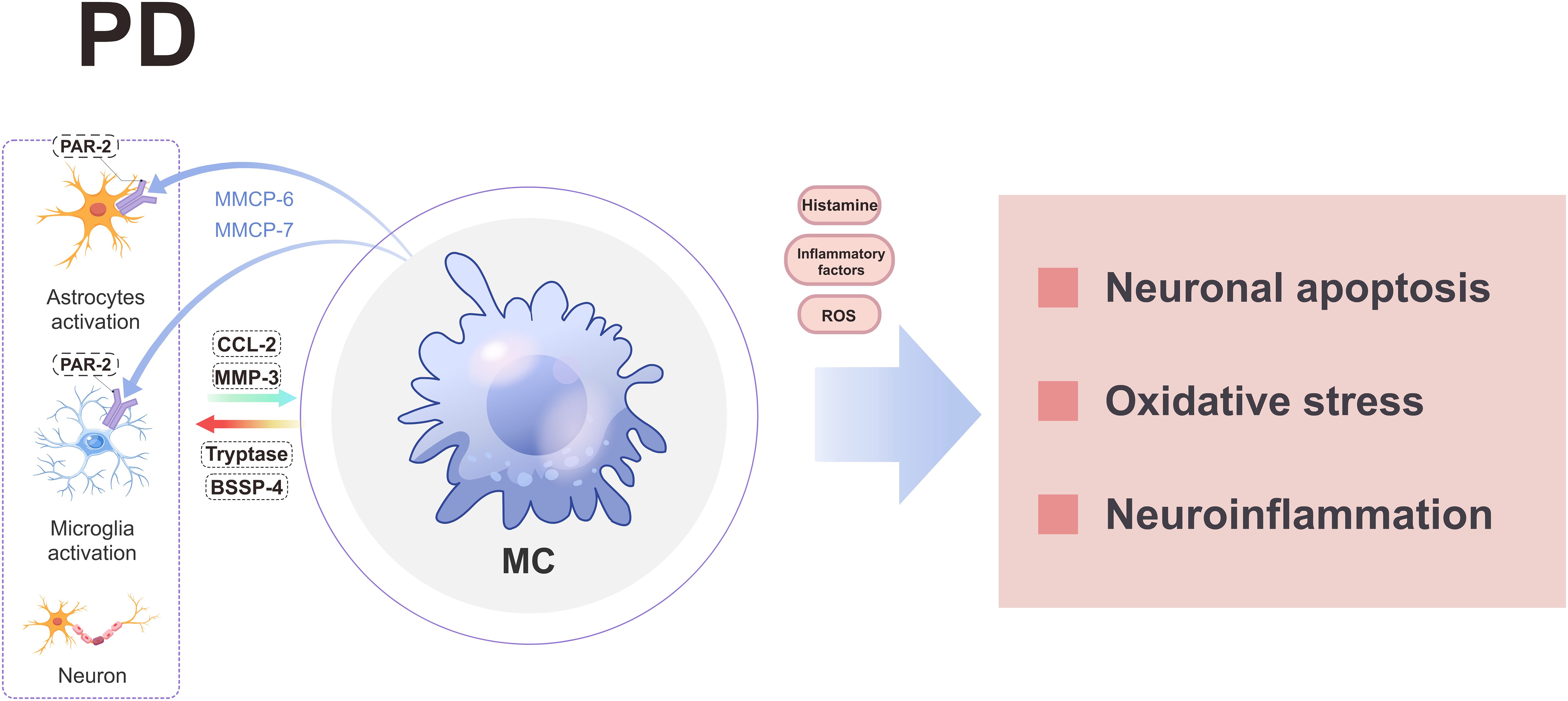
Figure 5. A schematic diagram illustrating the involvement of MCs in PD. MCs engage in interactions with neuroglial cells and neurons, leading to the release of various substances that ultimately induce neuroinflammation, oxidative stress, and neuronal apoptosis through the secretion of histamine, inflammatory factors, and ROS. BSSP-4, Brain-Specific Serine Protease-4; CCL-2, chemokine ligand 2; MC, mast cell; PD, parkinson’s disease; PAR-2, protease activated receptor 2; ROS, reactive oxygen species.
MCs may also contribute to the pathogenesis of PD by interacting with neuralglia cells and neurons. When MCs are co-cultured with neuralglia cells/neurons and exposed to glia activating factors, activated MCs can secrete tryptase and brain-specific serine protease-4 (BSSP-4) to stimulate the release of CC2L and MMP-3 by astrocytes and neurons, thereby contributing to the pathogenesis of PD (149). Meanwhile, MCs can enhance the expression of PAR-2 in neuroglia cells and neurons by releasing specific proteases (such as MMCP-6 and MMCP-7), leading to neuroinflammation and neuronal apoptosis (32). Conversely, neuroglia cells-secreted CCL2 can attract MCs to the substantia nigra region, triggering the release of diverse pro-inflammatory mediators and subsequent dopaminergic neuron death (150). Therefore, targeting MCs holds promise for novel therapeutic strategies in PD.
5.6 Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the degeneration of motor neurons and dysfunction of distal motor axons (151–153). Early studies have observed the activation of MCs in the degenerative regions of the central nervous system, such as the prefrontal cortex and pyramidal tract, as well as in the spinal cord of ALS patients (154, 155). Furthermore, Trials et al. discovered activated MCs in the skeletal muscles of ALS rats, which were found in the motor axons of the extensor digitorum longus muscle, sciatic nerve, and ventral root of the spinal cord (156, 157). These findings suggest that MCs play a role in ALS development by potentially inducing motor axon degeneration and accelerating neuromuscular junctions (NMJs) loss, thereby contributing to peripheral motor pathway degeneration in ALS patients (156, 157).
Studies indicate that MCs may also contribute to the vascular-related pathological mechanisms of ALS (158, 159). MCs play a crucial role in enhancing vascular permeability through the secretion of tryptase and chymase, which degrade adhesion protein complexes between endothelial cells (160), as well as connexin, procollagen, and type IV collagen in the extracellular matrix (161, 162). Furthermore, MCs release histamine and prostaglandin, further augmenting microvascular permeability (163), thereby facilitating the infiltration of peripheral inflammatory cells. These processes result in damage to microvascular endothelial cells and perivascular cells in the brains and spinal cords of ALS patients, leading to impairment of the BBB and blood-spinal cord barrier (164–166). Subsequently, MCs can breach the blood-spinal cord barrier (167, 168) and release neuropeptides, proteases, cytokines, and histamine, inducing local neuroinflammation and neuronal dysfunction (9).
MCs may also facilitate disease progression through interactions with other immune cells and neurons. They secrete chymase that promote neutrophil infiltration, contributing to the advancement of ALS paralysis (157). Furthermore, MCs can engage with microglia by secreting IL-6 and tryptase, establishing a positive feedback loop that continuously amplifies ALS neuroinflammation (9). Additionally, reactive astrocytes in ALS may express stem cell factor (SCF), which induces c-kit+ precursor MC differentiation and drives their migration from the periphery through the microvasculature to the spinal cord, where they localize and exacerbate the disorder (159). Under the influence of 75-kD neurotrophin receptors (p75NTR) abnormally expressed by damaged motor neurons, MCs can also induce apoptotic signaling (169). Therefore, these interactions between MCs and immune cells as well as neurons represent an important link in the pathogenesis of ALS.
Research has demonstrated that the MC inhibitor matsitini can decrease the population of MCs in the extensor digitorum longus muscle, thereby reducing the occurrence of NMJ denervation and motor deficits in ALS rats, as well as ameliorating ALS neurological symptoms (156, 157). This substantiates the specific involvement of MCs in ALS, and the positive outcomes from a phase III clinical trial of matsitini in ALS further support its therapeutic potential, indicating promising prospects for targeting MCs as a treatment strategy for ALS (170). Nevertheless, there is still a need for comprehensive research to delve into the specific mechanisms through which MCs are implicated in ALS.
6 Conclusion and future perspectives
In recent years, with the continuous deepening of research, we have gained a more profound comprehension of the neuroprotective function of MCs and its significance in various brain disorders. On one hand, MCs can induce neuroinflammation and oxidative stress, promote BBB damage, vascular edema, and hemorrhage formation, as well as recruit other immune cells to exacerbate inflammatory responses. On the other hand, the potentially beneficial effects of activated MCs on the brain cannot be disregarded. MCs can exert neuroprotective effects by secreting histamine, NGF, and angiogenic factors. Similar to microglia and macrophages, brain MCs may contribute to neuroprotection and repair in the advanced stages of brain disorders.
It is important to acknowledge that numerous obstacles must be overcome in order to propel research in this domain. First, the activation of MCs in the brain is a complex process that may give rise to both neuroprotective and pathogenic effects. Our current understanding of the transcriptional and epigenetic dynamics governing MC activation in the brain is still in its early stages, necessitating further exploration into the regulation of MC activation. Second, previous studies have predominantly focused on directly inhibiting MC activation in brain disorder models without delving into specific underlying mechanisms. Therefore, gaining a deeper comprehension of the cell-to-cell interactions between MCs and other immune cells in the brain is essential for effectively modulating the brain’s immune response network. Third, while targeted interventions for MCs have demonstrated promising therapeutic potential in most brain disorders during preclinical studies, there remains controversy surrounding the role, mechanism, and potential therapeutic value of MCs in certain brain disorders. This highlights an urgent need to investigate the precise functional roles of MCs within brain pathology. Finally, Intestinal MCs can engage with neurons and endocrine elements via the brain-gut axis, thereby modulating immune responses and neuroinflammation linked to neurodegenerative conditions in the brain. From this perspective, targeting MCs as a therapeutic approach holds significant promise; employing strategies to inhibit their activity may offer a novel perspective for treating brain disorders as disorder-modifying therapy.
In conclusion, MCs are progressively emerging as a novel target for the treatment of brain disorders. A comprehensive exploration of the underlying molecular mechanisms through mechanistic research and identification of potential therapeutic interventions is poised to catalyze breakthroughs in the management of brain disorders.
Author contributions
XH: Writing – original draft, Writing – review & editing. ZL: Conceptualization, Investigation, Writing – review & editing. ZH: Conceptualization, Investigation, Methodology, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant numbers 82271367).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Conti P, Ronconi G, Lauritano D, Mastrangelo F, Caraffa A, Gallenga CE, et al. Impact of TNF and IL-33 cytokines on mast cells in neuroinflammation. Int J Mol Sci. (2024) 25:3248. doi: 10.3390/ijms25063248
2. St John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol (Baltimore Md: 1950). (2013) 190:4458–63. doi: 10.4049/jimmunol.1203420
3. Traina G. Mast cells in the brain - Old cells, new target. J Integr Neurosci. (2017) 16:S69–s83. doi: 10.3233/JIN-170068
4. Kempuraj D, Ahmed ME, Selvakumar GP, Thangavel R, Raikwar SP, Zaheer SA, et al. Mast cell activation, neuroinflammation, and tight junction protein derangement in acute traumatic brain injury. Mediators inflammation. (2020) 2020:4243953. doi: 10.1155/2020/4243953
5. Silver R, Curley JP. Mast cells on the mind: new insights and opportunities. Trends neurosciences. (2013) 36:513–21. doi: 10.1016/j.tins.2013.06.001
6. Dong H, Zhang X, Qian Y. Mast cells and neuroinflammation. Med Sci monitor basic Res. (2014) 20:200–6. doi: 10.12659/MSMBR.893093
7. Fitzpatrick CJ, Morrow JD. Thalamic mast cell activity is associated with sign-tracking behavior in rats. Brain behavior immunity. (2017) 65:222–9. doi: 10.1016/j.bbi.2017.05.003
8. Lenz KM, Pickett LA, Wright CL, Davis KT, Joshi A, McCarthy MM. Mast cells in the developing brain determine adult sexual behavior. J neuroscience: Off J Soc Neurosci. (2018) 38:8044–59. doi: 10.1523/JNEUROSCI.1176-18.2018
9. Jones MK, Nair A, Gupta M. Mast cells in neurodegenerative disease. Front Cell Neurosci. (2019) 13:171. doi: 10.3389/fncel.2019.00171
10. Ransohoff RM, Schafer D, Vincent A, Blachère NE, Bar-Or A. Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics: J Am Soc Exp NeuroTherapeutics. (2015) 12:896–909. doi: 10.1007/s13311-015-0385-3
11. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. (2016) 173:692–702. doi: 10.1111/bph.13125
12. Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA. Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev. (2017) 79:119–33. doi: 10.1016/j.neubiorev.2017.05.001
13. Skaper SD, Facci L, Zusso M, Giusti P. Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist: Rev J bringing neurobiology Neurol Psychiatry. (2017) 23:478–98. doi: 10.1177/1073858416687249
14. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. (2014) 14:478–94. doi: 10.1038/nri3690
15. Skaper SD, Facci L, Kee WJ, Strijbos PJ. Potentiation by histamine of synaptically mediated excitotoxicity in cultured hippocampal neurones: a possible role for mast cells. J neurochemistry. (2001) 76:47–55. doi: 10.1046/j.1471-4159.2001.00008.x
16. McKittrick CM, Lawrence CE, Carswell HV. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2015) 35:638–47. doi: 10.1038/jcbfm.2014.239
17. Dong H, Zhang W, Zeng X, Hu G, Zhang H, He S, et al. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol neurobiology. (2014) 49:1487–500. doi: 10.1007/s12035-014-8697-6
18. Zhou Q, Wang YW, Ni PF, Chen YN, Dong HQ, Qian YN. Effect of tryptase on mouse brain microvascular endothelial cells via protease-activated receptor 2. J neuroinflammation. (2018) 15:248. doi: 10.1186/s12974-018-1287-1
19. Ocak U, Ocak PE, Wang A, Zhang JH, Boling W, Wu P, et al. Targeting mast cell as a neuroprotective strategy. Brain injury. (2019) 33:723–33. doi: 10.1080/02699052.2018.1556807
20. Zhang S, Zeng X, Yang H, Hu G, He S. Mast cell tryptase induces microglia activation via protease-activated receptor 2 signaling. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2012) 29:931–40. doi: 10.1159/000171029
21. Rocha SM, Saraiva T, Cristóvão AC, Ferreira R, Santos T, Esteves M, et al. Histamine induces microglia activation and dopaminergic neuronal toxicity via H1 receptor activation. J neuroinflammation. (2016) 13:137. doi: 10.1186/s12974-016-0600-0
22. Kempuraj D, Thangavel R, Selvakumar GP, Ahmed ME, Zaheer S, Raikwar SP, et al. Mast cell proteases activate astrocytes and glia-neurons and release interleukin-33 by activating p38 and ERK1/2 MAPKs and NF-κB. Mol neurobiology. (2019) 56:1681–93. doi: 10.1007/s12035-018-1177-7
23. Mele T, Jurič DM. Identification and pharmacological characterization of the histamine H3 receptor in cultured rat astrocytes. Eur J Pharmacol. (2013) 720:198–204. doi: 10.1016/j.ejphar.2013.10.028
24. Ferreira R, Santos T, Gonçalves J, Baltazar G, Ferreira L, Agasse F, et al. Histamine modulates microglia function. J neuroinflammation. (2012) 9:90. doi: 10.1186/1742-2094-9-90
25. Zhang H, Yang H, He S. TNF increases expression of IL-4 and PARs in mast cells. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2010) 26:327–36. doi: 10.1159/000320556
26. Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. (2015) 85:703–9. doi: 10.1016/j.neuron.2015.01.013
27. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab investigation; J Tech Methods pathology. (2007) 87:971–8. doi: 10.1038/labinvest.3700663
28. Moulin D, Donzé O, Talabot-Ayer D, Mézin F, Palmer G, Gabay C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine. (2007) 40:216–25. doi: 10.1016/j.cyto.2007.09.013
29. Hagiyama M, Furuno T, Hosokawa Y, Iino T, Ito T, Inoue T, et al. Enhanced nerve-mast cell interaction by a neuronal short isoform of cell adhesion molecule-1. J Immunol (Baltimore Md: 1950). (2011) 186:5983–92. doi: 10.4049/jimmunol.1002244
30. Goda Y. Cadherins communicate structural plasticity of presynaptic and postsynaptic terminals. Neuron. (2002) 35:1–3. doi: 10.1016/S0896-6273(02)00765-1
31. Wilhelm M, Silver R, Silverman AJ. Central nervous system neurons acquire mast cell products via transgranulation. Eur J Neurosci. (2005) 22:2238–48. doi: 10.1111/j.1460-9568.2005.04429.x
32. Kempuraj D, Selvakumar GP, Thangavel R, Ahmed ME, Zaheer S, Kumar KK, et al. Glia maturation factor and mast cell-dependent expression of inflammatory mediators and proteinase activated receptor-2 in neuroinflammation. J Alzheimer’s disease: JAD. (2018) 66:1117–29. doi: 10.3233/JAD-180786
33. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. (2008) 123:398–410. doi: 10.1111/j.1365-2567.2007.02705.x
34. Hu WW, Chen Z. Role of histamine and its receptors in cerebral ischemia. ACS Chem Neurosci. (2012) 3:238–47. doi: 10.1021/cn200126p
35. Forsythe P, Bienenstock J. The mast cell-nerve functional unit: a key component of physiologic and pathophysiologic responses. Chem Immunol Allergy. (2012) 98:196–221. doi: 10.1159/000336523
36. Rizzi C, Tiberi A, Giustizieri M, Marrone MC, Gobbo F, Carucci NM, et al. NGF steers microglia toward a neuroprotective phenotype. Glia. (2018) 66:1395–416. doi: 10.1002/glia.23312
37. Kulka M, Fukuishi N, Metcalfe DD. Human mast cells synthesize and release angiogenin, a member of the ribonuclease A (RNase A) superfamily. J leukocyte Biol. (2009) 86:1217–26. doi: 10.1189/jlb.0908517
38. Subramanian V, Crabtree B, Acharya KR. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum Mol Genet. (2008) 17:130–49. doi: 10.1093/hmg/ddm290
39. Nautiyal KM, Dailey CA, Jahn JL, Rodriquez E, Son NH, Sweedler JV, et al. Serotonin of mast cell origin contributes to hippocampal function. Eur J Neurosci. (2012) 36:2347–59. doi: 10.1111/j.1460-9568.2012.08138.x
40. Yang J, Ran M, Li H, Lin Y, Ma K, Yang Y, et al. New insight into neurological degeneration: Inflammatory cytokines and blood-brain barrier. Front Mol Neurosci. (2022) 15:1013933. doi: 10.3389/fnmol.2022.1013933
41. Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. (2005) 280:9291–6. doi: 10.1074/jbc.M410396200
42. Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J neuroscience: Off J Soc Neurosci. (2001) 21:7724–32. doi: 10.1523/JNEUROSCI.21-19-07724.2001
43. Strbian D, Kovanen PT, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. An emerging role of mast cells in cerebral ischemia and hemorrhage. Ann Med. (2009) 41:438–50. doi: 10.1080/07853890902887303
44. Liu X, Su P, Meng S, Aschner M, Cao Y, Luo W, et al. Role of matrix metalloproteinase-2/9 (MMP2/9) in lead-induced changes in an in vitro blood-brain barrier model. Int J Biol Sci. (2017) 13:1351–60. doi: 10.7150/ijbs.20670
45. Zhao BQ, Tejima E, Lo EH. Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke. (2007) 38:748–52. doi: 10.1161/01.STR.0000253500.32979.d1
46. del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. (2009) 158:972–82. doi: 10.1016/j.neuroscience.2008.08.028
47. Qin B, Peng Y, Zhong C, Cai Y, Zhou S, Chen H, et al. Mast cells mediate inflammatory injury and aggravate neurological impairment in experimental subarachnoid hemorrhage through microglial PAR-2 pathway. Front Cell Neurosci. (2021) 15:710481. doi: 10.3389/fncel.2021.710481
48. Hsieh JT, Rathore APS, Soundarajan G, St John AL. Japanese encephalitis virus neuropenetrance is driven by mast cell chymase. Nat Commun. (2019) 10:706. doi: 10.1038/s41467-019-08641-z
49. Deli MA, Dehouck MP, Cecchelli R, Abrahám CS, Joó F. Histamine induces a selective albumin permeation through the blood-brain barrier in vitro. Inflammation research: Off J Eur Histamine Res Soc. (1995) 44 Suppl 1:S56–7. doi: 10.1007/BF01674394
50. Bañuelos-Cabrera I, Valle-Dorado MG, Aldana BI, Orozco-Suárez SA, Rocha L. Role of histaminergic system in blood-brain barrier dysfunction associated with neurological disorders. Arch Med Res. (2014) 45:677–86. doi: 10.1016/j.arcmed.2014.11.010
51. Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L, et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death disease. (2019) 10:487. doi: 10.1038/s41419-019-1716-9
52. Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol (Baltimore Md: 1950). (2006) 177:5574–84. doi: 10.4049/jimmunol.177.8.5574
53. Labus J, Häckel S, Lucka L, Danker K. Interleukin-1β induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods. (2014) 228:35–45. doi: 10.1016/j.jneumeth.2014.03.002
54. Farina M, Vieira LE, Buttari B, Profumo E, Saso L. The nrf2 pathway in ischemic stroke: A review. Molecules (Basel Switzerland). (2021) 26:5001. doi: 10.3390/molecules26165001
55. Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. (2018) 315:C343–c56. doi: 10.1152/ajpcell.00095.2018
56. Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants Redox Signaling. (2011) 14:1505–17. doi: 10.1089/ars.2010.3576
57. Paschen W. Glutamate excitotoxicity in transient global cerebral ischemia. Acta neurobiologiae experimentalis. (1996) 56:313–22. doi: 10.55782/ane-1996-1136
58. Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol. (2015) 5:1147–60. doi: 10.1002/cphy.c140057
59. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J neuroimmunology. (2007) 184:53–68. doi: 10.1016/j.jneuroim.2006.11.014
60. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog neurobiology. (2014) 115:157–88. doi: 10.1016/j.pneurobio.2013.11.006
61. Trendelenburg G. Molecular regulation of cell fate in cerebral ischemia: role of the inflammasome and connected pathways. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2014) 34:1857–67. doi: 10.1038/jcbfm.2014.159
62. Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea JF, Damante G, et al. Blood-brain barrier dysfunction in disorders of the developing brain. Front Neurosci. (2015) 9:40. doi: 10.3389/fnins.2015.00040
63. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
64. Parrella E, Porrini V, Benarese M, Pizzi M. The role of mast cells in stroke. Cells. (2019) 8:437. doi: 10.3390/cells8050437
65. Strbian D, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2006) 26:605–12. doi: 10.1038/sj.jcbfm.9600228
66. Mattila OS, Strbian D, Saksi J, Pikkarainen TO, Rantanen V, Tatlisumak T, et al. Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke. (2011) 42:3600–5. doi: 10.1161/STROKEAHA.111.632224
67. Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. (1995) 703:151–5. doi: 10.1016/0006-8993(95)01089-0
68. Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. (2002) 39:279–91. doi: 10.1002/glia.10108
69. Barone FC, Hillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, et al. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem neuropathology. (1995) 24:13–30. doi: 10.1007/BF03160109
70. Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J neurological Sci. (1994) 125:3–10. doi: 10.1016/0022-510X(94)90234-8
71. Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. (2008) 39:1121–6. doi: 10.1161/STROKEAHA.107.500868
72. Arac A, Grimbaldeston MA, Nepomuceno AR, Olayiwola O, Pereira MP, Nishiyama Y, et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am J pathology. (2014) 184:2493–504. doi: 10.1016/j.ajpath.2014.06.003
73. Beridze M, Sanikidze T, Shakarishvili R, Intskirveli N, Bornstein NM. Selected acute phase CSF factors in ischemic stroke: findings and prognostic value. BMC neurology. (2011) 11:41. doi: 10.1186/1471-2377-11-41
74. Impellizzeri D, Cordaro M, Bruschetta G, Siracusa R, Crupi R, Esposito E, et al. N-palmitoylethanolamine-oxazoline as a new therapeutic strategy to control neuroinflammation: neuroprotective effects in experimental models of spinal cord and brain injury. J neurotrauma. (2017) 34:2609–23. doi: 10.1089/neu.2016.4808
75. Cordaro M, Siracusa R, Crupi R, Impellizzeri D, Peritore AF, D’Amico R, et al. 2-pentadecyl-2-oxazoline reduces neuroinflammatory environment in the MPTP model of parkinson disease. Mol neurobiology. (2018) 55:9251–66. doi: 10.1007/s12035-018-1064-2
76. Fusco R, Scuto M, Cordaro M, D’Amico R, Gugliandolo E, Siracusa R, et al. N-palmitoylethanolamide-oxazoline protects against middle cerebral artery occlusion injury in diabetic rats by regulating the SIRT1 pathway. Int J Mol Sci. (2019) 20:4845. doi: 10.3390/ijms20194845
77. Ganesh BP, Nelson JW, Eskew JR, Ganesan A, Ajami NJ, Petrosino JF, et al. Prebiotics, probiotics, and acetate supplementation prevent hypertension in a model of obstructive sleep apnea. Hypertension (Dallas Tex: 1979). (2018) 72:1141–50. doi: 10.1161/HYPERTENSIONAHA.118.11695
78. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann gastroenterology. (2015) 28:203–9.
79. Blasco MP, Chauhan A, Honarpisheh P, Ahnstedt H, d’Aigle J, Ganesan A, et al. Age-dependent involvement of gut mast cells and histamine in post-stroke inflammation. J neuroinflammation. (2020) 17:160. doi: 10.1186/s12974-020-01833-1
80. Chapman KZ, Dale VQ, Dénes A, Bennett G, Rothwell NJ, Allan SM, et al. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2009) 29:1764–8. doi: 10.1038/jcbfm.2009.113
81. Conesa MPB, Blixt FW, Peesh P, Khan R, Korf J, Lee J, et al. Stabilizing histamine release in gut mast cells mitigates peripheral and central inflammation after stroke. J neuroinflammation. (2023) 20:230. doi: 10.1186/s12974-023-02887-7
82. Yuan K, Jin X, Mo X, Zeng R, Zhang X, Chen Q, et al. Novel diagnostic biomarkers of oxidative stress, ferroptosis, immune infiltration characteristics and experimental validation in ischemic stroke. Aging. (2024) 16:746–61. doi: 10.18632/aging.205415
83. Sembolini A, Romoli M, Pannacci U, Gambaracci G, Floridi P, Acciarresi M, et al. Acute hematoma expansion after spontaneous intracerebral hemorrhage: risk factors and impact on long-term prognosis. Neurological sciences: Off J Ital Neurological Soc Ital Soc Clin Neurophysiology. (2020) 41:2503–9. doi: 10.1007/s10072-020-04356-y
84. Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global brain inflammation in stroke. Lancet Neurology. (2019) 18:1058–66. doi: 10.1016/S1474-4422(19)30078-X
85. Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog neurobiology. (2019) 178:101610. doi: 10.1016/j.pneurobio.2019.03.003
86. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. (2011) 42:1781–6. doi: 10.1161/STROKEAHA.110.596718
87. Haupenthal D, Kuramatsu JB, Volbers B, Sembill JA, Mrochen A, Balk S, et al. Disability-adjusted life-years associated with intracerebral hemorrhage and secondary injury. JAMA network Open. (2021) 4:e2115859. doi: 10.1001/jamanetworkopen.2021.15859
88. Yehya M, Torbey MT. The role of mast cells in intracerebral hemorrhage. Neurocritical Care. (2018) 28:288–95. doi: 10.1007/s12028-017-0416-5
89. Strbian D, Tatlisumak T, Ramadan UA, Lindsberg PJ. Mast cell blocking reduces brain edema and hematoma volume and improves outcome after experimental intracerebral hemorrhage. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2007) 27:795–802. doi: 10.1038/sj.jcbfm.9600387
90. Theoharides TC, Donelan J, Kandere-Grzybowska K, Konstantinidou A. The role of mast cells in migraine pathophysiology. Brain Res Brain Res Rev. (2005) 49:65–76. doi: 10.1016/j.brainresrev.2004.11.006
91. Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. New Engl J Med. (2001) 344:1450–60. doi: 10.1056/NEJM200105103441907
92. Dirnagl U, Lauritzen M. A new home for the Journal of Cerebral Blood Flow and Metabolism. J Cereb Blood Flow metabolism: Off J Int Soc Cereb Blood Flow Metab. (2016) 36:277. doi: 10.1177/0271678X15621576
93. Manaenko A, Lekic T, Ma Q, Zhang JH, Tang J. Hydrogen inhalation ameliorated mast cell-mediated brain injury after intracerebral hemorrhage in mice. Crit Care Med. (2013) 41:1266–75. doi: 10.1097/CCM.0b013e31827711c9
94. Akyol GY, Manaenko A, Akyol O, Solaroglu I, Ho WM, Ding Y, et al. IVIG activates FcγRIIB-SHIP1-PIP3 Pathway to stabilize mast cells and suppress inflammation after ICH in mice. Sci Rep. (2017) 7:15583. doi: 10.1038/s41598-017-15455-w
95. Yang Z, Huang J, Liao Y, Gan S, Zhu S, Xu S, et al. ER stress is involved in mast cells degranulation via IRE1α/miR-125/lyn pathway in an experimental intracerebral hemorrhage mouse model. Neurochemical Res. (2022) 47:1598–609. doi: 10.1007/s11064-022-03555-7
96. Lu W, Huang J, Flores J, Li P, Wang W, Liu S, et al. GW0742 reduces mast cells degranulation and attenuates neurological impairments via PPAR(β/δ)/CD300a/SHP1 pathway after GMH in neonatal rats. Exp neurology. (2024) 372:114615. doi: 10.1016/j.expneurol.2023.114615
97. Huang C, Hu D, Li K. Identification of biomarkers in intracranial aneurysm and their immune infiltration characteristics. World neurosurgery. (2022) 166:e199–214. doi: 10.1016/j.wneu.2022.06.138
98. Laurent D, Small C, Lucke-Wold B, Dodd WS, Chalouhi N, Hu YC, et al. Understanding the genetics of intracranial aneurysms: A primer. Clin Neurol neurosurgery. (2022) 212:107060. doi: 10.1016/j.clineuro.2021.107060
99. Ferreira M, Nahed BV, Babu MA, Walcott BP, Ellenbogen RG, Sekhar LN. Trapped fourth ventricle phenomenon following aneurysm rupture of the posterior circulation: case reports. Neurosurgery. (2012) 70:E253–8; discussion E8. doi: 10.1227/NEU.0b013e31822abf95
100. van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet (London England). (2007) 369:306–18. doi: 10.1016/S0140-6736(07)60153-6
101. Tsuruda T, Kato J, Hatakeyama K, Kojima K, Yano M, Yano Y, et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res. (2008) 102:1368–77. doi: 10.1161/CIRCRESAHA.108.173682
102. Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. (2007) 115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472
103. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J neuroinflammation. (2012) 9:222. doi: 10.1186/1742-2094-9-222
104. Ollikainen E, Tulamo R, Frösen J, Lehti S, Honkanen P, Hernesniemi J, et al. Mast cells, neovascularization, and microhemorrhages are associated with saccular intracranial artery aneurysm wall remodeling. J neuropathology Exp neurology. (2014) 73:855–64. doi: 10.1097/NEN.0000000000000105
105. Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr neurovascular Res. (2010) 7:113–24. doi: 10.2174/156720210791184916
106. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke. (2007) 38:162–9. doi: 10.1161/01.STR.0000252129.18605.c8
107. Fukuda S, Hashimoto N, Naritomi H, Nagata I, Nozaki K, Kondo S, et al. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation. (2000) 101:2532–8. doi: 10.1161/01.CIR.101.21.2532
108. Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke. (2003) 34:2980–4. doi: 10.1161/01.STR.0000102556.55600.3B
109. Ribatti D, Levi-Schaffer F, Kovanen PT. Inflammatory angiogenesis in atherogenesis–a double-edged sword. Ann Med. (2008) 40:606–21. doi: 10.1080/07853890802186913
110. Syväranta S, Helske S, Laine M, Lappalainen J, Kupari M, Mäyränpää MI, et al. Vascular endothelial growth factor-secreting mast cells and myofibroblasts: a novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arteriosclerosis thrombosis Vasc Biol. (2010) 30:1220–7. doi: 10.1161/ATVBAHA.109.198267
111. Bot I, van Berkel TJ, Biessen EA. Mast cells: pivotal players in cardiovascular diseases. Curr Cardiol Rev. (2008) 4:170–8. doi: 10.2174/157340308785160624
112. Kaartinen M, Penttilä A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis. (1996) 123:123–31. doi: 10.1016/0021-9150(95)05794-3
113. Furukawa H, Wada K, Tada Y, Kuwabara A, Sato H, Ai J, et al. Mast cell promotes the development of intracranial aneurysm rupture. Stroke. (2020) 51:3332–9. doi: 10.1161/STROKEAHA.120.030834
114. Hoh BL, Hosaka K, Downes DP, Nowicki KW, Wilmer EN, Velat GJ, et al. Stromal cell-derived factor-1 promoted angiogenesis and inflammatory cell infiltration in aneurysm walls. J neurosurgery. (2014) 120:73–86. doi: 10.3171/2013.9.JNS122074
115. Tada Y, Wada K, Shimada K, Makino H, Liang EI, Murakami S, et al. Roles of hypertension in the rupture of intracranial aneurysms. Stroke. (2014) 45:579–86. doi: 10.1161/STROKEAHA.113.003072
116. Shimada K, Furukawa H, Wada K, Korai M, Wei Y, Tada Y, et al. Protective role of peroxisome proliferator-activated receptor-γ in the development of intracranial aneurysm rupture. Stroke. (2015) 46:1664–72. doi: 10.1161/STROKEAHA.114.007722
117. Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J neuroinflammation. (2014) 11:77. doi: 10.1186/1742-2094-11-77
118. Peña-Silva RA, Chalouhi N, Wegman-Points L, Ali M, Mitchell I, Pierce GL, et al. Novel role for endogenous hepatocyte growth factor in the pathogenesis of intracranial aneurysms. Hypertension (Dallas Tex: 1979). (2015) 65:587–93. doi: 10.1161/HYPERTENSIONAHA.114.04681
119. Xie Z, Huang L, Enkhjargal B, Reis C, Wan W, Tang J, et al. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARγ/NFκB signaling pathway after subarachnoid hemorrhage in rats. Brain behavior immunity. (2018) 69:190–202. doi: 10.1016/j.bbi.2017.11.012
120. Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, et al. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int J Mol Sci. (2016) 17:497. doi: 10.3390/ijms17040497
121. Romenskaya T, Longhitano Y, Piccolella F, Berger JM, Artico M, Taurone S, et al. Cerebral vasospasm: practical review of diagnosis and management. Rev Recent Clin trials. (2023) 18:12–8. doi: 10.2174/1574887117666220810121048
122. Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, et al. The A3 adenosine receptor: history and perspectives. Pharmacol Rev. (2015) 67:74–102. doi: 10.1124/pr.113.008540
123. Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. (2018) 282:121–50. doi: 10.1111/imr.12634
124. Kuwabara A, Liu J, Kamio Y, Liu A, Lawton MT, Lee JW, et al. Protective effect of mesenchymal stem cells against the development of intracranial aneurysm rupture in mice. Neurosurgery. (2017) 81:1021–8. doi: 10.1093/neuros/nyx172
125. Liu J, Kuwabara A, Kamio Y, Hu S, Park J, Hashimoto T, et al. Human mesenchymal stem cell-derived microvesicles prevent the rupture of intracranial aneurysm in part by suppression of mast cell activation via a PGE2-dependent mechanism. Stem Cells (Dayton Ohio). (2016) 34:2943–55. doi: 10.1002/stem.2448
126. Zhang T, Huang L, Peng J, Zhang JH, Zhang H. LJ529 attenuates mast cell-related inflammation via A(3)R-PKCϵ-ALDH2 pathway after subarachnoid hemorrhage in rats. Exp neurology. (2021) 340:113686. doi: 10.1016/j.expneurol.2021.113686
127. Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology. (2014) 71:505–8. doi: 10.1001/jamaneurol.2013.5847
128. Zlomuzica A, Dere D, Binder S, De Souza Silva MA, Huston JP, Dere E. Neuronal histamine and cognitive symptoms in Alzheimer’s disease. Neuropharmacology. (2016) 106:135–45. doi: 10.1016/j.neuropharm.2015.05.007
129. Maslinska D, Laure-Kamionowska M, Maslinski KT, Gujski M, Maslinski S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflammation research: Off J Eur Histamine Res Soc. (2007) 56 Suppl 1:S17–8. doi: 10.1007/s00011-006-0508-8
130. Harcha PA, Vargas A, Yi C, Koulakoff AA, Giaume C, Sáez JC. Hemichannels are required for amyloid β-peptide-induced degranulation and are activated in brain mast cells of APPswe/PS1dE9 mice. J neuroscience: Off J Soc Neurosci. (2015) 35:9526–38. doi: 10.1523/JNEUROSCI.3686-14.2015
131. Shaik-Dasthagirisaheb YB, Conti P. The role of mast cells in alzheimer’s disease. Adv Clin Exp medicine: Off Organ Wroclaw Med University. (2016) 25:781–7. doi: 10.17219/acem/61914
132. Bhuiyan P, Wang YW, Sha HH, Dong HQ, Qian YN. Neuroimmune connections between corticotropin-releasing hormone and mast cells: novel strategies for the treatment of neurodegenerative diseases. Neural regeneration Res. (2021) 16:2184–97. doi: 10.4103/1673-5374.310608
133. d’Errico P, Meyer-Luehmann M. Mechanisms of pathogenic tau and Aβ Protein spreading in alzheimer’s disease. Front Aging Neurosci. (2020) 12:265. doi: 10.3389/fnagi.2020.00265
134. Liu J, Liu S, Zeng L, Tsilioni I. Amyloid beta peptides lead to mast cell activation in a novel 3D hydrogel model. Int J Mol Sci. (2023) 24:12002. doi: 10.3390/ijms241512002
135. Lin CJ, Herisson F, Le H, Jaafar N, ChEtal K, Oram MK, et al. Mast cell deficiency improves cognition and enhances disease-associated microglia in 5XFAD mice. Cell Rep. (2023) 42:113141. doi: 10.1016/j.celrep.2023.113141
136. Dubois B, López-Arrieta J, Lipschitz S, Doskas T, Spiru L, Moroz S, et al. Masitinib for mild-to-moderate Alzheimer’s disease: results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimer’s Res Ther. (2023) 15:39. doi: 10.1186/s13195-023-01169-x
137. Tolosa E, Garrido A, Scholz SW, Poewe W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurology. (2021) 20:385–97. doi: 10.1016/S1474-4422(21)00030-2
138. Bernardino L. Histamine in the crosstalk between innate immune cells and neurons: relevance for brain homeostasis and disease. Curr topics Behav neurosciences. (2022) 59:261–88. doi: 10.1007/7854_2021_235
139. Azadmanesh J, Lutz WE, Coates L, Weiss KL, Borgstahl GEO. Direct detection of coupled proton and electron transfers in human manganese superoxide dismutase. Nat Commun. (2021) 12:2079. doi: 10.1038/s41467-021-22290-1
140. Houldsworth A. Role of oxidative stress in neurodegenerative disorders: a review of reactive oxygen species and prevention by antioxidants. Brain Commun. (2024) 6:fcad356. doi: 10.1093/braincomms/fcad356
141. Belluzzi E, Bisaglia M, Lazzarini E, Tabares LC, Beltramini M, Bubacco L. Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: possible implications for Parkinson’s disease. PloS One. (2012) 7:e38026. doi: 10.1371/journal.pone.0038026
142. Nowak P, Noras L, Jochem J, Szkilnik R, Brus H, Körossy E, et al. Histaminergic activity in a rodent model of Parkinson’s disease. Neurotoxicity Res. (2009) 15:246–51. doi: 10.1007/s12640-009-9025-1
143. Rinne JO, Anichtchik OV, Eriksson KS, Kaslin J, Tuomisto L, Kalimo H, et al. Increased brain histamine levels in Parkinson’s disease but not in multiple system atrophy. J neurochemistry. (2002) 81:954–60. doi: 10.1046/j.1471-4159.2002.00871.x
144. Anichtchik OV, Rinne JO, Kalimo H, Panula P. An altered histaminergic innervation of the substantia nigra in Parkinson’s disease. Exp neurology. (2000) 163:20–30. doi: 10.1006/exnr.2000.7362
145. Panula P, Nuutinen S. The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci. (2013) 14:472–87. doi: 10.1038/nrn3526
146. Park HJ, Kim HJ, Park HK, Chung JH. Protective effect of histamine H2 receptor antagonist ranitidine against rotenone-induced apoptosis. Neurotoxicology. (2009) 30:1114–9. doi: 10.1016/j.neuro.2009.08.005
147. Zhou P, Homberg JR, Fang Q, Wang J, Li W, Meng X, et al. Histamine-4 receptor antagonist JNJ7777120 inhibits pro-inflammatory microglia and prevents the progression of Parkinson-like pathology and behaviour in a rat model. Brain behavior immunity. (2019) 76:61–73. doi: 10.1016/j.bbi.2018.11.006
148. Fang Q, Xicoy H, Shen J, Luchetti S, Dai D, Zhou P, et al. Histamine-4 receptor antagonist ameliorates Parkinson-like pathology in the striatum. Brain behavior immunity. (2021) 92:127–38. doi: 10.1016/j.bbi.2020.11.036
149. Kempuraj D, Selvakumar GP, Zaheer S, Thangavel R, Ahmed ME, Raikwar S, et al. Cross-talk between glia, neurons and mast cells in neuroinflammation associated with parkinson’s disease. J neuroimmune pharmacology: Off J Soc NeuroImmune Pharmacol. (2018) 13:100–12. doi: 10.1007/s11481-017-9766-1
150. Hong GU, Cho JW, Kim SY, Shin JH, Ro JY. Inflammatory mediators resulting from transglutaminase 2 expressed in mast cells contribute to the development of Parkinson’s disease in a mouse model. Toxicol Appl Pharmacol. (2018) 358:10–22. doi: 10.1016/j.taap.2018.09.003
151. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J neurology. (2020) 27:1918–29. doi: 10.1111/ene.14393
152. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. New Engl J Med. (2001) 344:1688–700. doi: 10.1056/NEJM200105313442207
153. Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp neurology. (2004) 185:232–40. doi: 10.1016/j.expneurol.2003.10.004
154. Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F, et al. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotrophic lateral sclerosis other motor neuron disorders: Off Publ World Fed Neurology Res Group Motor Neuron Diseases. (2004) 5:213–9. doi: 10.1080/14660820410020286
155. Fiala M, Chattopadhay M, La Cava A, Tse E, Liu G, Lourenco E, et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J neuroinflammation. (2010) 7:76. doi: 10.1186/1742-2094-7-76
156. Trias E, Ibarburu S, Barreto-Núñez R, Varela V, Moura IC, Dubreuil P, et al. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight. (2017) 2(20). doi: 10.1172/jci.insight.95934
157. Trias E, King PH, Si Y, Kwon Y, Varela V, Ibarburu S, et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight. (2018) 3(19). doi: 10.1172/jci.insight.123249
158. Cheng LE, Hartmann K, Roers A, Krummel MF, Locksley RM. Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity. (2013) 38:166–75. doi: 10.1016/j.immuni.2012.09.022
159. Kovacs M, Alamón C, Maciel C, Varela V, Ibarburu S, Tarragó L, et al. The pathogenic role of c-Kit+ mast cells in the spinal motor neuron-vascular niche in ALS. Acta neuropathologica Commun. (2021) 9:136. doi: 10.1186/s40478-021-01241-3
160. Dai H, Korthuis RJ. Mast cell proteases and inflammation. Drug Discovery Today Dis models. (2011) 8:47–55. doi: 10.1016/j.ddmod.2011.06.004
161. Dell’Italia LJ, Collawn JF, Ferrario CM. Multifunctional role of chymase in acute and chronic tissue injury and remodeling. Circ Res. (2018) 122:319–36. doi: 10.1161/CIRCRESAHA.117.310978
162. Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood. (2011) 118:5383–93. doi: 10.1182/blood-2011-07-358432
163. Ashina K, Tsubosaka Y, Nakamura T, Omori K, Kobayashi K, Hori M, et al. Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PloS One. (2015) 10:e0132367. doi: 10.1371/journal.pone.0132367
164. Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. (2012) 1469:114–28. doi: 10.1016/j.brainres.2012.05.056
165. Garbuzova-Davis S, Sanberg PR. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front Cell Neurosci. (2014) 8:21. doi: 10.3389/fncel.2014.00021
166. Yamadera M, Fujimura H, Inoue K, Toyooka K, Mori C, Hirano H, et al. Microvascular disturbance with decreased pericyte coverage is prominent in the ventral horn of patients with amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis frontotemporal degeneration. (2015) 16:393–401. doi: 10.3109/21678421.2015.1011663
167. Ribatti D. The crucial role of mast cells in blood-brain barrier alterations. Exp Cell Res. (2015) 338:119–25. doi: 10.1016/j.yexcr.2015.05.013
168. Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR, et al. Neurovascular aspects of amyotrophic lateral sclerosis. Int Rev neurobiology. (2012) 102:91–106. doi: 10.1016/B978-0-12-386986-9.00004-1
169. Pehar M, Vargas MR, Cassina P, Barbeito AG, Beckman JS, Barbeito L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neuro-degenerative Dis. (2005) 2:139–46. doi: 10.1159/000089619
170. Mora JS, Genge A, Chio A, Estol CJ, Chaverri D, Hernández M, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotrophic lateral sclerosis frontotemporal degeneration. (2020) 21:5–14. doi: 10.1080/21678421.2019.1632346
Keywords: mast cell, brain disorders, neuroinflammation, blood-brain barrier, neuroprotective effect
Citation: Huang X, Lan Z and Hu Z (2024) Role and mechanisms of mast cells in brain disorders. Front. Immunol. 15:1445867. doi: 10.3389/fimmu.2024.1445867
Received: 08 June 2024; Accepted: 12 August 2024;
Published: 26 August 2024.
Edited by:
Axel Lorentz, University of Hohenheim, GermanyReviewed by:
Jeannette Hübener-Schmid, University of Tübingen, GermanyElias Toubi, Technion Israel Institute of Technology, Israel
Copyright © 2024 Huang, Lan and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiping Hu, emhpcGluZ2h1QGNzdS5lZHUuY24=; Ziwei Lan, TGFueml3ZWlAY3N1LmVkdS5jbg==