 Fabíola Santos Zambon Robertoni1
Fabíola Santos Zambon Robertoni1 Ana Paula Pereira Velosa1
Ana Paula Pereira Velosa1 Luana de Mendonça Oliveira2
Luana de Mendonça Oliveira2 Francine Maria de Almeida3
Francine Maria de Almeida3 Lizandre Keren Ramos da Silveira1
Lizandre Keren Ramos da Silveira1 Zelita Aparecida de Jesus Queiroz1
Zelita Aparecida de Jesus Queiroz1 Thays de Matos Lobo1Vitória Elias Contini1
Thays de Matos Lobo1Vitória Elias Contini1 Camila Machado Baldavira4
Camila Machado Baldavira4 Solange Carrasco1Sandra de Morais Fernezlian4
Solange Carrasco1Sandra de Morais Fernezlian4 Maria Notomi Sato2
Maria Notomi Sato2 Vera Luiza Capelozzi4
Vera Luiza Capelozzi4 Fernanda Degobbi Tenorio Quirino dos Santos Lopes3
Fernanda Degobbi Tenorio Quirino dos Santos Lopes3 Walcy Paganelli Rosolia Teodoro1*
Walcy Paganelli Rosolia Teodoro1*- 1Division of Rheumatology, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 2Laboratory of Dermatology and Immunodeficiencies, Laboratório de Investigação Médica (LIM)-56, Department of Dermatology, Tropical Medicine Institute of São Paulo, University of São Paulo Medical School, São Paulo, Brazil
- 3Department of Clinical Medicine, Laboratory of Experimental Therapeutics, Laboratório de Investigação Médica (LIM)-20, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 4Department of Pathology, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Background: Chronic obstructive pulmonary disease (COPD) has been linked to immune responses to lung-associated self-antigens. Exposure to cigarette smoke (CS), the main cause of COPD, causes chronic lung inflammation, resulting in pulmonary matrix (ECM) damage. This tissue breakdown exposes collagen V (Col V), an antigen typically hidden from the immune system, which could trigger an autoimmune response. Col V autoimmunity has been linked to several lung diseases, and the induction of immune tolerance can mitigate some of these diseases. Evidence suggests that autoimmunity to Col V might also occur in COPD; thus, immunotolerance to Col V could be a novel therapeutic approach.
Objective: The role of autoimmunity against collagen V in COPD development was investigated by analyzing the effects of Col V-induced tolerance on the inflammatory response and lung remodeling in a murine model of CS-induced COPD.
Methods: Male C57BL/6 mice were divided into three groups: one exposed to CS for four weeks, one previously tolerated for Col V and exposed to CS for four weeks, and one kept in clean air for the same period. Then, we proceeded with lung functional and structural evaluation, assessing inflammatory cells in bronchoalveolar lavage fluid (BALF) and inflammatory markers in the lung parenchyma, inflammatory cytokines in lung and spleen homogenates, and T-cell phenotyping in the spleen.
Results: CS exposure altered the structure of elastic and collagen fibers and increased the pro-inflammatory immune response, indicating the presence of COPD. Col V tolerance inhibited the onset of emphysema and prevented structural changes in lung ECM fibers by promoting an immunosuppressive microenvironment in the lung and inducing Treg cell differentiation.
Conclusion: Induction of nasal tolerance to Col V can prevent inflammatory responses and lung remodeling in experimental COPD, suggesting that autoimmunity to Col V plays a role in COPD development.
Highlights
● Autoimmunity to type V collagen is involved in COPD physiopathology.
● Immunotolerance to type V collagen can prevent emphysema development.
1 Introduction
Chronic obstructive pulmonary disease (COPD) is a highly prevalent disease and one of the most important public health problems worldwide (1). It is characterized by extracellular matrix remodeling in the airways and alveolar parenchyma due to a chronic inflammatory process (2) and is mainly caused by exposure to cigarette smoke (CS) (3). COPD patients suffer from dyspnea and fatigue, which are due to impaired respiratory mechanics, gas exchange abnormalities, and deconditioning-related dysfunction of the peripheral muscles that affect their capacity to accomplish daily activities as well as their overall quality of life and life expectancy. (3). The frequent progressive worsening of the disease (4) eventually leads to exacerbations and hospitalizations that are associated with a poor prognosis (5). Pharmacotherapy can alleviate COPD symptoms, reduce exacerbations, and enhance overall health and exercise tolerance (3). Existing therapies, aside from smoke cessation, cannot consistently prevent disease progression and can cause many adverse health effects (6), such as cardiac rhythm disturbance, dryness of mouth, headaches, insomnia and nausea caused by bronchodilators, oral candidiasis, hoarse voice, skin bruising and pneumonia caused by inhaled corticosteroids (3), Therefore, better understanding of the mechanisms that underlie the development and progression of COPD is essential to help improve the management of this highly challenging and complex disease.
COPD is a multifactorial disease, and its pathophysiology has been linked to a number of mechanistic ideas (7), with increasing evidence that autoimmunity plays an essential role in its pathogenesis (8). Breakdown products from the extracellular matrix (such as collagen and elastin fragments) can activate adaptive immune responses in COPD patients’ lungs, including cytotoxic CD8+ T cells, T helper (Th)1 and Th17 CD4+ T cells, and B-cell responses leading to autoantibodies (7). Recent research has demonstrated other autoimmunity components associated with COPD. The serum antibody titer against carbonyl-modified proteins, resulting from the oxidation of various amino acid residues, is significantly elevated in COPD patients (GOLD 3) compared with healthy controls, which could lead to an autoimmune response (9). B cell-activating factor of tumor necrosis factor family (BAFF) overexpression is associated with autoimmune diseases, and it was found to be up-regulated in B cells in pulmonary lymphoid follicles (LFs), in blood and bronchoalveolar lavage samples from COPD patients (10). Blood samples collected from 2,396 individuals in the COPDGene study, indicated that interferon (IFN)-stimulated genetic signatures from bronchial brushings and peripheral blood were associated with exacerbations, lung function, and airway wall thickness. Since these samples were obtained during periods of stable disease, this genetic signature identified is likely the result of sustained autoimmune responses (11).
Damage caused directly by cigarette smoke and inflammation results in the degradation of collagen and the release of its fragments into the systemic circulation, (12) which could trigger autoimmune responses. In this sense, researchers have conducted studies to evaluate serological markers as potential diagnostic and prognostic markers of COPD (13–17). It has been proposed that fragments of types I, III, and V are the most effective in distinguishing between mild COPD patients and healthy controls. (12) A previous study showed that immunity against type V collagen (Col V) may play a role in COPD, as markers related to this process have been found in the serum of patients, especially during disease exacerbations (13). Furthermore, the presence of anti-collagen V antibody in an ELISA assay, and collagen V-specific Th1 immune responses (IFNγ) (EliSpot assay performed on isolated peripheral blood mononuclear cells) has been demonstrated in the peripheral blood of patients with COPD and smokers, suggesting that Col V-specific autoimmunity is associated with cigarette smoking history and may be involved in the pathogenesis of COPD (17).
Col V is a fibrillar collagen that is a minor component of the ECM and is found in the lung interstitium and capillary basement membranes (18). Usually, it is embedded within heterotypic collagen I/III fibrils (19–21). Due to its location and immunogenic and antigenic properties, Col V is considered a sequestered antigen that remains hidden from the immune system under normal conditions (22). However, in cases of tissue damage, such as CS-induced ECM lung injury, Col V epitopes may be exposed to the immune system, which can turn them into potential self-antigens (23, 24).
The role of autoimmunity against Col V has been explored in various diseases in recent decades, both in murine models and humans. Col V-specific autoimmunity is involved in several pathological processes, including some lung diseases, such as idiopathic pulmonary fibrosis (IPF) and asthma (23, 25–29). Conversely, different approaches to inducing immunological tolerance to Col V have proven effective in decreasing lung allograft rejection (23, 24, 30–32), reducing atherosclerotic plaque (33), preventing induced airway hyperresponsiveness (22), and mitigating pulmonary fibrosis (27, 34).
Taken together, this evidence suggests that in COPD, autoimmunity against Col V may play a role in the pathophysiology of the disease, and the induction of tolerance to this collagen may protect against ECM lung damage. Thus, our objective was to investigate this hypothesis by assessing whether Col V-induced tolerance interferes with the course of emphysema development in a short-term murine model of CS-induced COPD.
2 Methods
2.1 Experimental groups
Male C57BL/6 mice (aged 6–8 weeks and weighing 20–25 g) were divided at random into three groups: first, exposed to CS for four weeks (CS group); second, previously tolerated for Col V and exposed to CS for the same four weeks (CS/Tol group); and third, maintained under room air conditions for the same time intervals as the controls (CT group). All the animals received proper care according to the National Research Council’s Guide for the Care and Use of Laboratory Animals (2011). Our protocol was approved by the Ethics Committee on the Use of Animals from the Faculty of Medicine of the University of São Paulo under protocol number 1200/2018 (São Paulo, Brazil). To properly process the biological samples, the animals were divided into two groups: first, functional and morphological analyses of the lungs were performed; second, cell and cytokine profile analyses were performed.
2.2 Type V collagen nasal tolerance protocol
Induction of tolerance to Col V was achieved by daily nasal administration of 20 µl of Col V solution (0.5 mg/ml; Human Placenta Collagen, Bornstein and Traub Type V powder, Sigma-Aldrich, USA) to each animal (33), which was repeated for five days during the week before beginning the exposure to cigarette smoke protocol. Boosters at the same dosage were given three times a week for the entire duration of the cigarette smoke exposure protocol. Nasal administration of Col V was performed at least 30 minutes before the first CS exposure of the day, to ensure permeability of the nasal cavity of mice to CS.
2.3 CS exposure protocol
The animals were exposed to CS as previously described, in a in a full body inhalation chamber (28 L) (35), with slight modifications according to a more recent evaluation (36). Using a whole-body exposure chamber, animals were exposed to the CS of twenty commercially filtered cigarettes per day (Souza Cruz – BAT, Brazil; 10 mg of tar, 0.8 mg of nicotine, and 10 mg of CO per cigarette) (35). A 60-minute full body exposure to CS from 10 cigarettes was performed twice a day for five days/week for four weeks. The flow rate was set such that the carbon monoxide (CO) levels inside the chamber were maintained at approximately 350 parts per million (ppm). The control groups were maintained under clean room air conditions.
2.4 Respiratory mechanics evaluation
On the day following the last exposure to cigarette smoke, the first group of mice was deeply anesthetized by an intraperitoneal injection of 50 mg/kg sodium thiopental, tracheostomized and mechanically ventilated (n=7–10 per group) using a flexiVent small animal ventilator (SCIREQ, Montreal, QC, Canada). Pulmonary function assessment was performed using the forced oscillatory technique and a constant phase model (37), and values for the parameters of airway resistance (Raw), tissue damping (Gtis), and tissue elastance (Htis) were obtained.
2.5 Total cell and macrophage measurements in BALF
Immediately after lung mechanics assessment, the mice were euthanized by abdominal aortic exsanguination, and bronchoalveolar lavage fluid (BALF) samples were collected. The animals’ lungs were washed two times with 1 ml of phosphate-saline buffer (PBS) and infused into the lungs through the tracheal cannula, after which the fluid was recovered for subsequent analyses. The fluid was centrifuged at 1000 rpm for 8 min at 5°C, and the cell pellet was resuspended in 300 μl of physiological saline for total cell counting using a Neubauer hemocytometer chamber. Aliquots of the cell suspension were centrifuged on glass slides using a cytocentrifuge and stained with Diff-Quik (Medion Diagnostics, Dündingen, Switzerland). Differential cells (300 cells/slide) were evaluated by light microscopy with an immersion objective (38).
2.6 Lung preparation for histological analyses
After BALF recovery, the lungs were removed and fixed at constant pressure (20 cmH2O) using 10% buffered formalin infused through the trachea for 24 h (35). Then, the lungs were paraffin-embedded and cut into 5-μm-thick sections.
2.6.1 Morphometry
Lung tissue sections were stained with hematoxylin and eosin (H&E) to evaluate the mean linear intercept (Lm), which is used as an indicator of the mean diameter of airspaces, as described previously (39). The Lm was analyzed in a blinded fashion, and 20 randomly selected nonoverlapping fields of lung parenchyma were assessed for each slide at 200x magnification via standard light microscopy using an eyepiece with a known area attached to the ocular lens of the microscope (40). The same lung slides were scanned using a high-resolution digital slide scanner (Pannoramic 250 SCAN, 3DHISTECH Ltd., Budapest, HU). Subsequently, using SlideViewer 2.5 software (3DHISTECH Ltd., Budapest, Hungary), 5 to 10 random fields in the region of the peribronchovascular axis were acquired at a 20x zoom level. To quantify edema, the stereological method of counting points was applied (41) using image analysis software (Image-Pro Plus 6.0 for Windows, Media Cybernetics, Inc., Silver Spring, MS, USA). The number of positive hits for edema was counted and divided by the respective peribronchovascular total area calculated by the software (the results are expressed as +/μm2).
2.6.2 Histochemistry
For the identification of total elastic fibers on the lung tissue, sections were stained using the modified Weigert resorcin-fuchsin method, with prior oxidation by Oxone, as previously described (42). This method stains in purple all components of the elastic system, including fully developed elastic, oxytalan, and eulanin fibers, not allowing the identification of the components separately.
2.6.3 Immunohistochemistry
Lung tissue sections were immunostained using the biotin-streptavidin peroxidase method. The following primary antibodies were used: rat monoclonal anti-MAC-2 (CL8942AP, Cedarlane Labs, 1:25000), rabbit polyclonal anti-TGFβ1 (sc-146, Santa Cruz Biotechnology, 1:1500), rabbit polyclonal anti-FOXP3 (sc-28705, Santa Cruz Biotechnology, 1:100), rabbit polyclonal anti-IL-10 (BS20373R, Bioss Antibodies Inc., 1:3000) and rabbit polyclonal anti-IL-17 (sc-7927, Santa Cruz Biotechnology, 1:100). To complete the reactions, specific secondary antibodies (VECTASTAIN® Elite® ABC Kit - Vector Laboratories, Burlingame, CA, USA) were used. Then, the sections were stained with 3,3’-diaminobenzidine (DAB; Sigma-Aldrich Chemie, Steinheim, Germany) and counterstained with Harris’s hematoxylin (Merck, Darmstadt, Germany). Some slices were not incubated with primary antibodies and served as negative controls.
2.6.4 Immunofluorescence
The lung slices were incubated with a rabbit anti-collagen type I antibody (600-401-103-0.1, Rockland, Limerick, PA, USA, 1:2500) overnight at 4°C and then with a secondary antibody (Alexa 488-conjugated goat anti-rabbit IgG, Invitrogen, Life Technologies, Eugene, OR, USA, 1:200) at room temperature. As a control, PBS was used in place of the primary antibody.
2.6.5 Immunofluorescence for double staining
The lung slices were incubated with rabbit polyclonal anti-FOXP3 (sc-28705, 1:50, Santa Cruz Biotechnology, CA, USA) and mouse monoclonal anti-IL-10 (sc-8438, 1:50, Santa Cruz Biotechnology, CA, USA) and then with Alexa 488-conjugated goat anti-rabbit IgG (1:200, Invitrogen, Eugene, OR, USA) and Alexa 546-conjugated donkey anti-mouse IgG (1:200, Invitrogen, Eugene, OR, USA) at room temperature. The nuclei were then counterstained with DAPI (Molecular Probes, Invitrogen, Eugene, OR, USA).
2.6.6 Image analysis
Images of all stained slices were acquired with a photographic camera (QColor 5, Olympus Co., St. Laurent, Quebec, Canada) coupled to a microscope (Olympus BX-51, Olympus Corporation, Tokyo, Japan) and sent to a computer through a scanning system (Oculus TCX, Coreco, Inc., St. Laurent, Quebec, Canada). For all the stains performed, ten random and nonoverlapping fields were evaluated for each animal by a researcher blinded to the study groups. To quantify the elastic fibers (400x magnification) and immunostained cells (1000x magnification), the stereological method of counting points was used (41). The results for elastic fibers are given as the proportion of points with positive staining to the total number of points touching the parenchyma, and the results for immunostaining are given as the proportion of points with positive staining to the total number of points touching the parenchyma. The number of type I collagen fibers (400x magnification) was determined as the ratio of the immunostained area to the total parenchyma area. Measurements were made using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Silver Spring, MS, USA). The results of each slide are presented as the average of the percentages of all evaluated fields.
2.7 Immunophenotyping of T cells from mouse spleen by flow cytometry
From the second group of mice, the spleens were harvested for T-cell immunophenotyping. Mice were first anesthetized and euthanized, and the spleen was removed using previously washed and autoclaved dissection tools. Each spleen was dissociated into a single-cell suspension by maceration on a 40 µm nylon cell strainer (BD, San Diego, CA, USA) and placed in Petri dishes containing RPMI 1640 medium (Sigma, USA) supplemented with 10% fetal bovine serum (FBS). The samples were transferred to tubes, centrifuged, washed twice with RPMI 1640 and resuspended in 1 ml of PBS. After splenic cell harvesting, the samples were incubated with Fc blocking solution and stained with a surface antibody mixture containing anti-CD3 (PercP-Cy5; BD Pharmingen, clone 17A2), anti-CD4 (FITC; BD Pharmingen, clone H129.19), anti-CD8 (APC-Cy7; BD Pharmingen, clone 53-6.7), anti-CD25 (APC; BD Pharmingen, clone PC61), and anti-CD44 (BV 605; BD Horizon, clone IM7). Then, the cells were fixed/permeabilized and incubated with anti-FOXP3 V450 (BD Horizon, clone MF23) for intracellular staining. After 30 min, the cells were washed twice with staining buffer, resuspended, and analyzed by flow cytometry. A total of 500,000 events per sample were acquired on an LSRFortessa cytometer (BD Biosciences, USA), and the data were analyzed with FlowJo software (BD, OR, USA).
2.8 Preparation of lung and spleen homogenates for cytokine dosage
Spleens were harvested from the first group of animals and lungs from the second group after anesthesia and euthanasia as described above and stored in a -80°C freezer for later preparation of homogenates. After removal, an average of 44 mg of left lung tissue was homogenized in 750 μl of PBS supplemented with a protease inhibitor (SIGMA P2714-1BTL, 1:10 solution), and an average of 33 mg of spleen tissue was homogenized in 500 μl of the same solution with a tissue homogenizer (PowerLyzer® 24, QIAGEN). The homogenates were kept at 4°C and centrifuged at 4000 RCF for 10 min. The supernatants were collected and stored at −80°C before cytokine levels were assessed.
2.8.1 Cytokine dosage by flow cytometry
The levels of IFN-γ, IL-10, IL-17A, IL-6, and TNF cytokines in lung and spleen homogenates were determined by flow cytometry using a commercial kit specific for mice (BD Cytometric Bead Array (CBA) Mouse Enhanced Sensitivity Master Buffer Kit, BD Biosciences, San Diego, CA, USA), and procedures were followed according to the manufacturer’s instructions. The analysis was conducted in a flow cytometer (LSR Fortessa, BD), and the levels were determined using CBA analysis software.
2.8.2 Protein dosage
As fragments of different sizes were used for tissue homogenates, the total protein dosage of each sample was used to correct the cytokine levels found, increasing the accuracy of the analysis. The total protein concentration in the lung and spleen homogenates was measured by using a commercial kit based on bicinchoninic acid (Bicinchoninic Acid Protein Assay Kit, BCA1, Sigma-Aldrich, USA) following the manufacturer’s instructions on a spectrophotometer (Evolution 60S UV-Visible spectrophotometer, Thermo Scientific, Madison, WI, USA).
2.9 Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, Inc.). The normality of the data distribution was verified with the Shapiro-Wilk test. To compare the three experimental groups and detect significant differences, one-way ANOVA was performed, followed by the Holm-Sidak multiple comparisons test or Kruskal-Wallis test, followed by Dunn’s multiple comparisons test, depending on the normality of the variables. A p-value of <0.05 was considered significant.
3 Results
3.1 Immune tolerance to Col V protects the lungs from cigarette smoke-induced harm
3.1.1 Lung morphometry and respiratory mechanics
Lm was significantly greater in the Cigarette Smoke (CS) group than in the Control (CT) group (Figure 1A, p=0.0064), suggesting enlargement of the air spaces and loss of alveolar units. Compared with those in the CS group, the Lm in the Cigarette Smoke/Tolerance (CS/Tol) group significantly decreased (Figure 1A, p=0.0032), with values comparable to those in the CT group. Analysis revealed significantly greater edema in the peribronchovascular region in the CS group than in the CT group (Figure 1B; p=0.0082), and the level of edema was significantly lower in the CS/Tol group than in the CS group. (Figure 1B; p=0.043). Both groups exposed to cigarette smoke had some degree of subacute changes in the lung parenchyma and bronchovascular area, including inflammatory cell infiltration. However, these changes were diffuse and severe only in the CS group (Figure 1C). Despite the identified structural changes, respiratory system mechanics evaluation revealed no differences between the experimental groups for any of the evaluated parameters (airway resistance - Raw, tissue damping - Gtis, and tissue elastance - Htis; Figure 1D).
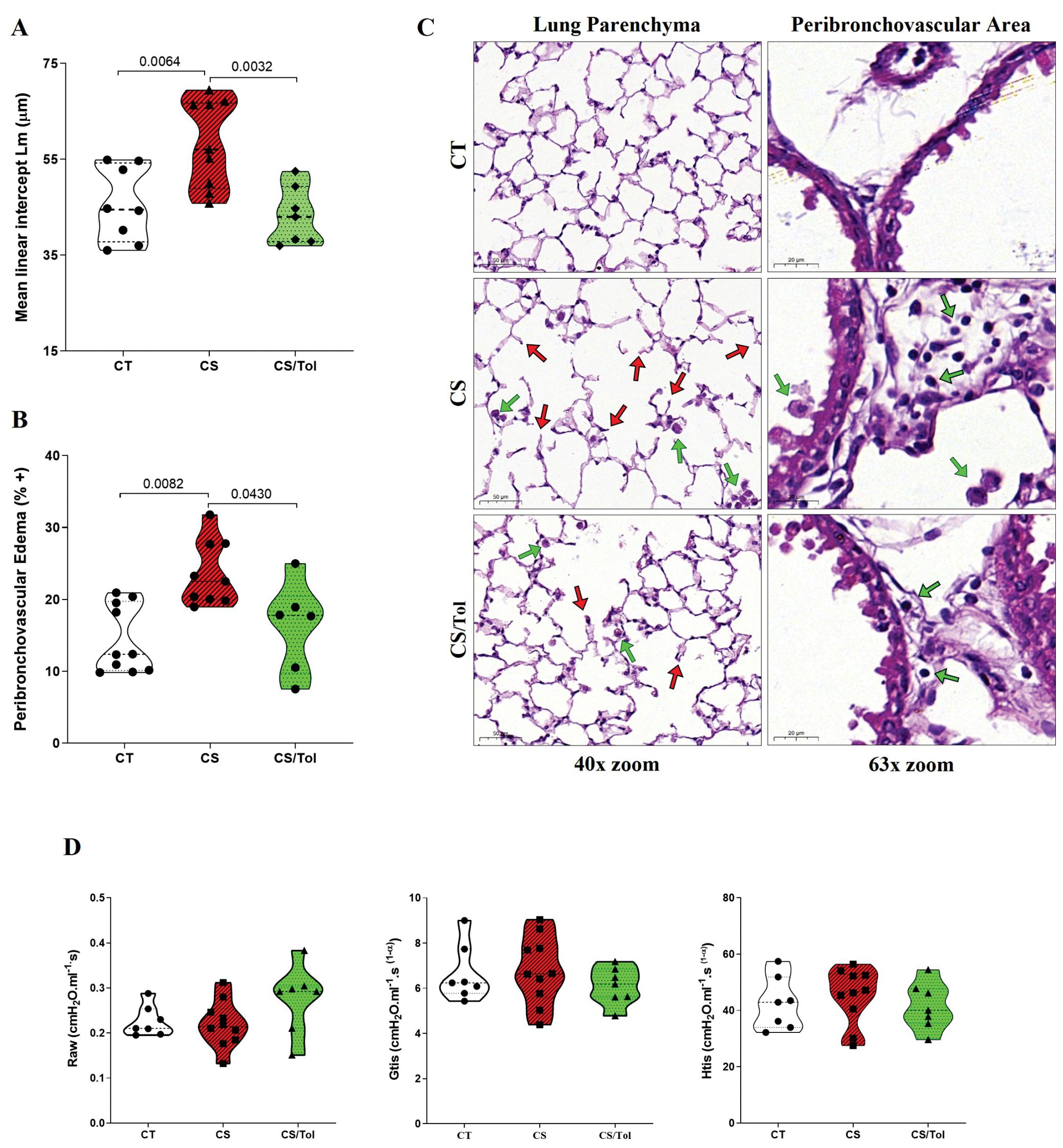
Figure 1. Lung Morphometry and Respiratory Mechanics (A) Increased Lm in the Cigarette Smoke (CS) group compared to that in the Control (CT) and Cigarette Smoke/Tolerance (CS/Tol) groups (p=0.0064 and p=0.0032, respectively; one-way ANOVA and Holm-Sidak multiple comparisons test; CT n=8, CS n=9, CS/Tol n=7). (B) Increased peribronchovascular edema in the CS group compared to the CT and CS/Tol groups (p=0.0082 and p=0.043, respectively, Kruskal-Wallis and Dunn´s multiple comparisons test; CT n=10, CS n=9, CS/Tol n=6). Violin plot graphs show the frequency distribution of the analyzed data, the median, and the interquartiles. (C) Photomicrographs of the lung parenchyma in the CT, CS, and CS/Tol groups (40x magnification) and the peribronchovascular area (63x magnification). H&E staining. Red arrows indicate tissue damage with alveolar enlargement, and green arrows indicate inflammatory cells. (D) There was no difference in airway resistance (Raw), tissue damping (Gtis), or tissue elastance (Htis) among the experimental, CT, CS, and CS/Tol groups (one-way ANOVA and Holm-Sidak multiple comparisons test; CT n=7, CS n=10, CS/Tol n=7). Violin plot graphs show the frequency distribution of the analyzed data, the median, and the interquartiles.
3.1.2 Lung matrix composition
Histomorphometric evaluation revealed a significant increase in elastic fiber staining in the lung parenchyma only in the CS group compared with the CT group (Figure 2A; p=0.0343). There was a significant reduction in the proportion of type I collagen fibers in the CS group compared to that in the CT group (Figure 2B; p=0.0012), and there was a tendency toward an increase in the proportion of type I collagen fibers in the CS/Tol group compared to that in the CS group (p= 0.0513). Although the elastic fibers suffered damage in both groups exposed to cigarette smoke, the significant increase in their staining intensity in the CS group reflects greater elastic fiber breakdown in the lung parenchyma in this group, as represented in Figure 2C. In Figure 2D, the photomicrographs show a decrease in type I collagen immunofluorescence staining in the CS group compared to the CT group.
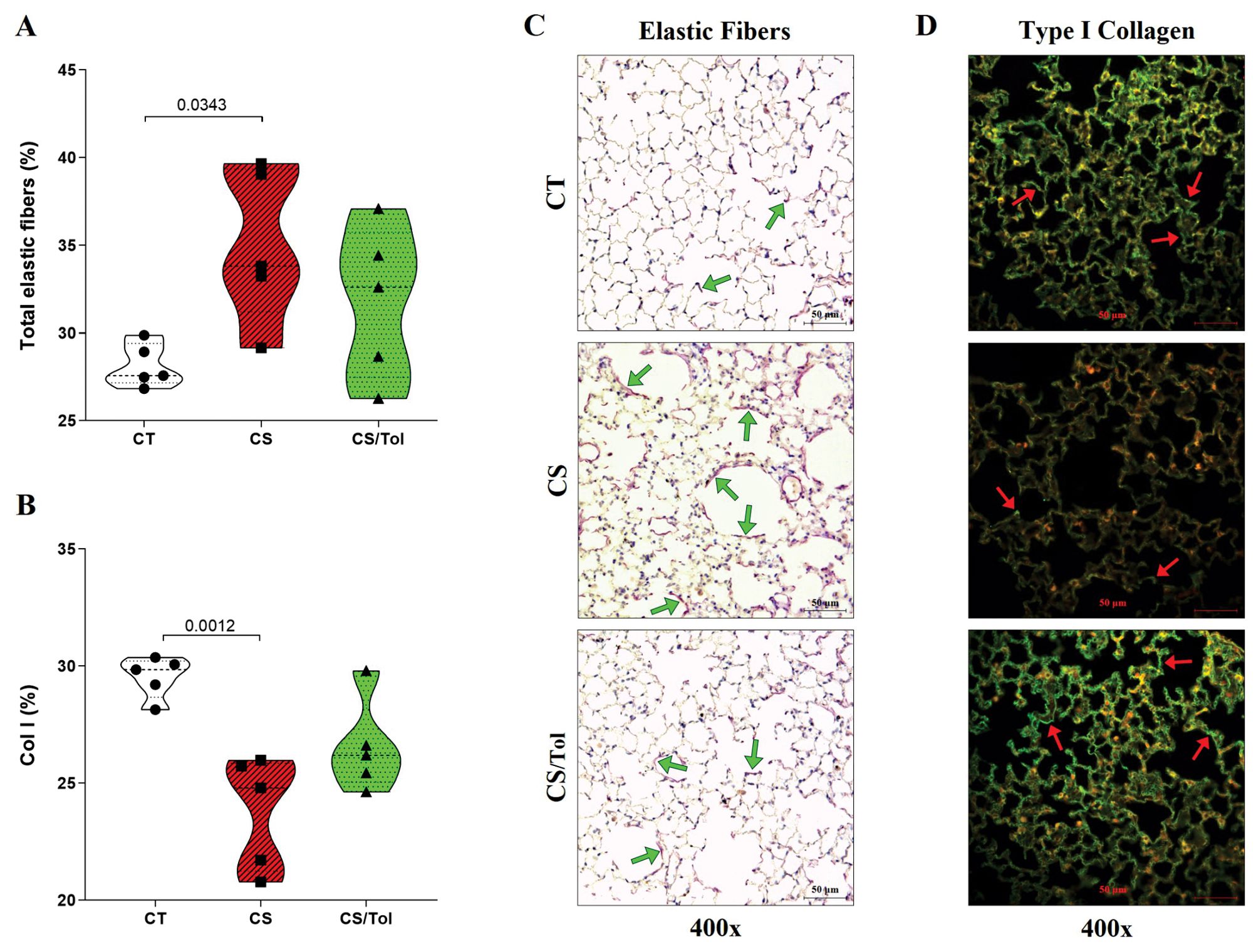
Figure 2. Identification of type I collagen and elastic fibers in the lung parenchyma. (A) The proportion of elastic fibers in the CS group was significantly greater than in the CT group (p=0.0393, one-way ANOVA and Holm-Sidak multiple comparisons test; n=6 in each experimental group). (B) The proportion of collagen type I fibers was significantly lower in the CS group than in the CT group (p=0.0012, one-way ANOVA and Holm-Sidak multiple comparisons test; n=5 in each experimental group). Violin plot graphs showing the frequency distribution of the analyzed data, the median and the interquartiles. (C) Photomicrographs of alveolar parenchyma with modified resorcin-fuchsin immunostaining (400x magnification) in the CT, CS, and CS/Tol groups, showing an increase in elastic fiber breakdown in the CS group reflected by increased staining in purple (green arrows show examples of positive staining). (D) Photomicrographs of alveolar parenchyma with type I collagen immunofluorescence staining (400x magnification) in the CT, CS, and CS/Tol groups, show a decrease in those fibers in the CS group (red arrows show examples of positive staining in green).
3.2 Regulatory cell profile induced by Col V tolerance
3.2.1 Macrophages in BALF and lung parenchyma
There was a significant increase in the total number of inflammatory cells in the BALF of the groups exposed to cigarette smoke, CS or CS/Tol compared to that in the BALF of the CT group (Figure 3A; p=0.0027 and p=0.0035, respectively). Differential evaluation revealed a predominance of macrophages, whose total number was significantly greater in the CS and CS/Tol groups than in the CT group (Figure 3B; p=0.0017 and p=0.0093, respectively). After four weeks of exposure to cigarette smoke, we observed an increase in Galectin-3+ cells distributed throughout the lung parenchyma in animals from the CS and CS/Tol groups, but statistical analysis revealed a significant increase in this marker only in the CS group compared to that in the CT group (Figure 3C; p=0.0020) and a tendency toward a decrease in this marker in the CS/Tol group in comparison to that in the CS group (p= 0.0721). In Figures 3D, E, the photomicrographs illustrate the increase in cells in the BALF and the galectin-3 immunostaining in the lung parenchyma, respectively.
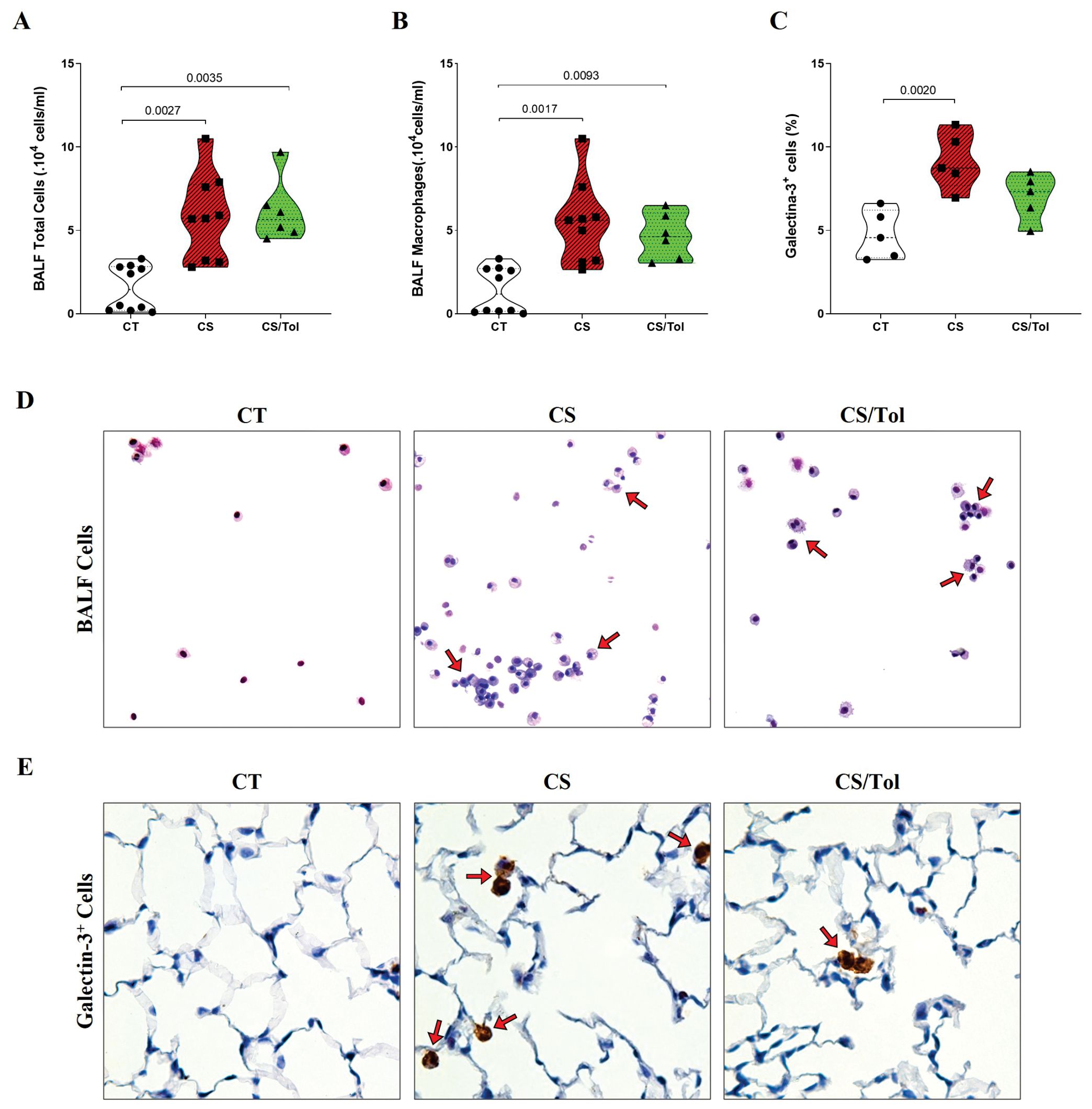
Figure 3. Total cell and macrophage counts in BALF, the proportion of Galectin-3+ cells in the lung parenchyma, and representative photomicrographs of total cells in BALF and immunostaining for Galectin-3+ cells. (A) There was a significant increase in the number of total BALF cells in the CS and CS/Tol groups compared to that in the CT group (p=0.0027 and p=0.0035, respectively; Kruskal-Wallis test and Dunn’s multiple comparisons test; CT, n=10; CS, n=9; CS/Tol, n=6). (B) Differential cell counts revealed a predominance of macrophages among the BALF cells, with significantly greater numbers in the CS and CS/Tol groups than in the CT group (p=0.0017 and p=0.0093, respectively; Kruskal-Wallis and Dunn’s multiple comparisons test; CT, n=10; CS, n=9; CS/Tol, n=6). (C) The number of Galectin-3+ cells was significantly greater in the CS group than in the C group (p=0.002, one-way ANOVA and Holm-Sidak’s multiple comparisons tests; n=5 in each experimental group). The violin plots show the frequency distributions of the analyzed data. (D) Photomicrographs of BALF cells in the CT, CS, and CS/Tol groups (200x magnification; Diff-Quik staining; red arrows show the predominance of macrophages in BALF). (E) Photomicrographs of galectin-3 immunostaining in the lung parenchyma in the CT, CS and CS/Tol groups (1000x magnification; red arrows show positive cells in brown).
3.2.2 IL-17+, IL-10+, TGFβ+ and FOXP3+ cells in the lung parenchyma
Cigarette smoke caused an increase in the number of IL-17+ cells in both exposed groups, but statistical analysis revealed a significant increase in the number of these cells only in the CS group compared to the CT group (Figure 4A; p=0.0044). In contrast, the quantification of IL-10+ in the alveolar parenchyma showed a significant increase only in the CS/Tol group compared to the CT group (Figure 4B; p=0.0034), although this cell type was increased in both the CS and CS/Tol groups. In Figures 4C, D, the photomicrographs ilustrate IL-17 and IL-10 immunostaining in the lung parenchyma, respectively.
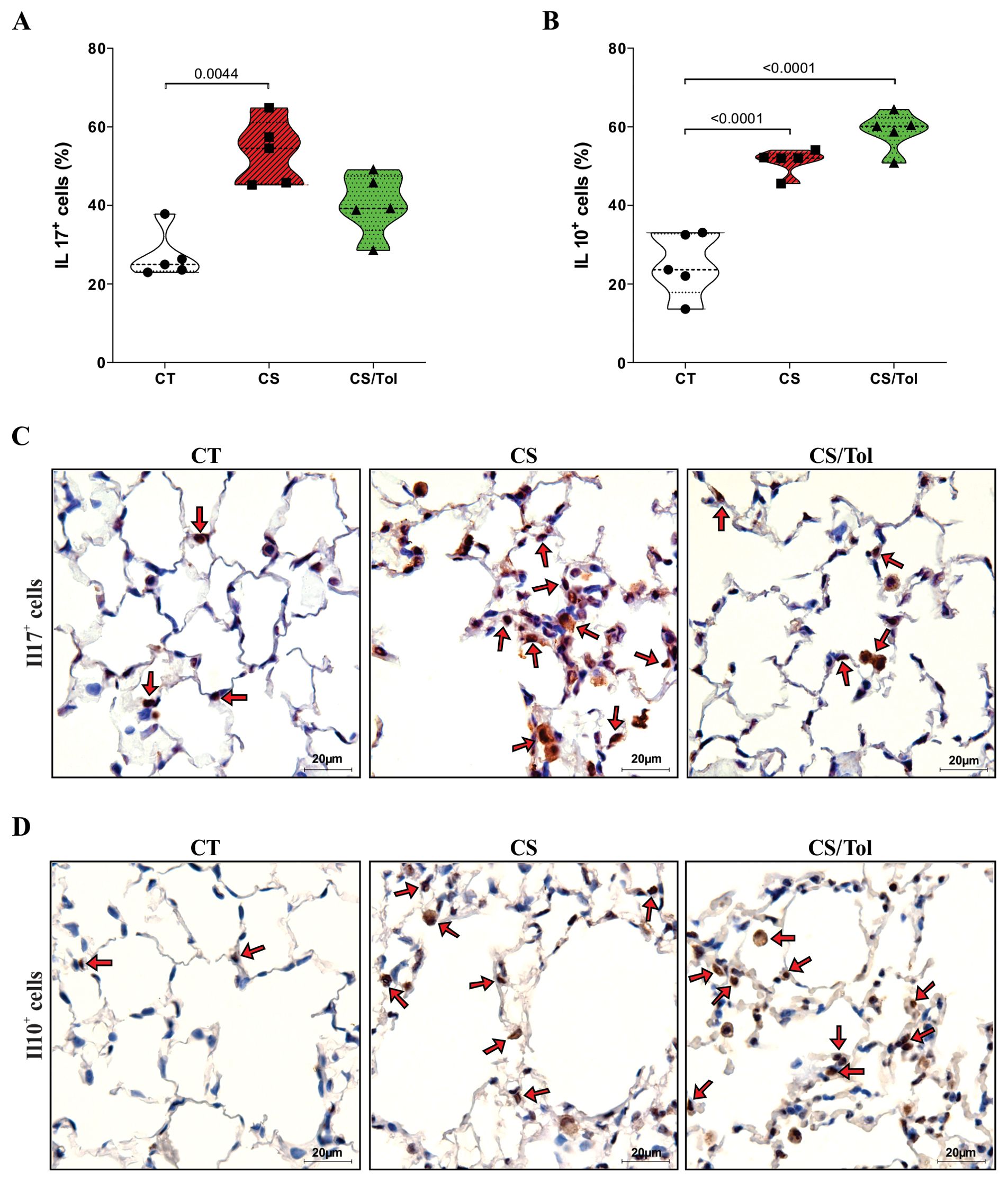
Figure 4. Proportion of IL-17+ and IL-10+ cells in the lung parenchyma and representative photomicrographs of immunostaining for IL-17+ and IL-10+ cells. (A) There was a significant increase in the number of IL-17+ cells in the CS group compared to that in the CT group (p=0.0044; Kruskal-Wallis and Dunn’s multiple comparisons tests; n=5 for each experimental group). (B) There was a significant increase in the number of IL-10+ cells in the CS group and the CS/Tol group compared to that in the CT group (p<0.0001, both; one-way ANOVA and Holm-Sidak’s multiple comparisons test; n=5 for each experimental group). The violin plots show the frequency distributions of the analyzed data. (C) Photomicrographs of IL-17 immunostaining in the lung parenchyma in the CT, CS and CS/Tol groups (1000x magnification; red arrows show positive cells in brown). (D) Photomicrographs of IL-17 immunostaining in the lung parenchyma in the CT, CS and CS/Tol groups (1000x magnification; red arrows show positive cells in brown).
Immunostaining of TGFβ+ cells did not show a significant difference between the groups for this marker, although there was a slight increase in the groups exposed to cigarette smoke CS and CS/Tol compared to the CT group (Figure 5A). Statistical analysis revealed a significant increase in FOXP3+ cells in the groups exposed to cigarette smoke CS or CS/Tol compared to those in the CS group (Figure 5B; p=0.0209 and p=0.0005, respectively). There was also a significant increase in these cells in the CS/Tol group compared to those in the CS group (Figure 5B; p=0.034). In Figures 5C, D, the photomicrographs ilustrate TGFβ and Foxp3 immunostaining in the lung parenchyma, respectively.
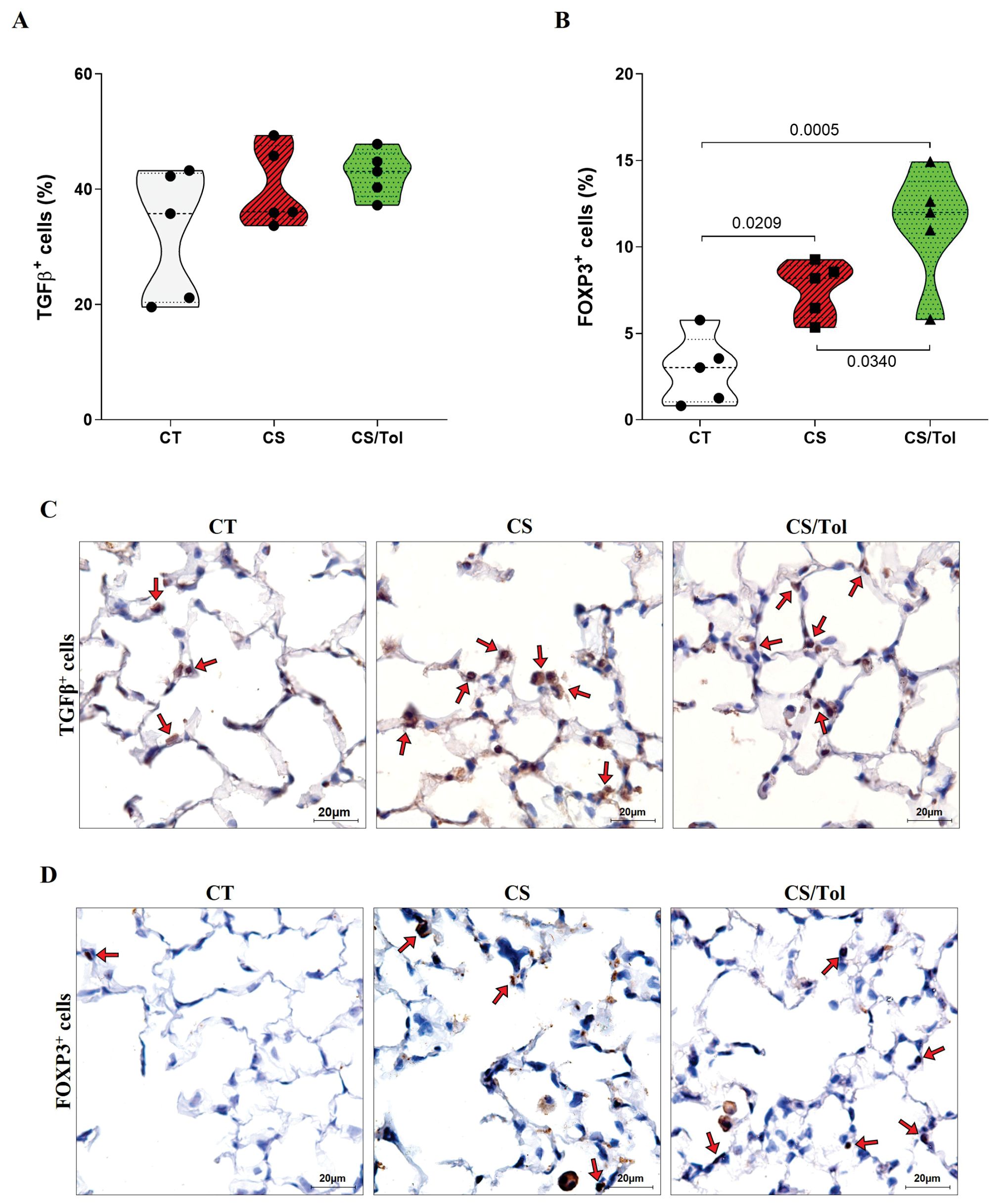
Figure 5. Proportion of TGFβ+ and FOXP3+ cells in the lung parenchyma and representative photomicrographs of immunostaining for TGFβ+ and FOXP3+ cells. (A) There was no difference in the number of TGFβ+ cells between the experimental groups (one-way ANOVA and Holm-Sidak multiple comparisons test, n=5 for each experimental group). (B) There was a significant increase in the number of FOXP3+ cells in the CS and CS/Tol groups compared to that in the CT group (p=0.0209 and p=0,0005, respectively), and a significant increase in the number of FOXP3+ cells in the CS/Tol group compared to that in the CS group (p=0,034, one-way ANOVA and Holm-Sidak’s multiple comparisons test, n=5 for each experimental group). The violin plots show the frequency distributions of the analyzed data. (C) Photomicrographs of TGFβ immunostaining in the lung parenchyma in the CT, CS and CS/Tol groups (1000x magnification; red arrows show positive cells in brown). (D) Photomicrographs of Foxp3 immunostaining in the lung parenchyma in the CT, CS and CS/Tol groups (1000x magnification; red arrows show positive cells in brown).
3.3 T lymphocyte autoimmunity response to cigarette smoke and Treg differentiation upon Col V-induced tolerance
3.3.1 Cigarette smoke-induced T lymphocyte activation
Analysis of the frequency of systemic CD4+ and CD8+ T lymphocytes, which are spleen lymphocytes, was performed according to the gating strategy depicted in Figure 6A. We first assessed CD44hi expression in CD4+ and CD8+ T cells. CD44hi expression in the CD4+ T lymphocytes of the groups exposed to cigarette smoke CS or CS/Tol was significantly greater than the CT group (Figures 6B, C; p=0.0015 and p=0.006, respectively). Analysis of the CD44hi T-cell subpopulations revealed greater CD44hi expression in the CS and CS/Tol groups than in the CT group (Figures 6B, C; both p<0.0001).
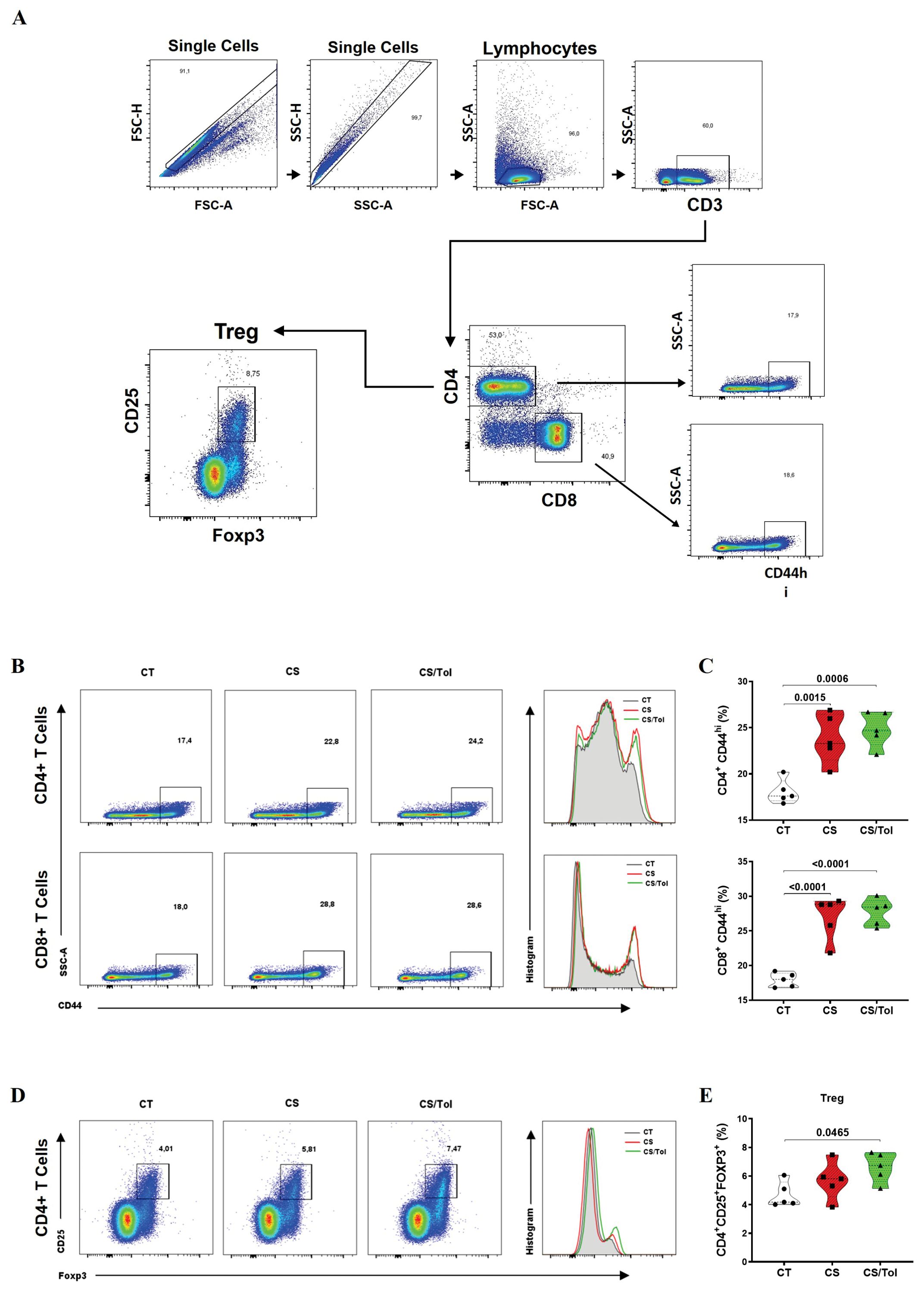
Figure 6. Frequencies of T lymphocyte phenotypes in the spleen (CD4+CD44hi, CD8+CD44hi, and CD4+CD25+FOXP3+). (A) Gating strategy used for flow cytometry: single cells, CD3+ cells, CD4+ cells or CD8+ cells, CD44hi cells, and CD4+ cells, CD25+ cells and FOXP3+ cells. (B) Percentage of CD44hi cells among CD4+ and CD8+ T cells. (C) There was an increase in the frequency of CD4+CD4hi cells in the CS and CS/Tol groups (p=0.0015 and p=0.006, respectively) compared to that in the CT group, and there was an increase in the frequency of CD8+CD4hi cells in the CS and CS/Tol groups (p< 0.0001, both). (D) Percentage of CD25+FOXP3+ CD4+ T cells markers. (E) There was an increase in the Treg (CD4+CD25+FOXP3+) frequency in the CS/Tol group (p=0,0465) compared to that in the CT group. (One-way ANOVA and Holm-Sidak multiple comparisons test, n=5 for each experimental group). The violin plots in the graphs show the frequency distributions of the analyzed data.
3.3.2 Tolerance-induced FOXP3+ Treg cells
Analysis of the Treg (CD4+CD25+FOXP3+) lymphocyte frequency in the spleen was performed according to the gating strategy depicted in Figure 6A. There was a significant increase in the frequency of Tregs in the spleen in the CS/Tol group compared to that in the CT group (Figures 6D, E; p=0.0465).
3.4 Col V-induced tolerance contributes to a regulatory microenvironment in lung tissue
3.4.1 Cytokine suppression in the lungs of tolerant model mice
The levels of inflammatory cytokines (IFNγ, IL-6, TNF, IL-10, and IL-17A) in the spleen and lung homogenates were determined by flow cytometry (CBA) and corrected for protein dosage. CBA analysis of the cytokine profile in lung homogenates revealed a greater inflammatory profile in the CS group than in the CT group and a significantly lower inflammatory profile in the CS/Tol group than in the CS group.
In the lung, there was a significant reduction in IFNγ (p=0.0073) and IL-6 (p=0.0009) in the CS/Tol group compared to those in the CS group (Figures 7A, B). TNF-α levels were significantly greater in the CS group than in the CT group (Figure 7C; p=0.0169). Additionally, there was a significant reduction in IL-10 (Figure 7D; p=0.0309) and IL-17A (Figure 7E; p=0.002) in the CS/Tol group compared to those in the CS group. The levels of the cytokines IL-6 and IL-17A in the lung were greatly reduced in the CS/Tol group and were significantly lower than those in the CT group (Figures 7B, E; p=0.0287 and p=0.0499, respectively). CBA analysis of spleen homogenates revealed no differences between the experimental groups for any of the measured inflammatory cytokines, as graphically demonstrated in Figure 7F.
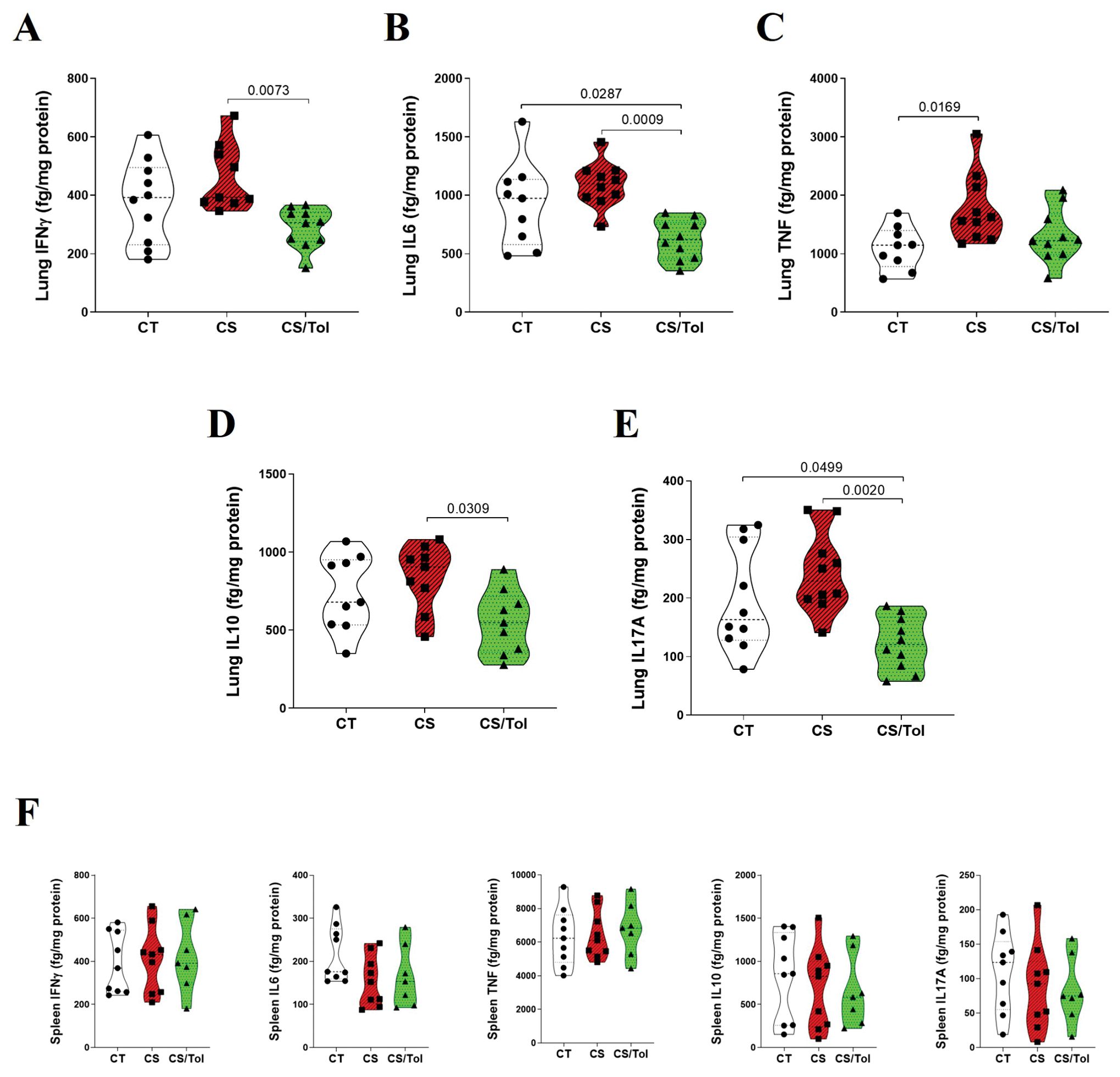
Figure 7. Levels of cytokines in the lung and spleen. (A) There was a significant decrease in lung IFNγ in the CS/Tol group compared to the CS group (p=0.0073). (B) There was a significant decrease in lung IL-6 in the CS/Tol group compared to the CS group (p=0.0009) and the CT group (p=0.0287). (C) There was a significant increase in lung TNF-α in the CS group compared to the CT group (p=0.0169). (D) There was a significant decrease in lung IL-10 in the CS/Tol group compared to that in the CS group (p=0.0309). (E) There was a significant decrease in lung IL-17A in the CS/Tol group compared to that in the CS group (p=0.002) and CT group (p=0.0499) (one-way ANOVA and Holm-Sidak’s multiple comparisons test, n=9-10 for all experimental groups). (F) There were no differences between groups for any of the inflammatory cytokines (one-way ANOVA and Holm-Sidak’s multiple comparisons test, n=9 for CT and CS, n=7 for the CS/Tol group). The violin plots show the frequency distributions of the analyzed data.
3.4.2 M2-polarized macrophage induced by immune tolerance
Immunofluorescence double staining for Foxp3 and IL-10 showed an increase in macrophages positive for these markers in animals from both the CS and CS/Tol groups. However, the tolerant animal from the CS/Tol group had a considerably higher predominance of IL-10 macrophages than the animal from the CS group, implying that immunological tolerance enhanced the polarization towards the M2 profile (Figure 8).
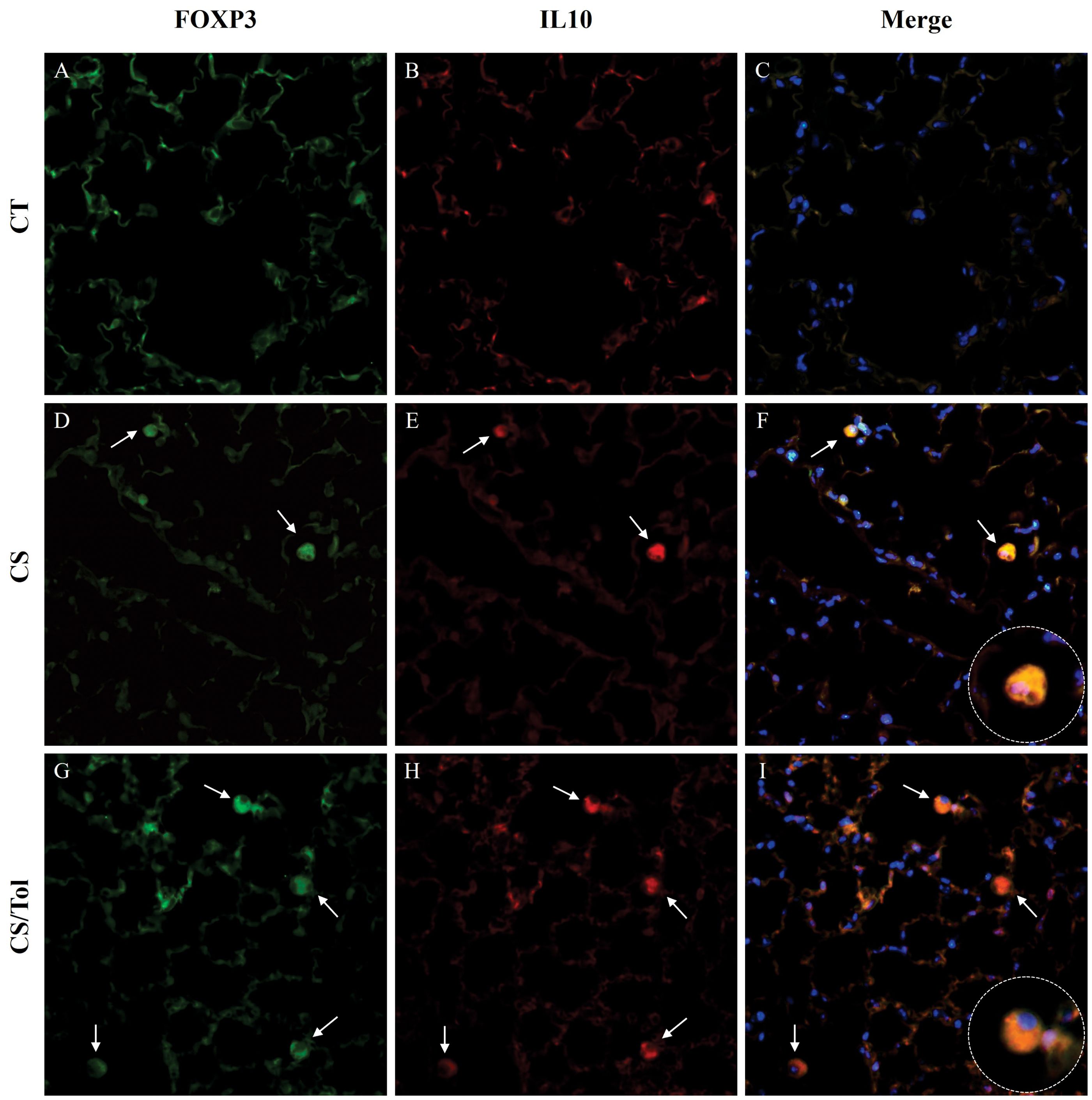
Figure 8. Immunofluorescence images of the lung parenchyma in all experimental groups showing FOXP3 (green) and IL-10 (red) immunostaining and merged images showing colocalization of both markers. The expression of FOXP3 (green) and IL-10 (red) in cells that infiltrated the lung parenchyma of the control, cigarette smoke, and cigarette smoke tolerance groups was assessed by double-label immunofluorescence. Nuclei were stained with DAPI (blue), and white arrows indicate positive cells. 400× magnification.
4 Discussion
We hypothesized that if autoimmunity to Col V contributes to COPD development, then inducing tolerance could mitigate lung damage and remodeling caused by exposure to cigarette smoke. Indeed, we prevented lung ECM changes in a short-term COPD experimental model by inducing nasal tolerance to Col V. We found that mice that received tolerance treatment exhibited notably less structural damage to the alveolar parenchyma and peribronchovascular axis following a month of cigarette smoke exposure. Col V-induced tolerance led to Treg cell accumulation in the alveolar parenchyma. This resulted in suppressive microenvironments in the lungs that hindered the proinflammatory activity of innate and adaptive immune cells in this tissue, preventing emphysema in the tolerant mice.
Regarding structural changes, tolerant mice in the CS/Tol group had Lm values comparable to those of control animals and significantly lower than those of untreated animals in the CS group. Early structural changes induced by cigarette smoke in mice have been previously demonstrated, (43, 44) including an increase in Lm in a 4-week COPD cigarette smoke induction model. (45) The increase in Lm observed in the CS group was accompanied by peribronchovascular edema. Studies conducted in animal models have shown that brief exposure to cigarette smoke can predispose the lungs to inflammation and edema. (46–49) According to previous studies, exposure to cigarette smoke leads to pulmonary endothelial barrier dysfunction and increased permeability in the epithelial layer of the alveolar-capillary membrane, which are likely the causes of the peribronchovascular edema observed. (50, 51) Edema was significantly lower in animals from the CS/Tol group than in those from the CS group, revealing a reduction in this sign of tissue inflammation promoted by Col V-induced tolerance.
An investigation of the lung ECM composition revealed that Col V-induced tolerance mitigated the damage caused by cigarette smoke to collagen fibers, leading to a tendency for an increase in the proportion of Col I in tolerant animals compared to that in untreated animals. However, tolerance treatment did not prevent damage to elastic fibers, although the increase in elastic fiber staining in the alveolar parenchyma was significant only in the untreated group compared to the control group. A reduction in collagen fibers occurs in the large and small airways of patients with mild and moderate COPD and the small airways of unobstructed smokers, (52) and appears to be caused directly by cigarette smoke. (53) Additionally, many studies have shown that elastolysis significantly contributes to the onset of pulmonary emphysema, (54, 55) and an increase in the fraction area of elastic fibers is found in all lung compartments of smokers. (52) Unlike what we observed in the CS group, the pulmonary ECM composition in the CS/Tol group did not change significantly, and the alveolar parenchyma structure remained mostly preserved. Tolerance appears to have had a regulatory influence on ECM turnover (56).
Despite the structural changes observed in the pulmonary ECM of the CS group, we did not observe differences in the respiratory mechanics of these animals, as observed in a previous study. (44) The CS/Tol group also did not show any changes in respiratory mechanics. This is not surprising since functional parameters of respiratory mechanics do not always reflect lung histological changes in animal models, and morphometric parameters are often considered more reliable in detecting the presence of emphysema (57).
An increase in the total number of alveolar inflammatory cells, mostly macrophages, was detected in the BALF from the airways of all animals exposed to cigarette smoke. Tolerance treatment had no impact in this regard, perhaps due to the emphysema induction model we applied, which is marked by repeated aggression caused by cigarette smoke. In experimental models of emphysema, inflammatory infiltration is a frequent finding. (35, 58) Macrophages are the main cell group present in the BALF of mice exposed to cigarette smoke and are the first cell population to increase in number one day after the beginning of exposure. (59) Additionally, in humans, the increase in macrophages in the sputum and lungs of patients with COPD is widely recognized (60).
Moreover, despite not preventing the influx of inflammatory cells, Col V-induced tolerance influenced the progression of the inflammatory process caused by cigarette smoke in the alveolar parenchyma. Immunohistochemical evaluation revealed a trend toward reduced lung macrophage activation (galectin-3+ macrophages) in tolerant animals compared to in untreated animals. Galectin-3 is highly expressed in immune cells of all human tissues, (61) is considerably increased in differentiated macrophages (62) and is used mainly as a marker of macrophage activation. (63) Furthermore, intracellular galectin-3 may contribute to the persistence of inflammation by acting as an antiapoptotic factor and promoting the survival of inflammatory cells.Clique ou toque aqui para inserir o texto (64).
Along with interfering with macrophage activation, Col V-induced tolerance suppressed the release of proinflammatory cytokines in the lungs compared to that in untreated mice. In this sense, the tolerant mice did not show the significant increase in TNF-α secretion observed in the CS group. The role of TNF-α in the pathophysiology of COPD is well known. (65) In response to cigarette smoke and other irritants, macrophages release several inflammatory mediators, including TNF-α. (66) Mice without TNF-α receptors do not develop inflammatory infiltrates or ECM breakdown after acute exposure to cigarette smoke, which further reinforces the importance of this factor in the development of emphysema. (67) The observed reductions in macrophage activation and TNF-α release likely contributed to tolerance-mediated preservation of the lung parenchyma.
Macrophages are recognized for their importance in keeping airways free of inhaled particles/pathogens and acting as initiators of the innate immune response. (60) Depending on the stimuli present in the lung microenvironment, macrophages can be polarized into M1 or M2 phenotypes to perform different functions. (68) Exposure to IFN-γ induces the proliferation of M1 macrophages, with roles equivalent to those of T helper (Th1) cells. (69) However, Th2 cytokines such as IL4 promote polarization toward the M2 phenotype. (70) Several prior investigations, both in animal models and humans, have indicated that cigarette smoke exposure causes the differentiation of both M1 and M2 macrophages. (71, 72) Our qualitative evaluation demonstrated that macrophages in both groups of mice exposed to cigarette smoke expressed IL-10 and IL-17. However, the prevalence of IL-10 macrophages was greater in tolerant animals, which suggests that polarization toward the M2 profile is promoted by Col V-induced tolerance, possibly due to the immunosuppressive milieu resulting from immunological tolerance. To corroborate this theory, further investigations, including quantitative assessments of M1 and M2 macrophages in this experimental model, are needed.
Furthermore, Col V-induced tolerance did not affect IL-17 expression in the lung parenchyma; however, the Th17 response in the CS/Tol group was significantly reduced, as evidenced by a marked decrease in IL-17 release in the lung tissue of tolerant animals. Most evidence points to IL-17 as fundamental for mediating inflammation and tissue immunity, acting at the interface between the innate and adaptive systems. (73) Indeed, the activation of innate cellular sources of IL-17A is essential for regulating macrophage accumulation during lung inflammation in mice exposed to cigarette smoke. (74) In contrast, anti-IL-17 antibodies have been shown to reduce inflammation and airway remodeling in a COPD animal model. (75) Furthermore, in our study, tolerance significantly suppressed the release of other proinflammatory cytokines, including IL-6 and IFNγ. In prior work, our group reported that Col V-induced tolerance effectively decreased the inflammatory cellular response in a murine model of bronchiolitis obliterans. (56) Other researchers have found a decrease in systemic levels of proinflammatory cytokines following Col V-induced tolerance via mucosal routes, (27, 33) corroborating our findings.
Along with the suppression of inflammatory responses discussed previously, we observed that regulatory responses were favored both in the lung tissue and systemically in tolerant mice. Col V-induced tolerance promoted a significant increase in the number of regulatory FOXP3+ cells and an increase in the number of IL10+ cells in the lung parenchyma. To a lesser extent, FOXP3+ and IL10+ cells were also elevated following subacute exposure to cigarette smoke. Previous studies have demonstrated an increase in Treg cells (CD4+CD25+Foxp3+) in the lungs of mice after four weeks of CS exposure. (76, 77) Additionally, subacute exposure to cigarette smoke can promote an increase in serum IL-10 in mice. (76) These findings suggest that in the early stages of COPD, the immune system attempts to maintain tissue homeostasis by differentiating Treg cells and producing IL-10, thus balancing the inflammatory response. Our findings indicate that tolerance treatment played a substantial role in enhancing this mechanism since in the lung parenchyma, the increase in the IL-10+ cell profile was significant only in the tolerant group, and the FOXP-3+ cell profile was significantly greater in tolerant animals than in untreated animals. The prevalence of regulatory cells in tolerant animals appears to be critical for protecting the lung parenchyma from injury resulting from cigarette smoke exposure. Moreover, the induction of Treg cell differentiation promoted by Col V-induced tolerance was evidenced by a significant increase in Treg lymphocytes (CD4+CD25+FOXP3+) in the spleens of tolerant animals. FOXP3+ regulatory T cells (Tregs) are essential for preserving immunological homeostasis and maintaining self-tolerance (78).
It has been demonstrated that local microenvironments play an essential role in maintaining immune tolerance through tolerogenic cytokines, such as IL-10. (79) However, contrary to our assumptions, the dose of IL-10 in the lung homogenate did not increase the secretion of this cytokine in the lungs of tolerant animals. This finding suggested that immunoregulation was restricted to the microenvironment of the lung parenchyma. Furthermore, peripheral tolerance-induced Tregs may affect the suppression of effector T cells through cell-to-cell contact, (80) not only through cytokine-dependent pathways. In addition to the local effects, immunological tolerance results in a systemic response with peripheral Treg cell induction.
In our experimental model, we considered that the pulmonary inflammation and extracellular degradation triggered by cigarette smoke could expose the immunogenic Col V. By the other hand, the Col V-induced tolerance could protect against ECM lung damage. In this context, would be interesting to considered other extracellular proteins non-collagen as control of the collagen V-intranasal instillation, such as elastin, one of the main proteins in the lung parenchyma degraded as a result of chronic exposure to cigarette smoke. Furthermore, as induced collagen V tolerance-control we could consider the ovalbumin, which induce a murine model of allergic airway disease (81). In addition, we could consider to use as induced tolerance-control the well-known autoimmunity-related protein such as myelin oligodendrocyte glycoprotein involved in autoimmunity in the experimental allergic encephalomyelitis (82). We consider these points a limitation of our study to be considered to future evaluations.
In summary, we found that autoimmunity against Col V may contribute to COPD pathophysiology, as inducing tolerance prevents emphysema progression. As evidence suggests, Col V-induced tolerance promoted Treg cell differentiation systemically and appeared to promote an immunosuppressive microenvironment in lung tissue. The stimulation of Treg cell clonal expansion overcame the effects of Th1 and Th17 cells. Concomitant with these events, the immunosuppressive environment seems to favor M2 macrophage polarization. This contributed to suppressing inflammation in lung tissue, thus preventing injury to the pulmonary ECM caused by exposure to cigarette smoke and, as a result, preventing emphysema development. Our findings indicate that macrophages are critical for this process, but further research is needed to confirm whether Col V-induced tolerance affects macrophage polarization. Although this study has limitations, such as the short duration of cigarette smoke exposure, the results are promising, and further research is needed to confirm our findings and explore the mechanisms underlying the effects of Col V tolerance on COPD lung remodeling. Additionally, further research is needed to explore the applicability of immune tolerance to Col V as a therapeutic approach for COPD.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by CEUA - Ethics Committee on the Use of Animals from the Faculty of Medicine of the University of São Paulo under protocol number 1200/2018. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
FR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition, Resources. AV: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LO: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Writing – review & editing. FA: Formal Analysis, Investigation, Methodology, Software, Supervision, Writing – review & editing. LS: Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Writing – review & editing. ZQ: Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Writing – review & editing. TL: Data curation, Formal Analysis, Investigation, Software, Writing – review & editing. VC: Methodology, Software, Validation, Writing – review & editing. CB: Data curation, Formal Analysis, Investigation, Software, Writing – review & editing. SC: Investigation, Resources, Supervision, Validation, Writing – review & editing. SF: Investigation, Methodology, Resources, Validation, Writing – review & editing. MS: Conceptualization, Investigation, Resources, Supervision, Validation, Writing – review & editing. VC: Conceptualization, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. FL: Conceptualization, Investigation, Methodology, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. WT: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the São Paulo Research Foundation (FAPESP), protocol number 2021/13220-5 and by the Coordination for the Improvement of Higher Education Personnel, protocol number 88882.376547/2019-01. National Council for Scientific and Technological Development (CNPq), protocol number 302957/2021-9.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BALF, bronchoalveolar lavage fluid; Col V, type V collagen; COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; ECM, extracellular matrix; Foxp3, Forkhead box protein 3; Treg, Regulatory T cell.
References
1. Varmaghani M, Dehghani M, Heidari E, Sharifi F, Saeedi Moghaddam S, Farzadfar F. Global prevalence of chronic obstructive pulmonary disease: systematic review and meta-analysis. Eastern Mediterr Health J. (2019) 25:47–57. doi: 10.26719/emhj.18.014
2. Karakioulaki M, Papakonstantinou E, Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev. (2020) 29:190124. doi: 10.1183/16000617.0124-2019
3. Venkatesan P. GOLD COPD report: 2024 update. Lancet Respir Med. (2024) 12:15–6. doi: 10.1016/S2213-2600(23)00461-7
4. Ito JT, Lourenço JD, Righetti RF, Tibério IFLC, Prado CM, Lopes FDTQS. Extracellular matrix component remodeling in respiratory diseases: what has been found in clinical and experimental studies? Cells. (2019) 8:342. doi: 10.3390/cells8040342
5. Hoogendoorn M, Hoogenveen RT, Rutten-van Molken MP, Vestbo J, Feenstra TL. Case fatality of COPD exacerbations: a meta-analysis and statistical modelling approach. Eur Respir J. (2011) 37:508–15. doi: 10.1183/09031936.00043710
6. Janjua S, Fortescue R, Poole P. Phosphodiesterase-4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database Systematic Rev. (2020) 5(5):CD002309. doi: 10.1002/14651858.CD002309.pub6
7. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. (2011) 378:1015–26. doi: 10.1016/S0140-6736(11)60988-4
8. Kheradmand F, Shan M, Xu C, Corry DB. Autoimmunity in chronic obstructive pulmonary disease: clinical and experimental evidence. Expert Rev Clin Immunol. (2012) 8:285–92. doi: 10.1586/eci.12.7
9. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress–induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:796–802. doi: 10.1164/rccm.201010-1605OC
10. Polverino F, Cosio BG, Pons J, Laucho-Contreras M, Tejera P, Iglesias A, et al. B cell–activating factor. An orchestrator of lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:695–705. doi: 10.1164/rccm.201501-0107OC
11. Yun JH, Lee S, Srinivasa P, Morrow J, Chase R, Saferali A, et al. An interferon-inducible signature of airway disease from blood gene expression profiling. Eur Respir J. (2022) 59:2100569. doi: 10.1183/13993003.00569-2021
12. Leeming DJ, Sand JM, Nielsen MJ, Genovese F, Martinez FJ, Hogaboam CM, et al. Serological investigation of the collagen degradation profile of patients with chronic obstructive pulmonary disease or idiopathic pulmonary fibrosis. biomark Insights. (2012) 7:BMI.S9415. doi: 10.4137/BMI.S9415
13. Schumann DM, Leeming D, Papakonstantinou E, Blasi F, Kostikas K, Boersma W, et al. Collagen degradation and formation are elevated in exacerbated COPD compared with stable disease. Chest. (2018) 154:798–807. doi: 10.1016/j.chest.2018.06.028
14. Bihlet AR, Karsdal MA, Sand JMB, Leeming DJ, Roberts M, White W, et al. Biomarkers of extracellular matrix turnover are associated with emphysema and eosinophilic-bronchitis in COPD. Respir Res. (2017) 18:22. doi: 10.1186/s12931-017-0509-x
15. Sand JMB, Knox AJ, Lange P, Sun S, Kristensen JH, Leeming DJ, et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir Res. (2015) 16:69. doi: 10.1186/s12931-015-0225-3
16. Sand JMB, Martinez G, Midjord A-K, Karsdal MA, Leeming DJ, Lange P. Characterization of serological neo-epitope biomarkers reflecting collagen remodeling in clinically stable chronic obstructive pulmonary disease. Clin Biochem. (2016) 49:1144–51. doi: 10.1016/j.clinbiochem.2016.09.003
17. Rinaldi M, Lehouck A, Heulens N, Lavend’Homme R, Carlier V, Saint-Remy JM, et al. Antielastin B-cell and T-cell immunity in patients with chronic obstructive pulmonary disease. Thorax. (2012) 67:694–700. doi: 10.1136/thoraxjnl-2011-200690
18. Madri J, Furthmayr H. Collagen polymorphism in the lungAn immunochemical study of pulmonary fibrosis. Hum Pathol. (1980) 11:353–66. doi: 10.1016/S0046-8177(80)80031-1
19. Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci. (1990) 95:649–57. doi: 10.1242/jcs.95.4.649
20. Chanut-Delalande H, Bonod-Bidaud C, Cogne S, Malbouyres M, Ramirez F, Fichard A, et al. Development of a functional skin matrix requires deposition of collagen V heterotrimers. Mol Cell Biol. (2004) 24:6049–57. doi: 10.1128/MCB.24.13.6049-6057.2004
21. Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. (2008) 20:495–501. doi: 10.1016/j.ceb.2008.06.008
22. Lott JM, Sehra S, Mehrotra P, Mickler EA, Fisher A, Zhang W, et al. Type V collagen-induced tolerance prevents airway hyperresponsiveness. Am J os Respir Crit Care Med. (2013) 187:454–7. doi: 10.1164/ajrccm.187.4.454
23. Haque MA, Mizobuchi T, Yasufuku K, Fujisawa T, Brutkiewicz RR, Zheng Y, et al. Evidence for immune responses to a self-antigen in lung transplantation: role of type V collagen-specific T cells in the pathogenesis of lung allograft rejection. J Immunol. (2002) 169:1542–9. doi: 10.4049/jimmunol.169.3.1542
24. Yasufuku K, Heidler KM, O’donnell PW, Smith GN, Cummings OW, Foresman BH, et al. Oral tolerance induction by type V collagen downregulates lung allograft rejection. Am J Respir Cell Mol Biol. (2001) 25:26–34. doi: 10.1165/ajrcmb.25.1.4431
25. Dart ML, Jankowska-Gan E, Huang G, Roenneburg DA, Keller MR, Torrealba JR, et al. Interleukin-17-dependent autoimmunity to collagen type v in atherosclerosis. Circ Res. (2010) 107:1106–16. doi: 10.1161/CIRCRESAHA.110.221069
26. Sumpter TL, Wilkes DS. Role of autoimmunity in organ allograft rejection: a focus on immunity to type V collagen in the pathogenesis of lung transplant rejection. Am J Physiology-Lung Cell Mol Physiol. (2004) 286:L1129–39. doi: 10.1152/ajplung.00330.2003
27. Vittal R, Mickler EA, Fisher AJ, Zhang C, Rothhaar K, Gu H, et al. Type V collagen induced tolerance suppresses collagen deposition, TGF-β and associated transcripts in pulmonary fibrosis. PloS One. (2013) 8:e76451. doi: 10.1371/journal.pone.0076451
28. Bezerra MC, Teodoro WR, de Oliveira CC, Velosa APP, Ogido LTI, Gauditano G, et al. Scleroderma-like remodeling induced by type V collagen. Arch Dermatol Res. (2006) 298:51–7. doi: 10.1007/s00403-006-0645-5
29. Velosa APP, Brito L, de Jesus Queiroz ZA, Carrasco S, Tomaz de Miranda J, Farhat C, et al. Identification of autoimmunity to peptides of collagen V α1 chain as newly biomarkers of early stage of systemic sclerosis. Front Immunol. (2021) 11. doi: 10.3389/fimmu.2020.604602
30. Mares DC, Heidler KM, Smith GN, Cummings OW, Harris ER, Foresman B, et al. Type V collagen modulates alloantigen-induced pathology and immunology in the lung. Am J Respir Cell Mol Biol. (2000) 23(1):62–70. doi: 10.1165/ajrcmb.23.1.3924
31. Yamada Y, Sekine Y, Yoshida S, Yasufuku K, Petrache I, Benson HL, et al. Type V collagen-induced oral tolerance plus low-dose cyclosporine prevents rejection of MHC class I and II incompatible lung allografts. J Immunol. (2009) 183:237–45. doi: 10.4049/jimmunol.0804028
32. Braun RK, Molitor-Dart M, Wigfield C, Xiang Z, Fain SB, Jankowska-Gan E, et al. Transfer of tolerance to collagen type V suppresses T-helper-cell-17 lymphocyte-mediated acute lung transplant rejection. Transplantation. (2009) 88:1341–8. doi: 10.1097/TP.0b013e3181bcde7b
33. Park AC, Huang G, Jankowska-Gan E, Massoudi D, Kernien JF, Vignali DA, et al. Mucosal administration of collagen V ameliorates the atherosclerotic plaque burden by inducing interleukin 35-dependent tolerance. J Biol Chem. (2016) 291:3359–70. doi: 10.1074/jbc.M115.681882
34. Wilkes DS, Chew T, Flaherty KR, Frye S, Gibson KF, Kaminski N, et al. Oral immunotherapy with type V collagen in idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1393–402. doi: 10.1183/09031936.00105314
35. Toledo AC, Magalhaes RM, Hizume DC, Vieira RP, Biselli PJC, Moriya HT, et al. Aerobic exercise attenuates pulmonary injury induced by exposure to cigarette smoke. Eur Respir J. (2012) 39:254–64. doi: 10.1183/09031936.00003411
36. Buccheri R, Duarte-Neto AN, Silva FLB, Haddad GC, Silva da LBR, Azevedo Netto R, Ledesma FL, et al. Chronic exposure to cigarette smoke transiently worsens the disease course in a mouse model of pulmonary paracoccidioidomycosis. Rev Inst Med Trop Sao Paulo. (2022) 64:e71. doi: 10.1590/s1678-9946202264071
37. Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ, Dar~czy B, et al. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. (1992) 72:168–78. doi: 10.1152/jappl.1992.72.1.168
38. Ramos DS, Olivo CR, Quirino Santos Lopes FDT, Toledo AC, Martins MA, Lazo Osório RA, et al. Low-intensity swimming training partially inhibits lipopolysaccharide-induced acute lung injury. Med Sci Sports Exerc. (2010) 42:113–9. doi: 10.1249/MSS.0b013e3181ad1c72
39. Takubo Y, Guerassimov A, Ghezzo H, Triantafillopoulos A, Bates JHT, Hoidal JR, et al. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med. (2002) 166:1596–603. doi: 10.1164/rccm.2202001
40. Weibel ER, Kistler GS, Scherle WF. Practical stereological methods for morphometric cytology. J Cell Biol. (1966) 30:23–38. doi: 10.1083/jcb.30.1.23
41. Gundersen HJG, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, et al. The new stereological tools: Disector, fractionator, mucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. (1988) 96:857–81. doi: 10.1111/j.1699-0463.1988.tb00954.x
42. Fullmer HM, Smeetz JH, Narkates AJ. Oxytalan connective tissue fibers A review. J Oral Pathol. (1974) 3:291–316. doi: 10.1111/j.1600-0714.1974.tb01724.x
43. He S, Li L, Sun S, Zeng Z, Lu J, Xie L. A novel murine chronic obstructive pulmonary disease model and the pathogenic role of microRNA-21. Front Physiol. (2018) 9. doi: 10.3389/fphys.2018.00503
44. Junqueira JJM, Lourenço JD, da Silva KR, Cervilha de DA B, da Silveira LKR, Correia AT, et al. Decreased bone type I collagen in the early stages of chronic obstructive pulmonary disease (COPD). COPD: J Chronic Obstructive Pulmonary Dis. (2020) 17:575–86. doi: 10.1080/15412555.2020.1808605
45. Ito JT, Cervilha de DA B, Lourenço JD, Gonçalves NG, Volpini RA, Caldini EG, et al. Th17/Treg imbalance in COPD progression: A temporal analysis using a CS-induced model. PloS One. (2019) 14:e0209351. doi: 10.1371/journal.pone.0209351
46. Sakhatskyy P, Wang Z, Borgas D, Lomas-Neira J, Chen Y, Ayala A, et al. Double-hit mouse model of cigarette smoke priming for acute lung injury. Am J Physiology-Lung Cell Mol Physiol. (2017) 312:L56–67. doi: 10.1152/ajplung.00436.2016
47. Witten ML, Lemen RJ, Quan SF, Sobonya RE, Magarelli JL, Bruck DC. Acute cigarette smoke exposure causes lung injury in rabbits treated with ibuprofen. Exp Lung Res. (1987) 13:113–26. doi: 10.3109/01902148709064313
48. Li XY, Rahman I, Donaldson K, MacNee W. Mechanisms of cigarette smoke induced increased airspace permeability. Thorax. (1996) 51:465–71. doi: 10.1136/thx.51.5.465
49. Bhavsar TM, Cerreta JM, Cantor JO. Short-term cigarette smoke exposure predisposes the lung to secondary injury. Lung. (2007) 185:227–33. doi: 10.1007/s00408-007-9013-2
50. Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, et al. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiology-Lung Cell Mol Physiol. (2011) 301:L847–57. doi: 10.1152/ajplung.00178.2011
51. Rounds S, Lu Q. Cigarette smoke alters lung vascular permeability and endothelial barrier function (2017 Grover Conference Series). Pulm Circ. (2018) 8:1–10. doi: 10.1177/2045894018794000
52. Annoni R, Lanças T, Yukimatsu Tanigawa R, de Medeiros Matsushita M, de Morais Fernezlian S, Bruno A, et al. Extracellular matrix composition in COPD. Eur Respir J. (2012) 40:1362–73. doi: 10.1183/09031936.00192611
53. Gugatschka M, Darnhofer B, Grossmann T, Schittmayer M, Hortobagyi D, Kirsch A, et al. Proteomic analysis of vocal fold fibroblasts exposed to cigarette smoke extract: exploring the pathophysiology of reinke’s edema*. Mol Cell Proteomics. (2019) 18:1511–25. doi: 10.1074/mcp.RA119.001272
54. Cardoso WV, Sekhon HS, Hyde DM, Thurlbeck WM. Collagen and elastin in human pulmonary emphysema. Am Rev Respir Dis. (1993) 147:975–81. doi: 10.1164/ajrccm/147.4.975
55. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. (2003) 22:672–88. doi: 10.1183/09031936.03.00040703
56. Garippo A, Parra E, Teodoro W, Rivero D, Souza F, Yoshinari N, et al. Nasal tolerance with collagen V protein reverts bronchovascular axis remodeling in experimental bronchiolitis obliterans. Clinics. (2007) 62:499–506. doi: 10.1590/S1807-59322007000400018
57. Anciães AM, Olivo CR, Prado CM, Kagohara KH, Pinto da T S, Moriya HT, et al. Respiratory mechanics do not always mirror pulmonary histological changes in emphysema. Clinics (Sao Paulo). (2011) 66:1797–803. doi: 10.1590/s1807-59322011001000020
58. Awji EG, Seagrave JC, Tesfaigzi Y. Correlation of cigarette smoke-induced pulmonary inflammation and emphysema in C3H and C57Bl/6 mice. Toxicological Sci. (2015) 147:75–83. doi: 10.1093/toxsci/kfv108
59. Campos KKD, Manso RG, Gonçalves EG, Silva ME, de Lima WG, Menezes CAS, et al. Temporal analysis of oxidative effects on the pulmonary inflammatory response in mice exposed to cigarette smoke. Cell Immunol. (2013) 284:29–36. doi: 10.1016/j.cellimm.2013.07.002
60. Finicelli M, Digilio FA, Galderisi U, Peluso G. The emerging role of macrophages in chronic obstructive pulmonary disease: the potential impact of oxidative stress and extracellular vesicle on macrophage polarization and function. Antioxidants. (2022) 11:464. doi: 10.3390/antiox11030464
61. Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, et al. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med. (2018) 41(2):599–614. doi: 10.3892/ijmm.2017.3311
62. Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR. Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol. (1995) 147:1016–28.
63. Elliott MJ, Strasser A, Metcalf D. Selective up-regulation of macrophage function in granulocyte-macrophage colony-stimulating factor transgenic mice. J Immunol. (1991) 147:2957–63. doi: 10.4049/jimmunol.147.9.2957
64. Dumic J, Dabelic S, Flögel M. Galectin-3: An open-ended story. Biochim Biophys Acta (BBA) - Gen Subj. (2006) 1760:616–35. doi: 10.1016/j.bbagen.2005.12.020
65. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. (2014) 35:71–86. doi: 10.1016/j.ccm.2013.10.004
66. Vlahos R, Bozinovski S, Gualano RC, Ernst M, Anderson GP. Modelling COPD in mice. Pulm Pharmacol Ther. (2006) 19:12–7. doi: 10.1016/j.pupt.2005.02.006
67. Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-α Drives 70% of cigarette smoke–induced emphysema in the mouse. Am J Respir Crit Care Med. (2004) 170:492–8. doi: 10.1164/rccm.200404-511OC
68. Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey B-G, O’Connor TP, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. (2009) 183:2867–83. doi: 10.4049/jimmunol.0900473
69. Goerdt S, Orfanos CE. Other functions, other genes. Immunity. (1999) 10:137–42. doi: 10.1016/S1074-7613(00)80014-X
70. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflammation. (2015) 2015:1–16. doi: 10.1155/2015/816460
71. Kohler JB, Cervilha de DA B, Riani Moreira A, Santana FR, Farias TM, Alonso Vale MIC, et al. Microenvironmental stimuli induce different macrophage polarizations in experimental models of emphysema. Biol Open. (2019) 8(4):bio040808. doi: 10.1242/bio.040808
72. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
73. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. (2008) 28:454–67. doi: 10.1016/j.immuni.2008.03.004
74. Bozinovski S, Seow HJ, Chan SPJ, Anthony D, McQualter J, Hansen M, et al. Innate cellular sources of interleukin-17A regulate macrophage accumulation in cigarette- smoke-induced lung inflammation in mice. Clin Sci. (2015) 129:785–96. doi: 10.1042/CS20140703
75. Shen N, Wang J, Zhao M, Pei F, He B. Anti-interleukin-17 antibodies attenuate airway inflammation in tobacco-smoke-exposed mice. Inhal Toxicol. (2011) 23:212–8. doi: 10.3109/08958378.2011.559603
76. Wang H, Peng W, Weng Y, Ying H, Li H, Xia D, et al. Imbalance of Th17/Treg cells in mice with chronic cigarette smoke exposure. Int Immunopharmacol. (2012) 14:504–12. doi: 10.1016/j.intimp.2012.09.011
77. Demoor T, Bracke KR, Joos GF, Brusselle GG. Increased T-regulatory cells in lungs and draining lymph nodes in a murine model of COPD. Eur Respir J. (2010) 35:688–9. doi: 10.1183/09031936.00158509
78. Regateiro FS, Chen Y, Kendal AR, Hilbrands R, Adams E, Cobbold SP, et al. Foxp3 expression is required for the induction of therapeutic tissue tolerance. J Immunol. (2012) 189:3947–56. doi: 10.4049/jimmunol.1200449
79. Weiner HL. The mucosal milieu creates tolerogenic dendritic cells and TR1 and TH3 regulatory cells. Nat Immunol. (2001) 2:671–2. doi: 10.1038/90604
80. Bertolini TB, Biswas M, Terhorst C, Daniell H, Herzog RW, Piñeros AR. Role of orally induced regulatory T cells in immunotherapy and tolerance. Cell Immunol. (2021) 359:104251. doi: 10.1016/j.cellimm.2020.104251
81. Natarajan P, Singh A, McNamara JT, Secor ER Jr, Guernsey LA, Thrall RS, et al. Regulatory B cells from hilar lymph nodes of tolerant mice in a murine model of allergic airway disease are CD5+, express TGF-β, and co-localize with CD4+Foxp3+ T cells. Mucosal Immunol. (2012) 5:691–701. doi: 10.1038/mi.2012.42
Keywords: chronic obstructive pulmonary disease, cigarette smoking, pulmonary emphysema, animal models, autoimmunity, collagen type V, regulatory T cell, immune tolerance
Citation: Robertoni FSZ, Velosa APP, Oliveira LM, Almeida FM, Silveira LKR, Queiroz ZAJ, Lobo TM, Contini VE, Baldavira CM, Carrasco S, Fernezlian SM, Sato MN, Capelozzi VL, Lopes FDTQS and Teodoro WPR (2024) Type V collagen-induced nasal tolerance prevents lung damage in an experimental model: new evidence of autoimmunity to collagen V in COPD. Front. Immunol. 15:1444622. doi: 10.3389/fimmu.2024.1444622
Received: 05 June 2024; Accepted: 16 August 2024;
Published: 05 September 2024.
Edited by:
Andrew W. Taylor, Chobanian & Avedisian School of Medicine, United StatesReviewed by:
Junsuk Josh Ko, Duke-NUS Medical School, SingaporeWayne Robert Thomas, University of Western Australia, Australia
Copyright © 2024 Robertoni, Velosa, Oliveira, Almeida, Silveira, Queiroz, Lobo, Contini, Baldavira, Carrasco, Fernezlian, Sato, Capelozzi, Lopes and Teodoro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walcy Paganelli Rosolia Teodoro, d2FsY3kudGVvZG9yb0BmbS51c3AuYnI=