 Alexander J. Dwyer1†
Alexander J. Dwyer1† Zachary R. Shaheen
Zachary R. Shaheen Brian T. Fife
Brian T. Fife- 1Center for Immunology, Department of Medicine, Division of Rheumatic and Autoimmune Diseases, University of Minnesota Medical School, Minneapolis, MN, United States
- 2Center for Immunology, Department of Pediatrics, Pediatric Rheumatology, Allergy, & Immunology, University of Minnesota Medical School, Minneapolis, MN, United States
Autoimmune diabetes is a disease characterized by the selective destruction of insulin-secreting β-cells of the endocrine pancreas by islet-reactive T cells. Autoimmune disease requires a complex interplay between host genetic factors and environmental triggers that promote the activation of such antigen-specific T lymphocyte responses. Given the critical involvement of self-reactive T lymphocyte in diabetes pathogenesis, understanding how these T lymphocyte populations contribute to disease is essential to develop targeted therapeutics. To this end, several key antigenic T lymphocyte epitopes have been identified and studied to understand their contributions to disease with the aim of developing effective treatment approaches for translation to the clinical setting. In this review, we discuss the role of pathogenic islet-specific T lymphocyte responses in autoimmune diabetes, the mechanisms and cell types governing autoantigen presentation, and therapeutic strategies targeting such T lymphocyte responses for the amelioration of disease.
1 Introduction
Autoimmune diabetes is characterized by the destruction of the insulin-producing β-cells within the pancreatic islets of Langerhans. Although the events preceding the onset of disease are not entirely understood, many putative precipitating risk factors have been investigated. A complex interplay among risk factors including host genetic susceptibility, environmental exposures (particularly viral infection), gastrointestinal microbiome composition, and dietary contributions lead to tissue destruction in a manner principally driven by T cells (summarized in Figure 1). This review will focus on the major β-cell antigens and associated autoreactive T lymphocyte populations that are responsible for mediating diabetes pathogenesis. This review will focus primarily on mechanisms of diabetes pathogenesis elucidated in the non-obese diabetic (NOD) mouse, henceforth referred to as autoimmune diabetes, and will correlate these findings to human diabetes when possible, denoted as type 1 diabetes (T1D).
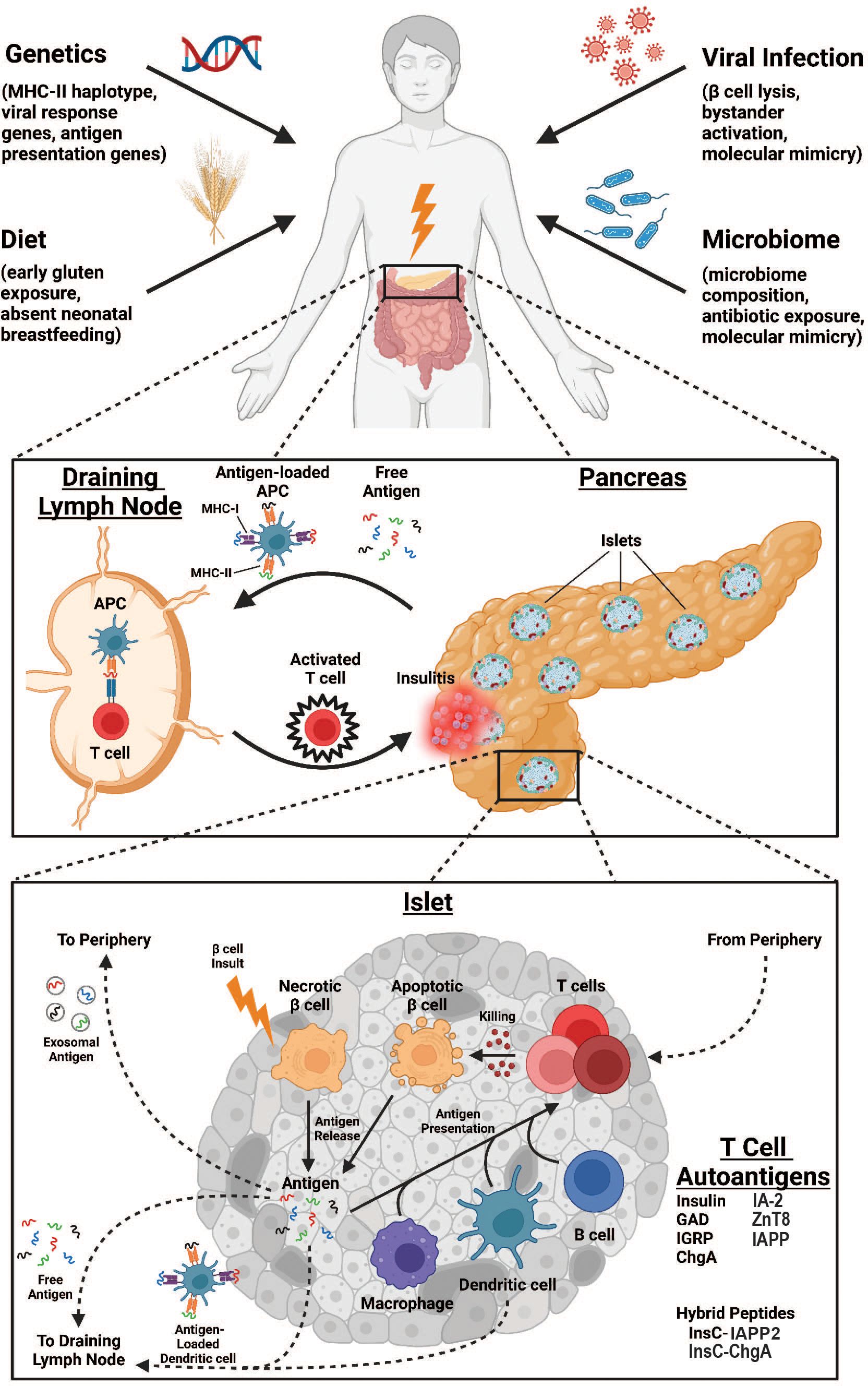
Figure 1. Overview of immunologic processes leading to enhanced antigen-specific T cell responses in autoimmune diabetes. Environmental factors such as genetic predisposition, viral infections, dietary exposures, and microbiome composition are all risk factors for AD development (top). At the organ level, destruction of pancreatic islets releases self-antigen that drains to pancreatic lymph nodes either as free antigen or loaded to antigen presenting cells (APCs) where it is presented to antigen-specific T cells that subsequently traffic to pancreatic islets to propagate further damage (middle). At the islet level, an unknown insult induces βcell necrosis and antigen release, initiating several pathways to T cell antigen presentation. Antigen may be drained to local lymph nodes either as free antigen or loaded to dendritic cells as above. Alternatively, antigen may enter the vasculature as exosomes for antigen presentation peripherally. Finally, T cell antigen presentation can occur within the islet, mediated by macrophages, dendritic cells, or B cells. Ultimately, T cells enter the islet proper from the periphery to promote killing of βcells through apoptotic mechanisms, further increasing the pool of available self-antigen.
1.1 Genetic risk
Extensive epidemiologic and genetic studies have demonstrated that certain genetic variations afford significant risk to the development of T1D. The overall risk for developing T1D is estimated around 0.4%, and this risk increases to approximately 6% in children of diabetic parents, demonstrating a heritable genetic component to T1D risk (1). Per genome-wide association studies (GWAS), over 50 distinct genetic loci have been identified in humans that are associated with T1D (2, 3). Generally, these susceptibility genes may be approximately grouped by the gene product’s characterized role in coordinating anti-viral responses, autoantigen formation or presentation, or T cell signaling responses, and have been discussed in detail elsewhere (1, 4).
Of these gene products, the human leukocyte antigen (HLA) region on chromosome 6p21 represents the strongest genetic association for inherited risk of T1D development, as elucidated by both GWAS and linkage studies (1–3). HLA regions correspond to major histocompatibility complex (MHC) molecules, which are cell surface receptors that form complexes with principally either endogenous peptides (class I) or extracellular, phagocytosed peptides (class II) that are then displayed to T cells for recognition. T cell receptor (TCR) binding to these MHC:peptide complexes is essential for induction of central tolerance (via deletion of auto-reactive T cells in the thymus), continuous maintenance of peripheral tolerance (via suppression of autoreactive T cells that escaped thymic deletion), and for appropriate activation when foreign peptides are detected during infection and cancers [reviewed in (5)]. The highest risk HLA haplotypes are MHC class-II molecules colloquially termed “DR4-DQ8,” specifically DR4 (DRB1 allele)-DQA1*03:01-DQB1*03:02 (4). These high-risk haplotypes are thought to confer diabetes risk through different amino acid residues in the MHC peptide binding pocket which alters both the register in which peptides bind to MHC and the binding affinity of the MHC:peptide complex when presented to T cells.
Genetic and environmental factors play a key role in susceptibility to T1D. Importantly, genetic risk alone does not account for T1D. Indeed, in surveillance studies that examined the risk of discordant monozygotic twins to both develop diabetes (e.g. long-term follow-up of a patient whose identical twin was diagnosed with T1D), the risk of identical twins to both develop diabetes is estimated to be 39–65% (6–10). Therefore, there are likely additional environmental factors that confer susceptibility to diabetes development.
1.2 Viral infection
Viral infections are hypothesized to be both environmental triggers and accelerators of T1D pathogenesis [reviewed in (11–13)] and are well described contributors to initial β-cell damage and the initiation of a selective, T cell dependent autoimmune response. In humans, several viruses have been associated with the development of T1D by serological methods, of which coxsackieviruses and other enteroviruses are the most well described (11–14). Notably, enteroviral RNA and capsid proteins have been detected in the pancreatic islets of living patients with new autoimmune diabetes diagnosis (15). Coxsackievirus has also been isolated from the pancreas of a deceased patient following new-onset diabetic ketoacidosis.
Coxsackievirus virus strain (coxsackievirus B4 – CVB4) isolated from new-onset diabetic patients has also been shown to stimulate the rapid development of diabetes in inoculated mice (16), demonstrating a causative effect of acute viral infections with the triggering of β-cell autoimmunity. Since, viral infections in mouse and rat strains have since been extensively used to better elucidate the underlying mechanisms governing β-cell destruction and the onset of autoimmune diabetes, including coxsackievirus infection in the NOD mouse (17), encephalomyocarditis virus (EMCV) in certain susceptible mouse strains (18, 19), and Kilham’s Rat Virus (KRV) in the diabetes-resistant BB-rat (20). There are multiple mechanisms by which viruses may lead to the development and acceleration of pancreatic β-cell autoimmunity, including direct infection of β-cells (leading to cell lysis and antigen release in addition to direct loss of β-cell mass), bystander activation (β-cell destruction via a robust inflammatory cytokine response), and molecular mimicry (viral antigens which share sufficient homology with autoantigens to stimulate an autoimmune response) (11–13, 21, 22). Together, these mechanisms may contribute to diabetes initiation through the induction of an inflammatory environment and enhanced autoantigen presentation.
1.3 Gastrointestinal microbiome and diet
Gastrointestinal homeostasis is critical for proper immune system function, and derangements of the intestinal microbiome and diet may afford increased risk for T1D development (23). Several studies have demonstrated that administration of antibiotics or probiotic bacterial cocktails to diabetes-susceptible mice are capable of altering gastrointestinal microbiome diversity with a concomitant decrease in autoimmune diabetes incidence, suggesting that various bacterial species differentially contribute to influencing self-tolerance (24–27). Supporting this notion, principle component analysis of autoantibody-positive prediabetic patients have fewer butyrate- and lactate-producing bacteria such as bifidobacterium, which are hypothesized to reduce gut permeability and inflammation, and an increased proportion of Bacteroides species (4.3% compared to 2.0% autoantibody negative controls) (28), and the diversity of these gastrointestinal bacteria changes throughout time in seroconverter and diabetic children (28–32). Further, neonates born by Caesarean section delivery have an altered microbiome, and these infants have a 20% increased rate of T1D (33, 34). However, this relationship may not be causal, as diabetes-susceptible mice born by Caesarean section have no change in diabetes incidence, despite having increased Bacteroides and Lachnospiraceae species and fewer T regulatory cells (Tregs) as adults (35). Importantly, microbiome-specific T cell responses are thought to be induced by a combination of mechanisms, including molecular mimicry [e.g. magnesium transporter protein (Mgt)267-275 stimulation of islet-specific CD8+ T cell responses (36)] and diminished Treg quantity (37, 38). Given these observations, we posit that simultaneous stimulation of autoreactive effector T cells via molecular mimicry and decreased Treg-mediated suppression results in enhanced autoimmunity.
Diet is thought to influence both gastrointestinal homeostasis and microbiome composition in the context of T1D development. Neonatal feeding method is one component of diet thought to influence T1D with evidence suggesting that any amount of breastfeeding in the neonatal and infant periods decreases the risk for T1D development (39, 40). Further, a link between dietary gluten consumption and T1D has been suggested given that diabetes-susceptible mice with reduced or absent wheat and barley proteins in their diets develop delayed diabetes onset, and in humans approximately 1.7–16.4% of patients with T1D also have celiac disease, an autoimmune disease that causes immune intolerance to gluten (41, 42). In humans, this association is more complex given that effects on autoantibody levels and disease state depend upon the age at which exposure occurs (43–47).
1.4 The non-obese diabetic mouse
The NOD mouse was derived from Cataract Shionogi (CTS) mice in 1980, and identified incidentally in a female mouse exhibiting polyuria, glucosuria, rapid weight loss, and lymphocytic infiltration of pancreatic islets (48). Approximately 60–80% of female and 20–30% of male mice become spontaneously diabetic within 12–14 weeks of life when maintained in germ-free environments (49–51). Interestingly, this rate of disease progression is much lower when maintained in dirty animal housing facilities (49), and viral infection in the NOD mouse may either confer protection from or accelerate diabetes onset depending on the age of the mouse (17), suggesting that the timing of infection and local cellular environment may dictate whether infection provokes an autoimmune response or whether peripheral tolerance to autoantigens are maintained.
The NOD mouse has remained a dominant animal model for the study of autoimmune diabetes over the last four decades given that it shares a number of similarities with human diabetes [reviewed in (50, 51)]. First, numerous autoantigens have been identified in both humans and the NOD mouse, including insulin, glutamic acid decarboxylase (GAD65), islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), insulinoma antigen-2 (IA-2), phogrin (IA-2β), chromogranin A (ChgA), islet amyloid polypeptide (IAPP), and islet zinc transporter (ZnT8) (52–60). Second, the genetic susceptibility of the NOD mouse mirrors that of humans such that MHC-II molecules (termed I-Ag7 in the NOD mouse; DQ8 in human) afford the highest risk for associated disease (1, 4, 61), which we posit allows for insufficient central tolerance to key pathogenic peptides. Functionally, human DQ8 and mouse I-Ag7 have been shown to share similar peptide binding and registers (62) and replacing transgenic mice with DQ8 maintains islet reactivity and diabetes incidence (63), highlighting the central role of antigen presentation in autoimmune disease risk. There are additionally over 40 other non-MHC genes associated with diabetes risk that are involved in anti-viral responses, auto-antigen formation or presentation, or T cell signaling responses (1, 4). Third, self-antigen-reactive CD4+ T cells are essential for both early and late stages of disease development (50, 51, 64, 65). Additionally, macrophages, dendritic cells, CD8+ T cells, and B cells have all also been identified infiltrating islets in humans and mice and have characterized roles with disease initiation and acceleration (50, 51). In line with these similarities, the study of NOD mice has revealed many mechanistic insights into T1D disease pathology; the focus of this review will be to discuss antigenic targets in the NOD mouse as well as the cellular mechanisms by which presentation of these antigens to T cells result in the loss of tolerance and induction of autoimmune diabetes.
2 Antigenic targets of T cells
2.1 Insulin
Although many β-cell protein self-antigens are known targets in T1D (summarized in Figure 1), several are thought to be more pathogenic than others and will be discussed here. Among these, insulin is perhaps the most well-known. Specifically, the InsB9-23 epitope is likely the critical antigenic portion of insulin given the large number of islet-infiltrating T cell clones that are reactive to InsB9-23 peptide (66–68). Supporting these data, it was subsequently found that NOD.ins1-/-ins2-/- mice expressing a transgene encoding hormonally functional insulin but with a tyrosine-to-alanine point mutation at position 16 of the insulin B-chain (NOD.Y16A mice) prevents I-Ag7 binding and antigen presentation and as a result, these mice do not develop autoimmune diabetes (53, 69, 70). Interestingly, transfer of splenocytes from NOD.Y16A mice to NOD.scid recipients induces disease but at a rate slower than splenocyte transfer from NOD mice, suggesting that InsB9-23-specific T cells may participate in disease initiation but require a latency period of several weeks to recruit participation of other islet antigen-specific responses (53).
Both murine I-Ag7 and human DQ8 MHC-II molecules bind peptide for antigen presentation using a core of nine amino acids, meaning that the fifteen amino acid InsB9-23 peptide could interact with MHC-II in multiple different positions, termed “registers” (71, 72). Of the nine amino acids comprising core binding to I-Ag7 and DQ8, binding of a negatively charged residue at position 9 (P9) is most important for determining stability of the peptide:MHC-II interaction (62). This is due to a polymorphism at position 57 of the β-chain of all HLA-II diabetes susceptibility alleles where a negatively charged aspartic acid residue is replaced by a neutral amino acid, leaving an exposed positively charged surface that alters the P9 pocket binding parameters (72, 73). The P9 pocket is thought to be important in determining the binding affinity of different insulin peptide registers to murine and human MHC-II (74).
Three insulin binding registers have been studied most frequently: InsB12-20 (register 1), InsB13-21 (register 2) and InsB14-22 (register 3). Registers 1 and 2 bind weakly to I-Ag7, with register 2 satisfying the P9 position negative charge condition through its glutamic acid while register 1 has a neutral glycine at this position (74). It has been suggested that I-Ag7 and other β57 polymorphic MHC-II molecules select for TCRs with aspartic acid and glutamic acid residues within the CDR3β region, allowing neutralization of the positively charged MHC-II surface following binding of peptides with neutral amino acids at P9 in a process termed the “P9 switch” (75, 76). This mechanism would explain how register 1 can bind I-Ag7 with low affinity to allow InsB12-20-specific CD4+ T cells to escape negative selection and home to pancreatic islets (76). Using InsB12-20-TCR specific transgenic 8F10 CD4+ T cells, it has been shown that these populations encounter insulin antigen directly in pancreatic islets without a need for initial antigen presentation at 4 weeks in the draining pancreatic lymph nodes (pLN) (77). Importantly, CD4+ T cells specific for register 1 are thought to recognize antigen only when antigen presenting cells (APCs) uptake InsB9-23 peptide and subsequently expand in the periphery (78). In contrast, CD4+ T cells specific for register 2 recognize antigen after APC uptake of full-length insulin and are deleted in the thymus (78). These studies suggest that development of autoreactive T-lymphocytes to insulin occurs in a manner dependent on recognition of InsB9-23 in register 1 but not 2; indeed, register 1-specific pLN-derived CD4+ T cell clones transfer disease to NOD.scid recipients (78, 79).
Insulin register 3 binds poorly to I-Ag7 due to a positively charged arginine residue occupying the P9 position, yet despite this, several insulin-specific CD4+ T cell clones have been found to recognize InsB9-23 bound in register 3 but not registers 1 or 2 (56). Further, the majority of InsB9-23-specific CD4+ T cells in NOD mice target register 3 with an arginine to glutamic acid mutation at P9 of the insulin chain (R22E) which enhances binding stability and segregates into two distinct populations: those with responses inhibited by additionally mutating B:21 glutamic acid to glycine (“P8E”) and those with augmented responses (“P8G”) (80–82). Interestingly, R22E register 3-specific PBMCs are also found in human patients with T1D (83). Additionally, agonistic R22E register 3 insulin peptide converts naïve CD4+ T cells into FoxP3+ Tregs and prevents autoimmune diabetes in NOD mice, supporting a crucial role for register 3 binding MHC-II in stimulating diabetogenesis (84). Similarly, NOD mice treated with a monoclonal antibody targeting R22E register 3 in the context of I-Ag7 delays autoimmune diabetes (85). Although the mechanism underlying natural formation of the P8E and P8G insulin mimotopes is unknown, a possible candidate may be ligation of InsB14-20 to a separate unidentified peptide comprising the relevant amino acids necessary to bind P8 and P9 to form a hybrid peptide neoantigen (see below). These data together suggest that presentation of InsB12-23 peptide via diabetes susceptibility MHC-II alleles can occur through insulin binding in three registers, but further investigation is required to determine whether T cell responses directed against one register dominate over the others.
2.2 Glutamic acid decarboxylase
Glutamic acid decarboxylase (GAD) is a β-cell self-antigen expressed as the GAD65 and GAD67 isoforms in human and murine islets respectively (86, 87). In contrast to insulin, no single critically targeted GAD epitope has been identified, and contributions of GAD-specific T cell responses to disease are not well-understood. Many newly diagnosed patients produce GAD65-specific antibodies, and these antibodies can be used to predict T1D disease status with marginal specificity and positive predictive value (88–90). Additionally, GAD65555-565-reactive CD4+ T cells can be found in the peripheral blood of diabetic patients, and peripheral GAD65-reactive T cells are known to mount TH1-like IFNγ secretion responses (91, 92). In NOD mice, GAD suppression prevents disease, suggesting that GAD-specific responses are required for autoimmune diabetes progression (93). A large number of GAD65-specific T cell hybridomas recognize GAD65206-220, likely due in part to an acidic glutamic acid residue occupying the positively charged P9 pocket of I-Ag7, but GAD65206-220-specific T cells have not been shown to be pathogenic (72, 94, 95). IFNγ secretion by GAD65509-528- and GAD65524-543-specific T cells is observed by 4 weeks in NOD mice, and analogous responses by coxsackievirus mimotope GAD65246-266-specific T cells emerge by 7 weeks (96). The GAD65524-543 peptide in particular has been further implicated as a pathogenic epitope given that adoptive transfer of a TH1-like GAD65524-543-specific CD4+ T cell clone induces diabetes in NOD.scid mice (97).
In spite of this evidence, GAD65524-543 T cell proliferative responses are also observed in the diabetes-resistant H-2g7-restricted B10.H-2g7 and NOD.B6Il2-Tshb mouse strains, suggesting that GAD524-543 reactive T cells alone are insufficient to stimulate autoimmune diabetes (98). Further, TH1-like CD4+ T cell clones targeting murine GAD65524-543 or human GAD65247-266 could not accelerate disease in NOD mice or cause disease in NOD.scid mice, though this may be due to a low-affinity TCR on generated hybridomas given successful disease induction in other settings (99). Additionally, a GAD65515-524-specific CD8+ T cell clone was unable to induce disease despite secreting large amounts of IFNγ and accelerating insulitis upon transfer to NOD mice (100). Although these studies do not collectively clearly explain the role of antigen-specific T cell responses in autoimmune diabetes it is likely that GAD65-specific T cell responses evolve over time and synergize with other antigen-specific T cell populations to orchestrate destruction of pancreatic β-cells.
2.3 Insulinoma antigen
Receptor-type tyrosine-protein phosphatase-like N, also known as insulinoma antigen-2 (IA-2) and IA-2β (also termed phogrin or ICA512) are transmembrane proteins expressed on the secretory granules of neuroendocrine cells, including pancreatic islets and neurons. IA-2 was first identified as an autoantigen by analyzing sera from patients with T1D, in which the autoantibodies preferentially bound to the intracellular domain of IA-2 (101, 102). The presence of IA-2 autoantibodies, along with insulin and GAD, strongly correlated with the incidence and rapidity of type 1 diabetes onset (103). Three observations in human patients suggest that IA-2, similar to GAD and ZnT8 (discussed next), may be pathogenic but not an early, primary initiating antigen of autoimmunity. First, T cells isolated from patients were more likely to be responsive to IA-2 antigen (104), demonstrating their pathogenic potential. Second, while the presence of both IA-2 and GAD antibodies in childhood-onset T1D were associated with MHC-II, this association was with the DRB1 and DQB1 alleles, respectively, and not highest risk allele DR4-DQ8 (105). Third, IA-2 and GAD antibodies are more likely to appear after anti-insulin antibodies and correlate with an older age of T1D diagnosis (105, 106), suggesting that their appearance may occur during epitope spreading and disease progression rather than the primary insult that triggers autoimmunity to β-cells.
2.4 Zinc transporter ZnT8
The Zinc Transporter ZnT8 is an islet-specific protein located on the membrane of insulin secretory granules, that mediates enrichment of zinc within the granules to store insulin as tightly packaged hexamers (107). Additionally, ZnT8 is detectable on cell surface of β-cell cultures following glucose-stimulated insulin secretion (108), providing a mechanism by which this islet-specific antigen is accessible to generate an immunologic response. ZnT8 was first identified as an associated autoantigen in human T1D by microarray expression profiling of human and rodent islet cells (109). Autoantibodies to ZnT8 are detected in 60–80% of new onset diabetics as compared to <2% of healthy controls (109). More recent studies show that the presence of ZnT8 autoantibodies portends a higher risk of diabetes independent of other islet cell autoantibodies (110), and when combined with assessment of other islet-specific autoantibodies, dramatically improves the risk of progression to diabetes (103) and autoimmunity detection rates at diabetes onset (109). These studies demonstrate clinical utility in screening for autoantibodies to ZnT8.
Given that ZnT8 autoantibodies are rarely identified in isolation and instead are more likely to be present in patients just before or at diagnosis of T1D (109), this suggests that ZnT8 autoreactivity may occur during epitope spreading and participate in acceleration or disease onset, rather than initiation of autoimmunity. Inconsistent and variable roles for both CD4+ and CD8+ T cells reactive to ZnT8 favor this observation. Isolated CD4+ T-cells from T1D patients exhibit significantly higher reactivity as measured by cytokine release when using a library of ZnT8-derived peptides (111). However, genome wide association studies have failed to identify consistent MHC-II genetic associations with ZnT8 antibody positivity (112). Rather, MHC class I regions, specifically HLA-A2, were associated with positive ZnT8 antibodies in patients with T1D (112, 113), and these CD8+ T cells are preferentially found in the pancreas of T1D patients compared to controls (113). Additionally, ZnT8 (186-194)-reactive CD8+ T cell clonotypes were found to also recognize a Bacteroides mimotope (113), providing an environmental link to the acceleration of autoimmunity. Limited studies in NOD mice also corroborate the hypothesis that ZnT8 participates during epitope spreading and not disease initiation. CD4+ T cells reactive to ZnT8 are unable to stimulate disease during transfer into NOD or NOD.Rag1-/- mice, and are only found in the pancreas or accelerate diabetes if significant islet damage is already present (114).
While detection of insulin, GAD, IA-2, and ZnT8 antibodies have provided immense benefit in the immunological surveillance of autoimmune diabetes risk and in formal diagnosis following symptom onset, these antigens were all first detected by identification of autoantibodies in T1D patients. Given that the pathogenesis of autoimmune diabetes is driven by autoreactive T cells, there are likely to be many yet unidentified autoantigens that play fundamental roles in the initiation of autoimmunity that lack recognition by islet autoantibodies or have titers well below detection levels. In this context, work in animal models have afforded an alternative screening method, by identifying the molecular targets of diabetogenic T cell clones:
2.5 Islet-specific glucose-6-phosphatase catalytic subunit-related protein
Islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) is another β-cell self-antigen that was identified as the source of the peptide ligand responsible for activating the diabetogenic NY8.3 CD8+ T cell clone (115). The NY8.3 clone was isolated from the islets of a diabetic NOD mouse and was found to induce disease upon adoptive transfer of IGRP-specific CD4+ T cells (116, 117). It was further discovered that this clone used a TCRα CDR3 sequence that was similar to sequences used by many islet-infiltrating β-cell-specific CD8+ T cells in NOD mice, suggesting that the NY8.3 antigen is a common target of cytotoxic CD8+ T cells in murine autoimmune diabetes (118, 119). For these reasons, the NOD.NY8.3 mouse was developed through introduction of transgenes encoding Kd-restricted TCR sequences derived from the NY8.3 clone and found to develop accelerated diabetes relative to littermate non-transgenic controls (120). When similar mice are depleted of macrophages, disease is entirely prevented and TH1-skewing of CD4+ T cells is impaired, demonstrating a requirement of macrophages for activation of CD8+ T cells in NOD.NY8.3 mice (121). Using high-performance liquid chromatography on cellular extracts from the pancreatic β-cell line NIT-1, fractions corresponding to IGRP206-214 were discovered to specifically stimulate NY8.3 cells, and tetramer staining identified endogenous IGRP206-214-specific CD8+ T cells in the islets and blood of NOD mice (115, 122). These experiments confirmed IGRP206-214 as the antigen of diabetogenic NY8.3 CD8+ T cells.
Following identification of IGRP206-214 as the NY8.3 CD8+ T cell antigen, several studies in NOD mice and humans have furthered knowledge regarding the pathogenicity of this epitope. Interestingly, complete tolerance to IGRP does not prevent disease in NOD mice despite elimination of IGRP206-214-specific CD8+ T cells (122). However, when depletion of IGRP206-214-specific CD8+ T cells occurs in a manner that spares low-avidity clones, disease in NOD mice is prevented. These data suggest that low-avidity IGRP206-214-specific clones are non-pathogenic and actively suppress disease in a manner dependent on suppression of autoreactive T cell clones that target additional self-peptides beyond IGRP itself (123). T cell IGRP antigen is encountered selectively in the pLN, because CFSE-labeled NY8.3 CD8+ T cells proliferate in the pLN but not the inguinal lymph node (iLN) following adoptive transfer to NOD mice (122). Interestingly, peripheral IGRP206-214-specific CD8+ T cell cytotoxicity can be used to predict disease, as NOD mice with splenocyte effector activity above a certain threshold develop disease while those without effector activity do not (124). When transgenic NOD mice express human HLA-A*0201 in place of murine MHC-I, islet-infiltrating CD8+ T cells were found to respond to IGRP228-236, IGRP265-273 and IGRP337-345, implicating these epitopes as potentially relevant to human disease (125). Of these epitopes, IGRP265-273 is a likely target of antigen-specific CD8+ T cell responses in patients with T1D because it is entirely conserved between mice and humans (126).
Supporting a role for the pathogenic potential of the IGRP265-273 epitope in humans, in vitro expanded IGRP265-273-specific CD8+ T cells derived from blood of a diabetic patient effectively lyse target cells in a peptide-specific manner (127). Further, IGRP265-273-specific CD8+ T cells utilize shared TCRα-chains across multiple patients (127). Despite this, IGRP265-273-specific CD8+ T cells from diabetic patients are incapable of producing IFNγ, suggesting an inability of these cells to amplify their own inflammatory response (126, 127). This lack of IFNγ secretion is likely epitope-specific, as peripheral CD8+ T cell IFNγ responses are observed against IGRP215-223 and IGRP222-230, two peptides that were computationally predicted to bind well to HLA-A*0201 (126). Detection of peripheral CD8+ T cell responses directed against IGRP265-273 and IGRP228-236 is also observed in recent-onset diabetic patients but not healthy controls (128). Importantly, IGRP-specific T cell responses appear to be most pathogenic when mediated by CD8+ T cells, as CD4+ T cells specific for a number of IGRP epitopes exist in similar proportions in the periphery of diabetic and healthy subjects and produce similar amounts of IFNγ and IL-10 (129). Altogether, these data support pathogenic contributions to autoimmune diabetes development by IGRP-specific CD8+ T cells in both NOD mice and humans.
2.6 Chromogranin A
Chromogranin A (ChgA) is a neuroendocrine protein with diverse tissue expression including pancreatic β-cells where it is critical for regulating intracellular vesicle trafficking dynamics and insulin secretion (130–133). The discovery of ChgA as an T1D self-antigen is related to its ability to stimulate the diabetogenic BDC-2.5 CD4+ T cell clone, whose cognate antigen went unrecognized for many years (57). The BDC-2.5 clone was identified among a panel of islet-specific CD4+ T cell clones expanded from the pancreas of a diabetic NOD mouse and was capable of inducing rapid diabetes in prediabetic NOD mice after only 2-weeks post-adoptive transfer in a manner partially dependent upon CD8+ T cell cytotoxicity (134–137). A transgenic NOD mouse harboring the BDC-2.5 TCRα- and β-chains on CD4+ T cells (NOD.BDC-2.5) was subsequently developed and found to become diabetic with higher penetrance than non-transgenic NOD mice despite a similar age of disease onset (138). Experiments involving transfer of diabetogenic CD4+ T cell clones of different antigen specificities to NOD.scid mice demonstrated that BDC-2.5-specific cells promoted disease earlier than other clones (139). Initially, GAD65 was the suggested antigen targeted by BDC-2.5 clones given the sequence similarity between GAD65528-539 and several synthetic peptides stimulatory to BDC-2.5 CD4+ T cells (140). However, the BDC-2.5 CD4+ T cell proliferation induced by this peptide was low and a similar peptide (GAD521-535) failed to cause BDC-2.5 IFNγ secretion, so this hypothesis was quickly abandoned (140, 141). Other CD4+ T cells responsive to synthetic peptides with BDC-2.5 CD4+ T cell stimulatory capacity were investigated through tetramer analysis and found in NOD mouse islets and spleens, but these experiments similarly were not able to elucidate the BDC-2.5 antigen (142).
The first clue that ChgA is an antigenic source for BDC-2.5 CD4+ T cells was the observation that the isolated secretory granule fraction from subcellular fractionation of β-cells stimulated BDC-2.5 T cells (143, 144). Further experiments involving chromatographic fractionation of β-cell membrane lysate followed by mass spectrometry of fractionated peptides identified ChgA as a candidate antigen for the BDC-2.5 clone due to its presence in a series of highly stimulatory elution fractions (57). Additionally, it was shown that NOD.ChgA-/- mouse islets do not stimulate BDC-2.5 CD4+ T cells to produce IFNγ, and disease was completely prevented, and only slight insulitis was observed in these mice (57, 145). While these data implicated ChgA-specific autoreactive T cells in autoimmune diabetes, genetic deficiency of chromogranin also likely affects disease trajectory via other β-cell intrinsic mechanisms. Specifically, genetic deletion of ChgA in the NOD mouse may result in β-cell secretory granule deficits given the contributions of ChgA to normal vesicular trafficking (131, 132). This is supported by results showing that NOD.ChgA-/- mice produce less insulin on glucose challenge, and several insulin-specific hybridomas are less responsive to NOD.ChgA-/- islets relative to NOD islets (133).
Additional experiments further implicated ChgA as a potential source of BDC-2.5-stimulatory peptides: the high-affinity pS3 BDC-2.5 antigen mimotope was found to have sequence similarity to the ChgA cleavage product WE14, and WE14 was capable of weak BDC-2.5 CD4+ T cell stimulation (57). WE14-reactive CD4+ T cells have also been detected in diabetic patients (146). Confusingly, WE14 could not elicit the same level of IFNγ secretion from BDC-2.5 CD4+ T cells as β-cell membrane or the pS3 mimotope, and the N-terminus of WE14 is predicted to occupy the C-terminal half of the I-Ag7 binding groove, leaving positions 1–4 unfilled (57). Vasostatin-1-derived ChgA29-42 peptide was found to stimulate BDC-2.5 proliferation better than WE14, but even this peptide did not stimulate as robustly as the pS3 mimotope (147). These data suggested that, while ChgA-derived peptides are clearly implicated in BDC-2.5 CD4+ T cell activation, an unknown alteration likely changes the amino acid sequence of a ChgA-derived peptide in some manner to increase its propensity for diabetogenic T cell activation. However, the details of this process and the identity of the natural BDC-2.5 antigen remained elusive.
2.7 Islet amyloid polypeptide
Similar to the identification of ChgA as the peptide targeted by the BDC-2.5 clone, islet amyloid polypeptide (IAPP) was identified via investigating the cognate antigen of the diabetogenic BDC-6.9 CD4+ T cell clone (135–137, 148). The BDC-6.9 clone was isolated from the pancreas of a diabetic NOD mouse and caused disease upon transfer to prediabetic mice after a 2-week latency period, similar to the BDC-2.5 clone (135–137). Interestingly, adoptive transfer of BDC-6.9 CD4+ T cells to NOD.scid recipients induces rapid disease while BDC-2.5 CD4+ T cells require co-transfer with CD8-enriched splenocytes, suggesting that the BDC-6.9 clone harbors intrinsic β-cell destructive properties independent of CD8+ T cell cytotoxicity in this setting (137). However, BDC-6.9 and BDC-2.5 CD4+ T cell clones (derived from T cell lines) transfer diabetes with equal onset and incidence to NOD mice, implying equivalent diabetogenic potential in spontaneous disease (137).
Identification of the antigenic source of the BDC-6.9 clone occurred quickly due to an early observation that transfer of BDC-6.9 CD4+ T cells to F1 progeny of NODxBALB/c breeding pairs did not cause diabetes, yet BDC-2.5 CD4+ T cell transfer was capable of disease induction (149). These F1 progeny were subsequently back-crossed to BALB/c mice (BC1) and patterns of BDC-6.9 islet antigen inheritance were statistically assessed (148). This demonstrated that approximately 50% of the BC1 mice bear the BDC-6.9 antigen, which therefore mapped to a single genetic locus given the Mendelian inheritance pattern. BC1 mice were then genotyped to determine linkage of a known genetic locus with the ability of islets from each mouse to stimulate BDC-6.9 CD4+ T cell proliferation. This process identified a telomeric region on chromosome 6 that encoded IAPP as the only exclusively islet-expressed gene strongly and therefore implicated IAPP as the antigen recognized by BDC-6.9 CD4+ T cells.
IAPP was further implicated as a diabetogenic antigen when NOD.IAPP-/- islets were found incapable of stimulating diabetogenic BDC-5.2.9 CD4+ T cell IFNγ responses, and BDC-5.2.9 T cells could not cause disease upon transfer to NOD.IAPP-/- mice (150). Further, the KS20 IAPP peptide segment could stimulate BDC-5.2.9 CD4 T cell IFNγ responses, and tetramer-specific KS20-reactive CD4+ T cells found in the pancreata of diabetic NOD mice could be cloned and expanded to rapidly transfer disease to prediabetic NOD recipients (59, 150). These results suggested that certain CD4+ T cell responses in the NOD mouse target unmodified epitopes derived from the IAPP protein. However, despite successes with IAPP stimulation of the BDC-5.2.9 clone, no group was able to demonstrate stimulation of the BDC-6.9 clone with IAPP antigen, framing an unidentified amino acid sequence modification of IAPP as the putative source of antigen for BDC-6.9 CD4+ T cells.
2.8 Hybrid peptides
In recent years, hybrid peptide-specific T cell responses associated with autoimmune diabetes pathogenesis have been described in both animal models and human samples (151–155). Given that these novel peptide targets are thought to originate in the periphery as post-translational modifications (PTMs), one might predict that hybrid peptide-specific T cells would therefore escape clonal deletion in the thymus and evade central tolerance mechanisms (156). Hybrid peptide-specific T cells that recognize their cognate antigen in an inflammatory context would then be expected to become activated and expand. This is because autoimmune regulator (AIRE)-expressing medullary thymic epithelial cells (mTECs) presumably do not express post-translationally modified peptides as tissue-restricted antigens and hybrid peptide antigens are predicted to be sufficiently limiting such that it could not be transported by APCs or solubilized to freely enter thymic tissue (157). This assumption is theoretical, and no such evidence has been identified that central tolerance is either occurring or not occurring has been published to date.
Evidence for the relevance of hybrid peptides as antigens in autoimmune diabetes initially emerged with further study of the BDC-2.5 CD4+ T cell clone. Because the ChgA-derived peptide sequence WE14 is only slightly stimulatory to BDC-2.5 CD4+ T cells, transglutamination of WE14 was explored as a possible mechanism of ChgA PTM and increased antigenicity given the known role of transglutaminase (TGase) enzymes in various autoimmune processes (158, 159). TGase treatment of WE14 was found to increase antigenicity towards BDC-2.5 CD4+ T cells via covalent crosslinking and isopeptide bond formation and TGase-treated WE14 stimulated PBCMs from certain diabetic patients better than WE14 (146, 158). However, in vivo identification of transglutaminated WE14 peptides was never described, so this hypothesis was soon abandoned. Given the prediction that unmodified WE14 optimally binds with its N-terminus in position 5 of the I-Ag7 peptide binding groove leaving positions 1–4 unfilled, it was soon discovered that the addition of four amino acids to the N-terminus of WE14 corresponding to positions 1–4 of the high-affinity pS3 BDC-2.5 antigen mimotope (RLGL-WE14) robustly enhanced its ability to stimulate BDC-2.5 CD4+ T cell IL-2 secretion (57, 160). This peculiar finding suggested that a post-translational process may generate a fusion peptide product between WE14 and another peptide at its N-terminus as the autoantigen for the BDC-2.5 clone.
Direct evidence for hybrid peptide sequences functioning as autoantigens was provided with mass spectrometric analyses performed on antigenic β-cell extract fractions, which showed that fractions corresponding to fusion peptide sequences between the insulin C-chain (InsC) and WE14 (DLQTLAL-WSRMD; InsC-ChgA) and InsC and IAPP (DQTLAL-NAARD; InsC-IAPP) stimulate IFNγ production from BDC-2.5 and BDC-6.9 CD4+ T cells respectively (152, 161). These results were recapitulated with direct in vitro culture of BDC-2.5 and BDC-6.9 CD4+ T cells with InsC-ChgA and InsC-IAPP peptide respectively (152, 161). Interestingly, a second unidentified β-cell extract fraction peak was found to stimulate BDC-2.5 CD4+ T cells even more intensely than the InsC-ChgA peak, suggestive of BDC-2.5 CD4+ T cell antigenic promiscuity (152). These data offer strong support for InsC-ChgA and InsC-IAPP peptides as physiologic agonists for the BDC-2.5 and BDC-6.9 CD4+ TCRs.
Identification of endogenous hybrid peptide-specific T cells has been possible with tetramer-tracking technologies (152, 153, 155, 161, 162). Polyclonal tetramer-specific InsC-ChgA-, InsC-IAPP- and InsB9-23 mimotope-reactive CD4+ T cells can be found in the pLN, spleen, and pancreas of prediabetic and diabetic NOD mice, and InsC-ChgA-specific CD4+ T cells are the most frequent of the three in pancreatic islets (152, 153, 161). Approximately 80% of InsC-ChgA-specific CD4+ T cells are CD44highCD62Llow in the pLN by 3-weeks and this proportion is persistent out to 10-weeks, much higher than the 20–40% expression by InsC-IAPP- and InsB9-23-specific CD4+ T cells (153). Diabetic NOD splenocytes also produce more IFNγ when cultured with InsC-ChgA peptide than InsC-IAPP or InsB9-23 peptides, and a greater proportion of pancreas and spleen InsB9-23-specific CD4+ T cells are FoxP3+ Tregs than InsC-ChgA- or InsC-IAPP-specific CD4+ T cells. These data implicate InsC-ChgA-specific CD4+ T cells as potent TH1-like effectors whereas a larger proportion of InsB9-23-specific CD4+ T cells may adopt a suppressive Treg phenotype. The phenotype of InsC-IAPP-specific CD4+ T cells is less clear from this study, and there were fewer cells of this specificity. However, it was also shown that the total frequency and proportion of CD44highCD62Llow InsC-ChgA- and InsC-IAPP-specific CD4+ T cells in peripheral blood of NOD mice increases over time with disease onset much more robustly than InsB9-23-specific CD4+ T cells, strongly implicating these two hybrid peptide-reactive populations in diabetes pathogenesis. Interestingly, it was also shown that a bimodal distribution of high- and low-affinity InsC-ChgA-specific CD4+ T cells exist in the islets of diabetic NOD mice, suggesting that two functionally different populations of these cells may contribute to disease in different ways (162). For example, high-affinity cells may be more likely to initiate disease pathology and epitope spreading while low-affinity cells may help maintain a long-term proinflammatory state later in disease (163). Regardless of the precise mechanism of hybrid peptide-specific CD4+ T cell antigen encounter, these cells are prime suspects for critical mediators of disease progression.
Hybrid peptide antigens are not limited to only murine InsC-ChgA and InsC-IAPP, as multiple hybrid peptides derived from various islet peptide fusion partners have been observed in both NOD mice and diabetic patients (151, 152, 154, 155). For example, both InsC-ChgA- and InsC-IAPP-specific CD4+ T cells have been identified in peripheral blood of patients with T1D (151, 154). Insulin-insulin hybrid peptides have also been frequently found, with InsC-InsC and InsC-InsA hybrids being the most common in diabetic patients relative to healthy controls (151, 154, 155). InsB left-half hybrid peptides have also been identified. InsB-neuropeptide Y-specific CD4+ T cells found within the islets of a diabetic patient secrete IFNγ upon in vitro stimulation, suggesting that these cells may be pathogenic effectors late in the disease process (152). Additionally, peripheral blood InsB:secretogranin I-specific CD4+ T cells have been identified in diabetic patients that possess a CD45RA-CCR7- effector memory phenotype, implicating this hybrid peptide fusion product as a potential contributor to the development of a long-term autoimmune response (155). Examples of hybrid peptide fusions to InsB are particularly interesting because they suggest a mechanism whereby InsB9-23 may be cleaved at amino acid 21 to allow formation of a fusion protein that would essentially accomplish a R22E or R22D amino acid substitution required for InsB9-23 to favorably bind to diabetes-susceptibility MHC molecules in register 3 (82). Given the sizeable contribution of hybrid peptides to the total peptide pool in NOD mouse islets, it is likely that additional roles for (and different antigen specificities of) hybrid peptide-specific T cells will continue to emerge as the field progresses (164).
Although hybrid peptide fusion products are relevant to T1D pathogenesis, their mechanism of production is unknown (151–155). Fusion of disparate protein segments into hybrid peptides may proceed through a transpeptidation reaction known to occur in other physiological settings (165–167). This transpeptidation process would be expected to occur optimally under conditions supporting exceedingly high protein concentrations, such as within β-cell dense-core insulin secretory granules that contain over 200,000 insulin molecules alone per secretory granule (168). Supporting this idea, β-cells from diabetic patients express peptide:HLA-I complexes loaded with several different transpeptidation products (169). While it has been shown that cathepsin L can mediate the transpeptidation of insulin C-chain (InsC) and chromogranin A (ChgA) peptide fragments to form an InsC-ChgA hybrid peptide that is highly stimulatory to the diabetogenic murine BDC-2.5 CD4+ T cell clone, this cathepsin subtype is not normally found at high levels within pancreatic islets (170–172). This implicates other β-cell secretory granule enzymes as putative key mediators of the transpeptidation reaction.
2.9 Alternative neoantigen products
Importantly, PTMs other than hybrid peptides are also likely relevant to T1D pathogenesis, as deamidation, transglutamination, and citrullination of critical pathogenic epitopes recognized by CD4+ and CD8+ T cells have all been observed (151, 173, 174). Another class of neoantigens that may contribute to disease progression includes defective ribosomal products, a key example of which is an alternative open reading frame within human insulin mRNA recognized by CD8+ T cells capable of directly killing β-cells (175, 176). Additionally, mRNA splice variants may function as T cell neoantigenic targets given that 35% of human islet genes undergo alternative splicing, and splice patterns are altered following exposure to the proinflammatory cytokines IL-1β and IFNγ (177). Indeed, splice variants of IA-2 are uniquely expressed in islets relative to the thymus and spleen, and β-cells from diabetic patients express peptide:HLA-I complexes loaded with alternative mRNA splice products of preproinsulin and secretogranin V (169, 178). Although many examples of diabetes-related neoantigens have been identified, the role of each class in stimulating diabetogenic T cell responses is still unclear and requires further investigation.
2.10 Epitope spreading
Once pancreatic islets become inflamed and β-cell destruction begins, additional antigen exposure occurs through subsequent activation of more diverse antigen-specific T cell responses in a process termed epitope spreading (179, 180). Because CD4+ and CD8+ T cell numbers within pancreatic islets increase as disease progresses in diabetic patients, it is likely that epitope spreading occurs early in disease and rapidly accelerates thereafter (58, 181, 182). T cells from patients with T1D target a greater variety of epitopes from GAD65 and proinsulin proteins compared to partially HLA-matched healthy controls, demonstrating intra-protein epitope spreading in disease (179). Additionally, PBMC responses from autoantibody-positive individuals target an increasing number of islet proteins throughout time, suggesting that a temporal hierarchy of pathogenic epitopes may exist both within and between different β-cell-derived proteins (180).
Supporting a model of antigenic hierarchy, insulin appears to be an earlier target of antigen-specific immune responses given that tetramer staining of recent-onset diabetic patient islets shows mostly insulin-specific CD8+ T cells, whereas responses against IGRP, IA-2, GAD65, IAPP and preproinsulin are present in islets from patients with longstanding disease (58). Similarly, InsB9-23-coupled splenocytes induce tolerance in young NOD mice, but tolerance is not achieved using epitopes derived from IGRP or GAD (183, 184). In contrast, TH1 CD4+ T cell responses have been shown to target GAD509-528 and GAD524-543 before spreading to insulin in NOD mice, though this may simply be due to a lack of targeting the pathogenic InsB9-23 epitope (96). Proinsulin-specific T cell responses appear to be a prerequisite for IGRP-specific responses, since proinsulin-tolerant NOD mice do not develop IGRP-specific CD8+ T cell expansion even though IGRP-tolerant NOD mice develop insulitis and AD (122).
Taken together, it is apparent that a complex interplay between T cell responses of multiple antigen specificities evolves as disease proceeds, though the details of these relationships and their origins during the earliest phases of diabetes initiation require further inquiry. Future studies in mice that can target the endogenous polyclonal T cell populations selective to each autoantigens will help define which antigen(s) determine disease initiation and progression. Given that hybrid peptides are neoantigens formed in the periphery, we posit that hybrid peptide-specific T cells are the principal early drivers of autoimmune diabetes.
3 Presentation of autoantigen to T cells
3.1 Dendritic cells
Activation of self-reactive T cells in the context of T1D is potentially driven by the antigen presenting activity of several APC subtypes including dendritic cells (DCs), macrophages, B cells and islet β-cells mediated through loading self-antigen on MHC-I or MHC-II surface molecules. DCs represent particularly compelling candidates given their increased presence in the islets of T1D patients as disease progresses and their production of the proinflammatory cytokines TNFα and IL-1β (185). This increase in islet-localized DCs throughout time is likely mediated at least partially through the attractive actions of chemotactic cytokines released by islet macrophages, given that macrophage depletion in NOD mice prevents the accumulation of islet DCs (186). However, the presence of DC precursors with proliferative potential in fetal NOD pancreata suggests a possible inherent residence and self-renewal capacity as well (187).
Irrespective of the origin of pancreatic DCs, islet DCs have been shown to phagocytose dense-core, insulin-like granules and produce β-cell protein-derived peptide:MHC complexes, with similar DCs also appearing in the pLN (188, 189). Further, NOD islet DCs pulsed with β-cell secretory granules are capable of direct presentation of insulin peptides, and this process is dependent upon the intrinsic affinity of I-Ag7 for pathogenic insulin peptides in both type 1 (XCR1+) and type 2 (SIRPα+) conventional DCs (cDC1s and cDC2s) (79, 190). This process is thought to be facilitated by widespread β-cell death early in life in NOD mice that increases β-cell antigen loading by CD11b+CD11c+CD8α- DCs that subsequently traffic to the pLN for antigen presentation (191). Importantly, cDC1s are required for the development of autoimmune diabetes given that NOD.Batf3-/- mice do not develop islet immune cell infiltration or diabetes (batf3 plays a central role in the development of conventional DCs) (192). Interestingly, adult NOD.Batf3-/- splenocytes transfer disease to NOD.Rag1-/- mice but not as efficiently as diabetic NOD splenocytes, suggesting a partial decrease in the pathogenic capacity of cDC1-deprived NOD T cells. This decrease in pathogenic capacity may be due to decreased CD8+ T cell effectors, because CD8+XCR1+BATF3+ cDC1s are also professional cross-presenting cells and thus also responsible for activation of diabetogenic CD8+ T cells (193, 194). These studies together support a model whereby islet cDC1s acquire antigen within the intra-islet space and subsequently traffic to the draining lymph node to induce activation of autoreactive T cells.
3.2 Macrophages
A role for macrophages in the pathogenesis of autoimmune diabetes has long been suspected given that macrophage depletion in NOD mice prevents disease, though the extent to which their diabetogenic properties are due to T cell-directed antigen presentation has been difficult to elucidate (186, 195, 196). Similar to islet DCs, resident islet macrophages produce TNFα and IL-1β; however, unlike DCs, there is not a need for replenishment by circulating monocytes, given that parabiosis experiments have demonstrated that islet macrophages undergo self-renewing proliferation (185, 197). Their location embedded within pancreatic islets permits CX3CR1+F4/80+ macrophages to regularly sample antigen derived from both intravascular sources in adjacent blood vessels as well as dense core secretory granules directly within β-cells, offering a mechanism by which macrophages might acquire auto-antigen for presentation (198). Additionally, it has been shown that macrophages are capable of insulin uptake from β-cell-derived secretory granules, a subset of which are likely obtained by sampling exosomes released by β-cells (189, 190). Using a model of macrophage depletion through administration of liposomal dichloromethyl diphosphonate to NOD.NY8.3 mice, insulitis and autoimmune diabetes were prevented and splenocytes were found to downregulate FasL and perforin, suggesting a role for macrophages in activating peripheral CD8+ T cells (121). Despite the evidence supporting macrophage antigen presentation as important for disease, it is crucial to note that macrophage depletion in NOD mice at late-stage prediabetic timepoints is just as effective at preventing disease onset as intervention at 3 weeks of age (186). Given that many autoreactive T cells have likely already become activated at late-stage diabetic timepoints, this result implies that macrophages exert the brunt of their pathogenicity through mechanisms distinct from direct antigen presentation such as expression of pro-inflammatory cytokine and nitric oxide release that results in direct β-cell damage, as well as enhancing further T cell recruitment once an autoimmune attack is underway.
One important mechanism by which tissue-resident macrophages may mediate β-cell destruction is via the local production inflammatory cytokines. Macrophages produce several inflammatory mediators upon viral infection and following recognition of other danger-associated molecular patterns (DAMPs), including IL-1β, TNFα, nitric oxide, and prostaglandins (22, 199, 200). Surprisingly, macrophage expression of inflammatory genes in response to diabetogenic viral infections is dependent on activation of the chemokine receptor CCR5 and not controlled by viral dsRNA sensors, suggesting novel and distinct signaling pathways in macrophages that may be implicated in β-cell damage (22, 201, 202). In vitro studies using isolated islets and β-cell lines show the activated macrophages inhibit insulin secretion, stimulate ER stress, cause DNA damage, and can result in β-cell death in a manner dependent on intra-islet macrophage cytokine release (203–209). Specifically, macrophage expression of IL-1β stimulates β-cell production of micromolar levels of nitric oxide, which results in these potentially deleterious (but reversible) effects on β-cell function and viability (203–209). Indeed, selective pharmacological blockade, antibody neutralization, or genetic deletion of inflammatory mediators such as IL-1β and inducible nitric oxide synthase (iNOS; the enzyme in β-cells responsible for nitric oxide production) can attenuate or prevent diabetes in response to encephalomyocarditis virus infection in genetically susceptible mice (210, 211), Kilham rat virus infection in the Bio-Breeding rat (20, 212–214), spontaneous diabetes in the NOD mouse (121, 215), and in other models of diabetes (216–218).
3.3 B cells
B cells are found in inflamed islets and are critical for autoimmune diabetes given that B cell-depleted NOD mice do not develop spontaneous disease (219–222). B cell self-antigen specificity is likely fundamental to influencing disease trajectory since B cells are not only known to bind the diabetogenic InsB9-23 epitope, but NOD mice also develop accelerated disease with VH transgene insertion forcing insulin specificity in 1–3% of mature B cells (190, 223). At least a portion of B cell disease contributions are governed by β-cell autoantigen capture by the B cell receptor (BCR) given that transgenic BCR fixation to target a hen egg lysozyme (HEL) epitope on a B cell-depleted NOD background (NOD.IgHEL.Igμnull mice) prevents stimulation of GAD-specific T cell responses in vitro despite adequate stimulation in the presence of only native B cells as APCs (224). Further, NOD.IgHEL.Igμnull mice develop delayed onset of diabetes similar to NOD.Igμnull mice, suggesting that B cell antigen presentation may actively promote disease progression (224). However, antigen identity is critical as artificial presentation of InsC-ChgA peptide on either MHC class I- or class II-expressing B cells prevents disease in a NOD.scid model of diabetes (225).
In support of a pathogenic role for B cell antigen presentation, islet-infiltrating B cells have enhanced expression of CD80 and CD86, and CD80/86 blockade to impair co-stimulation of T cells prevents in vitro NOD T cell proliferation when cultured with B cells as APCs (226). This study also demonstrated that co-transfer of diabetic NOD splenocytes with BCR-stimulated B cells to NOD.scid mice induces disease, yet disease can be nearly prevented when B cells are first incubated with anti-CD80 and anti-CD86 monoclonal antibodies (226). Importantly, NOD mice engineered with a deficiency in I-Ag7 expression confined to the B cell compartment are protected from disease but still develop peri-insulitis, demonstrating that antigen presentation by B cells to CD4+ T cells is crucial for disease development, yet other APCs likely also contribute (227). A role for B cell activation of CD8+ T cells has also been suggested since B cell-deficient NOD.Igμnull mice reconstituted with NOD B cells expectedly develop disease, but reconstitution with B cells from NOD.β2m-/- mice does not induce diabetes (β2 microglobulin is an essential component of MHC-I class molecules) (228). Taken together, these results strongly implicate B cells as important conduits for antigen presentation to both CD4+ and CD8+ T cells.
3.4 β-cells
Although it is generally thought that the primary role for β-cells in T1D pathogenesis involves functioning as a source of antigen for presentation by other cell types, evidence has emerged over time suggesting an additional role in direct antigen presentation as well (190, 229, 230). It is known that human β-cells express MHC-I, and β-cell class I peptidomes express peptide fragments derived from β-cell proteins (169, 229). Additionally, elevated glucose concentrations increase β-cell preproinsulin antigen presentation and death by cytotoxic T cells in vitro, unsurprisingly supporting the assertion that β-cells present antigen directly to cytotoxic CD8+ T cells (231). It has also been shown that deceased autoimmune diabetic children express HLA-DR in islets containing only insulin-producing cells, suggesting that β-cells are an islet endocrine cell type that uniquely expresses MHC-II (230). Notably, human β-cells also express class II transactivator mRNA whose protein product increases MHC-II levels, and its expression increases as islet infiltration proceeds (232). β-cell expression of MHC-II has also been demonstrated in the β-cells of infiltrated islets of transgenic NOD.NY4.1 (specific for an unknown islet antigen) mice; these MHC-II-expressing β-cells independently induce proliferation of diabetogenic BDC-2.5 CD4+ T cells in vitro (233). MHC-II expression in β-cells is context-specific, with proinflammatory cytokines (e.g. IFNγ and TNFα) capable of promoting MHC-II expression in human islets (233, 234). However, it remains controversial whether wild type NOD mice express MHC-II and whether MHC-II expression on human β-cells occurs in healthy conditions or only following cytokine exposure.
Given that expression of CD80 and CD86 is required for T cell activation during concurrent antigen presentation, it is surprising that no studies have demonstrated isolated expression of these molecules to β-cells (235, 236). Despite this, it has been shown that transgenic expression of CD80 in β-cells of C57BL/6 x NOD mice or C57BL/6 x DQ8+/mII- (global expression of DQ8 without murine MHC-II) induces rapid development of autoimmune diabetes, suggesting that β-cells are indeed capable of direct antigen presentation (237, 238). Transgenic β-cell expression of CD80 does not require CD4+ T cell participation for β-cell destruction as diabetes-susceptible mice engineered to express the co-stimulatory molecule CD80 under control of the rat insulin promoter (RIP) within pancreatic β-cells (C57BL/6.RIP-B7.1 mice) develop disease in the absence of human or murine MHC-II, emphasizing the limitations of these artificial models (239). These studies in combination are challenging to interpret given the lack of evidence for an in vivo role of β-cell antigen presentation and the use of contrived murine study systems. Because of this, direct antigen presentation by β-cells, if relevant, likely comprises only a subtle portion of antigen presentation events and likely are responsible for direct cytotoxic T cell killing after T cell autoreactivity has already been firmly established.
4 Antigen-specific therapies in autoimmune diabetes
4.1 Unmodified antigen
The discovery of multiple diabetes-relevant antigens throughout time has laid the groundwork for the design of disease mitigating therapeutics targeting antigen-specific immune responses (summarized in Table 1). The simplest therapeutic strategy studied involves introduction of antigen to induce tolerance. Clinical trials evaluating full-length insulin protein administered to humans either subcutaneously (240), intranasally (241), or orally (242) found there to be no effect on delaying or preventing T1D onset. Despite the failure of this protein immunization strategy, peptide-based approaches have had more favorable outcomes. For example, administration of InsB9-23 or an altered peptide ligand version termed NBI-6024 to NOD mice significantly delays disease (84, 243), though no effect on islet autoantibodies or C-peptide levels is observed when NBI-6024 is given subcutaneously to diabetic patients (244). However, both proinsulin C19-A3 peptide (245) and GAD65-alum treatment (246–248) given to recently diagnosed T1D patients maintain C-peptide levels over time with variable success. The mechanism of peptide therapy protection appears to be in part Treg-mediated given that treatment of NOD mice and in vitro prediabetic human CD4+ T cells with InsB9-23 and proinsulin peptide promotes conversion of naïve T cells to FoxP3+ Tregs (84, 245, 249). Protein/peptide approaches can be augmented through coupling of toxins to diabetes-relevant antigens, with oral and nasal administration of cholera toxin B-coupled insulin reducing islet inflammation and disease onset in NOD mice through a mechanism that involves downregulation of CD86 and increased IL-10 expression by DCs (250–253). Saporin-coupled IGRP-specific MHC-I tetramers can delete IGRP-reactive CD8+ T cells and delay autoimmune diabetes as well (254).
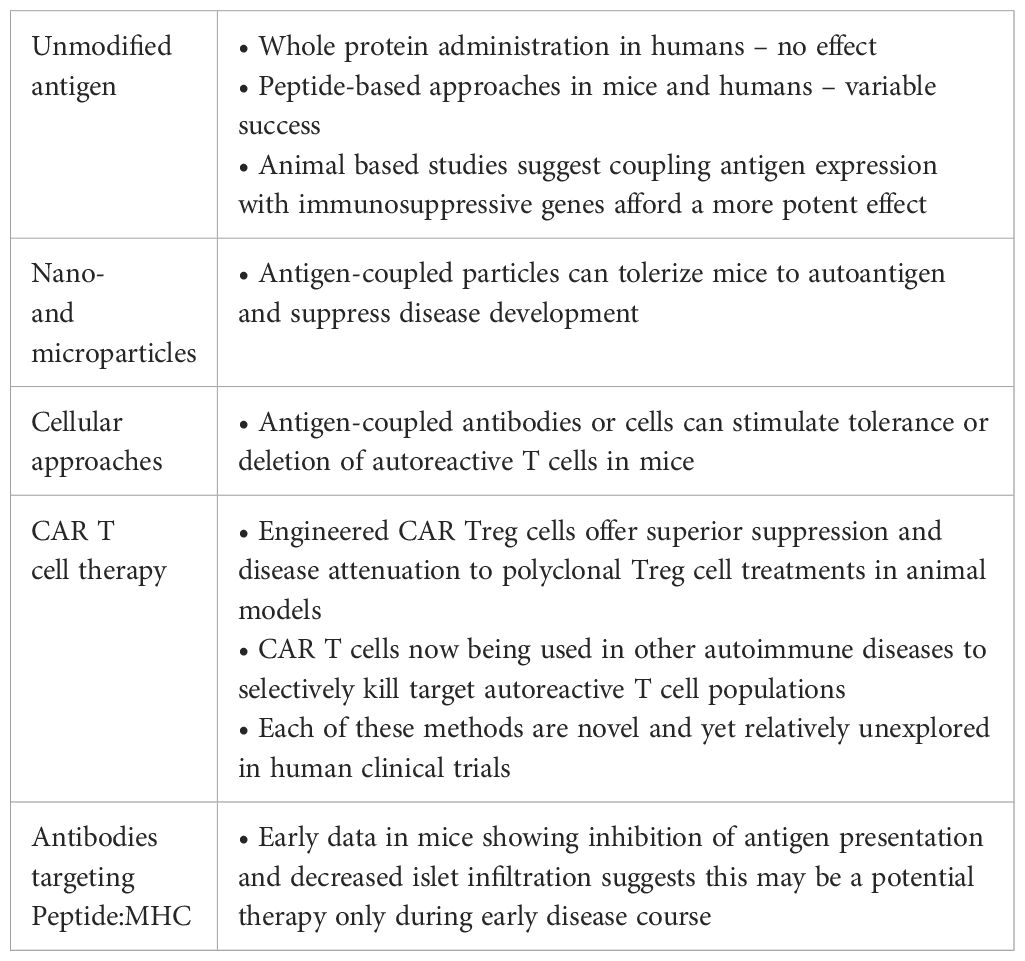
Table 1. Key findings and potential interpretations for antigen-specific therapies in the treatment and prevention of autoimmune diabetes.
DNA delivery techniques have also been employed, with a proinsulin plasmid improving C-peptide levels and decreasing proinsulin-specific CD8+ T cell numbers in diabetic patients (255). Plasmid therapies have been enhanced by encoding diabetogenic epitopes with immunosuppressive genes in one or more DNA vectors, as shown in studies combining GAD65 with IL-10 or the tolerogenic apoptosis-inducing molecule BAX to suppress autoimmune diabetes in NOD mice (256, 257). Extending this concept, other groups discovered that encoding multiple epitopes in one or more plasmids augments diabetes protection in NOD mice. For example, diabetes is suppressed with administration of a plasmid encoding ChgA, IGRP, GAD65 and insulin antigens, and targeting DβH233-241, ZnT8158-166, ZnT8282-290 and proinsulin with a plasmid approach provides better disease protection than proinsulin alone (258, 259). Another method of tolerogenic antigen administration involves oral delivery of Lactococcus lactis bacteria expressing IL-10 and proinsulin or GAD65 with simultaneous intravenous injection of αCD3 mAbs (monoclonal antibodies) to recently diabetic NOD mice (260–262). These therapies result in reversal of disease accompanied by increased FoxP3+ Tregs in the pancreas, blood, spleen, and pLN with suppressive activity dependent upon CTLA-4 and TGFβ signaling.
4.2 Nanoparticles and microparticles
Antigen-coupled nanoparticles (NPs) and microparticles (MPs) have also been explored as a possible tolerogenic therapeutic approach. Both insulin coupled to poly(lactic-co-glycolic acid) (PLGA) MPs and proinsulin coupled to gold methoxypolyethylene glycol-SH NPs prevent autoimmune diabetes when administered to prediabetic NOD mice, with mechanisms dominated by PD-1 upregulation on CD4+ and CD8+ T cells and increased splenic and pLN Tregs respectively (263, 264). Proinsulin peptide coupled to gold NPs injected into human breast skin samples are taken up by Langerhans cells and reduce antigen presentation, suggesting that induction of DC tolerance may contribute to proinsulin/insulin-coupled NP and MP disease inhibition (265).
Antigens other than insulin also demonstrate NP and MP therapeutic potential. For example, IGRP peptide-MHC-coupled iron oxide NPs prevent autoimmune diabetes in NOD mice by inducing suppression and killing of APCs and expansion of autoregulatory CD4+ and CD8+ T cells, and IGRP peptide-coupled PLGA NPs suppress disease in NOD.scid recipients of diabetogenic transgenic NY8.3 CD8+ T cells (266–268). Similarly, disease is inhibited in NOD.scid recipients following transfer of diabetogenic transgenic BDC-2.5 CD4+ T cells when mice are given p31- or InsC-ChgA-coupled PLGA NPs, and this result is recapitulated by co-treatment with p31-coupled acetylated dextran MPs and rapamycin (268–270). Importantly, spontaneous disease in NOD mice is delayed by subcutaneous co-injection of liposomal particles encapsulating a BDC-2.5 mimotope and vitamin D3, and disease is prevented with administration of BDC-2.5 mimotope-coupled iron oxide NPs (267, 271). Much like IGRP-specific NP and MP therapy, InsC-ChgA-specific treatments induce increased Treg numbers and a decrease in TH1-like effector cells as contributory mechanisms to disease suppression (267, 269–271). Given these consistent results in achieving disease prevention with various diabetes-relevant antigens independent of the specific NP or MP coupling formulation employed, successful translation of this technology to human T1D may be possible with prudent selection of autoantigen targets to maintain minimal toxicity.
4.3 Cellular approaches
Cell-based strategies are additional methodologies for autoimmune diabetes therapy. Similar to antigen-coupled NP and MP approaches, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (ECDI) coupling of intact insulin or InsB9-23 to splenocytes and sortase-mediated coupling of InsB9-23 to red blood cells (RBCs) both generate tolerogenic apoptotic coupled cells capable of preventing diabetes in NOD mice (184, 272). Beyond direct chemical crosslinking of antigen to apoptotic splenocytes or RBCs, p31 can be targeted to RBCs using antigen constructs specific for the RBC surface marker glycophorin A to completely prevent BDC-2.5-transferred disease in NOD mice (273).
To directly target APCs for tolerance induction, antigen delivery to CD205+CD8+ DCs using αCD205 antibodies coupled to antigen has been performed and shown to promote antigen internalization (274, 275). This model has shown promise as a tolerogenic therapy given its consistent conversion of naïve T cells to Tregs through enhanced TGFβ secretion by CD205+ DCs across multiple experimental systems (276–278). Antigen-coupled αCD205 antibodies have found variable success across multiple diabetes-relevant antigens in the NOD mouse model: 1) Co-transfer of insulin mimotope-αCD205 and insulin-specific AI4 CD8+ T cells causes antigen-specific T cell deletion, and spontaneous disease is suppressed with proinsulin-αCD205 administration (279, 280); 2) Co-transfer of p63-αCD205 and transgenic BDC-2.5 CD4+ T cells enhances Treg conversion despite not affecting disease incidence (280); and 3) IGRP206-214-αCD205 reduces the number of endogenous IGRP-specific CD8+ T cells and promotes deletion of transgenic NY8.3 CD8+ T cells in an adoptive transfer system (281). Interestingly, despite a lack of disease prevention with p63-αCD205 and transgenic BDC-2.5 CD4+ T cell co-transfer, p63-αDCIR2 targeting CD11b+ cDC2s could suppress BDC-2.5 CD4+ T cell-mediated disease, implicating multiple DC populations presenting various antigens as potential therapeutic foci (280, 282). While use of DC antigen-specific therapies in clinic has only recently been explored for T1D one study found that proinsulin pulsed tolerogenic DCs injected intradermally did not affect HbA1c, C peptide, hypoglycemic events or insulin dose required for symptom maintenance (283). However, the treatment was safe and caused only minor injection site symptoms, paving the way for future DC-based clinical applications (283).
4.4 Ex vivo and chimeric antigen receptor Treg
Work in animal models of autoimmunity have demonstrated the potent therapeutic potential that antigen-specific T-regulatory cells may have on diabetes prevention. The use of Tregs are appealing not only because of their specificity to auto-antigens, but because they are also capable of bystander (antigen-independent) suppression of other, local, autoreactive T-cells (284). Expanded transgenic BDC-2.5 Tregs were shown to attenuate BDC2.5 T-cell transfer-induced diabetes in NOD.scid mice (285, 286), demonstrating the feasibility of ex vivo Treg expansion in inhibiting effector T-cell populations that share similar specificities. These same expanded BDC-2.5 Tregs were also capable of attenuating spontaneous diabetes in the following diabetes models: NOD.CD28-/- mice (285), NOD.scid mice that were treated with splenocytes from diabetic NOD mice (286), or spontaneous diabetes in wild-type, pre-diabetic (13 weeks age) NOD mice (287). Importantly, BDC-2.5 Tregs inhibit disease in Treg-deficient NOD.CD28-/- mice better than anti-CD3ϵ-coupled bead-expanded polyclonal Tregs, suggesting that Tregs specific for diabetogenic antigens may be clinically applicable and much more potent than polyclonal Tregs (288). Besides treatment with expanded antigen-specific Tregs, chimeric antigen receptor (CAR) Tregs have recently been explored as a potential autoimmune diabetes therapy with variable success. Long-lived murine CAR Tregs expressing an insulin-specific single-chain variable fragment (scFv) proliferate and generate IL-2 upon in vitro stimulation and suppress proliferation of allogeneic CD8+ T cells, but have no effect on diabetes progression in NOD mice (289). These results highlight the robust bystander suppressive effect that Treg cells of single islet-derived antigen specificity may have on limiting autoimmune progression even when autoreactivity to numerous epitopes has occurred. Additionally, these data may illustrate why, to date, all clinical trials that used expanded polyclonal Treg cells have only demonstrated safety but not efficacy for disease prevention (290).
A number of limitations exist with ex vivo conversion and expansion of patient auto-reactive T-cells into Treg cells that can then be autologously transplanted. Relevant autoreactive T cell populations are rare in peripheral circulation, and there may be wide variability in TCR affinity and specificity, limiting therapeutic potential. Two alternative methods can be conducted that could confer greater specificity, activation potential, and manipulability as opposed to expanding T cells with antigen followed by enrichment of antigen-specific T cells. First, T cells may be engineered to become Treg cells with the desired antigen specificity (engTregs) by combining genetic editing to force express FOXP3 with lentiviral transduction of a specified TCR. The therapeutic potential of this approach was recently demonstrated by using BDC2.5+ engTreg cells to prevent diabetes pathogenesis in mice receiving either BDC2.5+ T cells which demonstrated antigen-specific repression, or diabetic NOD splenocytes which demonstrated bystander suppressive capacity (291). The second approach is to develop chimeric antigen receptors (CARs) composed of an extracellular single-chain antigen-specific antibody (scFv) linked to modular intracellular co-stimulatory signaling domains. Our laboratory designed an scFv CAR Treg specific to InsB chain peptide 10-23 in the context of MHC-II; this CAR suppressed diabetes in NOD.RAG-/- mice with co-transfer of BDC2.5 T cells and suppressed spontaneous diabetes in wild type NOD mice (292). These studies highlight the therapeutic potential for engineered and CAR Treg cells therapy in the prevention of autoimmune diabetes in humans.
CAR T cell therapy has revolutionized cancer therapy by facilitating antigen-specific immune activation against tumor antigens (293). These same treatments that have demonstrated remarkable efficacy in clearing lymphomas and other tumors refractory to conventional cancer therapies are now being repurposed to prevent or cure autoimmunity by targeting pathogenic immune cell populations. For example, systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by autoantibodies that mediate tissue damage; B cell depletion using rituximab, an anti-CD20 antibody is a common treatment used to treat SLE (294). However, SLE disease may be severe and refractory despite anti-CD20 depletion. CD8+ CAR T cells specific to CD19 were shown to effectively deplete B cells, reverse autoantibody production and tissue damage, and extend the life span of multiple murine models of lupus (295). Remarkably, initial clinical trials in humans have demonstrated that in patients with severe SLE refractory to conventional treatments, anti-CD19 CAR T cells were able to achieve complete remission in all 5 patients within 3 months of treatment, and these patients continue to be seronegative for autoantibodies and without symptoms for months after B cells repopulated (296). However, CAR T cell therapies that broadly target entire immune subsets, such as CD19 expressing B cells, results in broad immunosuppression risk. To circumvent this, CAR T cells are now being developed that selectively deplete autoantibody-expressing B cells. For example, NMDAR-specific chimeric autoantibody receptor (CAAR) T cells, comprised of NMDAR auto-antigen fused to CD8 hinge and intracellular signaling domains, were able to selectively kill anti-NMDAR cell lines in vivo (297). The selective targeting of autoreactive TCR populations is a compelling method to extrapolate to both mouse models of diabetes and genetically at-risk humans with the goal of disease prevention.
4.5 Peptide: MHC
Peptide: MHC-specific approaches represent another potential autoimmune diabetes therapy, first suggested by results demonstrating a delay in diabetes progression and induction of antigen-specific antibodies following immunization of 4-week-old NOD mice with recombinant InsB12-22 register 3 (I-Ag7-B:RE#3) peptide (298). Extending this approach, an anti-InsB12-22RE#3:I-Ag7 monoclonal antibody was found to delay autoimmune diabetes in NOD mice by broadly decreasing pancreatic islet infiltration of both CD4+ and CD8+ T cells (85). Additionally, an I-Ag7-B:RE#3-specific CAR CD8+ T cell treatment administered to NOD mice homed well to the pLN and delayed disease after one infusion, suggesting that the mechanism of these diverse therapies relies upon inhibition of the antigen presenting capabilities of relevant APCs (299). However, the utility of this approach is not limited to the I-Ag7-B:RE#3 antigen, as immunization of NOD mice at 4- and 8-weeks with an anti-InsC-ChgA:I-Ag7 delays the onset of autoimmune diabetes as well (300). To improve the production of anti-peptide:MHC monoclonal antibodies, a novel method for peptide:MHC mAb generation using a magnetic enrichment protocol was developed and resulted in generation of an anti-p63:I-Ag7 monoclonal antibody with exceptionally high affinity that could prevent tolerance induction in NOD mice following transfer of p63-coupled cells (301). Although disease prevention was not shown in this study, this methodology will likely be helpful for the future production of peptide:MHC-specific monoclonal antibodies (301). The use of such antigen-specific therapies in clinic has not yet gained momentum, but the success of the various approaches detailed here using in vitro and murine models offer compelling evidence that further refinement of these techniques may yield novel T1D therapies for humans.
5 Conclusions
T1D is a disease characterized by T cell-mediated destruction of the insulin-producing β-cells within the pancreatic islets of Langerhans, so determining the antigenic targets of these T cell responses is critical to understanding disease pathogenesis and devising therapeutic approaches. Several T cell islet autoantigens have been identified as key contributors to disease progression, including insulin, GAD, IGRP, ChgA and IAPP; hybrid peptide formation among these various epitopes may provide an additional essential mechanism by which central tolerance is bypassed during the early development of autoimmunity. DCs, macrophages, B cells and β-cells may all participate in islet self-antigen presentation to autoreactive T cells, though the precise sites of antigen acquisition and presentation have not been globally determined. Critically, islet antigen-specific T cells have been successfully targeted for autoimmune diabetes prophylaxis and therapy in NOD mice and humans using several treatment modalities.
Despite tremendous progress in understanding the roles of different antigen-specific T cell responses in autoimmune diabetes, several critical outstanding objectives remain. First, while HLA haplotype, other genetic risk factors, viral infection, and gut microbiome are all well described risk factors that lead to the induction of autoimmune diabetes, the exact mechanism by which each of these factors lead to emergence of a sustained autoreactive T cell response is still incompletely elucidated. Epitope discovery using methods such as mass spectrometry on size-eluted microbiome antigenic fractions found to stimulate pathogenic T cells is necessary to characterize the cross-reactive T cell responses that might occur between similar epitopes derived from non-self vs. pancreatic β-cell proteins. Second, it is necessary to further characterize the exact timing that each antigen-specific T cell develops as well as its related pathologic contribution in autoimmune diabetes. Determining the temporal and pathogenic hierarchy of antigen-specific T cell responses will assist in identifying important therapeutic targets for the amelioration of disease.
Third, additional effort is required to address differences in T cell antigenic targets between the NOD mouse and humans with autoimmune diabetes. This will be especially important for hybrid peptides given that T cell responses target alternate peptide fusion products in humans compared to NOD mice (151, 152, 154, 155), and different mechanisms of hybrid peptide formation may underly this disparity. Fourth, successful therapeutic strategies in NOD mice must be effectively translated to clinic. Although numerous antigen-specific treatments demonstrate success in NOD mice, many rely upon frequent dosing and/or therapeutic administration prior to symptom onset (84, 85, 243, 258, 263, 264, 280, 298, 300). Further, many clinical therapeutics administered to recent onset T1D patients demonstrate negligible impact on disease trajectory even with alterations in laboratory markers (244–248, 255, 283). To overcome these challenges, it is necessary to devise approaches that accurately identify likely antigen-specific T cell responses in at-risk patients. Further, therapies that reduce treatment frequency (e.g. CAR Treg infusion) should be considered to enhance patient compliance and affordability.
Development of an accurate autoimmune diabetes pathogenesis model requires detailed understanding and characterization of the myriad antigen-specific T cell responses underlying disease. Although many unanswered questions remain, substantial progress has been made in elucidating the identity of key autoantigens and the mechanisms permitting recognition and activation of their cognate T cells. Given the remarkable success of antigen-specific treatments in NOD mice, it is not unreasonable to expect further therapeutic application of antigen-specific approaches in clinical settings in the future.
Author contributions
ZS: Writing – review & editing, Writing – original draft, Supervision, Conceptualization. AD: Writing – review & editing, Writing – original draft, Visualization, Conceptualization. BF: Writing – review & editing, Supervision, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors for this work were supported by the National Institutes of Health (NIH) R01 AI106791 (to BF), R01 AI156276 (to BF), P01AI35296 (to BF) and T32 AI007313 (to AD), The Leona M. and Harry B. Helmsley Charitable Trust (G-2303-06775 and G-2405-06927 to BF), the Minnesota Lions Diabetes Foundation and University of Minnesota Foundation Fund 11724 (Diabetes Cure Research Using Immune Regulation and Tolerance).
Acknowledgments
The authors thank the members of the Fife lab for scientific discussions including: Justin Spanier, Jason Mitchell, Tijana Martinov, and Christopher Tucker.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Redondo MJ, Steck AK, Pugliese A. Genetics of type 1 diabetes. Pediatr Diabetes. (2018) 19:346–53. doi: 10.1111/pedi.12597
2. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. (2009) 41:703–7. doi: 10.1038/ng.381
3. Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. (2015) 47:381–6. doi: 10.1038/ng.3245.Fine
4. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harbor Perspect Med. (2012) 2:1–15. doi: 10.1101/cshperspect.a007732
5. Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harbor Perspect Biol. (2012) 4:1–15. doi: 10.1101/cshperspect.a006957
6. Olmos P, A'Hern R, Heaton DA, Millward BA, Risley D, Pyke DA, et al. The significance of the concordance rate for type 1 (insulin-dependent) diabetes in identical twins. Diabetologia. (1988) 31:747–50. doi: 10.1007/BF00274777
7. Verge CF, Gianani R, Yu L, Pietropaolo M, Smith T, Jackson RA, et al. Late progression to diabetes and evidence for chronic β-cell autoimmunity in identical twins of patients with type I diabetes. Diabetes. (1995) 44:1176–9. doi: 10.2337/diab.44.10.1176
8. Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. New Engl J Med. (2008) 359:2849–50. doi: 10.1056/NEJMc0805398
9. Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, et al. Heterogeneity of Type I diabetes: Analysis of monozygotic twins in Great Britain and the United States. Diabetologia. (2001) 44:354–62. doi: 10.1007/s001250051626
10. Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young finnish twin pairs. Diabetes. (2003) 52:1052–5. doi: 10.2337/diabetes.52.4.1052
11. Jun HS, Yoon JW. The role of viruses in Type I diabetes: Two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia. (2001) 44:271–85. doi: 10.1007/s001250051614
12. Isaacs SR, Foskett DB, Maxwell AJ, Ward EJ, Faulkner CL, Luo JYX, et al. Viruses and type 1 diabetes: from enteroviruses to the virome. Microorganisms. (2021) 9(7). doi: 10.3390/microorganisms9071519
13. Lloyd RE, Tamhankar M, Lernmark A. Enteroviruses and type 1 diabetes: multiple mechanisms and factors? Annu Rev Med. (2022) 73:483–99. doi: 10.1146/annurev-med-042320-015952
14. Yeung WCG, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ. (2011) 342. doi: 10.1136/bmj.d35
15. Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes. (2015) 64:1682–7. doi: 10.2337/db14-1370
16. Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. New Engl J Med. (1979) 300:1173–9. doi: 10.1056/NEJM197905243002102
17. Serreze DV, Ottendorfer EW, Elliss TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. (2000) 49:708–11. doi: 10.2337/diabetes.49.5.708
18. Craighead JE, McLane MF. Diabetes mellitus: induction in mice by encephalomyocarditis virus. Science. (2022) 162:913–4. doi: 10.1126/science.162.3856.913
19. Carocci M, Bakkali-Kassimi L. The encephalomyocarditis virus. Virulence. (2012) 3:351–67. doi: 10.4161/viru.20573
20. Guberski DL, Thomas VA, Shek WR, Like AA, Handler ES, Rossini AA, et al. Induction of Type I Diabetes by Kilham ' s Rat Virus in Diabetes-Resistant BB / Wor Rats Vol. 254. American Association for the Advancement of Science Stable (1991) p. 1010–3. Available at: https://www.jstor.org/stable/2879714. American Association for the Advancement of Scien. doi: 10.1126/science.1658938
21. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann New York Acad Sci. (2013) 1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x
22. Shaheen ZR, Corbett JA. Macrophage expression of inflammatory genes in response to EMCV infection. Biomolecules. (2015) 5:1938–54. doi: 10.3390/biom5031938
23. Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol. (1963) 42:471–83.
24. Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia. (2005) 48:1565–75. doi: 10.1007/s00125-005-1831-2
25. Hansen CHF, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sørensen SJ, et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. (2012) 55:2285–94. doi: 10.1007/s00125-012-2564-7
26. Candon S, Perez-Arroyo A, Marquet C, Valette F, Foray AP, Pelletier B, et al. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PloS One. (2015) 10:1–16. doi: 10.1371/journal.pone.0125448
27. Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol. (2015) 195:4176–84. doi: 10.4049/jimmunol.1500884
28. De Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes. (2013) 62:1238–44. doi: 10.2337/db12-0526
29. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PloS One. (2011) 6:1–9. doi: 10.1371/journal.pone.0025792
30. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. (2014) 5:678. doi: 10.3389/fmicb.2014.00678
31. De Goffau MC, Fuentes S, Van Den Bogert B, Honkanen H, De Vos WM, Welling GW, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. (2014) 57:1569–77. doi: 10.1007/s00125-014-3274-0
32. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression towards type 1 diabetes. Cell Host Microbe. (2015) 17:260–73. doi: 10.1086/203459
33. Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. (2019) 574:117–21. doi: 10.1038/s41586-019-1560-1
34. Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: A meta-analysis of observational studies. Diabetologia. (2008) 51:726–35. doi: 10.1007/s00125-008-0941-z
35. Hansen CHF, Andersen LSF, Krych Ł, Metzdorff SB, Hasselby JP, Skov S, et al. Mode of delivery shapes gut colonization pattern and modulates regulatory immunity in mice. J Immunol. (2014) 193:1213–22. doi: 10.4049/jimmunol.1400085
36. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med. (2016) 213:2129–46. doi: 10.1084/jem.20160526
37. Krych L, Nielsen DS, Hansen AK, Hansen CHF. Gut microbial markers are associated with diabetes onset, regulatory imbalance, and IFN-γ level in NOD Mice. Gut Microbes. (2015) 6:101–9. doi: 10.1080/19490976.2015.1011876
38. Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
39. Lund-Blix NA, Stene LC, Rasmussen T, Torjesen PA, Andersen LF, Rønningen KS. Infant feeding in relation to islet autoimmunity and type 1 diabetes in genetically susceptible children: The MIDIA study. Diabetes Care. (2015) 38:257–63. doi: 10.2337/dc14-1130
40. Lund-Blix NA, Sander SD, Størdal K, Nybo Andersen AM, Rønningen KS, Joner G, et al. Infant feeding and risk of type 1 diabetes in two large scandinavian birth cohorts. Diabetes Care. (2017) 40:920–7. doi: 10.2337/dc17-0016
41. Schmid S, Koczwara K, Schwinghammer S, Lampasona V, Ziegler AG, Bonifacio E. Delayed exposure to wheat and barley proteins reduces diabetes incidence in non-obese diabetic mice. Clin Immunol. (2004) 111:108–18. doi: 10.1016/j.clim.2003.09.012
42. Schober E, Rami B, Granditsch G, Crone J. Coeliac disease in children and adolescents with type 1 diabetes mellitus: to screen or not, to treat or not? Hormone Res. (2002) 57:97–100. doi: 10.1297/cpe.7.125
43. Antvorskov JC, Halldorsson TI, Josefsen K, Svensson J, Granström C, Roep BO, et al. Association between maternal gluten intake and type 1 diabetes in offspring: national prospective cohort study in Denmark. BMJ. (2018) 362:1–9. doi: 10.1136/bmj.k3547
44. Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-Associated autoantibodies. J Am Med Assoc. (2003) 290:1721–8. doi: 10.1001/jama.290.13.1721
45. Norris JM, Barriga K, Klingensmith G, Hoffman M, Eisenbarth GS, Erlich HA, et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. J Am Med Assoc. (2003) 290:1713–20. doi: 10.1001/jama.290.13.1713
46. Pastore MR, Bazzigaluppi E, Belloni C, Arcovio C, Bonifacio E, Bosi E. Six months of gluten-free diet do not influence autoantibody titers, but improve insulin secretion in subjects at high risk for type 1 diabetes. J Clin Endocrinol Metab. (2003) 88:162–5. doi: 10.1210/jc.2002-021177
47. Fuchtenbusch M, Ziegler AG, Hummel M. Elimination of dietary gluten and development of type 1 diabetes in high risk subjects. Rev Diabetic Stud. (2004) 1:39–41. doi: 10.1900/RDS.2004.1.39
48. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken dobutsu. Exp Anim. (1980) 29:1–13. doi: 10.1538/expanim1978.29.1_1
49. Pozzilli P, Signore A, Williams AJK, Beales PE. NOD mouse colonies around the world - recent facts and figures. Immunol Today. (1993) 14:193–6. doi: 10.1016/0167-5699(93)90160-M
50. Anderson MS, Bluestone JA. The NOD mouse: A model of immune dysregulation. Annu Rev Immunol. (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
51. Pearson JA, Wong FS, Wen L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
52. Naik RG, Beckers C, Wentwoord R, Frenken A, Duinkerken G, Brooks-Worrell B, et al. Precursor frequencies of T-cells reactive to insulin in recent onset type 1 diabetes mellitus. J Autoimmun. (2004) 23:55–61. doi: 10.1016/j.jaut.2004.04.002
53. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. (2005) 435:220–3. doi: 10.1038/nature03523
54. Standifer NE, Burwell EA, Gersuk VH, Greenbaum CJ, Nepom GT. Changes in autoreactive T cell avidity during type 1 diabetes development. Clin Immunol. (2009) 132:312–20. doi: 10.1016/j.clim.2009.04.013.Changes
55. Stadinski B, Kappler J, Eisenbarth GS. Molecular targeting of islet autoantigens. Immunity. (2010) 32:446–56. doi: 10.1016/j.immuni.2010.04.008
56. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA. (2010) 107:10978–83. doi: 10.1073/pnas.1006545107
57. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. (2010) 11:225–32. doi: 10.1038/ni.1844
58. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TWH, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. (2012) 209:51–60. doi: 10.1084/jem.20111187
59. Baker RL, Delong T, Barbour G, Bradley B, Nakayama M, Haskins K. Cutting Edge: CD4 T cells reactive to an IAPP peptide accumulate in the pancreas and contribute to disease pathogenesis in NOD mice. J Immunol. (2013) 191:3990–4. doi: 10.4049/jimmunol.1301480
60. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. (2017) 127:2881–91. doi: 10.1172/JCI94549
61. Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. (2011) 57:176–85. doi: 10.1373/clinchem.2010.148221
62. Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest. (2005) 115:2268–76. doi: 10.1172/JCI25350
63. Wen L, Wong FS, Sherwin R, Mora C. Human DQ8 can substitute for murine I-Ag7 in the selection of diabetogenic T cells restricted to I-Ag7. J Immunol. (2002) 168:3635–40. doi: 10.4049/jimmunol.168.7.3635
64. Shizuru JA, Taylor-Edwards C, Banks BA, Gregory AK, Fathman CG. Immunotherapy of the nonobese diabetic mouse: treatment with an antibody to T-helper lymphocytes. Science. (1988) 240:659–62. doi: 10.4135/9781412963954.n6
65. Lejon KC, Fathman G. Isolation of self antigen-reactive cells from inflamed islets of nonobese diabetic mice using CD4high expression as a marker. J Immunol. (1999) 163:5708–14. doi: 10.4049/jimmunol.163.10.5708
67. Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. (1995) 25:1056–62. doi: 10.1002/eji.1830250430
68. Zhang L, Jasinski JM, Kobayashi M, Davenport B, Johnson K, Davidson H, et al. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9–23 alpha chains. J Autoimmun. (2009) 33:42–9. doi: 10.1016/j.jaut.2009.02.003
69. Paronen J, Moriyama H, Abiru N, Sikora K, Melanitou E, Babu S, et al. Establishing insulin 1 and insulin 2 knockout congenic strains on NOD genetic background. Ann New York Acad Sci. (2003) 1005:205–10. doi: 10.1196/annals.1288.027
70. Nakayama M, Moriyama H, Abiru N, Babu SR, Sikora K, Li M, et al. Establishment of native insulin-negative NOD mice and the methodology to distinguish specific insulin knockout genotypes and a B:16 alanine preproinsulin transgene. Ann New York Acad Sci. (2004) 1037:193–8. doi: 10.1196/annals.1337.031
71. Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. (2000) 12:699–710. doi: 10.1016/S1074-7613(00)80220-4
72. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. (2000) 288:505–11. doi: 10.1126/SCIENCE.288.5465.505/SUPPL_FILE/1049210S3_THUMB.GIF
73. Todd JA, Bell JI, McDevitt HO. HLA-DQβ gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. (1987) 329:599–604. doi: 10.1038/329599a0
74. Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol. (2007) 178:6051–7. doi: 10.4049/jimmunol.178.10.6051
75. Yoshida K, Corper AL, Herro R, Jabri B, Wilson IA, Teyton L. The diabetogenic mouse MHC class II molecule I-Ag7 is endowed with a switch that modulates TCR affinity. J Clin Invest. (2010) 120:1578–90. doi: 10.1172/JCI41502
76. Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, et al. Position β57 of I-Ag7 controls early anti-insulin responses in NOD mice, linking an MHC susceptibility allele to type 1 diabetes onset. Sci Immunol. (2019) 4:6329–9. doi: 10.1126/SCIIMMUNOL.AAW6329/SUPPL_FILE/AAW6329_TABLE_S3.XLSX
77. Mohan JF, Calderon B, Anderson MS. Pathogenic CD4+ T cells recognizing an unstable peptide of insulin are directly recruited into islets bypassing local lymph nodes. J Exp Med. (2013) 210:2403–14. doi: 10.1084/jem.20130582
78. Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide–MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. (2011) 208:2375–83. doi: 10.1084/jem.20111502
79. Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. (2010) 11:350–4. doi: 10.1038/ni.1850
80. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA. (2011) 108:16729–34. doi: 10.1073/pnas.1113954108
81. Wang Y, Sosinowski T, Novikov A, Crawford F, Neau DB, Yang J, et al. C-terminal modification of the insulin B:11–23 peptide creates superagonists in mouse and human type 1 diabetes. Proc Natl Acad Sci U.S.A. (2018) 115:162–7. doi: 10.1073/pnas.1716527115
82. Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, et al. How C-terminal additions to insulin B-chain fragments create super-agonists for T cells in mouse and human type 1 diabetes. Sci Immunol. (2019) 4:1–12. doi: 10.1126/sciimmunol.aav7517
83. Yang J, et al. Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci USA. (2014) 111:14840–5. doi: 10.1073/PNAS.1416864111/-/DCSUPPLEMENTAL
84. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med. (2011) 208:1501–10. doi: 10.1084/jem.20110574
85. Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, et al. Monoclonal antibody blocking the recognition of an insulin peptide–MHC complex modulates type 1 diabetes. Proc Natl Acad Sci. (2014) 111:2656–61. doi: 10.1073/pnas.1323436111
86. Baekkeskov S, Aanstoot HJ, Christgai S, Reetz A, Solimena M, Cascalho M, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. (1990) 347:151–6. doi: 10.1038/347151a0
87. Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. (1993) 42:1799–808. doi: 10.2337/diab.42.12.1799
88. Baekkeskov S, Landin M, Kristensen JK, Srikanta S, Bruining GJ, Mandrup-Poulsen T, et al. Antibodies to a 64,000 Mr human islet cell antigen precede the clinical onset of insulin-dependent diabetes. J Clin Invest. (1987) 79:926–34. doi: 10.1172/JCI112903
89. Atkinson MA, Kaufman DL, Newman D, Tobin AJ, Maclaren NK. Islet cell cytoplasmic autoantibody reactivity to glutamate decarboxylase in insulin-dependent diabetes. J Clin Invest. (1993) 91:350–6. doi: 10.1172/JCI116192
90. Pollanen PM, Harkonen T, Ilonen J, Toppari J, Veijola R, Siljander H, et al. Autoantibodies to N-terminally truncated GAD65(96–585): HLA associations and predictive value for type 1 diabetes. J Clin Endocrinol Metab. (2022) 107:935–46.
91. Reijonen H, Mallone R, Heninger AK, Laughlin EM, Kochik SA, Falk B, et al. GAD65-specific CD4+ T-cells with high antigen avidity are prevalent in peripheral blood of patients with type 1 diabetes. Diabetes. (2004) 53:1987–94. doi: 10.2337/diabetes.53.8.1987
92. Kotani R, Kaufman DL, Newman D, Tobin AJ, Maclaren NK. Detection of GAD65-reactive T-cells in type 1 diabetes by immunoglobulin-free ELISPOT assays. Diabetes Care. (2002) 25:1390–7. doi: 10.2337/diacare.25.8.1390
93. Yoon JW, Harkonen T, Ilonen J, Toppari J, Veijola R, Siljander H, et al. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in β cells. Science. (1999) 284:1183–7. doi: 10.1126/science.284.5417.1183
94. Chao CC, McDevitt HO. Identification of immunogenic epitopes of GAD 65 presented by Ag7 in non-obese diabetic mice. Immunogenetics. (1997) 46:29–34. doi: 10.1007/s002510050238
95. Chao C-C, Sytwu HK, Chen EL, Toma J, McDevitt HO. The role of MHC class II molecules in susceptibility to type I diabetes: Identification of peptide epitopes and characterization of the T cell repertoire. Proc Natl Acad Sci USA. (1999) 96:9299–304. doi: 10.1073/pnas.96.22.12970
96. Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GSP, Robinson P, et al. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. (1993) 366:69–72. doi: 10.1038/366069a0.Spontaneous
97. Zekzer D, Wong FS, Ayalon O, Millet I, Altieri M, Shintani S, et al. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. J Clin Invest. (1998) 101:68–73. doi: 10.1172/JCI119878
98. Chen S-L, Whiteley PJ, Freed DC, Rothbard JB, Peterson LB, Wicker LS, et al. Responses of NOD congenic mice to a glutamic acid decarboxylase-derived peptide. J Autoimmun. (1994) 7:635–41. doi: 10.1006/jaut.1994.1048
99. Schloot NC, Daniel D, Norbury-Glaser M, Wegmann DR. Peripheral T cell clones from NOD mice specific for GAD65 peptides: Lack of islet responsiveness or diabetogenicity. J Autoimmun. (1996) 9:357–63. doi: 10.1006/jaut.1996.0048
100. Videbæk N, Harach S, Phillips J, Hutchings P, Ozegbe P, Michelsen BK, et al. An islet-homing NOD CD8+ cytotoxic T cell clone recognizes GAD65 and causes insulitis. J Autoimmun. (2003) 20:97–109. doi: 10.1016/S0896–8411(03)00003–9
101. Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-M(r) tryptic fragments of islet antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like molecule IA-2 (ICA512). J Clin Invest. (1995) 96:1506–11. doi: 10.1172/JCI118188
102. Kawasaki E, Eisenbarth GS, Wasmeier C, Hutton JC. Autoantibodies to protein tyrosine phosphatase-like proteins in type I diabetes. Overlapping specificities to phogrin and ICA512/IA-2. Diabetes. (1996) 45:1344–9. doi: 10.2337/diab.45.10.1344
103. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. Jama. (2013) 309:2473–9. doi: 10.1001/jama.2013.6285
104. Hawkes CJ, Schloot NC, Marks J, Willemen SJ, Drijfhout JW, Mayer EK, et al. T-cell lines reactive to an immunodominant epitope of the tyrosine phosphatase-like autoantigen IA-2 in type 1 diabetes. Diabetes. (2000) 49:356–66. doi: 10.2337/diabetes.49.3.356
105. Howson JM, Stevens H, Smyth DJ, Walker NM, Chandler KA, Bingley PJ, et al. Evidence that HLA class I and II associations with type 1 diabetes, autoantibodies to GAD and autoantibodies to IA-2, are distinct. Diabetes. (2011) 60:2635–44. doi: 10.2337/db11-0131
106. Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, et al. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care. (2011) 34:1397–9. doi: 10.2337/dc10-2088
107. Williams CL, Long AE. What has zinc transporter 8 autoimmunity taught us about type 1 diabetes? Diabetologia. (2019) 62:1969–76. doi: 10.1007/s00125-019-04975-x
108. Huang Q, Merriman C, Zhang H, Fu D. Coupling of insulin secretion and display of a granule-resident zinc transporter znT8 on the surface of pancreatic beta cells. J Biol Chem. (2017) 292:4034–43. doi: 10.1074/jbc.M116.772152
109. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA. (2007) 104:17040–5. doi: 10.1073/pnas.0705894104
110. Yu L, Boulware DC, Beam CA, Hutton JC, Wenzlau JM, Greenbaum CJ, et al. Zinc transporter-8 autoantibodies improve prediction of type 1 diabetes in relatives positive for the standard biochemical autoantibodies. Diabetes Care. (2012) 35:1213–8. doi: 10.2337/dc11-2081
111. Dang M, Rockell J, Wagner R, Wenzlau JM, Yu L, Hutton JC, et al. Human type 1 diabetes is associated with T cell autoimmunity to zinc transporter 8. J Immunol. (2011) 186:6056–63. doi: 10.4049/jimmunol.1003815
112. Howson JM, Krause S, Stevens H, Smyth DJ, Wenzlau JM, Bonifacio E, et al. Genetic association of zinc transporter 8 (ZnT8) autoantibodies in type 1 diabetes cases. Diabetologia. (2012) 55:1978–84. doi: 10.1007/s00125-012-2540-2
113. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol. (2018) 3(20). doi: 10.1126/sciimmunol.aao4013
114. Nayak DK, Calderon B, Vomund AN, Unanue ER. ZnT8-reactive T cells are weakly pathogenic in NOD mice but can participate in diabetes under inflammatory conditions. Diabetes. (2014) 63:3438–48. doi: 10.2337/db13-1882
115. Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA. (2003) 100:8384–8. doi: 10.1073/pnas.0932778100
116. Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon JW. Evidence for the role of CD8+ cytotoxic T cells in the destruction of pancreatic β-cells in nonobese diabetic mice. J Immunol. (1994) 152:2042–50. doi: 10.4049/jimmunol.152.4.2042
117. Utsugi T, Yoon J-W, Park B-J, Imamura M, Averill N, Kawazu S, et al. Major histocompatibility complex class I–restricted infiltration and destruction of pancreatic islets by NOD mouse-derived β-cell cytotoxic CD8+ T-cell clones in vivo. Diabetes. (1996) 45:1121–31. doi: 10.2337/diab.45.8.1121
118. Santamaria P, Utsugi T, Park BJ, Averill N, Kawazu S, Yoon JW, et al. Beta-cell-cytotoxic CD8+ T cells from nonobese diabetic mice use highly homologous T cell receptor α-chain CDR3 sequences. J Immunol. (1995) 154:2494–503. doi: 10.4049/jimmunol.154.5.2494
119. Verdaguer J, Yoon JW, Anderson B, Averill N, Utsugi T, Park BJ, et al. Acceleration of spontaneous diabetes in TCR-β-transgenic nonobese diabetic mice by β-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-α chains. J Immunol. (1996) 157:4726–35. doi: 10.4049/jimmunol.157.10.4726
120. Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med. (1997) 186:1663–76. doi: 10.1084/jem.186.10.1663
121. Jun H-S, Santamaria P, Lim H-W, Zhang ML, Yoon J-W. Absolute requirement of macrophages for the development and activation of β-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes. (1999) 48:34–42. doi: 10.2337/diabetes.48.1.34
122. Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest. (2006) 116:3258–65. doi: 10.1172/JCI29602
123. Han B, Serra P, Amrani A, Yamanouchi J, Marée AFM, Edelstein-Keshet L, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med. (2005) 11:645–52. doi: 10.1038/nm1250
124. Ko H-J, Chee J, Sutherland RM, Thomas HE, Zhan Y, Krishnamurthy B, et al. Functional cytotoxic T lymphocytes against IGRP206–214 predict diabetes in the non-obese diabetic mouse. Immunol Cell Biol. (2014) 92:640–4. doi: 10.1038/icb.2014.29
125. Takaki T, Marron MP, Mathews CE, Guttmann ST, Bottino R, Trucco M, et al. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol. (2006) 176:3257–65. doi: 10.4049/jimmunol.176.5.3257
126. Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol. (2008) 127:359–65. doi: 10.1016/j.clim.2008.01.015
127. Fuchs YF, Eugster A, Dietz S, Sebelefsky C, Kühn D, Wilhelm C, et al. CD8+ T cells specific for the islet autoantigen IGRP are restricted in their T cell receptor chain usage. Sci Rep. (2017) 7:44661–1. doi: 10.1038/srep44661
128. Mallone R, Martinuzzi E, Blancou P, Novelli G, Afonso G, Dolz M, et al. CD8+ T-cell responses identify β-cell autoimmunity in human type 1 diabetes. Diabetes. (2007) 56:613–21. doi: 10.2337/db06-1419
129. Yang J, Danke NA, Berger D, Reichstetter S, Reijonen H, Greenbaum C, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol. (2006) 176:2781–9. doi: 10.4049/jimmunol.176.5.2781
130. Gleeson CM, Curry WJ, Johnston CF, Buchanan KD. Occurrence of WE-14 and chromogranin A-derived peptides in tissues of the human and bovine gastro-entero-pancreatic system and in human neuroendocrine neoplasia. J Endocrinol. (1996) 151:409–20. doi: 10.1677/joe.0.1510409
131. Portela-Gomes GM, Gayen JR, Grimelius L, Stridsberg M, Mahata SK. The importance of chromogranin A in the development and function of endocrine pancreas. Regul peptides. (2008) 151:19–25. doi: 10.1016/j.regpep.2008.07.005
132. Wollam J, Mahata S, Riopel M, Hernandez-Carretero A, Biswas A, Bandyopadhyay GK, et al. Chromogranin A regulates vesicle storage and mitochondrial dynamics to influence insulin secretion. Cell Tissue Res. (2017) 368:487–501. doi: 10.1007/s00441-017-2580-5
133. Srivastava N, Hu H, Vomund AN, Peterson OJ, Baker RL, Haskins K, et al. Chromogranin A deficiency confers protection from autoimmune diabetes via multiple mechanisms. Diabetes. (2021) 70:2860–70. doi: 10.2337/db21-0513
134. Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. (1988) 37:1444–8. doi: 10.2337/diab.37.10.1444
135. Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA. (1989) 86:8000–4. doi: 10.1073/pnas.86.20.8000
136. Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. (1990) 249:1433–6. doi: 10.1126/science.2205920
137. Peterson JD, Haskins K. Transfer of diabetes in the NOD-scid mouse by CD4 T-cell clones: Differential requirement for CD8 T-cells. Diabetes. (1996) 45:328–36. doi: 10.2337/diab.45.3.328
138. Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. (1993) 74:1089–100. doi: 10.1016/0092-8674(93)90730-E
139. Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes. (2008) 57:1321–30. doi: 10.2337/db07-1129
140. Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. (2001) 166:908–17. doi: 10.4049/jimmunol.166.2.908
141. Yoshida K, Martin T, Yamamoto K, Dobbs C, Münz C, Kamikawaji N, et al. Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones. Int Immunol. (2002) 14:1439–47. doi: 10.1093/intimm/dxf106
142. You S, Chen C, Lee W-H, Wu C-H, Judkowski V, Pinilla C, et al. Detection and characterization of T cells specific for BDC2.5 T cell-stimulating peptides. J Immunol. (2003) 170:4011–20. doi: 10.4049/jimmunol.170.8.4011
143. Bergman B, Haskins K. Islet-specific T-cell clones from the NOD mouse respond to β-granule antigen. Diabetes. (1994) 43:197–203. doi: 10.2337/diab.43.2.197
144. Bergman B, McManaman JL, Haskins K. Biochemical characterization of a beta cell membrane fraction antigenic for autoreactive T cell clones. J Autoimmun. (2000) 14:343–51. doi: 10.1006/jaut.2000.0377
145. Baker RL, Bradley B, Wiles TA, Lindsay RS, Barbour G, Delong T, et al. Cutting edge: nonobese diabetic mice deficient in chromogranin A are protected from autoimmune diabetes. J Immunol. (2016) 196:39–43. doi: 10.4049/jimmunol.1501190
146. Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun. (2014) 50:38–41. doi: 10.1016/j.jaut.2013.10.003.Chromogranin
147. Nikoopour E, Sandrock C, Huszarik K, Krougly O, Lee-Chan E, Masteller EL, et al. Cutting edge: vasostatin-1–derived peptide chgA29–42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol. (2011) 186:3831–5. doi: 10.4049/jimmunol.1003617
148. Dallas-Pedretti A, McDuffie M, Haskins K. A diabetes-associated T-cell autoantigen maps to a telomeric locus on mouse chromosome 6. Proc Natl Acad Sci USA. (1995) 92:1386–90. doi: 10.1073/pnas.92.5.1386
149. Peterson JD, Pike B, McDuffie M, Haskins K. Islet-specific T cell clones transfer diabetes to nonobese diabetic (NOD) F1 mice. J Immunol. (1994) 153:2800–6. doi: 10.4049/jimmunol.153.6.2800
150. Delong T, Baker RL, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes. (2011) 60:2325–30. doi: 10.2337/db11-0288
151. Babon JAB, Denicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med. (2016) 22:1482–9. doi: 10.1038/nm.4203
152. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. (2016) 351:711–4. doi: 10.1126/science.aad2791
153. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes. (2018) 67:1836–46. doi: 10.2337/db18-0200
154. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes. (2019) 68:1830–40. doi: 10.2337/DB19–0128/-/DC1
155. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C, et al. Hybrid insulin peptides are recognized by human T cells in the context of DRB1*04:01. Diabetes. (2020) 69:1492–502. doi: 10.2337/db19-0620
156. Raposo B, Merky P, Lundqvist C, Yamada H, Urbonaviciute V, Niaudet C, et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat Commun. (2018) 9:1–11. doi: 10.1038/s41467-017-02763-y
157. Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones PJ, et al. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J Biol Chem. (1999) 274:22321–7. doi: 10.1074/jbc.274.32.22321
158. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K, et al. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. (2012) 61:3239–46. doi: 10.2337/db12-0112
159. Arentz-Hansen H, Körner R, Molberg Ø, Quarsten H, Vader W, Kooy YMC, et al. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. (2000) 191:603–12. doi: 10.1084/jem.191.4.603
160. Jin N, Wang Y, Crawford F, White J, Marrack P, Dai S, et al. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc Natl Acad Sci U.S.A. (2015) 112:13318–23. doi: 10.1073/pnas.1517862112
161. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun. (2017) 78:11–8. doi: 10.1016/j.jaut.2016.10.007
162. Liu B, Hood JD, Kolawole EM, Woodruff DM, Vignali DA, Bettini M, et al. A hybrid insulin epitope maintains high 2D affinity for diabetogenic T cells in the periphery. Diabetes. (2020) 69:381–91. doi: 10.2337/db19-0399
163. Tian J, Gregori S, Adorini L, Kaufman DL. The frequency of high avidity T cells determines the hierarchy of determinant spreading. J Immunol. (2001) 166:7144–50. doi: 10.4049/jimmunol.166.12.7144
164. Aaron Wiles T, Powell R, Michel CR, Scott Beard K, Hohenstein A, Bradley B, et al. Identification of hybrid insulin peptides (HIPs) in mouse and human islets by mass spectrometry. J Proteome Res. (2019) 18:814–25. doi: 10.1021/acs.jproteome.8b00875
165. Lorand L, Konishi K, Jacobsen A. Transpeptidation mechanism in blood clotting. Nature. (1962) 194:1148–9. doi: 10.1038/1941148a0
166. Hanada KI, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. (2004) 427:252–6. doi: 10.1038/nature02240
167. Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science. (2004) 304:587–90. doi: 10.1126/SCIENCE.1095522/SUPPL_FILE/VIGNERON.SOM.PDF
168. Howell SL. The mechanism of insulin secretion. Diabetologia. (1984) 26:319–27. doi: 10.1007/BF00266030
169. Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, et al. Conventional and neo-antigenic peptides presented by β Cells are targeted by circulating naïve CD8+ T cells in type 1 diabetic and healthy donors. Cell Metab. (2018) 28:946–60. doi: 10.1016/j.cmet.2018.07.007
170. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal cathepsin creates chimeric epitopes for diabetogenic CD4 T cells via transpeptidation. J Exp Med. (2021) 218:1–17. doi: 10.1084/jem.20192135
171. Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. (2006) 20:543–56. doi: 10.1101/gad.1407406
172. Brindle NR, Joyce JA, Rostker F, Lawlor ER, Swigart-Brown L, Evan G, et al. Deficiency for the cysteine protease cathepsin l impairs Myc-induced tumorigenesis in a mouse model of pancreatic neuroendocrine cancer. PloS One. (2015) 10:1–17. doi: 10.1371/journal.pone.0120348
173. Van Lummel M, Duinkerken G, Van Veelen PA, De Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes. (2014) 63:237–47. doi: 10.2337/DB12–1214/-/DC1
174. McGinty JW, et al. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes. (2014) 63:3033–40. doi: 10.2337/db13-1952
175. Kracht MJL, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med. (2017) 23:501–7. doi: 10.1038/nm.4289
176. Laban S, Suwandi JS, Van Unen V, Pool J, Wesselius J, Höllt T, et al. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PloS One. (2018) 13:1–15. doi: 10.1371/journal.pone.0200818
177. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PloS Genet. (2012) 8:1–17. doi: 10.1371/journal.pgen.1002552
178. Diez J, Park Y, Zeller M, Brown D, Garza D, Ricordi C, et al. Differential splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive genetic mechanism for autoimmunity against the IA-2 type 1 diabetes autoantigen. Diabetes. (2001) 50:895–900. doi: 10.2337/diabetes.50.4.895
179. Ott PA, Dittrich MT, Herzog BA, Guerkov R, Gottlieb PA, Putnam AL, et al. T cells recognize multiple GAD65 and proinsulin epitopes in human type 1 diabetes, suggesting determinant spreading. J Clin Immunol. (2004) 24:327–39. doi: 10.1023/B:JOCI.0000029120.77824.41
180. Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol. (2001) 166:5265–70. doi: 10.4049/jimmunol.166.8.5265
181. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. (2009) 155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x
182. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes. (2016) 65:719–31. doi: 10.2337/DB15–0779/-/DC1
183. Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J Autoimmun. (2012) 39:347–53. doi: 10.1016/j.jaut.2012.04.005.Pathogenesis
184. Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. (2006) 203:2737–47. doi: 10.1084/jem.20061577
185. Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-α in patients with recent-onset type 1 diabetes. Diabetologia. (2007) 50:596–601. doi: 10.1007/s00125-006-0569-9
186. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci. (2017) 114:E10418–27. doi: 10.1073/pnas.1713543114
187. Welzen-Coppens JMC, Van Helden-Meeuwsen CG, Drexhage HA, Versnel MA. Abnormalities of dendritic cell precursors in the pancreas of the NOD mouse model of diabetes. Eur J Immunol. (2012) 42:186–94. doi: 10.1002/eji.201141770
188. Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present β cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci USA. (2008) 105:6121–6. doi: 10.1073/pnas.0801973105
189. Vomund AN, Zinselmeyer BH, Hughes J, Calderon B, Valderrama C, Ferris ST, et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci. (2015) 112:E5496–502. doi: 10.1073/pnas.1515954112
190. Wan X, Zinselmeyer BH, Zakharov PN, Vomund AN, Taniguchi R, Santambrogio L, et al. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature. (2018) 560:107–11. doi: 10.1038/s41586-018-0341-6
191. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological β Cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. (2003) 198:1527–37. doi: 10.1084/jem.20030966
192. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER, et al. A minor subset of Batf3-dependent antigen presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity. (2014) 41:657–69. doi: 10.1016/j.immuni.2014.09.012
193. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. (2008) 322:1097–100. doi: 10.1126/science.1164206
194. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Güttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity. (2009) 31:823–33. doi: 10.1016/j.immuni.2009.08.027
195. Lee K, Amano K, Yoon J. Evidence for initial involvement of macrophage in development of insulitis in NOD mice. Diabetes. (1988) 37:989–91. doi: 10.2337/diab.37.7.989
196. Hutchings P, Rosen H, O'Reilly L, Simpson E, Gordon S, Cooke A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature. (1990) 348:639–41. doi: 10.1038/348639a0
197. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med. (2015) 212:1497–512. doi: 10.1084/jem.20150496
198. Zinselmeyer BH, Vomund AN, Saunders BT, Johnson MW, Carrero JA, Unanue ER, et al. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia. (2018) 61:1374–83. doi: 10.1007/s00125-018-4592-4
199. Janeway CA Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
200. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
201. Shaheen ZR, Christmann BS, Stafford JD, Moran JM, Buller RML, Corbett JA, et al. CCR5 is a required signaling receptor for macrophage expression of inflammatory genes in response to viral double-stranded RNA. Am J Physiol Regulatory Integr Comp Physiol. (2019) 316:R525–r534. doi: 10.1152/ajpregu.00019.2019
202. Shaheen ZR, Stafford JD, Voss MG, Oleson BJ, Stancill JS, Corbett JA. The location of sensing determines the pancreatic β-cell response to the viral mimetic dsRNA. J Biol Chem. (2020) 295:2385–97. doi: 10.1074/jbc.RA119.010267
203. Corbett JA, Lancaster JR Jr., Sweetland MA, McDaniel ML. Interleukin-1 beta-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 beta-induced inhibition of insulin secretion. J Biol Chem. (1991) 266:21351–4. doi: 10.1016/S0021-9258(18)54642-1
204. Corbett JA, McDaniel ML. Intraislet release of interleukin 1 inhibits beta cell function by inducing beta cell expression of inducible nitric oxide synthase. J Exp Med. (1995) 181:559–68. doi: 10.1084/jem.181.2.559
205. Arnush M, Scarim AL, Heitmeier MR, Kelly CB, Corbett JA. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J Immunol. (1998) 160:2684–91. doi: 10.4049/jimmunol.160.6.2684
206. Oleson BJ, Corbett JA. Dual role of nitric oxide in regulating the response of beta cells to DNA damage. Antioxidants Redox Signaling. (2017) 29(14):1432–45. doi: 10.1089/ars.2017.7351
207. Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest. (1998) 102:516–26. doi: 10.1172/JCI844
208. Welsh N, Eizirik DL, Bendtzen K, Sandler S. Interleukin-1 beta-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology. (1991) 129:3167–73. doi: 10.1210/endo-129-6-3167
209. Southern C, Schulster D, Green IC. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. (1990) 276:42–4. doi: 10.1016/0014-5793(90)80502-A
210. Hirasawa K, Jun HS, Maeda K, Kawaguchi Y, Itagaki S, Mikami T, et al. Possible role of macrophage-derived soluble mediators in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J Virol. (1997) 71:4024–31. doi: 10.1128/jvi.71.5.4024-4031.1997
211. Lee YS, Li N, Shin S, Jun HS. Role of nitric oxide in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J Virol. (2009) 83:8004–11. doi: 10.1128/JVI.00205-09
212. Ellerman KE, Richards CA, Guberski DL, Shek WR, Like AA. Kilham rat triggers T-cell-dependent autoimmune diabetes in multiple strains of rat. Diabetes. (1996) 45:557–62. doi: 10.2337/diab.45.5.557
213. Mendez II, Chung YH, Jun HS, Yoon JW. Immunoregulatory role of nitric oxide in Kilham rat virus-induced autoimmune diabetes in DR-BB rats. J Immunol. (2004) 173:1327–35. doi: 10.4049/jimmunol.173.2.1327
214. Chung YH, Jun HS, Kang Y, Hirasawa K, Lee BR, Van Rooijen N, et al. Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. J Immunol. (1997) 159:466–71. doi: 10.4049/jimmunol.159.1.466
215. Jun HS, Yoon CS, Zbytnuik L, van Rooijen N, Yoon JW. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med. (1999) 189:347–58. doi: 10.1084/jem.189.2.347
216. Wright JR Jr., Lefkowith JB, Schreiner G, Lacy PE. Essential fatty acid deficiency prevents multiple low-dose streptozotocin-induced diabetes in CD-1 mice. Proc Natl Acad Sci USA. (1988) 85:6137–41. doi: 10.1073/pnas.85.16.6137
217. OsChilewski U, Kiesel U, Kolb H. Administration of silica prevents diabetes in BB-rats. Diabetes. (1985) 34:197–9. doi: 10.2337/diab.34.2.197
218. Lefkowith J, Schreiner G, Cormier J, Handler ES, Driscoll HK, Greiner D, et al. Prevention of diabetes in the BB rat by essential fatty acid deficiency. Relationship between physiological and biochemical changes. J Exp Med. (1990) 171:729–43. doi: 10.1084/jem.171.3.729
219. Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PloS One. (2013) 8:e59701–1. doi: 10.1371/journal.pone.0059701
220. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: Analysis of a new "speed congenic" stock of NOD.Igμnull mice. J Exp Med. (1996) 184:2049–53. doi: 10.1084/jem.184.5.2049
221. Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A, et al. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. (1997) 46:941–6. doi: 10.2337/diab.46.6.941
222. Akashi M, Nagafuchi S, Anzai K, Kondo S, Kitamura D, Wakana S, et al. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol. (1997) 9:1159–64. doi: 10.1093/intimm/9.8.1159
223. Hulbert C, Riseili B, Rojas M, Thomas JW. Cutting edge: B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol. (2001) 167:5535–8. doi: 10.4049/jimmunol.167.10.5535
224. Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV, et al. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol. (2002) 32:3657–66. doi: 10.1002/1521–4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E
225. Chen D, Kakabadse D, Fishman S, Weinstein-Marom H, Davies J, Boldison J, et al. Novel engineered B lymphocytes targeting islet-specific T cells inhibit the development of type 1 diabetes in non-obese diabetic Scid mice. Front Immunol. (2023) 14:1227133. doi: 10.3389/fimmu.2023.1227133
226. Hussain S, Delovitch TL. Dysregulated B7–1 and B7–2 expression on nonobese diabetic mouse B cells is associated with increased T cell costimulation and the development of insulitis. J Immunol. (2005) 174:680–7. doi: 10.4049/jimmunol.174.2.680
227. Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. (1999) 163:743–50. doi: 10.4049/jimmunol.163.2.743
228. Mariño E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes. (2012) 61:2893–905. doi: 10.2337/db12-0006
229. Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PGF, Gamble DR, et al. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. New Engl J Med. (1985) 313:353–60. doi: 10.1056/NEJM198508083130604
230. Foulis AK, Farquharson MA. Aberrant expression of HLA-DR antigens by insulin-containing β-cells in recent-onset type I diabetes mellitus. Diabetes. (1986) 35:1215–24. doi: 10.2337/diabetes.35.11.1215
231. Skowera A, Ellis RJ, Varela-Calviño R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are targeted to kill β cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest. (2008) 118:3390–402. doi: 10.1172/JCI35449
232. Russell MA, Redick SD, Blodgett DM, Richardson SJ, Leete P, Krogvold L, et al. HLA class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet β-cells from donors with type 1 diabetes. Diabetes. (2019) 68:988–1001. doi: 10.2337/db18-0686
233. Zhao Y, Scott NA, Quah HS, Krishnamurthy B, Bond F, Loudovaris T, et al. Mouse pancreatic beta cells express MHC class II and stimulate CD4+ T cells to proliferate. Eur J Immunol. (2015) 45:2494–503. doi: 10.1002/eji.201445378
234. Soldevila G, Doshi M, James R, Lake SP, Sutton R, Gray D, et al. HLA DR, DP, DQ induction in human islet β cells by the cytokine combination IFN-γ + TNF-α. Autoimmunity. (1990) 6:307–17. doi: 10.3109/08916939008998422
235. Mueller DL, Jenkins MK, Schwartz RH. An accessory cell-derived costimulatory signal acts independently of protein kinase C activation to allow T cell proliferation and prevent the induction of unresponsiveness. J Immunol. (1989) 142:2617–28. doi: 10.4049/jimmunol.142.8.2617
236. Imagawa A, Hanafusa T, Itoh N, Miyagawa JI, Nakajima H, Namba M, et al. Islet-infiltrating T lymphocytes in insulin-dependent diabetic patients express CD80 (B7–1) and CD86 (B7–2). J Autoimmun. (1996) 9:391–6. doi: 10.1006/jaut.1996.0053
237. Wong S, Guerder S, Visintin I, Reich EP, Swenson KE, Flavell RA, et al. Expression of the co-stimulator molecule B7–1 in pancreatic β-cells accelerates diabetes in the NOD mouse. Diabetes. (1995) 44:326–9. doi: 10.2337/diab.44.3.326
238. Wen L, Wong FS, Tang J, Chen NY, Altieri M, David C, et al. In vivo evidence for the contribution of human histocompatibility leukocyte antigen (HLA)-DQ molecules to the development of diabetes. J Exp Med. (2000) 191:97–104. doi: 10.1084/jem.191.1.97
239. Havari E, Lennon-Dumenil AM, Klein L, Neely D, Taylor JA, McInerney MF, et al. Expression of the B7.1 costimulatory molecule on pancreatic β Cells abrogates the requirement for CD4 T cells in the development of type 1 diabetes. J Immunol. (2004) 173:787–96. doi: 10.4049/jimmunol.173.2.787
240. The Diabetes Prevention Trial-Type 1 Study, G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. New Engl J Med. (2002) 346:1685–1691b. doi: 10.1056/NEJMoa012350
241. Harrison LC, Honeyman MC, Steele CE, Stone NL, Sarugeri E, Bonifacio E, et al. Pancreatic β-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care. (2004) 27:2348–55. doi: 10.2337/diacare.27.10.2348
242. The Diabetes Prevention Trial-Type 1 Study, G. Effects of oral insulin in relatives of patients with type 1 diabetes. Diabetes Care. (2005) 28:1068–76. doi: 10.2337/diacare.28.5.1068
243. Alleva DG, Gaur A, Jin L, Wegmann D, Gottlieb PA, Pahuja A, et al. Immunological characterization and therapeutic activity of an altered-peptide ligand, NBI-6024, based on the immunodominant type 1 diabetes autoantigen insulin B-chain (9–23) peptide. Diabetes. (2002) 51:2126–34. doi: 10.2337/diabetes.51.7.2126
244. Walter M, Philotheou A, Bonnici F, Ziegler A-G, Jimenez R. No effect of the altered peptide ligand NBI-6024 on β-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care. (2009) 32:2036–40. doi: 10.2337/dc09-0449
245. Ali MA, Liu Y-f, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci Trans Med. (2017) 9:1–9.
246. Hannelius U, Beam CA, Ludvigsson J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia. (2020) 63:2177–81. doi: 10.1007/s00125-020-05227-z
247. Dietrich F, Barcenilla H, Tavira B, Wahlberg J, Achenbach P, Ludvigsson J, et al. Immune response differs between intralymphatic or subcutaneous administration of GAD-alum in individuals with recent onset type 1 diabetes. Diabetes/Metabolism Res Rev. (2022) 38:1–10. doi: 10.1002/dmrr.3500
248. Ludvigsson J, Krisky D, Casas R, Battelino T, Castaño L, Greening J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. New Engl J Med. (2012) 366:433–42. doi: 10.1089/dia.2013.1510
249. Serr I, Fürst RW, Achenbach P, Scherm MG, Gökmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3+ Treg induction in humanized mice. Nat Commun. (2016) 7:1–18. doi: 10.1038/ncomms10991
250. Bergerot I, Ploix C, Petersen J, Moulin V, Rask C, Fabien N, et al. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc Natl Acad Sci USA. (1997) 94:4610–4. doi: 10.1073/pnas.94.9.4610
251. Arakawa T, Yu J, Chong DKX, Hough J, Engen PC, Langridge WHR. A plant-based cholera toxin B subunit-insulin fusion protein protects against the developinent of autoimmune diabetes. Nat Biotechnol. (1998) 16:934–8. doi: 10.1038/nbt1098-934
252. Aspord C, Thivolet C. Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice. Clin Exp Immunol. (2002) 130:204–11. doi: 10.1046/j.1365-2249.2002.01988.x
253. Odumosu O, Payne K, Baez M, Jutzy J, Wall N, Langridge W. Suppression of dendritic cell activation by diabetes autoantigens linked to the cholera toxin B subunit. Immunobiology. (2011) 216:447–56. doi: 10.1021/nn2045246.Multifunctional
254. Vincent BG, Young EF, Buntzman AS, Stevens R, Kepler TB, Tisch RM, et al. Toxin-coupled MHC class I tetramers can specifically ablate autoreactive CD8+ T cells and delay diabetes in nonobese diabetic mice. J Immunol. (2010) 184:4196–204. doi: 10.4049/jimmunol.0903931
255. Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Leonard C, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci Trans Med. (2013) 5:1–20. doi: 10.1126/scitranslmed.3006103.Plasmid-Encoded
256. Liu X, Zhang S, Li X, Zheng P, Hu F, Zhou Z. Vaccination with a co-expression DNA plasmid containing GAD65 fragment gene and IL-10 gene induces regulatory CD4+ T cells that prevent experimental autoimmune diabetes. Diabetes/Metabolism Res Rev. (2016) 32:522–33. doi: 10.1002/dmrr
257. Alleva DG, Rezaee M, Yip L, Ren G, Rosenberg J, Concepcion W, et al. Reversal of hyperglycemia and suppression of type 1 diabetes in the NOD mouse with apoptotic DNA immunotherapyTM (ADiTM), ADi-100. Biomedicines. (2020) 8:1–13. doi: 10.3390/biomedicines8030053
258. Postigo-Fernandez J, Creusot RJ. A multi-epitope DNA vaccine enables a broad engagement of diabetogenic T cells for tolerance in Type 1 diabetes. J Autoimmun. (2019) 98:13–23. doi: 10.1016/j.jaut.2018.11.003.A
259. Yu C, Burns JC, Robinson WH, Utz PJ, Ho PP, Steinman L, et al. Identification of candidate tolerogenic CD8+ T cell epitopes for therapy of type 1 diabetes in the NOD mouse model. J Diabetes Res. (2016) 2016:1–12. doi: 10.1155/2016/9083103
260. Takiishi T, Cook DP, Korf H, Sebastiani G, Mancarella F, Cunha JPMCM, et al. Reversal of diabetes in NOD Mice by clinical-grade proinsulin and IL-10-secreting Lactococcus lactis in combination with low-dose Anti-CD3 depends on the induction of Foxp3-Positive T Cells. Diabetes. (2017) 66:448–59. doi: 10.2337/db15-1625
261. Cook DP, Cunha JPMCM, Martens PJ, Sassi G, Mancarella F, Ventriglia G, et al. Intestinal Delivery of Proinsulin and IL-10 via Lactococcus lactis Combined With Low-Dose Anti-CD3 Restores Tolerance Outside the Window of Acute Type 1 Diabetes Diagnosis. Front Immunol. (2020) 11:1103. doi: 10.3389/fimmu.2020.01103
262. Robert S, Gysemans C, Takiishi T, Korf H, Spagnuolo I, Sebastiani G, et al. Oral delivery of glutamic acid decarboxylase (GAD)-65 and IL10 by lactococcus lactis reverses diabetes in recent-onset NOD mice. Diabetes. (2014) 63:2876–87. doi: 10.2337/db13-1236
263. Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model. ACS Biomaterials Sci Eng. (2019) 5:2631–46. doi: 10.1021/acsbiomaterials.9b00332
264. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signaling. (2016) 9:1–11. doi: 10.1126/scisignal.aad0612
265. Dul M, Nikolic T, Stefanidou M, McAteer MA, Williams P, Mous J, et al. Conjugation of a peptide autoantigen to gold nanoparticles for intradermally administered antigen specific immunotherapy. Int J Pharmaceutics. (2019) 562:303–12. doi: 10.1016/j.ijpharm.2019.03.041
266. Tsai S, Shameli A, Yamanouchi J, Clemente-casares X, Wang J, Serra P, et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity. (2010) 32:568–80. doi: 10.1016/j.immuni.2010.03.015
267. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
268. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Lonnie D, et al. Tolerogenic ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun. (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010.Tolerogenic
269. Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, et al. Nanoparticles containing an insulin–chgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J Immunol. (2019) 203:48–57. doi: 10.4049/JIMMUNOL.1900127/-/DCSUPPLEMENTAL
270. Chen N, Kroger CJ, Tisch RM, Bachelder EM, Ainslie KM. Prevention of type 1 diabetes with acetalated dextran microparticles containing rapamycin and pancreatic peptide P31. Advanced Healthcare Materials. (2018) 7:1–12. doi: 10.1002/adhm.201800341
271. Bergot A-S, Buckle I, Cikaluru S, Naranjo JL, Wright CM, Talekar M, et al. Regulatory T cells induced by single-peptide liposome immunotherapy suppress islet-specific T cell responses to multiple antigens and protect from autoimmune diabetes. J Immunol. (2020) 204:1787–97. doi: 10.4049/jimmunol.1901128
272. Pishesha N, Bilate AM, Wibowo MC, Huang N-J, Li Z, Deshycka R, et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc Natl Acad Sci USA. (2017) 114:3157–62. doi: 10.1073/pnas.1705149114
273. Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci. (2013) 110:E60–8. doi: 10.1073/pnas.1216353110
274. Dudziak D, Kampfhorst A, Heidkamp G, Buhcholz V, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. (2007) 315:107–12. doi: 10.1126/science.1136080
275. Schloss J, Ali R, Babad J, Guerrero-Ros I, Pongsachai J, He L-Z, et al. Development and characterization of a preclinical model for the evaluation of CD205-mediated antigen delivery therapeutics in type 1 diabetes. ImmunoHorizons. (2019) 3:236–53. doi: 10.4049/immunohorizons.1900014
276. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. (2005) 6:1219–27. doi: 10.1038/ni1265
277. Kretschmer K, Heng TSP, von Boehmer H. De novo production of antigen-specific suppressor cells in vivo. Nat Protoc. (2006) 1:653–61. doi: 10.1038/nprot.2006.105
278. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol. (2008) 181:1–24. doi: 10.4049/jimmunol.181.10.6923
279. Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, et al. Selective delivery of β cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc Natl Acad Sci USA. (2008) 105:6374–9. doi: 10.1073/pnas.0802644105
280. Petzold C, Riewaldt J, Koenig T, Schallenberg S, Kretschmer K. Dendritic cell-targeted pancreatic β-cell antigen leads to conversion of self-reactive CD4+ T cells into regulatory T cells and promotes immunotolerance in NOD mice. Rev Diabetic Stud. (2010) 7:47–61. doi: 10.1900/RDS.2010.7.47
281. Mukherjee G, Geliebter A, Babad J, Santamaria P, Serreze DV, Freeman GJ, et al. DEC-205-mediated antigen targeting to steady-state dendritic cells induces deletion of diabetogenic CD8+ T cells independently of PD-1 and PD-L1. Int Immunol. (2013) 25:651–60. doi: 10.1093/intimm/dxt031
282. Price JD, Hotta-Iwamura C, Zhao Y, Beauchamp NM, Tarbell KV. DCIR2+ cDC2 DCs and Zbtb32 restore CD4+ T-cell tolerance and inhibit diabetes. Diabetes. (2015) 64:3521–31. doi: 10.2337/db14-1880
283. Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot H-J, et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide—for type 1 diabetes. Lancet Diabetes Endocrinol. (2020) 8:470–2. doi: 10.1016/S2213-8587(20)30104-2
284. Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med. (1991) 174:791–8. doi: 10.1084/jem.174.4.791
285. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
286. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. (2004) 199:1467–77. doi: 10.1084/jem.20040180
287. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. (2007) 204:191–201. doi: 10.1084/jem.20061631
288. Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+ CD25+ Regulatory T cells from nonobese diabetic mice. J Immunol. (2005) 175:3053–9. doi: 10.4049/jimmunol.175.5.3053
289. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun. (2019) 103:1–10. doi: 10.1016/j.jaut.2019.05.017
290. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Hellerstein MK, Herold KC, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med. (2015) 7:1–34. doi: 10.1126/scitranslmed.aad4134.Type
291. Yang SJ, Singh AK, Drow T, Tappen T, Honaker Y, Barahmand-Pour-Whitman F, et al. Pancreatic islet-specific engineered Tregs exhibit robust antigen-specific and bystander immune suppression in type 1 diabetes models. Sci Transl Med. (2022) 14:eabn1716. doi: 10.1126/scitranslmed.abn1716
292. Spanier JA, Fung V, Wardell CM, Alkhatib MH, Chen Y, Swanson LA, et al. Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J Clin Invest. (2023) 133(18). doi: 10.1172/JCI168601
293. Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. (2020) 6:eaaz3223. doi: 10.1126/sciadv.aaz3223
294. Alshaiki F, Obaid E, Almuallim A, Taha R, El-Haddad H, Almoallim H. Outcomes of rituximab therapy in refractory lupus: A meta-analysis. Eur J Rheumatol. (2018) 5:118–26. doi: 10.5152/eurjrheumatol.
295. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Trans Med. (2019) 11(482). doi: 10.1126/scitranslmed.aav1648
296. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
297. Reincke SM, von Wardenburg N, Homeyer MA, Kornau HC, Spagni G, Li LY, et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell. (2023) 186:5084–5097.e5018. doi: 10.1016/j.cell.2023.10.001
298. Zhang L, Stadinski BD, Michels A, Kappler JW, Eisenbarth GS. Immunization with an insulin peptide-MHC complex to prevent type 1 diabetes of NOD mice. Diabetes/Metabolism Res Rev. (2011) 27:784–9. doi: 10.1002/dmrr
299. Zhang L, Sosinowski T, Cox RA, Cepeda JR, Sekhar NS, Hartig SM, et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J Autoimmun. (2019) 96:50–8. doi: 10.1016/j.jaut.2018.08.004.Chimeric
300. Matsumoto Y, Kishida K, Matsumoto M, Matsuoka S, Kohyama M, Suenaga T, et al. A TCR-like antibody against a proinsulin-containing fusion peptide ameliorates type 1 diabetes in NOD mice. Biochem Biophys Res Commun. (2021) 534:680–6. doi: 10.1016/j.bbrc.2020.11.019
Keywords: autoimmune diabetes, autoantigens, T cells, hybrid insulin peptides, antigen specific therapy
Citation: Dwyer AJ, Shaheen ZR and Fife BT (2024) Antigen-specific T cell responses in autoimmune diabetes. Front. Immunol. 15:1440045. doi: 10.3389/fimmu.2024.1440045
Received: 28 May 2024; Accepted: 23 July 2024;
Published: 15 August 2024.
Edited by:
Monica Neagu, Victor Babes National Institute of Pathology (INCDVB), RomaniaReviewed by:
Saeed Mohammadi, Golestan University of Medical Sciences, IranAaron Michels, University of Colorado, United States
Copyright © 2024 Dwyer, Shaheen and Fife. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zachary R. Shaheen, c2hhaGUwMDFAdW1uLmVkdQ==
†These authors have contributed equally to this work