 Yuanhui Wu
Yuanhui Wu Jiao Luo
Jiao Luo Lihua Duan
Lihua Duan- 1Jiangxi Province Key Laboratory of Immunity and Inflammation, Jiangxi Provincial People's Hospital, Nanchang, China
- 2Department of Rheumatology and Clinical Immunology, Jiangxi Provincial People’s Hospital, The First Affiliated Hospital of Nanchang Medical College, Nanchang, China
- 3JXHC Key Laboratory of Rheumatology and Immunology, Jiangxi Provincial People’s Hospital, Nanchang, China
Idiopathic inflammatory myopathies (IIMs) encompass a spectrum of autoimmune diseases characterized by muscle inflammation and systemic involvement. This review aimed to synthesize current evidence on the clinical significance and pathogenic mechanisms underlying autoantibodies associated with IIMs. Autoantibodies targeting aminoacyl-tRNA synthetases (ARS) play a pivotal role in antisynthetase syndrome (ASS), highlighting associations with interstitial lung disease (ILD) and distinctive clinical features. Anti-Mi-2 antibodies in dermatomyositis (DM) are hallmarked by characteristic cutaneous manifestations and favorable prognostic outcomes. Conversely, anti-TIF1 antibodies are correlated with DM and a higher risk of malignancies, implicating CD8+ T cells in its pathogenesis. Anti-MDA5 antibodies signify clinically amyopathic DM (CADM) with severe ILD, linked to dysregulated neutrophil extracellular trap (NET) formation. In immune-mediated necrotizing myopathies (IMNMs), anti-SRP and anti-HMGCR antibodies induce complement-mediated myopathy, typically following statin exposure. Additionally, anti-TRIM72 antibodies emerge as potential diagnostic markers in IIMs. Anti-cN1A autoantibodies are linked to inclusion body myositis (IBM) and play a decisive role in muscle protein degradation. Meanwhile, anti-FHL1 autoantibodies are associated with severe disease manifestations and muscle damage, as established in experimental models. Anti-eIF3 autoantibodies, recently identified in polymyositis (PM) patients, are rarely detected (<1%) and associated with a favorable prognosis. Elucidating these autoantibodies is anticipated to not only assist in early diagnosis and disease stratification but also inform targeted therapeutic interventions, emphasizing the intricate interplay between autoimmunity, cellular dysfunction, and clinical outcomes in IIMs.
Introduction
Idiopathic inflammatory myopathies (IIMs) are heterogeneous autoimmune diseases, including dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), inclusion body myositis (IBM), and anti-synthetase syndrome (ASS), that are characterized by inflammation of the proximal arm and leg muscles, as well as other organs, such as the skin, lung, and joint (1–4).
Autoantibodies are detectable in up to 80% of the patients with IIMs. They can be categorized into two major groups, namely myositis-specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs), based on their specificity and clinical correlations. The former is typically characterized by a high level of specificity and holds significant value for both diagnosis and prognosis, particularly in categorizing patients into distinct or uniform subgroups for treatment purposes (2, 5–8). MSAs identified so far are autoantibodies against aminoacyl transfer RNA synthetases (ARS) including histidyl (Jo 1), alanyl (PL 12), threonyl (PL 7), glycyl (EJ) (9), isoleucyl (OJ), asparaginyl (KS), tyrosyl (Ha) and phenylalanyl (Zo) (10), signal recognition particle (SRP), melanoma differentiation-associated (MDA) 5/CADM 140, transcription intermediary factor 1 (TIF1), nuclear matrix protein (NXP) 2/MJ, Mi 2, 3-hydroxy-3-methylglutaryl-coA reductase (HMGCR) and small ubiquitin-like-modifier activating enzyme (SAE) (8, 11–13). MAAs are autoantibodies that are also present in other conditions that may culminate in myositis, such as systemic sclerosis (SSc) and systemic lupus erythematosus. MAAs include antibodies to SSA/Ro52, PM/Scl75, PM/Scl100, U1RNP, U1RNP, Ku and La (1, 6, 12, 14–21). The different types of MSAs and MAAs described in IIM have varying relevance for disease stratification, prognosis, and management.
The pathophysiology of IIMs is intricate and multifaceted and remains elusive. Its pathogenesis has been hypothesized to be influenced by genetic, environmental, and immunological factors. Notably, an increasing number of autoantibodies have been identified in recent years. Several autoantibodies play a clear pathogenic role in autoimmune disorders such as Grave’s disease, wherein thyrotropin receptor autoantibodies stimulate the overproduction of thyroid hormones, ultimately leading to hyperthyroidism. In myasthenia gravis, autoantibodies bind to acetylcholine receptors and inhibit the neurotransmitter acetylcholine from reaching muscle fibers, thereby inducing muscle weakness and fatigue (22). Nonetheless, the role of autoantibodies targeting endogenous antigens in disease pathogenesis or their presence as an epiphenomenon remains controversial (7, 23–26).
The specific clinical phenotypes associated with each MSA have raised questions on the role of these autoantibodies in the pathogenesis of IIM. Thus, this review examined the distinct clinical features associated with MSAs that potentially play a pathogenic role in the disease mechanisms. Thereafter, existing evidence from clinical longitudinal studies and experimental studies that might validate the pathogenic role of MSAs was outlined. However, the role of these antibodies in IIM is controversial. Our discussion focused on MSAs associated with the clinical subgroups of IIM and the possible pathogenic mechanism of autoantibody in myositis (Table 1). However, given the paucity of data suggesting that MAAs play a role in the pathogenesis of IIM, they were not comprehensively discussed in this review.
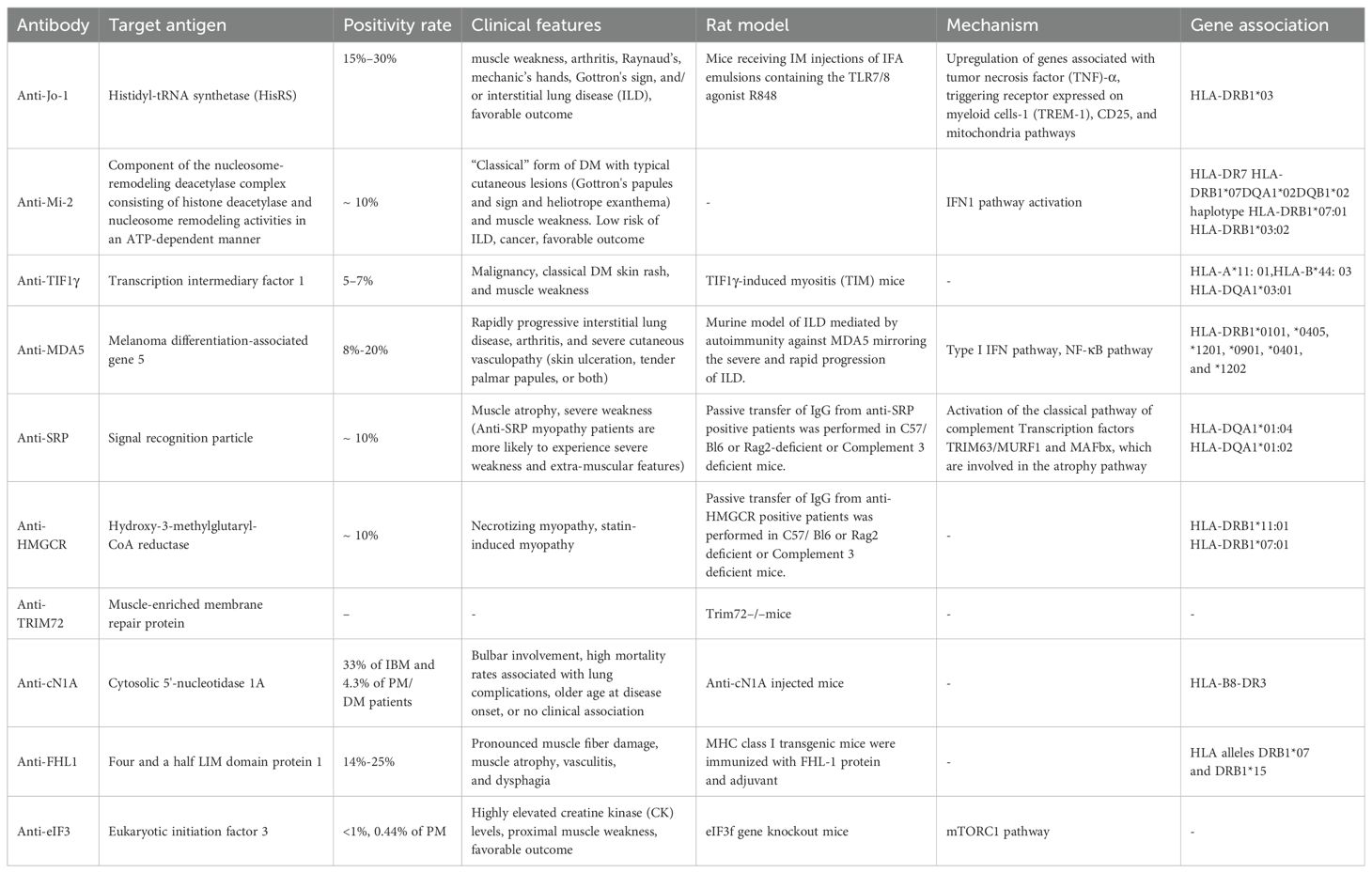
Table 1. MSAs in idiopathic inflammatory myopathies.
Anti-aminoacyl-tRNA synthetases autoantibodies
As is well documented, aminoacyl-transfer RNA synthetases (ARSs) are enzymes responsible for the first step of protein synthesis, attaching amino acids to their corresponding cognate transfer RNA (tRNA) sequences (17, 21, 27). Autoantibodies targeting ARSs, the most frequent MSAs detected in patients with IIMs, are associated with a distinct clinical phenotype termed “antisynthetase syndrome (ASS)”, characterized by the presence of one anti-ARS antibody plus one or more of the following manifestations: interstitial lung disease (ILD), myositis, arthritis, Raynaud’s phenomenon, fever, or mechanic’s hands (28, 29). Notably, ARS has been detected in 25%-35% of patients with ASS. To date, autoantibodies targeting 8 out of 21 ARSs have been identified and associated with ASS. While other less common antisynthetase autoantibodies have been reported in the literature, they are infrequently studied in the clinical setting. Known targets are histidyl (Jo1), threonyl (PL-7), alanyl (PL12), glycyl (EJ), isoleucyl (OJ), tyrosyl (Ha/YRS), asparagyl (KS), phenylalanyl (Zo), lysyl (SC), glutaminyl (JS), and tryptophanyl (WRS) tRNA synthetases (30–33).
Anti-Jo-1
Anti-Jo1 [anti-histidyl-tRNA synthetase (HisRS)] is the most common MSA that is present in 20-30% of IIM patients (2, 21, 34). A meta-analysis involving 27 studies investigating the clinical characteristics of ASA concluded that anti-Jo1 is linked to a higher risk of mechanic’s hands, arthritis, and myositis. Patients with anti-Jo-1 positive antibodies may experience Raynaud’s phenomenon before developing myositis (27). Worldwide epidemiological studies have documented that up to 90% of patients with anti-Jo1 autoantibodies develop ILD (4, 9, 26, 31, 34, 35). In the American and European Network of Antisynthetase Syndrome (AENEAS) cohort study recruiting anti-Jo1-positive patients, ILD was present in 50% of cases at disease onset and in 84% of patients after an 80-month follow-up period (19, 28). Moreover, anti-Jo-1-positive ASA patients have higher 5- and 10-year survival rates compared to non-Jo-1 patients, potentially ascribed to earlier diagnosis facilitated by the increased availability of anti-Jo1 testing (33). MHC class II alleles have been reported to be associated with certain MSAs, such as anti-Jo1 autoantibodies and the HLA 8.1 ancestral haplotype containing HLA-DRB1*03:01 (36). In addition, environmental factors can also induce ASS. Indeed, a strong association was noted between anti-Jo1 antibodies and HLA-DRB1*03:01 is strongest in patients with a smoking history.
Several experiments in human sera and passive transfer to mice indicate an immune response toward HisRS. In vitro studies have demonstrated that the N-terminal domain serves as a chemoattractant for naïve lymphocytes and immature dendritic cells through interaction with CCR5 (37). However, it is unknown whether or how the anti-Jo-1 antibodies regulate the chemokine activity of Jo-1. Instead, anti-Jo1 can form an immune complex (IC) that stimulates the synthesis and release of type I interferon by plasmacytoid dendritic cells (38). Furthermore, T-cell stimulation assays using a peptide from the HisRS N-terminal domain elicited an inflammatory response in blood and bronchoalveolar T-cells (29, 39, 40). Additionally, germinal center-like structures were identified in the lung tissue of anti-Jo1-positive patients, supporting the hypothesis of the lungs as a potential site for immune activation and production of anti-Jo1 autoantibodies. Numerous mice experiments have been performed to explore the role of HisRS in the pathogenesis of the disease. Initially, mice immunized with HisRS and adjuvant generated anti-HisRS antibodies but did not develop myositis, implying a species-specific antigenic immune response. In contrast, cDNA inoculation of human HisRS in mice induced inflammatory infiltrates in muscle along with detectable anti-HisRS antibodies, especially when using a truncated gene containing the N-terminal domain of the HisRS coding region, indicating different immune responses upon antigen presentation. Moreover, in an adjuvant-based model, the administration of murine N-terminal HisRS was capable of breaking tolerance and promoting T-cell proliferation and epitope spreading, causing muscle and lung inflammation similar to the antisynthetase syndrome (39). Histological studies of muscle tissues revealed diverse infiltration patterns, with perimysial/epimysial inflammation in a perivascular distribution, endomysial inflammation, and muscle fiber invasion/degeneration. High levels of anti-HisRS antibodies in bronchoalveolar lavage fluid and serum were detected in this mouse model (39). Furthermore, in an antigen-driven model, several strains of mice immunized with murine N-terminal HisRS displayed early T-cell infiltration in muscle and IgG class-switched autoantibody responses that persisted for at least 7 weeks (39). Further mouse experiments have explored the role of the MyD88 signaling pathways and highlighted the contributions of TLR2 and TLR4 (41). C3H/HeJ (TLR4-KO) mice deficient in anti-HisRS antibodies exhibited muscle inflammation induced by immunization with HisRS. These findings signal a key role for innate immune responses, but not HisRS-specific autoimmunity, in a HisRS-induced model of myositis (16). These findings collectively suggest that HisRS may induce myositis, but these models do not fully unravel the link between innate and adaptive immune responses. Taken together, additional in vitro and in vivo experiments are warranted to assess the mechanism of anti-Jo-1 antibodies in myositis.
Anti-Mi-2 autoantibody
Anti-Mi-2 is a dermatomyositis-specific autoantibody that targets antigens such as Mi-2a (240 kDa) and Mi-2b (218 kDa), thereby forming a protein complex with histone deacetylases, referred to as the nucleosome remodeling deacetylase (NuRD) complex. This autoantibody has been detected in patients with hallmark cutaneous DM lesions, including Gottron’s papules, heliotrope rash, cuticular overgrowth, and rashes on the neck and upper back or shoulders (V neck and shawl sign) (2, 6, 42, 43). The prevalence of anti-Mi-2 autoantibodies in DM patients varies from 5-10% in adults and 4-10% in juveniles. Furthermore, anti-Mi-2-positive patients typically have relatively mild muscle involvement, fewer complications such as ILD or cardiac disease, and are generally responsive to treatment with a favorable prognosis (43, 44).
Previous studies have established that anti-Mi-2 DM is associated with prominent pathological muscle involvement hallmarked by marked inflammatory cell infiltration. Tanboon et al. (45) concluded that anti-Mi-2 DM patients had a higher level of CD3- and CD20+ cell infiltration in the endomysium and CD68+ cell infiltration in the perimysium compared to non-Mi-2 DM patients. Anti-Mi-2 DM was also more frequently associated with CD20+ cell aggregation and ACP/CD68 cell infiltration in non-necrotic fibers. These findings, together with higher CK levels, may account for the more severe clinical muscle involvement observed in anti-Mi-2 DM patients (46).
Meanwhile, a higher level of CD68+ cell infiltration in the perimysium may explain the more frequent perimysial connective tissue alkaline phosphatase (ALP) activity in anti-Mi-2 DM patients. Considering that the expression of tissue nonspecific alkaline phosphatases can be up-regulated by cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), a subset of CD68+ cells in the perimysium has been speculated to secrete these cytokines, resulting in increased perimysial connective tissue ALP activity in anti-Mi-2 DM patients. On the contrary, perimysial connective tissue fragmentation could be attributed to inflammation and edema, which may account for the comparable percentages of perimysial connective tissue fragmentation between the anti-Mi-2 and non-Mi-2 DM groups.
In individuals with myositis autoantibodies, antibodies accumulate within myofibers in the same subcellular compartment as their corresponding autoantigens. Each autoantibody exerted effects that were in line with the malfunction of its corresponding autoantigen, such as internalization of antibodies from anti-Mi2 patients causing the derepression of Mi2/NURD-regulated genes (47). The study further evinced that anti-Mi2 autoantibodies exerted pathogenic effects by infiltrating damaged myofibers and inhibiting the CHD4/NuRD complex (48).
Besides, several studies have examined the relationship between exposure to ultraviolet (UV) light and the risk of anti-Mi-2-positive dermatomyositis. Love et al. determined that UV radiation intensity was correlated with the incidence of DM and anti-Mi-2-positivity (30, 49, 50). Burd et al. described that UV light upregulated Mi-2 expression in human keratinocytes (51). Given the relationship between anti-Mi-2 and HLA-DRB1*07DQA1*02DQB1*02 haplotype, HLA-DRB1*07:01 and HLA-DRB1*03:02, genetic background, as well as environmental factors, may influence the development of anti-Mi-2-positive DM.
Anti-TIF1 autoantibody
Anti-TIF1 targets TIF1γ of 155 kDa with or without TIF1α, formerly referred to as anti-p155 (52) and as anti-155/140 (53). Anti-TIF1 autoantibodies are specifically present in DM (15-20% of adult DM and 20% of juvenile DM cases). Anti-TIF1γ has a positive association with malignancy, especially when co-occurring with anti-TIF1α and a negative association with ILD (7). A meta-analysis involving 6 studies and 312 adult DM patients reported that the pooled sensitivity, specificity, and diagnostic OR of anti-p155 for the diagnosis of cancer-associated DM was 78% (95%, CI 45%–94%), 89% (95%, CI 82%–93%), and 27.26% (95%, CI 6.59%–112.82%), respectively (2, 49, 54, 55). Lastly, genetic susceptibility studies established that anti-TIF1 antibody was associated with HLA-DQA1*03:01 (52).
Anti-TIF1γ antibody-positive DM is linked to a higher risk of malignancy, especially in older patients (56). The presence of TIF1γ is significant in both cancerous tissues and during pregnancy, suggesting that it serves as a potential trigger for autoimmunity against TIF1γ (57). TIF1γ is frequently mutated or over-expressed in tumors and is over-expressed in embryonic and mammary epithelial cells during pregnancy. Therefore, cancer and pregnancy have been postulated to trigger autoimmunity against TIF1γ, which, in turn, contributes to the development of myositis. Konishi et al. established a TIF1γ-induced myositis model (TIM) in B6 mice via weekly subcutaneous injections of recombinant human TIF1γ protein emulsified in CFA four times, along with an intraperitoneal injection of pertussis toxin (PT) (16). As anticipated, the immunized mice developed TIF1γ-specific T cells and anti-human and murine TIF1γ antibodies, resulting in myositis in the hamstrings and quadriceps two weeks after the last immunization. Histological studies revealed atrophy and necrosis of muscle fibers, accompanied by infiltrating mononuclear cells in the perifascicular and endomysial sites of muscle tissues. Immunohistochemistry assays illustrated that CD8+ T cells predominantly infiltrated and adhered to muscle fibers, which upregulated the expression of MHC class I and type I IFN-responsive molecule Mx1. Beta 2 microglobulin-KO mice lacking MHC class I expression, perforin-KO mice, and anti-CD8 depleting antibody-treated mice rarely develop TIM (58). Meanwhile, adoptive transfer experiments demonstrated that CD8+ T cells derived from TIM mice could induce myositis in recipient B6 mice, whereas CD4+ T cells did not exert this effect. These findings identify CD8+ T cells as the primary pathogenic cells in TIM. In contrast, μMT mice, which completely lack B-cell lineages, developed myositis, whereas the adoptive transfer of IgGs collected from TIM mice failed to induce myositis in recipient mice. These results collectively indicate that B cells and autoantibodies are not essential for developing TIM. In other words, anti-TIF1γ antibodies detected in patients with DM may be a diagnostic biomarker but not a direct pathogenic factor. Accordingly, TIM, which is dependent on autoimmunity against TIF1γ, was mediated by TIF1γ-specific CD8+ T cells but not TIF1γ-specific CD4+ T cells, B cells, and autoantibodies.
The study evinced that B cells and autoantibodies are not essential for the development of TIF1γ-induced myositis (TIM), establishing that CD8+ T cells play a primary pathogenic role. Thus, the presence of anti-TIF1γ antibodies in patients with DM primarily serves as a diagnostic biomarker rather than a direct cause of the disease. The involvement of type I interferons in the pathogenesis of TIM and the effectiveness of tofacitinib treatment highlight potential therapeutic targets. The TIM model thus provides a robust framework for studying DM and pioneering targeted treatments.
Anti-MDA5 autoantibody
Anti-MDA5, reported to be a specific autoantibody for clinically amyopathic DM (CADM), was first named anti-CADM-140 in 2005 (59, 60). Subsequently, the target autoantigen was identified as melanoma differentiation-associated gene 5 (MDA5), also known as interferon-induced with helicase C domain protein 1 (IFIH1). Of note, MDA5 is a cytoplasmic retinoic acid-inducible gene-I (RIG-I)-like receptor that, upon the recognition of viral RNA, up-regulates the expression of type 1 interferon and other inflammatory cytokines (2, 49, 61).
Furthermore, the anti-MDA5 autoantibody is detected in 20-50% of adult DM patients (including CADM) and is associated with relatively lower creatine kinase (CK) levels, a high frequency of ILD (90-95%), especially rapidly progressive ILD (RP-ILD) (50-80%), and poor prognosis due to respiratory failure (62–65). However, lower frequencies of the antibody and RP-ILD were noted in American and European cohorts (49). Such clinical discrepancies might be explained by differences in ethnicity or environmental background. Anti-MDA5 antibody is also associated with HLA-DRB1*01:01/*04:05 (66).
Increased serum IL-6, IL-8, and IL-10 levels were associated with RP-ILD in PM/DM patients, whilst high concentrations of IFN-α and soluble CD163 and the upregulation of IFN-inducible genes have been detected in anti-MDA5-positive patients. These findings suggest the upregulation of the type 1 IFN system through the activation of monocytes, macrophages, or other immunocompetent cells in the pathophysiology of anti-MDA5-positive DM with RP-ILD (59). Depending on disease severity, autoAbs profiles differ among anti-MDA5+ patients, with autoAbs from B cells of patients directly stimulating IFN-gamma production in the peripheral blood. Mounting evidence indicates that dysregulated NET formation participates in the pathogenic process of IIM, particularly in anti-MDA5-positive disease (67, 68). Circulating NET levels were increased while the plasma DNase I activity was impaired, resulting in the failure to degrade the aberrant NETs in patients with anti-MDA5 autoantibodies, especially in those with ILD. Enhanced NET formation was observed in affected organs, including the skin, muscle, and lungs, of anti-MDA5-positive patients but not in those with MSA-negative IIM (67).
A recent study also delineated that anti-MDA5 autoantibodies isolated from patients significantly enhanced NET formation in neutrophils isolated from healthy controls compared with control IgG. Furthermore, NETs purified from IIM neutrophils have been observed to induce myotube damage. Another study described that peripheral NET levels were associated with calcinosis, ICs, and IL-8 levels in patients with JDM (49, 69). In particular, JDM patients with anti-MDA5 autoantibodies exhibited impaired NET clearance. Indeed, anti-MDA5 autoantibodies play a critical role in inducing NET formation. The study emphasizes the need for further investigation into the mechanisms underlying NET dysregulation and its contributions to autoimmune responses and tissue damage in IIM.
In addition, an earlier study found that a murine model of ILD mediated by autoimmunity against MDA5 mirrors the severe and rapid progression of ILD observed in patients with anti-MDA5 antibody-positive DM. Key findings highlighted the vital role of CD4+ T cells and IL-6 in the development and severity of fibrotic ILD. The results suggest that targeting IL-6 could be a potential therapeutic approach for the management of ILD in patients with anti-MDA5 antibody-positive DM (70). Animal models are valuable for studying disease mechanisms, given that they allow for controlled experimentation and manipulation of specific immune components. The evidence from these models convincingly unveiled that T cells can induce disease independently of autoantibodies, emphasizing their role in disease pathogenesis. Despite the compelling evidence from animal models, there are limitations that cannot be overlooked. Animal models may not fully reflect the complexity of human disease, and findings in animals may not always translate directly to humans. Besides, the presence of autoantibodies in patients with DM is associated with specific clinical features and outcomes, suggesting they still play a role in the disease, albeit not as primary inducers.
Anti-SRP and anti-HMGCR autoantibodies
Anti-signal recognition particle (anti-SRP) antibody was initially identified in a subgroup of polymyositis patients in 1986 (71). In 2002, muscle the presence of necrotic muscle fibers without significant muscle inflammation was detected in biopsies from anti-SRP antibody-positive patients (72). In 2003, a group of immune-mediated necrotizing myopathies (IMNMs) was recognized for the first time as a separate entity, based on pathological criteria showing predominant muscle fiber necrosis with no or mild muscle infiltrates, and these patients generally have a poor prognosis (73–75). A novel myositis-specific antibody targeting the hydroxy-3-methylglutaryl-CoA reductase (HMGCR) protein was discovered in a subset of IMNM patients thereafter (6, 7, 76–78).
Anti-SRP patients exhibit more severe muscle weakness and atrophy with substantial muscle damage in magnetic resonance imaging studies (79–81). Anti-SRP is detected in 2% of patients with adult dermatomyositis. Moreover, approximately 10-20% of anti-SRP patients develop extramuscular symptoms, especially ILD (80). Anti-SRP was associated with HLA-DQA1*01:04 and HLA-DQA1*01:02. Conversely, anti-HMGCR patients are often linked to statin exposure (82, 83) and were detected in 6% of adult dermatomyositis cases. Importantly, it was associated with HLA-DRB1*11:01 and HLA-DRB1*07:01. For both autoantibodies, a high correlation between CK levels and MAC (C5b-9) deposits with the percentage of myofiber necrosis was reported (r=0.6, p < 0.01 and r=0.4, p < 0.01, respectively) (84). The titer of these auto-antibodies correlates with disease activity in anti-SRP and anti-HMGCR-positive patients (49). Histopathological analysis of muscle biopsies from anti-HMGCR-positive patients revealed muscle fiber degeneration and regeneration, as well as up-regulation of MHC-I on occasional non-necrotic muscle fibers with rare or absent inflammatory infiltrates (22, 76, 83, 85, 86). Furthermore, necrotic fibers were largely associated with CD68+ macrophage infiltration and membrane attack complex (MAC) deposition on scattered non-necrotic fibers, suggesting the presence of an antibody-dependent cell-mediated toxicity pathway (87).
These autoantibodies were associated with an increased secretion of proinflammatory cytokines (IL-6 and TNF), reduction in the levels of anti-inflammatory cytokines (IL-4 and IL-13), production of reactive oxygen species, and upregulation of genes encoding atrophic factors. This decrease of IL-4 and IL-13 suppressed myotube formation by impairing myoblasts fusion. In in vitro experiments on myotubes, incubation with anti-SRP antibodies, anti-HMGCR antibodies, or total IgG from patients’ plasmapheresis induced atrophy and was associated with increased expression of the transcription factors TRIM63/MURF1 and MAFbx, which are involved in the atrophy pathway. In vitro experiments showed that purified anti-SRP and anti-HMGCR autoantibodies could recognize their cognate autoantigens and activate the classical complement pathway (87). Experimental studies in animal models further support the pathogenic role of the anti-SRP and anti-HMGCR autoantibodies (88, 89). Purified IgG from anti-HMGCR and anti-SRP-positive patients was injected in mice, provoking muscle deficiency and myofiber necrosis, similar to human disease (89). Interestingly, muscle deficiency tends to be more severe in mice receiving IgGs from anti-SRP antibody-positive patients compared to those receiving IgGs from anti-HMGCR antibody-positive patients. Immunization with SRP and HMGCR protein drove the production of specific antibodies, indicating a pathogenic association with these proteins. Myopathy in IMNM was alleviated in IgG-transferred complement C3-KO mice, whereas supplementation with human complement reversed this effect (16). The study implied that patient-derived anti-SRP and anti-HMGCR antibodies are pathogenic toward muscles in vivo through a complement-mediated mechanism. However, treatment with a C5 complement inhibitor was not effective in anti-SRP-positive and anti-HMGCR-positive patients (90).
The research highlights the dual diagnostic and pathogenic role of anti-SRP and anti-HMGCR autoantibodies in IMNMs, as well as their potential impact on muscle pathology through cytokine modulation, macrophage infiltration, and complement activation.
Anti-TRIM72 autoantibody
Several tripartite motif (TRIM) family proteins (Ro52, TIF1α, TIF1β, and TIF1γ) are well-established autoantigens associated with IIM (91–93). a novel TRIM family protein termed TRIM72 (also known as MG53) and its function were identified in IIM. TRIM72 is an integral component of the sarcolemmal repair process in striated muscle (94–96). ELISA analysis uncovered elevated TRIM72 autoantibody levels in IIM, with 11.5% of DM sera and 11.8% of PM sera tested presenting with high levels of anti-TRIM72 (97). Trim72–/– mice develop significant skeletal muscle myopathy and cardiovascular defects due to defective sarcolemmal repair (98–100). In an adoptive transfer mouse model, sarcolemmal resealing defects were detected at 1 and 4 weeks, indicating that in a Treg-deficient/dysfunctional environment, a marginal increase in anti-TRIM72 levels is correlated with reduced sarcolemmal resealing capacity. Additionally, exogenous delivery of a polyclonal antibody against TRIM72 significantly reduced sarcolemmal resealing capacity in flexor digitorum brevis (FDB) muscles from healthy C57BL mice regardless of anti-TRIM72 levels. Overall, the findings suggest that a defect in sarcolemmal resealing may precede skeletal muscle degeneration and inflammation associated with IIM (97). Taken together, these findings highlight the essential role of TRIM72 in muscle membrane repair and its potential involvement in the pathogenesis of IIM through autoantibody production and impaired membrane resealing.
Anti-cN1A autoantibody
Anti-cytosolic 5’- nucleotidase 1A (cN1A) was described in 2011 by Salajegheh et al. as an autoantibody against a 43 kDa protein associated with inclusion body myositis (IBM) (1, 7, 101, 102). The corresponding autoantigen was identified as cN1A expressed in skeletal muscle (103). This autoantigen is implicated in the hydrolysis of adenosine monophosphate, leading to physiological energy homeostasis, metabolic regulation, and cell replication (103, 104). Anti-cN1A is present in about 33-34% of IBM, 4-5% of PM, and 3-4% of DM cases. Considering that Herbert et al. concluded that this autoantibody is present in 36% of patients with Sjogren’s syndrome and 20% of SLE cases, the specificity of this autoantibody for myositis is likely low (105, 106). it might assist in differentiating between myositis subgroups (49, 106–108).
A recent study demonstrated the pathogenic role of anti-cN1A in IBM both in vivo and in vitro using a passive immunization model. The anti-cN1A autoantibody potentially affects protein degradation in myofibers (109). Several experimental in vivo and in vitro passive immunization studies in mice were performed. The results similarly identified that anti-cN1A antibodies could impact muscle protein degradation and fiber size with small angulated fibers in mice injected with anti-cN1A-positive sIBM IgG. However, these experiments failed to demonstrate changes in motor activities. The findings conjointly indicate that anti-cN1A autoantibodies play a role in the pathology of IBM by altering muscle protein degradation and muscle fiber morphology. Nonetheless, the lack of changes in observed motor activity suggests further research is warranted to fully elucidate the clinical impact of these autoantibodies.
Anti-FHL1 autoantibody
Four-and-a-half-LIM-domain 1 (FHL1) is a muscle-specific antigen abundantly expressed in skeletal and cardiac muscle. Mutations in the FHL1 gene have been detected in diverse X-linked myopathies (1, 110, 111). The prevalence of anti-FHL1 autoantibodies has been reported to range between 14 and 25% of IIM patients and has been associated with poor prognostic characteristics such as pronounced muscle fiber damage, muscle atrophy, vasculitis, and dysphagia. In an independent cohort, anti-FHL1 autoantibody frequency was higher in PM and IBM patients and was frequently observed in those with MSA-negative IIM.
To explore the role of FHL1, MHC class I transgenic mice were immunized with FHL1 protein and adjuvant, which resulted in muscle inflammation, weakness, and weight loss nine weeks after the immunization in double-transgenic mice but not in single-transgenic mice. The presence of anti-FHL1 autoantibody was detected in both immunized mice groups. HT mice had significantly lower survival rates, and histopathological examination exposed prominent muscle damage with IgM depositions, suggesting a link between anti-FHL1 responses and muscle damage. Anti-FHL1 autoantibodies serve as markers for more severe disease in IIM, and the experimental data reinforce their pathogenic role, underscoring the importance of FHL1 in the disease mechanism.
In total, the presence of anti-FHL1 autoantibodies in IIM is indicative of more severe disease manifestations. The experimental evidence from transgenic mice corroborates the pathogenic role of anti-FHL1 responses, emphasizing the role of FHL1 in muscle damage and inflammation in IIM patients.
Anti-eIF3 autoantibody
The anti-eukaryotic initiation factor 3 (eIF3) autoantibody was recently identified in the sera of three Caucasian patients with PM. Noteworthily, the level of this autoantibody was low (<1%) and was associated with a good prognosis and a favorable response to treatment (1). The depletion of eIF3 in mouse models was associated with reduced skeletal muscle mass, indicating that this protein might play a paramount role in muscle growth and skeletal muscle homeostasis (1, 112) However, these associations remain to be investigated in further studies.
Conclusion
This review summarizes the classical and novel MSAs in IIM identified over the past years. These autoantibodies are preferentially expressed in disease-associated tissues and play a major role in disease initiation and propagation. For some of the MSAs, experimental data support their potential role in the pathogenesis of IIM. Understanding the functional role of these autoantigens and their corresponding autoantibodies in disease initiation, propagation, and expression is fundamental for providing further insights into pathogenic pathways, which in turn may facilitate the development of new therapeutic targets.
Author contributions
YW: Writing – original draft, Writing – review & editing. JL: Writing – original draft, Writing – review & editing. LD: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. It was supported by National Natural Science Foundation of China (82360325, 82371773, 62363028), Jiangxi Province Key Laboratory of Immunology and Inflammation (No. 2024SSY06251, Science and Technology Research Project of Jiangxi Provincial Department of Education (GJJ218901), Jiangxi Provincial Health Technology Key Project (2024ZD003), Jiangxi Provincial Science and Technology Department key research and development plan (NO. 20232BBG70026).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Galindo-Feria AS, Wang G, Lundberg IE. Autoantibodies: pathogenic or epiphenomenon. Best Pract Res Clin Rheumatol. (2022) 36:101767. doi: 10.1016/j.berh.2022.101767
2. Kang EH, Ha YJ, Lee YJ. Autoantibody biomarkers in rheumatic diseases. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21041382
3. Dorph C, Lundberg IE. Idiopathic inflammatory myopathies - myositis. Best Pract Res Clin Rheumatol. (2002) 16:817–32. doi: 10.1053/berh.2002.0261
4. Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. (2010) 138:1464–74.
5. Gunawardena H, Betteridge ZE, McHugh NJ. Newly identified autoantibodies: relationship to idiopathic inflammatory myopathy subsets and pathogenesis. Curr Opin Rheumatol. (2008) 20:675–80. doi: 10.1097/BOR.0b013e328313bff4
6. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatol (Oxford). (2009) 48:607–12. doi: 10.1093/rheumatology/kep078
7. Liu Y, Zheng Y, Hao H, Yuan Y. Narrative review of autoantibodies in idiopathic inflammatory myopathies. Ann Transl Med. (2023) 11:291. doi: 10.21037/atm
8. McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol. (2018) 14:290–302. doi: 10.1038/nrrheum.2018.56
9. Targoff IN, Trieu EP, Plotz PH, Miller FW. Antibodies to glycyl-transfer RNA synthetase in patients with myositis and interstitial lung disease. Arthritis Rheum. (1992) 35:821–30. doi: 10.1002/art.1780350718
10. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatol (Oxford). (2007) 46:1005–8. doi: 10.1093/rheumatology/kem045
11. Hamaguchi Y, Fujimoto M, Matsushita T, Kaji K, Komura K, Hasegawa M, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PloS One. (2013) 8:e60442. doi: 10.1371/journal.pone.0060442
12. Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol. (2000) 12:475–81. doi: 10.1097/00002281-200011000-00001
13. Watanabe K, Handa T, Tanizawa K, Hosono Y, Taguchi Y, Noma S, et al. Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias. Respir Med. (2011) 105:1238–47. doi: 10.1016/j.rmed.2011.03.022
14. Babu AK, Mizaj Z, Thomas J, Chacko M. Clinical significance of myositis-specific and myositis-associated antibody profiles in dermatomyositis. Indian Dermatol Online J. (2023) 14:55–60. doi: 10.4103/idoj.idoj_188_22
15. Sloupenska K, Koubkova B, Horak P, Hutyrova B, Racansky M, Mares J, et al. Myositis autoantibodies in patients with suspected post-treatment lyme disease syndrome. Life (Basel). (2023) 13. doi: 10.3390/life13020527
16. Konishi R, Ichimura Y, Okiyama N. Murine models of idiopathic inflammatory myopathy. Immunol Med. (2023) 46:9–14. doi: 10.1080/25785826.2022.2137968
17. Fujikawa K, Kawakami A, Kaji K, Fujimoto M, Kawashiri S, Iwamoto N, et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: a single-centre, cross-sectional study. Scand J Rheumatol. (2009) 38:263–7. doi: 10.1080/03009740802687455
18. Marguerie C, Bunn CC, Beynon HL, Bernstein RM, Hughes JM, So AK, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. (1990) 77:1019–38. doi: 10.1093/qjmed/77.1.1019
19. Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, Albayda J, Paik JJ, Johnson C, et al. A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatol (Oxford). (2017) 56:999–1007. doi: 10.1093/rheumatology/kex021
20. Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. (1980) 23:881–8. doi: 10.1002/art.1780230802
21. Bunn CC, Bernstein RM, Mathews MB. Autoantibodies against alanyl-tRNA synthetase and tRNAAla coexist and are associated with myositis. J Exp Med. (1986) 163:1281–91. doi: 10.1084/jem.163.5.1281
22. Rosen A, Casciola-Rosen L. Autoantigens in systemic autoimmunity: critical partner in pathogenesis. J Intern Med. (2009) 265:625–31. doi: 10.1111/j.1365-2796.2009.02102.x
23. Gherardi RK. Pathogenic aspects of dermatomyositis, polymyositis and overlap myositis. Presse Med. (2011) 40:e209–18. doi: 10.1016/j.lpm.2010.12.013
24. Mammen AL, Allenbach Y, Stenzel W, Benveniste O, EtWS Group. 239th ENMC international workshop: classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 december 2018. Neuromuscul Disord. (2020) 30:70–92. doi: 10.1016/j.nmd.2019.10.005
25. Pinal-Fernandez I, Amici DR, Parks CA, Derfoul A, Casal-Dominguez M, Pak K, et al. Myositis autoantigen expression correlates with muscle regeneration but not autoantibody specificity. Arthritis Rheumatol. (2019) 71:1371–6. doi: 10.1002/art.40883
26. Bartoloni E, Gonzalez-Gay MA, Scire C, Castaneda S, Gerli R, Lopez-Longo FJ, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: Results from a multicenter, international and retrospective study. Autoimmun Rev. (2017) 16:253–7. doi: 10.1016/j.autrev.2017.01.008
27. Lega JC, Fabien N, Reynaud Q, Durieu I, Durupt S, Dutertre M, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. (2014) 13:883–91. doi: 10.1016/j.autrev.2014.03.004
28. Cavagna L, Nuno L, Scire CA, Govoni M, Longo FJL, Franceschini F, et al. Clinical spectrum time course in anti jo-1 positive antisynthetase syndrome: results from an international retrospective multicenter study. Med (Baltimore). (2015) 94:e1144. doi: 10.1097/MD.0000000000001144
29. Ramos-Martinez E, Falfan-Valencia R, Perez-Rubio G, Mejia M, Buendia-Roldan I, Gonzalez-Perez MI, et al. Anti-aminoacyl transfer-RNA-synthetases (Anti-tRNA) autoantibodies associated with interstitial lung disease: pulmonary disease progression has a persistent elevation of the th17 cytokine profile. J Clin Med. (2020) 9. doi: 10.3390/jcm9051356
30. Betteridge ZE, Gunawardena H, McHugh NJ. Pathogenic mechanisms of disease in myositis: autoantigens as clues. Curr Opin Rheumatol. (2009) 21:604–9. doi: 10.1097/BOR.0b013e328331638a
31. Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. (2012) 12:210–7. doi: 10.1016/j.autrev.2012.06.006
32. Yoshifuji H, Fujii T, Kobayashi S, Imura Y, Fujita Y, Kawabata D, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. (2006) 39:233–41. doi: 10.1080/08916930600622884
33. Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. (2014) 73:227–32. doi: 10.1136/annrheumdis-2012-201800
34. Hirakata M, Suwa A, Takada T, Sato S, Nagai S, Genth E, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. (2007) 56:1295–303. doi: 10.1002/art.22506
35. Richards TJ, Eggebeen A, Gibson K, Yousem S, Fuhrman C, Gochuico BR, et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. (2009) 60:2183–92. doi: 10.1002/art.24631
36. O’Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Med (Baltimore). (2006) 85:111–27. doi: 10.1097/01.md.0000217525.82287.eb
37. Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. (2002) 196:781–91. doi: 10.1084/jem.20020186
38. Eloranta ML, Helmers Barbasso S, Ulfgren AK, Ronnblom L, Alm GV, Lundberg IE. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthritis Rheum. (2007) 56:3112–24. doi: 10.1002/art.22860
39. Katsumata Y, Ridgway WM, Oriss T, Gu X, Chin D, Wu Y, et al. Species-specific immune responses generated by histidyl-tRNA synthetase immunization are associated with muscle and lung inflammation. J Autoimmun. (2007) 29:174–86. doi: 10.1016/j.jaut.2007.07.005
40. Galindo-Feria AS, Albrecht I, Fernandes-Cerqueira C, Notarnicola A, James EA, Herrath J, et al. Proinflammatory histidyl-transfer RNA synthetase-specific CD4+ T cells in the blood and lungs of patients with idiopathic inflammatory myopathies. Arthritis Rheumatol. (2020) 72:179–91.
41. Soejima M, Kang EH, Gu X, Katsumata Y, Clemens PR, Ascherman DP. Role of innate immunity in a murine model of histidyl-transfer RNA synthetase (Jo-1)-mediated myositis. Arthritis Rheum. (2011) 63:479–87. doi: 10.1002/art.30113
42. Hengstman GJ, Egberts Vree WT, Seelig HP, Lundberg IE, Moutsopoulos HM, Doria A, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis. (2006) 65:242–5. doi: 10.1136/ard.2005.040717
43. Komura K, Fujimoto M, Matsushita T, Kaji K, Kondo M, Hirano T, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatol Sci. (2005) 40:215–7. doi: 10.1016/j.jdermsci.2005.09.004
44. Liang L, Zhang YM, Chen H, Ye LF, Li SS, Lu X, et al. Anti-Mi-2 antibodies characterize a distinct clinical subset of dermatomyositis with favourable prognosis. Eur J Dermatol. (2020). doi: 10.1684/ejd.2020.3750
45. Tanboon J, Inoue M, Hirakawa S, Tachimori H, Hayashi S, Noguchi S, et al. Pathologic features of anti-mi-2 dermatomyositis. Neurology. (2021) 96:e448–59. doi: 10.1212/WNL.0000000000011269
46. Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. (1985) 28:796–803. doi: 10.1002/art.1780280711
47. Pinal-Fernandez I, Munoz-Braceras S, Casal-Dominguez M, Pak K, Torres-Ruiz J, Musai J, et al. Pathogenic autoantibody internalization in myositis. medRxiv. (2024). doi: 10.1101/2024.01.15.24301339
48. Pinal-Fernandez I, Milisenda JC, Pak K, Munoz-Braceras S, Casal-Dominguez M, Torres-Ruiz J, et al. Transcriptional derepression of CHD4/NuRD-regulated genes in the muscle of patients with dermatomyositis and anti-Mi2 autoantibodies. Ann Rheum Dis. (2023) 82:1091–7. doi: 10.1136/ard-2023-223873
49. Nakashima R. Clinical significance of myositis-specific autoantibodies. Immunol Med. (2018) 41:103–12. doi: 10.1080/25785826.2018.1531188
50. Love LA, Weinberg CR, McConnaughey DR, Oddis CV, Medsger TA Jr, Reveille JD, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. (2009) 60:2499–504. doi: 10.1002/art.24702
51. Burd CJ, Kinyamu HK, Miller FW, Archer TK. UV radiation regulates Mi-2 through protein translation and stability. J Biol Chem. (2008) 283:34976–82. doi: 10.1074/jbc.M805383200
52. Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O'Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. (2006) 54:3682–9. doi: 10.1002/art.22164
53. Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with Malignancy. Rheumatol (Oxford). (2007) 46:25–8. doi: 10.1093/rheumatology/kel161
54. Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. (2012) 64:513–22. doi: 10.1002/art.33403
55. Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, Balada E, Martinez MA, Milisenda JC, et al. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatol (Oxford). (2018) 57:388–96. doi: 10.1093/rheumatology/kex413
56. Aussy A, Boyer O, Cordel N. Dermatomyositis and immune-mediated necrotizing myopathies: A window on autoimmunity and cancer. Front Immunol. (2017) 8:992. doi: 10.3389/fimmu.2017.00992
57. Oya K, Inoue S, Saito A, Nakamura Y, Ishitsuka Y, Fujisawa Y, et al. Pregnancy triggers the onset of anti-transcriptional intermediary factor 1gamma antibody-positive dermatomyositis: a case series. Rheumatol (Oxford). (2020) 59:1450–1. doi: 10.1093/rheumatology/kez527
58. Uruha A, Nishikawa A, Tsuburaya RS, Hamanaka K, Kuwana M, Watanabe Y, et al. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology. (2017) 88:493–500. doi: 10.1212/WNL.0000000000003568
59. Nombel A, Fabien N, Coutant F. Dermatomyositis with anti-MDA5 antibodies: bioclinical features, pathogenesis and emerging therapies. Front Immunol. (2021) 12:773352. doi: 10.3389/fimmu.2021.773352
60. Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Fujita T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. (2009) 60:2193–200. doi: 10.1002/art.24621
61. Li X, Liu Y, Cheng L, Huang Y, Yan S, Li H, et al. Roles of biomarkers in anti-MDA5-positive dermatomyositis, associated interstitial lung disease, and rapidly progressive interstitial lung disease. J Clin Lab Anal. (2022) 36:e24726. doi: 10.1002/jcla.24726
62. Chen Z, Cao M, Plana MN, Liang J, Cai H, Kuwana M, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res (Hoboken). (2013) 65:1316–24. doi: 10.1002/acr.21985
63. Labrador-Horrillo M, Martinez MA, Selva-O'Callaghan A, Trallero-Araguas E, Balada E, Vilardell-Tarres M, et al. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J Immunol Res. (2014) 2014:290797. doi: 10.1155/2014/290797
64. Chino H, Sekine A, Baba T, Iwasawa T, Okudela K, Takemura T, et al. Radiological and pathological correlation in anti-MDA5 antibody-positive interstitial lung disease: rapidly progressive perilobular opacities and diffuse alveolar damage. Intern Med. (2016) 55:2241–6. doi: 10.2169/internalmedicine.55.5774
65. Tsuji H, Nakashima R, Hosono Y, Imura Y, Yagita M, Yoshifuji H, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. (2020) 72:488–98. doi: 10.1002/art.41105
66. Gono T, Kawaguchi Y, Kuwana M, Sugiura T, Furuya T, Takagi K, et al. Brief report: Association of HLA-DRB1*0101/*0405 with susceptibility to anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis in the Japanese population. Arthritis Rheum. (2012) 64:3736–40. doi: 10.1002/art.34657
67. Seto N, Torres-Ruiz JJ, Carmona-Rivera C, Pinal-Fernandez I, Pak K, Purmalek MM, et al. Neutrophil dysregulation is pathogenic in idiopathic inflammatory myopathies. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.134189
68. Peng Y, Zhang S, Zhao Y, Liu Y, Yan B. Neutrophil extracellular traps may contribute to interstitial lung disease associated with anti-MDA5 autoantibody positive dermatomyositis. Clin Rheumatol. (2018) 37:107–15. doi: 10.1007/s10067-017-3799-y
69. Duvvuri B, Pachman LM, Morgan G, Khojah AM, Klein-Gitelman M, Curran ML, et al. Neutrophil extracellular traps in tissue and periphery in juvenile dermatomyositis. Arthritis Rheumatol. (2020) 72:348–58. doi: 10.1002/art.41078
70. Ichimura Y, Konishi R, Shobo M, Tanaka R, Kubota N, Kayama H, et al. Autoimmunity against melanoma differentiation-associated gene 5 induces interstitial lung disease mimicking dermatomyositis in mice. Proc Natl Acad Sci U.S.A. (2024) 121:e2313070121. doi: 10.1073/pnas.2313070121
71. Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci U.S.A. (1986) 83:9507–11.
72. Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry. (2002) 73:420–8. doi: 10.1136/jnnp.73.4.420
73. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep. (2018) 20:21. doi: 10.1007/s11926-018-0732-6
74. Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. (2004) 50:209–15. doi: 10.1002/art.11484
75. Allenbach Y, Keraen J, Bouvier AM, Jooste V, Champtiaux N, Hervier B, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. (2016) 139:2131–5. doi: 10.1093/brain/aww054
76. Allenbach Y, Benveniste O. Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Curr Opin Rheumatol. (2018) 30:655–63. doi: 10.1097/BOR.0000000000000547
77. Anquetil C, Boyer O, Wesner N, Benveniste O, Allenbach Y. Myositis-specific autoantibodies, a cornerstone in immune-mediated necrotizing myopathy. Autoimmun Rev. (2019) 18:223–30. doi: 10.1016/j.autrev.2018.09.008
78. Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. (2010) 62:2757–66.
79. Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, Lahouti AH, Basharat P, Albayda J, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis. (2017) 76:681–7. doi: 10.1136/annrheumdis-2016-210198
80. Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. (1990) 33:1361–70. doi: 10.1002/art.1780330908
81. Allenbach Y, Benveniste O, Goebel HH, Stenzel W. Integrated classification of inflammatory myopathies. Neuropathol Appl Neurobiol. (2017) 43:62–81. doi: 10.1111/nan.12380
82. Tiniakou E, Pinal-Fernandez I, Lloyd TE, Albayda J, Paik J, Werner JL, et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatol (Oxford). (2017) 56:787–94. doi: 10.1093/rheumatology/kew470
83. Chung T, Christopher-Stine L, Paik JJ, Corse A, Mammen AL. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve. (2015) 52:189–95. doi: 10.1002/mus.24642
84. Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. (2012) 64:4087–93. doi: 10.1002/art.34673
85. Afzali AM, Ruck T, Wiendl H, Meuth SG. Animal models in idiopathic inflammatory myopathies: How to overcome a translational roadblock? Autoimmun Rev. (2017) 16:478–94. doi: 10.1016/j.autrev.2017.03.001
86. Alshehri A, Choksi R, Bucelli R, Pestronk A. Myopathy with anti-HMGCR antibodies: Perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e124. doi: 10.1212/NXI.0000000000000124
87. Rojana-udomsart A, Mitrpant C, Bundell C, Price L, Luo YB, Fabian V, et al. Complement-mediated muscle cell lysis: a possible mechanism of myonecrosis in anti-SRP associated necrotizing myopathy (ASANM). J Neuroimmunol. (2013) 264:65–70. doi: 10.1016/j.jneuroim.2013.08.008
88. Osaki Y, Nakagawa Y, Miyahara S, Iwasaki H, Ishii A, Matsuzaka T, et al. Skeletal muscle-specific HMG-CoA reductase knockout mice exhibit rhabdomyolysis: A model for statin-induced myopathy. Biochem Biophys Res Commun. (2015) 466:536–40. doi: 10.1016/j.bbrc.2015.09.065
89. Cole RN, Reddel SW, Gervasio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol. (2008) 63:782–9. doi: 10.1002/ana.21371
90. Bergua C, Chiavelli H, Allenbach Y, Arouche-Delaperche L, Arnoult C, Bourdenet G, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis. (2019) 78:131–9. doi: 10.1136/annrheumdis-2018-213518
92. Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. (2005) 201:591–601. doi: 10.1084/jem.20041367
93. Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, et al. The Sjogren’s syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol. (2006) 176:6277–85. doi: 10.4049/jimmunol.176.10.6277
94. Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. (2009) 11:56–64. doi: 10.1038/ncb1812
95. Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. (2012) 4:139ra85. doi: 10.1126/scitranslmed.3003921
96. He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol Ther. (2012) 20:727–35. doi: 10.1038/mt.2012.5
97. McElhanon KE, Young N, Hampton J, Paleo BJ, Kwiatkowski TA, Beck EX, et al. Autoantibodies targeting TRIM72 compromise membrane repair and contribute to inflammatory myopathy. J Clin Invest. (2020) 130:4440–55. doi: 10.1172/JCI131721
98. Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. (2010) 121:2565–74. doi: 10.1161/CIRCULATIONAHA.110.954628
99. Wang X, Xie W, Zhang Y, Lin P, Han L, Han P, et al. Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ Res. (2010) 107:76–83. doi: 10.1161/CIRCRESAHA.109.215822
100. Zhang Y, Lv F, Jin L, Peng W, Song R, Ma J, et al. MG53 participates in ischaemic postconditioning through the RISK signalling pathway. Cardiovasc Res. (2011) 91:108–15. doi: 10.1093/cvr/cvr029
101. Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PloS One. (2011) 6:e20266. doi: 10.1371/journal.pone.0020266
102. Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, et al. Cytosolic 5’-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. (2013) 73:408–18.
103. Careddu MG, Allegrini S, Pesi R, Camici M, Garcia-Gil M, Tozzi MG. Knockdown of cytosolic 5’-nucleotidase II (cN-II) reveals that its activity is essential for survival in astrocytoma cells. Biochim Biophys Acta. (2008) 1783:1529–35. doi: 10.1016/j.bbamcr.2008.03.018
104. Lucchini M, Maggi L, Pegoraro E, Filosto M, Rodolico C, Antonini G, et al. Anti-cN1A antibodies are associated with more severe dysphagia in sporadic inclusion body myositis. Cells. (2021) 10. doi: 10.3390/cells10051146
105. Herbert MK, Stammen-Vogelzangs J, Verbeek MM, Rietveld A, Lundberg IE, Chinoy H, et al. Disease specificity of autoantibodies to cytosolic 5’-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis. (2016) 75:696–701. doi: 10.1136/annrheumdis-2014-206691
106. Felice KJ, Whitaker CH, Wu Q, Larose DT, Shen G, Metzger AL, et al. Sensitivity and clinical utility of the anti-cytosolic 5’-nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: Report of 40 patients from a single neuromuscular center. Neuromuscul Disord. (2018) 28:660–4. doi: 10.1016/j.nmd.2018.06.005
107. Paul P, Liewluck T, Ernste FC, Mandrekar J, Milone M. Anti-cN1A antibodies do not correlate with specific clinical, electromyographic, or pathological findings in sporadic inclusion body myositis. Muscle Nerve. (2021) 63:490–6. doi: 10.1002/mus.27157
108. Lilleker JB, Rietveld A, Pye SR, Mariampillai K, Benveniste O, Peeters MT, et al. Cytosolic 5’-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis. (2017) 76:862–8. doi: 10.1136/annrheumdis-2016-210282
109. Tawara N, Yamashita S, Zhang X, Korogi M, Zhang Z, Doki T, et al. Pathomechanisms of anti-cytosolic 5’-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol. (2017) 81:512–25. doi: 10.1002/ana.24919
110. Cowling BS, McGrath MJ, Nguyen MA, Cottle DL, Kee AJ, Brown S, et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J Cell Biol. (2008) 183:1033–48. doi: 10.1083/jcb.200804077
111. Wilding BR, McGrath MJ, Bonne G, Mitchell CA. FHL1 mutants that cause clinically distinct human myopathies form protein aggregates and impair myoblast differentiation. J Cell Sci. (2014) 127:2269–81.
Keywords: autoantibodies, idiopathic inflammatory myopathies, dermatomyositis, polymyositis, autoantibodies targeting aminoacyl-tRNA synthetases (ARS)
Citation: Wu Y, Luo J and Duan L (2024) Pathogenic mechanisms of disease in idiopathic inflammatory myopathies: autoantibodies as clues. Front. Immunol. 15:1439807. doi: 10.3389/fimmu.2024.1439807
Received: 28 May 2024; Accepted: 08 August 2024;
Published: 30 August 2024.
Edited by:
Jianan Zhao, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Han Xu, Shandong University of Traditional Chinese Medicine, ChinaYu Shan, Shanghai University of Traditional Chinese Medicine, China
Ping Jiang, Shanghai Jiao Tong University, China
José Jiram Torres-Ruiz, National Institute of Medical Sciences and Nutrition Salvador Zubirán, Mexico
Copyright © 2024 Wu, Luo and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiao Luo, bHVvamlhbzAyQDE2My5jb20=; Lihua Duan, bGgtZHVhbkAxNjMuY29t