 Mohamed M. Shamseldin1,2,3†
Mohamed M. Shamseldin1,2,3† Kaitlin A. Read1†
Kaitlin A. Read1† Jesse M. Hall1†
Jesse M. Hall1† Jasmine A. Tuazon1
Jasmine A. Tuazon1 Jessica M. Brown1
Jessica M. Brown1 Myra Guo1Yash A. Gupta1
Myra Guo1Yash A. Gupta1 Rajendar Deora1,2
Rajendar Deora1,2 Kenneth J. Oestreich1,4
Kenneth J. Oestreich1,4 Purnima Dubey1,4,5*
Purnima Dubey1,4,5*- 1Departments of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, United States
- 2Departments of Microbiology, The Ohio State University, Columbus, OH, United States
- 3Department of Microbiology and Immunology, Faculty of Pharmacy, Helwan University-Ain Helwan, Helwan, Egypt
- 4Pelotonia Institute for Immuno-Oncology, The Ohio State University, Columbus, OH, United States
- 5Comprehensive Cancer Center, The Ohio State University, Columbus, OH, United States
Introduction: Adjuvants added to subunit vaccines augment antigen-specific immune responses. One mechanism of adjuvant action is activation of pattern recognition receptors (PRRs) on innate immune cells. Bordetella colonization factor A (BcfA); an outer membrane protein with adjuvant function, activates TH1/TH17-polarized immune responses to protein antigens from Bordetella pertussis and SARS CoV-2. Unlike other adjuvants, BcfA does not elicit a TH2 response.
Methods: To understand the mechanism of BcfA-driven TH1/TH17 vs. TH2 activation, we screened PRRs to identify pathways activated by BcfA. We then tested the role of this receptor in the BcfA-mediated activation of bone marrow-derived dendritic cells (BMDCs) using mice with germline deletion of TLR4 to quantify upregulation of costimulatory molecule expression and cytokine production in vitro and in vivo. Activity was also tested on human PBMCs.
Results: PRR screening showed that BcfA activates antigen presenting cells through murine TLR4. BcfA-treated WT BMDCs upregulated expression of the costimulatory molecules CD40, CD80, and CD86 and produced IL-6, IL-12/23 p40, and TNF-α while TLR4 KO BMDCs were not activated. Furthermore, human PBMCs stimulated with BcfA produced IL-6. BcfA-stimulated murine BMDCs also exhibited increased uptake of the antigen DQ-OVA, supporting a role for BcfA in improving antigen presentation to T cells. BcfA further activated APCs in murine lungs. Using an in vitro TH cell polarization system, we found that BcfA-stimulated BMDC supernatant supported TFH and TH1 while suppressing TH2 gene programming.
Conclusions: Overall, these data provide mechanistic understanding of how this novel adjuvant activates immune responses.
Introduction
Many approved vaccines are comprised of purified antigens admixed with an adjuvant, referred to as subunit vaccines. The inclusion of immune stimulatory adjuvants in vaccine formulations bolsters immune responses versus antigens alone, supports dose sparing, reduced frequency of administration (1, 2), and improves the stability and pharmacokinetics of the antigens leading to an increased in vivo half-life (2). Another notable advantage is the ability of the adjuvant to shape the phenotype of the resulting cellular and humoral responses (1).
Alum (aluminum hydroxide or aluminum phosphate) was the first adjuvant to be licensed for human use more than a century ago. For seven decades, it was the only adjuvant used in FDA- and EMA-approved vaccines administered to protect against a multitude of infectious diseases including hepatitis, pertussis, and diphtheria (3, 4). In the late 1990s, other adjuvants were approved for addition to previously licensed human vaccines (3). The first of these was the oil-in-water emulsion MF59 added to a trivalent influenza vaccine, FLUAD (5, 6). The more recently described AS01-AS04 family of adjuvants are TLR4 ligands and aim to maximize the immune response while maintaining tolerability using a mix of classical adjuvants and other immunostimulatory molecules (3). These are included in vaccines against shingles (7), malaria (8), pandemic influenza (9), HBV and HPV (10). The oligonucleotide cytosine phosphoguanosine 1018 (CpG 1018), is a TLR9 agonist included in a hepatitis B vaccine (11–13).
These approved adjuvants have different mechanisms of action and are safe and effective at generating T cell and antibody-mediated responses. Notably, however, they all elicit mixed TH1/TH2-polarized immunity (14). In contrast, natural immune responses to infection with most viral and bacterial pathogens elicit TH1/TH17-polarized T cell and antibody responses, which are correlated with sustained protection against disease and infections in both humans and mouse models (15–19). Recent studies (20) also highlight the importance of tissue-resident memory T (TRM) and B cell responses which provide sustained protection at barrier sites (21–24). Therefore, there is a need for safe novel adjuvants that elicit TH1/TH17-polarized immune responses, and also generate tissue-resident memory. To rationally design such adjuvants, it is critical to understand their mechanism of action. We previously identified Bordetella colonization factor A (BcfA), a bacterial protein of the Gram-negative pathogen Bordetella bronchiseptica, and we have reported that it functions as an adjuvant in vivo (25). Systemic administration of BcfA-containing vaccines elicited TH1- and TH17-polarized CD4+ and TH1-polarized CD8+ T cell responses (25), T follicular helper (TFH) cells, and TH1-skewed antibody responses (20) alone and in combination with alum. Mucosal immunization with antigens mixed with BcfA elicited CD4+IL-17+ TRM cells in the lungs and nose (20, 26). Importantly, when BcfA was mixed with alum-containing approved and experimental vaccines, TH2 responses were attenuated while TH1 and TH17 responses were sustained or amplified (20, 25), suggesting that BcfA can override the TH2 polarized responses elicited by alum. This unique property therefore supports the potentially broad applicability of BcfA as an adjuvant for bacterial and viral pathogens where TH1 and TH17 responses are important for protection against infection and disease.
Here, we investigated the mechanism of BcfA activation of immune responses. We report that BcfA activates bone marrow-derived dendritic cells (BMDCs) through the pattern-recognition receptor TLR4. We observed dose-dependent upregulation of costimulatory molecules CD40, CD80 and CD86 on wildtype (WT) BMDCs and the production of innate cytokines IL-6, TNFα and IL-12/23 p40. BcfA-activated BMDCs more efficiently processed and presented the model antigen DQ-OVA. Furthermore, BcfA-stimulated BMDC conditioned medium supported differentiation of TFH-like and TH1 CD4+ T cells in an in vitro culture system, demonstrating the ability of BcfA to shape therapeutically beneficial T cell responses. These results provide insight regarding the TLR-dependent mechanism of action of this novel adjuvant and suggest that inclusion of BcfA in next-generation vaccine formulations may represent a promising avenue to enhance protective long-term immunity.
Materials and methods
BcfA formulation
BcfA was produced and purified as described previously (27). Residual LPS was removed using MustangQ filters (Cytiva Life Sciences, Inc, Marlborough, MA, catalog no. MSTGXT25Q16) or polymyxin B agarose (Sigma-Aldrich, St. Louis MO, catalog no. P1411). Endotoxin was quantified [(LAL Chromogenic Endotoxin Quantitation Kit, Thermo Fisher Scientific, catalog no. 88282) and was ≤150 pg/µg protein.
PRR screening
HEK-Blue cells overexpressing murine TLR4 (catalog no. hkb-mtlr4) or murine TLR2 (catalog no. hkb-mtlr2) (InvivoGen, Inc. San Diego, CA) were stimulated with 1 or 5 µg/mL BcfA for 24hr in duplicate wells. Production of the co-expressed secreted embryonic alkaline phosphatase (SEAP) reporter was quantified. Minimal LPS (minLPS, 0.2ng/mL) in the BcfA preparation was used as negative control and purified LPS (100 µg/mL) (eBioscience, San Diego, CA catalog. no 00-4976-93) was used as positive control for stimulation.
Mice
All experiments were reviewed and approved by The Ohio State University (OSU) Institutional Animal Care and Use Committee (Protocol number 2017A00000090). C57BL/6J, TLR4 knockout mice on the C57BL/6 background were obtained from Jackson Laboratories and bred in our facility. Tail DNA from mice was genotyped by Transnetyx, Inc. (Cordova, TN) using validated probe sets to confirm genotypes.
Differentiation of murine bone marrow derived dendritic cells
Murine BMDCs were prepared according to published protocols (28). Briefly, bone marrow was isolated, dissociated into a single cell suspension, and red blood cells were lysed with ACK lysing buffer (Gibco Ref A10492). The cell suspension was resuspended in RPMI1640 (Gibco) + 10% fetal bovine serum (FBS, Sigma-Aldrich F4220), 10µg/mL gentamicin, 5x10-5M ß-mercaptoethanol and 40 ng/mL GM-CSF (R&D Systems, Minneapolis, MN, catalog no. 415-ML-020)) and seeded in 10 cm2 non-tissue culture treated petri dishes (VWR catalog no. 25384-342) at a density of 5-10 x106 cells/plate. Half of the medium was replaced every 2 days with the addition of fresh GM-CSF. On day 6-7 post-differentiation, BMDCs were transferred to 6-well tissue culture-treated plates (Falcon catalog no. 353046) at a density of 0.5-1 x106 cells/well. The next day, the cells were stimulated with various concentrations of BcfA for 20-24hr. Supernatant was collected for ELISA analysis and the cells were harvested for flow cytometry.
Collection of human PBMCs and stimulation with BcfA
Peripheral blood was collected from adult human donors under a protocol approved by The Ohio State University Institutional Review Board (Protocol numbers 2020H0404 and 2021H0179). Whole blood was collected in EDTA treated tubes. PBMCs were separated from whole blood on a Percoll gradient (Cytiva, catalog no. 17144003), and cryo-preserved at -80°C. Cells were thawed and cultured in RPMI + 10% human AB serum (Sigma Aldrich, catalog no. H4522). To determine TLR4 expression, PBMCs were stained with α-human TLR4 (Thermo Fisher Scientific catalog no. 12-9041-80) antibody. Cells were analyzed on a Cytek Aurora spectral flow cytometer. Fluorescence minus one (FMO) controls were used for gating. To test cytokine production, 1-2x106 cells from individual donors were stimulated for 20-24hr and the supernatant was tested by ELISA.
Enzyme-linked immunosorbent assay for innate cytokines
The production of murine TNF-α, IL-6, IL-12/23 p40 common γ chain and IL-12 was quantified by a sandwich ELISA according to the manufacturer’s instructions (Life Technologies or BioLegend). Human cytokines IL-6 and TNF-α were quantified using Quantikine ELISA kits (R&D Systems). Plates were read at A450 on a SpectraMax i3x® plate reader and concentrations were calculated based on the standard curve.
DQ-OVA uptake and processing
BMDCs were plated in 6 well plates at 1 x106 cells/well and stimulated with BcfA or cultured with medium alone. At 24 hr post-stimulation, DQ-OVA (InvivoGen, Inc. catalog no. D12053) was added at a concentration of 10 µg/mL for 45 min. The cells were then washed and harvested to quantify expression of CD11c, MHC-Class II and DQ-OVA by flow cytometry.
Immunization of mice
Mice were lightly anesthetized with 2.5% isoflurane/O2 for immunization. To stimulate APCs in the lungs, mice were immunized intranasally with 10 µg BcfA or LPS-EB VacciGrade™ (InvivoGen, catalog no. vac-3pelps) (a TLR4 agonist used as a positive control) in 50 µL divided between both nares. Lungs were harvested 24 hr post-inoculation.
Tissue dissociation and flow cytometry
BMDCs were harvested at 20-24hr post-stimulation and were washed with cold PBS prior to staining with Live/Dead Zombie NIR fixable viability dye (BioLegend, catalog no. 423105) for 30 min at 4°C. Cells were then washed twice with PBS supplemented with 1% heat-inactivated FBS (1% FBS) (FACS buffer) and resuspended in Fc Block (α-CD16/CD32 antibody, clone 93) (eBioscience, catalog no. 14-0161-86) at 4°C for 5 min before staining with a mixture of the following Abs for 20 min at 4°C: CD11c e450 (clone N418; Invitrogen, catalog no.48-0114-82), MHC Class II I-A/I-E BV785 (clone M5/114.15.2; Biolegend, catalog no.107645), CD40-APC (clone 1C10), CD80 PerCP-Cy5.5 (clone 16-10A1; Biolegend, catalog no.104722) and CD86 FITC (clone GL1; eBioscience, catalog no.11-0862-85).
Lungs were processed, digested (mouse lung dissociation kit, Miltenyi Biotec, catalog no. 130-095-927), and mechanically disrupted (gentleMACS) into a single-cell suspension followed by RBC lysis.Cells were washed with cold PBS and stained with Live/Dead Zombie NIR fixable viability dye (BioLegend, catalog no. 423105) for 30 min at 4°C, then washed with PBS/1% FBS and resuspended in Fc Block at 4°C for 5 min. Lung cells were stained with a mixture of the following antibodies for 20 min at 4°C: CD11b APC (clone M1/70; Invitrogen, catalog no.17-0112-82), CD11c e450 (clone N418; Invitrogen, catalog no.48-0114-82), MHC Class II I-A/I-E BV785 (clone M5/114.15.2; Biolegend, catalog no.107645), CD45 PE (clone 30-F11; BD Biosciences, catalog no.553081), CD40 PE-CF594 (clone 3/23; BD Biosciences, catalog no.562847), CD80 PerCP-Cy5.5 (clone 16-10A1; Biolegend, catalog no.104722) and CD86 FITC (clone GL1; eBioscience, catalog no.11-0862-85).
For intracellular cytokine staining, cells were first incubated in complete IMDM (cIMDM; IMDM [Life Technologies], 10% FBS [26140079, Life Technologies], 1% Penicillin-Streptomycin [Life Technologies], and 0.05% (50 mM) 2-ME [Sigma-Aldrich]) with eBioscience protein transport inhibitors (PTI; catalog no. 00-4980-93, Invitrogen) for 4 hr. For cell surface marker staining, samples were pre-incubated for 5 min at 4°C with TruStain FcX™ (α-mouse CD16/32) Fc block (clone 93; catalog no. 101320, BioLegend). Samples were then stained for extracellular markers in the presence of Fc block for 30 min at 4°C protected from light using the following antibodies: α-CD4 (PerCP-Cy5.5; 1:100; clone GK1.5; catalog no. 100434, BioLegend) and Ghost Dye (V510; 1:400; catalog no. 50-105-2992, Tonbo Biosciences). Cells were then washed twice with FACS buffer and were fixed and permeabilized using the eBioscience Foxp3 transcription factor staining kit (Thermo Fisher Scientific, catalog no. 00-8333-56) overnight at 4°C.
Following fixation, cells were washed once with the 1X eBioscience permeabilization buffer (Thermo Fisher Scientific). Recombinant Mouse IL-21R Fc Chimera primary antibody (catalog no. 596-MR, R&D) was diluted 1:5 in 1X eBioscience permeabilization buffer and 100 µL was added per well to incubate for 30 min at 4°C protected from light. Cells were washed once with 1X eBioscience permeabilization buffer. Goat F(ab’)2 Anti-Human IgG - Fc secondary antibody (PE, catalog no. ab98596, Abcam) was diluted 1:31.25 in 1X eBioscience permeabilization buffer and 100 µL was added per well to incubate for 30 min at 4°C protected from light. For staining the remaining intracellular markers, cells were washed once with 1X eBioscience permeabilization buffer and then stained with the following antibodies in 1X eBioscience permeabilization buffer for 1hr at room temperature protected from light: α-Bcl6 (AF488; 1:20; clone K112-91; catalog no. BDB561524, BD Biosciences); α-Gata3 (PE-Cy7; 1:20; clone TWAJ; catalog no. 25-9966-42, Invitrogen); α-IL-4 (APC; 1:50; clone 11B11; catalog no. 504106, BioLegend); α-IFN-γ (APC-Cy7; 1:300; clone XMG1.2; catalog no. 505850, BioLegend), and α-T-bet (PacBlue; 1:50; clone 4B10; catalog no. 644808, BioLegend). Cells were washed twice with 1X permeabilization buffer and resuspended in FACS buffer for analysis.
Fluorescence minus one or isotype control antibodies were used as negative controls. After two washes, cells were resuspended in PBS/1% FBS and samples were collected on or BD FACS Symphony or Cytek Aurora spectral flow cytometer (Cytek Biosciences).
Analysis was performed using FlowJo software version 10.8.0. The number of cells within each population was calculated by multiplying the frequency of live singlets in the population of interest by the total number of cells in each sample.
Preparing and assaying CD4+ T cells for lineage-defining transcription factors and effector cytokines
Naïve CD4+ T cells were purified from the spleen and lymph nodes of 5-8-week-old WT C57BL/6J mice using the BioLegend Mojosort kit. Cells were plated in 24-well plates on plate-bound anti-CD3 (5 μg/mL) and anti-CD28 (2μg/mL) in the presence or absence of IL-4 neutralizing antibody (11B11, BioLegend, 5μg/mL). After 16-20hr, medium from non-stimulated control (NS Ctrl) or BcfA-treated BMDCs was added to the well, with or without IL-4-neutralizing antibody. Where indicated, IL-6-neutralizing antibody (MP5-20F3, BioLegend, 10µg/mL) was also added. Cells were cultured for an additional 48hr before harvest and subsequent analysis.
RNA was isolated using the Macherey-Nagel Nucleospin kit per the manufacturer’s instructions, and complementary DNA (cDNA) was synthesized using the SYBR Superscript IV First Strand Synthesis System with oligo dT primers (Thermo Fisher). qRT-PCR reactions were run on the CFX Connect (BioRad) with 5-20ng of cDNA, using the SYBR Select Mastermix for CFX (ThermoFisher) and the following primers:
Bcl6 forward: 5’-CCAACCTGAAGACCCACACTC-3’, Bcl6 reverse: 5’-GCGGACATGGCTCTTCAGAGTC-3’; Il21 forward: 5’-TGGATCCTGAACTTCTATCAGCTCC -3’, Il21 reverse: 5’- AGGCAGCCTCCTCCTGAGC -3’; Tbx21 forward: 5’- GTGACTGCCTACCAGAACGC -3’, Tbx21 reverse: 5’- AGGGGACACTCGTATCAACAG -3’; Ifng forward: 5’- CTACCTTCTTCAGCAACAGC -3’, Ifng reverse: 5’- GCTCATTGAATGCTTGGCGC -3’. Data were normalized to Rps18 and are presented relative to either Rps18 or the control sample, as noted.
Statistical analysis
Data were analyzed using GraphPad Prism by the methods described in each figure legend.
Results
Activation of murine BMDCs by BcfA is dependent on TLR4
We hypothesized that BcfA activates antigen-presenting cells (APCs) through a PRR and conducted an empirical PRR screening using the HEK-Blue system, where human embryonic kidney (HEK) cells were transfected with the SEAP (secreted embryonic alkaline phosphatase) reporter gene under the control of a promoter inducible by NF-κB and activator protein-1 (AP-1) and expressing murine TLR (mTLR) or NOD receptors. SEAP reporter production was quantified using a colorimetric assay. The screen showed activation of mTLR4 and mTLR2 (Supplementary Figure S1A). We then stimulated cells expressing TLR4 (HEK-mTLR4) and TLR2 (HEK-mTLR2) with 1 or 5 µg/mL BcfA. Maximal SEAP release was detected from HEK cells expressing murine TLR4 (HEK-mTLR4) following stimulation with 1 µg/mL BcfA (Supplementary Figure S1B) at levels comparable to purified LPS used as a positive control. SEAP production from HEK-mTRL2 cells was nearly maximal at 5 µg/mL BcfA stimulation (Supplementary Figure S1C) with lower SEAP production detected with 1 µg/mL BcfA stimululation. To mitigate any confounding effects of BcfA produced in E. coli as a bacterial recombinant protein, we utilized a stringent purification procedure to remove LPS (27). HEK-mTLR4 or HEK-mTLR2 cells incubated with this minimal LPS (≤150 pg/µg protein) (min LPS) did not produce SEAP.
We then evaluated the BcfA stimulatory activity in a more physiologically relevant system. We isolated bone marrow from WT C57BL/6 mice and TLR4 KO mice and differentiated BMDCs in vitro. The immature DCs were treated with BcfA (1 or 5 µg/mL) for 24 hr and analyzed for the expression of costimulatory molecules by flow cytometry. BMDCs were identified as a CD11c+ MHC-IIhi population (Supplementary Figure S2) (29) and were evaluated for the expression of costimulatory molecules by flow cytometry. Representative flow plots are shown in Figure 1A. The mean fluorescence intensity (MFI) expression of CD40 (Figure 1B), CD80 (Figure 1C), and CD86 (Figure 1D) and percentage of cells expressing CD40 (Figure 1E) CD80 (Figure 1F) and CD86 (Figure 1G) was upregulated in response to BcfA stimulation compared to no stimulation (NS) control. LPS-EB (de-O-acelylated lipooligosaccharide) (5 µg/mL) that has been used as an adjuvant in vivo (30) and purified E. coli LPS (100 ng/mL) were included as positive controls for TLR4, and PAM3Csk was used as a positive control to confirm that TLR4 KO BMDCs respond to stimulation. Both the MFI and percentage of cells that upregulated costimulatory molecule expression was reduced to background in BcfA-stimulated TLR4 KO BMDCs, suggesting that BcfA primarily functions through this PRR. We also tested activation of TLR2 KO BMDCs which showed that costimulatory molecule expression was not reduced by the absence of TLR2 (Supplementary Figure S3), suggesting that BcfA primarily functions through TLR4. Thus, we focused on TLR4 mediated activity of BcfA in the following studies.
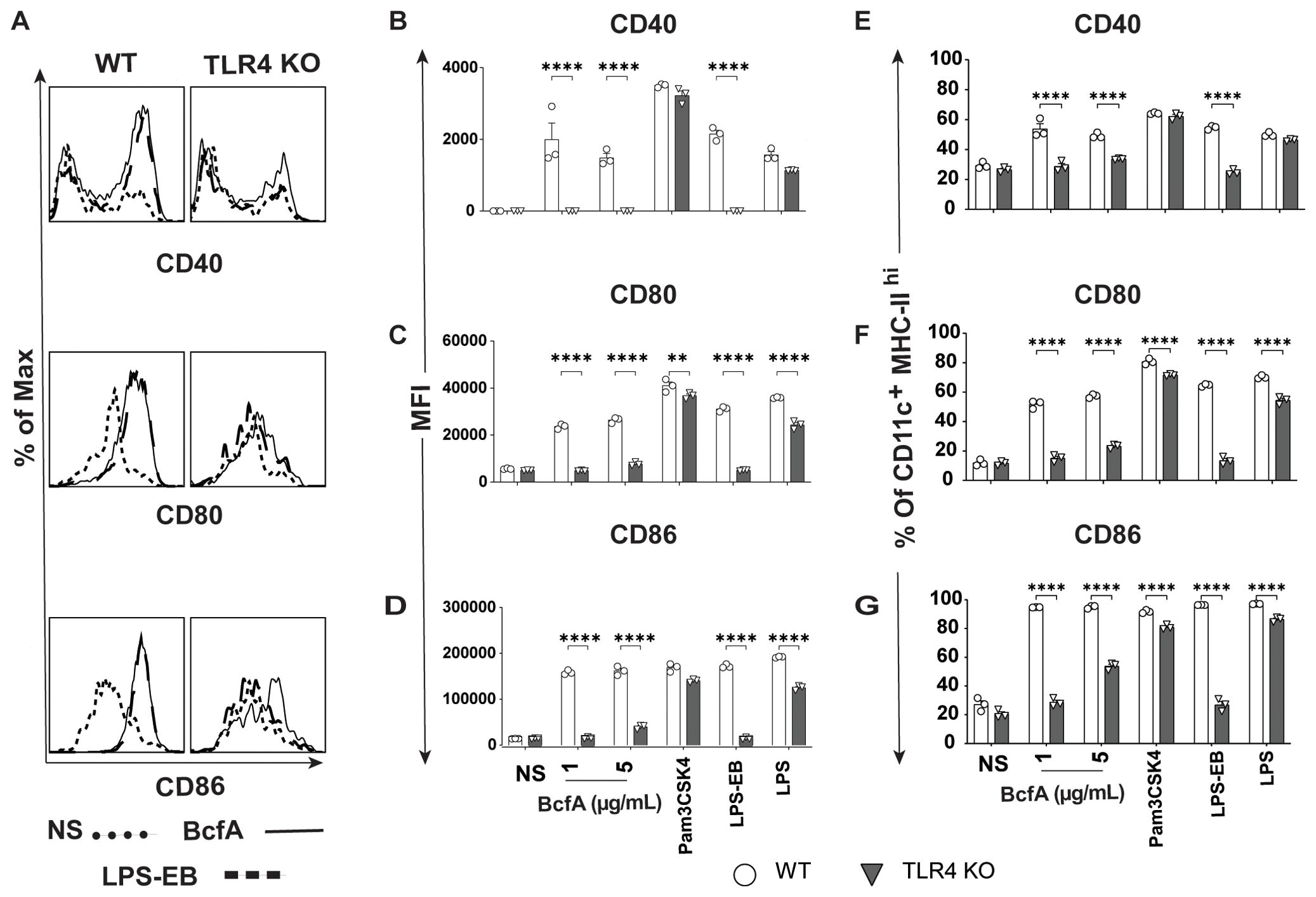
Figure 1. Costimulatory molecule expression is upregulated on BMDCs following BcfA stimulation. (A). Representative overlays of CD40, CD80 and CD86 expression on WT and TLR4 KO BMDCs stimulated with 5 µg/mL BcfA. Median fluorescence intensity (MFI) expression of (B). CD40, (C). CD80 and (D). CD86 and percentage of cells expressing (E). CD40, (F). CD80 and (G). CD86 on WT and TLR4 KO BMDCs stimulated with 1 µg/mL and 5 µg/mL BcfA for 20-24 hr. LPS, Pam3CSK4 and LPS-EB were used as positive controls. Mean ± SEM of triplicate wells is shown. *, p<0.05, **, p<0.01, ***, p<0.001, ****, p<0.0001 by ANOVA. One experiment of 2.
BcfA induces the production of TFH/TH1/TH17-polarizing cytokines by BMDCs
To determine the T cell responses that may be supported by APCs activated by BcfA, we quantified cytokine production from BcfA-stimulated BMDCs by ELISA. WT BMDCs produced IL-6 (Figure 2A), TNF-α (Figure 2B), IL-12/23 p40 (Figure 2C) and IL-12 (Figure 2D) at levels comparable to the positive control LPS. Production of all four cytokines was significantly reduced in TLR4 KO BMDCs. These results show that innate immune responses elicited by BcfA are mediated through TLR4 and may support TH1/TH17-polarized immune responses.
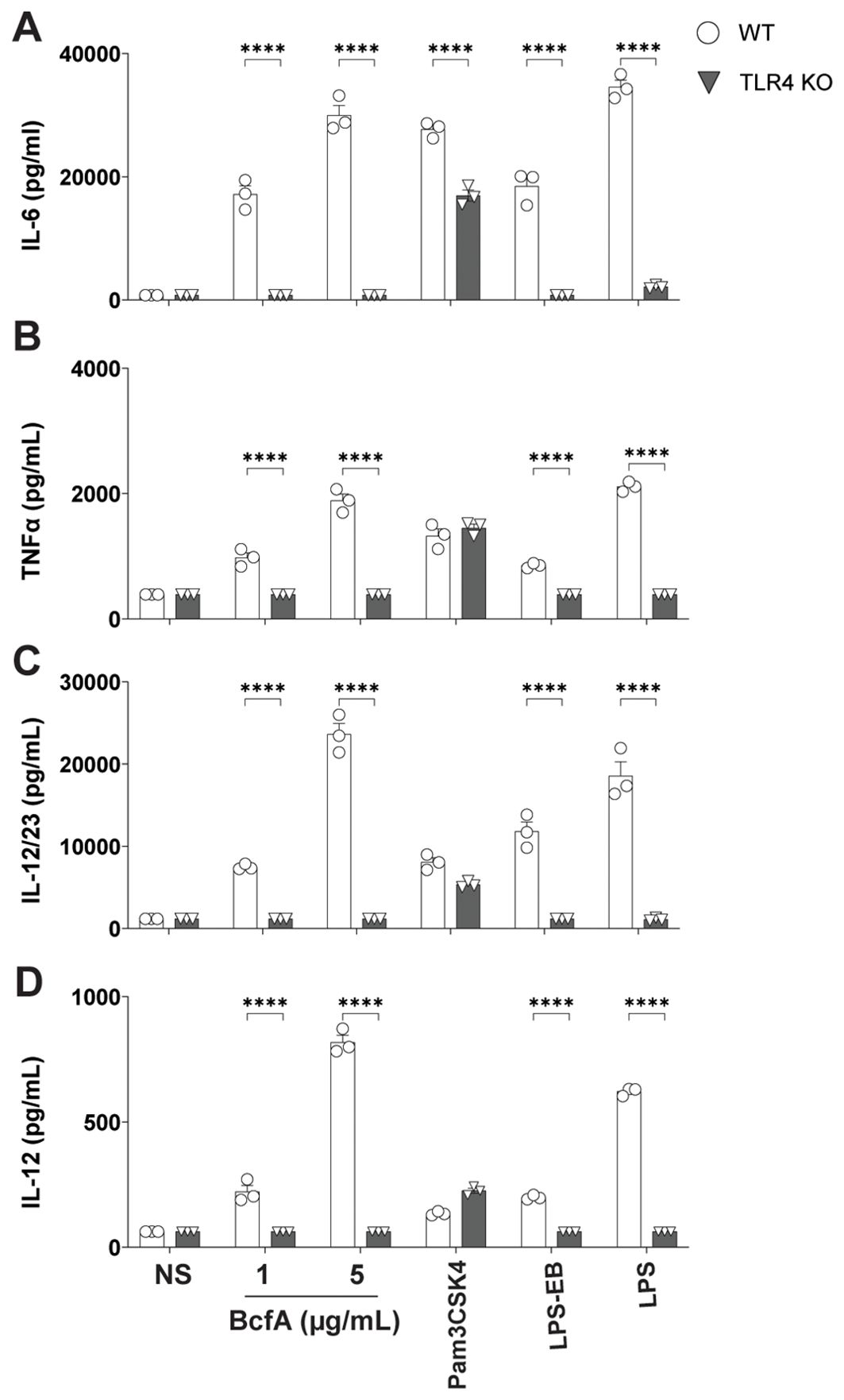
Figure 2. Production of TFH/TH1-polarizing innate cytokines following BcfA stimulation is dependent on TLR4. Expression of (A). IL-6, (B). TNF-α, (C). IL-12/23 p40 and (D). IL-12 by WT and TLR4 KO BMDCs stimulated with BcfA for 20-24 hr. LPS, Pam3CSK4 and LPS-EB were used as positive controls. Mean ± SEM of triplicate wells is shown. *, p<0.05, ***, p<0.001, ****, p<0.0001 by ANOVA. One experiment of 2.
We then tested whether BcfA activated human PBMCs. First, we confirmed expression of TLR4 on adult human PBMCs by flow cytometry (Figure 3A). We then stimulated PBMCs in vitro with BcfA and measured the levels of innate cytokines IL-6 and TNF-α present in the cell culture supernatant after 24 hr. We found that IL-6 was significantly induced in response to stimulation with 5 µg/mL or 25 µg/mL of BcfA (Figure 3B). Although TNF-α production was substantially increased upon stimulation with BcfA, differences were not statistically significant compared to unstimulated cells (Figure 3C). These data show that BcfA stimulation elicits TH1/TH17-polarizing innate cytokines by human cells.
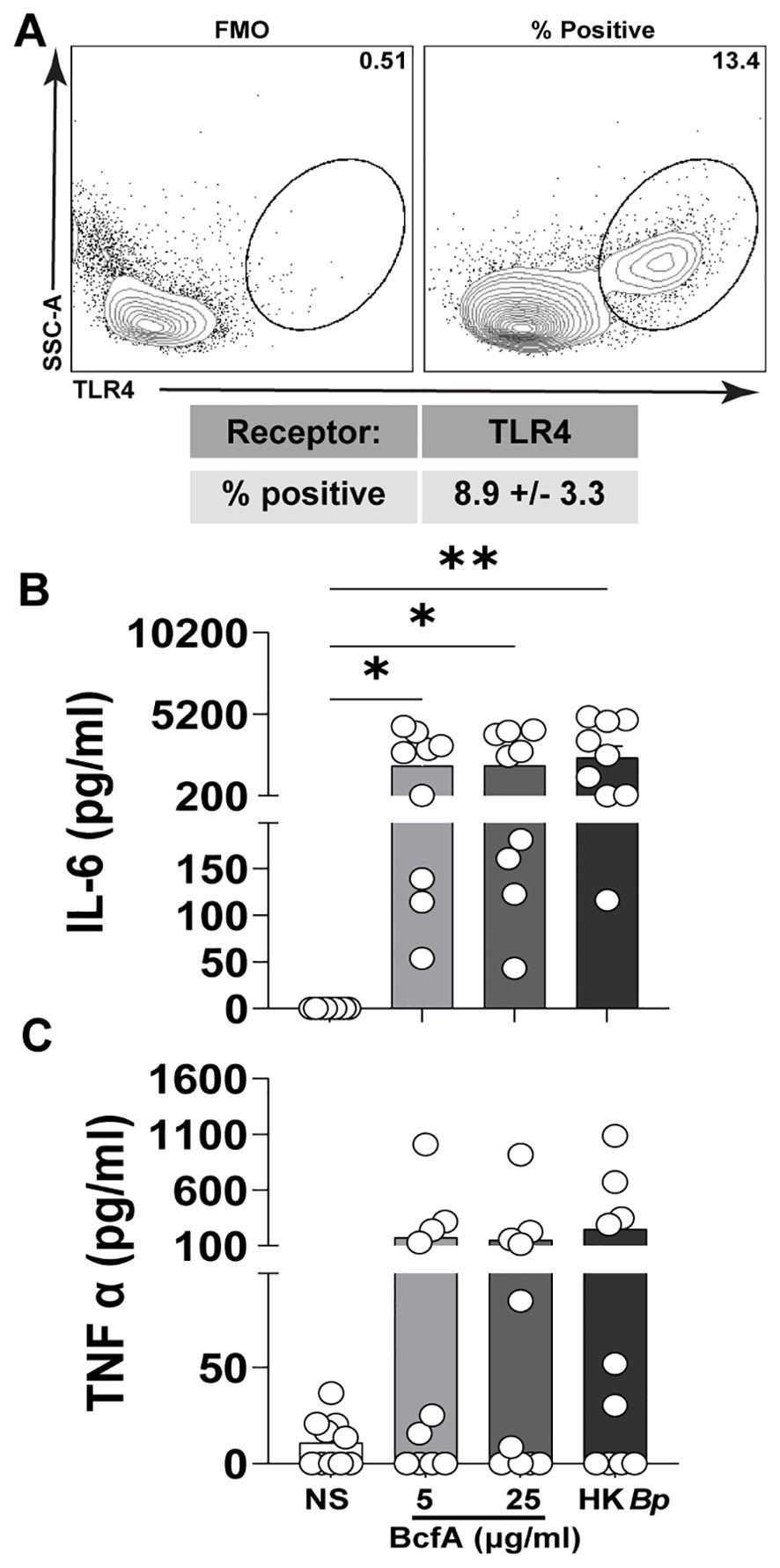
Figure 3. Human PBMCs produce IL-6 following stimulation with BcfA. (A). The percentage of live PBMCs expressing TLR4 (N=5 adult donors) (B). PBMCs from 10 adult donors were stimulated for 24 hr with the indicated concentrations of BcfA or heat-killed B. pertussis control (HK Bp). IL-6 and (C). TNF-α in culture supernatant was evaluated by ELISA). *, p<0.05, **, p<0.01 by ANOVA.
BcfA stimulation of BMDCs supports antigen uptake and processing
Next, we evaluated whether BcfA stimulation of BMDCs would increase antigen uptake and processing of DQ-OVA, a chicken ovalbumin (OVA) conjugate that displays a bright green fluorescence only after proteolytic processing. We stimulated BMDCs with 5 µg/mL BcfA or media alone as a negative control for 24 hr at 37°C. The next day, cells were treated with DQ-OVA for 60 min. at 37°C. The level of green fluorescence was then quantified by flow cytometry as a proxy for antigen uptake and processing. BcfA-treated BMDCs took up more DQ-OVA than unstimulated BMDCs (Figure 4A). We also observed an increase in the percentage of BMDCs with green fluorescence (Figure 4B) compared to NS controls. Together, these data suggest that BcfA-activated BMDCs have an increased ability to uptake and process antigen.
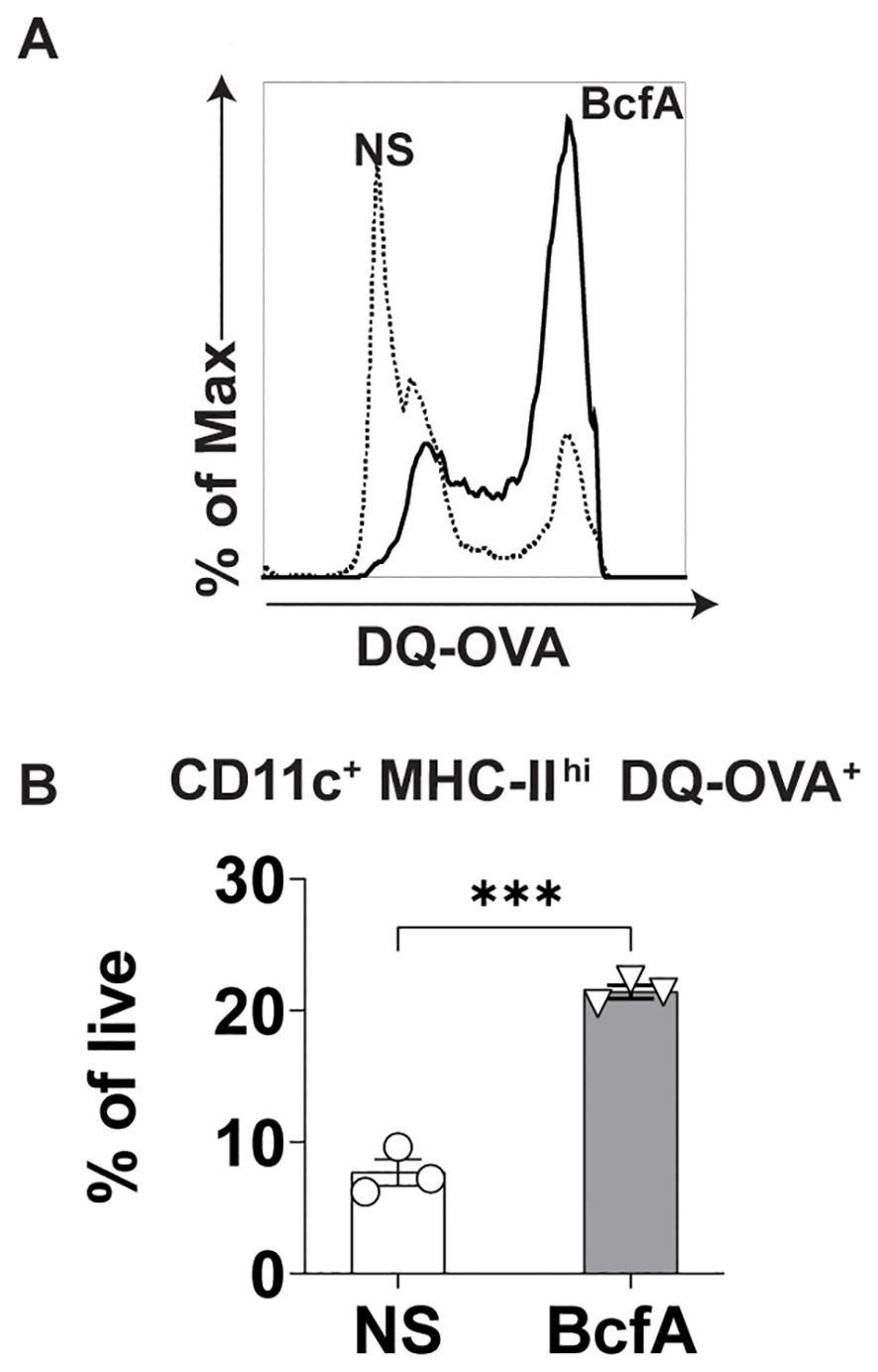
Figure 4. Increased uptake of DQ-OVA by BcfA-stimulated BMDCs. (A). Representative overlays of non-stimulated (NS) and BMDCs stimulated with 5 µg/mL BcfA. (B). CD11c+ MHC-II+ DQ-OVA+ BMDCs as % of live cells. Mean ± SEM of triplicate wells is shown. ***, p<0.001 by paired Student’s t-test. One experiment of 2.
BcfA upregulates costimulatory molecule expression on CD11b+CD11c+ cells via TLR4 in vivo
To test whether BcfA stimulates lung cells, we administered 10 µg BcfA or LPS-EB intranasally to C57BL/6 and TLR4 KO mice. Lungs were harvested 24 hr later and evaluated for the expression of MHC Class II, CD40, CD80, and CD86 on the CD11b+ CD11c+ population (31) (Figure 5A). The gating strategy is shown in Supplementary Figure S4. MFI expression of MHC Class II (Figure 5B), CD40 (Figure 5C), CD80 (Figure 5D) and CD86 (Figure 5E) and the percentage of cells that upregulated expression of MHC Class II (Figure 5F), CD40 (Figure 5G), CD80 (Figure 5H) and CD86 (Figure 5I) increased on this cell population, demonstrating that BcfA efficiently activates putative antigen presented cells in the lungs. The MFI of costimulatory molecule expression was not upregulated on CD11b+ CD11c+ cells in TLR4 KO lungs (Figures 5B–E). The percentage of cells expressing MHC Class II compared to no stimulation increased in WT and TLR4 KO lungs (Figure 5F). In contrast the percentage of TLR4 KO lung cells expressing the costimulatory molecules CD40 (Figure 5G), CD80 (Figure 5H) and CD86 (Figure 5I) did not increase compared to no stimulation. These data further confirm TLR4 as the primary bona fide PRR for BcfA mediated activity.
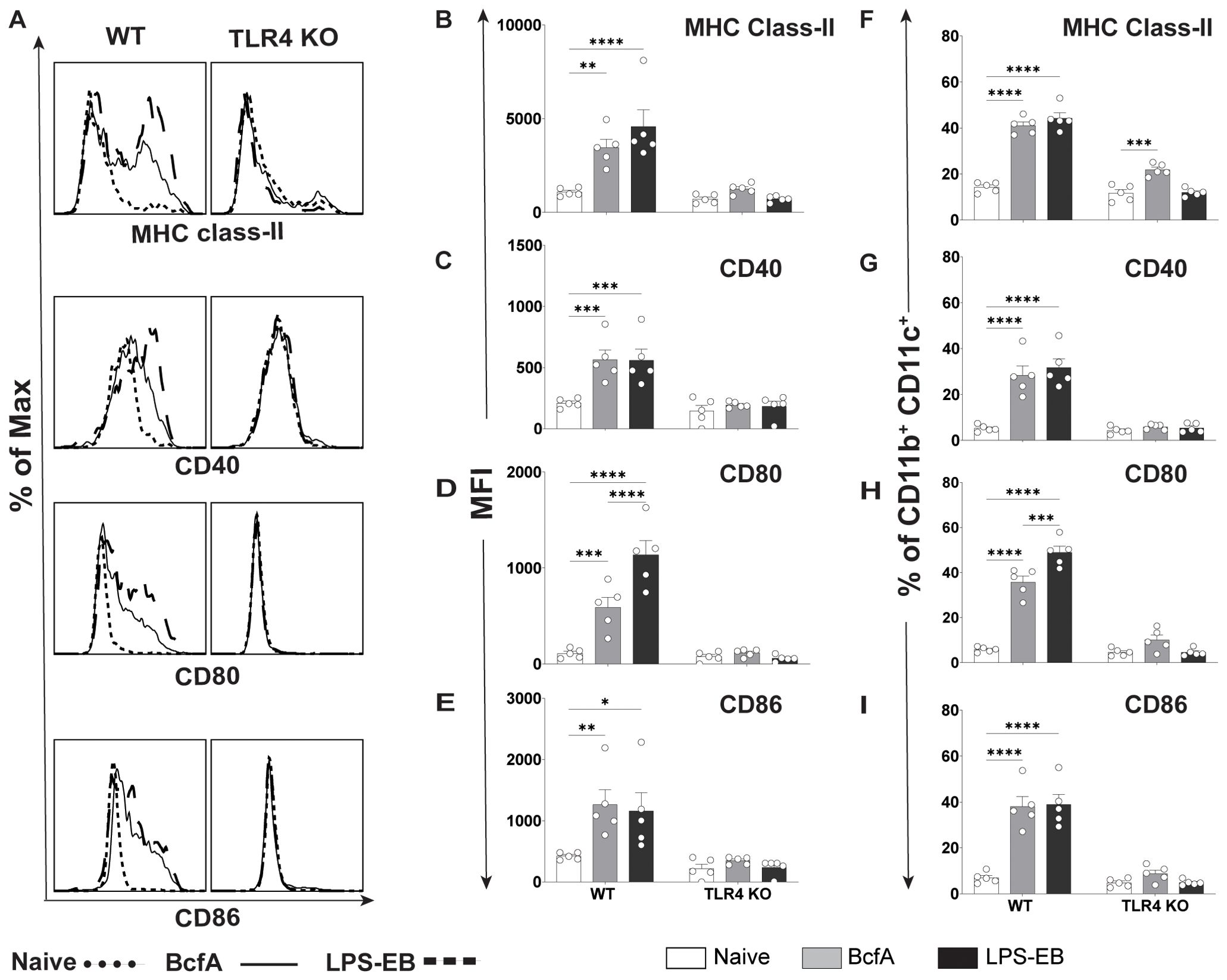
Figure 5. Upregulation of costimulatory molecule expression on lung CD11b+CD11c+ cells by BcfA is dependent on TLR4. (A). Representative overlays of MHC class-II, CD40, CD80 and CD86 expression on lung cells from WT and TLR4 KO mice. Median fluorescence intensity (MFI) of (B). MHC class-II, (C). CD40, (D). CD80 and (E). CD86 and percentage of cells expressing (F). MHC class-II, (G). CD40, (H). CD80, and (I). CD86 on CD11b+ CD11c+ lung cells from WT and TLR4 KO mice, analyzed 24 hr post intranasal immunization with 10 µg of BcfA or LPS-EB. Mean ± SEM of 5 mice is shown. *, p<0.05, **, p<0.01, ***, p<0.001, ****, p<0.0001 by ANOVA. One experiment of 2.
BcfA-stimulated BMDC-conditioned medium supports TFH and TH1 cell programming
As part of pathogen-specific immune responses, naïve CD4+ T cells differentiate into effector subsets which perform specialized activities to orchestrate immune-mediated clearance of infection. Particularly critical for responses to intracellular pathogens like SARS-CoV-2 and primarily extracellular pathogens like B. pertussis are T helper 1 (TH1) and T follicular helper (TFH) populations. Of these, TH1 cells activate other immune populations by secreting pro-inflammatory cytokines such as IFN-γ, while TFH cells produce IL-21 and provide help to B cells to promote antibody generation and humoral immunity (32, 33). We previously reported that intramuscular (i.m.) immunization with an acellular pertussis vaccine (aPV) containing BcfA elicited systemic TH1 responses in vivo (25). In our recent work (20), we established that immunization of mice with BcfA-adjuvanted SARS-CoV-2 spike protein supported production of IFN-γ-driven IgG2c spike protein-specific antibodies. Thus, we hypothesized that BcfA may support both TH1 and TFH cell differentiation programs, and consequently productive anti-bacterial and anti-viral cell-mediated and humoral immune responses.
Effector CD4+ T cell differentiation is directed by a coordinated interplay between cell-intrinsic transcriptional networks and environmental cytokine signals that are often produced by antigen-presenting cells (34). To test whether the IL-6, IL-12, and IL-12/23 common chain cytokines in BcfA-stimulated supernatants would be sufficient to induce TFH and/or TH1 differentiation, we stimulated naïve CD4+ T cells on plate-bound α-CD3/α-CD28 in the presence of cell-free supernatant from untreated BMDCs (NS) or those treated with 5 µg/mL BcfA for 72 hr. We then assessed the expression of key TFH and TH1/genes via qRT-PCR. Consistent with our hypothesis, expression of genes for lineage-defining transcription factors and effector cytokines associated with TFH (Bcl6, Il21; Figure 6A) and TH1 (Tbx21 (T-bet), Ifng; Figure 6B) cells were elevated in cells cultured in the presence of BcfA-conditioned medium relative to NS controls.
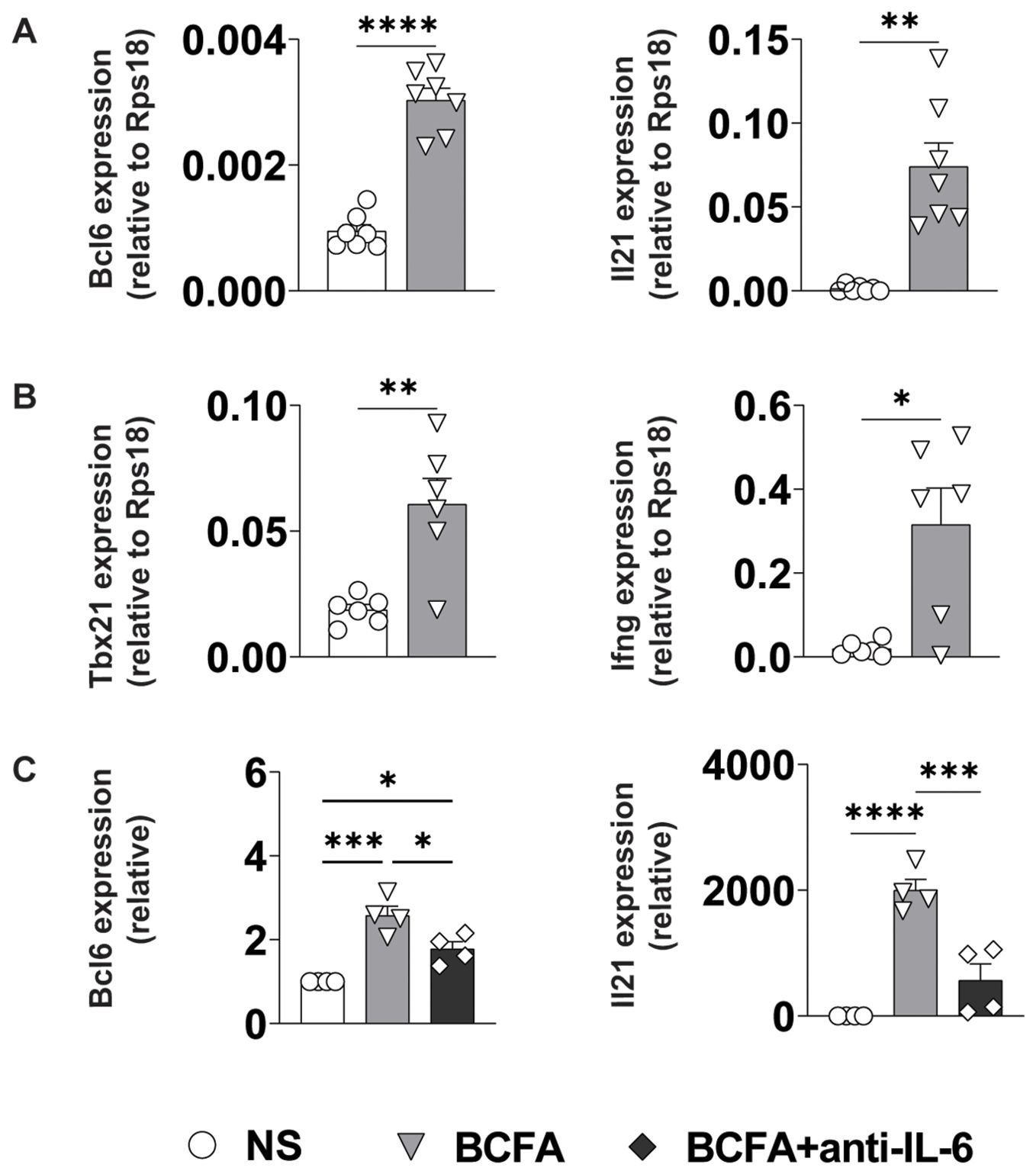
Figure 6. BcfA conditioned medium supports TFH and TH1 cell polarization in vitro. qRT-PCR of T cells that were unstimulated (NS) or stimulated with BcfA conditioned medium with the addition of IL-4-neutralizing antibody. Data are presented relative to housekeeping gene Rps18. Data pooled from individual experiments with 4-7 samples per group. (A). Bcl6 and IL21 expression, (B). Tbx21 and IFN-γ expression, (C). Bcl6 and IL21 expression alone or with anti-IL-6 neutralizing antibody. Mean ± SEM of 4 experiments is shown. *, p<0.05, **, p<0.01, ***, p<0.001, ****, p<0.0001 by paired Student’s t-test.
As IL-6/STAT3 signaling is an established driver of TFH differentiation, we determined whether the IL-6 in the culture medium was responsible for driving TFH differentiation. We cultured cells in BcfA-treated or NS control medium in the presence or absence of IL-6-neutralizing antibody. Consistent with a role for IL-6 in promoting TFH gene expression, we observed a significant reduction in the expression of both Bcl6 and Il21 when IL-6 was neutralized (Figure 6C).
As standard TFH-like and TH1 culture conditions include the addition of anti-IL-4 neutralizing antibody which prevents inherent TH2 polarization (35), we also tested whether BcfA elicited TFH or TH1 gene programming in the absence of IL-4-neutralization. Naïve T cells were differentiated in vitro without the addition of anti-IL-4 blocking antibody. We evaluated the expression of T cell subset-specific lineage-defining transcription factors and key cytokines by qRT-PCR and flow cytometry. Interestingly, BcfA reduced the RNA (Figure 7A) expression of the TH2 lineage-defining transcription factor Gata-3, while the protein level was unchanged (Figure 7B). Similarly, the RNA (Figure 7C) level of the TH2 effector cytokine IL-4 was reduced, while the protein (Figure 7D) expression was unchanged. In contrast, while transcript abundance for the TFH lineage-defining transcription factor Bcl-6 was unchanged (Figure 7E), its protein expression was elevated (Figure 7F). Yet, transcript levels for the TFH cytokine IL-21 were significantly elevated (Figure 7G), while protein expression was not significantly increased (Figure 7H). Finally, TH1 differentiation was elevated, as the RNA expression of the TH1-defining factor Tbx21 (T-bet) was increased (Figure 7I), and the protein level was slightly but not significantly elevated (Figure 7J). RNA for the TH1 cytokine IFN-γ was unchanged (Figure 7K) while protein expression was increased (Figure 7L). Overall, these data suggest that BcfA both negatively regulates TH2 differentiation and positively regulates TFH and TH1 programming.
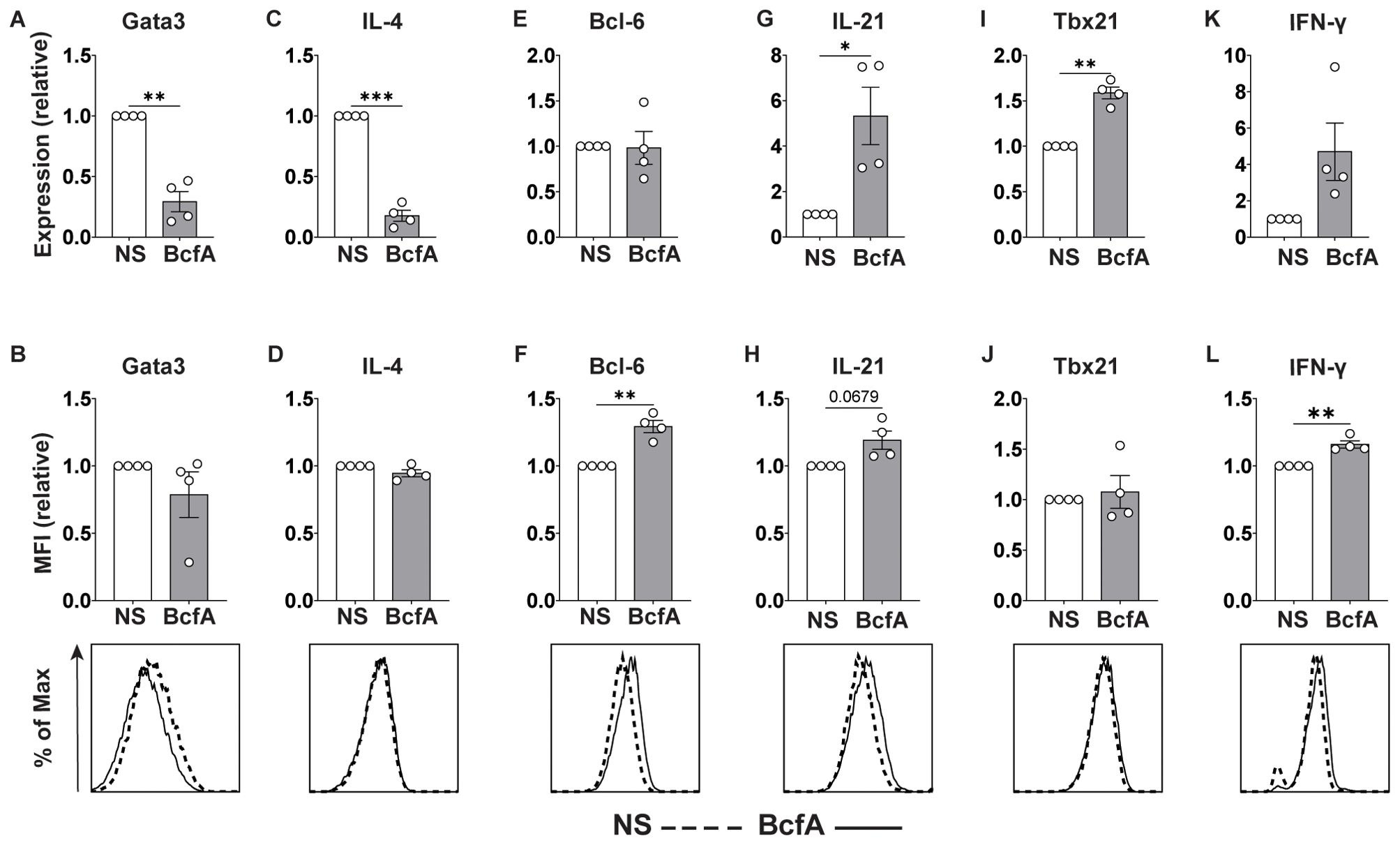
Figure 7. BcfA conditioned medium represses TH2 cell polarization in vitro. qRT-PCR and flow cytometry analysis of unstimulated (NS) T cells or stimulated with BcfA conditioned medium in the absence of IL-4-neutralizing antibody. qRT-PCR data are presented relative to housekeeping gene Rps18. Mean ± SEM of 4 experiments is shown. Expression of Gata3 (A, B), IL-4 (C, D), Bcl-6 (E, F), IL-21 (G, H), T-bet (I, J), and IFN-γ (K, L). *, p<0.05, **, p<0.01, ***, p<0.001 by paired Student’s t-test.
Discussion
The development and characterization of novel adjuvants is an active area of research, with the goal of defining adjuvants that elicit strong, sustained vaccine-mediated immune responses. Despite the decades-long use of alum as an adjuvant and the recent development of newer formulations, there remains a need for safe and effective adjuvants that elicit systemic and mucosal immunity.
Here, we investigated the mechanism of action of the adjuvant BcfA, which was discovered by our group and has demonstrated adjuvant activity in experimental vaccines against respiratory pathogens B. pertussis (25, 26) and SARS CoV-2 (20). The hallmark feature of BcfA that distinguishes it from FDA-approved and other experimental adjuvants is the absence of TH2 responses when BcfA is used as the single adjuvant (25) and strong attenuation of TH2 responses when BcfA is added to alum-adjuvanted vaccines (20, 26).
Furthermore, BcfA elicits TH1 and TH17 responses which are critical for protection against both bacterial and viral pathogens. Attenuation of TH2 immune responses may also reduce the risk of vaccine related adverse events such as antibody dependent enhancement of disease (ADE) and vaccine associated enhancement of respiratory disease (VAERD).
As BcfA is a protein, we hypothesized that it may activate immune responses through PRRs expressed on APCs. A PRR/NOD receptor screen identified both murine TLR4 and TLR2 as the receptors triggered on HEK-293 reporter cell lines following BcfA stimulation (Supplementary Figure S1). When the activity of BcfA was tested in murine BMDCs as a more physiological system, upregulation of costimulatory molecule expression (Figure 1) and production of innate cytokines (Figure 2) was significantly reduced in TLR4 KO BMDCs compared with WT BMDCs, but not in TLR2 KO BMDCs (Supplementary Figure S3). These data show that BcfA acts primarily through TLR4. BcfA-stimulated BMDCs showed uptake and processing of the model antigen DQ-OVA (Figure 4), suggesting that BcfA may also support T cell activation by amplifying antigen presentation. We pretreated BMDCs with BcfA prior to testing uptake of DQ-OVA. Coadministration of antigen and adjuvant may change the kinetics or magnitude of antigen uptake and processing and is a limitation of our experimental design.
To determine the potential utility of BcfA as an adjuvant for human vaccines, we tested cytokine production by human PBMCs stimulated with BcfA for 24 hr (Figure 3). Production of IL-6 was detected in supernatants of all samples tested, suggesting that human PBMCs stimulated with BcfA may support TFH polarization of CD4+ T cells. Expression of the inflammatory cytokine TNF-α was variable between donors, and not significantly increased compared with unstimulated cells. It is important to note that we collected supernatant at 24 hr post-stimulation, which may be beyond the time of maximal TNF-α production in this context. Alternately, there may be autocrine consumption of TNF-α produced by the monocytes (36). Thus, the ability of BcfA to activate human cells suggests that BcfA will have adjuvant function when included in human vaccines.
Although bacterial LPS is the canonical ligand for TLR4, this PRR may also be activated by a wide array of bacterial proteins (37). Interestingly, triggering of the same receptor may elicit disparate immune phenotypes. The pneumococcal proteins DnaJ and Ply (38, 39) and the M. tuberculosis derived proteins RpfE and Rv0652 are TLR4 ligands that activate DCs and polarize the immune response towards the TH1/TH17 phenotype (40, 41), but elicit different innate cytokines. Collectively, these findings suggest that the type of immune response elicited by adjuvants depends both on the responding PRR as well as the properties of the ligand.
Adjuvants also activate the adaptive immune response by directly triggering PRRs on lymphocytes (42). Murine and human T cells express TLR4 (43). Thus, BcfA may directly activate T cells when delivered in vivo as part of a vaccine formulation, and thereby amplify the vaccine-elicited response. This potential function of BcfA is an area of future study for our group.
Using an in vitro T cell polarization system, we showed that the innate cytokines produced by BcfA-stimulated BMDCs support the differentiation of TH1 and TFH cells while preventing TH2 differentiation in parallel (Figure 6 and Figure 7). This result is in accordance with our previous report of the ability of BcfA to attenuate alum-induced TH2 responses when combined in vivo (20, 26). In addition, the IL-6-dependent polarization of TFH cells we observed also provides a potential mechanistic explanation for the induction of IgG2c antibody production we observed in the serum and lungs of mice immunized with BcfA-containing vaccines.
Generation of mucosal immunity to respiratory pathogens is critical for providing sustained protection against infection (44, 45) and preventing subsequent transmission (46). For B. pertussis, the CD4+IL-17+ T cells generated during natural infection are essential for reducing bacterial colonization of the nose (45, 46). Immunization of baboons with the whole cell pertussis vaccine (wPV) also clears the infection from the nose (47). In contrast, acellular pertussis (aPV) vaccine immunization does not reduce nasal bacterial burden (48). Our recent paper (26) showed that mucosal delivery of BcfA-adjuvanted acellular pertussis vaccines elicited CD4+IL-17+ tissue-resident memory T cells (TRM) in the nose and reduced nasal bacterial burden. Although we detected production of the IL-12/23 p40 common γ chain by BcfA-stimulated BMDCs, suggesting that IL-17 may also be produced, we did not detect TH17 polarization in vitro (data not shown), suggesting that either the amount of IL-12/23 produced in BcfA supernatant is insufficient, or that additional factors such as such as TGF-β (49) that support TH17 generation are not replicated in the culture system.
In summary, this work provides a mechanistic understanding of the function of BcfA as an adjuvant and supports the utility of BcfA for use in human vaccines. Continued identification and validation of novel adjuvants that are safe, effective, and generate mucosal immunity is key to improving vaccine-mediated protection against infectious diseases.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ohio State University Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by Ohio State University Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MS: Writing – original draft, Investigation, Methodology. KR: Investigation, Writing – review & editing. JH: Investigation, Writing – review & editing, Writing – original draft. JT: Investigation, Writing – review & editing. JB: Investigation, Writing – review & editing. MG: Investigation, Writing – review & editing. YG: Investigation, Writing – review & editing. RD: Writing – review & editing, Funding acquisition. KO: Funding acquisition, Writing – review & editing, Supervision. PD: Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by 1R01AI125560 and 1R01AI153829 (to PD and RD) and 1R01AI134972 (to KO). MS was supported by a doctoral fellowship from the Egyptian Bureau of Higher Education. KR was supported by The Ohio State University College of Medicine Advancing Research in Infection and Immunity Fellowship Program. JH is supported by NIAID 1T32AI165391-01. JT is supported by NIAID 1F30AI172189-01A1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1439418/full#supplementary-material
Supplementary Figure 1 | BcfA activates BMDCs and TLR2 and TLR4 expressing reporter cell lines. (A). HEK-Blue™ cells expressing murine TLRs, and NOD1/2 were stimulated with BcfA (1µg/mL). SEAP reporter expression was detected using HEK-Blue™ Detection media. The average of duplicate wells is shown. (B). HEK-Blue-mTLR4 and (C). HEK-Blue-mTLR2 cells were stimulated with 1 and 5µg/mL of BcfA. NT = no treatment; min LPS = minimal LPS present in the BcfA preps. PAM3CSK4 and LPS were used as positive controls for TLR2 and TLR4 respectively. Mean ± SEM of triplicate wells is shown. ****, p<0.0001 by ANOVA. One experiment of 2.
Supplementary Figure 2 | Gating strategy to identify activated BMDCs. Differentiated BMDCs were stained with antibodies specific for CD11c, MHC Class II, CD40, CD80 and CD86. Live, single cells were gated as CD11c+MHC-IIhigh cells. This double positive population from WT and TLR4 KO cells was evaluated for expression of CD40, CD80 and CD86 as shown.
Supplementary Figure 3 | Upregulation of costimulatory molecule expression is unchanged in TLR2 KO BMDCs. (A). Representative overlays of CD40, CD80 and CD86 expression on WT and TLR2 KO BMDCs stimulated with 5 µg/mL BcfA. Median fluorescence intensity (MFI) expression of (B). CD40, (C). CD80 and (D). CD86 and percentage of cells expressing (E). CD40, (F). CD80 and (G). CD86 on WT and TLR2 KO BMDCs stimulated with 1 µg/mL and 5 µg/mL BcfA for 20-24 hr. Heat-killed B. pertussis (HKBp) were used as positive controls. Mean ± SEM of triplicate wells is shown. *, p<0.05, ** by ANOVA. One experiment of 2.
Supplementary Figure 4 | Gating strategy to identify lung CD11b+CD11c+ cells. Single cell suspensions from lungs were stained with antibodies specific for CD11b, CD11c, MHC Class II, CD40, CD80 and CD86. Live, single cells were gated as CD11b+CD11c+. This double positive population from WT and TLR4 KO lungs was evaluated for expression of MHC Class II, CD40, CD80 and CD86 as shown.
References
1. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. (2010) 33:492–503. doi: 10.1016/j.immuni.2010.10.002
2. Facciola A, Visalli G, Lagana A, Di Pietro A. An overview of vaccine adjuvants: current evidence and future perspectives. Vaccines (Basel). (2022) 10:819. doi: 10.3390/vaccines10050819
3. Pulendran B, S Arunachalam P, O'Hagan DT. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discovery. (2021) 20:454–75. doi: 10.1038/s41573-021-00163-y
4. Ghimire TR. The mechanisms of action of vaccines containing aluminum adjuvants: an in vitro vs in vivo paradigm. Springerplus. (2015) 4:181. doi: 10.1186/s40064-015-0972-0
5. Ott G, Barchfeld GL, Chernoff D, Radhakrishnan R, van Hoogevest P, Van Nest G. MF59. Design and evaluation of a safe and potent adjuvant for human vaccines. Pharm Biotechnol. (1995) 6:277–96.
6. De Donato S, Granoff D, Minutello M, Lecchi G, Faccini M, Agnello M, et al. Safety and immunogenicity of MF59-adjuvanted influenza vaccine in the elderly. Vaccine. (1999) 17:3094–101. doi: 10.1016/S0264-410X(99)00138-3
7. Chlibek R, Bayas JM, Collins H, de la Pinta ML, Ledent E, Mols JF, et al. Safety and immunogenicity of an AS01-adjuvanted varicella-zoster virus subunit candidate vaccine against herpes zoster in adults >=50 years of age. J Infect Dis. (2013) 208:1953–61. doi: 10.1093/infdis/jit365
8. Laurens MB. RTS,S/AS01 vaccine (Mosquirix): an overview. Hum Vaccin Immunother. (2020) 16:480–9. doi: 10.1080/21645515.2019.1669415
9. Canelle Q, Dewe W, Innis BL, van der Most R. Evaluation of potential immunogenicity differences between Pandemrix and Arepanrix. Hum Vaccin Immunother. (2016) 12:2289–98. doi: 10.1080/21645515.2016.1168954
10. Ebensen T, Delandre S, Prochnow B, Guzman CA, Schulze K. The combination vaccine adjuvant system alum/c-di-AMP results in quantitative and qualitative enhanced immune responses post immunization. Front Cell Infect Microbiol. (2019) 9:31. doi: 10.3389/fcimb.2019.00031
11. Krieg AM. An innate immune defense mechanism based on the recognition of CpG motifs in microbial DNA. J Lab Clin Med. (1996) 128:128–33. doi: 10.1016/S0022-2143(96)90004-9
12. Chatzikleanthous D, O'Hagan DT, Adamo R. Lipid-based nanoparticles for delivery of vaccine adjuvants and antigens: Toward multicomponent vaccines. Mol Pharm. (2021) 18:2867–88. doi: 10.1021/acs.molpharmaceut.1c00447
13. Wagner H. Bacterial CpG DNA activates immune cells to signal infectious danger. Adv Immunol. (1999) 73:329–68. doi: 10.1016/S0065-2776(08)60790-7
14. Del Giudice G, Rappuoli R, Didierlaurent AM. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin Immunol. (2018) 39:14–21. doi: 10.1016/j.smim.2018.05.001
15. Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramirez P, et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care. (2009) 13:R201. doi: 10.1186/cc8208
16. Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. (2009) 2:403–11. doi: 10.1038/mi.2009.100
17. Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, et al. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. (2009) 5:e1000703. doi: 10.1371/journal.ppat.1000703
18. Lyadova IV, Panteleev AV. Th1 and th17 cells in tuberculosis: Protection, Pathology, and Biomarkers. Mediators Inflammation. (2015) 2015:854507. doi: 10.1155/2015/854507
19. Ross PJ, Sutton CE, Higgins S, Allen AC, Walsh K, Misiak A, et al. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. (2013) 9:e1003264. doi: 10.1371/journal.ppat.1003264
20. Shamseldin MM, Kenney A, Zani A, Evans JP, Zeng C, Read KA, et al. Prime-Pull immunization of mice with a bcfA-adjuvanted vaccine elicits sustained mucosal immunity that prevents SARS-CoV-2 infection and pathology. J Immunol. (2023) 210:1–15. doi: 10.4049/jimmunol.2200297
21. Lee CM, Oh JE. Resident memory B cells in barrier tissues. Front Immunol. (2022) 13:953088. doi: 10.3389/fimmu.2022.953088
22. Mettelman RC, Allen EK, Thomas PG. Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity. (2022) 55:749–80. doi: 10.1016/j.immuni.2022.04.013
23. Poon MML, Rybkina K, Kato Y, Kubota M, Matsumoto R, Bloom NI, et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci Immunol. (2021) 6:eabl9105. doi: 10.1126/sciimmunol.abl9105
24. Zazara DE, Belios I, Lucke J, Zhang T, Giannou AD. Tissue-resident immunity in the lung: a first-line defense at the environmental interface. Semin Immunopathol. (2022) 44:827–54. doi: 10.1007/s00281-022-00964-2
25. Jennings-Gee J, Quataert S, Ganguly T, D'Agostino R Jr., Deora R, Dubey P. The adjuvant bordetella colonization factor A attenuates alum-induced th2 responses and enhances bordetella pertussis clearance from mouse lungs. Infect Immun. (2018) 86. doi: 10.1128/IAI.00935-17
26. Yount KS, Hall JM, Caution K, Shamseldin MM, Guo M, Marion K, et al. Systemic priming and intranasal booster with a BcfA-adjuvanted acellular pertussis vaccine generates CD4+ IL-17+ nasal tissue resident T cells and reduces B. pertussis nasal colonization. Front Immunol. (2023) 14:1181876. doi: 10.3389/fimmu.2023.1181876
27. Sukumar N, Love CF, Conover MS, Kock ND, Dubey P, Deora R. Active and passive immunizations with Bordetella colonization factor A protect mice against respiratory challenge with Bordetella bronchiseptica. Infect Immun. (2009) 77:885–95. doi: 10.1128/IAI.01076-08
28. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
29. Wang W, Li J, Wu K, Azhati B, Rexiati M. Culture and identification of mouse bone marrow-derived dendritic cells and their capability to induce T lymphocyte proliferation. Med Sci Monit. (2016) 22:244–50. doi: 10.12659/MSM.896951
30. Han JE, Wui SR, Kim KS, Cho YJ, Cho WJ, Lee NG. Characterization of the structure and immunostimulatory activity of a vaccine adjuvant, de-O-acylated lipooligosaccharide. PLoS One. (2014) 9:e85838. doi: 10.1371/journal.pone.0085838
31. Suzuki Y, Suda T, Furuhashi K, Shibata K, Hashimoto D, Enomto N, et al. Mouse CD11bhigh lung dendritic cells have more potent capability to induce IgA than CD103+ lung dendritic cells in vitro. Am J Respir Cell Mol Biol. (2012) 46:773–80. doi: 10.1165/rcmb.2011-0329OC
32. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
33. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. (2008) 112:1557–69. doi: 10.1182/blood-2008-05-078154
34. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
35. Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat Immunol. (2000) 1:199–205. doi: 10.1038/79734
36. Gane JM, Stockley RA, Sapey E. TNF-alpha autocrine feedback loops in human monocytes: The pro- and anti-inflammatory roles of the TNF-alpha receptors support the concept of selective TNFR1 blockade in vivo. J Immunol Res. (2016) 2016:1079851. doi: 10.1155/2016/1079851
37. Kumar S, Sunagar R, Gosselin E. Bacterial protein toll-like-receptor agonists: A novel perspective on vaccine adjuvants. Front Immunol. (2019) 10:1144. doi: 10.3389/fimmu.2019.01144
38. Su Y, Li D, Xing Y, Wang H, Wang J, Yuan J, et al. Subcutaneous immunization with fusion protein DnaJ-DeltaA146Ply without additional adjuvants induces both humoral and cellular immunity against pneumococcal infection partially depending on TLR4. Front Immunol. (2017) 8:686. doi: 10.3389/fimmu.2017.00686
39. Wu Y, Cui J, Zhang X, Gao S, Ma F, Yao H, et al. Pneumococcal DnaJ modulates dendritic cell-mediated Th1 and Th17 immune responses through Toll-like receptor 4 signaling pathway. Immunobiology. (2017) 222:384–93. doi: 10.1016/j.imbio.2016.08.013
40. Choi HG, Kim WS, Back YW, Kim H, Kwon KW, Kim JS, et al. Mycobacterium tuberculosis RpfE promotes simultaneous Th1- and Th17-type T-cell immunity via TLR4-dependent maturation of dendritic cells. Eur J Immunol. (2015) 45:1957–71. doi: 10.1002/eji.201445329
41. Lee SJ, Shin SJ, Lee MH, Lee MG, Kang TH, Park WS, et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One. (2014) 9:e104351. doi: 10.1371/journal.pone.0104351
42. Fan J, Jin S, Gilmartin L, Toth I, Hussein WM, Stephenson RJ. Advances in infectious disease vaccine adjuvants. Vaccines (Basel). (2022) 10. doi: 10.3390/vaccines10071120
43. Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. (2007) 19:39–45. doi: 10.1016/j.coi.2006.11.007
44. Cheon IS, Son YM, Sun J. Tissue-resident memory T cells and lung immunopathology. Immunol Rev. (2023). doi: 10.1111/imr.13201
45. Wilk MM, Mills KHG. CD4 T(RM) cells following infection and immunization: implications for more effective vaccine design. Front Immunol. (2018) 9:1860. doi: 10.3389/fimmu.2018.01860
46. Chasaide CN, Mills KHG. Next-generation pertussis vaccines based on the induction of protective T cells in the respiratory tract. Vaccines (Basel). (2020) 316(1):63–83. doi: 10.3390/vaccines8040621
47. Warfel JM, Zimmerman LI, Merkel TJ. Comparison of three whole-cell pertussis vaccines in the baboon model of pertussis. Clin Vaccine Immunol. (2016) 23:47–54. doi: 10.1128/CVI.00449-15
48. Warfel JM, Zimmerman LI, Merkel TJ. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc Natl Acad Sci U.S.A. (2014) 111:787–92. doi: 10.1073/pnas.1314688110
Keywords: adjuvants, antigen presenting cells, BcfA, T cell polarization, pattern recognition receptors
Citation: Shamseldin MM, Read KA, Hall JM, Tuazon JA, Brown JM, Guo M, Gupta YA, Deora R, Oestreich KJ and Dubey P (2024) The adjuvant BcfA activates antigen presenting cells through TLR4 and supports TFH and TH1 while attenuating TH2 gene programming. Front. Immunol. 15:1439418. doi: 10.3389/fimmu.2024.1439418
Received: 27 May 2024; Accepted: 09 August 2024;
Published: 29 August 2024.
Edited by:
Jiae Kim, Henry M Jackson Foundation for the Advancement of Military Medicine (HJF), United StatesReviewed by:
Erica Louise Stewart, Royal Prince Alfred Hospital, AustraliaOscar Badillo-Godinez, Faculty of Medicine, Uppsala University, Sweden
Copyright © 2024 Shamseldin, Read, Hall, Tuazon, Brown, Guo, Gupta, Deora, Oestreich and Dubey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Purnima Dubey, cHVybmltYS5kdWJleUBvc3VtYy5lZHU=
†These authors share first authorship