 Alevtina Gall1
Alevtina Gall1 Marita Bosticardo2
Marita Bosticardo2 Stacey Ma3
Stacey Ma3 Karin Chen1Kayla Amini2
Karin Chen1Kayla Amini2 Francesca Pala2
Francesca Pala2 Ottavia M. Delmonte2Tara Wenger4Michael Bamshad4,5
Ottavia M. Delmonte2Tara Wenger4Michael Bamshad4,5 John Sleasman6Matthew Blessing7
John Sleasman6Matthew Blessing7 Nicolai S. C. van Oers8
Nicolai S. C. van Oers8 Luigi D. Notarangelo2
Luigi D. Notarangelo2 M. Teresa de la Morena1*
M. Teresa de la Morena1*- 1Division of Immunology, Department of Pediatrics, University of Washington and Seattle Children’s Hospital, Seattle, WA, United States
- 2Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Bethesda, MD, United States
- 3Division of Allergy and Infectious Diseases, Department of Internal Medicine, University of Washington, Seattle, WA, United States
- 4Division of Genetic Medicine, Department of Pediatrics, University of Washington and Seattle Children’s Hospital, Seattle, WA, United States
- 5Brotman Baty Institute for Precision Medicine, University of Washington, Seattle, WA, United States
- 6Department of Pediatrics, Duke University Medical Center, Durham, NC, United States
- 7Division of Craniofacial Medicine, Department of Pediatrics, University of Washington and Seattle Children’s Hospital, Seattle, WA, United States
- 8Department of Immunology, Pediatrics and Microbiology, University of Texas Southwestern Medical Center, Dallas, TX, United States
Pathogenic variants in the transcription factor TP63 are associated with clinically overlapping syndromes including ectrodactyly-ectodermal dysplasia clefting (EEC) and ankyloblepharon-ectodermal defects-cleft lip/palate (AEC). T cell lymphopenia has rarely been described in individuals with TP63 variants and the cause of the T cell defect is unclear. Here, we present a case of a female infant born with TP63-related syndrome and profound T cell lymphopenia, first uncovered through newborn screening. Flow cytometry analysis revealed low CD4+ naïve T cells and nearly absent CD8+ T cells with intact B and NK cell compartments. A de novo heterozygous pathogenic variant c.1040 G>A (C347Y) in exon 8 of TP63 was identified. An artificial thymic organoid system, to assess the intrinsic ability of the patient’s hematopoietic cells to develop into T cells, was performed twice using separate peripheral blood samples. Ex vivo T cell differentiation was evident with the artificial organoid system, suggesting that a thymic stromal cell defect may be the cause of the T cell lymphopenia. Consistent with this, interrogation of publicly available data indicated that TP63 expression in the human thymus is restricted to thymic epithelial cells. Based on these data, congenital athymia was suspected and the patient received an allogenic cultured thymus tissue implant (CTTI). This is the first report of suspected congenital athymia and attempted treatment with CTTI associated with TP63 variant. At 9 months post-implant, peripheral lymphocyte analysis revealed measurable T cell receptor excision circles and presence of CD4+ recent thymic emigrants suggestive of early thymopoiesis. She will continue regular monitoring to ensure restoration of T cell immunity.
1 Introduction
Newborn screening for severe combined immunodeficiency (SCID) utilizes polymerase chain reaction to quantify T cell receptor excision circles (TRECs), a measure of normal T cell output from the thymus (1). SCID results from pathogenic variants in genes directly involved in the hematopoietic derived lymphocyte development and function (2). In addition to SCID, the TREC assay also identifies infants with hematopoietic extrinsic causes of T cell lymphopenia (TCL). Chromosome 22q11.2 deletion syndrome and pathogenic variants in CHD7, TBX1, TBX2, FOXI3, FOXN1 and PAX1 genes are known to be associated with thymic stromal cell defects (3–10). Recently, an ex vivo T cell differentiation assay using artificial thymic organoids (ATO) (11) has been described, which allows delineation of specific developmental blocks in T cell differentiation associated with SCID. This assay can help distinguish between hematopoietic intrinsic versus extrinsic causes of TCL (12, 13).
Pathogenic variants in TP63, a member of the p53-family of transcription factors, have been associated with TCL (Table 1). TP63 encodes the transcription factor p63, which has several functions including epithelial development and progenitor cell regulation. TP63 is comprised of five structural domains including the transactivation (TA) domain, DNA-binding domain (DBD), oligomerization domain (OD) (14), sterile-α-motif (SAM) domain and the transactivation inhibitory domain (TID) (15). Isoforms that either contain (TAp63) or lack (ΔNp63) the N-terminally located TA domain (Figure 1A) are differentially expressed in human tissues (18). ΔNp63 proteins are also expressed in cortical and medullary thymic epithelial cells (TECs) (Figures 1B, C) and in stratified epithelia where they play a central role in keratinocyte differentiation, proliferation, and cell adhesion (19). Mice that lack all isoforms of p63 do not develop limbs or stratified epithelia and have small, hypoplastic thymuses (20–22).
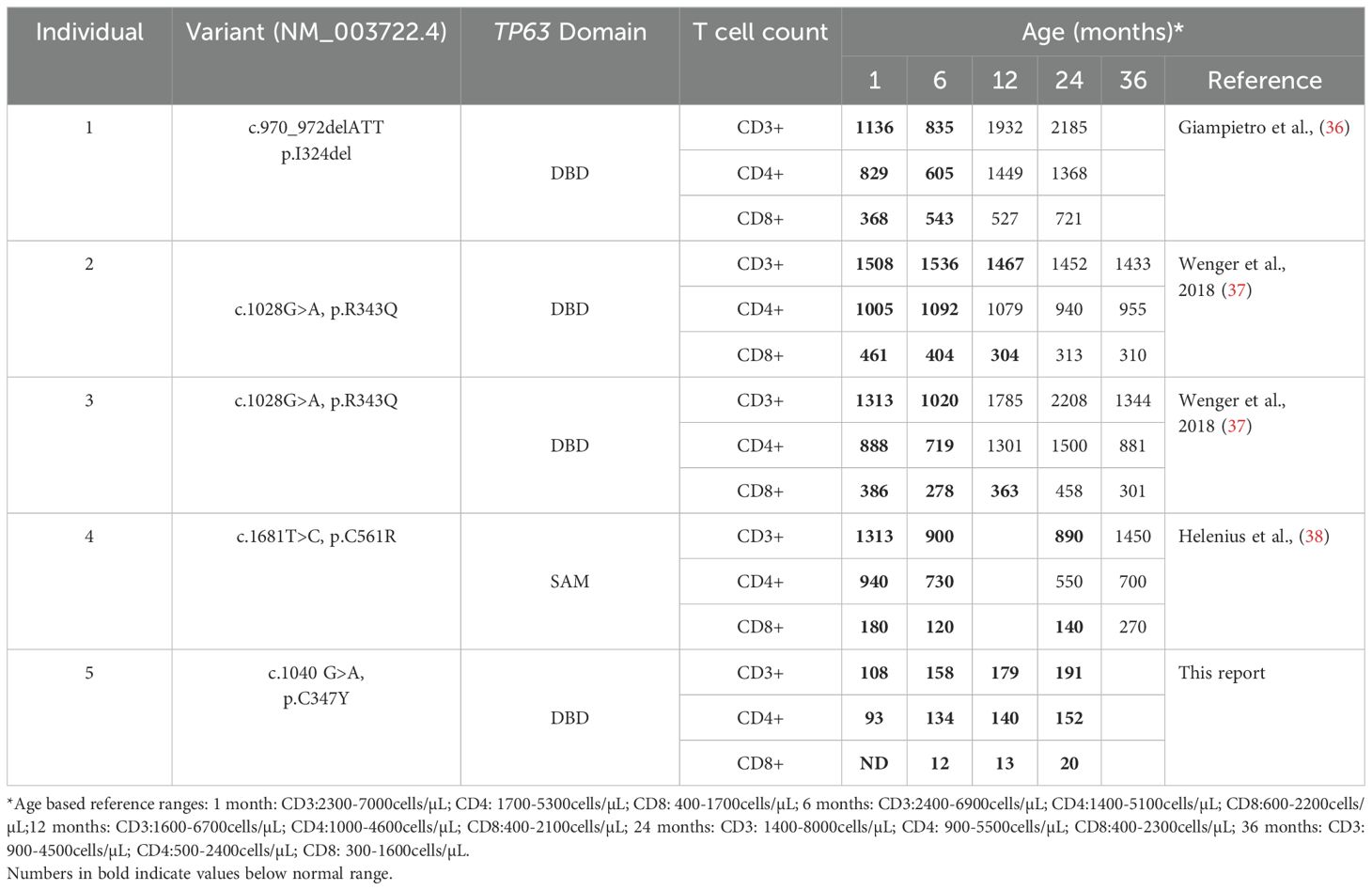
Table 1. Lymphocyte counts of individuals with TP63 variant associated with T cell lymphopenia.
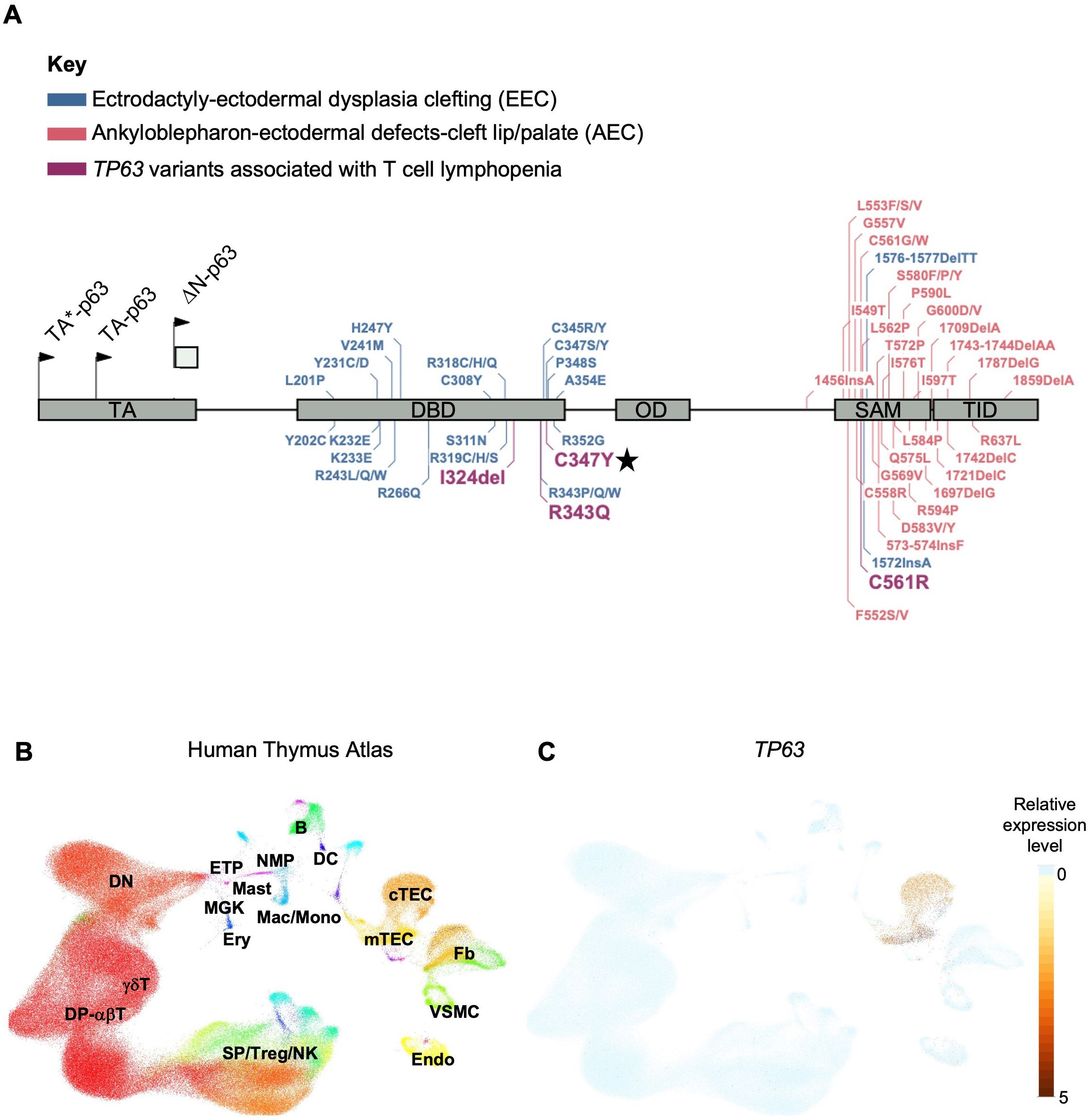
Figure 1. (A) Gene map of previously described TP63 pathogenic variants and associated syndromes as indicated by color key. Given that there is overlap and variability in the features of AEC and EEC, variants have been categorized based on the most prevalent disease features that were reported. Numbering of pathogenic variants in TA*-p63 isoform is based on reference sequence NP_003713.3. Start codons for TA*-p63, TA-p63 and ΔN-p63 isoforms are indicated by arrows. TA Transactivation domain; DBD DNA binding domain; OD oligomerization domain; SAM sterile-α-motif domain; TID transcriptional inhibitory domain. Figure adapted from Osterburg et al. (16). The TP63 variant identified in our patient is highlighted with a star. (B) Uniform manifold approximation and projection (UMAP) visualization showing the cellular composition of the human thymus obtained from the collection of single-cell datasets available on the UCSC Cell Browser [https://fetal-thymus.cells.ucsc.edu] (17). (C) Expression level of TP63 gene in the human thymus atlas is restricted to cTEC and mTEC subsets. DN, CD4/CD8 double negative T cells; DP, CD4/CD8 double positive T cells; SP, CD4/CD8 single positive T cells; Treg, regulatory T cells; NK, natural killer cells; Endo, endothelial cells; VSMC, vascular smooth muscle cells; Fb, fetal bone; mTEC, medullary thymic epithelial cells; cTEC, cortical thymic epithelial cells; DC, dendritic cells; B, B cells; NMP, neutrophil myeloid progenitors; Mac/Mono, macrophages/monocytes; Mast, mast cells; MGK, megakaryocytes; ETP, early thymic progenitor; Ery, erythrocytes.
TP63 variants are associated with 6 overlapping clinical phenotypes - ectrodactyly-ectodermal dysplasia clefting (EEC), ankyloblepharon-ectodermal defects-cleft lip/palate (AEC), limb-mammary syndrome (LMS) and acro–dermato–ungual–lacrimal–tooth (ADULT) syndromes, as well as split-hand/foot malformation type 4 (SHFM4) and non-syndromic cleft lip (NSCL) (16, 23). Major clinical features of this group of disorders includes ectodermal dysplasia, cleft lip or palate, as well as hand and foot malformations (24). EEC is typically characterized by limb malformations including ectrodactyly, syndactyly, as well as thin, dry skin, nail dysplasia and sparse hair. AEC is characterized by more severe skin erosions, nail and dental abnormalities, in addition to ankyloblepharon (partial or complete adhesion of the upper and lower eyelid). Some genotype-phenotype correlation has been suggested for this group of disorders. Variants in the DBD are more frequently associated with EEC, whereas variants in the SAM domain and TID are more frequently associated with AEC (25). TCL is not commonly reported in the majority of described TP63 variants. However, three out of four TP63 variants associated with TCL localize to the DBD (Figure 1A).
Here we present an infant with overlapping features of EEC and AEC, as well as undetectable TRECs on newborn screen who was found to have a pathogenic TP63 variant. With the use of the ATO co-culture system, we were able to determine that the patient’s TCL was likely due to a thymic, rather than hematopoietic, defect.
2 Case presentation
The patient is a 3-year-old girl born at term to healthy, non-consanguineous parents. No pertinent family history was identified. She required continuous positive airway pressure, then intubation, at birth due to micrognathia and tongue-base airway obstruction. Multiple congenital anomalies were noted including features of AEC, such as bilateral blepharophimosis, cloudy corneas, diffuse areas of denuded skin, particularly on her scalp and feet and cleft palate. In addition to her cleft palate, features consistent with EEC included complex hand anomalies (i.e. anterior polydactyly; 3, 4 syndactyly; unilateral ectrodactyly), hypoplastic nails and foot anomalies (i.e. bilateral ectrodactyly, posterior polydactyly, hypoplastic nails) (Figures 2A–H). She was also noted to have an atrial septal defect (ASD), imperforate anus and rectovestibular fistula. Heart defects and anorectal malformations have rarely been reported in individuals with TP63 variants (26, 27). Newborn screens were notable for 0 TREC copy number/μL of blood at 14 hours, 7, 19 and 30 days of life (Table 2). Lymphocyte subsets at 2 weeks of life revealed low CD3+ cells (30 cells/μl), low CD4+ cells (30 cells/μl), absent CD8+ cells, normal CD19+ (1029 cells/μl), CD16+ (418 cells/μl) and CD56+ (239 cells/μl), which was consistent with a T-B+NK+ severe combined immunodeficiency (SCID)-like phenotype (Table 2).
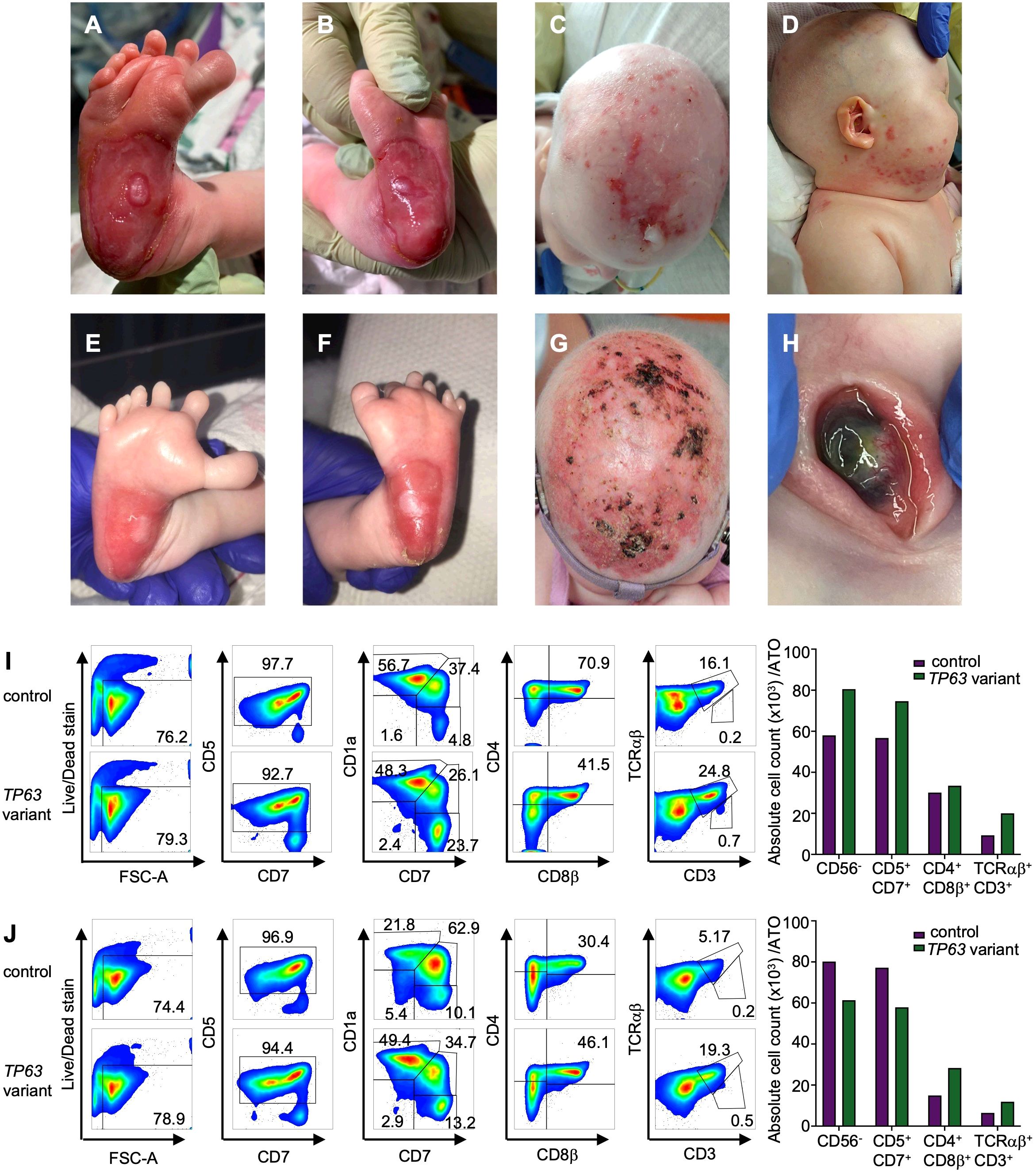
Figure 2. Clinical findings in a patient with TP63 variant and T cell lymphopenia. (A) Right foot with widely spaced toes, polysyndactyly and severe skin erosion at the heel at 1 week of age (B) left foot with skin erosions at the heel at 1 week of age. (C) Severe skin erosions on scalp at 4 months of age (D) skin erosions on the right cheek at 4 months of age. (E) Right foot with improved skin erosions at 1 month of age (F) left foot with widely spaced toes, polysyndactyly and improved skin erosions at 1 month of age. (G) Scalp with improved skin erosions at 13 months of age (H) Left eye with central thinning at 13 months of age. (I, J) T cell differentiation induced by artificial thymic organoids culturing CD34+ cells from individual with TP63 pathogenic variant (case) and unaffected age-matched control in the Artificial Thymic Organoid (ATO) system. Representative analysis of T cell differentiation in case and control at 4 months of age (I) and 26 months of age (J) after 6 weeks in culture in the ATO system. Cells were gated on LIVE/DEAD–CD45+CD14–CD56– cells and the expression of early and late T cell commitment markers (CD5, CD7, CD1α, CD4, CD8β, CD3, TCRαβ. Absolute cell counts per artificial thymic organoid and indicated early and late T cell commitment markers in TP63 variant (purple) and control (green) at 4 months (I) and 26 months of age (J).
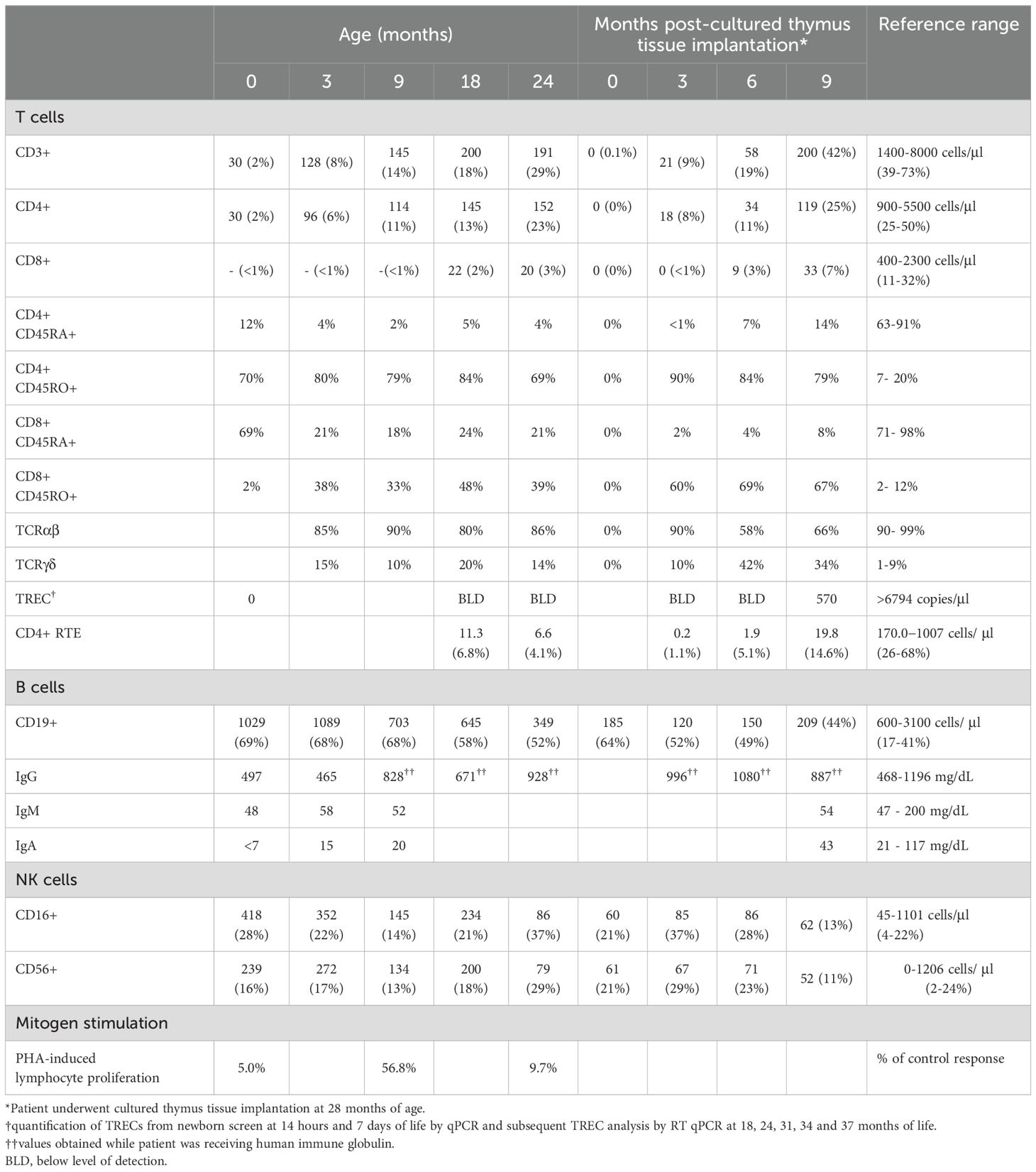
Table 2. Patient immune studies over time.
Initial testing demonstrated normal adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) enzymes and normal double-strand DNA break repair in lymphocytes 24 hours post-irradiation. No maternal engraftment was detected. Genetic studies included microarray analysis, which revealed three copy number variations at 1q44 (36kb copy gain), 3q26.1 (74kb copy gain) and 18p11.32 (66kb copy loss). However, all three represent population variants that are likely nonpathogenic. She underwent rapid whole genome sequencing through the SeqFirst research program at the University of Washington (IRB # STUDY00008810), which revealed a heterozygous de novo variant in exon 8 of TP63 corresponding to c.1040 G>A with predicted protein change C347Y in the DBD of p63 (Figure 1). This variant has been previously reported in patients with syndromic features consistent with AEC and with EEC syndrome (25, 28, 29). An additional heterozygous variant of uncertain significance (VUS) was identified in PRKDC (c.6901C>G; Q2301E), which was also present in the patient’s father who was healthy. Given that mutations in PRKDC have been associated with immunodeficiency (30–32), targeted RNA sequencing to assess for altered splicing or expression levels was performed. No significant changes in PRKDC transcript levels or splicing patterns were detected (data not shown). Additionally, the patient’s normal DNA repair studies and normal B cell counts further argued against the contribution of PRKDC associated immunodeficiency.
To determine whether the cause of the patient’s T cell lymphopenia was due to a thymic or hematopoietic defect, we took advantage of the recently developed artificial thymic organoid (ATO) system (11, 12). Upon parental informed consent, the patient was enrolled in protocol NCT03610802 approved by the National Institute of Health’s Institutional Review Board. CD34+CD3- cells were isolated from the patient’s peripheral blood mononuclear cells (PBMCs) and cultured in the ATO system. T cell maturation was analyzed after 6 weeks in culture in the ATO system both at 4 and at 26 months of age. Figures 2I, J show the flow cytometry gating strategy and absolute T cell counts of cells at different stages of intrathymic development. We found equivalent numbers of total cells (CD56-CD14-), T cell lineage committed cells (CD5+CD7+), double positive (DP) cells (CD4+CD8β+) and mature CD3+TCRαβ+ cells in the patient as compared to an unaffected control. These data suggested that the patient’s TCL was extrinsic to the hematopoietic cell lineage and most likely due to a thymic stromal cell defect in the setting of her TP63 variant. Consistent with this, interrogation of a publicly available database of gene expression profile in the human thymus demonstrated that TP63 expression is restricted to cortical and medullary thymic epithelial cells, and is not observed in thymic hematopoietic cell types (Figures 1B, C) (17).
Over time, the patient remained with persistently low numbers of T cells, high percentage of TCRγδ+ cells and low absolute counts of TCRαβ+ cells (Table 2). TRECs assessed at 18 and 24 months of age remained below the level of assay detection (Table 2). Within the T cell subset, cells displayed a predominantly memory CD4+CD45RO+ phenotype. The patient was started on IgG replacement at 4 weeks of life; thus, IgG levels reflect IgG replacement. However, she had normal levels of IgM, as well as normal levels of IgA and IgE suggesting adequate class-switching, presumably through B-cell interaction with γδT cells (33). Mitogen-induced lymphocyte proliferation was assessed at 2 weeks, 9, and 21 months of age and demonstrated a reduced response to phytohemagglutinin (PHA) stimulation when compared to control lymphocytes, which may reflect the patient’s low lymphocyte count (34). Recent thymic emigrants were reported as 11.3 CD4+ cells/μl (5.6%) and 6.6 CD4+ cells/μl (4.4%) [reference range 170-1007 cells/μl (25.8-68%)] suggesting no recovery of T cell counts by 18 and 24 months of age, respectively.
The patient’s clinical course (Supplementary Figure 1) was characterized by extubation to high flow nasal cannula by 1 month of age and subsequent weaning to room air. She had severe obstructive sleep apnea resulting in the need for supplemental oxygen during sleep. Due to her dysphagia and faltering growth, she required post-pyloric feeds, followed by gastrostomy tube feeds at 4 months of age. In the setting of her anorectal malformation, she initially had a colostomy placed and then underwent an anorectoplasty at 7 months of age. Eye exams demonstrated neovascularization and thinning of bilateral corneas (Figure 2H), which led to corneal perforation of both eyes requiring Gundersen conjunctival flap repair and corneal transplant. She has hearing impairment requiring hearing aids, as well as chronic otorrhea. Her cleft palate was closed at 16 months of age. Echocardiogram demonstrated normal systolic function and spontaneous closure of ASD by 18 months of age. She requires daily emollient application to her skin, as well as antibacterial and antifungal topical ointments. Given her TCL, she receives immunoglobulin replacement therapy, trimethoprim/sulfamethoxazole, fluconazole, azithromycin, and acyclovir prophylaxis. She has been kept isolated and has not had significant infectious complications.
In light of her persistent TCL and suspected athymia, she underwent cultured thymic tissue implantation (CTTI) at 28 months of age. Because her total CD3+ T cell counts were > 100 cells/µl and she had normal proliferation responses to PHA mitogen prior to implantation, she received immunosuppression with anti-thymocyte globulin and cyclosporin to reduce the risk of allograft rejection (35). She has been maintained on cyclosporin following implant without complications. At 6 months post-CTTI no significant improvement in thymic output was appreciated. Encouragingly, at 9 months post-CTTI evidence of measurable TRECs and CD4+ recent thymic emigrants (RTE) were noted, along with a 25% increase in total circulating CD4+ T cells (Table 2) suggesting early thymopoiesis. Thymopoiesis typically occurs between 6 and 12 months post-CTTI (10, 35). She will continue regular monitoring to ensure thymic recovery continues and circulating T cells are functional.
3 Discussion
Our patient’s case adds to a growing body of literature suggesting that individuals with TP63 variants can present with T cell lymphopenia. Four such individuals have been described previously, including a set of monozygotic twins (36–38). As in our patient, the other four individuals were identified on newborn screen as having low or absent TRECs. However, in all the published reports, the T cell lymphopenia was not severe and resolved by 1-3 years of age (Table 1). The individual described by Giampietro et al. initially demonstrated mild T cell lymphopenia, but intact lymphocyte response to PHA stimulation. His course was complicated by Pseudomonas aeruginosa periorbital cellulitis at 2 months of age, and he remained on intravenous immune globulin replacement (IVIG) and did not receive live vaccines until his lymphocyte counts spontaneously recovered at 14 months of age (36). Two monozygotic twins reported by Wenger et al. had low overall CD3, CD4 and CD8 counts that recovered by 12 months of age. Their lymphocytes also responded appropriately to PHA stimulation. Around the time of T cell recovery, both twins demonstrated normal effector and memory CD4+ and CD8+ T cell distribution. Both twins had mildly low IgG and IgM levels in infancy that recovered, and this was thought to be due to transient hypogammaglobulinemia of infancy. They did not require IVIG, and tolerated live vaccines once their T lymphocyte counts normalized. At 19 months of age, Twin 1 was hospitalized with a skin abscess caused by Methicillin Resistant Staphylococcus aureus (MRSA) complicated by toxin-mediated Staphylococcal Scalded Skin Syndrome. Twin 2 had MRSA orbital cellulitis around the same time, but did not require hospitalization. In addition, Twin 2 had E. coli pyelonephritis in infancy and was found to have vesicoureteral reflux (37). Helenius et al. described a preterm infant with overlapping features of EEC/AEC and lymphopenia. Interestingly, while all of the other known individuals with T cell lymphopenia have a TP63 variant in the DNA-binding domain, this individual’s TP63 variant is in the SAM domain (Figure 1). She had low CD3, CD4, and CD8 counts that recovered by age 3. She initially had undetectable IgA/IgM and low IgG and was started on IVIG and trimethoprim/sulfamethoxazole prophylaxis in the neonatal period and continued through age 30 months. She developed Staphylococcal sepsis on four separate occasions in the first four months of life, but then continued without additional serious bacterial infections (38). Finally, two years before the genetic cause of EEC and AEC was identified (39), Frick et al. reported a newborn with an undefined genetic variant resulting in EEC who was born at 35 weeks due to maternal eclampsia, and who died shortly after birth. Post-mortem examination revealed a small rudimentary thymus and lack of Hassall’s corpuscles (40). Compared to previously published cases, our patient demonstrated severe and persistent T cell lymphopenia (Table 1), prompting intervention with CTTI. To our knowledge, she is the first patient with TP63 variant and suspected congenital athymia in whom definitive treatment with cultured thymic tissue implantation has been attempted.
Variants in the DBD of TP63 have been more frequently associated with EEC. Our patient highlights the clinical variability and phenotypic overlap among TP63-related disorders. The presence of hand and foot malformations, as well as imperforate anus aligns with features found in EEC. However, the severity of her skin erosions and the presence of bilateral blepharophimosis is consistent with features of AEC. Interestingly, another infant with the same TP63 variant (c.1040G > A, p.C347Y) was also reported to have overlapping features of EEC and AEC. Unfortunately, she died at 2 months of age due to sepsis and it is unknown whether she had TCL (28). TP63 variant c.1039T>A, p.C347S has been reported in an individual with features of EEC and recurrent pulmonary infections (29). With the wide adoption of the TREC assay in NBS, more individuals with TP63 variants and TCL will likely be identified in the neonatal period when initiation of immunoglobulin replacement therapy and antimicrobial prophylaxis can prevent infectious complications.
It remains unclear why some individuals with TP63 variants develop T cell lymphopenia and why it is more severe and persistent in some and spontaneously resolves in others. In mouse models, the T cell lymphopenia appears to be linked to the loss of the ΔNp63. The p63-deficiency blocks the proliferation of thymic epithelial stem cells prenatally (21, 22). In the postnatal period, a recent study using conditional knockout of p63 in TECs showed that mice develop profound thymic hypoplasia, lack normal thymic architecture and have T cell differentiation defects (41). Based on these data, it is possible that TCL seen in individuals with TP63 variants is due to a direct effect of those variants on development and function of TECs. Furthermore, as demonstrated in FOXN1 variants, the variability in clinical phenotype in TP63 variants may be determined based on how the variant impacts transcriptional activity, nuclear localization and/or dominant negative effects (5). Indeed, two recent studies shed light on the mechanism of specific variants on transcriptional activity of TP63. Russo et al. show that several TP63 variants associated with AEC and that localize to the SAM domain cause decreased DNA binding and transcriptional activity due to TP63 protein aggregation (23). In a separate study, this group performed a comprehensive analysis of variants underlying EEC on DNA binding. Interestingly, they show that the C347Y variant found in our patient maps to a TP63 region responsible for direct DNA contact and does not bind DNA as demonstrated in their DNA pull down, p53 displacement and surface plasmon resonance assays evaluating protein-DNA interaction. Thus, the C347Y substitution is very damaging. In contrast, the C343S variant demonstrated wildtype-like DNA binding. Similarly, R343Q variant, identified in the monozygotic twins (Table 1), localizes to the TP63 protein interface with direct DNA contact and shows absent transcriptional activity and reduced Zinc binding (16). More studies are needed to understand why variants that similarly impact DNA binding to TP63 result in such heterogenous clinical phenotypes and varying impact on T cell development.
With regard to persistence of TCL, homeostatic expansion of T cells in the periphery may explain the spontaneous resolution of TCL in some patients. It is possible that this could lead to underreporting of TCL in patients with TP63 variants. Individuals with thymic defects due to 22q11.2 deletion syndrome and FOXN1 haploinsufficiency can have a marked T cell lymphopenia early in life with a gradual normalization of CD4+ and CD8+ T cell numbers possibly due to homeostatic expansion (42, 43). It has been proposed that in the absence of normal thymic development, homeostatic proliferation may result in a restricted T cell receptor repertoire and, subsequently, suboptimal T cell responses in the setting of infection or expansion of cells that react to self-antigen, leading to autoimmunity (44).
Infants with TCL who are identified on NBS can have a hematopoietic or non-hematopoietic cause of their lymphopenia and choosing the correct and timely therapy can be clinically challenging. With the wide adaptation of SCID NBS, an increasing number of patients are being recognized as having thymic defects or genetically undefined causes of SCID. Early recognition and prompt referral for CTTI leads to better clinical outcomes (45). Our case highlights the difficulty of making therapeutic decisions for treating SCID when a genetic variant is identified, but there is very little known about the effect of that gene variant on immune system functioning. The ATO assay demonstrated that the patient’s T cell precursors were capable of fully differentiating ex vivo into single positive CD4 and CD8 T cells suggesting that CTTI may provide definitive treatment for her T cell lymphopenia. The ATO co-culture system also has some limitations. For example, some metabolic causes of T cell lymphopenia, such as in adenosine deaminase deficiency, are not accurately captured by the assay resulting in T cell differentiation ex vivo (46). Our case also highlights the time needed for additional investigation, as well as a period of observation to determine if the TCL may resolve without intervention, as was seen with other individuals with TP63 variants. Nevertheless, the ATO co-culture system is a promising diagnostic tool that can help identify infants with thymic defects and novel genetic causes of SCID (46).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Institutional Review Board, National Institutes of Health. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
AG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Writing – review & editing. KC: Investigation, Writing – review & editing. SM: Writing – review & editing. KA: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. FP: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. OD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Writing – review & editing. TW: Investigation, Writing – review & editing. MiB: Investigation, Writing – review & editing. JS: Investigation, Writing – review & editing. MaB: Writing – review & editing. NO: Formal analysis, Investigation, Writing – review & editing. LN: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – review & editing. MM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing, Visualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. LN is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (grant AI001222).
Acknowledgments
We thank Dr. Kjell Helenius and Dr. Leena Kainulainen for their critical review of the manuscript and providing additional clinical laboratory data from a patient with TP63 variant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1438383/full#supplementary-material
Supplementary Figure 1 | Visual representation of the clinical time course and treatment of patient with TP63 variant and T cell lymphopenia. NBS, newborn screen; GT, Gastrostomy tube; OSA, obstructive sleep apnea; PSARP, Posterior Sagittal Anorectoplasty; ASD, atrial septal defect; ATG, anti-thymocyte globulin.
References
1. Puck JM. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol Rev. (2019) 287:241–52. doi: 10.1111/imr.12729
2. Dvorak CC, Haddad E, Heimall J, Dunn E, Cowan MJ, Pai S-Y, et al. The diagnosis of severe combined immunodeficiency: Implementation of the PIDTC 2022 Definitions. J Allergy Clin Immunol. (2023) 151:547–555.e5. doi: 10.1016/j.jaci.2022.10.021
3. Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aax1036
4. Du Q, Huynh LK, Coskun F, Molina E, King MA, Raj P, et al. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans. J Clin Investig. (2019) 129:4724–38. doi: 10.1172/jci127565
5. Moses A, Bhalla P, Thompson A, Lai L, Coskun FS, Seroogy CM, et al. Comprehensive phenotypic analysis of diverse FOXN1 variants. J Allergy Clin Immunol. (2023) 152:1273–1291.e15. doi: 10.1016/j.jaci.2023.06.019
6. Kubala SA, Sandhu A, Palacios-Kibler T, Ward B, Harmon G, DeFelice ML, et al. Natural history of infants with non-SCID T cell lymphopenia identified on newborn screen. Clin Immunol. (2022) 245:109182. doi: 10.1016/j.clim.2022.109182
7. Liu N, Schoch K, Luo X, Pena LDM, Bhavana VH, Kukolich MK, et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet. (2018) 27:2454–65. doi: 10.1093/hmg/ddy146
8. Bernstock JD, Totten AH, Elkahloun AG, Johnson KR, Hurst AC, Goldman F, et al. Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J Allergy Clin Immunol. (2020) 145:358–367.e2. doi: 10.1016/j.jaci.2019.09.020
9. Bhalla P, Wysocki CA, van Oers NSC. Molecular insights into the causes of human thymic hypoplasia with animal models. Front Immunol. (2020) 11:830. doi: 10.3389/fimmu.2020.00830
10. Collins C, Sharpe E, Silber A, Kulke S, Hsieh EWY. Congenital athymia: genetic etiologies, clinical manifestations, diagnosis, and treatment. J Clin Immunol. (2021) 41:881–95. doi: 10.1007/s10875-021-01059-7
11. Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods. (2017) 14:521–30. doi: 10.1038/nmeth.4237
12. Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. (2020) 4:2611–6. doi: 10.1182/bloodadvances.2020001730
13. Bifsha P, Leiding JW, Pai S-Y, Colamartino ABL, Hartog N, Church JA, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv. (2020) 4:2606–10. doi: 10.1182/bloodadvances.2020001736
14. Levrero M, Laurenzi VD, Costanzo A, Sabatini S, Gong J, Wang JYJ, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. (2000) 113:1661–70. doi: 10.1242/jcs.113.10.1661
15. Ghioni P, Bolognese F, Duijf PHG, van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains†. Mol Cell Biol. (2002) 22:8659–68. doi: 10.1128/mcb.22.24.8659-8668.2002
16. Osterburg C, Ferniani M, Antonini D, Frombach A-S, D’Auria L, Osterburg S, et al. Disease-related p63 DBD mutations impair DNA binding by distinct mechanisms and varying degree. Cell Death Dis. (2023) 14:274. doi: 10.1038/s41419-023-05796-y
17. Speir ML, Bhaduri A, Markov NS, Moreno P, Nowakowski TJ, Papatheodorou I, et al. UCSC cell browser: visualize your single-cell data. Bioinformatics. (2021) 37:btab503. doi: 10.1093/bioinformatics/btab503
18. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, et al. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol Cell. (1998) 2:305–16. doi: 10.1016/s1097-2765(00)80275-0
19. Dotto J, Pelosi G, Rosai J. Expression of p63 in thymomas and normal thymus. Am J Clin Pathol. (2007) 127:415–20. doi: 10.1309/2gaykpddm85p2vew
20. Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. (1999) 398:714–8. doi: 10.1038/19539
21. Candi E, Rufini A, Terrinoni A, Giamboi-Miraglia A, Lena AM, Mantovani R, et al. [amp]]Delta;Np63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc Natl Acad Sci. (2007) 104:11999–2004. doi: 10.1073/pnas.0703458104
22. Senoo M, Pinto F, Crum CP, McKeon F. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell. (2007) 129:523–36. doi: 10.1016/j.cell.2007.02.045
23. Russo C, Osterburg C, Sirico A, Antonini D, Ambrosio R, Würz JM, et al. Protein aggregation of the p63 transcription factor underlies severe skin fragility in AEC syndrome. Proc Natl Acad Sci USA. (2018) 115:E906–15. doi: 10.1073/pnas.1713773115
24. Sutton VR, van Bokhoven H. TP63-Related Disorders. (1993). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle.
25. Rinne T, Brunner HG, van Bokhoven H. p63-associated disorders. Cell Cycle. (2007) 6:262–8. doi: 10.4161/cc.6.3.3796
26. Sharma D, Kumar C, Bhalerao S, Pandita A, Shastri S, Sharma P. Ectrodactyly, ectodermal dysplasia, cleft lip, and palate (EEC syndrome) with tetralogy of fallot: A very rare combination. Front Pediatr. (2015) 3:51. doi: 10.3389/fped.2015.00051
27. Ruml J, Cuturilo G, Lukac M, Peters H. Ectodermal defects and anal atresia in a child with a TP63 mutation-expanding the phenotypic spectrum. Pediatr Dermatol. (2015) 32:421–2. doi: 10.1111/pde.12395
28. Celik TH, Buyukcam A, Simsek-Kiper PO, Utine GE, Ersoy-Evans S, Korkmaz A, et al. A newborn with overlapping features of AEC and EEC syndromes. Am J Méd Genet Part A. (2011) 155:3100–3. doi: 10.1002/ajmg.a.34328
29. van Bokhoven H, Hamel BCJ, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PHG, et al. p63 gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand–split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. (2001) 69:481–92. doi: 10.1086/323123
30. Mathieu A-L, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator–dependent autoimmunity. J Allergy Clin Immunol. (2015) 135:1578–1588.e5. doi: 10.1016/j.jaci.2015.01.040
31. van der Burg M, IJspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T–B– SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Investig. (2009) 119:91–8. doi: 10.1172/jci37141
32. Esenboga S, Akal C, Karaatmaca B, Erman B, Dogan S, Orhan D, et al. Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature. Clin Immunol. (2018) 197:1–5. doi: 10.1016/j.clim.2018.08.002
33. Rampoldi F, Ullrich L, Prinz I. Revisiting the interaction of γδ T-cells and B-cells. Cells. (2020) 9:743. doi: 10.3390/cells9030743
34. Mustillo PJ, Sullivan KE, Chinn IK, Notarangelo LD, Haddad E, Davies EG, et al. Clinical practice guidelines for the immunological management of chromosome 22q11.2 deletion syndrome and other defects in thymic development. J Clin Immunol. (2023) 43:247–70. doi: 10.1007/s10875-022-01418-y
35. Markert ML, Gupton SE, McCarthy EA. Experience with cultured thymus tissue in 105 children. J Allergy Clin Immunol. (2022) 149:747–57. doi: 10.1016/j.jaci.2021.06.028
36. Giampietro PF, Baker MW, Basehore MJ, Jones JR, Seroogy CM. Novel mutation in TP63 associated with ectrodactyly ectodermal dysplasia and clefting syndrome and T cell lymphopenia. Am J Méd Genet Part A. (2013) 161:1432–5. doi: 10.1002/ajmg.a.35885
37. Wenger T, Li D, Harr MH, Tan W, Pellegrino R, Stark Z, et al. Expanding the phenotypic spectrum of TP63-related disorders including the first set of monozygotic twins. Am J Méd Genet Part A. (2018) 176:75–81. doi: 10.1002/ajmg.a.38516
38. Helenius K, Ojala L, Kainulainen L, Peltonen S, Hietala M, Pohjola P, et al. Overlap between EEC and AEC syndrome and immunodeficiency in a preterm infant with a TP63 variant. Eur J Méd Genet. (2023) 66:104735. doi: 10.1016/j.ejmg.2023.104735
39. Celli J, Duijf P, Hamel BCJ, Bamshad M, Kramer B, Smits APT, et al. Heterozygous Germline Mutations in the p53 Homolog p63 Are the Cause of EEC Syndrome. Cell. (1999) 99:143–53. doi: 10.1016/s0092-8674(00)81646-3
40. Frick H, Münger DM, Fauchère JC, Stallmach T. Hypoplastic thymus and T-cell reduction in EECUT syndrome. Am J Méd Genet. (1997) 69:65–8. doi: 10.1002/(ISSN)1096-8628
41. Stefanski HE, Xing Y, Nicholls J, Jonart L, Goren E, Taylor PA, et al. P63 targeted deletion under the FOXN1 promoter disrupts pre-and post-natal thymus development, function and maintenance as well as induces severe hair loss. PloS One. (2022) 17:e0261770. doi: 10.1371/journal.pone.0261770
42. Piliero LM, Sanford AN, McDonald-McGinn DM, Zackai EH, Sullivan KE. T-cell homeostasis in humans with thymic hypoplasia due to chromosome 22q11.2 deletion syndrome. Blood. (2004) 103:1020–5. doi: 10.1182/blood-2003-08-2824
43. Bosticardo M, Yamazaki Y, Cowan J, Giardino G, Corsino C, Scalia G, et al. Heterozygous FOXN1 variants cause low TRECs and severe T cell lymphopenia, revealing a crucial role of FOXN1 in supporting early thymopoiesis. Am J Hum Genet. (2019) 105:549–61. doi: 10.1016/j.ajhg.2019.07.014
44. Surh CD, Sprent J. Homeostatic T cell proliferation. J Exp Med. (2000) 192:F9–F14. doi: 10.1084/jem.192.4.f9
45. Howley E, Golwala Z, Buckland M, Barzaghi F, Ghosh S, Hackett S, et al. Impact of newborn screening for SCID on the management of congenital athymia. J Allergy Clin Immunol. (2023) 153:330–4. doi: 10.1016/j.jaci.2023.08.031
Keywords: TP63, T cell lymphopenia, artificial thymic organoids, ectrodactyly-ectodermal dysplasia clefting (EEC), ankyloblepharon-ectodermal defects-cleft lip/palate (AEC), cultured thymus tissue implantation (CTTI)
Citation: Gall A, Bosticardo M, Ma S, Chen K, Amini K, Pala F, Delmonte OM, Wenger T, Bamshad M, Sleasman J, Blessing M, van Oers NSC, Notarangelo LD and de la Morena MT (2024) Case report: Artificial thymic organoids facilitate clinical decisions for a patient with a TP63 variant and severe persistent T cell lymphopenia. Front. Immunol. 15:1438383. doi: 10.3389/fimmu.2024.1438383
Received: 25 May 2024; Accepted: 27 August 2024;
Published: 18 September 2024.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Isabella Quinti, Sapienza University of Rome, ItalyWinnie Ip, Great Ormond Street Hospital, United Kingdom
Alexandra Yema Kreins, Great Ormond Street Hospital, United Kingdom
Copyright © 2024 Gall, Bosticardo, Ma, Chen, Amini, Pala, Delmonte, Wenger, Bamshad, Sleasman, Blessing, van Oers, Notarangelo and de la Morena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Teresa de la Morena, bWFpdGUuZGVsYW1vcmVuYUBzZWF0dGxlY2hpbGRyZW5zLm9yZw==