 Edward J. Filippone
Edward J. Filippone Rakesh Gulati1
Rakesh Gulati1 John L. Farber
John L. Farber- 1Division of Nephrology, Department of Medicine, Sidney Kimmel Medical College at Thomas Jefferson University, Philadelphia, PA, United States
- 2Department of Pathology, Sidney Kimmel Medical College at Thomas Jefferson University, Philadelphia, PA, United States
IgA nephropathy (IgAN) is considered the most common primary glomerulonephritis worldwide with a predilection for Asian-Pacific populations and relative rarity in those of African descent. Perhaps 20%–50% of patients progress to kidney failure. The pathogenesis is incompletely understood. Mesangial deposition of immune complexes containing galactose-deficient IgA1 complexed with anti-glycan IgG or IgA antibodies results in mesangial cell activation and proliferation, inflammatory cell recruitment, complement activation, and podocyte damage. Diagnosis requires a biopsy interpreted by the Oxford criteria. Additional pathologic features include podocytopathy, thrombotic microangiopathy, and C4d staining. Biomarkers predicting adverse outcomes include proteinuria, reduced GFR, hypertension, and pathology. Acceptable surrogate endpoints for therapeutic trials include ongoing proteinuria and rate of eGFR decline. The significance of persisting hematuria remains uncertain. The mainstay of therapy is supportive, consisting of lifestyle modifications, renin–angiotensin inhibition (if hypertensive or proteinuric), sodium-glucose-transporter 2 inhibition (if GFR reduced or proteinuric), and endothelin-receptor antagonism (if proteinuric). Immunosuppression should be considered for those at high risk after maximal supportive care. Corticosteroids are controversial with the most positive results observed in Chinese. They carry a high risk of serious side effects. Similarly, mycophenolate may be most effective in Chinese. Other immunosuppressants are of uncertain benefit. Tonsillectomy appears efficacious in Japanese. Active areas of investigation include B-cell inhibition with agents targeting the survival factors BAFF and APRIL and complement inhibition with agents targeting the alternate pathway (Factors B and D), the lectin pathway (MASP-2), and the common pathway (C3 and C5). Hopefully soon, the who and the how of immunosuppression will be clarified, and kidney failure can be forestalled.
Introduction
Initially described by Berger and Hinglais in 1968 (1), IgA nephropathy (IgAN) is characterized by the biopsy finding of dominant or codominant deposition of IgA-containing immune complexes with a variable inflammatory and/or sclerosing response (2). The clinical presentation is variable, ranging from recurrent bouts of macroscopic hematuria, following either an upper respiratory infection (synpharyngitic nephritis) or gastrointestinal infection, urinary sediment abnormalities (microscopic hematuria and/or proteinuria), to more serious presentations (frank nephrotic syndrome or rapidly progressive glomerulonephritis).
IgAN is typically referred to as the most common form of primary glomerulonephritis worldwide. However, variability exists among different populations with especially high relative frequencies in the Asia-Pacific region (China and Japan) and most Western European counties (up to 50% or more of biopsies with primary glomerulonephritis). IgAN is found less frequently in other areas, e.g., Africa and India (3). Such variability may result from multiple factors, including genetic susceptibility, the availability of immunofluorescence microscopy, differences in mass urinary screening programs, nephrology referral practices, and the willingness of nephrologists to biopsy cases with only mild urinary sediment abnormalities (3).
Although initially considered relatively benign, perhaps 25%–50% of patients with primary IgAN develop end-stage kidney disease (ESKD) if followed for 20–30 years (4). Pitcher et al. followed 2,439 IgAN patients (2,299 adults) referred to the UK National Registry of Rare Kidney Diseases (RaDaR) with either >0.5 g/day of proteinuria or eGFR <60 (all eGFR herein expressed in ml/min/1.73 m2) and found median kidney survival of 11.4 years with a mean age of kidney failure/death of 48 years (5). Given age at onset and baseline eGFR, it was estimated that almost all patients could progress to ESKD in their expected lifetimes, unless the average decline in eGFR was <1/year. Mortality is increased with IgAN. A Korean study of 1,364 IgAN patients revealed overall mortality to be 43% greater than the general population, but significantly so only in men (6). A nationwide Swedish cohort study estimated a reduced life expectancy of 6 years (7).
Demographic features affect the clinical presentation and course of IgAN. Originally thought to affect predominantly young adults in the second to fourth decade, the age of diagnosis has been increasing. In the RaDaR study, the median age at diagnosis in adults increased from 40 years in 2013 to 45 years in 2020 (5). The average age of presentation among 2,961 Spanish patients increased from 37.6 years in 1994–1997 to 44.9 years in 2010–2013 with approximately 10% >65 years old (8). Whereas younger patients were more likely to present with gross hematuria and urinary sediment abnormalities, the elderly were more likely to present with reduced eGFR, hypertension, acute kidney injury (AKI), and higher levels of proteinuria.
Racial and ethnic factors play a role in the development and progression of IgAN. Pacific-Asian populations show a greater prevalence than European–Caucasians, have more severe clinical presentations, more active lesions on biopsy, and have an increased rate of progression to ESKD (9, 10). IgAN is much less common in African Americans but does occur (11). Furthermore, racial/ethnic differences in response to therapy are reported with Chinese apparently more responsive to corticosteroids than Caucasians and Japanese more responsive to tonsillectomy (vide infra).
The genetic predisposition to IgAN has been extensively investigated (12). Kiryluk et al. performed genome-wide association study (GWAS) and meta-analysis of 17 international cohorts and identified 30 risk loci, which explained 11% of disease risk (13). Asian populations are enriched in susceptibility alleles (14). Additionally, epigenetic factors, such as altered DNA methylation (15) and interference from micro RNAs (16, 17), may contribute to risk.
The current understanding of the pathogenesis of IgAN is thought to involve four sequential hits (18). Elevated serum levels of primarily polymeric IgA1 with galactose-deficient hinge regions (Gd-IgA1) represent Hit 1 but are clearly not sufficient, as unaffected, healthy, first-degree relatives of patients with IgAN can have elevated levels. Gd-IgA1 forms polymers by self-aggregating. Hit 2 involves production of anti-Gd-IgA1 autoantibodies, predominantly IgG (but also IgA1). Hit 3 involves formation in the serum of immune complexes of polymeric IgA1 and anti-Gd-IgA1 of sufficient size to escape hepatic clearance and deposit in the glomerular mesangium. Finally, mesangial cells are activated, proliferate, recruit inflammatory cells, and secrete cytokines affecting podocyte structure and function (Hit 4). Importantly, both Gd-IgA1 and anti-GdIg-A1 antibodies are found in normal serum, albeit at lower levels than in IgAN patients (19). The naturally occurring anti-Gd-IgA1 may have been initially induced by various microorganisms (viruses and bacteria) that express the N-acetylgalactosamine residue characteristically overexposed by galactose deficiency in the Gd-IgA1 of IgAN patients (20).
Biomarkers and surrogate endpoints in IgAN
A biomarker in IgAN is a clinical or laboratory feature that predicts future development of a hard adverse outcome (ESKD, cardiovascular events, death). A valid surrogate endpoint is a biomarker whose alteration with a given therapy predicts a subsequent reduction in future hard endpoints with that therapy. Given the slowly progressive course of IgAN, hard clinical endpoints would not be feasible for therapeutic trials, and surrogate endpoints are needed. Surrogate endpoints may be validated or “reasonably likely.” A therapeutic benefit on a “reasonably likely” surrogate endpoint allows for accelerated FDA approval, although post-marketing trials are required to verify benefit. This is most germane when considering potentially toxic immunosuppressive therapy.
No biomarker is acceptable to diagnose IgAN, and biopsy is required. The most established biomarkers for predicting progression of IgAN at presentation include proteinuria, reduced eGFR, and hypertension clinically (21, 22) and the Oxford criteria pathologically. Potential clinical biomarkers requiring verification include genetic features, hematuria, levels of Gd-IgA1 and anti-Gd-IgA1 antibodies, and the IgA/C3 ratio. Additional pathologic findings indicating progression include thrombotic microangiopathy and positive C4d staining. Proteinuria, declining eGFR, persisting hematuria, and histologic changes on repeat biopsy are potential surrogate endpoints for clinical trials.
The level of Gd-IgA1 is a potential diagnostic tool and biomarker. Galactose-deficient IgA1 can be determined by various methods, including Western blotting, mass spectrometry, and liquid chromatography, but these are not readily available. Several lectins can bind to the unglycosylated N-acetylgalactosamine on Gd-IgA1, including lectins derived from the snail Helix aspersa (HAA) and the plant Vicia velosa. Moldoveanu et al. developed an HAA-based quantitative enzyme-linked immunosorbent assay (ELISA) for determining serum levels of Gd-IgA1 (23). Yasutake et al. developed an ELISA based on a rat monoclonal antibody (KM55) against human Gd-IgA1 correlating with the HAA-based assay (24).
As noted above, Gd-IgA1 is normally present in serum. Moldoveanu et al. found the 90th percentile using the HAA ELISA to be 1,076 U/ml in 150 healthy Caucasian adults (23). Sun et al. did a systematic review and meta-analysis of studies using lectin-based assays and showed that patients with IgAN have significantly higher mean levels Gd-IgA1 compared to normal but not different than first-degree relatives of IgAN patients or patients with IgA vasculitis. Levels in IgAN patients did not correlate with disease severity (25). Wada et al. used KM55 and found significantly higher serum levels of Gd-IgA1 in IgAN patients versus patients with lupus nephritis, ANCA-vasculitis, or minimal change disease, but similar levels to patients with IgA-vasculitis (26).
Similarly, anti-Gd-IgA1 antibodies are found in normal serum but at lower levels than in IgAN patients. Yanagawa et al. found that 41% of Japanese IgAN patients had serum levels of Gd-IgA1 above the 90th percentile of normal, and 91% had anti-Gd-IgA1 IgG antibodies above the 90th percentile of normal, as did up to 25% of patients with other kidney diseases, especially immune-mediated diseases such as lupus nephritis (27). The level of anti-Gd-IgA1 IgG performed well in separating IgAN patients from normal controls but less so compared to other immune-mediated kidney diseases.
Vaz de Castro et al. performed a systematic review and meta-analysis of observational studies and clinical trials where Gd-IgA1 levels were measured (29 studies involving 8,159 participants) (28). Levels were not significantly associated with hypertension, hematuria, or proteinuria. Results relating Gd-IgA1 levels to chronic kidney disease (CKD) stage or progression to ESKD were inconsistent. Currently, measurement of serum levels of Gd-IgA1 and/or corresponding antibodies are not sufficiently sensitive or specific to preclude biopsy and are not recommended as standard biomarkers of progressive disease.
Several studies assessed the serum IgA level/C3 ratio as a biomarker for diagnosis, grading severity, and/or determining prognosis. Tomino et al. found that the ratio could distinguish IgAN from other glomerulopathies in Japanese patients (29). Kawasaki et al. found that an elevated IgA/C3 ratio combined with more intense glomerular C3 staining predicted greater proteinuria, hematuria, and creatinine at follow-up, as well as more severe pathology scores on a second biopsy in Japanese pediatric IgAN patients (30). Mizerska-Wasiak et al. found more severe pathologic changes associated with IgA/C3 in pediatric Polish IgAN patients (31). By studying 1,219 Chinese adult patients, Chen et al. looked specifically at Gd-IgA1 and found a linear relationship between the serum Gd-IgA1/C3 ratio and kidney disease progression (32). More data are needed before general recommendations can be made regarding these ratios. Multiple other biomarkers in serum and urine have been proposed, but none are sufficiently validated for routine use (33, 34).
Proteinuria
Proteinuria is the best studied baseline biomarker and surrogate endpoint. Reich et al. followed 542 patients from the Toronto Glomerulonephritis Registry and found that 30% reached ESKD; follow-up proteinuria was the most important predictor by multivariable analysis (35). The rate of eGFR decline was 25-fold faster if the average proteinuria was >3 g/day versus <1 g/day. Reduction from >3 to <1 g/day produced a similar course to having <1 g/day throughout. Inker et al. performed an individual patient level meta-analysis of 11 randomized trials (830 patients) evaluating four interventions and found that decline in proteinuria at 9 months was significantly associated with reduced risk of doubling of serum creatinine, ESKD, or death [hazard ratio (HR) 0.40, 95% confidence interval (CI) 0.32–0.48] (36).
The Kidney Health Initiative extended this analysis to include 13 randomized controlled trials (RCTs) and 7 observational cohorts restricted to patients with IgAN (37) confirming the association of proteinuria reduction with hard clinical outcomes (doubling of serum creatinine, ESKD, or death). Proteinuria reduction is now considered a “reasonably likely” surrogate endpoint (see https://www.fda.gov/downloads/Drugs/Guidances/UCM358301.pdf) (37) that allowed for accelerated FDA approval of budesonide and sparsentan (vide infra).
Although Kidney Disease Improving Global Outcomes (KDIGO) considers proteinuria >0.75–1 g/day as high risk and reduction to <1 g/day as a reasonable target (38), patients with lower levels clearly may still progress. In the RaDaR study, approximately 30% of patients with time-averaged proteinuria <0.88 g/g creatinine (approximately <1 g/day) reached ESKD by 10 years, as did approximately 20% with <0.44 g/g (approximately <0.5 g/day) (5).
The nephrotic syndrome (NS), defined as nephrotic range proteinuria (NRP) with hypoalbuminemia, is found in approximately 10%–15% of primary IgAN patients (39, 40). The prognosis for IgAN is worse with NS. In one study, 24% of NS patients had doubling of serum creatinine over 45 months versus 7% of 885 patients without NS (40), and achieving remission significantly improved survival from that endpoint. Interestingly, 25% of nephrotic patients underwent spontaneous remission without relapse. In another study of 24 nephrotic IgAN patients, 5 had spontaneous remission, again with no relapse. In contrast, Huang et al. studied 1,413 Chinese IgAN patients and found no significant difference in the doubling of serum creatinine or ESKD in the 57 with NS (11).
Four studies of Chinese IgAN patients compared the prognosis of NS to NRP with normoalbuminemia (41–44). Three of these specifically excluded cases of IgAN with biopsies otherwise consistent with minimal change disease (MCD), the so-called MCD–IgAN (vide infra) (41, 42, 44). Li et al. compared 59 NS patients to 57 patients with NRP and found greater endocapillary inflammation and crescents in the former, but greater segmental sclerosis/adhesions to Bowman’s capsule in the latter. However, there was no significant difference in long-term outcome (41).
Chen et al. studied 119 patients with >2 g/day of proteinuria that were divided into 33 with NS, 34 with NRP but normoalbuminuria, and 52 controls with >2 but <3.5 g/day of proteinuria. Patients with NS had greater hematuria, more likely endocapillary inflammation, and more severe foot-process effacement (42). Survival free from a combined endpoint of doubling of serum creatinine and 50% reduction of eGFR or ESKD was surprisingly significantly better with NS versus NRP, and both were significantly worse than the controls.
Jiang et al. compared 85 patients with NS to 85 patients with only NRP and found the former had baseline higher eGFR, more severe hematuria, more severe foot process effacement, lower serum C3 levels, and lower body mass index (BMI) despite similar pathologic scores (44). Those with NS had double the risk of ≥30% decline in eGFR or ESKD on follow-up by Cox regression, although the rate of decline in eGFR was similar.
These three studies that excluded MCD–IgAN provide no clear evidence of worse outcome with NS versus NRP. The study not excluding MCD–IgAN compared 111 patients with NS to 111 with NRP and found no difference in composite outcome unless patients with complete remission were excluded in which case NS portended a worse outcome (43).
eGFR
Reduced baseline eGFR is a strong biomarker for an unfavorable outcome in IgAN. The rate of decline in eGFR has been extensively studied as a potential surrogate across a spectrum of kidney diseases. Inker et al. performed a meta-analysis of 47 RCTs assessing 12 interventions in over 60,000 patients and found a significant relationship between slope difference and hard clinical outcomes (45). For example, a difference in slope in the range of 0.5–1.0 ml/min/1.73 m2 from baseline to 3 years would have >97% chance of reducing adverse outcomes. Shorter time frames (e.g., 1 year) produce less certainty due to generally opposing short-term acute effects of many therapies. Similar results were obtained when analyzing observational data (46).
When analyzing 1-year data from 13 RCTs of IgAN therapy, Lafayette et al. found that both 1-year slope of eGFR decline and 1-year reduction of proteinuria were significantly associated with a composite end-point (eGFR reduction, serum creatinine doubling, ESKD, and/or death) (47). However, by multivariable analysis, only eGFR slope remained highly significantly associated with the composite endpoint, but proteinuria change did not.
Pathology: biomarker and surrogate?
The Oxford classification of IgAN (see Table 1), was published in 2009 to both standardize the pathologic description of IgAN and, importantly, to provide prognostic information (49, 50). Measures of both activity (mesangial hypercellularity, M, and endocapillary hypercellularity, E) and chronicity (segmental sclerosis, S, and tubular atrophy/interstitial fibrosis, T) were included, termed the MEST score. The prognostic ability has been substantiated multiple times [19 studies reviewed in (48)] including in both Asian (51) and European populations (52, 53). It was estimated that adding the MEST score to clinical features at biopsy would provide an equivalent predictive ability to 2 years of following clinical variables (53).

Table 1. Mest-C score (48).
Unsurprisingly, the T lesion has been most consistent in independently predicting adverse renal outcomes [in 14 of 19 studies by multivariable analysis (48)] since the endpoint in these studies was often ESKD. The E lesion was not predictive in most studies, but this was influenced by immunosuppression, as E1 biopsies were more likely to be immunosuppressed. The E lesion was predictive when analyzing only un-immunosuppressed patients suggesting it may be responsive to therapy. The S lesion may arise from healed necrotic or inflammatory lesions or be a result of primary podocyte damage. At least one study showed that either podocyte hypertrophy or the tip lesion was associated with an adverse outcome that was improved with immunosuppression (54). The presence or absence of both should be noted in the pathology report.
Crescents were not found to significantly predict adverse outcomes in the original Oxford classification presumably because rapidly progressive cases with eGFR <30 were excluded. The median number with crescents was only 9% also limiting predictive ability (49). This issue was addressed by Katafuchi et al. in 702 Japanese patients, including 254 with low-level proteinuria, 32 with eGFR <30, and 416 who met original Oxford inclusion criteria (55). Crescents were significantly associated with kidney survival overall and in the 286 not meeting Oxford criteria, but not in the 416 meeting those criteria. The predictive ability of crescents was validated in two pediatric studies (56, 57).
Haas et al. retrospectively analyzed 3,096 patients from four cohorts and found that crescents significantly predicted >50% decline in eGFR or ESKD overall, although only in patients not immunosuppressed when considered separately. However, having crescents in >25% of glomeruli was predictive regardless of immunosuppression. In 2016, crescent scores were added (now the MEST-C score) (48).
Additional pathologic changes not considered in the MEST-C score have a significant impact on the clinical presentation, prognosis, and response to therapy, most notably podocytpathy, thrombotic microangiopathy, and positive C4d staining (see Table 2).
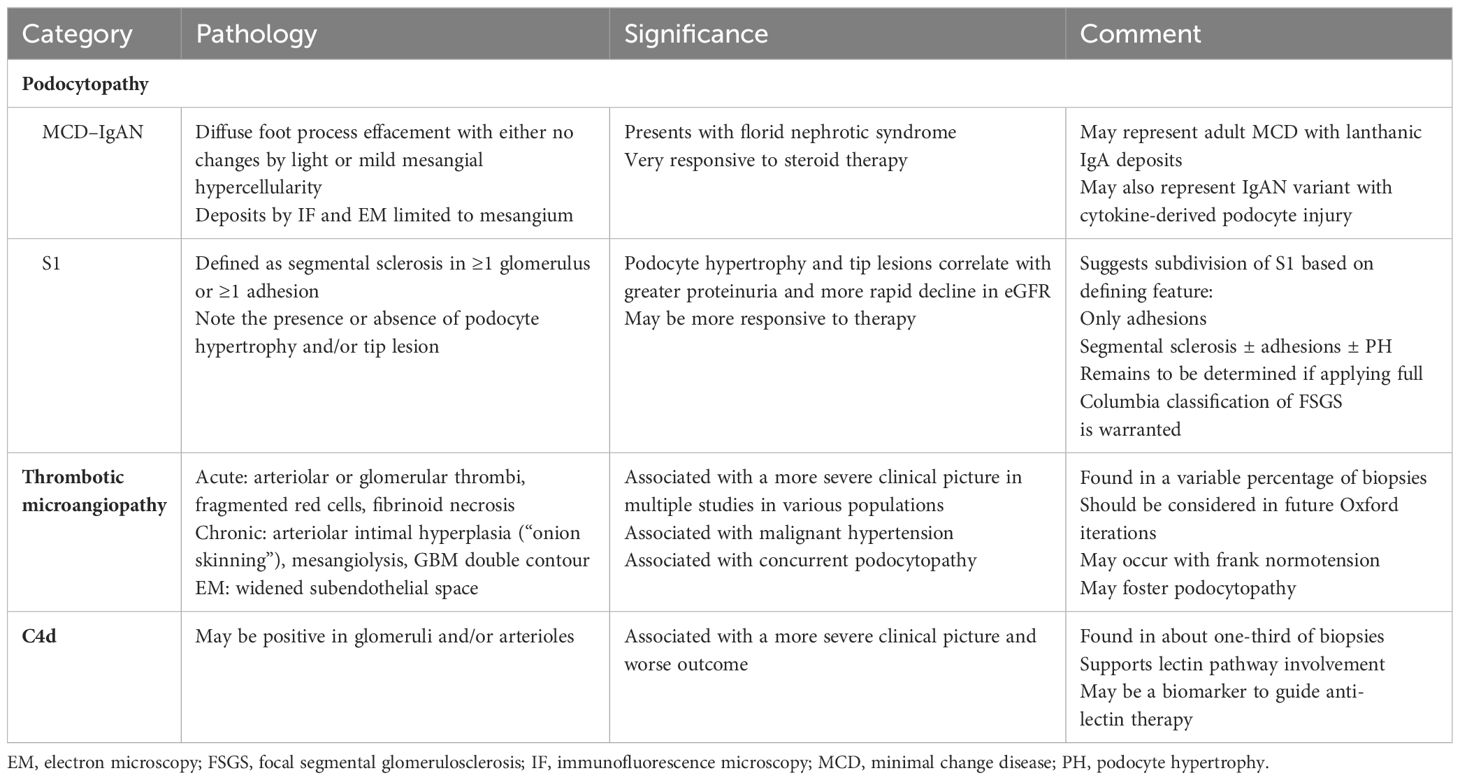
Table 2. Beyond MEST-C: additional pathological considerations.
Podocytopathy in IgA nephropathy
Proteinuria is the feature most indicative of podocyte injury or loss. Podocyte foot process effacement is well described in primary IgAN, the degree of which directly correlates with the amount of proteinuria (58, 59). Podocytopenia has been found in IgAN with an apparent threshold of 250 podocytes per glomerulus below which proteinuria increases and GFR decreases (60). In pediatric IgAN, cumulative urinary podocyte excretion over time significantly correlated with progressive glomerulosclerosis on serial biopsies (61).
The underlying pathology and associated clinical course of NS in IgAN patients is quite variable. Early reports centered on biopsies of MCD–IgAN and were characterized by either no light microscopic changes or only mild mesangial proliferation. Both the IgA deposits by immunofluorescence and the electron dense deposits by electron microcopy (EM) were restricted to the mesangium (62–69). Clinically, these patients typically had florid NS, normotension, preserved eGFR, and displayed marked steroid sensitivity with little tendency to progress. MCD–IgAN is not rare. For example, Han et al. reported a series of 1,336 Chinese IgAN patients that included 171 with MCD–IgAN and 171 others also with NS and pathology other than minimal glomerular abnormalities/diffuse foot process effacement (70). In the series of 57 Chinese IgAN patients of Huang et al. noted above, 13 (22.8%) had MCD–IgAN (11).
The pathophysiology of MCD–IgAN is uncertain. Some posit that it simply represents the coexistence of two glomerulopathies, MCD with a mild IgAN (67). It is also possible that these cases are simply MCD with lanthanic IgA deposits of otherwise no consequence. Asymptomatic IgA deposits are not rare in the general population. Waldherr et al. found IgA deposits in 12 of 250 consecutive autopsy cases without known kidney disease; only one had C3 deposits (71). Suzuki et al. found latent IgA deposits in 82 (16%) of 510 Japanese allografts at the time of implantation along with C3 in 16/82. These 16 had a significantly higher incidence of mesangial proliferative GN (50%) and greater macrophage infiltration but only a minority had increased urinary erythrocytes (72). Similarly, Sofue et al. found latent IgA deposits in 20 of 68 consecutive Japanese live-kidney donors of which 14 had mesangial expansion. There was no significant association of IgA deposits (± mesangial expansion) with allograft survival or function, and a repeat biopsy at 1 year showed that 14/20 no longer had IgA deposits (73). These data support the idea that some MCD–IgAN patients simply have MCD that responds to treatment as such, and that the IgA deposits are latent and otherwise of no consequence.
Additionally, Guo et al. compared 51 MCD–IgAN Chinese patients to both 51 consecutive IgAN patients without MCD and healthy participants as controls. Gd-IgA1 levels were lower in MCD–IgAN than in non-MCD–IgAN cases but higher than in controls, with only rare mesangial deposition of Gd-IgA1 in the MCD–IgAN patients (7/51, 13.7%) compared to non-MCD–IgAN patients (42/51, 82.4%).
Alternatively, MCD–IgAN may represent a primary IgAN podocytopathy. Given that the primary site of immune complex deposition and inflammation is the mesangium in IgAN, the mechanism of podocyte injury requires clarification. Potential mechanisms include toxicity of Gd-IgA1 aggregates directly, mesangial cell production of cytokines injurious to podocytes, or toxicity from C5b-9 generated in the mesangium. Wang et al. demonstrated reduced in vitro nephrin expression in podocytes exposed to aggregated IgA1 from IgAN patients and less so with IgA1 from normals, a finding that suggests a direct podocyte effect of abnormal IgA1 (74). Others found neither binding of IgA1 to podocytes nor podocyte expression of mRNA of known IgA receptors, and IgA1 from patients did not stimulate podocyte production of cytokines (75). However, cytokines produced from exposing mesangial cells in vitro to IgAN sera resulted in production of cytokines capable of directly activating podocytes (74–78). Candidate cytokines included TNF-α (75), TGF-β (76), angiotensin II (74, 77), and PAF (78).
Other patients with variable degrees of proteinuria, including NRP with or without NS, have more significant inflammatory and/or sclerotic glomerular lesions. Pathological findings may include necrosis, E1, C1/2, and S1 MEST-C scores, as well as varying types of focal and segmental glomerulosclerosis (FSGS), as defined in the Columbia classification (79). The clinical picture may include hypertension, variable proteinuria (up to and including NRP and NS), and reduced eGFR that worsens over time.
The Oxford S1 lesion requires special consideration and is found most frequently with sub-nephrotic range proteinuria. The original Oxford definition of S1 required at least one glomerulus with any degree of segmental sclerosis or the presence of a capsular adhesion. S1 defined as such correlated with clinical severity at diagnosis (blood pressure, eGFR, and proteinuria), as well as rate of decline of kidney function (49). Validation of S1 in the VALIGA cohort of patients from 55 European centers concurred (52). Segmental sclerosis in IgAN may develop by one of three mechanisms: healed segmental inflammatory or necrotic lesions, hyperfiltration from nephron reduction (typically associated with glomerulomegaly and perihilar sclerosis), or as a primary podocytopathic injury as outlined above for MCD–IgAN (54).
Hill et al. found that 92 of 128 French IgAN patients (72%) had capsular adhesions, including 53 with no underlying glomerular lesions, although there was loss of mature podocyte markers (synaptopodin, nephrin, VEGF) surrounding the adhesions. In comparison, 100% of primary FSGS patients, a well-known primary podocytopathy, had adhesions, but only 8% of 100 lupus patients, an immune-complex glomerulopathy like IgAN, had any (80). Applying the Columbia classification of FSGS to the 128 biopsies, 11 had collapsing FSGS, 27 the cellular variant, 4 tip lesions, 7 perihilar, 52 not otherwise specified (NOS), 11 non-definable as FSGS, and 16 had no lesions (81). Those with FSGS lesions associated with underlying proliferative lesions (M1, E1, or necrosis) did the worst long term, but even those with pure FSGS lesions without associated glomerular inflammatory lesions fared worse than those without FSGS.
To determine if subclassification of S1 is warranted, Bellur et al. reanalyzed the original Oxford cohort with available slides and found that 137/202 qualified as S1, of which 38% had podocyte hypertrophy (PH), 7% tip lesions, 3% perihilar sclerosis, and 2% endocapillary foam cells (54). PH and tip lesions were significantly associated with initial proteinuria and demonstrated significantly improved survival free from ≥50% decline in eGFR or ESKD when immunosuppressed. The 2016 Oxford update recommended not subclassifying S1, however, but rather simply specifying whether PH or tip lesions are present (48).
Bellur et al. attempted to validate S1 subclassification by reanalyzing the VALIGA cohort of 1,147 European patients regarding the association of specific podocyte lesions with proteinuria, outcome, and response to immunosuppression (82). They found that 69% satisfied S1 criteria, and the 796 were divided into 502 with any segmental sclerosis (NOS) as a qualifying criterion and 294 only having capsular adhesions with or without PH. The NOS+ group was further divided into those also having adhesions with or without PH to yield four groups: S1 without NOS sclerosis (adhesions only by definition), S1 with NOS but without adhesions or PH, NOS with adhesions without PH, and NOS with both adhesions and PH. They showed progressively worse proteinuria and kidney survival across the four groups. A benefit to immunosuppression was only found in the latter two groups. They advocate subclassifying S1 into active lesions more likely to respond to immunosuppression, i.e., those with adhesions or PH.
Thrombotic microangiopathy in IgAN
Thrombotic microangiopathy (TMA) of the kidney is defined pathologically as acute or chronic. The former includes cell swelling/subintimal edema, thrombus-occluding arterioles or glomerular capillaries with or without fragmented red cells, and fibrinoid necrosis of vessel walls. Chronic changes include arterial intimal proliferation (“onion skinning”), mesangiolysis, and/or double contours of the glomerular basement membrane by light microscopy, with glomerular capillary basement membrane duplication on EM. There may or may not be an associated microangiopathic hemolytic anemia (MAHA), characterized by anemia, schistocytes, thrombocytopenia, absent haptoglobin, and elevated LDH. Clinically, patients may have various diagnoses based on associated features, including thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS), antiphospholipid antibody syndrome, malignant hypertension, scleroderma renal crisis, radiation nephritis, infections, or drug/medication reactions. Although reported in IgAN, TMA is not considered in the Oxford classification.
A well-described cause of TMA, malignant hypertension, may develop in patients with IgAN with reported incidences ranging from 0.5% of Chinese patients to 15% of Spanish patients with IgAN. Chen et al. compared 45 cases of malignant hypertension complicating IgAN (1.2% of all IgAN cases) to 26 cases of malignant hypertension complicating primary hypertension. They found the IgAN patients had more severe proteinuria and hematuria, lower serum creatinine, and better kidney survival; however, kidney survival was not different when initial creatinine was considered (83). Chang et al. reviewed pathology reports and identified 10 cases of TMA out of 435 cases of IgAN; 6 had malignant hypertension and 3 others severe hypertension (84).
El Karoui et al. studied 128 French patients with IgAN and found that 63 (53%) had pathologic TMA, including 8 with evidence of MAHA (85). Only 18 had malignant hypertension (100% of which had TMA), and 47 had uncontrolled hypertension (66% had TMA), but 63 were normotensive, including 19 on no antihypertensive medications (16% had TMA). No difference was found between those with and without TMA in terms of antiphospholipid antibodies or complement gene mutations, but proteinuria in those with TMA was significantly higher (only 5% had <0.5 g/24 h), as were glomerular lesions and Oxford scores. Notably, 16% of TMA cases had eGFR > 60. Hence, TMA can be found in IgAN with normotension and preserved renal function. Prognosis was worse with TMA overall, but significantly so only in those with MAHA.
Cai et al. evaluated 944 Chinese IgAN patients and found that 194 (20.6%) had arterial/arteriolar TMA lesions compared to 0 of 60 with other glomerulopathies, and 69 of the 194 (37.3%) had glomerular subendothelial changes by EM (86). Patients with TMA had higher blood pressure and proteinuria, lower eGFR, and higher MEST-C scores. Similar to El Karoui et al., only 9.8% had malignant hypertension and 33% did not have hypertension. Seventy-five of 194 (38.7%) with arterial/arteriolar TMA progressed to ≥50% decline in eGFR, ESKD, or death versus 29 of 313 (9.3%) with arterial intimal fibrosis (arteriosclerosis) or arteriolar hyalinosis (arteriolosclerosis) alone versus 54 of 437 (12.4%) with no vascular lesions. By multivariable analysis, TMA conferred a 1.95 hazard ratio for the composite outcome.
Neves et al. found TMA in 21 of 118 Brazilian IgAN patients (17.8%), and TMA patients had more frequent hypertension and hematuria, lower eGFR, a greater E1 MEST-C score, and a significantly greater risk of ESKD. Similarly, Puapatanakul et al. found that 21 of 166 Thai patients (13%) with IgAN had TMA with significant associations with hypertension (81% versus 57% without TMA) and malignant hypertension (38% versus 6%), as well as lower eGFR, higher proteinuria, and a higher T2 MEST-C score. TMA was an independent predictor of ESKD.
In the report of FSGS lesions in IgAN patients mentioned above (81), TMA lesions were found in 6/7 with perihilar FSGS, 19/27 with the cellular variant, 29/52 with FSGS NOS, and 10/11 with collapse. In contrast, only 2/16 with no lesions and 2/11 with lesions not definable as FSGS had TMA. These findings suggest a link between TMA and progressive podocytopathy in IgAN.
The original Oxford classification, and subsequently others (86), did not find a significant association between arteriolar hyalinosis and/or arterial intimal fibrosis with outcome. Thus, vascular changes are not included in MEST-C scoring. In contrast, Nasri found that only 2 of 136 Pakistani IgAN patients had a TMA but did find a significant correlation of both arteriolar hyalinosis and arterial intimal fibrosis with serum creatinine and between arteriolar hyalinosis and amount of proteinuria (87).
Importantly, the lesions of TMA were not considered in the Oxford analyses. Given the significantly positive associations of such lesions with outcome noted above, TMA should be considered in future Oxford iterations.
C4d
C4d staining by immunofluorescence or immunohistochemistry reflects complement activation by either the classic or lectin pathways. In IgAN, lectin pathway activation is presumed to be the cause of positive staining, since mannose-binding lectin (MBL), L-ficolin, and MBL-associated serine proteases (MASPs) are found (88), whereas C1q is infrequently positive. Multiple studies assessed glomerular C4d staining as a biomarker for adverse outcomes. Jiang et al. performed a systematic review and meta-analysis and identified 12 studies (cohort and cross-sectional) with 1,251 patients and found a 34% prevalence of glomerular C4d staining. Significant heterogeneity existed, and definitions of positivity ranged from any non-sclerotic glomeruli to >75% positive. Asian populations had numerically higher positivity rates than Caucasian populations (46% versus 30%, p = NS). Patients with C4d deposition had lower eGFR, higher urinary protein, more hypertension, and higher Oxford scores. Glomerular C4d staining was independently associated with adverse kidney outcomes including ESKD.
Faria et al. assessed the prevalence and significance of arteriolar and glomerular C4d staining in 126 Portuguese adults and found arteriolar staining (defined as >25% of arterioles) in 17% that was associated with blood pressure, arterial intimal fibrosis, and chronic microangiopathy (89). Such staining remained a significant biomarker for progressive kidney dysfunction (>50% decline in eGFR or ESKD) by multivariable analysis, more so than glomerular staining.
The lectin pathway may be activated when MBL binds to IgM. Like C4d, IgM fluorescence is positive in a variable percentage of IgAN cases, ranging up to 70% (90). Moriyama et al. found IgM deposition to correlate with tuft adhesions but not with renal outcome in Japanese patients (90). Heybeli et al. found IgM to be significantly associated with doubling of serum creatinine or ESKD in Turkish patients (91), and Stefan found such deposition predicted shorter kidney survival in Romanian patients (92). Based on available evidence, the presence of neither C4d nor IgM staining should prompt immunosuppression. More study is needed to determine if therapy directed at the lectin pathway of complement activation should be considered.
Repeat biopsy in IgAN
The use of repeat biopsy histology as a surrogate endpoint or guide to ongoing immunosuppression has not been adequately studied (see Table 3). Two pediatric trials from a single center in Japan were reported (93, 94); one an RCT comparing 2 years of steroids and azathioprine plus standard therapy (heparin–warfarin and dipyridamole) versus standard alone in 86 patients who showed clinical and histologic improvement, including disappearance of mesangial IgA deposits in 7/33 but only with immunosuppression (94). After a mean of 77 months, Hotta et al. rebiopsied 35 adult Japanese patients with initial hematuria and proteinuria that had resolution of hematuria following treatment with warfarin, antiplatelet agents, steroids, and tonsillectomy. They found a significant reduction of mesangial proliferation and disappearance of endocapillary proliferation, necrosis, and cellular crescents, as well as complete loss of IgA deposits in 8 (95). Tumlin et al. found resolution of endocapillary proliferation, cellular crescents, and karyorrhexis after 6 months of steroids and cyclophosphamide in 12 American patients with rapidly progressive, crescentic IgAN that coincided with improved eGFR and reduced proteinuria (96).
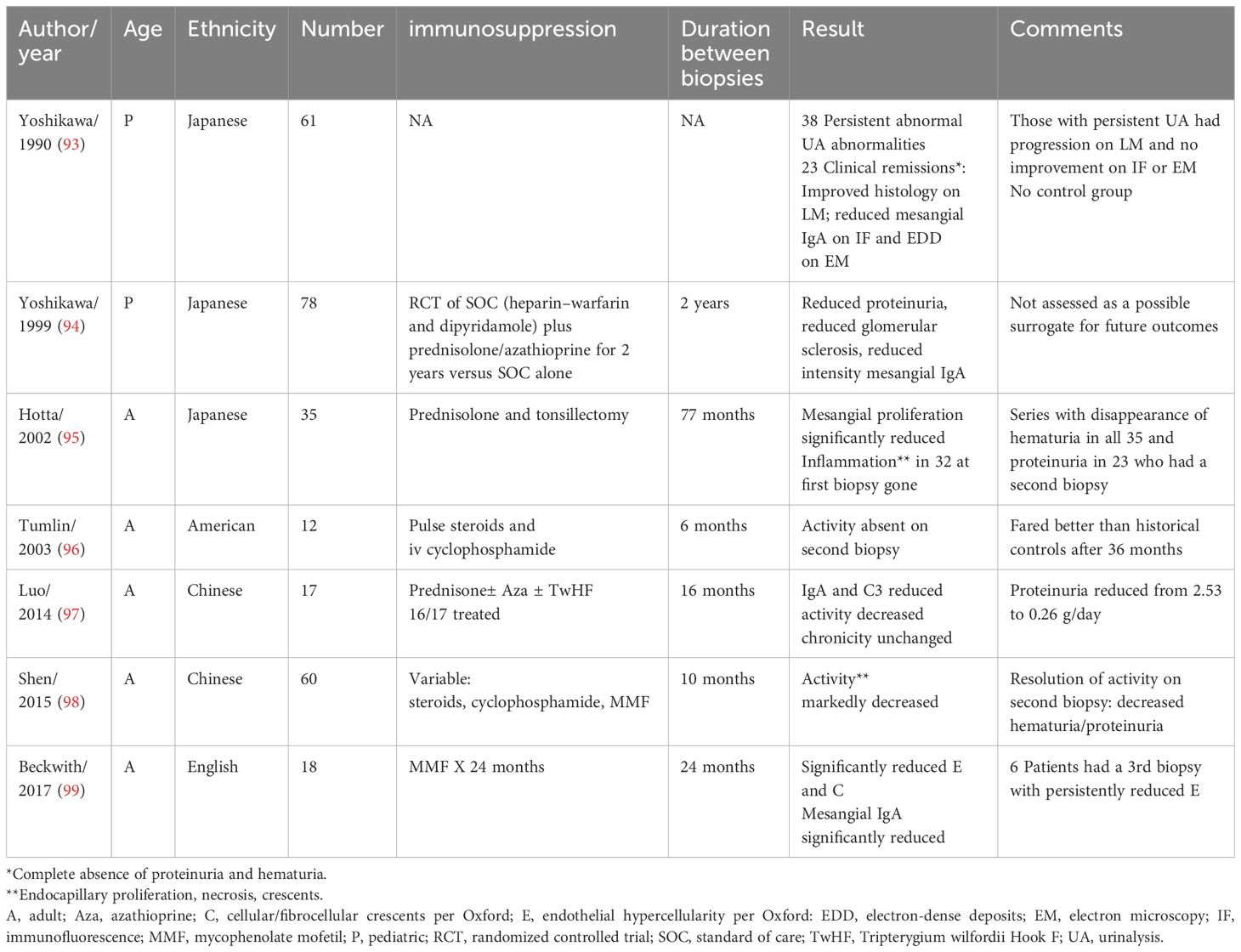
Table 3. Studies with repeat biopsies.
Luo et al. rebiopsied 17 Chinese patients (16 immunosuppressed) after 16 months and found reduced intensity of IgA and C3 staining and reduced activity index without worsening chronicity, which coincided with reduced urinary protein (2.5 g/day to 0.26) (97). Shen et al. rebiopsied 60 immunosuppressed Chinese adults after a mean of 10 months and found significantly reduced endocapillary hypercellularity, crescents, and necrosis coinciding with reduced hematuria/proteinuria only in those with resolution of activity (98). Only interstitial fibrosis/tubular atrophy on the second biopsy was significantly associated with 35% reduction in eGFR or ESKD after a 32-month follow-up. Finally, Beckwith et al. rebiopsied 18 English patients using the Oxford classification after immunosuppression with mycophenolate (99). A significant reduction in the E MEST-C score and cellular/fibrocellular crescents was found along with reduction of mesangial IgA deposition. Notably, after stopping mycophenolate, six patients had additional biopsies that showed persistence of the improved E score.
Although baseline biopsy is an adequate biomarker for prognosis, the currently available data are not sufficient to warrant repeat biopsy as a surrogate endpoint for therapeutic clinical trials. Intuitively, however, ongoing activity suggests the need for active immunosuppression. More study is needed.
The IgAN risk prediction tool
Barbour et al. derived and externally validated the IgAN Risk Prediction tool by retrospective analysis of 3,927 patients from Europe, North America, China, and Japan which included two prediction models (one with race/ethnicity and one without) with reasonably accurate risk prediction for >50% decline in eGFR or ESKD over a 5-year window (100). Features in the full models included age, sex, race/ethnicity, BMI, renin–angiotensin system (RAS) inhibition (RASi), immunosuppression, eGFR, proteinuria, mean arterial pressure (MAP), the MEST score, and interactions (proteinuria with MAP and with the T score). This tool is available on-line with a mobile app at https://qxmd.com/calculate-by-qxmd. Risk subgroups include low, intermediate, higher, and highest. This has subsequently been retrospectively validated in adult (101, 102) and pediatric cohorts (103). Additionally, since the original analysis evaluated data available around the time of biopsy, the prediction tool was updated to be applied at 1- or 2-year post-biopsy, also available on-line (104). Importantly, the tool has not been validated prospectively nor has it been shown to improve outcomes. The 2021 KDIGO guidelines recommend its use as a guide to inform patients of their risk but not to guide therapy (38).
Hematuria
The prognostic implication of hematuria depends on the type (macroscopic or microscopic) and the clinical context. Recurrent bouts of gross hematuria have been associated with episodes of AKI. Although initially reported to be benign (105–107) with complete return to baseline kidney function, later studies indicated up to 25% of cases had incomplete recovery with risk factors including age, duration of macroscopic hematuria, red blood cell (RBC) casts, tubular necrosis, and interstitial fibrosis (108). Trujillo published a multicenter series of 26 patients with anticoagulant-related hematuria who developed AKI and found underling biopsy-proven IgAN in 19 of 26 patients with a poor outcome. Only 5 of the 19 had complete recovery and 5 remained dialysis dependent (109).
Whereas gross hematuria is relatively easy to define, the assessment of microscopic hematuria is more problematic and varies between studies. Manual evaluation is often used, with typical cutoffs of 3 or 5 RBCs/high-power field (RBCs/hpf). Microscopic hematuria can be further categorized into lower versus higher levels (110, 111). An automated particle analyzer is available (UF-1000, Sysmex Corporation, Hyogo, Japan), with a proposed cut-off of 28 RBCs/µl. Excellent correlation was found compared to manual evaluation (r = 0.948, RBCs/µl = 5.6 × RBCs/hpf) (112).
Microscopic hematuria is emerging as an important prognostic risk factor, although it was omitted from the International Risk-Prediction Tool in IgAN due to missing data (100). Microscopic hematuria significantly associates with ongoing glomerular inflammation on biopsy (including mesangial hypercellularity, endocapillary proliferation, and crescents) (111). It has been proposed as a signal to initiate immunosuppressive therapy if unremitting and if significant proteinuria coexists (113). In addition to inter-patient variability, there can be significant intra-patient variability over time. Time-averaged hematuria can be used to offset such variability (112, 114–116). Whereas some studies assessed the prognosis of baseline hematuria, others evaluated persistence or remission over time (see Table 4).
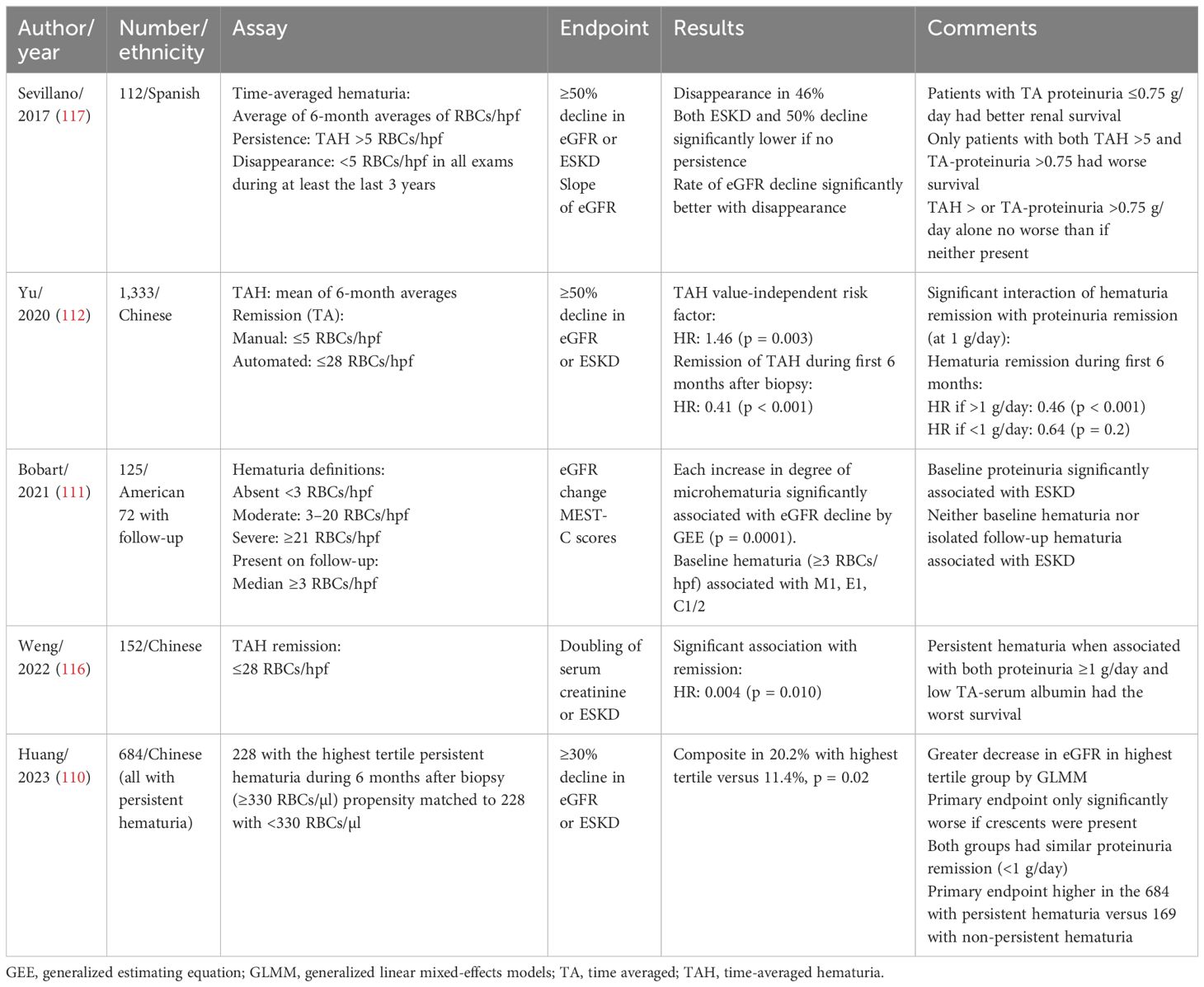
Table 4. Significance of persisting hematuria.
In IgAN patients with minimal proteinuria, hematuria at presentation is of uncertain significance. Earlier studies of Chinese (118–120), European (121), or Turkish (122) patients found adverse outcomes in some such patients, including increasing proteinuria, hypertension, impaired kidney function, and ESKD. In contrast, more recent studies showed a favorable outlook. Gutierrez et al. evaluated 141 Caucasian patients with microscopic hematuria at baseline, eGFR >60, and proteinuria <0.5 g/day with a 106-month follow-up (123). After 10 years, 96.7% had <50% increase in serum creatinine, as did 99% with <100% increase. After 20 years, the percentages were 91.9% and 99%, respectively. Clinical remission (absence of hematuria, proteinuria <0.2 g/day, normal renal function, normal BP) occurred in 53 (37.5%) by a mean of 60 months with only 12/17 receiving RAS blockers. Proteinuria >0.5 g/day developed in 21.
In Japanese patients with low-level proteinuria (<0.5 g/day), more severe baseline hematuria (>20 RBCs/hpf) fared like those with less severe hematuria regarding increasing proteinuria and kidney survival (124). More severe baseline hematuria did not predict adverse outcomes in Japanese patients even with higher levels of proteinuria (>1 g/day) (125).
He et al. performed a systematic review and meta-analysis of studies regarding the association of hematuria (at disease onset or at biopsy) with adverse outcomes (ESKD, 50% decline in eGFR, or doubling of serum creatinine) and identified 13 studies (5,660 patients) (126). Overall, initial hematuria (gross and microscopic together) was not significantly associated with ESKD (seven studies total), whereas microscopic hematuria considered separately was significantly associated with increased risk (four studies, relative risk 1.21). Macroscopic hematuria was associated with reduced risk (seven studies, RR 0.68). Three studies found no significant association between the magnitude of hematuria and adverse outcomes (111, 124, 125).
Several studies evaluated the significance of persistence or remission of hematuria over time. Le et al. retrospectively analyzed 1,155 Chinese patients with a median follow-up of 5.4 years (114). Time-averaged proteinuria was the strongest predictor of kidney failure, but time-averaged hematuria (defined as the ratio of the area-under-the-curve during follow-up to the duration of follow-up) was also significant by multivariable analysis. For every 10-fold increase, the risk for >50% decline in GFR or ESKD increased by 210%. An optimal cutoff was not established.
Sevillano et al. retrospectively studied 112 Caucasian patients with baseline hematuria for a mean of 14 years and compared those with minimal/negative hematuria (time averaged <5 RBCs/hpf) to those with persistent hematuria (>5 RBCs/hpf); time-averaged proteinuria >0.75 g/day was also compared to <0.75 g/day (117). Multivariable analysis revealed that both time-averaged hematuria and time-averaged proteinuria independently predicted ESKD or 50% eGFR reduction. Notably, 46% had disappearance of hematuria (<5 RBCs/hpf in all urinalyses over final 3 years) with a significant reduction in the rate of eGFR decline. Only patients with both elevated time-average hematuria and proteinuria had significantly worse kidney survival. Neither proteinuria in the absence of hematuria nor the reverse fared worse than having neither.
Using both a manual examination and an automated analyzer, Yu et al. evaluated 1,333 Chinese patients and found by multivariable analysis that time-averaged hematuria (the mean of all 6-month mean levels of hematuria) predicted 50% decline in eGFR or ESKD. Remission of hematuria during the first 6 months (average of <5 RBCs/hpf or <28 RBC/µl) occurred in 19% and significantly predicted a better outcome (112). A significant interaction with proteinuria remission existed such that the benefit of hematuria remission was only seen in those with persistent proteinuria (>1 g/day during the first 6 months) supporting Sevillano’s finding (117) that persisting hematuria is most adverse when coexisting with proteinuria.
Using an automated analyzer, Weng et al. studied 152 Chinese patients and found that persistence of time-averaged hematuria in 96 (defined as area-under-the-curve divided by time of >28 RBCs/µl) independently associated with worse renal outcome (doubling of serum creatinine or ESKD) (116). Receiver operator characteristic curve analysis revealed the optimal cutoff for predicting the adverse outcome was 37 RBCs/µl in males and 201 RBCs/µl in females.
Bobart et al. studied 125 American patients and defined moderate microscopic hematuria as a median of >3–20 RBCs/hpf and severe as >21 RBCs/hpf during follow-up (111). After multivariable adjustment, an increase in the degree of hematuria was significantly associated with decline of eGFR (−0.81, p < 0.011), especially comparing severe hematuria with absent/moderate (−3.99).
Using an automated analyzer, Huang et al. studied 861 Chinese patients and found that 79% had persistent hematuria (median during first 6 months >24 RBCs/µl). After propensity matching, the 228 with high-level hematuria (>330/µl, the upper tertile) to 228 with lower persistent hematuria, they found that the former had a significantly greater risk for the composite end-point (>30% decline in eGFR or ESKD) and a greater decline in eGFR, despite no difference in proteinuria remission (defined as <1 g/day) (110).
Altogether, there remains uncertainty regarding the prognostic significance of baseline hematuria over that provided by other clinical features (proteinuria, BP, eGFR) and the MEST-C score. No data support immunosuppressive therapy based solely on hematuria at baseline. In patients with persistent proteinuria despite 3–6 months of maximal supportive care, the persistence of hematuria indicates ongoing inflammation, portends a worse prognosis, and gives strong support to initiating immunosuppression. Remission of hematuria (repeatedly negative dipstick or <3 RBCs/hpf) despite persistent proteinuria suggests ongoing inflammation may not be present and immunosuppression may have no benefit. Before withholding such therapy, however, a repeat biopsy may be useful. A Japanese Study Group proposed that IgAN remission requires both hematuria remission (<5 RBCs/hpf) and proteinuria remission (<0.3 g/day) be present and persist for 6 months (127). Whereas persisting hematuria may be a potential biomarker, there is no significant evidence that hematuria remission is an adequate surrogate for treatment trials.
Therapy
As per KDIGO 2021, therapy for IgAN begins with optimizing supportive care. Lifestyle modifications include weight loss, regular exercise, smoking cessation, and sodium restriction. Blood pressure should be controlled with a KDIGO-recommended target of 120 systolic to maximize cardiovascular protection. All patients with proteinuria >0.5 g/day should be on maximally tolerated RASi regardless of blood pressure. At least three RCTs support RASi (128–130). A recent cohort study found proteinuria remission in 55%, including complete remission in 31% (131).
Importantly, KDIGO does not recommend dual RASi with angiotensin-converting enzyme inhibition (ACEi) plus angiotensin receptor antagonist (ARB), as available evidence in IgAN indicates no benefit (132). The potential for harm with ACEi/ARB dual therapy was shown in trials of patients with vascular disease or high-risk diabetes (133, 134). As shown for the nonsteroidal MRA finerenone in diabetic kidney disease (135, 136), whether dual blockade combining either an ACEi or an ARB with a mineralocorticoid antagonist (MRA) (or possibly an aldosterone synthase inhibitor) will benefit IgAN remains to be determined.
Additional antiproteinuric and renal protective therapies include sodium glucose transporter-2 inhibitors (SGLT2i) and endothelin receptor A (ETRA) antagonists (ETRAa). A prespecified analysis of 270 IgAN patients enrolled in the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease Trial revealed a hazard ratio of 0.29 (95% CI 0.12–0.73) for the primary end-point (50% decline in eGFR, ESKD, death from kidney or cardiovascular causes), a 26% reduction in albumin/creatinine ratio, and a lower rate of eGFR decline (137). Similarly, the Study of Heart and Kidney Protection with Empagliflozin (EMPA—KIDNEY) showed a 33% reduction in kidney disease progression (hazard ratio 0.67, 0.46–0.97) in 817 IgAN patients and a 28% reduced slope of decline of eGFR (138). Combining patients from both trials in meta-analysis, kidney disease progression (standardized to ≥50% decline in eGFR, ESKD, or death from kidney disease) was reduced by 51% [relative risk (RR) 0.49, 0.32–0.74] (139). Note that both trials excluded patients with eGFR >90, which does apply to many patients with IgAN.
Endothelin-1 is produced by all renal cell types, interacts with angiotensin II, and via activation of ETRA mediates multiple pathophysiological effects, such as vasoconstriction, inflammation, cell proliferation, and podocyte injury (140). The PROTECT study is an international, double-blind RCT, comparing the dual ETRAa and ARB sparsentan to the ARB irbesartan in 404 IgAN patients with ≥1 g/day of proteinuria, that found a 41% greater protein reduction at 36 weeks with sparsentan leading to accelerated FDA approval (141). After 2-years, proteinuria reduction was maintained (40%), along with significantly greater complete remission (proteinuria <300 mg/day, 31% versus 11%, RR 2.5, 1.6–4.1) and partial remission (<1.0 g/day, RR 1.5, 95% CI 1.1–1.9) (142). The chronic slope (from weeks 6 to 110) of eGFR decline was significantly lower by 1.1/year (p = 0.037), and the total slope (baseline to 110 weeks) was lower by 1.0/year (p = 0.058). The composite endpoint (≥40% reduction in eGFR, ESKD, death) occurred in 9% of sparsentan patients versus 13% irbesartan (RR 0.7, 0.4–1.2). Atrasentan is a selective ETRAa also being studied in IgAN (NCT04573478) (143).
Although not significant in PROTECT, salt and fluid retention are notable side effects of ETRAas. Given the effect of SGLT2is to cause sodium and volume loss, their combination with ETRAas makes intuitive sense. In the SONAR trial of atrasentan in diabetic nephropathy, the 14 patients receiving SGLT2is combined with atrasentan had decreased body weight and a greater decrease in albuminuria compared to 42 propensity match patients receiving only atrasentan (144). In the ZENITH-CKD trial, the ETRAa zibotentan (combined with dapagliflozin) was compared to placebo (plus dapagliflozin) in 449 patients with proteinuric CKD (including 18 IgAN patients) and was found to cause a greater reduction in urinary albumin without significant weight gain (145). The combination of SGLT2i with ETRAa is being evaluated in an extension of PROTECT.
In our opinion, patients with IgAN having either greater than 0.5 g/day proteinuria despite maximally tolerated RASi and eGFR <90 (or eGFR <45 regardless of proteinuria) should receive an SGLT2i. If proteinuria persists despite such dual therapy, an ETRAa should be added.
Immunosuppression in IgAN
Since IgAN is an immune-mediated disease, the use of immunosuppression is an obvious consideration. The two major questions are who to immunosuppress and how to. Options include corticosteroids and other anti-inflammatory agents, B-cell inhibition, and anti-complement agents. One size does not fit all, and therapy should be individualized.
As per KDIGO 2021, immunosuppression should only be considered in patients at high risk of progression (proteinuria >0.75–1 g/day) despite maximized supportive care. They do suggest a 6-month course of steroids in high-risk patients. Such patients should be counseled regarding the risks (especially with eGFR <50) and benefits of immunosuppression and offered enrollment in clinical trials. Neither the MEST-C score, the number of crescents, nor the IgAN prediction tool should be used to guide therapy.
Corticosteroids
Earlier corticosteroid trials targeted proteinuric IgAN but were hampered by a lack of standardized background therapy (RASi) and small sample sizes. Lv et al. performed a systematic review and meta-analysis of trials of steroid therapy for IgAN patients with >1 g/day proteinuria and preserved renal function published through March 2011 and identified nine trials with 532 patients (146). Steroids reduced kidney failure (relative risk 0.32, p = 0.002) and proteinuria (- 0.46 g/day, - 0.63 to – 0.29 g/day) without heterogeneity. By subgroup analysis, high-dose, short-term therapy showed benefit, but low-dose, long-term therapy did not. The quality of studies was low, and all had less than 100 patients. Generalizability was limited.
Subsequently, two large, multicenter, RCTs with conflicting results were published that addressed these concerns, the Supportive Versus Immunosuppressive Therapy for the Treatment of Progressive IgA Nephropathy trial (STOP-IgAN) Trial from Germany and the international Therapeutic Effects of Steroids in IgA Nephropathy Global (TESTING) Trial composed of predominantly (75%) Chinese patients.
In STOP-IgAN, 309 IgAN patients with >0.75 g/day proteinuria completed a 6-month run-in phase of optimized supportive care of which 106 (34%) were excluded due to reduction of proteinuria to <0.75 g/day prior to randomization. Subsequently, 162 were randomized to supportive care only versus immunosuppression with corticosteroids only if eGFR >60 [109 patients, using a regimen showing benefit in a RCT (147)] or corticosteroids plus oral cyclophosphamide for 3 months followed by azathioprine for 33 months if eGFR <60 [53 patients, also derived from a regimen showing benefit in a RCT (148)]. By combining immunosuppression groups, after 3 years, the co-primary endpoints of full clinical remission (protein/creatinine ratio <0.2 with preserved eGFR) was reached by 17% of immunosuppressed patients versus 5% of supportive care only (p = 0.01) and a decrease in eGFR ≥ 15 by 26% and 28%, respectively (p = nonsignificant). Furthermore, there was no significant difference in absolute difference in eGFR decline at 36 months (−4.2 versus −4.7), the slope of the reciprocal of serum creatinine per year, eGFR decline by ≥30, or ESKD between groups. Proteinuria was significantly reduced, but only at 12 months and not at 3 years. Microhematuria (by dipstick), present in 87% initially, remitted in 42% with immunosuppression versus 16% supportive only (p = 0.004).
A post-hoc analysis of STOP-IgAN considered the two immunosuppression protocols and their respective controls separately. The steroid-only group (eGFR ≥60) had greater full clinical remission but no difference in eGFR loss ≥15 versus its respective control group. Immunosuppression in the lower eGFR group had a significant difference in neither full remission nor eGFR decline ≥15 versus its respective control group (149). Severe infections, impaired glucose tolerance, and weight gain were significantly more common with immunosuppression.
In long-term follow-up of 149 patients of STOP-IgAN (median 7.4 years), the primary endpoint (>40% decline in eGFR, ESKD, total mortality) occurred in 45.5% with immunosuppression versus 50% with supportive care, a non-significant difference, with no significant differences when both immunosuppression regimens were compared with respective controls (150). Neither annual eGFR loss nor rate of eGFR loss >40% differed with immunosuppression.
In contrast, 503 patients (5% Caucasian/European) in TESTING were randomized to oral methylprednisolone (initially 0.6–0.8 mg/kg/day, maximum 48 mg/day, with a reduced dose protocol of 0.4 mg/kg, maximum 32 mg/day due to excessive infections with the higher dose) or placebo for 6–9 months with a 3.5-year follow-up. The primary endpoint (eGFR decline ≥40%, ESKD, or death due to kidney failure) was significantly reduced with steroids [hazard ratio (HR) 0.53, 0.39–0.72] with similar effects with both dosages. The rate of eGFR decline was significantly slower with steroids (−2.46/year, p = 0.002), as was development of ESKD (HR 0.59, p = 0.008). Time-averaged proteinuria was reduced by 0.69 g/day but, like STOP-IgAN, was no longer significantly different at 36 months. Serious adverse events (mostly hospitalizations and serious infections) were more frequent with steroids, especially the higher dose.
Since a major source of Gd-IgA1 production is gut-associated lymphoid tissue (Peyer’s patches) in the terminal ileum, a targeted-release formulation of the oral steroid budesonide (Nefecon) was developed that despite direct targeting of Peyer’s patches has first-pass hepatic clearance and less systemic steroid exposure. Nefecon demonstrated significant proteinuria reduction in a phase 2b trial (NEFIGAN) (151). Subsequently, part A of the phase 3 NefIgArd trial, a multicenter, double-blind trial of Nefecon 16 mg/day for 9 months, showed a 27% lower degree of proteinuria at 9 months versus placebo with better eGFR (3.78 difference).
The final results of NefIgArd included 364 primarily Caucasian patients with proteinuria >1 g/day or urine protein/creatinine ratio ≥0.8 g/g and eGFR 35–90 despite 3 months of RASi randomized to 16 mg/day Nefecon or placebo for 9 months with a 2-year follow-up. The primary endpoint, time-weighted average eGFR over 2 years, showed a significant benefit of 5.05 (p < 0.0001) with a 2-year total slope difference of 2.95/year favoring Nefecon (representing 50% less deterioration) and with no interactions based on baseline proteinuria. Proteinuria was reduced by 40% over months 12 to 24, but the maximal effect was at 12 months (~50%) that decreased by 24 months (~30%). Steroid-related side effects included edema, hypertension, muscle spasm, and headache. The risk of infection was no greater.
Overall, the best available data indicate that steroids are beneficial, especially in Chinese patients, but their effect is most pronounced while being given and appears to wane after discontinuation. It remains uncertain to what extent a relatively short course of therapy will affect the long-term progression of a disease evolving over decades in most patients. The dosages and therapy durations shown to be effective produce significant toxicity. Hence, case-by-case consideration is required. In our opinion, those demonstrating significant eGFR decline, persisting proteinuria, and hematuria while on maximal supportive therapy would gain the most benefit. Nefecon appears effective with less toxicity than prednisone but is much more expensive.
Targeting B-lineage cells
Central to the pathogenesis of IgAN is overproduction of Gd-IgA1 from B lymphocytes and plasma cells, the latter residing predominantly in mucosal-associated lymphoid tissues (MALT) of the gastrointestinal or respiratory tracts (156). Normal maturation of B-lineage cells to plasma cells, including class switching to IgA (157), can occur independent of T cells through interactions of the B-lineage survival factors B-cell-activating factor (BAFF) and a proliferation-induced ligand (APRIL) produced by dendritic cells and monocytes/macrophages in the lamina propria (157, 158). BAFF interacts with immature B cells through the BAFF receptor (BAFF-R), but also at later stages through interaction with transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) present on both mature naïve B cells and plasma cells and B-cell maturation antigen (BCMA) present on plasma cells (158). APRIL only interacts with TACI and BCMA, not BAFF-R, and is most important at later stages, i.e., memory B cells and plasma cells (158).
Normal B-cell maturation and class switching to IgA also occur in a T-cell-dependent manner in secondary lymphoid organ germinal centers (e.g., Peyer’s patches) through interaction with antigen-primed T cells (T-follicular/helper cells), with synergy from BAFF and APRIL (157). Mature IgA-secreting plasma cells normally home to survival niches in MALT but may also mishome to the bone marrow.
By targeting B cells, Lafayette et al. randomized 34 proteinuric IgAN patients to rituximab plus standard of care versus standard of care alone and found no difference after 1 year in proteinuria reduction or eGFR (Table 5). Serum levels of both Gd-IgA1 and anti-Gd-IgA1 did not change despite peripheral B-cell depletion, supporting a primary role of plasma cells in the production of these pathogenic antibodies (152). Whether more potent, second-generation, anti-CD20 monoclonal antibodies (ofatumumab or obinutzumab) will be more effective in IgAN remains to be determined. The utility of biologic agents targeting the later stages of B-cell development (i.e., plasma cells), such as the anti-CD38 monoclonal antibodies daratumumab and felzartamab (NCT05065970) remains to be determined. Proteosome inhibition with bortezomib is also under investigation (NCT01103778).
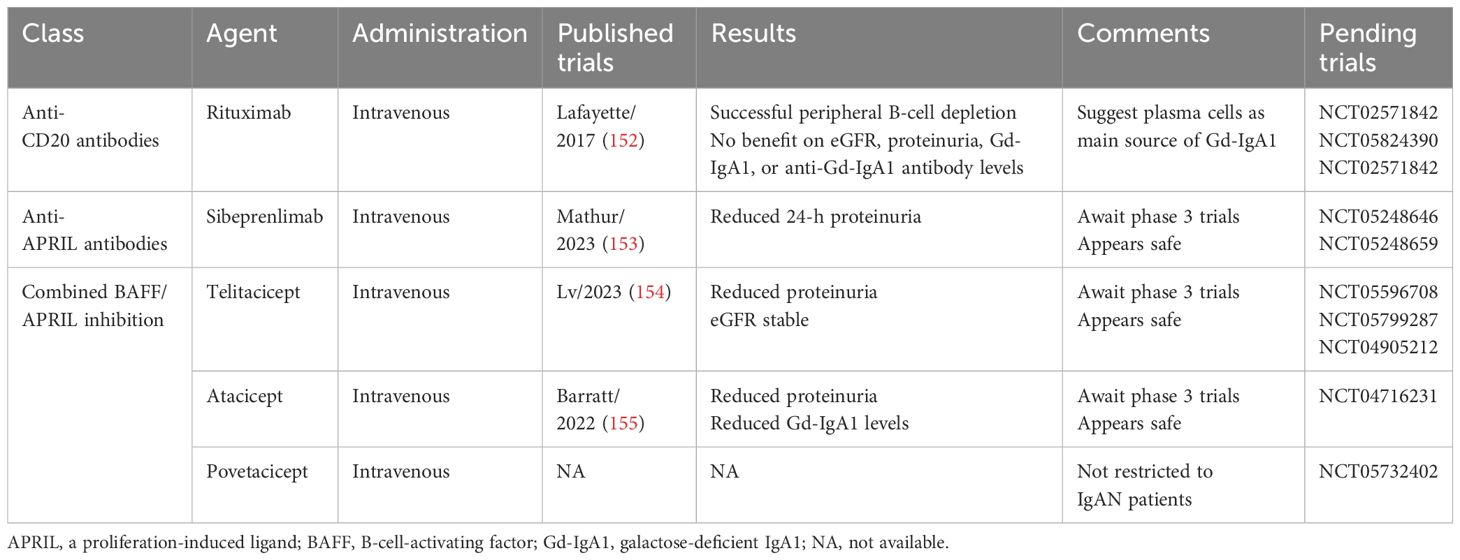
Table 5. Targeting B-lineage cells in IgAN.
Neutralizing BAFF and/or APRIL is germane to the pathophysiology of IgAN given their role in both T-cell-dependent and -independent class switching of B cells to produce IgA (Table 5). BAFF is involved in the earlier stages of B-cell maturation, and APRIL is involved in the later stages of B-cell maturation and plasma cell survival (158). Evidence indicates that both BAFF (159) and APRIL (160) are involved in experimental IgAN. In IgAN serum levels of BAFF are elevated and correlate with clinical and pathologic features (161). APRIL levels are also elevated and correlate with levels of Gd-IgA1 (162), as well as with progression to ESKD (163).
Multiple agents targeting BAFF and/or APRIL are under investigation (152–155). Blisibimod is a hybrid protein made in Escherichia coli targeting both membrane-bound and soluble BAFF. BRIGHT-SC (NCT02062684) was a study of subcutaneous blisibimod therapy in IgAN patients that had favorable results presented orally in 2016 but never published. A phase 3 trial was withdrawn.
Sibeprenlimab, a humanized IgG2-monoclonal antibody that neutralizes APRIL, was tested in a phase 2 trial (ENVISION) of 155 predominantly (~75%) Asian IgAN patients given sibeprenlimab 2, 4, 8 mg, or placebo subcutaneously monthly for 12 months (153). The geometric mean ratio reduction from baseline in 24-h urine protein/creatinine ratio was 49.6%, 56.7%, 62.8%, and 12.5%, respectively (p < 0.001 for linear trend). Clinical remission (<300 mg/day proteinuria) at 12 months occurred in 7.9%, 12.2%, 26.3%, and 2.6%, respectively. The slope of decline of eGFR was reduced in all three sibeprenlimab groups versus placebo. Serum levels of APRIL, IgA, IgM, IgG (less so), and Gd-IGA1 were suppressed during the 12 months but rebounded after 4 months. There were no safety concerns. A phase 3 trial is ongoing (VISIONARY, NCT05248646). Zigakibart (BION-1301) is a monoclonal antibody also targeting APRIL that showed efficacy (presented in 2021 but not published). A phase 3 trial is underway (BEYOND, NCT05852938).
Fusion proteins containing the TACI receptor inhibit both BAFF and APRIL. Telitacicept, a recombinant fusion protein of TACI and the Fc component of human IgG that binds and neutralizes both BAFF and APRIL, was studied for the treatment of lupus nephritis (164, 165). Lv et al. reported a phase 2 trial in 44 proteinuric Chinese IgAN patients given telitacicept 240 mg, telitacicept 160 mg, or placebo weekly for 24 weeks and found a 0.889-g/day (49%) reduction in mean proteinuria with 240 mg/week (least squared mean difference versus placebo −0.88, p = 0.013) but a lesser and nonsignificant reduction with 160 mg/week; eGFR remained stable in both telitacicept groups but fell significantly with placebo (154). IgA, IgG, and IgM levels decreased only with telitacicept.
Atacicept, also a human recombinant fusion protein of IgG1 and TACI under investigation for autoimmune diseases [systemic lupus (166)], was studied in the small phase 2a JANUS trial, comparing once weekly injections of atacicept 25 mg, atacicept 75 mg, and placebo in 16 proteinuric IgAN patients that was terminated early due to slow enrollment (155). No safety signals emerged, and immunoglobulin levels, including Gd-IgA1, were reduced. Early proteinuria reduction occurred that was maintained only with the 25-mg dose; eGFR was stable with atacicept but declined with placebo. The phase 2b ORIGIN trial (NCT04716231) randomized 116 patients with proteinuric IgAN to 150 mg of atacicept, 75 mg of atacicept, or placebo weekly for 36 weeks and found significantly greater reduction in proteinuria versus placebo at 24 weeks (25% combining both atacicept arms, p = 0.037), stability of eGFR, and a 60% reduction in Gd-IgA1 (167). A phase 3 trial comparing the 150-mg dose with placebo (ORIGIN 3) is ongoing. A third fusion protein dual BAFF/APRIL inhibitor, povitacicept, is under study for glomerulonephritis, including IgAN (NCT05732402).
Overall, APRIL and/or BAFF inhibition appear to be a reasonably well-tolerated approach to reducing the pathophysiologic driver of IgAN (Gd-IgA1 and anti-Gd-IgA1 antibodies). However, like steroids, the effect is likely to persist only while on therapy. Phase 3 trials demonstrating effectiveness and safety are required before recommendations can be made.
Complement inhibition in IgAN
The complement system is intricately involved in the pathogenesis of IgAN given the correlation of disease severity with both serum markers of activation and tissue deposition of components. Polymorphisms of complement-regulating genes are also linked to susceptibility to IgAN. The primary pathway of complement activation in IgAN is the alternate pathway. The lectin pathway may be involved in a significant minority of patients and appears to correlate with disease severity. Despite the frequent co-deposition of IgG and/or IgM, the classic pathway is apparently not involved as C1q is rarely found. The variability in complement activity and regulation from patient to patient allows for potential personalized therapeutic choices given the diversity of drugs currently under investigation and the pathways they target (Table 6) (168–172).
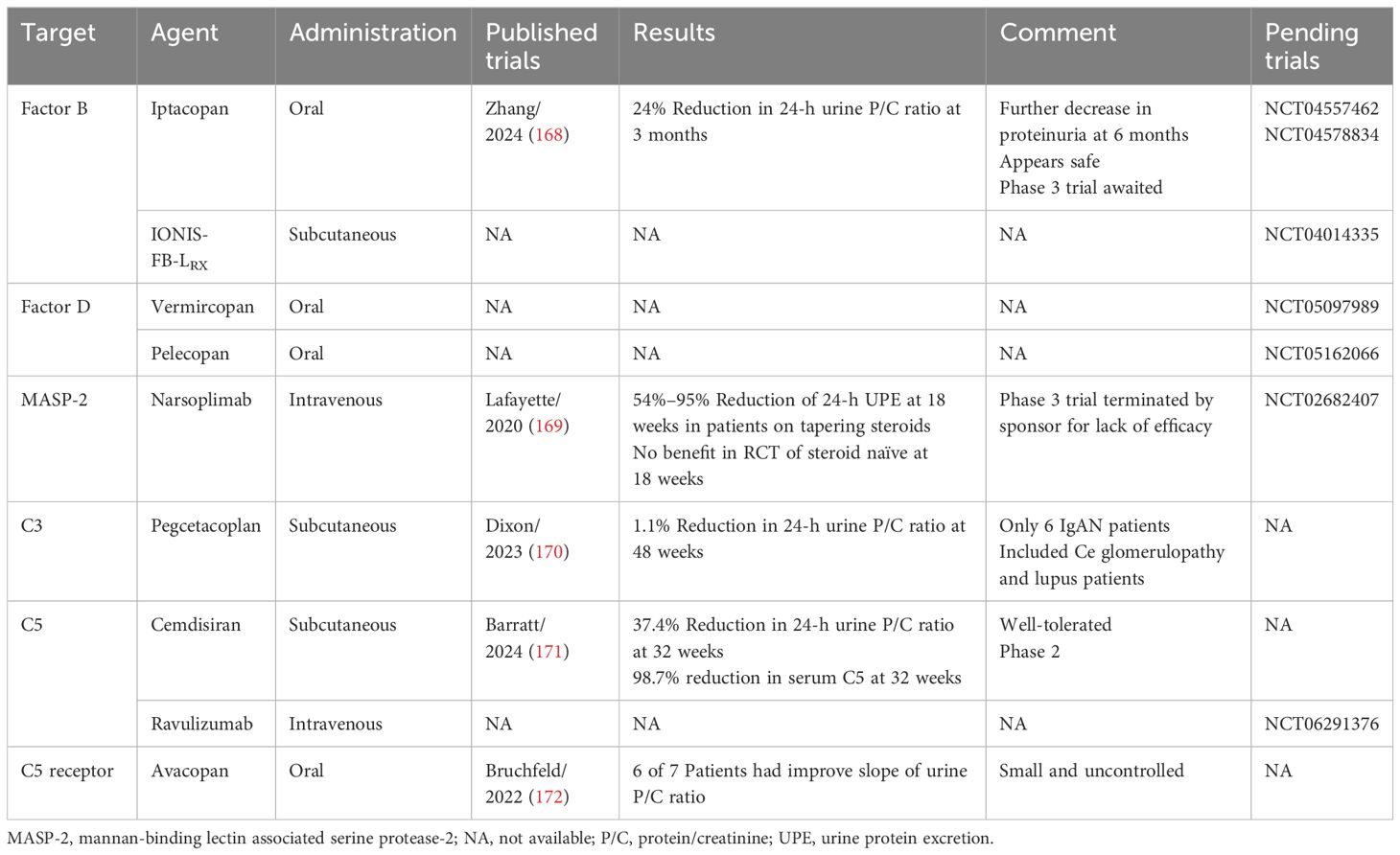
Table 6. Complement inhibition in IgAN.
Several inhibitors of the alternate pathway are being studied. A double-blind placebo-controlled phase 2 dose-finding study (10, 50, 100, or 200 mg bid) of the oral Factor B inhibitor iptacopan found a significant 23% reduction in 24-h urinary protein/creatinine ratio with the 200-mg bid dose at 3 months with further reduction at 6 months. Evidence of alternate pathway inhibition included significant biomarker reductions (plasma Bb levels, serum ex vivo alternate pathway activity by the Wieslab assay, and urinary C5b-9 levels) (168). A phase 3 trial of iptacopan in IgAN is ongoing (APPLAUSE-IgAN, NCT04578834). IONIS-FB-LRx, an antisense inhibitor of Factor B administered by subcutaneous injection to IgAN patients, had positive results to reduce urinary protein by 44% at week 29 as per company press release (Ionis Pharmaceuticals) in an unpublished phase 2 study. A phase 3 study is active (NCT04014335). Two oral Factor D inhibitors, pelecopan and vemircopan, are being studied for IgAN, but published results are not available.
Involvement of the lectin pathway of complement activation is based on glomerular staining of MBL, ficolins, and C4d in the absence of C1q. A humanized antibody targeting the effector enzyme MASP-2, narsoplimab, was found in a phase 2 study of 16 patients to be safe and well tolerated and reduced proteinuria up to 95% in patients on steroids and up to 61% in those not on steroids (169). However, the phase 3 trial of narsoplimab, ARTEMIS-IGAN (NCT03608033), was terminated by the sponsor, due to lack of efficacy in reducing proteinuria.
The common pathway has been targeted with multiple agents. Cleavage of C3 results in the anaphylatoxin C3a and C3b, the latter required for both amplifying all three pathways and completing their respective C5-convertases. Pegcetacoplan, extensively studied and approved for paroxysmal nocturnal hemaglobinuria (173), binds to C3 preventing its cleavage without interfering with the tickover pathway (similar to compstatin) (174). In a phase-2, 48-week, basket study in 21 patients with glomerulonephritis, including 6 with IgAN, pegcetacoplan was found to have a favorable safety profile, although proteinuria was only reduced from 1.3 g/day at baseline to 1.2 g/day at 48 weeks in IgAN patients (170).
Cemdisiran, a subcutaneous small interfering RNA therapeutic that inhibits C5 production, was studied in a 36-week, placebo-controlled, phase 2 trial of 31 IgAN patients. Findings included a placebo-adjusted 37.4% reduction of mean 24-h urine protein/creatinine ratio with serum C5 reduction of 98.7% (versus 25% with placebo), reduced dipstick-grade hematuria, and less decline of eGFR than placebo (171). Serum complement alternate pathway activity was reduced by 48% and classic pathway activity was reduced by 77% versus little change with placebo.
Eculizumab is a humanized monoclonal antibody targeting C5 approved for atypical HUS, which has shown transient benefit in case reports of aggressive IgAN (175, 176). Raviluzumab, another humanized anti-C5 monoclonal antibody, is being studied in a placebo-controlled trial in 450 projected IgAN patients (NCT06291376). Avacopan is an oral C5a receptor inhibitor approved for use in ANCA-vasculitis (177). In an open-label pilot study of seven IgAN patients, avacopan 30 mg bid for 12 weeks produced a 50% improvement in proteinuria in three patients (172). Terminal complement inhibition would seem to be most beneficial when TMA and/or strong glomerular C5b-9 staining are found on biopsy, but this remains to be determined.
Complement inhibition holds promise for the treatment of IgAN, but like BAFF and APRIL, phase 3 studies are required to demonstrate effectiveness and safety, and to better determine which patients would most likely respond to which agents. Given the effectiveness of complement inhibition in atypical hemolytic–uremic syndrome, it remains to be determined if the same applies to IgAN patients showing TMA on biopsy.
Other immunosuppressants in IgAN
Cyclophosphamide has been used together with steroids for clinically severe IgAN especially associated with necrosis, inflammation, and/or crescents. Nonrandomized studies have used oral (178, 179) or intravenous (96, 180) cyclophosphamide and compared outcomes to historical controls or contemporary untreated patients. Standardized definitions of either clinical course or pathologic severity were unavailable for inclusion into these studies for appropriate comparisons. Ballardie and Roberts performed an RCT in 38 patients with rapidly progressive primary IgAN defined as creatinine >130 µmol/L rising by ≥15% in the prior year with projected ESKD within 5 years. They compared no immunosuppression to tapering oral steroids plus oral cyclophosphamide 1.5 mg/kg/day for 3 months followed by oral azathioprine 1.5 mg/kg for up to 2 years (the regimen used in STOP-IgAN) and showed a reduced rate of renal function decline that was arrested in one-third with reductions in proteinuria and erythrocyturia (148). Per KDIGO, patients with a rapidly progressive course, defined as ≥50% decline in eGFR in ≤3 months in the absence of other causes with a biopsy typically showing a high percentage of crescents and focal necrosis, should be offered cyclophosphamide and steroids (38).
Multiple observational studies and RCTs assessed mycophenolate mofetil (MMF) in IgAN. Earlier meta-analyses (2008–2009) of RCTs found no significant benefit (181–183). Chen et al. performed a 2014 meta-analysis identifying eight RCTs (357 patients) and found no significant effects on proteinuria or renal function; however, studies employing short-term therapy had a benefit (184). Three of the eight trials (69 patients) found MMF superior to cyclophosphamide. A 2021 meta-analysis of nine trials (587 patients) comparing MMF ± steroids to placebo or full-dose steroids found no benefit on remission, proteinuria reduction, doubling of serum creatinine, or ERKD. Cushing’s syndrome was less, but infection was more with MMF compared to full-dose steroids (185).
The Effect of Mycophenolate Mofetil on Renal Outcomes in Advanced Immunoglobulin A Nephropathy trial (MAIN) was an RCT of MMF plus standard-of-care versus standard-of-care alone for 3 years in 170 Chinese IgAN patients with proteinuria >1 g/day. Treatment with MMF significantly reduced the two primary outcomes of a composite endpoint (doubling of serum creatinine, ESKD, renal or cardiovascular death, HR 0.23, 0.09–0.63) and progression of kidney disease (≥30% decline of eGFR from >60 to <60 or ≥50% decline if baseline eGFR <60, adjusted HR 0.23, 0.10–0.57) (186). In those patients stopping MMF at trial end, however, the eGFR decline during post-trial follow-up increased from −2.9/year during the trial to −6.1/year afterward. Based on these results, patients at high risk for both steroid side effects and progression can be considered for MMF, especially if Chinese.
Leflunomide, a dihydroorotate dehydrogenase inhibitor that suppresses lymphocyte proliferation, has been studied in multiple small trials. A systematic review and meta-analysis evaluated 44 studies of 1,802 patients (35 RCTs) and found both lower proteinuria and serum creatinine with leflunomide combined with steroids versus steroids alone (27 studies, 1,802 patients); 6 studies comparing leflunomide with RASi versus RASi alone (664 patients) also found significantly lower proteinuria and serum creatinine (187). A more recent meta-analysis restricted to low-dose steroids plus leflunomide versus full-dose steroids included three trials (342 patients) and found reduced urine protein with the combination, although there was no significant difference in renal function (188). No safety signals emerged from either meta-analysis.
Hydroxychloroquine, now standard of care for lupus nephritis, may also be effective in IgAN by inhibiting Toll-like receptor-9 upregulation of BAFF/APRIL and cytokine production, as well as by inhibiting antigen processing by antigen-presenting cells. A 2021 systematic review uncovered one RCT (60 patients), one prospective case-control study (28 patients), two retrospective case-control studies (236 patients), and one observational cohort study (180 patients) all restricted to Chinese patients (189). In the RCT, hydroxychloroquine plus losartan significantly reduced proteinuria versus losartan alone. The other studies supported proteinuria reduction overall. No effect on eGFR was detected.
He et al. performed a retrospective cohort study of 159 Chinese IgAN patients with moderate proteinuria (~0.8 g/day) given hydroxychloroquine + RASi, leflunomide + RASi, or RASi alone followed for 6 months (190). Compared with RASi alone, both active treatment groups had significantly lower proteinuria, with hydroxychloroquine having a significantly greater effect than leflunomide. Conversely, only leflunomide significantly reduced hematuria compared to either RASi alone or hydroxychloroquine. Overall, the data are insufficient to recommend either hydroxychloroquine or leflunomide for routine treatment of IgAN.
Tonsillectomy
Mucosal-associated lymphoid tissues, including Peyers patches and Waldeyer’s ring, are considered a likely source of Gd-IgA1 and associated antibodies (156). The clinical occurrence of gross hematuria concurrently or shortly following an upper respiratory infection (synpharyngitic nephritis) supports a prominent role of oral pharyngeal lymphoid tissue. By studying tonsillar plasma cells of IgAN patients, Bene et al. found a predominance of IgA- versus IgG-plasma cells, a reversal of the findings in patients with recurrent tonsillitis (191).
Tonsillectomy has received considerable attention for the treatment of IgAN (192). First reported from a Germen study in 1999 (193), tonsillectomy has gained wide acceptance in Japan. A 2011 meta-analysis reviewed 7 retrospective trials (858 patients, 6 from Japan, 1 from China) and found that remission was higher, but only when combined with pulse steroids, and not when used alone (194). A 2015 meta-analysis reviewed 14 mostly retrospective studies (1,794 patients), 12 from Japan and 1 from China, and found significantly greater odds of remission with tonsillectomy [10 studies, 1,431 patients, odds ratio (OR) 3.40, p < 0.001] and decreased odds of ESKD (9 studies, 873 patients, OR 0.25, p < 0.001) (195). A meta-analyzed 19 studies (15 from Japan, 2 from China) with 3,483 patients found a higher rate of remission (15 studies, 3,059 patients, OR 3.30, 2.47–4.40) and reduced ESKD (9 studies, 1,804 patients, OR 0.33, 0.16–0.69) with tonsillectomy (196). Most studies were observational and mostly retrospective.
There have been two RCTs of tonsillectomy. Kawamura and colleagues randomized 72 Japanese patients to steroid pulses with or without tonsillectomy and found significantly greater chance of proteinuria remission (OR 2.98, p = 0.049) but neither hematuria remission nor combined hematuria/proteinuria remission with tonsillectomy (197). On subsequent analysis, patients having tonsillectomy were more likely to have both proteinuria remission and combined remission versus no tonsillectomy, only if there were more advanced histological changes (198). Yang et al. randomized 98 Chinese IgAN patients to tonsillectomy plus drug therapy to drug therapy alone (the latter consisting of tapering Tripterygium glycosides in all patients and tapering prednisone if proteinuria >1 g/day) and found a significantly shorter time to proteinuria remission (2.5 versus 26.1 months, p < 0.001), hematuria remission (3.1 versus 24.9 months, p <0.001), greater cumulative remission rates for both hematuria and proteinuria, duration of remissions, and lower relapse rates with tonsillectomy (199).
Feehally et al. evaluated 1,147 European IgAN patients and found that 41 had tonsillectomy at some point for tonsillitis or obstructive sleep apnea, including 17 with the procedure performed after IgAN diagnosis (200). Comparing the 41 to 41 propensity-matched controls, there was no difference in either the rate of eGFR decline (−1.9/year versus −0.5/year, p = 0.105) or in the composite of ≥50% reduction in eGFR or ESKD (3/41 versus 8/41, p = 0.105); the same result obtained comparing the 17 with post-diagnosis tonsillectomy to 51 propensity-matched controls. These data do not support tonsillectomy for Caucasian patients, but tonsillectomy deserves consideration in Japanese patients.
Discussion
Primary IgAN displays variable clinical manifestations at presentation that progresses to ESKD in up to 50% of patients. Those of Pacific-Asian origin are at greater risk and those of African origin less so compared to Caucasians. There are no non-invasive biomarkers for diagnosis capable of replacing biopsy, which remains the gold standard. The Oxford classification should be used to standardize interpretation. Future iterations of the Oxford classification hopefully will address the significance of TMA, C4d staining, and podocyte abnormalities, including possible subclassification of S1.
The best available biomarkers for predicting progression at the time of presentation or initial biopsy include proteinuria, reduced eGFR, hypertension, and MEST-C score. Time-averaged proteinuria and slope of eGFR decline can further assess risk over time. Time-averaged hematuria requires more study. Other potential serum and urine biomarkers require verification. The IgAN Prediction tool can be used to better inform patients of their risk.
The backbone of treatment applicable to all patients is lifestyle modification, including sodium restriction, exercise, weight loss, smoking cessation, and strict control of blood pressure (<130/80 as a minimum, consider <120/70 as per KDIGO). RASi should be given to all patients with proteinuria (≥300 mg/g creatinine or ≥500 mg/day) regardless of blood pressure. SGLT2i are indicated for persisting albuminuria (albumin/creatinine ratio >200) or eGFR <45 regardless of albuminuria (based on inclusion criteria of EMPA—KIDNEY). ETRAa should be considered if proteinuria persists.
The use of immunosuppression should be individualized and considered after an adequate trial of supportive therapy (3–6 months). Exceptions requiring immediate therapy include nephrotic syndrome from MCD–IgAN, which should be treated with high-dose steroids as adult MCD, and those having a rapidly progressive course with necrosis and crescents on biopsy, where cyclophosphamide and steroids are recommended.
Otherwise, the most obvious case requiring immunosuppression is a younger patient with declining eGFR and persisting proteinuria despite maximal supportive therapy, especially with persisting hematuria as an indicator of ongoing inflammation. KDIGO suggests persisting proteinuria >0.75–1 g/day as the main indicator for high risk, but as shown in RaDaR, patients with <500 mg/day may still progress. Hence, all factors must be considered, especially declining eGFR and persisting hematuria even with lower levels of proteinuria.
Older patients with comorbidities capable of contributing to declining GFR and proteinuria (such as diabetes, hypertension, and vascular disease) favor not treating especially given the higher risk of immunosuppression in the elderly. Asian patients have a more aggressive course, and Chinese patients have shown significant responses to immunosuppression (steroids in TESTING and MMF in MAIN) supporting a more aggressive course in such patients. Further support for use of steroids and/or MMF in Chinese patients comes from a large observational database of 3,946 patients from which new users of immunosuppression were propensity-matched to patients treated only with supportive care that found a 40% reduction in hard endpoints with similar effect sizes with glucocorticoids alone, MMF alone, or both (201).
If a significant time has elapsed since diagnostic biopsy and hematuria is not persisting, rebiopsy may be indicated to determine if ongoing proteinuria is the result of chronic, irreversible changes (glomerular sclerosis and interstitial fibrosis) and not ongoing inflammation, in which case therapy would not be indicated.
The main alternatives include a 6-month course of high-dose oral steroids (with or without iv boluses) versus targeted-release budesonide. The latter reduces systemic steroid exposure but comes at significantly greater cost. Antibiotic prophylaxis and skeletal protection are required with oral steroids. Alternatively, in patients at higher risk of steroid toxicity, MMF can be considered, especially if Chinese. Tonsillectomy may be considered in Japanese patients, probably combined with bolus steroids. The roles of leflunomide and hydroxychloroquine remain uncertain.
Whereas such nonspecific immunosuppression may effectively quell inflammation while being administered, the underlying genetic tendency and pathophysiology are unlikely to change, i.e., enhanced Gd-IgA1 production and polymerization together with the enhanced production of anti-Gd-IgA1 antibodies, immune complex formation, mesangial deposition, complement activation, and mesangial cell activation/proliferation. High-dose immunosuppression cannot be continued for prolonged periods given obvious toxicity risks.
Multiple agents are being studied to offer a more targeted approach. Targeting APRIL alone or BAFF plus APRIL may reduce production of Gd-IgA1 by inhibiting class switching of developing B cells and reducing plasma cell survival. Phase 2 studies are promising, and phase 3 studies are awaited. It remains to be determined if the risk benefit ratio favors targeting APRIL alone or both BAFF and APRIL. Given the obvious role of complement, especially the alternate pathway but also the lectin pathway, multiple steps are potential targets with multiple agents under investigation in phase 2 and 3 studies. These include Factor B and D inhibitors, MMASP inhibitors, and C3 and C5 inhibitors. The best way to suppress complement in IgAN is uncertain and may vary from patient to patient. Perhaps serum, urine, and/or tissue biomarkers can help determine the best choice for an individual patient.
Ultimately, we suspect that combinations of agents will give the best results. It remains to be determined if they should be applied concurrently or sequentially and if multiple courses of an agent found to be effective in a given patient should be used. IgAN remains a fascinating disease under active study. Hopefully, IgAN can be relegated to a very uncommon cause of ESKD and shortened life expectancy.
Author contributions
EF: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. RG: Writing – review & editing. JF: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
EF is in the Speaker Bureau for Boehringer Ingelheim and Lilly. RG is in the Speaker Bureau for Travere and Astra Zeneca.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Berger J, Hinglais N. Intercapillary deposits of igA-igG. J Urol Nephrol (Paris). (1968) 74:694–5.
2. Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. (2013) 368:2402–14. doi: 10.1056/NEJMra1206793
3. Schena FP, Nistor I. Epidemiology of IgA nephropathy: A global perspective. Semin Nephrol. (2018) 38:435–42. doi: 10.1016/j.semnephrol.2018.05.013
4. Moriyama T, Tanaka K, Iwasaki C, Oshima Y, Ochi A, Kataoka H, et al. Prognosis in IgA nephropathy: 30-year analysis of 1,012 patients at a single center in Japan. PloS One. (2014) 9:e91756. doi: 10.1371/journal.pone.0091756
5. Pitcher D, Braddon F, Hendry B, Mercer A, Osmaston K, Saleem MA, et al. Long-term outcomes in IgA nephropathy. Clin J Am Soc Nephrol. (2023) 18(6):727–38. doi: 10.2215/CJN.0000000000000135
6. Lee H, Kim DK, Oh K, Joo KW, Kim YS, Chae D, et al. Mortality of IgA nephropathy patients: A single center experience over 30 years. PloS One. (2012) 7:e51225. doi: 10.1371/journal.pone.0051225
7. Jarrick S, Lundberg S, Welander A, Carrero J, Höijer J, Bottai M, et al. Mortality in IgA nephropathy: A nationwide population-based cohort study. J Am Soc Nephrol. (2019) 30(5):866–76. doi: 10.1681/ASN.2018101017
8. Gutiérrez E, Praga M, Rivera F, Sevillano A, Yuste C, Goicoechea M, et al. Changes in the clinical presentation of immunoglobulin A nephropathy: Data from the spanish registry of glomerulonephritis. Nephrol Dial Transplant. (2018) 33:472–7. doi: 10.1093/ndt/gfx058
9. Zhang H, Barratt J. Is IgA nephropathy the same disease in different parts of the world? Semin Immunopathol. (2021) 43:707–15. doi: 10.1007/s00281-021-00884-7
10. Barbour SJ, Cattran DC, Kim SJ, Levin A, Wald R, Hladunewich MA, et al. Individuals of pacific asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. (2013) 84:1017–24. doi: 10.1038/ki.2013.210
11. Huang N, Li J, Ai Z, Guo L, Chen W, Liu Q. Differences of clinicopathological characteristics and outcomes of IgA nephropathy patients with and without nephrotic syndrome. Int Urol Nephrol. (2024). doi: 10.1007/s11255-024-04040-6
12. Xu L, Zhou X, Zhang H. An update on the genetics of IgA nephropathy. J Clin Med. (2023) 13:123. doi: 10.3390/jcm13010123
13. Kiryluk K, Sanchez-Rodriguez E, Zhou X, Zanoni F, Liu L, Mladkova N, et al. Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat Genet. (2023) 55:1091–105. doi: 10.1038/s41588-023-01422-x
14. Li M, Yu X. Genetic determinants of IgA nephropathy: Eastern perspective. Semin Nephrol. (2018) 38:455–60. doi: 10.1016/j.semnephrol.2018.05.015
15. Sallustio F, Serino G, Cox SN, Dalla Gassa A, Curci C, De Palma G, et al. Aberrantly methylated DNA regions lead to low activation of CD4+ T-cells in IgA nephropathy. Clin Sci (Lond). (2016) 130:733–46. doi: 10.1042/CS20150711
16. Serino G, Sallustio F, Cox SN, Pesce F, Schena FP. Abnormal miR-148b expression promotes aberrant glycosylation of IgA1 in IgA nephropathy. J Am Soc Nephrol. (2012) 23:814–24. doi: 10.1681/ASN.2011060567
17. Yao X, Zhai Y, An H, Gao J, Chen Y, Zhang W, et al. MicroRNAs in igA nephropathy. Ren Fail. (2021) 43:1298–310. doi: 10.1080/0886022X.2021.1977320
18. Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. (2011) 22:1795–803. doi: 10.1681/ASN.2011050464
19. Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. (1997) 52:509–16. doi: 10.1038/ki.1997.361
20. Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. (1999) 104:73–81. doi: 10.1172/JCI5535
21. Goto M, Wakai K, Kawamura T, Ando M, Endoh M, Tomino Y. A scoring system to predict renal outcome in IgA nephropathy: A nationwide 10-year prospective cohort study. Nephrol Dial Transplant. (2009) 24:3068–74. doi: 10.1093/ndt/gfp273
22. Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L. Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol. (2011) 22:752–61. doi: 10.1681/ASN.2010040355
23. Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. (2007) 71:1148–54. doi: 10.1038/sj.ki.5002185
24. Yasutake J, Suzuki Y, Suzuki H, Hiura N, Yanagawa H, Makita Y, et al. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol Dial Transplant. (2015) 30:1315–21. doi: 10.1093/ndt/gfv221
25. Sun Q, Zhang Z, Zhang H, Liu X. Aberrant IgA1 glycosylation in IgA nephropathy: A systematic review. PloS One. (2016) 11:e0166700. doi: 10.1371/journal.pone.0166700
26. Wada Y, Matsumoto K, Suzuki T, Saito T, Kanazawa N, Tachibana S, et al. Clinical significance of serum and mesangial galactose-deficient IgA1 in patients with IgA nephropathy. PloS One. (2018) 13:e0206865. doi: 10.1371/journal.pone.0206865
27. Yanagawa H, Suzuki H, Suzuki Y, Kiryluk K, Gharavi AG, Matsuoka K, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PloS One. (2014) 9:e98081. doi: 10.1371/journal.pone.0098081
28. Vaz de Castro PAS, Amaral AA, Almeida MG, Selvaskandan H, Barratt J, Simões E Silva AC. Examining the association between serum galactose-deficient IgA1 and primary IgA nephropathy: A systematic review and meta-analysis. J Nephrol. (2024). doi: 10.1007/s40620-023-01874-8
29. Tomino Y, Suzuki S, Imai H, Saito T, Kawamura T, Yorioka N, et al. Measurement of serum IgA and C3 may predict the diagnosis of patients with IgA nephropathy prior to renal biopsy. J Clin Lab Anal. (2000) 14:220–3. doi: 10.1002/1098-2825(2000)14:5
30. Kawasaki Y, Maeda R, Ohara S, Suyama K, Hosoya M. Serum IgA/C3 and glomerular C3 staining predict severity of IgA nephropathy. Pediatr Int. (2018) 60:162–7. doi: 10.1111/ped.13461
31. Mizerska-Wasiak M, Małdyk J, Rybi-Szumińska A, Wasilewska A, Miklaszewska M, Pietrzyk J, et al. Relationship between serum IgA/C3 ratio and severity of histological lesions using the oxford classification in children with IgA nephropathy. Pediatr Nephrol. (2015) 30:1113–20. doi: 10.1007/s00467-014-3024-z
32. Chen P, Yu G, Zhang X, Xie X, Wang J, Shi S, et al. Plasma galactose-deficient IgA1 and C3 and CKD progression in IgA nephropathy. Clin J Am Soc Nephrol. (2019) 14(10):1458–65. doi: 10.2215/CJN.13711118
33. Schena FP, Cox SN. Biomarkers and precision medicine in IgA nephropathy. Semin Nephrol. (2018) 38:521–30. doi: 10.1016/j.semnephrol.2018.05.022
34. Neprasova M, Maixnerova D, Sparding N, Genovese F, Karsdal MA, Koprivova H, et al. Serum and urine biomarkers related to kidney fibrosis predict kidney outcome in czech patients with IgA nephropathy. Int J Mol Sci. (2023) 24:2064. doi: 10.3390/ijms24032064
35. Reich HN, Troyanov S, Scholey JW, Cattran DC. Toronto Glomerulonephritis Registry. Remission of proteinuria improves prognosis in IgA nephropathy. J Am Soc Nephrol. (2007) 18:3177–83. doi: 10.1681/ASN.2007050526
36. Inker LA, Mondal H, Greene T, Masaschi T, Locatelli F, Schena FP, et al. Early change in urine protein as a surrogate end point in studies of IgA nephropathy: An individual-patient meta-analysis. Am J Kidney Dis. (2016) 68:392–401. doi: 10.1053/j.ajkd.2016.02.042
37. Thompson A, Carroll K, Inker L, Floege J, Perkovic V, Boyer-Suavet S, et al. Proteinuria reduction as a surrogate end point in trials of IgA nephropathy. Clin J Am Soc Nephrol. (2019) 14(3):469–81. doi: 10.2215/CJN.08600718
38. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. (2021) 100:S1–S276. doi: 10.1016/j.kint.2021.05.021
39. Han SH, Kang EW, Park JK, Kie JH, Han DS, Kang S. Spontaneous remission of nephrotic syndrome in patients with IgA nephropathy. Nephrol Dial Transplant. (2011) 26:1570–5. doi: 10.1093/ndt/gfq559
40. Kim J, Kim JH, Lee SC, Kang EW, Chang TI, Moon SJ, et al. Clinical features and outcomes of IgA nephropathy with nephrotic syndrome. Clin J Am Soc Nephrol. (2012) 7(3):427–36. doi: 10.2215/CJN.04820511
41. Li H, Wang F, Jia J, Yan T, Liu Y, Lin S. The difference between patients with nephrotic syndrome and nephrotic-range proteinuria in IgA nephropathy: A propensity score matched cohort study. BMC Nephrol. (2022) 23:163. doi: 10.1186/s12882-022-02799-3
42. Chen Y, Yang A, Hou Y, Liu L, Lin J, Huang X, et al. Comparison between outcomes of IgA nephropathy with nephrotic-range proteinuria and nephrotic syndrome: Do podocytes play a role? Ren Fail. (2022) 44(1):1443–53. doi: 10.1080/0886022X.2022.2113796
43. Hu Y, Huang Z, Cao Q, Chen B, Xu S, Qiu W, et al. Clinical significance of massive proteinuria in primary IgA nephropathy with and without nephrotic syndrome: A single center cohort study. Ren Fail. (2023) 45(2):2267138. doi: 10.1080/0886022X.2023.2267138
44. Jiang Y, Chen P, Zhao W, Liu L, Shi S, Lv J, et al. Distinct characteristics and prognosis of IgA nephropathy patients with nephrotic syndrome: A propensity score-matched cohort study. Front Med (Lausanne). (2024) 11:1344219. doi: 10.3389/fmed.2024.1344219
45. Inker LA, Heerspink HJL, Tighiouart H, Levey AS, Coresh J, Gansevoort RT, et al. GFR slope as a surrogate end point for kidney disease progression in clinical trials: A meta-analysis of treatment effects of randomized controlled trials. J Am Soc Nephrol. (2019) 30(9):1735–45. doi: 10.1681/ASN.2019010007
46. Grams ME, Sang Y, Ballew SH, Matsushita K, Astor BC, Carrero JJ, et al. Evaluating glomerular filtration rate slope as a surrogate end point for ESKD in clinical trials: An individual participant meta-analysis of observational data. J Am Soc Nephrol. (2019) 30(9):1735–45. doi: 10.1681/ASN.2019010008
47. Lafayette RA, Reich HN, Stone AM, Barratt J. One-year estimated GFR slope independently predicts clinical benefit in immunoglobulin A nephropathy. Kidney Int Rep. (2022) 7:2730–3. doi: 10.1016/j.ekir.2022.09.017
48. Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, et al. Oxford classification of IgA nephropathy 2016: An update from the IgA nephropathy classification working group. Kidney Int. (2017) 91:1014–21. doi: 10.1016/j.kint.2017.02.003
49. A Working Group of the International AWG, Network IN, Cattran DC, Coppo R, Cook HT, Feehally J, et al. The oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int. (2009) 76:534–45. doi: 10.1038/ki.2009.243
50. A Working Group of the International AWG, Network IN, Roberts IS, Cook HT, Troyanov S, Alpers CE, et al. The oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int. (2009) 76:546–56. doi: 10.1038/ki.2009.168
51. Zeng C, Le W, Ni Z, Zhang M, Miao L, Luo P, et al. A multicenter application and evaluation of the oxford classification of IgA nephropathy in adult chinese patients. Am J Kidney dis. (2012) 60:812–20. doi: 10.1053/j.ajkd.2012.06.011
52. Coppo R, Troyanov S, Bellur S, Cattran D, Cook HT, Feehally J, et al. Validation of the oxford classification of IgA nephropathy in cohorts with different presentations and treatments. Kidney Int. (2014) 86:828–36. doi: 10.1038/ki.2014.63
53. Barbour SJ, Espino-Hernandez G, Reich HN, Coppo R, Roberts ISD, Feehally J, et al. The MEST score provides earlier risk prediction in lgA nephropathy. Kidney Int. (2016) 89:167–75. doi: 10.1038/ki.2015.322
54. Bellur SS, Lepeytre F, Vorobyeva O, Troyanov S, Cook HT, Roberts IS, et al. Evidence from the oxford classification cohort supports the clinical value of subclassification of focal segmental glomerulosclerosis in IgA nephropathy. Kidney Int. (2017) 91:235–43. doi: 10.1016/j.kint.2016.09.029
55. Katafuchi R, Ninomiya T, Nagata M, Mitsuiki K, Hirakata H. Validation study of oxford classification of IgA nephropathy: The significance of extracapillary proliferation. Clin J Am Soc Nephrol. (2011) 6:2806–13. doi: 10.2215/CJN.02890311
56. Halling E, Söderberg MP, Berg UB. Predictors of outcome in paediatric IgA nephropathy with regard to clinical and histopathological variables (oxford classification). Nephrol Dialysis Transplant. (2011) 27(2):715–22. doi: 10.1093/ndt/gfr339
57. Shima Y, Nakanishi K, Hama T, Mukaiyama H, Togawa H, Hashimura Y, et al. Validity of the oxford classification of IgA nephropathy in children. Pediatr Nephrol. (2012) 27:783–92. doi: 10.1007/s00467-011-2061-0
58. Choi SY, Suh KS, Choi DE, Lim BJ. Morphometric analysis of podocyte foot process effacement in IgA nephropathy and its association with proteinuria. Ultrastruct Pathol. (2010) 34:195–8. doi: 10.3109/01913121003648402
59. Lee J, Jang S, Cho N, Heo NH, Gil H, Lee EY, et al. Severity of foot process effacement is associated with proteinuria in patients with IgA nephropathy. Kidney Res Clin Pract. (2020) 39:295–304. doi: 10.23876/j.krcp.20.017
60. Lemley KV, Lafayette RA, Safai M, Derby G, Blouch K, Squarer A, et al. Podocytopenia and disease severity in IgA nephropathy. Kidney Int. (2002) 61:1475–85. doi: 10.1046/j.1523-1755.2002.00269.x
61. Hara M, Yanagihara T, Kihara I. Cumulative excretion of urinary podocytes reflects disease progression in IgA nephropathy and schönlein-henoch purpura nephritis. Clin J Am Soc Nephrol. (2007) 2:231–8. doi: 10.2215/CJN.01470506
62. Sinnassamy P, O'Regan S. Mesangial IgA deposits with steroid responsive nephrotic syndrome: Probable minimal lesion nephrosis. Am J Kidney Dis. (1985) 5:267–9. doi: 10.1016/S0272-6386(85)80120-7
63. Association of IgA nephropathy with steroid-responsive nephrotic syndrome. Am J Kidney Dis. (1985) 5:157–64. doi: 10.1016/S0272-6386(85)80044-5
64. Cheng IKP, Chan K, Chan M. Mesangial IgA nephropathy with steroid-responsive nephrotic syndrome: Disappearance of mesangial IgA deposits following steroid-induced remission. Am J Kidney Dis. (1989) 14:361–4. doi: 10.1016/S0272-6386(89)80168-4
65. Qin J, Yang Q, Tang X, Chen W, Li Z, Mao H, et al. Clinicopathologic features and treatment response in nephrotic IgA nephropathy with minimal change disease. Clin Nephrol. (2013) 79:37–44. doi: 10.5414/CN107682
66. Wang J, Juan C, Huang Q, Zeng C, Liu Z. Corticosteroid therapy in IgA nephropathy with minimal change-like lesions: A single-centre cohort study. Nephrol Dial Transplant. (2013) 28:2339–45. doi: 10.1093/ndt/gft211
67. Herlitz LC, Bomback AS, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. IgA nephropathy with minimal change disease. Clin J Am Soc Nephrol. (2014) 9:1033–9. doi: 10.2215/CJN.11951113
68. Li X, Liang S, Le W, Cheng S, Zeng C, Wang J, et al. Long-term outcome of IgA nephropathy with minimal change disease: A comparison between patients with and without minimal change disease. J Nephrol. (2016) 29:567–73. doi: 10.1007/s40620-015-0242-9
69. Guo W, Sun L, Dong H, Wang G, Xu X, Cheng W, et al. Characterization of patients with IgA nephropathy with and without associated minimal change disease. Front Nephrol. (2023) 3:1105933. doi: 10.3389/fneph.2023.1105933
70. Han X, Xiao Y, Tang Y, Zheng X, Anwar M, Qin W. Clinical and pathological features of immunoglobulin A nephropathy patients with nephrotic syndrome. Clin Exp Med. (2019) 19:479–86. doi: 10.1007/s10238-019-00580-9
71. Waldherr R, Rambausek M, Duncker WD, Ritz E. Frequency of mesangial IgA deposits in a non-selected autopsy series*. Nephrol Dial Transplant. (1989) 4:943–6. doi: 10.1093/ndt/4.11.943
72. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. (2003) 63:2286–94. doi: 10.1046/j.1523-1755.63.6s.2.x
73. Sofue T, Inui M, Hara T, Moritoki M, Nishioka S, Nishijima Y, et al. Latent IgA deposition from donor kidneys does not affect transplant prognosis, irrespective of mesangial expansion. Clin Transplant. (2013) 27:14–21. doi: 10.1111/ctr.12158
74. Wang C, Ye Z, Peng H, Tang H, Liu X, Chen Z, et al. Effect of aggregated immunoglobulin A1 from immunoglobulin A nephropathy patients on nephrin expression in podocytes. Nephrol (Carlton). (2009) 14:213–8. doi: 10.1111/j.1440-1797.2008.01025.x
75. Lai KN, Leung JCK, Chan LYY, Saleem MA, Mathieson PW, Lai FM, et al. Activation of podocytes by mesangial-derived TNF-alpha: Glomerulo-podocytic communication in IgA nephropathy. Am J Physiol Renal Physiol. (2008) 294:945. doi: 10.1152/ajprenal.00423.2007
76. Lai KN, Leung JCK, Chan LYY, Saleem MA, Mathieson PW, Tam KY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol Dial Transplant. (2009) 24:62–72. doi: 10.1093/ndt/gfn441
77. Ye Z, Wang C, Tang Y, Liu X, Peng H, Zhang H, et al. Serum IgA1 from patients with IgA nephropathy up-regulates integrin-linked kinase synthesis and inhibits adhesive capacity in podocytes through indirect pathways. Clin Invest Med. (2009) 32:20. doi: 10.25011/cim.v32i1.5083
78. Coppo R, Fonsato V, Balegno S, Ricotti E, Loiacono E, Camilla R, et al. Aberrantly glycosylated IgA1 induces mesangial cells to produce platelet-activating factor that mediates nephrin loss in cultured podocytes. Kidney Int. (2010) 77:417–27. doi: 10.1038/ki.2009.473
79. D'Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am J Kidney Dis. (2004) 43:368–82. doi: 10.1053/j.ajkd.2003.10.024
80. Hill GS, Karoui KE, Karras A, Mandet C, Van Huyen JD, Nochy D, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. I. immunohistochemical studies. Kidney Int. (2011) 79:635–42. doi: 10.1038/ki.2010.466
81. El Karoui K, Hill GS, Karras A, Moulonguet L, Caudwell V, Loupy A, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. II. light microscopic and clinical studies. Kidney Int. (2011) 79:643–54. doi: 10.1038/ki.2010.460
82. Bellur SS, Troyanov S, Vorobyeva O, Coppo R, Roberts IS, Cattran D, et al. Evidence from the large VALIGA cohort validates the subclassification of focal segmental glomerulosclerosis in IgA nephropathy. Kidney Int. (2024) 105(2):1279–90. doi: 10.1016/j.kint.2024.03.011
83. Chen Y, Tang Z, Yang G, Shen S, Yu Y, Zeng C, et al. Malignant hypertension in patients with idiopathic IgA nephropathy. Kidney Blood Press Res. (2005) 28:251–8. doi: 10.1159/000090058
84. Chang A, Kowalewska J, Smith KD, Nicosia RF, Alpers CE. A clinicopathologic study of thrombotic microangiopathy in the setting of IgA nephropathy. Clin Nephrol. (2006) 66:397–404. doi: 10.5414/CNP66397
85. El Karoui K, Hill GS, Karras A, Jacquot C, Moulonguet L, Kourilsky O, et al. A clinicopathologic study of thrombotic microangiopathy in IgA nephropathy. J Am Soc Nephrol. (2012) 23:137–48. doi: 10.1681/ASN.2010111130
86. Cai Q, Shi S, Wang S, Ren Y, Hou W, Liu L, et al. Microangiopathic lesions in IgA nephropathy: A cohort study. Am J Kidney Dis. (2019) 74:629–39. doi: 10.1053/j.ajkd.2019.03.416
87. Nasri H, Mubarak M. Significance of vasculopathy in IgA nephropathy patients with regard to oxford classification and immunostaining findings: A single center experience. J Renal Injury Prev. (2013) 2(2):41–5. doi: 10.12861/jrip.2013.16
88. Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. (2006) 17:1724–34. doi: 10.1681/ASN.2005090923
89. Faria B, Canão P, Cai Q, Henriques C, Matos AC, Poppelaars F, et al. Arteriolar C4d in IgA nephropathy: A cohort study. Am J Kidney Dis. (2020) 76:669–78. doi: 10.1053/j.ajkd.2020.03.017
90. Moriyama T, Shimizu A, Takei T, Uchida K, Honda K, Nitta K. Characteristics of immunoglobulin A nephropathy with mesangial immunoglobulin G and immunoglobulin M deposition. Nephrology. (2010) 15:747–54. doi: 10.1111/j.1440-1797.2010.01296.x
91. Heybeli C, Oktan MA, Yıldız S, Arda HÜ, Ünlü M, Çavdar C, et al. Clinical significance of mesangial IgM deposition in patients with IgA nephropathy. Clin Exp Nephrol. (2019) 23:371–9. doi: 10.1007/s10157-018-1651-6
92. Stefan G, Stancu S, Zugravu A, Terinte-Balcan G. Prognostic role of mesangial IgM deposition in IgA nephropathy: A long-term cohort study. Ren Fail. (2024) 46:2313179. doi: 10.1080/0886022X.2024.2313179
93. Yoshikawa N, Iijima K, Matsuyama S, Suzuki J, Kameda A, Okada S, et al. Repeat renal biopsy in children with IgA nephropathy. Clin Nephrol. (1990) 33:160–7.
94. Yoshikawa N, Ito H, Sakai T, Takekoshi Y, Honda M, Awazu M, et al. A controlled trial of combined therapy for newly diagnosed severe childhood IgA nephropathy. J Am Soc Nephrol. (1999) 10(1):101–9. doi: 10.1681/ASN.V101101
95. Hotta O, Furuta T, Chiba S, Tomioka S, Taguma Y. Regression of IgA nephropathy: A repeat biopsy study. Am J Kidney Dis. (2002) 39:493–502. doi: 10.1053/ajkd.2002.31399
96. Tumlin JA, Lohavichan V, Hennigar R. Crescentic, proliferative IgA nephropathy: Clinical and histological response to methylprednisolone and intravenous cyclophosphamide. Nephrol Dial Transplant. (2003) 18:1321–9. doi: 10.1093/ndt/gfg081
97. Luo M, Yao C, Xu B, Xu Y, Liu WJ, Feng Y, et al. Continuation of immunosuppressive treatment may be necessary in IgA nephropathy patients with remission of proteinuria: Evaluation by repeat renal biopsy. Exp Ther Med. (2014) 7:553–9. doi: 10.3892/etm.2013.1467
98. Shen X, Liang S, Chen H, Le W, Jiang S, Zeng C, et al. Reversal of active glomerular lesions after immunosuppressive therapy in patients with IgA nephropathy: A repeat-biopsy based observation. J Nephrol. (2015) 28:441–9. doi: 10.1007/s40620-014-0165-x
99. Beckwith H, Medjeral-Thomas N, Galliford J, Griffith M, Levy J, Lightstone L, et al. Mycophenolate mofetil therapy in immunoglobulin A nephropathy: Histological changes after treatment. Nephrol Dial Transplant. (2017) 32:i123–8. doi: 10.1093/ndt/gfw326
100. Barbour SJ, Coppo R, Zhang H, Liu Z, Suzuki Y, Matsuzaki K, et al. Evaluating a new international risk-prediction tool in IgA nephropathy. JAMA Intern Med. (2019) 179:942–52. doi: 10.1001/jamainternmed.2019.0600
101. Zhang J, Huang B, Liu Z, Wang X, Xie M, Guo R, et al. External validation of the international IgA nephropathy prediction tool. Clin J Am Soc Nephrol: CJASN. (2020) 15:1112. doi: 10.2215/CJN.16021219
102. Zhang Y, Guo L, Wang Z, Wang J, Er L, Barbour SJ, et al. External validation of international risk-prediction models of IgA nephropathy in an asian-caucasian cohort. Kidney Int Rep. (2020) 5:1753–63. doi: 10.1016/j.ekir.2020.07.036
103. Barbour SJ, Coppo R, Er L, Russo ML, Liu Z, Ding J, et al. Updating the international IgA nephropathy prediction tool for use in children. Kidney Int. (2021) 99:1439–50. doi: 10.1016/j.kint.2020.10.033
104. Barbour SJ, Coppo R, Zhang H, Liu Z, Suzuki Y, Matsuzaki K, et al. Application of the international IgA nephropathy prediction tool one or two years post-biopsy. Kidney Int. (2022) 102:160–72. doi: 10.1016/j.kint.2022.02.042
105. Kincaid-Smith P, Bennett WM, Dowling JP, Ryan GB. Acute renal failure and tubular necrosis associated with hematuria due to glomerulonephritis. Clin Nephrol. (1983) 19:206–10.
106. Praga M, Gutierrez-Millet V, Navas JJ, Ruilope LM, Morales JM, Alcazar JM, et al. Acute worsening of renal function during episodes of macroscopic hematuria in IgA nephropathy. Kidney Int. (1985) 28:69–74. doi: 10.1038/ki.1985.120
107. Delclaux C, Jacquot C, Callard P, Kleinknecht D. Acute reversible renal failure with macroscopic haematuria in IgA nephropathy. Nephrol Dial Transplant. (1993) 8:195–9.
108. Moreno JA, Martín-Cleary C, Gutiérrez E, Toldos O, Blanco-Colio L, Praga M, et al. AKI associated with macroscopic glomerular hematuria: Clinical and pathophysiologic consequences. Clin J Am Soc Nephrol. (2012) 7(1):175–84. doi: 10.2215/CJN.01970211
109. Trujillo H, Sandino J, Cavero T, Caravaca-Fontán F, Gutiérrez E, Sevillano ÁM, et al. IgA nephropathy is the most common underlying disease in patients with anticoagulant-related nephropathy. Kidney Int Rep. (2022) 7:831–40. doi: 10.1016/j.ekir.2022.01.1048
110. Huang Z, Zhang J, Chen B, Li D, You X, Zhou Y, et al. Clinical significance of persistent hematuria degrees in primary IgA nephropathy: A propensity score-matched analysis of a 10-year follow-up cohort. Am J Nephrol. (2023) 54:62–73. doi: 10.1159/000529650
111. Bobart SA, Alexander MP, Shawwa K, Vaughan LE, Ghamrawi R, Sethi S, et al. The association of microhematuria with mesangial hypercellularity, endocapillary hypercellularity, crescent score and renal outcomes in immunoglobulin A nephropathy. Nephrol Dial Transplant. (2021) 36:840–7. doi: 10.1093/ndt/gfz267
112. Yu G, Guo L, Dong J, Shi S, Liu L, Wang J, et al. Persistent hematuria and kidney disease progression in IgA nephropathy: A cohort study. Am J Kidney Dis. (2020) 76:90–9. doi: 10.1053/j.ajkd.2019.11.008
113. El Karoui K, Fervenza FC, De Vriese AS. Treatment of IgA nephropathy: A rapidly evolving field. J Am Soc Nephrol. (2024) 35(1):103–16. doi: 10.1681/ASN.0000000000000242
114. Le W, Liang S, Hu Y, Deng K, Bao H, Zeng C, et al. Long-term renal survival and related risk factors in patients with IgA nephropathy: Results from a cohort of 1155 cases in a chinese adult population. Nephrol Dial Transplant. (2012) 27:1479–85. doi: 10.1093/ndt/gfr527
115. Ma F, Liu L, Dong R, Yang X, Wei L, Li L, et al. Renal survival and risk factors in IgA nephropathy with crescents. Int Urol Nephrol. (2020) 52:1507–16. doi: 10.1007/s11255-020-02457-3
116. Weng M, Lin J, Chen Y, Zhang X, Zou Z, Chen Y, et al. Time-averaged hematuria as a prognostic indicator of renal outcome in patients with IgA nephropathy. J Clin Med. (2022) 11:6785. doi: 10.3390/jcm11226785
117. Sevillano AM, Gutiérrez E, Yuste C, Cavero T, Mérida E, Rodríguez P, et al. Remission of hematuria improves renal survival in IgA nephropathy. J Am Soc Nephrol. (2017) 28(10):3089–99. doi: 10.1681/ASN.2017010108
118. Shen P, He L, Li Y, Wang Y, Chan M. Natural history and prognostic factors of IgA nephropathy presented with isolated microscopic hematuria in chinese patients. Nephron Clin Pract. (2007) 106(4):c157–61. doi: 10.1159/000104426
119. Shen P, He L, Huang D. Clinical course and prognostic factors of clinical early IgA nephropathy. Neth J Med. (2008) 66:242–7.
120. Szeto CC, Lai FM, To KF, Wong TY, Chow KM, Choi PC, et al. The natural history of immunoglobulin a nephropathy among patients with hematuria and minimal proteinuria. Am J Med. (2001) 110:434–7. doi: 10.1016/S0002-9343(01)00659-3
121. Manno C, Strippoli GFM, D’Altri C, Torres D, Rossini M, Schena FP. A novel simpler histological classification for renal survival in IgA nephropathy: A retrospective study. Am J Kidney Dis. (2007) 49:763–75. doi: 10.1053/j.ajkd.2007.03.013
122. Harmankaya O, Oztürk Y, Baştürk T, Obek A, Kiliçarslan I. Efficacy of immunosuppressive therapy in IgA nephropathy presenting with isolated hematuria. Int Urol Nephrol. (2002) 33:167–71. doi: 10.1023/A:1014424723466
123. Gutiérrez E, Zamora I, Ballarín JA, Arce Y, Jiménez S, Quereda C, et al. Long-term outcomes of IgA nephropathy presenting with minimal or no proteinuria. J Am Soc Nephrol. (2012) 23(10):1753–60. doi: 10.1681/ASN.2012010063
124. Tanaka K, Moriyama T, Iwasaki C, Takei T, Nitta K. Effect of hematuria on the outcome of IgA nephropathy with mild proteinuria. Clin Exp Nephrol. (2015) 19(5):815–21. doi: 10.1007/s10157-014-1068-9
125. Iwasaki C, Moriyama T, Tanaka K, Takei T, Nitta K. Effect of hematuria on the outcome of immunoglobulin A nephropathy with proteinuria. J Nephropathol. (2016) 5:72. doi: 10.15171/jnp.2016.12
126. He P, Wang H, Huang C, He L. Hematuria was a high risk for renal progression and ESRD in immunoglobulin a nephropathy: A systematic review and meta-analysis. Ren Fail. (2021) 43:488–99. doi: 10.1080/0886022X.2021.1879852
127. Suzuki Y, Matsuzaki K, Suzuki H, Sakamoto N, Joh K, Kawamura T, et al. Proposal of remission criteria for IgA nephropathy. Clin Exp Nephrol. (2014) 18:481–6. doi: 10.1007/s10157-013-0849-x
128. Praga M, Gutie E, Gonza E, Morales E, Herna E. Treatment of IgA nephropathy with ACE inhibitors: A randomized and controlled trial. J Am Soc Nephrol. (2003) 14:1578–83. doi: 10.1097/01.ASN.0000068460.37369.DC
129. Li PK, Leung CB, Chow KM, Cheng YL, Fung SK, Mak SK, et al. Hong kong study using valsartan in IgA nephropathy (HKVIN): A double-blind, randomized, placebo-controlled study. Am J Kidney Dis. (2006) 47:751–60. doi: 10.1053/j.ajkd.2006.01.017
130. Coppo R, Peruzzi L, Amore A, Piccoli A, Cochat P, Stone R, et al. IgACE: A placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgA nephropathy and moderate proteinuria. J Am Soc Nephrol. (2007) 18:1880–8. doi: 10.1681/ASN.2006040347
131. Bagchi S, Mani K, Swamy A, Barwad A, Singh G, Bhowmik D, et al. Supportive management of IgA nephropathy with renin-angiotensin blockade, the AIIMS primary IgA nephropathy cohort (APPROACH) study. Kidney Int Rep. (2021) 6:1661–8. doi: 10.1016/j.ekir.2021.02.018
132. Lennartz DP, Seikrit C, Wied S, Fitzner C, Eitner F, Hilgers R, et al. Single versus dual blockade of the renin-angiotensin system in patients with IgA nephropathy. J Nephrol. (2020) 33:1231–9. doi: 10.1007/s40620-020-00836-8
133. ONTARGET Investigators, Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. (2008) 358:1547–59. doi: 10.1056/NEJMoa0801317
134. Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, Duckworth W, et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. (2013) 369:1892–903. doi: 10.1056/NEJMoa1303154
135. Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. (2020) 383:2219–29. doi: 10.1056/NEJMoa2025845
136. Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, et al. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N Engl J Med. (2021) 385:2252–63. doi: 10.1056/NEJMoa2110956
137. Wheeler DC, Toto RD, Stefánsson BV, Jongs N, Chertow GM, Greene T, et al. A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy. Kidney Int. (2021) 100:215–24. doi: 10.1016/j.kint.2021.03.033
138. Judge PK, Staplin N, Mayne KJ, Wanner C, Green JB, Hauske SJ, et al. Impact of primary kidney disease on the effects of empagliflozin in patients with chronic kidney disease: Secondary analyses of the EMPA-KIDNEY trial. Lancet Diabetes Endocrinol. (2024) 12:51–60. doi: 10.1016/S2213-8587(23)00322-4
139. Baigent C, Emberson J, Haynes R, Herrington WG, Judge P, Landray MJ, et al. Impact of diabetes on the effects of sodium glucose co-transporter-2 inhibitors on kidney outcomes: Collaborative meta-analysis of large placebo-controlled trials. Lancet. (2022) 400:1788–801. doi: 10.1016/S0140-6736(22)02074-8
140. Kohan DE, Barratt J, Heerspink HJL, Campbell KN, Camargo M, Ogbaa I, et al. Targeting the endothelin A receptor in IgA nephropathy. Kidney Int Rep. (2023) 8:2198–210. doi: 10.1016/j.ekir.2023.07.023
141. Heerspink HJ, Radhakrishnan J, Alpers CE, Barratt J, Bieler S, Diva U, et al. Sparsentan in patients with IgA nephropathy: A prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial. Lancet. (2023) 401:1584–94. doi: 10.1016/S0140-6736(23)00569-X
142. Rovin BH, Barratt J, Heerspink HJL, Alpers CE, Bieler S, Chae D, et al. Efficacy and safety of sparsentan versus irbesartan in patients with IgA nephropathy (PROTECT): 2-year results from a randomised, active-controlled, phase 3 trial. Lancet. (2023) 402:2077–90. doi: 10.1016/S0140-6736(23)02302-4
143. Heerspink HL, Jardine M, Kohan D, Lafayette R, Levin A, Liew A, et al. POS-527 A phase 3, randomized, double-blind, placebo-controlled study of atrasentan in patients with IgA nephropathy (the ALIGN study). Kidney Int Rep. (2022) 7:S229–30. doi: 10.1016/j.ekir.2022.01.558
144. Heerspink HJL, Kohan DE, de Zeeuw D. New insights from SONAR indicate adding sodium glucose co-transporter 2 inhibitors to an endothelin receptor antagonist mitigates fluid retention and enhances albuminuria reduction. Kidney Int. (2021) 99:346–9. doi: 10.1016/j.kint.2020.09.026
145. Heerspink HJL, Kiyosue A, Wheeler DC, Lin M, Wijkmark E, Carlson G, et al. Zibotentan in combination with dapagliflozin compared with dapagliflozin in patients with chronic kidney disease (ZENITH-CKD): A multicentre, randomised, active-controlled, phase 2b, clinical trial. Lancet. (2023) 402:2004–17. doi: 10.1016/S0140-6736(23)02230-4
146. Lv J, Xu D, Perkovic V, Ma X, Johnson DW, Woodward M, et al. Corticosteroid therapy in IgA nephropathy. J Am Soc Nephrol. (2012) 23:1108–16. doi: 10.1681/ASN.2011111112
147. Pozzi C, Andrulli S, Del Vecchio L, Melis P, Fogazzi GB, Altieri P, et al. Corticosteroid effectiveness in IgA nephropathy: Long-term results of a randomized, controlled trial. J Am Soc Nephrol. (2004) 15:157–63. doi: 10.1097/01.ASN.0000103869.08096.4F
148. Ballardie FW, Roberts ISD. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol. (2002) 13:142–8. doi: 10.1681/ASN.V131142
149. Rauen T, Fitzner C, Eitner F, Sommerer C, Zeier M, Otte B, et al. Effects of two immunosuppressive treatment protocols for IgA nephropathy. J Am Soc Nephrol. (2018) 29(1):317–25. doi: 10.1681/ASN.2017060713
150. Rauen T, Wied S, Fitzner C, Eitner F, Sommerer C, Zeier M, et al. After ten years of follow-up, no difference between supportive care plus immunosuppression and supportive care alone in IgA nephropathy. Kidney Int. (2020) 98:1044–52. doi: 10.1016/j.kint.2020.04.046
151. Fellström BC, Barratt J, Cook H, Coppo R, Feehally J, de Fijter JW, et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet. (2017) 389:2117–27. doi: 10.1016/S0140-6736(17)30550-0
152. Lafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, et al. A randomized, controlled trial of rituximab in IgA nephropathy with proteinuria and renal dysfunction. J Am Soc Nephrol. (2017) 28(4):1306–13. doi: 10.1681/ASN.2016060640
153. Mathur M, Barratt J, Chacko B, Chan TM, Kooienga L, Oh K, et al. A phase 2 trial of sibeprenlimab in patients with IgA nephropathy. N Engl J Med. (2024) 390:20–31. doi: 10.1056/NEJMoa2305635
154. Lv J, Liu L, Hao C, Li G, Fu P, Xing G, et al. Randomized phase 2 trial of telitacicept in patients with IgA nephropathy with persistent proteinuria. Kidney Int Rep. (2022) 8:499–506. doi: 10.1016/j.ekir.2022.12.014
155. Barratt J, Tumlin J, Suzuki Y, Kao A, Aydemir A, Pudota K, et al. Randomized phase II JANUS study of atacicept in patients with IgA nephropathy and persistent proteinuria. Kidney Int Rep. (2022) 7:1831–41. doi: 10.1016/j.ekir.2022.05.017
156. Gesualdo L, Di Leo V, Coppo R. The mucosal immune system and IgA nephropathy. Semin Immunopathol. (2021) 43(5):657–68. doi: 10.1007/s00281-021-00871-y
157. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. (2008) 8:421–34. doi: 10.1038/nri2322
158. Cheung CK, Barratt J, Liew A, Zhang H, Tesar V, Lafayette R. The role of BAFF and APRIL in IgA nephropathy: Pathogenic mechanisms and targeted therapies. Front Nephrol. (2024) 3:1346769. doi: 10.3389/fneph.2023.1346769
159. McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest. (2011) 121:3991–4002. doi: 10.1172/JCI45563
160. Kim YG, Alvarez M, Suzuki H, Hirose S, Izui S, Tomino Y, et al. Pathogenic role of a proliferation-inducing ligand (APRIL) in murine IgA nephropathy. PloS One. (2015) 10:e0137044. doi: 10.1371/journal.pone.0137044
161. Xin G, Shi W, Xu L, Su Y, Yan L, Li K. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J Nephrol. (2013) 26:683–90. doi: 10.5301/jn.5000218
162. Zhai Y, Zhu L, Shi S, Liu L, Lv J, Zhang H. Increased APRIL expression induces IgA1 aberrant glycosylation in IgA nephropathy. Med (Baltimore). (2016) 95:e3099. doi: 10.1097/MD.0000000000003099
163. Han SS, Yang SH, Choi M, Kim H, Kim K, Lee S, et al. The role of TNF superfamily member 13 in the progression of IgA nephropathy. J Am Soc Nephrol. (2016) 27:3430–9. doi: 10.1681/ASN.2015060677
164. Ji L, Geng Y, Zhang X, Deng X, Song Z, Tan M, et al. B cell pathway dual inhibition for systemic lupus erythematosus: A prospective single-arm cohort study of telitacicept. MedComm (2020). (2024) 5:e515. doi: 10.1002/mco2.515
165. Chen R, Fu R, Lin Z, Huang C, Huang W. The efficacy and safety of telitacicept for the treatment of systemic lupus erythematosus: A real life observational study. Lupus. (2023) 32:94–100. doi: 10.1177/09612033221141253
166. Wallace DJ, Isenberg DA, Morand EF, Vazquez-Mateo C, Kao AH, Aydemir A, et al. Safety and clinical activity of atacicept in the long-term extension of the phase 2b ADDRESS II study in systemic lupus erythematosus. Rheumatol (Oxford). (2021) 60:5379–89. doi: 10.1093/rheumatology/keab115
167. Lafayette R, Barbour S, Israni R, Wei X, Eren N, Floege J, et al. A phase 2b, randomized, double-blind, placebo-controlled, clinical trial of atacicept for treatment of IgA nephropathy. Kidney Int. (2024) 105:1306–15. doi: 10.1016/j.kint.2024.03.012
168. Zhang H, Rizk DV, Perkovic V, Maes B, Kashihara N, Rovin B, et al. Results of a randomized double-blind placebo-controlled phase 2 study propose iptacopan as an alternative complement pathway inhibitor for IgA nephropathy. Kidney Int. (2024) 105:189–99. doi: 10.1016/j.kint.2023.09.027
169. Lafayette RA, Rovin BH, Reich HN, Tumlin JA, Floege J, Barratt J. Safety, tolerability and efficacy of narsoplimab, a novel MASP-2 inhibitor for the treatment of IgA nephropathy. Kidney Int Rep. (2020) 5:2032–41. doi: 10.1016/j.ekir.2020.08.003
170. Dixon BP, Greenbaum LA, Huang L, Rajan S, Ke C, Zhang Y, et al. Clinical safety and efficacy of pegcetacoplan in a phase 2 study of patients with C3 glomerulopathy and other complement-mediated glomerular diseases. Kidney Int Rep. (2023) 8:2284–93. doi: 10.1016/j.ekir.2023.08.033
171. Barratt J, Liew A, Yeo SC, Fernström A, Barbour SJ, Sperati CJ, et al. Phase 2 trial of cemdisiran in adult patients with IgA nephropathy: A randomized controlled trial. Clin J Am Soc Nephrol. (2024) 19:452–62.
172. Bruchfeld A, Magin H, Nachman P, Parikh S, Lafayette R, Potarca A, et al. C5a receptor inhibitor avacopan in immunoglobulin A nephropathy-an open-label pilot study. Clin Kidney J. (2022) 15:922–8. doi: 10.1093/ckj/sfab294
173. Versino F, Fattizzo B. Complement inhibition in paroxysmal nocturnal hemoglobinuria: From biology to therapy. Int J Lab Hematol. (2024) 46(S1):43–54. doi: 10.1111/ijlh.14281
174. Kolev M, Barbour T, Baver S, Francois C, Deschatelets P. With complements: C3 inhibition in the clinic. Immunol Rev. (2023) 313:358–75. doi: 10.1111/imr.13138
175. Rosenblad T, Rebetz J, Johansson M, Békássy Z, Sartz L, Karpman D. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr Nephrol. (2014) 29:2225–8. doi: 10.1007/s00467-014-2863-y
176. Ring T, Pedersen BB, Salkus G, Goodship THJ. Use of eculizumab in crescentic IgA nephropathy: Proof of principle and conundrum? Clin Kidney J. (2015) 8:489–91. doi: 10.1093/ckj/sfv076
177. Jayne DRW, Merkel PA, Schall TJ, Bekker P, ADVOCATE Study Group. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. (2021) 384:599–609. doi: 10.1056/NEJMoa2023386
178. Roccatello D, Ferro M, Cesano G, Rossi D, Berutti S, Salomone M, et al. Steroid and cyclophosphamide in IgA nephropathy. Nephrol Dial Transplant. (2000) 15:833–5. doi: 10.1093/ndt/15.6.833
179. McIntyre CW, Fluck RJ, Lambie SH. Steroid and cyclophosphamide therapy for IgA nephropathy associated with crescenteric change: An effective treatment. Clin Nephrol. (2001) 56:193–8.
180. Beck N, Walz G, Schneider J. Effect of cyclophosphamide and glucocorticoid therapy in IgA nephropathy: A single-center retrospective analysis. Kidney360. (2022) 3:506–15. doi: 10.34067/KID.0006702021
181. Navaneethan SD, Viswanathan G, Strippoli GF. META-ANALYSIS OF MYCOPHENOLATE MOFETIL IN iga NEPHROPATHY. Nephrology. (2008) 13:90. doi: 10.1111/j.1440-1797.2007.00868.x
182. Tan CHR, Loh PT, Yang WS, Chan CM. Mycophenolate mofetil in the treatment of IgA nephropathy: A systematic review. Singapore Med J. (2008) 49:780–5.
183. Xu G, Tu W, Jiang D, Xu C. Mycophenolate mofetil treatment for IgA nephropathy: A meta-analysis. Am J Nephrol. (2009) 29:362–7. doi: 10.1159/000168483
184. Chen Y, Li Y, Yang S, Li Y, Liang M. Efficacy and safety of mycophenolate mofetil treatment in IgA nephropathy: A systematic review. BMC Nephrol. (2014) 15:193–3. doi: 10.1186/1471-2369-15-193
185. Peng X, Zheng W, Fu R, Huang Y, Deng M, Tao S, et al. Efficacy and safety of mycophenolate mofetil in the treatment for IgA nephropathy: A meta-analysis of randomized controlled trials. Clin Exp Nephrol. (2021) 25:788–801. doi: 10.1007/s10157-021-02028-5
186. Hou FF, Xie D, Wang J, Xu X, Yang X, Ai J, et al. Effectiveness of mycophenolate mofetil among patients with progressive IgA nephropathy: A randomized clinical trial. JAMA Netw Open. (2023) 6:e2254054. doi: 10.1001/jamanetworkopen.2022.54054
187. Yi J, He Z, Xu S, Feng S. Efficacy and safety of leflunomide in IgA nephropathy: A systematic review and meta-analysis. Int Urol Nephrol. (2019) 51:1987–98. doi: 10.1007/s11255-019-02255-6
188. Zhang D, Xia B, Zhang X, Liang P, Hu X. Efficacy and safety of low-dose corticosteroids combined with leflunomide for progressive IgA nephropathy: A systematic review and meta-analysis. BMC Urol. (2024) 24:56–3. doi: 10.1186/s12894-024-01438-3
189. Stefan G, Mircescu G. Hydroxychloroquine in IgA nephropathy: A systematic review. Ren Fail. (2021) 43:1520–7. doi: 10.1080/0886022X.2021.2000875
190. He W, Wang J, Liu N, Li G, Zhu X, Yao L, et al. The efficacy and safety of hydroxychloroquine versus leflunomide in patients with IgA nephropathy: A single-center experience. J Nephrol. (2024) 37:933–40. doi: 10.1007/s40620-023-01839-x
191. Bene MC, Faure G, De Ligny BH, Kessler M, Duheille J. Immunoglobulin A nephropathy. quantitative immunohistomorphometry of the tonsillar plasma cells evidences an inversion of the immunoglobulin A versus immunoglobulin G secreting cell balance. J Clin Invest. (1983) 71:1342–7. doi: 10.1172/JCI110886
192. Xie Y, Chen X, Nishi S, Narita I, Gejyo F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney Int. (2004) 65:1135–44. doi: 10.1111/j.1523-1755.2004.00486.x
193. Rasche FM, Schwarz A, Keller F. Tonsillectomy does not prevent a progressive course in IgA nephropathy. Clin Nephrol. (1999) 51:147–52.
194. Wang Y, Chen J, Wang Y, Chen Y, Wang L, Lv Y. A meta-analysis of the clinical remission rate and long-term efficacy of tonsillectomy in patients with IgA nephropathy. Nephrol Dial Transplant. (2011) 26:1923–31. doi: 10.1093/ndt/gfq674
195. Liu L, Wang L, Jiang Y, Yao L, Dong L, Li Z, et al. Tonsillectomy for IgA nephropathy: A meta-analysis. Am J Kidney Dis. (2015) 65:80–7. doi: 10.1053/j.ajkd.2014.06.036
196. Duan J, Liu D, Duan G, Liu Z. Long-term efficacy of tonsillectomy as a treatment in patients with IgA nephropathy: A meta-analysis. Int Urol Nephrol. (2017) 49:103–12. doi: 10.1007/s11255-016-1432-7
197. Kawamura T, Yoshimura M, Miyazaki Y, Okamoto H, Kimura K, Hirano K, et al. A multicenter randomized controlled trial of tonsillectomy combined with steroid pulse therapy in patients with immunoglobulin A nephropathy. Nephrol Dial Transplant. (2014) 29:1546–53. doi: 10.1093/ndt/gfu020
198. Katafuchi R, Kawamura T, Joh K, Hashiguchi A, Hisano S, Shimizu A, et al. Pathological sub-analysis of a multicenter randomized controlled trial of tonsillectomy combined with steroid pulse therapy versus steroid pulse monotherapy in patients with immunoglobulin A nephropathy. Clin Exp Nephrol. (2016) 20:244–52. doi: 10.1007/s10157-015-1159-2
199. Yang D, He L, Peng X, Liu H, Peng Y, Yuan S, et al. The efficacy of tonsillectomy on clinical remission and relapse in patients with IgA nephropathy: A randomized controlled trial. Ren Fail. (2016) 38:242–8. doi: 10.3109/0886022X.2015.1128251
200. Feehally J, Coppo R, Troyanov S, Bellur SS, Cattran D, Cook T, et al. Tonsillectomy in a european cohort of 1,147 patients with IgA nephropathy. Nephron. (2016) 132:15–24. doi: 10.1159/000441852
Keywords: IgA nephropathy, glomerulonephritis, hematuria, proteinuria, complement inhibition, thrombotic microangiopathy, podocytopathy, immunossuppression
Citation: Filippone EJ, Gulati R and Farber JL (2024) Contemporary review of IgA nephropathy. Front. Immunol. 15:1436923. doi: 10.3389/fimmu.2024.1436923
Received: 22 May 2024; Accepted: 02 July 2024;
Published: 12 August 2024.
Edited by:
Kutty Selva Nandakumar, Karolinska Institutet (KI), SwedenReviewed by:
Jiri Mestecky, University of Alabama at Birmingham, United StatesGianmarco Sabiu, University of Milan, Italy
Copyright © 2024 Filippone, Gulati and Farber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward J. Filippone, a2lkbmV5c0Bjb21jYXN0Lm5ldA==