 Sebastian J. Theobald
Sebastian J. Theobald Tony A. Müller1,2†
Tony A. Müller1,2† Dinah Lange
Dinah Lange Katharina Keck
Katharina Keck Jan Rybniker
Jan Rybniker- 1Department I of Internal Medicine, University of Cologne, Cologne, Germany
- 2Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany
- 3German Center for Infection Research (DZIF), Partner Site Bonn-Cologne, Cologne, Germany
Mycobacterium tuberculosis (Mtb) infection represents a global health problem and is characterized by formation of granuloma with a necrotic center and a systemic inflammatory response. Inflammasomes have a crucial role in the host immune response towards Mtb. These intracellular multi-protein complexes are assembled in response to pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). Inflammasome platforms activate caspases, leading to the maturation of the proinflammatory cytokines interleukin (IL)-1 and 18 and the cleavage of gasdermin D (GSDMD), a pore-forming protein responsible for cytokine release and pyroptotic cell death. Recent in vitro and in vivo findings have highlighted the importance of inflammasome signaling and subsequent necrotic cell death in Mtb-infected innate immune cells. However, we are just beginning to understand how inflammasomes contribute to disease or to a protective immune response in tuberculosis (TB). A detailed molecular understanding of inflammasome-associated pathomechanisms may foster the development of novel host-directed therapeutics or vaccines with improved activity. In this mini-review, we discuss the regulatory and molecular aspects of inflammasome activation and the associated immunological consequences for Mtb pathogenesis.
1 Introduction
Despite global control efforts, tuberculosis (TB) remains a leading cause of death worldwide with approximately 10.6 million new cases and 1.4 million deaths in 2022 (1). The causative agent of the disease, Mycobacterium tuberculosis (Mtb), is mainly transmitted via aerosols leading to pulmonary infection as well as various extrapulmonary manifestations (2). The course of the disease is characterized by long-lasting persistence, pointing towards various pathogenic strategies that have evolved in order to evade control by the immune system (3). As an intracellular pathogen, Mtb induces notable alterations in the cell death mechanisms of alveolar macrophages, a highly relevant host cell of this pathogen (4). Macrophage phagocytosis can trigger necrotic cell death, resulting in inflammatory tissue damage and bacterial spread.
There is evidence that innate immune responses activated by Mtb are regulated by large multimeric protein complexes called inflammasomes. These consist of a cytosolic sensor, an adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and an effector caspase pro-caspase-1 (5–7). Inflammasome assembly is triggered by the recognition of exogenous or endogenous danger signals by various sensor molecules. Subsequent cleavage and activation of the pro-caspase-1 results in the secretion of the pro-inflammatory cytokines interleukin 1β (IL-1β) and interleukin 18 (IL-18), as well as the induction of pyroptosis. Pyroptosis is a pro-inflammatory form of programmed cell death, mediated by the cleavage of a pore-forming protein known as gasdermin D (GSDMD).
Several types of inflammasomes have been described, including the nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family members NLRP1, NLRP3 and NLRC4, as well as the proteins absent in melanoma 2 (AIM2) and pyrin. These inflammasome types are mainly characterized by the specificity of the sensor protein towards a certain stimulus (6). For instance, the AIM2 inflammasome acts as a sensor for (cytosolic) double-stranded DNA and the NLRC4 inflammasome responds to bacterial flagellin and type III secretion system components (8–11). In contrast, the NLRP3 inflammasome responds to a diverse set of microbial, endogenous and environmental stimuli. Priming of the inflammasome induces transcriptional upregulation of NLRP3 complex members as well as posttranslational modifications. This process is initiated by cytokine receptors or Toll-like receptors (TLRs) after recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), ultimately inducing nuclear factor-κB (NF-κB) activation (12). The second step towards full activation of the NLRP3 inflammasome can be triggered by a large variety of unrelated signals such as ATP or ion influx (12–14).
2 Mtb mediated modulation of the NLRP3 inflammasome in vitro and ex vivo
Several reports indicate that Mtb infection of phagocytes in vitro or ex vivo leads to NLRP3 inflammasome activation, in combination with hallmarks of pyroptosis, including caspase-1 and GSDMD cleavage and the secretion of IL-1β and IL-18. This induction of NLRP3 inflammasome signaling by Mtb has been associated with multiple cell types including macrophage cell lines as well as murine and human-derived primary cells (THP-1 macrophages, monocyte-derived macrophages, bone marrow derived macrophages (BMDMs), bone marrow derived dendritic cells (BMDCs), murine retinal pigment epitheliums and primary murine microglial cells) (15–22). Using chemical inhibition and knock-out cell lines, we and others found several lines of evidence that key inflammasome components such as NLRP3, caspase-1 and GSDMD are required to mediate Mtb-induced pyroptotic cell death and the release of IL-1β and IL-18. In addition, several reports linked NLRP3 inflammasome activation with a functional ESX-1 secretion system (17–19, 22). This key virulence factor of Mtb secretes a pore forming protein (EsxA) into the cytosol of infected host cells and chemical inhibition of ESX-1 strongly impairs IL-1β release (23). However, the molecular mechanism by which EsxA or other co-secreted effector proteins mediate NLRP3 inflammasome activation is not known. Interestingly, EST-12, another secreted Mtb effector protein, binds to the cellular receptor RACK-1 leading to NLRP3/caspase-1/GSDMD mediated pyroptosis in vitro and in vivo. Consequently, an Mtb strain lacking EST-12 exhibited increased infectivity in the murine model, indicating that inflammasome activation can be protective in early stages of the infection (24). However, EST-12 is not secreted in an ESX-1 dependent manner, indicating the potential involvement of other secretory machineries of Mtb in NLRP3 inflammasome activation. Intriguingly, Chai et al. recently demonstrated that Mtb interferes with cell death mechanisms by secreting the phospholipid phosphatase (PtpB), which dephosphorylates plasma membrane phosphoinositides by binding to ubiquitin. PtpB enzymatic activity impairs binding of GSDMD to the plasma membrane subsequently leading to inhibition of pyroptosis (25). In line with this finding, we were able to show that IL-1β secretion of Mtb-infected macrophages is independent of GSDMD, but not NLRP3, indicating that alternative pore forming mechanisms are used to circumvent PtpB-dependent abrogation of GSDMD pore formation (17) (Figure 1).
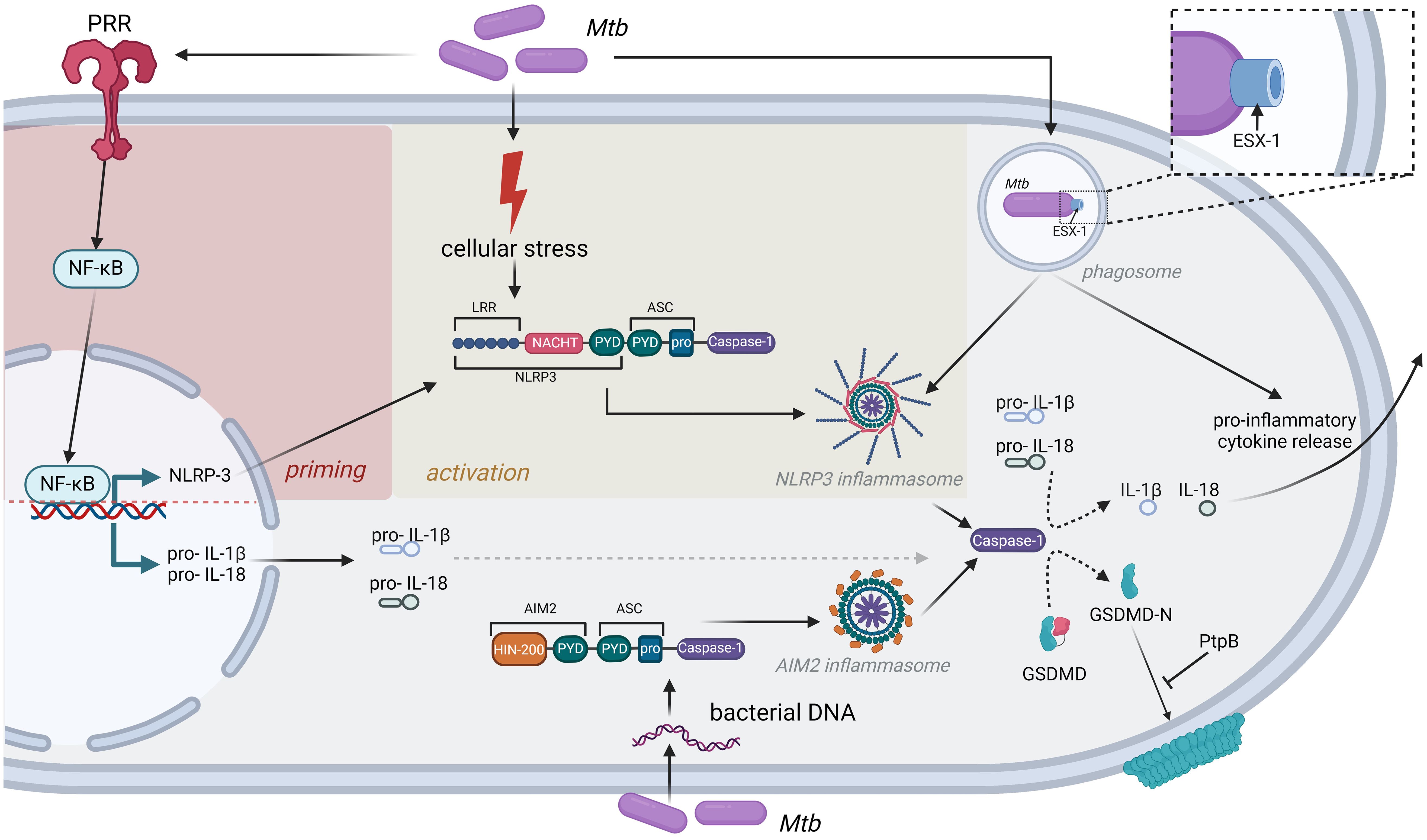
Figure 1. Inflammasomes as central mediators of inflammatory cytokine release after Mtb infection. Detection of Mtb by PRR induces cellular stress, leading to priming and activation of the NLRP3 inflammasome. In addition, Mtb DNA can be directly recognized by the AIM2 inflammasome. Both inflammasomes induce caspase-1 activation and subsequent release of inflammatory cytokines like IL-1β or IL-18. Mtb has evolved strategies to interfere with this immune response, exemplified by the bacterial PtpB phosphatase, which interferes with GSDMD binding to the cell membrane, or the ESX-1 secretion system that is strictly required to induce NLRP3 inflammasome formation.
While some of the studies discussed above point towards a role for Mtb in inducing inflammasome activation in vitro, some reports indicate that certain Mtb proteins might also have a suppressive effect on NLRP3 inflammasome activation. Rastogi et al. reported that Mtb infection of BMDMs could inhibit NLRP3 inflammasome activation in an ESX-1 independent mechanism. However, deletion of the bacterial phosphokinase PknF reversed this phenotype due to increased intracellular ROS production (26). Another study identified the bacterial protein PE12 as an inhibitor of NLRP3 inflammasome formation by PE12 binding to TLR-4 and subsequently silencing downstream signaling cascades (27). These in vitro findings require confirmation in suitable in vivo infection models that mirror necrotic granuloma formation observed in tissues of infected humans.
3 The role of IL-1β secretion and NLRP3 inflammasome activation in in vivo infection models
Despite numerous in vitro studies deciphering inflammasome-associated cytokine release and cell death pathways such as pyroptosis in the context of Mtb infection, there are contradictory data regarding the in vivo relevance of these findings.
Using mouse models of Mtb infection, several reports illustrate that knock-out of Nlrp3 or caspase-1 has little impact on survival, bacterial burden or the inflammatory responses (19, 28–30), whereas Pycard (the gene coding for ASC) knock-out mice show decreased survival associated with impaired granuloma formation (28). Recently, Stutz et al. reported that non-apoptotic functions of caspase-8, as well as caspase-1 or GSDMD do not contribute to the control of Mtb growth or Mtb-specific immune responses in vivo. In contrast, Mtb infection of their Bax/Bak knock-out model led to upregulation of multiple cytokines in lung tissue including IL-1β together with exacerbated lung lesions and higher bacterial loads, again indicating that GSDMD plays a minor role in secretion of this cytokine (31). Recently, Thomas et al. reported higher susceptibility towards Mtb infection in mice lacking phagocyte oxidase (Cybb) and caspase-1, however in their single knock-out for caspase-1 they were not able to show any differences compared to wild-type mice (30). In contrast, Chai et al. recently elucidated, as mentioned above, the role of PtpB in abrogation of GSDMD binding to the plasma membrane. In their work, they proposed that inflammatory cytokine–mediated granuloma formation is mediated by GSDMD and further showed that PtpB is an essential component of Mtb to evade host GSDMD-mediated immune responses in vivo (25).
In a C3HeB/FeJ mouse model, which better reflects human granuloma formation compared to conventional mouse models, Lovey et al. were able to show that alveolar macrophages strongly upregulate genes such as NLRP3, IL-1 α/β, TNF and several CCL`s and CXCL`s. Using an IL-1R1 blocking antibody, they demonstrated that bacterial control, Mtb pathogenicity and Mtb-specific T cell priming is dependent on IL-1R1 signaling (32). In line with these findings, we detected increased expression of NLRP3 in lung granuloma tissue derived from IL-13tg mice, another mouse model exhibiting human-like granuloma structures (17). Similar to the mentioned studies, Mayer-Barber et al. identified a crucial role for IL-1α and IL-1β, the proteins ultimately secreted after inflammasome activation, in conferring host resistance. Furthermore, they detected higher expression levels of both cytokines in lung macrophages and dendritic cells upon Mtb infection (33). The same group confirmed the importance of IL-1β, IL1R1 and type I interferons for Mtb pathology, in particular deciphering the immune crosstalk between both signaling pathways (34). Furthermore, the researchers propose that both targets are suitable for host-directed therapy approaches to control the outcome of TB (34). Taken together, there is clear evidence that IL-1β and the IL1R1 play important roles in Mtb pathogenicity in vivo, however, no consensus on the in vivo relevance of NLRP3 inflammasome components (NLRP3, caspase 1 and GSDMD) has been be achieved so far. Importantly, future research should focus on animal models that better reflect human granuloma formation and immunology.
4 The role of the AIM2 inflammasome during Mtb-infection
The first report highlighting the role of the AIM2 inflammasome during Mtb infection was published in 2012 by Saiga and colleagues (35). They detected increased susceptibility of Aim2-/- mice to intratracheal infection, which they related to decreased caspase-1 cleavage as well as lower IL-1β and IL-18 levels in the lungs. Mtb-antigen-specific T cell responses were also severely reduced in CD4+ T cells derived from Aim2-/- mice. In vitro data similarly revealed that depletion of AIM2 or ASC in murine macrophage cell lines diminishes IL-1β release following infection with Mtb or Mycobacterium bovis (36, 37). In addition, Mtb genomic DNA was confirmed to bind and activate AIM2 (35). Yan et al. revealed in in vitro experiments, that recognition of Mtb genomic DNA by AIM2 inhibits stimulator of IFN genes (STING) via interaction with ASC. This mechanism induces overreactive IFN-β and depressive IFN-γ responses in M. bovis-BCG infected Aim2-/- mice, translating into higher bacterial burden and more severe pathology (38). However, it remains unclear whether these results are of relevance for a better understanding of Mtb pathogenicity as the M. bovis -BCG vaccine strain lacks important virulence factors such as the ESX-1 secretion system, which is also crucial for the activation of AIM2 inflammasome components (39). Ex vivo studies using Aim2-deficient murine BMDMs or BMDCs have produced some contrasting results. Mtb as well as virulent M. bovis were reported to be capable of inducing AIM2 expression in BMDCs (36, 40). However, the impact of the increased expression remains unclear, as neither deficiency nor siRNA-medicated knockdown of Aim2 had an influence on IL-1β release in Mtb-infected BMDMs (39, 40). In a further step, Shah et al. were addressing, whether Mtb could actively inhibit AIM2 inflammasome activation originating from other sources, such as simultaneous infection of BMDMs with Mycobacterium smegmatis. Indeed, they found that Mtb is able to diminish the AIM2-dependent IL-1β production caused by this non-pathogenic fast-growing mycobacterium (39). In line with the host-protective role of AIM2 in in vivo experiments, it was also reported that IL-1β production is strongly reduced in Aim2-/-/Nlrp3-/- BMDMs infected with different clinical Mtb strains. However, it was not addressed, how much of this effect can be attributed to the individual inflammasome types (41).
Together, these studies indicate that the exact role of the AIM2 inflammasome pathway in Mtb infection remains not fully understood and that experimental findings may be influenced by the nature of the infected host cell (mouse versus human) or the mycobacterial strain under investigation. Some data point towards Mtb inhibition of AIM2 to be a potential evolutionary mechanism allowing immune evasion of the pathogen.
5 Concluding remarks and discussion
Although some light has been shed on Mtb host-pathogen interactions in recent years, the precise role of inflammasomes in this context is not fully understood. Here we systematically reviewed the current literature on the impact of NLRP3 and AIM2 inflammasome activation for Mtb pathogenicity. In summary, there is strong evidence from in vitro experiments that both inflammasome types are being modulated in Mtb infected macrophages, which leads to secretion of several pro-inflammatory cytokines as well as pyroptotic cell death (Figure 1). Consistent with this notion, there have been reports of NRLP3 gain-of-function variants in TB patients that confer some degree of resistance towards Mtb infection (42, 43). Interestingly, inflammasome activation is also increased in tuberculosis-associated immune reconstitution inflammatory syndrome (TB-IRIS), a hyperinflammatory state that is frequently observed in patients co-infected with HIV (44). In contrast, conventional knock-out mouse models failed to demonstrate any impact of NLRP3 or caspase-1 in Mtb pathogenicity, whereas mice deficient for AIM2 or PYCARD seemed to be more susceptible to Mtb infection. However, it remains doubtful whether results from these mouse models can be translated to Mtb infection in humans, as necrotic granuloma formation is not observed in these mice. A potential consequence might be that the immunological signaling pathways triggered by Mtb in these models differ from the human setting. Therefore, we and others have explored mouse models that reflect the human situation more closely, such as C3HeB/FeJ mice and IL-13tg mice, where NLRP3 and IL-1 signaling seems to play a more relevant role, which needs to be confirmed in future studies. At this point, it is important to note that the usage of more human-like in vivo models is essential to decipher the importance of inflammasome signaling in TB. Despite the contradictory results regarding the role of NLRP3 and AIM2, murine and human in vitro and murine in vivo studies consent that IL-1α/β and IL1R1 play an important role in Mtb pathogenicity. The relevance of these pathways has particularly been highlighted by a report connecting IL-1 signaling with type 1 interferon signaling, which is known to be involved in Mtb immunobiology (45). Therefore, IL-1R1 blockade represents a potential candidate for host-directed therapy (HDT) approaches. The development of novel and potent HDTs for the treatment of TB is of particular importance with the emergence of more multi-drug resistant strains. In general, inflammasomes represent a highly interesting target for HDTs, as a pharmacological intervention would potentially inhibit the excessive secretion of pro-inflammatory cytokines and, secondly, suppress tissue damage and necrosis mediated by host cell death.
Along these lines, we and others have shown in vitro activity for the selective NLRP3 inflammasome inhibitor MCC-950, a small-molecule, which has been used in a clinical study for rheumatoid arthritis (46). MCC-950 potently inhibits Mtb-induced cell death and further abrogates IL-1β and IL-18 secretion (17, 22). Similar results have been obtained for the small-molecule Vx-765, which blocks caspase-1 (17, 22). Further specific inhibitors of the inflammasome pathway are currently under development (47). However, for all potential inhibitors, in vivo efficacy studies are missing. Notably, we believe that the clinical success of such HDTs relies on a personalized treatment approach, which identifies the ideal time-point for the intervention and reduces potential side effects.
For this, a better understanding of the exact timing of inflammasome and IL-1 signaling during Mtb infection is crucial (i) to gain new insights into the immunopathology of Mtb during acute and chronic disease in suitable in vivo and ex vivo models and (ii) to identify patients that may benefit from inhibitors within host directed therapies.
Author contributions
ST: Conceptualization, Funding acquisition, Validation, Visualization, Writing – original draft, Writing – review & editing. TM: Conceptualization, Project administration, Validation, Writing – original draft, Writing – review & editing. DL: Visualization, Writing – review & editing. KK: Visualization, Writing – review & editing. JR: Conceptualization, Funding acquisition, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author’s research is funded by the European Union Innovative Medicines Initiative 2 Joint Undertaking program grant no. 853989 (ERA4TB to JR), the German Federal Ministry of Education and Research (BMBF; grant IdEpiCo to JR); the German Research Council (DFG; CRC 1403 to JR), and the German Center for Infection Research (DZIF; TTU 02.814 and 02.913 to JR). ST and JR are supported by a research grant of the CMMC (B10), and ST by stipends from the Imhoff-Stiftung and the Koeln Fortune Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bagcchi S. WHO’s global tuberculosis report 2022. Lancet Microbe. (2023) 4:e20. doi: 10.1016/S2666-5247(22)00359-7
2. Carabali-Isajar ML, Rodriguez-Bejarano OH, Amado T, Patarroyo MA, Izquierdo MA, Lutz JR, et al. Clinical manifestations and immune response to tuberculosis. World J Microbiol Biotechnol. (2023) 39:206. doi: 10.1007/s11274-023-03636-x
3. Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. (2010) 8:668–74. doi: 10.1038/nrmicro2387
4. Ahmad F, Rani A, Alam A, Zarin S, Pandey S, Singh H, et al. Macrophage: A cell with many faces and functions in tuberculosis. Front Immunol. (2022) 13:747799. doi: 10.3389/fimmu.2022.747799
5. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
6. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
7. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
8. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. (2011) 477:592–5. doi: 10.1038/nature10394
9. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. (2011) 477:596–600. doi: 10.1038/nature10510
10. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
11. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. (2009) 458:509–13. doi: 10.1038/nature07710
12. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
13. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20(13):3328. doi: 10.3390/ijms20133328
14. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
15. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. (2010) 12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x
16. Wong KW, Jacobs WR Jr. Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol. (2011) 13:1371–84. doi: 10.1111/j.1462-5822.2011.01625.x
17. Theobald SJ, Grab J, Fritsch M, Suarez I, Eisfeld HS, Winter S, et al. Gasdermin D mediates host cell death but not interleukin-1beta secretion in Mycobacterium tuberculosis-infected macrophages. Cell Death Discovery. (2021) 7:327. doi: 10.1038/s41420-021-00716-5
18. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front Immunol. (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
19. Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol. (2012) 42:374–84. doi: 10.1002/eji.201141548
20. Basu S, Fowler BJ, Kerur N, Arnvig KB, Rao NA. NLRP3 inflammasome activation by mycobacterial ESAT-6 and dsRNA in intraocular tuberculosis. Microb Pathog. (2018) 114:219–24. doi: 10.1016/j.micpath.2017.11.044
21. Lee HM, Kang J, Lee SJ, Jo EK. Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia. (2013) 61:441–52. doi: 10.1002/glia.22448
22. Beckwith KS, Beckwith MS, Ullmann S, Saetra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. (2020) 11:2270. doi: 10.1038/s41467-020-16143-6
23. Gries R, Chhen J, van Gumpel E, Theobald SJ, Sonnenkalb L, Utpatel C, et al. Discovery of dual-active ethionamide boosters inhibiting the Mycobacterium tuberculosis ESX-1 secretion system. Cell Chem Biol. (2024) 31:699–711 e6. doi: 10.1016/j.chembiol.2023.12.007
24. Qu Z, Zhou J, Zhou Y, Xie Y, Jiang Y, Wu J, et al. Mycobacterial EST12 activates a RACK1-NLRP3-gasdermin D pyroptosis-IL-1beta immune pathway. Sci Adv. (2020) 6(43):eaba4733. doi: 10.1126/sciadv.aba4733
25. Chai Q, Yu S, Zhong Y, Lu Z, Qiu C, Yu Y, et al. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin. Science. (2022) 378:eabq0132. doi: 10.1126/science.abq0132
26. Rastogi S, Ellinwood S, Augenstreich J, Mayer-Barber KD, Briken V. Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase PknF. PloS Pathog. (2021) 17:e1009712. doi: 10.1371/journal.ppat.1009712
27. Zhang J, Cui Y, Zang X, Feng T, Chen F, Wang H, et al. PE12 interaction with TLR4 promotes intracellular survival of Mycobacterium tuberculosis by suppressing inflammatory response. Int J Biol Macromol. (2023) 253:127547. doi: 10.1016/j.ijbiomac.2023.127547
28. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PloS One. (2010) 5:e12320. doi: 10.1371/journal.pone.0012320
29. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. (2010) 184:3326–30. doi: 10.4049/jimmunol.0904189
30. Thomas SM, Olive AJ. High lethality of mycobacterium tuberculosis infection in mice lacking the phagocyte oxidase and caspase1/11. Infect Immun. (2023) 91:e0006023. doi: 10.1128/iai.00060-23
31. Stutz MD, Allison CC, Ojaimi S, Preston SP, Doerflinger M, Arandjelovic P, et al. Macrophage and neutrophil death programs differentially confer resistance to tuberculosis. Immunity. (2021) 54:1758–71 e7. doi: 10.1016/j.immuni.2021.06.009
32. Lovey A, Verma S, Kaipilyawar V, Ribeiro-Rodrigues R, Husain S, Palaci M, et al. Early alveolar macrophage response and IL-1R-dependent T cell priming determine transmissibility of Mycobacterium tuberculosis strains. Nat Commun. (2022) 13:884. doi: 10.1038/s41467-022-28506-2
33. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. (2011) 35:1023–34. doi: 10.1016/j.immuni.2011.12.002
34. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. doi: 10.1038/nature13489
35. Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol. (2012) 24:637–44. doi: 10.1093/intimm/dxs062
36. Yang Y, Zhou X, Kouadir M, Shi F, Ding T, Liu C, et al. the AIM2 inflammasome is involved in macrophage activation during infection with virulent Mycobacterium bovis strain. J Infect Dis. (2013) 208:1849–58. doi: 10.1093/infdis/jit347
37. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe. (2015) 17:799–810. doi: 10.1016/j.chom.2015.05.003
38. Yan S, Shen H, Lian Q, Jin W, Zhang R, Lin X, et al. Deficiency of the AIM2-ASC signal uncovers the STING-driven overreactive response of type I IFN and reciprocal depression of protective IFN-gamma immunity in mycobacterial infection. J Immunol. (2018) 200:1016–26. doi: 10.4049/jimmunol.1701177
39. Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, et al. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol. (2013) 191:3514–8. doi: 10.4049/jimmunol.1301331
40. Arnett E, Wolff J, Leopold Wager CM, Simper J, Badrak JL, Ontiveros CO, et al. Cutting Edge: Cytosolic Receptor AIM2 Is Induced by Peroxisome Proliferator-activated Receptor gamma following Mycobacterium tuberculosis Infection of Human Macrophages but Does Not Contribute to IL-1beta Release. J Immunol. (2024) 212:765–70. doi: 10.4049/jimmunol.2300418
41. Subbarao S, Sanchez-Garrido J, Krishnan N, Shenoy AR, Robertson BD. Genetic and pharmacological inhibition of inflammasomes reduces the survival of Mycobacterium tuberculosis strains in macrophages. Sci Rep. (2020) 10:3709. doi: 10.1038/s41598-020-60560-y
42. Souza De Lima D, Bomfim CCB, Leal VNC, Reis EC, Soares JLS, Fernandes FP, et al. Combining host genetics and functional analysis to depict inflammasome contribution in tuberculosis susceptibility and outcome in endemic areas. Front Immunol. (2020) 11:550624. doi: 10.3389/fimmu.2020.550624
43. Souza de Lima D, Ogusku MM, Sadahiro A, Pontillo A. Inflammasome genetics contributes to the development and control of active pulmonary tuberculosis. Infect Genet Evol. (2016) 41:240–4. doi: 10.1016/j.meegid.2016.04.015
44. Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol. (2016) 196:4052–63. doi: 10.4049/jimmunol.1502203
45. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med. (2018) 215:1273–85. doi: 10.1084/jem.20180325
46. Li H, Guan Y, Liang B, Ding P, Hou X, Wei W, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur J Pharmacol. (2022) 928:175091. doi: 10.1016/j.ejphar.2022.175091
Keywords: Mycobacterium tuberculosis, inflammasome, tuberculosis, drug resistance, interleukin-1, NLRP3 inflammasome, AIM2 inflammasome, gasdermin
Citation: Theobald SJ, Müller TA, Lange D, Keck K and Rybniker J (2024) The role of inflammasomes as central inflammatory hubs in Mycobacterium tuberculosis infection. Front. Immunol. 15:1436676. doi: 10.3389/fimmu.2024.1436676
Received: 22 May 2024; Accepted: 26 August 2024;
Published: 11 September 2024.
Edited by:
Bart Tummers, King’s College London, United KingdomReviewed by:
André Alves Dias, Oswaldo Cruz Foundation (Fiocruz), BrazilCopyright © 2024 Theobald, Müller, Lange, Keck and Rybniker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Rybniker, amFuLnJ5Ym5pa2VyQHVrLWtvZWxuLmRl
†These authors have contributed equally to this work