 Jeanette A. Maier1*
Jeanette A. Maier1* Sara Castiglioni1
Sara Castiglioni1 Alessandra Petrelli2
Alessandra Petrelli2 Rosanna Cannatelli3Francesca Ferretti3
Rosanna Cannatelli3Francesca Ferretti3 Greta Pellegrino4
Greta Pellegrino4 Piercarlo Sarzi Puttini1,4
Piercarlo Sarzi Puttini1,4 Paolo Fiorina1
Paolo Fiorina1 Sandro Ardizzone3
Sandro Ardizzone3- 1Department of Biomedical and Clinical Sciences, Università di Milano, Milano, Italy
- 2Department of Clinical Sciences and Community Health, University of Milan, Milano, Italy
- 3Gastroenterology Unit, ASST Fatebenefratelli-Sacco, Milano, Italy
- 4IRCCS Ospedale Galeazzi-Sant’Ambrogio, Milano, Italy
Patients with Immune-Mediated Inflammatory Diseases (IMIDs) are known to have an elevated risk of developing cancer, but the exact causative factors remain subject to ongoing debate. This narrative review aims to present the available evidence concerning the intricate relationship between these two conditions. Environmental influences and genetic predisposition lead to a dysregulated immune response resulting in chronic inflammation, which is crucial in the pathogenesis of IMIDs and oncogenic processes. Mechanisms such as the inflammatory microenvironment, aberrant intercellular communication due to abnormal cytokine levels, excessive reparative responses, and pathological angiogenesis are involved. The chronic immunosuppression resulting from IMIDs treatments further adds to the complexity of the pathogenic scenario. In conclusion, this review highlights critical gaps in the current literature, suggesting potential avenues for future research. The intricate interplay between IMIDs and cancer necessitates more investigation to deepen our understanding and improve patient management.
1 Introduction
The term Immune-Mediated Inflammatory Diseases (IMIDs) groups apparently unrelated multifactorial and polygenic diseases with multi-organ involvement, all sharing an aberrant, severe and continuous immune dysregulation associated with high levels of inflammatory cytokines (1). IMIDs affect a variety of organs and tissues, including the skin (e.g., psoriasis, atopic dermatitis), eyes (uveitis), joints [e.g., rheumatoid arthritis (RA)], internal lumens (e.g., inflammatory bowel disease (IBD), i.e. Crohn’s disease (CD) and ulcerative colitis (UC), asthma) (2), white and gray matter of the central nervous system [e.g., multiple sclerosis, neuromyelitis and autoimmune epilepsy (3)], and endocrine glands (e.g., type 1 diabetes (T1D), Addison’s disease). Some of these conditions arise from autoimmune mechanisms, while others result from hypersensitivity reactions. These aberrant immune responses lead to inflammation, tissue damage, and functional impairment of the involved organs or systems. IMIDs are prevalent in 5–7% of populations in developed Western countries, and, as globalization continues to expand, their occurrence is becoming more common in developing countries and among immigrant populations (4). IMIDs reduce the quality of life, are potentially disabling and represent an economic burden for health care systems. In addition, they increase the risk of developing cancer and the big question is why.
In this narrative review, we begin with a brief overview of the common etiological factors in IMIDs and cancer and then focus on the association between cancer and three common IMIDs, such as IBD, RA and T1D, and cancer. We performed a worldwide review of studies on IMIDs and cancer using three electronic medical databases, i.e. PubMed, EMBASE, and Web of Science. We selected the following keywords: “Immune-Mediated Inflammatory Diseases”, “IMIDs”, “IBD”, “RA”, “rheumatic diseases”, “T1D”, “cancer”, and “malignancy”. We included studies published in English, with available abstracts, and excluded case reports.
2 Common etiologic factors in IMIDs and cancer
Environmental factors play a role in the etiology of IMIDs in genetically predisposed individuals. Epidemiological studies have highlighted smoking, dietary habits, drugs, microbial dysbiosis, pollution and emotional stress (5) as common risk factors in the onset of IMIDs. Intriguingly, these environmental factors have a role also in tumorigenesis (6) (Figure 1).
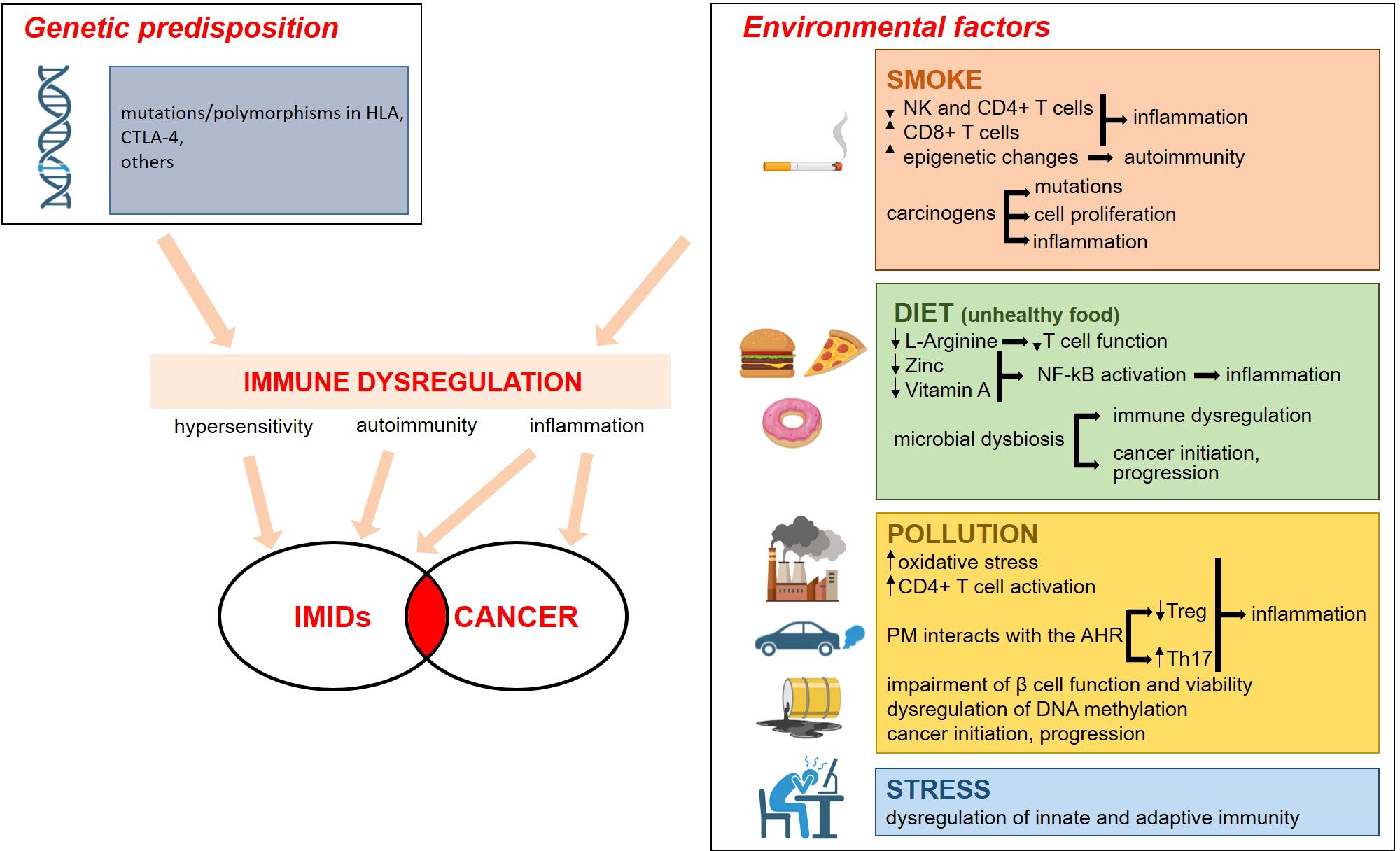
Figure 1. Genetic and environmental factors contribute to the etiopathogenesis of IMID. The environmental factors that promote IMIDs are the same as those that cause cancer. The boxes on the right summarize the mechanisms involved. Everything converges on a significant dysregulation of the immune system, with inflammation being a key event. For details see the text. Image created with BioRender.com.
A clear association has been reported between smoke and RA, CD, and psoriasis (6) mainly because of immune dysregulation. Indeed, active tobacco smoking impacts the immune system by decreasing circulating natural killer and CD4+ T cells and increasing CD8+ and CD8+ memory lymphocytes (7). Moreover, smoke endorses epigenetic changes to trigger the development of autoimmunity (8). The causal relation between smoke and cancer is well established, and is due to the fact that several of its components are carcinogens that cause permanent somatic mutations while others promote cell proliferation and/or prompt inflammation (9).
Processed foods, additives as well as deficiencies in micro- and macronutrients trigger inflammatory responses and disrupt immune response (10). For example, specific nutrients play key immunoregulatory roles. L-Arginine, for instance, acts as a critical nutrient and signaling molecule that shapes immune responses through the production of nitric oxide, T cell activation, immune cell proliferation, and modulation of immune suppression (11). Accordingly, L-Arginine depletion impairs anti-tumor immune responses, primarily by compromising T cell function (12). Additionally, zinc and vitamin A exhibit anti-inflammatory effects through the inhibition of the nuclear factor (NF)-kB pathway and also control the rate of antibody synthesis (13). Indeed, zinc and vitamin A depletion disrupts normal cellular processes, impairs immune surveillance, and promotes environments conducive to cancer development and progression. Nutrition also shapes the microbiota, the community of microorganisms that colonize the skin, the gut, and the respiratory mucosa (14). The microbiota influences tissue homeostasis and metabolism as well as the development of several immune cells including Th1, Th2, Th17, and immunosuppressive Treg cells (15). Not unexpectedly, gut microbiota is significantly different in IMIDs’ patients vs healthy individuals (16) and, while each specific disease has its own characteristic microbic signature, there are common dysbiotic alterations in different IMIDs (16). Considering that the microbiota also affects cell proliferation and death, it is not surprising that its composition in cancer-associated areas is different from the neighboring healthy tissue (17, 18). Accordingly, the microbiota is mechanistically involved in cancer initiation, progression, metastasis, and response to therapy (19). While some microorganisms produce genotoxins and reactive oxygen species, leading to oxidative stress and direct DNA damage (20), others release metabolites that promote tumor cell proliferation (21, 22). Moreover, dysbiosis disrupts the immune system balance by regulating immune cell activity, including T-cells and regulatory cells, thus impairing the body’s ability to fight malignancies (23). It is also relevant to highlight that some bacteria metabolize chemotherapeutic drugs, thus inducing resistance (24), and that dysbiosis impairs immunotherapy by creating an immunosuppressive environment (25). Therefore, studies should be encouraged to individuate microbial drivers in the progression from IMIDs to cancer.
Pollution is emerging as a novel player in IMIDs (26). Beyond inducing oxidative stress and long-term inflammation, pollutants dysregulate DNA methylation and Particulate Matter (PM) interacts with the Aryl hydrocarbon receptor (AHR) pathway, known to be involved in inflammatory processes and adaptive immune responses. In particular, the binding to AHR decreases Treg while augmenting Th17. Pollutants activate CD4+ T lymphocytes with the consequent increased production of pro-inflammatory cytokines, and also induce epigenetic modifications in T cells. For instance, the incidence of RA is higher in urban than in rural areas and this is due to high concentrations of PM2.5 and nitrogen dioxide (NO2) (27). Increased exposure to NO2, PM2.5 and ozone (O3) during childhood is also associated with increased risk of overall IBD (28). Persistent organic pollutants might play a role in T1D etiology, because they impair β cell function and viability (29).
Pollutants are also implicated in tumorigenesis. Some of them damage DNA (30), thereby activating oncogenic mutations, while others, such as PM2.5, promote cancer by inducing the expansion of cells with pre-existing oncogenic mutations (31). It is intriguing that lung malignancy is more frequent in patients with RA (32, 33), an issue that raises the question about the links between PM2.5 and the onset of the disease eventually complicated with the development of lung cancer.
Also chronic psychological stress accounts for a place in the pathogenesis of IMIDs (34–36), since it dysregulates innate and adaptive immune responses (37, 38). Moreover, the diagnosis of IMIDs engenders chronic psychological stress that, on one side, might generate mental health concerns (39), on the other one sustain chronic inflammation.
Both in IMIDs and in cancer, genetic predisposition is pivotal. Over the last twenty years, the genetic landscape of IMIDs has been intensely explored. The human leukocyte antigen (HLA) complex, located on chromosome 6, encodes proteins that play a crucial role in regulating the immune system by presenting antigens to T cells. Variations or polymorphisms in HLA genes are strongly associated with susceptibility to many IMIDs, because they influence how the immune system recognizes self versus non-self, leading to immune dysregulation and chronic inflammation in IMIDs. Moreover, hundreds of non-HLA genetic variants have been unraveled (40). For example, the locus containing T lymphocyte-associated antigen (CTLA)-4 is associated with many IMIDs (41). Accordingly, targeting the CTLA-4 pathway, often used in tumor immunotherapy, leads to multi-organ autoimmune reactions (42, 43). Genome-wide association studies have demonstrated a remarkable overlap in the loci predisposing to IMIDs (44, 45). It is known that, differently from autoantibody-negative IMIDs, such as psoriasis and CD, autoantibody-positive IMIDs, among which RA, strictly cluster with each other, as demonstrated by the evidence that, among the 150 genetic loci associated with RA, only a few are specific to the disease (46). All this knowledge has disclosed novel pathways implicated in the pathogenesis of IMIDs and has pinpointed the involvement of genes that might offer insights into the higher risk of developing cancer in IMIDs patients.
3 Immune dysregulation in IMIDs: an overview
Innate and adaptive immune dysregulation, driven by environmental factors in genetically predisposed individuals, is central to generate cytokine dysregulation, the decisive event in the pathophysiology of IMIDs. Cytokine signature hubs have been described in single IMIDs (2). However, an appraisal of the distinct mechanisms involved in each individual disease is beyond the scope of this article. Our aim is to provide a synthetic overview on the common aspects of the complex immune dysregulation occurring in IMIDs and the connections with cancer.
In addition to the classical pro-inflammatory cytokines interleukin (IL)-1, IL-6 and Tumor Necrosis Factor (TNF)α, whose role in IMIDs has been amply described (47), the IL-23/17 axis is emerging as a common feature in several IMIDs among which IBD, psoriasis, uveitis, psoriatic arthritis and evidence are accumulating about its role in RA (48, 49). Upon exposure to a pro-inflammatory milieu, IL-23 is synthesized by several types of cells, including dendritic cells and macrophages, and acts on IL-23 responsive cells, which include neutrophils, natural killer lymphocytes, mast cells, macrophages, memory T cells, all localized at the barrier surface, and also cells involved in transmitting biomechanical forces (50–52). IL-23 promotes the release of IL-17, which induces pro-inflammatory mediators and cooperates with other molecules in triggering and chronicizing inflammation (53). By transcriptional and post-transcriptional regulation, IL-17 stimulates the release of TNFα, a downstream effector common to many IMIDs (2), IL-1, IL-6, IL-8 and other cytokines and chemokines, thus unbalancing the complex communication network that, tightly tuned (54) in physiological conditions, is radically deregulated in IMIDs. As an example, IL-1 acts synergistically with IL-23 to perpetuate the continuous high production of IL-17 (55) and, consequently, chronic inflammation (55). Therefore, one would expect the inhibition of IL-17 to be a success, but this is true in psoriasis and ankylosing spondylitis, and not in CD (56, 57) where, paradoxically, the clinical course is aggravated. Similarly, in spite of the fact that TNFα is upregulated in most IMIDs, not all the patients respond to anti-TNFα therapy, and many of the initially responders lose response over time (58). It is likely that anti-TNFα antibodies promote a change in innate and immune cell infiltrates so that TNFα independent inflammatory pathways emerge and keep the disease active.
It is worth mentioning that chemokines are important actors in several IMIDs, from RA (59) to IBD (60), from LSE (61) to psoriasis (62) and T1D (63), as they recruit immune cells into the tissues and regulate their reciprocal interactions. Of note, the chemokine/chemokine receptor axis is also implicated in tumorigenesis, because it controls cell proliferation, stemness, survival and neovascularization, and contributes to the generation of an immunosuppressive tumor microenvironment (64).
Dysfunction of immune checkpoints in IMIDs is beginning to draw some attention. An imbalance between co-stimulators, such as CD28 and CD40, and co-inhibitors, among which CTLA-4 and programmed cell death (PD)-1, contributes to immune deregulation and inflammation (65), as demonstrated by immune-related adverse effects, which include IBD and dermatitis (66), experienced by individuals receiving checkpoint inhibitors to treat malignancies (67). CTLA-4, expressed by activated and regulatory T lymphocytes, has a relevant role in maintaining immune homeostasis. Abatacept, a fusion protein consisting of the extracellular domain of CTLA-4 and a genetically engineered fragment of the Fc region of human immunoglobulin G1 (IgG1), is efficacious in RA, because it inhibits the co-stimulation of T cells (68). In T1D it modifies the pattern of immune cells and enhances insulin secretion, but it does not delay the progression to glucose intolerance (69). In IBD (70) and psoriasis (71), abatacept is not effective, thus highlighting on one side the complexity of approaching IMIDs, on the other the current gaps in our knowledge. PD-1 is another immune checkpoint receptor which is expressed predominantly by T lymphocytes (72). When PD-1 interacts with its ligands PD-L1 or PD-L2, it elicits an inhibitory response by targeting T cell receptor signaling (73). Since many malignant cells overexpress the PD-1 and its ligands, this pathway is a target for immunotherapy. PD-1 is upregulated in peripheral T lymphocytes in RA (74) and in the professional immune cells of the lamina propria in IBD (75). However, a reduced binding of PD-1 by PD-L1 may down-regulate pathogenic immune responses. Indeed, a phase 2 trial in patients with RA has recently shown that the PD-1 agonist monoclonal antibody peresolimab is safe and improves the clinical course of the disease (76).
4 Chronic inflammation in IMIDs and cancer
The connection between IMIDs and some cancers is well known, but the mechanisms involved remain unclear and are often controversial. As mentioned above, infection, diet, environment are common to IMIDs and cancer, and lead to chronic inflammation that plays the lion’s share in the development of malignancies.
A link between inflammation and cancer has been appreciated for a long time, since when Rudolf Virchow wrote that “chronic irritation and inflammatory hyperplasia predispose to cancer development” (77). Nowadays, prolonged inflammation is considered one of the hallmarks of cancer (78). An inflammatory microenvironment can contribute to tumorigenesis by increasing oxidative stress, which damages DNA, by activating prosurvival pathways and promoting growth, migration, invasion of tumor cells, and also angiogenesis, thereby supporting tumor progression locally and at metastatic sites (79–81).
The altered intercellular communication due to the upheaval of the cytokine network has a prominent role in the progression toward neoplasia. Inflammatory cytokines activate the transcription factor NF-kB, which on one side fuels cytokine production, on the other inhibits epithelial apoptosis. They also induce another transcription factor, i.e. STAT3, which not only contributes to the maintenance of an inflammatory environment but also acts on the epithelium stimulating growth and protecting from apoptosis (82). In addition to the prototypical inflammatory cytokines such as TNFα, IL-1s and IL-6 whose role in cancer has been largely described, novel players are entering the scene. The IL-23/17 pathway, which is implicated in several IMIDs, not only promotes and maintains inflammation, but also weakens the barrier function of the skin, gut and lung, and reduces CD8+ lymphocyte antitumor immunosurveillance, both factors that contribute to cancerogenesis (53). A seminal finding is that IL-17A is necessary and sufficient to activate the hypoxia inducible factor (HIF)1α (83), thus demonstrating the coupling of inflammatory, metabolic, and migratory programs as well as angiogenesis (84), all events clearly involved in cancer. IL-17 also stimulates epithelial stem cell proliferation after injuring the tissue with a carcinogenic agent (85) and the inhibition of IL-17 prevents colon cancer in an experimental murine model of colitis (86).
Attention has been devoted also to IL-36, a member of the IL-1 superfamily, which is upregulated in the synovium of patients with RA, in psoriatic skin, in the mucosa of patients with IBD, in the sera of patients with SLE (87–89). Its role in cancer is controversial, as it displays both anti and pro tumor properties depending on the type of neoplasm and its level of expression (90). IL-36 markedly increases and exerts pro-tumorigenic effects in lung and colorectal cancers (91, 92). Of interest, colon cancer cells without the IL-36 receptor grow slower and express lower amounts of Ki-67 than controls (91). A recent study shows that increased IL-36 expression is associated with a decrease of 5 year survival rates in colon cancer patients (92).
5 Common cues in IMIDs and cancer
While in the early stages of cancer the immune system identifies and controls the tumor cells, in the later stages anti-tumor immune cells are corrupted into tumor-promoting immune cells that sustain survival, growth, invasiveness of tumor cells and generate chronic inflammation. In the end, tumor cells escape immune surveillance through the activation of various anti-detection pathways (93). These events are orchestrated by the cytokine storm generated within the tumor microenvironment (94–96). Among the various mechanisms of evasion of anticancer immunity, a light is shed on myeloid-derived suppressor cells (MDSC), which derive from neutrophils and monocytes in response to high levels of inflammatory cytokines (66, 97). MDSC are potent inhibitors of immune responses mediated by natural killer cells, B and T lymphocytes, thus facilitating the escape of tumor cells. The number of MDSC rises in various pathological conditions, such as cancer, inflammation, and transplantation (98, 99). Notably, MDSC accumulate in the lesions occurring in IMIDs and their number is proportional to the severity of the disease (100). The number and activity of MDSC are increased in the blood of T1D, RA and IBD patients (101–103). To the best of our knowledge, there are no studies correlating MDCS, IMIDs and cancer risk, albeit this issue is very challenging and deserves further investigations. The link between autoimmunity and neoplasia is further supported by increased risk of lymphoma and gastric cancer in individuals with mutated CTLA-4 and, consequently, with dysregulated immune responses (104).
It should also be underscored that tissue damage in IMIDs triggers a reparative response that represents a double edged sword. Whereas the primary aim is to heal the injured tissue, an exuberant and unleashed production of growth factors together with the myriad of inflammatory mediators overstimulates cell proliferation, bolsters transformation, and promotes the development of dysplasia that can progress into malignancy (105). Also stromal cells play a role in the attempt of healing the tissue and, eventually, in the onset of cancer. Fibroblasts and myofibroblasts deposit collagen, fibronectin, laminin and continuously remodel the extracellular matrix by releasing proteases in IMIDs and, even more, in cancer (106).
Another common event in IMIDs and cancer is pathological angiogenesis. Inflammation and cancer share molecules that support the formation of a new vascular network, such as prostaglandins, cytokines, chemokines and growth factors, with a prominent role of the members of the vascular endothelial growth factor (VEGF) family (107), secreted by platelets, activated T lymphocytes, neutrophils, macrophages, dendritic cells and tumor cells.
The inflammatory environment suffices to explain the strong association existing between organ-specific immune-mediated diseases and the risk of local cancers, in agreement with the idea of tumors as wound that do not heal (108). However, IMIDs moderately increase oncologic risk also in distant organs and in different systems (see below). Again, lifelong immune dysregulation and altered cytokine profile are likely to be implicated.
Another interesting, albeit overlooked, issue is that cytokines also activate the hypothalamic–pituitary–adrenal axis (109), significantly shaping immune function and consequently inhibiting antitumor immune responses. This effect is further magnified by the common anxiety or depression experienced by patients with IMIDs, who must live with a chronic, recurrent, and disabling disease (110).
6 IMIDs and cancer: the example of IBD
It is well known that IBD predispose to intestinal cancers, a finding that does not surprise because the persistent activation of the transcription factors NF-kB and STAT3 in the lesions fuels inflammation and upregulates genes implicated in tumor cell survival, proliferation and invasion. Intestinal inflammation can also affect the brain through the brain-gut axis, resulting in the activation the hypothalamic–pituitary–adrenal system, which impairs the antitumor immune defenses and promotes cancer occurrence. Indeed, malignancies are the second most common cause of death in IBD patients after cardiovascular diseases both in male and female (111).
Long-standing UC and CD colitis cause an approximately 2–3-fold increased risk of colorectal cancer (112). Typically, neoplasms develop from dysplasia originating on inflamed areas, in the sequence inflammation-dysplasia-adenocarcinoma differently from the adenoma-carcinoma sequence described in sporadic colon cancer (Figure 2). Accordingly, dysplasia is the most reliable marker of increased risk of colon cancer in IBD (113). Of note, mutant cells bearing genomic and epigenomic alterations are detectable even before the onset of dysplasia. Apart from the low rate of KRAS mutations, in IBD associated colon cancer driver genes are the same as in sporadic colon cancer, but the timing of the mutations is different. P53 mutation or silencing occurs very early in the process, eventually before the onset of dysplasia, whereas APC is mutated or lost later and less frequently than in sporadic colon cancer (114). It is noteworthy that genome wide studies demonstrate an important increase in mutations in the 5’ untranslated region of p53 in IBD associated colon cancer (115). The same study individuates as a unique feature in IBD colon cancer the hypermethylation and consequent loss of function of the polymeric immunoglobulin receptor (PIGR), which is responsible for the transport of IgA and IgM through the epithelium (116). This event can be interpreted as a loss of the epithelial properties of tumor cell, as further supported by the downregulation of genes promoting epithelial differentiation. In parallel, genes involved in modeling the extracellular matrix are upregulated. All together these alterations facilitate the acquisition of a mesenchymal colon cancer subtype. This subtype is linked to drug resistance (117), reduced survival and is characterized by the presence of a high number of Treg, indicating that the microenvironment is highly immunosuppressive (118). In the case of colon cancer arising in patients with UC, a systems biology approach (119) revealed the upregulation of two chemokines, namely CXCL1 and IL-8, the matrix metalloproteinase (MMP)-7, the serine protein urokinase-type plasminogen activator, the tissue inhibitor of metalloproteinase (TIMP)-1, and the solute carrier 16 member 9 (SLC16A9) which transports monocarboxylic acids. This molecular signature is proposed as specific for colon cancer in UC and further corroborates the dominant role of inflammation and matrix remodeling pathways.
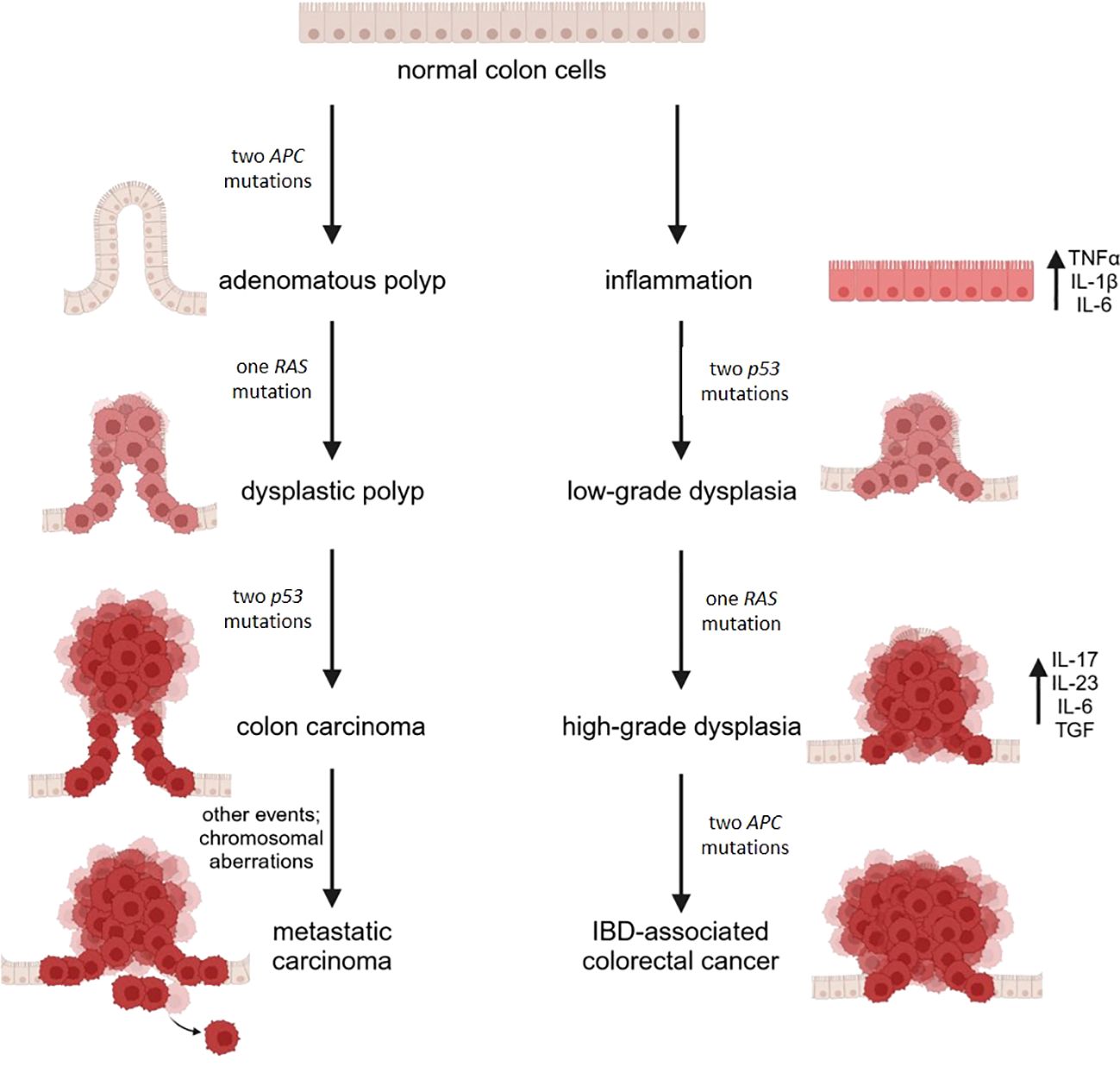
Figure 2. A schematic diagram illustrating the molecular mechanisms in sporadic (CRC) and IBD-related colorectal cancer. CRC arises from the accumulation of mutations in oncogenes and tumor suppressor genes, driving the progression from single preneoplastic cells to adenoma and ultimately to carcinoma. In contrast, IBD-related colorectal cancer is driven by chronic inflammation, which leads to the production of proinflammatory cytokines and the buildup of reactive oxygen and nitrogen species, promoting genomic aberrations and instability. This process results in mutations in oncogenes and tumor suppressor genes. Mutations in p53 lead to low-grade dysplastic mucosa, with subsequent mutations in KRAS contributing to the progression from low-grade to high-grade dysplasia. Finally, mutations in the APC gene culminate in the development of cancer. Image created with BioRender.com.
CD is also associated with a high risk of developing small bowel adenocarcinoma (120), which occurs mainly in young adults and has a poor prognosis (121). KRAS mutation and p53 loss of function are common characteristics of sporadic and CD related small bowel cancer, whereas p16 positivity, the nuclear accumulation of β-catenin and mutations of isocitrate dehydrogenase (IDH)-1 seem to be specific in CD small bowel cancer (122).
Although much rarer, intestinal B cell lymphoma, which is uncommon in the general population, is a dreadful complication in IBD patients, mainly in male older than 65, after a mean average of 12 years from the diagnosis (123). Chronic inflammation, therapy with the immunosuppressant thiopurine and Epstein Barr virus positive lymphocytes are the main driving factors for uncontrolled B cell proliferation ultimately progressing to lymphoma.
Patients with long lasting IBD have a slightly increased risk of extralocal malignancies (124), among which cholangiocarcinoma (125, 126). It frequently develops in the context of primary sclerosing cholangitis, a chronic inflammatory disease, according to the inflammation-dysplasia-cancer sequence. Even though the pathogenic mechanisms remain unclear, it is reasonable to propose the involvement of altered microbiota and metabolism in the inflamed gut, along with an altered bile acid profile due to impairment of the enterohepatic axis, as described in IBD (125). Moreover, a recent metanalysis of cohort studies confirms previous reports about the increased risk of prostate cancer in IBD, in particular in European UC patients (127). In addition to the tumor promoting role of the aberrant microbiome in UC, chronic intestinal inflammation fosters genetic instability and the upregulation of pro-cancer signaling pathways in the prostate (128).
7 IMIDs and cancer: the example of rheumatic and musculoskeletal diseases
The association between rheumatic and musculoskeletal diseases (RMDs) and neoplasm is a dynamic and continuously evolving field of scientific research. While certain RMDs, such as RA, SLE, Systemic Sclerosis (SSc), Sjögren’s syndrome, and inflammatory myopathies are recognized to have a heightened association with an increased risk of cancer (129), the underlying mechanisms remain multifaceted. Undoubtedly, a significant factor contributing to the increased cancer risk is the presence of chronic inflammation and associated tissue damage (130). For instance, the risk of lymphoma in patients with Sjogren’s syndrome is known to correlate with disease activity and severity (131). Similarly, in the case of RA, elevated markers of inflammation, such as erythrocyte sedimentation rate and C-reactive protein, have been associated with an increased risk of neoplastic disease (129, 132).
Another hypothesis suggests that natural immune responses against cancer cells may trigger autoimmunity and rheumatic disease (133). This theory is supported by cases where cancer rapidly develops in patients with dermatomyositis and SSc, often occurring within three years of the onset of autoimmune disease. Moreover, the presence of specific autoantibodies, such as anti-RNA polymerase III and anti-RNA-binding region-containing protein 3 (RNPC3) in SSc (134, 135), or anti- transcription intermediary factor 1 (TIF1)-γ and anti-nuclear matrix protein 2 (NXP2) in dermatomyositis, could help to stratify patients at higher risk of cancer, as a strong association has been demonstrated (136, 137). In addition to inflammation and immune response, the inability to clear viral infection, as observed in SLE, can also elevate the risk of certain cancer. For example, the higher susceptibility of SLE patients to human papillomavirus infection is believed to contribute the increased risk of cervical cancer in this population (138).
Sjögren’s syndrome is primarily associated with an elevated risk of developing lymphoma, notably non-Hodgkin lymphoma (with a prevalence of around 5%). The risk of lymphoma is estimated to be 5-10 times greater than that of the general population (139). However, a recent meta-analysis by Zongh et al. showed that patients with Sjögren’s syndrome also have an increased risk of solid tumors, such as lung, thyroid, and non-melanoma skin cancers (140).
Individuals diagnosed with inflammatory myopathies have long been recognized as having a higher likelihood of developing specific cancer types, with adenocarcinoma being the predominant histological tumor type (141, 142). The period of greatest cancer susceptibility occurs within three to five years before and after the diagnosis of myositis, and cancer risk appears to be contingent on the specific subtype of inflammatory myopathy. Individuals with dermatomyositis exhibit a 5.5-fold increased cancer risk, while those with polymyositis display a 1.6-fold elevation (142–144).
SSc is characterized by an increased age- and sex-adjusted risk of developing cancer, often ranging from 1.5 to 4 times higher than that of the general population (145, 146). The relationship between SSc and cancer risk is thought to be related to the damage caused by SSc in various body sites, potentially predisposing individuals to malignant transformation. This may explain why esophageal and lung cancers are more frequently observed in these patients, given the association of gastroesophageal reflux and interstitial lung disease with the pathology (147, 148).
There is evidence suggesting association between SLE and an increased susceptibility to certain malignancies (149, 150). Epidemiological studies have indicated that individuals with SLE face a moderately elevated risk of cancer, particularly hematological malignancies such as non-Hodgkin lymphoma (150). Additionally, a higher prevalence of cervical dysplasia and cervical cancer has been observed in women with SLE (151).
A recent meta-analysis indicated that individuals with RA may have a slightly increased risk of cancer (152). In particular, lymphoma and lung cancer are the most commonly observed types of neoplasm in this group of patients (153). This elevated risk is believed to be influenced by shared risk factors. such as smoking, in addition to the mechanism of chronic, persistent inflammation (154, 155).
In conclusion, the intricate relationship between neoplastic diseases and RMDs is an ongoing subject of scientific investigation. This complex connection highlights the need for more comprehensive data and further research to elucidate the underlying biological mechanisms. A deeper understanding of these mechanisms is essential for improving the management and care of patients with both RMDs and cancer.
8 IMIDs and cancer: the example of type 1 diabetes
Type 1 diabetes (T1D) is a chronic autoimmune condition characterized by the destruction of insulin-producing β cells in the pancreas (156). Emerging evidence suggests a link between T1D and cancer risk, but the relationship is multifaceted and not fully understood. Numerous studies have investigated the standardized mortality ratio for cancers among patients with T1D compared to the general population. These reports have yielded conflicting results (157–159), often due to limitations in statistical power, which affect the precision of risk estimates for specific cancer types.
In 2016, Carstensen et al. conducted an extensive study analyzing cancer incidence in individuals with T1D using population-based registries in five countries (160). Their findings revealed that hazard ratios (HRs) for all cancers combined were slightly elevated in both men (HR 1.01) and women (HR 1.07) with T1D. Notably, elevated HRs were observed for cancers of the liver, pancreas, and kidney. Conversely, prostate cancer (HR 0.56) and breast cancer (HR 0.90) exhibited reduced risks in men and women with T1D, respectively. Interestingly, the risk of some cancers in individuals with T1D appears to resemble that in people with type 2 diabetes. Factors such as high blood sugar levels may contribute to the elevated cancer risk in both types of diabetes. Additionally, emerging contributors, such as obesity and insulin resistance - conditions for which growing evidence indicates increased incidence in symptomatic and pre-symptomatic T1D individuals (161–163) - may also play a significant role in cancer development.
An intriguing explanation of the link between T1D and cancers lies in the relationship between daily insulin dose and cancer risk. A recent study revealed that higher daily insulin doses are associated with an increased risk of cancer, even after adjusting for age and sex (164). Both in vitro and in vivo studies have highlighted the pivotal role of insulin and the insulin receptor in cancer biology (165). Hyperinsulinemic states contribute to increased hepatic insulin-like growth factor (IGF)-1 production through the upregulation of the growth hormone receptor (GHR) and enhanced GHR signaling (166), demonstrating the potential to induce cancer cell proliferation and their capacity to spread to secondary sites (167). However, epidemiological data on the link between disease duration and cancer in T1D are inconsistent, with some studies indicating that cancer risk is highest at the time of diabetes diagnosis and decreases over time (160), while others report cancer development in patients with a mean diabetes duration of 25 years (164).
Hyperglycemia promotes tumorigenesis through the “Warburg effect,” which involves increased glucose uptake by cancer cells to fuel their proliferation (168, 169). This can contribute to the cancer predisposition associated with diabetes (170). Furthermore, hyperglycemia stimulates the production of advanced glycation end products (AGEs), which interact with their receptor, RAGE, to activate NF-kB and generate reactive oxygen species (171). This cascade accelerates oxidative stress, leading to increased proinflammatory signaling and potentially promoting transformation (172, 173).
In conclusion, the relationship between T1D and cancer risk is intricate and influenced by various factors, including insulin dose, disease duration, and the complex interplay of metabolic pathways. As research continues, a deeper understanding of these mechanisms may shed light on strategies for cancer prevention and improved care for individuals with T1D.
9 IMIDs therapeutics: a role in cancer?
The relationship between immunosuppressive drugs and risk of malignancies has been widely explored, but the results are still conflicting (174).
Historically, immunomodulators such as thiopurines and methotrexate were the milestone of treatment of IMIDs. In this setting, the longstanding experience in transplanted patients showed an increased risk of skin cancer such non-melanoma skin cancers (NMSC), and lymphoproliferative diseases associated to Epstein-Barr virus infection (175). In the past, the large use of azathioprine and 6-mercaptopurine in IBD patients confirmed this association (176–178), and in 2009 the large prospective study by Beaugerie et al. (176) found that IBD patients receiving thiopurines showed an hazard ratio (HR) of 5.28 (2.01-13.9, p=0.0007) of developing lymphoproliferative disorders compared to other IBD patients.
Data on methotrexate are controversial: a systematic review in 2010 found an increased risk of melanoma in RA and NMSC in patients with psoriasis (179), but other studies did not confirm it (180, 181). Recent evidence suggests a higher rate of NMSC associated with the use of methotrexate, demonstrating a dose-response pattern (182). Similarly, cyclosporine can also increase the risk of skin cancer in patients with psoriasis (183). Studies in IBD patients are not available; however, effects are likely to be the same.
On one hand, these drugs may promote the development of malignancies through direct DNA modifications and by altering immunosurveillance of tumor cells or mutagenic viruses (184–186). On the other hand, controlling inflammation with these drugs is one of the primary strategies for cancer prevention in some gastrointestinal malignancies among IBD patients (187).
In the late 1990s, the introduction of biologic therapies revolutionized treatment approaches. Initially, their ‘targeted’ effects were considered to ensure safety, and short-term cancer risks were thought to be minimal. However, long-term effects were unpredictable at that time and remain controversial today. Early studies on the association between anti-TNFα therapies and malignancies reported a possible link to higher rates of lymphoma and melanoma (188, 189). However, more recent studies on anti-TNFα therapies appear to exclude a link with increased risk of cancer in RA (190), psoriasis (191), and IBD (192).
According to the phase 2/3 studies, the use of selective agents such as the anti-integrin vedolizumab and the anti IL-12/23 ustekinumab does not carry any risk of cancer development (193, 194) and they are considered safe in patients with a prior history of cancer (195, 196). Regarding antibodies targeting IL-23, risankizumab modestly increased oncologic events, with a clear prevalence in men (197), while cancer occurred in a small number of patients treated with mirikizumab (198). Due to the quite recent introduction of these drugs, long-term effects are still unknown.
Recently, small molecules such the Janus kinase (JAK) inhibitor tofacitinib were approved as therapeutic option in IMIDs patients (199). Clinical trials showed a higher incidence of malignancies compared to anti-TNFα or general population, especially in RA, not confirmed in real life studies and in clinical practice (200, 201). An increased short term risk for NMSC was described in patients with RA or psoriatic arthritis initiating treatment with the JAK inhibitors tofacitinib, baricitinib (202), filgotinib (203) and upadacitinib (204). This latter molecule increases the risk of NMSC in a dose dependent manner (204).
Thus, as available data are still limited and controversial, more long-term studies are needed to confirm this association.
10 Gaps and future directions in research on IMIDs and cancer
In spite of the significant advances in our understanding of the link between IMIDs and cancer, there is still a long way to go for researchers and clinicians.
Studies should be fostered to highlight whether sex or ethnicity impact the progression from IMIDs to local and distant malignancies. It is also important to identify molecular markers involved in this progression, as recently demonstrated by a system biology approach in UC patients [108]. These markers can function as prognostic tools and therapeutic targets. It is clear that intercellular communication goes awry in IMIDs and until now attention has been devoted mainly to soluble molecules and far less to exosomes. These 30-150 nm sized vesicles are released by many cells and contain lipids, proteins and nucleic acids (including non coding RNAs) that can be delivered locally as well as to distant districts. They regulate immune system, remodel the extracellular matrix and other crucial biological processes (205). Exosomes can also promote chronic inflammation, facilitate immune evasion, and contribute to tumor progression (206). While their role in cancer is well established, very little is known about potential alterations of exosome characteristics in IMIDs. Therefore, it would be relevant to individuate differences in exosomes between IMIDs patients and the general population, and to investigate whether their cargo changes when malignancies arise.
Interesting insights may emerge from studies on organoids derived from biopsies or induced pluripotent stem cells of IMIDs patients. Organoids are three-dimensional, cell-based in vitro models that replicate the complex structure and function of tissues (207). They are useful to answer fundamental questions about disease modeling, gene expression, drug response (208) with the final aim of personalizing medical approaches. Organoids have been successfully developed from pluripotent stem cells of UC patients and shown to recap colitic reactivity (209), thus underscoring that this approach may advance diagnostics and therapy at the individual level. The question is: can organoids be helpful to predict the potential progression in neoplasia?
Author contributions
JM: Conceptualization, Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. PF: Writing – original draft, Writing – review & editing. AP: Writing – original draft, Writing – review & editing. RC: Writing – original draft, Writing – review & editing. FF: Writing – original draft, Writing – review & editing. GP: Writing – original draft, Writing – review & editing. PP: Writing – original draft, Writing – review & editing. SA: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors acknowledge the support of the APC central fund of the University of Milan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Monteleone G, Moscardelli A, Colella A, Marafini I, Salvatori S. Immune-mediated inflammatory diseases: Common and different pathogenic and clinical features. Autoimmun Rev. (2023) 22:103410. doi: 10.1016/j.autrev.2023.103410
2. Schett G, McInnes IB, Neurath MF. Reframing immune-mediated inflammatory diseases through signature cytokine hubs. N Engl J Med. (2021) 385:628–39. doi: 10.1056/NEJMra1909094
3. Bhagavati S. Autoimmune disorders of the nervous system: pathophysiology, clinical features, and therapy. Front Neurol. (2021) 12:664664. doi: 10.3389/fneur.2021.664664
4. Agrawal M, Shah S, Patel A, Pinotti R, Colombel J-F, Burisch J. Changing epidemiology of immune-mediated inflammatory diseases in immigrants: A systematic review of population-based studies. J Autoimmun. (2019) 105:102303. doi: 10.1016/j.jaut.2019.07.002
5. Rahman P, Inman RD, El-Gabalawy H, Krause DO. Pathophysiology and pathogenesis of immune-mediated inflammatory diseases: commonalities and differences. J Rheumatol Suppl. (2010) 85:11–26. doi: 10.3899/jrheum.091462
6. GBD 2019 Cancer Risk Factors Collaborators. The global burden of cancer attributable to risk factors, 2010-19: a systematic analysis for the Global Burden of Disease Study 2019. Lancet (London England). (2022) 400:563–91. doi: 10.1016/S0140-6736(22)01438-6
7. Piaggeschi G, Rolla S, Rossi N, Brusa D, Naccarati A, Couvreur S, et al. Immune trait shifts in association with tobacco smoking: A study in healthy women. Front Immunol. (2021) 12:637974. doi: 10.3389/fimmu.2021.637974
8. Perricone C, Versini M, Ben-Ami D, Gertel S, Watad A, Segel MJ, et al. Smoke and autoimmunity: The fire behind the disease. Autoimmun Rev. (2016) 15:354–74. doi: 10.1016/j.autrev.2016.01.001
9. Hecht SS. Lung carcinogenesis by tobacco smoke. Int J Cancer. (2012) 131:2724–32. doi: 10.1002/ijc.27816
10. Whelan K, Bancil AS, Lindsay JO, Chassaing B. Ultra-processed foods and food additives in gut health and disease. Nat Rev Gastroenterol Hepatol. (2024) 21:406–27. doi: 10.1038/s41575-024-00893-5
11. Martí I Líndez A-A, Reith W. Arginine-dependent immune responses. Cell Mol Life Sci. (2021) 78:5303–24. doi: 10.1007/s00018-021-03828-4
12. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
13. Munteanu C, Schwartz B. The relationship between nutrition and the immune system. Front Nutr. (2022) 9:1082500. doi: 10.3389/fnut.2022.1082500
14. Leeming ER, Johnson AJ, Spector TD, Le Roy CI. Effect of diet on the gut microbiota: rethinking intervention duration. Nutrients. (2019) 11:2862. doi: 10.3390/nu11122862
15. Lee N, Kim W-U. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med. (2017) 49:e340. doi: 10.1038/emm.2017.36
16. Forbes JD, Chen C-Y, Knox NC, Marrie R-A, El-Gabalawy H, de Kievit T, et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome. (2018) 6:221. doi: 10.1186/s40168-018-0603-4
17. Hou K, Wu Z-X, Chen X-Y, Wang J-Q, Zhang D, Xiao C, et al. Microbiota in health and diseases. Signal Transduct Target Ther. (2022) 7:135. doi: 10.1038/s41392-022-00974-4
18. Khan AA, Sirsat AT, Singh H, Cash P. Microbiota and cancer: current understanding and mechanistic implications. Clin Transl Oncol Off Publ Fed Spanish Oncol Soc Natl Cancer Inst Mex. (2022) 24:193–202. doi: 10.1007/s12094-021-02690-x
19. Ivleva EA, Grivennikov SI. Microbiota-driven mechanisms at different stages of cancer development. Neoplasia. (2022) 32:100829. doi: 10.1016/j.neo.2022.100829
20. Lopez LR, Bleich RM, Arthur JC. Microbiota effects on carcinogenesis: initiation, promotion, and progression. Annu Rev Med. (2021) 72:243–61. doi: 10.1146/annurev-med-080719-091604
21. Charles A, Thomas RM. The Influence of the microbiome on the innate immune microenvironment of solid tumors. Neoplasia. (2023) 37:100878. doi: 10.1016/j.neo.2023.100878
22. DiPalma MP, Blattman JN. The impact of microbiome dysbiosis on T cell function within the tumor microenvironment (TME). Front Cell Dev Biol. (2023) 11:1141215. doi: 10.3389/fcell.2023.1141215
23. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
24. Li S, Zhu S, Yu J. The role of gut microbiota and metabolites in cancer chemotherapy. J Adv Res. (2023) 26:S2090–1232(23)00366–1. doi: 10.1016/j.jare.2023.11.027
25. Guo C, Kong L, Xiao L, Liu K, Cui H, Xin Q, et al. The impact of the gut microbiome on tumor immunotherapy: from mechanism to application strategies. Cell Biosci. (2023) 13:188. doi: 10.1186/s13578-023-01135-y
26. Zhao C-N, Xu Z, Wu G-C, Mao Y-M, Liu L-N, Qian-Wu, et al. Emerging role of air pollution in autoimmune diseases. Autoimmun Rev. (2019) 18:607–14. doi: 10.1016/j.autrev.2018.12.010
27. Bernatsky S, Smargiassi A, Joseph L, Awadalla P, Colmegna I, Hudson M, et al. Industrial air emissions, and proximity to major industrial emitters, are associated with anti-citrullinated protein antibodies. Environ Res. (2017) 157:60–3. doi: 10.1016/j.envres.2017.04.035
28. Elten M, Benchimol EI, Fell DB, Kuenzig ME, Smith G, Chen H, et al. Ambient air pollution and the risk of pediatric-onset inflammatory bowel disease: A population-based cohort study. Environ Int. (2020) 138:105676. doi: 10.1016/j.envint.2020.105676
29. Bresson SE, Isom S, Jensen ET, Huber S, Oulhote Y, Rigdon J, et al. Associations between persistent organic pollutants and type 1 diabetes in youth. Environ Int. (2022) 163:107175. doi: 10.1016/j.envint.2022.107175
30. Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A compendium of mutational signatures of environmental agents. Cell. (2019) 177:821–836.e16. doi: 10.1016/j.cell.2019.03.001
31. Hill W, Lim EL, Weeden CE, Lee C, Augustine M, Chen K, et al. Lung adenocarcinoma promotion by air pollutants. Nature. (2023) 616:159–67. doi: 10.1038/s41586-023-05874-3
32. Chatzidionysiou K, di Giuseppe D, Soderling J, Catrina A, Askling J. Risk of lung cancer in rheumatoid arthritis and in relation to autoantibody positivity and smoking. RMD Open. (2022) 8:e002465 doi: 10.1136/rmdopen-2022-002465
33. Fragoulis GE, Chatzidionysiou K. Lung cancer in rheumatoid arthritis. Is there a need for better risk assessment and screening? Clin Rheumatol. (2020) 39:957–61. doi: 10.1007/s10067-019-04882-x
34. Brzozowski B, Mazur-Bialy A, Pajdo R, Kwiecien S, Bilski J, Zwolinska-Wcislo M, et al. Mechanisms by which stress affects the experimental and clinical inflammatory bowel disease (IBD): role of brain-gut axis. Curr Neuropharmacol. (2016) 14:892–900. doi: 10.2174/1570159x14666160404124127
35. Ortega MA, García-Montero C, Fraile-Martinez O, Alvarez-Mon MA, Gómez-Lahoz AM, Lahera G, et al. Immune-mediated diseases from the point of view of psychoneuroimmunoendocrinology. Biol (Basel). (2022) 11:973. doi: 10.3390/biology11070973
36. Rampton DS. The influence of stress on the development and severity of immune-mediated diseases. J Rheumatol Suppl. (2011) 88:43–7. doi: 10.3899/jrheum.110904
37. Dhabhar FS. Effects of stress on immune function: the good, the bad, and the beautiful. Immunol Res. (2014) 58:193–210. doi: 10.1007/s12026-014-8517-0
38. Liu Y-Z, Wang Y-X, Jiang C-L. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. (2017) 11:316. doi: 10.3389/fnhum.2017.00316
39. Blaney C, Sommer J, El-Gabalawy R, Bernstein C, Walld R, Hitchon C, et al. Incidence and temporal trends of co-occurring personality disorder diagnoses in immune-mediated inflammatory diseases. Epidemiol Psychiatr Sci. (2020) 29:e84. doi: 10.1017/S2045796019000854
40. Wu X, Chen H, Xu H. The genomic landscape of human immune-mediated diseases. J Hum Genet. (2015) 60:675–81. doi: 10.1038/jhg.2015.99
41. Demela P, Pirastu N, Soskic B. Cross-disorder genetic analysis of immune diseases reveals distinct gene associations that converge on common pathways. Nat Commun. (2023) 14:2743. doi: 10.1038/s41467-023-38389-6
42. Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. (2003) 349:1907–15. doi: 10.1056/NEJMoa035075
43. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033
44. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. (2016) 48:510–8. doi: 10.1038/ng.3528
45. Saurabh R, Fouodo CJK, König IR, Busch H, Wohlers I. A survey of genome-wide association studies, polygenic scores and UK Biobank highlights resources for autoimmune disease genetics. Front Immunol. (2022) 13:972107. doi: 10.3389/fimmu.2022.972107
46. Padyukov L. Genetics of rheumatoid arthritis. Semin Immunopathol. (2022) 44:47–62. doi: 10.1007/s00281-022-00912-0
47. Möller B, Villiger PM. Inhibition of IL-1, IL-6, and TNF-alpha in immune-mediated inflammatory diseases. Springer Semin Immunopathol. (2006) 27:391–408. doi: 10.1007/s00281-006-0012-9
48. Schinocca C, Rizzo C, Fasano S, Grasso G, La Barbera L, Ciccia F, et al. Role of the IL-23/IL-17 pathway in rheumatic diseases: an overview. Front Immunol. (2021) 12:637829. doi: 10.3389/fimmu.2021.637829
49. Yuan N, Yu G, Liu D, Wang X, Zhao L. An emerging role of interleukin-23 in rheumatoid arthritis. Immunopharmacol Immunotoxicol. (2019) 41:185–91. doi: 10.1080/08923973.2019.1610429
50. Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. (2006) 203:2577–87. doi: 10.1084/jem.20060244
51. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
52. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. (2006) 116:1310–6. doi: 10.1172/JCI21404
53. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol. (2019) 20:1594–602. doi: 10.1038/s41590-019-0514-y
54. Jansen JE, Aschenbrenner D, Uhlig HH, Coles MC, Gaffney EA. A method for the inference of cytokine interaction networks. PloS Comput Biol. (2022) 18:e1010112. doi: 10.1371/journal.pcbi.1010112
55. Dayer J-M, Oliviero F, Punzi L. A brief history of IL-1 and IL-1 ra in rheumatology. Front Pharmacol. (2017) 8:293. doi: 10.3389/fphar.2017.00293
56. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet (London England). (2013) 382:1705–13. doi: 10.1016/S0140-6736(13)61134-4
57. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PDR, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
58. Roda G, Jharap B, Neeraj N, Colombel J-F. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol. (2016) 7:e135. doi: 10.1038/ctg.2015.63
59. Elemam NM, Hannawi S, Maghazachi AA. Role of chemokines and chemokine receptors in rheumatoid arthritis. ImmunoTargets Ther. (2020) 9:43–56. doi: 10.2147/ITT.S243636
60. Camba-Gómez M, Arosa L, Gualillo O, Conde-Aranda J. Chemokines and chemokine receptors in inflammatory bowel disease: Recent findings and future perspectives. Drug Discovery Today. (2022) 27:1167–75. doi: 10.1016/j.drudis.2021.12.004
61. Zeng Y, Lin Q, Yu L, Wang X, Lin Y, Zhang Y, et al. Chemokine CXCL1 as a potential marker of disease activity in systemic lupus erythematosus. BMC Immunol. (2021) 22:82. doi: 10.1186/s12865-021-00469-x
62. Purzycka-Bohdan D, Nedoszytko B, Zabłotna M, Gleń J, Szczerkowska-Dobosz A, Nowicki RJ. Chemokine profile in psoriasis patients in correlation with disease severity and pruritus. Int J Mol Sci. (2022) 23:13330. doi: 10.3390/ijms232113330
63. Pan X, Kaminga AC, Kinra S, Wen SW, Liu H, Tan X, et al. Chemokines in type 1 diabetes mellitus. Front Immunol. (2021) 12:690082. doi: 10.3389/fimmu.2021.690082
64. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
65. Liu S, Xu J, Wu J. The role of co-signaling molecules in psoriasis and their implications for targeted treatment. Front Pharmacol. (2021) 12:717042. doi: 10.3389/fphar.2021.717042
66. Matza Porges S, Shamriz O. Genetics of immune dysregulation and cancer predisposition: two sides of the same coin. Clin Exp Immunol. (2022) 210:114–27. doi: 10.1093/cei/uxac089
67. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
68. Herrero-Beaumont G, Martínez Calatrava MJ, Castañeda S. Abatacept mechanism of action: concordance with its clinical profile. Reumatol Clin. (2012) 8:78–83. doi: 10.1016/j.reuma.2011.08.002
69. Russell WE, Bundy BN, Anderson MS, Cooney LA, Gitelman SE, Goland RS, et al. Abatacept for delay of type 1 diabetes progression in stage 1 relatives at risk: A randomized, double-masked, controlled trial. Diabetes Care. (2023) 46:1005–13. doi: 10.2337/dc22-2200
70. Sandborn WJ, Colombel J-F, Sands BE, Rutgeerts P, Targan SR, Panaccione R, et al. Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology. (2012) 143:62–69.e4. doi: 10.1053/j.gastro.2012.04.010
71. Harris KM, Smilek DE, Byron M, Lim N, Barry WT, McNamara J, et al. Effect of costimulatory blockade with abatacept after ustekinumab withdrawal in patients with moderate to severe plaque psoriasis: the PAUSE randomized clinical trial. JAMA Dermatol. (2021) 157:1306–15. doi: 10.1001/jamadermatol.2021.3492
72. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. (2020) 6:eabd2712. doi: 10.1126/sciadv.abd2712
73. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki I-M, et al. PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol. (2019) 10:630. doi: 10.3389/fimmu.2019.00630
74. Luo Q, Ye J, Zeng L, Luo Z, Deng Z, Li X, et al. Elevated expression of PD−1 on T cells correlates with disease activity in rheumatoid arthritis. Mol Med Rep. (2018) 17:3297–305. doi: 10.3892/mmr.2017.8204
75. Chulkina M, Beswick EJ, Pinchuk IV. Role of PD-L1 in gut mucosa tolerance and chronic inflammation. Int J Mol Sci. (2020) 21:9165. doi: 10.3390/ijms21239165
76. Tuttle J, Drescher E, Simón-Campos JA, Emery P, Greenwald M, Kivitz A, et al. A phase 2 trial of peresolimab for adults with rheumatoid arthritis. N Engl J Med. (2023) 388:1853–62. doi: 10.1056/NEJMoa2209856
77. Virchow R. Aetiologie der neoplastischen Geschwülste. In: Die krankhaften Geschwülste: Erster Band: Dreissig Vorlesungen, gehalten während des Wintersemesters 1862–1863 an der Universität zu Berlin. Springer Berlin Heidelberg, Berlin, Heidelberg (1978). p. 57–71. doi: 10.1007/978-3-642-66491-5_4
78. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
79. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
80. Piotrowski I, Kulcenty K, Suchorska W. Interplay between inflammation and cancer. Rep Pract Oncol Radiother J Gt Cancer Cent Pozn Polish Soc Radiat Oncol. (2020) 25:422–7. doi: 10.1016/j.rpor.2020.04.004
81. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5
82. Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to inflammatory and fibrotic diseases and prospects for its targeting for treatment. Int J Mol Sci. (2018) 19:2299. doi: 10.3390/ijms19082299
83. Konieczny P, Xing Y, Sidhu I, Subudhi I, Mansfield KP, Hsieh B, et al. Interleukin-17 governs hypoxic adaptation of injured epithelium. Science. (2022) 377:eabg9302. doi: 10.1126/science.abg9302
84. Majumder S, McGeachy MJ. IL-17 in the pathogenesis of disease: good intentions gone awry. Annu Rev Immunol. (2021) 39:537–56. doi: 10.1146/annurev-immunol-101819-092536
85. Chen X, Cai G, Liu C, Zhao J, Gu C, Wu L, et al. IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1(+) stem cells. J Exp Med. (2019) 216:195–214. doi: 10.1084/jem.20171849
86. Zepp JA, Zhao J, Liu C, Bulek K, Wu L, Chen X, et al. IL-17A-induced PLET1 expression contributes to tissue repair and colon tumorigenesis. J Immunol. (2017) 199:3849–57. doi: 10.4049/jimmunol.1601540
87. Boutet M-A, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, et al. Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol. (2016) 184:159–73. doi: 10.1111/cei.12761
88. Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, et al. IL-36α expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. (2016) 9:1193–204. doi: 10.1038/mi.2015.134
89. Wang X-R, Xiao J-P, Wang D-G. Elevated levels of serum IL-36α in patients with systemic lupus erythematosus. BioMed Rep. (2021) 15:76. doi: 10.3892/br.2021.1452
90. Byrne J, Baker K, Houston A, Brint E. IL-36 cytokines in inflammatory and Malignant diseases: not the new kid on the block anymore. Cell Mol Life Sci. (2021) 78:6215–27. doi: 10.1007/s00018-021-03909-4
91. Baker K, O’Donnell C, Bendix M, Keogh S, Byrne J, O’Riordain M, et al. IL-36 signalling enhances a pro-tumorigenic phenotype in colon cancer cells with cancer cell growth restricted by administration of the IL-36R antagonist. Oncogene. (2022) 41:2672–84. doi: 10.1038/s41388-022-02281-2
92. Baker KJ, Brint E, Houston A. Transcriptomic and functional analyses reveal a tumour-promoting role for the IL-36 receptor in colon cancer and crosstalk between IL-36 signalling and the IL-17/IL-23 axis. Br J Cancer. (2023) 128:735–47. doi: 10.1038/s41416-022-02083-z
93. Kim SK, Cho SW. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharmacol. (2022) 13:868695. doi: 10.3389/fphar.2022.868695
94. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
95. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. (2021) 21:345–59. doi: 10.1038/s41568-021-00347-z
96. Kartikasari AER, Huertas CS, Mitchell A, Plebanski M. Tumor-induced inflammatory cytokines and the emerging diagnostic devices for cancer detection and prognosis. Front Oncol. (2021) 11:692142. doi: 10.3389/fonc.2021.692142
97. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
98. Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol. (2012) 3:208. doi: 10.3389/fimmu.2012.00208
99. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1004983. doi: 10.1080/2162402X.2015.1004983
100. Wegner A, Verhagen J, Wraith DC. Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology. (2017) 151:26–42. doi: 10.1111/imm.12718
101. Grohová A, Dáňová K, Adkins I, Šumník Z, Petruželková L, Obermannová B, et al. Myeloid - derived suppressor cells in Type 1 diabetes are an expanded population exhibiting diverse T-cell suppressor mechanisms. PloS One. (2020) 15:e0242092. doi: 10.1371/journal.pone.0242092
102. Guo C, Hu F, Yi H, Feng Z, Li C, Shi L, et al. Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann Rheum Dis. (2016) 75:278–85. doi: 10.1136/annrheumdis-2014-205508
103. Haile LA, von Wasielewski R, Gamrekelashvili J, Krüger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology. (2008) 135:871–81, 881.e1–5. doi: 10.1053/j.gastro.2008.06.032
104. Li CM, Chen Z. Autoimmunity as an etiological factor of cancer: the transformative potential of chronic type 2 inflammation. Front Cell Dev Biol. (2021) 9:664305. doi: 10.3389/fcell.2021.664305
105. Svrcek M, Piton G, Cosnes J, Beaugerie L, Vermeire S, Geboes K, et al. Small bowel adenocarcinomas complicating Crohn’s disease are associated with dysplasia: a pathological and molecular study. Inflammation Bowel Dis. (2014) 20:1584–92. doi: 10.1097/MIB.0000000000000112
106. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. (2015) 33:230–6. doi: 10.1016/j.tibtech.2015.01.004
107. Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, Lopez-Camarillo C, Hernadez de la Cruz ON, Lopez-Gonzalez JS. Contribution of angiogenesis to inflammation and cancer. Front Oncol. (2019) 9:1399. doi: 10.3389/fonc.2019.01399
108. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
109. Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. (2000) 917:608–17. doi: 10.1111/j.1749-6632.2000.tb05426.x
110. Marrie RA, Bernstein CN. Psychiatric comorbidity in immune-mediated inflammatory diseases. World Psychiatry. (2021) 20:298–9. doi: 10.1002/wps.20873
111. Nieminen U, Färkkilä M. Malignancies in inflammatory bowel disease. Scand J Gastroenterol. (2015) 50:81–9. doi: 10.3109/00365521.2014.992041
112. Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. (2022) 162:715–730.e3. doi: 10.1053/j.gastro.2021.10.035
113. Goldman H. Significance and detection of dysplasia in chronic colitis. Cancer. (1996) 78:2261–3. doi: 10.1002/(sici)1097-0142(19961201)78:11<2261::aid-cncr1>3.0.co;2-q
114. Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. (2015) 372:1441–52. doi: 10.1056/NEJMra1403718
115. Rajamäki K, Taira A, Katainen R, Välimäki N, Kuosmanen A, Plaketti R-M, et al. Genetic and epigenetic characteristics of inflammatory bowel disease-associated colorectal cancer. Gastroenterology. (2021) 161:592–607. doi: 10.1053/j.gastro.2021.04.042
116. Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. (2005) 206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x
117. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. (2017) 17:79–92. doi: 10.1038/nrc.2016.126
118. Picard E, Verschoor CP, Ma GW, Pawelec G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol. (2020) 11:369. doi: 10.3389/fimmu.2020.00369
119. Shahnazari M, Afshar S, Emami MH, Amini R, Jalali A. Novel biomarkers for neoplastic progression from ulcerative colitis to colorectal cancer: a systems biology approach. Sci Rep. (2023) 13:3413. doi: 10.1038/s41598-023-29344-y
120. Yu J, Refsum E, Perrin V, Helsingen LM, Wieszczy P, Løberg M, et al. Inflammatory bowel disease and risk of adenocarcinoma and neuroendocrine tumors in the small bowel. Ann Oncol Off J Eur Soc Med Oncol. (2022) 33:649–56. doi: 10.1016/j.annonc.2022.02.226
121. Palascak-Juif V, Bouvier AM, Cosnes J, Flourié B, Bouché O, Cadiot G, et al. Small bowel adenocarcinoma in patients with Crohn’s disease compared with small bowel adenocarcinoma de novo. Inflammation Bowel Dis. (2005) 11:828–32. doi: 10.1097/01.mib.0000179211.03650.b6
122. Liao X, Li G, McBride R, Houldsworth J, Harpaz N, Polydorides AD. Clinicopathological and molecular characterisation of Crohn’s disease-associated small bowel adenocarcinomas. J Crohns Colitis. (2020) 14:287–94. doi: 10.1093/ecco-jcc/jjz135
123. Muller M, Broséus J, Feugier P, Thieblemont C, Beaugerie L, Danese S, et al. Characteristics of lymphoma in patients with inflammatory bowel disease: A systematic review. J Crohns Colitis. (2021) 15:827–39. doi: 10.1093/ecco-jcc/jjaa193
124. Chang M, Chang L, Chang HM, Chang F. Intestinal and extraintestinal cancers associated with inflammatory bowel disease. Clin Colorectal Cancer. (2018) 17:e29–37. doi: 10.1016/j.clcc.2017.06.009
125. Gulamhusein AF, Eaton JE, Tabibian JH, Atkinson EJ, Juran BD, Lazaridis KN. Duration of inflammatory bowel disease is associated with increased risk of cholangiocarcinoma in patients with primary sclerosing cholangitis and IBD. Am J Gastroenterol. (2016) 111:705–11. doi: 10.1038/ajg.2016.55
126. Yadlapati S. Cholangiocarcinoma surveillance in patients with Inflammatory Bowel Disease and Primary Sclerosing Cholangitis. Adv Res Gastroenterol Hepatol. (2021) 17. doi: 10.19080/argh.2021.17.555954
127. Zhou B-G, Yu Q, Jiang X, Mei Y-Z, Ding Y-B, Wang M. Association between inflammatory bowel disease and risk of incident prostate cancer: a systematic review and meta-analysis of cohort studies. Int J Colorectal Dis. (2023) 38:168. doi: 10.1007/s00384-023-04465-y
128. Desai AS, Sagar V, Lysy B, Weiner AB, Ko OS, Driscoll C, et al. Inflammatory bowel disease induces inflammatory and pre-neoplastic changes in the prostate. Prostate Cancer Prostatic Dis. (2022) 25:463–71. doi: 10.1038/s41391-021-00392-7
129. Cappelli LC, Shah AA. The relationships between cancer and autoimmune rheumatic diseases. Best Pract Res Clin Rheumatol. (2020) 34:101472. doi: 10.1016/j.berh.2019.101472
130. Egiziano G, Bernatsky S, Shah AA. Cancer and autoimmunity: Harnessing longitudinal cohorts to probe the link. Best Pract Res Clin Rheumatol. (2016) 30:53–62. doi: 10.1016/j.berh.2016.03.001
131. Baldini C, Pepe P, Luciano N, Ferro F, Talarico R, Grossi S, et al. A clinical prediction rule for lymphoma development in primary Sjögren’s syndrome. J Rheumatol. (2012) 39:804–8. doi: 10.3899/jrheum.110754
132. Llorca J, Lopez-Diaz MJ, Gonzalez-Juanatey C, Ollier WER, Martin J, Gonzalez-Gay MA. Persistent chronic inflammation contributes to the development of cancer in patients with rheumatoid arthritis from a defined population of northwestern Spain. Semin Arthritis Rheum. (2007) 37:31–8. doi: 10.1016/j.semarthrit.2007.01.002
133. Shah AA, Casciola-Rosen L, Rosen A. Review: cancer-induced autoimmunity in the rheumatic diseases. Arthritis Rheumatol (Hoboken NJ). (2015) 67:317–26. doi: 10.1002/art.38928
134. Igusa T, Hummers LK, Visvanathan K, Richardson C, Wigley FM, Casciola-Rosen L, et al. Autoantibodies and scleroderma phenotype define subgroups at high-risk and low-risk for cancer. Ann Rheum Dis. (2018) 77:1179–86. doi: 10.1136/annrheumdis-2018-212999
135. Shah AA, Xu G, Rosen A, Hummers LK, Wigley FM, Elledge SJ, et al. Brief report: anti-RNPC-3 antibodies as a marker of cancer-associated scleroderma. Arthritis Rheumatol (Hoboken NJ). (2017) 69:1306–12. doi: 10.1002/art.40065
136. Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. (2013) 65:2954–62. doi: 10.1002/art.38093
137. Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, Balada E, Martínez MA, Milisenda JC, et al. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatol (Oxford). (2018) 57:388–96. doi: 10.1093/rheumatology/kex413
138. Santana IU, Gomes A do N, Lyrio LD, Rios Grassi MF, Santiago MB. Systemic lupus erythematosus, human papillomavirus infection, cervical pre-malignant and Malignant lesions: a systematic review. Clin Rheumatol. (2011) 30:665–72. doi: 10.1007/s10067-010-1606-0
139. Stefanski A-L, Tomiak C, Pleyer U, Dietrich T, Burmester GR, Dörner T. The diagnosis and treatment of Sjögren’s syndrome. Dtsch Arztebl Int. (2017) 114:354–61. doi: 10.3238/arztebl.2017.0354
140. Zhong H, Liu S, Wang Y, Xu D, Li M, Zhao Y, et al. Primary Sjögren’s syndrome is associated with increased risk of Malignancies besides lymphoma: A systematic review and meta-analysis. Autoimmun Rev. (2022) 21:103084. doi: 10.1016/j.autrev.2022.103084
141. Chen Y-J, Wu C-Y, Huang Y-L, Wang C-B, Shen J-L, Chang Y-T. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. (2010) 12:R70. doi: 10.1186/ar2987
142. Sigurgeirsson B, Lindelöf B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. (1992) 326:363–7. doi: 10.1056/NEJM199202063260602
143. Oldroyd A, Sergeant JC, New P, McHugh NJ, Betteridge Z, Lamb JA, et al. The temporal relationship between cancer and adult onset anti-transcriptional intermediary factor 1 antibody-positive dermatomyositis. Rheumatol (Oxford). (2019) 58:650–5. doi: 10.1093/rheumatology/key357
144. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and Malignancy risk: a metaanalysis study. J Rheumatol. (2015) 42:282–91. doi: 10.3899/jrheum.140566
145. Hill CL, Nguyen A-M, Roder D, Roberts-Thomson P. Risk of cancer in patients with scleroderma: a population based cohort study. Ann Rheum Dis. (2003) 62:728–31. doi: 10.1136/ard.62.8.728
146. Onishi A, Sugiyama D, Kumagai S, Morinobu A. Cancer incidence in systemic sclerosis: meta-analysis of population-based cohort studies. Arthritis Rheum. (2013) 65:1913–21. doi: 10.1002/art.37969
147. Abu-Shakra M, Guillemin F, Lee P. Cancer in systemic sclerosis. Arthritis Rheum. (1993) 36:460–4. doi: 10.1002/art.1780360405
148. Colaci M, Giuggioli D, Sebastiani M, Manfredi A, Vacchi C, Spagnolo P, et al. Lung cancer in scleroderma: results from an Italian rheumatologic center and review of the literature. Autoimmun Rev. (2013) 12:374–9. doi: 10.1016/j.autrev.2012.06.003
149. Song L, Wang Y, Zhang J, Song N, Xu X, Lu Y. The risks of cancer development in systemic lupus erythematosus (SLE) patients: a systematic review and meta-analysis. Arthritis Res Ther. (2018) 20:270. doi: 10.1186/s13075-018-1760-3
150. Zhang M, Wang Y, Wang Y, Bai Y, Gu D. Association between systemic lupus erythematosus and cancer morbidity and mortality: findings from cohort studies. Front Oncol. (2022) 12:860794. doi: 10.3389/fonc.2022.860794
151. Wadström H, Arkema EV, Sjöwall C, Askling J, Simard JF. Cervical neoplasia in systemic lupus erythematosus: a nationwide study. Rheumatol (Oxford). (2017) 56:613–9. doi: 10.1093/rheumatology/kew459
152. Simon TA, Thompson A, Gandhi KK, Hochberg MC, Suissa S. Incidence of Malignancy in adult patients with rheumatoid arthritis: a meta-analysis. Arthritis Res Ther. (2015) 17:212. doi: 10.1186/s13075-015-0728-9
153. De Cock D, Hyrich K. Malignancy and rheumatoid arthritis: Epidemiology, risk factors and management. Best Pract Res Clin Rheumatol. (2018) 32:869–86. doi: 10.1016/j.berh.2019.03.011
154. Coussens LM, Werb Z. Inflammation and cancer. Nature. (2002) 420:860–7. doi: 10.1038/nature01322
155. Sugiyama D, Nishimura K, Tamaki K, Tsuji G, Nakazawa T, Morinobu A, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. (2010) 69:70–81. doi: 10.1136/ard.2008.096487
156. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
157. Dawson SI, Willis J, Florkowski CM, Scott RS. Cause-specific mortality in insulin-treated diabetic patients: a 20-year follow-up. Diabetes Res Clin Pract. (2008) 80:16–23. doi: 10.1016/j.diabres.2007.10.034
158. Gordon-Dseagu VLZ, Shelton N, Mindell JS. Epidemiological evidence of a relationship between type-1 diabetes mellitus and cancer: a review of the existing literature. Int J Cancer. (2013) 132:501–8. doi: 10.1002/ijc.27703
159. Secrest AM, Becker DJ, Kelsey SF, Laporte RE, Orchard TJ. Cause-specific mortality trends in a large population-based cohort with long-standing childhood-onset type 1 diabetes. Diabetes. (2010) 59:3216–22. doi: 10.2337/db10-0862
160. Carstensen B, Read SH, Friis S, Sund R, Keskimäki I, Svensson A-M, et al. Cancer incidence in persons with type 1 diabetes: a five-country study of 9,000 cancers in type 1 diabetic individuals. Diabetologia. (2016) 59:980–8. doi: 10.1007/s00125-016-3884-9
161. Buzzetti R, Zampetti S, Pozzilli P. Impact of obesity on the increasing incidence of type 1 diabetes. Diabetes Obes Metab. (2020) 22:1009–13. doi: 10.1111/dom.14022
162. Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. (2004) 47:1661–7. doi: 10.1007/s00125-004-1507-3
163. Petrelli A, Giovenzana A, Insalaco V, Phillips BE, Pietropaolo M, Giannoukakis N. Autoimmune inflammation and insulin resistance: hallmarks so far and yet so close to explain diabetes endotypes. Curr Diabetes Rep. (2021) 21:54. doi: 10.1007/s11892-021-01430-3
164. Zhong W, Mao Y. Daily insulin dose and cancer risk among patients with type 1 diabetes. JAMA Oncol. (2022) 8:1356–8. doi: 10.1001/jamaoncol.2022.2960
165. Baxter RC, Bryson JM, Turtle JR. Somatogenic receptors of rat liver: regulation by insulin. Endocrinology. (1980) 107:1176–81. doi: 10.1210/endo-107-4-1176
166. Chen W, Wang S, Tian T, Bai J, Hu Z, Xu Y, et al. Phenotypes and genotypes of insulin-like growth factor 1, IGF-binding protein-3 and cancer risk: evidence from 96 studies. Eur J Hum Genet. (2009) 17:1668–75. doi: 10.1038/ejhg.2009.86
167. Yakar S, Leroith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: Lessons from animal models. Cytokine Growth Factor Rev. (2005) 16:407–20. doi: 10.1016/j.cytogfr.2005.01.010
168. Devic S. Warburg effect - a consequence or the cause of carcinogenesis? J Cancer. (2016) 7:817–22. doi: 10.7150/jca.14274
169. WARBURG O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
170. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. (2008) 8:915–28. doi: 10.1038/nrc2536
171. Abe R, Yamagishi S. AGE-RAGE system and carcinogenesis. Curr Pharm Des. (2008) 14:940–5. doi: 10.2174/138161208784139765
172. Kuniyasu H, Oue N, Wakikawa A, Shigeishi H, Matsutani N, Kuraoka K, et al. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J Pathol. (2002) 196:163–70. doi: 10.1002/path.1031
173. Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. (2009) 7:17. doi: 10.1186/1479-5876-7-17
174. Ferretti F, Cannatelli R, Maconi G, Ardizzone S. Therapeutic management of adults with inflammatory bowel disease and Malignancies: A clinical challenge. Cancers (Basel). (2023) 15:542. doi: 10.3390/cancers15020542
175. Martinez OM, de Gruijl FR. Molecular and immunologic mechanisms of cancer pathogenesis in solid organ transplant recipients. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. (2008) 8:2205–11. doi: 10.1111/j.1600-6143.2008.02368.x
176. Beaugerie L, Brousse N, Bouvier AM, Colombel JF, Lémann M, Cosnes J, et al. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet (London England). (2009) 374:1617–25. doi: 10.1016/S0140-6736(09)61302-7
177. Kandiel A, Fraser AG, Korelitz BI, Brensinger C, Lewis JD. Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut. (2005) 54:1121–5. doi: 10.1136/gut.2004.049460
178. Setshedi M, Epstein D, Winter TA, Myer L, Watermeyer G, Hift R. Use of thiopurines in the treatment of inflammatory bowel disease is associated with an increased risk of non-melanoma skin cancer in an at-risk population: a cohort study. J Gastroenterol Hepatol. (2012) 27:385–9. doi: 10.1111/j.1440-1746.2011.06865.x
179. Krathen MS, Gottlieb AB, Mease PJ. Pharmacologic immunomodulation and cutaneous Malignancy in rheumatoid arthritis, psoriasis, and psoriatic arthritis. J Rheumatol. (2010) 37:2205–15. doi: 10.3899/jrheum.100041
180. Buchbinder R, Barber M, Heuzenroeder L, Wluka AE, Giles G, Hall S, et al. Incidence of melanoma and other Malignancies among rheumatoid arthritis patients treated with methotrexate. Arthritis Rheum. (2008) 59:794–9. doi: 10.1002/art.23716
181. Salliot C, van der Heijde D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis. (2009) 68:1100–4. doi: 10.1136/ard.2008.093690
182. Polesie S, Gillstedt M, Schmidt SAJ, Egeberg A, Pottegård A, Kristensen K. Use of methotrexate and risk of skin cancer: a nationwide case-control study. Br J Cancer. (2023) 128:1311–9. doi: 10.1038/s41416-023-02172-7
183. Paul CF, Ho VC, McGeown C, Christophers E, Schmidtmann B, Guillaume J-C, et al. Risk of Malignancies in psoriasis patients treated with cyclosporine: a 5 y cohort study. J Invest Dermatol. (2003) 120:211–6. doi: 10.1046/j.1523-1747.2003.12040.x
184. Beaugerie L. Management of inflammatory bowel disease patients with a cancer history. Curr Drug Targets. (2014) 15:1042–8. doi: 10.2174/1389450115666140821113330
185. Münz C, Moormann A. Immune escape by Epstein-Barr virus associated Malignancies. Semin Cancer Biol. (2008) 18:381–7. doi: 10.1016/j.semcancer.2008.10.002
186. O’Donovan P, Perrett CM, Zhang X, Montaner B, Xu Y-Z, Harwood CA, et al. Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science. (2005) 309:1871–4. doi: 10.1126/science.1114233
187. Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. (2016) 22:4794–801. doi: 10.3748/wjg.v22.i20.4794
188. Narous M, Nugent Z, Singh H, Bernstein CN. Risks of melanoma and nonmelanoma skin cancers pre- and post-inflammatory bowel disease diagnosis. Inflammation Bowel Dis. (2023) 29:1047–56. doi: 10.1093/ibd/izac171
189. Chupin A, Perduca V, Meyer A, Bellanger C, Carbonnel F, Dong C. Systematic review with meta-analysis: comparative risk of lymphoma with anti-tumour necrosis factor agents and/or thiopurines in patients with inflammatory bowel disease. Aliment Pharmacol Ther. (2020) 52:1289–97. doi: 10.1111/apt.16050
190. Huss V, Bower H, Wadström H, Frisell T, Askling J. Short- and longer-term cancer risks with biologic and targeted synthetic disease-modifying antirheumatic drugs as used against rheumatoid arthritis in clinical practice. Rheumatol (Oxford). (2022) 61:1810–8. doi: 10.1093/rheumatology/keab570
191. Vaengebjerg S, Skov L, Egeberg A, Loft ND. Prevalence, incidence, and risk of cancer in patients with psoriasis and psoriatic arthritis: A systematic review and meta-analysis. JAMA Dermatol. (2020) 156:421–9. doi: 10.1001/jamadermatol.2020.0024
192. Muller M, D’Amico F, Bonovas S, Danese S, Peyrin-Biroulet L. TNF inhibitors and risk of Malignancy in patients with inflammatory bowel diseases: A systematic review. J Crohns Colitis. (2021) 15:840–59. doi: 10.1093/ecco-jcc/jjaa186
193. Sandborn WJ, Feagan BG, Danese S, O’Brien CD, Ott E, Marano C, et al. Safety of ustekinumab in inflammatory bowel disease: pooled safety analysis of results from phase 2/3 studies. Inflammation Bowel Dis. (2021) 27:994–1007. doi: 10.1093/ibd/izaa236
194. Colombel J-F, Sands BE, Rutgeerts P, Sandborn W, Danese S, D’Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut. (2017) 66:839–51. doi: 10.1136/gutjnl-2015-311079
195. Hasan B, Tandon KS, Miret R, Khan S, Riaz A, Gonzalez A, et al. Ustekinumab does not increase risk of new or recurrent cancer in inflammatory bowel disease patients with prior Malignancy. J Gastroenterol Hepatol. (2022) 37:1016–21. doi: 10.1111/jgh.15806
196. Hong SJ, Zenger C, Pecoriello J, Pang A, Vallely M, Hudesman DP, et al. Ustekinumab and vedolizumab are not associated with subsequent cancer in IBD patients with prior Malignancy. Inflammation Bowel Dis. (2022) 28:1826–32. doi: 10.1093/ibd/izac035
197. Calapai F, Mannucci C, Cardia L, Currò M, Calapai G, Esposito E, et al. Suspected oncologic adverse reactions associated with interleukin-23 inhibitors in EudraVigilance: Comparative study and gender distribution. Pharmacol Res Perspect. (2023) 11:e01130. doi: 10.1002/prp2.1130
198. D’Haens G, Dubinsky M, Kobayashi T, Irving PM, Howaldt S, Pokrotnieks J, et al. Mirikizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. (2023) 388:2444–55. doi: 10.1056/NEJMoa2207940
199. Ferretti F, Cannatelli R, Monico MC, Maconi G, Ardizzone S. An update on current pharmacotherapeutic options for the treatment of ulcerative colitis. J Clin Med. (2022) 11:2302. doi: 10.3390/jcm11092302
200. Deepak P, Alayo QA, Khatiwada A, Lin B, Fenster M, Dimopoulos C, et al. Safety of tofacitinib in a real-world cohort of patients with ulcerative colitis. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc. (2021) 19:1592–1601.e3. doi: 10.1016/j.cgh.2020.06.050
201. Chaparro M, Garre A, Mesonero F, Rodríguez C, Barreiro-de Acosta M, Martínez-Cadilla J, et al. Tofacitinib in ulcerative colitis: real-world evidence from the ENEIDA registry. J Crohns Colitis. (2021) 15:35–42. doi: 10.1093/ecco-jcc/jjaa145
202. Huss V, Bower H, Hellgren K, Frisell T, Askling J. Cancer risks with JAKi and biological disease-modifying antirheumatic drugs in patients with rheumatoid arthritis or psoriatic arthritis: a national real-world cohort study. Ann Rheum Dis. (2023) 82:911–9. doi: 10.1136/ard-2022-223636
203. Winthrop KL, Tanaka Y, Takeuchi T, Kivitz A, Matzkies F, Genovese MC, et al. Integrated safety analysis of filgotinib in patients with moderately to severely active rheumatoid arthritis receiving treatment over a median of 1.6 years. Ann Rheum Dis. (2022) 81:184–92. doi: 10.1136/annrheumdis-2021-221051
204. Rubbert-Roth A, Kakehasi AM, Takeuchi T, Schmalzing M, Palac H, Coombs D, et al. Malignancy in the upadacitinib clinical trials for rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis. Rheumatol Ther. (2024) 11:97–112. doi: 10.1007/s40744-023-00621-6
205. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367:eaau6977. doi: 10.1126/science.aau6977
206. Paskeh MDA, Entezari M, Mirzaei S, Zabolian A, Saleki H, Naghdi MJ, et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J Hematol Oncol. (2022) 15:83. doi: 10.1186/s13045-022-01305-4
207. Zhao Z, Chen X, Dowbaj AM, Sljukic A, Bratlie K, Lin L, et al. Organoids. Nat Rev Methods Prim. (2022) 2:94. doi: 10.1038/s43586-022-00174-y
208. Takahashi Y, Inoue Y, Sato S, Okabe T, Kojima H, Kiyono H, et al. Drug cytotoxicity screening using human intestinal organoids propagated with extensive cost-reduction strategies. Sci Rep. (2023) 13:5407. doi: 10.1038/s41598-023-32438-2
Keywords: Immune-Mediated Inflammatory Diseases, inflammation, immunosuppression, biologic therapy, malignancy, immune dysregulation
Citation: Maier JA, Castiglioni S, Petrelli A, Cannatelli R, Ferretti F, Pellegrino G, Sarzi Puttini P, Fiorina P and Ardizzone S (2024) Immune-Mediated Inflammatory Diseases and Cancer - a dangerous liaison. Front. Immunol. 15:1436581. doi: 10.3389/fimmu.2024.1436581
Received: 22 May 2024; Accepted: 29 August 2024;
Published: 18 September 2024.
Edited by:
Michele Maria Luchetti Gentiloni, Marche Polytechnic University, ItalyReviewed by:
Arianna Dal Buono, Humanitas Research Hospital, ItalyJacek M. Witkowski, Medical University of Gdansk, Poland
Copyright © 2024 Maier, Castiglioni, Petrelli, Cannatelli, Ferretti, Pellegrino, Sarzi Puttini, Fiorina and Ardizzone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeanette A. Maier, amVhbmV0dGUubWFpZXJAdW5pbWkuaXQ=