 Xiangmei Gong
Xiangmei Gong Shukang He
Shukang He Pengcheng Cai
Pengcheng Cai- Department of Clinical Laboratory, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Multiple factors contribute to the development of connective tissue diseases (CTD), often alongside a range of interstitial lung diseases (ILD), including Sjögren’s syndrome-associated ILD, systemic sclerosis-associated ILD, systemic lupus erythematosus-associated ILD, idiopathic inflammatory myositis-associated ILD. TRIM21(or Ro52), an E3 ubiquitin ligase, plays a vital role in managing innate and adaptive immunity, and maintaining cellular homeostasis, and is a focal target for autoantibodies in various rheumatic autoimmune diseases. However, the effectiveness of anti-TRIM21 antibodies in diagnosing CTD remains a matter of debate because of their non-specific nature. Recent studies indicate that TRIM21 and its autoantibody are involved in the pathogenesis of CTD-ILD and play an important role in diagnosis and prognosis. In this review, we focus on the contribution of TRIM21 in the pathogenesis of CTD-ILD, as well as the potential diagnostic value of its autoantibodies in different types of CTD-ILD for disease progression and potential as a novel therapeutic target.
1 Introduction
Connective tissue disease (CTD) is a group of autoimmune diseases that can involve multiple organ systems, among which interstitial lung diseases (ILD) are one of the common complications (1). ILD is a disease that affects the lung parenchyma, characterized by inflammation, fibrosis, and damage of the interstitium around the alveoli, including the alveolar walls and interstitial tissue (2). ILD can be associated with most types of CTD, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SS), Sjogren’s syndrome (SjS), and mixed connective tissue disease (MCTD); but it can be associated with other factors, such as medications and infections (3, 4). The general treatments include anti-inflammatory drugs such as corticosteroids, immunomodulators, and other supportive measures (5). However, the mortality rate of CTD-ILD is extremely high, and its pathophysiology is still not well understood. The molecular biomarkers for screening CTD-ILD are also not yet clear, leading to ongoing diagnostic and classification issues. In addition, intrinsic differences in the pathogenic mechanisms of autoimmune disease-associated disorders are probably reflected in different patterns of inflammation and tissue damage, and subtypes of individual diseases are correlated, and other potential markers might exist for CTD-ILD. It is worth noting that research has found a possible association between the presence of anti-tripartite motif-containing 21 (TRIM21 or Ro52) antibodies and poor quality of life and high mortality in ILD.
TRIM21, an autoantigen detected in 1988 in the sera of patients with autoimmune diseases (6), is a 52-kD protein as an antibody-binding protein of unusual structure by yeast two-hybrid analysis, whose molecular structural characterization and encoding genes indicate that it has no homology to the Ro60 protein, although both are considered to be the main antigenic targets of anti-Ro/SSA antibodies Figure 1 (7, 8). Autoantibodies are often considered to be a response to defects in the immune system that may result from excessive apoptosis or necrotic cell death, causing the immune system to mistakenly perceive its own cells and tissues as foreign invaders and mount an immune response against them (9). Generally, it is accepted that TRIM21 is involved in the occurrence and development of autoimmune diseases, and its autoantibodies have also been detected in patients with different types of autoimmune diseases (10). However, the significance of TRIM21 and its antibodies in the pathogenesis of CTD-ILD and their implications for the disease progression have not been fully elucidated in current studies. Therefore, this review will discuss the tissue and cellular expression distribution of TRIM21 and its antibodies in the pathophysiological process of CTD-ILD, as well as their impact on the diagnosis and prognosis of CTD-ILD.
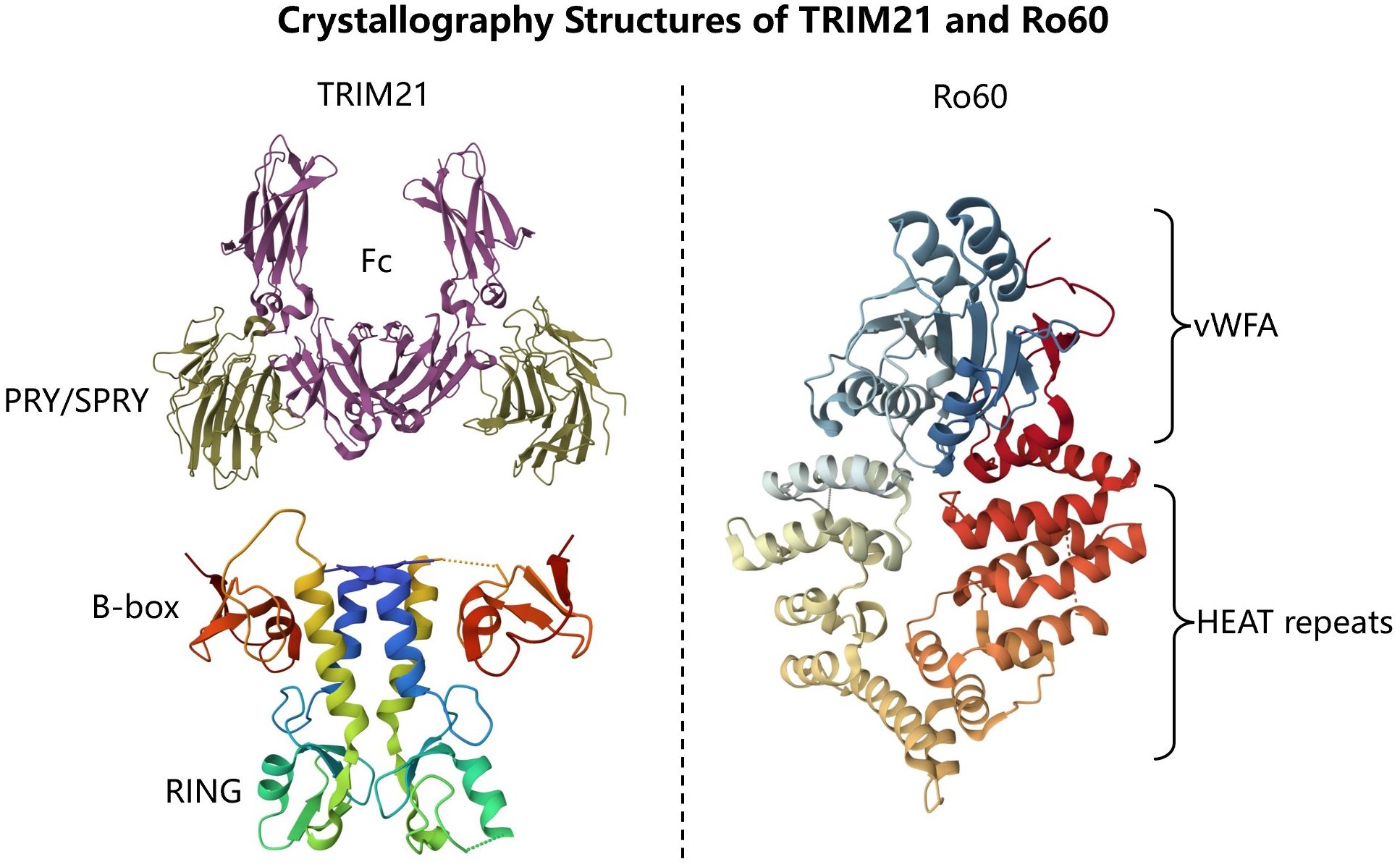
Figure 1 Crystallography Structures of TRIM21 and Ro60. TRIM21 consists of RING, B-box, coiled-coil and PRY/SPRY domains (PDB: 2IWG, 5OLM). Ro60 consists of a toroid of HEAT repeats and a von Willebrand factor A (vWFA) domain (PDB:1YVR).
2 TRIM21/Ro52
There are more than 80 known members of the TRIM family, the majority of which have E3 ligase activity, and TRIM21 is no exception (10, 11). TRIM21, as a member of the TRIM family, consists of N-terminal Really Interesting New Gene (RING), B-box, and a central coiled-coil (CC) domain involved in mediating TRIM21 dimerization, as well as a C-terminal PRY/SPRY domain, Figure 1 (10–13). The functions of the four different domains predict the remarkable biological potential of TRIM21.
The RING domain of TRIM21 is mainly involved in mediating Lysine 63(K63) and Lysine 48(K48)-linked ubiquitination (14). K63 is involved in the regulation of DNA repair and inflammation signaling, while K48-modified proteins are involved in targeted protein degradation via the proteasome (14, 15). Studies have shown that the RING domain of TRIM21 binds with the ubiquitin-conjugating enzyme E2W (Ube2W), thereby mediating the ubiquitination of K63-linked protein complexes bound by TRIM21 (16). In peripheral blood mononuclear cells (PBMCs), macrophages, and dendritic cells (DCs), intracellular TRIM21 mediates the ubiquitination and degradation of key regulatory proteins in the STING signaling pathway through K48 linkage, thereby affecting the production of pro-inflammatory cytokines during disease progression (17–19). It is worth noting that the intermolecular dimerization of the RING domain is one of the important mechanisms of TRIM21 ubiquitination (20). However, inhibiting RING domain does not affect the ubiquitination and degradation of substrates mediated by TRIM21 (21).
The B-box domain, as another important regulatory factor of TRIM21, plays a crucial role through its interaction with the RING domain (22). Research has observed that the B-box domain can act as an E2 mimic, inhibiting the ubiquitination function of TRIM21 by occupying the E2 binding site on the RING domain; however, TRIM21 could counteract the inhibition of the B-box by phosphorylating inhibitory kappa B kinase β (IKKβ) and transforming growth factor β-activated kinase 1(TBK1) at the LxxIS motif on the RING domain (22–24). The CC domain has a hydrophobic core that contributes to stabilizing protein conformation and mediates interactions between proteins (25). Recent studies have shown that the CC domain promotes TRIM21 self-association into homodimers through dimerization, thereby enhancing the E3 ubiquitin ligase activity of TRIM21’s RING domain (20, 26, 27).
TRIM21 could bind to most immunoglobulins thanks to the complex tertiary structure of its C-terminal B30.2 domain (also termed PRYSPRY) (28). The PRY/SPRY domain of TRIM21 contains a high-affinity immunoglobulin Fc-binding site interacting with immunoglobulins with specificity and extremely high affinity, which is highly conserved by binding to the CH2-CH3 interface, and does not overlap with the Fc domains of Fc gamma receptors (FcγR) and C1q (28). Besides attaching to IgG, TRIM21 is also capable of binding to IgA and IgM, albeit with a markedly reduced affinity in comparison to IgG (29–31). Therefore, the high affinity between PRY/SPRY domain of TRIM21 and IgG may be associated with promoting the pathogenic accumulation of immune complexes formed by anti-TRIM21 autoantibodies in autoimmune diseases. Moreover, recent study showed that histone deacetylase 6 (HDAC6) promotes the homodimerization of TRIM21 through its PRY/SPRY domain, indicating that the PRY/SPRY domain regulates the homodimerization of TRIM21 through acetylation, thereby indirectly affecting the E3 ubiquitin ligase activity mediated by TRIM21 (32).
It is now widely recognized that TRIM21 is primarily present in the cytoplasm, where it regulates cytoplasmic substrates through ubiquitination (33). The localization of TRIM21 is regulated by endogenous sequences, in particular the leucine zipper in the domain of the convoluted helix structure, which has been proven to play an important role in the cytoplasmic localization of TRIM21 (34, 35) When this region is deleted, TRIM21 rapidly translocates to the nucleus. Previous studies have demonstrated that TRIM21 can interact with E2s Ube2D1 located in the cytoplasm and Ube2E1 located in the nucleus (36, 37). Moreover, inflammatory mediators such as nitric oxide (NO) and interferon (IFN)-α induce TRIM21 and contribute to TRIM21 translocation to the nucleus (38–40). Therefore, TRIM21 exerts a broad intracellular defense by translocation with the help of cellular compartmentalization for the regulation of the topological distribution of molecules that are harmful or desirable to pathogens.
3 Role of TRIM21/Ro52 in immune and inflammatory responses
3.1 TRIM21 in intracellular immunity
Activation of the nuclear transcription factor kappaB (NF-κB) and interferon regulatory factor (IRF3) pathways occurs through intracellular delivery of antibodies that bind to specific pathogens in vivo during infection, which enhances innate immune signaling (31, 41). In addition to the dependence of intracellular pathogens on the proteasome for their degradation in the cell, TRIM21 of the cytoplasmic antibody receptor can also be involved in exerting the effects of intracellular antibody-mediated pathogen degradation (41). Studies have reported that TRIM21 acts primarily on viral particles that enter the cytoplasm and are bound by specific antibodies (31). In vitro experiments revealed that antibody-dependent intracellular neutralization of TRIM21 up-regulated by IFN-α was enhanced in adenovirus-infected Hela cells, and viral infection was inhibited. However, effective virus clearance also needs to be dependent on the E3 ubiquitin ligase activity of TRIM21 and degradation involving the proteasome.
Consequently, TRIM21 is also limiting for the clearance of antibody-bound pathogens, involving multiple important cofactors (42). TRIM21 requires recruitment of the E2 enzyme Ube2W and catalytic auto-monoubiquitination before exerting its effects and recruits the E2 heterodimer Ube2N/2V2h and the deubiquitinase Poh1, which promotes elongation of K63 polyubiquitin chains, Figure 2 (16, 43). Ubiquitinated TRIM21 serves as a substrate for the proteasome, recruiting proteasomes to facilitate the degradation of intracellular pathogens and thus, contributing to the initiation of innate immune signaling (16). In conclusion, in intracellular immunity, TRIM21 functions through antibody-dependent intracellular neutralization with antibody-bound pathogens associated with pathogens in infected cells, as well as intracellular antibody-mediated degradation of pathogens in conjunction with the proteasome.
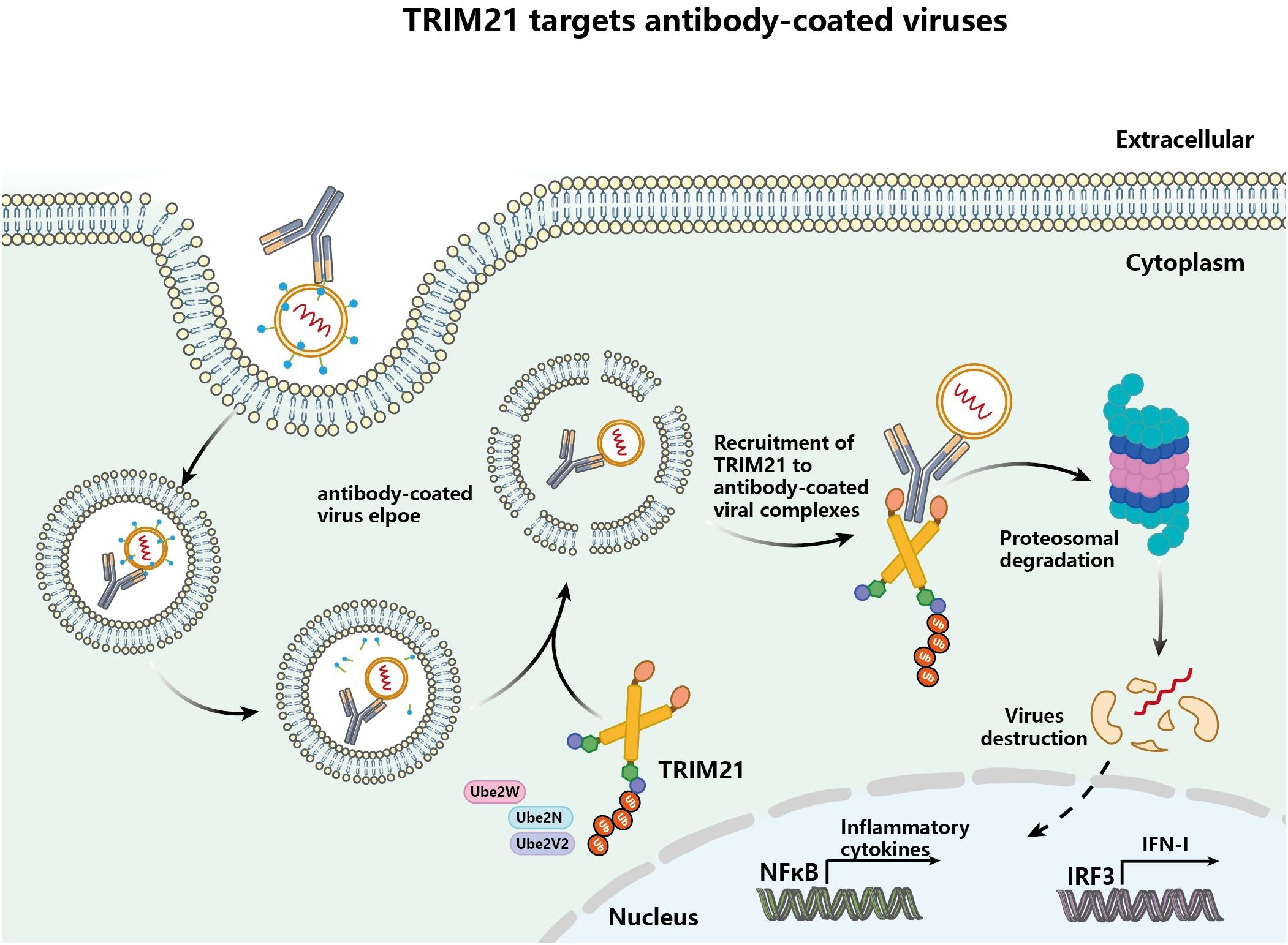
Figure 2 TRIM21 targets and destroys antibody-coated pathogens via ubiquitination and degradation (viruses as an example). Pathogens coated by antibodies are translocated into the cytoplasm, where the intracellular Fc receptor TRIM21 binds the antibody coating the pathogen and is activated to initiate ubiquitination and proteasome-dependent degradation of the targeted pathogen.
Moreover, in turn, gene expression of TRIM21 is regulated by immune signaling. Basic research has found that IFN-I and IFN-II can induce TRIM21 mRNA expression in various cells such as Hela cells, EL4 cells, and macrophages (44–46). Research showed that after IFN-I binds to cell surface receptors and stimulates the phosphorylation of JAK1 and TYK2, it promotes the phosphorylation of downstream molecules STAT1 and STAT2, which then form the IFN-stimulated gene factor 3 (ISGF3) with IRF9, thereby inducing the expression of IRF2; on the other hand, IFN-II stimulates downstream signaling molecules to produce IRF1 through a signaling cascade, Figure 3 (47). Both IRF1 and IRF2 can bind to the IFN-stimulated response elements (ISRE) site of the TRIM21 gene, thus upregulating the expression of TRIM21 (46). Of course, TRIM21 is expressed in various immune cells such as macrophages, DCs, neutrophils, B cells and T cells. TRIM21 influences disease outcomes and prognosis differently by regulating the expression of upstream and downstream molecules in immune cell signaling pathways and the production of cytokines in different immune cells.
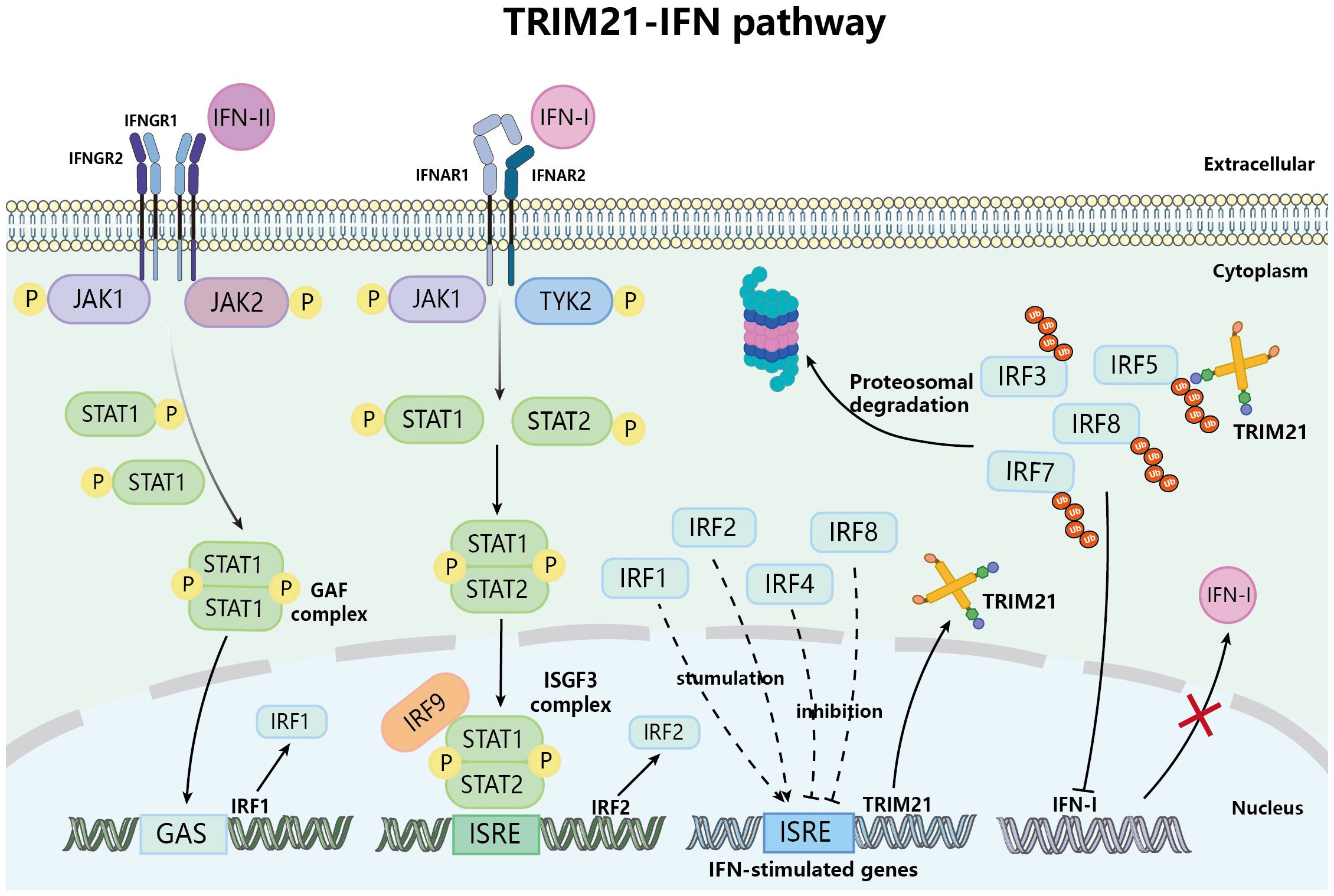
Figure 3 TRIM21 and IFN signaling pathway. Upon attaching to their specific interferon receptors, IFN-I and IFN-II facilitate the phosphorylation of the JAK/STAT pathway, resulting in the formation of ISGF3 and GAF complexes, respectively, initially triggering the expression of IRF1 and IRF2. IRF1 and IRF2 translocate to the nucleus and bind to ISRE, thereby inducing the expression of IFN-stimulated genes that include TRIM21, thus promoting the upregulation of TRIM21. However, the upregulated TRIM21 inhibits the expression of IFN-I by ubiquitinating IRF3/5/7/8.
3.2 TRIM21 and dysregulation of immune cell homeostasis
3.2.1 Macrophages
Macrophages are effective immune effector cells that play a crucial role in both tissue homeostasis and injury, such as promoting tissue damage and progression, as well as facilitating wound healing and tissue remodeling in various pathogenic conditions (48). TRIM21 in macrophages targets different regulatory proteins, exhibiting dual roles in the immune system’s defense and response. Research has shown that compared to monocytes, macrophages significantly express higher levels of TRIM21, indicating that it may be an important regulatory factor in macrophages’ resistance to infectious and autoimmune diseases (49). In vitro experiments also have shown that human THP1-derived macrophages and mouse macrophages are activated and upregulate the expression of TRIM21 mRNA through Toll-like receptor 3 (TLR3) and TLR4 ligands (50). IFN-I signaling-activated STAT1 promoted TRIM21 expression in macrophages, leading to Ube2M, an E2 NEDD8-binding enzyme involved in neddylation (a post-translational modification analogous to ubiquitination), ubiquitinated by TRIM21 and degraded by the proteasome (51). Interestingly, macrophage Ube2M was involved in the induced neddylation of TRIM21, enhancing the interaction between TRIM21 and Von Hippel-Lindau (VHL), promoting TRIM21-dependent ubiquitination and degradation of VHL (52). This inhibited the proteasomal degradation of hypoxia-inducible factor-1α (HIF-1α), further inducing the production of IL-1β in macrophages, thereby exacerbating obesity-induced inflammation and metabolic disorders, Figure 4. Similarly, inhibiting Ube2M-mediated neddylation of TRIM21 in IL-17A+Foxp3+ Treg cells suppressed the expression of IL-1β, thereby reducing the activity of DEPTOR-mTOR axis-related protein molecules, and consequently alleviating lipid accumulation and inflammation in hepatocytes (53, 54). Furthermore, lipopolysaccharide (LPS) induced macrophage polarization and enhanced the interaction between TRIM21 and SIRT5, Figure 4 (55). SIRT5, functioning as a desuccinylase, hinders the pro-inflammatory response mechanism of LPS-activated macrophages by impeding the ectopic movement of pyruvate kinase M2 (PKM) to the nucleus and augmenting its pyruvate kinase activity via deacetylation (56). However, TRIM21, which was upregulated in inflammatory macrophages, targeted SIRT5 by ubiquitination and degradation, thereby abrogating the role of SIRT5 in inhibiting the expression of the inflammatory factor IL-1β, Figure 4 (55). Studies have found that in the myocardial infarction (MI) mouse model, TRIM21 affects macrophage polarization through the PI3K/AKT pathway, promoting the transformation of macrophages into the pro-inflammatory M1 type, further exacerbating post-infarction cardiac dysfunction, Figure 4 (57). Conversely, TRPM2-dependent TRIM21 ubiquitination results in reduced production of reactive oxygen species (ROS) and inflammatory cytokines in macrophages, thereby promoting autophagy and apoptosis in inflammatory macrophages, which in turn reduces the pro-inflammatory activity of macrophages (58).
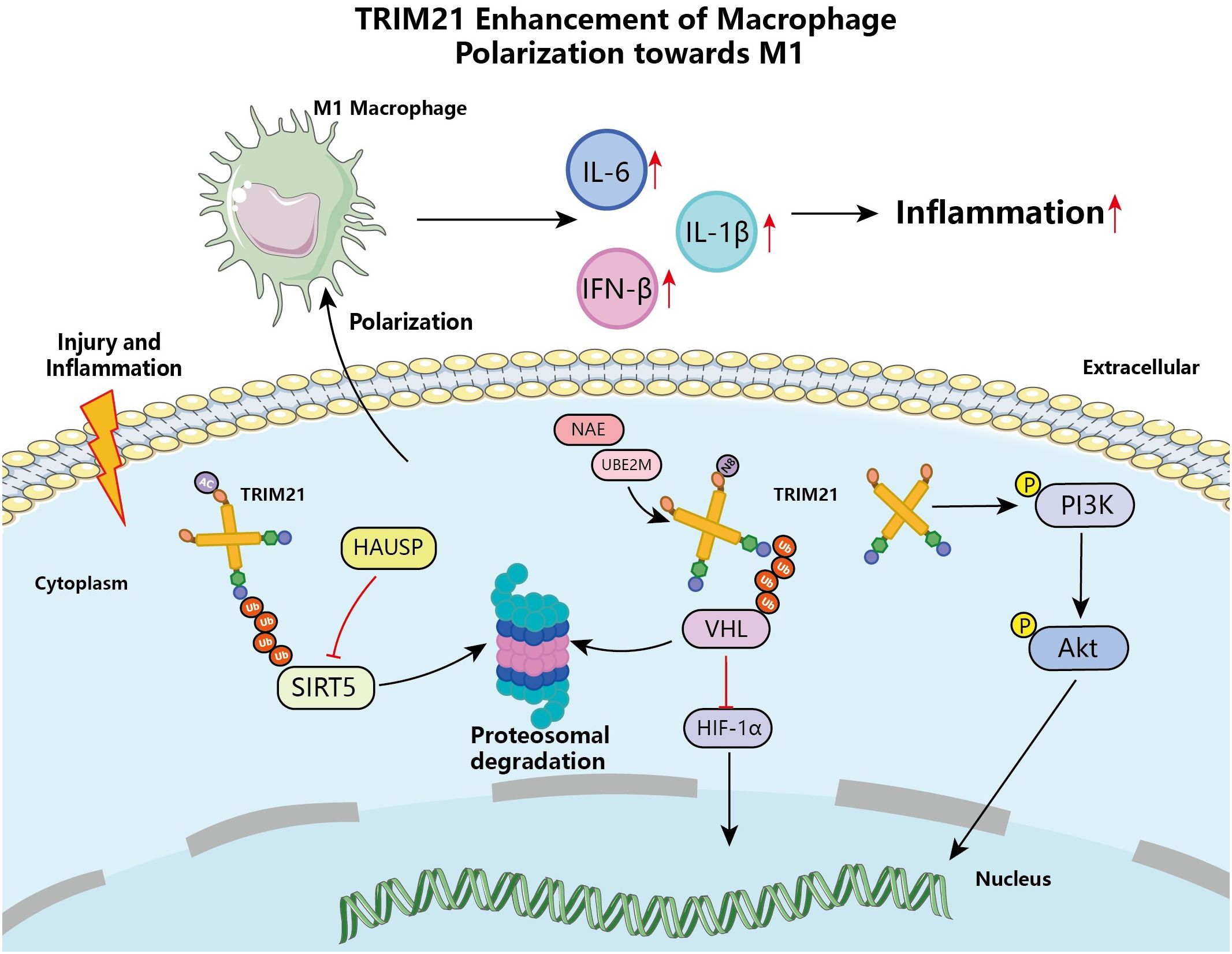
Figure 4 TRIM21 enhancement of macrophage polarization towards M1 macrophage. TRIM21-regulated signaling pathways are involved in macrophage polarization during injury or inflammation, involving the PI3K/Akt signaling pathway. Together with the proteasome, TRIM21 is involved in the degradation process of complexes of VHL-HIF-1α and SIRT5, which promotes the polarization of macrophages toward M1 macrophages, enhances the expression of pro-inflammatory cytokines, and exacerbates the inflammatory process.
Overall, the above studies indicated that TRIM21 in macrophages primarily influences disease progression by targeting specific proteins required for macrophage polarization for ubiquitination and degradation, indirectly determining the fate of macrophages. Current researches mainly focus on the regulation of inflammatory macrophages by TRIM21, but its role in the later stages of disease regulation is not significant. This is related to the functional crosstalk between various molecules and TRIM21 in the later stages of the disease, but the specific mechanisms still require further exploration.
3.2.2 Dendritic cells
TRIM21 not only regulates cell signaling in macrophages but also assumes a crucial role as a regulator in DCs, contributing to innate immune responses. DCs, which are responsible for delivering antigens, could trigger a more extensive immune response by taking in, processing, and displaying antigen-derived substances from pathogens to other immune cells (59). In the study of mDC, it has been found that TRIM21 mediates the ubiquitination and subsequent degradation of intracellular DDX41 (18). DDX41, as one of the members of the DEXDc helicase family, relies on the binding of pathogenic DNA by STING (the signaling adaptor of the cytosolic DNA sensor) to co-localize in the cytoplasm and initiate IFN-I and cytokine responses (60–62). Through the interaction between the PRY/SPRY domain and the Lys9 and Lys115 sites of the DDX41 DEADc domain, K48 subsequently mediates the ubiquitination and degradation of DDX41, thereby negatively regulating the expression of IFN-β and inhibiting the innate immune response of mDCs to intracellular dsDNA and DNA viruses (18). Additionally, it has been reported that in LPS/IFN-γ induced human moDCs, TRIM21 mediates the proteasomal degradation of IRF8, thereby promoting the production of IL-1β, IL-23, and IL-12 (63).
However, it has been reported that antibodies bound to pathogens enter cells as immune complexes, and antigens from immune complexes can further enhance the adaptive immune response to CD8+T cells by increasing affinity between Fc and TRIM21 to enhance antigen cross-presentation in DCs (64–66). CLEC-1 (C-type lectin) binds to conserved protein structures exposed during programmed cell death, promoting tumor progression by facilitating immunosuppressive tumor microenvironment and hindering DC from cross-presenting dead cell-associated antigens to CD8+T cells (67). However, Drouin et al. found that TRIM21 is an endogenous ligand of CLEC-1 and, as a self-antigen released by necrotic cells, induces necroptosis, but the Fc of CLEC-1 does not bind to the FcR of TRIM21 (67), yet this mechanism has not seen further explanation.
3.2.3 T cells
Research has indicated that TRIM21 plays a key role in the production of IL-2 in T-cells triggered by CD28 and is essential in managing the growth and diversification of T-cells (68). The primary expression of TRIM21 was observed in CD4+T cells within normal intestinal mucosal tissues, with a reduction noted in inflammatory tissues during IBD (69). Inhibiting the ubiquitination of TRIM21 and the degradation of IRF3 in colitis induced by TRIM21-/-mice caused an increase in IL-17+CD4+ and IFN-γ+CD4+T cells, accompanied by an intensified immune reaction from CD4+T cells, culminating in heightened inflammation of the intestinal mucosa (33, 70, 71). Similarly, low TRIM21 expression promotes the differentiation of CD4+T cells into Th17 cells in atherosclerotic plaques and increases IL-17A expression, further exacerbating the formation of atherosclerotic plaques (72), implying that the balance of T cell differentiation appears to be dependent on the regulation of TRIM21. Conversely, TRIM21 binds to ubiquitinated NF-κB through K63 in CD3+T cells in Oral lichen planus (OLP) tissue, resulting in a heightened release of pro-inflammatory factor in T cells, subsequently triggering the NF-κB signaling pathway and exacerbating tissue damage (73, 74).
3.2.4 B cells
Additionally, TRIM21 dysfunction is involved in promoting aberrant activation and proliferation of B cells (75). Researches indicate that T cells and DCs contribute to the activation and diversification of B cells, but in lupus mouse models, the absence of TRIM21 did not alter the status of T cells and DCs, due to the abnormal function of B cells themselves (76, 77). TRIM21 participates in the regulation of B cells function by regulating the ubiquitination and degradation of IRF5, an important regulator of B cells differentiation and the production of antibodies by self-reactive B cells, Figure 5 (78, 79). Overexpression of TRIM21 resulted in reduced IRF5 expression in B-cell lines during in vitro experiments, whereas the stimulation of B-cell growth lessened and encouraged apoptosis (79). Moreover, TLR7 enhances the nuclear localization of IRF5 in activated B cells and promotes the differentiation of B cells into plasma cells that secrete a variety of autoantibodies and thereby trigger tissue injury, among which TRIM21 has been found to be the target of secreted autoantibodies (80). TLR9 may affect B cell activation by indirectly regulating TLR7 through its signaling pathway, but the exact mechanism has not been elucidated, Figure 5 (81). The relevant study also revealed that reduced TRIM21 gene expression in diffuse large B-cell lymphoma leads to increased aggressiveness of lymphoma and is associated with poor prognosis (75). However, the differentiation and proliferation of TRIM21 into adaptive immune cells vary due to elements such as triggers, tissue location, and the severity of inflammation, resulting in diverse disease outcomes.
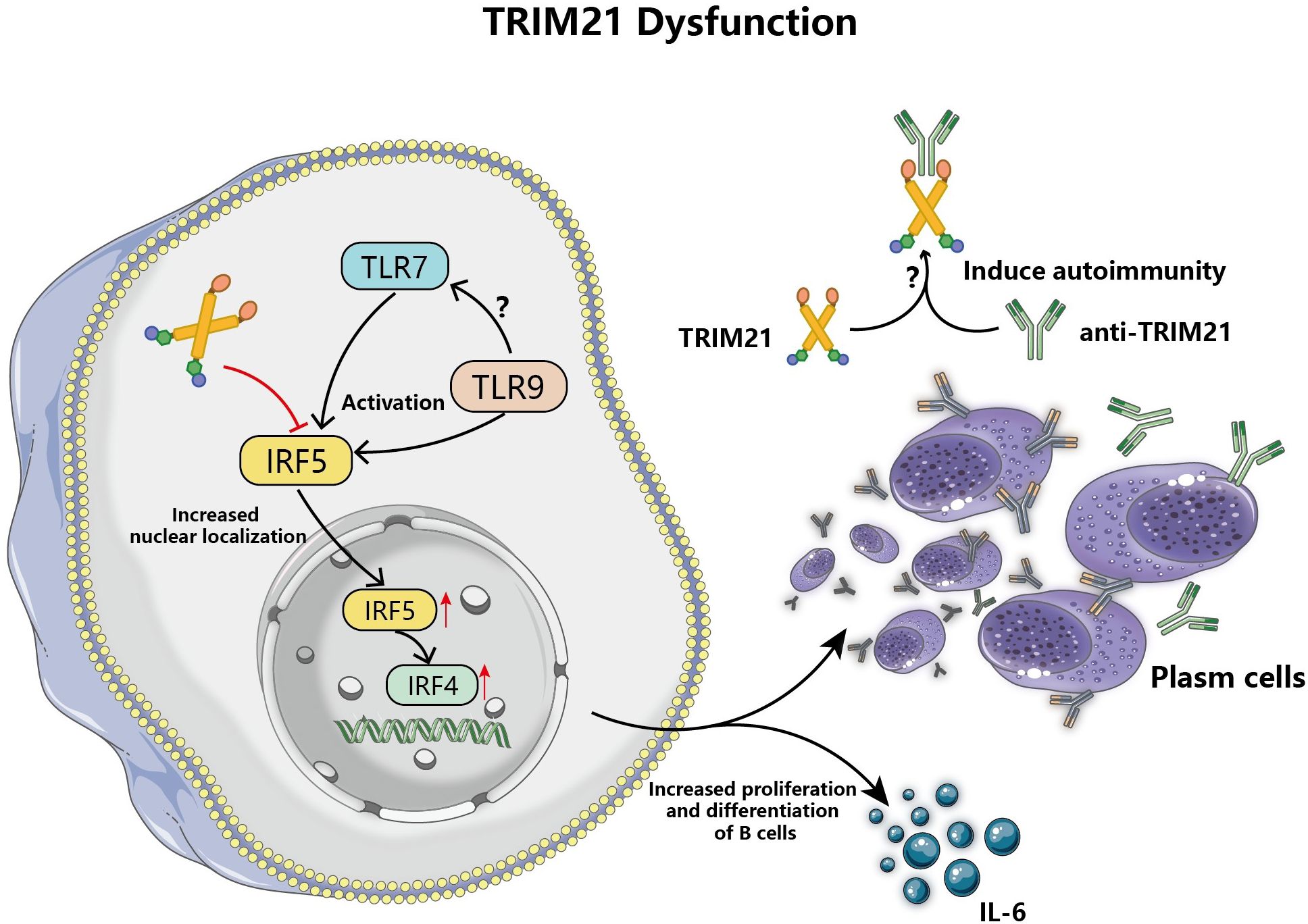
Figure 5 TRIM21 dysfunction leads to aberrant activation and proliferation of B cells. TRIM21 dysfunction attenuates the regulation of IRF5 ubiquitination, and TLR7 promotes IRF5 expression and nuclear localization in B cells to increase and enhance the expression of the downstream molecule, IRF4, and increase the differentiation of antibody-secreting plasma cells and the production of IL-6, which triggers autoimmune responses. In addition, TLR9 may balance the autoimmune damage caused by the abnormal proliferation of activated B cells stimulated by TLR7 through its regulatory function.
Together, the proteins targeted by TRIM21 in different immune cells vary depending on the disease, indicating that TRIM21, as an E3 ubiquitin ligase, has extensive interactions and common regulatory characteristics within the immune system. As it’s been described, proteins in the signal transduction pathways designed in both innate and adaptive immune cells in diseases may be directly or indirectly regulated by TRIM21. However, due to the different functions of the proteins targeted by TRIM21, the impact on the disease progression also varies.
Therefore, understanding the general rules of how TRIM21 interacts with related pathway proteins in various immune cells within the immune system will help identify potential therapeutic targets in diseases. This requires further research and exploration.
4 TRIM21/Ro52 and connective tissue disease-associated interstitial lung diseases
The immune system is an intricate network of cells that distinguishes between self and foreign materials to efficiently combat invading pathogens (82). Nevertheless, malfunctioning peripheral and central immune tolerance mechanisms may cause immune cells to react to self-antigens, resulting in the widespread inflammation and tissue damage observed in autoimmune diseases (83). CTD is a common combination of autoimmune diseases, while ILD is one of the primary causes of morbidity and mortality among CTD patients. TRIM21 has been confirmed as the main target of autoantibody responses in CTD, and the value of anti-TRIM21 antibodies, as common autoantibodies in CTD, is increasingly being recognized in CTD-associated ILD. This section will focus on the potential diagnostic value of TRIM21 and its autoantibodies in the development of CTD-ILD based on currently published clinical research data and preclinical studies, as well as the possible molecular mechanisms involved in the pathogenic process.
4.1 TRIM21/Ro52 in Sjögren’s syndrome-associated interstitial lung disease
SjS is a systemic chronic inflammatory autoimmune disease that manifests as a decrease in the production of glands in the body, such as tears, saliva, sweat, and mucus, and the main symptoms may extend from dry mucosal surfaces to systemic involvement (84). Pulmonary involvement is a common extra-glandular complication, with ILD being the most common complication, with a prevalence of 10%-20% reported in studies (85, 86). Despite SjS-ILD typically exhibiting a mild and self-limiting progression, there was a notable rise in mortality cases among SjS patients with ILD (87, 88). Therefore, choosing suitable biomarkers is crucial for forecasting the variances between patients with advancing illness and those who might progress to a slow or stable phase of the disease.
In the 1960s, the serum of patients with SjS revealed the presence of anti-Ro/SSA antibodies, identified as a defining feature of SjS (89). While anti-TRIM21 antibodies frequently correlate with anti-Ro60, anti-TRIM21 positivity alone is found in 20-30% of SjS patients (90, 91). Research indicates that patients with high anti-TRIM21 titration experience more severe clinical symptoms, such as an enlarged parotid gland, a positive rheumatoid factor, and diminished blood cells. Similarly, patients with SjS exhibiting only anti-TRIM21 positivity showed elevated disease activity and a higher frequency of simultaneous mixed cryoglobulinemia, in contrast to those with anti-Ro60/La positivity or patients lacking these autoantibodies (92). In a cohort study of 527 patients with primary SjS in China, the researchers found that the detection rate of anti-TRIM21 positivity in patients with concomitant ILD was 36% and that anti-TRIM21 was independently associated with ILD, Table 1 (95). In recent years other studies also have reported that up to 39-86% of patients with SjS-ILD exhibit anti-TRIM21 positivity, Table 1 (96–103). Consistent with previous studies, the positivity of anti-SSB and anti-TRIM21 antibodies was significantly higher than in patients without ILD and may be predictive of the extensive lung involvement seen in patients with SjS-ILD (96, 98, 101, 102). However, in contrast, it has been suggested that there is no difference between SjS-ILD and SjS patients without ILD (SjS-NILD) in the presence of anti-SSA, anti-TRIM21, rheumatoid factor (RF), and antinuclear antibody (ANA) (100, 103). In conclusion, these cohort studies suggest that SS-ILD patients with anti-TRIM21 antibodies have more disease activity compared to patients without these autoantibodies, and that anti-TRIM21 is a possible cause of lung involvement leading to ILD.
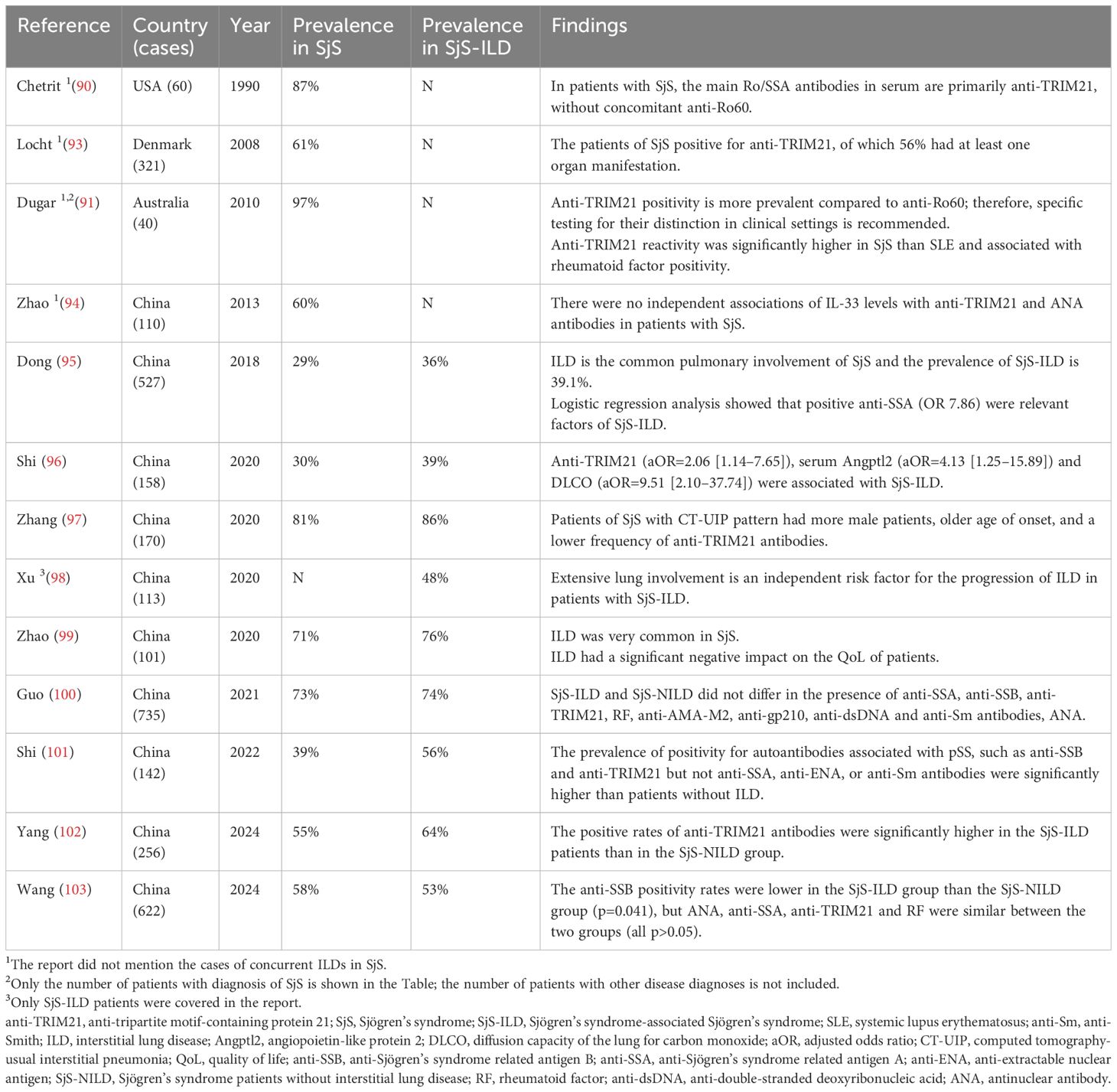
Table 1 Main studies’ results regarding the prevalence of anti-TRIM21 antibody in SjS-ILD.
Preclinical studies have shown that TRIM21 is significantly expressed in the ductal epithelium of focal tissues in salivary gland tissues of patients with primary SjS (pSjS), and the degree of TRIM21 expression in the ductal epithelium correlates with the degree of inflammation (104). Additionally, Aqrawi et al. suggested that Ro/SSA-specific memory B cells in the salivary glands of SjS patients are converted to plasma cells at the site of inflammation, thereby promoting autoantibody production (105). These data indicate that the overexpression of TRIM21 in the lesions of SjS patients is involved in the production of the specific autoantibody anti-TRIM21. Furthermore, during chronic inflammation, the overexpression of TRIM21 in secretory glands is involved in regulating immune cell infiltration, ultimately leading to tissue degeneration and salivary gland damage.
In SjS animal models, the passive transfer of TRIM21-positive serum from immunized mice to non-immunized model mice resulted in dysfunction of the salivary and lacrimal glands, along with local deposition of anti-TRIM21 antibodies in the glands (106), suggesting that anti-TRIM21 antibodies may be directly involved in the pathogenic process of SjS. Furthermore, in SjS, systemic B cell hyperactivity and high expression of IFN-I are the reasons for the elevated expression of TRIM21 on the surface of circulating plasmacytoid DCs (46, 107). Notably, TLR7-stimulated SjS salivary gland epithelial cells presented TRIM21 antigen via major histocompatibility complex (MHC) class I molecules (108). These results suggest that TRIM21 may function on the surface of immune cells to identify and destroy other immune cells.
Interestingly, patients with SjS appear to have autoantibody specificity for several different structural epitopes of the TRIM21 protein, including its RING, B-box, and CC domains (109). Sroka et al. demonstrated that anti-TRIM21 antibodies directed against this CC domain were involved in salivary gland dysfunction induced by E3 ubiquitin ligase activity and that IgG deposition in the glands was significantly increased (110). Moreover, in vitro experiments revealed that anti-TRIM21 antibodies from pSjS patients inhibited the E3 ligase activity of TRIM21 by binding to its RING domain (111). TRIM21 is widely expressed in all cells of the body, including those related to lung tissue. In patients with SjS associated with ILD, anti-TRIM21 antibodies appear to cause functional impairment of lung tissue through inhibition of TRIM21 ubiquitin ligase activity, leading to interstitial pneumonitis, but there are currently no reports on the mechanism of pathogenesis of SjS-ILD with TRIM21.
4.2 TRIM21/Ro52 in systemic sclerosis-associated interstitial lung disease
SSc is a rare autoimmune disease characterized by hyperplasia and hardening of connective tissue in the skin, internal organs, and blood vessels. Based on skin involvement levels, SSc is categorized into three types: limited cutaneous scleroderma, diffuse cutaneous scleroderma, and SSc sine scleroderma (112). Due to the limited treatment options, the advanced stages of SSc are often accompanied by life-threatening fibrotic complications, including SSc-ILD (113). The progression of SSc-ILD is believed to occur in three primary phases: first, abnormal activation of the immune system due to damage to the alveolar epithelium, endothelial cells, and vasculature, followed by abnormal activation of immune cells to release cytokines and autoantibodies, prompting fibroblasts to collect from lung and further promote its activation and adhesion, as well as the production of large amounts of extracellular matrix that eventually leads to normal pulmonary architecture being replaced by scar tissue (114).
Currently, autoantibodies have been identified in studies as specific serological biomarkers for SSc diagnosis and prognosis, including anti-centromere, anti-topoisomerase I (anti-Topo I), anti-RNA polymerase III, and two major types of SSA/Ro antibodies, anti-Ro60 and anti-TRIM21, which co-exist with them (115). Research indicates an increased likelihood of SSc-ILD in individuals with anti-Topo I, yet complications from ILD are rare in those with anti-centromere-positive SSc, despite anti-centromere being the most identifiable specific autoantibody in SSc, Table 2 (124). In 2012, the Canadian Scleroderma Multicenter Study Group’s comprehensive analysis of autoantibodies associated with 963 patients with SSc showed that anti-TRIM21 was detected in 20% of patients and was the second most common autoantibody in the patient cohort, except for anti-centromere antibodies (119). Additionally, patients with SSc exhibiting anti-TRIM21 positivity demonstrated an increased probability of concurrent ILD (118, 119). In 2021, the Canadian scleroderma research team once more reported positive rate results for anti-TRIM21 in 26% of 1,698 SSc patients, marking the most extensive cohort analysis of anti-TRIM21 in SSc to date, and crucially, this aligns closely with the UK’s reported rate of anti-TRIM21 positivity in 1,010 SSc patients (27%), Table 2 (117, 123). Interestingly, it was found that the probability of concurrent lung disease was relatively high among anti-TRIM21 positive patients relative to anti-TRIM21 negative patients, and there was an association with concurrent ILD (118, 125). For patients with SSc-ILD, the anti-TRIM21 antibodies were found to overlap with nearly all autoantibodies linked to SSc (119). Moreover, the levels of anti-TRIM21 were notably elevated in patients with SSc who tested positive for anti-Ro60 and anti-aminoacyl-tRNA synthetase antibodies (117). Additionally, the existence of anti-TRIM21 in patients with SSc-ILD was independently linked to lower survival rates (121). In conclusion, the data from the cohort studies robustly endorses both the function and predictive significance of anti-TRIM21 antibodies in diagnosing the condition in SSc-ILD.
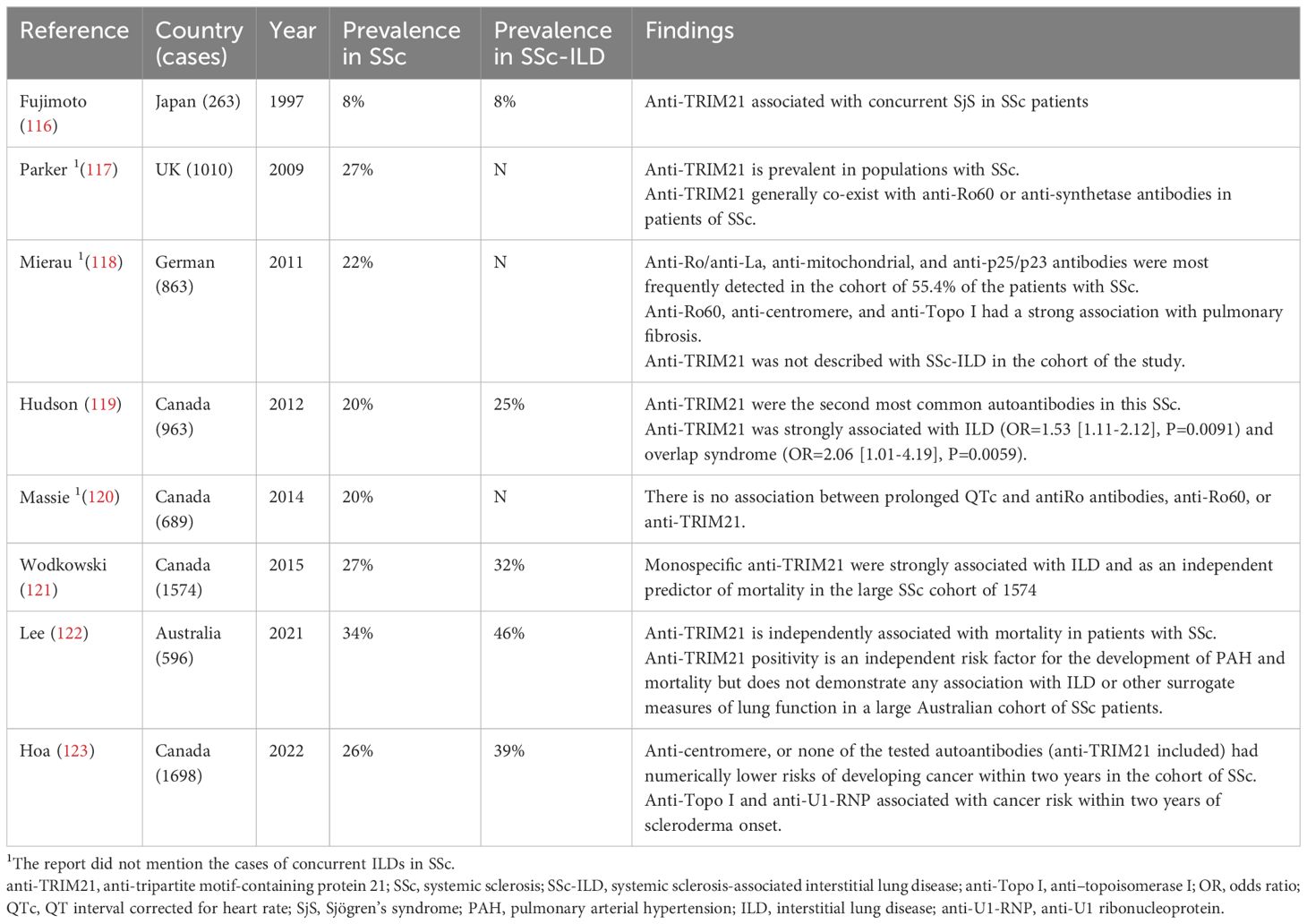
Table 2 Main studies’ results regarding the prevalence of anti-TRIM21 antibody in SSc-ILD.
Through immunohistochemical staining, studies have observed that TRIM21 is localized in alveolar M2 macrophages in the peripheral lung tissue of SSc-ILD patients and that the accumulation of anti-TRIM21 in bronchoalveolar lavage (BAL) fluid suggests a pathobiological link between TRIM21 and its autoantibodies and the progression of SSc-ILD (126). M2 macrophages are increased in both skin and peripheral blood in SSc patients, and despite the function of M2 in the disease process to inhibit the inflammatory process and participate in the repair of damaged tissues, SSc-ILD persists as a chronic inflammatory disease that continues to prolong, ultimately leading to persistent damage and irreversible dysfunction of lung tissue (127, 128). Recent study has found that TRIM21 targets Sohlh2 (a member of the basic helix-loop-helix transcription factor superfamily) for ubiquitination and degradation, inhibiting its high expression in M2 macrophages and its role in promoting macrophage polarization to the M2 phenotype (129). In the progression of SSc-ILD, TRIM21 may serve as a beneficial molecular regulator in the differentiation of macrophages into M1 and M2. However, the production of autoantibodies may impair the normal biological function of TRIM21, exacerbating disease progression. Therefore, TRIM21 may regulate the production of autoantibodies, and dysfunction of TRIM21 could lead to the advancement of autoimmunity.
B cells circulating in SSc-ILD patients were activated, resulting in an increased production of specific autoantibodies accumulating in the lung, causing localized tissue damage (130). Similarly, the tight-skin (TSK/+) mouse, used as an animal model for SSc, exhibited unusual B cell activation, heightened production of serum immunoglobulin, and increased autoantibody production (131, 132). These researches suggest that abnormally differentiated B cells might produce anti-TRIM21 antibodies, which could explain the presence of anti-TRIM21 detected in the serum of SSc-ILD patients. However, no study has yet reported the effect of TRIM21 and its autoantibodies in B-cell dysfunction on the SSc-ILD disease process in SSc animal models, and the possible mechanisms remain unclear.
4.3 TRIM21/Ro52 in systemic lupus erythematosus-associated interstitial lung disease
SLE, an autoimmune disorder impacting various systems and organs, leads to the development of diverse autoantibodies in patients, with clinical manifestations evolving from minor skin issues to critical organ failure (133). The lung is one of the common organs accumulated in SLE, and the main lung lesions are pleurisy, organizing pneumonia, pulmonary atrophy syndrome, occlusive fine bronchitis, pulmonary hypertension, small airway lesions, pulmonary embolism, and interstitial pneumonia (134, 135). ILD is a complication of SLE with an incidence of approximately 3-9% and is a key contributor to poor SLE prognosis (136). SLE-ILD might manifest at any phase of the illness, from its initial diagnosis to the disease’s 20-year progression, considering the patient’s age and the progression of concurrent diseases, with ILD being either clinically negligible in the early stages or exhibiting subclinical phases (137). Approximately 40-50% of SLE patients are positive for anti-TRIM21 antibodies, frequently combined with anti-Ro60 and anti-La antibodies; the occurrence of anti-TRIM21 antibody positivity by itself is uncommon in SLE, Table 3 (141, 146, 147). In addition, Xia et al. found that the positive detection rate of anti-TRIM21 in SLE-ILD patients was 30%, but no correlation between SLE-ILD and anti-TRIM21 has been identified (143).
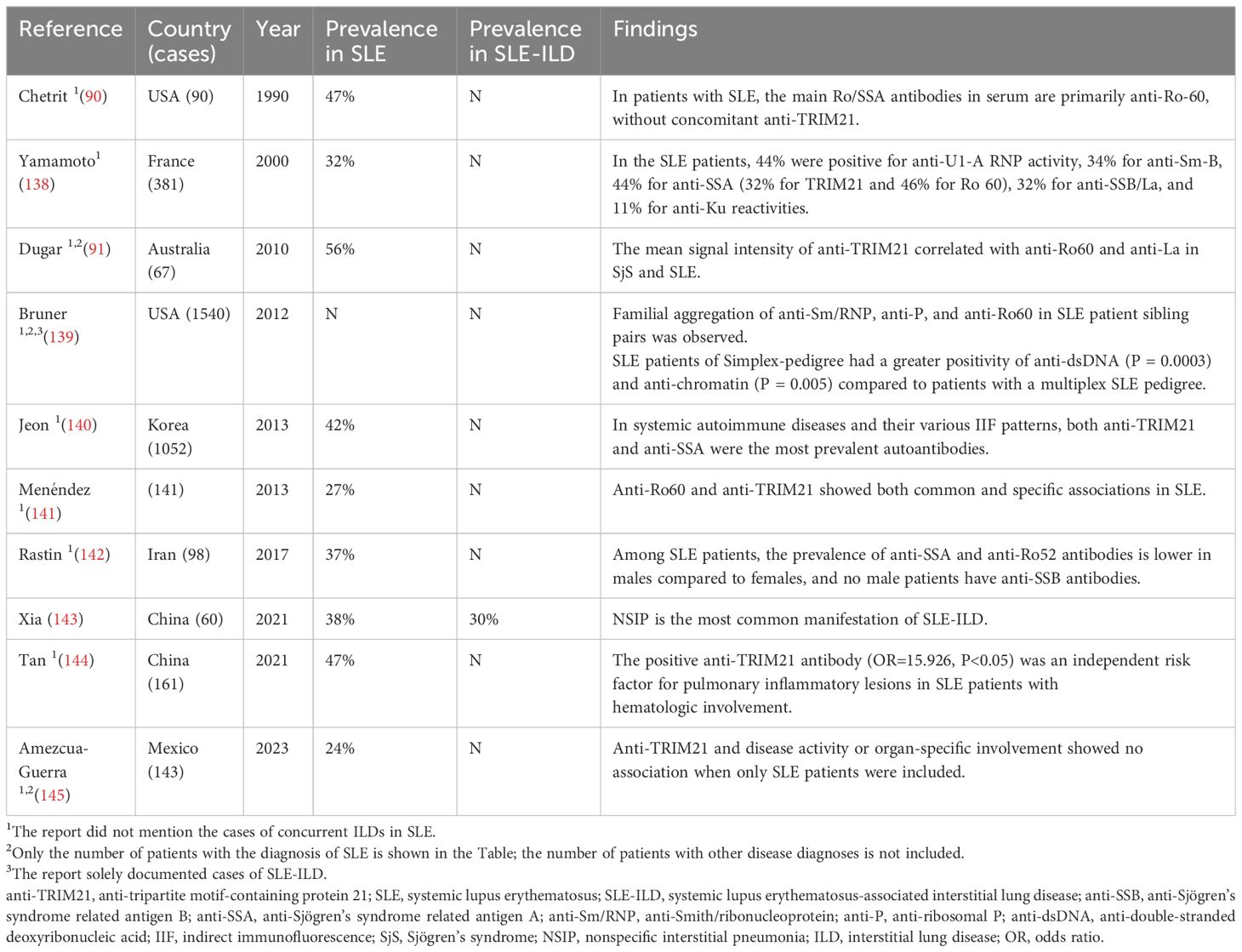
Table 3 Main studies’ results regarding the prevalence of anti-TRIM21 antibody in SLE-ILD.
Recent studies have found that IFN-I plays an important role in SLE pathogenesis, especially IFN-α (148). Studies have shown increased IFN-α expression and higher TRIM21 expression levels in PBMC from SLE patients compared to healthy controls (149). Animal model studies revealed that TRIM21-/- mice developed SLE-related clinical manifestations after skin lesions and abnormalities in the IL-23-Th17 pathway, which triggered tissue inflammation and systemic autoimmunity, suggesting that TRIM21 protects the organism by negatively regulating pro-inflammatory factor production in SLE (33). IRFs can be ubiquitinated by TRIM21 through E3 ubiquitination of ligase activity in its RING domain, thereby affecting IFN-I (33, 39, 150). In SLE, anti-TRIM21 antibodies inhibit the degradation of IRF, leading to an imbalance in IFN expression and an increase in TRIM21 expression; however, this is not the case in anti-TRIM21 antibodies negative populations, suggesting that anti-TRIM21 antibodies are involved in the progression of anti-TRIM21 positive SLE by interfering with the ubiquitination and degradation of IRF by TRIM21 (149). In MRL/lpr lupus susceptible mice, a dysfunction in TRIM21 was associated with the pathological progression of SLE, atypical B cell differentiation, elevated autoantibody levels, and proteinuria (151). Additionally, the research also revealed that circulating B cells derived from patients with TRIM21-positive SLE had increased potential to differentiate into plasmablast compared to healthy controls (151). In conclusion, preclinical studies indicate that the dysfunction of TRIM21 in SLE appears to be due to the immune system mistakenly recognizing the TRIM21 antigen as a foreign substance, thereby breaking immune tolerance.
The connection between anti-TRIM21 antibodies and SLE-ILD is still not well understood in the current study among a limited group of SLE-ILD patients, but anti-TRIM21 antibodies positivity in SLE-ILD is greatly likely to be associated with negative patient outcomes, and the specific mechanism needs to be further explored.
4.4 TRIM21/Ro52 in idiopathic inflammatory myositis-associated interstitial lung disease
Idiopathic inflammatory myopathy (IIM) manifests as muscular feebleness, inflammation, and discomfort (152). The primary identified variants of IIM, as per clinical and serological characteristics, include polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM). During the diagnosis of IIM, autoantibodies are deemed dependable for distinguishing various myopathy subtypes, primarily divided into myositis-specific autoantibodies (MSA) and myositis-associated autoantibodies (MAA) (153). MAA frequently appears in cases of myositis overlap syndrome, yet it’s a group of antibodies linked to the disease and lacks specificity, including anti-TRIM21, which is frequently linked to a heightened risk of concurrent ILD (153). It’s estimated that more than 40% of patients globally suffer from concurrent ILD in IIM, with regional differences in prevalence, peaking at 50% in Asia and 23% and 26% in North America and Europe, respectively (154). Clinical data collected from researches showed that anti-TRIM21 antibodies are one of the independent serological risk factors for different subtypes of IIM-ILD, with a positivity rate of up to 50%, which is similar to the frequency of anti-Jo-1 in IIM-ILD and are associated with the severity and prognosis of IIM-ILD, Table 4 (159, 161, 162, 164, 165, 168, 169). Within the PM subtypes, anti-synthetase syndrome (ASS) stands out, with anti-TRIM21 showing a greater likelihood of occurrence in ASS compared to other subtypes and frequently linked to the severity of the disease. It has been reported that patients with anti-TRIM21 positive ASS who are anti-Jo-1 and/or anti-PL7 positive have a significantly increased risk of concomitant ILD and exhibit more severe myositis and lower survival rates (170, 171). Similarly, patients with anti-MDA5 and anti-TRIM21 positive DM also tend to complicate more severe ILD and have a worse prognosis; Sabbagh et al. also concluded that anti-TRIM21 can be a predictive marker for complication of ILD in patients with juvenile myositis, and also emphasized that it is likely to be associated with the severity of the disease, a worse prognosis, and an increase in the dosage of immunosuppressant medications during the follow-up period (172, 173). In clinical practice, the main tools commonly used for ILD screening are pulmonary function tests (PETs). Vojinovic et al. and Gui et al. did not find progression of IIM-ILD and changes in PFTs over time in anti-TRIM21-positive patients with IIM, but found an increase in the patient’s lung diffusion capacity (DLCO) at five-year follow-up (162, 165). These clinical research data indicate that anti-TRIM21 antibodies seem to exist in IIM-ILD patients before any clinical symptoms appear, and they can be detected in the serum possibly even before IIM complicates with ILD, or even earlier. Therefore, the detection of anti-TRIM21 antibodies in IIM is of great significance for early prediction of the risk of concurrent ILD.
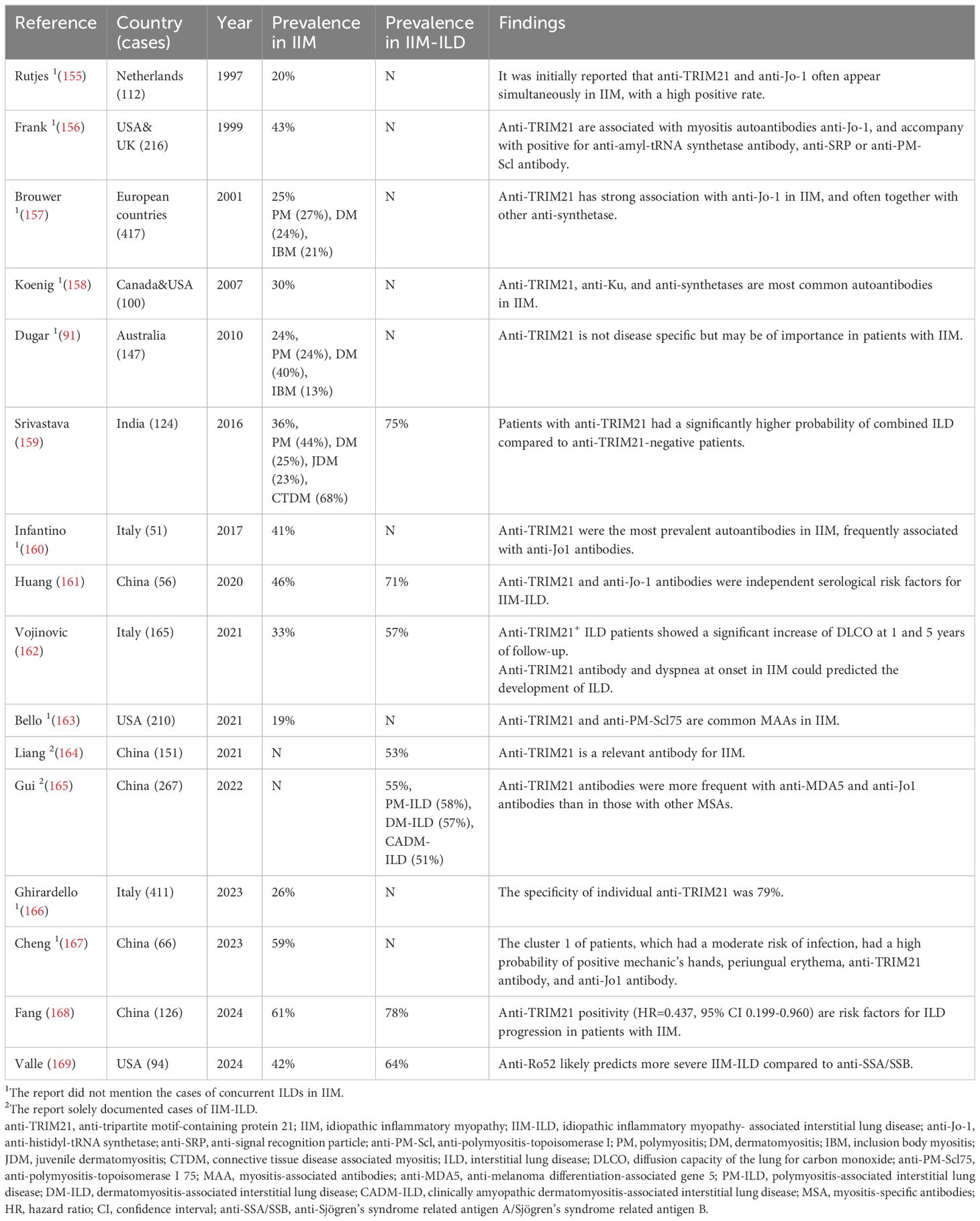
Table 4 Main studies’ results regarding the prevalence of anti-TRIM21 antibody in IIM-ILD.
Interestingly, in IIM-ILD, the presence of autoantibodies to TRIM21 disrupts immune homeostasis and is associated with poor prognosis, and the role played by TRIM21 antigens in the pathogenesis of IIM-ILD is also thought-provoking. TRIM21 expression was reduced in monocytes and CD4+T lymphocytes from IIM patients compared to healthy controls and resulted in decreased secretion of IL-6 and IFN-α from monocytes and IL-17 and TNF-α from CD4+T cells (17). Reports indicate that TRIM21 has the capability to suppress its E3 ligase function by engaging in anti-TRIM21 interactions with E2/E3 (111). However, Martín et al. showed no correlation between TRIM21 expression and anti-TRIM21 in IIM (17). Hassan et al. found that myositis patients with positive of anti-TRIM21 antibodies have an imbalance in TNF and IL-10 expression in serum (174). These findings may predict that during the pathogenesis of IIM, the reduced expression of TRIM21 weakens the ubiquitination process of related regulatory factors (such as IRFs), leading to an imbalance between pro-inflammatory and anti-inflammatory cytokines, thereby triggering a persistent chronic inflammatory process. Additionally, in muscle biopsy specimens from IIM patients positive for anti-TRIM21, the number of B cells increases, and the expression of BAFF (an important cytokine involved in B cell differentiation and maturation) and its receptors (BAFF-R, BCMA, and TACI) is upregulated, ultimately leading to the production of autoantibodies and the destruction of muscle tissue (175, 176).
Based on the existing clinical research data and the very limited preclinical study results, the detection of anti-TRIM21 antibodies appears to be a potential biological marker for detecting the occurrence, development, and prognosis of IIM-ILD. Additionally, the expression of TRIM21 in muscle tissue and the dysfunction of its E3 ubiquitin ligase significantly affect the disease outcomes of IIM-ILD. However, the specific molecular mechanisms require further research exploration.
4.5 TRIM21/Ro52 in rheumatoid arthritis-associated interstitial lung disease
ILD is a common extra-articular manifestation in patients with RA, often worsens the prognosis of the disease, and leads to considerable illness and increased death rates in those with RA (177). Research indicates that the occurrence of RA-ILD varies between 10% and 61% (178). Multiple risk elements contribute to RA-ILD, encompassing advanced age, active synovitis, seropositivity for RF and anti-cyclic citrullinated peptide (anti-CCP), along with MUC5B polymorphism. RA-ILD is mostly asymptomatic in its early stages, making early diagnosis difficult. Clinically, lung involvement in RA-ILD patients is often discovered after joint damage has occurred, but it is worth noting that the lungs may already be affected in the early stages of RA, remaining in a state of mild chronic inflammation. Paulin et al. predicted aberrant activation of myofibroblasts and alveolar epithelial cells in the lungs early in RA-ILD, initiation of a systemic immune response at the level of the pulmonary mucosa by the process of citrullination, and subsequent migration of anti-CCP antibodies from the lungs to the synovium, which exacerbates joint damage (179).
It is noteworthy that studies have reported a lower positivity rate of anti-TRIM21 in RA compared to other CTD, Table 5 (180, 182). Nonetheless, RA patients lacking RA-related risk factors may continue to experience concurrent ILD (178). Pertinent studies have reported that among females with preparatory RA, RF-positive patients were more positive for anti-TRIM21 than RF-negative patients, and both were concurrently positive for anti-Ro60 (183). Recently, researchers have proposed another possible pathway: the immune response of RA-ILD is located in the synovial tissue, where Th cells produce specific cytokines that spread the inflammation to the lungs, causing fibroblasts to differentiate into myofibroblasts, ultimately leading to ILD (179). Moreover, current studies have shown that lung tissue macrophages express TRIM21 and increased TRIM21 expression in monocytes has been found to promote Th1 and Th17 differentiation in other diseases, which seems to predict that TRIM21 positively regulates the disease process of RA-ILD in the course of RA-ILD, but the specific mechanism has not yet been reported in any study (126, 184).
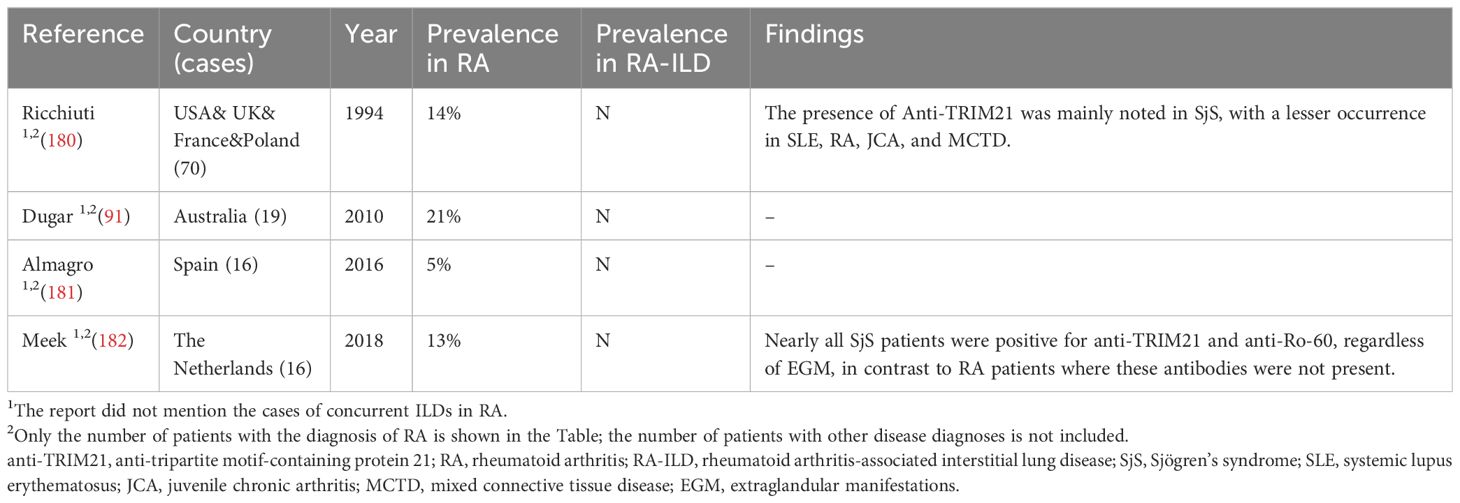
Table 5 Main studies’ results regarding the prevalence of anti-TRIM21 antibody in RA-ILD.
RA patients with anti-TRIM21 positivity often have concurrent with other subtypes of CTD and ILD. The presence of anti-TRIM21 antibodies may indicate the occurrence and progression of ILD, but the TRIM21 antigen may only indirectly influence RA in the context of other concurrent CTD (185). However, there are currently no reports on TRIM21 in RA and RA-ILD, and the diagnostic efficacy of anti-TRIM21 in diagnosing RA-ILD remains controversial. The molecular mechanisms executed by TRIM21 in RA-ILD are still a mystery.
5 Conclusions and future directions
Differences in the structural domains and functions with which TRIM21 interacts in immune activity give it multiple roles in the immune response. TRIM21, as an intracellular receptor whose PRY/SPRY domain binds to the Fc region of the antibody complex, is of great significance for the intracellular detection of pathogens that have escaped neutralization by extracellular antibodies and have entered into the host cell. Subsequently, TRIM21 is involved in the regulation of immune signaling through its E3 ubiquitin ligase activity in the ubiquitination and degradation of key proteins and pathogen complexes within immune cells.
Differences in the regulation of different molecules by TRIM21 lead to its high requirement for target specificity in different immune cells. Variations in how TRIM21 regulates distinct molecules during inflammation result in it displaying contrasting immune responses, either anti-inflammatory or pro-inflammatory, across various infectious diseases. It’s a recognized fact that increased levels of IFN enhance TRIM21 expression, yet, paradoxically, heightened TRIM21 levels lead to the ubiquitination of IRF, thereby attenuating the immune effects produced by IFN. Therefore, TRIM21 may act as a lever to maintain and regulate immune homeostasis through interactions with endogenous proteins, though its underlying mechanisms still require further investigation. Additionally, whether TRIM21’s interactions with different signaling proteins play a decisive role in the maturation and differentiation of immune cells also needs more demonstration.
Dysfunctions in TRIM21 structure or function have been associated with autoimmune imbalances in the body. Mutated TRIM21 may be recognized by the immune system as an intracellular antigen and produce anti-TRIM21 antibodies, but it is not known whether TRIM21 can be located on the cell surface. In addition, impairment of TRIM21 ubiquitination activity may also lead to the production of anti-TRIM21 antibodies, which may result in relevant cellular dysfunction in the body, followed by apoptosis that exposes TRIM21 to the cell surface and induces a stronger immune response leading to autoimmune diseases such as CTD-ILD, (Figure 6).
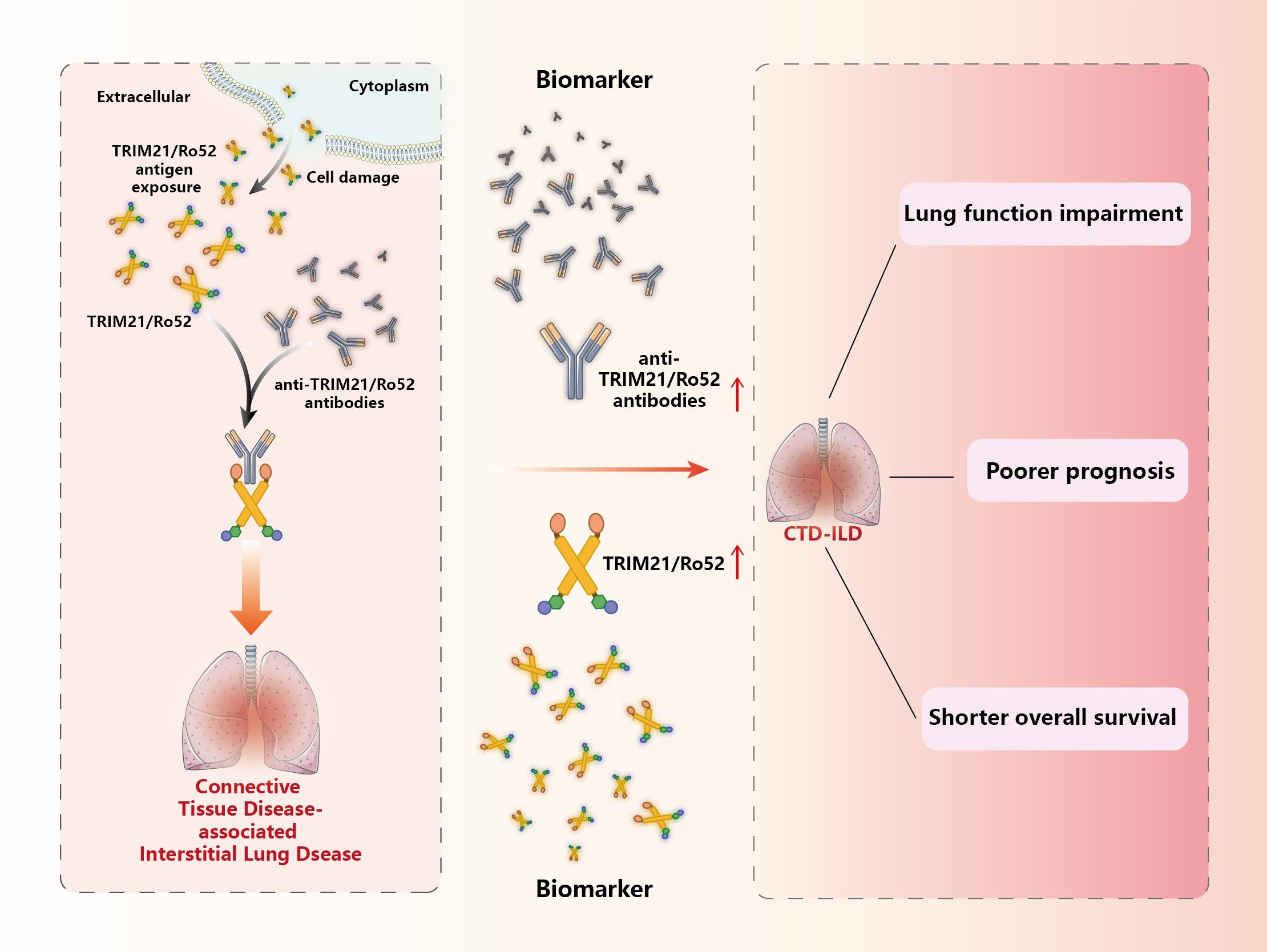
Figure 6 TRIM21/Ro52 and autoantibodies with CTD-ILD. TRIM21/Ro52, as the antigen targeted by specific anti-TRIM21/Ro52 antibodies, exists as an antigen that disrupts innate immunity, triggering CTD-associated ILD. TRIM21/Ro52 and anti-TRIM21/Ro52 antibodies, as potential biomarkers for CTD-ILD, may be related to lung tissue and functional damage, poorer prognosis, and shorter survival in CTD-ILD patients.
TRIM21 is highly expressed in lungs compared to other tissues due to its high antigenicity (186). Additionally, viral infections are a trigger for various types of CTD, and TRIM21, as a cytosolic receptor for IgG antibodies, plays a crucial role in combating viral infections (187). Therefore, more preclinical studies are needed to explore whether TRIM21 and its autoantibodies accumulate in target organs before the onset of CTD-ILD and why they only affect specific tissue organs. In addition, the fact that TRIM21 maintains the anti-inflammatory properties of lung endothelial cells may be partial evidence for the existence of a correlation between anti-TRIM21 and CTD-ILD, but the role of TRIM21 in other lung tissue constituents remains an unsolved mystery and needs to be further investigated. Furthermore, TRIM21 maintains the anti-inflammatory characteristics of lung endothelial cells, potentially contributing to the proof of a link between anti-TRIM21 and CTD-ILD. However, the function of TRIM21 in additional lung tissue elements is still a mystery and requires more research.
Anti-TRIM21 has a high rate of seropositivity for autoantibodies in patients with CTD, and preclinical research indicates its potential role in CTD progression. Nonetheless, the anti-TRIM21 may yield positive results in serum samples from individuals with different infections or tumors. Consequently, its limited specificity has sparked clinical debate regarding its effectiveness as a diagnostic biomarker for CTD-ILD. Current clinical evidence suggests that the presence of anti-TRIM21 antibodies is associated with an increased incidence of concomitant ILD in SjS, SSc, and IIM, with a higher probability of ILD occurring and being more severe, usually predicting poorer outcomes and survival. Intriguingly, most of the relevant published studies are retrospective, and retrospective studies suffer from flaws such as potential bias, impact of treatment, and the lack of adjustment for person-years at risk. Moreover, anti-TRIM21 antibody detection is mostly qualitative or semi-quantitative, and the actual level of autoantibodies may be another important factor in diagnosing the probability of occurrence of CTD-ILD. The above influencing factors limit our ability to correctly assess the diagnostic value of anti-TRIM21 in CTD-ILD. Therefore, more studies are needed to verify whether anti-TRIM21 is a major biological marker for assessing the probability of CTD-ILD occurrence. Nonetheless, the available studies appear to be persuasive that patients with CTD who tests positive for anti-TRIM21 have a higher probability of concurrent ILD, and a high titer of anti-TRIM21 correlates with worse outcomes and shorter overall survival in CTD-ILD patients. However, a more detailed investigation into the connection between anti-TRIM21 and CTD-ILD is required.
Furthermore, the reason why anti-TRIM21 antibodies target the lungs rather than other organs and the significant variability among CTD-ILD patients, seems to suggest a link to immunogenetic factors. Therefore, the influence of genetic factors on TRIM21 and anti-TRIM21 remains a question to be explored.
In general, there are very limited preclinical studies on TRIM21 and anti-TRIM21 antibodies in CTD and CTD-ILD, mostly published data from clinical cohort studies. In this review, we attempt to speculate on the potential molecular mechanisms of TRIM21 and anti-TRIM21 antibodies in CTD-ILD based on the very limited existing foundational research. However, CTD-ILD affects multiple organs and involves complex interactions between molecules, posing significant challenges for studying TRIM21 in CTD-ILD. Therefore, unraveling the molecular mechanisms of TRIM21 in the pathogenesis of CTD-ILD still has a long way to go.
Author contributions
XG: Conceptualization, Investigation, Methodology, Visualization, Writing – original draft. SH: Investigation, Writing – review & editing, Methodology, Resources. PC: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mira-Avendano I, Abril A, Burger CD, Dellaripa PF, Fischer A, Gotway MB, et al. Interstitial lung disease and other pulmonary manifestations in connective tissue diseases. Mayo Clin Proc. (2019) 94:309–25. doi: 10.1016/j.mayocp.2018.09.002
2. Azadeh N, Limper AH, Carmona EM, Ryu JH. The role of infection in interstitial lung diseases: A review. Chest. (2017) 152:842–52. doi: 10.1016/j.chest.2017.03.033
3. Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ. (2016) 352:h6819. doi: 10.1136/bmj.h6819
4. England BR, Hershberger D. Management issues in rheumatoid arthritis-associated interstitial lung disease. Curr Opin Rheumatol. (2020) 32:255–63. doi: 10.1097/BOR.0000000000000703
5. Vacchi C, Sebastiani M, Cassone G, Cerri S, Della Casa G, Salvarani C, et al. Therapeutic options for the treatment of interstitial lung disease related to connective tissue diseases. A narrative review. J Clin Med. (2020) 9:407. doi: 10.3390/jcm9020407
6. Ben-Chetrit E, Chan EK, Sullivan KF, Tan EM. A 52-kD protein is a novel component of the SS-A/Ro antigenic particle. J Exp Med. (1988) 167:1560–71. doi: 10.1084/jem.167.5.1560
7. Itoh K, Itoh Y, Frank MB. Protein heterogeneity in the human Ro/SSA ribonucleoproteins. The 52- and 60-kD Ro/SSA autoantigens are encoded by separate genes. J Clin Invest. (1991) 87:177–86. doi: 10.1172/JCI114968
8. Yang YS, Yang MC, Wang B, Weissler JC. Autoantigen Ro52 directly interacts with human IgG heavy chain in vivo in mammalian cells. Mol Immunol. (2000) 37:591–602. doi: 10.1016/s0161-5890(00)00068-7
9. Plotz PH. The autoantibody repertoire: searching for order. Nat Rev Immunol. (2003) 3:73–8. doi: 10.1038/nri976
10. Yang W, Gu Z, Zhang H, Hu H. To TRIM the immunity: from innate to adaptive immunity. Front Immunol. (2020) 11:2157. doi: 10.3389/fimmu.2020.02157
11. Di Rienzo M, Romagnoli A, Antonioli M, Piacentini M, Fimia GM. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. (2020) 27:887–902. doi: 10.1038/s41418-020-0495-2
12. Dahl MLN, Mikkelsen JH, Hvid M, Korsholm TL, Nielsen KO, Andersen CBF, et al. Validation of an indirect ELISA assay for assessment of autoantibodies against full-length TRIM21 and its individual domains. Scand J Clin Lab Invest. (2023) 83:309–17. doi: 10.1080/00365513.2023.2221862
13. Chan EK, Hamel JC, Buyon JP, Tan EM. Molecular definition and sequence motifs of the 52-kD component of human SS-A/Ro autoantigen. J Clin Invest. (1991) 87:68–76. doi: 10.1172/JCI115003
14. Ohtake F, Tsuchiya H. The emerging complexity of ubiquitin architecture. J Biochem. (2017) 161:125–33. doi: 10.1093/jb/mvw088
15. Kiss L, Clift D, Renner N, Neuhaus D, James LC. RING domains act as both substrate and enzyme in a catalytic arrangement to drive self-anchored ubiquitination. Nat Commun. (2021) 12:1220. doi: 10.1038/s41467-021-21443-6
16. Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF, James LC. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc Natl Acad Sci. (2015) 112:10014–9. doi: 10.1073/pnas.1507534112
17. Gomez-Martin D, Galindo-Feria AS, Barrera-Vargas A, Merayo-Chalico J, Juarez-Vega G, Torres-Ruiz J, et al. Ro52/TRIM21-deficient expression and function in different subsets of peripheral blood mononuclear cells is associated with a proinflammatory cytokine response in patients with idiopathic inflammatory myopathies. Clin Exp Immunol. (2017) 188:154–62. doi: 10.1111/cei.12914
18. Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol. (2013) 14:172–8. doi: 10.1038/ni.2492
19. Chen Y, Bian H, Lv J, Song W, Xing C, Hui C, et al. Gelsevirine is a novel STING-specific inhibitor and mitigates STING-related inflammation in sepsis. Front Immunol. (2023) 14:1190707. doi: 10.3389/fimmu.2023.1190707
20. Zeng J, Santos AF, Mukadam AS, Osswald M, Jacques DA, Dickson CF, et al. Target-induced clustering activates Trim-Away of pathogens and proteins. Nat Struct Mol Biol. (2021) 28:278–89. doi: 10.1038/s41594-021-00560-2
21. Kiss L, Rhinesmith T, Luptak J, Dickson CF, Weidenhausen J, Smyly S, et al. Trim-Away ubiquitinates and degrades lysine-less and N-terminally acetylated substrates. Nat Commun. (2023) 14:2160. doi: 10.1038/s41467-023-37504-x
22. Wallenhammar A, Anandapadamanaban M, Lemak A, Mirabello C, Lundstrom P, Wallner B, et al. Solution NMR structure of the TRIM21 B-box2 and identification of residues involved in its interaction with the RING domain. PloS One. (2017) 12:e0181551. doi: 10.1371/journal.pone.0181551
23. Dickson C, Fletcher AJ, Vaysburd M, Yang JC, Mallery DL, Zeng J, et al. Intracellular antibody signalling is regulated by phosphorylation of the Fc receptor TRIM21. Elife. (2018) 7:e32660. doi: 10.7554/eLife.32660
24. Esposito D, Koliopoulos MG, Rittinger K. Structural determinants of TRIM protein function. Biochem Soc Trans. (2017) 45:183–91. doi: 10.1042/BST20160325
25. Ottosson L, Hennig J, Espinosa A, Brauner S, Wahren-Herlenius M, Sunnerhagen M. Structural, functional and immunologic characterization of folded subdomains in the Ro52 protein targeted in Sjogren’s syndrome. Mol Immunol. (2006) 43:588–98. doi: 10.1016/j.molimm.2005.04.013
26. Fiorentini F, Esposito D, Rittinger K. Does it take two to tango? RING domain self-association and activity in TRIM E3 ubiquitin ligases. Biochem Soc Trans. (2020) 48:2615–24. doi: 10.1042/BST20200383
27. Dawidziak DM, Sanchez JG, Wagner JM, Ganser-Pornillos BK, Pornillos O. Structure and catalytic activation of the TRIM23 RING E3 ubiquitin ligase. Proteins. (2017) 85:1957–61. doi: 10.1002/prot.25348
28. Rhodes DA, Trowsdale J. TRIM21 is a trimeric protein that binds IgG Fc via the B30.2 domain. Mol Immunol. (2007) 44:2406–14. doi: 10.1016/j.molimm.2006.10.013
29. Keeble AH, Khan Z, Forster A, James LC. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci U.S.A. (2008) 105:6045–50. doi: 10.1073/pnas.0800159105
30. Bidgood SR, Tam JC, McEwan WA, Mallery DL, James LC. Translocalized IgA mediates neutralization and stimulates innate immunity inside infected cells. Proc Natl Acad Sci U.S.A. (2014) 111:13463–8. doi: 10.1073/pnas.1410980111
31. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc Natl Acad Sci U.S.A. (2010) 107:19985–90. doi: 10.1073/pnas.1014074107
32. Xie S, Zhang L, Dong D, Ge R, He Q, Fan C, et al. HDAC6 regulates antibody-dependent intracellular neutralization of viruses via deacetylation of TRIM21. J Biol Chem. (2020) 295:14343–51. doi: 10.1074/jbc.RA119.011006
33. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med. (2009) 206:1661–71. doi: 10.1084/jem.20090585
34. Harbers M, Nomura T, Ohno S, Ishii S. Intracellular localization of the Ret finger protein depends on a functional nuclear export signal and protein kinase C activation. J Biol Chem. (2001) 276:48596–607. doi: 10.1074/jbc.M108077200
35. Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. (1995) 82:463–73. doi: 10.1016/0092-8674(95)90435-2
36. Plafker SM, Plafker KS, Weissman AM, Macara IG. Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J Cell Biol. (2004) 167:649–59. doi: 10.1083/jcb.200406001
37. Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun. (2006) 339:415–21. doi: 10.1016/j.bbrc.2005.11.029
38. Nobuhara Y, Kawano S, Kageyama G, Sugiyama D, Saegusa J, Kumagai S. Is SS-A/Ro52 a hydrogen peroxide-sensitive signaling molecule? Antioxid Redox Signal. (2007) 9:385–91. doi: 10.1089/ars.2006.1480
39. Strandberg L, Ambrosi A, Espinosa A, Ottosson L, Eloranta ML, Zhou W, et al. Interferon-alpha induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J Clin Immunol. (2008) 28:220–31. doi: 10.1007/s10875-007-9157-0
40. Espinosa A, Oke V, Elfving A, Nyberg F, Covacu R, Wahren-Herlenius M. The autoantigen Ro52 is an E3 ligase resident in the cytoplasm but enters the nucleus upon cellular exposure to nitric oxide. Exp Cell Res. (2008) 314:3605–13. doi: 10.1016/j.yexcr.2008.09.011
41. McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat Immunol. (2013) 14:327–36. doi: 10.1038/ni.2548
42. McEwan W. Surveillance for intracellular antibody by cytosolic fc receptor TRIM21. Antibodies. (2016) 5:21. doi: 10.3390/antib5040021
43. Fletcher AJ, James LC, Tsai B. Coordinated neutralization and immune activation by the cytosolic antibody receptor TRIM21. J Virol. (2016) 90:4856–9. doi: 10.1128/jvi.00050-16
44. Rhodes DA, Ihrke G, Reinicke AT, Malcherek G, Towey M, Isenberg DA, et al. The 52 000 MW Ro/SS-A autoantigen in Sjogren’s syndrome/systemic lupus erythematosus (Ro52) is an interferon-gamma inducible tripartite motif protein associated with membrane proximal structures. Immunology. (2002) 106:246–56. doi: 10.1046/j.1365-2567.2002.01417.x
45. Rajsbaum R, Stoye JP, O’Garra A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol. (2008) 38:619–30. doi: 10.1002/eji.200737916
46. Sjostrand M, Ambrosi A, Brauner S, Sullivan J, Malin S, Kuchroo VK, et al. Expression of the immune regulator tripartite-motif 21 is controlled by IFN regulatory factors. J Immunol. (2013) 191:3753–63. doi: 10.4049/jimmunol.1202341
47. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct inhibition of IRF-dependent transcriptional regulatory mechanisms associated with disease. Front Immunol. (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
48. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. (2014) 26:192–7. doi: 10.1016/j.cellsig.2013.11.004
49. Lenart M, Rutkowska-Zapala M, Szatanek R, Weglarczyk K, Stec M, Bukowska-Strakova K, et al. Alterations of TRIM21-mRNA expression during monocyte maturation. Immunobiology. (2017) 222:494–8. doi: 10.1016/j.imbio.2016.10.016
50. Jiang MX, Hong X, Liao BB, Shi SZ, Lai XF, Zheng HY, et al. Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci Rep. (2017) 7:42781. doi: 10.1038/srep42781
51. Kong X, Lu X, Wang S, Hao J, Guo D, Wu H, et al. Type I interferon/STAT1 signaling regulates UBE2M-mediated antiviral innate immunity in a negative feedback manner. Cell Rep. (2023) 42:112002. doi: 10.1016/j.celrep.2023.112002
52. Lu X, Kong X, Wu H, Hao J, Li S, Gu Z, et al. UBE2M-mediated neddylation of TRIM21 regulates obesity-induced inflammation and metabolic disorders. Cell Metab. (2023) 35:1390–1405 e8. doi: 10.1016/j.cmet.2023.05.011
53. Serrano-Macia M, Simon J, Gonzalez-Rellan MJ, Azkargorta M, Goikoetxea-Usandizaga N, Lopitz-Otsoa F, et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol Metab. (2021) 53:101275. doi: 10.1016/j.molmet.2021.101275
54. Bhaskaran N, Faddoul F, Paes da Silva A, Jayaraman S, Schneider E, Mamileti P, et al. IL-1beta-myD88-mTOR axis promotes immune-protective IL-17A(+)Foxp3(+) cells during mucosal infection and is dysregulated with aging. Front Immunol. (2020) 11:595936. doi: 10.3389/fimmu.2020.595936
55. Yao P, Chen T, Jiang P, Li L, Du W. Functional skewing of TRIM21-SIRT5 interplay dictates IL-1beta production in DSS-induced colitis. EMBO Rep. (2022) 23:e54391. doi: 10.15252/embr.202154391
56. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
57. Li Z, Liu X, Zhang X, Zhang W, Gong M, Qin X, et al. TRIM21 aggravates cardiac injury after myocardial infarction by promoting M1 macrophage polarization. Front Immunol. (2022) 13:1053171. doi: 10.3389/fimmu.2022.1053171
58. Li X, Wang W, Shao Y, Zhou J, Huang J, Xu F, et al. LncTRPM2-AS inhibits TRIM21-mediated TRPM2 ubiquitination and prevents autophagy-induced apoptosis of macrophages in asthma. Cell Death Dis. (2021) 12:1153. doi: 10.1038/s41419-021-04437-6
59. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. (2012) 12:557–69. doi: 10.1038/nri3254
60. Zhong B, Zhang L, Lei C, Li Y, Mao A-P, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
61. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. (2011) 12:959–65. doi: 10.1038/ni.2091
62. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
63. Agod Z, Pazmandi K, Bencze D, Vereb G, Biro T, Szabo A, et al. Signaling lymphocyte activation molecule family 5 enhances autophagy and fine-tunes cytokine response in monocyte-derived dendritic cells via stabilization of interferon regulatory factor 8. Front Immunol. (2018) 9:62. doi: 10.3389/fimmu.2018.00062
64. Ng PML, Kaliaperumal N, Lee CY, Chin WJ, Tan HC, Au VB, et al. Enhancing antigen cross-presentation in human monocyte-derived dendritic cells by recruiting the intracellular fc receptor TRIM21. J Immunol. (2019) 202:2307–19. doi: 10.4049/jimmunol.1800462
65. Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. (1999) 189:371–80. doi: 10.1084/jem.189.2.371
66. Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci U.S.A. (2011) 108:9927–32. doi: 10.1073/pnas.1019037108
67. Drouin M, Saenz J, Gauttier V, Evrard B, Teppaz G, Pengam S, et al. CLEC-1 is a death sensor that limits antigen cross-presentation by dendritic cells and represents a target for cancer immunotherapy. Sci Adv. (2022) 8:eabo7621. doi: 10.1126/sciadv.abo7621
68. Ishii T, Ohnuma K, Murakami A, Takasawa N, Yamochi T, Iwata S, et al. SS-A/Ro52, an autoantigen involved in CD28-mediated IL-2 production. J Immunol. (2003) 170:3653–61. doi: 10.4049/jimmunol.170.7.3653
69. Zhou G, Wu W, Yu L, Yu T, Yang W, Wang P, et al. Tripartite motif-containing (TRIM) 21 negatively regulates intestinal mucosal inflammation through inhibiting T(H)1/T(H)17 cell differentiation in patients with inflammatory bowel diseases. J Allergy Clin Immunol. (2018) 142:1218–1228 e12. doi: 10.1016/j.jaci.2017.09.038
70. Bolland S, Garcia-Sastre A. Vicious circle: systemic autoreactivity in Ro52/TRIM21-deficient mice. J Exp Med. (2009) 206:1647–51. doi: 10.1084/jem.20091507
71. Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. (2008) 181:1780–6. doi: 10.4049/jimmunol.181.3.1780
72. Brauner S, Jiang X, Thorlacius GE, Lundberg AM, Ostberg T, Yan ZQ, et al. Augmented Th17 differentiation in Trim21 deficiency promotes a stable phenotype of atherosclerotic plaques with high collagen content. Cardiovasc Res. (2018) 114:158–67. doi: 10.1093/cvr/cvx181
73. Wei W, Wang Y, Sun Q, Jiang C, Zhu M, Song C, et al. Enhanced T-cell proliferation and IL-6 secretion mediated by overexpression of TRIM21 in oral lesions of patients with oral lichen planus. J Oral Pathol Med. (2020) 49:350–6. doi: 10.1111/jop.12938
74. Yao Y, Pan L, Wei Y, Feng M, Li X, Sun L, et al. TRIM21 promotes inflammation by ubiquitylating NF-kappaB in T cells of oral lichen planus. J Oral Pathol Med. (2023) 52:448–55. doi: 10.1111/jop.13428
75. Brauner S, Zhou W, Backlin C, Green TM, Folkersen L, Ivanchenko M, et al. Reduced expression of TRIM21/Ro52 predicts poor prognosis in diffuse large B-cell lymphoma patients with and without rheumatic disease. J Intern Med. (2015) 278:323–32. doi: 10.1111/joim.12375
76. Grammer AC, Lipsky PE. B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther. (2003) 5 Suppl 4:S22–7. doi: 10.1186/ar1009
77. Jego G, Pascual V, Palucka AK, Banchereau J. Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun. (2005) 8:124–39. doi: 10.1159/000082101
78. Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PloS One. (2014) 9:e103609. doi: 10.1371/journal.pone.0103609
79. Savitsky DA, Yanai H, Tamura T, Taniguchi T, Honda K. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proc Natl Acad Sci U.S.A. (2010) 107:10154–9. doi: 10.1073/pnas.1005599107
80. Bracken SJ, Suthers AN, DiCioccio RA, Su H, Anand S, Poe JC, et al. Heightened TLR7 signaling primes BCR-activated B cells in chronic graft-versus-host disease for effector functions. Blood Adv. (2024) 8:667–80. doi: 10.1182/bloodadvances.2023010362
81. Cosgrove HA, Gingras S, Kim M, Bastacky S, Tilstra JS, Shlomchik MJ. B cell-intrinsic TLR7 expression drives severe lupus in TLR9-deficient mice. JCI Insight. (2023) 8:e172219. doi: 10.1172/jci.insight.172219
82. Yang SH, Gao CY, Li L, Chang C, Leung PSC, Gershwin ME, et al. The molecular basis of immune regulation in autoimmunity. Clin Sci. (2018) 132:43–67. doi: 10.1042/cs20171154
83. Strand V, Goncalves J, Isaacs JD. Immunogenicity of biologic agents in rheumatology. Nat Rev Rheumatol. (2020) 17:81–97. doi: 10.1038/s41584-020-00540-8
84. Ramos-Casals M, Tzioufas AG, Font J. Primary Sjogren’s syndrome: new clinical and therapeutic concepts. Ann Rheum Dis. (2005) 64:347–54. doi: 10.1136/ard.2004.025676
85. Natalini JG, Johr C, Kreider M. Pulmonary involvement in sjogren syndrome. Clin Chest Med. (2019) 40:531–44. doi: 10.1016/j.ccm.2019.05.002
86. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjogren’s syndrome. Eur Respir Rev. (2016) 25:110–23. doi: 10.1183/16000617.0011-2016
87. Palm O, Garen T, Berge Enger T, Jensen JL, Lund MB, Aalokken TM, et al. Clinical pulmonary involvement in primary Sjogren’s syndrome: prevalence, quality of life and mortality–a retrospective study based on registry data. Rheumatol (Oxford). (2013) 52:173–9. doi: 10.1093/rheumatology/kes311
88. Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, et al. Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Med (Baltimore). (2008) 87:210–9. doi: 10.1097/MD.0b013e318181e6af
89. Anderson JR, Gray KG, Beck JS, Kinnear WF. Precipitating autoantibodies in Sjogren’s disease. Lancet. (1961) 2:456–60. doi: 10.1016/s0140-6736(61)92430-8
90. Ben-Chetrit E, Fox RI, Tan EM. Dissociation of immune responses to the SS-A (Ro) 52-kd and 60-kd polypeptides in systemic lupus erythematosus and Sjogren’s syndrome. Arthritis Rheum. (1990) 33:349–55. doi: 10.1002/art.1780330307
91. Dugar M, Cox S, Limaye V, Gordon TP, Roberts-Thomson PJ. Diagnostic utility of anti-Ro52 detection in systemic autoimmunity. Postgrad Med J. (2010) 86:79–82. doi: 10.1136/pgmj.2009.089656
92. Lee AYS, Putty T, Lin MW, Swaminathan S, Suan D, Chataway T, et al. Isolated anti-Ro52 identifies a severe subset of Sjogren’s syndrome patients. Front Immunol. (2023) 14:1115548. doi: 10.3389/fimmu.2023.1115548
93. Locht H, Pelck R, Manthorpe R. Diagnostic and prognostic significance of measuring antibodies to alpha-fodrin compared to anti-Ro-52, anti-Ro-60, and anti-La in primary Sjögren’s syndrome. J Rheumatol. (2008) 35:845–9.
94. Zhao L, Yao L, Yuan L, Xia L, Shen H, Lu J. Potential contribution of interleukin-33 to the development of interstitial lung disease in patients with primary Sjogren’s Syndrome. Cytokine. (2013) 64:22–4. doi: 10.1016/j.cyto.2013.07.006
95. Dong X, Zhou J, Guo X, Li Y, Xu Y, Fu Q, et al. A retrospective analysis of distinguishing features of chest HRCT and clinical manifestation in primary Sjogren’s syndrome-related interstitial lung disease in a Chinese population. Clin Rheumatol. (2018) 37:2981–8. doi: 10.1007/s10067-018-4289-6
96. Shi L, Fu Q, Chen N, Liu R, Zheng Y. Angiopoietin-like protein 2 as a novel marker for patients with primary Sjogren’s syndrome-related interstitial lung disease. Clin Exp Med. (2020) 20:393–9. doi: 10.1007/s10238-020-00623-6
97. Zhang T, Yuan F, Xu L, Sun W, Liu L, Xue J. Characteristics of patients with primary Sjogren’s syndrome associated interstitial lung disease and relevant features of disease progression. Clin Rheumatol. (2020) 39:1561–8. doi: 10.1007/s10067-019-04906-6
98. Xu Y, Zhou J, Dong X, Guo X, Lu Y, Zheng Y. Risk factors for progression and prognosis of primary Sjogren’s syndrome-associated interstitial lung disease in a Chinese population. Int J Rheum Dis. (2020) 23:1734–40. doi: 10.1111/1756-185X.14023
99. Zhao R, Wang Y, Zhou W, Guo J, He M, Li P, et al. Associated factors with interstitial lung disease and health-related quality of life in Chinese patients with primary Sjogren’s syndrome. Clin Rheumatol. (2020) 39:483–9. doi: 10.1007/s10067-019-04753-5
100. Guo T, Long Y, Shen Q, Guo W, Duan W, Ouyang X, et al. Clinical profiles of SS-ILD compared with SS-NILD in a Chinese population: a retrospective analysis of 735 patients. Ann Med. (2021) 53:1340–8. doi: 10.1080/07853890.2021.1965205
101. Shi L, Wang J, Guo HX, Han XL, Tang YP, Liu GY. Circulating Th2 cell reduction and Th1/Th2 imbalance are correlated with primary Sjogren’s syndrome-associated interstitial lung disease. Arthritis Res Ther. (2022) 24:121. doi: 10.1186/s13075-022-02811-z
102. Yang Z, Zhao H, Shan L, Wang D. Clinical features and risk factors for primary Sjogren’s syndrome combined with interstitial lung disease: a retrospective study. Acta Biochim Pol. (2024) 71:12461. doi: 10.3389/abp.2024.12461
103. Wang Y, Jia Y, Qin Y, Feng M, Liang Z, Zhao X, et al. Predicting the risk of interstitial lung disease in patients with primary Sjogren’s syndrome: Novel nomogram and elevated Th2 cells. Mol Immunol. (2024) 168:25–37. doi: 10.1016/j.molimm.2024.02.008
104. Aqrawi LA, Kvarnstrom M, Brokstad KA, Jonsson R, Skarstein K, Wahren-Herlenius M. Ductal epithelial expression of Ro52 correlates with inflammation in salivary glands of patients with primary Sjogren’s syndrome. Clin Exp Immunol. (2014) 177:244–52. doi: 10.1111/cei.12341
105. Aqrawi LA, Skarstein K, Oijordsbakken G, Brokstad KA. Ro52- and Ro60-specific B cell pattern in the salivary glands of patients with primary Sjogren’s syndrome. Clin Exp Immunol. (2013) 172:228–37. doi: 10.1111/cei.12058
106. Szczerba BM, Kaplonek P, Wolska N, Podsiadlowska A, Rybakowska PD, Dey P, et al. Interaction between innate immunity and Ro52-induced antibody causes Sjogren’s syndrome-like disorder in mice. Ann Rheum Dis. (2016) 75:617–22. doi: 10.1136/annrheumdis-2014-206297
107. Hillen MR, Urso K, Koppe E, Lopes AP, Blokland SLM, Pandit A, et al. Autoantigen TRIM21/Ro52 is expressed on the surface of antigen-presenting cells and its enhanced expression in Sjogren’s syndrome is associated with B cell hyperactivity and type I interferon activity. RMD Open. (2020) 6:e001184. doi: 10.1136/rmdopen-2020-001184
108. Nishihata SY, Shimizu T, Umeda M, Furukawa K, Ohyama K, Kawakami A, et al. The toll-like receptor 7-mediated ro52 antigen-presenting pathway in the salivary gland epithelial cells of sjogren’s syndrome. J Clin Med. (2023) 12:4423. doi: 10.3390/jcm12134423
109. Bozic B, Pruijn GJ, Rozman B, van Venrooij WJ. Sera from patients with rheumatic diseases recognize different epitope regions on the 52-kD Ro/SS-A protein. Clin Exp Immunol. (1993) 94:227–35. doi: 10.1111/j.1365-2249.1993.tb03436.x
110. Sroka M, Bagavant H, Biswas I, Ballard A, Deshmukh US. Immune response against the coiled coil domain of Sjögren’s syndrome associated autoantigen Ro52 induces salivary gland dysfunction. Clin Exp Rheumatol. (2018) 112:41–6.
111. Espinosa A, Hennig J, Ambrosi A, Anandapadmanaban M, Abelius MS, Sheng Y, et al. Anti-Ro52 autoantibodies from patients with Sjogren’s syndrome inhibit the Ro52 E3 ligase activity by blocking the E3/E2 interface. J Biol Chem. (2011) 286:36478–91. doi: 10.1074/jbc.M111.241786
112. Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. (2020) 8:304–20. doi: 10.1016/S2213-2600(19)30480-1
113. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. (2007) 66:940–4. doi: 10.1136/ard.2006.066068
114. Akter T, Silver RM, Bogatkevich GS. Recent advances in understanding the pathogenesis of scleroderma-interstitial lung disease. Curr Rheumatol Rep. (2014) 16:411. doi: 10.1007/s11926-014-0411-1
115. Denton CP, Khanna D. Systemic sclerosis. Lancet. (2017) 390:1685–99. doi: 10.1016/S0140-6736(17)30933-9
116. Fujimoto M, Shimozuma M, Yazawa N, Kubo M, Ihn H, Sato S, et al. Prevalence and clinical relevance of 52-kDa and 60-kDa Ro/SS-A autoantibodies in Japanese patients with systemic sclerosis. Ann Rheum Dis. (1997) 56:667–70. doi: 10.1136/ard.56.11.667
117. Parker JC, Burlingame RW, Bunn CC. Prevalence of antibodies to Ro-52 in a serologically defined population of patients with systemic sclerosis. J Autoimmune Dis. (2009) 6:2. doi: 10.1186/1740-2557-6-2
118. Mierau R, Moinzadeh P, Riemekasten G, Melchers I, Meurer M, Reichenberger F, et al. Frequency of disease-associated and other nuclear autoantibodies in patients of the German Network for Systemic Scleroderma: correlation with characteristic clinical features. Arthritis Res Ther. (2011) 13:R172. doi: 10.1186/ar3495
119. Hudson M, Pope J, Mahler M, Tatibouet S, Steele R, Baron M, et al. Clinical significance of antibodies to Ro52/TRIM21 in systemic sclerosis. Arthritis Res Ther. (2012) 14:R50. doi: 10.1186/ar3763
120. Massie C, Hudson M, Tatibouet S, Steele R, Huynh T, Fritzler MJ, et al. Absence of an association between anti-Ro antibodies and prolonged QTc interval in systemic sclerosis: a multicenter study of 689 patients. Semin Arthritis Rheum. (2014) 44:338–44. doi: 10.1016/j.semarthrit.2014.07.001
121. Wodkowski M, Hudson M, Proudman S, Walker J, Stevens W, Nikpour M, et al. Monospecific anti-Ro52/TRIM21 antibodies in a tri-nation cohort of 1574 systemic sclerosis subjects: evidence of an association with interstitial lung disease and worse survival. Clin Exp Rheumatol. (2015) 33:S131–5.
122. Lee A, Patterson KA, Tan DJ, Wilson ME, Proudman SM, Stevens W, et al. Anti-Ro52/TRIM21 is independently associated with pulmonary arterial hypertension and mortality in a cohort of systemic sclerosis patients. Scand J Rheumatol. (2021) 50:469–74. doi: 10.1080/03009742.2021.1887927
123. Hoa S, Lazizi S, Baron M, Wang M, Fritzler MJ, Hudson M, et al. Association between autoantibodies in systemic sclerosis and cancer in a national registry. Rheumatol (Oxford). (2022) 61:2905–14. doi: 10.1093/rheumatology/keab735
124. Jung E, Suh CH, Kim HA, Jung JY. Clinical characteristics of systemic sclerosis with interstitial lung disease. Arch Rheumatol. (2018) 33:322–7. doi: 10.5606/ArchRheumatol.2018.6630
125. Ghillani P, Andre C, Toly C, Rouquette AM, Bengoufa D, Nicaise P, et al. Clinical significance of anti-Ro52 (TRIM21) antibodies non-associated with anti-SSA 60kDa antibodies: results of a multicentric study. Autoimmun Rev. (2011) 10:509–13. doi: 10.1016/j.autrev.2011.03.004
126. Hamberg V, Sohrabian A, Volkmann ER, Wildt M, Lofdahl A, Wuttge DM, et al. Anti-Ro52 positivity is associated with progressive interstitial lung disease in systemic sclerosis-an exploratory study. Arthritis Res Ther. (2023) 25:162. doi: 10.1186/s13075-023-03141-4
127. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
128. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res. (2018) 19:186. doi: 10.1186/s12931-018-0891-z
129. Zhang R, Shen Y, Zhang Q, Feng X, Liu X, Huo X, et al. TRIM21-mediated Sohlh2 ubiquitination suppresses M2 macrophage polarization and progression of triple-negative breast cancer. Cell Death Dis. (2023) 14:850. doi: 10.1038/s41419-023-06383-x
130. Lafyatis R, O’Hara C, Feghali-Bostwick CA, Matteson E. B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum. (2007) 56:3167–8. doi: 10.1002/art.22847
131. Saito E, Fujimoto M, Hasegawa M, Komura K, Hamaguchi Y, Kaburagi Y, et al. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J Clin Invest. (2002) 109:1453–62. doi: 10.1172/JCI15078
132. Asano N, Fujimoto M, Yazawa N, Shirasawa S, Hasegawa M, Okochi H, et al. B Lymphocyte signaling established by the CD19/CD22 loop regulates autoimmunity in the tight-skin mouse. Am J Pathol. (2004) 165:641–50. doi: 10.1016/S0002-9440(10)63328-7
133. Dorner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. (2019) 393:2344–58. doi: 10.1016/S0140-6736(19)30546-X
134. Keane MP, Lynch JP 3rd. Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax. (2000) 55:159–66. doi: 10.1136/thorax.55.2.159
135. Narvaez J, Borrell H, Sanchez-Alonso F, Rua-Figueroa I, Lopez-Longo FJ, Galindo-Izquierdo M, et al. Primary respiratory disease in patients with systemic lupus erythematosus: data from the Spanish rheumatology society lupus registry (RELESSER) cohort. Arthritis Res Ther. (2018) 20:280. doi: 10.1186/s13075-018-1776-8
136. Shin JI, Lee KH, Park S, Yang JW, Kim HJ, Song K, et al. Systemic lupus erythematosus and lung involvement: A comprehensive review. J Clin Med. (2022) 11:6714. doi: 10.3390/jcm11226714
137. Aguilera-Pickens G, Abud-Mendoza C. Pulmonary manifestations in systemic lupus erythematosus: pleural involvement, acute pneumonitis, chronic interstitial lung disease and diffuse alveolar hemorrhage. Reumatol Clin (Engl Ed). (2018) 14:294–300. doi: 10.1016/j.reuma.2018.03.012
138. Yamamoto AM, Amoura Z, Johannet C, Jeronimo AL, Campos H, Koutouzov S, et al. Quantitative radioligand assays using de novo–synthesized recombinant autoantigens in connective tissue diseases: New tools to approach the pathogenic significance of anti-RNP antibodies in rheumatic diseases. Arthritis Rheumatism. (2000) 43:689. doi: 10.1002/1529-0131(200003)43:3<689::aid-anr27>3.0.co;2-u
139. Bruner BF, Guthridge JM, Lu R, Vidal G, Kelly JA, Robertson JM, et al. Comparison of autoantibody specificities between traditional and bead-based assays in a large, diverse collection of patients with systemic lupus erythematosus and family members. Arthritis Rheum. (2012) 64:3677–86. doi: 10.1002/art.34651
140. Jeon YL, Kim MH, Lee WI, Kang SY. Comparison of indirect immunofluorescence and line immunoassay for autoantibody detection. Clin Exp Rheumatol. (2013) 31:84–90.
141. Menendez A, Gomez J, Caminal-Montero L, Diaz-Lopez JB, Cabezas-Rodriguez I, Mozo L. Common and specific associations of anti-SSA/Ro60 and anti-Ro52/TRIM21 antibodies in systemic lupus erythematosus. ScientificWorldJournal. (2013) 2013:832789. doi: 10.1155/2013/832789
142. Rastin M, Mahmoudi M, Sahebari M, Tabasi N. Clinical & immunological characteristics in systemic lupus erythematosus patients. Indian J Med Res. (2017) 146:224–9. doi: 10.4103/ijmr.IJMR_1356_15
143. Xia FF, Lu FA, Lv HM, Yang GA, Liu Y. [Clinical characteristics and related factors of systemic lupus erythematosus with interstitial pneumonia]. Beijing Da Xue Xue Bao Yi Xue Ban. (2021) 53:266–72. doi: 10.19723/j.issn.1671-167X.2021.02.006
144. Tan L, Zhao Y. Analysis of multiple organ damage and clinical immunological characteristics in systemic lupus erythematosus patients with hematologic involvement. Int J Med Sci. (2021) 18:2624–9. doi: 10.7150/ijms.48997
145. Amezcua-Guerra LM, Perez-Garcia LF, Jimenez-Rojas V, Marquez-Velasco R, Silveira LH. Anti-Ro52/TRIM21 antibodies are associated with aberrant inflammatory circuits in patients with systemic autoimmune rheumatic diseases. Gac Med Mex. (2023) 159:55–64. doi: 10.24875/GMM.M22000739
146. Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. (2009) 8:632–7. doi: 10.1016/j.autrev.2009.02.010
147. Menendez A, Gomez J, Escanlar E, Caminal-Montero L, Mozo L. Clinical associations of anti-SSA/Ro60 and anti-Ro52/TRIM21 antibodies: Diagnostic utility of their separate detection. Autoimmunity. (2013) 46:32–9. doi: 10.3109/08916934.2012.732131
148. Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. (2012) 24:499–505. doi: 10.1097/BOR.0b013e3283562c3e
149. Kamiyama R, Yoshimi R, Takeno M, Iribe Y, Tsukahara T, Kishimoto D, et al. Dysfunction of TRIM21 in interferon signature of systemic lupus erythematosus. Mod Rheumatol. (2018) 28:993–1003. doi: 10.1080/14397595.2018.1436028
150. Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J Neuroinflamm. (2014) 11:24. doi: 10.1186/1742-2094-11-24
151. Kunishita Y, Yoshimi R, Kamiyama R, Kishimoto D, Yoshida K, Hashimoto E, et al. TRIM21 dysfunction enhances aberrant B-cell differentiation in autoimmune pathogenesis. Front Immunol. (2020) 11:98. doi: 10.3389/fimmu.2020.00098
152. Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. (2021) 7:86. doi: 10.1038/s41572-021-00321-x
153. Gasparotto M, Franco C, Zanatta E, Ghirardello A, Zen M, Iaccarino L, et al. The interferon in idiopathic inflammatory myopathies: Different signatures and new therapeutic perspectives. A literature review Autoimmun Rev. (2023) 22:103334. doi: 10.1016/j.autrev.2023.103334
154. Sun KY, Fan Y, Wang YX, Zhong YJ, Wang GF. Prevalence of interstitial lung disease in polymyositis and dermatomyositis: A meta-analysis from 2000 to 2020. Semin Arthritis Rheum. (2021) 51:175–91. doi: 10.1016/j.semarthrit.2020.11.009
155. Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol. (1997) 109:32–40. doi: 10.1046/j.1365-2249.1997.4081308.x
156. Frank MB, McCubbin V, Trieu E, Wu Y, Isenberg DA, Targoff IN. The association of anti-Ro52 autoantibodies with myositis and scleroderma autoantibodies. J Autoimmun. (1999) 12:137–42. doi: 10.1006/jaut.1998.0265
157. Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. (2001) 60:116–23. doi: 10.1136/ard.60.2.116
158. Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senecal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. (2007) 9:R78. doi: 10.1186/ar2276
159. Srivastava P, Dwivedi S, Misra R. Myositis-specific and myositis-associated autoantibodies in Indian patients with inflammatory myositis. Rheumatol Int. (2016) 36:935–43. doi: 10.1007/s00296-016-3494-3
160. Infantino M, Manfredi M, Grossi V, Benucci M, Morozzi G, Tonutti E, et al. An effective algorithm for the serological diagnosis of idiopathic inflammatory myopathies: The key role of anti-Ro52 antibodies. Clin Chim Acta. (2017) 475:15–9. doi: 10.1016/j.cca.2017.10.002
161. Huang HL, Lin WC, Yeh CC, Sun YT. Serological risk factors for concomitant interstitial lung disease in patients with idiopathic inflammatory myopathy. J Clin Neurosci. (2020) 74:32–5. doi: 10.1016/j.jocn.2020.01.060
162. Vojinovic T, Cavazzana I, Ceruti P, Fredi M, Modina D, Berlendis M, et al. Predictive features and clinical presentation of interstitial lung disease in inflammatory myositis. Clin Rev Allergy Immunol. (2021) 60:87–94. doi: 10.1007/s12016-020-08814-5
163. Gonzalez-Bello Y, Garcia-Valladares I, Reyes-Perez IV, Garcia-Cerda D, Medrano-Ramirez G, Navarro-Zarza JE, et al. Myositis-specific antibodies and myositis-associated antibodies in patients with idiopathic inflammatory myopathies from the PANLAR myositis study group. J Clin Rheumatol. (2021) 27:e302–6. doi: 10.1097/RHU.0000000000001350
164. Liang J, Cao H, Yang Y, Ke Y, Yu Y, Sun C, et al. Efficacy and tolerability of nintedanib in idiopathic-inflammatory-myopathy-related interstitial lung disease: A pilot study. Front Med (Lausanne). (2021) 8:626953. doi: 10.3389/fmed.2021.626953
165. Gui X, Shenyun S, Ding H, Wang R, Tong J, Yu M, et al. Anti-Ro52 antibodies are associated with the prognosis of adult idiopathic inflammatory myopathy-associated interstitial lung disease. Rheumatol (Oxford). (2022) 61:4570–8. doi: 10.1093/rheumatology/keac090
166. Ghirardello A, Gatto M, Franco C, Zanatta E, Padoan R, Ienna L, et al. Detection of myositis autoantibodies by multi-analytic immunoassays in a large multicenter cohort of patients with definite idiopathic inflammatory myopathies. Diagnostics (Basel). (2023) 13:3080. doi: 10.3390/diagnostics13193080
167. Cheng L, Li Y, Wu Y, Luo Y, Zhou Y, Liao Z, et al. Risk of early infection in idiopathic inflammatory myopathies: cluster analysis based on clinical features and biomarkers. Inflammation. (2023) 46:1036–46. doi: 10.1007/s10753-023-01790-w
168. Fang X, Zou L, Zhao H, Liu B. Progression prediction in idiopathic inflammatory myopathy-associated interstitial lung disease: a combination of initial high attenuation areas and anti-melanoma differentiation-associated gene 5-positive. Clin Exp Rheumatol. (2024) 42:253–61. doi: 10.55563/clinexprheumatol/5h7bsu
169. Valle A, Narain S, Barilla-Labarca ML, Marder G. The differential role of SSa/SSb and Ro52 antibodies in defining clinical phenotypes in idiopathic inflammatory myopathies. Semin Arthritis Rheum. (2024) 65:152407. doi: 10.1016/j.semarthrit.2024.152407
170. Bauhammer J, Blank N, Max R, Lorenz HM, Wagner U, Krause D, et al. Rituximab in the treatment of jo1 antibody-associated antisynthetase syndrome: anti-ro52 positivity as a marker for severity and treatment response. J Rheumatol. (2016) 43:1566–74. doi: 10.3899/jrheum.150844
171. Shi J, Li S, Yang H, Zhang Y, Peng Q, Lu X, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol. (2017) 44:1051–7. doi: 10.3899/jrheum.161480
172. Xing X, Li A, Li C. Anti-Ro52 antibody is an independent risk factor for interstitial lung disease in dermatomyositis. Respir Med. (2020) 172:106134. doi: 10.1016/j.rmed.2020.106134
173. Sabbagh S, Pinal-Fernandez I, Kishi T, Targoff IN, Miller FW, Rider LG, et al. Anti-Ro52 autoantibodies are associated with interstitial lung disease and more severe disease in patients with juvenile myositis. Ann Rheum Dis. (2019) 78:988–95. doi: 10.1136/annrheumdis-2018-215004
174. Hassan AB, Fathi M, Dastmalchi M, Lundberg IE, Padyukov L. Genetically determined imbalance between serum levels of tumour necrosis factor (TNF) and interleukin (IL)-10 is associated with anti-Jo-1 and anti-Ro52 autoantibodies in patients with poly- and dermatomyositis. J Autoimmun. (2006) 27:62–8. doi: 10.1016/j.jaut.2006.06.003
175. Krystufkova O, Barbasso Helmers S, Venalis P, Malmstrom V, Lindroos E, Vencovsky J, et al. Expression of BAFF receptors in muscle tissue of myositis patients with anti-Jo-1 or anti-Ro52/anti-Ro60 autoantibodies. Arthritis Res Ther. (2014) 16:454. doi: 10.1186/s13075-014-0454-8
176. Lopez De Padilla CM, McNallan KT, Crowson CS, Bilgic H, Bram RJ, Hein MS, et al. BAFF expression correlates with idiopathic inflammatory myopathy disease activity measures and autoantibodies. J Rheumatol. (2013) 40:294–302. doi: 10.3899/jrheum.120555
177. Dai Y, Wang W, Yu Y, Hu S. Rheumatoid arthritis-associated interstitial lung disease: an overview of epidemiology, pathogenesis and management. Clin Rheumatol. (2021) 40:1211–20. doi: 10.1007/s10067-020-05320-z
178. Koduri G, Solomon JJ. Identification, monitoring, and management of rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. (2023) 75:2067–77. doi: 10.1002/art.42640
179. Paulin F, Doyle TJ, Fletcher EA, Ascherman DP, Rosas IO. Rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis: shared mechanistic and phenotypic traits suggest overlapping disease mechanisms. Rev Investigacion Clinica-Clinical Trans Invest. (2015) 67:280–6.
180. Ricchiuti V, Briand JP, Meyer O, Isenberg DA, Pruijn G, Muller S. Epitope mapping with synthetic peptides of 52-kD SSA/Ro protein reveals heterogeneous antibody profiles in human autoimmune sera. Clin Exp Immunol. (1994) 95:397–407. doi: 10.1111/j.1365-2249.1994.tb07010.x
181. Menor Almagro R, Jurado Roger A, Rodriguez Gutierrez FJ, Solis Diaz R, Cardiel MH, Salaberri Maestrojuan JJ. Association of anti-Ro52, anti-Ro60 and anti-La antibodies with diagnostic, clinical and laboratory features in a referral hospital in Jerez, Spain. Reumatol Clin. (2016) 12:256–62. doi: 10.1016/j.reuma.2015.10.010
182. Meek B, Kelder JC, Claessen AME, van Houte AJ, Ter Borg EJ. Rheumatoid factor isotype and Ro epitope distribution in primary Sjogren syndrome and rheumatoid arthritis with keratoconjunctivitis sicca. Rheumatol Int. (2018) 38:1487–93. doi: 10.1007/s00296-018-4090-5
183. Smeele HTW, Schreurs MWJ, Costedoat-Chalumeau N, Cornette JMJ, Dolhain RJEM. Low prevalence of anti-SSA (anti-Ro) and anti-SSB (anti-La) autoantibodies in female patients with rheumatoid arthritis with a wish to conceive. RMD Open. (2021) 7:e001727. doi: 10.1136/rmdopen-2021-001727
184. Yang L, Zhang T, Zhang C, Xiao C, Bai X, Wang G. Upregulated E3 ligase tripartite motif-containing protein 21 in psoriatic epidermis ubiquitylates nuclear factor-kappaB p65 subunit and promotes inflammation in keratinocytes. Br J Dermatol. (2021) 184:111–22. doi: 10.1111/bjd.19057
185. Elhai M, Avouac J, Kahan A, Allanore Y. Systemic sclerosis at the crossroad of polyautoimmunity. Autoimmun Rev. (2013) 12:1052–7. doi: 10.1016/j.autrev.2013.05.002
186. Burbelo PD, Ching KH, Han BL, Bush ER, Reeves WH, Iadarola MJ. Extraordinary antigenicity of the human Ro52 autoantigen. Am J Trans Res. (2010) 2:145–55.
Keywords: TRIM21/Ro52, autoantibody, autoimmune disease, connective tissue disease, interstitial lung disease
Citation: Gong X, He S and Cai P (2024) Roles of TRIM21/Ro52 in connective tissue disease-associated interstitial lung diseases. Front. Immunol. 15:1435525. doi: 10.3389/fimmu.2024.1435525
Received: 20 May 2024; Accepted: 19 July 2024;
Published: 06 August 2024.
Edited by:
Alenka Gagro, Children’s Hospital Zagreb, CroatiaReviewed by:
Yasuhiro Shimojima, Shinshu University, JapanShui Lian Yu, The Second Affiliated Hospital of Guangzhou Medical University, China
Copyright © 2024 Gong, He and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengcheng Cai, Y2FpcGVuZ2NoZW5nQGh1c3QuZWR1LmNu
†These authors have contributed equally to this work