 Qingliang Meng
Qingliang Meng Kai Wei
Kai Wei Yu Shan
Yu Shan- 1Department of Rheumatism, Henan Province Hospital of Traditional Chinese Medicine (TCM), Zhengzhou, Henan, China
- 2Department of Rheumatology and Immunology, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, China
- 3Department of Rheumatology, Shanghai Guanghua Hospital of Integrative Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 4Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 5Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease characterized by synovitis, degradation of articular cartilage, and bone destruction. Fibroblast-like synoviocytes (FLS) play a central role in RA, producing a significant amount of inflammatory mediators such as tumor necrosis factor(TNF)-α and IL-6, which promote inflammatory responses within the joints. Moreover, FLS exhibit tumor-like behavior, including aggressive proliferation and enhanced anti-apoptotic capabilities, which collectively drive chronic inflammation and joint damage in RA. TNF is a major pro-inflammatory cytokine that mediates a series of signaling pathways through its receptor TNFR1, including NF-κB and MAPK pathways, which are crucial for inflammation and cell survival in RA. The abnormal proliferation and anti-apoptotic characteristics of FLS in RA may result from dysregulation in TNF-mediated cell death pathways such as apoptosis and necroptosis. Ubiquitination is a critical post-translational modification regulating these signaling pathways. E3 ubiquitin ligases, such as cIAP1/2, promote the ubiquitination and degradation of target proteins within the TNF receptor complex, modulating the signaling proteins. The high expression of the BIRC3 gene and its encoded protein, cIAP2, in RA regulates various cellular processes, including apoptosis, inflammatory signaling, immune response, MAPK signaling, and cell proliferation, thereby promoting FLS survival and inflammatory responses. Inhibiting BIRC3 expression can reduce the secretion of inflammatory cytokines by RA-FLS under both basal and inflammatory conditions and inhibit their proliferation. Although BIRC3 inhibitors show potential in RA treatment, their possible side effects must be carefully considered. Further research into the specific mechanisms of BIRC3, including its roles in cell signaling, apoptosis regulation, and immune evasion, is crucial for identifying new therapeutic targets and strategies.
1 Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune condition marked by synovitis, degradation of articular cartilage, and bone destruction. A notable pathological feature is the abnormal proliferation of fibroblast-like synovial cells (FLS), a common occurrence in RA. Under normal conditions, molecular mechanisms in human joints regulate FLS turnover, ensuring apoptotic clearance without excessive accumulation (1). Conversely, pathological conditions can disrupt these processes, leading to carcinoid-like deposits of FLS due to aberrant cell death dynamics. Notably, apoptosis and necroptosis serve as the principal cellular death pathways in this context (2). Apoptosis acts as a regulatory mechanism for necrotic cell death, potentially triggered in its absence, whereas necroptosis represents a form of regulated necrosis, similarly inducible without apoptotic pathways (3). Tumor necrosis factor (TNF), an inaugural cytokine capable of inducing cellular necrosis, exerts pro-inflammatory effects that enhance cell activation, proliferation, and survival (4). As progenitors of a broad family of ligands, their diverse functions have a significant impact on cellular fate. It has been determined that receptor-interacting serine/threonine kinase 1 (RIPK1) mediates inflammation and cell death, facilitating both apoptosis and necroptosis in TNF-stimulated cells (5, 6). Moreover, TNF and nuclear factor-kappa B (NF-kB) signaling pathway performs vital functions in various cancers and immune responses (7, 8). RIPK1-regulated inflammatory signaling, apoptosis, and programmed necrosis play crucial roles in RA-FLS. The interaction of RIPK1 with TNF receptor 1 (TNFR1) activates the NF-κB inflammatory signaling cascade, leading to RA inflammation. Modulation of RIPK1 activity affects FLS survival and death, thus influencing the intensity and duration of inflammation (9). RIPK1 may further regulate the physiological and pathological functions of RA-FLS by interacting with other signaling molecules, such as MAPK. These interactions influence cell proliferation, migration, and secretion of inflammatory mediators, thereby playing a key role in the pathogenesis of RA (10). Although ubiquitination is an integral component of these signaling cascades, the ubiquitination-mediated proteasomal degradation pathway (UPP) has a vital part in the pathophysiology of RA by controlling the degradation of proteins associated with the inflammatory process. This pathway could affect the levels of inflammatory factors and other proteins, thereby influencing the inflammatory and immune responses in RA (11–14).
Ubiquitination, a vital post-translational modification, includes ubiquitin (Ub) and its target protein forming a covalent binding. Three different enzymes must function in succession for this process to occur, E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) (15, 16), culminating in the formation of an isopeptide bond between the C-terminal glycine (Gly76) of Ub and a lysine residue on the target protein (16). In rheumatoid arthritis (RA), ubiquitination influences immune cells and inflammatory responses by altering critical inflammatory signaling pathways, including NF-κB. For example, synoviolin 1 (SYVN1) is a novel pathogenic factor in RA, inherently an E3 ubiquitin ligase, highly expressed in the synovial tissues of RA patients and mice (17). SYVN1 elevates quantities of inflammatory cytokines like TNF-α and IL-1β, inhibits apoptosis of synovial cells in collagen-induced arthritis in mice, and significantly promotes synovial tissue proliferation (18). Furthermore, midline 1 facilitates the progression of RA by regulating the proliferation, migration, invasion, and inflammatory activities of FLS through ubiquitin-mediated proteasomal degradation of DPP4 (19). While TNF-α inhibitors have markedly revolutionized the management of RA, not all patients exhibit responsiveness, and a significant majority experience relapse upon cessation of therapy (20). The clinical use of TNF inhibitors still needs to be further improved, in which case ubiquitination could potentially be used as an adjuvant therapeutic strategy. Ubiquitination can directly affect specific signaling proteins within the TNF receptor complex, promoting their degradation and thereby mitigating TNF-induced inflammatory responses. It can also specifically degrade excessively active inflammation-related proteins without completely suppressing the immune system, potentially reducing side effects of TNF inhibitors, such as immunosuppression and infection risk (21).
As E3 ubiquitin ligase, cellular inhibitor of apoptosis protein 1 (cIAP2), and cellular inhibitor of apoptosis protein 2 (cIAP2) are involved in the proliferation and survival of FLS by regulating TNF receptor signaling and inflammatory cytokine production to influence the immune response to RA, and interacting with apoptosis-related signaling pathways to prevent apoptosis (22). In addition, cIAP1 and cIAP2 play roles in the assembly of inflammasomes, regulating the release of inflammatory mediators and affecting the function of immune cells (23). Notably, cIAP2 is encoded by the BIRC3 gene. Studies have found that BIRC3 mediates NF-κB activation in various regions of RA synovium, promoting inflammation and disease progression. BIRC3 also regulates inflammation and apoptosis in RA-FLS, indicating its dual role in promoting FLS survival and inflammatory responses (24). This review comprehensively analyzes the involvement of E3 ubiquitin ligases cIAP1/2 in RA-FLS, delving into the significance of cIAP1/2 in the regulation of TNF-induced cell death pathways and RA pathophysiology, emphasizing their roles as E3 ubiquitin ligases. Additionally, we highlight the potential clinical significance of the BIRC3 gene, which encodes cIAP2 protein, as a therapeutic target, offering new perspectives for RA treatment.
2 Inhibitors of apoptosis proteins
CIAP1 and cIAP2 are members of the inhibitor of apoptosis protein (IAPs) family and play critical roles in regulating various cellular processes (25). IAPs, akin to their counterparts in baculovirus-infected mammalian cells, play pivotal roles in cellular regulation, including apoptosis, proliferation, and differentiation. This protein family includes X-linked IAP (XIAP), cellular IAPs 1 and 2 (cIAP1 and cIAP2), neuronal apoptosis inhibitory protein (NAIP), and baculoviral IAP repeat containing 5(BIRC5) (26, 27). IAPs are central to the modulation of several cellular processes, including signal transduction, cytokine production, and cell survival, thereby influencing both innate and adaptive immune responses. The E3 ubiquitin ligase activity of XIAP and cIAP1 primarily governs immune regulatory functions by targeting key signaling pathways such as NF-κB and mitogen-activated protein kinase (MAPK). Additionally, NAIP, cIAP1, and cIAP2 orchestrate inflammasome assembly, which is crucial for innate immune response (28). The hallmark of the IAP family is the baculovirus IAP repeat (BIR) domain, which is essential for protein-protein interactions (29, 30). In addition to the BIR domain, IAPs also possess other significant domains, including the C-terminal ubiquitin-conjugated (UBC) domain, caspase recruitment domain (CARD), and C-terminal RING zinc-finger domain, facilitating a range of functional interactions (29, 31). Originally characterized as caspase inhibitors, mammalian IAPs (cIAP1, cIAP2, and XIAP) also modulate apoptosis via E3 ubiquitin ligase activity (27). Specifically, cIAP1 and cIAP2 are integral to the receptor complexes of the TNF receptor family members, modulating signal transduction via ubiquitination of associated proteins. This includes their recruitment to TNFR1 through the TNF receptor-associated death domain (TRADD)/TNF receptor-associated factor 2 (TRAF2) pathway, leading to the ubiquitination of RIPK1 within the TNFR1 complex (32, 33). The absence or inhibition of cIAP1 and cIAP2, particularly by the second mitochondrial activator of caspases (SMAC), renders cells vulnerable to TNF-induced apoptosis by disrupting NF-κB-mediated survival signals and facilitating the formation of pro-apoptotic TNFR1-induced complex II or RIPK1/RIPK3-dependent apoptosis (34, 35).
3 TNF-related pathway of cell death induced by cIAP1/2
3.1 TNFR1-induced complex
Binding of TNF to its receptor TNFR1 catalyzes the immediate assembly of the TNFR1 signal complex (TNFR1-SC), previously known as the TNF receptor 1 signaling complex (TNF-RSC). This complex incorporates TNF, TNFR1, TRADD, RIPK1, TNF receptor-associated factor 2 (TRAF2), cIAP1/2, linear ubiquitin chain assembly complex (LUBAC), inhibitor of kappa B kinase (IKK), and TGF-beta activated kinase 1 (TAK1) binding protein (TAB)-TAK complex (32). The integration of TAB-TAK and IKK complexes into TNFR1-SC relies on the recognition of lysine 63 (K63) and methionine 1 (M1) ubiquitin bonds by TAB2/3 and the NF-κB essential modulator (NEMO), respectively (36–38). The TAB-TAK complex requires only a K63 chain for recruitment, whereas the IKK complex requires both the K63 and M1 linkages. cIAP1/2, which serves as a crucial intermediary, facilitates this process by ubiquitinating several components of Complex I with K63-linked chains, including RIPK1. Subsequent recruitment of LUBAC enhances the M1-linked chain modification on RIPK1 (39–41). These modifications recruit the TAK1 and IKK complexes, activating the MAPK and NF-κB pathways. Additionally, cIAP1 enhances IKK complex recruitment through the K11-linked chain modifications of RIPK1 (42). Complex I represents the primary assembly in this signaling cascade. However, if RIPK1 is deubiquitinated by CYLD lysine 63 deubiquitinase (CYLD) or remains unubiquitinated during Complex I formation (e.g., due to IAP depletion) (43), Complex IIa or IIb is generated, leading to apoptosis or necroptosis based on cellular conditions (44). Contrary to cIAP1/2’s supportive role in TNFR1 signaling, its function in other TNF receptor family members, such as the tumor necrosis factor-like weak apoptosis inducer (TWEAK) and CD40 ligand (CD40L), is inhibitory. cIAP1/2, in conjunction with TRAF2 and TRAF3, suppresses alternative NF-κB pathways through ubiquitination of lysine 48 (K48) linkages and degradation of NF-κB-inducing kinase (NIK), in the absence of ligand stimulation (28) (Figure 1).
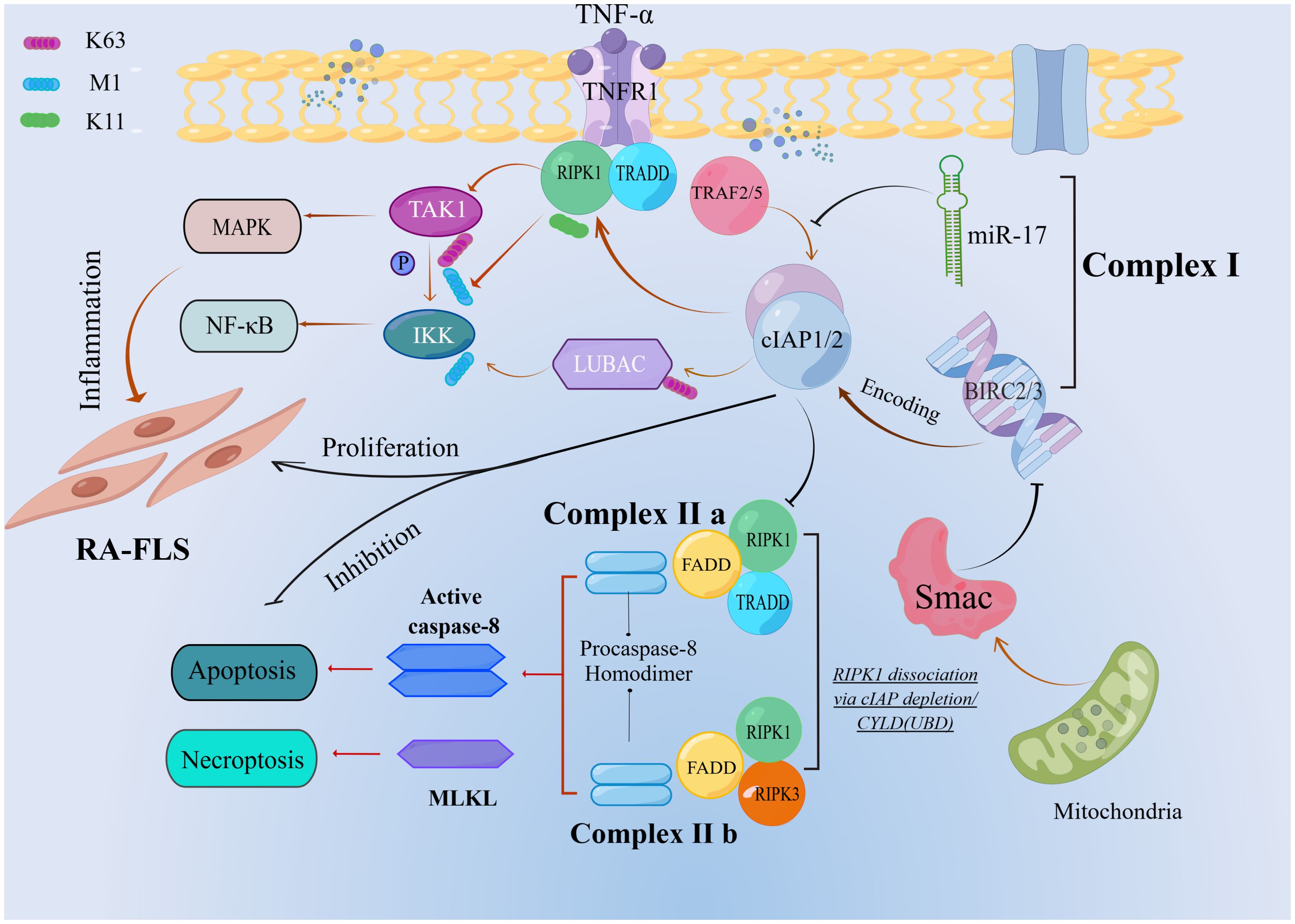
Figure 1. Ubiquitin-mediated regulation of TNFR1 apoptotic signaling. The cIAP1/2 mediates a series of ubiquitination in complex I. When RIPK1 dissociation via cIAP depletion or CYLD is deubiquitinated, complexIIb is formed, which leads to apoptosis.
3.2 TNF-related apoptosis-inducing ligand receptor–induced complex
TRAIL (tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), also known as the APO-2 Ligand (APO2L), belongs to the family of tumor necrosis factors. The activation of TRAIL-R has been shown to induce the formation of complexes containing proteins similar to those induced by TNFR1, which are regulated by ubiquitin. In TRAIL-R signal transduction, pro-apoptotic proteins (FADD and Caspase-8) are first recruited to TRAIL-R and can be used as scaffolds for anti-apoptotic protein recruitment (RIPK1, TRAF2, cIAP1/2, LUBAC, TAK1, and IKK complexes) (45–47). The compound mentioned earlier is called complex I. The composition of complex II is very similar to that of complex I. It is generally believed that the former dissociates from activated TRAIL-R and forms cytoplasmic complexes (48). TRAIL-R-induced complexes I and II can activate NF-κB, MAPK pathway, and apoptosis, but only complex II can activate necroptosis (45). E3 ligase not only regulates apoptosis, but also the gene activation output of the TRAIL signal by participating in the two core signal components of TRAIL-induced gene activation, caspase-8 and RIPK1 (47, 49). This process involved cIAP1/2. The E3 ligase cIAP1/2 is recruited to two TRAIL signaling complexes in a FADD-caspase-8-dependent manner. Furthermore, TRAF2 and cIAP1/2 both promote TRAIL- and CD95L-mediated gene activation (47, 49–51). In the context of TNF signaling, TRAF2-cIAP1/2-mediated ubiquitination of RIPK1 promotes NF-kB activation (32, 42, 52, 53), the TRAF2’s gene activation function is dependent on its ability to recruit cIAP1/2 (33). Consistent with the activation of TRAF2 as a TRAIL signal transduction scaffold, cIAP1/2 depletion strongly reduced RIPK1 ubiquitin, IKK recruitment, NF-kB activation, and inactivation of TRAIL-mediated cytokine secretion, whereas TRAF2 recruitment remained unaffected (49, 54). As previously demonstrated for TNF signal transduction, TRAF2 may promote TRAIL-induced cytokine production by acting as a recruitment platform for cIAPs (33). Through an unknown mechanism, cIAP1/2 is also required downstream of TRAF2 to recruit LUBAC to track complex I (49). LUBAC mediates TNFR1-SC stabilization via linear ubiquitination of TRADD, RIPK1, NEMO, and TNFR1 and is critical for TNFR1-induced gene activation signaling (40, 55). TRAF2 depletion strongly reduced RIPK1 ubiquitination, IKK recruitment, NF-kB activation, and TRAIL-mediated cytokine secretion, whereas TRAF2 recruitment was unaffected (49, 56) (Figure 2).
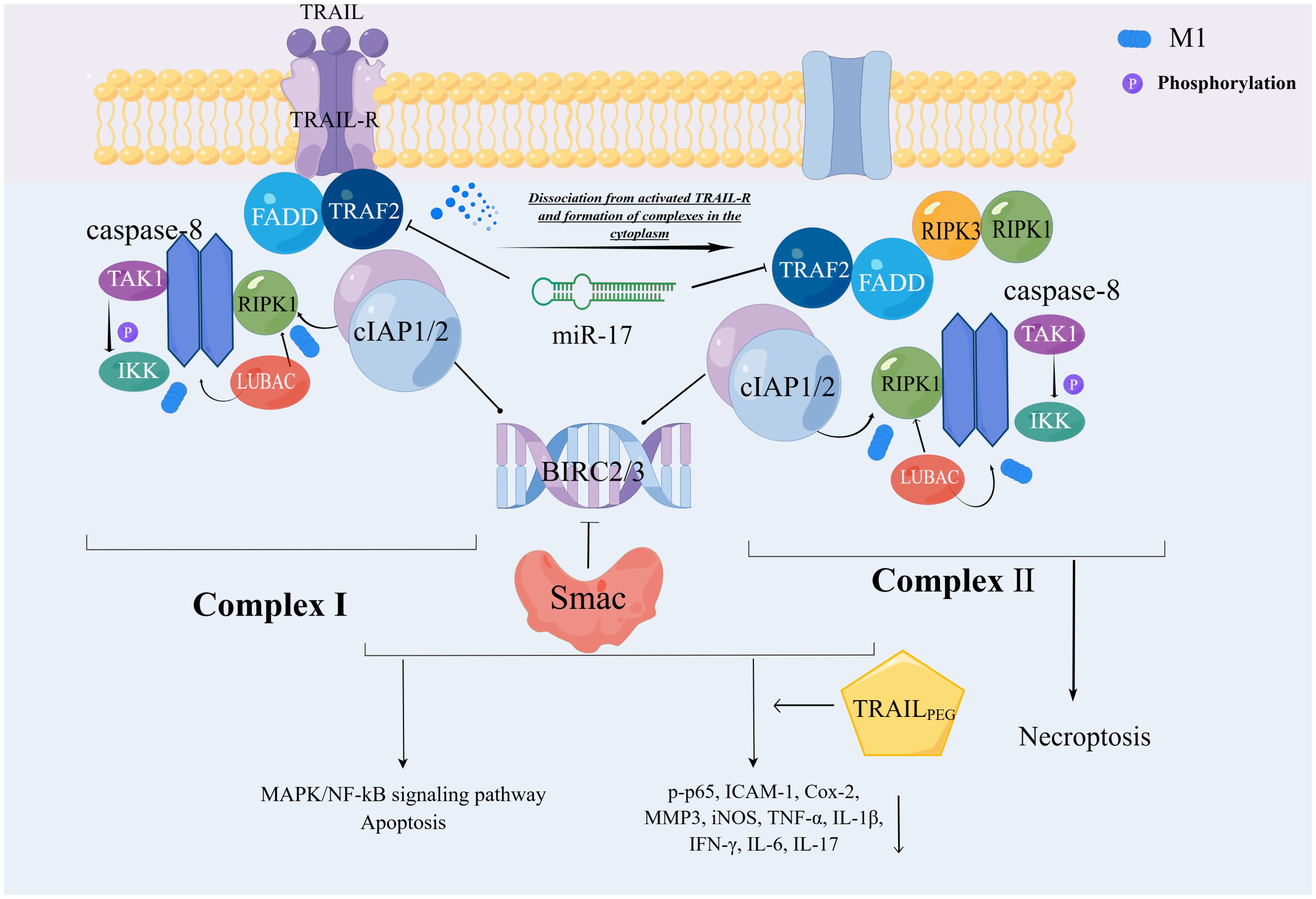
Figure 2. Ubiquitin-mediated regulation of TRAIL-R cell death signaling pathway. The composition of complexes I and II is relatively similar, with complexII derived from I detached from activated TRAIL-R. CIAP1/2 is involved in the ubiquitination of this process. Through the M1 chain, cIAP1/2 indirectly catalyzes the functional activation of caspase-8, which ultimately leads to the MAPK/NF-kB signaling pathway or apoptosis. This series of reactions is manifested in both complexes, but only complex 2 causes necroptosis.
Although several recent studies have demonstrated that the mechanism by which TRAIL promotes RA inflammation does not depend exclusively on cell death, TRAIL regulation of RA inflammation is mainly due to its ability to promote apoptosis of synoviocytes and infiltrating lymphocytes (57). TRAIL produces various signals. TRAIL can induce migration, proliferation, and cytokine production in both cancerous and non-cancerous cells, in addition to inducing cell death via apoptosis and necroptosis. As a result, uncovering the mechanisms that regulate the complex balance between these various outputs can help us better understand TRAIL’s role of TRAIL in tissue homeostasis, immunity, and cancer. Previous animal experiments have confirmed that TRAILPEG ameliorates arthritis severity and significantly reduces the accumulation of inflammatory molecules (p-p65, ICAM-1, Cox-2, MMP3 and iNOS), pro-inflammatory cytokines (TNF-α, IL-1β, IFN-γ, IL-6, IL-17) and activated macrophages (58), all of which have been shown to play a cellular and cellular component in important role in the pathogenesis of RA. The role of TRAIL in the mechanism of RA-FLS apoptosis has also been confirmed in several studies, and this role correlates with disease severity in a cell cycle-dependent manner (59–61). Blocking the expression of TRAIL and cIAP can significantly promote the apoptosis of FLS (59–61). In addition, ubiquitin and deubiquitination are key regulators of immune receptor signal transduction (45).
In comparison to TRAIL-R, the precise space and time positions of proteins in the TRAIL-R signal cascade are not well defined in TNFR1. However, it is unclear what determines whether complex I/II induces non-apoptotic or apoptotic signal transduction. However, ubiquitination of the M1 connection of Caspase-8 by LUBAC inhibits its activity (49), which may promote the spread of survival and inflammatory pathways that are usually inhibited by Caspase-8 activity (47, 62).
4 MicroRNA inhibits cIAP1/2 expression
In RA, both serum and synovial fluid (SF) samples, as well as synovial tissues and the serum and joints of rats with adjuvant arthritis, exhibit notably reduced expression of miR-17. Investigations revealed that miR-17 diminishes the expression of TRAF2, a cellular inhibitor of cIAP1 and cIAP2, in RA SFs when stimulated by TNF-α. Moreover, restoration of miR-17 activity enhanced the polyubiquitination of lysine 48 (K48) linkages of TRAF2, cIAP1, and cIAP2 in these cells, as determined by immunoprecipitation analyses. This modulation by miR-17 leads to the destabilization of TRAF2, impairing its ability to associate with cIAP2. Consequently, TNF-α suppresses the nuclear translocation of NF-κB p65, c-Jun, and signal transducer and activator of transcription 3 (STAT3) prompted by TNF-α. This molecular alteration results in decreased production of IL-6, interleukin-8 (IL-8), matrix metalloproteinase-1 (MMP1), and matrix metalloproteinase-13 (MMP13) in human RA SF, underscoring the pivotal regulatory role of miR-17 in inflammatory and degradative pathways in RA (63).
Besides RA-FLS, miRNAs also affect cIAP2 expression in other diseases. Studies have shown that BIRC3 mRNA is upregulated in hepatocellular carcinoma (HCC) tissues, and the high expression of cIAP2 encoded by BIRC3 promotes HCC proliferation and migration. Although miR-124 was underexpressed in HCC tissues and cell lines, BIRC3 was identified as its target gene. Overexpression of miR-124 could target BIRC3, thereby decreasing cIAP2 protein levels and inhibiting HCC proliferation and migration through regulating the NF-κB signaling pathway (64). Circular RNA (circRNAs) are also involved in gene regulation. The oncogenic circular RNA hsa_circ_0070039 (circNUP54) is significantly upregulated in HCC. This interaction stabilizes downstream BIRC3 mRNA by binding to the 3’-UTR region, thereby promoting HCC progression via the HuR/BIRC3/NF-κB axis (65). Furthermore studies on oral squamous cell carcinoma (OSCC) have shown that silencing circDOCK1 and upregulating miR-196a-5p results in increased apoptosis and decreased BIRC3 formation (66).
5 Smac mimetics mediate cell death through degradation of cIAP1/2
IAPs, especially cellular cIAP1/2 and XIAP, can prevent cell death by preventing the activation of caspase-8 or inhibiting the activity of caspases-9,-3 and-7 (67–69), respectively. The E3 ubiquitin ligase domain of cIAP1/2 promotes cIAP1/2 and proteasome-dependent degradation (70, 71). A second mitochondria-derived caspase activator (Smac/DIABLO), which, along with cytochrome C, is released from the mitochondria under the induction of an inherent apoptotic pathway in response to stimuli such as genotoxic stress, relieves the inhibition of apoptosis by IAPs (72, 73). Direct binding and blocking of the interaction of XIAP with caspase-9,-3, and-7, as well as inducing proteasomal degradation of cIAP1 and cIAP2, are the main mechanisms by which Smac mimetics cause cell death (34, 35, 74, 75). The degradation of cIAP1/2 allows the release of RIPK1 from TNFR1 and then incorporates the complex associated with caspase-8 and the fas-related death domain, which can promote cell death and cover the survival-promoting effect of NF-κB signaling (32, 76, 77). This means that any cell expressing TNFR1 should respond to Smac mimic/TNF-α therapy because the expression of TNFR1 is very common in all types of cancer cells (78). The E3 ubiquitin ligase activity of cIAP promotes cancer cell survival (32, 35, 74, 76). Furthermore, removing cIAP from cancer cells can alter cytokine signaling by converting pro-death signals into inflammatory signals, resulting in cell death via apoptosis or necroptosis (46, 79). BIRC3 (cIAP2) is thought to be a member of the IAP family of proteins that regulate cell death and survival, according to Bai et al. (80). Previous studies have shown that BIRC3 (cIAP2) promotes survival and anti-apoptosis in cancer cells and is a therapeutic target of the drug family known as “Smac mimic” (81), although not all cases support this view (81). Owing to its dual function in TNF, cIAP’s regulatory role of cIAP in cancer and the immune system is critical. Abnormal expression results in uncontrollable outcomes. In the field of cancer applications, the Smac simulator has been used in clinical trials of hematological and solid cancers, but unfortunately, the results so far are inconsistent and limited (82). The role of the Smac simulator in the treatment of RA is theoretically applicable (83). However, thus far, there has been little research on Smac simulations in RA-FLS. In vitro, it has been confirmed that BIRC3 has an abnormally high expression in RA-FLS compared to OA. Simultaneously, given the carcinoid proliferation of FLS and the similar TNFR1 signal transduction mechanism of cIAPs in RA, Smac mimetics may be of breakthrough significance in the treatment of RA.
6 Discussion
Rheumatoid arthritis is a chronic inflammatory autoimmune disease characterized by the infiltration of immune cells into the synovium, leading to the release of inflammatory cytokines and subsequent tissue damage (84). In addition to inflammation, fibroblast-like synoviocytes, the principal effector cells in RA, also exhibit tumor-like proliferation and invasiveness. Therefore, controlling inflammation and inhibiting FLS proliferation are crucial elements in the treatment of RA (85). CIAP1 and cIAP2, E3 ubiquitin ligases, play critical roles in the regulation of TNFR1 and TRAIL-R signal transduction, ensuring effective signal propagation. These proteins modulate various NF-κB pathways, which are pivotal for controlling diverse cellular inflammatory and immune responses. Although cIAP1 and cIAP2 are frequently discussed collectively because of their overlapping functions in ubiquitination across different signaling pathways, their specific roles in modulating cell death in distinct cell types and tissues remain poorly understood (86). In certain pathological conditions, cIAP1 has been shown to mediate disease progression through its regulatory effects on TNF signaling, whereas cIAP2 does not participate in this process (42, 87). Conversely, cIAP2 exhibits a unique therapeutic mechanism distinct from that of cIAP1 in the context of RA, highlighting the differential contribution of these proteins to disease pathology and treatment (88). In addition, within the array of death receptor (DR) ligands, TRAIL has garnered significant interest because of its structural homology with CD95L (89, 90) and its distinct ability to selectively eliminate cancer cells without notable systemic toxicity (91, 92). This characteristic has spurred the development of TRAIL receptor agonists (TRAs), although their clinical trials have been discontinued because of issues with their efficacy (93). Consequently, the targeted modulation of BIRC3 gene expression and ubiquitination processes in TNFR1 or TRAIL signaling mediated by BIRC3 (cIAP2) present promising strategies for enhancing treatments for related disorders, including autoimmune diseases.
The BIRC3 gene encodes the multifunctional protein cIAP2, which plays an important role in the regulation of various cellular processes including caspases, apoptosis, inflammatory signaling, immunity, mitogen-activated protein kinase signaling, and cell proliferation (94). We speculate that increased expression of BIRC3 in RA promotes FLS survival and contributes to the inflammatory response. Inhibition of BIRC3 expression can reduce the secretion of inflammatory cytokines by RA FLSs under both basal and inflammatory conditions, and impede their proliferation. Existing evidence has demonstrated that BIRC3 plays a crucial role in promoting cancer cell survival and inhibiting apoptosis (95). The interaction between BIRC3, MAP3K14, and the NF-κB pathway highlights the impact of BIRC3 inactivation on tumor cells dependent on this pathway (96). For example, in colorectal cancer cells, overexpression of BIRC3 can lead to the activation of receptor-interacting serine/threonine protein kinase 2 (RIPK2), which promotes the ubiquitination of IKBKG, thereby inhibiting IKBKG protein expression and enhancing the expression of NF-κB subunits p50 and p65 (97). Recent studies have established a correlation between Fusobacterium nucleatum (Fn) and the occurrence and development of colorectal cancer (CRC), particularly noting that Fn infection upregulates BIRC3 in CRC cells via the TLR4/NF-κB pathway (98). Moreover, the upregulation of BIRC3 reduces the responsiveness of CRC cells to 5-fluorouracil (5-Fu), suggesting that targeting BIRC3 may offer a promising strategy to alleviate chemotherapy resistance in advanced CRC (99).
Additionally, BIRC2/BIRC3, in complex with TNFR2 and TRAF2, activates the NF-κB and MAPK signaling pathways, which are crucial for inflammation and cell survival (24). Combining anti-tumor necrosis factor therapy with cIAP1/2 inhibitors by blocking the BIRC2/BIRC3-mediated apoptotic pathway has the potential to improve therapeutic efficacy and effectively reduce symptoms and disease progression in rheumatoid arthritis. According to researches, glioblastoma (GBM) is a highly malignant brain tumor characterized by elevated BIRC3 expression, which is associated with tumor progression from low to high differentiation and resistance to temozolomide (TMZ) and radiation therapy, ultimately leading to reduced patient survival rates (100). Inhibiting the E3 ubiquitin ligase activity of BIRC3 can increase tumor cell sensitivity to radiotherapy and chemotherapy, and developing BIRC3 inhibitors may offer a promising approach for treating glioblastoma. Currently, small molecule inhibitors or antibodies targeting BIRC3 are being developed as potential treatments to disrupt its E3 ubiquitin ligase activity and induce tumor cell apoptosis (101). A comprehensive study of the specific mechanisms of BIRC3, including its role in cell signaling, regulation of apoptosis, and evasion of immune responses, is crucial for identifying new therapeutic targets and strategies.
Although BIRC3 inhibitors show promise in treating rheumatoid arthritis, potential side effects must be carefully considered, such as inducing apoptosis in normal cells dependent on BIRC3-regulated pathways and triggering adverse immune responses due to its role in immune regulation. FLS may activate other anti-apoptotic mechanisms, such as upregulating myeloid cell leukemia1(MCL1), thereby reducing sensitivity to BIRC3 inhibitors (102). Furthermore, changes in the surrounding microenvironment, such as alterations in immunogenicity and extracellular matrix composition, may also affect the efficacy of these inhibitors.
This review discusses the critical roles of the E3 ubiquitin ligases cIAP1/2 in mediating cell death pathways induced by TNF and promoting abnormal proliferation of fibroblast-like synoviocytes in RA, emphasizing the importance of the BIRC3-mediated ubiquitination process and its potential as a therapeutic target. However, the review has several limitations. Primarily, it focuses on molecular and cellular mechanisms due to the scarcity of clinical studies on BIRC3 in RA, lacking support from large-scale, multicentric clinical trials. Additionally, it does not cover longitudinal studies to track the long-term effects of BIRC3 inhibition on RA, which could provide deeper insights into the durability of treatment effects and potential development of resistance or adverse reactions. Although the article mentions several signaling pathways affected by BIRC3, it does not sufficiently explore alternative pathways that might compensate for BIRC3 inhibition, which could aid in understanding potential resistance mechanisms and refining therapeutic strategies. Due to the limited research on targeting BIRC3, there is a lack of discussion on the potential side effects and safety concerns of BIRC3-targeted therapies. Finally, a more robust comparative analysis is needed, especially comparing the role of BIRC3 in RA with other autoimmune diseases, to provide a broader perspective on its specific functions and potential as a therapeutic target.
Author contributions
QM: Investigation, Methodology, Writing – original draft. KW: Investigation, Methodology, Writing – original draft. YS: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Henan Province Special Major Project in Traditional Chinese Medicine, studying the mechanism by which Gui Zhi Shao Yao Zhi Mu Tang, formulated with Gui Zhi in different environments, regulates the intestinal flora-Th17/Treg cell balance to prevent and treat bone erosion in rheumatoid arthritis (20-21ZYZD05), and the Henan Province Traditional Chinese Medicine Top Talents Training Project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol. (2021) 5652. doi: 10.3389/fimmu.2021.809806
2. Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. (2000) 407:802–9. doi: 10.1038/35037739
3. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
4. Carswell E, Old LJ, Kassel R, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci. (1975) 72:3666–70. doi: 10.1073/pnas.72.9.3666
5. Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discovery. (2020) 19:553–71. doi: 10.1038/s41573-020-0071-y
6. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
7. Barrow M. An Overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun Rev. (2021) 20:102741. doi: 10.1016/j.autrev.2020.102741
8. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
9. Wang J, Li C, Liu Y, Mei W, Yu S, Liu C, et al. JAB1 determines the response of rheumatoid arthritis synovial fibroblasts to tumor necrosis factor-alpha. Am J pathology. (2006) 169:889–902. doi: 10.2353/ajpath.2006.051161
10. Wang Q, Ye Q, Xi X, Cao X, Wang X, Zhang M, et al. KW2449 ameliorates collagen-induced arthritis by inhibiting RIPK1-dependent necroptosis. Front Immunol. (2023) 14:1135014. doi: 10.3389/fimmu.2023.1135014
11. Kanarek N, London N, Schueler-Furman O, Ben-Neriah Y. Ubiquitination and degradation of the inhibitors of NF-κB. Cold Spring Harbor Perspect Biol. (2010) 2:a000166. doi: 10.1101/cshperspect.a000166
12. Wertz D-U. Ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. (2004) 430:694–9. doi: 10.1038/nature02794
13. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
14. Verboom L, Hoste E, van Loo G. OTULIN in NF-κB signaling, cell death, and disease. Trends Immunol. (2021) 42:590–603. doi: 10.1016/j.it.2021.05.003
15. Schlesinger DH, Goldstein G, Niall HD. Complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry. (1975) 14:2214–8. doi: 10.1021/bi00681a026
16. Hershko A. Lessons from the discovery ofthe ubiquitin system. Trends Biochem Sci. (1996) 21:445–9. doi: 10.1016/S0968-0004(96)10054-2
17. Aratani S, Fujita H, Yagishita N, Yamano Y, Okubo Y, Nishioka K, et al. Inhibitory effects of ubiquitination of synoviolin by PADI4. Mol Med Rep. (2017) 16:9203–9. doi: 10.3892/mmr.2017.7764
18. Xu Y, Fang D. Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J autoimmunity. (2020) 109:102423. doi: 10.1016/j.jaut.2020.102423
19. Lin L, Huang Z, Li W, Liu X, Li X, Gao S, et al. Mid1 promotes synovitis in rheumatoid arthritis via ubiquitin-dependent post-translational modification. Pharmacol Res. (2024) 205:107224. doi: 10.1016/j.phrs.2024.107224
20. Kawalkowska JZ, Ogbechi J, Venables PJ, Williams RO. cIAP1/2 inhibition synergizes with TNF inhibition in autoimmunity by down-regulating IL-17A and inducing Tregs. Sci Adv. (2019) 5:eaaw5422. doi: 10.1126/sciadv.aaw5422
21. Qian G, Guo J, Vallega KA, Hu C, Chen Z, Deng Y, et al. Membrane-associated RING-CH 8 functions as a novel PD-L1 E3 ligase to mediate PD-L1 degradation induced by EGFR inhibitors. Mol Cancer research: MCR. (2021) 19:1622–34. doi: 10.1158/1541-7786.MCR-21-0147
22. Bai W, Huo S, Li J, Shao J. Advances in the study of the ubiquitin-editing enzyme A20. Front Pharmacol. (2022) 13:845262. doi: 10.3389/fphar.2022.845262
23. Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. (2012) 33:535–45. doi: 10.1016/j.it.2012.06.004
24. Ge L, Wang T, Shi D, Geng Y, Fan H, Zhang R, et al. ATF6α contributes to rheumatoid arthritis by inducing inflammatory cytokine production and apoptosis resistance. Front Immunol. (2022) 13:965708. doi: 10.3389/fimmu.2022.965708
25. Estornes Y, Bertrand MJ. IAPs, regulators of innate immunity and inflammation. Semin Cell Dev Biol. (2015) 39:106–14. doi: 10.1016/j.semcdb.2014.03.035
26. Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat Rev Cancer. (2010) 10:561–74. doi: 10.1038/nrc2889
27. Silke J, Vucic D. IAP family of cell death and signaling regulators. Methods enzymology. (2014) 545:35–65. doi: 10.1016/B978-0-12-801430-1.00002-0
28. Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. (2012) 33:535–45. doi: 10.1016/j.it.2012.06.004
29. Liang J, Zhao W, Tong P, Li P, Zhao Y, Li H, et al. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med Genomics. (2020) 13:1–13. doi: 10.1186/s12920-020-0661-x
30. Wang D, Berglund A, Kenchappa RS, Forsyth PA, Mulé JJ, Etame AB. BIRC3 is a novel driver of therapeutic resistance in Glioblastoma. Sci Rep. (2016) 6:1–13. doi: 10.1038/srep21710
31. Cossu F, Milani M, Mastrangelo E, Lecis D. Targeting the BIR domains of inhibitor of apoptosis (IAP) proteins in cancer treatment. Comput Struct Biotechnol J. (2019) 17:142–50. doi: 10.1016/j.csbj.2019.01.009
32. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. (2008) 30:689–700. doi: 10.1016/j.molcel.2008.05.014
33. Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (TNF) to efficiently activate NF-κB and to prevent TNF-induced apoptosis. J Biol Chem. (2009) 284:35906–15. doi: 10.1074/jbc.M109.072256
34. Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL-and TNFα-mediated cell death. Science. (2004) 305:1471–4. doi: 10.1126/science.1098231
35. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. (2007) 131:669–81. doi: 10.1016/j.cell.2007.10.030
36. Wu C-J, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nat Cell Biol. (2006) 8:398–406. doi: 10.1038/ncb1384
37. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell. (2009) 136:1098–109. doi: 10.1016/j.cell.2009.03.007
38. Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell. (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
39. Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Biophys Res Commun. (2016) 474:452–61. doi: 10.1016/j.bbrc.2016.04.141
40. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. (2011) 471:591–6. doi: 10.1038/nature09816
41. Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker-Jensen S, et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol Cell. (2013) 50:818–30. doi: 10.1016/j.molcel.2013.06.004
42. Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izraelvav A, Phu L, et al. cl.ael and UbcH5 promote K11moteav) polyubiquitination of RIP1 in TNF signalling. EMBO J. (2010) 29:4198–209. doi: 10.1038/emboj.2010.300
43. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. (2003) 424:801–5. doi: 10.1038/nature01802
44. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
45. Lafont E, Hartwig T, Walczak H. Paving TRAIL’s path with ubiquitin. Trends Biochem Sci. (2018) 43:44–60. doi: 10.1016/j.tibs.2017.11.002
46. Dickens LS, Powley IR, Hughes MA, MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. (2012) 318:1269–77. doi: 10.1016/j.yexcr.2012.04.005
47. Henry CM, Martin SJ. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol Cell. (2017) 65:715–29. e5. doi: 10.1016/j.molcel.2017.01.022
48. Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. (2005) 280:40599–608. doi: 10.1074/jbc.M509560200
49. Lafont E, Kantariont C, Draber P, De Miguel D, Hartwig T, Reichert M, et al. The linear ubiquitin chain assembly complex regulates TRAILatest9): gene activation and cell death. EMBO J. (2017) 36:1147–66. doi: 10.15252/embj.201695699
50. Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell. (2013) 49:1034–48. doi: 10.1016/j.molcel.2013.01.025
51. Trauzold A, Röder C, Sipos B, Karsten K, Arlt A, Jiang P, et al. CD95 and TRAF2 promote invasiveness of pancreatic cancer cells. FASEB J. (2005) 19:1–24. doi: 10.1096/fj.04-2984fje
52. Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. (2006) 22:245–57. doi: 10.1016/j.molcel.2006.03.026
53. Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J Biol Chem. (2008) 283:24295–9. doi: 10.1074/jbc.C800128200
54. Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol Cell. (2017) 65:730–42. e5. doi: 10.1016/j.molcel.2017.01.021
55. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
56. Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem. (2003) 278:25534–41. doi: 10.1074/jbc.M303399200
57. Chyuan I-T, Tsai H-F, Liao H-J, Wu C-S, Hsu P-N. An apoptosis-independent role of TRAIL in suppressing joint inflammation and inhibiting T-cell activation in inflammatory arthritis. Cell Mol Immunol. (2018) 15:846–57. doi: 10.1038/cmi.2017.2
58. Park J-S, Oh Y, Park O, Foss CA, Lim SM, Jo D-G, et al. PEGylated TRAIL ameliorates experimental inflammatory arthritis by regulation of Th17 cells and regulatory T cells. J Controlled Release. (2017) 267:163–71. doi: 10.1016/j.jconrel.2017.10.004
59. Xu B, Li J, Wu C, Liu C, Yan X, Chang X. CXCL10 and TRAIL are upregulated by TXNDC5 in rheumatoid arthritis fibroblast-like synoviocytes. J Rheumatol. (2018) 45:335–40. doi: 10.3899/jrheum.170170
60. Audo R, Combe B, Hahne M, Morel J. The two directions of TNF-related apoptosis-inducing ligand in rheumatoid arthritis. Cytokine. (2013) 63:81–90. doi: 10.1016/j.cyto.2013.04.011
61. Dharmapatni AA, Smith MD, Findlay DM, Holding CA, Evdokiou A, Ahern MJ, et al. Elevated expression of caspase-3 inhibitors, survivin and xIAP correlates with low levels of apoptosis in active rheumatoid synovium. Arthritis Res Ther. (2009) 11:R13. doi: 10.1186/ar2603
62. Kreuz S, Siegmund D, Rumpf J-J, Samel D, Leverkus M, Janssen O, et al. NFκB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol. (2004) 166:369–80. doi: 10.1083/jcb.200401036
63. Akhtar N, Singh AK, Ahmed S. MicroRNA-17 suppresses TNF-α signaling by interfering with TRAF2 and cIAP2 association in rheumatoid arthritis synovial fibroblasts. J Immunol. (2016) 197:2219–28. doi: 10.4049/jimmunol.1600360
64. Cao J, Qiu J, Wang X, Lu Z, Wang D, Feng H, et al. Identification of microRNA-124 in regulation of Hepatocellular carcinoma through BIRC3 and the NF-κB pathway. J Cancer. (2018) 9:3006–15. doi: 10.7150/jca.25956
65. Tang C, Zhuang H, Wang W, Wang Q, Ma X, Wang B, et al. CircNUP54 promotes hepatocellular carcinoma progression via facilitating HuR cytoplasmic export and stabilizing BIRC3 mRNA. Cell Death disease. (2024) 15:191. doi: 10.1038/s41419-024-06570-4
66. Wang L, Wei Y, Yan Y, Wang H, Yang J, Zheng Z, et al. CircDOCK1 suppresses cell apoptosis via inhibition of miR−196a−5p by targeting BIRC3 in OSCC. Oncol Rep. (2018) 39:951–66. doi: 10.3892/or.2017.6174
67. Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. (2000) 408:1004–8. doi: 10.1038/35050006
68. Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. (2001) 410:112–6. doi: 10.1038/35065125
69. Wu G, Chai J, Suber TL, Wu J-W, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. (2000) 408:1008–12. doi: 10.1038/35050012
70. Morizane Y, Honda R, Fukami K, Yasuda H. X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J Biochem. (2005) 137:125–32. doi: 10.1093/jb/mvi029
71. Suzuki Y, Nakabayashi Y, Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci. (2001) 98:8662–7. doi: 10.1073/pnas.161506698
72. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell. (2000) 102:33–42. doi: 10.1016/S0092-8674(00)00008-8
73. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. cell. (2000) 102:43–53. doi: 10.1016/S0092-8674(00)00009-X
74. Vince JE, Wong WW-L, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell. (2007) 131:682–93. doi: 10.1016/j.cell.2007.10.037
75. Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. (2008) 68:9384–93. doi: 10.1158/0008-5472.CAN-08-2655
76. Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al. Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. (2007) 12:445–56. doi: 10.1016/j.ccr.2007.08.029
77. Wang L, Du F, Wang X. TNF-α induces two distinct caspase-8 activation pathways. Cell. (2008) 133:693–703. doi: 10.1016/j.cell.2008.03.036
78. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differentiation. (2003) 10:45–65. doi: 10.1038/sj.cdd.4401189
79. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug discovery. (2012) 11:109–24. doi: 10.1038/nrd3627
80. Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther. (2014) 144:82–95. doi: 10.1016/j.pharmthera.2014.05.007
81. Frazzi R. BIRC3 and BIRC5: multi:izzi):8 inhibitors in cancer. Cell Bioscience. (2021) 11:1–14. doi: 10.1186/s13578-020-00521-0
82. Morrish E, Brumatti G, Silke J. Future therapeutic directions for Smac-mimetics. Cells. (2020) 9:406. doi: 10.3390/cells9020406
83. Jensen S, Seidelin JB, LaCasse EC, Nielsen OH. SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci Signaling. (2020) 13:eaax8295. doi: 10.1126/scisignal.aax8295
84. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (London England). (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
85. Tsaltskan V, Firestein GS. Targeting fibroblast-like synoviocytes in rheumatoid arthritis. Curr Opin Pharmacol. (2022) 67:102304. doi: 10.1016/j.coph.2022.102304
86. Silke J, Vaux DL. IAP gene deletion and conditional knockout models. Semin Cell Dev Biology;. (2015) 39:97–105. doi: 10.1016/j.semcdb.2014.12.004
87. Grabinger T, Bode KJ, Demgenski J, Seitz C, Delgado ME, Kostadinova F, et al. Inhibitor of apoptosis protein-1 regulates tumor necrosis factor–mediated destruction of intestinal epithelial cells. Gastroenterology. (2017) 152:867–79. doi: 10.1053/j.gastro.2016.11.019
88. Xia L, Shen H, Lu J, Xiao W. TRAF2 and cIAP2 involve in TWEAK-induced MMP-9 production in fibroblast-like synoviocytes. Rheumatol Int. (2012) 32:281. doi: 10.1007/s00296-010-1640-x
89. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang C-P, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. (1995) 3:673–82. doi: 10.1016/1074-7613(95)90057-8
90. Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. (1996) 271:12687–90. doi: 10.1074/jbc.271.22.12687
91. Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat Med. (1999) 5:157–63. doi: 10.1038/5517
92. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. (1999) 104:155–62. doi: 10.1172/JCI6926
93. Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. (2013) 169:1723–44. doi: 10.1111/bph.12238
94. Vasilikos L, Spilgies LM, Knop J, Wong WW. Regulating the balance between necroptosis, apoptosis and inflammation by inhibitors of apoptosis proteins. Immunol Cell Biol. (2017) 95:160–5. doi: 10.1038/icb.2016.118
95. Wellinger LC, Hogg SJ, Newman DM, Friess T, Geiss D, Michie J, et al. BET inhibition enhances TNF-mediated antitumor immunity. Cancer Immunol Res. (2022) 10:87–107. doi: 10.1158/2326-6066.CIR-21-0224
96. Frazzi R. BIRC3 and BIRC5: multi-faceted inhibitors in cancer. Cell bioscience. (2021) 11:8. doi: 10.1186/s13578-020-00521-0
97. Zhang Z, Pan Y, Zhao Y, Ren M, Li Y, Lu G, et al. RIPK2 promotes the progression of colon cancer by regulating BIRC3-mediated ubiquitination of IKBKG. Exp Cell Res. (2023) 429:113644. doi: 10.1016/j.yexcr.2023.113644
98. Wang S, Liu Y, Li J, Zhao L, Yan W, Lin B, et al. Fusobacterium nucleatum acts as a pro-carcinogenic bacterium in colorectal cancer: from association to causality. Front Cell Dev Biol. (2021) 9:710165. doi: 10.3389/fcell.2021.710165
99. Zhang S, Yang Y, Weng W, Guo B, Cai G, Ma Y, et al. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J Exp Clin Cancer research: CR. (2019) 38:14. doi: 10.1186/s13046-018-0985-y
100. Wang D, Berglund AE, Kenchappa RS, MacAulay RJ, Mulé JJ, Etame AB. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Sci Rep. (2017) 7:9350. doi: 10.1038/s41598-017-09503-8
101. Finlay D, Teriete P, Vamos M, Cosford NDP, Vuori K. Inducing death in tumor cells: roles of the inhibitor of apoptosis proteins. F1000Research. (2017) 6:587. doi: 10.12688/f1000research
102. Sancho M, Leiva D, Lucendo E, Orzáez M. Understanding MCL1: from cellular function and regulation to pharmacological inhibition. FEBS J. (2022) 289:6209–34. doi: 10.1111/febs.16136
Glossary
RA: rheumatoid arthritis
BIRC3: baculoviral IAP Repeat Containing 3
TNF: tumor necrosis factor
CIAP1: cellular inhibitor of apoptosis protein 1
CIAP2: cellular inhibitor of apoptosis protein 2
FLS: fibroblast-like synoviocytes
RIPK1: receptor interacting serine/threonine kinase 1
Ub: ubiquitin
Gly76: C-terminal glycine
TNF-α: tumor necrosis factor alpha
IAPs: Inhibitors of apoptosis proteins
FADD: X-linked IAP
NAIP: neuronal apoptosis inhibitory protein
BIRC5: baculoviral IAP repeat containing 5
NF-kB: nuclear factor-kappa B
MAPK: mitogen-activated protein kinase
BIR: baculovirus IAP repeat
UBC: C-terminal ubiquitin-conjugated
CARD: caspase recruitment domain
TRADD: TNF receptor-associated death domain
SMAC: second mitochondrial activator of caspases
TNFR1-SC: TNFR1 signal complex
TNF-RSC: TNF receptor 1 signaling complex
TRAF2: TNF receptor-associated factor 2
LUBAC: linear ubiquitin chain assembly complex
IKK: inhibitor of kappa B kinase
TAK1: TGF-beta activated kinase 1
K63: lysine 63
NEMO: NF-κB essential modulator
CYLD: CYLD lysine 63 deubiquitinase
CD40L: CD40 ligand
TWEAK: tumor necrosis factor-like weak apoptosis inducer
K48: lysine 48
RING: really interesting new gene
NIK: NF-kB-induced kinase
TRAIL-R: TNF-related apoptosis-inducing ligand receptor
APO2L: APO-2 Ligand
TRAIL: tumour necrosis factor-related apoptosis-inducing ligand
ICAM-1: intercellular cell adhesion molecule-1
MMP3: matrix metallopeptidase 3
iNOS: inducible nitric oxide synthase
IL-1β: interleukin 1 beta
IFN-γ: interferon gamma
IL-6: interleukin 6
IL-8: interleukin-8
IL-17: interleukin 17
STAT3: signal transducer and activator of transcription 3
MMP-1: matrix metalloproteinase-1
MMP-13: matrix metalloproteinase-13
DR: death receptor TRAs, TRAIL receptor agonists
MCL1: myeloid cell leukemia1
GBM: glioblastoma
Keywords: BIRC3/cIAP2, tumor necrosis factor, rheumatoid arthritis, ubiquitination, MAPK/NF-κB
Citation: Meng Q, Wei K and Shan Y (2024) E3 ubiquitin ligase gene BIRC3 modulates TNF-induced cell death pathways and promotes aberrant proliferation in rheumatoid arthritis fibroblast-like synoviocytes. Front. Immunol. 15:1433898. doi: 10.3389/fimmu.2024.1433898
Received: 16 May 2024; Accepted: 21 August 2024;
Published: 05 September 2024.
Edited by:
Wenyi Jin, City University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Zheng Liu, Xiangtan Central Hospital, ChinaXiaomao Li, Jiangsu Food and Pharmaceutical Science College, China
Runyue Huang, Guangzhou University of Chinese Medicine, China
Copyright © 2024 Meng, Wei and Shan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Shan, c2hhbnl1MDAxMkAxMjYuY29t
†These authors have contributed equally to this work