 Tomas Mustelin
Tomas Mustelin Felipe Andrade
Felipe Andrade- 1Division of Rheumatology, Department of Medicine, University of Washington, Seattle, WA, United States
- 2Division of Rheumatology, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
Cracks in the ‘loss of tolerance’ concept
Chapters and text books about autoimmunity typically begin with the statement that autoimmunity arises as a consequence of the ‘loss of tolerance’, which is described as intrinsic to T cells (1) and/or B cells (2), which, therefore, begin to react against self-antigens. On the surface this seems perfectly logical: the difference between a healthy young person with full immunological tolerance to ‘self’ and the same individual a decade later diagnosed with an autoimmune disease, is, clearly, that the latter has a devastating reactivity of their immune system against ‘self’. The unspoken assumption here is that ‘self’ in the diagnosed patient is identical to ‘self’ in the same individual at earlier time points in their life before symptoms developed. This is the first of four problem areas with the ‘loss of tolerance’ concept that we will discuss.
Dilemma 1 – the unchanging self?
Rheumatoid arthritis (RA) represents a well-known exception to the notion that all self-derived peptides that T cells develop central and peripheral tolerance for during childhood and adolescence remain the same throughout life. In this disease, a substantial portion of self-reactive T cells recognize epitopes in which arginine residues have been post-translationally modified into citrulline by deimination (3–6). In concert with the activated T cells, B cells generate autoantibodies against numerous citrullinated proteins to the extent that these antibodies have become diagnostic of the disease (7). Is this ‘loss of tolerance’? In our stricter molecular definition of the concept, it is not. Rather, it is the response to antigens never seen by the immune system before. Physiological citrullination is limited and does not provoke an immune response, while the much more extensive citrullination in RA of many proteins (8, 9) that are not normally citrullinated creates ‘neo-autoantigens’ that are indistinguishable from pathogen-derived antigens (10–12). Hence, the immune system will do exactly what it is supposed to: activate and make every effort to eliminate the danger.
Numerous other examples of post-translationally generated neoantigens in autoimmune diseases have accumulated in recent years (13–15). In type 1 diabetes (T1D), autoantigens include citrullinated β-cell antigens (16–18), insulin peptides covalently cross-linked to other peptides by transglutaminase (17), and peptides from abnormally spliced transcripts (19). This is also true in multiple sclerosis, SLE, scleroderma, Sjögren’s syndrome, and autoimmune myositis (15, 20).
Dilemma 2 – features of clinical disease, autoantibody repertoires, and responses to therapy
The second shortcoming of the loss of tolerance dogma is its inability to explain many of the basic aspects of autoimmune diseases as we observe them in human patients. If autoimmunity was initiated by a molecular mechanism causing T cell-intrinsic loss of tolerance, the antigens seen by these T cells would likely be stochastic, resulting in a broad spectrum of autoimmune syndromes against a myriad of autoantigens. This is not what we see in the clinic. Rather, we observe a modest number of distinct disease entities, each with a characteristic prevalence, gender ratio, peak age of onset, clinical course and prognosis, a set of unique autoantibodies, a spectrum of typical symptoms and organ manifestations, and response to different medications. Clinically, autoimmune diseases can be remarkably different from each other, and they are managed by different specialists: rheumatologists, gastroenterologists, endocrinologists, neurologists etc. It is difficult to believe that all these diseases would be initiated by the same type of molecular mechanism, i.e., loss of tolerance in T and/or B cells.
Although currently used drugs are generally immunosuppressive (largely because of the reliance on mouse models for their discovery and development (21)), it is remarkable how differently autoimmune diseases respond to specific medications. For example, psoriasis improves dramatically after interleukin 17 blockade (22, 23), while RA does not (24, 25). Instead, most RA patients respond well to TNF blockers (26, 27), while patients with systemic lupus erythematosus (SLE) do not (28, 29). B cell depletion by rituximab works well in RA (30, 31) and multiple sclerosis (32), but not in SLE (33). Interestingly, however, these therapeutic effects match our understanding of different flavors of immune responses to different classes of antigens: bacterial, viral, carbohydrates, and nucleic acids. On the other hand, the loss-of-tolerance hypothesis struggles to provide an explanation for what becomes an autoantigen. Evidence is lacking for ideas like antigen overexpression (e.g. in tumors), molecular mimicry, autoimmune ‘pre-disease’, or medication. Mass action does not apply to the immune system, which seeks low-abundance antigen and is tolerized or anergized by higher amounts of antigen. Molecular mimicry, which postulates that a foreign antigen that is sufficiently similar to a self-antigen can trigger an immune response against this self-antigen, may explain tendencies towards autoimmunity during severe infection, e.g. by SARS-CoV2, but tends to fade as soon as the exogenous antigen is cleared. To the best of our knowledge there are no cases of proven molecular mimicry driving chronic autoimmune disease. The concept of pre-autoimmune disease is interesting and in line with what we propose as the neoantigen hypothesis.
There is also a wealth of evidence in support of environmental factors (34–36), such as infectious agents, the gut microbiome, cigarette smoking, and ultraviolet light exposure, influencing the pathogenesis and/or triggering exacerbations in human autoimmune conditions. It is not obvious how one would fit all of these into a model where T cells have lost the tolerance for self-antigens.
Dilemma 3 – genome-wide association studies
The genetic underpinnings of autoimmune disorders have been explored over the last two decades, particularly through GWAS (37), which rely on common single-nucleotide polymorphisms, i.e., the minor allele is present in sufficient frequencies in the general population. An allele is deemed disease-associated if its frequency is higher in the disease than in the healthy control group. Importantly, the sufficiently high frequency of the disease-associated allele in the population means that it has been positively selected for over evolutionary time, presumably because it conferred a survival advantage to the carrier. Known examples of this include the role of the autoimmune-associated allele of PTPN22 (38, 39) in resistance to viral infection (40) and tuberculosis (41). Apparently, the immune system in carriers of the disease-associated PTPN22*W620 allele respond more appropriately to certain foreign antigens. Similarly, autoimmune-associated HLA haplotypes, such as HLA-DR4/DQ8, DR2/DQ6 and DR3/DQ2, may have been selected for their ability to present microbial peptides and promote the production of anti-microbial cytokines (42). Consequently, the disease-associated alleles do not tell us that the immune system has gone awry or that autoimmunity starts by loss of tolerance. They do, however, support the notion that the sensitivity of the immune system is an important factor.
Another category of genes associated with autoimmune disease are those that are not clearly immune-related. For example, several genes associated with autoimmune diseases, particularly SLE, are involved in the clearance of DNA (43). Another example is the association of RA with the genes PADI2 (44, 45) and PADI4 (46), which encode the two citrullinating enzymes that participate, alone or in combination, in the production of citrullinated neoantigen in this disease.
Dilemma 4 – recent findings through deep immune profiling
Recent advances in ultra-sensitive DNA sequencing technology now allow researchers to profile immune cells at the single-cell level for transcriptional repertoire and whole-genome epigenetic landscapes. Publications built on this new capability are now frequent in top journals and they promise new drug targets and the identification of the pathogenic cell. Unfortunately, nearly all these studies compared immune cells between disease tissue and healthy tissue, one disease at a time. While skewing of immune cell lineages are observed, and large sets of genes are expressed differently, the main take-home message is that the immune system is activated. As these studies were conducted in more and more diseases, it became apparent that the findings were quite similar in each disease. Particularly striking was the observation that the immune response in COVID-19 patients is very similar to that in autoimmune disease. Despite the differences between an exogenous virus like SARS-CoV2 and the unknown causes of RA or SLE, the immune response is very similar. We reported that changes in gene expression is near-identical in neutrophils from COVID-19 and SLE; only one single gene was statistically significantly regulated in opposite directions (47). No smoking gun. All the immune cells observed in autoimmune conditions represent lineages and phenotypes present in healthy individuals, particularly during viral or bacterial infections. Despite the frequent use of the term ‘dysregulated’, it seems to us that all the observations reflect a normal (‘eu-regulated’) immune activation with precious few, if any, disease-specific abnormalities that would illuminate the cause or pathogenesis of the disease in question. This is precisely what you would expect to see if autoimmune disease was the result of neoantigens.
The neoantigen hypothesis
Given the above shortcomings of the loss of tolerance concept, we propose another model for the initiation and progression of autoimmune disorders. We posit that it is not a malfunction of the immune system that leads to autoimmunity, but that aberrantly generated neoantigens are the cause of an immune response that may be asymptomatic at first, but then gradually escalates over a longer period of time into a clinically manifest condition that can be given a specific diagnosis. By ‘aberrantly generated’ we mean that these neoantigens are not present during normal development, so tolerance for them cannot develop, but that they are produced later in life by molecular mechanisms unique to a specific autoimmune disease in individuals who subsequently develop that disease. These currently poorly understood molecular mechanisms can be considered the upstream and real cause of the disease, while the immune system that reacts to the neoantigen is executing its preprogrammed function, even if that is driving the inflammation that underlies the symptoms and organ manifestations of the disease. The immune system is responding exactly as it would against any ‘foreign’ antigen and it will stay activated until the antigen is eliminated. The problem is that the source of the neoantigen is not under the immune system’s control.
Unlike the loss of tolerance concept, the neoantigen hypothesis postulates that ‘self’ indeed can change. It also fits the basic immunological concept that the nature of an immune response depends on the type of antigen. The molecular mechanism that generates a particular kind of neoantigen determines when and where that occurs. It may be restricted to a cell type or organ, such as the salivary gland in Sjögren’s syndrome, or skeletal muscle in myositis, or it may be widespread. Neoantigen production could be episodic, resulting a waxing and waning immune response, or chronic and stable. It may start at a certain age, it may have a sex bias, and it may be influenced by the risk factors of autoimmunity, such as smoking. Carriers of disease-associated alleles – like PTPN22*W620, DR2 (DRβ1*1501)/DQ6 (DQβ1*0602), DR3 (DRβ1*0301)/DQ2 (DQβ1*0201), and DR4 (DRβ1*0401)/DQ8 (DQβ1*0302) – may respond more robustly to neoantigens. Non-immune genes with disease-associated alleles may have a role in the generation of neoantigens. Monogenic SLE caused by DNASE1L3 deficiency (42, 48) is an excellent example of how a non-immune gene involved in the clearance of an antigen (i.e., DNA) has a dominant effect in the development of an autoimmune disease in the context of a normal immune system. The neoantigen hypothesis also predicts that an immune response to neoantigen should look like a normal immune response to exogenous antigens, just as we observe in the deep immune profiling of patient immune cells and tissues.
Whether the neoantigens are present episodically or in a more continuous manner, the immune system may become frustrated by its persistence and escalate to levels of activity that include tissue damage and epitope spreading. The latter may broaden the set of involved antigens and may encompass bona fide autoantigens, as occurs during more severe and longer lasting COVID-19. However, we speculate that, just as in COVID-19, this broader reactivity would subside over some period of time if the neoantigens vanish.
Sources and molecular mechanisms of neoantigen production
Research of anti-tumor immunity was greatly stimulated by the introduction of checkpoint inhibitors, such as the antibodies to CTLA-4 (ipilimumab) and PD-1 (pembrolizumab), over a decade ago. Anti-tumor immunity is a form of autoimmunity, in which the immune system is recognizing and responding to neoantigens derived from altered ‘self’. These antigens are now well-accepted in the immuno-oncology community and generally fall into four distinct categories, schematically shown in Figure 1: (i) epitopes generated by point-mutations or other genetic changes in the malignant cells; (ii) the translation of transcripts resulting from aberrant mRNA splicing (49, 50); (iii) peptides and proteins encoded by retroelements and endogenous retroviruses (51–53), many of which are expressed at elevated levels in malignant cells; (iv) aberrant post-translational modifications.
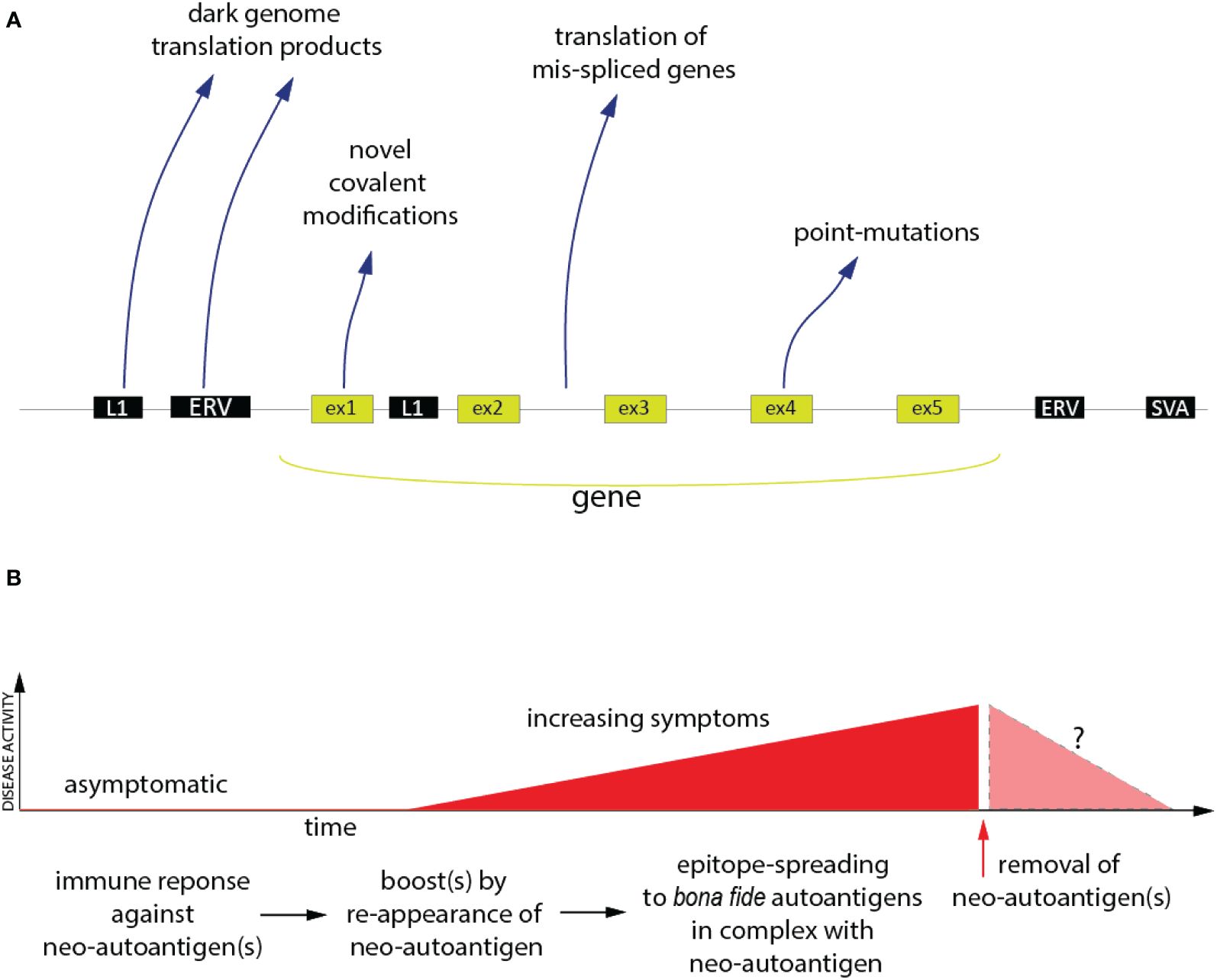
Figure 1 The Neoantigen Hypothesis. (A) The genomic and post-translational sources of the four principal categories of neo(auto)antigens. Novel covalent modifications are not restricted to exon-encoded proteins, but could, in principle, also affect the other classes of neoantigen polypeptides. (B) Proposed time-course of events in the pathogenesis of an autoimmune disease (red) driven by neo(auto)antigens and the hypothetical consequence of removal of the neoantigens (pink).
While these antigens are instrumental for the success of cancer immunotherapy, a causative role of similar neoantigens in non-cancer autoimmunity remains speculative. In fact, they are generally ignored by the autoimmunity research community. Nevertheless, there are recent publications showing that all four categories of tumor neoantigens are also present in patients with non-cancer autoimmune disorders. There is also a tantalizing co-occurrence of certain rheumatic disease with tumors, both characterized by shared autoantibodies (54).
Point-mutations have been reported in Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic (VEXAS) syndrome, in clonal hematopoiesis (55), and in RA (56–58). So far, these studies have focused on point-mutations altering the function of genes, not as neoepitopes. There are several reports of dysregulated mRNA splicing in SLE (59, 60), which can lead to translation products predicted (61) to contain stretches of novel amino acids due to frame shifts or retained parts of introns. We have shown that mRNA splicing is particularly aberrant in SLE neutrophils (Najjar et al., 2024)1, in which 80 events resulted in novel amino acid sequences. Every patient in our study (n=15) had at least one such event, while some had many, one as many as 36 of them (Najjar et al., 2024)1. In another example, a subset of autoantibodies against Ro52 were found to be directed towards a unique domain due to a frameshift in a splice variant found in SLE neutrophils (62). Proteins and peptides from non-exonic regions of our genome are present in increased quantities in several rheumatic diseases (61, 63, 64) and autoantibodies against them (65–68) are prevalent.
The best example of neoantigens created by post-translational modification is in RA (3), but there are many other examples of post-translational modifications creating neoantigens in autoimmune diseases, such as carbamylation (69), acetylation, cysteine carboxyethylation (70) and covalent metabolite attachment (71). As is well-documented, self-proteins covalently modified by a small-molecule drug, termed a hapten, can be very immunogenic and drive a dangerous immune response (72).
Post-translational neoantigens can also be generated in the context of infection through the interaction of viral and host products, as well as during the killing of infected cells by cytotoxic cells. For example, the autoantigen La binds to virus-associated (VA) RNA in adenovirus-infected cells (73), as well as EBV-encoded RNAs (EBER1 and EBER2) during EBV infection (74). Moreover, numerous autoantigens are modified as result of proteolysis in target cells killed by cytotoxic cells (15). Interestingly, the autoantigen Ro60 is in complex with endogenous Alu retroelements and anti-Ro60-Alu RNA immune complexes are found in circulation in SLE (75).
Drug development
The neoantigen hypothesis predicts that immunosuppressive drugs will reduce disease activity in autoimmune conditions. This is largely the case but comes at the cost of reducing immune responses against exogenous antigens to a similar extent. The neoantigen hypothesis also opens a completely new avenue for drug development, namely the opportunity to stop the process responsible for neoantigen production. Since this process likely is active only in patients with the disease, blocking it should be very safe. At present, we have an incomplete understanding of the mechanisms that generate neoantigens, and most of what we know is in the context of tumors, where the desire is to increase them for the benefit of immunotherapy. In non-cancer autoimmunity, the goal will be to reduce or eliminate these processes.
Concluding remarks and future perspectives
The neoantigen hypothesis seeks to stimulate a critical rethinking and re-examination of the current loss of tolerance concept. We do not wish to state that the loss of tolerance concept is wrong, but we believe it is obvious that other mechanisms must also be at play. Defining the primary role of neoantigens vs. loss of tolerance in autoimmune diseases has critical implications for pathogenesis, prevention and treatment. Progress in understanding the molecular pathways that generate neoantigens will enable the design of new preventive and therapeutic approaches, which could be applied prior to the preclinical detection of autoantibodies or once the disease has been established without interfering with the normal function of the immune system. Eventually, this approach will demonstrate in clinical trials whether neoantigens are important in precipitating or perpetuating autoimmune conditions and their exacerbations.
Author contributions
TM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FA: Conceptualization, Formal analysis, Funding acquisition, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health grants R01 AR074939, R01 AR081654, R01 AI186337, R21 AR077266, R21 AR075134, R21AG087517 to TM, and R01 AR079404, R21 AI169851, R21 AI176766 to FA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ Najjar R, Wang X, Pineda JMB, Alessi H, Bays A, Bradley RK, et al. Altered protein structures and neoepitopes in lupus neutrophils from dysregulated splicing of messenger RNA. ACR Open Rheumatol. (2024). in press.
References
1. Zhang P, Lu Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell Mol Immunol. (2018) 15:575–85. doi: 10.1038/cmi.2017.137
2. Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, et al. B-cell-intrinsic type 1 interferon signaling is crucial for loss of tolerance and the development of autoreactive B cells. Cell Rep. (2018) 24:406–18. doi: 10.1016/j.celrep.2018.06.046
3. Darrah E, Andrade F. Rheumatoid arthritis and citrullination. Curr Opin Rheumatol. (2018) 30:72–8. doi: 10.1097/BOR.0000000000000452
4. Moon JS, Younis S, Ramadoss NS, Iyer R, Sheth K, Sharpe O, et al. Cytotoxic CD8(+) T cells target citrullinated antigens in rheumatoid arthritis. Nat Commun. (2023) 14:319. doi: 10.1038/s41467-022-35264-8
5. Rims C, Uchtenhagen H, Kaplan MJ, Carmona-Rivera C, Carlucci P, Mikecz K, et al. Citrullinated aggrecan epitopes as targets of autoreactive CD4+ T cells in patients with rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:518–28. doi: 10.1002/art.40768
6. Gerstner C, Dubnovitsky A, Sandin C, Kozhukh G, Uchtenhagen H, James EA, et al. Functional and structural characterization of a novel HLA-DRB1*04:01-restricted alpha-enolase T cell epitope in rheumatoid arthritis. Front Immunol. (2016) 7:494. doi: 10.3389/fimmu.2016.00494
7. Kroot EJ, de Jong BA, van Leeuwen MA, Swinkels H, van den Hoogen FH, van't Hof M, et al. The prognostic value of anti-cyclic citrullinated peptide antibody in patients with recent-onset rheumatoid arthritis. Arthritis Rheumatol. (2000) 43:1831–5. doi: 10.1002/(ISSN)1529-0131
8. De Rycke L, Nicholas AP, Cantaert T, Kruithof E, Echols JD, Vandekerckhove B, et al. Synovial intracellular citrullinated proteins colocalizing with peptidyl arginine deiminase as pathophysiologically relevant antigenic determinants of rheumatoid arthritis-specific humoral autoimmunity. Arthritis Rheumatol. (2005) 52:2323–30. doi: 10.1002/art.21220
9. Tutturen AE, Fleckenstein B, de Souza GA. Assessing the citrullinome in rheumatoid arthritis synovial fluid with and without enrichment of citrullinated peptides. J Proteome Res. (2014) 13:2867–73. doi: 10.1021/pr500030x
10. Gomez-Banuelos E, Konig MF, Andrade F. Microbial pathways to subvert host immunity generate citrullinated neoantigens targeted in rheumatoid arthritis. Curr Opin Struct Biol. (2022) 75:102423. doi: 10.1016/j.sbi.2022.102423
11. Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheumatol. (2010) 62:2662–72. doi: 10.1002/art.27552
12. Gully N, Bright R, Marino V, Marchant C, Cantley M, Haynes D, et al. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS One. (2014) 9:e100838. doi: 10.1371/journal.pone.0100838
13. Nguyen H, Arribas-Layton D, Chow IT, Speake C, Kwok WW, Hessner MJ, et al. Characterizing T cell responses to enzymatically modified beta cell neo-epitopes. Front Immunol. (2022) 13:1015855. doi: 10.3389/fimmu.2022.1015855
14. Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, et al. Modifying enzymes are elicited by ER stress, generating epitopes that are selectively recognized by CD4(+) T cells in patients with type 1 diabetes. Diabetes. (2018) 67:1356–68. doi: 10.2337/db17-1166
15. Rosen A, Casciola-Rosen L. Autoantigens as partners in initiation and propagation of autoimmune rheumatic diseases. Annu Rev Immunol. (2016) 34:395–420. doi: 10.1146/annurev-immunol-032414-112205
16. Buitinga M, Callebaut A, Marques Camara Sodre F, Crevecoeur I, Blahnik-Fagan G, Yang ML, et al. Inflammation-induced citrullinated glucose-regulated protein 78 elicits immune responses in human type 1 diabetes. Diabetes. (2018) 67:2337–48. doi: 10.2337/db18-0295
17. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes. (2014) 63:3033–40. doi: 10.2337/db13-1952
18. James EA, Pietropaolo M, Mamula MJ. Immune recognition of beta-cells: neoepitopes as key players in the loss of tolerance. Diabetes. (2018) 67:1035–42. doi: 10.2337/dbi17-0030
19. Rodriguez-Calvo T, Johnson JD, Overbergh L, Dunne JL. Neoepitopes in type 1 diabetes: etiological insights, biomarkers and therapeutic targets. Front Immunol. (2021) 12:667989. doi: 10.3389/fimmu.2021.667989
20. Martin Monreal MT, Hansen BE, Iversen PF, Enevold C, Odum N, Sellebjerg F, et al. Citrullination of myelin basic protein induces a Th17-cell response in healthy individuals and enhances the presentation of MBP85-99 in patients with multiple sclerosis. J Autoimmun. (2023) 139:103092. doi: 10.1016/j.jaut.2023.103092
21. Hegen M, Keith JC Jr., Collins M, Nickerson-Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis. (2008) 67:1505–15. doi: 10.1136/ard.2007.076430
22. Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. (2010) 2:52ra72. doi: 10.1126/scitranslmed.3001107
23. Papp KA, Langley RG, Sigurgeirsson B, Abe M, Baker DR, Konno P, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. (2013) 168:412–21. doi: 10.1111/bjd.12110
24. Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis. (2013) 72:863–9. doi: 10.1136/annrheumdis-2012-201601
25. Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, Erondu N. A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J Rheumatol. (2015) 42:912–9. doi: 10.3899/jrheum.141271
26. Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. (1997) 337:141–7. doi: 10.1056/NEJM199707173370301
27. Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheumatol. (1998) 41:1552–63. doi: 10.1002/(ISSN)1529-0131
28. Shakoor N, Michalska M, Harris CA, Block JA. Drug-induced systemic lupus erythematosus associated with etanercept therapy. Lancet. (2002) 359:579–80. doi: 10.1016/S0140-6736(02)07714-0
29. Almoallim H, Al-Ghamdi Y, Almaghrabi H, Alyasi O. Anti-tumor necrosis factor-alpha induced systemic lupus erythematosus(). Open Rheumatol J. (2012) 6:315–9. doi: 10.2174/1874312901206010315
30. Leandro MJ, Edwards JC, Cambridge G. Clinical outcome in 22 patients with rheumatoid arthritis treated with B lymphocyte depletion. Ann Rheum Dis. (2002) 61:883–8. doi: 10.1136/ard.61.10.883
31. De Vita S, Zaja F, Sacco S, De Candia A, Fanin R, Ferraccioli G. Efficacy of selective B cell blockade in the treatment of rheumatoid arthritis: evidence for a pathogenetic role of B cells. Arthritis Rheumatol. (2002) 46:2029–33. doi: 10.1002/art.10467
32. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
33. Oon S, Huq M, Godfrey T, Nikpour M. Systematic review, and meta-analysis of steroid-sparing effect, of biologic agents in randomized, placebo-controlled phase 3 trials for systemic lupus erythematosus. Semin Arthritis Rheumatol. (2018) 48:221–39. doi: 10.1016/j.semarthrit.2018.01.001
34. Cooper GS, Dooley MA, Treadwell EL, St Clair EW, Parks CG, Gilkeson GS. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheumatol. (1998) 41:1714–24. doi: 10.1002/(ISSN)1529-0131
35. Horton DB, Shenoi S. Review of environmental factors and juvenile idiopathic arthritis. Open Access Rheumatol. (2019) 11:253–67. doi: 10.2147/OARRR
36. Wheeler AM, Baker JF, Poole JA, Ascherman DP, Yang Y, Kerr GS, et al. Genetic, social, and environmental risk factors in rheumatoid arthritis-associated interstitial lung disease. Semin Arthritis Rheumatol. (2022) 57:152098. doi: 10.1016/j.semarthrit.2022.152098
37. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. (2019) 47:D1005–D12. doi: 10.1093/nar/gky1120
38. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. (2004) 36:337–8. doi: 10.1038/ng1323
39. Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. (2006) 18:207–13. doi: 10.1016/j.smim.2006.03.008
40. Orozco RC, Marquardt K, Pratumchai I, Shaikh AF, Mowen K, Domissy A, et al. Autoimmunity-associated allele of tyrosine phosphatase gene PTPN22 enhances anti-viral immunity. PloS Pathog. (2024) 20:e1012095. doi: 10.1371/journal.ppat.1012095
41. Gomez LM, Anaya JM, Martin J. Genetic influence of PTPN22 R620W polymorphism in tuberculosis. Hum Immunol. (2005) 66:1242–7. doi: 10.1016/j.humimm.2005.11.008
42. Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol. (2013) 190:513–8. doi: 10.4049/jimmunol.1201891
43. Keyel PA. Dnases in health and disease. Dev Biol. (2017) 429:1–11. doi: 10.1016/j.ydbio.2017.06.028
44. Too CL, Murad S, Dhaliwal JS, Larsson P, Jiang X, Ding B, et al. Polymorphisms in peptidylarginine deiminase associate with rheumatoid arthritis in diverse Asian populations: evidence from MyEIRA study and meta-analysis. Arthritis Res Ther. (2012) 14:R250. doi: 10.1186/ar4093
45. Chang X, Xia Y, Pan J, Meng Q, Zhao Y, Yan X. PADI2 is significantly associated with rheumatoid arthritis. PLoS One. (2013) 8:e81259. doi: 10.1371/journal.pone.0081259
46. Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. (2003) 34:395–402. doi: 10.1038/ng1206
47. Najjar R, Rogel N, Pineda JMB, Wang X, Tran M, Bays A, et al. Large overlap in neutrophil transcriptome between lupus and COVID-19 with limited lupus-specific gene expression. Lupus Sci Med. (2024) 11:e001059. doi: 10.1136/lupus-2023-001059
48. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
49. Bigot J, Lalanne AI, Lucibello F, Gueguen P, Houy A, Dayot S, et al. Splicing patterns in SF3B1-mutated uveal melanoma generate shared immunogenic tumor-specific neoepitopes. Cancer Discovery. (2021) 11:1938–51. doi: 10.1158/2159-8290.CD-20-0555
50. Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell. (2021) 184:4032–47.e31. doi: 10.1016/j.cell.2021.05.038
51. Bonte PE, Arribas YA, Merlotti A, Carrascal M, Zhang JV, Zueva E, et al. Single-cell RNA-seq-based proteogenomics identifies glioblastoma-specific transposable elements encoding HLA-I-presented peptides. Cell Rep. (2022) 39:110916. doi: 10.1016/j.celrep.2022.110916
52. Burbage M, Rocanin-Arjo A, Baudon B, Arribas YA, Merlotti A, Rookhuizen DC, et al. Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements. Sci Immunol. (2023) 8:eabm6360. doi: 10.1126/sciimmunol.abm6360
53. Merlotti A, Sadacca B, Arribas YA, Ngoma M, Burbage M, Goudot C, et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci Immunol. (2023) 8:eabm6359. doi: 10.1126/sciimmunol.abm6359
54. Shah AA, Casciola-Rosen L, Rosen A. Review: cancer-induced autoimmunity in the rheumatic diseases. Arthritis Rheumatol. (2015) 67:317–26. doi: 10.1002/art.38928
55. Heimlich JB, Bhat P, Parker A, Jenkins MT, Vlasschaert C, Ulloa J, et al. Multiomic profiling of human clonal hematopoiesis reveals genotype and cell-specific inflammatory pathway activation. Blood Adv. (2024). doi: 10.1182/bloodadvances.2023011445
56. Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A. (1997) 94:10895–900. doi: 10.1073/pnas.94.20.10895
57. Reme T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. (1998) 111:353–8. doi: 10.1046/j.1365-2249.1998.00508.x
58. Qiu C, Chan JTW, Zhang DW, Wong IN, Zeng Y, Law BYK, et al. The potential development of drug resistance in rheumatoid arthritis patients identified with p53 mutations. Genes Dis. (2023) 10:2252–5. doi: 10.1016/j.gendis.2023.02.007
59. Kosmara D, Papanikolaou S, Nikolaou C, Bertsias G. Extensive alternative splicing patterns in systemic lupus erythematosus highlight sexual differences. Cells. (2023) 12:2678. doi: 10.3390/cells12232678
60. Papanikolaou S, Bertsias GK, Nikolaou C. Extensive changes in transcription dynamics reflected on alternative splicing events in systemic lupus erythematosus patients. Genes (Basel). (2021) 12:1260. doi: 10.3390/genes12081260
61. Ukadike KC, Najjar R, Ni K, Laine A, Wang X, Bays A, et al. Expression of L1 retrotransposons in granulocytes from patients with active systemic lupus erythematosus. Mob DNA. (2023) 14:5. doi: 10.1186/s13100-023-00293-7
62. Gomez-Banuelos E, Wahadat MJ, Li J, Paz M, Antiochos B, Celia AI, et al. Alternative exon usage in TRIM21 determines the antigenicity of Ro52/TRIM21 in systemic lupus erythematosus. JCI Insight. (2022) 7:e163795. doi: 10.1172/jci.insight.163795
63. Reynier F, Verjat T, Turrel F, Imbert PE, Marotte H, Mougin B, et al. Increase in human endogenous retrovirus HERV-K (HML-2) viral load in active rheumatoid arthritis. Scand J Immunol. (2009) 70:295–9. doi: 10.1111/j.1365-3083.2009.02271.x
64. Laine A, Wang X, Ni K, Smith SEB, Najjar R, Whitmore LS, et al. Expression of envelope protein encoded by endogenous retrovirus K102 in rheumatoid arthritis neutrophils. Microorganisms. (2023) 11:1310. doi: 10.3390/microorganisms11051310
65. Mameli G, Erre GL, Caggiu E, Mura S, Cossu D, Bo M, et al. Identification of a HERV-K env surface peptide highly recognized in Rheumatoid Arthritis (RA) patients: a cross-sectional case-control study. Clin Exp Immunol. (2017) 189:127–31. doi: 10.1111/cei.12964
66. Carter V, LaCava J, Taylor MS, Liang SY, Mustelin C, Ukadike KC, et al. High prevalence and disease correlation of autoantibodies against p40 encoded by long interspersed nuclear elements in systemic lupus erythematosus. Arthritis Rheumatol. (2020) 72:89–99. doi: 10.1002/art.41054
67. Wang E, Pineda JMB, Kim WJ, Chen S, Bourcier J, Stahl M, et al. Modulation of RNA splicing enhances response to BCL2 inhibition in leukemia. Cancer Cell. (2023) 41:164–80.e8. doi: 10.1016/j.ccell.2022.12.002
68. Khadjinova AI, Wang X, Laine A, Ukadike K, Eckert M, Stevens A, et al. Autoantibodies against the envelope proteins of endogenous retroviruses K102 and K108 in patients with systemic lupus erythematosus correlate with active disease. Clin Exp Rheumatol. (2022) 40:1306–12. doi: 10.55563/clinexprheumatol/2kg1d8
69. Verheul MK, van Erp SJ, van der Woude D, Levarht EW, Mallat MJ, Verspaget HW, et al. Anti-carbamylated protein antibodies: a specific hallmark for rheumatoid arthritis. Comparison to conditions known for enhanced carbamylation; renal failure, smoking and chronic inflammation. Ann Rheum Dis. (2016) 75:1575–6. doi: 10.1136/annrheumdis-2016-209248
70. Zhai Y, Chen L, Zhao Q, Zheng ZH, Chen ZN, Bian H, et al. Cysteine carboxyethylation generates neoantigens to induce HLA-restricted autoimmunity. Science. (2023) 379:eabg2482. doi: 10.1126/science.abg2482
71. Zhai Y, Zhu P. Post-translationally modified neoantigens: Promising targets for diagnostic strategy of autoimmune diseases. Clin Transl Med. (2023) 13:e1373. doi: 10.1002/ctm2.1373
72. Trejo-Zambrano MI, Gomez-Banuelos E, Andrade F. Redox-mediated carbamylation as a hapten model applied to the origin of antibodies to modified proteins in rheumatoid arthritis. Antioxid Redox Signal. (2022) 36:389–409. doi: 10.1089/ars.2021.0064
73. Lerner MR, Boyle JA, Hardin JA, Steitz JA. Two novel classes of small ribonucleoproteins detected by antibodies associated with lupus erythematosus. Science. (1981) 211:400–2. doi: 10.1126/science.6164096
74. Lerner MR, Andrews NC, Miller G, Steitz JA. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci U S A. (1981) 78:805–9. doi: 10.1073/pnas.78.2.805
Keywords: neoantigen, autoimmunity, autoantibodies, citrullination, rheumatoid arthritis, systemic lupus erythematosus, transposable elements
Citation: Mustelin T and Andrade F (2024) Autoimmunity: the neoantigen hypothesis. Front. Immunol. 15:1432985. doi: 10.3389/fimmu.2024.1432985
Received: 15 May 2024; Accepted: 17 June 2024;
Published: 27 June 2024.
Edited by:
Mihaela Adriana Ilie, Länssjukhuset i Kalmar, SwedenReviewed by:
Sabrina Patzelt, University of Lübeck, GermanyCopyright © 2024 Mustelin and Andrade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomas Mustelin, dG9tYXMyQHV3LmVkdQ==