 Guangtan Du
Guangtan Du Cunmiao Dou
Cunmiao Dou Peng Sun
Peng Sun Shasha Wang
Shasha Wang Jia Liu
Jia Liu Leina Ma
Leina Ma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 29 July 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1431211
This article is part of the Research TopicRegulators of the Immune-Tumor Microenvironment: A New Frontier for Cancer ImmunotherapyView all 18 articles
Liver cancer, which most commonly manifests as hepatocellular carcinoma (HCC), is the sixth most common cancer in the world. In HCC, the immune system plays a crucial role in the growth and proliferation of tumor cells. HCC achieve immune escape through the tumor microenvironment, which significantly promotes the development of this cancer. Here, this article introduces and summarizes the functions and effects of regulatory T cells (Tregs) in the tumor microenvironment, highlighting how Tregs inhibit and regulate the functions of immune and tumor cells, cytokines, ligands and receptors, etc, thereby promoting tumor immune escape. In addition, it discusses the mechanism of CAR-T therapy for HCC and elaborate on the relationship between CAR-T and Tregs.
HCC is a complex malignancy, with its tumor microenvironment playing a crucial role in promoting tumor growth, invasion, metastasis, and immune escape. This microenvironment is composed of a variety of cellular and molecular components, including tumor cells, cancer-associated fibroblasts (CAFs), vascular cells, and a range of immune cells such as tumor-associated macrophages, bone marrow-derived suppressor cells, Tregs, and liver-specific Kupffer cells. While many of these components contribute to tumor growth and metastasis, certain immune cells, including dendritic cells, regulatory B cells, and tumor-associated neutrophils, exhibit tumor-suppressing effects. Additionally, the tumor microenvironment contains stromal tissue that provides structural support, as well as metabolic products and circulating DNA.
In the immune escape mechanism of HCC, Tregs inhibit the immune cells’ ability to kill HCC through multiple pathways, thereby promoting immune escape of HCC cells. Tregs are closely associated with the poor prognosis of HCC patients. They not only suppress excessive immune responses to prevent impairment of normal functions but also promote the formation of immune tolerance, thus protecting liver cancer cells from systemic attacks. Additionally, studies have shown that an excess of Tregs in liver cancer patients is linked to poor treatment outcomes because Tregs limit the effectiveness of immunotherapy and weaken the efficacy of immune checkpoint inhibitors.
Unlike many other tumors, liver cancer often develops from chronic liver disease and possesses a unique dual blood supply from the hepatic artery and portal vein. This dual supply facilitates the invasion and metastasis of liver cancer. Additionally, the liver functions as an immune regulatory center, aggregating various types of immune cells and creating distinct immune characteristics. These properties significantly impact the physiological state of the liver and cause Tregs in liver cancer to exhibit different expression patterns compared to other tumors. For instance, in liver cancer, Tregs frequently express high levels of Programmed Death-Ligand 1 (PD-L1) and Cytotoxic T-lymphocyte-associated protein 4 (CTLA4). These unique features significantly increase the complexity and challenge of researching and treating liver cancer.
The immune microenvironment in HCC is predominantly influenced by Tregs, which play a crucial role in immune suppression and regulation, significantly affecting the progression of liver cancer. This relationship prompts an in-depth exploration of the interaction between regulatory T cells and liver cancer.
Regulatory T cells, a subpopulation of T lymphocytes, possess immunosuppressive capabilities. They regulate immune responses by directly inhibiting activated immune cells to prevent an overactive immune response. Additionally, they achieve immunosuppression by releasing cytokines and other substances that inhibit the activation of immune cells.
Treg research involves multiple fields, including the pathogenesis and treatment of various autoimmune diseases, cancer and allergy, as well as transplantation research and tissue engineering. Here, it focuses on regulatory T cells in hepatocellular carcinoma. It is found following the development of liver cancer, a significant increase in Tregs is observed in the tumor microenvironment of the cancerous part, which affect the development and treatment outcomes of hepatocellular carcinoma through various mechanisms. This raises the question: what is the origin of these abundant Tregs in liver cancer? After the onset of liver cancer, dendritic cells in the lesion site recognize and present antigens, activating a large number of T cell responses. Studies have shown that transforming growth factor-beta (TGF-β) has been shown to promote the plasticity of Th17 cells in a dose-dependent manner facilitating their conversion into Treg cells (1). Additionally, it has been demonstrated that when activated T cells are co-cultured with exosomes derived from HCC, there is an induced conversion of CD4+ T cells into Tregs (2). Regulatory T cells express high levels of chemokine receptors such as Characteristic Chemokine Receptor 4 (CCR4), Characteristic Chemokine Receptor 8 (CCR8), Characteristic Chemokine Receptor 10 (CCR10), and C-X-C Motif Chemokine Receptor 3 (CXCR3). In liver cancer, chemokines such as C-C Motif Chemokine Ligand 17 (CCL17), C-C Motif Chemokine Ligand 22 (CCL22), C-C Motif Chemokine Ligand 28 (CCL28), and C-X-C Motif Chemokine Ligand 9 (CXCL9) are produced, which bind to these receptors, and consequently recruit a substantial number of Tregs to the liver cancer site (3). Furthermore, interleukin-10 secreted by tumor-associated macrophages (TAMs) also activate regulatory T cells (4).
Previous studies have shown that regulatory T cells play an inhibitory role in hepatocellular carcinoma. In this section, it mainly discusses how Treg cells exert their inhibitory effects in the tumor microenvironment of hepatocellular carcinoma.
Immune cells are vital components of the tumor microenvironment (TME), with different immune cells playing distinct roles in the tumor microenvironment. Some immune cells infiltrate the TME to exert cytotoxic effects on HCC, including T cells, Natural killer cell (NK cell), B cells, Dendritic cells (DCs) and Neutrophils, etc.
As shown in the Supplementary Table, in the tumor microenvironment of HCC, infiltrating T cells include CD8+ cytotoxic T lymphocytes (CTLs), natural killer T (NKT) cells, regulatory T cells, and helper T cells. CD8+ CTLs target tumor cells by recognizing specific antigens and secreting cytotoxic substances, while NKT cells link the innate and adaptive immune systems, with type I NKT cells exerting anti-tumor effects and type II NKT cells promoting tumor growth through immunosuppressive actions (5). Different levels of CD40 expression alter T cell responses through different mechanisms: first, CD40 signaling affects the process of antigen processing and presentation, and second, different levels of CD40 produce different levels of IL-12 and IL-10 (6).
NK cells activate their cytotoxicity by recognizing stress molecules on the surface of tumor cells, subsequently destroying the tumor cell membrane and inducing apoptosis through the release of perforins and granzymes. For B cells, B cells primarily include activated B cells, depleted B cells, and plasma cells, with plasma cells producing and secreting antibodies. Additionally, the heightened glucose and oxygen consumption by activated B cells and plasma cells depletes the tumor microenvironment of essential nutrients and energy, thereby impairing the effector function of immune cells (7, 8).
In HCC, the two main subtypes of dendritic cells (DCs) are conventional DCs, which maintain self-tolerance and induce specific immune responses by presenting liver-acquired antigens to T cells (9, 10), and plasmacytoid DCs, which circulate in the bloodstream and is either tolerogenic when immature or immunogenic and cytokine-secreting when mature (11, 12). CD40 activation on dendritic cells enhances cytokine and chemokine production, induces costimulatory molecules, and facilitates antigen cross-presentation, thus improving DC-T cell interactions and promoting anti-tumor effects. As the most abundant type of granulocytes in innate immunity, neutrophils are crucial immune cells active during liver inflammation and injury. Regarding neutrophils, neutrophils are divided into type 1 and type 2 subsets. Type 1 neutrophils exert a pro-inflammatory effect by producing interleukin 12 (IL-12) and C-C Motif Chemokine Ligand 3 (CCL3), while Type 2 neutrophils are immunosuppressive secreting IL-10 and C-C Motif Chemokine Ligand 12 (CCL2) (13). Additionally, myeloid-derived suppressor cells (MDSCs) inhibit T cell proliferation and activation by depleting essential amino acids through elevated arginase activity (14).
Tumor-associated macrophages (TAMs) exhibit two distinct phenotypes: the M1 phenotype, which expresses TGF-α, IL-12, and other factors that exert pro-inflammatory and anti-tumor effects, and the M2 phenotype, which expresses IL-10, TGF-β, and other factors that exert anti-inflammatory and pro-tumor effects (15). CD40 signaling also shifts macrophages from the M2 to the more tumoricidal M1 phenotype (16). In the liver, Kupffer cells, which are resident macrophages, play a key role in the formation and development of HCC by mediating inflammatory responses, fibrosis, and immune cell recruitment, thereby affecting the balance between pro-fibrotic and anti-fibrotic processes. Additionally, liver cell death induces the formation of apoptotic bodies, which are engulfed by Kupffer cells, leading to a pro-fibrotic response that promotes the development of HCC (17).
Resting Treg cells express and release TGF-β on their surface, which then bind to TGF-β receptors on the surface of resting NK cells. This interaction inhibits the proliferation and cytotoxic functions of NK cells (18). Naive B cells contribute to enhancing immune tolerance and facilitating the differentiation of Treg cells (19). Additionally, B cells support their own differentiation into IgA-producing plasma cells by stimulating the proliferation of Treg cells (20). In the case of macrophages, they recruit mature Treg cells into HCC and promote the transformation of naive T cells into Treg cells by secreting cytokines such as CCL22, CCL20, TGF-β, and IL-10 (21). Pro-tumor neutrophils recruit Treg and anti-inflammatory M2 macrophages and help further inhibit the function of cytotoxic T cells (22). In addition, LPS-stimulated Treg has been found to induce IL-10 production in neutrophils in a cellular contact-dependent manner. Additionally, MDSCs stimulate the differentiation and development of Tregs during tumor progression and inhibit NK and DC cell functions through TGF-β to promote tumor immune escape (23).
In HCC, in addition to immune cells, stromal cells are present, forming part of the extracellular matrix. These stromal cells include cancer-associated fibroblasts (CAFs) and hepatic stellate cells (HSCs). CAFs participate in aerobic glycolysis and secrete lactate, which promote the metabolism of cancer cells (24). Increased secretion of metabolites by CAFs along with reorganization of Extracellular Matrix (ECM) proteins, enhances collagen deposition, leading to ECM stiffening (25). This stiffened extracellular matrix not only enhances tumor cell adhesion but also disrupts intercellular contact, preventing contact inhibition between cells, thereby promoting the growth and survival of cancer cells. Additionally, the stiffened extracellular matrix impedes drug uptake into the tumor area, enhancing the survival of tumor cells, reducing cancer cell death and the release of cancer cell antigens, and weakening immune therapy for cancer.
During HCC development, HSCs are activated by fibrosis in liver stromal spaces, transitioning from a quiescent state to an activated myofibroblast phenotype with the ability to release collagen and ECM remodeling factors. In addition, activated HSCs secrete angiogenesis growth factors to form new vasculature in the TME (26, 27). These activities of activated HSCs establish connections with the circulatory system, providing tumors with the nutrients needed for growth and metastasis.
Tregs interact with these stromal cells, influencing the progression of HCC. Tregs promote the activation and transformation of CAFs by secreting inhibitory cytokines IL-10 and TGF-β. This causes CAFs to produce more collagen and extracellular matrix proteins, thereby facilitating the invasion and metastasis of tumor cells (28). Regarding HSCs, studies have shown that activated HSCs significantly upregulate the expression of Treg cells, thereby promoting the growth of HCC in the spleen, bone marrow, and other tissues (29) (Figure 1).
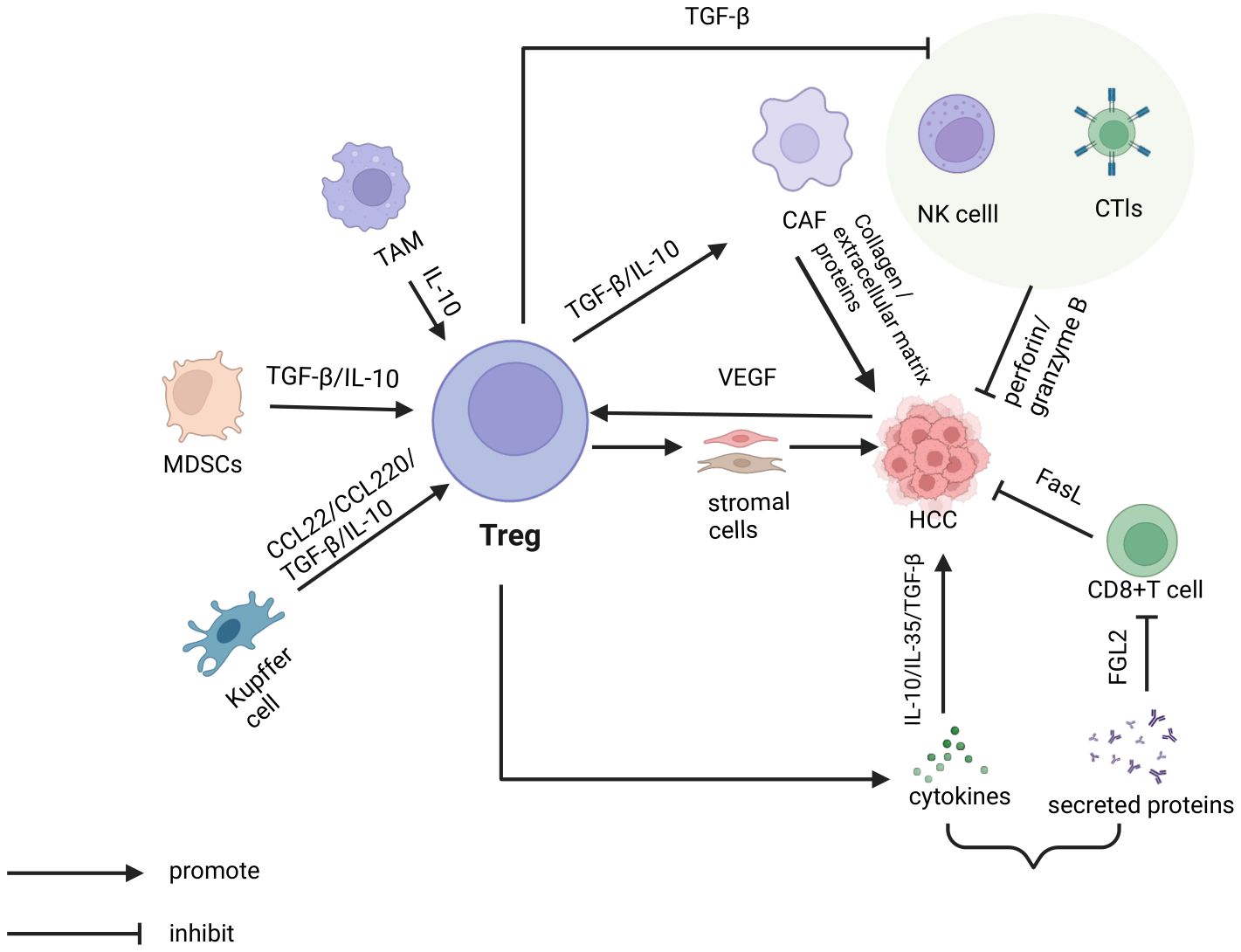
Figure 1 In the tumor microenvironment of hepatocellular carcinoma, various cells indirectly promote the escape of tumor cells by regulating T cells. This includes tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and Kupffer cells, which release cytokines such as TGF-β and IL-10, as well as chemokines like CCL22. Additionally, Tregs directly promote tumor cell escape by secreting immunosuppressive cytokines, including TGF-β, IL-10, and IL-35. On one hand, tregs secrete TGF-β and IL-10 to stimulate cancer-associated fibroblasts (CAFs). These CAFs then produce collagen and extracellular matrix components, which facilitate tumor cell escape. On the other hand, tregs inhibit the cytotoxic activity of natural killer (NK) cells, cytotoxic T lymphocytes (CTLs), and CD8+T cells against tumor cells through the secretion of TGF-β and proteins such as FGL2.
In previous studies, it discussed that Treg cells exert inhibitory effects either through direct cell-to-cell contact or by releasing cytokines that act on other cells. This article focuses primarily on the role of cytokines within the immune microenvironment.
In the immune microenvironment, enzymes secreted by various cells influence the progression of HCC through distinct pathways. In the tumor microenvironment, enzymes like intracellular glucose-6-phosphate dehydrogenase (G6PD) are involved in various metabolic pathways, such as the pentose phosphate pathway (PPP). NADPH primarily functions in defending against reactive oxygen species and in lipid biosynthesis, while ribose, produced as a result, provides a saccharide phosphate backbone essential for nucleotide synthesis (30). The defensive role of NADPH against reactive oxygen species enhances the ability of cancer cells to withstand oxidative stress (31), thereby facilitating their evasion from immune system clearance. Ribose, as a metabolite of glucose, provide the energy needed for the growth and metabolism of tumor cells. In addition, ribose is also an essential substrate for DNA synthesis, which promote the proliferation and growth of tumor cells (32). In normal tissues, the tumor suppressor gene p53 binds to G6PD, preventing the formation and activation of G6PD dimers, and thus inhibiting tumor development. However, in cancer, TAp73, a member of the p53 family, is often overexpressed, leading to the induction of G6PD expression which promotes tumor growth (33, 34).
Tumor cells secrete matrix metalloproteinases (MMPs), enzymes that degrade the extracellular matrix, enabling the tumor cells to penetrate the basement membrane and invade surrounding tissues and blood vessels, thereby promoting liver cancer metastasis (35).
Ecto-nucleotidases CD39 and CD73 are expressed on the surface of regulatory T cells. Under normal conditions, ATP is strictly retained within the cell. However, research has shown that following cellular damage, intracellular ATP is released into the extracellular space through vesicle exocytosis and membrane transporters. CD39 on the surface of Treg cells then metabolizes ATP into ADP and AMP, and CD73 subsequently converts AMP into adenosine. The adenosine produced binds to adenosine receptors, inhibiting the function of effector T cells (36).
In the TME, cancer-associated fibroblasts (CAFs) secrete vascular endothelial growth factor (VEGF) to stimulate the formation of new blood vessels, enabling tumor cells to metastasize through these newly formed blood vessels. Additionally, VEGF inhibits the differentiation of dendritic cells into their mature cells, thus suppressing the antigen-presenting function of mature dendritic cells and the cytotoxic effects of effector T cells, favoring immune evasion by tumor cells (37).
Tumor cells, inflammatory cells, and damaged tissues all produce VEGF (vascular endothelial growth factor). Elevated levels of VEGF stimulate the proliferation and accumulation of Tregs. Tumor-derived VEGF serve as a chemotactic agent, attracting Tregs to the tumor microenvironment (38). Furthermore, VEGF enhance the immunosuppressive function of Tregs by regulating signaling pathways, such as STAT3, to promote Treg differentiation and stability (39).
In the tumor microenvironment, various cells secrete transforming growth factor-beta (TGF-β). For example, TGF-β secreted by cancer-associated fibroblasts (CAFs), induces hepatocellular carcinoma cells to undergo epithelial-mesenchymal transition (EMT), where the cells transition from an epithelial to a mesenchymal phenotype. This EMT process causes liver cancer cells to lose their adhesive ability and polarity, increasing their migration and invasion capabilities, thereby promoting immune evasion in liver cancer (40).
Kupffer cells, a type of macrophage, exhibit both M1 and M2 phenotypes. As liver cancer progresses, Kupffer cells transition from an intermediate phenotype to the M1 phenotype, characterized by an anti-inflammatory profile. When exposed to factors like IL-3 and IL-4, M1 cells secrete TGF-β. With the increasing levels of TGF-β in the TME, Kupffer cells gradually transition from an anti-inflammatory to an immune-suppressive phenotype, promoting immune evasion (17, 41).
Myeloid-derived suppressor cells (MDSCs) release TGF-β to inhibit the activity and function of natural killer (NK) cells, leading to immune evasion in tumors. For instance, TGF-β inhibits the secretion of IFN-λ by NK cells and reduces the expression of activation receptors on their surfaces, thereby impairing the recognition and elimination of target cells. TGF-β also act back on MDSCs, inducing them to secrete inhibitory molecules such as IDO, further suppressing NK cell function (42).
In the immune microenvironment, various cells secrete transforming growth factor-beta (TGF-β). For instance, regulatory T cells secrete TGF-β, which diminish the cytotoxic functions of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs). This process induces NK cells to transform into type 1 innate lymphoid cells within the tumor microenvironment, reducing their ability to suppress tumor growth and metastasis (43).
In HCC, IFN-γ is primarily produced by activated T cells and NK cells, playing a crucial role in immune regulation. Research has shown that in HCC, IFN-γ synergize with IL-1β to enhance the expression of IRF-1, which subsequently leads to an increase in PD-L1 expression (44). Moreover, IFN-γ induce a state in tumor-derived Tregs where they exhibit a reduction in their suppressive activity (45). Moreover, IFN-γ induce a state in tumor-derived Tregs where they exhibit a reduction in their suppressive activity.
Regulatory T cells secrete inhibitory factors for immune suppression, such as IL-10 and IL-35. IL-10 has broad immunosuppressive and anti-inflammatory effects. Its main mechanisms include regulating the activity of inflammatory cells. For example, IL-10 upregulate mature protein-1 induced by B lymphocytes to inhibit CD28 tyrosine phosphorylation and induce exhaustion of CD8+ T cells (46). It also reduces the production of inflammatory mediators, lowers the intensity of inflammatory reactions, inhibits antigen presentation and co-stimulatory signal provision, and promotes the formation of immune tolerance (47). IL-10 has a potent immunosuppressive effect on APC and effector T cells, and convert DCs to tolerant DCs. In addition, local production of IL-10 lead to the exclusion of APC from the tumor mass (48). IL-35 has extensive immunosuppressive effects and is considered a negative regulatory factor. It suppress the immune response through pathways such as inhibiting cell proliferation, reducing the production of inflammatory mediators, promoting the development and function of Treg cells, assisting immune escape. For instance, IL-35 induces T cells to express inhibitory receptors like PD-1, TIM3, LAG3, facilitating T cell exhaustion and contributing to immune escape (49). IL-35 has also been found to inhibit the production of IFN-γ, thus suppressing the development of inflammation through this pathway (50).
One integral protein expressed on the surface of regulating T cells is Leukocyte Function-Associated Antigen-1 (LFA-1). Under normal conditions, LFA-1 on regulatory T cells facilitates interaction with dendritic cells, supporting the initiation and regulation of immune responses, supporting T cells in recognizing and responding to tumor cells. However, regulatory T cells, upon stimulation of antigen receptors, form aggregates with DCs through LFA-1, inhibiting the co-stimulatory process between DCs and effector T cells (51).
VISTA, a B7 family member, crucially maintains T cell quiescence and regulates bone marrow cell populations. In HCC, higher VISTA levels correlate with CD8+ tumor-infiltrating lymphocytes (TILs) (52). VISTA is more expressed in tumor-infiltrating Tregs compared to those in peripheral lymph nodes, indicating its role in suppressing tumor-specific immunity within the tumor microenvironment (TME) (53). VISTA-Ig fusion protein has been shown to inhibit T cell activation by blocking proliferation and cytokine production (54). Moreover, VISTA inhibits iTreg transformation to Th1 and Th17 in inflammatory environments, while promoting iTreg differentiation under TGF-β induction in vitro (55). On the other hand, VISTA-Ig promotes the differentiation of iTregs in response to in vitro TGF-β induction (53).
Regulatory T cells assist in immune suppression by secreting various immune-related proteins. For example, CTLA-4, expressed on Tregs, negatively regulates T cell activation by binding to CD80/CD86 on APCs, thus blocking the stimulatory effects of CD28 binding to these molecules. Both CD28 and CTLA-4 have affinities for CD80/CD86, but they have different effects on co-stimulatory signal transduction. Binding of CD28 to CD80/CD86 provides a second signal that enhances T cell activation, proliferation, and function. In contrast, when CTLA-4 binds to CD80/CD86, it competitively antagonizes the signaling of CD28, inhibiting its signal transduction. Through this mechanism, Tregs suppress T cell activation, contributing to immune escape of cancer cells (56).
Chemokines are a class of small cytokines knowns for their similar structures, functions, and chemotactic properties. They directly act on tumor cells and non-immune cells such as vascular endothelial cells, regulating the proliferation, invasion, and metastasis of tumor cells, thereby promoting the progression of cancer in the tumor microenvironment (57).
CCR8+Treg cells are recruited to tumors by C-C Motif Chemokine Ligand 1 (CCL1), which promotes immune escape. CCL1 also induces STAT3-dependent upregulation of FOXP3, CD39, and IL-10, thus enhancing the immunosuppressive activity of Tregs (58). Another way chemical factors promote tumor development is by mediating the entry of Tregs into the tumor microenvironment. Treg cells express CCR4 and are recruited to the tumor microenvironment by CCL22 induction. In addition to the CCL22-CCR4 pathway, Treg cells express CCR10 and respond to migration cues like CCL28 in hypoxic areas of the tumor microenvironment (59, 60).
Bone marrow, a common site for tumor metastasis, hosts numerous Treg cells with a memory phenotype and functional C-X-C Motif Chemokine Ligand 4 (CXCR4) expression. Treg cells move from bone marrow to peripheral blood through the action of granulocyte colony-stimulating factor (G-CSF), which promotes the degradation of C-X-C Motif Chemokine Ligand 12 (CXCL12) in the bone marrow. The abundance of Treg cells in the bone marrow may provide an immune “shield,” favoring tumor metastasis to this site. Treg cells producing C-X-C Motif Chemokine Ligand 8 (CXCL8) and IL-17 not only suppress T cells but also promote inflammation within the cancer microenvironment.
Substances produced by the metabolism of components in the tumor microenvironment of liver cancer also impact the formation and development of tumor cells. Metabolites in the microenvironment interact with various cells to exert their effects. In hepatocellular carcinoma, the Warburg effect leads tumor cells to utilize glucose via aerobic glycolysis—even under oxygen-rich conditions—to produce ATP, concurrently increasing lactate production and lowering pH in the tumor microenvironment (61). High lactate production in the tumor microenvironment increases FOXP3 concentration. Under normal circumstances, FOXP3 promotes the growth of Tregs and limits the differentiation of Tregs into other immune cells, maintaining the stability and function of Treg cells (62). The metabolic processes of tumor cells transform the tumor microenvironment into a nutrient-restricted, hypoxic, and lactate-rich environment. In tumors, Tregs convert pyruvate into acetyl-CoA via mitochondria, utilizing the tricarboxylic acid cycle to sustain survival (63). Furthermore, Tregs have a unique fatty acid metabolism mechanism where lipid oxidation in Tregs reduces their glucose requirements and mitigates the cytotoxic effects induced by fatty acids. Even under nutrient-restricted conditions, Tregs utilize fatty acids for proliferation and to perform their immunosuppressive functions (64).
Exosomes are small vesicles containing a variety of biomolecules that mediate intercellular communication, thus regulating the microenvironment and immune system interactions. Research has shown that exosomes not only transmit downstream signals to target cells but also transfer genetic material to downstream cells, thus facilitating intercellular communication (65).
Apart from their communicative role, exosomes participate in various processes in hepatocellular carcinoma, including tumor survival, growth, angiogenesis, and invasion. Exosomes transfer biological molecules such as miRNAs, lncRNAs, and circRNAs, which are primarily involved in regulating interactions between HCC cells and endothelial cells, thereby stimulating angiogenesis to supply oxygen and nutrients to the tumor. Exosomes from cancer-associated fibroblasts (CAFs) modulate liver cancer cell phenotypes by transferring miRNAs, proteins, and miRNA-mediated RNA oxidases, thus activating signaling pathways like Wnt/β-catenin and promoting epithelial-mesenchymal transition (EMT), which enhances migration and invasion capabilities (66). Tumor-derived exosomes promote M2 polarization and increase PD1 expression on T cells, while concurrently inhibiting cytokines that activate M1 macrophages and their phagocytic activity (67, 68). In addition to tumor sources, exosomes secreted by macrophages express specific CD11b/CD18 proteins to promote MMP-9 expression, thereby promoting HCC migration (69).Furthermore, exosomes alter the stromal environment surrounding the tumor by regulating the synthesis and degradation of matrix components and matrix adhesion factors, thereby creating a suitable tumor microenvironment and enhancing the metastatic ability of hepatocellular carcinoma (70). Certain molecules within exosomes regulate the function and immune response of immune cells. They suppress the activity of immune cells, inhibit the cytotoxic effects of natural killer cells and T cells, thereby helping liver cancer cells evade immune attacks and metastasis (71). In the tumor microenvironment, exosomes package circular RNAs that suppress immunity in HCC, for instance, by increasing CD39 expression in macrophages, which contributes to resistance against anti-PD-1 therapy (72).
Common treatments for hepatocellular carcinoma include drug therapy, radiation therapy, and surgical interventions, such as donafenib, apatinib, durvazumab, and sorafenib. However, these traditional therapies often result in low cure rates and poor prognosis, whether used alone or in combination (73). This underscores the urgent need for innovative treatments. Consequently, to improve treatment outcomes in hepatocellular carcinoma, researchers have developed a novel immunological approach—CAR-T therapy (Figure 2). CAR-T therapy is chimeric antigen receptor T-cell immunotherapy, which is also known as Chimeric Antigen Receptor T-Cell Immunotherapy. CAR-T therapy, a precise form of targeted tumor treatment, has recently made significant advancements in liver cancer treatment. A CAR-T cell is a special type of cell in T cells. CAR consists of three parts: an extracellular antigen recognition domain with or without hinge/spacer domains, a transmembrane domain, and an intracellular signal domain. The hinge/spacer ensures the accessibility of CAR-T cell epitopes, making CAR expression very stable. The transmembrane domain affect the expression of CAR on the cell surface and signal transduction in T cells. The intracellular signaling domain comprises two parts: the costimulatory and signal transduction domains. The costimulatory domain typically includes either the CD28 receptor family (CD28,ICOS) or the tumor necrosis factor receptor family (4–1BB,OX40,CD27). The role of the costimulatory domain is to achieve synergistic stimulation of the dual activation of molecular and intracellular signals, and improve T cell action and anti-tumor ability. However, CD28 and 4–1BB have different effects, and CD28 cause CAR-T cells to be metabolized by glycolysis, which promote the differentiation of CAR-T cells into effector T cells. 4–1BB promote mitochondrial production, enhance respiration and fatty acid oxidation, and CAR-T cells will differentiate into central memory T cells after exposure to antigen stimulation (74–76). In addition to this, CD28 exhibits faster and stronger signaling activity, while 4–1BB is comparatively slower and gentler, and studies have found that 4–1BBnbetter prolong T cell lifespan and maintain its anti-cancer effects (77). Based on the number of costimulatory molecules, CAR proteins are classified into three generations (78). Each of the three generations of CAR proteins plays a crucial role in treating hepatocellular carcinoma. A key advantage of CAR-T therapy in treating hepatocellular carcinoma is that its efficacy does not rely on antigen presentation by major histocompatibility complex (MHC) molecules, thus circumventing MHC-related restrictions and addressing tumor immune escape issues due to MHC downregulation (79). The treatment of hepatocellular carcinoma through CAR-T start with the types of CAR-T, treatment targets for hepatocellular carcinoma, and substances secreted by CAR-T.
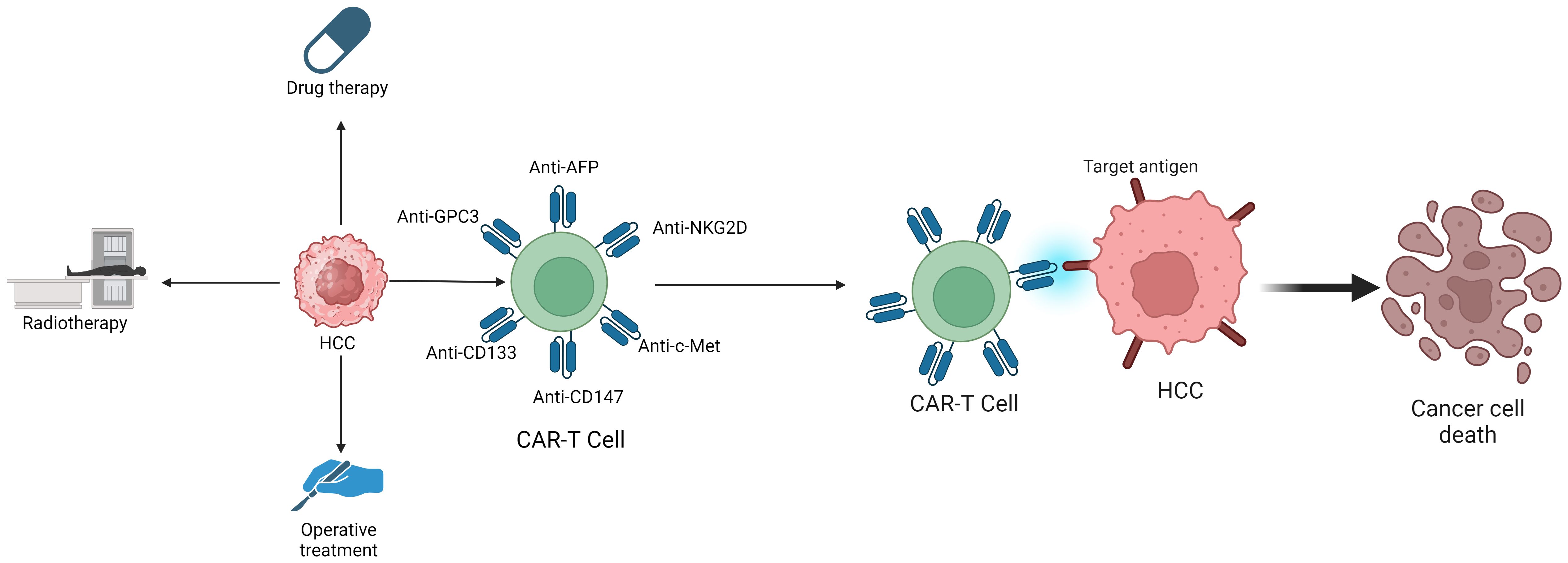
Figure 2 The treatment methods for HCC include Drug therapy, Radiotherapy, Operative treatment and CAR-T Cell therapy.
There are many types of CAR-T cells, among which GPC3 CAR-T cells, CD133 CAR-T cells, c-Met CAR-T cells, NKG2D CAR-T cells, and other CAR-T cells effectively treat hepatocellular carcinoma (80).
Glypican-3 (GPC3) is a carcinoembryonic heparan sulfate proteoglycan that attaches to cell membranes via glycophosphatidylinositol anchors (81). It is highly expressed in hepatocellular carcinoma but absent in normal tissues, potentially playing a critical role in regulating cell division and growth. Numerous studies have demonstrated that higher GPC3 expression in hepatocellular carcinoma correlates with poorer prognosis. This suggests GPC3’s potential as a prognostic marker for hepatocellular carcinoma. Notably, GPC3 is highly expressed in hepatocellular carcinoma and squamous non-small cell lung cancer but is expressed at low levels in other cancers. This indicates that GPC3 serve as an important target for the treatment of hepatocellular carcinoma. Therefore, GPC3 CAR-T cells have emerged (82). GPC3 CAR-T cells secrete IL15 and IL21, enhancing the proliferation of poorly differentiated GPC3-CAR T cells, maintaining TCF-1 expression, and increasing both the persistence and anti-tumor activity of these cells. This greatly enhances the treatment of hepatocellular carcinoma by GPC3 CAR-T cells (83). In addition, Lin28 is also overexpressed in hepatocellular carcinoma, and regulating Lin28B enhance the anti-tumor activity of GPC3 CAR-T cells, enabling GPC3 CAR-T cells to better treat hepatocellular carcinoma (84). Researchers have found that generating GPC3-specific CAR-T cells through simultaneous electroporation of plasmid DNA encoding the piggyBac (PB) transposon and overactive piggyBac transposase, rather than using common viral vectors, results in cells with enhanced proliferation and cytokine secretion (85). This discovery has propelled further development of GPC3 CAR-T cell therapy for hepatocellular carcinoma. However, GPC3 CAR-T cell therapy for hepatocellular carcinoma remains in its early stages, with its methods and effects yet to meet clinical needs. Therefore, a large amount of research is still needed to make this treatment method better treat hepatocellular carcinoma.
CD133, which is highly expressed in hepatocellular carcinoma, presents an attractive therapeutic target (86). Studies indicate that CD133 CAR-T cells exhibit effective anti-tumor activity and manageable safety profiles in treating advanced hepatocellular carcinoma. In fact, CD133 is also highly expressed in endothelial progenitor cells of tumors. CD133 promotes angiogenesis in hepatocellular carcinoma; thus, combining CD133 CAR-T cells with anti-angiogenic drugs effectively inhibit new blood vessel formation in tumors. Unlike GPC3 CAR-T cells, CD133 CAR-T cells not only block the nutritional supply to tumor cells but also are used in combination with other drugs to treat hepatocellular carcinoma (87). It should be noted that CD133 CAR-T treatment for hepatocellular carcinoma in patients with biliary obstruction requires more caution (86).
c-Met, a hepatocyte growth factor receptor (also known as HGFR) and a cell surface protein tyrosine kinase, is overexpressed in hepatocellular carcinoma. It plays a crucial role in tumor cell proliferation, invasion, and apoptosis. Overexpression of c-Met is an indicator of increased tumor invasiveness and poor prognosis (88), and because of antigen escape effect and tumor microenvironment and other factors, ordinary CAR-T cells have poor therapeutic effect on hepatocellular carcinoma (89). Therefore, c-Met is an important target for immunotherapy of hepatocellular carcinoma. c-Met CAR-T cells bind to c-Met targets in hepatocellular carcinoma and specifically kill tumor cells in an antigen-dependent manner (88). However, the immunosuppressive tumor microenvironment of hepatocellular carcinoma induce the expression of PD-1 on CAR-T cells, and make PD-1 combine with PD-L1 on tumor cells to inhibit the role of CAR-T cells, so that tumor cells avoid immune surveillance (90, 91). Consequently, constructing CAR-T cells that target both c-Met and PD-1 could enhance therapeutic effects in hepatocellular carcinoma. c-Met/PD-1 CAR-T cells block the interaction between PD-1 and PD-L1, displaying lower levels of inhibitory receptors, reduced differentiation, enhanced anti-tumor activity, and extended survival compared to conventional CAR-T cells (92). Therefore, c-Met/PD-1 CAR-T cells have broad application prospects for better treatment of hepatocellular carcinoma. In addition, various c-Met CAR-T cells such as c-Met/PD-L1 CAR-T cells are being studied to better treat hepatocellular carcinoma.
NKG2D is a type II transmembrane C-type lectin-like protein receptor expressed on natural killer cells, CD8+ T cells, and some autoreactive CD4+ T cells (93, 94). NKG2DL, the ligand for NKG2D, is not expressed in normal cells but is highly expressed in tumor cells, with expression further inducible by radiotherapy and chemotherapy (95). Therefore, NKG2DL is an important target for the treatment of hepatocellular carcinoma (96). Combining NKG2D CAR-T cells with NKG2DL targets in hepatocellular carcinoma eradicate tumor neovascularization, ameliorate the tumor microenvironment, and augment immunotherapy efficacy (97). Moreover, NKG2D CAR-T cells target multiple ligands expressed in hepatocellular carcinoma to cope with tumor immune escape caused by tumor heterogeneity. Therefore, NKG2D CAR-T cells have the possibility of radical treatment of hepatocellular carcinoma (98). In addition, NKG2D CAR-T cells have good effects and broad application prospects in the treatment of multiple myeloma, glioblastoma, osteosarcoma and other tumors (96).
CD147, a glycoprotein, acts as a regulator of matrix metalloproteinases (MMPs). Numerous studies have shown that CD147 regulate cell proliferation, drug resistance, and cell matrix adhesion characteristics through cell matrix and cell interactions. CD147 is overexpressed in cancer cells and contributes to angiogenesis by regulating vascular endothelial growth factor production in tumor and stromal cells, thus promoting tumor progression. Therefore, CD147 has become a potential therapeutic target for treating hepatocellular carcinoma (99). However, CD147 is not only expressed in cancer cells, but also in small amounts in other tissues, which may result in CD147 CAR-T cells not accurately reaching the CD147 target of hepatocellular carcinoma and may be toxic to normal cells. To minimize the toxicity of CD147 CAR-T cells, researchers have employed the Tet-On 3G system. Tet-On 3G is the third-generation tetracycline induced gene expression system, which use Doxycycline to turn on or off gene expression reversibly. This system allows for the controlled induction of Tet-CD147 CAR-T cells at specific times and locations, optimizing the treatment of hepatocellular carcinoma. Furthermore, in the presence of Doxycycline, Tet-CD147 CAR-T cell activity is significantly enhanced in hepatocellular carcinoma, potentially improving treatment outcomes (100).
Alpha fetoprotein (AFP), a glycoprotein of the albumin family, is primarily synthesized by fetal liver cells and the yolk sac. The content of AFP in normal adult serum is extremely low. Research indicates that AFP promotes cell proliferation and inhibits apoptosis; thus, elevated levels are commonly associated with tumors. Alpha fetoprotein (AFP) is the most commonly used biomarker in hepatocellular carcinoma, often used for detection, diagnosis, and prognosis (101). Therefore, AFP is a good target for the treatment of hepatocellular carcinoma. Traditional CAR-T cell therapy for hepatocellular carcinoma cause serious side effects, including cytokine release syndrome (CRS), and the AFP peptide-MHC complex is less expressed on the surface of tumor cells. AFP CAR-T cells effectively kill tumor cells without inducing cytokine release syndrome. Therefore, AFP CAR-T cells are safer to use. Moreover, studies have shown that local injection of AFP CAR-T cells into tumors produce deeper, faster, and more lasting anti-tumor responses. Thus, the appropriate route of administration is crucial for optimizing the efficacy of AFP CAR-T cells (102).
However, CAR-T cell therapy for hepatocellular carcinoma still has significant limitations. When using CAR-T cell therapy, serious adverse reactions may occur, leading to poor treatment effectiveness. In addition, CAR-T cell therapy for hepatocellular carcinoma is still in its early stages, and research in this field is still insufficient. Therefore, it is necessary to be very cautious when using CAR-T cells to treat hepatocellular carcinoma (78). In order to reduce adverse reactions and improve efficacy, researchers have found that using Treg help improve CAR-T treatment for hepatocellular carcinoma.
During the CAR-T treatment of hepatocellular carcinoma, rapid immune activation reactions are induced, leading to cytokine release syndrome (CRS). CRS causes severe therapeutic toxicity in the body, causing fever, hypoxia, and hypotension, and in severe cases, it is life-threatening (103). Tregs play a good role in eliminating excessive inflammation, maintaining self-immune tolerance, and immune homeostasis (104). The therapeutic effect of Treg cells in metabolic diseases has been proven, and they maintain metabolic balance and prevent inflammation well (105). So, research has found that Treg cells derived from allogeneic umbilical cord blood (UCB) effectively eliminate the inflammatory response caused by CAR-T cells in the treatment of hepatocellular carcinoma. UCB Treg cells may preferentially accumulate at inflammatory sites and interact with antigen-presenting cells, rapidly reducing inflammation. However, it should be noted that UCB Treg cell adjuvant therapy should be administered 3 days after CAR-T cell infusion to minimize the interference of CAR-T cells in the treatment of hepatocellular carcinoma (106).
Although the inflammatory response caused by CAR-T cells in the treatment of hepatocellular carcinoma has been resolved, excessive immune suppression of Treg cells leading to the promotion of tumor proliferation and metastasis remains a major issue. Treg cells exhibit two primary immunosuppressive mechanisms: one involves the release of transforming growth factor-beta (TGF-β) to perform immune suppression. The second mechanism is achieved when the ligand CCL1 binds to its receptor, C-C chemokine receptor 8 (CCR8) (107). CCR8 is highly expressed in tumors and tumor infiltrating Treg cells. CCR8 is the only receptor of CCL1, so CCL1 activation of the CCR8 receptor on tumor cells promotes their proliferation and metastasis. CCL1 also recruits a large number of Treg cells to the tumor site for immune suppression, promoting tumor growth (108, 109). Utilizing the CCL1-CCR8 axis, which recruits Treg cells to tumor sites, researchers have developed CCR8-CAR-T cells. These CCR8-CAR-T cells are recruited to tumor sites via the CCL1-CCR8 axis, potentially enhancing the efficacy of CAR-T cell therapy. Although CAR-T cells reach tumor cells through this axis, the immunosuppressive effect produced by a large number of Treg cells at the site of tumor infiltration still reduces the therapeutic effect of CAR-T. For this purpose, researchers constructed new CAR-T cells, namely CCR8-DNR-CAR-T cells, to resist the immune suppression produced by Treg. DNR, a truncated form of TGF-β receptor 2, lacks the intracellular signaling components necessary for TGF-β signal transduction. It competes with TGF-β receptor 2 to bind TGF-β, allowing DNR-transduced T cells to clear TGF-β and reduce immunosuppressive reactions (110, 111). Studies have shown that CCR8-DNR-CAR-T cells are recruited in large quantities to tumor sites, but not to other normal organ tissues. CCR8-DNR-CAR-T cells have a long survival and good proliferation ability at the tumor site, and the therapeutic effect on the tumor has also been significantly improved (109) (Figure 3).
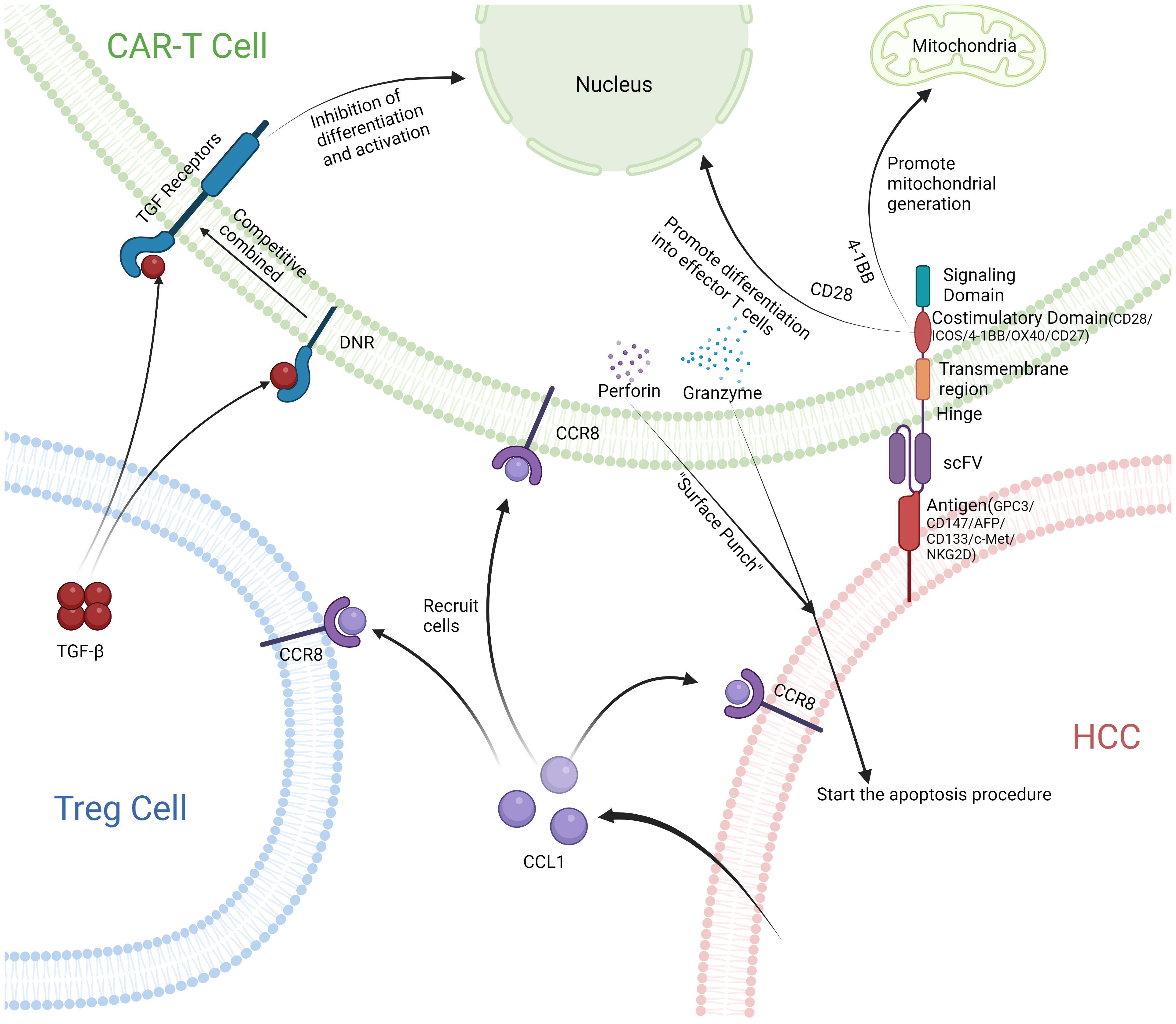
Figure 3 After releasing CCL1 from HCC, Treg cells and CAR-T cells with CCR8 receptor are recruited together. CAR-T cells combine with HCC and release perforin and granzyme to initiate apoptosis of HCC. DNR receptors on the surface of CAR-T cells compete well for binding to TGF-β, Prevent immunosuppression.
Additionally, modifying CAR-T cells to evade Treg cell inhibition is another effective strategy to enhance tumor treatment efficacy. T cells secrete interleukin-2 (IL-2) when stimulated by antigens, and IL-2 promotes the production of Tregs. It has been found that ordinary CAR-T cells promote the production of Treg cells with the help of the IL-2 axis through the co-stimulatory signal CD28 (112). In order to eliminate IL-2 secretion, the researchers replaced two amino acids in the CAR transgene to prevent lymphocyte-specific tyrosine kinase (Lck) from attaching to the CD28 cytoplasmic tail (113), which eliminated IL-2 secretion but also impaired the role of CAR-T cells in vivo. However, 4–1BB is a costimulatory glycoprotein on activated immune cells, including lytic and helper T lymphocytes, with its ligand 4–1BBL primarily on antigen-presenting cells like DCs, macrophages, and B cells (114). This interaction enhances T cell proliferation, differentiation, and effector function (115, 116). Cross-linking of 4–1BB in T cells promotes proliferation, memory cell formation, and increases viability and cytokine production, such as IFN-γ and IL-2 (117). In addition, 4–1BB converts T cells with reprogramming tolerance potential into effector T cells with antitumor activity (118), so the introduction of a 4–1BB signaling domain within CAR-T cells made up for this deficiency. 4–1BB CAR-T cells that inhibit Lck activation are not only active at tumor sites but also resist Treg-mediated immunosuppression. Studies have shown that 4–1BB is targeted by Treg cells in various cancers and plays a crucial role in regulating effector T cell responses, demonstrating significant therapeutic effects within the immune microenvironment (119, 120).
Although improvements have been made in the effects and adverse reactions of CAR-T cells, significant challenges remain in developing standardized, safe, and reliable CAR-T cells (121). To address this, the construction process has been optimized to enhance the efficacy of CAR-T cells against tumors. For instance, incorporating an apoptosis switch in CAR-T cells that activates caspase9 (iCasp9) is beneficial. Activation of iCasp9 by a synthetic dimerizer induces apoptosis in CAR-T cells, allowing for controlled regulation of their activity based on specific situations. This improves the safety of CAR-T cell therapy. Additionally, gene editing is employed to develop multifunctional CAR-T cells. In hepatocellular carcinoma, PD-L1 is upregulated and expressed by various activated immune cells including macrophages, B cells, dendritic cells, and T cells (122). PD-1 is expressed on immature thymocytes, activated CD4 and CD8+ T cells, B cells, dendritic cells, and natural killer (NK) cells. PD-L1, when expressed on hepatocellular carcinoma cells, bind to PD-1 on T cells, inducing tolerance by inhibiting their ability to mount an effective immune response against specific cancer antigens (123). Regulatory T cells express PD-L1, which bind to inhibitory receptors on NK cells, thereby suppressing their cytotoxic function and preventing the direct killing of tumor cells (124). The interaction of PD-L1 with PD-1 on immune cells transmits an inhibitory signal that decreases the cytotoxic activity of effector T cells, aiding the survival of tumor cells (125). Activated PD-1/PD-L1 complexes modulate T cell receptor and CD28 co-stimulatory signaling pathways by recruiting SHP1/2, which dephosphorylates these receptors, leading to reduced T cell activation and increased apoptosis, promoting immune escape (126). SHP1/2 remove phosphate groups, inhibit T cell activation, increase cytokine production, promote expression of apoptotic molecules, ultimately leading to T cell apoptosis and non-responsiveness, driving tumor immune escape and progression (127). Both PD-1 and CD276 are members of the B7 family, making their effects on the tumor microenvironment similar. The binding of CD276/PD-1 and the corresponding receptor exert a synergistic effect, thereby jointly inhibiting the proliferation of T cells and the secretion of related cytokines (128, 129). Therefore, by utilizing gene editing technology to disrupt genes like PD-1 and CD276 in CAR-T cells, researchers inhibit the tumor microenvironment and enhance the therapeutic efficacy of CAR-T cells (130).
For Treg cells, researchers have found that the tumor microenvironment significantly induces high expression of histone demethylase JMJD1C in tumor Tregs. Inhibiting JMJD1C expression does not affect the development and function of peripheral Tregs but reduces tumor-associated Tregs, consequently slowing tumor growth. This effect is primarily due to JMJD1C’s role in promoting PD-1 expression and inhibiting AKT signaling and IFN-g production in tumor-associated Tregs. The use of JMJD1C inhibitors significantly inhibits tumor growth and has good application prospects (131).
Tumor-associated macrophages (TAMs) inhibit the anti-tumor immune response within the tumor microenvironment, thereby promoting tumor growth through various mechanisms and diminishing the efficacy of CAR-T therapy (132). First, TAMs express the inhibitory molecule PD-L1, which binds to PD-1 on CAR-T cells, reducing their cytotoxicity and efficacy. Secondly, TAMs secrete several immunosuppressive cytokines such as IL-10 and TGF-β. IL-10 weakens the response of CD8+ T cells (133), while TGF-β excludes T cells, including CAR-T cells, from the tumor, significantly impairing their function. Thirdly, TAMs inhibit the recruitment of T cells to tumor sites by producing peroxynitrites and nitrifying CCL2 and CCL5, thereby preventing chemotaxis of CAR-T and other T cells to the tumor, reducing their effectiveness (134). Finally, TAMs produce VEGF and other growth factors that support blood vessel and tumor survival.
Cancer-associated fibroblasts (CAFs) inhibit the infiltration and function of T cells, including CAR-T cells, within the immune microenvironment, thereby reducing the efficacy of CAR-T therapy. As a significant component of the tumor microenvironment, CAFs promote the production of TAMs, which in turn inhibit CAR-T cell function (135). Additionally, CAFs secrete immunosuppressive cytokines like IL-10 and TGF-β, further suppressing T cell activity. CAFs also enhance the recruitment and differentiation of Tregs by producing chemokines (136), indirectly inhibiting CAR-T cell function. Moreover, CAFs interfere with T cell-dependent immune responses by regulating myeloid-derived suppressor cells (MDSCs). By secreting CXCL12/SDF1, CAFs recruit monocytes to the tumor site, which are then induced to differentiate into MDSCs through IL-6-mediated STAT3 activation, thus altering CAR-T cell proliferation and function and diminishing their therapeutic effect (137).
The unique metabolic profile of tumor cells creates a nutrient-restricted, hypoxic, and lactate-rich environment in the tumor microenvironment. Hypoxia is closely associated with cancer cell resistance to radiotherapy and chemotherapy, as well as impaired immune cell function, thus impacting the efficacy of CAR-T therapy. Researchers observed that hypoxia inhibits CAR-T cell proliferation and differentiation and reduces granzyme B and cytokine production without affecting their frequency and cytotoxicity (138). To enable CAR-T cells to function in hypoxic conditions, researchers integrated hypoxia-induced transcription factor-1α (HIF-1α) hypoxia response element (HRE) into CAR-T cells, creating a hypoxia-induced transcription amplification system that significantly enhances anti-tumor efficacy under hypoxia (139).
Tumor proliferation also results in increased glucose consumption and lactate production, causing metabolic acidosis and a low pH state in the tumor microenvironment. Acidosis inhibits glycolysis and oxidative phosphorylation, affecting CAR-T cell metabolism. Low pH induces V-domain Ig suppressor of T cell activation (VISTA), which inhibits T cell activation, proliferation, and cytokine production, thus affecting CAR-T therapy (140). Researchers found that pretreating T cells with lactate in vitro enhance CAR-T therapy by improving their stemness and anti-tumor immunity (141).
PD-1/PD-L1 interactions assist tumor cells in immune evasion and impact CAR-T therapy by reducing T cell glycolysis, impairing T cell effector functions, and limiting glucose availability for T cells (142). Initially, researchers used monoclonal antibodies against PD-1 to block the PD-1/PD-L1 axis, but this approach had serious adverse reactions and increased the risk of immune tolerance breakdown. To enhance CAR-T therapy for solid tumors like HCC, researchers combined CAR-T cells with PD-1 knockout (KO) therapy to improve anti-tumor activity and safety (143).
Exosomes are extracellular vesicles that carry bioactive molecules and influence recipient cells’ pathophysiological processes. Tumor cells release small extracellular vesicles (sEVs) after CAR-T therapy, promoting PD-L1 expression and inhibiting CAR-T cell efficacy. Exosome inhibitors, such as GW4869 and Nexinhib20, have been shown to enhance CAR-T therapy by improving CD8+ T cell infiltration and activation (144).
In HCC, excessive collagen and hyaluronic acid in the ECM lead to matrix hardening, poor diffusion, decreased oxygen content, and delayed material exchange in the tumor microenvironment(TME) (145).ECM stiffness suppresses T lymphocyte-mediated anti-tumor responses and reduces CAR-T cell infiltration (146). Researchers have incorporated strategies to inhibit hyaluronan synthesis and enhance degradation into CAR-T therapy to decompose ECM and promote T cell penetration (147). Additionally, combining CAR-T therapy with tumor vaccines targeting ECM components softens the ECM and improve T cell infiltration (148).
Although CAR-T therapy is effective in treating liver cancer cells, there are still many deficiencies and challenges. Firstly, various substances in the immune microenvironment weaken the efficacy of CAR-T in hepatocellular carcinoma. Among these, several obstacles affecting the function of CAR-T in the tumor microenvironment (TME) include hypoxia, high lactate levels, activation of the PD-1/PD-L1 axis, and accumulation of the extracellular matrix (ECM) (138, 140, 142, 145).Secondly, CAR-T therapy also faces limitations such as long treatment cycles, high costs, and the need for standardization, safety, and reliability (130). Additionally, CAR-T therapy has certain hepatotoxicity, which may damage the normal function and proliferation of hepatocytes. Currently, CAR-T cells are still insufficiently infiltrated in the environment of hepatocellular carcinoma, so CAR-T cell therapy still needs improvement.
However, with technological advancements, these issues are gradually being addressed. For example, anti-hypoxia CAR-T cells have been developed by introducing hypoxia-inducible factor elements (139), and immune checkpoint molecules such as PD-1/PD-L1 have been knocked out using CRISPR/Cas9 gene editing technology (143). Combining CAR-T cell therapy with anti-PD-1/PD-L1 monoclonal antibodies to enhance the anti-tumor response is a promising research direction. Additionally, combined therapy is used to integrate anticancer drugs with CAR-T therapy to improve the efficacy of CAR-T cells by enhancing the tumor microenvironment. Finally, with future research directions and the use of technological advancements, the field of CAR-T treatment for liver cancer progresses towards more effective and lasting treatments, ultimately improving patient prognosis.
This review has highlighted the significant role of various components of the tumor microenvironment in aiding immune escape in hepatocellular carcinoma, particularly emphasizing the importance of regulating T cells in the growth and spread of cancer cells. To counteract the immune escape mechanisms of hepatocellular carcinoma and enhance T cell therapy efficacy, researchers have explored the potential of CAR-T cell therapy. In conclusion, a thorough understanding of how T cells regulate immune escape in hepatocellular carcinoma offers valuable insights for theoretical and clinical advancements in developing more effective immunotherapies for this cancer. We hope that our research will lead to more treatment options and improve the quality of life for patients with hepatocellular carcinoma.
GD: Writing – original draft. CD: Writing – original draft. PS: Writing – review & editing. SW: Writing – review & editing, Funding acquisition. JL: Writing – review & editing. LM: Writing – review & editing, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the National Natural Science Foundation of China (grant nos.82372806), the Natural Science Foundation of Shandong Province, China (ZR2021MC039) and Qingdao Key Clinical Specialty Elite Discipline.
All the figures are created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1431211/full#supplementary-material
1. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. (2015) 523:221–5. doi: 10.1038/nature14452
2. Huang M, Huang X, Huang N. Exosomal circGSE1 promotes immune escape of hepatocellular carcinoma by inducing the expansion of regulatory T cells. Cancer Sci. (2022) 113:1968–83. doi: 10.1111/cas.15365
3. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–9. doi: 10.1111/cas.14069
4. Huang J, Wu Q, Geller DA, Yan Y. Macrophage metabolism, phenotype, function, and therapy in hepatocellular carcinoma (HCC). J Trans Med. (2023) 21:815. doi: 10.1186/s12967-023-04716-0
5. Bian J, Lin J, Long J, Yang X, Yang X, Lu X, et al. T lymphocytes in hepatocellular carcinoma immune microenvironment: insights into human immunology and immunotherapy. Am J Cancer Res. (2020) 10:4585–606.
6. Murugaiyan G, Martin S, Saha B. Levels of CD40 expression on dendritic cells dictate tumour growth or regression. Clin Exp Immunol. (2007) 149:194–202. doi: 10.1111/j.1365-2249.2007.03407.x
7. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab. (2020) 2:1001–12. doi: 10.1038/s42255-020-00280-9
8. Roe K. NK-cell exhaustion, B-cell exhaustion and T-cell exhaustion-the differences and similarities. Immunology. (2022) 166:155–68. doi: 10.1111/imm.13464
9. Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. (2007) 8:1217–26. doi: 10.1038/ni1522
10. Tan JK, O'Neill HC. Maturation requirements for dendritic cells in T cell stimulation leading to tolerance versus immunity. J Leukocyte Biol. (2005) 78:319–24. doi: 10.1189/jlb.1104664
11. Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells–virus experts of innate immunity. Semin Immunol. (2005) 17:253–61. doi: 10.1016/j.smim.2005.05.008
12. Penna G, Vulcano M, Roncari A, Facchetti F, Sozzani S, Adorini L. Cutting edge: differential chemokine production by myeloid and plasmacytoid dendritic cells. J Immunol (Baltimore Md 1950). (2002) 169:6673–6. doi: 10.4049/jimmunol.169.12.6673
13. Yang F, Feng C, Zhang X, Lu J, Zhao Y. The diverse biological functions of neutrophils, beyond the defense against infections. Inflammation. (2017) 40:311–23. doi: 10.1007/s10753-016-0458-4
14. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
15. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatol (Baltimore Md). (2020) 71:1247–61. doi: 10.1002/hep.30889
16. Richards DM, Sefrin JP, Gieffers C, Hill O, Merz C. Concepts for agonistic targeting of CD40 in immuno-oncology. Hum Vaccines Immunotherapeut. (2020) 16:377–87. doi: 10.1080/21645515.2019.1653744
17. Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. (2020) 9. doi: 10.3390/cells9040875
18. Dasgupta S, Bhattacharya-Chatterjee M, O'Malley BW Jr., Chatterjee SK. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol (Baltimore Md 1950). (2005) 175:5541–50. doi: 10.4049/jimmunol.175.8.5541
19. Chien CH, Chiang BL. Regulatory T cells induced by B cells: a novel subpopulation of regulatory T cells. J Biomed Sci. (2017) 24:86. doi: 10.1186/s12929-017-0391-3
20. Wang L, Ray A, Jiang X, Wang JY, Basu S, Liu X, et al. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol. (2015) 8:1297–312. doi: 10.1038/mi.2015.20
21. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
22. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. (2019) 133:2159–67. doi: 10.1182/blood-2018-11-844548
23. Sajid M, Liu L, Sun C. The dynamic role of NK cells in liver cancers: role in HCC and HBV associated HCC and its therapeutic implications. Front Immunol. (2022) 13:887186. doi: 10.3389/fimmu.2022.887186
24. Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab. (2021) 3:21–32. doi: 10.1038/s42255-020-00317-z
25. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
26. Coulouarn C, Corlu A, Glaise D, Guénon I, Thorgeirsson SS, Clément B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. (2012) 72:2533–42. doi: 10.1158/0008-5472.CAN-11-3317
27. Heindryckx F, Gerwins P. Targeting the tumor stroma in hepatocellular carcinoma. World J Hepatol. (2015) 7:165–76. doi: 10.4254/wjh.v7.i2.165
28. Ruf B, Heinrich B, Greten TF. Immunobiology and immunotherapy of HCC: spotlight on innate and innate-like immune cells. Cell Mol Immunol. (2021) 18:112–27. doi: 10.1038/s41423-020-00572-w
29. Zhao W, Zhang L, Xu Y, Zhang Z, Ren G, Tang K, et al. Hepatic stellate cells promote tumor progression by enhancement of immunosuppressive cells in an orthotopic liver tumor mouse model. Lab Investigat J Tech Methods Pathol. (2014) 94:182–91. doi: 10.1038/labinvest.2013.139
30. Chen J, Wu H, Zhang W, Mu W. Ribose-5-phosphate isomerases: characteristics, structural features, and applications. Appl Microbiol Biotechnol. (2020) 104:6429–41. doi: 10.1007/s00253-020-10735-4
31. Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxidants Redox Signaling. (2012) 16:476–95. doi: 10.1089/ars.2011.4289
32. Li R, Wang W, Yang Y, Gu C. Exploring the role of glucose−6−phosphate dehydrogenase in cancer (Review). Oncol Rep. (2020) 44:2325–36. doi: 10.3892/or
33. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. (2013) 15:991–1000. doi: 10.1038/ncb2789
34. Jiang P, Du W, Yang X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle (Georgetown Tex). (2013) 12:3720–6. doi: 10.4161/cc.27267
35. Santi A, Kugeratski FG, Zanivan S. Cancer associated fibroblasts: the architects of stroma remodeling. Proteomics. (2018) 18:e1700167. doi: 10.1002/pmic.201700167
36. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
37. Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. (2017) 20:185–204. doi: 10.1007/s10456-017-9552-y
38. Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. (2012) 209:2001–16. doi: 10.1084/jem.20111497
39. Zhu AX, Abbas AR, de Galarreta MR, Guan Y, Lu S, Koeppen H, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. (2022) 28:1599–611. doi: 10.1038/s41591-022-01868-2
40. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol CB. (2020) 30:R921–r5. doi: 10.1016/j.cub.2020.06.081
41. Ramachandran P, Iredale JP, Fallowfield JA. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin liver Dis. (2015) 35:119–31. doi: 10.1055/s-00000069
42. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (Baltimore Md 1950). (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
43. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
44. Numata Y, Akutsu N, Ishigami K, Koide H, Wagatsuma K, Motoya M, et al. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem Biophys Rep. (2022) 30:101270. doi: 10.1016/j.bbrep.2022.101270
45. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ Drives T(reg) fragility to promote anti-tumor immunity. Cell. (2017) 169:1130–41.e11. doi: 10.1016/j.cell.2017.05.005
46. Scott EN, Gocher AM, Workman CJ, Vignali DAA. Regulatory T cells: barriers of immune infiltration into the tumor microenvironment. Front Immunol. (2021) 12:702726. doi: 10.3389/fimmu.2021.702726
47. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
48. Koh YT, Higgins SA, Weber JS, Kast WM. Immunological consequences of using three different clinical/laboratory techniques of emulsifying peptide-based vaccines in incomplete Freund's adjuvant. J Transl Med. (2006) 4:42. doi: 10.1186/1479-5876-4-42
49. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity. (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
50. Wang W, Guo H, Li H, Yan Y, Wu C, Wang X, et al. Interleukin-35 gene-modified mesenchymal stem cells protect concanavalin A-induced fulminant hepatitis by decreasing the interferon gamma level. Hum Gene Ther. (2018) 29:234–41. doi: 10.1089/hum.2017.171
51. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci United States America. (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
52. Villarroel-Espindola F, Yu X, Datar I, Mani N, Sanmamed M, Velcheti V, et al. Spatially resolved and quantitative analysis of VISTA/PD-1H as a novel immunotherapy target in human non-small cell lung cancer. Clin Cancer Res. (2018) 24:1562–73. doi: 10.1158/1078-0432.CCR-17-2542
53. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
54. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
55. Wang Q, He J, Flies DB, Luo L, Chen L. Programmed death one homolog maintains the pool size of regulatory T cells by promoting their differentiation and stability. Sci Rep. (2017) 7:6086. doi: 10.1038/s41598-017-06410-w
56. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Sci (New York NY). (2011) 332:600–3. doi: 10.1126/science.1202947
57. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
58. Kryczek I, Wang L, Wu K, Li W, Zhao E, Cui T, et al. Inflammatory regulatory T cells in the microenvironments of ulcerative colitis and colon carcinoma. Oncoimmunology. (2016) 5:e1105430. doi: 10.1080/2162402X.2015.1105430
59. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
60. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
61. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. (2004) 4:891–9. doi: 10.1038/nrc1478
62. Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. (2022) 39:110986. doi: 10.1016/j.celrep.2022.110986
63. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
64. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight. (2017) 2:e89160. doi: 10.1172/jci.insight.89160
65. Li X, Li C, Zhang L, Wu M, Cao K, Jiang F, et al. The significance of exosomes in the development and treatment of hepatocellular carcinoma. Mol Cancer. (2020) 19:1. doi: 10.1186/s12943-019-1085-0
66. Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer. (2021) 20:154. doi: 10.1186/s12943-021-01463-y
67. Li X, Lei Y, Wu M, Li N. Regulation of macrophage activation and polarization by HCC-derived exosomal lncRNA TUC339. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19102958
68. Yin C, Han Q, Xu D, Zheng B, Zhao X, Zhang J. SALL4-mediated upregulation of exosomal miR-146a-5p drives T-cell exhaustion by M2 tumor-associated macrophages in HCC. Oncoimmunology. (2019) 8:1601479. doi: 10.1080/2162402X.2019.1601479
69. Wu J, Gao W, Tang Q, Yu Y, You W, Wu Z, et al. M2 macrophage-derived exosomes facilitate HCC metastasis by transferring α(M) β(2) integrin to tumor cells. Hepatol (Baltimore Md). (2021) 73:1365–80. doi: 10.1002/hep.31432
70. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci CMLS. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
71. Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer-Associated Fibroblasts-Derived Exosomes Suppress Immune Cell Function in Breast Cancer via the miR-92/PD-L1 Pathway. Front Immunol. (2020) 11:2026. doi: 10.3389/fimmu.2020.02026
72. Lu JC, Zhang PF, Huang XY, Guo XJ, Gao C, Zeng HY, et al. Amplification of spatially isolated adenosine pathway by tumor-macrophage interaction induces anti-PD1 resistance in hepatocellular carcinoma. J Hematol Oncol. (2021) 14:200. doi: 10.1186/s13045-021-01207-x
73. Yang C, Zhang H, Zhang L, Zhu AX, Bernards R, Qin W, et al. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2023) 20:203–22. doi: 10.1038/s41575-022-00704-9
74. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/s1074-7613(02)00323-0
75. Rial Saborido J, Volkl S, Aigner M, Mackensen A, Mougiakakos D. Role of CAR T cell metabolism for therapeutic efficacy. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14215442
76. Singh R, Kim YH, Lee SJ, Eom HS, Choi BK. 4-1BB immunotherapy: advances and hurdles. Exp Mol Med. (2024) 56:32–9. doi: 10.1038/s12276-023-01136-4
77. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-003354
78. Chen Y, CY E, Gong ZW, Liu S, Wang ZX, Yang YS, et al. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat Dis Int. (2018) 17:301–9. doi: 10.1016/j.hbpd.2018.05.005
79. Liu Z, Liu X, Liang J, Liu Y, Hou X, Zhang M, et al. Immunotherapy for hepatocellular carcinoma: current status and future prospects. Front Immunol. (2021) 12:765101. doi: 10.3389/fimmu.2021.765101
80. Guizhen Z, Guanchang J, Liwen L, Huifen W, Zhigang R, Ranran S, et al. The tumor microenvironment of hepatocellular carcinoma and its targeting strategy by CAR-T cell immunotherapy. Front Endocrinol (Lausanne). (2022) 13:918869. doi: 10.3389/fendo.2022.918869
81. Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11091339
82. Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin Cancer Res. (2020) 26:3979–89. doi: 10.1158/1078-0432.CCR-19-3259
83. Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. (2020) 8:309–20. doi: 10.1158/2326-6066.CIR-19-0293
84. Patra T, Cunningham DM, Meyer K, Toth K, Ray RB, Heczey A, et al. Targeting Lin28 axis enhances glypican-3-CAR T cell efficacy against hepatic tumor initiating cell population. Mol Ther. (2023) 31:715–28. doi: 10.1016/j.ymthe.2023.01.002
85. Wang P, Qin W, Liu T, Jiang D, Cui L, Liu X, et al. PiggyBac-engineered T cells expressing a glypican-3-specific chimeric antigen receptor show potent activities against hepatocellular carcinoma. Immunobiology. (2020) 225:151850. doi: 10.1016/j.imbio.2019.09.009
86. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis Malignancies: A phase I trial. Oncoimmunology. (2018) 7:e1440169. doi: 10.1080/2162402X.2018.1440169
87. Dai H, Tong C, Shi D, Chen M, Guo Y, Chen D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. Oncoimmunology. (2020) 9:1846926. doi: 10.1080/2162402X.2020.1846926
88. Huang X, Guo J, Li T, Jia L, Tang X, Zhu J, et al. c-Met-targeted chimeric antigen receptor T cells inhibit hepatocellular carcinoma cells in vitro and in vivo. J Biomed Res. (2022) 36:10–21. doi: 10.7555/JBR.35.20200207
89. Jiang W, Li T, Guo J, Wang J, Jia L, Shi X, et al. Bispecific c-met/PD-L1 CAR-T cells have enhanced therapeutic effects on hepatocellular carcinoma. Front Oncol. (2021) 11:546586. doi: 10.3389/fonc.2021.546586
90. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. (2016) 7:550. doi: 10.3389/fimmu.2016.00550
91. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. (2013) 39:61–73. doi: 10.1016/j.immuni.2013.07.005
92. Yuan X, Sun Z, Yuan Q, Hou W, Liang Q, Wang Y, et al. Dual-function chimeric antigen receptor T cells targeting c-Met and PD-1 exhibit potent anti-tumor efficacy in solid tumors. Invest New Drugs. (2021) 39:34–51. doi: 10.1007/s10637-020-00978-3
93. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
94. Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol. (2015) 6:97. doi: 10.3389/fimmu.2015.00097
95. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. (2018) 51:55–61. doi: 10.1016/j.coi.2018.02.004
96. Sun B, Yang D, Dai H, Liu X, Jia R, Cui X, et al. Eradication of hepatocellular carcinoma by NKG2D-based CAR-T cells. Cancer Immunol Res. (2019) 7:1813–23. doi: 10.1158/2326-6066.CIR-19-0026
97. Zhang T, Sentman CL. Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D-modified T cells. J Immunol. (2013) 190:2455–63. doi: 10.4049/jimmunol.1201314
98. Soriani A, Fionda C, Ricci B, Iannitto ML, Cippitelli M, Santoni A. Chemotherapy-elicited upregulation of NKG2D and DNAM-1 ligands as a therapeutic target in multiple myeloma. Oncoimmunology. (2013) 2:e26663. doi: 10.4161/onci.26663
99. Huang D, Rao D, Jin Q, Lai M, Zhang J, Lai Z, et al. Role of CD147 in the development and diagnosis of hepatocellular carcinoma. Front Immunol. (2023) 14:1149931. doi: 10.3389/fimmu.2023.1149931
100. Zhang RY, Wei D, Liu ZK, Yong YL, Wei W, Zhang ZY, et al. Doxycycline inducible chimeric antigen receptor T cells targeting CD147 for hepatocellular carcinoma therapy. Front Cell Dev Biol. (2019) 7:233. doi: 10.3389/fcell.2019.00233
101. Hu X, Chen R, Wei Q, Xu X. The landscape of alpha fetoprotein in hepatocellular carcinoma: where are we? Int J Biol Sci. (2022) 18:536–51. doi: 10.7150/ijbs.64537
102. Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res Off J Am Assoc Cancer Res. (2017) 23:478–88. doi: 10.1158/1078-0432.CCR-16-1203
103. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. (2019) 25:e123–e7. doi: 10.1016/j.bbmt.2018.12.756
104. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
105. Becker M, Dirschl SM, Scherm MG, Serr I, Daniel C. Niche-specific control of tissue function by regulatory T cells-Current challenges and perspectives for targeting metabolic disease. Cell Metab. (2024) 36:229–39. doi: 10.1016/j.cmet.2023.12.019
106. Zeng K, Huang M, Lyu MA, Khoury JD, Ahmed S, Patel KK, et al. Adjunct therapy with T regulatory cells decreases inflammation and preserves the anti-tumor activity of CAR T cells. Cells. (2023) 12. doi: 10.3390/cells12141880
107. Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, et al. CCR8(+)FOXp3(+) T(reg) cells as master drivers of immune regulation. Proc Natl Acad Sci U.S.A. (2017) 114:6086–91. doi: 10.1073/pnas.1621280114
108. Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska-Bosiacka I. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 ligands. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21207619
109. Cadilha BL, Benmebarek MR, Dorman K, Oner A, Lorenzini T, Obeck H, et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci Adv. (2021) 7:eabi5781. doi: 10.1126/sciadv.abi5781
110. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
111. Brand T, MacLellan WR, Schneider MD. A dominant-negative receptor for type beta transforming growth factors created by deletion of the kinase domain. J Biol Chem. (1993) 268:11500–3. doi: 10.1016/S0021-9258(19)50226-5
112. Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. (2011) 19:760–7. doi: 10.1038/mt.2011.9
113. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. (2005) 6:152–62. doi: 10.1038/ni1160
114. DeBenedette MA, Shahinian A, Mak TW, Watts TH. Costimulation of CD28- T lymphocytes by 4-1BB ligand. J Immunol (Baltimore Md 1950). (1997) 158:551–9. doi: 10.4049/jimmunol.158.2.551
115. Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol (Baltimore Md 1950). (2000) 165:6193–204. doi: 10.4049/jimmunol.165.11.6193
116. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
117. Snell LM, Lin GH, McPherson AJ, Moraes TJ, Watts TH. T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy. Immunol Rev. (2011) 244:197–217. doi: 10.1111/j.1600-065X.2011.01063.x
118. Akhmetzyanova I, Zelinskyy G, Littwitz-Salomon E, Malyshkina A, Dietze KK, Streeck H, et al. CD137 agonist therapy can reprogram regulatory T cells into cytotoxic CD4+ T cells with antitumor activity. J Immunol (Baltimore Md 1950). (2016) 196:484–92. doi: 10.4049/jimmunol.1403039
119. Suryadevara CM, Desai R, Farber SH, Choi BD, Swartz AM, Shen SH, et al. Preventing lck activation in CAR T cells confers treg resistance but requires 4-1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res an Off J Am Assoc Cancer Res. (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
120. Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest. (2020) 130:1405–16. doi: 10.1172/JCI128672
121. Fritsche E, Volk HD, Reinke P, Abou-El-Enein M. Toward an optimized process for clinical manufacturing of CAR-treg cell therapy. Trends Biotechnol. (2020) 38:1099–112. doi: 10.1016/j.tibtech.2019.12.009
122. Ke MY, Xu T, Fang Y, Ye YP, Li ZJ, Ren FG, et al. Liver fibrosis promotes immune escape in hepatocellular carcinoma via GOLM1-mediated PD-L1 upregulation. Cancer Lett. (2021) 513:14–25. doi: 10.1016/j.canlet.2021.05.007
123. Zhao L, Pei R, Ding Y, Su Z, Li D, Zhu S, et al. LOXL4 shuttled by tumor cells-derived extracellular vesicles promotes immune escape in hepatocellular carcinoma by activating the STAT1/PD-L1 axis. J Immunother. (Hagerstown Md 1997). (2024) 47:64–76. doi: 10.1097/CJI.0000000000000496
124. Bozward AG, Warricker F, Oo YH, Khakoo SI. Natural killer cells and regulatory T cells cross talk in hepatocellular carcinoma: exploring therapeutic options for the next decade. Front Immunol. (2021) 12:643310. doi: 10.3389/fimmu.2021.643310
125. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci United States America. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
126. Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. (2009) 15:971–9. doi: 10.1158/1078-0432.CCR-08-1608
127. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
128. Ma J, Ma P, Zhao C, Xue X, Han H, Liu C, et al. B7-H3 as a promising target for cytotoxicity T cell in human cancer therapy. Oncotarget. (2016) 7:29480–91. doi: 10.18632/oncotarget.v7i20
129. Loos M, Hedderich DM, Friess H, Kleeff J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol. (2010) 2010:683875. doi: 10.1155/2010/683875
130. Luo M, Zhang H, Zhu L, Xu Q, Gao Q. CAR-T cell therapy: challenges and optimization. Crit Rev Immunol. (2021) 41:77–87. doi: 10.1615/CritRevImmunol.2021037253
131. Long X, Zhang S, Wang Y, Chen J, Lu Y, Hou H, et al. Targeting JMJD1C to selectively disrupt tumor T(reg) cell fitness enhances antitumor immunity. Nat Immunol. (2024) 25:525–36. doi: 10.1038/s41590-024-01746-8
132. Sánchez-Paulete AR, Mateus-Tique J, Mollaoglu G, Nielsen SR, Marks A, Lakshmi A, et al. Targeting macrophages with CAR T cells delays solid tumor progression and enhances antitumor immunity. Cancer Immunol Res. (2022) 10:1354–69. doi: 10.1158/2326-6066.CIR-21-1075
133. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
134. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
135. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
136. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
137. Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. (2018) 9:414. doi: 10.3389/fimmu.2018.00414
138. Berahovich R, Liu X, Zhou H, Tsadik E, Xu S, Golubovskaya V, et al. Hypoxia selectively impairs CAR-T cells in vitro. Cancers. (2019) 11. doi: 10.3390/cancers11050602
139. He H, Liao Q, Zhao C, Zhu C, Feng M, Liu Z, et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-002755
140. Tu VY, Ayari A, O'Connor RS. Beyond the lactate paradox: how lactate and acidity impact T cell therapies against cancer. Antibodies (Basel Switzerland). (2021) 10. doi: 10.3390/antib10030025
141. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun. (2022) 13:4981. doi: 10.1038/s41467-022-32521-8
142. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
143. McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomedicine Pharmacother. (2020) 121:109625. doi: 10.1016/j.biopha.2019.109625
144. Zhong W, Xiao Z, Qin Z, Yang J, Wen Y, Yu Z, et al. Tumor-derived small extracellular vesicles inhibit the efficacy of CAR T cells against solid tumors. Cancer Res. (2023) 83:2790–806. doi: 10.1158/0008-5472.CAN-22-2220
145. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. (2019) 574:565–70. doi: 10.1038/s41586-019-1674-5
146. Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, et al. CAR T cell therapy of non-hematopoietic Malignancies: detours on the road to clinical success. Front Immunol. (2018) 9:2740. doi: 10.3389/fimmu.2018.02740
147. Whatcott CJ, Han H, Posner RG, Hostetter G, Von Hoff DD. Targeting the tumor microenvironment in cancer: why hyaluronidase deserves a second look. Cancer Discovery. (2011) 1:291–6. doi: 10.1158/2159-8290.CD-11-0136
Keywords: hepatocellular carcinoma (HCC), tumor microenvironment, CAR-T, regulatory T cells(Treg), immunotherapy
Citation: Du G, Dou C, Sun P, Wang S, Liu J and Ma L (2024) Regulatory T cells and immune escape in HCC: understanding the tumor microenvironment and advancing CAR-T cell therapy. Front. Immunol. 15:1431211. doi: 10.3389/fimmu.2024.1431211
Received: 11 May 2024; Accepted: 12 July 2024;
Published: 29 July 2024.
Edited by:
Xiaohua Lu, Johannes Gutenberg University Mainz, GermanyCopyright © 2024 Du, Dou, Sun, Wang, Liu and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leina Ma, bGVpbmFtYUBnbWFpbC5jb20=; Shasha Wang, d3NzXzA1MjhAcWR1LmVkdS5jbg==; Jia Liu, ZGFkYWxpdWppYUBxZHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.