
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 24 July 2024
Sec. Primary Immunodeficiencies
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1430938
This article is part of the Research TopicCommunity Series in Primary Immunodeficiencies Worldwide, volume IIView all 11 articles
 Franz-Martin Fink1*
Franz-Martin Fink1* Reinhard Höpfl2Martina Witsch-Baumgartner3Gabriele Kropshofer4
Reinhard Höpfl2Martina Witsch-Baumgartner3Gabriele Kropshofer4 Sabine Martin1
Sabine Martin1 Valentin Fink1
Valentin Fink1 Maximilian Heeg5
Maximilian Heeg5 Christina Peters6,7
Christina Peters6,7 Johannes Zschocke3
Johannes Zschocke3 Oskar A. Haas8,9*†
Oskar A. Haas8,9*†Severe aplastic anemia (SAA) is a life-threatening bone marrow failure syndrome whose development can be triggered by environmental, autoimmune, and/or genetic factors. The latter comprises germ line pathogenic variants in genes that bring about habitually predisposing syndromes as well as immune deficiencies that do so only occasionally. One of these disorders is the autosomal dominant form of chronic mucocutaneous candidiasis (CMC), which is defined by germ line STAT1 gain-of-function (GOF) pathogenic variants. The resultant overexpression and constitutive activation of STAT1 dysregulate the Janus kinase/signal transducer and activator of transcription 1 (STAT) signaling pathway, which normally organizes the development and proper interaction of different components of the immunologic and hematopoietic system. Although SAA is an extremely rare complication in this disorder, it gained a more widespread interest when it became clear that the underlying causative pathomechanism may, in a similar fashion, also be instrumental in at least some of the idiopathic SAA cases. Based on these premises, we present herein what is the historically most likely first cord blood–transplanted SAA case in a CMC family with a documented STAT1 GOF pathogenic variant. In addition, we recapitulate the characteristics of the six CMC SAA cases that have been reported so far and discuss the significance of STAT1 GOF pathogenic variants and other STAT1 signaling derangements in the context of these specific types of bone marrow failure syndromes. Because a constitutively activated STAT1 signaling, be it driven by STAT1 GOF germ line pathogenic variants or any other pathogenic variant-independent events, is apparently important for initiating and maintaining the SAA disease process, we propose to acknowledge that SAA is one of the definite disease manifestations in STAT1-mutated CMC cases. For the same reason, we deem it necessary to also incorporate molecular and functional analyses of STAT1 into the diagnostic work-up of SAA cases.
Severe aplastic anemia (SAA) is a life-threatening bone marrow failure (BMF) syndrome that can be caused by a radiation-evoked damage of the hematopoietic system, an autoimmune- or infection-associated destruction of bone marrow cells either by hepatitis B, parvo-, cytomegalo-, or Epstein–Barr viruses or by other yet unknown causes (1). There are three genetic disorders that typically predispose affected children to the development of SAA, namely, the Fanconi anemia DNA repair and dyskeratosis congenita telomere maintenance systems and the Shwachman–Diamond syndrome, a disorder of defective ribosome biogenesis (2–5). Moreover, SAA may occasionally also develop in the context of four specific immune disorders. These include the adenosine deaminase 2 deficiency (due to homozygous or compound heterozygous variants in the ADA2/CERC gene) (6–8), the loss of function (LOF) of the cytotoxic T-lymphocyte–associated protein 4 (caused by autosomal recessive variants in the CTLA4 gene) (4, 9), the gain of function (GOF) of the Toll-like receptor 8 (10–12), and the signal transducer and activator of transcription 1 (STAT1), which all result from dominant heterozygous gene alterations (13–16).
Recurrent and persistent infections with various candida species are the hallmark of several genetically distinct immunodeficiency disorders (17–24). The autosomal dominant form of chronic mucocutaneous candidiasis (CMC) results from STAT1 GOF pathogenic variants (OMIM #614162 and orphan designation ORPHA1334) (18, 19, 22, 24–26). They designate neither a genetically nor clinically distinct entity but rather a form of combined immunodeficiency with recurrent or persistent infections of the nails, skin, and mucous membranes that are caused by various candida species and commonly accompanied by diverse other bacterial and viral infections as well as a smorgasbord of autoimmune and inflammatory disorders, cytopenias, aneurysms, and squamous cell carcinomas (19, 22, 26, 27). The most frequent autoimmune manifestations comprise thyroiditis and destructive blood cytopenias, such as autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and neutropenia. The few cases of SAA in carriers with STAT1 GOF variants gained a more widespread interest when it became clear that also pathogenic variant-independent modes of STAT1 activation can trigger the development of idiopathic SAA forms, an insight that is, nowadays, already exploited therapeutically with drugs that inhibit the Janus kinase (JAK) (13–16, 28–31).
We report herein a CMC family with three STAT1 pathogenic variant carriers, two brothers and their father, who had been transplanted as a child because of a SAA.
A 4-year-old boy (patient 1) presented with CMC-associated recurring oral candidiasis and candida-associated paronychia since infancy, body-wide scaly seborrheic eczemas that extended to the eyelids, and a dystrophic growth of both thumbnails due to a chronic inflammation of the nail folds (Figure 1). His 14-month-old brother (patient 2) developed a severe oral thrush 3 months later (Figure 1D). In 1991, 28 years earlier, their father (patient 3) developed a SAA when he was 4 years old. At that time, his white blood cell count was 3.6 × 109/L (age-adjusted normal range, 4.1 to 14.6 × 109/L) with an absolute neutrophil count of 324 × 106/L (normal, 200 × 106/L) and a red blood cell count of 1.08 × 1012/L (normal, 3.98 to 5.33 × 109/L). His hemoglobin was 3.6 g/dL (normal, 10.7 g/dL to 14.2 g/dL) and his platelets were 10 × 109/L (normal, 168 to 453 × 109/L) (32). Eleven months earlier, he had a hepatitis A infection; 6 months earlier, a bronchitis; and 2 months, earlier a whooping cough. When he was 3 years old, he had developed a mild psoriasis-like seborrheic eczema but none of the more specific CMC signs. The patient became transfusion-dependent 2 months after first presentation and was, therefore, treated unsuccessfully with high-dose corticosteroids and anti-lymphocyte globulin. Six months later, he was transplanted with cord blood cells from his newly born brother. The conditioning consisted of cyclophosphamide at 200 mg/kg and 5 Gy of total nodal irradiation and the graft-versus-host prophylaxis of ciclosporin A and methotrexate. Stem cell engraftment was delayed, but, following G-CSF treatment, his white blood cell count rose over 1.0 × 103/µL on day +39. He received his last platelet transfusion on day +40 and the last unit of packed red blood cells on day +47. Since then, the now 37-year-old father has normal blood cell counts, had never experienced any CMC symptoms, and remained healthy. Also, the clinical course of the two affected boys is so far benign. They require only occasionally symptomatic antifungal treatment. Fluorescence activated cell sorting (FACS) analyses of peripheral blood mononuclear cells of the two brothers and their transplanted father revealed a normal distribution of lymphocyte subpopulations. Their levels of immunoglobulins and immunoglobulin G (IgG) subclasses were normal and none of them had developed autoantibodies or endocrine deficiencies.
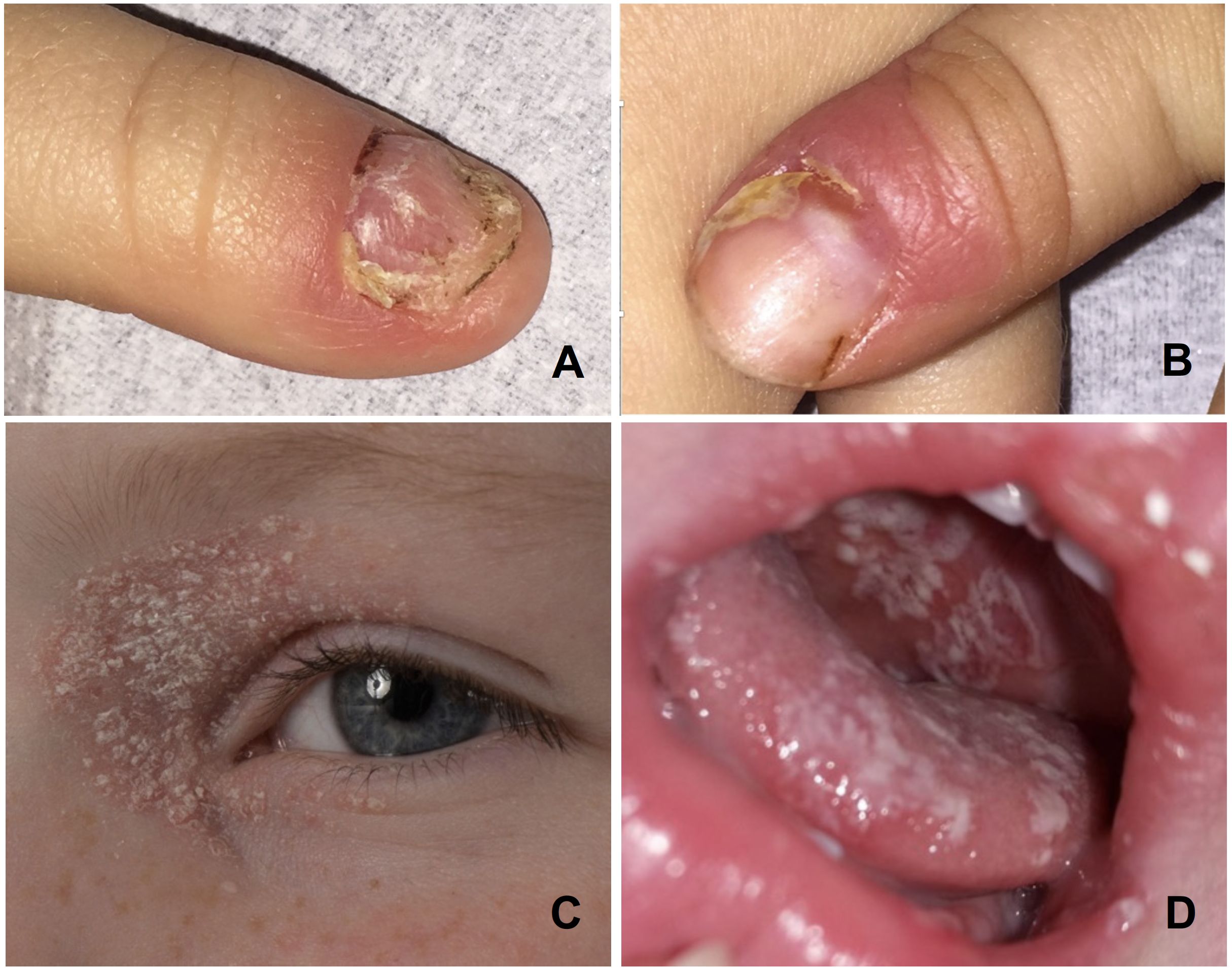
Figure 1 Photographs of the CMC manifestations in the two boys: Candida paronychias and onychodystrophic thumbnails (A, B) and periocular seborrheic eczema (C) in the index patient 1, and oral candidiasis in his brother (D).
To identify the genetic cause of this familial disorder, we used the Illumina’s TruSight™ One Panel to sequence DNA that was extracted from the older boy’s peripheral blood cells and focused our screening efforts on the three most likely responsible genes, namely, STAT1, IL17RA, and AIRE. This approach uncovered a hitherto undescribed heterozygous STAT1 missense variant c.1013G>T (p.Gly338Val) that affects the DNA-binding domain (Figure 2) (19). Based on the criteria put forward by the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants, we initially classified it as variant of unknown significance (33). Twenty-eight years after the father had been transplanted, we succeeded to identify this variant in his skin fibroblasts with an allele frequency of 50%. Neither the children’s parental grandparents nor their mother carried the pathogenic variant. The fact that we found the same variant in both diseased brothers and their father enabled us to reclassify it as likely pathogenic.
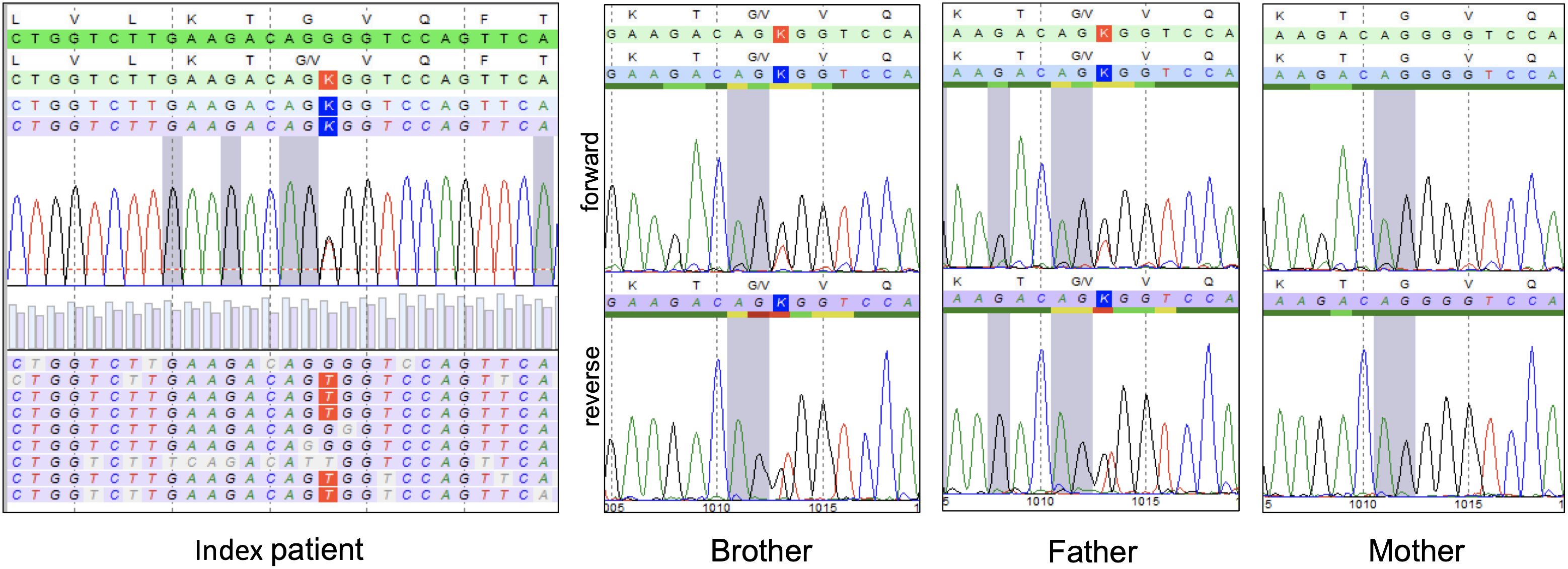
Figure 2 Result of the pathogenic variant screening in our CMC family. In the index patient, targeted pathogenic variant screening revealed a heterozygous c.1013G>T (p.Gly338Val) missense pathogenic variant in the STAT1 gene, which was subsequently also identified in the peripheral blood of his brother and in the fibroblasts of his father with Sanger sequencing the respective PCR-amplified Exon 11, but not in the healthy mother.
To further verify the functional relevance of this novel STAT1 missense variant, we stimulated peripheral blood mononuclear cells (PB-MNCs) of the two brothers with interferon (IFN)–α and IFN-γ, which led to a significant increase in the phosphorylation of STAT1 (Figure 3). Conversely, incubating PB-MNCs with the “nuclear factor ‘kappa-light-chain enhancer’ of activated B cells” (NF-κB) activators phorbol myristate acetate and ionomycin disclosed an impaired expression of interleukin-17 (IL-17) as well as IFN-γ in the patient’s CD45RO+CD4+ T cells (Figure 3). Together, these results clearly proved the GOF nature of this c.1013G>T STAT1 missense variant.
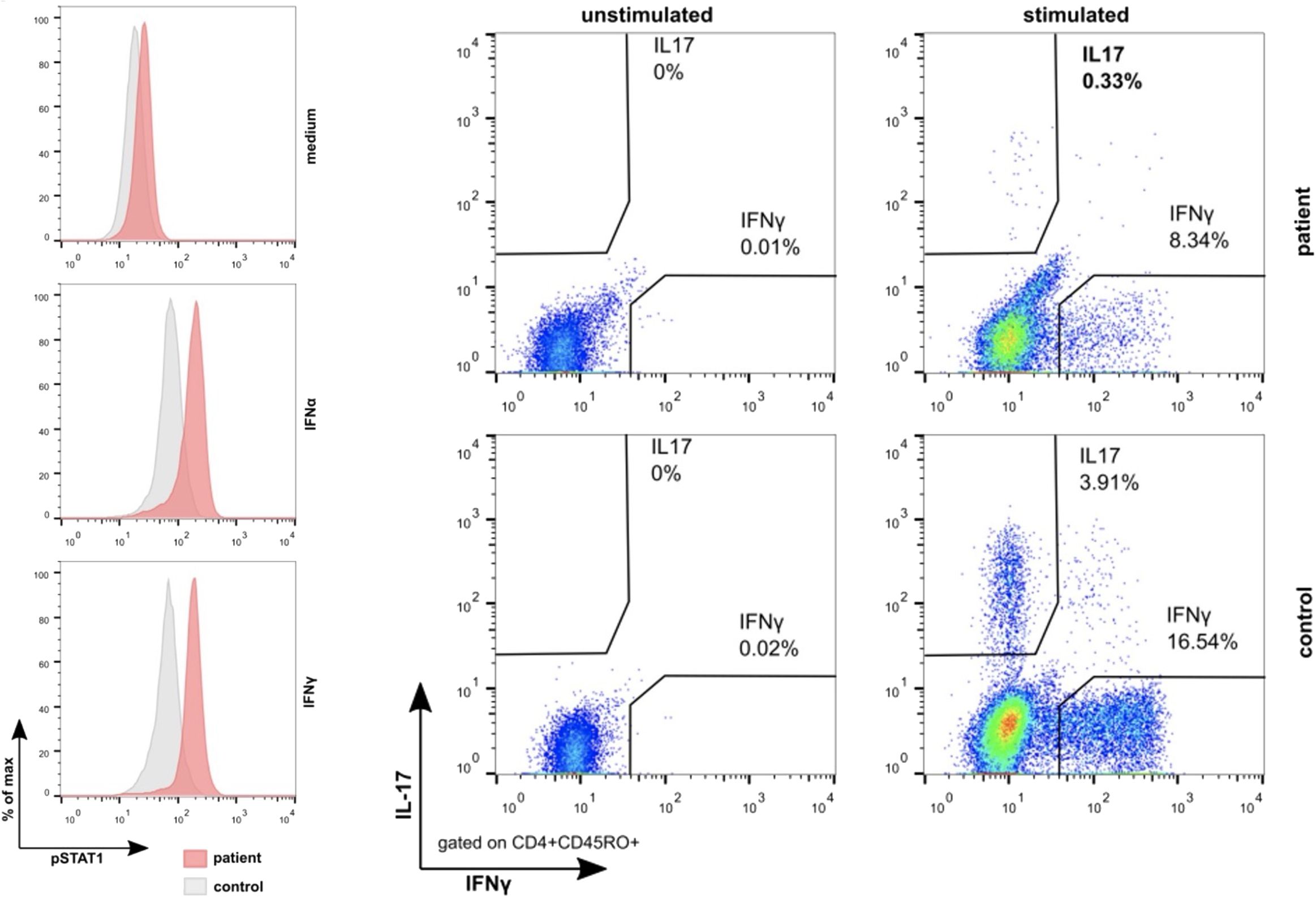
Figure 3 Results of the analyses of the functional consequences of the STAT1 c.1013G>T (p.Gly338Val) missense pathogenic variant. Compared to healthy controls, stimulation with interferon-α as well as interferon-γ leads to the hyperphosphorylation of STAT1 in the monocytes (left) of the index patient and his brother (not shown). Conversely, stimulation with the NF-κB activator phorbol myristate acetate and ionomycin revealed the impaired expression of interleukin-17 as well as interferon-γ in their CD45RO+CD4+ T cells. Together, these findings provide persuasive evidence for the GOF effect of this pathogenic variant.
The most remarkable finding in the CMC family reported herein is that the father was a STAT1 GOF variant carrier (case 7, Table 1). As a child, he was transplanted with his brother’s cord blood cells because of a presumably infection- or autoimmune-induced SAA. If it had not been for his diseased sons, then we would have never contemplated that he might be a STAT1 GOF variant carrier. Yet, 28 years later, we were able to secure the GOF variant in his skin fibroblasts and, by that, identify the historically first SAA case with a STAT1 GOF variant who had received a life-saving cord blood transplant.
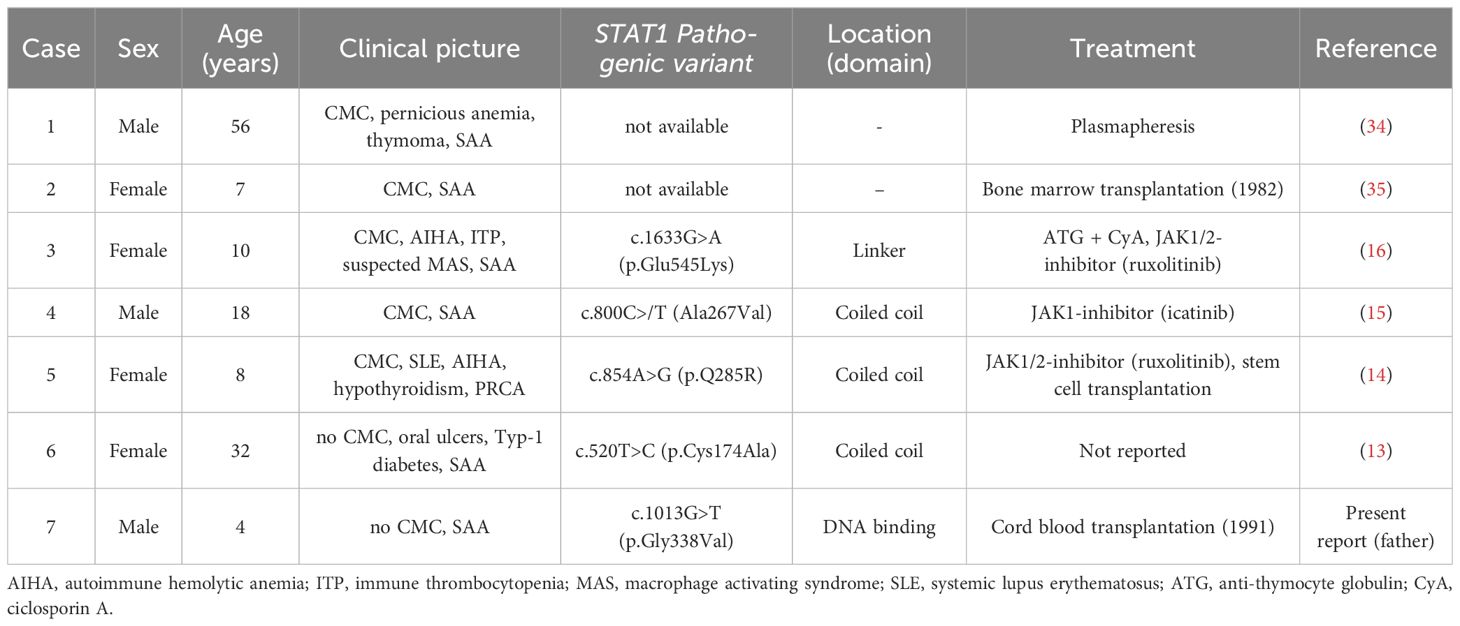
Table 1 Overview of seven cases with CMC, GOF-STAT1 mutations and SAA.
In individuals with CMC, genuine BMF syndromes, such as SAA and pure red cell aplasia (PRCA), seem either extremely rare or underreported (19, 22, 26, 36). There are only two such cases that were published earlier. The first one was reported in 1975 by Twomey et al. (case 1, Table 1) (34). An already 56-year-old man developed SAA and a small thymoma 9 years after mucocutaneous candidiasis was first noted. The second case concerned a 7-year-old girl with CMC and SAA, who was successfully treated with a bone marrow transplant (case 2, Table 1) (35).
In addition to these two cases and the one presented herein, only three other STAT1-associated CMC cases with SAA and one with a PRCA were reported (Table 1) (13–16). The first of these concerns a 10-year-old girl who originally suffered from a life-threatening Evans syndrome with episodes of AIHA and ITP (case 3, Table 1) (16). Later on, she developed a SAA together with a suspected macrophage activation syndrome. She was treated with steroids, intravenous immunoglobulins, the anti-CD20 antibody rituximab, the anti-complement C5 antibody eculizumab, and, thereafter, with anti-thymocyte globulin and ciclosporin A. Only the latter restored her bone marrow function to some extent but did not produce a complete remission. Following the detection of a STAT1 GOF pathogenic variant and the subsequent meticulous in vitro assessment of its functional consequences, she received the JAK1/2 inhibitor ruxolitinib. Continuous treatment with this inhibitor alone resolved all her autoimmune-mediated problems, and she was reported to be still in complete remission 18 months later.
The second case concerns an 18-year-old man with oral ulcers and CMC who developed SAA with an only moderately decreased number of platelets, but weekly transfusion-dependent anemia and several episodes of febrile neutropenia (case 4, Table 1) (15). He was treated with the JAK1 inhibitor itacitinib for 20 months without any adverse effects that resulted in a rapid and remarkable recovery of hematopoiesis, before he self-discontinued this treatment. Three years after initial presentation and approximately 12 months off itacitinib therapy, he was still in continuous hematologic remission. Based on the discovery and insight that a significant proportion of other patients with “idiopathic” forms of SAA share similar STAT1-mediated pathophysiologic changes, the authors were the first to suggest that such “idiopathic” cases might also benefit from treatment with JAK inhibitors in a similar manner.
The third case concerns a 32-year-old woman with oral ulcers, oral candidiasis, recurrent pneumonia, type 1 diabetes mellitus, and SAA with a relatively preserved thrombocyte level (case 5, Table 1) (13). The authors did not provide any further clinical information, but they also found that STAT1 can be overexpressed in patients with idiopathic SAA.
Because PRCA and SAA are BMF syndromes with a closely related etiology, we also include the girl with CMC and a PRCA (case 6, Table 1) (14). With 1 year, she already suffered from a systemic lupus erythematosus before she developed CMC-typical infections and eventually PRCA with 8 years. Because conventional immunosuppressive therapies were not effective, she received the JAK inhibitor ruxolitinib and was transplanted after she had transiently improved but succumbed to transplant-related complications.
STAT1 encodes one of the seven STAT family transcription factors. They are downstream components of the JAK signaling pathway, which regulates the expression of more than 50 cytokines, growth factors and IFNs (37). After binding to their cognate receptors, the respective ligands dimerize and activate JAKs, which phosphorylate the STAT proteins and, by that, turn on their individual transcriptional programs (15, 37, 38). An activated STAT1 not only impacts the expression of other transcription factors but also self-regulates its own expression (39).
The constantly enhanced STAT1 signaling in STAT1 GOF variant carriers creates two closely interlinked feedback loops that enhance the IFN-controlled and inhibit the IL-17–controlled immune defense (25, 40–47). Within this context, STAT1 acts both as a signal transducer and transcription activator for the IFN type I (IFN-α and IFN- β), type II (IFN-γ), and type III (IFN-λ) as well as for IL-27 (25, 40, 41, 48). The increased levels of these factors eventually cause all the varied inflammatory, autoimmune, and tissue destructive ailments that CMC patients have to endure (19, 22, 40, 41). The fact that STAT1 signaling occurs through both the type I and the type II IFN receptors prompted Largent et al. to examine the differences between these two transmission routes (43). They found that the deletion of the type I IFN receptors in mice with a Stat1 GOF pathogenic variant only partly protected them from STAT1-driven systemic inflammations, whereas deletion of the type II IFN receptors normalized total STAT1 expression, which safeguarded them from autoimmune manifestations (43). These findings suggest that IFN-γ is the relevant driver that upregulates STAT1 in a STAT1 GOF environment (41, 43). The long-term exposure of hematopoietic stem cells to high IFN-γ levels inhibits their self-renewal capacity and pushes them toward terminal differentiation (49–52). It is, therefore, conceivable that an exhaustion of the stem cell compartment is one of the reasons why, in some STAT1 GOF variant carriers, the bone marrow will fail. We presume that, in our SAA case, this process was most likely instigated and reinforced by the three infections that he had gone through beforehand.
Whether and to which extent STAT1 activity is primarily governed alone by an enhanced production, by the subsequent phosphorylation, and/or by an impaired dephosphorylation of the respective proteins is still a controversial and unresolved issue. While an in vitro IFN-α and IFN-γ stimulation–associated phosphorylation is a diagnostic hallmark of every STAT1 GOF variant (46), Scott et al. and others suggested that STAT1 increased phosphorylation might be a secondary event and only take place when the STAT1 level is already elevated. They, therefore, consider the overall amount of unphosphorylated STAT1 as probably being more relevant for the primary pathogenic effects than phosphorylation itself (39, 53, 54).
Recent in vitro experiments provided some evidence that specific STAT1 GOF variants may predetermine specific disease patterns and clinical phenotypes (25, 39, 55, 56). This notion derives from the variant-specific IFN-α and IFN-γ gene expression patterns that were caused by two different STAT1 GOF variants in a STAT1-deficient U3A fibrosarcoma cell line transfection model (56). These observations were later extended by Scott et al., who generated five heterozygous CRISPR/Cas9 base–edited STAT1 GOF variants in diploid HAP1 cells and found that baseline as well as IFN-α or IFN-γ induced expression levels of five investigated IFN-stimulated genes varied starkly depending on the specific pathogenic variant (39). Taken together, these widely divergent variant-specific transcriptional responses suffice at least to comprehend why the resultant disease spectra are so diverse and why it is virtually impossible to predict the overall clinical course in STAT1 GOF–mutated individuals.
The second feedback loop revolves around the highly versatile context- and tissue-dependent function of the proinflammatory cytokine IL-17 (42, 47, 57). IL-17 is primarily protective against fungal as well as, to a lesser extent, also other types of mucocutaneous infections, which is the reason why individuals with an insufficient IL-17 defense develop such otherwise uncommon diseases (21, 42, 46, 57, 58). However, even in cases with an intact IL-17 response, the local effects of an IFN-γ/STAT1–driven interferonopathy alone can be enough to promote mucocutaneous fungal infections by impairing the integrity of the epithelial barrier (42, 57, 59). Because, in STAT1 GOF–mutated individuals, both these immune signaling alterations usually concur, their effects may jointly contribute to their heightened susceptibility and the severity of fungal infections.
A hyperactive STAT1 downregulates the IL-17 signaling pathway by interfering with the STAT3-mediated differentiation program of Th17 cells, as already evidenced by the similar immunological and clinical effects of STAT3 LOF and STAT1 GOF variants (41, 46, 60–62). Activation of STAT1 and STAT3 by IL-6, IL-21, IL-27, and other factors affect the differentiation program of Th17 cells in opposing ways that range from strong induction (IL-6) to strong inhibition (IL-27) (21, 47, 61, 63, 64). This tightly STAT1- and STAT3-regulated cellular differentiation and interaction network relies on a well-balanced and appropriately adjusted intracellular ratio of the STAT1 and STAT3 transcription factors. Because STAT1 and STAT3 form homo- as well as heterodimers, an overabundance of STAT1 will upregulate the STAT1 transcription program either in a direct or indirect way, for instance, by competing with STAT3 for common receptor docking sites on their respective target promotors (41, 62, 65).
Noteworthy, IL-27 priming impairs also Th17 differentiation by upregulating the programmed cell death protein ligand 1 (PD-L1) in CD4-positive T cells (66, 67). Because this occurs in a STAT1-dependent manner, PD-L1 expression is markedly increased in STAT3 LOF and STAT1 GOF naïve CD4+ T cells. Although such Th17 differentiation defects might theoretically be overcome by blocking PD-L1 with checkpoint inhibitors, this is currently not a viable option in auto-immune diseases because of adverse effects of the respective drugs (66, 68, 69).
Patients with CMC with a STAT1 GOF pathogenic variant are at risk to develop a broad spectrum of infectious and non-infectious complications. The first sign in infants is usually a recurrent or persisting oral candidiasis (thrush) alone or in combination with chronic nail-fold inflammation. As long as the course of CMC is mild and remains controllable, symptomatic treatment with antifungal, antibacterial, antiviral, and perhaps immune-dampening remedies is generally sufficient (17, 70). The only potential curative option for cases with more severe, exacerbating, and life-threatening disease forms was, for a long time, a hematopoietic stem cell transplantation. With a high rate of secondary graft failures and survival rates of only up to 40%, the overall outcome of this procedure is still poor (26, 36, 71). The main arguments that are commonly put forward to explain these disappointing outcome data are the advanced disease stage and consequently also the poorer health state of patients who are selected for transplantation (36, 71). Thus, some of the factors, which assured that the transplantation went smoothly and without complications in our patient, were his young age, his overall good clinical condition, and the lack of any noteworthy additional ailments before he received his cord blood graft.
The identification of STAT1 pathogenic variants together with their subsequent meticulous functional assessment paved the way for the development for well-defined targeted treatment approaches (17). The currently best explored ones are JAK inhibitors, such as the JAK1/2 inhibitor ruxolitinib. It downregulates JAK signaling and thereby also reduces STAT1 activity (14–16, 31, 72–78). Hence, JAK inhibitors help to manage the disease by suppressing autoimmune processes and by improving host defense mechanisms. Although sufficient data on the risk-benefit ratio, especially on the long-term application are still pending, JAK inhibitors are already increasingly used in experienced treatment centers in an off-label setting for bridging or as an alternative for stem cell transplantation (72, 73, 77). Another potentially interesting drug that could become helpful in the pretransplant preparation of patients with STAT1 GOF pathogenic variants is the IFN-γ blocker emapalumab (79).
Taken together, we presented herein what is the historically first cord blood–transplanted SAA case in a CMC family with a STAT1 GOF pathogenic variant. We reviewed the characteristics of all available six cases with CMC and a BMF and briefly discussed the role of STAT1 GOF pathogenic variants and other STAT1 signaling derangements that are encountered in pathogenic variant-driven and sporadic SAA cases, respectively. Based on this information, we deem it necessary to acknowledge that SAA is one of the definitive hematologic manifestations of STAT1 GOF–associated CMC and to perform molecular and functional analyses of STAT1 in all unclear cases of BMFs in the future.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding authors.
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements because all the molecular-genetic analyses, functional studies and therapeutic interventions were part of the advanced standard care of the involved patients. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
F-MF: Writing – original draft, Writing – review & editing, Supervision, Project administration, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. RH: Writing – review & editing, Supervision, Investigation, Data curation, Funding acquisition. MW-B: Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation. GK: Writing – review & editing, Data curation. SM: Writing – review & editing, Data curation. VF: Writing – review & editing, Data curation. MH: Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation. CP: Writing – review & editing, Data curation. JZ: Writing – review & editing, Supervision, Resources, Methodology, Investigation, Funding acquisition. OH: Writing – review & editing, Writing – original draft, Supervision, Formal analysis, Data curation, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We are grateful for the financial support provided by the St. Anna Children’s Cancer Research Institute (CCRI) Vienna, the Department of Dermatology and Venerology and the Institute of Human Genetics, Medical University Innsbruck, to cover the publication costs.
We acknowledge the excellent support of the CCI Advanced Diagnostic Unit (Ilka Fuchs) and the CCI Clinical Research Unit/Biobank (Alexandra Nieters).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Fiesco-Roa MO, Garcia-de Teresa B, Leal-Anaya P, van 't Hek R, Wegman-Ostrosky T, Frias S, et al. Fanconi anemia and dyskeratosis congenita/telomere biology disorders: Two inherited bone marrow failure syndromes with genomic instability. Front Oncol. (2022) 12:12949435. doi: 10.3389/fonc.2022.949435
3. Dokal I, Tummala H, Vulliamy T. Inherited bone marrow failure in the pediatric patient. Blood. (2022) 140:556–70. doi: 10.1182/blood.2020006481
4. Kallen ME, Dulau-Florea A, Wang W, Calvo KR. Acquired and germline predisposition to bone marrow failure: Diagnostic features and clinical implications. Semin Hematol. (2019) 56:69–82. doi: 10.1053/j.seminhematol.2018.05.016
5. Kelaidi C, Makis A, Petrikkos L, Antoniadi K, Selenti N, Tzotzola V, et al. Bone marrow failure in fanconi anemia: clinical and genetic spectrum in a cohort of 20 pediatric patients. J Pediatr Hematol Oncol. (2019) 41:612–7. doi: 10.1097/MPH.0000000000001549
6. Albalawi R, Hanafy E, Alnafea H, Altowijiry M, Riyad S, Abufara F, et al. Novel adenosine deaminase 2 (ADA2) mutations associated with hematological manifestations. J Investig Med High Impact Case Rep. (2021) 9:923247096211056770. doi: 10.1177/23247096211056770
7. Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38:569–78. doi: 10.1007/s10875-018-0525-8
8. Michniacki TF, Hannibal M, Ross CW, Frame DG, DuVall AS, Khoriaty R, et al. Hematologic manifestations of deficiency of adenosine deaminase 2 (DADA2) and response to tumor necrosis factor inhibition in DADA2-associated bone marrow failure. J Clin Immunol. (2018) 38:166–73. doi: 10.1007/s10875-018-0480-4
9. Solhaug TS, Tjønnfjord GE, Bjørgo K, Kildahl-Andersen O. A family with cytotoxic T-lymphocyte-associated protein 4 haploinsufficiency presenting with aplastic anaemia. BMJ Case Rep. (2022) 15:e247653. doi: 10.1136/bcr-2021-247653
10. Bleesing J. Gain-of-function defects in toll-like receptor 8 shed light on the interface between immune system and bone marrow failure disorders. Front Immunol. (2022) 13:13935321. doi: 10.3389/fimmu.2022.935321
11. Aluri J, Bach A, Kaviany S, Chiquetto Paracatu L, Kitcharoensakkul M, Walkiewicz MA, et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood. (2021) 137:2450–62. doi: 10.1182/blood.2020009620
12. Boisson B, Casanova JL. TLR8 gain of function: a tall surprise. Blood. (2021) 137:2420–2. doi: 10.1182/blood.2020010463
13. Solimando AG, Desantis V, Palumbo C, Marasco C, Pappagallo F, Montagnani M, et al. STAT1 overexpression triggers aplastic anemia: a pilot study unravelling novel pathogenetic insights in bone marrow failure. Clin Exp Med. (2023). doi: 10.1007/s10238-023-01017-0
14. Xie Y, Shao F, Lei J, Huang N, Fan Z, Yu H. Case report: A STAT1 gain-of-function mutation causes a syndrome of combined immunodeficiency, autoimmunity and pure red cell aplasia. Front Immunol. (2022) 13:13928213. doi: 10.3389/fimmu.2022.928213
15. Rosenberg JM, Peters JM, Hughes T, Lareau CA, Ludwig LS, Massoth LR, et al. JAK inhibition in a patient with a STAT1 gain-of-function variant reveals STAT1 dysregulation as a common feature of aplastic anemia. Med. (2022) 3:42–57 e45. doi: 10.1016/j.medj.2021.12.003
16. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. (2017) 139:1629–1640 e1622. doi: 10.1016/j.jaci.2016.11.022
17. Jing D, Liang G, Li X, Liu W. Progress in molecular diagnosis and treatment of chronic mucocutaneous candidiasis. Front Immunol. (2024) 15:1343138. doi: 10.3389/fimmu.2024.1343138
18. Wang X, Netea MG, van de Veerdonk FL. Genetics of Chronic Mucocutaneous Candidiasis. In: Immunogenetics of Fungal Diseases. Springer International Publishing (2017). p. 85–103.
19. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. (2016) 36:73–84. doi: 10.1007/s10875-015-0214-9
20. Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci USA. (2016) 113:E8277–85. doi: 10.1073/pnas.1618300114
21. Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunol. (2016) 5:e114. doi: 10.1038/cti.2016.71
22. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
23. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. (2013) 39:676–86. doi: 10.1016/j.immuni.2013.09.002
24. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
25. Guo L, Lian D, Gu Y, Lu X, Zhang Y, Li X, et al. Dysregulated STAT1 gain-of-function: Pathogen-free autoimmunity and fungal infection. hLife. (2024). doi: 10.1016/j.hlife.2024.03.002
26. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical relevance of gain- and loss-of-function germline mutations in STAT1: A systematic review. Front Immunol. (2021) 12:12654406. doi: 10.3389/fimmu.2021.654406
27. Hambleton S. When the STATs are against you. Blood. (2016) 127:3109–10. doi: 10.1182/blood-2016-05-715029
28. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
29. Notarangelo LD, Fleisher TA. Targeted strategies directed at the molecular defect: Toward precision medicine for select primary immunodeficiency disorders. J Allergy Clin Immunol. (2017) 139:715–23. doi: 10.1016/j.jaci.2017.01.004
30. Mossner R, Diering N, Bader O, Forkel S, Overbeck T, Gross U, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis. (2016) 62:951–3. doi: 10.1093/cid/ciw020
31. Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. (2015) 135:551–3. doi: 10.1016/j.jaci.2014.12.1867
32. Zierk J, Arzideh F, Rechenauer T, Haeckel R, Rascher W, Metzler M, et al. Age- and sex-specific dynamics in 22 hematologic and biochemical analytes from birth to adolescence. Clin Chem. (2015) 61:964–73. doi: 10.1373/clinchem.2015.239731
33. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
34. Twomey JJ, Waddell CC, Krantz S, O'Reilly R, L'Esperance P, Good RA. Chronic mucocutaneous candidiasis with macrophage dysfunction, a plasma inhibitor, and co-existent aplastic anemia. J Lab Clin Med. (1975) 85:968–77.
35. Deeg HJ, Lum LG, Sanders J, Levy GJ, Sullivan KM, Beatty P, et al. Severe aplastic anemia associated with chronic mucocutaneous candidiasis. Immunologic and hematologic reconstitution after allogeneic bone marrow transplantation. Transplantation. (1986) 41:583–6. doi: 10.1097/00007890-198605000-00006
36. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. (2018) 141:704–717 e705. doi: 10.1016/j.jaci.2017.03.049
37. Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. (2000) 95:19–29. doi: 10.1182/blood.V95.1.19
38. Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R. STAT mutations as program switchers: turning primary immunodeficiencies into autoimmune diseases. J Leukoc Biol. (2017) 101:29–38. doi: 10.1189/jlb.5RI0516-237RR
39. Scott O, Lindsay K, Erwood S, Mollica A, Roifman CM, Cohn RD, et al. STAT1 gain-of-function heterozygous cell models reveal diverse interferon-signature gene transcriptional responses. NPJ Genom Med. (2021) 6:34. doi: 10.1038/s41525-021-00196-7
40. Casanova JL, MacMicking JD, Nathan CF. Interferon-gamma and infectious diseases: Lessons and prospects. Science. (2024) 384:eadl2016. doi: 10.1126/science.adl2016
41. Asano T, Noma K, Mizoguchi Y, Karakawa S, Okada S. Human STAT1 gain of function with chronic mucocutaneous candidiasis: A comprehensive review for strengthening the connection between bedside observations and laboratory research. Immunol Rev. (2024) 322:81–97. doi: 10.1111/imr.13300
42. Huangfu L, Li R, Huang Y, Wang S. The IL-17 family in diseases: from bench to bedside. Signal Transduct Target Ther. (2023) 8:402. doi: 10.1038/s41392-023-01620-3
43. Largent AD, Lambert K, Chiang K, Shumlak N, Liggitt D, Oukka M, et al. Dysregulated IFN-gamma signals promote autoimmunity in STAT1 gain-of-function syndrome. Sci Transl Med. (2023) 15:eade7028. doi: 10.1126/scitranslmed.ade7028
44. Conti HR, Gaffen SL. IL-17-mediated immunity to the opportunistic fungal pathogen candida albicans. J Immunol. (2015) 195:780–8. doi: 10.4049/jimmunol.1500909
45. Netea MG, Joosten LA, van der Meer JW, Kullberg BJ, van de Veerdonk FL. Immune defence against Candida fungal infections. Nat Rev Immunol. (2015) 15:630–42. doi: 10.1038/nri3897
46. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. (2011) 208:1635–48. doi: 10.1084/jem.20110958
47. Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O'Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. (2010) 21:425–34. doi: 10.1016/j.cytogfr.2010.10.006
48. d'Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I interferonopathies in children: an overview. Front Pediatr. (2021) 9:9631329. doi: 10.3389/fped.2021.631329
49. Serio B, Giudice V, Selleri C. All roads lead to interferon-gamma: from known to untraveled pathways in acquired aplastic anemia. Medicina (Kaunas). (2023) 59:2170. doi: 10.3390/medicina59122170
50. Patel BA, Giudice V, Young NS. Immunologic effects on the haematopoietic stem cell in marrow failure. Best Pract Res Clin Haematol. (2021) 34:101276. doi: 10.1016/j.beha.2021.101276
51. Morales-Mantilla DE, King KY. The role of interferon-gamma in hematopoietic stem cell development, homeostasis, and disease. Curr Stem Cell Rep. (2018) 4:264–71. doi: 10.1007/s40778-018-0139-3
52. Smith JN, Kanwar VS, MacNamara KC. Hematopoietic stem cell regulation by type I and II interferons in the pathogenesis of acquired aplastic anemia. Front Immunol. (2016) 7:7330. doi: 10.3389/fimmu.2016.00330
53. Zimmerman O, Olbrich P, Freeman AF, Rosen LB, Uzel G, Zerbe CS, et al. STAT1 gain-of-function mutations cause high total STAT1 levels with normal dephosphorylation. Front Immunol. (2019) 10:101433. doi: 10.3389/fimmu.2019.01433
54. Bernasconi AR, Yancoski J, Villa M, Oleastro MM, Galicchio M, Rossi JG. Increased STAT1 amounts correlate with the phospho-STAT1 level in STAT1 gain-of-function defects. J Clin Immunol. (2018) 38:745–7. doi: 10.1007/s10875-018-0557-0
55. Giovannozzi S, Demeulemeester J, Schrijvers R, Gijsbers R. Transcriptional profiling of STAT1 gain-of-function reveals common and mutation-specific fingerprints. Front Immunol. (2021) 12:12632997. doi: 10.3389/fimmu.2021.632997
56. Ovadia A, Sharfe N, Hawkins C, Laughlin S, Roifman CM. Two different STAT1 gain-of-function mutations lead to diverse IFN-gamma-mediated gene expression. NPJ Genom Med. (2018) 3:323. doi: 10.1038/s41525-018-0063-6
57. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
58. Mengesha BG, Conti HR. The role of IL-17 in protection against mucosal candida infections. J Fungi (Basel). (2017) 3:52. doi: 10.3390/jof3040052
59. Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science. (2021) 371:371. doi: 10.1126/science.aay5731
60. Hiller J, Hagl B, Effner R, Puel A, Schaller M, Mascher B, et al. STAT1 gain-of-function and dominant negative STAT3 mutations impair IL-17 and IL-22 immunity associated with CMC. J Invest Dermatol. (2018) 138:711–4. doi: 10.1016/j.jid.2017.09.035
61. Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, Pot C. IL-27 induces th17 differentiation in the absence of STAT1 signaling. J Immunol. (2015) 195:4144–53. doi: 10.4049/jimmunol.1302246
62. Zheng J, van de Veerdonk FL, Crossland KL, Smeekens SP, Chan CM, Al Shehri T, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol. (2015) 45:2834–46. doi: 10.1002/eji.201445344
63. Huber M, Steinwald V, Guralnik A, Brustle A, Kleemann P, Rosenplanter C, et al. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol. (2008) 20:223–34. doi: 10.1093/intimm/dxm139
64. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. (2011) 12:247–54. doi: 10.1038/ni.1995
65. Mogensen TH. IRF and STAT transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol. (2018) 9: 93047. doi: 10.3389/fimmu.2018.03047
66. Zhang Y, Ma CA, Lawrence MG, Break TJ, O'Connell MP, Lyons JJ, et al. PD-L1 up-regulation restrains Th17 cell differentiation in STAT3 loss- and STAT1 gain-of-function patients. J Exp Med. (2017) 214:2523–33. doi: 10.1084/jem.20161427
67. Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol. (2013) 131:1691–3. doi: 10.1016/j.jaci.2013.01.004
68. Mohammadi P, Hesari M, Chalabi M, Salari F, Khademi F. An overview of immune checkpoint therapy in autoimmune diseases. Int Immunopharmacol. (2022) 107:107108647. doi: 10.1016/j.intimp.2022.108647
69. Zhang K, Kong X, Li Y, Wang Z, Zhang L, Xuan L. PD-1/PD-L1 inhibitors in patients with preexisting autoimmune diseases. Front Pharmacol. (2022) 13: 13854967. doi: 10.3389/fphar.2022.854967
70. van de Veerdonk FL, Netea MG. Treatment options for chronic mucocutaneous candidiasis. J Infect. (2016) 72 Suppl:S56–60. doi: 10.1016/j.jinf.2016.04.023
71. Kiykim A, Charbonnier LM, Akcay A, Karakoc-Aydiner E, Ozen A, Ozturk G, et al. Hematopoietic stem cell transplantation in patients with heterozygous STAT1 gain-of-function mutation. J Clin Immunol. (2019) 39:37–44. doi: 10.1007/s10875-018-0575-y
72. Fischer M, Olbrich P, Hadjadj J, Aumann V, Bakhtiar S, Barlogis V, et al. JAK inhibitor treatment for inborn errors of JAK/STAT signaling: An ESID/EBMT-IEWP retrospective study. J Allergy Clin Immunol. (2024) 153:275–86. doi: 10.1016/j.jaci.2023.10.018
73. Deya-Martinez A, Riviere JG, Roxo-Junior P, Ramakers J, Bloomfield M, Guisado Hernandez P, et al. Impact of JAK inhibitors in pediatric patients with STAT1 gain of function (GOF) mutations-10 children and review of the literature. J Clin Immunol. (2022) 42:1071–82. doi: 10.1007/s10875-022-01257-x
74. Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova JL, et al. Human STAT1 gain-of-function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol. (2020) 40:1065–81. doi: 10.1007/s10875-020-00847-x
75. Moriya K, Suzuki T, Uchida N, Nakano T, Katayama S, Irie M, et al. Ruxolitinib treatment of a patient with steroid-dependent severe autoimmunity due to STAT1 gain-of-function mutation. Int J Hematol. (2020) 112:258–62. doi: 10.1007/s12185-020-02860-7
76. Vargas-Hernandez A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2018) 141:2142–55 e2145. doi: 10.1016/j.jaci.2017.08.040
77. Bloomfield M, Kanderova V, Parackova Z, Vrabcova P, Svaton M, Fronkova E, et al. Utility of ruxolitinib in a child with chronic mucocutaneous candidiasis caused by a novel STAT1 gain-of-function mutation. J Clin Immunol. (2018) 38:589–601. doi: 10.1007/s10875-018-0519-6
78. Baris S, Alroqi F, Kiykim A, Karakoc-Aydiner E, Ogulur I, Ozen A, et al. Severe early-onset combined immunodeficiency due to heterozygous gain-of-function mutations in STAT1. J Clin Immunol. (2016) 36:641–8. doi: 10.1007/s10875-016-0312-3
Keywords: chronic mucocutaneous candidiasis (CMC), severe aplastic anemia (SAA), STAT1, gain-of-function pathogenic variant, transplantation
Citation: Fink F-M, Höpfl R, Witsch-Baumgartner M, Kropshofer G, Martin S, Fink V, Heeg M, Peters C, Zschocke J and Haas OA (2024) Retrospective identification of the first cord blood–transplanted severe aplastic anemia in a STAT1-associated chronic mucocutaneous candidiasis family: case report, review of literature and pathophysiologic background. Front. Immunol. 15:1430938. doi: 10.3389/fimmu.2024.1430938
Received: 10 May 2024; Accepted: 13 June 2024;
Published: 24 July 2024.
Edited by:
Maurizio Miano, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Blachy Javier Dávila Saldaña, Children’s National Hospital, United StatesCopyright © 2024 Fink, Höpfl, Witsch-Baumgartner, Kropshofer, Martin, Fink, Heeg, Peters, Zschocke and Haas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franz-Martin Fink, Zmlua0BraHNqLmF0; Oskar A. Haas, b3NrYXIuaGFhc0BnbWFpbC5jb20=
†Retired former member of these institutions
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.