 Keith Alan Nair II1,2
Keith Alan Nair II1,2 Bei Liu1,2*
Bei Liu1,2*- 1Division of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, Columbus, OH, United States
- 2The Pelotonia Institute for Immuno-Oncology, The Ohio State University Comprehensive Cancer Center, Columbus, OH, United States
Endoplasmic reticulum stress occurs due to large amounts of misfolded proteins, hypoxia, nutrient deprivation, and more. The unfolded protein is a complex intracellular signaling network designed to operate under this stress. Composed of three individual arms, inositol-requiring enzyme 1, protein kinase RNA-like ER kinase, and activating transcription factor-6, the unfolded protein response looks to resolve stress and return to proteostasis. The CD8+ T cell is a critical cell type for the adaptive immune system. The unfolded protein response has been shown to have a wide-ranging spectrum of effects on CD8+ T cells. CD8+ T cells undergo cellular stress during activation and due to environmental insults. However, the magnitude of the effects this response has on CD8+ T cells is still understudied. Thus, studying these pathways is important to unraveling the inner machinations of these powerful cells. In this review, we will highlight the recent literature in this field, summarize the three pathways of the unfolded protein response, and discuss their roles in CD8+ T cell biology and functionality.
1 Introduction
The endoplasmic reticulum (ER) is a complex and robust cellular organelle that specializes in the synthesis and folding of proteins, the biogenesis of lipids, and calcium metabolism (1). The ER is composed of two different parts: the rough, sheet-like ER and the smooth, tubule ER (2). The rough ER is characterized by the large amounts of ribosomes on the surface of its sheets, while the smooth ER is characterized by the non-appearance of ribosomes (3). While the smooth ER is known for lipid production and calcium storage, the rough ER is the primary location of protein synthesis and modification (1, 3). The ER is also the regulator of the translation of secretory proteins as well, which commonly occurs on the ER membrane (4, 5), whereafter they translocate into the ER lumen and are properly folded (6, 7).
Proteostasis, the maintenance of the balance of proteins in a cell, is an integral component of maintaining the health of the cell (8). Proteostasis is maintained by protein chaperones, needed for conformational changes in developing proteins, and both the ubiquitin-proteasome and lysosome-autophagy systems, necessary for degrading un- or misfolded proteins (9). However, an accumulation of misfolded proteins can occur, leading to ER stress and thus activating the unfolded protein response (UPR) (10). This can be caused by several factors, such as hypoxia, nutrient deprivation, reduced glycosylation, ineffective degradation of proteins, errors in post-translation modifications, lipid bilayer alterations, and calcium level shifts (11, 12).
The UPR was initially hypothesized in a work published by Kozutsumi et al. in 1988, in which they deduced the presence of two major chaperone proteins induced by ER stress: GRP78 and GRP94 (13). It was eventually revealed that when unfolded proteins reach a certain concentration in the lumen of the ER, aggregation is considered significant, leading to imbalance and activation of the UPR (14, 15). To detect shifts in proteostasis, the UPR uses three unique sensors: inositol-requiring protein 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor-6 (ATF6). The UPR has three general methods of clearance using these sensors and their downstream functions: depletion, upregulation, and apoptosis (16) (Figure 1).
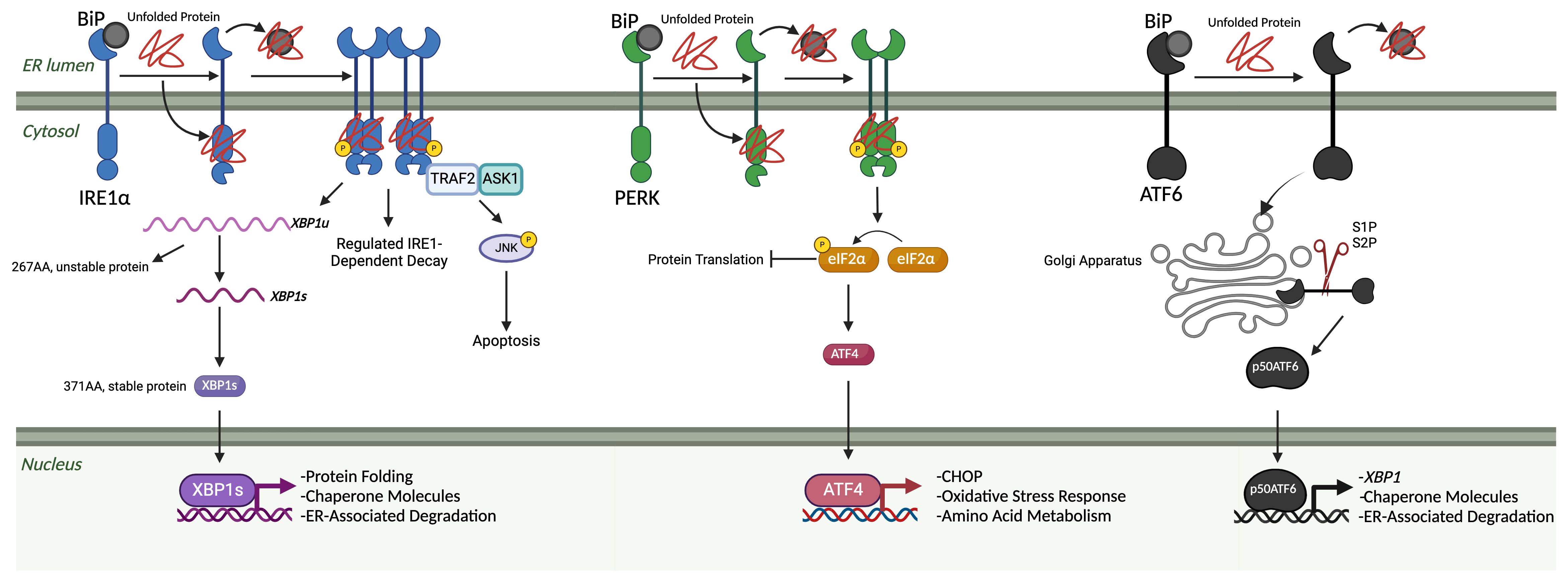
Figure 1 The unfolded protein response. The three arms of the UPR are IRE1α, PERK, and ATF6. Under homeostatic, non-stressed conditions, these three arms are held in an inactivated state by BiP. Once misfolded proteins accumulate in the ER lumen, BiP binds to those and releases them from the three arms, leading to their subsequent activation. Once free, IRE1α begins to oligomerize and auto-phosphorylate, leading to the start of its endoribonuclease activity. Two primary outcomes associated with IRE1α endoribonuclease activity: splicing of XBP1 mRNA into the XBP1s transcription factor and regulated RIDD. XBP1s translocates to the nucleus, where it upregulates genes related to protein folding, chaperone molecules, and ER-associated degradation elements. IRE1α also associates with a TNF-receptor associated factor 2 (TRAF2) – apoptosis signal-regulating kinase 1 (ASK1) complex, which results in the phosphorylation of c-Jun N-terminal kinase (JNK) and downstream apoptosis. When PERK is unbound, it activates in a similar oligomerization and auto-phosphorylation manner as IRE1. Once activated, it acts primarily through the phosphorylation of eIF2α. Then, eIF2α can lead to both protein translation inhibition and the favoring of the translation of ATF4. ATF4 then translocates to the nucleus and can upregulate CHOP, oxidative stress response elements, and amino acid metabolism elements. Once unbound from BiP, ATF6 will travel to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases. Once cleaved into a 50-kDa protein, p50ATF6 translocates to the nucleus, which can upregulate XBP1, chaperone molecules, and ER-associated degradation elements. (Created via BioRender).
CD8+ T cells are one of the most extensively studied immune cell subsets, often being compared to soldiers, as they are the body’s targeted weapon against insults (17). These cells are primed through three distinct mechanisms: 1) T cell receptor being presented with an antigen by major histocompatibility complex-I (MHC-I) from an antigen-presenting cell; 2) co-stimulation with various ligands; and 3) cytokines supporting the activation and differentiation (18). Once primed, they traffic from the lymph nodes to the site of insult, wherein they gain their effector program upon re-engagement of the TCR-MHC complex and co-stimulation molecules (19, 20). However, the role of the UPR in CD8+ T cells has been shown to have a wide-ranging spectrum of effects on these processes. In this review, we will further explore the functions of these pathways and their roles in CD8+ T cell biology and functionality.
2 The IRE1 pathways
2.1 Overview of the IRE1/XBP1 pathway
IRE1 was discovered as a transmembrane kinase in Saccharomyces cerevisiae in 1992 and speculated to play a role in signal transduction (21). It wasn’t until one year later that its role was deduced when it was shown that IRE1 is able to transduce signals from the ER to the nucleus (22, 23). Then, IRE1 was characterized as a serine-threonine protein kinase initiated by unfolded protein accumulation, where it then oligomerizes and is activated by trans-autophosphorylation (24, 25). Also, IRE1 has endoribonuclease activity that cleaves the HAC1 mRNA into a transcription factor (TF) that translocates to the nucleus and controls the UPR. Previously, this was all shown in yeast, but a breakthrough came when a yeast IRE1 homolog was found in mammalian cells and had similar trans-autophosphorylation and endoribonuclease activity. It also splices yeast HAC1, revealing a homologous protein may exist in mammalian cells (26). The same group found that the IRE1 endoribonuclease activity is autoregulatory to its own mRNA and theorized the presence of two human isoforms of IRE1 (27). These two forms of human IRE1 were identified and characterized as IRE1α and IRE1β, each with distinct functions. IRE1α was shown to be ER stress-related, while IRE1β was found to cleave 28s ribosomal RNA, causing translational repression (28). The disparities in function were later attributed to structural differences in the RNase domain of the isoforms (29).
The primary splicing target of IRE1α in mammals was later discovered to be X-box binding protein 1 (XBP1) (30, 31). XBP1 was first identified in 1996 as a basic region leucine zipper (bZIP) TF (32) that acts on several transcriptional elements, later identified as the unfolded protein response element (UPRE) and the endoplasmic reticulum stress element (ERSE) I and II (31, 33). XBP1 binds at CCACG sequences in the ERSE regions and the palindrome sequence (CGTGC) in the UPRE regions (30, 32). XBP1 was shown to be two separate protein products named based on their form: an unstable, unspliced XBP1u or a stable, spliced XBP1s (34). IRE1α excises a 26 nucleotide intron from the XBP1 mRNA that is then re-ligated and results in a frameshift mutation, extending the open reading frame and becoming a stable 376 amino acid protein (34, 35). The RNA ligase RtcB is responsible for ligating the cleaved XBP1 mRNA together after IRE1α excision (36, 37). RtcB is regulated via phosphorylation by ABL1 and PTP1B (38, 39). XBP1s has several functions, such as upregulating the ER degradation-enhancing α-mannosidase-like protein (EDEM), which is critical for ER-associated degradation (ERAD), as well as several other proteins involved in relieving ER stress, such as chaperones (40, 41). It also plays an important role in glucose and lipid metabolism, inflammatory pathways, collagen secretion, pro-insulin synthesis in β-cells, and several other processes (41–46).
2.1.1 Activation of yeast IRE1
The method of activation of IRE1 has undergone much debate, but three models have been proposed: a direct or indirect model and one in which it’s a synergistic combination. In 2000, it was shown that a protein called Kar2p (also known as GRP78 or BiP) is bound to the ER lumen region of IRE1, interacts and binds with unfolded proteins, and is released, causing the sequence of activating events for IRE1 in yeast (47), which led to the initial hypothesis that the UPR is via an indirect model in which BiP binding to unfolded proteins leads to its disassociation with the monomeric IRE1, thus activating the downstream IRE1 pathways. There was a challenge when Kimata et al. showed that BiP binding to IRE1 is not required to prevent constitutive activation of IRE1, demonstrating that BiP may play a role as an adjustor of the UPR (48). Crystallography analysis of IRE1 revealed the structure of the luminal domain (LD) as containing a deep groove similar to MHC, suggesting that activation may be dependent on that domain being occupied by unfolded proteins, leading to a proposed direct activation model (49). It was later hypothesized that BiP disassociation leads to monomeric IRE1 binding to unfolded proteins via the LD, thus inducing a conformational change, oligomerization, and activation. Once stress is resolved, BiP can rebind to IRE1, leading to deactivation via monomerization (50). Furthermore, studies showed that the LD of yeast IRE1 directly bound to unfolded proteins caused oligomerization and activation of the UPR functions (51).
2.1.2 Activation of mammalian IRE1
BiP was discovered at a similar time in mammalian cells as in yeast and hypothesized to directly repress IRE1 functions (52). The indirect model was proposed in which unfolded proteins sequester BiP away from IRE1, which allows the hydrophobic regions to dimerize and trans-autophosphorylation to occur (53). The activation of IRE1 was later determined to be reliant on at least four IRE1 monomers forming an oligomer (54), which was confirmed later when an IRE1α mutant was generated in mammalian cells with a low-affinity BiP binding site, showing that in the absence of BiP binding, IRE1α is activated under homeostatic conditions (55). Although it was proposed that BiP is tapered from the LD of IRE1 during ER stress (16), there was still the question of the sensitivity of the UPR. Importantly, even under low amounts of ER stress, the pathway can still be activated (49). One study introduced a new aspect of BiP that can explain this phenomenon. There is a non-canonical interaction between IRE1 LD and BiP’s ATPase domain that retains it in the monomeric state that is detached once unfolded protein binds to the substrate binding domain of BiP, indicating an allosteric regulatory role (56). This was further explored by the discovery of ER-localized J-protein 4 (ERdj4) and its role in drawing BiP towards dimerized IRE1 (57). BiP has a low affinity for the LD of IRE1. Thus, the complex is conserved through an ATP-mediated process that involves a release via nucleotide exchange, such as with a protein like GRP170, and then reunification with the help of a J-protein like ERdj4 (57, 58). ERdj4 also plays an active role in the suppression of IRE1. ERdj4 binds to IRE1 dimers via the C-terminal targeting domain and recruits BiP to break apart the dimers via ATP hydrolysis with its J domain, in which ERdj4 is then released (57). Karagöz et al. showed that mammalian IRE1 can be activated through unfolded proteins binding to the LD of IRE1 and hypothesize that BiP functions as a tuning mechanism. Importantly, they also show that BiP and IRE1 LD prefer different subsets of peptides. BiP prefers serine and threonine residues, and IRE1 LD prefers prolines and histidines. The group hypothesizes that unfolded protein binding in the LD leads to rearrangements that allow stabilization of the oligomeric form of IRE1, then allows trans-autophosphorylation and the activation of downstream functions (59). Work done in this field is still ongoing, and a clear consensus has not been reached yet. Considering the data, the true model seems to lie in the synergized combination. BiP appears to disassociate with IRE1 once unfolded proteins interact with it, which induces a conformational change in the LD of IRE1 that enables unfolded protein binding. This allows for the domains of IRE1 to interact and oligomerize, trans-autophosphorylate, and thus become active. When ER stress is resolved, IRE1 begins to disassociate from its oligomeric form, and ERdj4 binds to it, which then recruits BiP and leads to an ATP hydrolysis reaction that binds BiP to the LD of IRE1 and releases ERdj4, causing IRE1 to become monomeric and inactivated.
2.1.3 Alternative activation pathways
The phosphorylation of IRE1α is also vital, with three regions needing to be phosphorylated: the activation loop, the linker region, and the RNase domain (60). The activation loop phosphorylation is necessary for the enhancement of the splicing of XBP1 (60) and the locking of IRE1α into an active state for oligomerization (61). Alternatively, several non-canonical methods can activate IRE1. For example, membrane aberrancy (62) and changes in lipid composition (63) activate the IRE1 pathway. Also, IRE1 is sensitive to the thickness and the amount of lipid packing in the ER membrane through an amphipathic helix (64, 65). HSP47, a collagen chaperone, was discovered to bind to the IRE1α LD and reduce BiP binding, engaging the UPR (66), which provides a link to the UPR and collagen synthesis and trafficking, as TANGO1, a protein that assists in the secretion of collagen, is upregulated by XBP1s (44).
2.2 Non-canonical IRE1 functions
IRE1 has various physiological functions that primarily center around regulating cell death and re-establishing homeostasis after ER stress. One of the first functions described was the ability of IRE1 to regulate caspase-12 during ER stress. If ER stress reaches a critical level, an IRE1α-TRAF2-ASK1 complex can induce apoptosis through the JNK signaling pathway that causes caspase activation (67–69). Pro-apoptotic proteins Bax and Bak also directly interact with IRE1, wherein Bax and Bak appear to be necessary for the lessening of ER stress in pancreatic islet cells, and in a double knockout of these two proteins, IRE1 and XBP1s were both significantly upregulated (70). Sustained expression of IRE1α can also destabilize miRNA-17, which leads to increased thioredoxin-interacting protein (TXNIP) levels. When this occurs, the NLRP3 inflammasome is activated, leading to downstream cleavage of caspase-1, IL-1β secretion, and cell death (71, 72). An important discovery was made in Drosophila when it was discovered that, besides cleaving XBP1, the IRE1α endoribonuclease domain is also vital for cleaving and causing degradation of mRNAs for secretory proteins in a process called regulated IRE1-dependent decay (RIDD) (73). This process was later shown to be conserved in mammalian studies (74), with one study specifically showing that RIDD appears to be activated during more severe ER stress, whereas XBP1 splicing occurs during low-level stress (75). Although the consensus sequence was CUGCAG and a stem-loop structure for canonical cleavage targets like XBP1 and CD59 (76), it was unknown what this sequence was for RIDD targets. In 2021, Le Thomas et al. described a promiscuous form of RIDD, titled RIDDLE or RIDD lacking endomotif. RIDD requires dimerization of IRE1α and is shown by this group to recognize and cleave the CNGCAGN sequence and stem-loop structure, whereas RIDDLE requires a higher-order phospho-oligomerization of IRE1α and is promiscuous. Specifically, it can degrade non-optimal or non-existent stem-loop endomotifs and consensus RIDD substrates (77). The ability of IRE1 to perform RIDD is a direct effector function for it to relieve ER stress by lowering the amount of transcripts present. RIDD also has other unique roles, such as cleaving a group of miRNAs that repress the pro-apoptotic protein caspase-2 (78) and cleaving miRNAs that suppress angiopoietin, leading to better wound healing in a diabetic model (79). IRE1α is vital in the placenta; in a knockout model, mice died at the embryonic stage for reasons that appear to be linked to VEGF-A (80) and β-cell proinsulin folding (81). Related to the topic of β-cell proinsulin, it has also been reported that IRE1α splicing of XBP1 mRNA is required for accelerating the synthesis of proinsulin in response to elevated blood glucose levels (45). Acosta-Alvear later showed that IRE1 interacts with a signal recognition particle on the surface of ribosomes, arriving at the ER translocon in two unique ways: preemptive mode and surveillance mode. In the preemptive mode, IRE1 is able to degrade the mRNA before it enters the ER, and in the surveillance mode, it can degrade proteins that have folding issues as it is translated and pushed through the translocon (82). Targeting this pathway therapeutically has led to positive results in diseases such as cancers, renal failure, and diabetes (Table 1).
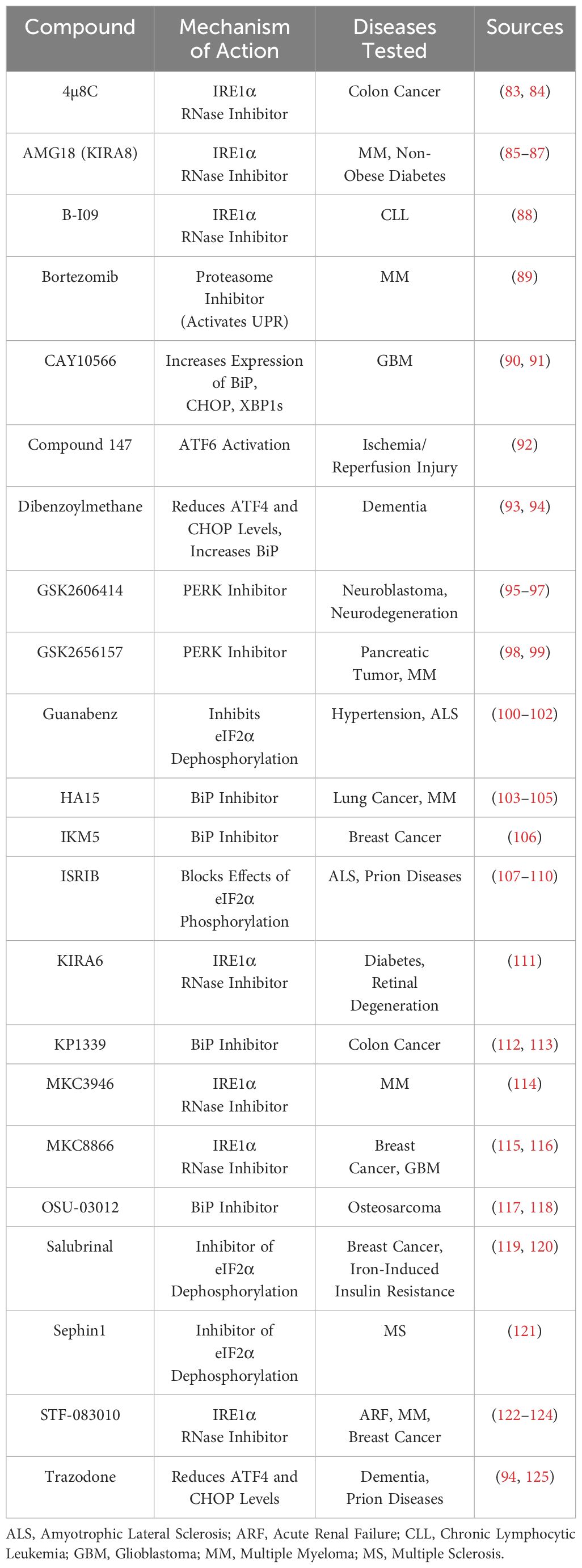
Table 1 Potential therapeutic compounds for the treatment of UPR-related disorders.
2.3 IRE1/XBP1s pathway in CD8+ T cells
Initially, using an IRE1-GFP reporter model, a previous study showed that IRE1 was upregulated during the double-positive thymic development stage yet only remained upregulated in CD8 single-positive T cells in both the thymus and spleen (126). However, the role of IRE1 in T cell development is still an area of active research, as the loss of both IRE1 and XBP1 in T cells doesn’t affect their development or frequency (127, 128). IRE1 may play a role in regulating TCR arrangement, although more research is needed in this area. Interestingly, one group showed that IL-2 can induce XBP1u transcripts, while TCR ligation initiates IRE1α activation to cleave XBP1u into XBP1s (129). In mice infected with ovalbumin-expressing Listeria monocytogenes (LM-OVA), both ERN1 (IRE1) and ATF6 mRNA were shown to increase 12 hours post-infection via a transcriptome analysis (130). It was later revealed that TCR ligation activates protein kinase C, which leads to the induction of the UPR, specifically BiP levels increasing. Interestingly, it was also shown that the phorbol 12-myristate 13-acetate (PMA) and ionomycin, common CD8+ T cell activating compounds in vitro, activate the UPR also due to protein kinase C activation (131). This indicates a regulatory system between calcium signaling, T cell activation, and the UPR that further research is required to understand fully. Similarly, store-operated calcium entry (SOCE) is a critical step for T cell activation (132) and is promoted via IRE1 and STIM1 interactions to increase ER and plasma membrane contact sites (133).
In both LM-OVA and LCMV models, XBP1s supports CD8+ T cell differentiation into end-stage effector cells via enhanced KLRG1 expression, although it is dispensable for the differentiation state (129). IRE1α inhibition in CD8+ T cells was also shown to result in immunosuppression due to lower effector functionality and memory commitment, thus the rejection of the transplant was lessened (134). While studying hereditary sensory and autonomic neuropathy type I (HSAN-I), one group found that serine palmitoyltransferase long chain base subunit 2 (SPTLC2) dysfunction results in increased mTORC1 activation and ER stress, specifically XBP1s and CHOP upregulation, thus resulting in CD8+ T cell apoptosis (135). Human CD8+ T cells isolated from ovarian cancer ascites also showed increased XBP1s expression compared to healthy donor cells, and the knockout of XBP1s in CD8+ T cells in a murine model showed higher perforin and IFN-γ production (136). Another example of the role of XBP1s in CD8+ T cells was revealed when a research group showed that increased cholesterol in the TME disrupts lipid metabolism, leading to ER stress, higher XBP1s and immune checkpoint expression, and fewer effector molecules (137). Recently, a group published that in a comparison between healthy donor and multiple myeloma CD8+ T cells, there is increased XBP1s that they show bind to and decrease SLC38A2, a gene encoding for the glutamine transporter SNAT2 (138). Within 24 hours of antigen activation in a murine model of LCMV in vitro (139), it was found that protein synthesis tripled in CD8+ T cells, indicating that there may be a need for a protein regulation system such as the UPR. An incomplete image of how IRE1 and XBP1s regulate CD8+ T cells is still present, and further research will need to focus on the mechanisms of how these molecules regulate CD8+ T cells. Currently, it appears that this pathway regulates CD8+ T cells via multiple mechanisms: managing protein burden, calcium signaling and activation, metabolism, and exhaustion. How, or if, these pathways intersect is unknown, although it is plausible that the activation of CD8+ T cells upregulates the UPR via calcium signaling, which results in the downregulation of metabolite transporters, like SNAT2, and thus T cell exhaustion (Figure 2A).
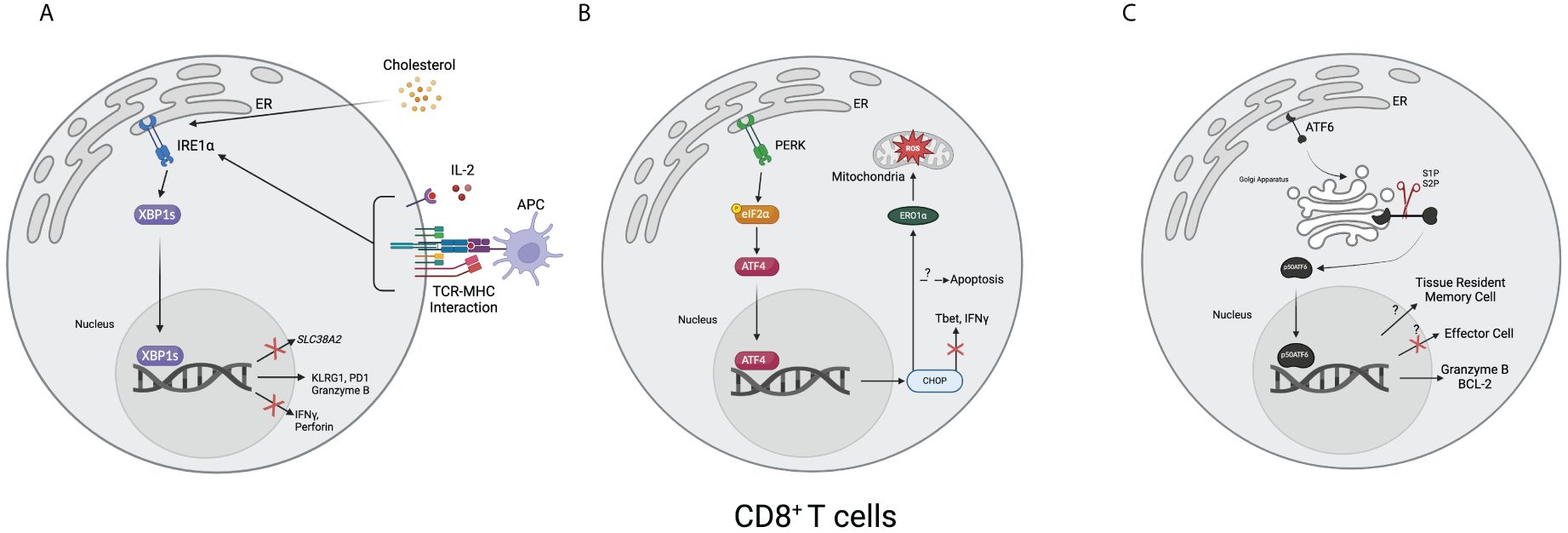
Figure 2 Overview of the unfolded protein response in CD8+ T cells. (A) T cell receptors and MHC combined with interleukin-2 result in the upregulation of XBP1s. Cholesterol, such as from the tumor microenvironment, can also lead to upregulating XBP1s. Once upregulated, XBP1s can increase the expression of KLRG1, PD1, and granzyme B and decrease the expression of IFNγ, perforin, and SLC38A2, a gene encoding for a glutamine transporter. (B) After activating, ATF4 translocates to the nucleus and upregulates CHOP, resulting in decreased Tbet and IFNγ. It can also result in apoptosis. Downstream, CHOP increases endoplasmic reticulum oxidoreductase 1α (ERO1α), leading to an accumulation of mitochondrial reactive oxygen species (ROS). (C) After the 50-kDa portion of activating transcription factor 6 (p50ATF6) translocates to the nucleus, it has hypothesized effects on enforcing tissue-resident memory cell formation and downregulating effector cell formation. It has also been shown to bind to and upregulate granzyme B and B-cell lymphoma 2 (BCL-2). (Created via BioRender).
3 The PERK pathway
3.1 Overview of the PERK pathway
PERK was identified as a type I transmembrane-ER-resident protein that is similar to IRE1. It contains an ER-stress-sensing luminal domain and a cytoplasmic region that is structurally similar to the eukaryotic initiation factor 2 (eIF2α) kinases (140). Once activated by ER stress, PERK autophosphorylates and begins its protein kinase activity by phosphorylating eIF2α on serine 51, which inhibits the translation of select mRNA (139, 140). Phosphorylation of Thr980 of PERK stabilizes the activation loop and the C-terminal helix αG, which was shown to be important for preparing it for binding to eIF2α (141, 142). The conformational change of PERK was also shown to be vital for increasing the binding affinity of the kinase loop to eIF2α (143). After phosphorylation, eIF2α was discovered to increase the translation of activating transcription factor 4 (ATF4), which induces the protein C/EBP-homologous protein (CHOP) (144). ATF4 plays an important role in promoting resistance to oxidative stress, mediating amino acid metabolism, and regulating angiogenesis (144–146). The activation of PERK was later discovered to be linked to BiP disassociating from it, in which, after disassociation, PERK will oligomerize, trans-autophosphorylate, and then activate similarly to IRE1 (52). It was also hypothesized that PERK can bind unfolded proteins in its luminal domain through an MHC-like groove, which will cause the molecules to line up and form dimers and oligomers (52, 147). PERK activation can result in the disassociation of Nrf2/Keap1 complexes, which leads to Nrf2 translocating to the nucleus and increasing cell survival pathways in an eIF2α phosphorylation-independent manner (148).
3.2 PERK functions
In an early experiment, PERK was knocked out in murine cells, and it was shown that there was a decrease in the inhibition of protein synthesis, phosphorylation of eIF2α, and cell survival when exposed to ER-stress-inducing agents (149). It has also been shown that cells can be caught in G1 arrest in the cell cycle when PERK is activated due to eIF2α phosphorylation and the loss of cyclin D1 (150). PERK plays an important role in ER-mitochondria contact sites and mitochondrial-associated ER membranes (MAMs) by transducing crosstalk that can induce reactive oxygen species cell death and regulate Ca2+ stores (151). Then, Muñoz et al. discovered that the key GTPase in the MAM site is mitofusin 2 (Mfn2), which regulates the transfer of Ca2+ between the ER and mitochondria. Mfn2 is a regulator of PERK that can keep PERK inactive under homeostatic conditions and can also detect cellular stress to coordinate the activation of PERK. They also showed that PERK was able to control the morphology and function of mitochondria, as well as oxidative stress (152). Although it was previously believed that Ca2+ perturbations can activate PERK (16, 151), the link was not fully clear until van Vliet et al. discovered that Filamin A was the protein that interacts with dimerized PERK once Ca2+ levels rise in the cytosol to drive actin remodeling. These events lead to an increase in stromal interaction molecule 1 (STIM1), which increases the contact sites between the ER and plasma membrane to refill the ER Ca2+ stock (153). Similar to IRE1, PERK has also been implicated in playing an important role in β-cell pathways; when PERK is knocked out in mice, it leads to Wolcott-Rallison syndrome, a severe infantile diabetes mellitus (154). By repressing the levels of the NF-κB inhibitor IκBα, eIF2α plays an important role in regulating the wide-ranging effects of NF-κB in the cells, although a physiological link has not yet been shown (155). This phenomenon has been shown to occur through ultraviolet light exposure as well (156). eIF2α phosphorylation is also necessary for β-cell insulin trafficking and upregulation of various transcription factors; without it, it can lead to β-cell dysfunction and diabetes mellitus (157, 158). Additionally, the PERK pathway has been implicated in the field of neurology, including improving cognitive memory (107) and controlling cortical neurogenesis (159).
3.3 PERK pathway regulatory mechanisms
While the binding of PERK with BiP is the initial regulatory mechanism in the pathway, there are several others as well. One of the first mechanisms discovered that regulated the PERK pathway was uncovered by Novoa et al. and revolved around a protein called growth arrest and DNA damage-inducible 34 (GADD34) associating with protein phosphatase 1 (PP1c), which in turn reduced the levels of eIF2α phosphorylation. This results in lowered amounts of ATF4 and CHOP, lessening the effects of the PERK pathway (160). The constitutive repressor of eIF2α phosphorylation (CReP) was later discovered and was also shown to associate with PP1c, leading to dephosphorylated eIF2α and a lessened stress response (161). One group discovered that, despite their low amino acid similarity, GADD34 and CReP have a common function: both are vital for regulating eIF2α phosphorylation and the loss of both results in embryonic lethality (162). P58IPK is upregulated by ER stress and can inhibit PERK signaling and phosphorylation, which will reduce the activation of the pathway, resulting in a negative feedback loop (163, 164). Protein disulfide isomerase A6 (PDIA6) attenuates the response of the UPR by inhibiting the luminal domain of both IRE1 and PERK (165). An ER-localized transmembrane protein called transducin beta-like 2 (TBL2) was discovered to associate with activated PERK and to positively regulate the downstream post-transcriptional processing of ATF4 (166). Related to the aforementioned role of PERK in calcium homeostasis, calcineurin (CN), a Ca2+ and calmodulin-dependent phosphatase, binds to PERK and can increase autophosphorylation and thus the activity of the UPR, resulting in CN playing the role of a positive regulator of ER stress (167). Rheb, a protein involved in the mTORC1 protein synthesis pathway, has also been shown to interact with PERK, thus increasing the efficiency of PERK phosphorylation onto eIF2α (168). Interestingly, nitric oxide was discovered to affect PERK via S-nitrosylation by Nakato et al. S-nitrosylation of PERK results in higher phosphorylation of eIF2α, thus increasing the pro-apoptotic pathways (169).
CHOP, the prominent effector protein expressed by the PERK pathway, was first discovered by a group looking for DNA-damage-inducible genes. Instead, they found only one that was upregulated during both heat shock stress and DNA damage as well as growth-attenuating conditions: growth arrest and DNA damage-inducible 153 (GADD-153) (170, 171). Previously only shown in hamster cells, the homologous protein was later identified in murine cells as C/EBP-homologous protein 10 (CHOP-10), a protein that forms heterodimers with C/EBP-like proteins that inhibit their binding to their respective DNA enhancer elements (172). It was then shown that this heterodimer complex can also be phosphorylated, as well as activate genes known as downstream of CHOP (DOCs) (173). The first link to the UPR with CHOP was uncovered when, after cells were exposed to stress-causing conditions, CHOP was upregulated quickly yet was attenuated by overexpression of BiP (174). ATF4 was then discovered as the necessary TF that upregulates CHOP during ER stress, linking the PERK pathway and CHOP (175). Interestingly, in a positive feedback loop, CHOP is also able to interact with ATF4, which leads to increased protein synthesis as well as oxidative stress and cell death (176). The primary role of CHOP, apoptosis (177), was originally shown to be due to CHOP decreasing the levels of B-cell lymphoma 2 (BCL-2), causing cells to die (178). This was later shown to be unlikely; rather, the BCL-2 family member protein called Bim is the increased pro-apoptotic protein due to transcriptional induction from CHOP (179). Death receptor 5 (DR5), a protein involved in caspase 8-mediated apoptosis, was discovered to be upregulated by CHOP during ER stress by Lu et al. In an interesting interplay between UPR pathways, IRE1 cleaves the DR5 mRNA, leading to no apoptosis, but if ER stress continues, IRE1 levels begin to wane while PERK and CHOP remain consistent, leading to enhanced DR5 expression and apoptosis (180). Marciniak et al. discovered that CHOP activates GADD34, resulting in a negative feedback loop for the PERK pathway. They were also able to show that CHOP can directly activate endoplasmic reticulum oxidoreductase 1α (ERO1α), leading to oxidizing conditions in the ER and thus apoptosis (181). Similar to PERK, CHOP is vital to controlling β-cell failure in diabetes; when knocked out in a murine model, the mice had higher glycemic control and lower oxidative damage (182). Inhibitors of the PERK pathway have been used for diseases such as neuroblastomas, multiple myeloma, and multiple sclerosis (Table 1).
3.4 PERK pathway in CD8+ T cells
CHOP was shown to be induced when GTPase of the immunity-associated nucleotide-binding protein 5 (Gimap5) was lost, resulting in apoptosis of rat CD8+ T cells (183). Cao et al. discovered that CHOP was upregulated in CD8+ T cells from tumor-bearing mice, which resulted in negative regulation of Tbet. Deletion of CHOP from CD8+ T cells significantly reduces tumor burden via increased IFN-γ and Tbet expression (184). Another study by Hurst et al. zoomed out from CHOP, instead using both a PERK knockout and a PERK inhibitor to show that tumor burden, mitochondrial exhaustion, and reactive oxygen species are all decreased. ERO1α, a protein upregulated by CHOP and involved in protein folding through oxidation reactions, was shown to be a large contributor to their phenotype via the accumulation of reactive oxygen species and subsequent damage to the mitochondria. They also used combination therapy with anti-PD1 and the PERK inhibitor, showing significantly increased tumor clearance and survival (185). KDEL receptor 1 (KDELR1) was identified as an associate of protein phosphatase 1 (PP1). Thus, without KDELR1, PP1 does not perform its phosphatase activity on eIF2α, resulting in increased CHOP in CD8+ T cells and apoptosis (186). Solanki et al. showed that ribosomal protein L22 (RPL22) resulted in significant ER stress if lost, leading to the induction of the arresting protein p53. In rescue experiments, they found that the knockdown of PERK specifically reduces p53 induction and rescues the T cell, thus showing a link between the cell cycle and the UPR (187). Cytoplasmic polyadenylation element-binding protein 4 (CPEB4) regulates the UPR in CD8+ T cells. When CPEB4 is deleted, there is weaker anti-tumor immunity and significantly increased CHOP expression (188). Stimulators of IFN genes (STING) regulate ER stress and T cell survival primarily through calcium homeostasis (189) and CHOP (190). While the knowledge of how the PERK pathway regulates CD8+ T cells is still incomplete, the data indicates that PERK upregulation, and subsequent CHOP upregulation, are both negative regulators of T cell immunity via oxidative stress and the downregulation of effector transcription factors and cytokines (Figure 2B).
4 The ATF6 pathway
4.1 Overview of the ATF6 pathway
ATF6 was initially discovered in 1998 as a basic leucine zipper (bZIP) TF that binds ER stress response elements (ERSE) to resolve ER stress (191). ATF6 binds to the consensus CCAAT-N9-CCACG sequence, specifically binding to the CCACG sequence when the CCAAT sequence is bound by the NF-Y TF upstream (192). The TF YY-1 (193) and TBP (194) were also shown to be factors that can increase the upregulation of ERSE transcription via interactions with ATF6. p90ATF6 exists as a transmembrane protein in the ER, but it is proteolyzed during ER stress, leading to activated p50ATF6 that is translocated to the nucleus to upregulate the ERSE, such as BiP (194, 195). While the activation mechanism was unknown for some time, it was discovered that BiP plays an inhibitory role, wherein it binds to Golgi localization sites on ATF6 and unbinds during ER stress, allowing it to translocate (196). Shortly after the initial discovery, ATF6’s cleavage was shown to be mediated by site 1 and site 2 proteases (S1P and S2P) (197). The Golgi apparatus was discovered to be the site of this cleavage, with the LZIP protein being an important player in translocating ATF6 after recognizing ER stress via the luminal domain, where it is then cleaved and shuttled to the nucleus (198). The mechanism of translocation from ER to the Golgi was later shown to be mediated by COPII vesicles once ATF6 is unbound from BiP (199). ATF6, similar to IRE1, was also shown to exist in two distinct forms: ATF6α and ATF6β (200). Although it was initially shown that knocking out both forms was not vital to cell survival (41), it was disproven in a murine model when knockout of the two isoforms was shown to be embryonic lethal. The same study also showed that ATF6a is the isoform for the induction of ERSE and upregulating proteins such as ER chaperones. It can also heterodimerize with XBP1s to upregulate ERAD components (201).
Temporally, ATF6 was shown to be activated quickly in response to ER stress, while the IRE1 and PERK pathways appear to be slightly delayed (40). Although not required (34), ATF6 upregulates XBP1 mRNA, leading to higher activity of IRE1α and its pathway (30). XBP1s and ATF6 have very similar effector functions, including upregulating genes involved in correcting folding, trafficking, and degradation of misfolded proteins (202). Also similar to XBP1s, when a BiP inhibitor called subtilase cytotoxin was used, NFκB activation was reduced, showing the interplay between the UPR and NFκB disorders (203). They share a common function in upregulating ER expansion, but ATF6 does it independently through upregulating phosphatidylcholine and phospholipid synthesis (204). When ATF6α KO cells are cultured in stress conditions, they similarly handle stress as WT cells, but they do not return to equilibrium as quickly, nor do they survive chronic stress, instead showing increased CHOP, apoptosis, and lowered ER chaperones (205). ATF6 has been shown to bind to other proteins, such as calnexin and protein disulfide isomerases, allowing for increases in protein folding and ER chaperone functionality (206, 207). When only the absence of ATF6α, once challenged with tunicamycin, mice began to show signs of liver dysfunction and steatosis, showing the importance of ATF6α in lipid metabolism (208). Hepatic gluconeogenesis was also found to be related to ATF6 via the negative regulation of CREB-regulated transcription coactivation 2 (CRTC2), an important regulator of the aforementioned pathway (209). Another group also showed that a single knockout of ATF6α can impair spermatogenesis via the TSSK4 gene in mice (210).
ATF6 has relations to Wolfram syndrome and cancer. Mutations in WFS1 lead to the protein form transporting ATF6α to proteasomes for degradation, thus causing an aberrant UPR pathway in β cells (211). In regards to cancer, ATF6 was shown to promote proliferation and migration through MAPK signaling in cervical cancer (212). In colon cancer cells, ATF6 was found to activate mTOR, leading to sustained HSP90 expression and the prevention of apoptosis (213). Similarly, in colorectal cancer, GREM1, a protein involved in the epithelial-mesenchymal transition, was discovered to upregulate ATF6, resulting in metastasis promotion, linking this protein to the mTOR pathway (214). While therapeutically targeting ATF6 is not currently a large field, success has been seen in ischemia-reperfusion injuries (Table 1).
4.2 Alternative activation pathways of ATF6
ATF6 also has a non-canonical method of activation. During times of ER Ca2+ depletion, p90ATF6 can become partially glycosylated, ATF6(f), which reduces its binding with calreticulin, leading to increased export to the Golgi and under-glycosylated ATF6 becoming a sensing mechanism for ER stress (215). Sphingolipids, such as dihydrosphingosine and dihydroceramide, can also activate ATF6 in a non-canonical way, although they result in normal effector functions of ATF6 (216). In its inactive form in unstressed cells, ATF6 was shown to exist as monomers, dimers, and oligomers via disulfide bridges between the cysteine regions in the luminal domain, with only the monomeric version reaching the Golgi during stress (217). ERp18, an ER oxidoreductase, was shown to be a regulator of ATF6 release from the ER by associating with it and ensuring the monomeric form is released to the Golgi (218). To return to homeostasis, ATF6 can be negatively regulated by NUCB1, a Golgi-localized protein that is upregulated in response to ER stress (219). ATF4 has also been shown to have a regulatory role on ATF6; it can upregulate ATF6, as well as genes vital for the COPII vesicles, and when knocked out, it lessens ATF6 transport (220).
4.3 ATF6 pathway in CD8+ T cells
ATF6 is still relatively understudied for its impacts on CD8+ T cells. One group found that in a constitutive TNF-expressing model used for representing ileitis, ATF6, ATF4, and XBP1s can bind to the promoter regions of Granzyme B and BCL-2, an anti-apoptotic protein (221). In addition, using an in vivo CRISPR screen, ATF6 appears to be a restrictive element of T effector cell development, although further validation studies are needed (222). Similarly, a transcriptome study of tissue-resident memory chimeric antigen receptor T cells (CAR-TRM) uncovered that both ATF6 and ATF4 are upregulated, perhaps indicating that these two molecules may play a role in memory differentiation (223) (Figure 2C).
5 Conclusion
While recent work has highlighted the deeper intricacies of the UPR, more questions remain, particularly regarding the roles of each arm. For example, how does the UPR regulate the activation of each arm individually? There is also a lack of work regarding the exact function of ATF6. New technologies are consistently being refined that will allow for further exploration of these topics. As for CD8+ T cells, while there has been an effort to study the effects the UPR has on them, the exact mechanism by which they are regulated by the UPR is still unknown. The UPR is a broad system that encompasses three arms and several effector molecules. Research investigating the functionality of each molecule in each arm will be the goal, although this will take time. This leaves a broad space for researchers to investigate the UPR in CD8+ T cells due to several understudied aspects. For example, an outstanding question currently is what are the mechanisms that activate the UPR in CD8+ T cells. Some studies have uncovered those, but many are still needed to determine the exact effects of this activation and why it occurs. One study has shown in CD4+ T cells that XBP1 affects the abundance of glutamine transporters (136), although evidence in CD8+ T cells has been shown only by RNA-level changes (138). Further research is needed to validate the phenomenon and its effects on CD8+ T cells, albeit this evidence, as well as the evidence presented by the effects of PERK, indicate that metabolism, the UPR, and CD8+ T cells appear to be intricately linked. Work completed towards understanding the UPR has the potential to uncover novel ways to manipulate CD8+ T cells by using the UPR to increase their efficacy in the diseased setting.
Author contributions
KN: Conceptualization, Writing – original draft, Writing – review & editing. BL: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by NIH NCI (CA193939) and Ohio State University Comprehensive Cancer Center research fund.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci CMLS. (2016) 73:79–94. doi: 10.1007/s00018-015-2052-6
2. Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. Mechanisms determining the morphology of the peripheral ER. Cell. (2010) 143:774–88. doi: 10.1016/j.cell.2010.11.007
3. Shibata Y, Voeltz GK, Rapoport TA. Rough sheets and smooth tubules. Cell. (2006) 126:435–9. doi: 10.1016/j.cell.2006.07.019
4. Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein. J Cell Biol. (1981) 91:551–6. doi: 10.1083/jcb.91.2.551
5. Walter P, Ibrahimi I, Blobel G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J Cell Biol. (1981) 91:545–50. doi: 10.1083/jcb.91.2.545
6. Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. (2007) 450:663–9. doi: 10.1038/nature06384
7. Karamyshev AL, Tikhonova EB, Karamysheva ZN. Translational control of secretory proteins in health and disease. Int J Mol Sci. (2020) 21:2538. doi: 10.3390/ijms21072538
8. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. (2008) 319:916–9. doi: 10.1126/science.1141448
10. Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduct Target Ther. (2023) 8:352. doi: 10.1038/s41392-023-01570-w
11. Read A, Schröder M. The unfolded protein response: an overview. Biology. (2021) 10:384. doi: 10.3390/biology10050384
12. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci. (2019) 6:11. doi: 10.3389/fmolb.2019.00011
13. Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. (1988) 332:462–4. doi: 10.1038/332462a0
14. Stevens FJ, Argon Y. Protein folding in the ER. Semin Cell Dev Biol. (1999) 10:443–54. doi: 10.1006/scdb.1999.0315
15. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
16. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
17. Zhang N, Bevan MJ. CD8+ T cells: foot soldiers of the immune system. Immunity. (2011) 35:161–8. doi: 10.1016/j.immuni.2011.07.010
18. Giles JR, Globig AM, Kaech SM, Wherry EJ. CD8+ T cells in the cancer-immunity cycle. Immunity. (2023) 56:2231–53. doi: 10.1016/j.immuni.2023.09.005
19. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. (2019) 234:8509–21. doi: 10.1002/jcp.27782
20. Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. CD8+ T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity. (2023) 56:107–124.e5. doi: 10.1016/j.immuni.2022.12.002
21. Nikawa JI, Yamashita S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol. (1992) 6:1441–6. doi: 10.1111/j.1365-2958.1992.tb00864.x
22. Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. (1993) 73:1197–206. doi: 10.1016/0092-8674(93)90648-A
23. Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a cdc2+CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. (1993) 74:743–56. doi: 10.1016/0092-8674(93)90521-Q
24. Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. (1996) 15:3028–39. doi: 10.1002/embj.1996.15.issue-12
25. Welihinda AA, Kaufman RJ. The unfolded protein response pathway in. J Biol Chem. (1996) 271:18181–7. doi: 10.1074/jbc.271.30.18181
26. Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. (1998) 12:1812–24. doi: 10.1101/gad.12.12.1812
27. Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. (2000) 14:2725–36. doi: 10.1101/gad.839400
28. Iwawaki T, Hosoda A, Okuda T, Kamigori Y, Nomura-Furuwatari C, Kimata Y, et al. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat Cell Biol. (2001) 3:158–64. doi: 10.1038/35055065
29. Imagawa Y, Hosoda A, Tsuru A, Kohno K. RNase domains determine the functional difference between IRE1α and IRE1β. FEBS Lett. (2008) 582:656–60. doi: 10.1016/j.febslet.2008.01.038
30. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. (2001) 107:881–91. doi: 10.1016/S0092-8674(01)00611-0
31. Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans Dev Cell. (2001) 107:893–903. doi: 10.1016/S0092-8674(01)00612-2
32. Clauss IM, Chu M, Zhao JL, Glimcher LH. The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res. (1996) 24:1855–64. doi: 10.1093/nar/24.10.1855
33. Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem. (2001) 276:9199–205. doi: 10.1074/jbc.M010486200
34. Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. (2002) 16:452–66. doi: 10.1101/gad.964702
35. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. (2002) 415:92–6. doi: 10.1038/415092a
36. Lu Y, Liang FX, Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. (2014) 55:758–70. doi: 10.1016/j.molcel.2014.06.032
37. Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. (2014) 33:2922–36. doi: 10.15252/embj.201490332
38. Papaioannou A, Centonze F, Metais A, Maurel M, Negroni L, Gonzalez-Quiroz M, et al. Stress-induced tyrosine phosphorylation of RtcB modulates IRE1 activity and signaling outputs. Life Sci Alliance. (2022) 5:e202201379. doi: 10.26508/lsa.202201379
39. Mahdizadeh SJ, Stier M, Carlesso A, Lamy A, Thomas M, Eriksson LA. Multiscale in silico study of the mechanism of activation of the rtcB ligase by the PTP1B phosphatase. J Chem Inf Model. (2024) 64:1054. doi: 10.1021/acs.jcim.3c01600
40. Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. (2003) 4:265–71. doi: 10.1016/S1534-5807(03)00022-4
41. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. (2003) 23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003
42. Piperi C, Adamopoulos C, Papavassiliou AG. XBP1: A pivotal transcriptional regulator of glucose and lipid metabolism. Trends Endocrinol Metab. (2016) 27:119–22. doi: 10.1016/j.tem.2016.01.001
43. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. (2016) 16:469–84. doi: 10.1038/nri.2016.62
44. Maiers JL, Kostallari E, Mushref M, deAssuncao TM, Li H, Jalan-Sakrikar N, et al. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology. (2017) 65:983–98. doi: 10.1002/hep.28921
45. Hassler JR, Scheuner DL, Wang S, Han J, Kodali VK, Li P, et al. The IRE1α/XBP1s pathway is essential for the glucose response and protection of β Cells. PloS Biol. (2015) 13:e1002277. doi: 10.1371/journal.pbio.1002277
46. Park SM, Kang TI, So JS. Roles of XBP1s in transcriptional regulation of target genes. Biomedicines. (2021) 9:791. doi: 10.3390/biomedicines9070791
47. Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. (2000) 279:445–50. doi: 10.1006/bbrc.2000.3987
48. Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J Cell Biol. (2004) 167:445–56. doi: 10.1083/jcb.200405153
49. Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci. (2005) 102:18773–84. doi: 10.1073/pnas.0509487102
50. Pincus D, Chevalier MW, Aragón T, van AE, SE V, El-Samad H, et al. BiP binding to the ER-stress sensor ire1 tunes the homeostatic behavior of the unfolded protein response. PloS Biol. (2010) 8:e1000415. doi: 10.1371/journal.pbio.1000415
51. Gardner BM, Walter P. Unfolded proteins are ire1-activating ligands that directly induce the unfolded protein response. Science. (2011) 333:1891–4. doi: 10.1126/science.1209126
52. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. (2000) 2:326–32. doi: 10.1038/35014014
53. Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, et al. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci. (2006) 103:14343–8. doi: 10.1073/pnas.0606480103
54. Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci. (2010) 107:16113–8. doi: 10.1073/pnas.1010580107
55. Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. (2009) 315:2496–504. doi: 10.1016/j.yexcr.2009.06.009
56. Carrara M, Prischi F, Nowak PR, Kopp MC, Ali MM. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. eLife. (2015) 4:e03522. doi: 10.7554/eLife.03522
57. Amin-Wetzel N, Saunders RA, Kamphuis MJ, Rato C, Preissler S, Harding HP, et al. A J-protein co-chaperone recruits biP to monomerize IRE1 and repress the unfolded protein response. Cell. (2017) 171:1625–1637.e13. doi: 10.1016/j.cell.2017.10.040
58. Amin-Wetzel N, Neidhardt L, Yan Y, Mayer MP, Ron D. Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. eLife. (2019) 8:e50793. doi: 10.7554/eLife.50793.sa2
59. Karagöz GE, Acosta-Alvear D, Nguyen HT, Lee CP, Chu F, Walter P. An unfolded protein-induced conformational switch activates mammalian IRE1. eLife. (2017) 6:e30700. doi: 10.7554/eLife.30700
60. Prischi F, Nowak PR, Carrara M, Ali MMU. Phosphoregulation of Ire1 RNase splicing activity. Nat Commun. (2014) 5:3554. doi: 10.1038/ncomms4554
61. Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. (2009) 457:687–93. doi: 10.1038/nature07661
62. Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. (2011) 22:3520–32. doi: 10.1091/mbc.e11-04-0295
63. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci. (2013) 110:4628–33. doi: 10.1073/pnas.1217611110
64. Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hänelt I, et al. Activation of the unfolded protein response by lipid bilayer stress. Mol Cell. (2017) 67:673–684.e8. doi: 10.1016/j.molcel.2017.06.012
65. Kono N, Amin-Wetzel N, Ron D. Generic membrane-spanning features endow IRE1α with responsiveness to membrane aberrancy. Gilmore R editor Mol Biol Cell. (2017) 28:2318–32. doi: 10.1091/mbc.e17-03-0144
66. Sepulveda D, Rojas-Rivera D, Rodríguez DA, Groenendyk J, Köhler A, Lebeaupin C, et al. Interactome screening identifies the ER luminal chaperone hsp47 as a regulator of the unfolded protein response transducer IRE1α. Mol Cell. (2018) 69:238–252.e7. doi: 10.1016/j.molcel.2017.12.028
67. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. (2000) 287:664–6. doi: 10.1126/science.287.5453.664
68. Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. (2001) 276:13935–40. doi: 10.1074/jbc.M010677200
69. Zeng T, Peng L, Chao H, Xi H, Fu B, Wang Y, et al. IRE1α-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem Biophys Res Commun. (2015) 460:530–6. doi: 10.1016/j.bbrc.2015.03.064
70. White SA, Zhang LS, Pasula DJ, Yang YHC, Luciani DS. Bax and Bak jointly control survival and dampen the early unfolded protein response in pancreatic β-cells under glucolipotoxic stress. Sci Rep. (2020) 10:10986. doi: 10.1038/s41598-020-67755-3
71. Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1α Induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. (2012) 16:250–64. doi: 10.1016/j.cmet.2012.07.007
72. Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β Cell death through initiation of the inflammasome. Cell Metab. (2012) 16:265–73. doi: 10.1016/j.cmet.2012.07.005
73. Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. (2006) 313:104–7. doi: 10.1126/science.1129631
74. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. (2009) 186:323–31. doi: 10.1083/jcb.200903014
75. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1α Kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. (2009) 138:562–75. doi: 10.1016/j.cell.2009.07.017
76. Oikawa D, Tokuda M, Hosoda A, Iwawaki T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. (2010) 38:6265–73. doi: 10.1093/nar/gkq452
77. Le Thomas A, Ferri E, Marsters S, Harnoss JM, Lawrence DA, Zuazo-Gaztelu I, et al. Decoding non-canonical mRNA decay by the endoplasmic-reticulum stress sensor IRE1α. Nat Commun. (2021) 12:7310. doi: 10.1038/s41467-021-27597-7
78. Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. (2012) 338:818–22. doi: 10.1126/science.1226191
79. Wang JM, Qiu Y, Yang ZQ, Li L, Zhang K. Inositol-requiring enzyme 1 facilitates diabetic wound healing through modulating microRNAs. Diabetes. (2017) 66:177–92. doi: 10.2337/db16-0052
80. Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci. (2009) 106:16657–62. doi: 10.1073/pnas.0903775106
81. Tsuchiya Y, Saito M, Kadokura H, Miyazaki Ji, Tashiro F, Imagawa Y, et al. IRE1–XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J Cell Biol. (2018) 217:1287–301. doi: 10.1083/jcb.201707143
82. Acosta-Alvear D, Karagöz GE, Fröhlich F, Li H, Walther TC, Walter P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. eLife. (2018) 7:e43036. doi: 10.7554/eLife.43036.025
83. Stewart C, Estrada A, Kim P, Wang D, Wei Y, Gentile C, et al. Regulation of IRE1α by the small molecule inhibitor 4μ8c in hepatoma cells. Endoplasmic Reticulum Stress Dis. (2017) 4:1–10. doi: 10.1515/ersc-2017-0001
84. Cross BCS, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci. (2012) 109:869–78. doi: 10.1073/pnas.1115623109
85. Yamashita Y, Morita S, Hosoi H, Kobata H, Kishimoto S, Ishibashi T, et al. Targeting adaptive IRE1α Signaling and PLK2 in multiple myeloma: possible anti-tumor mechanisms of KIRA8 and nilotinib. Int J Mol Sci. (2020) 21:6314. doi: 10.3390/ijms21176314
86. Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, et al. Unfolded protein response in cancer: IRE1α Inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med Chem Lett. (2015) 6:68–72. doi: 10.1021/ml500315b
87. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1α Signaling spares ER-stressed pancreatic β Cells to reverse autoimmune diabetes. Cell Metab. (2017) 25:883–897.e8. doi: 10.1016/j.cmet.2017.03.018
88. Tang CHA, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, et al. Inhibition of ER stress–associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. (2014) 124:2585–98. doi: 10.1172/JCI73448
89. Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. (2006) 107:4907–16. doi: 10.1182/blood-2005-08-3531
90. Oatman N, Dasgupta N, Arora P, Choi K, Gawali MV, Gupta N, et al. Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci Adv. (2021) 7:eabd7459. doi: 10.1126/sciadv.abd7459
91. Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, et al. Stearoyl coA desaturase is essential for regulation of endoplasmic reticulum homeostasis and tumor growth in glioblastoma cancer stem cells. Stem Cell Rep. (2019) 12:712–27. doi: 10.1016/j.stemcr.2019.02.012
92. Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. (2019) 10:187. doi: 10.1038/s41467-018-08129-2
93. Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain. (2017) 140:1768–83. doi: 10.1093/brain/awx074
94. Frazier MC, Jackson KM, Jankowska-Stephens E, Anderson MG, Harris WB. Proteomic analysis of proteins altered by dibenzoylmethane in human prostatic cancer LNCaP cells. PROTEOMICS. (2004) 4:2814–21. doi: 10.1002/pmic.200400834
95. Gundu C, Arruri VK, Sherkhane B, Khatri DK, Singh SB. GSK2606414 attenuates PERK/p-eIF2α/ATF4/CHOP axis and augments mitochondrial function to mitigate high glucose induced neurotoxicity in N2A cells. Curr Res Pharmacol Drug Discovery. (2022) 3:100087. doi: 10.1016/j.crphar.2022.100087
96. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. (2013) 5:206ra138. doi: 10.1126/scitranslmed.3006767
97. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1 H -indol-5-yl)-7 H -pyrrolo[2,3- d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. (2012) 55:7193–207. doi: 10.1021/jm300713s
98. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. (2013) 73:1993–2002. doi: 10.1158/0008-5472.CAN-12-3109
99. Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, et al. Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med Chem Lett. (2013) 4:964–8. doi: 10.1021/ml400228e
100. Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. (2014) 71:317–24. doi: 10.1016/j.nbd.2014.08.010
101. Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. (2011) 332:91–4. doi: 10.1126/science.1201396
102. Meacham RH, Ruelius HW, Kick CJ, Peters JR, Kocmund SM, Sisenwine SF, et al. Relationship of guanabenz concentrations in brain and plasma to antihypertensive effect in the spontaneously hypertensive rat. J Pharmacol Exp Ther. (1980) 214:594–8.
103. Wu J, Wu Y, Lian X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol Open. (2020) 9:bio.053298. doi: 10.1242/bio.053298
104. Cerezo M, Rocchi S. New anti-cancer molecules targeting HSPA5/BIP to induce endoplasmic reticulum stress, autophagy and apoptosis. Autophagy. (2017) 13:216–7. doi: 10.1080/15548627.2016.1246107
105. Chen Y, Tao Y, Hu K, Lu J. GRP78 inhibitor HA15 increases the effect of Bortezomib on eradicating multiple myeloma cells through triggering endoplasmic reticulum stress. Heliyon. (2023) 9:e19806. doi: 10.1016/j.heliyon.2023.e19806
106. Nayak D, Katoch A, Sharma D, Faheem M, Chakraborty S, Sahu PK, et al. Indolylkojyl methane analogue IKM5 potentially inhibits invasion of breast cancer cells via attenuation of GRP78. Breast Cancer Res Treat. (2019) 177:307–23. doi: 10.1007/s10549-019-05301-0
107. Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife. (2013) 2:e00498. doi: 10.7554/eLife.00498
108. Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife. (2015) 4:e05033. doi: 10.7554/eLife.05033.016
109. Halliday M, Radford H, Sekine Y, Moreno J, Verity N, Le Quesne J, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. (2015) 6:e1672–2. doi: 10.1038/cddis.2015.49
110. Bugallo R, Marlin E, Baltanás A, Toledo E, Ferrero R, Vinueza-Gavilanes R, et al. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. (2020) 11:397. doi: 10.1038/s41419-020-2601-2
111. Ghosh R, Wang L, Wang ES, Perera BGK, Igbaria A, Morita S, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. (2014) 158:534–48. doi: 10.1016/j.cell.2014.07.002
112. Heffeter P, Böck K, Atil B, Reza Hoda MA, Körner W, Bartel C, et al. Intracellular protein binding patterns of the anticancer ruthenium drugs KP1019 and KP1339. JBIC J Biol Inorg Chem. (2010) 15:737–48. doi: 10.1007/s00775-010-0642-1
113. Wernitznig D, Kiakos K, Del Favero G, Harrer N, Machat H, Osswald A, et al. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics. (2019) 11:1044–8. doi: 10.1039/c9mt00051h
114. Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood. (2012) 119:5772–81. doi: 10.1182/blood-2011-07-366633
115. Logue SE, McGrath EP, Cleary P, Greene S, Mnich K, Almanza A, et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat Commun. (2018) 9:3267. doi: 10.1038/s41467-018-05763-8
116. Le Reste PJ, Pineau R, Voutetakis K, Samal J, Jégou G, Lhomond S, et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy in vivo. Cancer Lett. (2020) 494:73–83. doi: 10.1016/j.canlet.2020.08.028
117. Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. (2004) 64:4309–18. doi: 10.1158/0008-5472.CAN-03-4063
118. Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, et al. OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol. (2008) 73:1168–84. doi: 10.1124/mol.107.042697
119. Alsterda A, Asha K, Powrozek O, Repak M, Goswami S, Dunn AM, et al. Salubrinal exposes anticancer properties in inflammatory breast cancer cells by manipulating the endoplasmic reticulum stress pathway. Front Oncol. (2021) 11:654940. doi: 10.3389/fonc.2021.654940
120. Nguyen K, Tang J, Cho S, Ying F, Sung HK, Jahng JW, et al. Salubrinal promotes phospho-eIF2α-dependent activation of UPR leading to autophagy-mediated attenuation of iron-induced insulin resistance. Mol Metab. (2024) 83:101921. doi: 10.1016/j.molmet.2024.101921
121. Chen Y, Podojil JR, Kunjamma RB, Jones J, Weiner M, Lin W, et al. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain. (2019) 142:344–61. doi: 10.1093/brain/awy322
122. Liu L, Xu L, Zhang S, Wang D, Dong G, Chen H, et al. STF-083010, an inhibitor of XBP1 splicing, attenuates acute renal failure in rats by suppressing endoplasmic reticulum stress-induced apoptosis and inflammation. Exp Anim. (2018) 67:373–82. doi: 10.1538/expanim.17-0131
123. Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. (2011) 117:1311–4. doi: 10.1182/blood-2010-08-303099
124. Ming J, Ruan S, Wang M, Ye D, Fan N, Meng Q, et al. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget. (2015) 6:40692–703. doi: 10.18632/oncotarget.v6i38
125. Albert-Gasco H, Smith HL, Alvarez-Castelao B, Swinden D, Halliday M, Janaki-Raman S, et al. Trazodone rescues dysregulated synaptic and mitochondrial nascent proteomes in prion neurodegeneration. Brain. (2024) 147:649–64. doi: 10.1093/brain/awad313
126. Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, et al. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem. (2008) 283:17954–61. doi: 10.1074/jbc.M801395200
127. Kemp KL, Lin Z, Zhao F, Gao B, Song J, Zhang K, et al. The serine-threonine kinase inositol-requiring enzyme 1α (IRE1α) promotes IL-4 production in T helper cells. J Biol Chem. (2013) 288:33272–82. doi: 10.1074/jbc.M113.493171
128. Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. (2015) 16:829–37. doi: 10.1038/ni.3225
129. Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol Baltim Md 1950. (2008) 181:5433–41. doi: 10.4049/jimmunol.181.8.5433
130. Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, et al. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol. (2013) 14:404–12. doi: 10.1038/ni.2536
131. Pino SC, O’Sullivan-Murphy B, Lidstone EA, Thornley TB, Jurczyk A, Urano F, et al. Protein kinase C signaling during T cell activation induces the endoplasmic reticulum stress response. Cell Stress Chaperones. (2008) 13:421–34. doi: 10.1007/s12192-008-0038-0
132. Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. (2010) 28:491–533. doi: 10.1146/annurev.immunol.021908.132550
133. Carreras-Sureda A, Zhang X, Laubry L, Brunetti J, Koenig S, Wang X, et al. The ER stress sensor IRE1 interacts with STIM1 to promote store-operated calcium entry, T cell activation, and muscular differentiation. Cell Rep. (2023) 42:113540. doi: 10.1016/j.celrep.2023.113540
134. Shi Y, Lu Y, Zhu C, Luo Z, Li X, Liu Y, et al. Targeted regulation of lymphocytic ER stress response with an overall immunosuppression to alleviate allograft rejection. Biomaterials. (2021) 272:120757. doi: 10.1016/j.biomaterials.2021.120757
135. Wu J, Ma S, Sandhoff R, Ming Y, Hotz-Wagenblatt A, Timmerman V, et al. Loss of neurological disease HSAN-I-associated gene SPTLC2 impairs CD8+ T cell responses to infection by inhibiting T cell metabolic fitness. Immunity. (2019) 50:1218–1231.e5. doi: 10.1016/j.immuni.2019.03.005
136. Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. (2018) 562:423–8. doi: 10.1038/s41586-018-0597-x
137. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002
138. Wan Y, Chen M, Li X, Han X, Zhong L, Xiao F, et al. Single-cell RNA sequencing reveals XBP1-SLC38A2 axis as a metabolic regulator in cytotoxic T lymphocytes in multiple myeloma. Cancer Lett. (2023) 562:216171. doi: 10.1016/j.canlet.2023.216171
139. Howden AJM, Hukelmann JL, Brenes A, Spinelli L, Sinclair LV, Lamond AI, et al. Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat Immunol. (2019) 20:1542–54. doi: 10.1038/s41590-019-0495-x
140. Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol. (1998) 18:7499–509. doi: 10.1128/MCB.18.12.7499
141. Cui W, Li J, Ron D, Sha B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr D Biol Crystallogr. (2011) 67:423–8. doi: 10.1107/S0907444911006445
142. Wang P, Li J, Tao J, Sha B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J Biol Chem. (2018) 293:4110–21. doi: 10.1074/jbc.RA117.001294
143. Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol. (2006) 172:201–9. doi: 10.1083/jcb.200508099
144. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. (2000) 6:1099–108. doi: 10.1016/S1097-2765(00)00108-8
145. Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. (2008) 40:14–21. doi: 10.1016/j.biocel.2007.01.020
146. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. (2003) 11:619–33. doi: 10.1016/S1097-2765(03)00105-9
147. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. (1999) 397:271–4. doi: 10.1038/16729
148. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. (2003) 23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003
149. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. (2000) 5:897–904. doi: 10.1016/S1097-2765(00)80330-5
150. Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. (2000) 97:12625–30. doi: 10.1073/pnas.220247197
151. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. (2012) 19:1880–91. doi: 10.1038/cdd.2012.74
152. Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. (2013) 32:2348–61. doi: 10.1038/emboj.2013.168
153. Van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell. (2017) 65:885–899.e6. doi: 10.1016/j.molcel.2017.01.020
154. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk–/– mice reveals a role for translational control in secretory cell survival. Mol Cell. (2001) 7:1153–63. doi: 10.1016/S1097-2765(01)00264-7
155. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. (2004) 24:10161–8. doi: 10.1128/MCB.24.23.10161-10168.2004
156. Wu S, Tan M, Hu Y, Wang JL, Scheuner D, Kaufman RJ. Ultraviolet light activates NFκB through translational inhibition of IκBα Synthesis. J Biol Chem. (2004) 279:34898–902. doi: 10.1074/jbc.M405616200
157. Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. (2009) 10:13–26. doi: 10.1016/j.cmet.2009.06.002
158. Scheuner D, Mierde DV, Song B, Flamez D, Creemers JWM, Tsukamoto K, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. (2005) 11:757–64. doi: 10.1038/nm1259
159. Laguesse S, Creppe C, Nedialkova DD, Prévot PP, Borgs L, Huysseune S, et al. A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev Cell. (2015) 35:553–67. doi: 10.1016/j.devcel.2015.11.005
160. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. (2001) 153:1011–22. doi: 10.1083/jcb.153.5.1011
161. Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, et al. Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. (2003) 163:767–75. doi: 10.1083/jcb.200308075
162. Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2α) dephosphorylation in mammalian development. Proc Natl Acad Sci. (2009) 106:1832–7. doi: 10.1073/pnas.0809632106
163. Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, et al. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58 IPK. Proc Natl Acad Sci. (2002) 99:15920–5. doi: 10.1073/pnas.252341799
164. Van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α Signaling. J Biol Chem. (2003) 278:15558–64. doi: 10.1074/jbc.M212074200
165. Eletto D, Eletto D, Dersh D, Gidalevitz T, Argon Y. Protein disulfide isomerase A6 controls the decay of IRE1α Signaling via disulfide-dependent association. Mol Cell. (2014) 53:562–76. doi: 10.1016/j.molcel.2014.01.004
166. Tsukumo Y, Tsukahara S, Furuno A, Iemura Si, Natsume T, Tomida A. TBL2 is a novel PERK-binding protein that modulates stress-signaling and cell survival during endoplasmic reticulum stress. PloS One. (2014) 9:e112761. doi: 10.1371/journal.pone.0112761
167. Bollo M, Paredes RM, Holstein D, Zheleznova N, Camacho P, Lechleiter JD. Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PloS One. (2010) 5:e11925. doi: 10.1371/journal.pone.0011925
168. Tyagi R, Shahani N, Gorgen L, Ferretti M, Pryor W, Chen PY, et al. Rheb inhibits protein synthesis by activating the PERK-eIF2α Signaling cascade. Cell Rep. (2015) 10:684–93. doi: 10.1016/j.celrep.2015.01.014
169. Nakato R, Ohkubo Y, Konishi A, Shibata M, Kaneko Y, Iwawaki T, et al. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci Rep. (2015) 5:14812. doi: 10.1038/srep14812
170. Fornace AJ, Alamo I, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci. (1988) 85:8800–4. doi: 10.1073/pnas.85.23.8800
171. Fornace AJ, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, et al. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol Cell Biol. (1989) 9:4196–203. doi: 10.1128/mcb.9.10.4196-4203.1989
172. Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. (1992) 6:439–53. doi: 10.1101/gad.6.3.439
173. Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, et al. Identification of novel stress-induced genes downstream of chop. EMBO J. (1998) 17:3619–30. doi: 10.1093/emboj/17.13.3619
174. Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol. (1996) 16:4273–80. doi: 10.1128/MCB.16.8.4273
175. Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. (1999) 339:135–41. doi: 10.1042/bj3390135
176. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. (2013) 15:481–90. doi: 10.1038/ncb2738
177. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. (1998) 12:982–95. doi: 10.1101/gad.12.7.982
178. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating bcl2 and perturbing the cellular redox state. Mol Cell Biol. (2001) 21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001
179. Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein bim. Cell. (2007) 129:1337–49. doi: 10.1016/j.cell.2007.04.027
180. Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. (2014) 345:98–101. doi: 10.1126/science.1254312
181. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. (2004) 18:3066–77. doi: 10.1101/gad.1250704
182. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. (2008) 118:3378–89. doi: 10.1172/JCI34587
183. Pino SC, O’Sullivan-Murphy B, Lidstone EA, Yang C, Lipson KL, Jurczyk A, et al. CHOP mediates endoplasmic reticulum stress-induced apoptosis in gimap5-deficient T cells. PloS One. (2009) 4:e5468. doi: 10.1371/journal.pone.0005468
184. Cao Y, Trillo-Tinoco J, Sierra RA, Anadon C, Dai W, Mohamed E, et al. ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat Commun. (2019) 10:1280. doi: 10.1038/s41467-019-09263-1
185. Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, Thaxton JE. Endoplasmic reticulum stress contributes to mitochondrial exhaustion of CD8+ T cells. Cancer Immunol Res. (2019) 7:476–86. doi: 10.1158/2326-6066.CIR-18-0182
186. Kamimura D, Katsunuma K, Arima Y, Atsumi T, Jiang JJ, Bando H, et al. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat Commun. (2015) 6:7474. doi: 10.1038/ncomms8474
187. Solanki NR, Stadanlick JE, Zhang Y, Duc AC, Lee SY, Lauritsen JPH, et al. Rpl22 loss selectively impairs αβ T cell development by dysregulating endoplasmic reticulum stress signaling. J Immunol. (2016) 197:2280–9. doi: 10.4049/jimmunol.1600815
188. Fernández-Alfara M, Sibilio A, Martin J, Tusquets Uxó E, Malumbres M, Alcalde V, et al. Antitumor T-cell function requires CPEB4-mediated adaptation to chronic endoplasmic reticulum stress. EMBO J. (2023) 42:e111494. doi: 10.15252/embj.2022111494
189. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. (2019) 216:867–83. doi: 10.1084/jem.20182192
190. Quaney MJ, Pritzl CJ, Luera D, Newth RJ, Knudson KM, Saxena V, et al. STING controls T cell memory fitness during infection through T cell-intrinsic and IDO-dependent mechanisms. Proc Natl Acad Sci U S A. (2023) 120:e2205049120. doi: 10.1073/pnas.2205049120
191. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. J Biol Chem. (1998) 273:33741–9. doi: 10.1074/jbc.273.50.33741
192. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis -acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. (2000) 20:6755–67. doi: 10.1128/MCB.20.18.6755-6767.2000
193. Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. (2000) 20:5096–106. doi: 10.1128/MCB.20.14.5096-5106.2000
194. Luo R, Lu JF, Hu Q, Maity SN. CBF/NF-Y controls endoplasmic reticulum stress induced transcription through recruitment of both ATF6(N) and TBP. J Cell Biochem. (2008) 104:1708–23. doi: 10.1002/jcb.21736
195. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. (1999) 10:3787–99. doi: 10.1091/mbc.10.11.3787
196. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of biP/GRP78 binding and unmasking of golgi localization signals. Dev Cell. (2002) 3:99–111. doi: 10.1016/S1534-5807(02)00203-4
197. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. (2000) 6:1355–64. doi: 10.1016/S1097-2765(00)00133-7
198. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the golgi. J Biol Chem. (2002) 277:13045–52. doi: 10.1074/jbc.M110636200
199. Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci. (2009) 106:17775–80. doi: 10.1073/pnas.0910342106
200. Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J. (2001) 355:19–28. doi: 10.1042/bj3550019
201. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. (2007) 13:365–76. doi: 10.1016/j.devcel.2007.07.018
202. Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. (2013) 3:1279–92. doi: 10.1016/j.celrep.2013.03.024
203. Nakajima S, Hiramatsu N, Hayakawa K, Saito Y, Kato H, Huang T, et al. Selective Abrogation of BiP/GRP78 Blunts Activation of NF-κB through the ATF6 Branch of the UPR: Involvement of C/EBPβ and mTOR-Dependent Dephosphorylation of Akt. Mol Cell Biol. (2011) 31:1710–8. doi: 10.1128/MCB.00939-10
204. Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, et al. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. (2009) 122:1626–36. doi: 10.1242/jcs.045625
205. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6α Optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. (2007) 13:351–64. doi: 10.1016/j.devcel.2007.07.005
206. Sato Y, Nadanaka S, Okada T, Okawa K, Mori K. Luminal domain of ATF6 alone is sufficient for sensing endoplasmic reticulum stress and subsequent transport to the golgi apparatus. Cell Struct Funct. (2011) 36:35–47. doi: 10.1247/csf.10010
207. Higa A, Taouji S, Lhomond S, Jensen D, Fernandez-Zapico ME, Simpson JC, et al. Endoplasmic reticulum stress-activated transcription factor ATF6α Requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol. (2014) 34:1839–49. doi: 10.1128/MCB.01484-13
208. Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. (2010) 21:2975–86. doi: 10.1091/mbc.e09-02-0133
209. Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. (2009) 460:534–7. doi: 10.1038/nature08111
210. Yu R, Chen X, Zhu X, He B, Lu C, Liu Y, et al. ATF6 deficiency damages the development of spermatogenesis in male Atf6 knockout mice. Andrologia. (2022) 54:e14350. doi: 10.1111/and.14350
211. Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest. (2010) 120:744–55. doi: 10.1172/JCI39678
212. Liu F, Chang L, Hu J. Activating transcription factor 6 regulated cell growth, migration and inhibiteds cell apoptosis and autophagy via MAPK pathway in cervical cancer. J Reprod Immunol. (2020) 139:103120. doi: 10.1016/j.jri.2020.103120
213. Benedetti R, Romeo MA, Arena A, Gilardini Montani MS, Di Renzo L, D’Orazi G, et al. ATF6 prevents DNA damage and cell death in colon cancer cells undergoing ER stress. Cell Death Discovery. (2022) 8:295. doi: 10.1038/s41420-022-01085-3
214. Li R, Zhou H, Li M, Mai Q, Fu Z, Jiang Y, et al. Gremlin-1 promotes colorectal cancer cell metastasis by activating ATF6 and inhibiting ATF4 pathways. Cells. (2022) 11:2136. doi: 10.3390/cells11142136
215. Hong M, Luo S, Baumeister P, Huang JM, Gogia RK, Li M, et al. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem. (2004) 279:11354–63. doi: 10.1074/jbc.M309804200
216. Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, et al. The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev Cell. (2018) 46:327–343.e7. doi: 10.1016/j.devcel.2018.04.023
217. Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. (2007) 27:1027–43. doi: 10.1128/MCB.00408-06
218. Oka OB, Van Lith M, Rudolf J, Tungkum W, Pringle MA, Bulleid NJ. ERp18 regulates activation of ATF6a during unfolded protein response. EMBO J. (2019) 38:e100990. doi: 10.15252/embj.2018100990
219. Tsukumo Y, Tomida A, Kitahara O, Nakamura Y, Asada S, Mori K, et al. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem. (2007) 282:29264–72. doi: 10.1074/jbc.M705038200
220. Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. (2011) 22:4390–405. doi: 10.1091/mbc.e11-06-0510
221. Chang JS, Ocvirk S, Berger E, Kisling S, Binder U, Skerra A, et al. Endoplasmic reticulum stress response promotes cytotoxic phenotype of CD8αβ+ Intraepithelial lymphocytes in a mouse model for crohn’s disease-like ileitis. J Immunol. (2012) 189:1510–20. doi: 10.4049/jimmunol.1200166
222. Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, et al. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell. (2021) 184:1262–1280.e22. doi: 10.1016/j.cell.2021.02.019
Keywords: endoplasmic reticulum stress, unfolded protein response, IRE1, PERK, ATF6, CD8 T cell
Citation: Nair KA II and Liu B (2024) Navigating the landscape of the unfolded protein response in CD8+ T cells. Front. Immunol. 15:1427859. doi: 10.3389/fimmu.2024.1427859
Received: 05 May 2024; Accepted: 24 June 2024;
Published: 04 July 2024.
Edited by:
Cosima T. Baldari, University of Siena, ItalyReviewed by:
Matthew Staron, AbbVie, United StatesShoulong Deng, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2024 Nair and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bei Liu, YmVpLmxpdUBvc3VtYy5lZHU=