 Jiwon Lyu1,2Drew E. Narum2
Jiwon Lyu1,2Drew E. Narum2 Susan L. Baldwin3
Susan L. Baldwin3 Sasha E. Larsen3
Sasha E. Larsen3 Xiyuan Bai2,4
Xiyuan Bai2,4 David E. Griffith5
David E. Griffith5 Véronique Dartois6
Véronique Dartois6 Threnesan Naidoo7
Threnesan Naidoo7 Adrie J. C. Steyn8,9
Adrie J. C. Steyn8,9 Rhea N. Coler3,10,11
Rhea N. Coler3,10,11 Edward D. Chan2,4,12*
Edward D. Chan2,4,12*- 1Division of Pulmonary and Critical Medicine, Soon Chun Hyang University Cheonan Hospital, Seoul, Republic of Korea
- 2Department of Academic Affairs, National Jewish Health, Denver, CO, United States
- 3Center for Global Infectious Diseases, Seattle Children’s Research Institute, Seattle, WA, United States
- 4Division of Pulmonary Sciences and Critical Care Medicine, University of Colorado School of Medicine, Aurora, CO, United States
- 5Department of Medicine, National Jewish Health, Denver, CO, United States
- 6Center for Discovery and Innovation, Hackensack Meridian School of Medicine, Nutley, NJ, United States
- 7Departments of Forensic & Legal Medicine and Laboratory Medicine & Pathology, Faculty of Medicine & Health Sciences, Walter Sisulu University, Mthatha, South Africa
- 8Africa Health Research Institute, University of KwaZulu-Natal, Durban, South Africa
- 9Department of Microbiology and Centers for AIDS Research and Free Radical Biology, University of Alabama at Birmingham, Birmingham, AL, United States
- 10Department of Pediatrics, University of Washington School of Medicine, Seattle, WA, United States
- 11Department of Global Health, University of Washington, Seattle, WA, United States
- 12Department of Medicine, Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, CO, United States
Granulomas, organized aggregates of immune cells which form in response to Mycobacterium tuberculosis (Mtb), are characteristic but not exclusive of tuberculosis (TB). Despite existing investigations on TB granulomas, the determinants that differentiate host-protective granulomas from granulomas that contribute to TB pathogenesis are often disputed. Thus, the goal of this narrative review is to help clarify the existing literature on such determinants. We adopt the a priori view that TB granulomas are host-protective organelles and discuss the molecular and cellular determinants that induce protective granulomas and those that promote their failure. While reports about protective TB granulomas and their failure may initially seem contradictory, it is increasingly recognized that either deficiencies or excesses of the molecular and cellular components in TB granuloma formation may be detrimental to the host. More specifically, insufficient or excessive expression/representation of the following components have been reported to skew granulomas toward the less protective phenotype: (i) epithelioid macrophages; (ii) type 1 adaptive immune response; (iii) type 2 adaptive immune response; (iv) tumor necrosis factor; (v) interleukin-12; (vi) interleukin-17; (vii) matrix metalloproteinases; (viii) hypoxia in the TB granulomas; (ix) hypoxia inducible factor-1 alpha; (x) aerobic glycolysis; (xi) indoleamine 2,3-dioxygenase activity; (xii) heme oxygenase-1 activity; (xiii) immune checkpoint; (xiv) leukotriene A4 hydrolase activity; (xv) nuclear-factor-kappa B; and (xvi) transforming growth factor-beta. Rather, more precise and timely coordinated immune responses appear essential for eradication or containment of Mtb infection. Since there are several animal models of infection with Mtb, other species within the Mtb complex, and the surrogate Mycobacterium marinum – whether natural (cattle, elephants) or experimental (zebrafish, mouse, guinea pig, rabbit, mini pig, goat, non-human primate) infections – we also compared the TB granulomatous response and other pathologic lung lesions in various animals infected with one of these mycobacteria with that of human pulmonary TB. Identifying components that dictate the formation of host-protective granulomas and the circumstances that result in their failure can enhance our understanding of the macrocosm of human TB and facilitate the development of novel remedies – whether they be direct therapeutics or indirect interventions – to efficiently eliminate Mtb infection and prevent its pathologic sequelae.
1 Introduction
Granulomas are aggregates of immune cells that form in response to repetitive exposure to various stimuli that include infectious agents (e.g., bacteria, fungi, protozoa, helminths, and viruses), non-infectious foreign bodies (e.g., talc, starch, sutures), or an unknown inciting agent (e.g., sarcoidosis, Crohn’s enteritis, granulomatosis with polyangiitis, polyarteritis nodosa, etc.) (1–3). While granulomas are typically regarded as a host-protective response against microbial agents, they may also lead to pathologic consequences, especially if the granulomas are unable to eradicate the microbes (2). In the context of tuberculosis (TB), granulomas are well documented in both humans and experimental animals (4).
The objective of this narrative review is to describe the factors that provide a host-protective granulomatous response with TB and the circumstances which cause granulomas fail. To help shed light on determinants of protective and non-protective TB granulomas, we also compared the TB granulomatous response and other pathologic lung lesions in various animals naturally or experimentally infected with Mycobacterium tuberculosis (Mtb), other Mtb complex species, or a mycobacterial surrogate with that of human pulmonary TB.
2 Cellular composition and functions of the TB granuloma
2.1 Overall structure of granulomas
Granulomas are considered a key histopathologic feature of TB. Classically, the spatial organization of TB granulomas is comprised of a central region populated with macrophages (epithelioid, activated, and foamy macrophages) with an innermost zone that may become necrotic. Other myeloid cell types (dendritic cells, neutrophils, eosinophils, multinucleated giant cells, and/or mast cells) may be interspersed with the macrophages. Surrounding the macrophages are lymphocytes (T cells, B cells, natural killer cells, and innate lymphoid cells), forming a cuff in the outer zone of the granulomas (Figures 1A, B). Granulomas may also contain endothelial and epithelial cells from blood vessels and airways, respectively, as well as be encapsulated by collagen secreted by fibroblasts. However, granulomas may display great diversity with regards to certain immunological characteristics even within a single individual (5). In more protective granulomas, the concentric cell layers are gradually organized along with the presence of fibrosis, which suppresses necrosis, strengthens the lymphocytic rim, and leads to pathologic resolution (Figure 1A). These resolved granulomas can be identified radiologically and histologically by dystrophic calcification of the necrotic debris within these structures (6). In cases of active and progressive TB, granulomas have higher number of foamy macrophages and neutrophils as well as exhibit necrotic centers (Figure 1B).
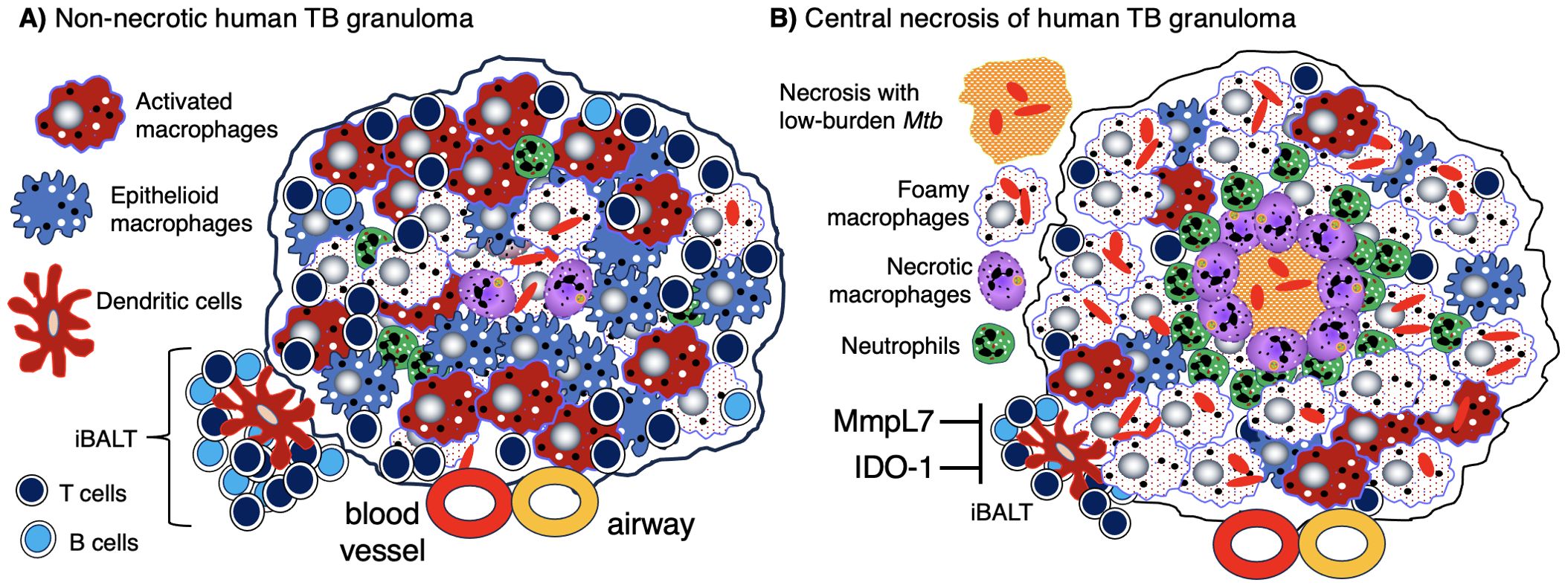
Figure 1. Human TB granulomas may be non-necrotic or necrotic. Several different fates of granulomas occur and such heterogeneity may present in the same individual. (A) A non-necrotic TB granuloma is characterized by the presence of epithelioid macrophages and activated macrophages as well as fewer foamy macrophages, necrotic macrophages, neutrophils, and more or less a rim of host-protective T cells and B cells along with relatively few bacilli. iBALT may be considered an “appendage” of granulomas typically comprised of dendritic cells, B cells, and T cells and may serve as a ready supply of immune cells for the neighboring granuloma. This granuloma is protective as shown by the relatively few Mtb (red-colored rod-shaped structures. (B) A granuloma with central necrosis characterized by fewer epithelioid and activated macrophages and more foamy macrophages, necrotic macrophages, and neutrophils along with an area of central necrosis. There are also fewer host-protective T cells and B cells. Due to the fewer number of host-protective innate and adaptive immune cells, there is overall a greater Mtb burden. However, in the central necrotic area of this human granuloma, there is active killing of Mtb resulting in fewer viable Mtb in this region. Higher levels of MmpL7 and IDO-1 inhibit iBALT formation. IDO-1, indoleamine 2,3-dioxygenase; iBALT, inducible bronchus-associated lymphoid tissue; MmpL7, Mycobacterial membrane protein Large 7; Mtb, Mycobacterium tuberculosis; TB, tuberculosis.
Granulomas can be classified by several different morphological characteristics. One is whether cells within the central regions of granulomas are necrotic, which may be found in human TB, in natural TB in animals, and experimental TB in some laboratory animals. Caseation is the gross pathological term which alludes to the “cheese-like” material found when sufficient number of macrophages in the granuloma core undergo necrotic cell death. Caseating granulomas typically are consisted of necrotic, acellular core surrounded by epithelioid macrophages, and an outer lymphocytic cuff comprised of B and T cells (7). While the presence or absence of granuloma necrosis is often linked, respectively, to infectious (e.g., TB) or non-infectious (e.g., sarcoidosis) etiology for the granuloma, this dichotomy is not absolute. For example, necrosis may be seen in some non-infectious granulomas (2) and TB granulomas may not show any necrosis (Figure 2) (8).
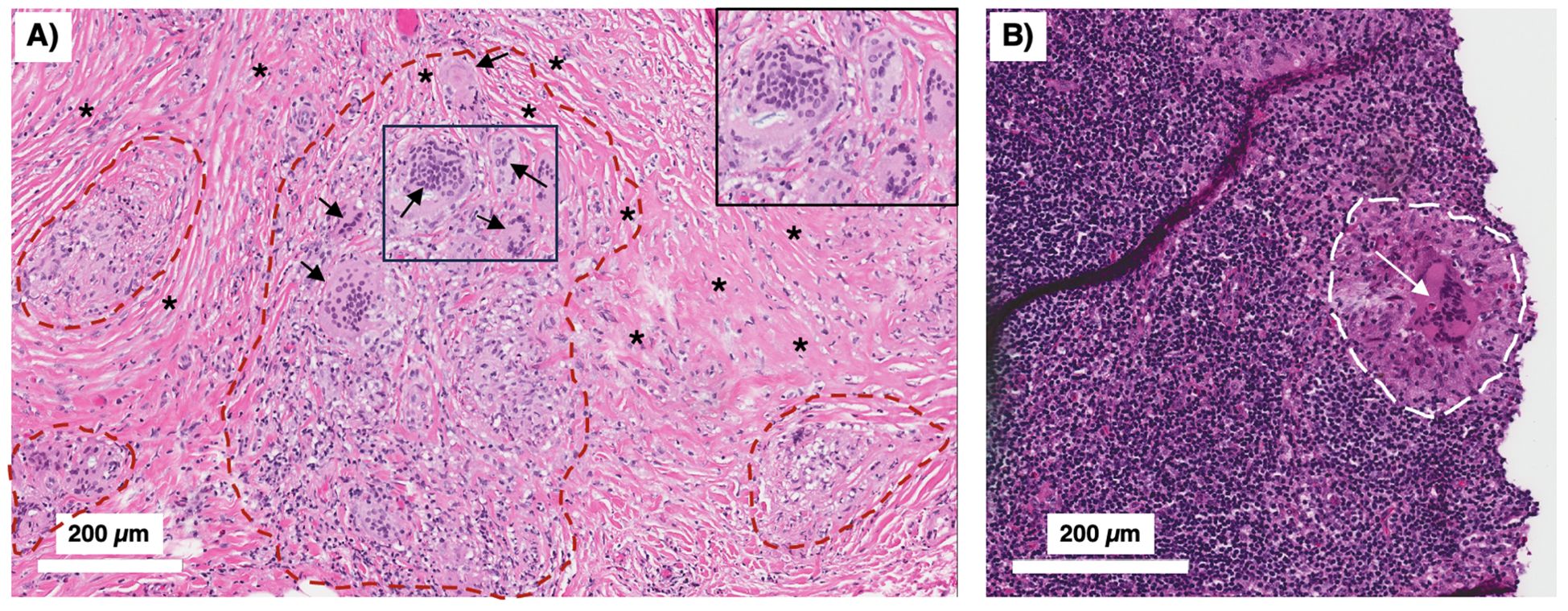
Figure 2. Peritoneal TB with non-necrotizing granulomas. A 51-year-old man from the Philippines presented with a two-year history of early satiety, intractable abdominal pain, and weight loss exceeding 60 pounds. Abdominal CT showed evidence of “peritoneal carcinomatosis.” (A) Peritoneal biopsy showed no evidence of malignancy. Instead, there were multiple non-necrotizing granulomas (demarcated by red dashed lines) with prominent Langhans-type multinucleated giant cells (arrows) and surrounding fibrosis (asterisks) (H&E). The boxed inset shows a magnified view of the smaller box demonstrating the multinucleated giant cells. (B) A peritoneal lymph node biopsy showed similar non-necrotizing granulomatous inflammation (white dashed line) with a conspicuous multinucleated giant cells (white arrow) (H&E). The acid-fast stain in both the peritoneal and lymph node biopsies were negative. With standard treatment for drug-susceptible TB, his symptoms abated and he regained his lost body weight. TB, tuberculosis.
Granulomas can show other morphological characteristics. Some granulomas may display fibrosis, characterized by collagen deposit either peripherally (those with a cuff of collagen surrounding them) or centrally (those with collagen throughout the entire granuloma) (9). Another subtype of granulomas is known as suppurative or neutrophil-rich granuloma. These granulomas arise when neutrophils infiltrate the center of the granuloma. Overall, high concentrations of neutrophils within Mtb granulomas have shown to be host destructive (enable spread of infection, higher levels of inflammation, and result in poorer host outcomes) (10). A calcified granuloma is typically regarded as the “final” stage of infection, and a sign of a successful host-immune response. The calcification (or mineralization) process typically begins within the caseous center (7, 11).
2.2 Selected immune cells that comprise granulomas
2.2.1 Macrophages
Lung macrophages are ubiquitous in granulomas and along with dendritic cells are likely the first cell types in the nascent granuloma. These phagocytes engulf Mtb and orchestrate the influx of other cell types (2). After being engulfed, Mtb are killed when Mtb-containing phagosomes fuse with lysosomes. However, the bacilli may avoid this fate by inhibiting phagosome and autophagosome maturation (12–16). Ingested Mtb may also escape from arrested phagosomes by disrupting the phagosome membrane – via the release of Mtb pore-forming protein ESAT-6 (early secreted antigenic target-6) utilizing the ESX-1 (type VII) secretion system – and translocating into the cytoplasm (17, 18). In the cytosol, Mtb replicates, precipitating both necrosis of the phagocytes and release of bacilli extracellularly. Neighboring macrophages then phagocytose the released Mtb and the infection cycle repeats. During this cycle of suboptimal control of infection, the infected and subsequent necrotic macrophages release both inflammatory chemokines and mycobacterial cell wall components that induce influx of neutrophils and monocytes to the infection site, forming the early granuloma (19).
Infected macrophages and dendritic cells also migrate to the regional mediastinal lymph nodes to help differentiate and recruit antigen-specific T cells (e.g., TH1 and TH17), which then migrate to the infection site, activate monocytes and macrophages, and participate in the formation of evolving granulomas (15, 16, 19–21). This delayed influx of recruited T cells to the site of the infection may allow Mtb to persist if the initial macrophages encountered are of the more “permissive” phenotype (22). However, with sufficient recruitment of Mtb-specific and activated T cells to the granulomas, containment or eradication of Mtb within the granulomas is possible. Macrophages in evolving granulomas may be comprised of various phenotypes, including “classically activated”/inflammatory (M1) macrophages and various subsets of “alternatively activated” (M2) macrophages as well as other morphologically distinct macrophages (epithelioid macrophages, foamy macrophages) that likely have overlapping features with M1 or M2 macrophages, and multinucleated Langhans giant cells (23, 24).
M1 macrophages are activated in response to TH1 T-cell signals (interferon-gamma [IFNγ] and tumor necrosis factor [TNF]) as well as lipopolysaccharide (LPS) (7). Once activated, M1 macrophages secrete pro-inflammatory cytokines (TNF, interleukin-1 [IL-1], IL-6) and inducible nitric oxide synthase (iNOS). Whereas the M1 response is important in TB control, an excessive response can lead to chronic inflammation as well as inflammatory diseases (25). On the other hand, M2 macrophages are known to inhibit the inflammatory response, playing an important role in wound healing and tissue repair. These macrophages are activated by TH2 T-cell cytokines (IL-4 and IL-13). In response, they produce anti-inflammatory cytokines (IL-10, transforming growth factor-beta [TGFβ]), IL-6, and arginase (7, 25). However, this binary categorization is likely an oversimplification to fully describe the function of these subtypes, especially M2 macrophages which are divided into at least three sub-phenotypes (26, 27). We expound below two distinct macrophage phenotypes given their preponderance in granulomas: “epithelioid macrophages” and “foamy macrophages” (2, 28).
2.2.1.1 Epithelioid macrophages
Epithelioid macrophages are so named due to their cellular characteristics that resemble epithelial cells: spread morphology, elongated nuclei, high cytoplasm-to-nucleus ratio, and contact interaction with neighboring macrophages. Epithelioid macrophages are also less phagocytic and more secretive than activated macrophages (29, 30). Additional characteristics of epithelioid macrophages are discussed in Table 1.

Table 1. Epithelioid and foamy macrophages in TB granulomas.
2.2.1.2 Foamy macrophages
Macrophages in some TB granulomas appear foamy due to the accumulation of various intracellular lipids (cholesterol, triglycerides, and phospholipids) (31). In foamy macrophages, bacilli are observed in proximity to lipid droplets, posited to be a nutrient source for Mtb (29). Foamy macrophages are typically found around the edge of the necrotic core of caseating granulomas and rarely found in non-necrotic granulomas (Figures 1, 3) (32). In human autopsy cases of TB, foamy DEC-205+ dendritic cells were more common in lipoid pneumonia and cavitary lesions than in granulomas (33). Additional characteristics of foamy macrophages are discussed in Table 1.
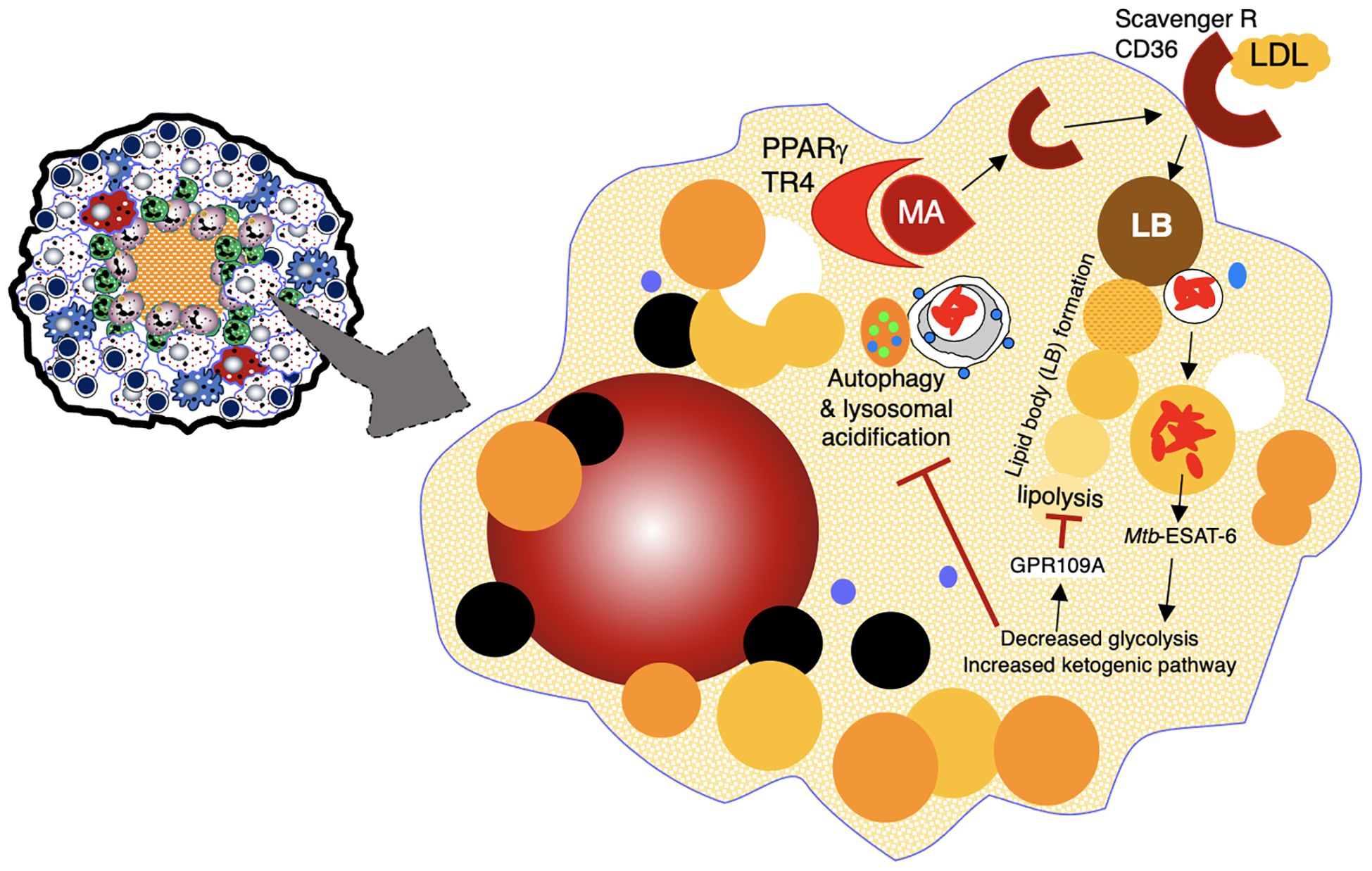
Figure 3. Formation of foamy macrophages and their role in TB pathogenesis. Foamy macrophages play a key role in the formation of caseous necrosis. They are formed when macrophages acquire cholesterol-containing LDL particles into lipid bodies. See text for discussion. ESAT-6, 6 kDa early secretory antigenic target; GPR109A, a specific G-protein coupled receptor; LB, lipid bodies; LDL, low density lipoprotein; MA, mycolic acids; PPARγ, peroxisome proliferator-gamma; TR4, testicular receiver 4.
2.2.1.3 Apoptotic vs. necrotic cell death of phagocytes in granulomas
Death of Mtb-containing phagocytes may either be a mechanism of restricting Mtb replication or spreading the infection. Apoptosis of Mtb-infected cells is a mechanism by which phagocytosed intracellular bacilli are killed (50–58). In addition, macrophage phagocytosis of apoptotic bodies – a process known as efferocytosis – can further enhance the killing of Mtb contained within the apoptotic bodies (59). Thus, apoptosis is a host protective mechanism to not only dispose of phagocytosed Mtb but to limit excessive inflammation associated with necrosis (58). In contrast, in the zebrafish-M. marinum model, phagocytosis of apoptotic macrophages infected with M. marinum promoted the spread of mycobacteria (60).
In contrast, cell necrosis permits survival and release of Mtb to infect neighboring cells (58, 61–63). Necrosis may be dictated, in part, by the Mtb strain and thus an immune evasive virulence phenomenon. Divangahi et al. (64) demonstrated that the more virulent Mtb H37Rv, in contrast to the less virulent Mtb H37Ra, induced the lipoxin A4 (LXA4) production, which then blocks both synthesis of prostaglandin E2 (PGE2) and activation of synaptotagmin 7. Since PGE2 prevents necrosis of Mtb-infected macrophages (61, 64–66) and synaptotagmin 7 (a calcium ion sensor) is involved with lysosome-mediated repair mechanism (64), LXA4 induced by the more virulent Mtb prevents repair of the injured plasma membrane caused by Mtb, resulting in cellular necrosis. Another mechanism by which Mtb induces necrosis is via activation of the cytosolic receptor-interacting protein kinase 3 (RIPK-3) pathway, preventing apoptosis of Mtb-infected macrophages through Bcl-xL and promoting reactive oxygen species (ROS)-mediated cell necrosis (67).
2.2.2 Neutrophils
Neutrophils can phagocytose Mtb as well as release neutrophil extracellular traps (NETs), which are comprised of chromatin (DNA) decorated with neutrophil-derived proteins (elastase, histones) that entrap the extracellular bacilli. NETs have antimicrobial activity and the NET-induced inflammatory response may promote migration of activated dendritic cells to the draining lymph nodes to activate naïve T cells; however, excessive NET-associated inflammation also causes tissue injury (68, 69). In addition, type 1 interferon-induced NET formation was found to promote Mtb growth and disease severity in mice genetically susceptible to TB (65). Neutrophils may also be present at the interface between the necrotic center and epithelioid macrophages as well be a source of immunoregulatory cytokines TNF and IL-10 (70). In C3HeB/FeJ mice, a fulminant necrotizing alveolitis with uncontrolled neutrophilic response in the lungs is associated with greater Mtb burden and mortality (34). Neutrophils may also provide a niche for mycobacterial growth, and this ability was associated with increased mitochondrial metabolic activity (71, 72).
2.2.3 Lymphocytes
2.2.3.1 CD4+ T effector cells
CD4+IFNγ+ T cells are critical in the formation of granulomas with early containment of Mtb (73–75). However, excessive amounts of CD4+IFNγ+ T cells, in conjunction with increased TNF, can also cause macrophage necrosis and poorer control of Mtb (76, 77). Similarly, either excessive or insufficient CD4+IL-13+ T cells (37, 78) impairs host control of Mtb (79–81). However, BALB/c mice with genetic disruption of IL-4 were more proficient in controlling Mtb (82), indicating that attenuating TH2 (IL-4, IL-13) immunity in more immunocompromised mice is host-protective. TGFβ produced by macrophages and T regulatory cells (Tregs) predisposes to Mtb in a computer simulated TB granuloma model, in experimental animals, and in humans (83–85). However, TGFβ combined with IL-23 promotes the expansion of TH17 cells, which are important in the control of Mtb during earlier stages of infection (86). IL-17 secreted by TH17 cells has other host-protective functions (limiting excessive granuloma hypoxia, inducing iBALT formation, recruiting CD4+IFNγ+ T cells) (87–90) but also host-deleterious properties (inducing excessive influx of neutrophils that may limit the formation of protective granulomas) (91, 92). Serum IL-9 (produced by TH9 cells) was found to be significantly higher in pulmonary TB subjects than individuals with sarcoidosis, a granulomatous disorder of unknown etiology (93).
2.2.3.2 CD8+ T effector cells
CD8+ T cells, through their cytotoxic response, help limit bacterial replication in the later stage of disease (6, 94). Following aerosol Mtb infection of mice, CD8+ cells were not considered to play a major protective role (95) whereas with high intravenous inoculum, they were more essential for protection (96–98). CD8+ T-cells are able to recognize and induce apoptosis of infected macrophages (99). In mice, CD8+ T cells were found to be important in maintaining stable latent phase of infection, in part through production of IFNγ (100). However, increased TGFβ and IL-10 expression and reduced granzyme B production by CD8+ T cells are associated with poorer control in patients with active TB (101).
2.2.3.3 T regulatory cells
Tregs are CD4+ T cells (or less frequently CD8+ marker) characterized by the phenotypic markers Foxp3+CD25hiCD127dim/–. Tregs develop from naïve T cells in the presence of TGFβ and dampen the function of M1 macrophages and T effector cells through expression of immunosuppressive cytokines TGFβ, IL-10, and IL-35 (102–104). Tregs were more commonly present in cavities than in the lipoid pneumonia or granulomas of human post-primary TB in the lungs (33) Within granuloma-infiltrating T cells of both Mtb-infected mice and rhesus macaques, TGFβ signaling antagonized activity of IFNγ-producing CD4+ T cells and impaired control of Mtb (83).
2.2.3.4 B cells
B cells also play an important role in the granulomatous and iBALT response against Mtb (105, 106). B cells found in granulomas produce the chemokine CXCL13, which recruits specific T cells to the infection site, promoting follicle-like structure formation within granulomas (107). B cells also function as antigen-presenting cells within granulomas, enhancing proliferation of a host-protective T cells and plasma cells against Mtb infection (108). Studies in mice have shown that B cells decrease neutrophil motility (109–111).
Three distinct populations of B cells exist: B effector 1 cells (Be1), B effector 2 cells (Be2), and regulatory IL-10 producing B cells (B10). Be1 cells are produced when naïve B cells are primed by TH1 cytokines. These cells then produce IFNγ, IL-12, TNF, IL-10, and IL-6. On the other hand, Be2 cells are differentiated in the presence of TH2 cytokines, and, in turn, are producers of IL-2, lymphotoxin, IL-4, IL-13, IL-10 and IL-6 (105, 112, 113). Once produced, Be1 and Be2 cells are able to influence the differentiation of naïve CD4+ T cells into TH1 or TH2, respectively (112). Regulatory B cells are known to downregulate the T cell response through production of IL-10 or TGFβ (112, 113). Thus, whereas effector B cells induce the inflammatory T cell response, regulatory B cells downregulate this pathway. Due to the crucial role that CD4+ T cells play in the formation of TB granulomas, the ability of B cells to induce or block this pathway illustrates their importance in granuloma formation. Indeed, clusters of B cells are found in human TB granulomas (114).
2.3 Inducible bronchus-associated lymphoid tissue, an accessory organoid to granulomas
Tertiary lymphoid organs (TLOs) are organized lymphoid structures that form in non-lymphoid tissues in response to injury, inflammation, or chronic infection (115). These structures have been shown to surround granulomas during mycobacterial infection (116, 117). While TLOs and granulomas are generally regarded as discrete structures, they have been shown to share similar biological processes (118). One such TLO that is associated with granuloma formation is inducible bronchus-associated lymphoid tissue (iBALT). iBALT are comprised of B cells, T cells, and antigen-presenting cells (e.g., dendritic cells) and are juxtaposed to granulomas (Figures 1A, B) (119–121). The immune cell inflammation that accompanies Mtb infection: (i) induces “stromal lymphoid tissue organizer cells” (fibroblasts, myofibroblasts, and endothelial cells) to transform into follicular dendritic cells; (ii) triggers them to produce chemokines (CXCL13, CXCL12, CCL21, CCL19), which recruit and organize T cells, B cells, and dendritic cells into iBALT; and (iii) supports high endothelial venule formation to allow lymphocyte trafficking into the infection site (122). Interleukin-17 (IL-17), IL-23, and group 3 innate lymphoid cells (ILC3s) also promote the differentiation and activation of lymphoid cells that participate in iBALT formation (89, 90, 123–125).
iBALT has been described with pulmonary TB in humans (75, 121, 126), non-human primates (NHP) (75, 121, 126), rabbits (127), guinea pigs (127), and mice (75, 125). iBALTs are considered to be protective against Mtb for several reasons: (i) they can serve as a site for local antibody production (128, 129); (ii) they can render support to neighboring granulomas as a ready supply of additional immune cells (119, 130); (iii) the presence of iBALT can be associated with the maintenance of latency and containment of infection, whereas its absence is associated with active disease (75, 121). However, in the context of sustained and unresolved TB, iBALT may become pathogenic and contribute to lung injury, especially with sustained IL-17 expression and neutrophil recruitment (119).
Certain molecules can have an impact on iBALT formation. For example, NHP infected with Mtb that lacked MmpL7 (Mycobacterial membrane protein Large 7) showed an increase in granulomas containing iBALT, suggesting that Mtb-derived MmpL7 prevents the formation of host-protective iBALT (Figure 1B) (131). Another study found in both in vitro (macrophage) and in vivo (murine and NHP) Mtb models that inhibition of indoleamine 2,3-dioxygenase (IDO-1) activity resulted in a greater host protective response as well as increased iBALT, indicating that IDO-1 also inhibits iBALT formation (Figure 1B) (132).
3 The granuloma paradox
3.1 Granulomas may be viewed from different perspectives
One overarching paradox of TB is that despite being the most lethal infectious disease globally, most who are infected with Mtb do not ever develop disease, consistent with pathologic studies that most human TB lesions are regressive (133, 134). Because humans and Mtb have co-existed for a long time, it is likely that the genetics of both humans and Mtb are important in determining who are at risk for developing latent infection or active TB disease (135, 136). The battle between Mtb and host immunity may be viewed from either the perspective of the bacilli or the host. When the host is unable to completely eradicate the Mtb upon initial infection – due to immune evasive strategies of Mtb such as arrest of phagosome maturation (137) – the next best recourse is to contain the infection through the formation of granulomas. While granulomas are generally regarded as a host-protective response by controlling the Mtb infection, they also provide a niche that allows Mtb survival by limiting the trafficking of immune cells (4). Some newly infected macrophages may also escape from the original granuloma to form a secondary granuloma, albeit this was observed in the M. marinum-zebrafish model (60).
Drawing from epidemiological evidence indicating that the majority of individuals with latent TB infection do not progress to active TB but remain at risk, our discussion framework centers on the host-protective nature of TB granulomas. We explore the multifaceted circumstances leading to their occasional failure in protecting the host. There are many factors that determine whether TB granulomas are protective or harmful. One plausible explanation for variability in the protective efficacy of granulomas lies in the existence of different granuloma phenotypes – a heterogenous spectrum of lesions – within the same individual. These distinct granuloma types emerge due to a complex interplay between the host immune response and Mtb components, reflecting varying endotypes (138). However, a glaring limitation emerges when assigning a specific granuloma phenotype as detrimental to the host: the challenge of discerning whether a particular granuloma phenotype fails to protect or if it arises as a consequence of such failure or an attempt to correct the failure. In essence, our comprehension of granulomas is significantly hindered by our inability to observe the temporal evolution of individual granulomas in the context of live bacilli burden. Nevertheless, we have organized below the descriptions of granulomas in the context of host-protection or not.
3.2 Evidence that granulomas serve host-protective function
3.2.1 Epidemiological and clinical observations
The notion that TB granulomas are host-protective is evinced indirectly from the observation that most individuals infected with Mtb – identified through positive tuberculin skin tests or IFNγ release assays – do not progress to active TB and commonly exhibit calcified granulomas. Indeed, global data show a much greater preponderance of individuals infected with Mtb without disease (by ~200-fold) than those with active TB (139).
A stronger piece of clinical evidence that granulomas serve a host-protective function is that individuals with severe immunodeficiency (such as those with AIDS or immune suppression by pharmacologic TNF blocking agents) are highly susceptible to Mtb and have poorly-formed or absent granulomas (140–145). On a cellular level, TNF is necessary for macrophage-mediated control of mycobacterial infection (146). The requirement of TNF in forming a host-protective TB granuloma is evinced by the finding that mice knocked out for TNF or neutralized of TNF function were unable to form granulomas in response to Mtb infection and showed greater Mtb burden (147). One novel function of TNF in granuloma formation is the induction of a T cell receptor (TCR) αβ-based recombinatorial immune receptor in subpopulations of monocytes/macrophages (148). As macrophage-TCRαβ was found to induce the release of CCL2 (monocyte chemoattractant protein 1), which recruits monocytes, dendritic cells, and memory T cells, deletion of this variable macrophage-TCRαβ or TNF results in structurally compromised TB granulomas (148). Yet, there are case reports of caseating granulomas (often a sign of high Mtb burden) found in active TB in the presence of TNF blocking agents, indicating that granulomas that form despite TNF deficiency are not protective (149, 150). One possible scenario for this finding is that latent TB granulomas previously existed but with the introduction of an anti-TNF agent, there was proliferation of dormant Mtb, causing disruption of the intact, protective granuloma with secondary necrosis. An alternative explanation is that during treatment with an anti-TNF agent, a new Mtb infection occurred (plausible in TB endemic countries), resulting in inadequate control of the initial infection and the recruitment of other cell types such as neutrophils, resulting in caseating necrosis of the poorly protective granulomas. The observation that most of the Mtb are extracellular within necrotic granulomas, with lesser number located in the macrophage-rich region surrounding the core would support either scenario (151).
3.2.2 HIF-1α, a response to granuloma hypoxia, is largely host-protective
In non-necrotic, cellular granulomas, Mtb exists mainly intracellularly in the infected macrophage population within the core of the intact granuloma (11, 151, 152). One likely factor that allows granulomas to remain intact and non-necrotic is the prevention of a hypoxic environment within granulomas. More specifically, if cellular hypoxia occurs, one thwarting response is through increased production of hypoxia inducible factor-1 alpha (HIF-1α) by innate immune cells (macrophages, dendritic cells, and neutrophils) and lymphocytes (153). HIF-1α, which combines with constitutive HIF-1β to form HIF1, increases angiogenesis to the granuloma via induction of vascular endothelial growth factor, which allows oxygen and nutrient transport to the cells within granulomas (154). HIF-1α also aids cell survival in the hypoxic environment by augmenting glycolysis to generate quickly available ATP (155–157).
Several studies have shown that inducible HIF-1α is host-protective against Mtb infection. HIF-1α enhances the Mtb-killing abilities of both phagocytes and monocytes by several effector mechanisms including apoptosis of Mtb-infected phagocytes and enhanced production of cellular anti-microbial elements (nitric oxide, granule proteases, and anti-microbial peptides) (154, 158–160). Live Mtb alone has been shown to induce HIF-1α (154, 161) but this effect was especially seen in conjunction with IFNγ (162). In turn, HIF-1α augments IFNγ-induced genes that play important roles in effector functions against Mtb such as inflammatory cytokines and chemokines (IL-1α, IL-1β, and IL-6) (154, 162, 163). Hypoxia-induced HIF-1α also induces expression of CXCR4 – the receptor for the lymphocyte chemokine (and pro-angiogenic) CXCL12; thus, HIF-1α maintains both the influx of potentially host-protective immune cells to granulomas and the viability of these cells (164, 165). These findings are supported by studies of mice with conditional genetic disruption of HIF-1α in myeloid cells wherein with Mycobacterium avium infection, there was greater granuloma necrosis (perhaps due in part to impaired neovascularization of the granulomas) and impaired clearance of the mycobacteria (166). In the M. marinum-zebrafish model, stabilization of HIF-1α at the early stages of infection enhanced the levels of nitric oxide and reduced mycobacterial burden (167). Furthermore, suppression of granuloma-associated angiogenesis – which would be expected to cause hypoxia and induce the expression of HIF-1α – decreased M. marinum burden (168). While the ability of HIF-1α to enhance host immune cells against mycobacteria may be mediated by NFκB activation (169), HIF-1α may also attenuate an excessive NFκB response to prevent injurious inflammation (170).
3.3 Evidence that granulomas serve to propagate Mtb
3.3.1 Is bacillary patience a survival factor for Mtb? A teleological perspective
The first observation that certain granulomas – depending on the model used – benefit Mtb growth is that Mtb has adapted to remain quiescent for long periods with the opportunity to propagate at later times. Unlike pathogens that do not develop latency in the host and evade host immunity by antigenic variation, Mtb is capable of surviving (in a latent form), paradoxically, because of its recognition by host immune cells. The notion that Mtb has exploited recognition by the host as a survival mechanism is supported by the extensive diversity of Mtb antigens and epitopes that are recognized by CD4+ and CD8+ T cells of both active TB subjects and healthy controls (171–173). Hence, a potential interpretation is that T-cell recruitment to the site of infection, although initially a host-protective mechanism, could inadvertently aid Mtb by facilitating the establishment of a latent infection within an organized granuloma. This arrangement extends the bacilli’s survival, enabling them to lay dormant until conditions conducive to their reactivation arise, such as during immune senescence, thereby facilitating their dissemination to other individuals (6, 174, 175). Therefore, a teleological perspective suggests that Mtb, by exhibiting remarkable “patience” in remaining quiescent after encountering host immune cells within granulomas, ensures a higher probability of long-term survival for its species.
3.3.2 The co-evolution of Mtb with its host to survive within granulomas
The second observation that granulomas benefit Mtb is that the bacilli have developed survival mechanisms within the host phagocytes. A denouement of such survival capability is that the bacilli can spread between phagocytes within either quiescent or disruptive stages of granulomas. Mechanisms for this intracellular survival and spread to other cells are Mtb-mediated rupture of the phagosome membrane to escape into the cytoplasm, evading phagosome-lysosome fusion (17) as well as apoptosis-mediated cell-to-cell spread (60, 176). This intracellular movement of mycobacteria is likely an immune evasive mechanism because pathogenic mycobacteria are more capable of translocating from the intra-phagosome compartment to the cytosol via pore-inducing mycobacterial ESAT-6 protein than non-pathogenic mycobacteria (discussed in Section II.2.1 above) (17, 18). A connection between this intracellular immune evasive mechanism and the earlier-discussed Mtb latency survival strategy within organized granulomas emerges. ESAT-6, known to contain multiple T-cell epitopes (both CD4 and CD8), plays a pivotal role (177). Interestingly, just before Mtb burden plateaued post-infection in mice, the mRNA levels of both ESAT-6 and Ag85B decreased by 10-fold (178). This decline, reflecting little turnover of Mtb during the stationary phase, allows for Mtb to be still recognized by T cells but prevented its complete eradication by the immune system.
ESAT-6 secretion systems (ESX), particularly ESX-1 and ESX-5, have been shown to be virulence factors during mycobacterial infection. The importance of ESX-1 in mycobacterial pathogenesis is evinced by the finding that Mtb strains lacking ESX-1 have decreased pathogenesis and intracellular replication compared to wild-type strains (179). In humans, ESX-1 induction of the chemokine fractalkine (CX3CL1) mediates the recruitment of monocytes to the expanding granuloma (180). In contrast, the ESX-1 of M. marinum induced expression of matrix metalloproteinase 9 (MMP-9), which facilitates influx of uninfected macrophages, expanding the granulomatous lesions that may contain viable mycobacteria (181, 182). ESX-5 of M. marinum has been shown to inhibit induction of proinflammatory cytokines (IL-12p40, TNF, IL-6) (183) as well as induce both inflammasome/IL-1β activation and caspase-independent cell death (184).
Mtb also secretes lipids that affect the host immune response and caseous granuloma formation. These include mycolic acids, specifically trehalose 6,6’-dimycolate (TDM, aka cord factor), a toxic lipid that induces epithelioid macrophage transformation, pro-inflammatory cytokine production, and necrosis of the granulomas (185–187). Experimentally, the largest oil droplets of oil-water emulsions containing TDM were the most granuloma-genic (188). In contrast, de-lipidated BCG – where TDM was stripped from the outer cell wall – induced more of an acute (neutrophilic-like) infiltration and reduced delayed hypersensitivity (granulomatous) response along with more rapid clearance of de-lipidated BCG in mice, indicating that TDM is both a virulence factor and responsible for granuloma formation (189). The programming Mtb into a dormancy state by the dormancy survival regulon (DosR) is induced by carbon monoxide and hydrogen sulfide as well as a local environment that is hypoxic, acidic, and contains high nitric oxide levels (190–195). This induced dormancy of Mtb results in reduced metabolic activities of the bacilli, but they are also able to dysregulate host-lipid synthesis resulting in the production of lipid bodies (39). This process transforms macrophages into the foamy phenotype capable of not only sustaining the dormant Mtb but contributing to the caseous necrosis when granulomas breakdown (31).
Mtb has the capacity to import and catabolize host-derived cholesterol (an integral part of cellular membranes) – via proteins encoded by the mycobacterial gene cluster mce4 and by the igr locus, respectively – and use it as a carbon and energy source to survive for prolonged periods in caseous granulomas (196, 197). The catabolism of cholesterol in host-cells requires a shift from aerobic to anaerobic metabolism within the hypoxic and necrotic granuloma core which contributes to the dormant state of Mtb. In this state, Mtb remodels the cell wall and produces intracellular triglycerides (comprised of three long-chain fatty acids esterified to glycerol). β-oxidation of even-chain fatty acids produces acetyl-Co-A, which can be used for either synthesizing triglycerides (a lipid relevant for dormancy) or fueling the tricarboxylic acid (TCA) cycle (for ATP production) (198). Odd chain fatty acids undergo β-oxidation to produce the 3-carbon propionyl-CoA, which can be further metabolized to products that enter the TCA cycle or produce methyl-branched lipids to synthesize the mycobacterial cell wall (198). The mycobacterial enzyme isocitrate lyase (Icl) serves two functions for Mtb: (i) it is essential for conversion of even-chain fatty acids under nutrient limitation; i.e., Icl metabolizes isocitrate (6C) to glyoxylate (2C) and succinate (4C) with subsequent combining of glyoxylate with acetyl-Co-A (2C, a product of β-oxidation of fatty acids) to form malate (4C), which enters and fuels the tricarboxylic acid cycle and (ii) by fueling the TCA cycle, it activates the events that occur downstream of the initial steps in the metabolism of the toxic propionyl-CoA. In addition, the gene that encodes the lipid transporter also plays an important role in Mtb growth (6, 199).
3.3.3 What causes necrosis and caseation of TB granulomas?
The root cause of the necrotic cell death and subsequent caseation in TB granulomas is not precisely known. Within TB granulomas, macrophage necrosis occurs by various and not necessarily mutually exclusive mechanisms. The first mechanism is dysregulation of the pro-inflammatory cytokine TNF. On the one hand, either deficiency or antagonism of TNF impairs macrophage control of Mtb (and of M. marinum), increases mycobacterial burden in macrophages, and results in necrotic macrophage death with subsequent tissue spread of Mtb (4, 76, 146, 200). This TNF deficiency may also occur endogenously in certain individuals due to decreased leukotriene A4 hydrolase (LTA4H) activity (CC genotype of LTA4H gene), resulting in a decrease ratio of the pro-inflammatory leukotriene B4 (LTB4) to the anti-inflammatory LXA4 (201, 202). On the other hand, TNF excess due to high LTA4H activity (TT genotype), resulting in increased LTB4:LXA4 ratio), leads to activation of the TNF receptor-interacting serine/threonine kinases 1 and 3 (RIP1 and RIP3), mitochondrial reactive oxygen species (ROS), and initiated programmed necrosis (necroptosis) of the macrophages (76). Thus, both the hypoinflammatory (CC) and hyperinflammatory (TT) genotypes of LTA4H were associated with poorer outcomes compared to the heterozygous (CT) genotype (201, 202).
The second mechanism for macrophage necrosis is the insufficient clearance of Mtb-infected apoptotic phagocytes. Mtb-infected macrophages that undergo apoptosis are further phagocytosed by (recruited) uninfected macrophages, a process known as efferocytosis (60). However, if the influx of these uninfected macrophages is insufficient, apoptotic Mtb-infected macrophages are not cleared, leading to necrosis (203). While apoptosis of Mtb-infected macrophages is known to kill the intracellular bacilli, any surviving bacilli in apoptotic bodies that are not efferocytosed are released during secondary necrosis of the apoptotic bodies (51, 204).
The third hypothesized mechanism for the development of caseous necrosis is related to a deficiency or an excess of CD4+ T cell response. Deficiency of CD4+IFNγ+ T cells and insufficient formation of host-protective granulomas (147, 205, 206) results in increased IL-17-mediated neutrophilic influx and overwhelming Mtb infection of the macrophages, with both events leading to necrosis of the phagocytes (Figure 4A). The mechanism by which an excess of CD4+ T cells contribute to necrosis is not clear, but based on experimental evidence, the exuberant IFNγ response induces TNF and other downstream inflammatory mediators (207), causing activation of the aforementioned RIP1 and RIP3, necrotic macrophage death (necroptosis), and release and proliferation of mycobacteria (Figure 4B) (76, 208–210). IFNγ also increases CXCR3 (a receptor for several angiostatic chemokines) that may result in granuloma hypoxia and necrosis (Figure 4B) (211). Another supporting clue that excess IFNγ (and its downstream effects) may contribute to a detrimental immune response to the host is based on a study showing that IFNγ production by TH1 cells is far more important for TB control in the spleen than in the lungs and that increasing the IFNγ-producing capacity by the TH1 cells actually exacerbated the lung infection and led to more rapid mortality (77). In contrast to these two extremes of CD4+IFNγ+ T cell deficiency and excess, we posit that a more precise CD4+ T cell response would be more optimal (Figure 4C).
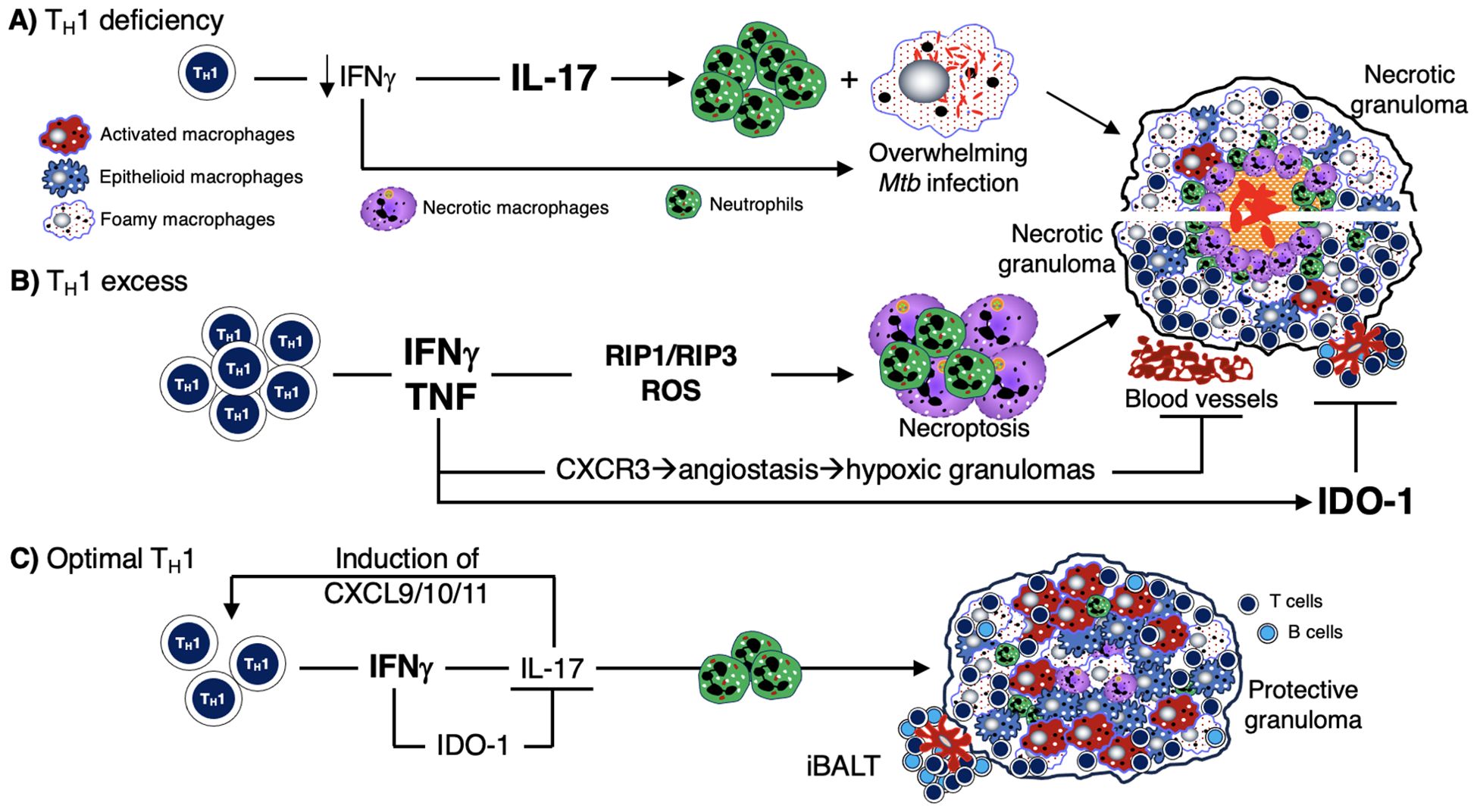
Figure 4. Mechanisms by which deficiency or excess CD4+IFNγ+ T cells predispose to TB. (A) Deficiency of TH1 (CD4+IFNγ+) T cells – as seen in advanced AIDS, genetic defects in the IFNγ-IL-12 axis, and in animal models – has been shown to increase the risk of TB and other mycobacterial infections. This IFNγ deficiency leads to an increase in IL-17 and neutrophilic influx. This combined with a lack of macrophage activation (due to insufficient number of CD4+IFNγ+ T cells) leads to an overwhelming Mtb infection of phagocytes, resulting in an unprotective, necrotic granuloma. (B) Excess TH1 cell activation and numbers lead to a secondary increase in TNF, which can then induce RIP1 and RIP3 activation, excessive ROS formation, and macrophage necroptosis. The sequence of these events leads to over-inflamed, necrotic, and unprotective granulomas. (C) There is ample evidence in both humans and experimental animal models that optimal quantity and temporal influx of Mtb-specific CD4+IFNγ+ T cells are necessary for control ± eradication of Mtb infection, in part through activation of macrophages. IFNγ also inhibits IL-17 production from TH17 cells which can induce chemokines for CD4+IFNγ+ T cells. In non-hematopoietic cells, IFNγ also induces IDO-1 production that inhibit excessive IL-17 production (via the catabolized products of tryptophan by the actions of IDO). Hence, IL-17 production is kept in check, limiting the amount of potentially harmful neutrophilic influx. IDO-1, indoleamine 2,3-dioxygenase; IFNγ, interferon-gamma; IL-12, interleukin-12; IL-17, interleukin-17; Mtb, Mycobacterium tuberculosis; RIP, receptor-interacting serine-threonine kinases; ROS, reactive oxygen species; TB, tuberculosis; TNF, tumor necrosis factor.
A fourth mechanism for the development of caseous necrosis, related to an excess of CD4+IFNγ+ T cells discussed above, is insufficient negative regulation of IFNγ. Mtb induction of IFNγ upregulates the immune checkpoint PD-1 (programmed cell death protein-1), which, in turn, inhibits IFNγ+ TH1 cells, a form of negative feedback mechanism to prevent excessive IFNγ production (212). The importance of dampening an excessive IFNγ production is evinced by studies showing that PD-1 knockout mice are paradoxically more susceptible to Mtb infection (213–216).
A fifth proposed mechanism for the development of necrosis in TB granulomas is the inability of the granuloma to overcome excessive hypoxia (described above), causing cell death and central necrosis in the granuloma. TB granulomas are hypoxic in guinea pigs, rabbits, and NHP (217). Employing dynamic PET imaging using the tracer [18F]- fluoromisonidazole, heterogenous degrees of tissue hypoxia were found in the TB pneumonia and around cavities (218). In contrast to the host-protective function of HIF-1α against TB (discussed in Section III.2.2 above), excessive HIF-1α may be harmful. Domingo-Gonzalez et al. (87) showed that when IL-17 was neutralized in Mtb-infected mice, there was higher production of HIF-1α, more hypoxic necrotic granulomas, exacerbated inflammation, and increased Mtb burden. They further showed that restricting HIF-1α activity in the mice by neutralizing IL-17 reversed both the excessive inflammation and the increased Mtb burden (87). While it is not clear how HIF-1α may induce hypoxic necrotic granulomas – since it has known properties that mitigates hypoxia and its detrimental sequelae such as induction angiogenesis and switch to glycolytic metabolism – one possible mechanism is that HIF-1α can induce mucin production and transepithelial resorption of sodium ion and water, resulting in increased mucus in the airways and thus indirectly worsening hypoxia to the lungs and the granulomas (219). Either Mtb or hypoxia-induced HIF-1α is known to individually and synergistically induce MMP-1, which can lead to lung tissue destruction and cavity formation via its collagenase activity (218). Baay-Guzman et al. (154) found a dual opposing roles for HIF-1α in mice: (i) a protective role in early stage TB (≤28 days after infection) through macrophage activation in conjunction with IFNγ and (ii) a detrimental role at a later stage of infection through HIF-1α inhibition of apoptosis of foamy macrophages, impairing an effector mechanism by which intracellular Mtb are killed.
Finally, type-1 interferons, produced by interstitial macrophages and plasmacytoid dendritic cells, have been found to exacerbate Mtb infection, recruit neutrophils, NETs formation, and promote tissue necrosis (220, 221). Not only have type-1 interferons been found to inhibit protective cytokines (IFNγ, TNF, IL-12, IL-1α, IL-1β), but they also contribute to spread of infection and lung inflammation by increasing accumulation of myeloid cells (66, 222). Type-1 interferons have been shown to contribute to necrosis through inhibition of PGE2. As noted above, PGE2 is known to prevent necrosis of Mtb-infected macrophages (61, 64–66). In turn, PGE2 (and IL-1) were found to inhibit expression of type-1 interferons (65, 223). It has also been shown that type-1 interferon induction of interleukin-1 receptor antagonist IL-1Ra mediated susceptibility to Mtb (224). Despite these host-harmful effects of type 1 interferons during Mtb infection, they have also been shown to exhibit host-protective properties in certain conditions; (i) co-administration with antimycobacterial chemotherapy; (ii) lack of IFNγ signaling from the host; (iii) BCG vaccination in certain animal models (65).
3.4 Lesion heterogeneity
The physical makeup and host-protective abilities of granulomas can vary greatly. When granulomas form in response to an infection, it gives rise to a trajectory that varies across a spectrum between complete bacterial clearance to uncontrolled infection. Distinct types of TB granulomas exist, all potentially coexisting within a single host infected with Mtb (Table 2) (138, 152, 225). Several factors can affect granuloma heterogeneity, such as host inflammatory/immune responses as well as differences in physiological-chemical properties of the different regions of the lungs (discussed in Section IV.3 below). Consequently, spatially grouped bacilli may encounter diverse granuloma environments depending on factors such as granuloma type, age, and regional immune response; in turn, Mtb can display different phenotypes depending on the microenvironment they are being enveloped (225). This variability leads to a dynamic landscape where some granulomas progress while others regress, even within a single infected individual. This notion of lesion heterogeneity is also supported by reports that sterile (mainly calcified) lesions may coexist with active TB lesions (226).
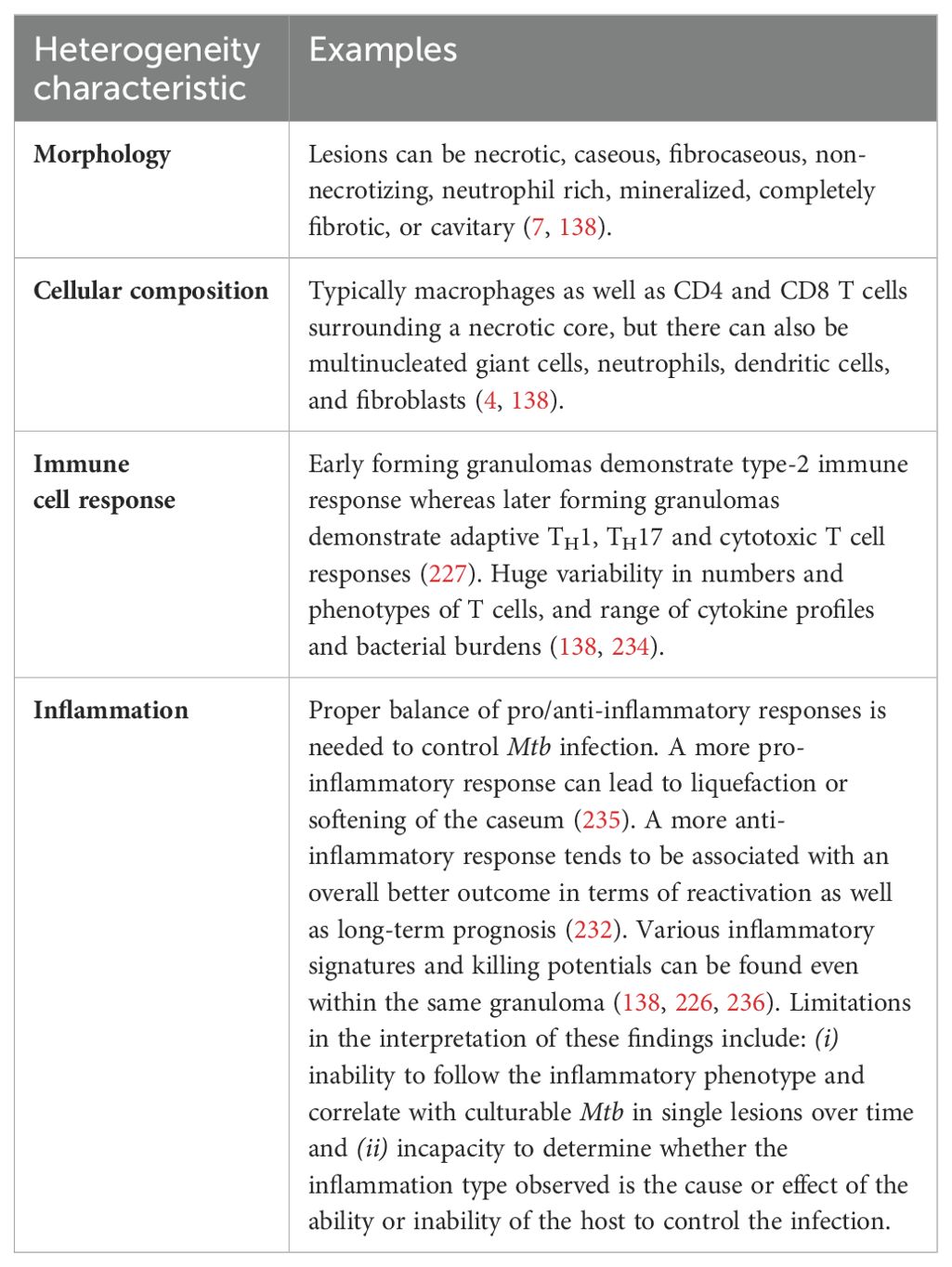
Table 2. Heterogeneity of granulomas and other TB lesions.
More supporting evidence for this claim comes from a study in which cynomolgus macaques were infected with Mtb and their developing granulomas were tracked using computed tomography (CT) combined with deoxy-2-[18F]-fluoro-D-glucose (FDG) positron emission tomography (PET) scans before necropsy was performed (226). Based on the PET-CT findings, the metabolic state of individual granulomas showed a dynamic pattern, even within the same animal. Remarkably, their findings revealed that individual granulomas mostly arise from one bacillus of Mtb (226). Furthermore, the number of culturable bacteria in the granulomas decreased significantly after time (~15-fold decrease from 4 to 11 weeks). Interestingly, compared to lesions of the NHP at 4 and 11 weeks after infection, animals with latent infection and those with active TB (defined as clinical disease and Mtb culture positivity ≥ 3 months after infection) had 10-100–fold less culturable bacteria (226). While the CFU count modestly correlated directly with FDG uptake – a sign of increased glycolysis – it does not necessarily implicate glycolysis as a cause of impaired disposal of Mtb; i.e., increased FDG uptake may be a marker for increases of either glycolysis alone or of both glycolysis plus mitochondrial oxidative phosphorylation. Another study using the cynomolgus macaque Mtb model found that the lesions showed different immune cell responses based on whether the lesions formed earlier vs later (227). Consistent with the prior Mtb culture results, earlier forming granulomas in Mtb-infected cynomolgus macaques displayed type-2 immune responses, which tend to allow for the survival of Mtb within the granuloma, whereas later forming granulomas demonstrate adaptive TH1, TH17 and cytotoxic T cell responses, which tend to be more effective at killing Mtb bacilli and controlling the infection (227). This heterogeneity not only underpins the diversity in infection outcomes between lesions but also complicates treatment efforts. Individual lesions may exhibit varying responses to antibiotics, posing challenges in achieving consistent therapeutic outcomes.
TB is classically divided into “active” vs. “latent” forms, although the current paradigm acknowledges a clinical continuum that also include incipient and subclinical TB (228). Perhaps reflecting these clinical phenotypes is the presence of a spectrum of pathologic lesions that may be seen in human and experimental TB. Following low-dose (~25 bacilli) infection of cynomolgus macaque, about 40% develop active disease whereas 60% develop latent infection (229, 230). In those macaques with active TB, some lesions showed replicating high-burden mycobacteria and necrotic granuloma whereas others showed fewer mycobacteria, more fibrosis, and resolved granulomas, mirrored by PET-CT imaging that demonstrated both metabolically active and metabolically inactive granulomas. Interestingly, even with culture confirmed negativity of NHP with latent infection, both PET-positive and PET-negative lesions may be seen with the caveat that the degree of PET-CT positivity correlates with glucose uptake by the cells and does not necessarily inform the level of control of the Mtb infection. In an antibiotic treatment study of pulmonary TB in cynomolgus macaques and in humans, decline in PET activity correlated with reduced bacterial load in both treatment groups and at necropsy in the NHP, with also a reduction in pulmonary pathology (231). Another study showed that a substantial number of humans treated for active drug-susceptible TB remained PET-positive along with positive Mtb mRNA in sputa, indicating either subclinical active disease or resolved TB with residual dead Mtb (232). These findings show that dividing clinical TB into “active” or “latent” is an oversimplification, and that the dominant type among a spectrum of lesions is likely to dictate the clinical disease (6, 152). Indeed, a recent review categorized TB into eight different states, ranging from elimination by various mechanisms to disseminated disease (233).
3.5 Does aerobic glycolysis in TB lesions–granulomas protect against or predispose to TB?
Answering this question is challenging because it is difficult to know in any distinct granuloma that is failing – defined as unopposed Mtb replication and worsening pathology – whether the presence of aerobic glycolysis is the cause of the failure or an attempt to rectify the failure. Furthermore, since it has been shown that seemingly distinct granulomas may in fact be different “branches” of the same granuloma (237, 238), the task in answering this question is even more daunting. Nevertheless, we will summarize the reported data.
On the one hand, there are indirect evidence that aerobic glycolysis increases host-susceptibility to TB: (i) PET-CT is commonly employed to detect TB lesions in NHP because this avoids the need to sacrifice the animal while obtaining more data in a temporal fashion. While PET-CT has been cited as a radiographic marker of aerobic glycolysis (a Warburg effect) (239), it is not specific for aerobic glycolysis alone since it is only a marker for glucose uptake, which can be seen with increased glycolysis (either aerobic or anaerobic) alone or with increase of both glycolysis and oxidative phosphorylation. Compared to the more TB-susceptible rhesus macaques, the more TB-resistant cynomolgus macaques infected with Mtb showed reduced dissemination of infectious granulomas and fewer PET-positive/necrotic lymph nodes (240). While this finding may suggest that aerobic glycolysis increases host susceptibility to TB in the rhesus macaques, one could contrarily argue that the increased PET-CT positivity (increased glycolysis) was an attempt to control the Mtb infection in the lymph nodes (240); (ii) in cancer cells, there is generally an inverse relationship between glycolysis and autophagy; i.e., increased glycolysis contributes to decreased autophagy (241) and loss of autophagy switches energy metabolism to glycolysis (242); in contrast to cancer cells, the glycolytic byproduct lactate increases autophagy in macrophages (see below); (iii) since the lung apices are more alkalotic in upright humans and alkalosis drives aerobic glycolysis (due to increased activity of hexokinase and phosphofructokinase activity (243, 244), key enzymes in the glycolytic pathway), one could also posit that perhaps an increase in glycolysis in the upper lung zones is (partly) responsible for the increased involvement of the upper lung zones in post-primary TB.
On the other hand, there is a collective strong argument that aerobic glycolysis of macrophages is host-protective against TB: (i) Mtb infection of macrophages inhibits aerobic glycolysis and increases fatty acid metabolism by mitochondria; this inhibition of aerobic glycolysis increased mycobacterial growth and decreased the production of certain pro-inflammatory mediators (40, 245–247); (ii) phagocytosis is augmented by glycolysis (40, 248); (iii) using live Mtb, HIF-1α switches macrophage metabolism to aerobic glycolysis which polarizes macrophages to the M1 phenotype (and secondarily to the TH1 phenotype) against Mtb (162, 163) (Figure 5); (iv) since the ability of mycobacterial ESX-1 to convert glycolysis to ketosis induces the formation of foamy macrophages – which are known be impaired in controlling intracellular mycobacteria due to decrease of both autophagy and lysosomal acidification (39) – lending credence that glycolysis is host-protective; (v) lactate, the byproduct of glycolysis, increases autophagy, possibly through hyperlactylation of PI3 kinase (39, 41, 42); (vi) while the aforementioned inverse relationship between protection and aerobic glycolysis in Mtb-infected macaques (240) suggests that aerobic glycolysis confers susceptibility, one could argue that the greater protection against TB in the cynomolgus macaques prevented the need for an increase in host-protective aerobic glycolysis; (vii) indirect evidence that aerobic glycolysis is host-protective is that calcification of granulomas – a sign of well-controlled TB lesions – is more likely to occur in an alkaline environment, which would also promote aerobic glycolysis; (viii) the central parts of necrotic granulomas are considered to be more hypoxic and have greater HIF-1α expression; since HIF-1α increases glycolysis, whose byproduct lactate augments autophagy, this may account for the low Mtb number in the central necrotic area of granulomas (Figure 5, left-hand side) (161, 163). Because immune cells at the granuloma periphery are less hypoxic and have less HIF-1α activity, there is greater burden of Mtb in the outer regions of the granulomas (Figure 5, right-hand side). Interestingly, increased aerobic glycolysis may participate in a positive feedback phenomenon as increased autophagy – due to greater macrophage activation – also promotes aerobic glycolysis (43, 44). However, analyzing for the presence of aerobic glycolysis is challenging and requires more testing than simply measuring lactate levels and transcriptional analysis of glycolytic enzymes (40).
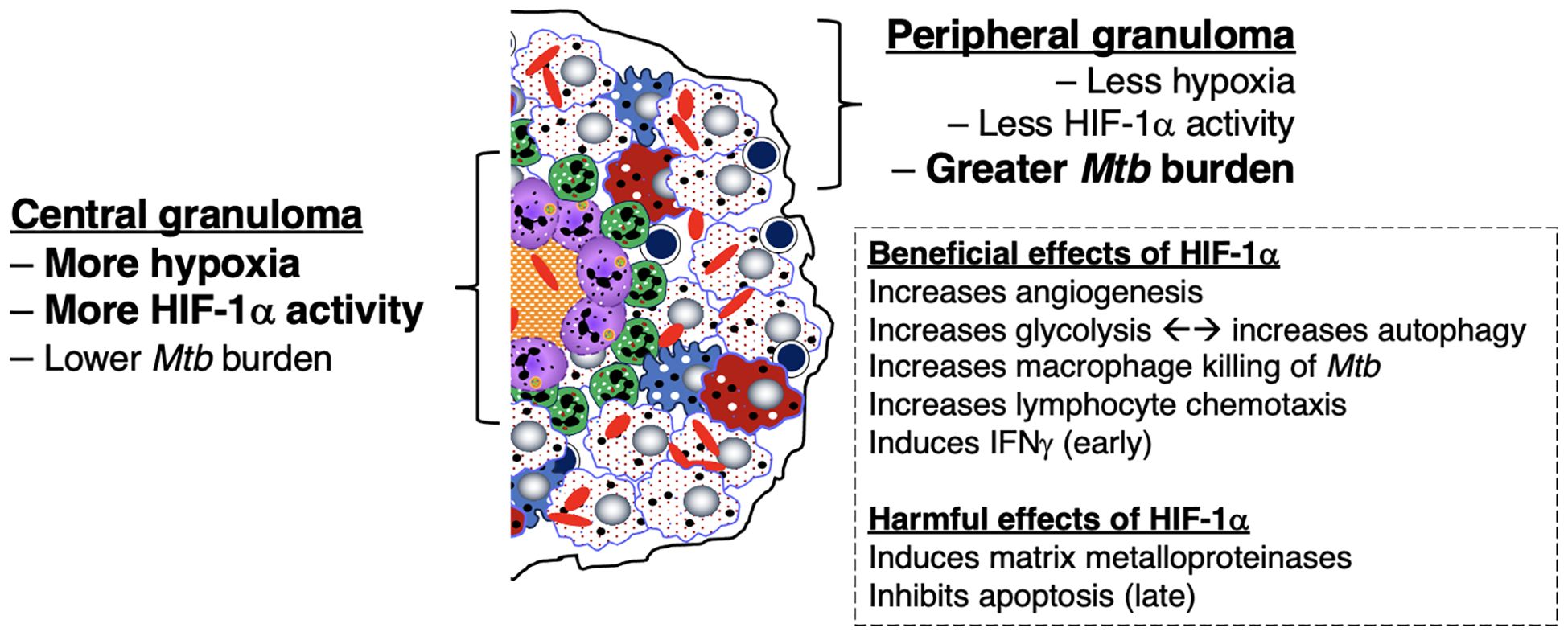
Figure 5. Differential expression of hypoxia, HIF-1α, and downstream effects in the central and peripheral regions of the necrotic granuloma. The central region of a necrotic granuloma is more hypoxic. As a result, there is greater HIF-1α expression with a panoply of downstream effects due to HIF-1 activity. One immune mechanism induced by HIF-1 is aerobic glycolysis, which in turn induces autophagy through the byproduct lactate. Hence, the central necrotic area of an intact human TB granuloma is usually sterile or with low bacterial burden. In contrast, the peripheral region of the granuloma less hypoxic with less HIF-1α induction. In the presence of macrophages that are less effective in killing Mtb (as shown by the foamy macrophages), there is less ability to kill Mtb, as shown by the greater bacilli number. However, in the presence of activated macrophages and T cells, the peripheral granuloma may also be efficient in either killing or controlling the Mtb infection. HIF-1α, hypoxia-inducible factor 1-alpha; IFNγ, interferon-gamma; Mtb, Mycobacterium tuberculosis; TB=tuberculosis;.
3.6 Either insufficient or excessive immune response impairs the ability of host granulomas/lesions in controlling Mtb infection
There is strong evidence that lack of CD4+IFNγ+ T cells or IFNγ signaling, as seen in advanced AIDS patients (73, 249) and individuals with genetic mutations of various components IFNγ-IL-12 axis (250–253), respectively are vulnerable to TB and non-tuberculous mycobacterial infections. These clinical evidences are supported experimentally by increased susceptibility of IFNγ-knockout mice to TB (254, 255). Thus, one would predict that genetic disruption of the immune checkpoint PD-1, which increases CD4+IFNγ+ T cell activation, would enhance the ability of the host to control an Mtb infection. Yet, three studies have shown that mice with the PD-1 gene knocked out, which would increase CD4+IFNγ+ T cell activation, are more susceptible to Mtb infection with not only more severe pathology but greater Mtb burden (213, 214, 216). In contrast to the findings with the PD-1 gene knockout mice, wild type mice that were administered blocking antibody to TIM3 (another immune checkpoint) or the TIM3 knockout mice were more resistant to Mtb with reduced the bacilli burden in the lungs compared to their respective control mice (256). Similarly, administration of an anti-CTLA-4 antibody decreased Mycobacterium bovis (M. bovis) Bacillus Calmette Guérin (BCG) burden by one-half log at 6 weeks post-infection (257). Plausible mechanistic explanations for these differences include: (i) differential capacity among the various immune checkpoints (PD-1 vs. TIM3) to inhibit T effector cell function; (ii) the disparity between partial inhibition of immune checkpoints with an antibody vs. complete genetic disruption of an immune checkpoint; and (iii) separate background strains of mice used (C57BL/6 vs. BALB/c) wherein mice that are intrinsically and relatively more immunosuppressed (e.g., the BALB/c background in the TIM3 gene knockout mice) may be more likely to benefit from immune checkpoint inhibitors.
Either deficiency or excess of other molecular and cellular components that impair the host’s ability to control Mtb infections are listed in Table 3. Thus, we posit that a “modest” activation of these components, at the appropriate times in the course of the Mtb infection, are necessary for optimal control of the infection. This “Goldilocks principle” – illustrating the concept of finding the “optimal middle ground” based on the English fairy tale Goldilocks and the Three Bears – is also shown graphically (Figure 6).
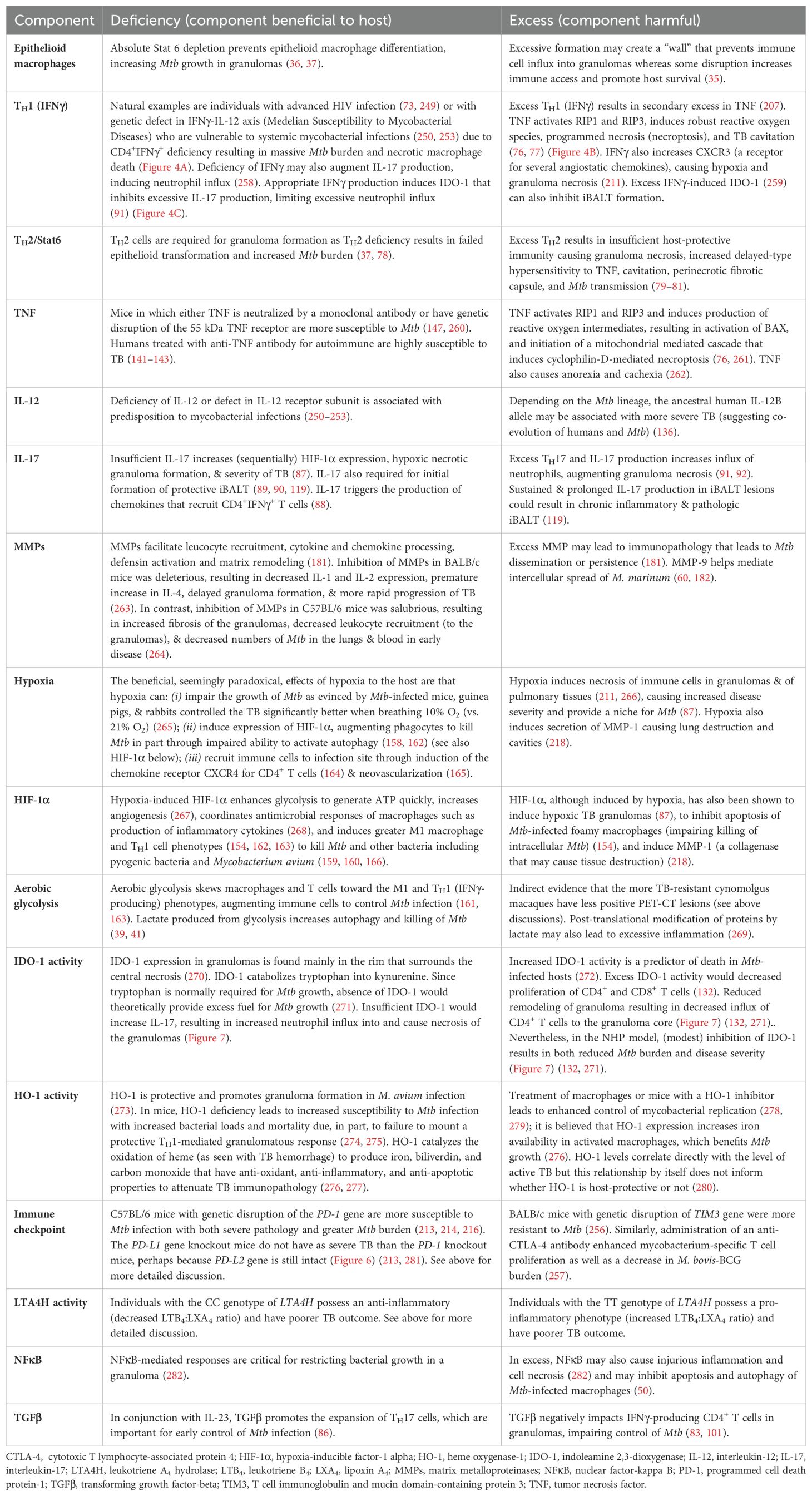
Table 3. Deficiency or excess of specific components that impairs host control of Mtb.
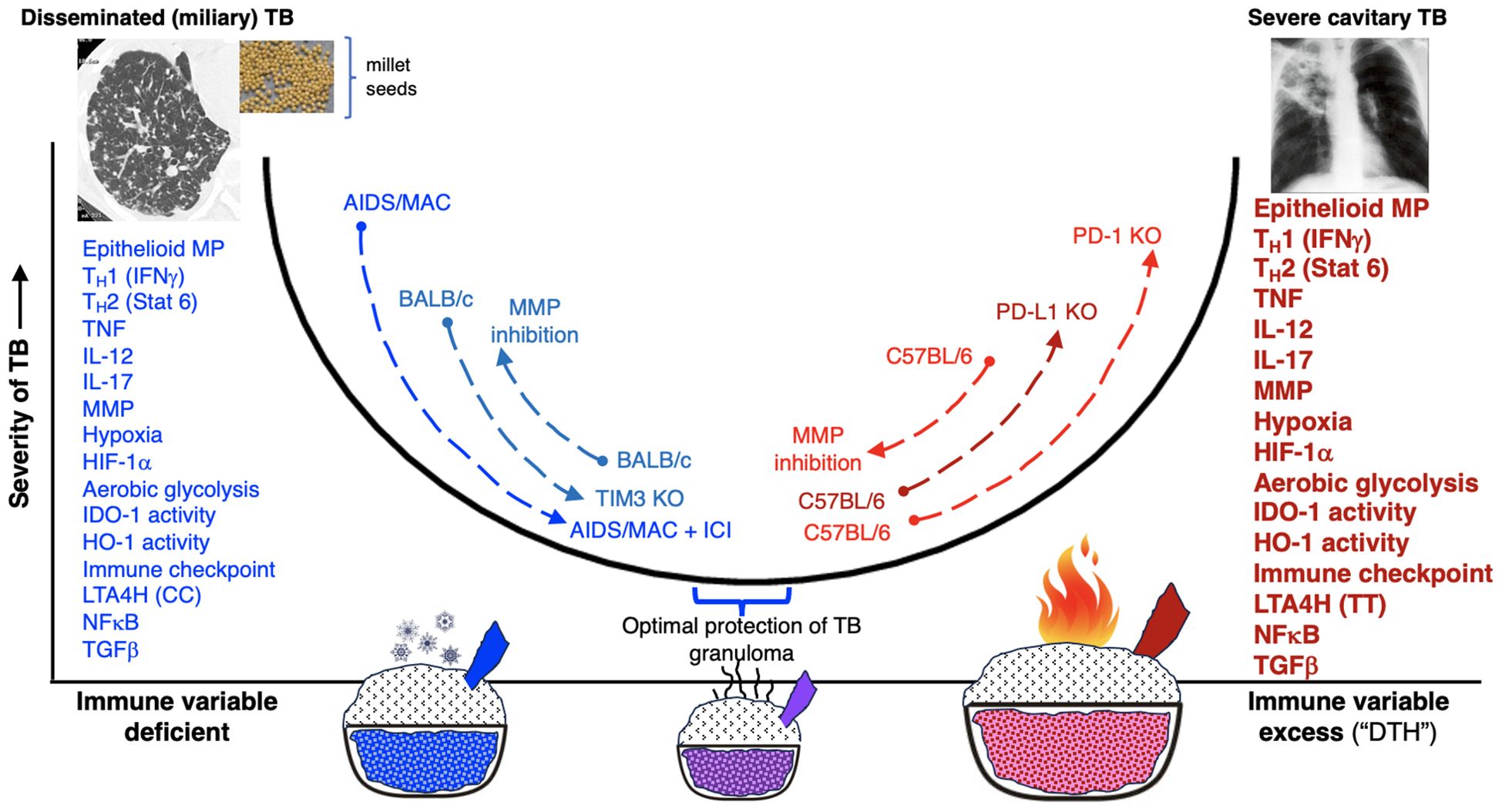
Figure 6. Either excessive or deficiency of molecular or cellular immune elements may predispose to unprotective granuloma or defense against Mtb. The diagram illustrates that either deficiency or excessive of specific immune components (x-axis) may result in increased susceptibility and severity of TB (y-axis). Using the immune checkpoint inhibitors as an example, we posit that in conditions where the host is either severely immunocompromised, immune checkpoint inhibition would be beneficial against mycobacterial infection, as exemplified by: (i) advanced AIDS patient with severe mycobacterial infection that improved with an anti-PD-1 antibody and (ii) TIM3 antagonism or TIM3 gene knockout mice are protected against mycobacteria in the less immunocompetent BALB/c mice. In contrast, in hosts with better preserved immune function, immune checkpoint inhibition results in a hyperinflammatory state, worsening control of mycobacterial infections, as exemplified by the mice with genetic disruption for PD-1 or PD-L1 gene. Perhaps the severity of TB is not as severe in the PD-L1 knockout mice than the PD-1 knockouts because in the PD-L1 knockout mice, PD-L2 is still intact and thus the enhanced immune activation with the genetic disruption of PD-L1 is not as severe as of PD-1. We have also shown other molecular and cellular components in which deficiency or excess predispose to TB. See also text and Table 2 for further discussion. AIDS, acquired immunodeficiency syndrome; HIF-1α, hypoxia-inducible factor 1-alpha; HO-1, heme oxygenase-1; ICI, immune checkpoint inhibitor; IDO-1, indoleamine 2,3-dioxygenase; IFNγ, interferon-gamma; IL-12, interleukin-12; IL-17, interleukin-17; LTA4H, leukotriene A4 hydrolase; MAC, Mycobacterium avium complex; MP, macrophages; MMP, matrix Mtb, Mycobacterium tuberculosis; NFκB, nuclear factor-kappa B; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand-1; TB, tuberculosis; TGFβ, transforming growth factor-beta; TNF, tumor necrosis factor.
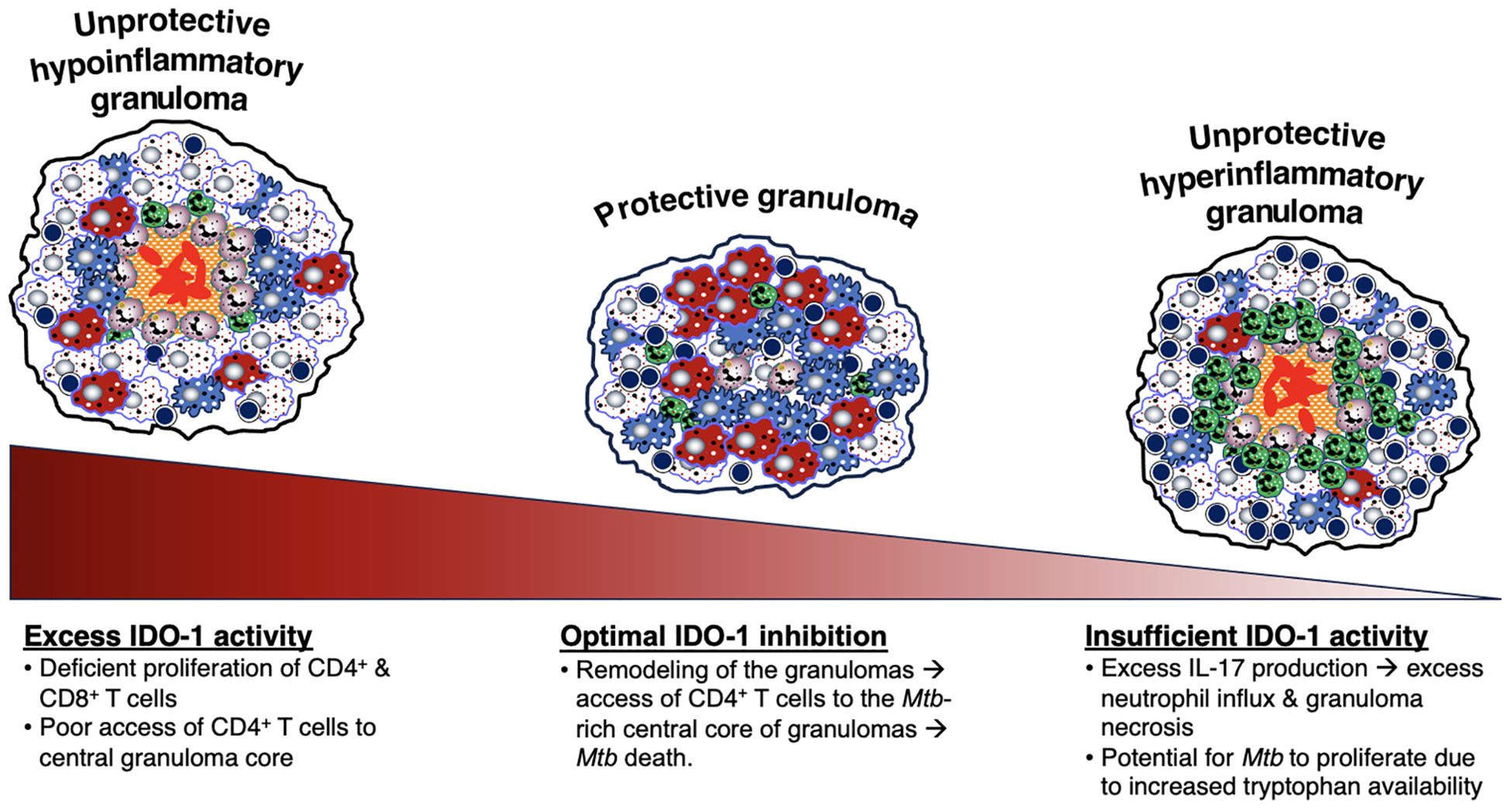
Figure 7. Relative IDO-1 activity helps dictate the level of protection by TB granulomas. IDO-1 activity can also be an indicator of the protectiveness of a granuloma. When there is an optimal level of IDO-1 inhibition, the remodeling of the granulomas allows access of CD4+ T cells to the central core of the granuloma, resulting in Mtb death. However, excessive or insufficient IDO-1 activity leads to an unprotective granuloma. An excess of IDO activity results in an unprotective, hypoinflammatory granuloma, characterized by deficient proliferation of CD4+ and CD8+ T cells and poor access of CD4+ T cells to the granuloma core, allowing Mtb to grow within the center. On the other hand, an insufficient level of IDO-1 activity leads to an unprotective, hyperinflammatory granuloma, characterized by excess IL-17 production and excess neutrophil influx and granuloma necrosis. IDO-1 is also responsible for the catabolization of tryptophan (a molecule necessary for Mtb growth) into kynurenine. A deficiency of IDO-1 would in turn increase levels of tryptophan and allow for more Mtb growth. IDO-1, indoleamine 2,3-dioxygenase; IL-17, interleukin-17; Mtb, Mycobacterium tuberculosis;.
4 Characteristics of human TB and granulomas
In humans, active TB is classically divided into primary TB and post-primary TB, the latter known as reactivation TB, usually located in the lungs after establishment of systemic host-protective immunity in primary TB (283). Since Mtb is acquired through inhalation of Mtb-containing droplet nuclei less than 5 µm that reach the terminal bronchioles and alveoli, the granulomas in primary TB are located more commonly in the lower lung zones where the ventilation is greater (284). In contrast, the location of post-primary TB is typically more prevalent in the upper lung zones, especially the lung apices but also the superior segments of the lower lobes. These differences suggest that the immunological correlates of protection of primary TB and post-primary TB may be quite different as perhaps evinced by BCG protection against primary TB but less successfully with post-primary TB (285).
4.1 Self-limiting primary TB and primary progressive TB
In immunocompetent individuals, primary TB – most commonly affecting the lungs but may present as extrapulmonary TB – typically remains asymptomatic and resolves spontaneously. It is most often detected radiographically as a Ghon lesion (a sequela of untreated primary TB characterized by a lung nodule comprised of caseous granulomas surrounded by a fibroblastic rim), a Ghon complex (the combination of a nodule with associated draining mediastinal lymphadenopathy), or a Ranke complex (characterized by calcification of the Ghon complex) (286). On imaging, such lesions may be suspicious for lung cancer prompting surgical removal, but diagnosed histopathologically as TB-related (287). A relatively small number of bacilli adapt to the granuloma micro-environment and persist as asymptomatic latent infection. Autopsy studies revealed that granulomas in latent TB tissues exhibited lower cellularity and inflammation but displayed more prominent sclerosis, fibrous encapsulation, and calcification compared to their active counterparts (152). Histological analysis of latent TB lesions from individuals who died of non-TB-related causes has revealed caseous and fibrocaseous characteristics (288).
Ghesani and co-workers (289) reported the PET-CT findings of five household contacts of active TB patients before and during treatment for latent TB infection. These five individuals had no evidence of active TB and asymptomatic (“latent”) TB infection was established based on positive QuantiFERON testing and negative chest radiographs. This report showed that: (i) FDG uptake was increased in the thoracic lymph nodes in four of the five contacts; (ii) on repeat PET-CT testing while on treatment for latent TB infection, the FDG uptake decreased or became negative in three of the four and no change in one with initial mild FDG uptake; (iii) in the one patient with initial negative FDG uptake in the chest, there was evidence of remote latent TB infection as evinced by calcified granuloma in the lung parenchyma and ipsilateral calcified hilar lymph node (Ranke complex); (iv) the QuantiFERON results correlated highly with the intensity of the FDG uptake, indicating the PET positivity likely reflected a type 1 (CD4+IFNγ+) immune response (289).
Some individuals, perhaps with sub-optimal and not necessarily severely immunocompromised immune function may develop primary progressive TB. Except for a history of a relatively recent active TB exposure and the location of disease (more likely to be mid to lower lung zones), such patients are essentially indistinguishable from those with post-primary TB.
4.2 Post-primary TB
Post-primary TB accounts for the majority (≥80%) of active TB cases. In the 19th and early 20th centuries, pathologic analyses of human TB tissues were commonly performed on subjects with varying profiles of disease severity. These studies were conducted prior to the discovery of effective antibiotic therapy and thus also reflect the natural history of TB (134, 288, 290–292). Dr. Robert Hunter and his colleagues have critically reviewed these older literature as well as microscopically examined treatment-naïve TB lung specimens (287, 293–297). Two characteristic lesions of human pulmonary TB emerged: caseating granulomas and tuberculous pneumonia (Figures 8A, B) (293). Caseating granulomas were reflective of primary TB, which may be self-limiting or progressive (Figure 8A). While post-primary TB may begin with degeneration of primary granulomas (Figure 8A), some contend that it starts as a nascent pneumonic process which then spreads endobronchially resulting in airway obstruction and lipoid pneumonia, the latter due to the presence of lipid-laden (“foamy”) macrophages and DEC-205+ dendritic cells infiltrating the airway lumina and alveolar spaces (Figure 8B) (33). However, because most surgically resected or autopsy TB cases represent advanced disease, it is challenging to determine with certainty whether endobronchial spread is a primary or secondary process, or a combination of both. The early inflammation that occurs with this post-primary pneumonic process was largely attributed to a Type IV hypersensitivity reaction to Mtb antigen and not to an increase in the number of viable Mtb since the early stages of this pneumonic process were very often paucibacillary (133, 298, 299). The formation of necrotic granulomas due to host-mediated bacillary destruction rather than proliferation is supported by recent RNAscope and immunohistochemistry studies (300). In this study, a significant amount of Mtb mRNA and Ag85B was detected in human necrotic granulomas in the lungs and lymph nodes of a confirmed TB patient, despite being Ziehl-Nielsen-negative (300).
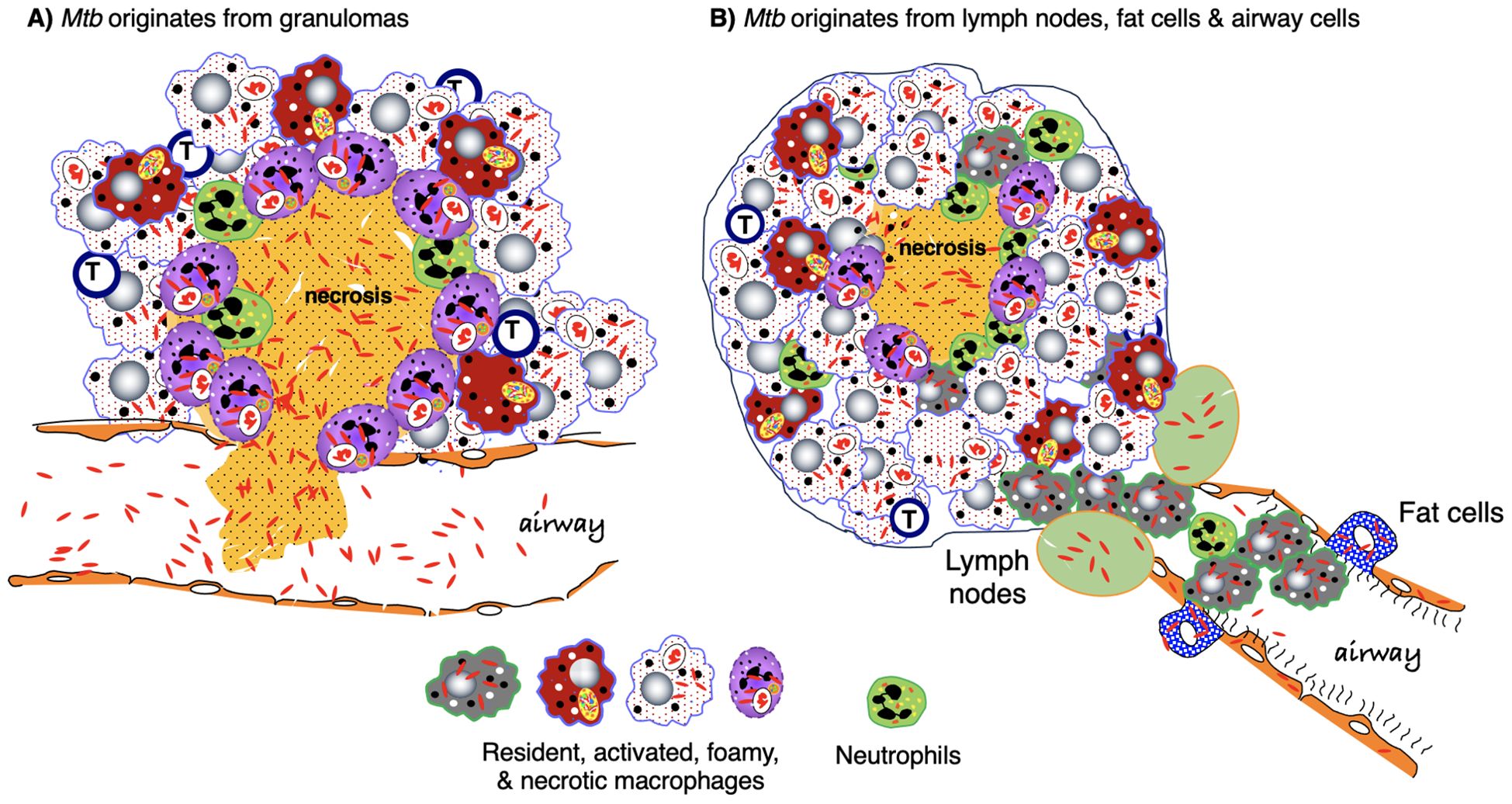
Figure 8. Origin of Mtb and early events in the development of post-primary TB. (A) An often-cited description for the evolution of post-primary (reactivation) TB is that immune dysfunction results in progressive necrosis and liquefaction of the granuloma, exuberant multiplication of dormant Mtb, disruption of the integrity of the granuloma wall, and spillage Mtb-filled necrotic granulomatous content into the airways. Pneumonia forms due to endobronchial spread of granuloma content. (B) An alternative description of post-primary TB is based on the paradigm chronic stable granulomas are mostly sterile and that the dormant Mtb that “reawaken” originated from lymph nodes. fat cells, and airway epithelial cells causing a bronchogenic lipoid pneumonia. Granulomas may form around pockets of necrotizing pneumonia. As discussed in the text, it is plausible that both reactivation models occur. Mtb, Mycobacterium tuberculosis; TB, tuberculosis.
These findings are further corroborated by another study that characterized the micro-anatomical environment of human TB lung tissues using micro-computed tomography (µCT), histopathology, and immunohistochemistry to determine the three-dimensional shape of the TB granulomas and their spatial relationship to airway and vascular structures (237, 238, 301). Consequently, unlike the typical two-dimensional description of TB granulomas as round discrete structures, they found that human necrotic TB granulomas demonstrated cylindrical, branched morphologies which are connected to the airways and shaped by the bronchi, supporting the bronchogenic spread of Mtb. Another clinically relevant finding is that the lack of vascularization within obstructed bronchi likely resulted in further necrosis poor antimicrobial penetration of affected lung segments, and the development of functional drug resistance (237). Additionally, utilizing histopathology and genome-wide transcriptomic analysis, immunological profiling of human lungs surgically resected for recalcitrant active TB showed considerable variation in T cell influx in different granulomas as well as significant differences in gene expression of the inflammatory response, immune cell trafficking, and cell mediated immune response (5).
The post-primary pneumonic process can either resolve spontaneously with resolution of the airway obstruction (thought to occur in the majority of cases) or develop necrosis and a caseating pneumonia (in ~5% of cases) (Figure 8B). One Mtb component that may be responsible for the caseating pneumonia is the glycolipid TDM (185, 188). If caseous necrosis develops – often linked to a rapid surge in Mtb numbers (133, 297) – the infection may progress in one of two ways: either through the formation of a post-primary granuloma around the caseous pneumonia, attempting to contain the infection, or through cavitation, which leads to even greater Mtb replication and facilitates their expulsion into the airways, enabling person-to-person transmission. The pathogenesis of cavitary disease may arise not only from the caseating necrosis associated with the pneumonic process but also from the degradation of fibrous tissue at the periphery of secondary granuloma by MMP-1 and MMP-9 (302–306). Additional factors for cavitation include pulmonary infarction resulting from vasculitis (287, 294), and an excessive immune response (285, 307, 308). Interestingly, the lack of necrotizing granulomas in most standard mouse strains may be linked to the absence of lung expression of the mouse ortholog of MMP-1 (303, 304).
4.3 Where do the Mtb originate from in post-primary TB?
The precise location of dormant Mtb before their reactivation, leading to post-primary TB, remains a topic of debate. Some argue that dormant Mtb residing within primary granulomas break out as the integrity of the granulomas declines due to waning immunity (Figure 9A). Subsequently, Mtb enters the airway lumina, leading to endobronchial spread. Alternative evidence suggests that Mtb originates from normal-appearing lung tissues, fat cells, and thoracic lymph nodes since primary granulomas are noted to be sterile after five years (21, 294, 309). However, unless all innumerable microscopic granulomas in the lungs of primary TB can be individually dissected and cultured – logistically an untenable task – it is impossible to exclude primary TB granulomas as potential sources of reactivated Mtb during development of post-primary TB. Furthermore, both hypotheses are not necessarily mutually exclusive, leading to a reasonable supposition that both quiescent lung granulomas and other tissues (fat cells, lymph nodes) are potential sources of reactivated Mtb. Following cavitation, endobronchial spread may occur to all other parts of the lungs (310).
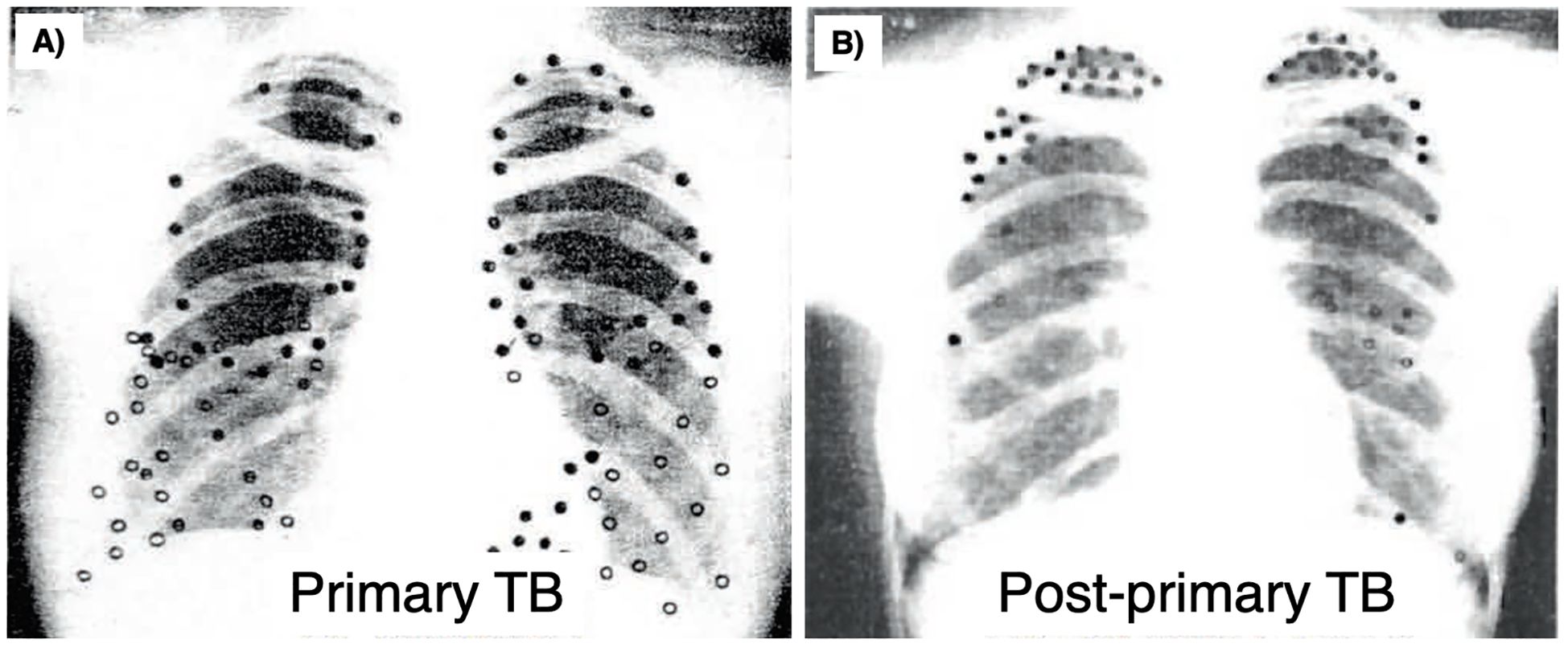
Figure 9. Distribution of lesions in primary and post primary TB. The localization of lesions of (A) primary and (B) post-primary TB of several people were plotted on an X-ray. Each dot marks the location of an individual’s lesion. The distribution of primary TB is consistent with chance distribution of air-borne infection while most post-primary lesions were in the upper lobes, especially the apical segments. Reprinted with permission of the American Thoracic Society. Copyright © 2024 American Thoracic Society. All rights reserved. Medlar E.M. The pathogenesis of minimal pulmonary tuberculosis: A study of 1,225 necropsies in cases of sudden and unexpected death. Am Rev Tuberc 1948; 58: 583-611. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society. TB, tuberculosis.
Most primary TB lesions reside in the lower lung zone (Figures 9A, 10A) because inhaled Mtb are preferentially deposited in the more ventilated lower lung zones. However, primary TB lesions can also form in the upper lung zones, albeit less frequently (Figure 9A) (294, 298). Nonetheless, post-primary TB is more common in the upper lobes (especially apices) and superior segments of the lower lobes (Figures 9B, 10A). The exact reason for the differing lung distribution between primary TB and post-primary TB remains unclear, as it seems unlikely to be solely attributed to the initial residence of dormant Mtb. We suggest that this locational disparity arises from the preferential reactivation of primary lesions in the upper lung zones, influenced by fundamental physiological and chemical principles.
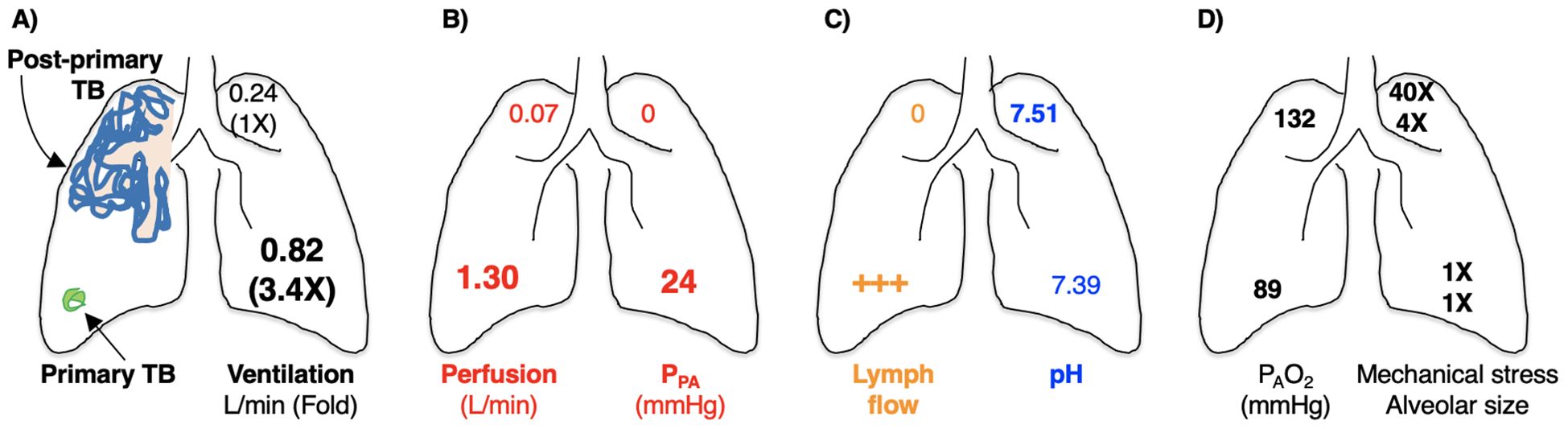
Figure 10. Anatomical-physiological-chemical mechanisms by which post-primary TB is more common in the upper lobes of the lungs. Based on differences in the physiological-chemical properties between the upper lobes and lower lobes, primary Mtb lesions located in the upper lobes, while fewer than that in the lower lobes, are more likely to reactivate. (A) Diagram showing that primary TB is located mostly in the lower lung zone (green circular structure that represents a calcified granuloma – Ghon complex). This distribution of primary TB is due to greater (3.4X) ventilation in the lower lobes than the upper lobes of an upright human. (B) In the upright human, there is decreased lung perfusion pressure due to lower pulmonary artery pressure in the upper lobes. This decreased perfusion will result in decreased influx of immune cells and soluble mediators to the upper lobes in the upright position. (C) Lymph formation and flow are also significantly decreased in the upper lobes, resulting in increased accumulation of mycobacteria and antigens in the upper lobes and contributing to increased hypersensitivity reactions and lung damage. pH in the upper lobes is more alkalotic which induces glycolysis and, in turn, increases autophagic clearance of Mtb by the macrophages. (D) The PAO2 is also significantly greater in the upper lobes than the lower lobes, which favors Mtb growth. The mechanical stress and alveolar size of the upper lobes are also greater (by 40X and 4X, respectively) than the lower lobes, predisposing this area to the development of cavities, especially with large inspiratory efforts. Mtb, Mycobacterium tuberculosis; PAO2, partial pressure of alveolar oxygen; TB, tuberculosis.
First, while there is less ventilation to the upper lung zones in the upright position in humans, there is much less perfusion in the upper lung zones (Figure 10B). The relative overall decrease in blood perfusion to the upper lung zones will also result in decreased influx of both immune cells and soluble factors such as antibodies and complement components (311, 312). Second, because the lung apices have decreased capillary perfusion pressure in the upright human, there is decreased lymphatic fluid formation in the upper lung zones, resulting in reduced lymphatic drainage of Mtb antigen/debris removal to lymph nodes and reduced T cell priming (Figure 10C) (313). Third, there is relative alkalosis in the upper lung zones compared to the lung bases (~pH 7.51 vs. 7.39, respectively) due to increased ventilation:perfusion ratio in the upper lung zones (314). Alkalosis promotes aerobic glycolysis over oxidative phosphorylation (315, 316) and this increase in glycolysis can hinder autophagic flux (241), impairing a key mechanism for killing intracellular Mtb. Supporting this hypothesis, experimental TB in rabbits predominantly affects the dorsal regions of the lungs; however, when the rabbits are kept in a vertical position, the infection primarily targets the superior parts of the lungs (317, 318). Fourth, the relatively greater O2 delivery to the upper lung zones (Figure 10D) creates a more hospitable environment for Mtb replication (313, 319). The reason that there is greater O2 tension in the upper lobes is that perfusion by deoxygenated blood arising from the pulmonary artery in a low pressure right ventricular circulation is surpassed by oxygenated blood flow from the bronchial artery, which is a high-pressure left ventricular circulation; i.e., compared to the lower lobes, there is relatively less O2 extracted from the upper lobe alveoli by the red blood cells in the upright human. This notion is supported by evidence that in marginal human donor lungs, there is significantly better oxygenation to the upper lobes than the remaining lungs, as measured by the ratio PaO2:FiO2 (partial pressure of O2 in the blood:inspired O2 concentration) (320). One piece of supporting evidence for this mechanism is that Mtb-infected guinea pigs breathing 10% oxygen exhibit less severe TB pathology compared to control animals breathing ambient air (265, 317). Conversely, Mtb-infected mice breathing 60% O2 had significantly higher burden of viable Mtb in the lungs than control mice breathing 20% O2 (317).
4.4 What causes pulmonary cavitation?
There are two prevailing models regarding the formation of TB cavities in the lungs, each corresponding to the two main models of post-primary TB development. It is likely that both models occur with varying frequencies within individuals. The first model suggests that a stable granuloma harboring dormant Mtb becomes destabilized due to “immunosuppression,” leading to liquefaction of the central caseous necrosis and subsequent multiplication of Mtb. The contents of the cavity then erode into an airway, allowing air to enter the emptying granuloma and expanding the space. The second model proposes that post-primary TB initially presents as bronchogenic lipoid pneumonia, which then undergoes necrosis. This triggers a secondary granulomatous reaction, which forms around the necrotizing pneumonia, resulting in cavitation due to the necrosis and expectoration of the necrotic material. One finding that challenges both models is that air analysis reveals most cavities contain lower oxygen levels and higher carbon dioxide levels than those measured in the airway lumen (321). However, it is plausible that intermittent or progressive obstruction of the airway emanating from the cavity could lead to hypoxia and hypercarbia within the cavity lumina. Once cavities form, further necrosis and disruption of the cavitary walls may allow extracellular mycobacteria to access the blood and lymphatic vessels, as well as the airways, facilitating their spread to other individuals through aerosolization (322). Tuberculous cavities typically harbor high concentrations of bacilli, and their presence heightens infectiousness due to their communication to the airways and subsequent aerosol transmission of Mtb. While necrosis is a prerequisite for cavitation, it alone is insufficient to ensure its development.
Recently, a study compared the cell-specific gene expression profiles of sarcoidosis granuloma (usually not associated with necrosis) and human TB granuloma (often associated with necrosis). Compared to sarcoidosis, the most upregulated gene in TB was the collagenase matrix metalloproteinase-1 (MMP-1) (305). MMP-1 is a collagenase that is essential for cavity formation through the degradation of the fibrous extracellular matrix that surrounds granulomas (304). Infection with Mtb induces MMP-1 and MMP-9 production in macrophages (302). During the early stages of granuloma formation, MMP-1 expression increases in the surrounding epithelial cells and leukocytes (181). While MMPs facilitate leucocyte recruitment, cytokine/chemokine processing, and defensin activation, excess MMP activity may lead to damaging immunopathology including granuloma destruction and pathogen dissemination or persistence (181, 303). Inhibition of MMP with a broad spectrum MMP inhibitor yielded conflicting results, but this may be due to timing of the inhibition in relation to the Mtb infection (263, 264). Early MMP inhibition (at the time of Mtb infection) with a broad spectrum MMP inhibitor (BB-94) in mice resulted in decrease in pro-inflammatory cytokines (IL-1 and IL-2), premature increase in IL-4, delayed and smaller granuloma formation, and more rapid progression of disease characterized by small lung nodules suggestive of miliary TB (263). In contrast, delayed administration of BB-94 (18 days after Mtb inoculation) resulted in increased collagen deposition within early granulomas with decreased Mtb burden (264).
Mechanical factors may also account for why TB cavities are more common the upper lung zones. In the upright human, there is significant increase in both physical stress and alveolar sizes in the upper lung zones during inspiration, ~40 times and ~4 times greater, respectively, than the lower lung zones (Figure 10D). These physical differences may subject the upper lung zones to increased physical stress and along with actions of various metalloproteinases produced by immune cells that degrade lung tissues, increase vulnerability to cavitary formation. Based on these plausible pathogenic mechanisms, it has been suggested that historical TB treatment of enforced bed rest may be an effective adjunctive remedy for TB (312).
Animal models are essential for studying TB pathogenesis and developing treatments and vaccines. As with all disease models, it is vital to select species that replicate key characteristics of human TB, including the development of caseous necrosis surrounded by a collagen rim, hypoxia, both intracellular and extracellular populations of bacilli, liquefaction, and cavity formation (152). In this context, we will first discuss select animals with natural Mtb complex infections, such as cattle and elephants, followed by a review of experimental animal models including zebrafish, mice, guinea pigs, rabbits, and non-human primates (152, 266, 323–327).
5 Characteristics of natural TB infections of animals
5.1 Cattle
Cattle are a natural host of Mycobacterium bovis, an Mtb complex organism (328). While cattle can be experimentally infected with Mtb, TB is less severe (329). M. bovis also infects many other domesticated and wild animal species (328, 330). Cattle have several advantages in TB vaccine research – the disease progresses slowly, the granulomatous reaction are similar to that seen in humans, and vaccination in neonatal calves confers protection but wanes over time (331–334). Disadvantages include their large size/high cost in experimental housing (326) and post-primary TB is not a characteristic of them (294).
Granulomas in tuberculin reacting cattle (due to M. bovis) may be uncommon to rare (1-2%) when tissues are examined in a slaughterhouse but are commonly found (>70%) when analyzed using 5 mm lung slices (335). Wangoo et al. (336) have classified bovine TB granulomas in experimentally infected calves with M. bovis into four separate stages of severity based on size and microscopic morphological characteristics such as cellular composition, necrosis and fibrosis. Similar to humans and NHP, all stages of granulomas in this M. bovis infection model may be seen simultaneously in the same animal (336, 337).
As in human TB, CD4+ and CD8+ T cells are important in host immunity to bovine TB. In most stage I/II lesions of bovine TB, CD8+ and CD25+ T cells were found in the rim of the granulomas whereas CD4+ T cells were distributed in both the lymphocytic rim and center of the granulomas (337). γδ T cells were interspersed among the macrophages, leading to the speculation that CD4+ and γδ T cells – observed in granulomas within the first 21 days after experimental M. bovis infection in cattle (338, 339) – both play a role in the maintenance and the maturation of the granulomas as well as a host-protective role in respiratory mucosal immunity (340, 341). In stage III/IV lesions, all the examined T cell populations were present among the mononuclear component of the granulomas. The clinical and pathologic features of M. bovis-infected cattle that mimic human pulmonary TB are described in Table 4.
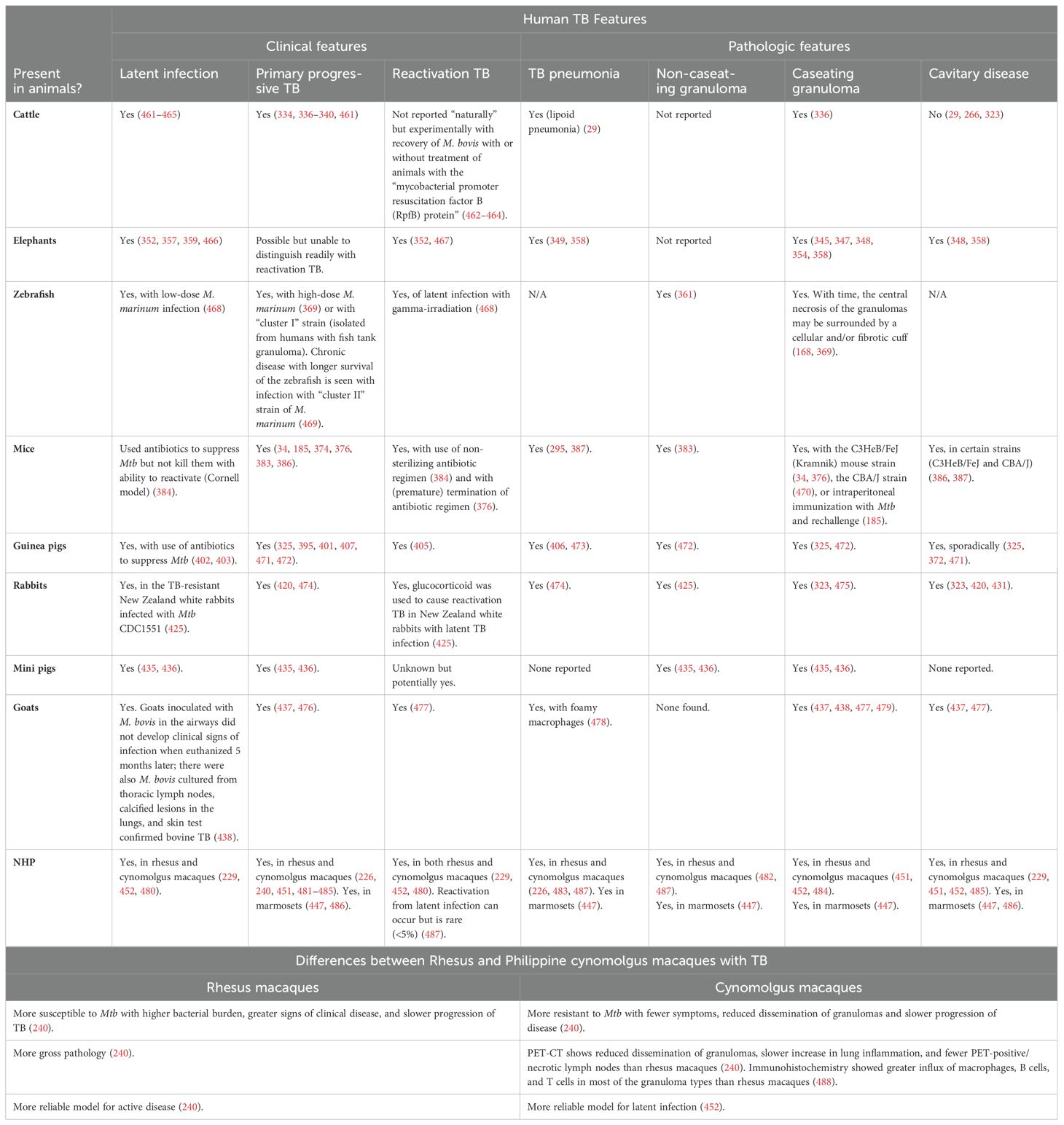
Table 4. Comparison of the features of natural and experimental animal TB with human pulmonary TB.
5.2 Elephants
Elephants are divided into African elephants (Loxodonta africana) and Asian (Indian) elephants (Elephas maximus). While TB in elephants is mostly due to Mtb, M. bovis (342–345) and M. caprae (346) have been reported. TB in elephants has been identified more commonly in Asian elephants than African elephants, possibly due to greater contact between the former and humans. However, it is not known whether Asian elephants are intrinsically more susceptible (347, 348).
TB has been observed in both wild (349–351) and captive elephants (344, 352). Spread among elephants and inter-species infection between humans and elephants (likely bidirectional) have been either strongly suspected or well-documented (353–357).
Heterogenous TB granulomas are also found in elephant TB. The main pathological lesions of elephant TB occur in the lungs and thoracic lymph nodes (358). Gross anatomical features include non-caseating and caseating granulomatous nodules as well as calcified caseous and cavitary lesions. Histopathologic features include epithelioid granulomas with giant cells in both lymph nodes and lung lesions as well as caseous and necrotizing granulomatous pneumonia in more severe cases (358). One study showed that of nine Asian elephants that died from TB, six had poorly-formed granulomas in the lungs with low to moderate number of lymphocytes, comprised mostly of B cells (359). However, the other two elephants had discrete and well-demarcated granulomas and greater number of T cells. The clinical and pathologic features of Mtb-infected elephants that mimic human pulmonary TB are described in Table 4.
6 TB Granulomas associated with experimental infections
6.1 Zebrafish
The primary limitations of zebrafish in experimental TB are that they do not have lungs and cannot be infected in a sustained fashion with Mtb due to lower temperature requirements of zebrafish. Instead, M. marinum is used as a surrogate because it naturally inhabits zebrafish, is genetically similar to Mtb, and causes TB-like diseases in poikilothermic animals (76, 261, 360, 361). Nevertheless, both the embryonic-larval and adult zebrafish models have several advantages in understanding mycobacterial pathogenesis: (i) large-scale screening studies can be attempted since they are small and easy to reproduce and raise; (ii) their transparency in the embryonic and larval stages allows their internal structures to be visualized temporally without the need to sacrifice them; (iii) zebrafish embryos have a temporal separation between the earlier development of innate immunity (macrophages and neutrophils) and later development of adaptive immunity (lymphocytes), allowing the study of only innate immunity during the initial stages of mycobacterial infections; (iv) hypoxic or non-hypoxic granulomas can be induced according to the infection site – via inoculation of M. marinum in poorly or richly vascularized areas, respectively – in the larva model; (v) in the advanced model, acute or chronic/latent infection may be induced based on the infectious dose (4, 362–370). The clinical and pathologic features of M. marinum-infected zebrafish that mimic human pulmonary TB are described in Table 4.
6.2 Mice
Mice are a tractable animal model for TB because they are relatively easy to handle, inexpensive, require little space, have similar immune components to humans, and there is an expansive array of immunological reagents available. Furthermore, there is the ability to genetically engineer mouse strains to elucidate the role of specific genes with Mtb infection; i.e., “knock-out” and “knock-in” mouse strains through targeted disruption of specific genes (266, 323, 326, 371, 372). These advantages allow for assessment of large-scale drug screening and vaccine efficacy testing, in addition to monitoring disease endpoints following Mtb infection.
While susceptibility to TB can be affected by the virulence of Mtb strain used, route of infection, and the inoculum dose (373), the genetic background of mice plays a critical role with Mtb infection. Many distinct mouse lineages exist with varying genetic backgrounds. Historically, these lineages are divided into two main categories: “resistant” or “susceptible” strains. Currently, the most common mouse lineages used in TB research are the BALB/c and C57BL/6 mice (“resistant” strains) and the C3HeB/FeJ, DBA/2, and 129/Sv mice (“susceptible” strains). The C3HeB/FeJ strain is regarded as a valuable resource for host-pathogen and drug response characterization since it better mimics histopathologic features of human TB: well-formed, hypoxic TB granulomas with central necrosis and liquefaction, elements not seen in other inbred strains (34, 326, 374, 375). Indeed, C3HeB/FeJ mice infected with Mtb may develop three morphologically distinct lesion types in the lungs (Figures 11A–C) (34). The unique susceptibility of C3HeB/FeJ mice to TB is due to de novo mutation and defective expression of the Ipr1 gene located within the sst1 (super-susceptibility-to-tuberculosis 1) locus (376, 377). The sst1 locus from C3HeB/FeJ has been engineered into a C57BL/6 background, termed congenic B6.C3H-sst1 which harbored a 10–fold higher bacterial burden at four weeks post-infection and displayed large necrotic lesions in histopathology evaluations (376, 378). The B6.C3H-sst1 strain has helped demonstrate that the sst1 locus independently defines necrotic lesion progression in the mouse model (376).
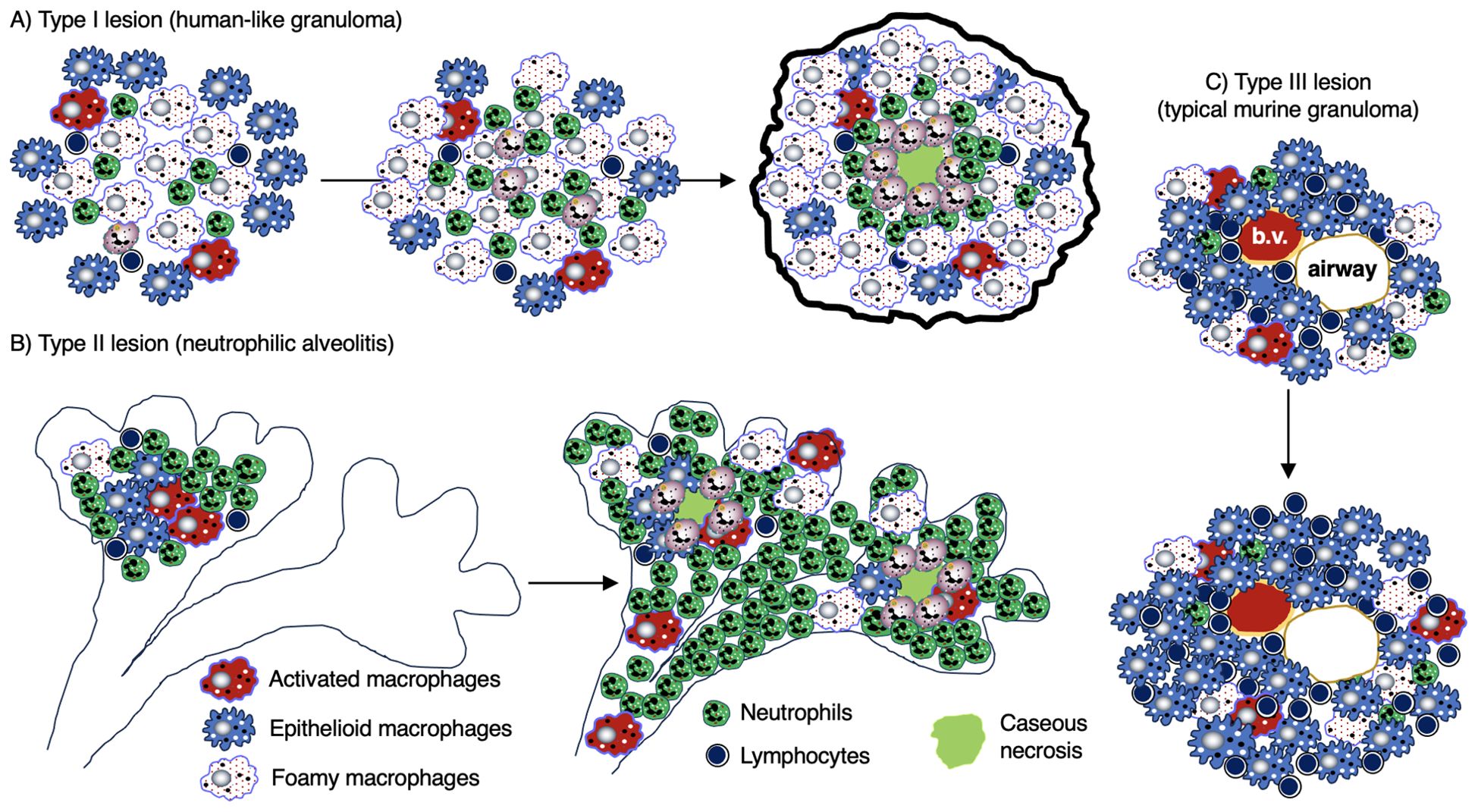
Figure 11. Different types of TB lung lesions in the C3HeB/FeJ mice. Three types of histopathologic TB lesions are seen in the C3HeB/FeJ mice. (A) Type 1 lesion is mainly comprised of foamy macrophages, few epithelioid macrophages and lymphocytes, and a caseous center surrounded by neutrophils. This lesion most resembles to that seen in human TB. (B) Type 2 lesion is the most severe form and is characterized by a neutrophilic, necrotizing alveolitis with occlusion of the terminal bronchioles and caseous necrosis. (C) Type 3 lesion is comprised mainly of infiltration of epithelioid macrophages and lymphocytes, smaller numbers of foamy and activated macrophages and neutrophils around the airway and blood vessel pair. This is the typical lesion seen in most other mouse strains. TB, tuberculosis.
The Collaborative Cross (CC) mice are a multi-parental mouse genetic reference population characterized by genetic polymorphism with a spectrum of sensitivity to Mtb infection (379–381). Bacterial burden after intravenous infection with Mtb ranges over 1000-fold across these strains (379). This CC strain catalog has advanced our understanding of the interplay of different genes on host susceptibility to TB by enabling the ability to reference and isolate key genetic loci influencing that sensitivity. For example, CC042 strains exhibit deficient IFNγ production which correlated with fewer macrophages and T cells being recruited to the lungs but enhanced neutrophil infiltration and necrotic lesions compared to C57BL/6 mice (380). In addition to informing which immune components provide host protection, the CC or diversity outbred (DO) mouse strains have also been shown to impact the effectiveness of TB vaccines and thus more likely to be more informative and a better alternative to inbred mouse strains. A DO model inherently lowers the power available to decipher key genetic components as each genotype is only present in a single animal, whereas the CC model retains diversity of the DO model while powering studies with repeatable and well powered “n” per genotype (382). Both CC and DO modeling better recapitulates the diversity and spectrum of TB disease observed in human clinical cases, including interrogation of pulmonary lesions and granuloma formation.
One notable disadvantage of the TB mouse model is that Mtb infection results in a slowly progressive infection that eventually leads to death, a model of primary progressive TB rather than post-primary TB or latent TB infection (323, 383). However, two models have been reported for latent TB in mice (with potential for reactivation): (i) the use of antibiotics to eliminate actively replicating Mtb but not sterilizing all Mtb (the Cornell model) (384) and (ii) intradermal (ear) inoculation of mice that produced a long-term clinical state characterized by the presence of Mtb-specific T cells but no overt signs of active TB (385). Major gaps in the field also include understanding subclinical TB disease and biological features that promote Mtb transmission, which remain outside the capacity of the present mouse models.
Other shortcomings of mice in TB research include: (i) their inability to form caseating granulomas and lung cavitation except in the C3HeB/FeJ mouse (386, 387) or as part of an experimental condition, such as mice that received intraperitoneal injection with TDM in oil prior to infection with Mtb (185, 295); (ii) predominance of loosely formed granulomatous lesion (Type III lesions mentioned in Figure 11), whereas humans form discrete granulomas (34, 266); (iii) absence of hypoxia in mouse granulomas (with the exception of the C3HeB/FeJ strain) and thus murine macrophages may be more likely to undergo apoptosis rather than necrosis (266, 388); (iv) localization of Mtb intracellularly in non-necrotic lesions in commonly used mouse strains (389), whereas in human and other animal models, Mtb primarily resides extracellularly (372, 387, 390); thus, the sterilizing activity of new TB drug/regimen seen in mice may not be observed in other animal models or human clinical trials (391); (v) alterations in how the CC mouse strains with genetic polymorphisms respond to TB vaccines (392); and (vi) differences in antigen clusters between mice and humans that may impact host immunity to Mtb; e.g., mice do not possess a homolog of human IL-32, a cytokine associated with host protection in human macrophages and in a transgenic murine model that expresses human IL-32γ (393, 394). The clinical and pathologic features of Mtb-infected mice that mimic human pulmonary TB are described in Table 4.
6.3 Guinea pigs
Guinea pigs were one of the first experimental animals used to study TB. Some of the earliest studies using this model were conducted by Robert Koch and Max Lurie in the late 19th and early 20th centuries, respectively (395–397). Currently, the outbred Dunkin-Hartley strain is the most common strain used in research (398). Guinea pigs display many features during Mtb infection that are similar to humans: (i) newborn guinea pigs have mature lympho-myeloid system like that found in human infants; (ii) their hormonal and immunological phenotypes; (iii) guinea pigs also do not synthesize vitamin C but must obtain it from their diet; (iv) guineas pigs are glucocorticoid-resistant (324); and (v) presence of necrotic primary granuloma after low-dose Mtb infection (323–325).
Guinea pigs are also highly susceptible to Mtb, making them good models for primary progressive disease. They develop pulmonary TB with very low doses of Mtb (as low as 2-4 CFU per animal) (399) as well as with exposure to exhaust ambient air from human TB patients (400) or to neighboring Mtb-infected guinea pigs within the same room but in different cages (397). Lurie also showed that concomitant respiratory and enteric infection with Mtb of guinea pigs resulted in more chronic respiratory TB whereas only respiratory inoculation with Mtb resulted in more acute respiratory TB (396). Although it has been argued for many years that a latent TB model and thus post-primary (reactivation) TB are not possible in guinea pigs due to their increased susceptibility (325, 395, 401), some have found it possible to do so (402, 403). Because guinea pigs are also well protected by the BCG vaccine and respond well to anti-TB antibiotics, they are useful for drug and vaccine studies (399, 404–411).
Disadvantages of guinea pigs in TB research include: (i) greater expense than mice; (ii) most Mtb infections of guinea pigs model primary progressive disease than post-primary TB; (iii) more limited immunologic reagents than for mice. However, successful cloning of various cytokines (IFNγ, IL-4, IL-10, and IL-17) from guinea pigs expands the ability to characterize the immune response to Mtb (412–415). The clinical and pathologic features of Mtb-infected guinea pigs that mimic human pulmonary TB are described in Table 4.
6.4 Rabbits
Nearly 100 years ago, Lurie and colleagues developed a rabbit model of TB, wherein rabbits with varying levels of Mtb resistance were developed (416–418). Rabbits are useful to model both human latent TB infection and active TB, developing caseating granulomas that may liquify and cavitate (323, 395, 419–425). It has also been used to distinguish between protective cellular immunity and potentially damaging mechanisms of “delayed type hypersensitivity” (426–428).
The severity of rabbit TB lung pathology can be influenced by two main factors: the specific Mtb strain used; e.g., Erdman Mtb was found to cause more severe pathology and worse outcomes than Mtb CDC1551 and H37Rv (424, 429) and the rabbit genotype; e.g., inbred rabbits are more susceptible to Mtb, resulting in the formation of lung granulomas and cavities, than outbred rabbit strains (395, 424).
Another way that rabbits mimic human TB infection is that they show a spectrum of disease outcome, including the development of latent TB infection in the TB-resistant outbred strain of New Zealand white rabbits (424, 425, 430). Lung cavities are also common with serial low-dose Mtb infection of rabbits (431–433). Serial CT scans demonstrated that the cavities developed from areas of necrotizing pneumonia in the dorsal-caudal region of the rabbit lungs – anatomically analogous to the apical region of the human lung – pulmonary segments with the greatest mechanical stress, decreased blood flow, and highest partial pressure of alveolar O2 in the normal position of rabbits (311–313, 319, 431). Disadvantages of rabbits include larger housing requirements, greater expense, more difficulty in handling, greater challenge in gene manipulation, and lesser repertoire of immunologic reagents compared to mouse models (323, 434). The clinical and pathologic features of Mtb-infected rabbits that mimic human pulmonary TB are described in Table 4.
6.5 Mini pigs
Mini pigs have been used to model primary progressive TB (435, 436). Like guinea pigs, spread of Mtb may occur between infected mini pigs and their previously uninfected neighbors (436). The advantage of the mini pig model is that the anatomical structure of the lungs is like that seen in humans, with the normal presence of intralobular septae that become thickened following encapsulation of granulomas, which prevents the development of new lesions (435). This encapsulation – present in humans but not in rodents or NHP – makes the mini pigs a promising model to study latency because drainage of latent bacilli into the airways or alveolar spaces (435, 436). However, mini pigs also have the disadvantage of being expensive and large (up to 150 pounds). The clinical and pathologic features of Mtb-infected mini pigs that mimic human pulmonary TB are described in Table 4.
6.6 Goats
Goats are natural hosts of M. caprae and M. bovis and have been used to study bacillary load, pathology, and vaccine efficiency. Their high susceptibility and the fact that they mimic human infection by developing caseous necrotizing granulomas as well as cavities make them good models for studying TB (437). Compared to other experimental animals, goats are also more like humans in terms of size, body weight, and respiratory anatomy (266, 438). In goats, it was shown that the immune response varied depending on the route of BCG vaccination; i.e., a negative intradermal or interferon-gamma release assay test for latent TB infection was more likely to be observed following oral administration of heat-inactivated M. bovis vaccine compared to the intramuscular route (439). Given that goats may develop TB lesions like that seen in humans and that either heat-inactivated M. bovis vaccine and BCG vaccine showed efficacy against M. caprae (440), goats are potential models to study vaccine efficacy. The clinical and pathologic features of Mtb-infected goats that mimic human pulmonary TB are described in Table 4.
6.7 Non-human primates
In early 1900’s, rhesus macaques were used in experimental TB by Dr. Gerald B. Webb and his colleagues at his home in Colorado Springs, Colorado (441, 442). Their work was involved subcutaneous injections of Mtb in the NHPs. Subsequent experimental TB work with rhesus macaques from ~1950-1970 also involved vaccination with intravenous BCG (443, 444). Since 1996, the Philippine cynomolgus macaque was utilized for experimental TB, showing that depending on the dose, either active TB or latent TB infection may develop (229, 445). Cynomolgus macaques primed with BCG and then boosted with the Mtb72F/AS02A vaccine showed long-term protection against TB (446). Whereas the control monkeys showed granulomas with caseous centers, extensive edema, and were fused, the granulomas of the NHP vaccinated sequentially with BCG and Mtb72F/AS02A were discrete lesions with no necrosis or edema (446). More recently, marmosets, which are much smaller than the macaques, have been used in experimental TB (447).
NHPs are likely the most accurate model in human TB research because of their similarities to humans genetically, immunologically (e.g., functional MHC regions, T-cell antigen receptors, and other cellular features that are relevant for vaccine testing), clinically (manifesting a spectrum of TB from latent infection to active disease), pathologically (including pneumonia, various forms of granulomas, and cavitary disease) and pharmacokinetically (with relevance for testing new drugs) (229, 326, 448–457). Limitations of the NHP model are more logistical – smaller sample sizes and time points due to cost and more difficulty in handling (456). However, serial radiographic imaging of a relatively large animal – namely, chest radiograph, CT, magnetic resistance imaging, and 18FDG-PET-CT to detect areas of increased metabolic activity to indicate host immune response to the infection (458–460) – have been highly informative. The clinical and pathologic features of Mtb-infected NHP that mimic human pulmonary TB are described in Table 4. The key clinical, radiographic, and pathological differences between the rhesus and cynomolgus macaques relevant for TB are shown in Table 4.
7 Summary and future perspective
Substantial progress has been made in our understanding of the host immune responses that either protect against TB or contribute to TB pathogenesis. Much of the work have focused on the granulomas and the molecular and cellular components that participate in their formation in experimental animals as well as re-analysis of archived and more contemporaneous human TB tissues. While descriptive analyses of the different cell types in the granuloma architecture have been dominating, the role of soluble mediators (cytokines, chemokines, growth factors) and metabolic studies (hypoxia, aerobic glycolysis, and activity of various cellular enzymes such as MMPs, IDO-1, and HO-1) have enriched our understanding of TB granulomas but have also raised new avenues of research. One emerging paradigm is that either a deficiency or excess of certain host-derived molecular and cellular components may be harmful to the host which, in turn, allow for the proliferation and persistence of Mtb. While new imaging techniques such as MIBI-TOF (multiplexed ion beam imaging by time of flight) and PET-CT using the radioactive tracers deoxy-2-[18F]-fluoro-D-glucose and [18F]- fluoromisonidazole have contributed to our understanding of TB, it is important to understand their limitations. For example, one overarching limitation is our inability to analyze immunological and microbiological data simultaneously in individual granulomatous TB lesions in vivo over time. Rather, “snapshots” of events limits our ability to interpret whether the myriad of molecular and cellular components that can be found in active TB are: (i) the main drivers of TB pathogenesis (causing loss of host protection), (ii) attempts to correct an inadequate initial immune response, or (iii) possibly both depending on the temporality of the responses. While some of these limitations can be overcome by targeted genetic disruption of specific molecular or genetic components, inherent limitations of these models to human TB remain. A better understanding of the immune correlates of protection may also help guide the development of antimicrobial agents that can target privileged sites of Mtb and of host-directed therapies. While “high-tech” approaches to better understand granulomas and other TB lesions are occurring, we ought not lose sight of the value of “low-tech” foundational principles of anatomy, physiology, and chemistry that should be included in both the experimental studies and care of TB patients.
Author contributions
JL: Writing – original draft, Writing – review & editing. DN: Writing – review & editing. SB: Writing – review & editing. SL: Writing – review & editing. XB: Writing – review & editing. DG: Writing – review & editing. VD: Writing – review & editing. TN: Writing – review & editing. AS: Writing – review & editing. RC: Writing – review & editing. EC: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00029.
Acknowledgments
We are grateful to Lorelenn Fornis for help with the initial literature search.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
AFB, acid-fast bacilli; BCG, Bacillus Calmette Guérin; CT, computed tomography; CTLA-4, cytotoxic T lymphocyte-associated protein 4; ESAT-6, early secreted antigenic target 6; FDG, deoxy-2-[18F]-fluoro-D-glucose; iBALT, inducible bronchus-associated lymphoid tissue; IDO-1, indoleamine 2,3-dioxygenase; IFNγ, interferon-gamma; HIF-1α, hypoxia inducible factor-1-alpha; HO-1, heme oxygenase-1; IL-12, interleukin-12; IL-17, interleukin-17; LDL, low density lipoprotein; LTA4H, leukotriene A4 hydrolase; LTA4, leukotriene A4; LTB4, leukotriene B4; LXA4, lipoxin A4; MGC, multinucleated giant cell; MIBI-TOF, multiplexed ion beam imaging by time-of-flight; μCT, micro-computed tomography; MMP-1, matrix metalloproteinase-1; MMP-9, matrix metalloproteinase-9; Mtb, Mycobacterium tuberculosis; NET, neutrophil extracellular trap; NFκB, nuclear factor kappa-B; NHP, non-human primates; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand 1; PET, positron emission tomography; PPARγ, peroxisome proliferator-gamma; RIP1, receptor-interacting serine/threonine kinase 1; RIP3, receptor-interacting protein interacting serine/threonine kinase 3; RIPK-3, receptor interacting protein kinase 3; ROS, reactive oxygen species; STAT6, signal transducer and activator of transcription 6; TB, tuberculosis; TCR, T cell receptor; TDM, trehalose 6,6’-dimycolate; TIM3, T cell immunoglobin and mucin domain-containing protein-3; TLO, tertiary lymphoid organs; TGFβ, transforming growth factor-beta; TNF, tumor necrosis factor; TR4, testicular receiver 4.
References
1. Mehta AC, Ali SR. Mnemonic for the differential diagnosis of non-caseating granulomas. Sarcoidosis Vasc Diffuse Lung Dis. (2017) 34:200–7. doi: 10.36141/svdld.v34i2.5674
2. Pagan AJ, Ramakrishnan L. The formation and function of granulomas. Annu Rev Immunol. (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
3. Shah KK, Pritt BS, Alexander MP. Histopathologic review of granulomatous inflammation. J Clin Tuberc Other Mycobact Dis. (2017) 7:1–12. doi: 10.1016/j.jctube.2017.02.001
4. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. (2012) 12:352–66. doi: 10.1038/nri3211
5. Subbian S, Tsenova L, Kim MJ, Wainwright HC, Visser A, Bandyopadhyay N, et al. Lesion-specific immune response in granulomas of patients with pulmonary tuberculosis: A pilot study. PloS One. (2015) 10:e0132249. doi: 10.1371/journal.pone.0132249
6. Martinot AJ. Microbial offense vs host defense: who controls the TB granuloma? Vet Pathol. (2018) 55:14–26. doi: 10.1177/0300985817705177
7. Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. (2011) 4:271–8. doi: 10.1038/mi.2011.14
8. Anonymous. Granulomatous Infections and Inflammations: Cellular and Molecular Mechanisms. Boros DL, editor. Washington, D.C: ASM Press (2003). doi: 10.1128/9781555817879
9. Warsinske HC, DiFazio RM, Linderman JJ, Flynn JL, Kirschner DE. Identifying mechanisms driving formation of granuloma-associated fibrosis during Mycobacterium tuberculosis infection. J Theor Biol. (2017) 429:1–17. doi: 10.1016/j.jtbi.2017.06.017
10. Hult C, Mattila JT, Gideon HP, Linderman JJ, Kirschner DE. Neutrophil dynamics affect mycobacterium tuberculosis granuloma outcomes and dissemination. Front Immunol. (2021) 12:712457. doi: 10.3389/fimmu.2021.712457
11. Cronan MR. In the thick of it: formation of the tuberculous granuloma and its effects on host and therapeutic responses. Front Immunol. (2022) 13:820134. doi: 10.3389/fimmu.2022.820134
12. Deretic V, Singh S, Master S, Harris J, Roberts E, Kyei G, et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. (2006) 8:719–27. doi: 10.1111/j.1462-5822.2006.00705.x
13. Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. (2001) 2:569–77. doi: 10.1038/35085034
14. Russell DG, Purdy GE, Owens RM, Rohde KH, Yates RM. Mycobacterium tuberculosis and the four-minute phagosome. ASM News. (2005) 71:459–63.
15. Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. (2009) 11:1170–8. doi: 10.1111/j.1462-5822.2009.01335.x
16. Meena LS. Rajni. Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. (2010) 277:2416–27. doi: 10.1111/j.1742-4658.2010.07666.x
17. Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. (2012) 14:1287–98. doi: 10.1111/j.1462-5822.2012.01799.x
18. Peng X, Sun J. Mechanism of ESAT-6 membrane interaction and its roles in pathogenesis of Mycobacterium tuberculosis. Toxicon. (2016) 116:29–34. doi: 10.1016/j.toxicon.2015.10.003
19. Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. (2011) 240:252–68. doi: 10.1111/j.1600-065X.2010.00984.x
20. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
21. Ganchua SKC, White AG, Klein EC, Flynn JL. Lymph nodes-The neglected battlefield in tuberculosis. PloS Pathog. (2020) 16:e1008632. doi: 10.1371/journal.ppat.1008632
22. McCaffrey EF, Donato M, Keren L, Chen Z, Delmastro A, Fitzpatrick MB, et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat Immunol. (2022) 23:318–29. doi: 10.1038/s41590-021-01121-x
23. Marakalala MJ, Martinez FO, Plüddemann A, Gordon S. Macrophage heterogeneity in the immunopathogenesis of tuberculosis. Front Microbiol. (2018) 9:1028. doi: 10.3389/fmicb.2018.01028
24. McClean CM, Tobin DM. Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog Dis. (2016) 74:ftw068. doi: 10.1093/femspd/ftw068
25. Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci. (2021) 23:144. doi: 10.3390/ijms23010144
26. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
27. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
29. Palmer MV, Kanipe C, Boggiatto PM. The bovine tuberculoid granuloma. Pathogens. (2022) 11:61. doi: 10.3390/pathogens11010061
30. Mariano M, Malucelli BE. Defective phagocytic ability of epithelioid cells reversed by levamisole. J Pathol. (1980) 130:33–6. doi: 10.1002/path.1711300105
31. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. (2009) 10:943–948. doi: 10.1038/ni.1781
32. Caceres N, Tapia G, Ojanguren I, Altare F, Gil O, Pinto S, et al. Evolution of foamy macrophages in the pulmonary granulomas of experimental tuberculosis models. Tuberculosis (Edinb). (2009) 89:175–82. doi: 10.1016/j.tube.2008.11.001
33. Welsh KJ, Risin SA, Actor JK, Hunter RL. Immunopathology of postprimary tuberculosis: increased T-regulatory cells and DEC-205-positive foamy macrophages in cavitary lesions. Clin Dev Immunol. (2011) 2011:307631. doi: 10.1155/2011/307631
34. Irwin SM, Driver E, Lyon E, Schrupp C, Ryan G, Gonzalez-Juarrero M, et al. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis. Dis Model Mech. (2015) 8:591–602. doi: 10.1242/dmm.019570
35. Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity. (2016) 45:861–76. doi: 10.1016/j.immuni.2016.09.014
36. Cohen SB, Gern BH, Urdahl KB. The tuberculous granuloma and preexisting immunity. Annu Rev Immunol. (2022) 40:589–614. doi: 10.1146/annurev-immunol-093019-125148
37. Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, et al. A non-canonical type 2 immune response coordinates tuberculous granuloma formation and epithelialization. Cell. (2021) 184:1757–1774.e14. doi: 10.1016/j.cell.2021.02.046
38. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PloS Pathog. (2008) 4:e1000204. doi: 10.1371/journal.ppat.1000204
39. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. (2012) 12:669 –681. doi: 10.1016/j.chom.2012.09.012
40. Cumming BM, Pacl HT, Steyn AJC. Relevance of the warburg effect in tuberculosis for host-directed therapy. Front Cell Infect Microbiol. (2020) 10:576596. doi: 10.3389/fcimb.2020.576596
41. Maoldomhnaigh C, Cox DJ, Phelan JJ, Mitermite M, Murphy DM, Leisching G, et al. Lactate alters metabolism in human macrophages and improves their ability to kill mycobacterium tuberculosis. Front Immunol. (2021) 12:663695. doi: 10.3389/fimmu.2021.663695
42. Sun W, Jia M, Feng Y, Cheng X. Lactate is a bridge linking glycolysis and autophagy through lactylation. Autophagy. (2023) 19:3240–1. doi: 10.1080/15548627.2023.2246356
43. Fan Q, Yang L, Zhang X, Ma Y, Li Y, Dong L, et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J Exp Clin Cancer Res. (2018) 37:9. doi: 10.1186/s13046-018-0673-y
44. Lee YR, Wu SY, Chen RY, Lin YS, Yeh TM, Liu HS. Regulation of autophagy, glucose uptake, and glycolysis under dengue virus infection. Kaohsiung J Med Sci. (2020) 36:911–9. doi: 10.1002/kjm2.v36.11
45. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
46. Dkhar HK, Nanduri R, Mahajan S, Dave S, Saini A, Somavarapu AK, et al. Mycobacterium tuberculosis keto-mycolic acid and macrophage nuclear receptor TR4 modulate foamy biogenesis in granulomas: a case of a heterologous and noncanonical ligand-receptor pair. J Immunol. (2014) 193:295–305. doi: 10.4049/jimmunol.1400092
47. Mahajan S, Dkhar HK, Chandra V, Dave S, Nanduri R, Janmeja AK, et al. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J Immunol. (2012) 188:5593–603. doi: 10.4049/jimmunol.1103038
48. Johansen MD, Kasparian JA, Hortle E, Britton WJ, Purdie AC, Oehlers SH. Mycobacterium marinum infection drives foam cell differentiation in zebrafish infection models. Dev Comp Immunol. (2018) 88:169–72. doi: 10.1016/j.dci.2018.07.022
49. Genoula M, Marín Franco JL, Dupont M, Kviatcovsky D, Milillo A, Schierloh P, et al. Formation of foamy macrophages by tuberculous pleural effusions is triggered by the interleukin-10/signal transducer and activator of transcription 3 axis through ACAT upregulation. Front Immunol. (2018) 9:459. doi: 10.3389/fimmu.2018.00459
50. Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su WL, Griffin L, et al. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PloS One. (2013) 8:e61925. doi: 10.1371/journal.pone.0061925
51. Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. (2010) 11:751–8. doi: 10.1038/ni.1904
52. Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. (1997) 65:298–304. doi: 10.1128/iai.65.1.298-304.1997
53. Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. (2000) 164:2016–20. doi: 10.4049/jimmunol.164.4.2016
54. Keane J, Shurtleff B, Kornfeld H. TNF-dependent BALB/c murine macrophage apoptosis following Mycobacterium tuberculosis infection inhibits bacillary growth in an IFN-gamma independent manner. Tuberculosis (Edinb). (2002) 82:55–61. doi: 10.1054/tube.2002.0322
55. Lam A, Prabhu R, Gross CM, Riesenberg LA, Singh V, Aggarwal S. Role of apoptosis and autophagy in tuberculosis. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L218–29. doi: 10.1152/ajplung.00162.2017
56. Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol. (1998) 160:5448–54. doi: 10.4049/jimmunol.160.11.5448
57. Rojas M, Barrera LF, Puzo G, Garcia LF. Differential induction of apoptosis by virulent Mycobacterium tuberculosis in resistant and susceptible murine macrophages: role of nitric oxide and mycobacterial products. J Immunol. (1997) 159:1352–61. doi: 10.4049/jimmunol.159.3.1352
58. Sia JK, Rengarajan J. Immunology of mycobacterium tuberculosis infections. Microbiol Spectr. (2019) 7:1–37. doi: 10.1128/microbiolspec.GPP3-0022-201
59. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. (2012) 12:289–300. doi: 10.1016/j.chom.2012.06.010
60. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. (2009) 136:37–49. doi: 10.1016/j.cell.2008.11.014
61. Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. (2008) 205:2791–801. doi: 10.1084/jem.20080767
62. Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol. (2006) 176:3707–16. doi: 10.4049/jimmunol.176.6.3707
63. Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. (2008) 9:1189–97. doi: 10.1038/ni.1654
64. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. (2009) 10:899–906. doi: 10.1038/ni.1758
65. Moreira-Teixeira L, Mayer-Barber K, Sher A, O'Garra A. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med. (2018) 215:1273–85. doi: 10.1084/jem.20180325
66. Mundra A, Yegiazaryan A, Karsian H, Alsaigh D, Bonavida V, Frame M, et al. Pathogenicity of type I interferons in mycobacterium tuberculosis. Int J Mol Sci. (2023) 24:3919. doi: 10.3390/ijms24043919
67. Zhao X, Khan N, Gan H, Tzelepis F, Nishimura T, Park SY, et al. Bcl-xL mediates RIPK3-dependent necrosis in M. tuberculosis-infected macrophages. Mucosal Immunol. (2017) 10:1553–68. doi: 10.1038/mi.2017.12
68. Soldevilla P, Vilaplana C, Cardona P-J. Mouse models for mycobacterium tuberculosis pathogenesis: show and do not tell. Pathogens. (2022) 12:49. doi: 10.3390/pathogens12010049
69. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2017) 18:134–47. doi: 10.1038/nri.2017.105
70. Gideon HP, Phuah J, Junecko BA, Mattila JT. Neutrophils express pro- and anti-inflammatory cytokines in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Mucosal Immunol. (2019) 12:1370–81. doi: 10.1038/s41385-019-0195-8
71. Lovewell RR, Baer CE, Mishra BB, Smith CM, Sassetti CM. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. (2021) 14:229–41. doi: 10.1038/s41385-020-0300-z
72. Andrews JT, Zhang Z, Prasad GVRK, Huey F, Nazarova EV, Wang J, et al. Metabolically active neutrophils represent a permissive niche for. Mycobacterium tuberculosis. Mucosal Immunol. (2024) 17:825–42. doi: 10.1016/j.mucimm.2024.05.007
73. Bhatt A, Quazi Syed Z, Singh H. Converging epidemics: A narrative review of tuberculosis (TB) and human immunodeficiency virus (HIV) coinfection. Cureus. (2023) 15:e47624. doi: 10.7759/cureus.47624
74. Saunders BM, Frank AA, Orme IM, Cooper AM. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol. (2002) 216:65–72. doi: 10.1016/S0008-8749(02)00510-5
75. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5+ T helper cells mediate protective immunity against tuberculosis. J Clin Invest. (2013) 123:712–26. doi: 10.1172/JCI65728
76. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. (2013) 153:521–34. doi: 10.1016/j.cell.2013.03.022
77. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-γ Plays a minimal role in control of pulmonary mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PloS Pathog. (2016) 12:e1005667. doi: 10.1371/journal.ppat.1005667
78. Hammarén MM, Oksanen KE, Nisula HM, Luukinen BV, Pesu M, Rämet M, et al. Adequate Th2-type response associates with restricted bacterial growth in latent mycobacterial infection of zebrafish. PloS Pathog. (2014) 26:e1004190. doi: 10.1371/journal.ppat.1004190
79. Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, Mckenzie ANJ, et al. The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J Pathol. (2014) 234:338–50. doi: 10.1002/path.2014.234.issue-3
80. Mazzarella G, Bianco A, Perna F, D'Auria D, Grella E, Moscariello E, et al. T lymphocyte phenotypic profile in lung segments affected by cavitary and non-cavitary tuberculosis. Clin Exp Immunol. (2003) 132:283–8. doi: 10.1046/j.1365-2249.2003.02121.x
81. Hernandez-Pando R, Aguilar D, Hernandez ML, Orozco H, Rook G. Pulmonary tuberculosis in BALB/c mice with non-functional IL-4 genes: changes in the inflammatory effects of TNF-alpha and in the regulation of fibrosis. Eur J Immunol. (2004) 34:174–83. doi: 10.1002/eji.200324253
82. Buccheri S, Reljic R, Caccamo N, Ivanyi J, Singh M, Salerno A, et al. IL-4 depletion enhances host resistance and passive IgA protection against tuberculosis infection in BALB/c mice. Eur J Immunol. (2007) 37:729–37. doi: 10.1002/eji.200636764
83. Gern BH, Adams KN, Plumlee CR, Stoltzfus CR, Shehata L, Moguche AO, et al. TGFβ restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe. (2021) 29:594–606.e6. doi: 10.1016/j.chom.2021.02.005
84. Hirsch CS, Hussain R, Toossi Z, Dawood G, Shahid F, Ellner JJ. Cross-modulation by transforming growth factor beta in human tuberculosis: suppression of antigen-driven blastogenesis and interferon gamma production. Proc Natl Acad Sci U.S.A. (1996) 93:3193–8. doi: 10.1073/pnas.93.8.3193
85. Warsinske HC, Pienaar E, Linderman JJ, Mattila JT, Kirschner DE. Deletion of TGF-β1 increases bacterial clearance by cytotoxic T cells in a tuberculosis granuloma model. Front Immunol. (2017) 8:1843. doi: 10.3389/fimmu.2017.01843
86. Basile JI, Kviatcovsky D, Romero MM, Balboa L, Monteserin J, Ritacco V, et al. Mycobacterium tuberculosis multi-drug-resistant strain M induces IL-17+ IFNγ- CD4+ T cell expansion through an IL-23 and TGF-β-dependent mechanism in patients with MDR-TB tuberculosis. Clin Exp Immunol. (2017) 187:160–73. doi: 10.1111/cei.12873
87. Domingo-Gonzalez R, Das S, Griffiths KL, Ahmed M, Bambouskova M, Gopal R, et al. Interleukin-17 limits hypoxia-inducible factor 1α and development of hypoxic granulomas during tuberculosis. JCI Insight. (2017) 2:e92973. doi: 10.1172/jci.insight.92973
88. Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. (2007) 8:369–77. doi: 10.1038/ni1449
89. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. (2011) 12:639–46. doi: 10.1038/ni.2053
90. Zhu M, Fu Y. Proinflammatory IL-17 induces iBALT development. Cell Mol Immunol. (2012) 9:101–2. doi: 10.1038/cmi.2011.46
91. Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. (2009) 31:974–85. doi: 10.1016/j.immuni.2009.10.007
92. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919
93. Jain R, Kumari R, Chakraborty S, Mitra DK, Mohan A, Hadda V, et al. T-cell signature cytokines distinguish pulmonary sarcoidosis from pulmonary tuberculosis. Eur J Immunol. (2023) 53:e2250255. doi: 10.1002/eji.202250255
94. Lin PL, Flynn JL. CD8 T cells and Mycobacterium tuberculosis infection. Semin Immunopathol. (2015) 37:239–49. doi: 10.1007/s00281-015-0490-8
95. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. (2001) 193:271–80. doi: 10.1084/jem.193.3.271
96. Flynn J, Goldstein MM, Triebold KJ, Koller B, Bloom B. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. (1992) 89:12013–7. doi: 10.1073/pnas.89.24.12013
97. Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med. (1999) 189:1973–80. doi: 10.1084/jem.189.12.1973
98. Sousa AO, Mazzaccaro RC, Russell RG, Lee FK, Turner OC, Hong S, et al. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc Natl Acad Sci USA. (2000) 97:4204–8. doi: 10.1073/pnas.97.8.4204
99. Lazarevic V, Flynn J. CD8+ T cells in tuberculosis. Am J Respir Crit Care Med. (2002) 166:1116–21. doi: 10.1164/rccm.2204027
100. van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P. Control of latent mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur J Immunol. (2000) 30:3689–98. doi: 10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4
101. Silva BDS, Trentini MM, da Costa AC, Kipnis A, Junqueira-Kipnis AP. Different phenotypes of CD8+ T cells associated with bacterial load in active tuberculosis. Immunol Lett. (2014) 160:23–32. doi: 10.1016/j.imlet.2014.03.009
102. Cavassani KA, Carson WF, Moreira AP, Wen H, Schaller MA, Ishii M, et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood. (2010) 115:4403–11. doi: 10.1182/blood-2009-09-241083
103. Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
104. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
105. Chan J, Mehta S, Bharrhan S, Chen Y, Achkar JM, Casadevall A, et al. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin Immunol. (2014) 26:588–600. doi: 10.1016/j.smim.2014.10.005:588-600
106. Achkar JM, Chan J, Casadevall A. B cells and antibodies in the defense against Mycobacterium tuberculosis infection. Immunol Rev. (2015) 264:167–81. doi: 10.1111/imr.2015.264.issue-1
107. Choreño-Parra JA, Bobba S, Rangel-Moreno J, Ahmed M, Mehra S, Rosa B, et al. Mycobacterium tuberculosis HN878 infection induces human-like B-cell follicles in mice. J Infect Dis. (2020) 221:1636–46. doi: 10.1093/infdis/jiz663
108. Stewart P, Patel S, Comer A, Muneer S, Nawaz U, Quann V, et al. Role of B cells in mycobacterium tuberculosis infection. Vaccines (Basel). (2023) 11:955. doi: 10.3390/vaccines11050955
109. Achkar JM, Chan J, Casadevall A. Role of B cells and antibodies in acquired immunity against Mycobacterium tuberculosis. Cold Spring Harb Perspect Med. (2014) 5:a018432. doi: 10.1101/cshperspect.a018432
110. Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. (2007) 178:7222–34. doi: 10.4049/jimmunol.178.11.7222
111. Kondratieva TK, Rubakova EI, Linge IA, Evstifeev VV, Majorov KB, Apt AS. B cells delay neutrophil migration toward the site of stimulus: tardiness critical for effective bacillus Calmette-Guérin vaccination against tuberculosis infection in mice. J Immunol. (2010) 184:1227–34. doi: 10.4049/jimmunol.0902011
112. Kozakiewicz L, Phuah J, Flynn J, Chan J. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Adv Exp Med Biol. (2013) 783:225–50. doi: 10.1007/978-1-4614-6111-1_12
113. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol. (2010) 10:236–47. doi: 10.1038/nri2729
114. Choi HS, Rai PR, Chu HW, Cool C, Chan ED. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. Am J Respir Crit Care Med. (2002) 166:178–86. doi: 10.1164/rccm.2201023
115. Bery AI, Shepherd HM, Li W, Krupnick AS, Gelman AE, Kreisel D. Role of tertiary lymphoid organs in the regulation of immune responses in the periphery. Cell Mol Life Sci. (2022) 79:359. doi: 10.1007/s00018-022-04388-x
116. Schinköthe J, Köhler H, Liebler-Tenorio EM. Characterization of tuberculous granulomas in different stages of progression and associated tertiary lymphoid tissue in goats experimentally infected with Mycobacterium avium subsp. hominissuis. Comp Immunol Microbiol Infect Dis. (2016) 47:41–51. doi: 10.1016/j.cimid.2016.05.006
117. Ogbonna E. Deep spatial characterization of tertiary lymphoid structures in pulmonary tuberculosis granulomas. Open Forum Infect Dis. (2023) 10:ofad500.1715. doi: 10.1093/ofid/ofad500.1715
118. Krausgruber T, Redl A, Barreca D, Doberer K, Romanovskaia D, Dobnikar L, et al. Single-cell and spatial transcriptomics reveal aberrant lymphoid developmental programs driving granuloma formation. Immunity. (2023) 56:289–306.e7. doi: 10.1016/j.immuni.2023.01.014
119. Marin ND, Dunlap MD, Kaushal D, Khader SA. Friend or foe: the protective and pathological roles of inducible bronchus-associated lymphoid tissue in pulmonary diseases. J Immunol. (2019) 202:2519–2526. doi: 10.4049/jimmunol.1801135
120. Silva-Sanchez A, Randall TD. Role of iBALT in respiratory immunity. Curr Top Microbiol Immunol. (2020) 426:21–43. doi: 10.1007/82_2019_191
121. Ulrichs T, Kosmiadi GA, Trusov V, Jorg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol. (2004) 204:217–28. doi: 10.1002/path.v204:2
122. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: It is all starting to come together. Front Immunol. (2016) 7:401. doi: 10.3389/fimmu.2016.00401
123. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature. (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
124. Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, et al. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol. (2013) 6:972–84. doi: 10.1038/mi.2012.135
125. Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, et al. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol. (2011) 187:5402–7. doi: 10.4049/jimmunol.1101377
126. Zhang M, Wang Z, Graner MW, Yang L, Liao M, Yang Q, et al. B cell infiltration is associated with the increased IL-17 and IL-22 expression in the lungs of patients with tuberculosis. Cell Immunol. (2011) 270:217–23. doi: 10.1016/j.cellimm.2011.05.009
127. Pabst R, Gehrke I. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am J Respir Cell Mol Biol. (1990) 3:131–5. doi: 10.1165/ajrcmb/3.2.131
128. Hughes CE, Benson RA, Bedaj M, Maffia P. Antigen-presenting cells and antigen presentation in tertiary lymphoid organs. Front Immunol. (2016) 7:481. doi: 10.3389/fimmu.2016.00481
129. Foo SY, Phipps S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol. (2010) 3:537–44. doi: 10.1038/mi.2010.52
130. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology. (2016) 147:141–51. doi: 10.1111/imm.2016.147.issue-2
131. Dunlap MD, Prince OA, Rangel-Moreno J, Thomas KA, Scordo JM, Torrelles JB, et al. Formation of lung inducible bronchus associated lymphoid tissue is regulated by mycobacterium tuberculosis expressed determinants. Front Immunol. (2020) 11:1325. doi: 10.3389/fimmu.2020.01325
132. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci U.S.A. (2018) 115:E62–71. doi: 10.1073/pnas.1711373114
133. Canetti G. The tubercle bacillus in the pulmonary lesion of man. In: Histobacteriology and Its Bearing on the Therapy of Pulmonary Tuberculosis. Springer Publishing Company Inc, New York (1955).
135. McHenry ML, Williams SM, Stein CM. Genetics and evolution of tuberculosis pathogenesis: New perspectives and approaches. Infect Genet Evol. (2020) 81:104204. doi: 10.1016/j.meegid.2020.104204
136. McHenry ML, Bartlett J, Igo RP Jr., Wampande EM, Benchek P, Mayanja-Kizza H, et al. Interaction between host genes and Mycobacterium tuberculosis lineage can affect tuberculosis severity: Evidence for coevolution? PloS Genet. (2020) 16:e1008728. doi: 10.1371/journal.pgen.1008728
137. Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by mycobacterium tuberculosis. Microbiol Spectr. (2014) 2:MGM2–0005-2013. doi: 10.1128/microbiolspec.MGM2-0005-2013
138. Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol. (2017) 17:691–702. doi: 10.1038/nri.2017.69
140. Diedrich CR, O'Hern J, Wilkinson RJ. HIV-1 and the Mycobacterium tuberculosis granuloma: A systematic review and meta-analysis. Tuberculosis (Edinb). (2016) 98:62–76. doi: 10.1016/j.tube.2016.02.010
141. Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol. (2010) 161:1–9. doi: 10.1111/j.1365-2249.2010.04146.x
142. Keane J. TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatol (Oxford). (2005) 44:714–20. doi: 10.1093/rheumatology/keh567
143. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. (2001) 345:1098–104. doi: 10.1056/NEJMoa011110
144. Horsburgh CR Jr., O'Donnell M, Chamblee S, Moreland JL, Johnson J, Marsh BJ, et al. Revisiting rates of reactivation tuberculosis: a population-based approach. Am J Respir Crit Care Med. (2010) 182:420–5. doi: 10.1164/rccm.200909-1355OC
145. Solovic I, Sester M, Gomez-Reino JJ, Rieder HL, Ehlers S, Milburn HJ, et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: a TBNET consensus statement. Eur Respir J. (2010) 36:1185–206. doi: 10.1183/09031936.00028510
146. Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. (2008) 29:283–94. doi: 10.1016/j.immuni.2008.06.011
147. Saunders BM, Cooper AM. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol Cell Biol. (2000) 78:334–41. doi: 10.1046/j.1440-1711.2000.00933.x
148. Beham AW, Puellmann K, Laird R, Fuchs T, Streich R, Breysach C, et al. A TNF-regulated recombinatorial macrophage immune receptor implicated in granuloma formation in tuberculosis. PloS Pathog. (2011) 7:e1002375. doi: 10.1371/journal.ppat.1002375
149. Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, Salavert M, Vidal R, Rodríguez Carballeira M, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis. (2005) 40:756–9. doi: 10.1086/427941
150. Iliopoulos A, Psathakis K, Aslanidis S, Skagias L, Sfikakis PP. Tuberculosis and granuloma formation in patients receiving anti-TNF therapy. Int J Tuberc Lung Dis. (2006) 10:588–90.
151. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. (2013) 191:773–84. doi: 10.4049/jimmunol.1300113
152. Lenaerts A, Barry CE 3rd., Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev. (2015) 264:288–307. doi: 10.1111/imr.2015.264.issue-1
153. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
154. Baay-Guzman GJ, Duran-Padilla MA, Rangel-Santiago J, Tirado-Rodriguez B, Antonio-Andres G, Barrios-Payan J, et al. Dual role of hypoxia-inducible factor 1 α in experimental pulmonary tuberculosis: its implication as a new therapeutic target. Future Microbiol. (2018) 13:785–98. doi: 10.2217/fmb-2017-0168
155. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1 alpha is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
156. Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, et al. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol. (2009) 182:7974–81. doi: 10.4049/jimmunol.0804216
157. Strehl C, Fangradt M, Fearon U, Gaber T, Buttgereit F, Veale DJ. Hypoxia: how does the monocyte-macrophage system respond to changes in oxygen availability? J Leukoc Biol. (2014) 95:233–41. doi: 10.1189/jlb.1212627
158. Li Q, Xie Y, Cui Z, Huang H, Yang C, Yuan B, et al. Activation of hypoxia-inducible factor 1 (Hif-1) enhanced bactericidal effects of macrophages to Mycobacterium tuberculosis. Tuberculosis (Edinb). (2021) 126:102044. doi: 10.1016/j.tube.2020.102044
159. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
160. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
161. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. (2015) 5:18176. doi: 10.1038/srep18176
162. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an essential mediator of IFN-γ-dependent immunity to mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. doi: 10.4049/jimmunol.1600266
163. Shi L, Eugenin EA, Subbian S. Immunometabolism in tuberculosis. Front Immunol. (2016) 7:150. doi: 10.3389/fimmu.2016.00150
164. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. (2003) 198:1391–402. doi: 10.1084/jem.20030267
165. Schutyser E, Su Y, Yu Y, Gouwy M, Zaja-Milatovic S, Van Damme J, et al. Hypoxia enhances CXCR4 expression in human microvascular endothelial cells and human melanoma cells. Eur Cytokine Netw. (2007) 18:59–70. doi: 10.1684/ecn.2007.0087
166. Cardoso MS, Silva TM, Resende M, Appelberg R, Borges M. Lack of the transcription factor hypoxia-inducible factor 1α (HIF-1α) in macrophages accelerates the necrosis of mycobacterium avium-induced granulomas. Infect Immun. (2015) 83:3534–44. doi: 10.1128/IAI.00144-15
167. Elks PM, Brizee S, van der Vaart M, Walmsley SR, van Eeden FJ, Renshaw SA, et al. Hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PloS Pathog. (2013) 9:e1003789. doi: 10.1371/journal.ppat.1003789
168. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. (2015) 517:612–5. doi: 10.1038/nature13967
169. de Oliveira Rezende A, Sabóia RS, da Costa AC, da Silva Monteiro DMP, Zagmignan A, Santiago LÂM, et al. Restricted activation of the NF-κB pathway in individuals with latent tuberculosis infection after HIF-1α Blockade. Biomedicines. (2022) 10:817. doi: 10.3390/biomedicines10040817
170. Bandarra D, Biddlestone J, Mudie S, Müller HA, Rocha S. HIF-1α restricts NF-κB-dependent gene expression to control innate immunity signals. Dis Model Mech. (2015) 8:169–81. doi: 10.1242/dmm.017285
171. Panda S, Morgan J, Cheng C, Saito M, Gilman RH, Ciobanu N, et al. Identification of differentially recognized T cell epitopes in the spectrum of tuberculosis infection. Nat Commun. (2024) 15:765. doi: 10.1038/s41467-024-45058-9
172. Lindestam Arlehamn CS, Paul S, Mele F, Huang C, Greenbaum JA, Vita R, et al. Immunological consequences of intragenus conservation of Mycobacterium tuberculosis T-cell epitopes. Proc Natl Acad Sci U.S.A. (2015) 112:E147–155. doi: 10.1073/pnas.1416537112
173. Lindestam Arlehamn CS, Lewinsohn D, Sette A, Lewinsohn D. Antigens for CD4 and CD8 T cells in tuberculosis. Cold Spring Harb Perspect Med. (2014) 4:a018465. doi: 10.1101/cshperspect.a018465
174. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. (2010) 42:498–503. doi: 10.1038/ng.590
175. Coscolla M, Copin R, Sutherland J, Gehre F, de Jong B, Owolabi O, et al. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe. (2015) 18:538–48. doi: 10.1016/j.chom.2015.10.008
176. Aguilo JI, Alonso H, Uranga S, Marinova D, Arbués A, de Martino A, et al. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol. (2013) 15:1994–2005. doi: 10.1111/cmi.12169
177. Mustafa AS. Immunological characterization of proteins expressed by genes located in mycobacterium tuberculosis-specific genomic regions encoding the ESAT6-like proteins. Vaccines (Basel). (2021) 9:27. doi: 10.3390/vaccines9010027
178. Rogerson BJ, Jung YJ, LaCourse R, Ryan L, Enright N, North RJ. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen specific T cells during stationary level lung infection in mice. Immunology. (2006) 118:195–201. doi: 10.1111/j.1365-2567.2006.02355.x
179. Gröschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol. (2016) 14:677–91. doi: 10.1038/nrmicro.2016.131
180. Hingley-Wilson SM, Connell D, Pollock K, Hsu T, Tchilian E, Sykes A, et al. ESX1-dependent fractalkine mediates chemotaxis and Mycobacterium tuberculosis infection in humans. Tuberculosis (Edinb). (2014) 94:262–70. doi: 10.1016/j.tube.2014.01.004
181. Elkington PT, O’Kane CM, Friedland JS. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol. (2005) 142:12–20. doi: 10.1111/j.1365-2249.2005.02840.x
182. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. (2010) 327:466–9. doi: 10.1126/science.1179663
183. Abdallah AM, Savage ND, van Zon M, Wilson L, Vandenbroucke-Grauls CM, van der Wel NN, et al. The ESX-5 secretion system of Mycobacterium marinum modulates the macrophage response. J Immunol. (2008) 181:7166–75. doi: 10.4049/jimmunol.181.10.7166
184. Abdallah AM, Bestebroer J, Savage ND, de Punder K, van Zon M, Wilson L, et al. Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J Immunol. (2011) 187:4744–53. doi: 10.4049/jimmunol.1101457
185. Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6'-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. (2006) 168:1249–61. doi: 10.2353/ajpath.2006.050848
186. Bekierkunst A. Acute granulomatous response produced in mice by trehalose-6,6-dimycolate. J Bacteriol. (1968) 96:958–61. doi: 10.1128/jb.96.4.958-961.1968
187. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of mycobacterium bovis bacillus calmette guerin is due principally to trehalose mycolates. J Immunol. (2005) 174:5007–15. doi: 10.4049/jimmunol.174.8.5007
188. Yarkoni E, Rapp HJ. Granuloma formation in lungs of mice after intravenous administration of emulsified trehalose-6,6'-dimycolate (Cord factor): reaction intensity depends on size distribution of the oil droplets. Infect Immun. (1977) 18:552–4. doi: 10.1128/iai.18.2.552-554.1977
189. Silva CL, Ekizlerian SM, Fazioli RA. Role of cord factor in the modulation of infection caused by mycobacteria. Am J Pathol. (1985) 118:238–47.
190. Chen T, He L, Deng W, Xie J. The Mycobacterium DosR regulon structure and diversity revealed by comparative genomic analysis. J Cell Biochem. (2013) 114:1–6. doi: 10.1002/jcb.v114.1
191. Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc Natl Acad Sci USA. (2001) 98:7534–9. doi: 10.1073/pnas.121172498
192. Shiloh MU, Manzanillo P, Cox JS. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe. (2008) 3:323–30. doi: 10.1016/j.chom.2008.03.007
193. Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. (2003) 198:705–13. doi: 10.1084/jem.20030205
194. Sevalkar RR, Glasgow JN, Pettinati M, Marti MA, Reddy VP, Basu S, et al. Mycobacterium tuberculosis DosS binds H2S through its Fe3+ heme iron to regulate the DosR dormancy regulon. Redox Biol. (2022) 52:102316. doi: 10.1016/j.redox.2022.102316
195. Saini V, Chinta KC, Reddy VP, Glasgow JN, Stein A, Lamprecht DA, et al. Hydrogen sulfide stimulates Mycobacterium tuberculosis respiration, growth and pathogenesis. Nat Commun. (2020) 11:557. doi: 10.1038/s41467-019-14132-y
196. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U.S.A. (2008) 105:4376–80. doi: 10.1073/pnas.0711159105
197. Miner MD, Chang JC, Pandey AK, Sassetti CM, Sherman DR. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol. (2009) 47:407–11.
198. Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev. (2012) 36:514–32. doi: 10.1111/j.1574-6976.2012.00331.x
199. Martinot AJ, Farrow M, Bai L, Layre E, Cheng TY, Tsai JH, et al. Mycobacterial metabolic syndrome: lprG and rv1410 regulate triacylglyceride levels, growth rate and virulence in mycobacterium tuberculosis. PloS Pathog. (2016) 12:e1005351. doi: 10.1371/journal.ppat.1005351
200. Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the zebrafish. Cell Host Microbe. (2007) 2:29–39. doi: 10.1016/j.chom.2007.06.004
201. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. (2012) 148:434–46. doi: 10.1016/j.cell.2011.12.023
202. Tobin DM, Vary JC Jr., Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. (2010) 140:717–30. doi: 10.1016/j.cell.2010.02.013
203. Pagan AJ, Yang CT, Cameron J, Swaim LE, Ellett F, Lieschke GJ, et al. Myeloid growth factors promote resistance to mycobacterial infection by curtailing granuloma necrosis through macrophage replenishment. Cell Host Microbe. (2015) 18:15–26. doi: 10.1016/j.chom.2015.06.008
204. Berg RD, Levitte S, O’Sullivan MP, O’Leary SM, Cambier CJ, Cameron J, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell. (2016) 165:139–52. doi: 10.1016/j.cell.2016.02.034
205. Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J Immunol. (1999) 162:5407–16. doi: 10.4049/jimmunol.162.9.5407
206. Kwan CK, Ernst JD. HIV and tuberculosis: A deadly human syndemic. Clin Microbiol Rev. (2011) 24:351–76. doi: 10.1128/CMR.00042-10
207. Vila-del Sol V, Punzón C, Fresno M. IFN-gamma-induced TNF-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol. (2008) 181:4461–70. doi: 10.4049/jimmunol.181.7.4461
208. Ahmed M, Tezera LB, Elkington PT, Leslie AJ. The paradox of immune checkpoint inhibition re-activating tuberculosis. Eur Respir J. (2022) 60:2102512. doi: 10.1183/13993003.02512-2021
209. Pagán AJ, Ramakrishnan L. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb Perspect Med. (2014) 5:a018499. doi: 10.1101/cshperspect.a018499
210. Tezera LB, Bielecka MK, Ogongo P, Walker NF, Ellis M, Garay-Baquero DJ, et al. Anti-PD-1 immunotherapy leads to tuberculosis reactivation via dysregulation of TNF-α. Elife. (2020) 9:e52668. doi: 10.7554/eLife.52668
211. Aly S, Laskay T, Mages J, Malzan A, Lang R, Ehlers S. Interferon-gamma-dependent mechanisms of mycobacteria-induced pulmonary immunopathology: the role of angiostasis and CXCR3-targeted chemokines for granuloma necrosis. J Pathol. (2007) 212:295–305. doi: 10.1002/path.v212:3
212. Jurado JO, Alvarez IB, Pasquinelli V, Martínez GJ, Quiroga MF, Abbate E, et al. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol. (2008) 181:116–25. doi: 10.4049/jimmunol.181.1.116
213. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. (2011) 186:1598–607. doi: 10.4049/jimmunol.1003304
214. Lázár-Molnár E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U.S.A. (2010) 107:13402–7. doi: 10.1073/pnas.1007394107
215. Sable SB. Programmed Death 1 lives up to its reputation in active tuberculosis. J Infect Dis. (2013) 208:541–3. doi: 10.1093/infdis/jit211
216. Tousif S, Singh Y, Prasad DV, Sharma P, Van Kaer L, Das G. T cells from Programmed Death-1 deficient mice respond poorly to Mycobacterium tuberculosis infection. PloS One. (2011) 6:e19864. doi: 10.1371/journal.pone.0019864
217. Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, et al. Tuberculous granulomas are hypoxic in Guinea pigs, rabbits, and nonhuman primates. Infect Immun. (2008) 76:2333–40. doi: 10.1128/IAI.01515-07
218. Belton M, Brilha S, Manavaki R, Mauri F, Nijran K, Hong YT, et al. Hypoxia and tissue destruction in pulmonary TB. Thorax. (2016) 71:1145–53. doi: 10.1136/thoraxjnl-2015-207402
219. Mikami Y, Grubb BR, Rogers TD, Dang H, Asakura T, Kota P, et al. Chronic airway epithelial hypoxia exacerbates injury in muco-obstructive lung disease through mucus hyperconcentration. Sci Transl Med. (2023) 15:eabo7728. doi: 10.1126/scitranslmed.abo7728
220. Kotov DI, Lee OV, Fattinger SA, Langner CA, Guillen JV, Peters JM, et al. Early cellular mechanisms of type I interferon-driven susceptibility to tuberculosis. Cell. (2023) 186:5536–5553.e22. doi: 10.1016/j.cell.2023.11.002
221. Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat Commun. (2020) 11:5566. doi: 10.1038/s41467-020-19412-6
222. Xu G, Wang J, Gao GF, Liu CH. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell. (2014) 5:728–36. doi: 10.1007/s13238-014-0077-5
223. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
224. Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, et al. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat Microbiol. (2019) 4:2128–35. doi: 10.1038/s41564-019-0578-3
225. Ryan GJ, Hoff DR, Driver ER, Voskuil MI, Gonzalez-Juarrero M, Basaraba RJ, et al. Multiple M. tuberculosis phenotypes in mouse and Guinea pig lung tissue revealed by a dual-staining approach. PloS One. (2010) 5:e11108. doi: 10.1371/journal.pone.0011108
226. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. (2014) 20:75–9. doi: 10.1038/nm.3412
227. Gideon HP, Hughes TK, Tzouanas CN, Wadsworth MH 2nd., Tu AA, Gierahn TM, et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity. (2022) 55:827–846.e10. doi: 10.1016/j.immuni.2022.04.004
228. Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, et al. Incipient and subclinical tuberculosis: a clinical review of early stages and progression of infection. Clin Microbiol Rev. (2018) 31:e00021–18. doi: 10.1128/CMR.00021-18
229. Capuano SV 3rd., Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, et al. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect Immun. (2003) 71:5831–44. doi: 10.1128/IAI.71.10.5831-5844.2003
230. Pena JC, Ho WZ. Monkey models of tuberculosis: lessons learned. Infect Immun. (2015) 83:852–62. doi: 10.1128/IAI.02850-14
231. Coleman MT, Chen RY, Lee M, Lin PL, Dodd LE, Maiello P, et al. PET/CT imaging reveals a therapeutic response to oxazolidinones in macaques and humans with tuberculosis. Sci Transl Med. (2014) 6:265ra167. doi: 10.1126/scitranslmed.3009500
232. Malherbe ST, Shenai S, Ronacher K, Loxton AG, Dolganov G, Kriel M, et al. Catalysis TB–Biomarker Consortium. Persisting positron emission tomography lesion activity and Mycobacterium tuberculosis mRNA after tuberculosis cure. Nat Med. (2016) 22:1094–100. doi: 10.1038/nm.4177
233. Zaidi SMA, Coussens AK, Seddon JA, Kredo T, Warner D, Houben RMGJ, et al. Beyond latent and active tuberculosis: a scoping review of conceptual frameworks. EClinicalMedicine. (2023) 66:102332. doi: 10.1016/j.eclinm.2023.102332
234. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PloS Pathog. (2015) 11:e1004603. doi: 10.1371/journal.ppat.1004603
235. Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun. (2003) 71:7099–108. doi: 10.1128/IAI.71.12.7099-7108.2003
236. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med. (2016) 22:531–8. doi: 10.1038/nm.4073
237. Wells G, Glasgow JN, Nargan K, Lumamba K, Madansein R, Maharaj K, et al. Micro-computed tomography analysis of the human tuberculous lung reveals remarkable heterogeneity in three-dimensional granuloma morphology. Am J Respir Crit Care Med. (2021) 204:583–95. doi: 10.1164/rccm.202101-0032OC
238. Wells G, Glasgow JN, Nargan K, Lumamba K, Madansein R, Maharaj K, et al. A high-resolution 3D atlas of the spectrum of tuberculous and COVID-19 lung lesions. EMBO Mol Med. (2022) 14:e16283. doi: 10.15252/emmm.202216283
239. Ohashi T, Terasawa K, Aoki M, Akazawa T, Shibata H, Kuze B, et al. The importance of FDG-PET/CT parameters for the assessment of the immune status in advanced HNSCC. Auris Nasus Larynx. (2020) 47:658–67. doi: 10.1016/j.anl.2020.01.002
240. Maiello P, DiFazio RM, Cadena AM, Rodgers MA, Lin PL, Scanga CA, et al. Rhesus macaques are more susceptible to progressive tuberculosis than cynomolgus macaques: a quantitative comparison. Infect Immun. (2018) 86:e00505–17. doi: 10.1128/IAI.00505-17
241. Chu Y, Chang Y, Lu W, Sheng X, Wang S, Xu H, et al. Regulation of autophagy by glycolysis in cancer. Cancer Manag Res. (2020) 12:13259–71. doi: 10.2147/CMAR.S279672
242. Clarke AJ, Simon A. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. (2019) 19:170–83. doi: 10.1038/s41577-018-0095-2
243. Dargent A, Wallet F, Friggeri A, Bohe J. Lactic alkalosis in intensive care: a red flag? Crit Care. (2023) 27:184. doi: 10.1186/s13054-023-04464-z
244. Quach CHT, Jung K-H, Lee JH, Park JW, Moon SH, Cho YS. Mild alkalization acutely triggers the warburg effect by enhancing hexokinase activity via voltage-dependent anion channel binding. PloS One. (2016) 11:e0159529. doi: 10.1371/journal.pone.0159529
245. Cumming BM, Addicott KW, Adamson JH, Steyn AJ. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife. (2018) 7:e39169. doi: 10.7554/eLife.39169
246. Hackett EE, Charles-Messance H, O'Leary SM, Gleeson LE, Muñoz-Wolf N, Case S, et al. Mycobacterium tuberculosis Limits Host Glycolysis and IL-1β by Restriction of PFK-M via MicroRNA-21. Cell Rep. (2020) 30:124–136.e4. doi: 10.1016/j.celrep.2019.12.015
247. Olson GS, Murray TA, Jahn AN, Mai D, Diercks AH, Gold ES, et al. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Rep. (2021) 35:109195. doi: 10.1016/j.celrep.2021.109195
248. Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. (1959) 234:1355–62. doi: 10.1016/S0021-9258(18)70011-2
249. Lähdevirta J, Maury CP, Teppo AM, Repo H. Elevated levels of circulating cachectin/tumor necrosis factor in patients with acquired immunodeficiency syndrome. Am J Med. (1988) 85:289–91. doi: 10.1016/0002-9343(88)90576-1
250. Chan ED. Vulnerability to nontuberculous mycobacterial lung disease or systemic infection due to genetic / heritable disorders. In: Griffith DE, editor. Nontuberculous Mycobacterial Disease: A Comprehensive Approach to Diagnosis and Management. Springer Nature, Switzerland (2019). p. 89–110.
251. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Med (Baltimore). (2010) 89:381–402. doi: 10.1097/MD.0b013e3181fdd832
252. Honda JR, Alper S, Bai X, Chan ED. Acquired and genetic host susceptibility factors and microbial pathogenic factors that predispose to nontuberculous mycobacterial infections. Curr Opin Immunol. (2018) 54:66–73. doi: 10.1016/j.coi.2018.06.001
253. Wu UI, Holland SM. Host susceptibility to non-tuberculous mycobacterial infections. Lancet Infect Dis. (2015) 15:968–80. doi: 10.1016/S1473-3099(15)00089-4
254. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. (1993) 178:2243–7. doi: 10.1084/jem.178.6.2243
255. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. (1993) 178:2249–54. doi: 10.1084/jem.178.6.2249
256. Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, et al. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PloS Pathog. (2016) 12:e1005490. doi: 10.1371/journal.ppat.1005490
257. Kirman J, McCoy K, Hook S, Prout M, Delahunt B, Orme I, et al. CTLA-4 blockade enhances the immune response induced by mycobacterial infection but does not lead to increased protection. Infect Immun. (1999) 67:3786–92. doi: 10.1128/IAI.67.8.3786-3792.1999
258. Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. (2009) 119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618
259. Watcharanurak K, Zang L, Nishikawa M, Yoshinaga K, Yamamoto Y, Takahashi Y, et al. Effects of upregulated indoleamine 2, 3-dioxygenase 1 by interferon γ gene transfer on interferon γ-mediated antitumor activity. Gene Ther. (2014) 21:794–801. doi: 10.1038/gt.2014.54
260. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. (1995) 2:561–72. doi: 10.1016/1074-7613(95)90001-2
261. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell. (2019) 178:1344–61. doi: 10.1016/j.cell.2019.08.004
262. Patel HJ, Patel BM. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. (2017) 170:56–63. doi: 10.1016/j.lfs.2016.11.033
263. Hernandez-Pando R, Orozco H, Arriaga K, Pavön L, Rook G. Treatment with BB-94, a broad spectrum inhibitor of zinc-dependent metalloproteinases, causes deviation of the cytokine profile towards type-2 in experimental pulmonary tuberculosis in Balb/c mice. Int J Exp Pathol. (2000) 81:199–209. doi: 10.1046/j.1365-2613.2000.00152.x
264. Izzo AA, Izzo LS, Kasimos J, Majka S. A matrix metalloproteinase inhibitor promotes granuloma formation during the early phase of Mycobacterium tuberculosis pulmonary infection. Tuberculosis (Edinb). (2004) 84:387–96. doi: 10.1016/j.tube.2004.07.001
265. Rich AR, Follis JH Jr. The effect of low oxygen tension upon the development of experimental tuberculosis. Am Rev Tuberc. (1942) 71:345–57.
266. Hunter L, Ruedas-Torres I, Agulló-Ros I, Rayner E, Salguero FJ. Comparative pathology of experimental pulmonary tuberculosis in animal models. Front Vet Sci. (2023) 10:1264833. doi: 10.3389/fvets.2023.1264833
267. Li L, Wang M, Mei Z, Cao W, Yang Y, Wang Y, et al. lncRNAs HIF1A-AS2 facilitates the up-regulation of HIF-1α by sponging to miR-153-3p, whereby promoting angiogenesis in HUVECs in hypoxia. BioMed Pharmacother. (2017) 96:165–72. doi: 10.1016/j.biopha.2017.09.113
268. Knight M, Stanley S. HIF-1α as a central mediator of cellular resistance to intracellular pathogens. Curr Opin Immunol. (2019) 60:111–6. doi: 10.1016/j.coi.2019.05.005
269. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. (2022) 7:305. doi: 10.1038/s41392-022-01151-3
270. Mehra S, Alvarez X, Didier PJ, Doyle LA, Blanchard JL, Lackner AA, et al. Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. J Infect Dis. (2013) 207:1115–27. doi: 10.1093/infdis/jis778
271. Lott JS. The tryptophan biosynthetic pathway is essential for Mycobacterium tuberculosis to cause disease. Biochem Soc Trans. (2020) 48:2029–37. doi: 10.1042/BST20200194
272. Suzuki Y, Suda T, Asada K, Miwa S, Suzuki M, Fujie M, et al. Serum indoleamine 2,3-dioxygenase activity predicts prognosis of pulmonary tuberculosis. Clin Vaccine Immunol. (2012) 19:436–42. doi: 10.1128/CVI.05402-11
273. Regev D, Surolia R, Karki S, Zolak J, Montes-Worboys A, Oliva O, et al. Heme oxygenase-1 promotes granuloma development and protects against dissemination of mycobacteria. Lab Invest. (2012) 92:1541–52. doi: 10.1038/labinvest.2012.125
274. Silva-Gomes S, Appelberg R, Larsen R, Soares MP, Gomes MS. Heme catabolism by heme oxygenase-1 confers host resistance to Mycobacterium infection. Infect Immun. (2013) 81:2536–45. doi: 10.1128/IAI.00251-13
275. Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. (2004) 165:1045–53. doi: 10.1016/S0002-9440(10)63365-2
276. Yang S, Ouyang J, Lu Y, Harypursat V, Chen Y. A dual role of heme oxygenase-1 in tuberculosis. Front Immunol. (2022) 13:842858. doi: 10.3389/fimmu.2022.842858
277. Chinta KC, Rahman MA, Saini V, Glasgow JN, Reddy VP, Lever JM, et al. Microanatomic distribution of myeloid heme oxygenase-1 protects against free radical-mediated immunopathology in human tuberculosis. Cell Rep. (2018) 25:1938–1952.e5. doi: 10.1016/j.celrep.2018.10.073
278. Abdalla MY, Ahmad IM, Switzer B, Britigan BE. Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol. (2015) 4:328–39. doi: 10.1016/j.redox.2015.01.012
279. Costa DL, Namasivayam S, Amaral EP, Arora K, Chao A, Mittereder LR, et al. Pharmacological inhibition of host heme oxygenase-1 suppresses mycobacterium tuberculosis infection in vivo by a mechanism dependent on T lymphocytes. mBio. (2016) 7:e01675–16. doi: 10.1128/mBio.01675-16
280. Rockwood N, Costa DL, Amaral EP, Du Bruyn E, Kubler A, Gil-Santana L, et al. Mycobacterium tuberculosis induction of heme oxygenase-1 expression is dependent on oxidative stress and reflects treatment outcomes. Front Immunol. (2017) 8:542. doi: 10.3389/fimmu.2017.00542
281. Vaddi A, Hulsebus HJ, O’Neill EL, Knight V, Chan ED. A narrative review of the controversy on the risk of mycobacterial infections with immune checkpoint inhibitor use: does Goldilocks have the answer? J Thorac Dis. (2024) 16:1601–16. doi: 10.21037/jtd-23-1395
282. Fallahi-Sichani M, Kirschner DE, Linderman JJ. NF-κB signaling dynamics play a key role in infection control in tuberculosis. Front Physiol. (2012) 3:170. doi: 10.3389/fphys.2012.00170
283. Iseman MD. A Clinician’s Guide to Tuberculosis. 1st ed. Philadelphia, PA: Lippincott Williams & Wilkins (2000).
284. Balasubramanian V, Wiegeshaus EH, Taylor BT, Smith DW. Pathogenesis of tuberculosis: pathway to apical localization. Tuber Lung Dis. (1994) 75:168–78. doi: 10.1016/0962-8479(94)90002-7
285. Elkington PT, Friedland JS. Permutations of time and place in tuberculosis. Lancet Infect Dis. (2015) 15:1357–60. doi: 10.1016/S1473-3099(15)00135-8
286. Donald PR, Diacon AH, Thee S. Anton ghon and his colleagues and their studies of the primary focus and complex of tuberculosis infection and their relevance for the twenty-first century. Respiration. (2021) 100:557–67. doi: 10.1159/000509522
287. Hunter RL, Jagannath C, Actor JK. Pathologyt of postprimary tuberculosis in humans and mice: Contradiction of long-held beliefs. Tuberculosis. (2007) 87:267–78. doi: 10.1016/j.tube.2006.11.003
288. Opie EL, Aronson JD. Tubercle bacilli in latent tuberculous lesions and in the lung tissue without tuberculous lesions. Arch Pathol Lab Med. (1927) 4:1–21.
289. Ghesani N, Patrawalla A, Lardizabal A, Salgame P, Fennelly KP. Increased cellular activity in thoracic lymph nodes in early human latent tuberculosis infection. Am J Respir Crit Care Med. (2014) 189:748–50. doi: 10.1164/rccm.201311-1976LE
290. Laennec R. A treatise on diseases of the chest in which they are described according to their anatomical characters, and their diagnosis established on a new principle by means of acoustick instruments. Birmingham AL., London: T&G Underwood (1821). reprinted 1979 by The Classics of Medicine Library.
291. Virchow R. Cellular pathology as based upon physiological and pathological histology. Birmingham, AL, London: John Churchill (1860). reprinted by The Classics of Medicine Library, 1978.
293. Hunter RL. Tuberculosis as a three-act play: A new paradigm for the pathogenesis of pulmonary tuberculosis. Tuberculosis (Edinb). (2016) 97:8–17. doi: 10.1016/j.tube.2015.11.010
294. Hunter RL, Actor JK, Hwang SA, Karev V, Jagannath C. Pathogenesis of post primary tuberculosis: immunity and hypersensitivity in the development of cavities. Ann Clin Lab Sci. (2014) 44:365–87.
295. Hunter RL, Actor JK, Hwang SA, Khan A, Urbanowski ME, Kaushal D, et al. Pathogenesis and animal models of post-primary (Bronchogenic) tuberculosis, A review. Pathogens. (2018) 7:19. doi: 10.3390/pathogens7010019
296. Hunter RL. On the pathogenesis of post primary tuberculosis: the role of bronchial obstruction in the pathogenesis of cavities. Tuberculosis (Edinb). (2011) 91 Suppl 1:S6–S10. doi: 10.1016/j.tube.2011.10.003
297. Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb). (2011) 91:497–509. doi: 10.1016/j.tube.2011.03.007
298. Medlar EM. The behavior of pulmonary tuberculous lesions; a pathological study. Am Rev Tuberc. (1955) 71:1e244.
299. Pagel W, Simmonds F, MacDonald N, Nassau E. Pulmonary tuberculosis, bacteriology, pathology, management, epidemiology and prevention. London: Oxford University Press (1964).
300. Nargan K, Glasgow JN, Nadeem S, Naidoo T, Wells G, Hunter RL, et al. Spatial distribution of Mycobacterium tuberculosis mRNA and secreted antigens in acid-fast negative human antemortem and resected tissue. EBioMedicine. (2024) 105:105196. doi: 10.1016/j.ebiom.2024.105196
301. Dartois V, Dick T. A ginger root or plum model for the tuberculosis "Granuloma"? Am J Respir Crit Care Med. (2021) 204:505–7. doi: 10.1164/rccm.202104-1052ED
302. Chang JC, Wysocki A, Tchou-Wong KM, Moskowitz N, Zhang Y, Rom WN. Effect of Mycobacterium tuberculosis and its components on macrophages and the release of matrix metalloproteinases. Thorax. (1996) 51:306–11. doi: 10.1136/thx.51.3.306
303. Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, et al. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest. (2011) 121:1827–33. doi: 10.1172/JCI45666
304. Elkington PT, Ugarte-Gil CA, Friedland JS. Matrix metalloproteinases in tuberculosis. Eur Respir J. (2011) 38:456–64. doi: 10.1183/09031936.00015411
305. Reichmann MT, Tezera LB, Vallejo AF, Vukmirovic M, Xiao R, Reynolds J, et al. Integrated transcriptomic analysis of human tuberculosis granulomas and a biomimetic model identifies therapeutic targets. J Clin Invest. (2021) 131:e148136. doi: 10.1172/JCI148136
306. Salgame P. MMPs in tuberculosis: granuloma creators and tissue destroyers. J Clin Invest. (2011) 121:1686–8. doi: 10.1172/JCI57423
307. Nedeltchev GG, Raghunand TR, Jassal MS, Lun S, Cheng QJ, Bishai WR. Extrapulmonary dissemination of Mycobacterium bovis but not Mycobacterium tuberculosis in a bronchoscopic rabbit model of cavitary tuberculosis. Infect Immun. (2009) 77:598–603. doi: 10.1128/IAI.01132-08
308. Kaufmann SH, Schaible UE. 100th anniversary of Robert Koch's Nobel Prize for the discovery of the tubercle bacillus. Trends Microbiol. (2005) 13:469–75. doi: 10.1016/j.tim.2005.08.003
309. Ganchua SKC, Cadena AM, Maiello P, Gideon HP, Myers AJ, Junecko BF, et al. Lymph nodes are sites of prolonged bacterial persistence during Mycobacterium tuberculosis infection in macaques. PloS Pathog. (2018) 14:e1007337. doi: 10.1371/journal.ppat.1007337
310. Chen RY, Yu X, Smith B, Liu X, Gao J, Diacon AH, et al. Radiological and functional evidence of the bronchial spread of tuberculosis: an observational analysis. Lancet Microbe. (2021) 2:e518–26. doi: 10.1016/S2666-5247(21)00058-6
311. Dock W. Apical localization of phthisis: its significance in treatment by prolonged rest in bed. Am Rev Tuberc. (1946) 53:297–305. doi: 10.1164/art.1946.53.4.297
312. Murray JF. Bill Dock and the location of pulmonary tuberculosis: how bed rest might have helped consumption. Am J Respir Crit Care Med. (2003) 168:1029–33. doi: 10.1164/rccm.200307-1016OE
313. Goodwin RA, Des Prez RM. Apical localization of pulmonary tuberculosis, chronic pulmonary histoplasmosis, and progressive massive fibrosis of the lung. Chest. (1983) 83:801–5. doi: 10.1378/chest.83.5.801
314. West JB, Luks AM. West’s Respiratory Physiology: The Essentials. 11th ed. Philadelphia, PA: Wolters Kluwer (2020).
315. Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HH, Elhassan GO, et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. (2014) 1:777–802. doi: 10.18632/oncoscience.v1i12
316. López-Lázaro M. A new view of carcinogenesis and an alternative approach to cancer therapy. Mol Med. (2010) 16:144–53. doi: 10.2119/molmed.2009.00162
317. Sever JL, Youmans G. The relation of oxygen tension to virulence of tubercle bacilli and to acquired resistance in tuberculosis. J Infect Dis. (1957) 101:193–202. doi: 10.1093/infdis/101.2.193
318. Medlar EM, Sasano KT. A study of the pathology of experimental pulmonary tuberculosis in the rabbit. Am Rev Tuberc. (1935) 34:456–75.
319. Dock W. Effect of posture on alveolar gas tension in tuberculosis: explanation for favored sites of chronic pulmonary lesions. Arch Intern Med. (1954) 94:700–8. doi: 10.1001/archinte.1954.00250050014003
320. Niikawa H, Okamoto T, Ayyat KS, Itoda Y, Farver CF, McCurry KR. Better oxygenation in upper lobes than in whole lungs in marginal human donor lungs utilized for cellular ex vivo lung perfusion. J Heart Lung Transplant. (2019) 38:S321. doi: 10.1016/j.healun.2019.01.808
321. Haapanen JH, Kass I, Gensini G, Middlebrook G. Studies on the gaseous content of tuberculous cavities. Am Rev Respir Dis. (1959) 80:1–5. doi: 10.1164/arrd.1959.80.1P1.1
322. Fayyazi A, Eichmeyer B, Soruri A, Schweyer S, Herms J, Schwarz P, et al. Apoptosis of macrophages and T cells in tuberculosis associated caseous necrosis. J Pathol. (2000) 191:417–25. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH664>3.0.CO;2-R
323. Dharmadhikari AS, Nardell EA. What animal models teach humans about tuberculosis. Am J Respir Cell Mol Biol. (2008) 39:503–8. doi: 10.1165/rcmb.2008-0154TR
324. Gupta UD, Katoch VM. Animal models of tuberculosis. Tuberculosis (Edinb). (2005) 85:277–93. doi: 10.1016/j.tube.2005.08.008
325. Sakamoto K. The pathology of Mycobacterium tuberculosis infection. Vet Pathol. (2012) 49:423–39. doi: 10.1177/0300985811429313
326. Williams A, Orme IM. Animal models of tuberculosis: an overview. Microbiol Spectr. (2016) 4:1–12. doi: 10.1128/microbiolspec.TBTB2-0004-2015
327. Bucsan AN, Mehra S, Khader SA, Kaushal D. The current state of animal models and genomic approaches towards identifying and validating molecular determinants of Mycobacterium tuberculosis infection and tuberculosis disease. Pathog Dis. (2019) 77:ftz037. doi: 10.1093/femspd/ftz037
328. Palmer MV. Mycobacterium bovis: characteristics of wildlife reservoir hosts. Transbound Emerg Dis. (2013) 60 Suppl 1:1–13. doi: 10.1111/tbed.12115
329. Villarreal-Ramos B, Berg S, Whelan A, Holbert S, Carreras F, Salguero FJ, et al. Experimental infection of cattle with Mycobacterium tuberculosis isolates shows the attenuation of the human tubercle bacillus for cattle. Sci Rep. (2018) 8:894. doi: 10.1038/s41598-017-18575-5
330. Pesciaroli M, Alvarez J, Boniotti MB, Cagiola M, Di Marco V, Marianelli C, et al. Tuberculosis in domestic animal species. Res Vet Sci. (2014) 97 Suppl:S78–85. doi: 10.1016/j.rvsc.2014.05.015
331. Cardona PJ, Williams A. Experimental animal modeling for TB vaccine development. Int J Infect Dis. (2017) 56:268–73. doi: 10.1016/j.ijid.2017.01.030
332. Siddiqui N, Price S, Hope J. BCG vaccination of neonatal calves: potential roles for innate immune cells in the induction of protective immunity. Comp Immunol Microbiol Infect Dis. (2012) 35:219–26. doi: 10.1016/j.cimid.2011.11.003
333. Buddle BM, Skinner MA, Wedlock DN, de Lisle GW, Vordermeier HM, Glyn Hewinson R. Cattle as a model for development of vaccines against human tuberculosis. Tuberculosis (Edinb). (2005) 85:19–24. doi: 10.1016/j.tube.2004.09.003
334. Villarreal-Ramos B, Berg S, Chamberlain L, McShane H, Hewinson RG, Clifford D, et al. Development of a BCG challenge model for the testing of vaccine candidates against tuberculosis in cattle. Vaccine. (2014) 32:5645–9. doi: 10.1016/j.vaccine.2014.08.009
335. McIlroy SG, Neill SD, McCracken RM. Pulmonary lesions and Mycobacterium bovis excretion from the respiratory tract of tuberculin reacting cattle. Vet Rec. (1986) 118:718–21. doi: 10.1136/vr.118.26.718
336. Wangoo A, Johnson L, Gough J, Ackbar R, Inglut S, Hicks D, et al. Advanced granulomatous lesions in Mycobacterium bovis-infected cattle are associated with increased expression of type I procollagen, gammadelta (WC1+) T cells and CD 68+ cells. J Comp Pathol. (2005) 133:223–34. doi: 10.1016/j.jcpa.2005.05.001
337. Liebana E, Marsh S, Gough J, Nunez A, Vordermeier HM, Whelan A, et al. Distribution and activation of T-lymphocyte subsets in tuberculous bovine lymph-node granulomas. Vet Pathol. (2007) 44:366–72. doi: 10.1354/vp.44-3-366
338. Cassidy JP, Bryson DG, Gutiérrez Cancela MM, Forster F, Pollock JM, Neill SD. Lymphocyte subtypes in experimentally induced early-stage bovine tuberculous lesions. J Comp Pathol. (2001) 124:46–51. doi: 10.1053/jcpa.2000.0427
339. Palmer MV, Wiarda J, Kanipe C, Thacker TC. Early Pulmonary Lesions in Cattle Infected via Aerosolized Mycobacterium bovis. Vet Pathol. (2019) 56:544–54. doi: 10.1177/0300985819833454
340. Rusk RA, Palmer MV, Waters WR, McGill JL. Measuring bovine γδ T cell function at the site of Mycobacterium bovis infection. Vet Immunol Immunopathol. (2017) 193-194:38–49. doi: 10.1016/j.vetimm.2017.10.004
341. McGill JL, Sacco RE, Baldwin CL, Telfer JC, Palmer MV, Waters WR. The role of gamma delta T cells in immunity to Mycobacterium bovis infection in cattle. Vet Immunol Immunopathol. (2014) 159:133–43. doi: 10.1016/j.vetimm.2014.02.010
342. Mikota SK. Tuberculosis in elephants. In: Fowler ME, Miller RE, editor. Zoo and wild animal medicine, current therapy, Sixth ed. Saunders/Elsevier, St Louis, MO (2008). p. 355–64.
343. Payeur JB, Jarnagin JL, Marquardt JG, Whipple DL. Mycobacterial isolations in captive elephants in the United States. Ann N Y Acad Sci. (2002) 969:256–8. doi: 10.1111/j.1749-6632.2002.tb04388.x
344. Rajbhandari RM, de la Fuente J, Karmacharya D, Mathema S, Maharjan B, Dixit SM, et al. Understanding Mycobacterium tuberculosis complex in elephants through a One Health approach: a systematic review. BMC Vet Res. (2022) 18:262. doi: 10.1186/s12917-022-03356-8
345. Miller MA, Kerr TJ, de Waal CR, Goosen WJ, Streicher EM, Hausler G, et al. Mycobacterium bovis infection in free-ranging african elephants. Emerg Infect Dis. (2021) 27:990–2. doi: 10.3201/eid2703.204729
346. Suga S, Mukai Y, Ishikawa S, Yoshida S, Paudel S, Wada T. Intensive treatment of A captive bornean elephant (Elephas maximus borneensis) infected with mycobacterium caprae in Japan. J Zoo Wildl Med. (2021) 51:1062–6. doi: 10.1638/2019-0152
347. Mikota SK, Peddie L, Peddie J, Isaza R, Dunker F, West G, et al. Epidemiology and diagnosis of Mycobacterium tuberculosis in captive Asian elephants (Elephas maximus). J Zoo Wildl Med. (2001) 32:1–16. doi: 10.1638/1042-7260(2001)032[0001:EADOMT]2.0.CO;2
348. Mikota SK, Larsen RS, Montali RJ. Tuberculosis in elephants in North America. Zoo Biol. (2000) 19:393e403. doi: 10.1002/1098-2361(2000)19:5<393::AID-ZOO9>3.0.CO;2-T
349. Miller MA, Buss P, Roos EO, Hausler G, Dippenaar A, Mitchell E, et al. Fatal tuberculosis in a free-ranging African elephant and one health implications of human pathogens in wildlife. Front Vet Sci. (2019) 6:18. doi: 10.3389/fvets.2019.00018
350. Perera BVP, Salgadu MA, de S. Gunawardena GSP, Smith NH, Jinadasa HRN. First confirmed case of fatal tuberculosis in a wild Sri Lankan elephant. Gajah. (2014) 41:28–31.
351. Zachariah A, Pandiyan J, Madhavilatha GK, Mundayoor S, Chandramohan B, Sajesh PK, et al. Mycobacterium tuberculosis in wild asian elephants, southern India. Emerg Infect Dis. (2017) 23:504–6. doi: 10.3201/eid2303.161741
352. Lewerin SS, Olsson SL, Eld K, Röken B, Ghebremichael S, Koivula T, et al. Outbreak of Mycobacterium tuberculosis infection among captive Asian elephants in a Swedish zoo. Vet Rec. (2005) 156:171e5. doi: 10.1136/vr.156.6.171
353. Obanda V, Poghon J, Yongo M, Mulei I, Ngotho M, Waititu K, et al. First reported case of fatal tuberculosis in a wild African elephant with past human-wildlife contact. Epidemiol Infect. (2013) 141:1476e80. doi: 10.1017/S0950268813000022
354. Michalak K, Austin C, Diesel S, Bacon JM, Zimmerman P, Maslow JN. Mycobacterium tuberculosis infection as a zoonotic disease: Transmission between humans and elephants. Emerg Infect Dis. (1998) 4:283–7. doi: 10.3201/eid0402.980217
355. Mikota SK, Maslow JN. Tuberculosis at the human-animal interface: an emerging disease of elephants. Tuberculosis (Edinb). (2011) 91:208–11. doi: 10.1016/j.tube.2011.02.007
356. Oh P, Granich R, Scott J, Sun B, Joseph M, Stringfield C, et al. Human exposure following Mycobacterium tuberculosis infection of multiple animal species in a Metropolitan zoo. Emerg Infect Dis. (2002) 8:1290–3. doi: 10.3201/eid0811.020302
357. Murphree R, Warkentin JV, Dunn JR, Schaffner W, Jones TF. Elephant-to-human transmission of tuberculosis, 2009. Emerg Infect Dis. (2011) 17:366–71. doi: 10.3201/eid1703.101668
358. Montali RJ, Mikota SK, Cheng LI. Mycobacterium tuberculosis in zoo and wildlife species. Rev Sci Tech. (2001) 20:291–303. doi: 10.20506/rst.issue.20.1.16
359. Landolfi JA, Terio KA, Miller M, Junecko BF, Reinhart T. Pulmonary tuberculosis in Asian elephants (Elephas maximus): histologic lesions with correlation to local immune responses. Vet Pathol. (2015) 52:535–42. doi: 10.1177/0300985814548517
360. Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. (2008) 18:729–41. doi: 10.1101/gr.075069.107
361. Tobin DM, Ramakrishnan L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol. (2008) 10:1027–39. doi: 10.1111/j.1462-5822.2008.01133.x
362. Berg RD, Ramakrishnan L. Insights into tuberculosis from the zebrafish model. Trends Mol Med. (2012) 18:689–90. doi: 10.1016/j.molmed.2012.10.002
363. Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. (2002) 17:693–702. doi: 10.1016/S1074-7613(02)00475-2
364. Langenau DM, Ferrando AA, Traver D, Kutok JL, Hezel JP, Kanki JP, et al. In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc Natl Acad Sci USA. (2004) 101:7369–74. doi: 10.1073/pnas.0402248101
365. Lesley R, Ramakrishnan L. Insights into early mycobacterial pathogenesis from the zebrafish. Curr Opin Microbiol. (2008) 11:277–83. doi: 10.1016/j.mib.2008.05.013
366. Meijer AH, Spaink HP. Host-pathogen interactions made transparent with the zebrafish model. Curr Drug Targets. (2011) 12:1000–17. doi: 10.2174/138945011795677809
367. Pozos TC, Ramakrishnan L. New models for the study of Mycobacterium-host interactions. Curr Opin Immunol. (2004) 16:499–505. doi: 10.1016/j.coi.2004.05.011
368. Ramakrishnan L. Looking within the zebrafish to understand the tuberculous granuloma. Adv Exp Med Biol. (2013) 783:251–66. doi: 10.1007/978-1-4614-6111-1_13
369. Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun. (2006) 74:6108–17. doi: 10.1128/IAI.00887-06
370. van Leeuwen LM, van der Sar AM, Bitter W. Animal models of tuberculosis: zebrafish. Cold Spring Harb Perspect Med. (2014) 5:a018580. doi: 10.1101/cshperspect.a018580
371. Kaufmann SH. Immune response to tuberculosis: experimental animal models. Tuberculosis (Edinb). (2003) 83:107–11. doi: 10.1016/S1472-9792(02)00063-X
372. Singh AK, Gupta UD. Animal models of tuberculosis: Lesson learnt. Indian J Med Res. (2018) 147:456–63. doi: 10.4103/ijmr.IJMR_554_18
373. Chackerian AA, Behar SM. Susceptibility to Mycobacterium tuberculosis: Lessons from inbred strains of mice. Tuberculosis. (2003) 83:279–85. doi: 10.1016/S1472-9792(03)00017-9
374. Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis. (2012) 205:595–602. doi: 10.1093/infdis/jir786
375. Kramnik I, Dietrich WF, Demant P, Bloom BR. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (2000) 97:8560–5. doi: 10.1073/pnas.150227197
376. Pichugin AV, Yan BS, Sloutsky A, Kobzik L, Kramnik I. Dominant role of the sst1 locus in pathogenesis of necrotizing lung granulomas during chronic tuberculosis infection and reactivation in genetically resistant hosts. Am J Pathol. (2009) 174:2190–201. doi: 10.2353/ajpath.2009.081075
377. Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. (2005) 434:767–72. doi: 10.1038/nature03419
378. Yan B-S, Kirby A, Shebzukhov YV, Daly MJ, Kramnik I. Genetic architecture of tuberculosis resistance in a mouse model of infection. Genes Immun. (2006) 7:201–10. doi: 10.1038/sj.gene.6364288
379. Smith CM, Baker RE, Proulx MK, Mishra BB, Long JE, Park SW, et al. Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife. (2022) 11:e74419. doi: 10.7554/eLife.74419
380. Smith CM, Proulx MK, Lai R, Kiritsy MC, Bell TA, Hock P, et al. Functionally overlapping variants control tuberculosis susceptibility in collaborative cross mice. mBio. (2019) 10:e02791–19. doi: 10.1128/mBio.02791-19
381. Noll KE, Ferris MT, Heise M. The collaborative cross: A systems genetics resource for studying host-pathogen interactions. Cell Host Microbe. (2019) 25:484–98. doi: 10.1016/j.chom.2019.03.009
382. Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, et al. Tuberculosis susceptibility and vaccine protection are independently controlled by host genotype. mBio. (2016) 7:e01516–16. doi: 10.1128/mBio.01516-16
383. Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. (1997) 78:57–66. doi: 10.1016/S0962-8479(97)90016-2
384. McCune RM, Feldmann FM, Lambert HP, McDermott W. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J Exp Med. (1966) 123:445–68. doi: 10.1084/jem.123.3.445
385. Nemeth J, Olson GS, Rothchild AC, Jahn AN, Mai D, Duffy FJ, et al. Contained Mycobacterium tuberculosis infection induces concomitant and heterologous protection. PloS Pathog. (2020) 16:e1008655. doi: 10.1371/journal.ppat.1008655
386. Ordonez AA, Tasneen R, Pokkali S, Xu Z, Converse PJ, Klunk MH, et al. Mouse model of pulmonary cavitary tuberculosis and expression of matrix metalloproteinase-9. Dis Model Mech. (2016) 9:779–88. doi: 10.1242/dmm.025643
387. Kramnik I, Beamer G. Mouse models of human TB pathology: Roles in the analysis of necrosis and the development of host-directed therapies. Semin Immunopathol. (2016) 38:221–37. doi: 10.1007/s00281-015-0538-9
388. Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. (2006) 8:218–32. doi: 10.1111/j.1462-5822.2005.00612.x
389. Hoff DR, Ryan GJ, Driver ER, Ssemakulu CC, De Groote MA, Basaraba RJ, et al. Location of intra- and extracellular M. tuberculosis populations in lungs of mice and Guinea pigs during disease progression and after drug treatment. PloS One. (2011) 6:e17550. doi: 10.1371/journal.pone.0017550
390. Yan BS, Pichugin AV, Jobe O, Helming L, Eruslanov EB, Gutiérrez-Pabello JA, et al. Progression of pulmonary tuberculosis and efficiency of bacillus Calmette-Guérin vaccination are genetically controlled via a common sst1-mediated mechanism of innate immunity. J Immunol. (2007) 179:6919–32. doi: 10.4049/jimmunol.179.10.6919
391. Dooley KE, Phillips PP, Nahid P, Hoelscher M. Challenges in the clinical assessment of novel tuberculosis drugs. Adv Drug Delivery Rev. (2016) 102:116–22. doi: 10.1016/j.addr.2016.01.014
392. Lai R, Gong DN, Williams T, Ogunsola AF, Cavallo K, Lindestam Arlehamn CS, et al. Host genetic background is a barrier to broadly effective vaccine-mediated protection against tuberculosis. J Clin Invest. (2023) 133:e167762. doi: 10.1172/JCI167762
393. Bai X, Shang S, Henao-Tamayo M, Basaraba RJ, Ovrutsky AR, Matsuda JL, et al. Human IL-32 expression protects mice against a hypervirulent strain of Mycobacterium tuberculosis. Proc Natl Acad Sci U.S.A. (2015) 112:5111–6. doi: 10.1073/pnas.1424302112
394. Bai X, Kim SH, Azam T, McGibney MT, Huang H, Dinarello CA, et al. IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol. (2010) 184:3830–40. doi: 10.4049/jimmunol.0901913
395. Myllymäki H, Niskanen M, Oksanen KE, Rämet M. Animal models in tuberculosis research - where is the beef? Expert Opin Drug Discovery. (2015) 10:871–83. doi: 10.1517/17460441.2015.1049529
396. Lurie MB. Experimental epidemiology of tuberculosis: the effect of eliminating exposure of enteric infection on the incidence and course of tuberculosis acquired by normal Guinea pigs confined with tuberculous cage mates. J Exp Med. (1930) 51:753–68. doi: 10.1084/jem.51.5.753
397. Lurie MB. Experimental epidemiology of tuberculosis: airborne contagion of tuberculosis in an animal room. J Exp Med. (1930) 51:743–51. doi: 10.1084/jem.51.5.743
398. Clark S, Hall Y, Williams A. Animal models of tuberculosis: Guinea pigs. Cold Spring Harb Perspect Med. (2014) 5:a018572. doi: 10.1101/cshperspect.a018572
399. Smith DW, Harding GE. Animal model of human disease. Pulmonary tuberculosis. Animal model: Experimental airborne tuberculosis in the Guinea pig. Am J Pathol. (1977) 89:273–6.
400. Dharmadhikari AS, Basaraba RJ, van der Walt ML, Weyer K, Mphahlele M, Venter K, et al. Natural infection of Guinea pigs exposed to patients with highly drug-resistant tuberculosis. Tuberculosis (Edinb). (2011) 91:329–38. doi: 10.1016/j.tube.2011.03.002
401. Padilla-Carlin DJ, McMurray DN, Hickey AJ. The Guinea pig as a model of infectious diseases. Comp Med. (2008) 58:324–40.
402. Kashino SS, Napolitano DR, Skobe Z, Campos-Neto A. Guinea pig model of Mycobacterium tuberculosis latent/dormant infection. Microbes Infect. (2008) 10:1469–76. doi: 10.1016/j.micinf.2008.08.010
403. Sugawara I, Udagawa T, Aoki T, Mizuno S. Establishment of a Guinea pig model of latent tuberculosis with GFP-introduced Mycobacterium tuberculosis. Tohoku J Exp Med. (2009) 219:257–62. doi: 10.1620/tjem.219.257
404. Shang S, Shanley CA, Caraway ML, Orme EA, Henao-Tamayo M, Hascall-Dove L, et al. Activities of TMC207, rifampin, and pyrazinamide against Mycobacterium tuberculosis infection in Guinea pigs. Antimicrob Agents Chemother. (2011) 55:124–31. doi: 10.1128/AAC.00978-10
405. Shang S, Shanley CA, Caraway ML, Orme EA, Henao-Tamayo M, Hascall-Dove L, et al. Drug treatment combined with BCG vaccination reduces disease reactivation in Guinea pigs infected with Mycobacterium tuberculosis. Vaccines (Basel). (2012) 30:1572–82. doi: 10.1016/j.vaccine.2011.12.114
406. Brandt L, Skeiky YA, Alderson MR, Lobet Y, Dalemans W, Turner OC, et al. The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-infected Guinea pigs. Infect Immun. (2004) 72:6622–32. doi: 10.1128/IAI.72.11.6622-6632.2004
407. Smith DW, McMurray DN, Wiegeshaus EH, Grover AA, Harding GE. Host-parasite relationships in experimental airborne tuberculosis. IV. early events in the course of infection in vaccinated and nonvaccinated Guinea pigs. Am Rev Respir Dis. (1970) 102:937–49. doi: 10.1164/arrd.1970.102.6.937
408. Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, et al. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. (2007) 51:3338–45. doi: 10.1128/AAC.00276-07
409. Baldwin SL, D'Souza C, Roberts AD, Kelly BP, Frank AA, Lui MA, et al. Evaluation of new vaccines in the mouse and Guinea pig model of tuberculosis. Infect Immun. (1998) 66:2951–9. doi: 10.1128/IAI.66.6.2951-2959.1998
410. Ordway DJ, Shanley CA, Caraway ML, Orme EA, Bucy DS, Hascall-Dove L, et al. Evaluation of standard chemotherapy in the Guinea pig model of tuberculosis. Antimicrob Agents Chemother. (2010) 54:1820–33. doi: 10.1128/AAC.01521-09
411. Tree JA, Smith S, Baker N, Clark S, Aldwell FE, Chambers M, et al. Method for assessing IFN-γ responses in Guinea pigs during TB vaccine trials. Lett Appl Microbiol. (2012) 55:295–300. doi: 10.1111/j.1472-765X.2012.03292.x
412. Jeevan A, Yoshimura T, Lee KE, McMurray DN. Differential expression of gamma interferon mRNA induced by attenuated and virulent Mycobacterium tuberculosis in Guinea pig cells after Mycobacterium bovis BCG vaccination. Infect Immun. (2003) 71:354–64. doi: 10.1128/IAI.71.1.354-364.2003
413. Jeevan A, Yoshimura T, Ly LH, Dirisala VR, McMurray DN. Cloning of Guinea pig IL-4: reduced IL-4 mRNA after vaccination or Mycobacterium tuberculosis infection. Tuberculosis (Edinb). (2011) 91:47–56. doi: 10.1016/j.tube.2010.11.006
414. Dirisala VR, Jeevan A, Bix G, Yoshimura T, McMurray DN. Molecular cloning and expression of the IL-10 gene from Guinea pigs. Gene. (2012) 498:120–7. doi: 10.1016/j.gene.2012.01.076
415. Dirisala VR, Jeevan A, Ramasamy SK, McMurray DN. Molecular cloning, expression, and in silico structural analysis of Guinea pig IL-17. Mol Biotechnol. (2013) 55:277–87. doi: 10.1007/s12033-013-9679-z
416. Lurie MB. The fate of human and bovine tubercle bacilli in various organs of the rabbit. J Exp Med. (1928) 48:155–82. doi: 10.1084/jem.48.2.155
417. Lurie MB. Heredity, constitution and tuberculosis: an experimental study. Am Rev Tuberc. (1941) 44:1–125.
418. Lurie MB, Abramson S, Heppleston AG. On the response of genetically resistant and susceptible rabbits to the quantitative inhalation of human-type tubercle bacilli and the nature of resistance to tuberculosis. J Exp Med. (1952) 95:119–34. doi: 10.1084/jem.95.2.119
419. Acosta A, Norazmi MN, Hernandez-Pando R, Alvarez N, Borrero R, Infante JF, et al. The importance of animal models in tuberculosis vaccine development. Malays J Med Sci. (2011) 18:5–12.
420. Converse PJ, Dannenberg AM Jr., Estep JE, Sugisaki K, Abe Y, Schofield BH, et al. Cavitary tuberculosis produced in rabbits by aerosolized virulent tubercle bacilli. Infect Immun. (1996) 64:4776–87. doi: 10.1128/iai.64.11.4776-4787.1996
421. Converse PJ, Dannenberg AM Jr., Shigenaga T, McMurray DN, Phalen SW, Stanford JL, et al. Pulmonary bovine-type tuberculosis in rabbits: bacillary virulence, inhaled dose effects, tuberculin sensitivity, and Mycobacterium vaccae immunotherapy. Clin Diagn Lab Immunol. (1998) 5:871–81. doi: 10.1128/CDLI.5.6.871-881.1998
422. Dannenberg AM Jr. Liquefaction and cavity formation in pulmonary TB: A simple method in rabbit skin to test inhibitors. Tuberculosis. (2009) 89:243–7. doi: 10.1016/j.tube.2009.05.006
423. Dannenberg AM Jr. Perspectives on clinical and preclinical testing of new tuberculosis vaccines. Clin Microbiol Rev. (2010) 23:781–94. doi: 10.1128/CMR.00005-10
424. Manabe YC, Dannenberg AM Jr., Tyagi SK, Hatem CL, Yoder M, Woolwine SC, et al. Different strains of Mycobacterium tuberculosis cause various spectrums of disease in the rabbit model of tuberculosis. Infect Immun. (2003) 71:6004–11. doi: 10.1128/IAI.71.10.6004-6011.2003
425. Subbian S, Tsenova L, O'Brien P, Yang G, Kushner NL, Parsons S, et al. Spontaneous latency in a rabbit model of pulmonary tuberculosis. Am J Pathol. (2012) 181:1711–24. doi: 10.1016/j.ajpath.2012.07.019
426. Dannenberg AM Jr. Cellular hypersensitivity and cellular immunity in the pathogensis of tuberculosis: specificity, systemic and local nature, and associated macrophage enzymes. Bacteriol Rev. (1968) 32:85–102. doi: 10.1128/br.32.2.85-102.1968
427. Dannenberg AM Jr. Pathogenesis of tuberculosis: local and systemic immunity and cellular hypersensitivity. Bull Int Union Tuberc. (1970) 43:177–8.
428. Dannenberg AM Jr. Delayed-type hypersensitivity and cell-mediated immunity in the pathogenesis of tuberculosis. Immunol Today. (1991) 12:228–33. doi: 10.1016/0167-5699(91)90035-R
429. Mantilla Galindo A, Ocampo M, Patarroyo MA. Experimental models used in evaluating anti-tuberculosis vaccines: the latest advances in the field. Expert Rev Vaccines. (2019) 18:365–77. doi: 10.1080/14760584.2019.1583558
430. Dorman SE, Hatem CL, Tyagi S, Aird K, Lopez-Molina J, Pitt ML, et al. Susceptibility to tuberculosis: clues from studies with inbred and outbred New Zealand White rabbits. Infect Immun. (2004) 72:1700–5. doi: 10.1128/IAI.72.3.1700-1705.2004
431. Ihms EA, Urbanowski ME, Bishai WR. Diverse cavity types and evidence that mechanical action on the necrotic granuloma drives tuberculous cavitation. Am J Pathol. (2018) 188:1666–75. doi: 10.1016/j.ajpath.2018.04.006
432. Helke KL, Mankowski JL, Manabe YC. Animal models of cavitation in pulmonary tuberculosis. Tuberculosis (Edinb). (2006) 86:337–48. doi: 10.1016/j.tube.2005.09.001
433. Leong FJ, Dartois V, Dick T. A Color Atlas of Comparative Pathology of Pulmonary Tuberculosis. Boca Raton, FL: CRC Press (2010).
434. Gong W, Liang Y, Wu X. Animal models of tuberculosis vaccine research: an important component in the fight against tuberculosis. BioMed Res Int. (2020) 2020:4263079. doi: 10.1155/2020/4263079
435. Gil O, Díaz I, Vilaplana C, Tapia G, Díaz J, Fort M, et al. Granuloma encapsulation is a key factor for containing tuberculosis infection in minipigs. PloS One. (2010) 5:e10030. doi: 10.1371/journal.pone.0010030
436. Ramos L, Obregon-Henao A, Henao-Tamayo M, Bowen R, Lunney JK, Gonzalez-Juarrero M. The minipig as an animal model to study Mycobacterium tuberculosis infection and natural transmission. Tuberculosis (Edinb). (2017) 106:91–8. doi: 10.1016/j.tube.2017.07.003
437. de Val Pérez B, López-Soria S, Nofrarías M, Martín M, Vordermeier HM, Villarreal-Ramos B, et al. Experimental model of tuberculosis in the domestic goat after endobronchial infection with Mycobacterium caprae. Clin Vaccine Immunol. (2011) 18:1872–81. doi: 10.1128/CVI.05323-11
438. Wedlich N, Figl J, Liebler-Tenorio EM, Köhler H, von Pückler K, Rissmann M, et al. Video endoscopy-guided intrabronchial spray inoculation of mycobacterium bovis in goats and comparative assessment of lung lesions with various imaging methods. Front Vet Sci. (2022) 9:877322. doi: 10.3389/fvets.2022.877322
439. Roy A, Risalde MA, Casal C, Romero B, de Juan L, Menshawy AM, et al. Oral vaccination with heat-inactivated mycobacterium bovis does not interfere with the antemortem diagnostic techniques for tuberculosis in goats. Front Vet Sci. (2017) 4:124. doi: 10.3389/fvets.2017.00124
440. Arrieta-Villegas C, Perálvarez T, Vidal E, Puighibet Z, Moll X, Canturri A, et al. Efficacy of parenteral vaccination against tuberculosis with heat-inactivated Mycobacterium bovis in experimentally challenged goats. PloS One. (2018) 13:e0196948. doi: 10.1371/journal.pone.0196948
442. Webb GB, Gilbert GB. Immunity in tuberculosis: Further experiments. J AMA. (1914) LXIII:1098–104. doi: 10.1001/jama.1914.02570130034009
443. Schmidt LH. Some observations on the utility of simian pulmonary tuberculosis in defining the therapeutic potentialities of isoniazid. Am Rev Tuberc. (1956) 74:138–53. doi: 10.1164/artpd.1956.74.2-2.138
444. Barclay WR, Anacker RL, Brehmer W, Leif W, Ribi E. Aerosol-induced tuberculosis in subhuman primates and the course of the disease after intravenous BCG vaccination. Infect Immun. (1970) 2:574–82. doi: 10.1128/iai.2.5.574-582.1970
445. Walsh GP, Tan EV, de la Cruz EC, Abalos RM, Villahermosa LG, Young LJ, et al. The Philippine cynomolgus monkey (Macaca fasicularis) provides a new nonhuman primate model of tuberculosis that resembles human disease. Nat Med. (1996) 2:430–6. doi: 10.1038/nm0496-430
446. Reed SG, Coler RN, Dalemans W, Tan EV, DeLa Cruz EC, Basaraba RJ, et al. Defined tuberculosis vaccine, Mtb72F/AS02A, evidence of protection in cynomolgus monkeys. Proc Natl Acad Sci U.S.A. (2009) 106:2301–6. doi: 10.1073/pnas.0712077106
447. Via LE, Weiner DM, Schimel D, Lin PL, Dayao E, Tankersley SL, et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus). Infect Immun. (2013) 81:2909–19. doi: 10.1128/IAI.00632-13
448. Bontrop RE, Otting N, de Groot NG, Doxiadis GG. Major histocompatibility complex class II polymorphisms in primates. Immunol Rev. (1999) 167:339–50. doi: 10.1111/j.1600-065X.1999.tb01403.x
449. Flynn JL, Tsenova L, Izzo A, Kaplan G. . Experimental animal models of tuberculosis. In: Kaufmann SHE, Britton WJ, editors. Handbook of Tuberculosis: Immunology and Cell Biology. Wiley-VCH, Weinheim (2008). p. 389–417.
450. Geluk A, Elferink DG, Slierendregt BL, van Meijgaarden KE, de Vries RR, Ottenhoff TH, et al. Evolutionary conservation of major histocompatibility complex-DR/peptide/T cell interactions in primates. J Exp Med. (1993) 177:979–87. doi: 10.1084/jem.177.4.979
451. Lin PL, Pawar S, Myers A, Pegu A, Fuhrman C, Reinhart TA, et al. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. (2006) 74:3790–803. doi: 10.1128/IAI.00064-06
452. Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun. (2009) 77:4631–42. doi: 10.1128/IAI.00592-09
453. McLeay SC, Morrish GA, Kirkpatrick CM, Green B. The relationship between drug clearance and body size: systematic review and meta-analysis of the literature published from 2000 to 2007. Clin Pharmacokinet. (2012) 51:319–30. doi: 10.2165/11598930-000000000-00000
454. McShane H, Williams A. A review of preclinical animal models utilised for TB vaccine evaluation in the context of recent human efficacy data. Tuberculosis (Edinb). (2014) 94:105–10. doi: 10.1016/j.tube.2013.11.003
455. Qiu CL, Yang GB, Yu K, Li Y, Li XL, Liu Q, et al. Characterization of the major histocompatibility complex class II DQB (MhcMamu-DQB1) alleles in a cohort of Chinese rhesus macaques (Macaca mulatta). Hum Immunol. (2008) 69:513–21. doi: 10.1016/j.humimm.2008.05.014
456. Scanga CA, Flynn JL. Modeling tuberculosis in nonhuman primates. Cold Spring Harb Perspect Med. (2014) 4:a018564. doi: 10.1101/cshperspect.a018564
457. Sharpe SA, McShane H, Dennis MJ, Basaraba RJ, Gleeson F, Hall G, et al. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin Vaccine Immunol. (2010) 17:1170–82. doi: 10.1128/CVI.00079-10
458. Verreck FA, Vervenne RA, Kondova I, van Kralingen KW, Remarque EJ, Braskamp G, et al. MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PloS One. (2009) 4:e5264. doi: 10.1371/journal.pone.0005264
459. Lewinsohn DM, Tydeman IS, Frieder M, Grotzke JE, Lines RA, Ahmed S, et al. High resolution radiographic and fine immunologic definition of TB disease progression in the rhesus macaque. Microbes Infect. (2006) 8:2587–98. doi: 10.1016/j.micinf.2006.07.007
460. Rayner EL, Pearson GR, Hall GA, Basaraba RJ, Gleeson F, McIntyre A, et al. Early lesions following aerosol infection of rhesus macaques (Macaca mulatta) with Mycobacterium tuberculosis strain H37RV. J Comp Pathol. (2013) 149:475–85. doi: 10.1016/j.jcpa.2013.05.005
461. Bekara Mel A, Courcoul A, Bénet JJ, Durand B. Modeling tuberculosis dynamics, detection and control in cattle herds. PloS One. (2014) 9:e108584. doi: 10.1371/journal.pone.0108584
462. Sabio Y García J, Bigi MM, Klepp LI, García EA, Blanco FC, Bigi F. Does Mycobacterium bovis persist in cattle in a non-replicative latent state as Mycobacterium tuberculosis in human beings? Vet Microbiol. (2020) 247:108758. doi: 10.1016/j.vetmic.2020.108758
463. Cassidy JP. The pathogenesis and pathology of bovine tuberculosis with insights from studies of tuberculosis in humans and laboratory animal models. Vet Microbiol. (2006) 112:151–61. doi: 10.1016/j.vetmic.2005.11.031
464. Capparelli R, De Chiara F, Nocerino N, Medaglia C, Di Costanzo R, Ramunno L, et al. Heterozygosity at the A625C polymorphic site of the MyD88 gene is associated with Mycobacterium bovis infection in cattle. Infect Immun. (2013) 81:2139–44. doi: 10.1128/IAI.01398-12
465. Neill SD, Cassidy J, Hanna J, Mackie DP, Pollock JM, Clements A, et al. Detection of Mycobacterium bovis infection in skin test-negative cattle with an assay for bovine interferon-gamma. Vet Rec. (1994) 135:134–5. doi: 10.1136/vr.135.6.134
466. Ong BL, Ngeow YF, Razak MF, Yakubu Y, Zakaria Z, Mutalib AR, et al. Tuberculosis in captive Asian elephants (Elephas maximus) in Peninsular Malaysia. Epidemiol Infect. (2013) 141:1481–7. doi: 10.1017/S0950268813000265
467. Lyashchenko KP, Greenwald R, Esfandiari J, Olsen JH, Ball R, Dumonceaux G, et al. Tuberculosis in elephants: antibody responses to defined antigens of Mycobacterium tuberculosis, potential for early diagnosis, and monitoring of treatment. Clin Vaccine Immunol. (2006) 13:722–32. doi: 10.1128/CVI.00133-06
468. Parikka M, Hammaren MM, Harjula SK, Halfpenny NJ, Oksanen KE, Lahtinen MJ, et al. Mycobacterium marinum causes a latent infection that can be reactivated by gamma irradiation in adult zebrafish. PloS Pathog. (2012) 8:e1002944. doi: 10.1371/journal.ppat.1002944
469. van der Sar AM, Abdallah AM, Sparrius M, Reinders E, Vandenbroucke-Grauls CM, Bitter W. Mycobacterium marinum strains can be divided into two distinct types based on genetic diversity and virulence. Infect Immun. (2004) 72:6306–12. doi: 10.1128/IAI.72.11.6306-6312.2004
470. Major S, Turner J, Beamer G. Tuberculosis in CBA/J mice. Vet Pathol. (2013) 50:1016–21. doi: 10.1177/0300985813482952
471. Kato-Maeda M, Shanley CA, Ackart D, Jarlsberg LGS, Shang S, Obregon-Henao A, et al. Beijing sublineages of Mycobacterium tuberculosis differ in pathogenicity in the Guinea pig. Clin Vaccine Immunol. (2012) 19:1227–37. doi: 10.1128/CVI.00250-12
472. Larenas-Muñoz F, Ruedas-Torres I, Hunter L, Bird A, Agulló-Ros I, Winsbury R, et al. Characterisation and development of histopathological lesions in a Guinea pig model of Mycobacterium tuberculosis infection. Front Vet Sci. (2023) 10:1264200. doi: 10.3389/fvets.2023.1264200
473. Willis HS, Woodruff CE. Tuberculosis in allergic and desensitized Guinea pigs: A study of histological changes. Am J Pathol. (1938) 14:337–46.
474. Jassal MS, Nedeltchev GG, Osborne J, Bishai WR. A modified scoring system to describe gross pathology in the rabbit model of tuberculosis. BMC Microbiol. (2011) 11:49. doi: 10.1186/1471-2180-11-49
475. Manabe YC, Kesavan AK, Lopez-Molina J, Hatem CL, Brooks M, Fujiwara R, et al. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis (Edinb). (2008) 88:187–96. doi: 10.1016/j.tube.2007.10.006
476. Gonzalez-Juarrero M, Bosco-Lauth A, Podell B, Soffler C, Brooks E, Izzo A, et al. Experimental aerosol Mycobacterium bovis model of infection in goats. Tuberculosis (Edinb). (2013) 93:558–64. doi: 10.1016/j.tube.2013.05.006
477. Sanchez J, Tomás L, Ortega N, Buendía AJ, del Rio L, Salinas J, et al. Microscopical and immunological features of tuberculoid granulomata and cavitary pulmonary tuberculosis in naturally infected goats. J Comp Pathol. (2011) 145:107–17. doi: 10.1016/j.jcpa.2010.12.006
478. Gutiérrez M, García Marín JF. Cryptococcus neoformans and Mycobacterium bovis causing granulomatous pneumonia in a goat. Vet Pathol. (1999) 36:445–8. doi: 10.1354/vp.36-5-445
479. Liebler-Tenorio EM, Heyl J, Wedlich N, Figl J, Köhler H, Krishnamoorthy G, et al. Vaccine-induced subcutaneous granulomas in goats reflect differences in host-mycobacterium interactions between BCG- and recombinant BCG-derivative vaccines. Int J Mol Sci. (2022) 23:10992. doi: 10.3390/ijms231910992
480. Mehra S, Golden NA, Dutta NK, Midkiff CC, Alvarez X, Doyle LA, et al. Reactivation of latent tuberculosis in rhesus macaques by coinfection with simian immunodeficiency virus. J Med Primatol. (2011) 40:233–43. doi: 10.1111/j.1600-0684.2011.00485.x
481. Langermans JA, Andersen P, van Soolingen D, Vervenne RA, Frost PA, van der Laan T, et al. Divergent effect of bacillus Calmette-Guérin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: implications for primate models in tuberculosis vaccine research. Proc Natl Acad Sci USA. (2001) 98:11497–502. doi: 10.1073/pnas.201404898
482. Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH 2nd., Hughes TK, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. (2020) 577:95–102. doi: 10.1038/s41586-019-1817-8
483. Pereira AHB, Lopes CAA, Pissinatti TA, Pinto ACA, Oliveira DRA, Leal GM, et al. Pulmonary granuloma is not always the tuberculosis hallmark: pathology of tuberculosis stages in new world and old world monkeys naturally infected with the mycobacterium tuberculosis complex. J Comp Pathol. (2022) 199:55–74. doi: 10.1016/j.jcpa.2022.09.011
484. Roodgar M, Ross CT, Tarara R, Lowenstine L, Dandekar S, Smith DG. Gene expression and TB pathogenesis in rhesus macaques: TR4, CD40, CD40L, FAS (CD95), and TNF are host genetic markers in peripheral blood mononuclear cells that are associated with severity of TB lesions. Infect Genet Evol. (2015) 36:396–409. doi: 10.1016/j.meegid.2015.10.010
485. Nogueira I, Català M, White AD, Sharpe SA, Bechini J, Prats C, et al. Surveillance of daughter micronodule formation is a key factor for vaccine evaluation using experimental infection models of tuberculosis in macaques. Pathogens. (2023) 12:236. doi: 10.3390/pathogens12020236
486. Beites T, O'Brien K, Tiwari D, Engelhart CA, Walters S, Andrews J, et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat Commun. (2019) 10:4970. doi: 10.1038/s41467-019-12956-2
487. Flynn JL, Gideon HP, Mattila JT, Lin PL. Immunology studies in non-human primate models of tuberculosis. Immunol Rev. (2015) 264:60–73. doi: 10.1111/imr.2015.264.issue-1
488. Hunter L, Hingley-Wilson S, Stewart GR, Sharpe SA, Salguero FJ. Dynamics of macrophage, T and B cell infiltration within pulmonary granulomas induced by mycobacterium tuberculosis in two non-human primate models of aerosol infection. Front Immunol. (2022) 12:776913. doi: 10.3389/fimmu.2021.776913
Keywords: tuberculosis, pathology, animal models, immunology, lung, mycobacteria, granuloma
Citation: Lyu J, Narum DE, Baldwin SL, Larsen SE, Bai X, Griffith DE, Dartois V, Naidoo T, Steyn AJC, Coler RN and Chan ED (2024) Understanding the development of tuberculous granulomas: insights into host protection and pathogenesis, a review in humans and animals. Front. Immunol. 15:1427559. doi: 10.3389/fimmu.2024.1427559
Received: 04 May 2024; Accepted: 18 November 2024;
Published: 09 December 2024.
Edited by:
Angelo Izzo, Royal Prince Alfred Hospital, AustraliaReviewed by:
Jayne Hope, University of Edinburgh, United KingdomMohd Saqib, Albany Medical College, United States
Taru S. Dutt, Colorado State University, United States
Copyright © 2024 Lyu, Narum, Baldwin, Larsen, Bai, Griffith, Dartois, Naidoo, Steyn, Coler and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward D. Chan, Y2hhbmVAbmpoZWFsdGgub3Jn