 Yiwen Wang
Yiwen Wang Lan Wu
Lan Wu Luc Van Kaer
Luc Van Kaer- Department of Pathology, Microbiology and Immunology, Vanderbilt University School of Medicine, Nashville, TN, United States
Autophagy is a regulated intracellular catabolic process by which invading pathogens, damaged organelles, aggregated proteins, and other macromolecules are degraded in lysosomes. It has been widely appreciated that autophagic activity plays an important role in regulating the development, fate determination, and function of cells in the immune system, including B lymphocytes. Autophagy encompasses several distinct pathways that have been linked to B cell homeostasis and function. While B cell presentation of major histocompatibility complex (MHC) class II-restricted cytosolic antigens to T cells involves both macroautophagy and chaperone-mediated autophagy (CMA), plasma cells and memory B cells mainly rely on macroautophagy for their survival. Emerging evidence indicates that core autophagy factors also participate in processes related to yet clearly distinct from classical autophagy. These autophagy-related pathways, referred to as noncanonical autophagy or conjugation of ATG8 to single membranes (CASM), contribute to B cell homeostasis and functions, including MHC class II-restricted antigen presentation to T cells, germinal center formation, plasma cell differentiation, and recall responses. Dysregulation of B cell autophagy has been identified in several autoimmune and autoinflammatory diseases such as systemic lupus erythematosus, rheumatoid arthritis, and inflammatory bowel disease. In this review, we discuss recent advances in understanding the role of canonical and noncanonical autophagy in B cells, including B cell development and maturation, antigen processing and presentation, pathogen-specific antibody responses, cytokine secretion, and autoimmunity. Unraveling the molecular mechanisms of canonical and noncanonical autophagy in B cells will improve our understanding of B cell biology, with implications for the development of autophagy-based immunotherapies.
1 Introduction
B lymphocytes are the source of pathogen-specific antibodies, and thus, are critical players of the vertebrate immune system. Their capacity to generate long-lasting humoral immunity against microbial pathogens enables B cells to protect the host against repeated infections, which provides the mechanistic basis for vaccine efficacy.
B cells can be partitioned into two major subpopulations, namely B1 and B2 cells, each containing several subsets (1–5). B1 cells develop from precursors during the prenatal period and are largely maintained by self-renewal after birth. They display limited immunoglobulin receptor diversity and are enriched in the peritoneal and pleural cavities. These cells are the major source of so-called natural IgM antibodies that are constitutively secreted without prior immune activation. B1 cells can be further divided into B1a and B1b cells based on the presence or absence of the inhibitory surface receptor CD5. B1a cells produce antibodies that react with molecular features common to many microbes and also contribute to the removal of senescent cells (4). B1b cells contribute to the generation of adaptive immune responses against bacterial pathogens without the assistance of T cells, thus contributing to T cell-independent (TI) antibody responses. Antibodies produced by B1 cells tend to be of low affinity and are largely limited to the IgM and IgG3 subclasses due to their scarcity of affinity maturation and class switching, respectively. The contribution of B1 cells to immune memory remains unclear (5).
The B2 B cell lineage includes marginal zone B (MZB) cells and follicular B (FOB) cells (1–3). MZB cells are derived from the bone marrow and share a common bone marrow precursor with FOB cells, from which they diverge at the transitional T2 stage (6). MZB cells are predominantly generated early in life. They are long-lived and are found within the marginal zone of the spleen where they can rapidly respond to incoming antigens from the circulation. Similar to B1 cells, MZB cells exhibit innate-like properties and functions. They display a restricted B cell receptor (BCR) repertoire, can rapidly generate TI antibody responses of the IgM and IgG isotypes against blood-borne invaders, and have limited capacity to generate memory responses (7).
FOB cells, hereafter usually referred to as B cells, constitute the majority of the B cell population. These cells develop in the bone marrow with successive steps of assembling and expressing functional antigen-receptor genes. With the surface expression of a unique BCR, immature B cells are tested for autoreactivity following exposure to self-antigens in their environment. They then migrate to peripheral lymphoid organs where they go through transitional stages (T1-T3) before becoming mature, antigen-inexperienced B cells (8, 9). Following interaction with cognate antigen, activated B cells may differentiate into short-lived, antibody-secreting cells (ASCs). However, robust antibody and B cell memory responses require the participation of a specialized subset of major histocompatibility complex (MHC) class II-restricted CD4+ T cells called T follicular helper (TFH) cells. This process involves B cell internalization of BCR-bound antigens, followed by processing in intracellular vesicles and presentation of antigen-derived peptides by MHC class II to CD4+ T cells. Naïve B cells activated by TFH cells proliferate and differentiate within a specialized anatomic structure of lymphoid organs called the germinal center (GC). Within GCs, B cells undergo somatic hypermutation that drives the generation of high-affinity antibody responses. B cells may also undergo class switch recombination (CSR), driven by CD4+ T cell produced cytokines, and this process may occur within or outside GCs. B cells eventually differentiate into antibody-secreting plasma cells or long-lived memory B cells (3, 10).
The life cycle of a B cell spans its genesis and maturation in the bone marrow, its survival as a mature naïve B cell in the periphery, its activation and proliferation in response to cognate antigen, its differentiation into an antibody-secreting plasma cell, and its acquisition of a memory phenotype. Each stage along the dynamic life cycle of a B cell is enabled by a bioenergetically unique profile of metabolites that facilitates its transition between non-cycling quiescent and rapidly proliferating states (1–3, 11). In dealing with these metabolic alterations and their associated stress, B cells have taken advantage of the fundamental cellular process called autophagy. Under the suppressive control of nutrient sensor mechanistic target of rapamycin (mTOR) and the activating control of the energy sensor AMP-activated protein kinase (AMPK), autophagy ensures the cellular availability of biomolecules required for B cell survival and function (12). Additionally, autophagy selectively degrades deleterious cytosolic entities and organelles (e.g., damaged mitochondria) during B cell activation (13–15). Interestingly, emerging evidence indicates that many autophagy factors also participate in processes that are related to yet clearly distinct from the classical autophagy pathway, including noncanonical autophagy (16). These noncanonical autophagy pathways are emerging as important contributors to B cell biology.
Here, we discuss how autophagy shapes B cell development, differentiation, fate, and function. We highlight exciting recent findings showing a critical role of noncanonical autophagy in B cell biology. We further aim to provide a perspective to reconcile and better understand discrepancies observed in early studies investigating the role of autophagy factors in B cell development and function.
While this review article focuses on B cells, autophagy plays a critical role in the development and function of many other immune cell types. We refer the readers to excellent prior articles focused on autophagy in T cells (12, 17, 18), macrophages (19, 20), and dendritic cells (21).
2 Autophagy
Autophagy is a conserved intracellular recycling process that delivers cytoplasmic material to lysosomes for degradation (22, 23). Classical or canonical autophagy processes are classified into three pathways depending on the type of cargo delivered to the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (24, 25). Macroautophagy (hereafter usually referred to as autophagy) is the best studied form that can be subdivided into five phases (Figure 1): a) initiation by nutrient stress, b) nucleation of the isolation membrane, c) elongation and sealing of the double membrane to sequester cargo, d) fusion of the autophagosome and lysosome, and e) lysosomal degradation of the vesicle substrates (26).
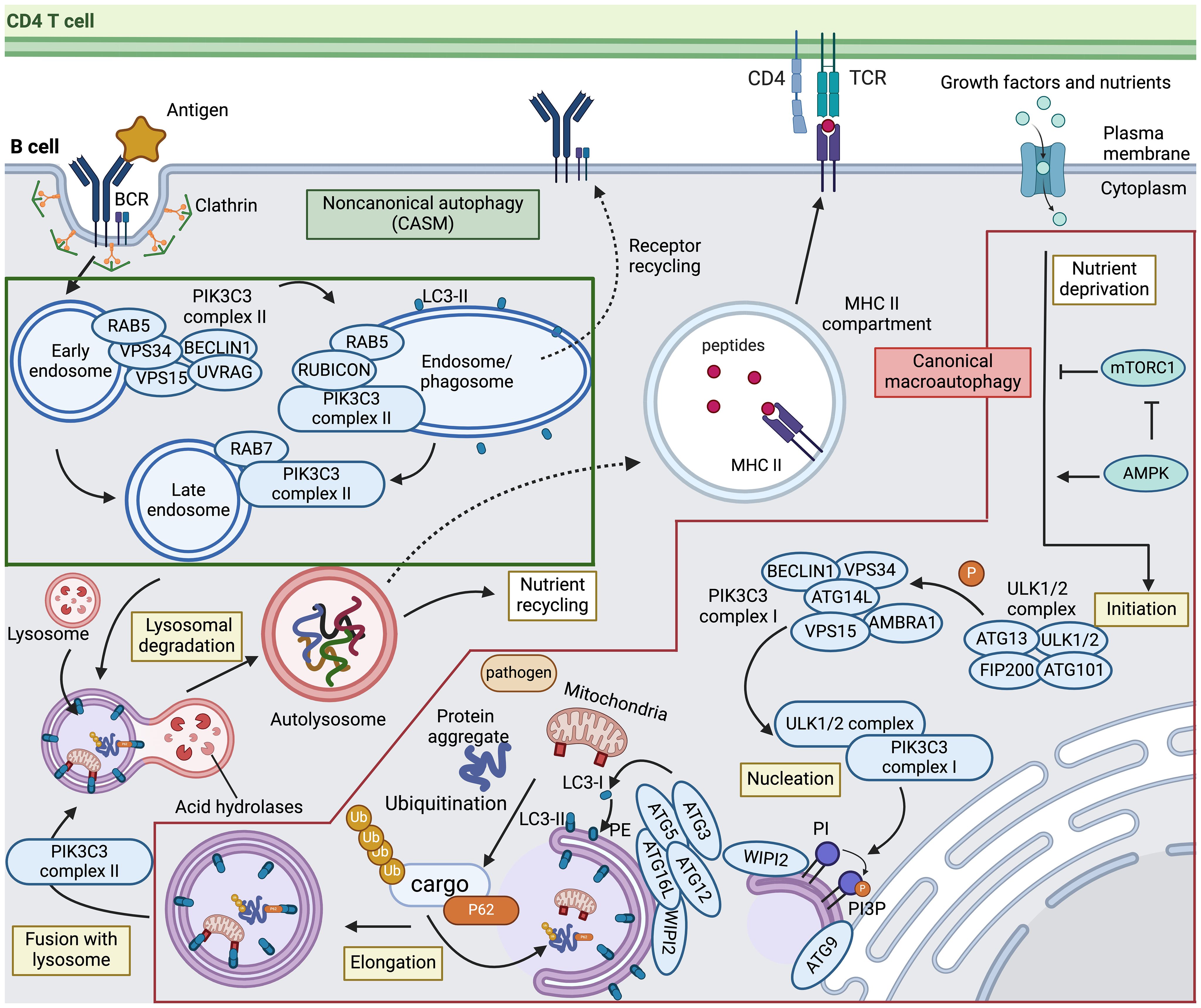
Figure 1 Canonical and noncanonical autophagy pathways in B cells. Canonical macroautophagy (indicated in the red box) is initiated when signals mediated by nutrient starvation or other stressors activate AMP-activated protein kinase (AMPK) and concurrently inhibit mechanistic target of rapamycin complex 1 (mTORC1), thereby prompting the formation of the ULK1/2 autophagy initiation complex. This complex then phosphorylates PIK3C3 complex I to promote phagophore nucleation. The ULK1/2 complex also recruits and activates transmembrane protein ATG9 to distribute lipids for autophagosome biogenesis from various membrane sources. Upon nucleation, the ATG12–ATG5–ATG16L1 conjugation system, recruited by WIPI2, functions as an E3-like ligase to mediate LC3 lipidation. The precursor of LC3 is cleaved into its mature form (LC3-I), followed by conjugation to phosphatidylethanolamine (PE) into LC3-PE (LC3-II). LC3 is critically involved in sequestering specifically labeled cargo into autophagosomes via cargo receptors. After maturation, autophagosomes fuse with the lysosome to form the autolysosome in a process involving PIK3C3 complex II. The sequestered cytoplasmic materials are then degraded via lysosomal hydrolases and recycled. The dashed line towards the MHC II compartment indicates the potential contribution of both macroautophagy and chaperone-mediated autophagy to the capacity of B cells to present antigens to CD4 T cells. Noncanonical autophagy, also called conjugation of ATG8 to single membranes (CASM) (indicated in the green box), is thought to play a critical role in antigen-activated B cells. Upon antigen engagement, the B cell receptor (BCR) undergoes clathrin-mediated internalization. PIK3C3 complex II that includes RUBICON is recruited to drive LC3 lipidation on the sealed phagosome and maturation of RAB5-positive early endosomes. Although the mechanisms of LC3-associated phagocytosis (LAP) and LC3-associated endocytosis (LANDO), indicated by dashed lines, remain unclear, these pathways may be involved in cargo degradation and recycling of receptors to the plasma membrane. TCR, T cell receptor; PI, phosphatidylinositol; PI3P, PI 3-phosphate. This figure was created by BioRender.com.
Bioenergetic stress imposed by starvation results in AMPK activation and mTORC1 inactivation, which directly or indirectly initiates autophagic cascades involving multiple protein complexes (27, 28). Starting with autophagy initiation, the UNC51-like autophagy-activating kinase (ULK)1/2 complex is a multiprotein complex consisting of ULK1, ULK2, FAK family-interactive-protein of 200 kDa (FIP200/RB1CC1), autophagy-related (ATG) protein 13 (ATG13) and ATG101. The ULK1/2 complex directly activates the class III phosphatidylinositol (PtdIns) 3-kinase (PI3K) catalytic subunit 3 (PIK3C3) complex I, which produces a pool of PtdIns 3-phosphate (PtdIns3P) that triggers nucleation of the phagophore. This PIK3C3 complex I consists of PIK3C3/vacuolar protein sorting 34 (VPS34), BECLIN1, protein kinase-like VPS15, ATG14L, nuclear receptor-binding factor 2 (NRBF2), and activating molecule in BECLIN1-regulated autophagy (AMBRA) (29–31). ULK1/2 also recruits and phosphorylates transmembrane proteins, including ATG9, to distribute lipids for autophagosome biogenesis from membrane sources such as the endoplasmic reticulum (ER), mitochondria, and plasma membrane (32). Following nucleation, two essential ubiquitin-like conjugation systems are activated for membrane expansion and fusion. First, WD repeat domain phosphoinositide-interacting 2 (WIPI2) recruits ATG16L1 and forms the ATG12-ATG5:ATG16L1 complex, sequentially catalyzed by the E1-like ubiquitin enzyme ATG7 and the E2-like ubiquitin enzyme ATG10. This complex then acts as an E3-like ubiquitin ligase that leads to the lipidation of ATG8/microtubule-associated protein light chain 3 (LC3) family proteins. ATG4, ATG7, and ATG3 cooperate to cleave the precursors of ATG8/LC3-like proteins into their mature forms (termed LC3-I), followed by conjugation to phosphatidylethanolamine (PE) into LC3-PE (termed LC3-II) and recruitment to autophagosomes with the support of WIPI2 (33–36). The conversion of LC3-I to LC3-II, measured in the presence of lysosomal protease inhibitors, is often employed as an approach to monitor autophagy (37, 38). While this process may non-selectively degrade all targets in response to stressors such as starvation, selective forms of autophagy may specifically target ubiquitinated cargo with the help of ATG8/LC3 and autophagy receptors such as P62/SQSTM1 (39). Upon completion of phagophore expansion and closure, the resulting double-membraned autophagosome vesicle fuses with a lysosome to generate an autolysosome, facilitated by regulators controlling endosome maturation and vesicle trafficking (29). Such regulators include cytoskeleton components and related motor proteins, endosomal sorting complexes required for transport (ESCRT) components, small guanosine triphosphatases (GTPases), including RAS-related protein RAB7, and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes (40–42). Once the enclosed contents are exposed to lysosomal products, lysosomal hydrolases mediate substrate degradation and metabolite recycling.
Employing different pathways to route cytoplasmic materials for lysosomal degradation, microautophagy directly sequesters small cytosolic cargos with endosomes or lysosomes, whereas CME is mediated by cytosolic chaperones to deliver cytosolic cargos to lysosomes for selective degradation (43).
3 Noncanonical autophagy
Recent research has provided evidence that classical autophagy factors are also engaged in several related yet distinct pathways, collectively called noncanonical autophagy (23, 43). Distinct from the double-membrane structures formed in canonical autophagy, one feature of noncanonical autophagy is the formation of single-membrane vesicles within the endolysosomal system for ATG8/LC3 conjugation and lysosomal degradation (Figure 1). These noncanonical autophagy pathways are now collectively referred to as conjugation of ATG8 to single membranes (CASM) (44). These two terms, noncanonical autophagy and CASM, will be used interchangeably in this review. Associated with this distinctive feature, CASM is independent of some of the upstream autophagy machinery, including ULK1/2, FIP200, ATG13, ATG9, WIPI2, and ATG14L, but requires the core ubiquitin-like conjugation system that supports ATG8/LC3 lipidation to the membrane, including ATG3, ATG4, ATG5, ATG7, ATG10, ATG12, and ATG16L1 (31, 45). Another pivotal dissimilarity is that CASM requires PIK3C3 complex II, with a different composition compared to the canonical autophagy PIK3C3 complex I. While both complexes employ PIK3C3/VPS34 and VPS15, PIK3C3 complex II is composed of ultraviolet radiation resistance-associated gene protein (UVRAG) and Run domain BECLIN1-interacting and cysteine-rich containing protein (RUBICON), instead of BECLIN1, AMBRA and ATG14L (30). Of note, two isoforms of RUBICON with distinguished size, ~130 kDa and ~100 kDa, have been identified and show opposing roles for autophagy regulation (46). In addition, PIK3C3 complex II locates to the phagophore/endosome after cargo encapsulation and vesicle sealing whereas PIK3C3 complex I is recruited to the ER for autophagosome biogenesis during canonical autophagy (30).
Several recent findings have deepened the understanding of CASM, especially the fundamental process involving autophagosome fusion with vesicles within the endocytic network of phagocytosis or endocytosis. Both pathways involve conjugation of ATG8/LC3 to single-membranes, and these processes are thus termed LC3-associated phagocytosis (LAP) and LC3-associated endocytosis (LANDO) (45, 47, 48). The latter pathways, as well as other CASM pathways, are discussed in more detail below.
3.1 LC3-associated phagocytosis and endocytosis
In LAP, LC3 lipidation on the outer face of the phagosome membrane requires the production of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 (NOX2)-induced reactive oxygen species (ROS). Under these conditions, LC3 promotes phagosome closure and phagosome-lysosome fusion, leading to degradation of the engulfed material (49). Macrophages with LAP-deficiency display impaired phagocytosis, highlighting the crucial role of LAP in the effective clearance of phagocytic cargo (50). In contrast to LAP, the specific role of LC3 lipidation in the context of LANDO is poorly defined. LANDO is characterized by conjugation of LC3 to clathrin- and RAB5-positive endosomes (51–53). Typically, endocytic pathways are categorized as clathrin-dependent or -independent, both relying on PIK3C3 complex II (30). In contrast to LAP, the absence of LC3 conjugation in LANDO does not result in defects in cargo degradation but instead causes reduced cargo receptor surface recycling (54). Subsequent sections will explore the impact of LAP and LANDO on B cell biology.
3.2 Other CASM pathways
In addition to its established role in heterophagy, CASM controls extracellular secretion. In the LC3-dependent extracellular vesicle loading (LDEL) pathway, LC3 and LC3-conjugation machinery mediate the loading of cytosolic cargos, including RNA binding proteins and RNA, into extracellular vesicles (EVs) for secretion outside cells (55, 56). Genetic evidence also implicates ATG factors to participate in unconventional secretion of acyl-CoA-binding protein Acb1 in yeast, several inflammatory cytokines in macrophages, and plasma membrane trafficking of membrane proteins in HEK293 cells (57–60). Despite limited evidence correlating CASM-mediated exocytosis to B cells, it is tempting to speculate that this secretory pathway may play a role in B cell function. The mechanism by which secretory CASM may relate to B cell function will be further explored in subsequent sections.
4 Autophagy in B cell biology
Autophagy in B cells is a dynamic process that varies based on the stage of B cell maturation and the particular B cell subset. Figure 2 summarizes the stages in the life of B cells that are impacted by canonical or noncanonical autophagy. Many of these studies were performed by analyzing the B cell phenotypes of mice genetically deficient in specific ATG factors, as summarized in Table 1. These studies are discussed in more detail in the following sections.
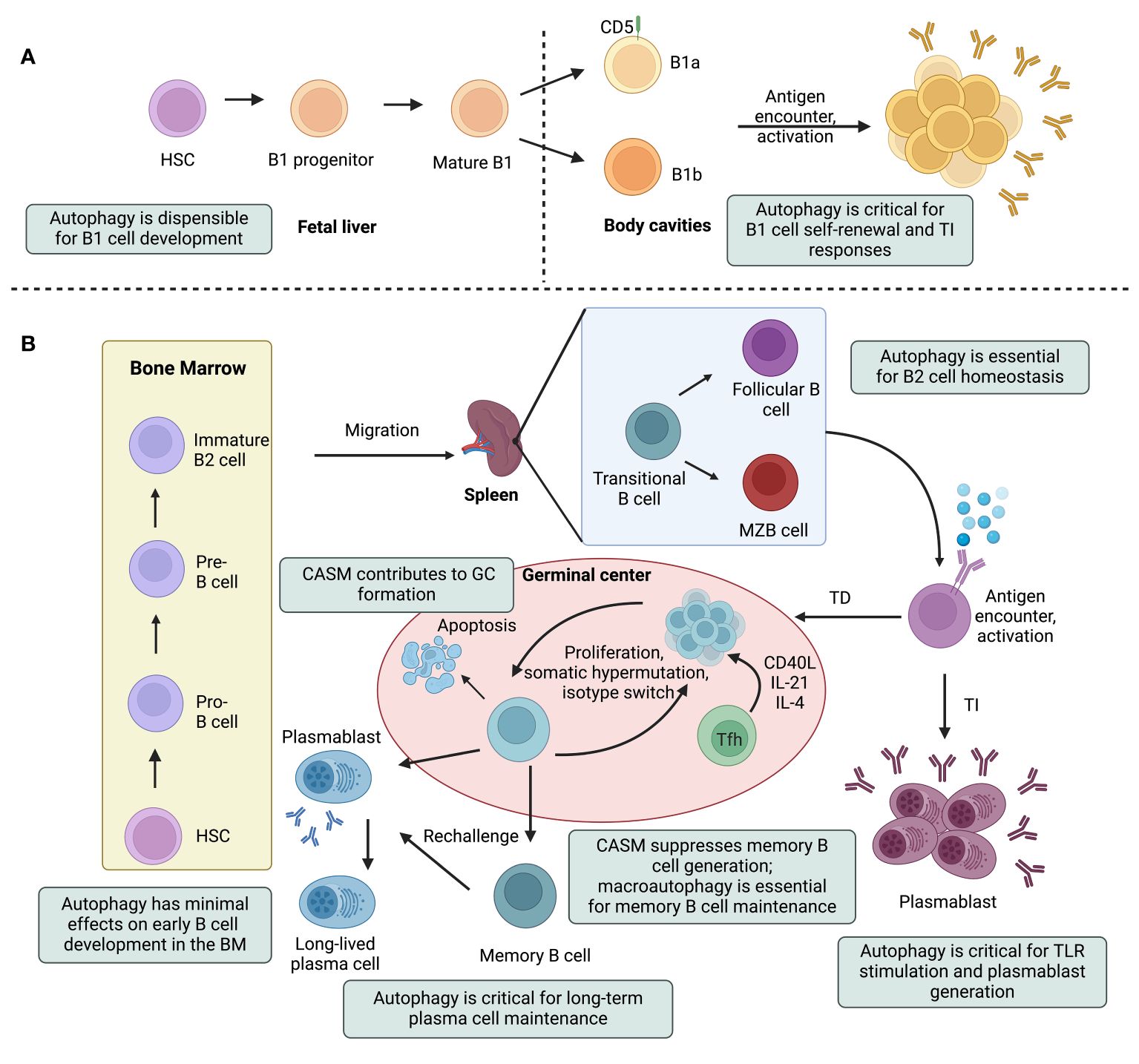
Figure 2 Canonical and noncanonical autophagy pathways impinge on multiple steps in the life of a B cell. A simplified outline of B cell lineage differentiation is depicted, with a focus on the effects of autophagy on B cell development, maturation, and effector functions. Hematopoietic stem cells (HSCs) give rise to the progenitors of two major subtypes of B lymphocytes: innate-like B1 cells (A) and B2 cells (B). B1 cell generation is restricted to fetal life and these cells then undergo self-renewal in the periphery. B1 cells can be further subdivided into B1a and B1b cells, based on the expression of the CD5 surface marker. Autophagy-deficient B1a cells fail to self-renew due to dysregulation of lipid and mitochondrial homeostasis. B2 cells, on the other hand, develop in the bone marrow (BM) and mature in peripheral lymphoid tissues. B2 cell development in the BM progresses through the pro-B cell, pre-B cell and immature B cell stages. Immature B cells that successfully express nonself-reactive B cell receptors leave the BM, become transitional B cells, and mature into follicular B (FOB) or marginal zone B (MZB) cells in secondary lymphoid tissues (i.e., lymph nodes and spleen). Depending on the antigen they encounter, mature B cells may engage in either a T cell-dependent (TD) or T cell-independent (TI) antibody response. B cells, mostly MZB cells, undergo TI responses and rapidly differentiate into short-lived plasmablasts that secrete IgM antibodies. Under certain activation conditions, these short-lived plasmablasts require autophagy to alleviate adverse effects of ER stress and mitochondrial damage. During TD responses, activated B cells form structures called germinal centers (GCs) and undergo differentiation, somatic hypermutation, and class switch recombination. Eventually, these cells will differentiate into antibody-secreting plasma cells or long-lived memory B cells. Conjugation of ATG8 to single membranes (CASM) promotes the formation of high-affinity GC B cells. The unfolded protein response in plasma cells induces autophagy, which is crucial for the maintenance of plasma cells and antibody production. Memory B cell generation is inhibited by CASM, but relies on canonical autophagy that protects against mitochondrial damage. Tfh, T follicular helper; TLR, toll-like receptor. This figure was created by BioRender.com.
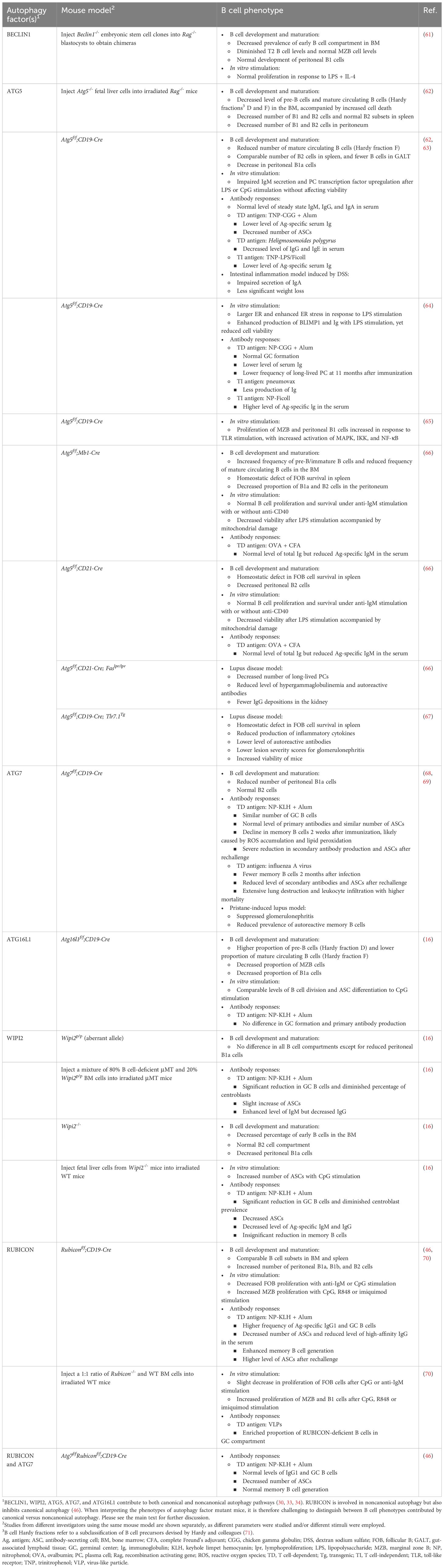
Table 1 Effects of B cell autophagy factor-deficiency on B cell development, homeostasis, differentiation and function.
4.1 B cell development
Autophagy is dynamically regulated to meet the metabolic demands during B cell development, with the maximal levels of metabolic activity occurring in the proliferative early B cell developmental stages, and a decline in quiescent mature B cells (1). Studies using chimeric mice generated by complementing recombination activating gene (Rag)-deficient blastocysts with Beclin1-/- embryonic stem cells or by transplanting fetal liver cells from Atg5-/- or Wipi2-/- mice revealed dysregulated pro- to pre-B cell transition (16, 61, 62). However, mice with B cell-specific ablation of Atg5, Atg7, Atg16l1, or Rubicon mediated by Mb1-Cre, CD19-Cre, or CD21-Cre demonstrated minimal effects on early B cell development in the bone marrow. The varying outcomes observed in these different models may be caused by the nuanced role of autophagy in hematopoietic cell development, affecting the global but not conditional knockout animals (16, 63, 66, 68). In secondary lymphoid tissues, the absence of ATG5, ATG7, or RUBICON had little impact on the homeostasis of the mature B cell population (46, 64, 68). Again, contrasting findings emerged from other investigators employing the same or different genetic models. A study with Atg5f/f;CD19-Cre mice observed a decrease in the total B cell population in the spleen, Peyer’s patches, and lamina propria (63). Additionally, similar reductions in spleen FOB cells were reported when Atg5 was deleted by Mb1-Cre that is expressed at an early B-lymphoid progenitor stage and by CD21-Cre that is expressed at the mature B cell stage. Notably, Atg5f/f;Mb1-Cre mice demonstrated more pronounced outcomes compared with Atg5f/f;CD21-Cre mice, indicating that the timing of deleting a core autophagy factor differentially influences B cell homeostasis (66). Unlike the discrepancies observed for total B cells and FOB cells, MZB cells remained unaffected in most of the mouse models examined, except for a decrease in their prevalence in Atg16l1f/f;CD19-Cre mice (16, 64, 68).
Innate-like B1 cells contain a unique metabolic profile characterized by enhanced glucose uptake and oxidative phosphorylation (72–74). Multiple studies have provided evidence that autophagy is required for peripheral self-renewal of B1a cells but not for the development of B1 progenitor cells (66, 75). Autophagy-deficient B1a cells fail to self-renew and contain substantial metabolic disturbances, including dysregulated expression of metabolic genes, decreased fatty acid uptake, and accumulation of intracellular lipid droplets (75). Interestingly, Rubiconf/f;CD19-Cre mice exhibited elevated counts of peritoneal B1a, B1b, and B2 cells, underscoring the distinct contribution of RUBICON to the development and/or maintenance of B1 cells compared with ATG5, ATG7 or ATG16L1 (46). Given the peritoneum-specific increases in both B1 and B2 cells in Rubiconf/f;CD19-Cre mice, it is possible that RUBICON inherently facilitates B cell adaptation within the peritoneal cavity.
4.2 BCR internalization and MHC class II-restricted antigen presentation
After migrating to secondary lymphoid organs, antigen-specific B cells may recognize antigens either as soluble structures or displayed at the surface of macrophages, dendritic cells or follicular dendritic cells, which can all present antigens to B cells (3). BCR engagement triggers the formation of BCR-dependent immunological synapses to internalize and process the antigen to generate peptide-MHC class II complexes for cognate T cell recognition (76–78). Recent research suggests that ATG5, likely in complex with ATG12 and ATG16L1, is responsible for BCR clustering and internalization of synapse-tethered particulate antigen to MHC class II-containing vesicles (79). This conclusion was supported by studies inhibiting PI3K activity with wortmannin, with the caveat that this reagent is not specific for PIK3C3. However, an ULK1 inhibitor did not impede this process, suggesting the involvement of CASM (79). Consistent with these findings, an additional study found that BCR-mediated antigen uptake utilizes the ATG8/LC3 lipidation machinery (80, 81). Collectively, these studies provide evidence that the ATG8/LC3 conjugation system is activated in response to BCR ligation and targets the internalized antigen for further processing. Antigen recognition also boosts lysosome exocytosis, releasing digestive hydrolases to facilitate antigen extraction from the surface of cells presenting antigens to B cells (82, 83). Upon their recruitment following immune synapse formation, lysosomes fuse with the plasma membrane and secrete their acidic vesicle contents, including the hydrolase cathepsin S, to facilitate antigen extraction and presentation to T cells (84). While the involvement of ATG8/LC3 lipidation in the generation of EVs is established in various mammalian cells, the effects of autophagy-related components on the secretion of B cell EVs are not yet fully understood. Nevertheless, a recent study showed that IL-4- and anti-CD40-stimulated B cells secrete LC3-II-containing EVs in a manner dependent on the small GTPase RAB27a (85). Together, the available studies suggest that autophagy regulators play a significant role in the extraction of B cell-associated antigens, potentially by altering the secretion and function of EVs.
The connection between macroautophagy and MHC class II-restricted antigen presentation was initially demonstrated by the significant proportion of MHC class II ligands sourced from intracellular antigens, including cytosolic and nuclear antigens, in human B lymphoblastoid cells (86–91). Interestingly, nutrient deprivation in these cells resulted in enhanced presentation of intracellular antigens, further indicating the role of macroautophagy in this process (86, 87, 91–93). Moreover, inhibiting PI3K or silencing Atg7 in human B cells disrupted the intracellular processing of Epstein-Barr virus (EBV) nuclear antigen EBNA1, thereby reducing the display of specific CD4+ T cell epitopes from EBNA1 on the B cell surface (94). However, the mechanisms by which such antigens are specifically targeted for presentation by MHC class II on B cells are not well understood. Several autophagy receptors, including P62, NDP52, NBR1, and optineurin, can direct ubiquitinated targets to the autophagosome (95, 96), and these receptors therefore represent possible candidates for targeting intracellular antigens to MHC class II-containing compartments. In addition to these studies providing evidence for macroautophagy-mediated MHC class II presentation of intracellular antigens by B cells, multiple reports have shown that fusing viral or tumor antigens with LC3 boosts their MHC class II presentation by human epithelial or melanoma cells and leads to stronger CD4+ T cell responses (93, 97, 98). Yet, these findings have not been explicitly tested in B cells, which will require further investigation.
In addition to macroautophagy, studies with human EBV-transformed B lymphoblastoid cells have provided evidence that CMA contributes to the presentation of cytosolic antigens on MHC class II (88, 99, 100). During CMA, lysosome-associated membrane protein 2A (LAMP-2A), with the assistance of heat shock protein 70 (HSC70), imports cytosolic proteins harboring the pentapeptide KFERQ sequence directly into lysosomal membranes for degradation (24, 101). Overexpression of LAMP-2A or HSC70 resulted in enhanced MHC class II presentation of a peptide derived from the cytoplasmic antigen glutamic acid decarboxylase (GAD) by human B lymphoblastoid cells (99). LAMP-2 isoforms also have a reciprocal modulatory mechanism that can influence the CMA pathway and MHC class II-restricted antigen presentation. Specifically, the LAMP-2C isoform selectively perturbs the MHC class II presentation of cytosolic antigen by blocking CMA-mediated peptide loading onto endosomal MHC class II molecules (102, 103).
4.3 B cell activation and differentiation
Autophagy not only influences B lymphocyte development but also controls B cell activation and differentiation. With appropriate stimulation, B cells in lymphoid follicles either differentiate into short-lived ASCs or enter GCs to undergo antibody affinity maturation, which generates either long-lived plasma cells or memory B cells (1–3). Early studies suggested no major involvement of autophagy in short-term B cell activation, as ATG5-deficient B cells displayed comparable proliferation and survival when activated by anti-IgM antibodies with or without anti-CD40 antibodies in vitro (66). Moreover, mice with B cell-specific deficiencies in ATG5 or ATG7 exhibited a similar capacity to generate GC B cells as compared to their wild-type counterparts following T cell-dependent (TD) antigen immunization (64, 68, 79). These mutant mice also demonstrated proficiency in undergoing CSR and somatic hypermutation (63, 68, 69). However, subsequent studies revealed a bafilomycin A1 (BafA1)-sensitive autophagy flux in response to BCR stimulation (16). Since BafA1 inhibits the vacuolar type ATPase (V-ATPase) and thus suppresses ATG8/LC3 lipidation during CASM, this finding suggests involvement of CASM in GC B cell activity (44, 104, 105). The engagement of receptors on B cells, most notably interaction of CD40 with T cell-expressed CD40L, triggers the activation of transcription factor NF-κB, which directly induces the expression of activation-induced cytidine deaminase (AID) that is crucial for CSR in the immunoglobulin heavy chain locus (106, 107). Co-localized with CD40, RAB7, a small GTPase that regulates endosome maturation and autolysosome formation, directly interacts with signal adaptor tumor necrosis factor receptor-associated factor 6 (TRAF6) E3 ubiquitin ligase to upregulate the activity of TRAF6 and enhance NF-κB activation (108). In particular, RAB7 also interacts with RUBICON to regulate both the endocytic and autophagic pathways, suggesting a role for CASM in B cell signaling (109). The possible involvement of noncanonical autophagy was further investigated using mice with B cell-specific RUBICON-deficiency. Immunization of mice transplanted with a 1:1 mixture of RUBICON-deficient and wild-type bone marrow cells with virus-like particles (VLPs) showed significant enrichment of mutant B cells in the VLP+ GC compartment, suggesting that RUBICON-deficiency provides a competitive advantage to GC B cells (70, 110). Additionally, Rubiconf/f;CD19-Cre mice displayed increased levels of antigen-specific IgG+ GCs following TD antigen immunization, suggesting a unique role for noncanonical autophagy in antibody affinity maturation and isotope switching. However, Atg7f/fRubiconf/f;CD19-Cre double knockout animals displayed normal GC formation, indicating that enhanced autophagy was responsible for the increase in antigen-specific IgG+ GCs in Rubiconf/f;CD19-Cre mice (46).
More distinct outcomes were observed in the primary antibody response to TD antigen immunization and in the context of primary infection. Atg5f/f;CD19-Cre mice exhibited reduced levels of both antigen-specific IgM and IgG antibodies following trinitrophenol (TNP)-chicken gamma globulin (CGG) and nitrophenol (NP)-CGG immunizations (63, 64), whereas Atg7f/f;CD19-Cre and Atg16l1f/f;CD19-Cre mice did not display such decreases after NP-keyhole limpet hemocyanin (KLH) immunization (16, 68). This variability may possibly have arisen due to the utilization of diverse antigens and/or adjuvants, or alternatively, due to distinct functions of the autophagy factors investigated. Moreover, mice with B cell-specific ATG7-deficiency exhibited diminished levels of antigen-specific antibodies as time progressed, suggesting the potential significance of ATG7 in maintaining ASC populations (68). These findings highlight the nuanced and factor-specific impact of autophagy-related gene ablation on antibody production in response to distinct antigens. Particularly, compromised BCR clustering was noted exclusively upon stimulation with particulate antigens rather than soluble ones, illustrating that various antigens can elicit distinct responses (79, 111). In addition, the switch from canonical autophagy to noncanonical autophagy during the GC reaction may also explain the paradox that autophagy contributes to BCR trafficking and MHC class II-restricted antigen presentation but shows limited effects on TD antibody responses, suggesting that alternating autophagy routes can influence B cell responses to antigen exposure (16). Moreover, prior to their secretion, antibodies are routed from the ER to the Golgi apparatus where they are glycosylated (112, 113). Recent evidence indicates that one member of the SNARE family, SEC22B, is involved in this constitutive exocytosis pathway. Sec22bf/f;Mb1-Cre mice displayed reduced circulating antibodies at both steady state levels and after TI antigen immunization, exacerbated unfolded protein response (UPR), and morphologically disturbed ER and mitochondria, all of which suggests SEC22B as an essential regulator of plasma cell fitness and humoral immune responses (114).
In additional to BCR-mediated stimulation, B cells can be activated by engagement of toll-like receptors (TLRs) by pathogen-associated molecular patterns, which induce B cell proliferation and differentiation into ASCs (115). Stimulation of ATG5-deficient B cells isolated from Atg5f/f;CD19-Cre mice with a TLR4 agonist (lipopolysaccharide [LPS]) or a TLR9 agonist (CpG oligodeoxynucleotides) led to impaired differentiation into ASCs ex vivo. Consistent with these findings, these cells downregulated the expression of multiple genes, including Prdm1 (encoding the transcriptional repressor BLIMP1), Sdc-1 (encoding the regulator of cell behavior SYNDECAN-1/CD138), and Xbp-1 (encoding the stress-induced transcription factor XBP-1), that play critical roles in plasma cell differentiation (63). However, these findings were not recapitulated by all studies. Upon depletion of ATG5 in mice with different Cre drivers (Mb1-Cre and CD21-Cre) B cells showed decreased survival with normal expression of SYNDECAN-1/CD138 and increased mitochondrial content, suggesting autophagy participates in early plasma cell survival by limiting mitochondrial stress (66). Mediated by AMPK, ULK1 migrates to the mitochondria and triggers a signaling cascade that leads to the recruitment of the ATG8/LC3 machinery to support mitochondrial clearance and quality control in B cells (23, 66, 116–118). After LPS stimulation, plasma cells from Atg5f/f;CD19-Cre mice displayed ER enlargement, upregulation of Prdm1 and Xbp-1 gene expression, and increased IgM production, demonstrating that proficient autophagy confers a selective advantage on differentiating plasma cells (64). Another study further showed that this reduced proliferation of ATG5-deficient FOB cells occurs only in response to TLR7 or TLR9 stimulation but not TLR4 stimulation, whereas MZB cells with the same deficiency showed increased proliferation in response to activation by all three TLRs, with induction of mitogen-activated protein kinase (MAPK), IκB kinase (IKK), and NF-κB (65). Furthermore, MZB cells from RUBICON-deficient mice showed impaired LC3 lipidation and enhanced proliferation upon stimulation with TLR7 or TLR9 ligands, relative to control cells (70). This suggests that RUBICON plays a role similar to ATG5 in regulating MZB cell responses to specific TLR ligands (65, 70). This discrepancy between FOB and MZB cells may explain the discordance between different studies and underline the importance of distinguishing B cell subsets when exploring B cell autophagy (70). In vivo experiments with ATG5-deficient mice showed a reduction in serum antigen-specific IgM antibodies in response to immunization with the TI antigens TNP-LPS, TNP-Ficoll, or the pneumococcal vaccine pneumovax (63, 64). A notable exception was that immunization with NP-Ficoll resulted in higher titers of anti-NP immunoglobulin and ASCs two weeks after immunization, for reasons that remain unclear (64). While there has been limited exploration into the role of CASM during TLR stimulation in B cells, LANDO has been found to enhance TLR4 recycling in primary microglia, where it facilitates amyloid-beta (Aβ)-associated phagocytosis (52, 119). Whether similar processes are at play in B cells remains to be explored.
4.4 B cell memory and long-term humoral immunity
Two major components of long-term humoral immunity include long-lived plasma cells and memory B cells. Long lived plasma cells reside in the bone marrow and secrete antibodies to neutralize pathogens immediately upon reinfection (3). Atg5 ablation via CD19-Cre compromised long-term maintenance of plasma cells (64, 66). The available evidence indicates that ATG7 is dispensable for memory B cell generation (68). Transcriptionally, autophagy genes do not increase until 4 to 8 weeks after TD immunization, and high levels of ATG8/LC3 aggregation in memory B cells are not identified until 8 weeks after immunization (68). Nevertheless, ATG7-deficient memory B cells display reduced capacity to form ASCs upon antigen rechallenge. This was associated with increased death of mutant memory cells, which was prevented by inhibiting oxidative stress with NecroX-2 or α-Tocopherol, or by scavenging ROS with N-acetyl-L-cysteine (NAC) or Tempol, indicating the importance of autophagy in metabolic homeostasis of memory B cells (68). Members of the forkhead box (FOXO) family of transcription factors, especially FOXO1 and FOXO3, regulate autophagy at the epigenetic level, which is upregulated in memory B cells. Silencing FOXO3 in mice decreased the B cell expression of Beclin1 and Ulk1, and impaired memory responses upon antigen rechallenge (69). However, given the potential variations in results when depleting different autophagy factors, it is crucial to interpret findings obtained from B cell-specific ATG7-deficient mice with caution. Further investigation into the functions of other autophagy regulators in memory B cells is warranted.
A recent study suggested the potential role of CASM in memory B cell functions. Rubiconf/f;CD19-Cre mice contain significant increases in antigen-specific memory B cells and ASCs after antigen rechallenge, indicating that RUBICON suppresses the formation of memory B cells and ASCs (46). Similar to GC B cells, the increase in memory B cells was attenuated in Atg7f/fRubiconf/f;CD19-Cre double knockout mice, indicating that increased macroautophagy induced by RUBICON-deficiency is responsible for promoting memory B cell differentiation (46).
4.5 B cell cytokine secretion
In addition to their capacity to produce antibodies and present antigens to MHC class II-restricted T cells, B cells can produce immunomodulatory cytokines, including pro-inflammatory cytokines such as IL-6 and tumor necrosis factor (TNF)-α and immunosuppressive cytokines such as IL-10 that can promote or protect against disease (120).
While not yet investigated in B cells, autophagy suppresses inflammatory cytokine production by phagocytes via multiple mechanisms: a) preventing inflammasome activation by removing mitochondria (121, 122), b) targeting inflammasome components and cytokines for degradation (123, 124), c) preventing constitutive immune cell signaling by stalling cargo degradation in LAP or reducing receptor recycling in LANDO (54, 125), d) inhibiting the assembly of essential cytokine signal transducers, stimulators of interferon genes (STING), and TANK-binding kinase 1 (TBK1), that are required for cytosolic DNA-triggered innate immune responses (126), and e) enhancing the degradation of cytoplasmic DNA to prevent excessive cGAS-STING activation (127).
The regulatory capacity of B cells is highlighted in the context of IL-10-producing regulatory B cells (Bregs), known for their critical function in modulating immune responses (128). Studies have indicated that dual stimulation via TLR9 and CD40 for 48 hours is optimal for inducing and differentiating IL-10-producing Bregs (129). As autophagy enhances B cell differentiation and proliferation in response to TLR9 stimulation, autophagy might be expected to play a critical role in Breg cell differentiation (65). This notion is further supported by observations that, following BCR engagement, TLR9-containing endosomes are recruited to autophagosomes, which contain the internalized BCR-antigen complex. This co-localization is disrupted by the PI3K inhibitor 3-methyladenine (3-MA), suggesting potential interactions between BCR engagement, TLR9 signaling, and autophagic pathways in Bregs (80).
5 Impact of B cell autophagy on health and disease
Due to the pleiotropic role of autophagy in immune and non-immune cells, mutations in autophagy factors have been linked to multiple human diseases, including cancer, chronic inflammation, cardiovascular diseases, infection, neurodegeneration, immune system disorders, and aging (130).
The clinical significance of autophagy in B cells primarily manifests in its involvement in adaptive immunity, including protective immunity against infection and the development of autoimmunity (13, 14, 88, 92, 131–133). In models of influenza virus infection, deletion of Atg7 in B cells via CD19-Cre led to the loss of memory B cells and impaired secondary responses (66). Additionally, B cells from Atg5f/f;CD19-Cre mice exhibited reduced antibody production and impaired protection against the helminth parasite Heligmosomoides polygyrus and reduced severity of dextran sodium sulfate-induced colitis (63).
Compromised adaptive immunity is a major hallmark of aging (134). One recent study linked autophagy deficiency to impaired memory B cell responses and poor vaccination efficacy in elderly individuals (134). More importantly, treatment with the autophagy inducer spermidine was able to reverse memory B cell senescence in old mice (134), underscoring the therapeutic potential of autophagy intervention for age-related diseases.
Deletion of Atg5 via CD21-Cre or CD19-Cre causes a notable reduction in autoantibody production and kidney inflammation in genetic mouse models of systemic lupus erythematosus (SLE) (66, 67). Similarly, these hallmarks of lupus failed to manifest in Atg7f/f;CD19-Cre mice treated with the hydrocarbon oil pristane (67, 131). Consistent with these findings, dysregulation of macroautophagy and CMA have been described in B cells of lupus patients (133, 135). Autophagy has also been implicated in the pathogenesis of rheumatoid arthritis (RA). Classic pro-autophagic stimuli, such as starvation, facilitated B cell-associated MHC class II-mediated presentation of citrullinated peptides, which are prominently involved in RA (136). This enhanced presentation of citrullinated peptides was diminished by the autophagy inhibitor 3-MA or reduced Atg5 expression (136). Since the presentation of peptides derived from self-antigens is crucial for the development of self-tolerance, these findings suggested a protective role of autophagy in RA development (136, 137).
Autophagy’s role in adaptive immune responses offers therapeutic potential, particularly in enhancing vaccine efficacy in the elderly, and may hold promise for treating autoimmune disorders.
6 Conclusions and future directions
The studies reviewed here provide strong evidence that macroautophagy plays a critical role in B cell biology, intersecting with B cell development, BCR signaling, MHC class II antigen presentation, GC formation, antibody production, plasma and memory cell differentiation, and disease (summarized in Figure 2 and Table 1). It is now well appreciated that components of the macroautophagy machinery are utilized for other intracellular vesicle trafficking processes that are instrumental in the development and functionality of B cells (84). While offering explanations for inconsistencies found in previous studies, the newly identified role of CASM in B cells has generated further questions (138). ATG16L1-deficient mouse models could serve as valuable tools for differentiating between macroautophagy and CASM. In the process of macroautophagy, ATG16L1 associates with FIP200 and WIPI2 through its central FIP200-binding domain. Conversely, the WD40 domain of ATG16L1 is crucial for noncanonical autophagy but dispensable for macroautophagy (139). Future research may focus on comparing outcomes from these two models, particularly in the context of GC formation and primary immune responses, to unravel the distinct contributions of macroautophagy and CASM to B cell function. Furthermore, additional studies are needed to explore the role of canonical and noncanonical autophagy to the presentation of MHC class II-restricted antigens by B cells, particularly in relation to ATG8/LC3 lipidation. To date, only a small selection of antigens has been studied, and those investigations have predominantly utilized cell lines transformed with EBV, which modulates autophagy (94). Exploring antigen targeting through various autophagy regulators or ATG8/LC3 lipidation might be a fruitful avenue for future exploration, as initially indicated by reports employing ATG8/LC3 fusion antigens. Specifically, focusing on the intracellular expression of these targeted antigens in B cells might enhance the precision of T cell activation and antibody production against infectious agents and tumors (97, 98, 140). Lastly, autophagy has been implicated in the survival and functionality of self-reactive B cells in autoimmune diseases (13, 66, 141). Future research may concentrate on how B cell autophagy contributes to the MHC class II-mediated presentation of autoantigens to CD4+ T cells, the benefits of autophagy to B cell apoptotic or metabolic stress, and its impact on T cell evasion of tolerogenic controls. Collectively, these studies will be helpful for designing autophagy-based therapies of human disease that target B cells. Pharmacological agents that selectively target autophagy are under development (141).
Author contributions
YW: Writing – original draft, Visualization, Writing – review & editing, Conceptualization. LW: Writing – review & editing. LVK: Supervision, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by American Heart Association predoctoral fellowship 24PRE1191939 (to YW), National Institutes of Health grant RO1AI139046 (to LVK), and American Heart Association grant 19TPA34910078 (to LVK).
Conflict of interest
LVK is a member of the scientific advisory board of Isu Abxis Co., Ltd. South Korea.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boothby MR, Hodges E, Thomas JW. Molecular regulation of peripheral B cells and their progeny in immunity. Genes Dev. (2019) 33:26–48. doi: 10.1101/gad.320192.118
2. Boothby M, Rickert RC. Metabolic regulation of the immune humoral response. Immunity. (2017) 46:743–55. doi: 10.1016/j.immuni.2017.04.009
3. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
4. Popi AF, Longo-Maugéri IM, Mariano M. An overview of B-1 cells as antigen-presenting cells. Front Immunol. (2016) 7:138. doi: 10.3389/fimmu.2016.00138
5. Baumgarth N. A hard(y) look at B-1 cell development and function. J Immunol. (2017) 199:3387–94. doi: 10.4049/jimmunol.1700943
6. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. (2008) 112:1570–80. doi: 10.1182/blood-2008-02-078071
7. Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. (2013) 13:118–32. doi: 10.1038/nri3383
8. Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. (1989) 59:1035–48. doi: 10.1016/0092-8674(89)90760-5
9. The nobel prize in physiology or medicine 1960. NobelPrize.Org (2024). Available at: www.nobelprize.org/prizes/medicine/1960/burnet/facts/. Anonymous.
10. Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. (2015) 36:410–8. doi: 10.1016/j.it.2015.05.005
11. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. (2014) 159:1263–76. doi: 10.1016/j.cell.2014.11.006
12. Metur SP, Klionsky DJ. Adaptive immunity at the crossroads of autophagy and metabolism. Cell Mol Immunol. (2021) 18:1096–105. doi: 10.1038/s41423-021-00662-3
13. Arbogast F, Gros F. Lymphocyte autophagy in homeostasis, activation, and inflammatory diseases. Front Immunol. (2018) 9:1801. doi: 10.3389/fimmu.2018.01801
14. Raza IGA, Clarke AJ, Cell Metabolism B. and autophagy in autoimmunity. Front Immunol. (2021) 12:681105. doi: 10.3389/fimmu.2021.681105
15. Sandoval H, Kodali S, Wang J. Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion. (2018) 41:58–65. doi: 10.1016/j.mito.2017.11.005
16. Martinez-Martin N, Maldonado P, Gasparrini F, Frederico B, Aggarwal S, Gaya M, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. (2017) 355:641–7. doi: 10.1126/science.aal3908
17. Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J, et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function. Autophagy. (2021) 17:1193–204. doi: 10.1080/15548627.2020.1752979
18. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. (2007) 204:25–31. doi: 10.1084/jem.20061303
19. Wen JH, Li DY, Liang S, Yang C, Tang JX, Liu HF. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front Immunol. (2022) 13:946832. doi: 10.3389/fimmu.2022.946832
20. Wu MY, Lu JH. Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells. (2019) 9. doi: 10.3390/cells9010070
21. Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol. (2018) 15:944–52. doi: 10.1038/cmi.2018.2
22. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discovery. (2017) 16:487–511. doi: 10.1038/nrd.2017.22
23. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. (2019) 177:1682–99. doi: 10.1016/j.cell.2019.05.026
24. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
25. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. (2012) 69:1125–36. doi: 10.1007/s00018-011-0865-5
26. Levine B, Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
27. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. (2011) 12:21–35. doi: 10.1038/nrm3025
28. Nazio F, Cecconi F. Autophagy up and down by outsmarting the incredible ULK. Autophagy. (2017) 13:967–8. doi: 10.1080/15548627.2017.1285473
29. Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. (2017) 86:225–44. doi: 10.1146/annurev-biochem-061516-044820
30. Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. (2019) 20:515–34. doi: 10.1038/s41580-019-0129-z
31. Safaroghli-Azar A, Sanaei MJ, Pourbagheri-Sigaroodi A, Bashash D. Phosphoinositide 3-kinase (PI3K) classes: From cell signaling to endocytic recycling and autophagy. Eur J Pharmacol. (2023) 953:175827. doi: 10.1016/j.ejphar.2023.175827
32. Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. (2016) 7:12420. doi: 10.1038/ncomms12420
33. Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. (2015) 128:207–17. doi: 10.1242/jcs.146258
34. Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. (2020) 63:1–10. doi: 10.1016/j.ceb.2019.12.001
35. Martens S, Fracchiolla D. Activation and targeting of ATG8 protein lipidation. Cell Discovery. (2020) 6:23. doi: 10.1038/s41421-020-0155-1
36. Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. (2016) 354:1036–41. doi: 10.1126/science.aaf6136
37. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. (2007) 3:542–5. doi: 10.4161/auto.4600
38. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. (2021) 17:1–382. doi: 10.1080/15548627.2020.1797280
39. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. (2014) 16:495–501. doi: 10.1038/ncb2979
40. Abada A, Levin-Zaidman S, Porat Z, Dadosh T, Elazar Z. SNARE priming is essential for maturation of autophagosomes but not for their formation. Proc Natl Acad Sci U.S.A. (2017) 114:12749–54. doi: 10.1073/pnas.1705572114
41. Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. (2010) 188:253–69. doi: 10.1083/jcb.200907015
42. Zhou F, Wu Z, Zhao M, Murtazina R, Cai J, Zhang A, et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Biol. (2019) 218:1908–27. doi: 10.1083/jcb.201811173
43. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
44. Durgan J, Lystad AH, Sloan K, Carlsson SR, Wilson MI, Marcassa E, et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol Cell. (2021) 81:2031–2040.e2038. doi: 10.1016/j.molcel.2021.03.020
45. Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. (2011) 13:1335–43. doi: 10.1038/ncb2363
46. Tsai CY, Sakakibara S, Kuan YD, Omori H, El Hussien MA, Okuzaki D, et al. Opposing roles of RUBCN isoforms in autophagy and memory B cell generation. Sci Signal. (2023) 16:eade3599. doi: 10.1126/scisignal.ade3599
47. Inomata M, Xu S, Chandra P, Meydani SN, Takemura G, Philips JA, et al. Macrophage LC3-associated phagocytosis is an immune defense against. Proc Natl Acad Sci U.S.A. (2020) 117:33561–9. doi: 10.1073/pnas.2015368117
48. Heckmann BL, Teubner BJW, Boada-Romero E, Tummers B, Guy C, Fitzgerald P, et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer's disease. Sci Adv. (2020) 6:eabb9036. doi: 10.1126/sciadv.abb9036
49. Yang CS, Lee JS, Rodgers M, Min CK, Lee JY, Kim HJ, et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe. (2012) 11:264–76. doi: 10.1016/j.chom.2012.01.018
50. Levin R, Grinstein S, Schlam D. Phosphoinositides in phagocytosis and macropinocytosis. Biochim Biophys Acta. (2015) 1851:805–23. doi: 10.1016/j.bbalip.2014.09.005
51. Jülg J, Strohm L, Behrends C. Canonical and noncanonical autophagy pathways in microglia. Mol Cell Biol. (2021) 41:e0038920. doi: 10.1128/MCB.00389-20
52. Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine alzheimer's disease. Cell. (2019) 178:536–551.e514. doi: 10.1016/j.cell.2019.05.056
53. Magné J, Green DR. LC3-associated endocytosis and the functions of Rubicon and ATG16L1. Sci Adv. (2022) 8:eabo5600. doi: 10.1126/sciadv.abo5600
54. Peña-Martinez C, Rickman AD, Heckmann BL. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci Adv. (2022) 8:eabn1702. doi: 10.1126/sciadv.abn1702
55. Gardner JO, Leidal AM, Nguyen TA, Debnath J. LC3-dependent EV loading and secretion (LDELS) promotes TFRC (transferrin receptor) secretion. via extracellular vesicles. Autophagy. (2023) 19:1551–61. doi: 10.1080/15548627.2022.2140557
56. Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol. (2020) 22:187–99. doi: 10.1038/s41556-019-0450-y
57. Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. (2010) 188:527–36. doi: 10.1083/jcb.200911154
58. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
59. Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. (2011) 146:746–60. doi: 10.1016/j.cell.2011.07.021
60. Cadwell K, Debnath J. Beyond self-eating: The control of nonautophagic functions and signaling pathways by autophagy-related proteins. J Cell Biol. (2018) 217:813–22. doi: 10.1083/jcb.201706157
61. Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, et al. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol. (2011) 186:2201–9. doi: 10.4049/jimmunol.1002223
62. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. (2008) 4:309–14. doi: 10.4161/auto.5474
63. Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, et al. ATG5 regulates plasma cell differentiation. Autophagy. (2013) 9:528–37. doi: 10.4161/auto.23484
64. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. (2013) 14:298–305. doi: 10.1038/ni.2524
65. Acharya M, Sokolovska A, Tam JM, Conway KL, Stefani C, Raso F, et al. αv Integrins combine with LC3 and atg5 to regulate Toll-like receptor signalling in B cells. Nat Commun. (2016) 7:10917. doi: 10.1038/ncomms10917
66. Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. (2016) 23:853–64. doi: 10.1038/cdd.2015.149
67. Weindel CG, Richey LJ, Bolland S, Mehta AJ, Kearney JF, Huber BT. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy. (2015) 11:1010–24. doi: 10.1080/15548627.2015.1052206
68. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. (2014) 20:503–10. doi: 10.1038/nm.3521
69. Chen M, Kodali S, Jang A, Kuai L, Wang J. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol. (2015) 194:2607–15. doi: 10.4049/jimmunol.1403001
70. Raso F, Sagadiev S, Du S, Gage E, Arkatkar T, Metzler G, et al. αv Integrins regulate germinal center B cell responses through noncanonical autophagy. J Clin Invest. (2018) 128:4163–78. doi: 10.1172/JCI99597
71. Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. (1991) 173:1213–25. doi: 10.1084/jem.173.5.1213
72. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
73. Hayakawa K, Formica AM, Brill-Dashoff J, Shinton SA, Ichikawa D, Zhou Y, et al. Early generated B1 B cells with restricted BCRs become chronic lymphocytic leukemia with continued c-Myc and low Bmf expression. J Exp Med. (2016) 213:3007–24. doi: 10.1084/jem.20160712
74. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. (2011) 11:34–46. doi: 10.1038/nri2901
75. Clarke AJ, Riffelmacher T, Braas D, Cornall RJ, Simon AK. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J Exp Med. (2018) 215:399–413. doi: 10.1084/jem.20170771
76. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. (2015) 15:203–16. doi: 10.1038/nri3818
77. Kuokkanen E, Šuštar V, Mattila PK. Molecular control of B cell activation and immunological synapse formation. Traffic. (2015) 16:311–26. doi: 10.1111/tra.12257
78. Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity. (2011) 35:361–74. doi: 10.1016/j.immuni.2011.07.008
79. Arbogast F, Arnold J, Hammann P, Kuhn L, Chicher J, Murera D, et al. ATG5 is required for B cell polarization and presentation of particulate antigens. Autophagy. (2019) 15:280–94. doi: 10.1080/15548627.2018.1516327
80. Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. (2008) 28:799–809. doi: 10.1016/j.immuni.2008.03.019
81. Runsala M, Kuokkanen E, Uski E, Sustar V, Balci MO, Rajala J, et al. The small GTPase Rab7 regulates antigen processing in B cells in a possible interplay with autophagy machinery. Cells. (2023) 12. doi: 10.3390/cells12212566
82. Maeda FY, van Haaren JJ, Langley DB, Christ D, Andrews NW, Song W. Surface-associated antigen induces permeabilization of primary mouse B-cells and lysosome exocytosis facilitating antigen uptake and presentation to T-cells. Elife. (2021) 10. doi: 10.7554/eLife.66984
83. Yuseff MI, Lennon-Duménil AM. B cells use conserved polarity cues to regulate their antigen processing and presentation functions. Front Immunol. (2015) 6:251. doi: 10.3389/fimmu.2015.00251
84. Hämälistö S, Del Valle Batalla F, Yuseff MI, Mattila PK. Endolysosomal vesicles at the center of B cell activation. J Cell Biol. (2024) 223. doi: 10.1083/jcb.202307047
85. Kuan Y, Tsai C, Sakakibara S, Standley D, Kikutani H. External stimulation induces the secretion of autophagosome-like vesicles by B cells. Autophagy Rep. (2023) 2. doi: 10.1080/27694127.2023.2179287
86. Münz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. (2016) 37:755–63. doi: 10.1016/j.it.2016.08.017
87. Münz C. The macroautophagy machinery in MHC restricted antigen presentation. Front Immunol. (2021) 12:628429. doi: 10.3389/fimmu.2021.628429
88. Münz C. Canonical and non-canonical functions of the autophagy machinery in MHC restricted antigen presentation. Front Immunol. (2022) 13:868888. doi: 10.3389/fimmu.2022.868888
89. Hayward AP, Dinesh-Kumar SP. Special delivery for MHC II via autophagy. Immunity. (2010) 32:587–90. doi: 10.1016/j.immuni.2010.04.015
90. Valečka J, Almeida CR, Su B, Pierre P, Gatti E. Autophagy and MHC-restricted antigen presentation. Mol Immunol. (2018) 99:163–70. doi: 10.1016/j.molimm.2018.05.009
91. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U.S.A. (2005) 102:7922–7. doi: 10.1073/pnas.0501190102
92. Lei Y, Klionsky DJ. Transcriptional regulation of autophagy and its implications in human disease. Cell Death Differ. (2023) 30:1416–29. doi: 10.1038/s41418-023-01162-9
93. Menéndez-Benito V, Neefjes J. Autophagy in MHC class II presentation: sampling from within. Immunity. (2007) 26:1–3. doi: 10.1016/j.immuni.2007.01.005
94. Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U.S.A. (2010) 107:2165–70. doi: 10.1073/pnas.0909448107
95. Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. (2014) 53:167–78. doi: 10.1016/j.molcel.2013.12.014
96. Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, et al. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. (2015) 17:515–25. doi: 10.1016/j.chom.2015.02.008
97. Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. (2007) 26:79–92. doi: 10.1016/j.immuni.2006.10.018
98. Fonteneau JF, Brilot F, Münz C, Gannagé M. The tumor antigen NY-ESO-1 mediates direct recognition of melanoma cells by CD4+ T cells after intercellular antigen transfer. J Immunol. (2016) 196:64–71. doi: 10.4049/jimmunol.1402664
99. Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. (2005) 22:571–81. doi: 10.1016/j.immuni.2005.03.009
100. Deffit SN, Blum JS. A central role for HSC70 in regulating antigen trafficking and MHC class II presentation. Mol Immunol. (2015) 68:85–8. doi: 10.1016/j.molimm.2015.04.007
101. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. (2014) 24:92–104. doi: 10.1038/cr.2013.153
102. Qiao L, Hu J, Qiu X, Wang C, Peng J, Zhang C, et al. LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy. (2023) 19:2837–52. doi: 10.1080/15548627.2023.2235196
103. Pérez L, McLetchie S, Gardiner GJ, Deffit SN, Zhou D, Blum JS. LAMP-2C inhibits MHC class II presentation of cytoplasmic antigens by disrupting chaperone-mediated autophagy. J Immunol. (2016) 196:2457–65. doi: 10.4049/jimmunol.1501476
104. Oh-oka K, Nakatogawa H, Ohsumi Y. Physiological pH and acidic phospholipids contribute to substrate specificity in lipidation of Atg8. J Biol Chem. (2008) 283:21847–52. doi: 10.1074/jbc.M801836200
105. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. (1998) 23:33–42. doi: 10.1247/csf.23.33
106. Mai T, Pone EJ, Li G, Lam TS, Moehlman J, Xu Z, et al. Induction of activation-induced cytidine deaminase-targeting adaptor 14-3-3γ is mediated by NF-κB-dependent recruitment of CFP1 to the 5'-CpG-3'-rich 14-3-3γ promoter and is sustained by E2A. J Immunol. (2013) 191:1895–906. doi: 10.4049/jimmunol.1300922
107. Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL, et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep. (2013) 5:702–14. doi: 10.1016/j.celrep.2013.09.031
108. Yan H, Fernandez M, Wang J, Wu S, Wang R, Lou Z, et al. B cell endosomal RAB7 promotes TRAF6 K63 polyubiquitination and NF-κB activation for antibody class-switching. J Immunol. (2020) 204:1146–57. doi: 10.4049/jimmunol.1901170
109. Tabata K, Natsunaga K, Sakane A, Sadsaki T, Noda T, Yoshimori T. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway. via novel Rab7-binding domain. Mol Biol Cell. (2010) 21:4162–72. doi: 10.1091/mbc.e10-06-0495
110. Leventhal JS. Lose appetite, lose control: integrins and noncanonical autophagy regulate germinal center reactions. J Clin Invest. (2018) 128:3752–3. doi: 10.1172/JCI122766
111. Chen S, Pounraj S, Sivakumaran N, Kakkanat A, Sam G, Kabir MT, et al. Precision-engineering of subunit vaccine particles for prevention of infectious diseases. Front Immunol. (2023) 14:1131057. doi: 10.3389/fimmu.2023.1131057
112. Tartakoff AM, Vassalli P. Plasma cell immunoglobulin secretion: arrest is accompanied by alterations of the golgi complex. J Exp Med. (1977) 146:1332–45. doi: 10.1084/jem.146.5.1332
113. Reales E, Mora-Lopez F, Rivas V, Garcia-Poley A, Brieva JA, Campos-Caro A. Identification of soluble N-ethylmaleimide-sensitive factor attachment protein receptor exocytotic machinery in human plasma cells: SNAP-23 is essential for antibody secretion. J Immunol. (2005) 175:6686–93. doi: 10.4049/jimmunol.175.10.6686
114. Bonaud A, Gargowitsch L, Gilbert SM, Rajan E, Canales-Herrerias P, Stockholm D, et al. Sec22b is a critical and nonredundant regulator of plasma cell maintenance. Proc Natl Acad Sci U.S.A. (2023) 120:e2213056120. doi: 10.1073/pnas.2213056120
115. Pone EJ, Lou Z, Lam T, Greenberg ML, Wang R, Xu Z, et al. B cell TLR1/2, TLR4, TLR7 and TLR9 interact in induction of class switch DNA recombination: modulation by BCR and CD40, and relevance to T-independent antibody responses. Autoimmunity. (2015) 48:1–12. doi: 10.3109/08916934.2014.993027
116. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. (2007) 282:24131–45. doi: 10.1074/jbc.M702824200
117. Cadete VJJ, Vasam G, Menzies KJ, Burelle Y. Mitochondrial quality control in the cardiac system: An integrative view. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:782–96. doi: 10.1016/j.bbadis.2018.11.018
118. Brookens SK, Cho SH, Basso PJ, Boothby MR. AMPKα1 in B cells dampens primary antibody responses yet promotes mitochondrial homeostasis and persistence of B cell memory. J Immunol. (2020) 205:3011–22. doi: 10.4049/jimmunol.1901474
119. Doens D, Fernández PL. Microglia receptors and their implications in the response to amyloid β for Alzheimer's disease pathogenesis. J Neuroinflamm. (2014) 11:48. doi: 10.1186/1742-2094-11-48
120. Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol. (2015) 15:441–51. doi: 10.1038/nri3857
121. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
122. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
123. Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation - a two-way street. Mol Med. (2017) 23:188–95. doi: 10.2119/molmed.2017.00077
124. Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. (2019) 38. doi: 10.15252/embj.201899430
125. Tonnus W, Locke S, Meyer C, Maremonti F, Eggert L, von Massenhausen A, et al. Rubicon-deficiency sensitizes mice to mixed lineage kinase domain-like (MLKL)-mediated kidney ischemia-reperfusion injury. Cell Death Dis. (2022) 13:236. doi: 10.1038/s41419-022-04682-3
126. Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell. (2018) 175:429–441.e416. doi: 10.1016/j.cell.2018.08.061
127. Prabakaran T, Bodd C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. (2018) 37. doi: 10.15252/embj.201797858
128. Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillon JC, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol. (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
129. Bankó Z, Pozsgay J, Szili D, Toth M, Gati T, Nagy G, et al. Induction and differentiation of IL-10-producing regulatory B cells from healthy blood donors and rheumatoid arthritis patients. J Immunol. (2017) 198:1512–20. doi: 10.4049/jimmunol.1600218
130. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. (2021) 40:e108863. doi: 10.15252/embj.2021108863
131. Jang A, Sharp R, Wang JM, Feng Y, Wang J, Chen M. Dependence on autophagy for autoreactive memory B cells in the development of pristane-induced lupus. Front Immunol. (2021) 12:701066. doi: 10.3389/fimmu.2021.701066
132. Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. (2019) 19:170–83. doi: 10.1038/s41577-018-0095-2
133. Wang F, Muller S. Manipulating autophagic processes in autoimmune diseases: a special focus on modulating chaperone-mediated autophagy, an emerging therapeutic target. Front Immunol. (2015) 6:252. doi: 10.3389/fimmu.2015.00252
134. Zhang H, Alsaleh G, Feltham J, Sun Y, Napolitano G, Riffelmacher T, et al. Polyamines control eIF5A hypusination, TFEB translation, and autophagy to reverse B cell senescence. Mol Cell. (2019) 76:110–125.e119. doi: 10.1016/j.molcel.2019.08.005
135. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. (2015) 74:912–20. doi: 10.1136/annrheumdis-2013-204343
136. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. (2011) 208:2625–32. doi: 10.1084/jem.20110640
137. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. (2013) 39:211–27. doi: 10.1016/j.immuni.2013.07.017
138. Durgan J, Florey O. Many roads lead to CASM: Diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci Adv. (2022) 8:eabo1274. doi: 10.1126/sciadv.abo1274
139. Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. (2018) 37. doi: 10.15252/embj.201797840
140. Nedjic J, Aichinger M, Mizushima N, Klein L. Macroautophagy, endogenous MHC II loading and T cell selection: the benefits of breaking the rules. Curr Opin Immunol. (2009) 21:92–7. doi: 10.1016/j.coi.2009.01.013
Keywords: antibodies, autophagy, B cell biology, B lymphocytes, canonical autophagy, humoral immunity, noncanonical autophagy
Citation: Wang Y, Wu L and Van Kaer L (2024) Role of canonical and noncanonical autophagy pathways in shaping the life journey of B cells. Front. Immunol. 15:1426204. doi: 10.3389/fimmu.2024.1426204
Received: 30 April 2024; Accepted: 16 July 2024;
Published: 30 July 2024.
Edited by:
Leopoldo Santos-Argumedo, Center for Research and Advanced Studies, National Polytechnic Institute of Mexico (CINVESTAV), MexicoReviewed by:
Jelena Bezbradica, University of Oxford, United KingdomGabriela Lopez-Herrera, National Institute of Pediatrics, Mexico
Copyright © 2024 Wang, Wu and Van Kaer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luc Van Kaer, bHVjLnZhbi5rYWVyQHZ1bWMub3Jn