 Sulayman Mourabit1†
Sulayman Mourabit1† Sarah Römer2†
Sarah Römer2† Erin R. Bonner1Fabian Winter3Julian Tschollar1
Erin R. Bonner1Fabian Winter3Julian Tschollar1 Mladen V. Tzvetkov2
Mladen V. Tzvetkov2 Werner Weitschies3Stefan Engeli4‡*Werner Tschollar1‡*
Werner Weitschies3Stefan Engeli4‡*Werner Tschollar1‡*- 1AMYRA Biotech AG, Basel, Switzerland
- 2Department of General Pharmacology, Institute of Pharmacology Center of Drug Absorption and Transport (C_DAT), University Medicine Greifswald, Greifswald, Germany
- 3Department of Pharmaceutical Technology and Biopharmaceutics, Institute of Pharmacy, Center of Drug Absorption and Transport (C_DAT), University of Greifswald, Greifswald, Germany
- 4Department of Clinical Pharmacology, Institute of Pharmacology, Center of Drug Absorption and Transport (C_DAT), University Medicine Greifswald, Greifswald, Germany
Introduction: Celiac disease is a common autoimmune-like enteropathy caused by an aberrant response to incompletely digested dietary gluten. Gluten immunogenic peptides including the immunodominant 33-mer are thought to be resistant to proteolytic digestion by human gastrointestinal peptidases. We developed a novel enzyme therapy approach to support gluten peptide digestion using a combination of two tandem-acting exopeptidases, AMYNOPEP, that complement the intrinsic enzymatic activity of intestinal brush border enterocytes.
Methods: We evaluated the effects of AMYNOPEP supplementation on 33-mer degradation in vitro and in vivo. In a cross-over clinical study, healthy volunteers with no gastrointestinal disorders were given stable isotope (SI) labelled 33-mer peptides in the presence of varying peptide substrates and caloric loads, with and without AMYNOPEP. 33-mer degradation products (SI-labelled single amino acids) were measured in the blood plasma using LC-MS/MS.
Results: AMYNOPEP achieved rapid, complete amino-to-carboxyl terminal degradation of the 33-mer in vitro, generating single amino acids and dipeptides. In healthy volunteers, AMYNOPEP supplementation significantly increased 33-mer degradation and absorption of SI-labelled amino acids even in the presence of competing substrates. Specifically, we observed a 2.8-fold increase in the Cmax of stable isotope-labelled amino acids in the presence of wheat gluten. The absorption kinetics of labelled amino acids derived from 33-mer digestion with AMYNOPEP closely resembled that of SI-labelled X-Proline dipeptides administered without enzyme supplementation, highlighting the rapid hydrolytic activity of AMYNOPEP on polypeptides.
Conclusions: AMYNOPEP achieved complete degradation of the 33-mer into single amino acids and dipeptides in vitro and significantly improved 33-mer degradation kinetics in healthy volunteers, as measured by labelled amino acid detection, warranting further investigation into the potential therapeutic benefits of exopeptidase combinations for patients with gluten-related health disorders including celiac disease.
Introduction
Celiac disease (CeD) is a common autoimmune-like enteropathy affecting 1-1.4% of the global population (1, 2) with its incidence rising by 7.5% annually over the past several decades (3). CeD is caused by an aberrant response to specific peptide fragments released during dietary gluten digestion. A limited number of gluten immunogenic peptides (GIPs) are considered to be immunodominant, including the α2-gliadin derived 33-mer peptide which carries six overlapping T cell epitopes (4). The 33-mer is thought to be stable to proteolytic digestion by human gastric, pancreatic, and intestinal peptidases due to its abundance of proline residues (5), though studies of duodenal biopsies have shown the 33-mer to be almost fully degraded during intestinal transport in healthy individuals (6). Interestingly, the 33-mer peptide has also been shown to spontaneously form peptide self-aggregates in vitro (7, 8), which may further interfere with its digestion. In patients with CeD, GIP exposure reactivates a CD4+ T cell-driven immunological response resulting in a broad range of gastrointestinal (GI) and systemic symptoms (as reviewed elsewhere (9, 10)). A lifelong gluten-free diet (GFD) is seen as the only effective approach to prevent gastrointestinal symptoms in CeD patients. However, most patients do not experience complete mucosal healing on a GFD even with well-controlled symptoms (11, 12) and up to 80% of GFD-adhering patients experience inadvertent gluten contamination (13), highlighting a need for more effective CeD therapeutics.
Enzyme therapies currently in development aim to support the body’s natural digestion of gluten peptides using exogenous bacterial, fungal, or plant-derived peptidases. These therapies have almost exclusively focused on stomach-acting endopeptidases (ENPs), a class of enzyme that generates peptides of variable lengths by cleaving intra-chain residues (e.g., AN-PEP, Latiglutenase, TAK-062). While ENPs, such as pepsin, trypsin, and chymotrypsin, partially digest peptides into progressively smaller chains, exopeptidases (EXPs) complete digestion by systematically cleaving peptide bonds on either terminal end into absorbable lengths (e.g., single amino acids, dipeptides). The majority of EXPs are anchored to enterocytes at the brush border membrane (BBM) or released by BBM vesicles into the lumen of the small intestine (14, 15). Patients with CeD experience damage to the intestinal BBM and reduced activity of certain brush border EXPs (16, 17). For instance, activity of the endogenous proline-specific dipeptidyl peptidase-IV (DPP-IV) was shown to be reduced by an average of 70% in CeD patients compared to healthy individuals without gastrointestinal diseases (17), likely aggravating the indigestibility of GIPs and their accumulation.
Here, we investigated a novel enzyme therapy approach for the digestion of GIPs using a combination of two exopeptidases, AMYNOPEP, that complements the intrinsic exopeptidase activity of enterocytes. AMYNOPEP consists of two tandem-acting aminopeptidases (a monoaminopeptidase and dipeptidyl peptidase) that digest peptides from the amino- to carboxy-terminal to generate absorbable single amino acids and dipeptides. We assessed the action of AMYNOPEP enzymes on stable isotope (SI) labelled 33-mer peptide digestion using a quantitative LC-MS/MS method for near-real-time detection of SI-labelled amino acids in the blood of healthy volunteers.
Results
AMYNOPEP rapidly and completely degrades the 33-mer peptide into dipeptides and single amino acids in vitro
The degradation activity and efficacy of AMYNOPEP was assessed on the 33-mer in vitro (1:10 enzyme ratio, see Methods). Since both enzymes are aminopeptidases, the 33-mer was expected to be degraded through stepwise cleaving events beginning at the amino-terminal end and resulting in the release of single amino acids (leucine, L; glutamine, Q; phenylalanine, F) and X-proline (XP) dipeptides (QP, FP, LP, and (tyrosine, Y) YP, Figure 1A). Within 30 minutes of incubation with AMYNOPEP, less than 0.05% of the full 33-mer sequence was detectable (Figure 1B), and all anticipated degradation products (XP dipeptides and single amino acids) were detected (Figure 1C). We further analyzed the extent of 33-mer digestion by monitoring the appearance of degradation intermediates: 28-mer, 16-mer, 9-mer, and 5-mer peptides (Figure 1D). The degradation intermediates appeared in a time-dependent manner, in line with progressive N-to-C terminal degradation of 33-mer peptides. Accordingly, XP dipeptides and free amino acids increased in a time-dependent manner (Figure 1E). XP dipeptides appeared to be stable to enzymatic cleavage, as indicated by the lack of detectable free P or Y residues. Given the lack of XP cleavage, detection of free F residues indicated complete degradation of the 33-mer down to the carboxy-terminal-most F residue. Thus, the presence of detectable F within 30 minutes of AMYNOPEP incubation proved complete N-to-C terminal degradation of the 33-mer peptide in vitro. Using the terminal F, it could be estimated that 81% to 91% of analyzed 33-mer was degraded completely down to single amino acids and XP dipeptides after 60 minutes of incubation (Figure 1F). To further assess the complete degradation of the 33-mer peptide by AMYNOPEP in vitro, we monitored all theoretically possible 33-mer degradation intermediates expected to result from AMYNOPEP’s mode-of-action, revealing complete degradation of the measured peptide products by 60 minutes (Supplementary Figure 1). Faster 33-mer degradation in vitro was observed with an enzyme ratio of 1:1 (Supplementary Figure 2), thus a 1:1 enzyme ratio was used for all subsequent studies in healthy volunteers.
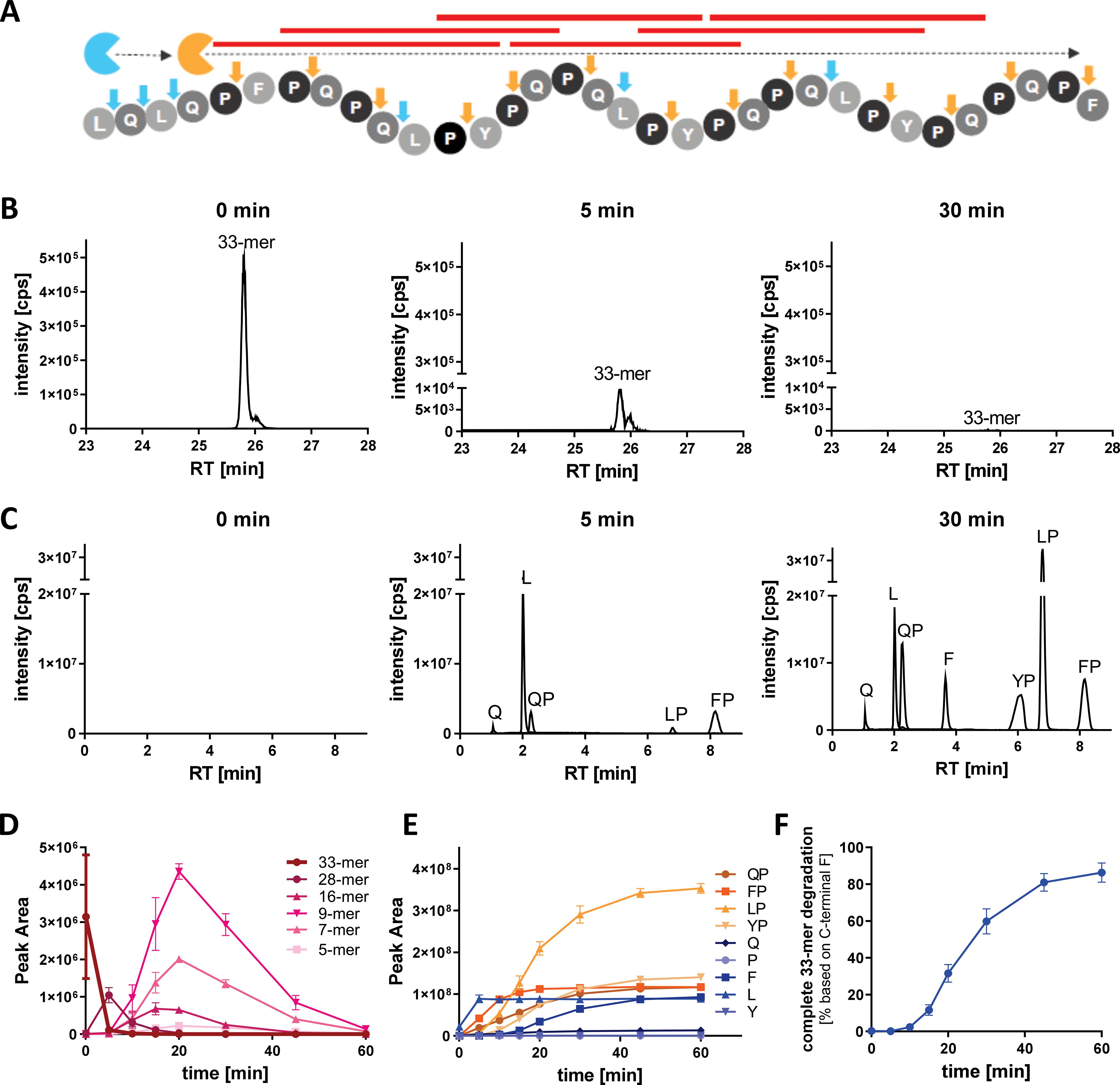
Figure 1. AMYNOPEP rapidly and completely degrades the 33-mer GIP into single amino acids and dipeptides in vitro. (A) Schematic representation of the AMYNOPEP mode-of-action. Tandem EXP activity ensures systematic degradation of the 33-mer through stepwise cleavage of single amino acids (L, Q, F), mediated by the aminopeptidase (blue), and cleavage of XP dipeptides (QP, FP, LP, YP) mediated by the dipeptidyl peptidase (orange). Cleaving sites for each enzyme are indicated by arrows. Red lines represent immunogenic HLA-DQ2/-DQ8 epitopes. (B) LC-MS/MS analysis of full length 33-mer degradation 0, 5, and 30 min after addition of AMYNOPEP. (C) Detection of XP dipeptides and amino acids during 33-mer degradation 0, 5, and 30 min after addition of AMYNOPEP. Peaks are labelled with their corresponding degradation product. (D) Time-dependent degradation of the 33-mer by AMYNOPEP and appearance of selected peptide intermediates, 28-mer (FPQPQLPYPQPQLPYPQPQLPYPQPQPF), 16-mer (LPYPQPQLPYPQPQPF), 9-mer (LPYPQPQPF), and 5-mer (QPQPF). (E) Corresponding time-dependent appearance of XP dipeptides and free amino acids. (F) Quantitative determination of complete 33-mer degradation based on the detection of F that is only cleaved once as single amino acid during the last cleavage step. (D–F) show means and SD of three technical replicates.
SI-labelled amino acids are detectable in the plasma of healthy volunteers
To evaluate the in vivo detection efficacy of labelled 33-mer degradation products, we performed a pilot study wherein SI-labelled XP dipeptides were orally administered to healthy volunteers (n=9 total) and subsequently measured in plasma and urine using HPLC-MS/MS (Figure 2, top panel pilot cohort). Volunteers (n=3) were given 50 mg of each SI-labelled XP dipeptide (F*P*, LP*, and L*P*) diluted in water, and the appearance of SI-labelled products was assessed over 72 hours in plasma and pooled urine samples. Only trace amounts (<LOQ) of XP dipeptides were detected in plasma (Figure 3A) and urine. In contrast, all three SI-labelled amino acids (L*, F*, and P*) that would result from the cleavage of their respective XP dipeptides were detectable in plasma in a time-dependent manner following oral administration (Figure 3A). Since SI-labelled amino acids were only partially excreted in urine with 0.002% to 0.017% of the orally administered amount (data not shown), subsequent in vivo studies did not include urine analyses.
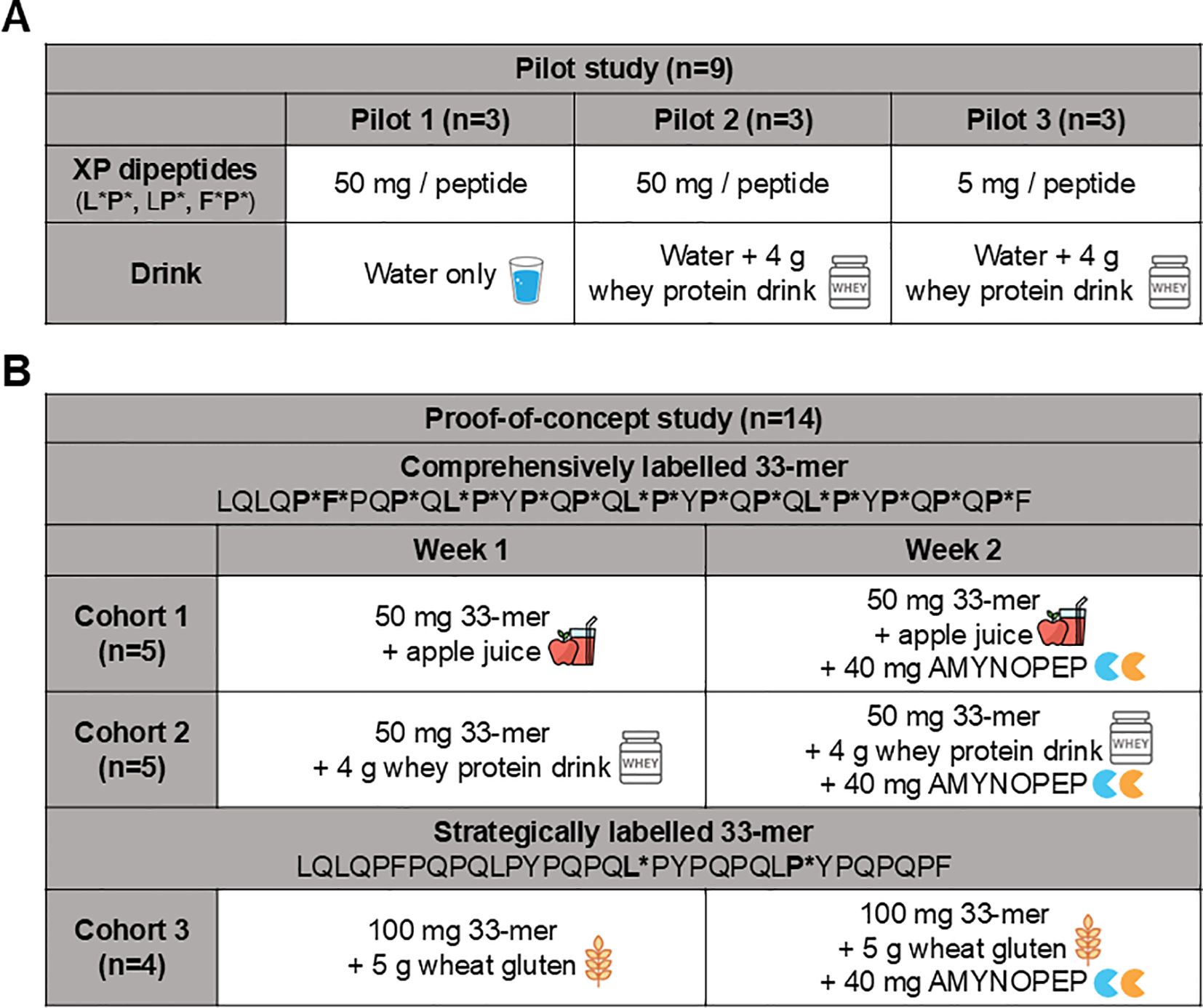
Figure 2. Overview of pilot and proof-of-concept study design assessing AMYNOPEP supplementation on 33-mer peptide digestion in healthy volunteers. Pilot study: To assess detection of orally administered amino acids in blood, a pilot study was conducted beforehand. (A) SI-labelled XP dipeptides were administered and resorption in the form of SI-labelled amino acids was measured in blood. (B) Proof-of-concept study (Cohorts 1-3): Two different isotope labelled peptides were used to evaluate 33-mer degradation: (1) a comprehensively labelled peptide with labels covering epitopes throughout the peptide, and (2) a strategically labelled peptide to evaluate end-to-end peptide digestion. A cross-over study design was utilized in which healthy volunteers followed a specified protocol on week one, followed by the same protocol with the addition of AMYNOPEP on week 2. In cohort 1, five volunteers were given 50 mg of comprehensively labelled peptide with apple juice. In cohort 2, five volunteers were given 50 mg of comprehensively labelled peptide with Peptamen. In cohort 3, four volunteers were given 100 mg of strategically labelled peptide with 5 g of wheat gluten. Blood was collected at regular intervals and 33-mer digestion products were measured as described in the Methods.
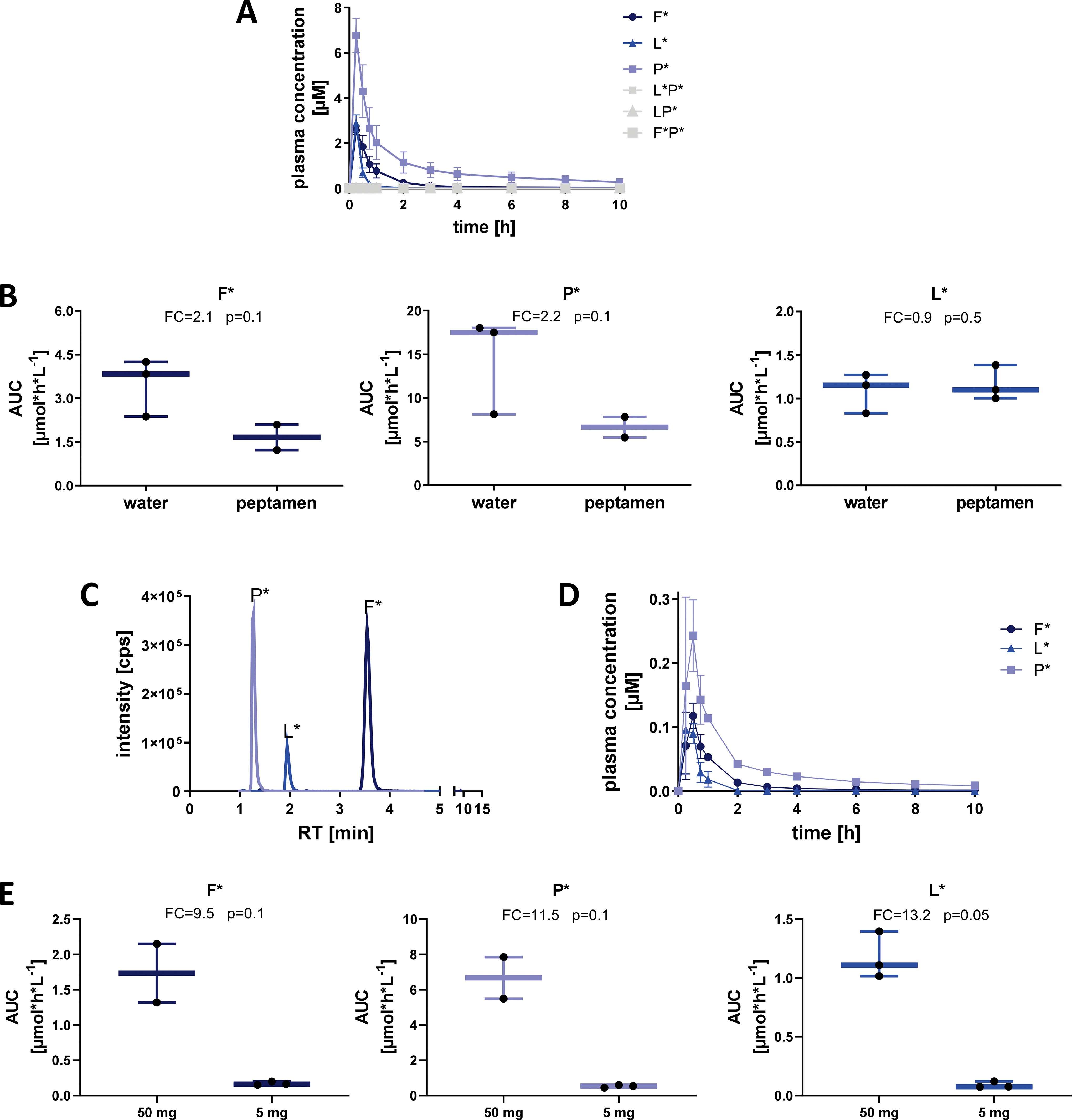
Figure 3. Detection of SI-labelled single amino acids in plasma after oral administration of XP dipeptides in healthy volunteers. (A) Plasma concentration [µM] of labelled dipeptides and amino acids after oral administration of L*P*, LP*, and F*P* (50 mg each) with water. Means and SD of three individuals are shown. (B) Influence of Peptamen (hydrolyzed protein) on absorption and detection of labelled amino acids after oral administration of dipeptides (50 mg each). Means and min/max of three individuals are shown. (C) LC-MS/MS detection intensity [cps] of labelled amino acids 1 hour after oral administration of dipeptides (5 mg each) taken with Peptamen. (D) Plasma concentration [µM] of labelled amino acids after administration of 5 mg of each labelled XP taken with Peptamen. (E) Influence of dose reduction from 50 mg to 5 mg of each labelled XP taken with Peptamen on plasma AUC of labelled amino acids. Box plots show the median, 25th to 75th percentiles and whisker show the min/max of three healthy individuals per cohort. FC, fold-change; AUC, area under the curve.
To assess differences in SI-labelled amino acid digestion and absorption in the presence of a caloric load and competing peptide substrate, we administered 50 mg of each XP dipeptide with 100 mL of Peptamen (n=3 volunteers, Figure 2 top panel). Peptamen did not influence the detection of XP dipeptides which were again only present in trace amounts in the plasma (data not shown). Peptamen resulted in decreased detection of F* and P* in plasma, as seen through a decrease in AUC of 2.1-fold and 2.4-fold, respectively (Figure 3B). In contrast, detection of L* was not significantly altered in the presence of Peptamen relative to water.
To assess the detection limit of our system, we lowered the dipeptide dose to 5 mg per XP, in combination with 100 mL Peptamen (n=3 volunteers, Figure 2 top panel). The 10-fold dipeptide dose reduction resulted in clearly detectable F*, P*, and L* in plasma (Figures 3C, D), with a proportional reduction in plasma AUC of 9.5-fold, 11.5-fold, and 13.2-fold, respectively (Figure 3E). Based on the XP dipeptide absorption data, we proceeded to administer at least 50 mg of labelled 33-mer peptides in subsequent studies.
AMYNOPEP supplementation rapidly increases the degradation of 33-mer peptides in healthy volunteers
To assess enzyme efficacy in healthy volunteers, we analyzed degradation of two different SI-labelled 33-mer peptides with and without AMYNOPEP (Figure 2, middle and lower panels). The first was a comprehensively labelled peptide with SI labels distributed throughout the sequence, covering all immunogenic epitopes with the goal of ensuring sufficient detection of enzyme activity in vivo (Figure 4A). In the first cohort (n=5 volunteers, Figure 2 middle panel), 50 mg of comprehensively labelled 33-mer peptide were administered with apple juice, a low-caloric fluid to facilitate rapid gastric emptying, and to avoid the introduction of competing peptides that could affect enzyme activity. Interestingly, 33-mer degradation was also observed in volunteers without AMYNOPEP, though AMYNOPEP supplementation resulted in significantly higher plasma Cmax of all labelled amino acids in the first 30 min post-administration compared to control (Figures 4B, C). Specifically, AMYNOPEP resulted in significant 1.8, 2.0, and 2.4-fold-change increases in maximum plasma concentrations (Cmax) of F*, P*, and L*, respectively (Figure 4C). In addition, AMYNOPEP supplementation resulted in significantly increased AUCs of F* (FC=1.3, p=0.025), P* (FC=1.2, p=0.006) and L* (FC=1.3, p=0.018) compared to control (Supplementary Figure 3).
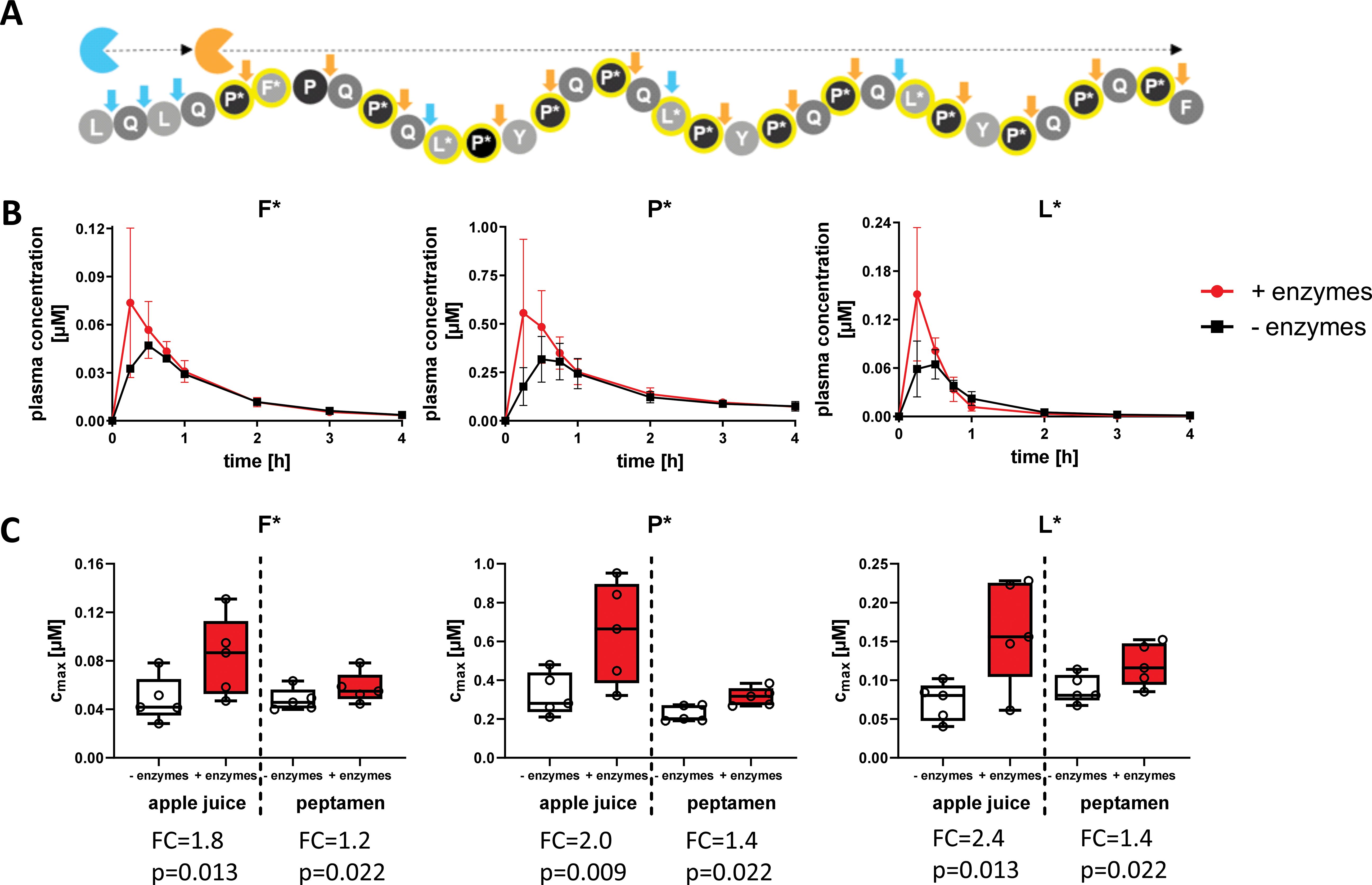
Figure 4. Rapid and enhanced degradation of the 33-mer peptide with AMYNOPEP supplementation. (A) Schematic overview of comprehensively labelled 33-mer peptide digestion by AMYNOPEP enzymes (represented by blue and orange sectors). Cleavage sites are indicated by arrows. (B) 50 mg of labelled 33-mer peptide was administered to five healthy volunteers with apple juice, preceded by administration of AMYNOPEP (visit 2) or plain water (visit 1). Plasma concentration of labelled amino acids (F*, P*, L*) was measured over the course of four hours. Means and SD of five healthy individuals are shown (C) Cmax of labelled amino acids in plasma, with and without AMYNOPEP supplementation, and co-administration of apple juice (left side of graphs) or Peptamen (right side of graphs). Box plots show the median, 25th to 75th percentiles and whisker show the min/max of five healthy individuals. FC, fold-change; Cmax, maximum concentration.
In the second cohort (n=5 volunteers, Figure 2 middle panel), we assessed the effect of competing peptide substrates in the form of 100 mL Peptamen on the degradation activity of 33-mer by AMYNOPEP. As expected, the addition of Peptamen decreased the efficacy of 33-mer digestion when compared to enzyme administration with apple juice, as shown by decreased Cmax (Figure 4C). However, even with Peptamen, significant increases in Cmax of F* (FC=1.2, p=0.022), P* (FC=1.4, p=0.022) and L* (FC=1.4, p=0.022) were observed with AMYNOPEP compared to control. Additionally, AMYNOPEP supplementation still resulted in significantly increased AUCs of F* (FC=1.1, p=0.022), P* (FC=1.2, p=0.022) and L* (FC=1.1, p=0.022) in the presence of Peptamen compared to controls.
AMYNOPEP supplementation significantly decreased time to Cmax (tmax) for F* (tmax= 22.5 min with AMYNOPEP vs 31.5 min without, p=0.048) and P* (tmax= 24 min with AMYNOPEP vs. 33 min without, p=0.032) when administered with apple juice (Supplementary Figure 4). No significant differences in tmax were observed with Peptamen (Supplementary Figure 4).
We further assessed AMYNOPEP enzyme efficacy using a second SI-labelled 33-mer peptide with strategic labeling of single L* and P* residues, to assess the extent of N-to-C terminal peptide digestion (Figure 2 bottom panel, Figure 5A). Detection of L* would indicate degradation of the first half of the 33-mer and disruption of four out of six immunogenic epitopes, while the downstream P* shows disruption of all immunogenic epitopes. In a third cohort (n=4 volunteers), we administered 100 mg of the strategically labelled 33-mer with 5 g of wheat gluten as a competitive substrate (Figure 2). AMYNOPEP supplementation resulted in significantly increased Cmax of P* (FC=2.8, p=0.034) and L* (FC=3.4, p=0.034) compared to control, even in the presence of gluten (Figures 5B, C). We observed a trend in which AMYNOPEP decreased tmax, which was reached at 22.5 min post-administration of AMYNOPEP for both P* and L*, compared to tmax of 45 min without enzyme (p=0.054 for both; Supplementary Figure 5).
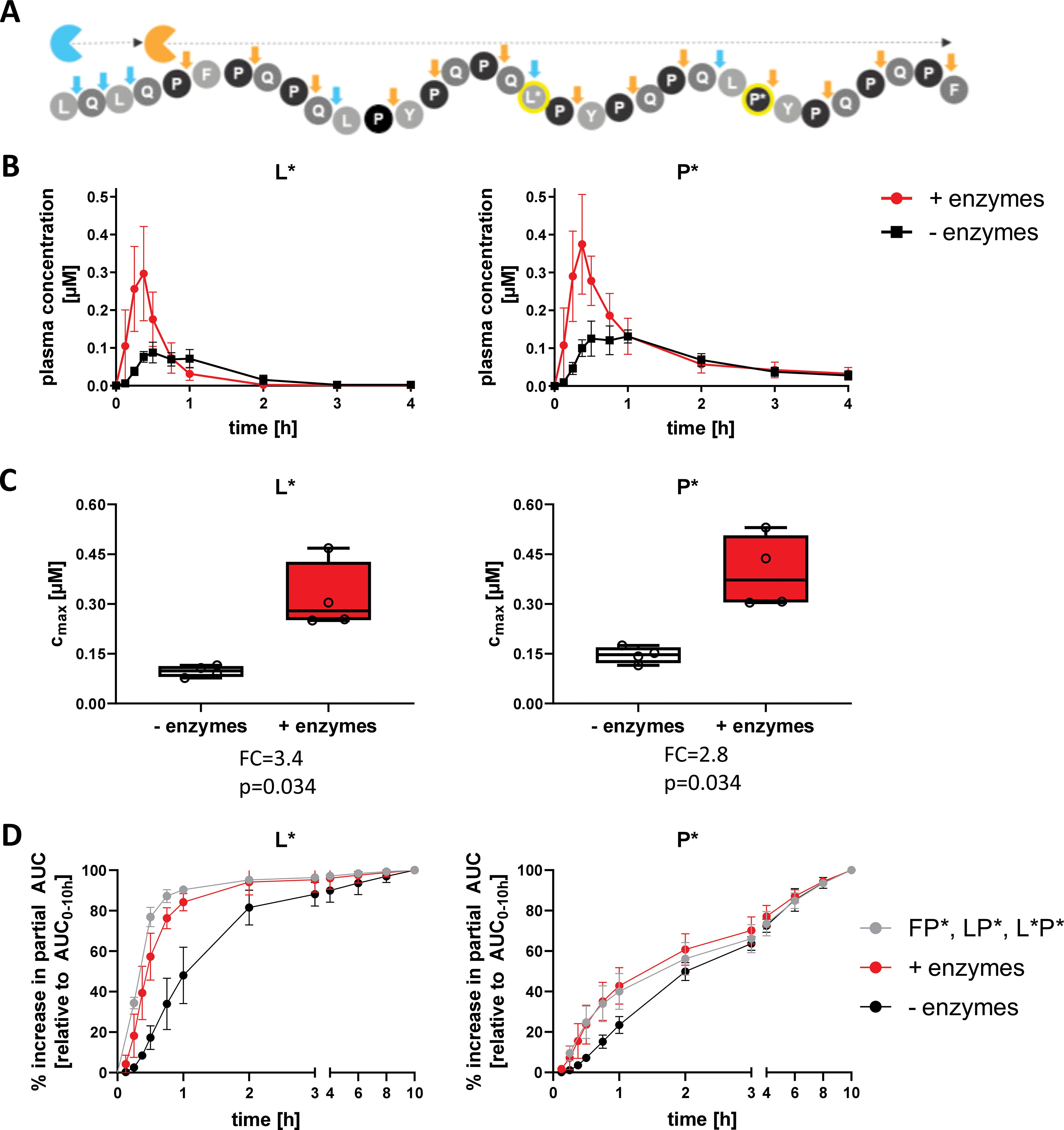
Figure 5. Comprehensive degradation of SI-labelled 33-mer by AMYNOPEP in the presence of gluten. (A) Schematic overview of strategically labelled 33-mer peptide digestion by AMYNOPEP enzymes (represented by blue and orange sectors). Cleavage sites are indicated by arrows. (B, C) 100 mg of strategically labelled 33-mer peptide was administered to four healthy volunteers with 5 g wheat gluten, with (red) or without (black) AMYNOPEP. Influence of enzyme addition on L* and P* plasma concentrations over time (B) and on maximum plasma concentrations (Cmax, C) were determined. (D) Comparison of amino acid absorption after administration of XP dipeptides (F*P*, LP*, L*P*) with water or 33-mer strategically labelled peptide with and without addition of AMYNOPEP. Plasma concentration-time plots show means and SD of four healthy individuals. Box plots show the median, 25th to 75th percentiles and whiskers show and min/max of four healthy individuals. Cmax, maximum concentration; AUC, area under the curve; FC, fold-change.
Most importantly, the absorption kinetics of single amino acids resolving from 33-mer degradation after AMYNOPEP addition closely resembled that of the amino acids when administered as XP dipeptides in the pilot study (Figure 5D). This finding reinforces the rapid action of AMYNOPEP, whereby the digestion and absorption of amino acids from a long (>30-amino acid) peptide was comparable to that of ingested dipeptides.
Discussion
We have demonstrated rapid, systematic N-to-C terminal degradation of 33-mer peptides by AMYNOPEP in vitro, with corresponding degradation of SI-labelled 33-mer peptides in healthy volunteers even in the presence of competing substrates (Peptamen, wheat gluten). While the study was limited by its focus on one representative SI-labelled GIP, the positive results described herein warrant further investigation into AMYNOPEP’s efficacy at digesting other proteolytically-resistant and immunogenic peptides. Given the observed end-to-end degradation mode of action of AMYNOPEP in vitro, the same mechanism very likely underlies the improved degradation of 33-mer peptides in vivo, with AMYNOPEP supporting endogenous digestive enzyme machinery. Our strategy to use SI-labelled 33-mer combined with quantitative LC-MS/MS detection allowed us to distinguish between SI-labelled 33-mer degradation products and naturally occurring amino acids in plasma samples, including those from competing substrates co-administered with SI-labelled 33-mer. Considering the N-to-C terminal degradation of peptides by AMYNOPEP, the two differently SI-labelled peptides provided complementary information. Whereas the comprehensively labelled peptide allowed for sensitive detection of even minor degradation, the strategically labelled peptide gave us an indication of the extent of 33-mer digestion. Following the mode-of-action demonstrated in vitro, detection of labelled L* would indicate degradation of half of the 33-mer and four out of six immunogenic epitopes, whereas detection of labelled P* would show further N-to-C terminal degradation and disruption of all six immunogenic epitopes.
We observed 33-mer degradation in healthy volunteers even without AMYNOPEP. These findings further support that the 33-mer is not indigestible in healthy humans, but that its digestion in the small intestine may be performed by rate-limited enzymes (e.g., brush border EXPs (18)). In line with this, research on duodenal biopsies from active CeD, GFD-managed CeD, and healthy volunteers showed that enterocytes of healthy individuals and GFD-managed, but not active CeD patients, can fully digest certain digestion-resistant gliadin peptides (6), suggesting defects in BBM peptidase activity in active CeD. As noted, compromised enterocyte functionality and reduced activity of certain BBM peptidases in CeD reinforce the need to supplement or replace BBM deficiencies. To date, there have been limited research and clinical studies aimed at developing EXP-based enzyme therapies, perhaps due to a limited understanding of the human BBM proteome and lack of suitable in vitro models to study the human BBM (19, 20), the source of most EXPs (14). Some exogenous EXPs have been studied in human and commercial settings, including DPP-IV and separately, leucyl aminopeptidase, demonstrating favorable safety profiles of these enzymes. DPP-IV is already available as an over-the-counter dietary supplement in several products, and a food enzyme leucyl aminopeptidase recently underwent safety evaluation showing no safety concerns under conditions of use in eight food manufacturing processes (21). Regarding the safety of AMYNOPEP, we collected data on adverse events during the course of study participation, and no volunteers reported adverse events related to the ingestion of study materials (AMYNOPEP or SI-labelled peptides, see Appendix I for more details on safety data). We demonstrated here that EXP supplementation via AMYNOPEP greatly increased 33-mer degradation and SI-labelled amino acid absorption in vivo, even in healthy volunteers with presumably intact BBM. Our approach, with quantitative LC-MS/MS measurement of SI-labelled 33-mer degradation products, allowed for sensitive, non-invasive monitoring of GIP degradation in near-real-time. We did not detect SI-labelled XP dipeptides in vivo, likely due to endogenous processing of dipeptides into single amino acids by enterocytes (as demonstrated in the pilot study).
Contrary to many ENP-based enzyme therapies under development for the treatment of CeD, our enzyme combination targets peptide degradation in the small intestine. The focus of most enzyme therapies on peptide digestion in the stomach likely stems from concerns that GIP digestion must occur before gastric emptying to prevent immune activation in the small intestine. However, recent research into the immunological timeline of events following gluten ingestion revealed a one-to-four-hour window before peak detection of immune markers in the blood and associated gastrointestinal symptoms (22), suggesting that fast-acting enteric enzymes may still digest gluten peptides in time to prevent adverse effects. Furthermore, enzyme approaches relying solely on gluten digestion in the stomach are likely to be ineffective due to limited mixing in the stomach (23, 24) and rapid gastric emptying of low-caloric fluids and small particles (23, 25). Stomach-targeting enzyme supplements may therefore be rapidly emptied into the small intestine, where they might experience greatly reduced activity or inactivation due to changes in pH or vulnerability to trypsin, thereby missing their window of opportunity to efficiently digest gluten peptides. Our novel mode-of-action is based on combinatorial EXP action in the small intestine, hence addressing these limitations, complementing the body’s EXP activity of luminal or brush border origin to achieve thorough digestion of GIPs into absorbable single amino acids and dipeptides. While our study was limited by a small sample size of healthy volunteers and did not include patients with CeD, our SI-labelled approach offers a highly sensitive and accurate detection method for assessing enzyme efficacy in near real-time. Future enzymatic approaches to gluten digestion should account for issues of motility and enzyme mode-of-action, with a particular focus on intestinal activity and EXP supplementation.
Materials and methods
Materials
All labelled peptides, XP dipeptides, and amino acids were synthesized using 13C and 15N stable isotopes (SI, labelling further indicates as *). Labelled 33-mer (LQLQP*F*PQP*QL*P*YP*QP*QL*P*YP*QP*QL*P*YP*QP*QP*F and LQLQPFPQPQLPYPQPQL*PYPQPQLP*YPQPQPF) and XP dipeptides phenylalanine-proline (F*P*), leucine-proline (LP*), and leucine-proline (L*P*) were synthesized by Intavis Peptide Services GmbH (Tübingen, Germany). Labelled and non-labelled amino acids (F, P, L, Y, Q) were obtained from Sigma-Aldrich (Taufkirchen, Germany). Non-labelled 33-mer was obtained from Ontores biotechnologies (Shanghai, China). Non-labelled XP dipeptides FP, LP, YP and QP as well as 28-mer, 16-mer, 9-mer and 5-mer were obtained as SpikeTides™ for method development from JPT Peptide Technology GmbH (Berlin, Germany). LC-MS/MS grade acetonitrile, methanol, and formic acid were obtained from Merck (Darmstad, Germany). LoBind 1.5mL reaction tubes were obtained from Sarstedt (Nümbrecht, Germany). Peptamen™ (Neutral SmartFlex) was obtained from Nestlé Health Science Gmbh (Frankfurt, Germany). Wheat gluten was obtained from Veganz Group AG (Berlin, Germany). Hydrogencarbonate (NaHCO3) was obtained from Arnold Holste Wwe. GmbH & Co. KG (Bielefeld, Germany).
In vitro digestion of 33-mer peptide by AMYNOPEP
In vitro digestion of 33-mer peptides was performed using a two-form enzyme combination, AMYNOPEP, which utilizes two tandem-acting aminopeptidases. The 33-mer digestion was done in Tris buffer at pH 7.0 using an enzyme mixture consisting of 0.86 mg/L dipeptidyl peptidase and 8.6 mg/L aminopeptidase (1:10 enzyme ratio). Enzyme mixture was incubated with 100 mg/L 33-mer in a final volume of 1.8 mL. Digestions were performed at 37°C and 350 rpm. Samples of 200 µL were collected at 0, 5, and 30 min and immediately transferred to new 1.5 mL reaction tubes prefilled with 1.6 mL 0.1% TFA to stop the reaction. Samples were stored at -80°C prior to LC-MS/MS measurement, for which samples were diluted 1:5 in 0.1% formic acid. 5 µL or 10 µL of samples were analyzed to detect the intact 33-mer, degradation intermediates (28-mer: FPQPQLPYPQPQLPYPQPQLPYPQPQPF; 16-mer: LPYPQPQLPYPQPQPF; 9-mer: LPYPQPQPF; 5-mer: QPQPF), and degradation products (dipeptides and amino acids). In order to determine the proportion of complete degradation, the amount of cleaved F, which is only generated in the last step of degradation, was quantified and set in relation to the theoretical maximum achievable concentration with complete degradation of all 33-mer molecules. Detailed description of LC-MS/MS method to detect in vitro degradation products can be found in Supplementary Tables 1, 2.
Pilot study demonstrating SI-labelled amino acid detection in plasma of healthy volunteers
Healthy volunteers were selected based on no previous history of CeD, NCGS, or wheat allergy. The study (German Register of Clinical Studies, DRKS00033099) was approved by the Ethics Committee of the University Medicine Greifswald, and was performed in adherence to the declaration of Helsinki (2013 Version), the Medical Association’s Professional code of conduct for Mecklenburg-Vorpommern (BOÄ M-V) and the data protection regulation of the EU (EU-DSGVO) and of Mecklenburg-Vorpommern (DSG-MV). Written informed consent was obtained from all volunteers prior to enrollment in the study.
A mixture of all three SI labelled XP dipeptides (F*P*, LP*, and L*P*) were orally administered to volunteers (n=9 total), divided into three cohorts of three volunteers each (see Figure 2). Cohort 1 received 50 mg of each dipeptide in 300 mL water. Cohort 2 received 100 mL Peptamen first, then 50 mg of each dipeptide in 150 mL water, followed by another 50 mL water. All drinks were taken within a minute. Cohort 3 received 100 mL Peptamen first, then a reduced dose of 5 mg of each dipeptide in 150 mL water, followed by another 50 mL water. All drinks were taken within a minute. Blood samples were collected after 15 min, 30 min, 45 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, 14 h, 48 h and 72 h after dosing. Urine was collected and pooled at 0-4 h, 4-6 h, and 6-10 h, and was collected as spot urine at 48 h and 72 h after dosing.
Proof-of-concept study evaluating 33-mer peptide degradation by AMYNOPEP in healthy volunteers
Healthy volunteers were selected based on no previous history of CeD, NCGS, or wheat allergy. The study (German Register of Clinical Studies, DRKS00033108) was approved by the Ethics Committee of the University Medicine Greifswald, and was performed in adherence to the declaration of Helsinki (2013 Version), the Medical Association’s Professional code of conduct for Mecklenburg-Vorpommern (BOÄ M-V) and the data protection regulation of the EU (EU-DSGVO) and of Mecklenburg-Vorpommern (DSG-MV). Written informed consent was obtained from all volunteers prior to enrollment in the study.
Volunteers were divided into three cohorts, utilizing differently labelled 33-mer peptides and different food intake conditions. Two differently SI-labelled 33-mer peptides were used: (1) a comprehensively labelled 33-mer (LQLQP*F*PQP*QL*P*YP*QP*QL*P*YP*QP*QL*P*YP*QP*QP*F), and (2) a strategically labelled 33-mer (LQLQPFPQPQLPYPQPQL*PYPQPQLP*YPQPQPF). Bold asterisked letters indicate SI-labelled amino acids. The study was designed as a cross-over study, with study visits one week apart. During visit 1, each volunteer received the labelled 33-mer with apple juice, Peptamen, or wheat gluten without AMYNOPEP. During visit 2, each volunteer received the labelled 33-mer with apple juice, Peptamen, or wheat gluten plus AMYNOPEP. Consistently across the study cohorts, AMYNOPEP was prepared in a 1:1 enzyme ratio (20 mg of each enzyme) dissolved in 40 mL water with 500 mg sodium bicarbonate to neutralize gastric acid.
In cohort 1, volunteers (n=5) received 40 mL water with 500 mg sodium bicarbonate, and 5 min later, 50 mg of comprehensively labelled 33-mer dissolved in 20 mL water and 180 mL apple juice (week 1). Apple juice was treated with 2 g sodium bicarbonate before administration to neutralize fruit acids. In week 2, volunteers first received 40 mg of AMYNOPEP in 40 mL water with 500 mg sodium bicarbonate, and 5 min later, received the same treatment conditions as in week 1. In cohort 2, volunteers (n=5) followed the same study protocol as in cohort 1, but apple juice was replaced with 100 mL of Peptamen (containing partially hydrolyzed whey protein, 100 kCal, 4 g peptides per 100 mL; caloric content approximately similar to the volume of apple juice in cohort 1) neutralized with 1 g sodium bicarbonate. In cohort 3, volunteers (n=4) received 100 mg of strategically labelled 33-mer together with 5 g of wheat gluten dissolved in 180 mL of water (week 1). Wheat gluten was previously neutralized by 2 g of sodium bicarbonate. In week 2, volunteers received 40 mg of AMYNOPEP, and after 5 min, received the same treatment conditions as in week 1.
Blood samples were collected after 15 min, 30 min, 45 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, and 24 h after treatments, and plasma amino acid levels were monitored at each time point by LC-MS/MS. In cohorts 2 and 3, additional blood samples were collected at 7.5 min and 22.5 min after treatments.
Sample preparation
Plasma samples were thawed on ice, mixed, and 100 µL of each sample was transferred to a 1.5 mL LoBind reaction tube. Plasma samples were precipitated after adding 200 µL of acetonitrile/methanol (10:1), and incubated on ice for 15 min. Samples were centrifuged at 16,000 g for 15 min at 4 °C. 100 µL supernatant was dried under nitrogen at 40 °C. Samples were reconstituted in 100 µL 0.1% formic acid and 5 µL was applied to the LC-MS/MS instrument.
Analysis of LC-MS/MS data
Labelled XP dipeptides and single amino acids (P*, L*, F*) in plasma samples were quantified by LC-MS/MS using Nexera LC40 HPLC system (Shimadzu, Duisburg, Germany) coupled to an API 6500+ ™ tandem mass spectrometer (AB Sciex, Darmstadt, Germany). For chromatographic separation an ACQUITY UPLC HSS PFP Column (100 Å, 1.8 µm, 2.1 mm X 150 mm; Waters, Eschborn, Germany) protected by a SecurityGuard C18 column (4x2 mm; Phenomenex, Aschaffenburg, Germany) was used. Chromatographic separation was performed at an oven temperature of 40°C and the solvent flow rate was set to 400 µL/min. Elution was achieved by varying composition of solvent A (90% acetonitrile + methanol (6 + 1), 0.1% formic acid in H2O) and solvent B (0.1% formic acid in H2O) using following LC conditions: 0-1.4 min 0% solvent A; 1.4-10.4 min 0-50% solvent A; 10.5-12.9 min 80% solvent A; 13-15 min 0% solvent A. To decrease the introduction of debris into the MS, a valve was directed to waste at 0.1-1 min and 9.5-14.9 min.
MS detection of labelled amino acids and XP dipeptides was performed in positive mode with curtain gas set to 40, gas 1 of 50, gas 2 of 70, temperature of 500°C and an IS of 3500. Detection was achieved using MS parameters and mass transitions listed in Supplementary Table 3.
The LC-MS/MS method was verified to be specific for the quantitative determination of P*, F*, L*, F*P*, LP* and L*P* in human plasma and urine. No interferences of the analytical signals with the biological matrix were observed. The calibration curves for labelled amino acids were linear between 0.002 µM and 5 µM for F*, P* and L*. This correlates to 0.24 to 605 ng/ml P*, 0.35 to 875 ng/ml F and 0.28 to 690 ng/ml L*. The calibration curves for F*P*, LP* and L*P* were linear between 0.005 µM and 5 µM. This correlates to 1.39 to 1390 ng/ml for F*P*, 1.17 to 1170 ng/ml for LP* and 1.21 to 1205 ng/ml for L*P*. The correlation coefficients for all analytes ranged between 0.9986 and 1.
Analysis of in vitro samples was performed semi quantitatively. Non-labelled XP dipeptides and single amino acids from in vitro samples were measured applying the method described above using mass transitions and MS parameters listed in Supplementary Table 1. To estimate total degradation, measurement of carboxy-terminal F that is only cleaved in the last step of 33-mer digestion by exopeptidases, was performed quantitatively. Detection of 33-mer and its degradation intermediates (28-mer, 16-mer, 9-mer, 7-mer and 5-mer; see Supplementary Table 2) was achieved using an ACQUITY UPLC BEH C18 Column (130Å, 1.7 µm, 2.1 mm X 50 mm (Waters) protected by a SecurityGuard C18 column (4x2 mm; Phenomenex, Aschaffenburg, Germany) for chromatographic separation. Chromatographic separation was performed at an oven temperature of 40°C and the solvent flow rate was set to 400 µL/min. Elution was achieved using following LC conditions: 0-4 min 0% solvent A; 4-6 min 0-15% solvent A; 6-20 min 15-30% solvent A; 20-26 min 30-40% solvent A, 26.1-28 min 60% solvent A, 28.1-31.2 min 0% solvent A. MS detection was performed in positive mode with curtain gas set to 40, gas 1 of 50, gas 2 of 50, temperature of 450°C and IS of 5500.
Pharmacokinetics analyses
LC-MS/MS-generated chromatograms were analyzed using Analyst 1.7.2 (Sciex, Darmstadt, Germany). The pharmacokinetics parameter AUC was calculated using GraphPad Prism version 8.0 (GraphPad Prism Software Inc., La Jolla, CA, USA). Partial AUCs were calculated manually using the trapezoidal rule.
Statistical analyses
Statistical analyses were performed using SPSS Statistics Version 28 (SPSS Inc., IBM, Chicago, IL, USA). To compare the effects between cohorts in the dipeptide resorption study the one-tailed Mann-Whitney-Test was applied. To compare the effects between enzyme supplementation and controls during the proof-of-concept study the one tailed Wilcoxon matched-pairs signed rank test was applied.
Data availability statement
The data generated during this study are available upon request from the corresponding authors. Access to the data will be considered on a case-by-case basis, and any requests will be evaluated in accordance with ethical guidelines and data sharing policies. Please contact the corresponding authors for further information.
Ethics statement
The studies involving humans were approved by Ethics Committee of the University Medicine Greifswald. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SM: Conceptualization, Project administration, Writing – original draft, Writing – review & editing. SR: Methodology, Investigation, Formal analysis, Visualization, Writing – review & editing. ERB: Writing – original draft, Writing – review & editing, Visualization. FW: Writing – review & editing. JT: Funding acquisition, Project administration, Writing – review & editing. MVT: Conceptualization, Supervision, Formal analysis, Writing – review & editing. WW: Conceptualization, Supervision, Writing – review & editing. SE: Conceptualization, Supervision, Investigation, Writing – review & editing. WT: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was sponsored by AMYRA Biotech AG.
Acknowledgments
The authors would like to thank all volunteers for their participation in the clinical study. The authors would also like to thank Intavis Peptide Services GmbH for synthesis of the stable isotope-labelled peptides used in the present study.
Conflict of interest
Authors SM and JT were employed by AMYRA Biotech AG. EB has a consulting agreement with AMYRA Biotech AG. SR, MT, WW, and SE have contract research agreements with AMYRA Biotech AG. WT is co-founder of and shareholder in AMYRA Biotech AG.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The authors declare that this study received funding from AMYRA Biotech AG. The funder(s) had the following involvement in the study: study design, decision to publish, and preparation of the manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1425982/full#supplementary-material
References
1. Mustalahti K, Catassi C, Reunanen A, Fabiani E, Heier M, McMillan S, et al. The prevalence of celiac disease in Europe: results of a centralized, international mass screening project. Ann Med. (2010) 42:587–95. doi: 10.3109/07853890.2010.505931
2. Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global prevalence of celiac disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2018) 16:823–836.e822. doi: 10.1016/j.cgh.2017.06.037
3. King JA, Jeong J, Underwood FE, Quan J, Panaccione N, Windsor JW, et al. Incidence of celiac disease is increasing over time: A systematic review and meta-analysis. Am J Gastroenterol. (2020) 115:507–25. doi: 10.14309/ajg.0000000000000523
4. Sollid LM, Tye-Din JA, Qiao SW, Anderson RP, Gianfrani C, Koning F. Update 2020: nomenclature and listing of celiac disease-relevant gluten epitopes recognized by CD4. Immunogenetics. (2020) 72:85–8. doi: 10.1007/s00251-019-01141-w
5. Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science. (2002) 297:2275–9. doi: 10.1126/science.1074129
6. Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology. (2003) 125:696–707. doi: 10.1016/s0016-5085(03)01049-7
7. Amundarain MJ, Herrera MG, Zamarreño F, Viso JF, Costabel MD, Dodero VI. Molecular mechanisms of 33-mer gliadin peptide oligomerisation. Phys Chem Chem Phys. (2019) 21:22539–52. doi: 10.1039/c9cp02338k
8. Herrera MG, Dodero VI. Gliadin proteolytical resistant peptides: the interplay between structure and self-assembly in gluten-related disorders. Biophys Rev. (2021) 13:1147–54. doi: 10.1007/s12551-021-00856-z
9. Anderson RP. Review article: Diagnosis of coeliac disease: a perspective on current and future approaches. Aliment Pharmacol Ther. (2022) 56 Suppl:1, S18–S37. doi: 10.1111/apt.16840
10. Anderson RP. Innate and adaptive immunity in celiac disease. Curr Opin Gastroenterol. (2020) 36:470–8. doi: 10.1097/MOG.0000000000000672
11. Daveson AJM, Popp A, Taavela J, Goldstein KE, Isola J, Truitt KE, et al. Baseline quantitative histology in therapeutics trials reveals villus atrophy in most patients with coeliac disease who appear well-controlled on gluten-free diet. GastroHep. (2020) 2:22–30. doi: 10.1002/ygh2.380
12. Adelman DC, Murray J, Wu TT, Mäki M, Green PH, Kelly CP. Measuring change in small intestinal histology in patients with celiac disease. Am J Gastroenterol. (2018) 113(3):339–47. doi: 10.1038/ajg.2017.480
13. Hollon JR, Cureton PA, Martin ML, Puppa EL, Fasano A. Trace gluten contamination may play a role in mucosal and clinical recovery in a subgroup of diet-adherent non-responsive celiac disease patients. BMC Gastroenterol. (2013) 13:40. doi: 10.1186/1471-230X-13-40
14. Hooton D, Lentle R, Monro J, Wickham M, Simpson R. The secretion and action of brush border enzymes in the mammalian small intestine. Rev Physiol Biochem Pharmacol. (2015) 168:59–118. doi: 10.1007/112_2015_24
15. McConnell RE, Benesh AE, Mao S, Tabb DL, Tyska MJ. Proteomic analysis of the enterocyte brush border. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G914–926. doi: 10.1152/ajpgi.00005.2011
16. Smith MW, Phillips AD. Abnormal expression of dipeptidylpeptidase IV activity in enterocyte brush-border membranes of children suffering from coeliac disease. Exp Physiol. (1990) 75:613–6. doi: 10.1113/expphysiol.1990.sp003439
17. Detel D, Persić M, Varljen J. Serum and intestinal dipeptidyl peptidase IV (DPP IV/CD26) activity in children with celiac disease. J Pediatr Gastroenterol Nutr. (2007) 45:65–70. doi: 10.1097/MPG.0b013e318054b085
18. Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G996–G1003. doi: 10.1152/ajpgi.00136.2002
19. Mamone G, Picariello G. Optimized extraction and large-scale proteomics of pig jejunum brush border membranes for use in in vitro digestion models. Food Res Int. (2023) 164:112326. doi: 10.1016/j.foodres.2022.112326
20. Verhoeckx K, Bøgh KL, Dupont D, Egger L, Gadermaier G, Larre C, et al. The relevance of a digestibility evaluation in the allergenicity risk assessment of novel proteins. Opinion of a joint initiative of COST action ImpARAS and COST action INFOGEST. Food Chem Toxicol. (2019) 129:405–23. doi: 10.1016/j.fct.2019.04.052
21. Efsa Panel on Food Contact Materials, Enzymes and Processing Aids (CEP), Lambré C, Baviera JMB, Bolognesi C, Cocconcelli PS, Crebelli R, et al. Safety evaluation of the food enzyme leucyl aminopeptidase from non-genetically modified Aspergillus oryzae strain NZYM-EX. EFSA J. (2023) 21:e8507. doi: 10.2903/j.efsa.2023.8507
22. Goel G, Tye-Din JA, Qiao SW, Russell AK, Mayassi T, Ciszewski C, et al. Cytokine release and gastrointestinal symptoms after gluten challenge in celiac disease. Sci Adv. (2019) 5:eaaw7756. doi: 10.1126/sciadv.aaw7756
23. Koziolek M, Grimm M, Schneider F, Jedamzik P, Sager M, Kuhn P, et al. Navigating the human gastrointestinal tract for oral drug delivery: Uncharted waters and new frontiers. Adv Drug Delivery Rev. (2016) 101:75–88. doi: 10.1016/j.addr.2016.03.009
24. Schulze K. Imaging and modelling of digestion in the stomach and the duodenum. Neurogastroenterol Motil. (2006) 18:172–83. doi: 10.1111/j.1365-2982.2006.00759.x
Keywords: gluten, gluten immunogenic peptide, celiac disease, exopeptidase, glutenase, 33-mer, enzyme therapy, enzyme supplementation
Citation: Mourabit S, Römer S, Bonner ER, Winter F, Tschollar J, Tzvetkov MV, Weitschies W, Engeli S and Tschollar W (2024) Exopeptidase combination enhances the degradation of isotopically labelled gluten immunogenic peptides in humans. Front. Immunol. 15:1425982. doi: 10.3389/fimmu.2024.1425982
Received: 30 April 2024; Accepted: 16 September 2024;
Published: 16 October 2024.
Edited by:
Simone Baldi, Department of Experimental and Clinical Medicine, ItalyReviewed by:
Maria Georgina Herrera, Ruhr University Bochum, GermanyDiego S. Vazquez, National Scientific and Technical Research Council (CONICET), Argentina
Copyright © 2024 Mourabit, Römer, Bonner, Winter, Tschollar, Tzvetkov, Weitschies, Engeli and Tschollar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Werner Tschollar, V2VybmVyLlRzY2hvbGxhckBhbXlyYS5jb20=; Stefan Engeli, U3RlZmFuLkVuZ2VsaUBtZWQudW5pLWdyZWlmc3dhbGQuZGU=
†These authors share first authorship
‡These authors share last authorship