
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 31 May 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1425282
This article is part of the Research TopicMechanisms and Challenges in Cancer Immunotherapy Resistance: Bridging Extrinsic and Intrinsic FactorsView all 3 articles
 Leire Arrizabalaga1,2
Leire Arrizabalaga1,2 Aline Risson1,2
Aline Risson1,2 Miriam Ezcurra-Hualde1,2
Miriam Ezcurra-Hualde1,2 Fernando Aranda1,2*
Fernando Aranda1,2* Pedro Berraondo1,2,3*
Pedro Berraondo1,2,3*Interleukin 33 (IL-33), once predominantly recognized for its pro-tumoral activities, has emerged as a multifunctional cytokine with antitumor properties. IL-33 pleiotropic activities include activation of Th1 CD4+ T cells, CD8+ T cells, NK cells, dendritic cells, eosinophils, as well as type 2 innate lymphoid cells. Regarding this immunomodulatory activity, IL-33 demonstrates synergistic interactions with various cancer therapies, including immune checkpoint blockade and chemotherapy. Combinatorial treatments leveraging IL-33 exhibit enhanced antitumor efficacy across different tumor models, promising novel avenues for cancer therapy. Despite its antitumor effects, the complex interplay of IL-33 within the tumor microenvironment underscores the need for further investigation. Understanding the mechanisms underlying IL-33’s dual role as both a promoter and inhibitor of tumor progression is essential for refining therapeutic strategies and fully realizing its potential in cancer immunotherapy. This review delves into the intricate landscape of IL-33 effects within the tumor microenvironment, highlighting its pivotal role in orchestrating immune responses against cancer.
Interleukin 33 (IL-33), initially identified as a nuclear factor in high endothelial venules (NF-HEV) (1), is structurally related to the cytokines of the IL-1 family and it is expressed by fibroblasts, endothelial and epithelial cells, among other cell types (2). The full-length human IL-33 precursor encompasses 270 amino acids, featuring an N-terminal region that includes a chromatin binding motif and a nuclear localization signal (amino acids 1–65), a sensor domain from amino acid 66 to 111, and a C-terminal IL-1-like cytokine domain (amino acids 112–270) that gives IL-33 its cytokine-like properties (3). The N-terminal region is formed by a homeodomain-like helix–turn–helix motif of 65 amino acids. The nuclear localization signal is located within amino acids 46–67 (4). The binding capacity to DNA determines the activities of full-length IL-33 in the nucleus as a transcription factor (5). Human and mouse IL-33 share 52% similarity at the amino acid level. Recombinant human IL-33 is as effective as recombinant mouse IL-33 in activating mouse lymphoid cells (6).
The IL-33 gene, situated on chromosome 9 (9p24.1) in humans and in mice, located at 19qC1 on the same chromosome (3), encodes the precursor protein for IL-33, known as full-length IL-33. Transcriptional regulation of the IL-33 gene is subject to various factors, including inflammatory stimuli, tissue damage, and cellular stress. Notably, epigenetic modifications, such as DNA methylation and histone acetylation, play crucial roles in modulating IL-33 expression, thereby influencing its physiological functions and pathological implications. Additionally, transcription factors, such as NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and AP-1 (activator protein 1), are implicated in the regulation of IL-33 gene expression, orchestrating its responsiveness to diverse extracellular cues and intracellular signaling cascades. In the steady state, full-length IL-33 is constitutively expressed, and upon cellular damage or necrosis, the expression of IL-33 is upregulated, and the full-length IL-33 is processed to mature IL-33 (7). Additionally, immune homeostasis depends on nuclear localization and sequestration of the full-length IL-33, which control IL-33’s release and pro-inflammatory effects (Figure 1) (3).
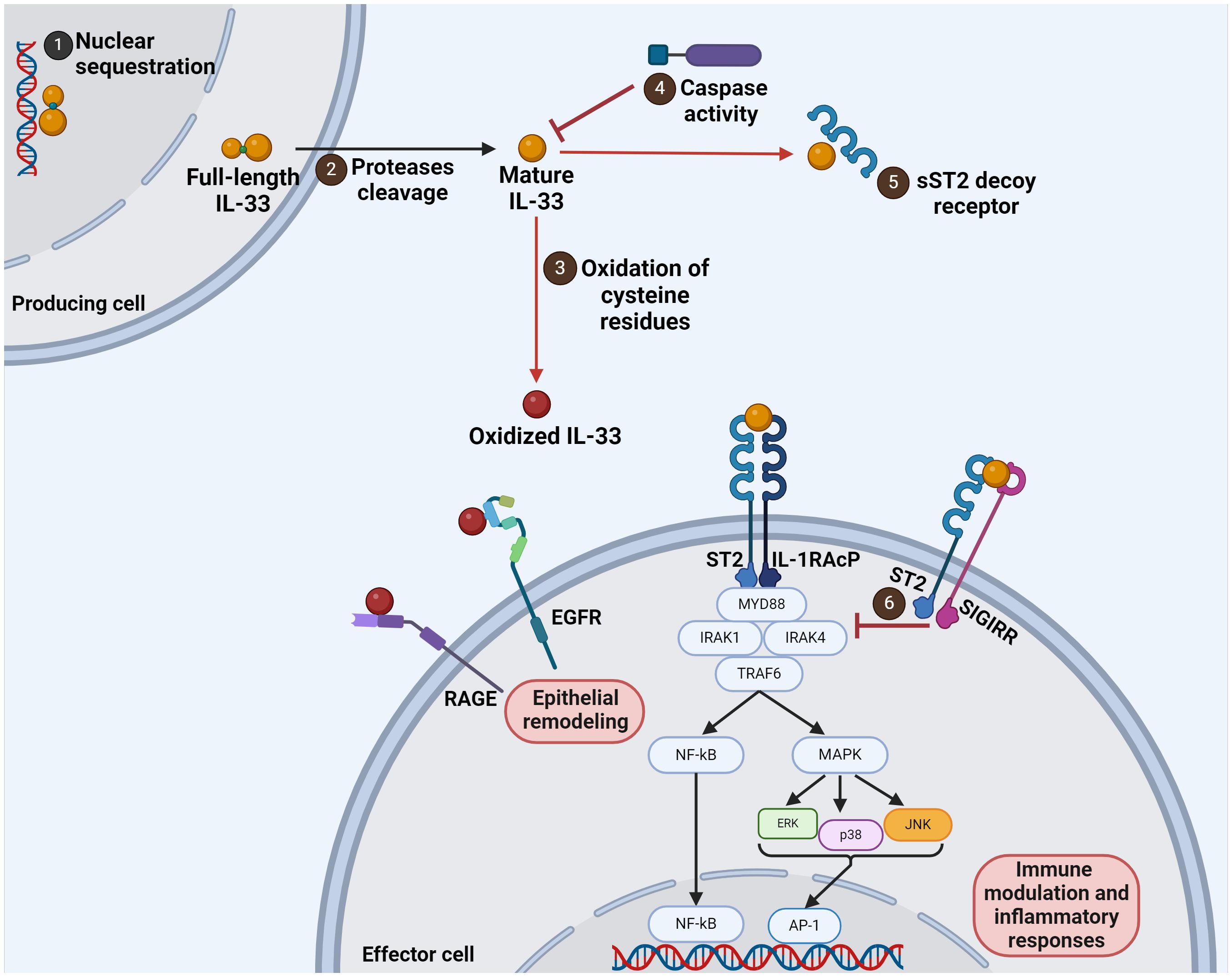
Figure 1 Interleukin 33 regulatory mechanisms and signaling pathways. The full-length IL-33 molecule of 33 kDa can be found in the nucleus and retained there (1). When cellular injury occurs, IL-33 is released and processed to a mature form by protease cleavage (2). The mature form of IL-33 has a high affinity to the ST2 receptor. This mature IL-33 can be modified by oxidation, which generates an oxidized form that mediates epithelial remodeling when recognized by RAGE and EGFR (3). However, the mature IL-33 can also be inactivated by caspases (4), a decoy receptor formed by a soluble form of ST2 (5), or by SIGIRR binding (6).
After cellular injury, the 33 kDa full-length IL-33 can be cleaved in the sensor domain by several endogenous or exogenous proteases (elastase, cathepsin G, chymase, tryptase, granzyme B) (8) to generate a 19 kDa version of the protein defined as the mature form (IL-3395-270, IL-3399-270, and IL-33109-270), with 10 to 30 times higher biological activity (9). This form of IL-33 elicits immune responses via its interactions with specific receptors, acting as an alarmin (10).
IL-33 exerts mainly its pro-inflammatory effects by engaging with specific cell surface receptors. The main receptor is composed of the ST2 (suppression of tumorigenicity 2) receptor (also known as IL-1 receptor-like 1 IL-1RL1, T1, DER4, or Fit-1) in association with the IL-1 receptor accessory protein (IL-1RAcP). The ST2 receptor exists in two isoforms: a membrane-bound form (ST2L) and a soluble form (sST2). While ST2L mediates the pro-inflammatory signaling cascades triggered by IL-33, sST2 acts as a decoy receptor, antagonizing IL-33-induced responses (11). Moreover, the pro-inflammatory activity of IL-33 is also regulated by oxidation. The four cysteines of the protein sequence can generate disulfide bridges, resulting in an extensive conformational change that disrupts the binding to the receptor ST2. Alternatively, IL-33 can be rendered inactive by proteolytic cleavage by caspases (5). The caspase cleavage site is located at amino acid 178 in the IL-1 cytokine domain and is not present in other members of the IL-1 superfamily (3). The inactivation of the IL-33/ST2 axis can also be mediated by the negative regulator SIGIRR (single immunoglobulin IL-1R-related molecule). It has been suggested that SIGIRR binds to ST2 in a complex formed upon IL-33 activation, restricting the recruitment of the IL-1RAcP adaptor protein (9).
Following ST2 receptor binding, IL-33 initiates intracellular signaling pathways, culminating in the activation of various downstream effectors involved in immune modulation and inflammatory responses (12). The canonical signaling cascade triggered by IL-33 involves the recruitment of MyD88 (myeloid differentiation primary response 88), IRAK (IL-1 receptor-associated kinase) 1 and 4, and TRAF6 (tumor necrosis factor receptor-associated factor 6), leading to the activation of NF-κB and MAPK (mitogen-activated protein kinase) signaling pathways. Subsequently, these signaling events orchestrate the transcriptional regulation of target genes associated with inflammation, tissue repair, and immune cell activation (Figure 1) (13). ST2-independent activities of IL-33 have been described, pointing to an unidentified receptor associating with IL-1RAcP (13, 14). Moreover, the oxidized forms of IL-33 bind to the receptor for advanced glycation end products (RAGE) and epidermal growth factor receptor (EGFR), providing another alternative pathway to ST2. Following ligand binding, RAGE activates downstream signaling pathways involving AP-1, NFAT, NF-κB, STAT3, and CREB transcription factors. Oxidized IL-33 leads to phosphorylation of signaling molecules (JNK, ERK1/2, and STAT5) downstream of EGFR, inducing a reduction in epithelial defense functions (15, 16). Tozorakimab (MEDI3506), a strong human anti-IL-33 monoclonal antibody, has recently been shown to prevent IL-33 from oxidizing and from acting through the RAGE/EGFR signaling pathway, causing a reduction in inflammation and epithelial dysfunction (17).
Despite the well-established pro-tumor activity of IL-33, this cytokine has been shown to potentiate Th1-mediated immune responses, the most relevant in tackling cancer. IL-33 synergizes with interleukin 12 (IL-12) to promote Th1 CD4+ T cells and the production of IFN-γ by CD8+ T lymphocytes and natural killer (NK) cells (18, 19). The ST2 receptor is not expressed in NK cells at baseline but is upregulated upon exposure to IL-12 or TNF-α (19, 20). In CD8+ T lymphocytes, the expression of ST2 is dependent on the transcription factor T-bet, expressed in effector CD8+ T cells (18). This capacity of IL-33 plays a pivotal role in shaping the tumor microenvironment (TME) and influencing tumor growth. Overexpression of IL-33 in B16 melanoma cells or 4T1 breast cancer cells highlighted the effect of IL-33 as a potent inhibitor of tumor progression, exerting its effects through the modulation of CD8+ T and NK cells. Notably, IL-33 not only promotes the functionality of CD8+ T cells but also induces robust type 1 antitumor immune responses. This induction leads to an upsurge in the production of critical type 1 effector molecules, including IFN-γ and granzymes, by both CD8+ T and NK cells within TME (Figure 2). As tumors grow, they trigger inflammatory responses and express tumor antigens, setting the stage for the dynamic interplay between the immune system and malignancy. Tumor progression, however, faces resistance from type 1 lymphocytes (Th1 CD4+ T cells, CD8+ T cells, NK cells, and γδ T cells) and collaborating innate cells (dendritic cells, M1 macrophages, and N1 neutrophils). Despite these defensive mechanisms, tumors manage to advance by recruiting immune suppressive cell types like T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSC) (21).
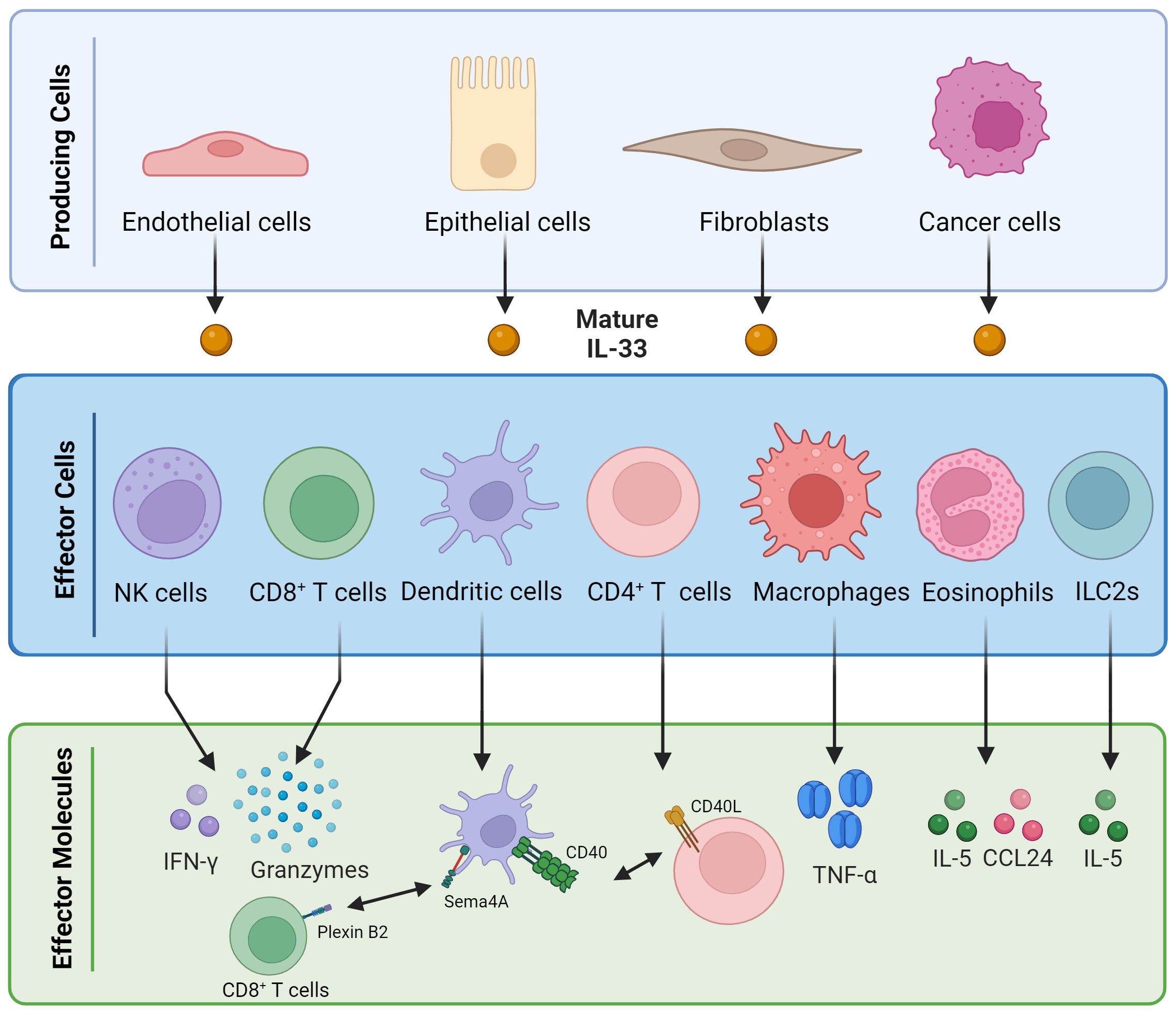
Figure 2 Pleiotropic immune activities of interleukin 33. Mature IL-33 can be released by various types of cells, including endothelial cells, epithelial cells, fibroblasts, and cancer cells. Once released, it can influence the activity of different types of immune cells, such as NK cells, CD8+ T cells, dendritic cells, CD4+ T cells, macrophages, eosinophils, and innate lymphoid cells, inducing the release of several soluble factors.
In addition to directly affecting T lymphocytes and NK cells, IL-33 orchestrates the antitumor immune response acting on dendritic cells. Dendritic cells (DCs) are critical for initiating effective cancer immune responses. In these cells, IL-33 induces the expression of semaphorin 4A (Sema4A) that regulates CD8+ T cell activation through the receptor plexin B2 (22, 23). Interestingly, the antitumor activity of recombinant IL-33 (rIL-33) in the Lewis lung carcinoma (LLC) model was totally abrogated in the Sema4A knock-out (KO) mouse, indicating the relevance of the activation of DCs in the IL-33-mediated antitumor activity (22). An alternative mechanism for activating DCs is the upregulation of CD40L on CD4+ T lymphocytes. CD40L binds to CD40 expressed on DCs, triggering the maturation and activation of these cells. Activated DCs exhibit enhanced antigen-presenting capabilities, upregulating the expression of co-stimulatory molecules and cytokines. This, in turn, facilitates the activation and differentiation of T cells. Interestingly, it has been demonstrated that IL-33 is a potent inductor of CD40L on CD4+ T lymphocytes (24).
By using ST2 KO mice, several groups have demonstrated the essential role of this receptor for the antitumor activity of IL-33 in different tumor models (20, 24, 25). Another key mediator of the IL-33 activity located downstream from this receptor is MyD88. This is a common adaptor utilized by many Toll-like receptors (TLRs) and interleukin 1 receptors (IL-1Rs), including the IL-33 receptor complex. Upon binding of IL-33 to ST2, MyD88 is recruited to the cytoplasmic domain of ST2. MyD88 acts as a bridge, connecting the receptor complex to downstream signaling components. Interestingly, the profound impact of rIL-33 on tumor immunity in the LLC lung cancer model was not replicated in MyD88-/- mice, underscoring the crucial role of the MyD88 pathway in mediating IL-33’s immunomodulatory effects. Further insight into the mechanism revealed that IL-33 required the MyD88 pathway to promote the activation of CD8+ T, NK cells, and DCs (26).
In certain tumor indications, other immune cell populations have been described as important mediators for the antitumor effect of IL-33. In the case of pulmonary metastasis of 4T1 breast cancer or B16 melanoma tumors, macrophages are the main mediator of the antitumor activity of rIL-33. These cells produce large amounts of TNF-α upon IL-33 inoculation. TNF-α induces the expression of ST2 in NK cells, sensitizing this effector immune effect to the activity of rIL-33 (20). In a pancreatic ductal adenocarcinoma (PDA) mouse model, Dixit A. et al. demonstrated that blocking the infiltration of bone marrow-derived macrophages through various CCR2 inhibition strategies significantly reduces TNF-α levels and elevates IL-33 levels. Interestingly, the earlier discovery that PDA patients with higher IL-33 levels have longer survival is supported by the increased presence of CD103+ dendritic cells and CD8+ cytotoxic T cells. This increase is attributed to the elevated IL-33 resulting from the reduction of immunosuppressive myeloid cells and TNF-α (27).
Another important immune cell population regarding IL-33 antitumor activity is eosinophils. In CT26 colon cancer cell engraftment models, IL-33 treatment demonstrated a notable reduction in tumor growth, a phenomenon intricately linked to eosinophils. This dependency was elucidated as eosinophil-deficient mice exhibited an absence of the growth reduction effect, which was subsequently restored upon the adoptive transfer of ex vivo-activated eosinophils. Mechanistically, IL-33 emerged as an eosinophil regulator, amplifying the expression of activation and homing markers (CD11b and Siglec-F) and degranulation markers (CD63 and CD107a). Moreover, IL-33 not only enhanced the viability, cytotoxic potential, and migration properties of eosinophils but also contributed to the heightened levels of IL-5 and CCL24 in tumors (Figure 2) (28). Blomberg et al. described that the patients with metastatic triple-negative breast cancer who responded to immune checkpoint blockade (ICB) therapy had increased eosinophils levels, and the accumulation of these cells was induced by CD4+ T cells, IL-5, and IL-33 (29).
Eosinophils and macrophages also participate in a multipronged immune response orchestrated by IL-33 to eradicate tumors in models of peritoneal carcinomatosis. Thus, depletion of CD4+ T cells, eosinophils, or macrophages significantly diminishes the benefits conferred by IL-33, highlighting the intricate interplay between these immune components and the cytokine’s protective effects. Surprisingly, the absence of B cells during IL-33 treatment takes an unexpected turn, as it leads to an increase in overall survival rates (30). Another report supported the relevant role of IL-33 in shaping an antitumor response in the peritoneal compartment. Overexpression of IL-33 emerged as a pivotal factor in prolonging survival and favoring the elimination of tumors in the peritoneum, but not when the ID8 ovarian cancer cell line was implanted subcutaneously. A crucial aspect of IL-33’s impact lies in its ability to inhibit MDSC differentiation, thereby promoting a robust tumor immune response within the ascitic environment (31). It has also been suggested that type 2 innate lymphoid cells (ILC2s) mediate the recruitment of eosinophils through IL-5 production in the case of melanoma models based on intravenous inoculation of B16.F10 cells to generate lung metastases (32) or subcutaneously implanted B16-F10 tumors (33). These cells express the ST2 receptor, and IL-33 is a potent factor that induces their proliferation and activation (34).
The combination of IL-33 and ICB represents a compelling strategy, as evidenced by multiple studies. The induction of IL-33 in tumor cells following treatment with ICB, such as cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1) monoclonal antibodies (mAbs), establishes a significant link between ICB immunotherapy and IL-33 expression within the TME (25).
One key aspect contributing to the effectiveness of ICB immunotherapy is the ST2 signaling in non-tumor cells, particularly CD8+ T cells. IL-33 plays a crucial role by augmenting the accumulation and effector function of tumor-resident CD103+ CD8+ T cells. Additionally, IL-33 contributes to increased numbers of CD103+ DCs in the TME, which are vital for both the antitumor effect of IL-33 and the accumulation of tumor-infiltrating CD8+ T cells (25). In pancreatic cancer, IL-33 has been shown to mediate the activation of ILC2s, which anti-PD-1 mAbs can further expand. These cells emerge as a potential strategy to enhance T cell priming within cancers infiltrated by ILC2s (21).
The key effect of endogenous IL-33 on the antitumor efficacy of ICB can be enhanced by exogenous administration of IL-33. This has been demonstrated through the overexpression of IL-33 by tumor cells that enhanced the antitumor activity of anti-CTLA-4 mAb or anti-PD-1 mAb in a B16 melanoma model (25, 35). Moreover, rIL-33 also enhanced the antitumor activity of anti-CTLA-4 mAb in a model of 4T1.2 lung metastases (16) or of anti-PD-1 mAb in a mouse model of pancreatic adenocarcinoma (36). In addition to this, intraperitoneal administration of IL-33 was combined with an anti-CSF1R or p38 inhibitor to treat peritoneal metastasis of gastric cancer. This promising approach prevents M2 polarization of macrophages by blocking the activation of the p38-GATA3 signaling pathway (37).
IL-33 synergy has also been demonstrated with chemotherapy. In colorectal cancer (CRC), the role of IL-33 emerges as a crucial factor in influencing the efficacy of 5-fluorouracil (5-FU) treatment. The presence of IL-33 not only enhances the sensitivity of CRC cells to 5-FU but also plays a pivotal role in creating an immune-active TME. This occurs through the orchestration of antitumoral T-cell responses, where IL-33 acts as a key mediator in activating T cells (38).
The field of adoptive cell transfer in cancer immunotherapy is witnessing promising advancements, and the ability of IL-33 to generate an antitumor immune response can be used to potentiate tumor-specific T cells. It has been evaluated the antitumor effect of chimeric antigen receptor T cells (CAR T) engineered to express the interleukin 2 (IL-2) superkine “Super2” and IL-33. Super2 is a variant of human IL-2 designed to bind to the intermediate IL-2 receptor complex expressed on naïve T cells and NK cells with 200-fold higher affinity than native IL-2. Super2 and IL-33 armored CAR T cells can reshape the TME, recruiting and activating a broad repertoire of immune cells, and promote antitumor immunity in multiple solid tumor models (39). A similar strategy was pursued by George Koukos and colleagues (40). OT1 ovalbumin-specific T cells were used as tumor-specific T cells, and these cells were engineered to express an IL-2 variant (IL-2v), which does not bind to the high-affinity IL-2 receptor α-chain (CD25), and IL-33. IL-2v was selected to promote CD8+ T-cell stemness, while IL-33 was introduced to potentiate the cross-priming potential of tumor-associated dendritic cells. The combination generated a unique effector state in the transferred T cells characterized by high expression of granzymes and downregulation of multiple transcription factors associated with T-cell dysfunction (40).
IL-33 is a pleiotropic cytokine with dual roles, modulating either pro-tumoral or anti-tumoral immune responses within the tumor microenvironment. Its pro-tumor potential is mediated through various mechanisms, including the accumulation of immune-suppressive cells, increased glucose uptake and glycolysis via the upregulation of the membrane glucose transporter 1 on tumor cells, macrophage polarization, and the release of cytokines that promote tumor invasion and migration, such as metalloproteases and TNF-α (41).
Several reports have highlighted IL-33’s potential as an immune checkpoint inhibitor. This concept was first proposed by Kevin Van der Jeught et al. (42). In a colon cancer model, they demonstrated that the anti-tumor effect of anti-PD-1 mAb was enhanced in ST2 knock-out mice. Moreover, they developed a fusion protein consisting of the extracellular part of ST2 and its coreceptor IL-1RAcP, linked by a flexible linker. This protein, known as IL-33trap, was able to neutralize IL-33 in the tumor microenvironment and efficiently reduced ST2-expressing tumor-associated macrophages, thereby limiting the infiltration of immune-suppressive cells (42). The role of IL-33 as an immune checkpoint was further confirmed by Mariana Z. Jovanovic et al. (43). They demonstrated in mouse models of breast and colon cancer an enhanced antitumor effect of anti-PD-1 mAb in ST2 knock-out mice. Interestingly, this effect was replicated with the dual blockade of the IL-33/ST2 and PD-L1/PD-1 axes using antibodies against IL-33 and PD-1. The dual blockade enhanced anti-tumor immunity and reduced tumor growth by boosting NK cells and increasing intratumoral CD8+ T cell frequencies and function (43).
As seen in this mini-review, numerous studies highlight the promising antitumor properties of IL-33, whether it is naturally released or externally administered. However, it is crucial to recognize that the impact of IL-33 on tumors can be contingent upon the specific tumor microenvironment. In certain scenarios, IL-33 might paradoxically facilitate tumor growth by fostering the expansion of T regulatory cells and myeloid suppressor cells, both known for their pro-tumoral effects on the immune system (43–45). Consequently, the activity of IL-33 could be envisioned as an amplifier of the pre-existing immune response within the tumor microenvironment.
This dual role underscores the complexity of leveraging IL-33 therapeutically, necessitating further investigation. One potential avenue lies in combining IL-33 with other immunotherapies that stimulate an antitumor immune response. Alternatively, engineering IL-33 could enhance its antitumor efficacy while mitigating its pro-tumoral effects. Hence, IL-33 emerges as a compelling target for cancer immunotherapy, awaiting refined therapeutic strategies to fully harness its considerable potential.
LA: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. AR: Writing – original draft, Writing – review & editing. ME-H: Writing – original draft, Writing – review & editing. FA: Writing – original draft, Writing – review & editing. PB: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Supervision, Visualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Instituto de Salud Carlos III (PI20/00203, and PI22/00147), co-financed by European Union and Gobierno de Navarra Proyecto ARNMUNE Ref.: 0011–1411–2023. Work produced with the support of a 2022 Leonardo Grant for Researchers and Cultural Creators (BBVA Foundation). FA receives a Miguel Servet I (CP19/00114) contract from ISCIII (Instituto de Salud Carlos III), cofinanced by FSE (Fondo Social Europeo). LA is the recipient of an FPU grant from The Spanish Ministry of Education and Professional training (FPU21/00042).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
2. Wang Z, Tang N. Unpacking the complexity of nuclear IL-33 (nIL-33): a crucial regulator of transcription and signal transduction. J Cell Commun Signal. (2023) 17:1131–43. doi: 10.1007/s12079-023-00788-1
3. Dwyer GK, D'Cruz LM, Turnquist HR. Emerging functions of IL-33 in homeostasis and immunity. Annu Rev Immunol. (2022) 40:15–43. doi: 10.1146/annurev-immunol-101320-124243
4. Luzina IG, Clerman A, Fishelevich R, Todd NW, Lockatell V, Atamas SP. Identification of the IL-33 protein segment that controls subcellular localization, extracellular secretion, and functional maturation. Cytokine. (2019) 119:1–6. doi: 10.1016/j.cyto.2019.02.015
5. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U.S.A. (2009) 106:9021–6. doi: 10.1073/pnas.0812690106
6. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
7. Guo H, Bossila EA, Ma X, Zhao C, Zhao Y. Dual immune regulatory roles of interleukin-33 in pathological conditions. Cells. (2022) 11:3237. doi: 10.3390/cells11203237
8. Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U.S.A. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
9. Cayrol C. IL-33, an alarmin of the IL-1 family involved in allergic and non allergic inflammation: focus on the mechanisms of regulation of its activity. Cells. (2021) 11:107. doi: 10.3390/cells11010107
10. Dai X, Utsunomiya R, Shiraishi K, Mori H, Muto J, Murakami M, et al. Nuclear IL-33 plays an important role in the suppression of FLG, LOR, keratin 1, and keratin 10 by IL-4 and IL-13 in human keratinocytes. J Invest Dermatol. (2021) 141:2646–2655 e6. doi: 10.1016/j.jid.2021.04.002
11. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. (2007) 282:26369–80. doi: 10.1074/jbc.M704916200
12. Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discovery. (2008) 7:827–40. doi: 10.1038/nrd2660
13. Nishizaki T. IL-33 suppresses GSK-3beta activation through an ST2-independent MyD88/TRAF6/RIP/PI3K/Akt pathway. Heliyon. (2018) 4:e00971. doi: 10.1016/j.heliyon.2018.e00971
14. Luzina IG, Lockatell V, Courneya JP, Mei Z, Fishelevich R, Kopach P, et al. Full-length IL-33 augments pulmonary fibrosis in an ST2- and Th2-independent, non-transcriptomic fashion. Cell Immunol. (2023) 383:104657. doi: 10.1016/j.cellimm.2022.104657
15. Strickson S, Houslay KF, Negri VA, Ohne Y, Ottosson T, Dodd RB, et al. Oxidised IL-33 drives COPD epithelial pathogenesis via ST2-independent RAGE/EGFR signalling complex. Eur Respir J. (2023) 62:2202210. doi: 10.1183/13993003.02210-2022
16. De Martinis M, Ginaldi L, Sirufo MM, Pioggia G, Calapai G, Gangemi S, et al. Alarmins in osteoporosis, RAGE, IL-1, and IL-33 pathways: A literature review. Medicina (Kaunas). (2020) 56:138. doi: 10.3390/medicina56030138
17. England E, Rees DG, Scott IC, Carmen S, Chan DTY, Chaillan Huntington CE, et al. Tozorakimab (MEDI3506): an anti-IL-33 antibody that inhibits IL-33 signalling via ST2 and RAGE/EGFR to reduce inflammation and epithelial dysfunction. Sci Rep. (2023) 13:9825. doi: 10.1038/s41598-023-36642-y
18. Yang Q, Li G, Zhu Y, Liu L, Chen E, Turnquist H, et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur J Immunol. (2011) 41:3351–60. doi: 10.1002/eji.201141629
19. Smithgall MD, Comeau MR, Yoon BRP, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK Cells. Int Immunol. (2008) 20:1019–30. doi: 10.1093/intimm/dxn060
20. Qi L, Zhang Q, Miao Y, Kang W, Tian Z, Xu D, et al. Interleukin-33 activates and recruits natural killer cells to inhibit pulmonary metastatic cancer development. Int J Cancer. (2020) 146:1421–34. doi: 10.1002/ijc.32779
21. Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. (2015) 194:438–45. doi: 10.4049/jimmunol.1401344
22. Suga Y, Nagatomo I, Kinehara Y, Koyama S, Okuzaki D, Osa A, et al. IL-33 induces sema4A expression in dendritic cells and exerts antitumor immunity. J Immunol. (2021) 207:1456–67. doi: 10.4049/jimmunol.2100076
23. Ito D, Kumanogoh A. mTOR complex signaling through the SEMA4A plexin 62 axis is required for optimal activation and differentiation of CD8+T cells. J Immunol. (2016) 195:934–43. doi: 10.4049/jimmunol.1403038
24. Luo P, Deng S, Ye H, Yu X, Deng Q, Zhang Y, et al. The IL-33/ST2 pathway suppresses murine colon cancer growth and metastasis by upregulating CD40 L signaling. BioMed Pharmacother. (2020) 127:110232. doi: 10.1016/j.biopha.2020.110232
25. Chen L, Sun R, Xu J, Zhai W, Zhang D, Yang M, et al. Tumor-derived IL33 promotes tissue-resident CD8(+) T cells and is required for checkpoint blockade tumor immunotherapy. Cancer Immunol Res. (2020) 8:1381–92. doi: 10.1158/2326-6066.CIR-19-1024
26. Xu LS, Zheng Y, Wang JY, Xu YF, Xie YH, Yang ZP. IL33 activates CD8+T and NK cells through MyD88 pathway to suppress the lung cancer cell growth in mice. Biotechnol Lett. (2020) 42:1113–21. doi: 10.1007/s10529-020-02815-2
27. Dixit A, Sarver A, Zettervall J, Huang H, Zheng K, Brekken RA, et al. Targeting TNF-alpha-producing macrophages activates antitumor immunity in pancreatic cancer via IL-33 signaling. JCI Insight. (2022) 7:e153242. doi: 10.1172/jci.insight.153242
28. Kienzl M, Hasenoehrl C, Valadez-Cosmes P, Maitz K, Sarsembayeva A, Sturm E, et al. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology. (2020) 9:1776059. doi: 10.1080/2162402X.2020.1776059
29. Blomberg OS, Spagnuolo L, Garner H, Voorwerk L, Isaeva OI, van Dyk E, et al. IL-5-producing CD4(+) T cells and eosinophils cooperate to enhance response to immune checkpoint blockade in breast cancer. Cancer Cell. (2023) 41:106–123 e10. doi: 10.1016/j.ccell.2022.11.014
30. Perales-Puchalt A, Svoronos N, Villarreal DO, Zankharia U, Reuschel E, Wojtak K, et al. IL-33 delays metastatic peritoneal cancer progression inducing an allergic microenvironment. Oncoimmunology. (2019) 8:e1515058. doi: 10.1080/2162402X.2018.1515058
31. Sekiya A, Suzuki S, Tanaka A, Hattori S, Shimizu Y, Yoshikawa N, et al. Interleukin−33 expression in ovarian cancer and its possible suppression of peritoneal carcinomatosis. Int J Oncol. (2019) 55:755–65. doi: 10.3892/ijo.2019.4845
32. Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol. (2012) 188:703–13. doi: 10.4049/jimmunol.1101270
33. Wagner M, Ealey KN, Tetsu H, Kiniwa T, Motomura Y, Moro K, et al. Tumor-derived lactic acid contributes to the paucity of intratumoral ILC2s. Cell Rep. (2020) 30:2743–2757 e5. doi: 10.1016/j.celrep.2020.01.103
34. Flamar AL, Klose CSN, Moeller JB, Mahlakoiv T, Bessman NJ, Zhang W, et al. Interleukin-33 induces the enzyme tryptophan hydroxylase 1 to promote inflammatory group 2 innate lymphoid cell-mediated immunity. Immunity. (2020) 52:606–619 e6. doi: 10.1016/j.immuni.2020.02.009
35. He H, Shi L, Meng D, Zhou H, Ma J, Wu Y, et al. PD-1 blockade combined with IL-33 enhances the antitumor immune response in a type-1 lymphocyte-mediated manner. Cancer Treat Res Commun. (2021) 28:100379. doi: 10.1016/j.ctarc.2021.100379
36. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
37. Che K, Luo Y, Song X, Yang Z, Wang H, Shi T, et al. Macrophages reprogramming improves immunotherapy of IL-33 in peritoneal metastasis of gastric cancer. EMBO Mol Med. (2024) 16:251–66. doi: 10.1038/s44321-023-00012-y
38. Song M, Yang J, Di M, Hong Y, Pan Q, Du Y, et al. Alarmin IL-33 orchestrates antitumoral T cell responses to enhance sensitivity to 5-fluorouracil in colorectal cancer. Theranostics. (2023) 13:1649–68. doi: 10.7150/thno.80483
39. Brog RA, Ferry SL, Schiebout CT, Messier CM, Cook WJ, Abdullah L, et al. Superkine IL-2 and IL-33 armored CAR T cells reshape the tumor microenvironment and reduce growth of multiple solid tumors. Cancer Immunol Res. (2022) 10:962–77. doi: 10.1158/2326-6066.CIR-21-0536
40. Corria-Osorio J, Carmona SJ, Stefanidis E, Andreatta M, Ortiz-Miranda Y, Muller T, et al. Orthogonal cytokine engineering enables novel synthetic effector states escaping canonical exhaustion in tumor-rejecting CD8(+) T cells. Nat Immunol. (2023) 24:869–83. doi: 10.1038/s41590-023-01477-2
41. Jiang W, Lian J, Yue Y, Zhang Y. IL-33/ST2 as a potential target for tumor immunotherapy. Eur J Immunol. (2021) 51:1943–55. doi: 10.1002/eji.202149175
42. Van der Jeught K, Sun Y, Fang Y, Zhou Z, Jiang H, Yu T, et al. ST2 as checkpoint target for colorectal cancer immunotherapy. JCI Insight. (2020) 5:e136073. doi: 10.1172/jci.insight.136073
43. Jovanovic MZ, Geller DA, Gajovic NM, Jurisevic MM, Arsenijevic NN, Jovanovic MM, et al. Dual blockage of PD-L/PD-1 and IL33/ST2 axes slows tumor growth and improves antitumor immunity by boosting NK cells. Life Sci. (2022) 289:120214. doi: 10.1016/j.lfs.2021.120214
44. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. (2014) 134:1669–82. doi: 10.1002/ijc.28481
Keywords: IL-33, cancer immunotherapy, tumor microenvironment, immune modulation, engineered cytokines
Citation: Arrizabalaga L, Risson A, Ezcurra-Hualde M, Aranda F and Berraondo P (2024) Unveiling the multifaceted antitumor effects of interleukin 33. Front. Immunol. 15:1425282. doi: 10.3389/fimmu.2024.1425282
Received: 29 April 2024; Accepted: 21 May 2024;
Published: 31 May 2024.
Edited by:
Laura Senovilla, Spanish National Research Council (CSIC), SpainReviewed by:
Samantha Sharma, Indiana University School of Medicine, United StatesCopyright © 2024 Arrizabalaga, Risson, Ezcurra-Hualde, Aranda and Berraondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Berraondo, cGJlcnJhb25kb2xAdW5hdi5lcw==; Fernando Aranda, ZmFyYW5kYUB1bmF2LmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.