 Ruochen Bao
Ruochen Bao Hongtao Qu
Hongtao Qu Baifeng Li
Baifeng Li Kai Cheng
Kai Cheng Yandong Miao
Yandong Miao Jiangtao Wang
Jiangtao Wang- 1Thyroid and Breast Surgery, Yantai Affiliated Hospital of Binzhou Medical University, The 2ndMedical College of Binzhou Medical University, Yantai, China
- 2Emergency Department of Yantai Mountain Hospital, Yantai, China
- 3Cancer Center, Yantai Affiliated Hospital of Binzhou Medical University, The 2nd Medical College of Binzhou Medical University, Yantai, China
Triple-negative breast cancer (TNBC) has become a thorny problem in the treatment of breast cancer because of its high invasiveness, metastasis and recurrence. Although immunotherapy has made important progress in TNBC, immune escape caused by many factors, especially metabolic reprogramming, is still the bottleneck of TNBC immunotherapy. Regrettably, the mechanisms responsible for immune escape remain poorly understood. Exploring the mechanism of TNBC immune escape at the metabolic level provides a target and direction for follow-up targeting or immunotherapy. In this review, we focus on the mechanism that TNBC affects immune cells and interstitial cells through hypoxia, glucose metabolism, lipid metabolism and amino acid metabolism, and changes tumor metabolism and tumor microenvironment. This will help to find new targets and strategies for TNBC immunotherapy.
1 Introduction
In 2020, breast cancer had the highest incidence worldwide and was the fifth leading cause of cancer-related deaths, accounting for one-quarter of female cancer cases and one-sixth of cancer deaths (1). Although the 5-year survival rate of patients with breast cancer has reached 80% (2), triple-negative breast cancer (TNBC) has emerged as a significant obstacle to improving the prognosis of patients with breast cancer due to its high invasiveness, propensity for metastasis, and recurrence (3). At present, surgery, radiotherapy and chemotherapy are still the main treatments for TNBC (4), but immunotherapy has shown promising results in the management of TNBC (5, 6), with a response rate of approximately 20% (7). Immune escape caused by many factors has become an important factor in the limited effect of immunotherapy for TNBC. Tumor heterogeneity and tumor metabolism play a pivotal role in this process.
Tumor heterogeneity is a key determinant of treatment efficacy and is associated with poor survival outcomes and prognosis (8, 9).There are various types of tumor heterogeneity. Foremost, heterogeneity may occur between individual tumor cells, known as intratumor heterogeneity. Intratumor heterogeneity can be further categorized into spatial and temporal heterogeneity, as it may be present in distinct regions of the primary tumor and the primary tumor may evolve over time. Secondly, the differences between different metastatic lesions in the same patient are called inter-metastatic heterogeneity. Thirdly, heterogeneity may also exist within a single metastatic cell. Finally, there is also heterogeneity between tumors of different patients, so personalized treatment is needed for each patient (10, 11).Tumor heterogeneity is ubiquitous in all cancers. In general, epigenetic modifications of cell characteristics such as tumor transcriptional changes, gene mutations, and changes in protein levels all show tumor heterogeneity. In addition to these intrinsic factors, other extrinsic factors such as pH, hypoxia, and crosstalk between tumor cells and other stromal cells in the TME can also affect tumor genotype and phenotype, further leading to tumor heterogeneity (12).
Metabolic reprogramming is a hallmark feature of tumors (13, 14), enabling tumor cells to meet the energy demands and biosynthetic requirements necessary for malignant proliferation, maintain redox homeostasis, proliferate rapidly in a nutrient-deficient tumor microenvironment (15), and facilitate the survival and metastasis of cancer cells (16). Alterations in metabolic reprogramming in tumor cells also impact immune cells and other cell types, contributing to tumor initiation and progression. In comparison to other subtypes of breast cancer, TNBC exhibits reduced mitochondrial oxidative phosphorylation, increased glycolytic activity, enhanced fatty acid synthesis, and altered amino acid metabolism (17–20).
Tumor heterogeneity plays an important role in tumor formation and development. Revealing tumor heterogeneity still faces many challenges. How to fully and comprehensively understand the changes in heterogeneity in patients is one of the future directions of tumor research. Due to the rich content of tumor heterogeneity and metabolic reprogramming, this review mainly analyzes the role of metabolic reprogramming in the immune escape of triple-negative breast cancer. This review starts from the mechanism of metabolic reprogramming in TNBC immune escape, and expounds that hypoxia, glucose metabolism, lipid metabolism and amino acid metabolism alter tumor metabolism and the tumor microenvironment by affecting immune cells, interstitial cells and other processes, resulting in immune escape. Indicating the importance of metabolic reprogramming in tumor progression, and then providing help for the discovery of new TNBC immunotherapy targets and strategies.
2 Hypoxia and immune escape in TNBC
Hypoxia is a prominent characteristic of many solid tumors (21), particularly in TNBC (22, 23). Hypoxia causes an increase in hypoxia-inducible factor (HIF), which affects the tumor microenvironment, changes the metabolic state of tumors, and even promotes tumor angiogenesis by promoting the production and aggregation of immunosuppressive cells, inhibiting the function of immune effector cells, activating the signaling axis of stromal cells, and enhancing the relationship between cytokines and immune cells. This leads to the invasion and progression of TNBC (Figure 1).
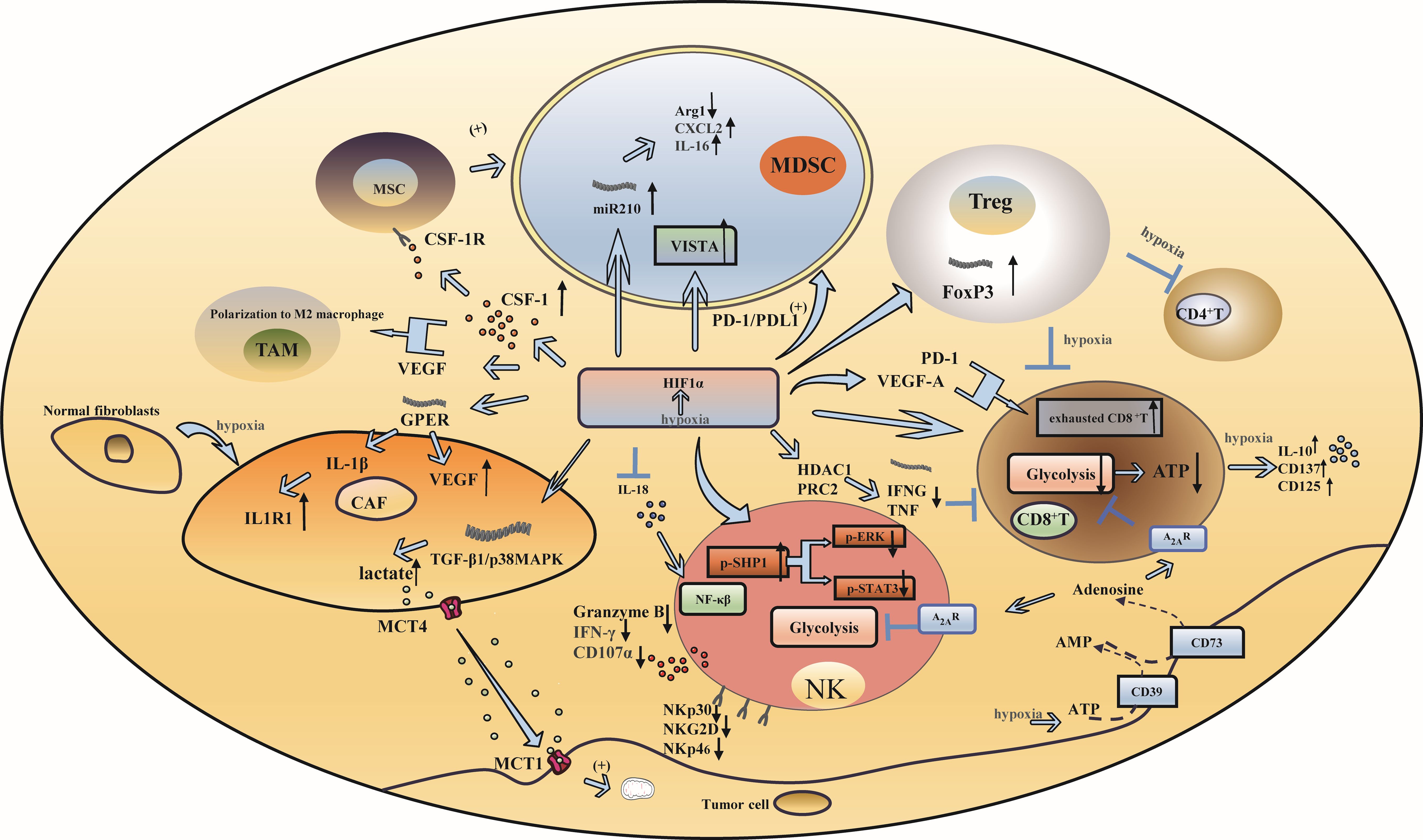
Figure 1. Hypoxia and immune escape in triple-negative breast cancer. (A) Hypoxia induces increased infiltration of immunosuppressive cells: Under hypoxic conditions, HIF-1α influences the function of MDSCs through microRNA-210, PD-L1 and VISTA. Additionally, HIF-1α enhances the generation of Tregs by upregulating FoxP3 transcription. (B) Hypoxia inhibits the activation and function of immune effector cells: The reduced expression of granzyme B, IFN-γ, and the degranulation marker CD107a, as well as the downregulation of activating receptors including NKP30, NKp46, and NKG2D on the surface of NK cells, resulted in a significant decrease in NK cell activity. The upregulation of HIF- α led to a reduction in glycolytic activity in CD8+ T cells. VEGF and PD-1, both regulated by HIF- α, influenced the anti-tumor response of CD8+ T cells. (C) Hypoxia can produce effects that affect mesenchymal cell function: CAF enhance the activity of cancer cell mitochondria by activating the TGF-β1/p38MAPK/MMP2/9 signaling axis to produce lactic acid under hypoxic conditions. (D) Hypoxia changes the relationship between cytokines and immune cells: HIF-1α has the ability to suppress the expression of interleukin-18 (IL-18), whereas hypoxia triggers the release of the immunosuppressive cytokine IL-10 and the upregulation of CD137 and CD25.
2.1 Hypoxia induces increased infiltration of immunosuppressive cells
Myeloid-derived suppressor cells (MDSCs) inhibit immune responses and promote tumor immune escape (24). Under hypoxic conditions, hypoxia-inducible factor 1 α (HIF-1α) can synergize with chemokine signals from mesenchymal stem cells to trigger colony-stimulating factor 1 (CSF-1) and chemokine receptor type 5 (CCR5) gene transcription in breast cancer cells. Chemokine (C-C motif) ligand 5(CCL5) and CCR5 signaling regulates the expression of CSF1 in breast cancer cells, while CSF1 and CSF1 receptor (CSF1R) signaling regulates the expression of CCL5 in mesenchymal stem cells. This signaling cascade ultimately facilitates the recruitment of MDSCs (25).In addition, it was found that HIF-1α directly interacts with the HRE located in the proximal promoter region of programmed death-ligand 1 (PD-L1) and selectively upregulates PD-L1 on MDSCs. Inhibition of PD-L1 can abrogate MDSC-mediated suppression of T cell suppression in part by modulating the production of cytokines interleukin-6 (IL-6) and interleukin-10 (IL-10) in hypoxic MDSCs (26). Concurrently, HIF-1α upregulates the expression of microRNA 210 in MDSCs and then regulates the activity of MDSCs by regulating arginase-1 (ARG1), C-X-C motif chemokine ligand 12 (Cxcl12) and interleukin-16 (IL16), thus enhancing the immunosuppressive function of MDSCs (27). Furthermore, the V-domain Ig suppressor of T-cell activation (VISTA) is a mediator of MDSC function, and HIF-1α binds to the conserved hypoxia response element in the T-cell-activated VISTA promoter to upregulate VISTA in myelocytes. Targeting VISTA with antibodies or gene ablation can mitigate MDSC-mediated T-cell inhibition (28).
The expression of HIF-1α can promote the transcription of forkhead boxP3 (FoxP3), which in turn promotes the production of regulatory T cells (Tregs), thus strengthening their inhibitory function (29–31). Hypoxia can induce Tregs to inhibit the proliferation and differentiation of CD4+ T cells and CD8+ T cells (32, 33). Hypoxia induces a variety of cell types, including Tregs. In addition, hypoxia induces the expression of CD73 in various types of cells, including Tregs, which has a negative effect on T-cell function by participating in the production of adenosine, an immunosuppressive metabolite (34).
Hypoxia leads to the upregulation of HIF-1, which in turn promotes tumor angiogenesis and invasiveness, while tumor-associated macrophages (TAMs) play an important role in promoting tumor angiogenesis; therefore, HIF-1 is considered to be the factor by which TAMs promote angiogenesis (35, 36).
2.2 Hypoxia inhibits the activation and function of immune effector cells
Although natural killer (NK) cells play an important role in tumor immunity, decreased expression of granzyme B, interferon-gamma (IFN-γ), and the degranulation marker CD107a and reduced expression of activated receptors on the surface of NK cells, such as NKP30, NKp46 and NKG2D, can significantly reduce the activity of NK cells. Some studies have indicated that the decreased phosphorylation of extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) induced by hypoxia is closely associated with the attenuation of NK cytotoxicity (37). Hypoxia can also promote the transformation of NK cells into tumor-resistant and immunosuppressive phenotypes. Because of the enrichment of the NF-kB pathway in NK cells, the antitumor activity of NK cells can be enhanced by inhibiting HIF-1α (38).
CD8+ T cells are critical antitumor immune cells. TNBC patients with high levels of CD8+ T cells exhibit improved clinical prognosis and a robust immune response (39). The immune-killing function of CD8+ T cells is inhibited in the hypoxic microenvironment, which is determined by the epigenetic mechanism of immune effector genes mediated by HIF-α (40). Tumor cells with elevated expression of HIF-α can consume large amounts of glucose and decrease the glycolytic activity of CD8+ T cells, resulting in a decrease in adenosine triphosphate (ATP), which in turn affects the function of CD8+ T cells (40, 41). The antitumor activity and infiltration level of CD8+ T cells are affected by vascular endothelial-derived growth factor (VEGF) and programmed cell death protein 1 (PD-1), which are regulated by HIF-α. VEGF-A can affect the transport and killing ability and promote the differentiation of PD-1+ TIM-3+ CXCR5+ terminally exhausted-like CD8+ T cells (42). An in vitro study of TNBC culture systems showed that HIF-1α inhibited the expression of immune effector genes such as interferon gamma (IFNG) and tumor necrosis factor (TNF) through histone modification mediated by histone deacetylase 1 (HDAC1) and polycomb repressive complex 2 (PRC2) during hypoxia and caused CD8+ T cells to dysfunction (43). Moreover, the amount of adenosine produced by tumor cells regulated by HIF-1α increased in the hypoxic microenvironment. Adenosine interacts with adenosine A2A receptors to promote T-cell apoptosis and inhibit proliferation, thereby suppressing antitumor immune function (44).
2.3 Hypoxia can affect mesenchymal cell function
Stromal cells include fibroblasts, astrocytes, adipocytes and other types, among which cancer-associated fibroblasts play an important role in hypoxia. Cancer-associated fibroblasts (CAFs) are highly sensitive to hypoxia, and fibroblast-specific HIF activates and promotes the arrangement and stiffness of the extracellular matrix (ECM), leading to morphological and migratory changes in breast cancer cells. During hypoxia, CAFs produce lactic acid, which is released by monocarboxylate transporter4 (MCT4) by activating the TGF-β1/p38MAPK/MMP2/9 signaling axis, and enters the cell with the help of the cancer cell monocarboxylate transporter 1 (MCT1). This process enhances the activity of cancer cell mitochondria and promotes cancer cell invasion (45). Hypoxia can also induce the transformation of normal fibroblasts into CAFs (46). In breast cancer, HIF-1α and the G protein-coupled estrogen receptor (GPER) signaling pathway stimulate the expression of VEGF in CAFs, while GPER in breast cancer CAFs induces interleukin-1β (IL-1β) to express interleukin-1 receptor 1 (IL1R1) in breast cancer cells (47). Therefore, by investigating the effect of GPER silencing on breast cancer caused by hypoxia, it was found that the expression of connective tissue growth factor (CTGF) could be inhibited by knocking down GPER in CAFs to inhibit the invasion of breast cancer cells induced by hypoxia (48).
2.4 Hypoxia changes the relationship between cytokines and immune cells
HIF-1α inhibits the production of interleukin-18 (IL-18), which is necessary for the activation of NF-κB and enhancement of the antitumor activity of NK cells (38). The oxygen tension during the activation of CD8+ T cells is decreased due to hypoxia, which in turn induces the secretion of the immunosuppressive factor IL-10 and the upregulation of CD137 and CD25. As a result, the phenotype of CD8+ T cells transitions from that of effector cells to that of nonproliferating cells (49). Simultaneously, hypoxia-induced production of VEGF, epithelial growth factor receptor (EGFR), C-C Motif Chemokine Ligand 5 (CCL5), CSF-1, oncostatin M (OSM), eotaxin, succinic acid and granulocyte-macrophage colony-stimulating factor (GM-CSF) can facilitate the recruitment of TAMs and promote their polarization toward the M2 phenotype (50–53).
3 Altered glycolysis and TNBC
In TNBC, tumor proliferation is closely related to glycolysis. When glycolysis is altered, the altered rate of glycolysis and the metabolites of glycolysis lead to changes in the nutrient supply to immune cells as well as to other cells, which can lead to problems such as the immune escape of tumor cells (54, 55) (Figure 2).
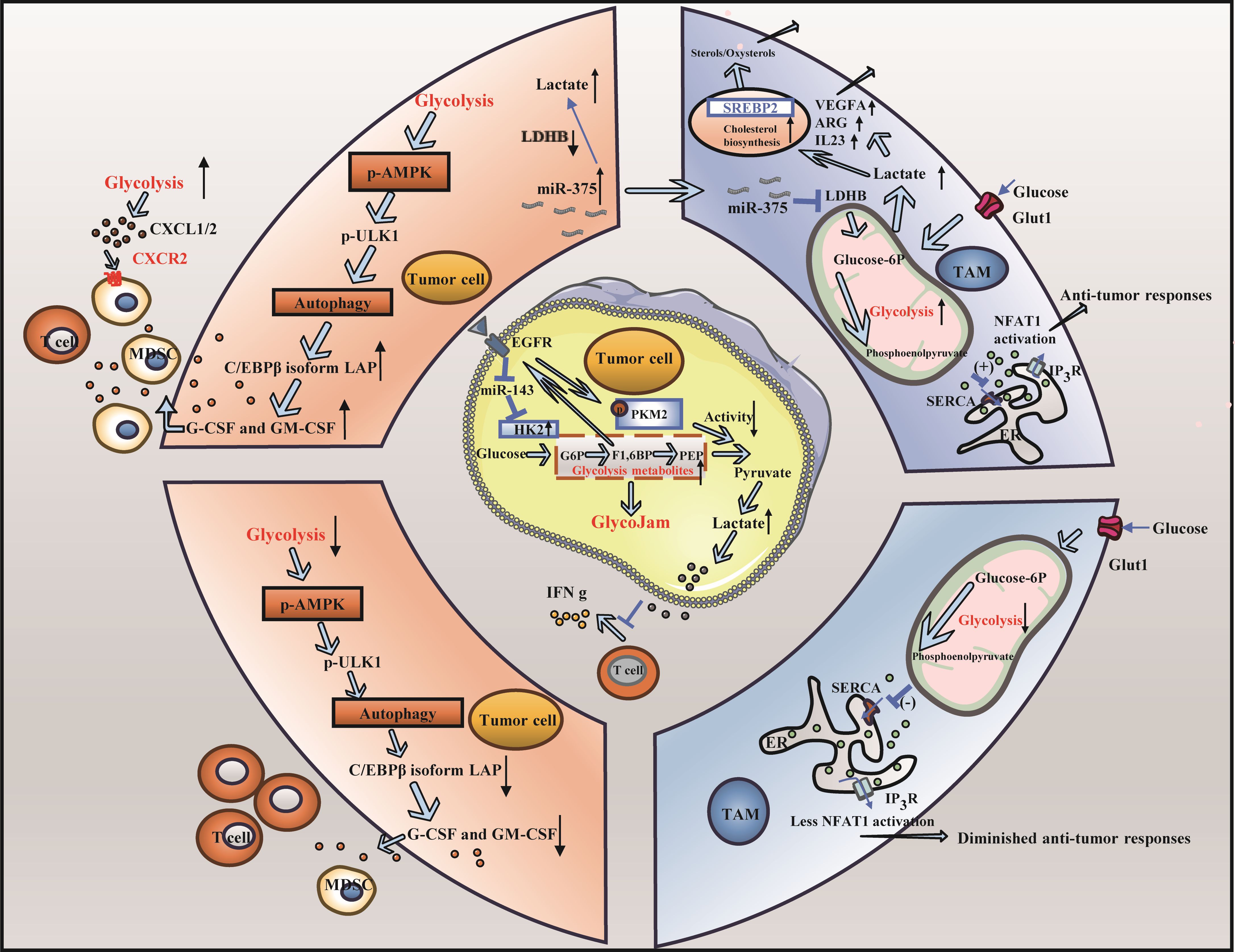
Figure 2. Altered glycolysis and triple-negative breast cancer. (A) The change of glycolysis rate is involved in immune escape of triple negative breast cancer: High rates of tumor cell glycolysis activate LAP leading to the formation of MDSCs and a decrease in the population of effector CD8+ T cells. Furthermore, T cells exhibited decreased glycolytic activity in the glucose-deficient tumor microenvironment, leading to impaired anti-tumor response. (B) Carbohydrate metabolite lactic acid participates in immune escape of triple negative breast cancer cells: Lactic acid produced by cancer cells induces the upregulation of VEGF and ARG1, thereby facilitating the differentiation of TAMs toward the M2 phenotype. Furthermore, the elevated lactic acid levels within TAMs result in reduced fatty acid levels, heightened cholesterol synthesis, and enhanced cancer cell proliferation.
3.1 Changes in the glycolysis rate are involved in immune escape in TNBC
The modulation of the glycolysis rate can facilitate tumor immune escape. High rates of tumor cell glycolysis can activate the autophagy signaling pathway and AMP-activated protein kinase ULK1 (AMPK-ULK1), resulting in inhibition of enhancer-binding protein beta (CEBPB) subtypes and activation of liver-enriched activator protein (LAP), thus controlling the expression of GM-CSF and granulocyte colony-stimulating factor (G-CSF), supporting the formation of MDSCs, reducing the number of effector CD8+ T cells, weakening antitumor immunity, and hastening the progression of TNBC. Conversely, when glycolysis is limited, the expression of G-CSF and GM-CSF is inhibited, which affects the formation of MDSCs and increases the number of CD8+ T cells (56). In addition, during high rates of tumor cell glycolysis, high glucose and high oxygen consumption cause a glucose-deficient tumor microenvironment, which decreases the glycolytic ability of T cells, reduces the production of the metabolite phosphoenolpyruvate (PEP), and weakens the ability to inhibit the activity of sarco/ERCa2 +-ATPase (SERCA), resulting in a decrease in the ability of SERCA to maintain T-cell receptor-mediated Ca-NFAT signal transduction and effector function, thereby attenuating the antitumor response of T cells. Conversely, if tumor cells do not consume glucose excessively, the glycolytic capacity of T cells remains intact, and PEP, a glycolytic metabolite, ensures the antitumor function of T cells by inhibiting SERCA activity (57). Some studies have demonstrated that by inducing the Warburg effect in the tumor microenvironment (TME), activated C-X-C motif chemokine ligand 1/2 (CXCL1/2) binds to C-X-C motif chemokine receptor 2 (CXCR2), recruiting MDSCs and other cells, thereby fostering tumor growth and immune escape (58). Notably, during tumor glycolysis, lactate dehydrogenase indirectly modulates the activity of CD4+ T cells via the PD-1 pathway, depriving these cells of sufficient energy and consequently inhibiting their function (59).
3.2 Carbohydrate metabolite lactic acid participates in the immune escape of TNBC cells
The increase in glycolysis in TNBC cells results in the production of a large amount of lactic acid, which accumulates in the TME and forms an acidic environment (60). The function of immune cells, especially immunosuppressive TAMs, is inhibited by high levels of lactic acid and an acid-stimulated TME in tumors, which blocks the immune surveillance of tumors, resulting in immune escape. TAMs can differentiate and proliferate into a pretumor phenotype in this acidic environment, facilitating tumor immune escape (61). First, lactic acid from cancer cells can stimulate the expression of VEGF and ARG1 and promote the polarization of TAMs to the M2 phenotype through histone lactoacylation, leading to tumor cell proliferation (62–64). In addition, a large amount of lactic acid in TAMs is induced by downregulating lactate dehydrogenase B (LDHB), resulting in a decrease in fatty acid synthesis, which activates sterol regulatory element binding transcription factor 2 (SREBP2), increases cholesterol synthesis in TAMs, and increases cholesterol levels that promote cancer cell proliferation (44). Lactic acid can also activate the lactate receptor GPR81, which is related to tumor growth in dendritic cells, thereby eliminating antigen presentation, reducing the secretion of the proinflammatory cytokines IL-6 and IL-12, and inhibiting T-cell function (65). Some studies have shown that in TNBC, pyruvate kinase isozyme type M2 (PKM2) is suppressed through the EGFR signal transduction axis, and glucose phosphorylation is catalyzed by upregulating hexokinase 2 (HK2) to form a “glycolytic jam”, thus reducing the expression of INF-γ and IL-2, affecting T-cell function and contributing to tumor growth and immune escape (66).
Notably, as an intermediate of glycolysis, glucose-6-phosphate affects the antitumor immune response of immune cells through the pentose phosphate pathway (PPP) (67). This process interferes with oxidative phosphorylation (OXPHOS) and the PPP of immune cells, ultimately suppressing immune function (68, 69).
4 Lipid metabolism and TNBC
During the occurrence and development of breast cancer, fatty acids can be absorbed by tumor cells, which in turn affects the absorption of fatty acids by immune cells. The activation, infiltration and effector function of immune cells are also disrupted by disordered lipid metabolism (70, 71), which in turn leads to immune escape (72–74) (Figure 3).
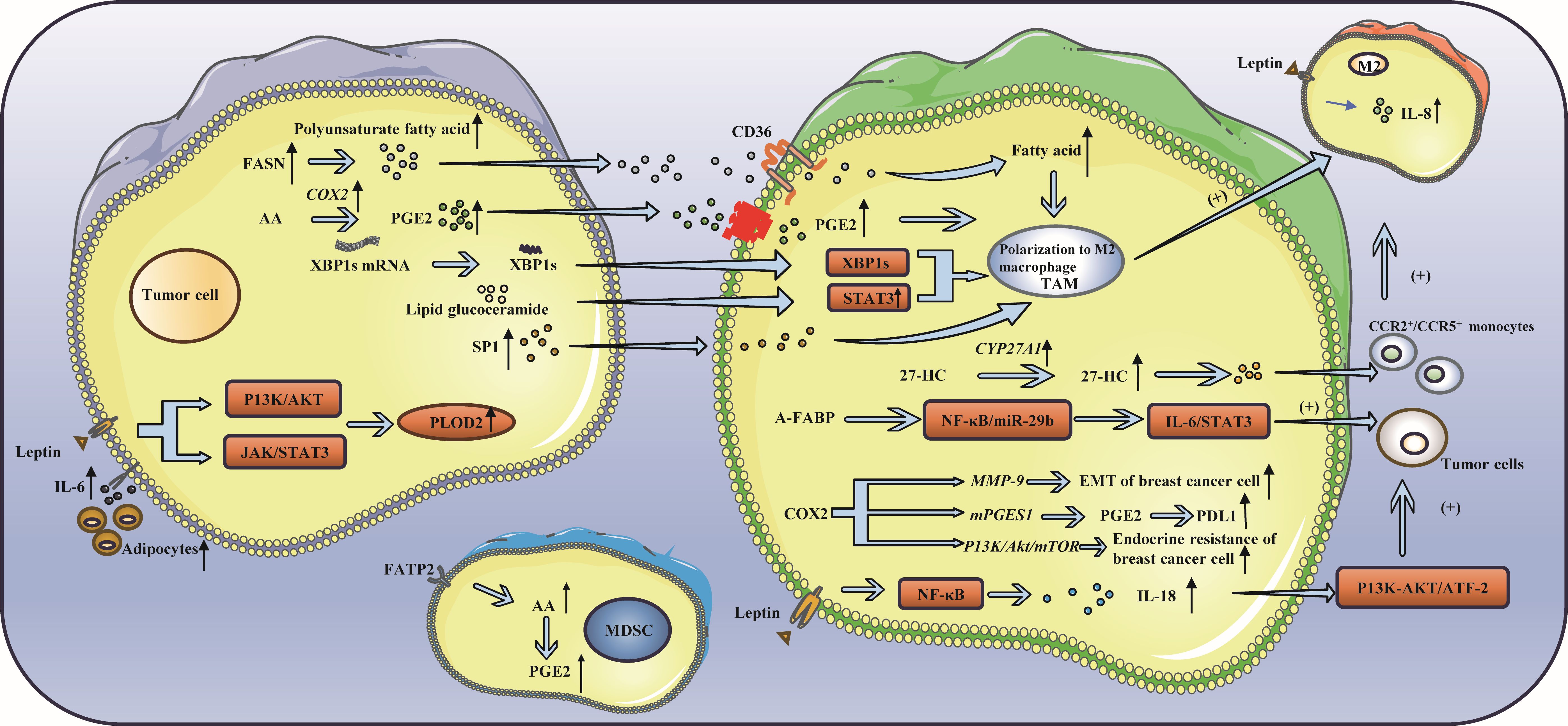
Figure 3. Lipid metabolism and triple-negative breast cancer. (A) Lipid metabolism can influence immune cell function: FASN and PGE2 in cancer cells have been shown to induce the polarization of TAMs toward the M2 phenotype. TAMs secrete 27-HC, which can enhance cancer cell proliferation and drive the differentiation of monocytes into M2 macrophages. Activation of IRE1 splicing pathway leads to the activation of signal transducer and activator of STAT3 and XBP1, thereby promoting the acquisition of a precancerous phenotype by TAMs. Additionally, S1P released from apoptotic tumor cells has been found to induce TAM-like polarization in macrophages. (B) Leptin, a fat factor in lipid metabolism, is involved in immune escape: Leptin has been shown to upregulate the expression of IL-8 in M2 macrophages and IL-18 in TAMs. Additionally, leptin has been found to increase the expression of PLOD2, thereby promoting cancer cell metastasis. Furthermore, leptin has the ability to suppress the function of CD8+ T cells, leading to immune escape.
4.1 Lipid metabolism can influence immune cell function
The metabolism of lipids impacts immunosuppressive cells, such as M2 macrophages, through various pathways (75). TAM are commonly classified into two main phenotypes: the anti-tumor M1 and the pro-tumor M2. M2 macrophages rely on fatty acids as a source of energy to produce ATP, thereby promoting fatty acid oxidation (FAO) (76).The TME contains many signaling molecules that can alter lipid metabolism in TAMs by increasing FAO and lipid uptake, promoting TAM polarization to a tumor-promoting M2 phenotype, thereby exerting an immunosuppressive effect and promoting tumor growth, metastasis, and angiogenesis (77). In cancer cells, fatty acid synthase (FASN) increases polyunsaturated fatty acids and promotes the polarization of TAMs to the M2 phenotype under the regulation of CD36 (76, 78). Prostaglandin E2 (PGE2), which is derived from arachidonic acid in cancer cells, can also promote the polarization of TAMs to the M2 phenotype (79–81). 27-HC, a metabolite of cholesterol (CHOL) secreted by TAMs, can not only promote the proliferation of cancer cells but also stimulate TAMs to secrete chemokines, causing CCR2+ and CCR5+ monocytes to migrate to the tumor site and polarize into M2 macrophages (82). Moreover, the endoplasmic reticulum of macrophages can be induced by lipids in cancer cells to induce stress and activate STAT3 and X-box binding protein 1 (XBP1) through inositol-requiring enzyme 1 (IRE1) splicing, thus promoting the pretumor phenotype of TAMs (83). Moreover, apoptosis-derived sphingosine-1-phosphate (S1P) promotes TAM-like polarization of macrophages (84). The tumor-promoting factor adipose fatty acid-binding protein (A-FABP) is highly expressed in TAMs. A-FABP enhances IL6/STAT3 signal transduction, promoting cancer growth and metastasis by regulating the NF-κB/miR-29b pathway. The PGE2-COX2 (cyclooxygenase-2) axis plays an important role in the lipid metabolism of TAMs (85, 86). COX-2 in TAMs promotes epithelial–mesenchymal transition of BCCs by triggering the expression of matrix metalloproteinase 9 (MMP-9) (87). TAMs increase the expression of COX-2 through the PI3K/Akt/mTOR pathway, thus enhancing endocrine drug resistance in breast cancer (88), and the expression of PDL1 in TAMs can be regulated through the COX2/mPGES1/PGE2 pathway (89). Moreover, fatty acid transport protein 2 (FATP2) promoted the accumulation of arachidonic acid, resulting in increased prostaglandin E2 synthesis in MDSCs, thus enhancing the immunosuppressive activity of MDSCs (90). Due to the metabolism of tumor cells, the tumor microenvironment lacks nutrients and accumulates immunosuppressive metabolites, which leads to obvious inhibition of the tumor microenvironment, resulting in obvious inhibition of immune effector cells such as NK cells, even in a resting state (91, 92). In this state, lipid metabolism is enhanced (93, 94), and peroxisome proliferator-activated receptors drive lipid proliferation in NK cells, leading to complete “paralysis of cellular metabolism and transport (95, 96). Therefore, preventing lipids from entering the mitochondria can reverse the metabolic paralysis of NK cells and restore cell activity (75). Additionally, senescent CD8+ T cells accumulate continuously by the binding of cytosolic phospholipase A2 alpha (cPLA2a) to MAPK/STAT signals, which induces TNBC cells to undergo immune escape (97). Moreover, the ability of CAFs to take up exogenous lipids can be enhanced by the upregulation of FATP1, thus promoting the metastasis of cancer cells (98, 99).
4.2 Leptin, a fat factor involved in lipid metabolism, is involved in immune escape
There are more adipocytes around the tumor in patients with breast cancer, particularly in obese patients. Adipocytes can produce numerous adipokines, such as leptin, thus promoting tumor progression (100). First, leptin mediates the bidirectional interaction between cancer cells and TAMs (101). Leptin increases the expression of IL-8 in M2 macrophages and leads to the migration and invasion of cancer cells. Leptin can also promote the expression and secretion of IL-18 in TAMs through NF-κB/NF-κB1, thereby fostering the expression and proliferation of cancer cells through the PI3K-AKT/ATF-2 pathway (102). In addition, leptin and adipocyte-derived IL-6 enhance the expression of lysyl hydroxylase (PLOD2) by activating the JAK/STAT3 and PI3K/AKT signaling pathways, ultimately facilitating cancer cell metastasis (103–106). Some studies have indicated that leptin can be secreted by tumor-stromal adipocytes (TSAs) stimulated by external conditions and bind to receptors on tumor cells, resulting in proliferation, invasion and other effects (107, 108). Moreover, leptin can also accumulate in breast adipocytes and adipose tissue, activate the STAT3-FAO axis, reduce glycolysis, inhibit the function of CD8+ T cells, and cause immune escape (100).
Targeting fatty acid receptor CD36 may be effective against multiple cancers (109, 110).Inhibition of fatty acid binding proteins (FABPs) has demonstrated potential anti-tumor effects. In the prostate cancer, FABP5 has emerged as a novel therapeutic target (111).Of course, inhibition of FATP2 can also be considered (112).FASN inhibitors show significant anti-tumor effects in multiple cancers (113).Upstream regulators SREBPs are key to lipid synthesis and can be considered as potential therapeutic targets (114).Due to the plasticity of lipid metabolism, cancer cells can switch to another metabolic pathway when one pathway is blocked, which to some extent hinders the anti-tumor efficacy of monotherapy (115).Therefore, we can consider combining treatments to enhance the therapeutic effect.
5 Amino acid metabolism is involved in immune escape in TNBC
Amino acids are essential nutrients for cells, and the demand for amino acids in normal cells is lower than that in tumor cells (116). Like glucose, amino acids are important fuels for the development of tumor cells. For example, glutamine metabolism in breast cancer can maintain the balance between Tregs and effector T cells (Teffs), change the function and state of immune cells in the TME, control the level of reactive oxygen species (ROS), provide the energy needed for immune cell progression, and lead to immune escape (117). Arginine is the basis of protein synthesis and the premise of metabolism and is mainly involved in the activation and functional activation of immune cells, especially T cells. In addition, tryptophan is an essential amino acid for cell proliferation, and its catabolism can inhibit T-cell proliferation (118). Thus, they are particularly important in the metabolism of TNBC (Figure 4).
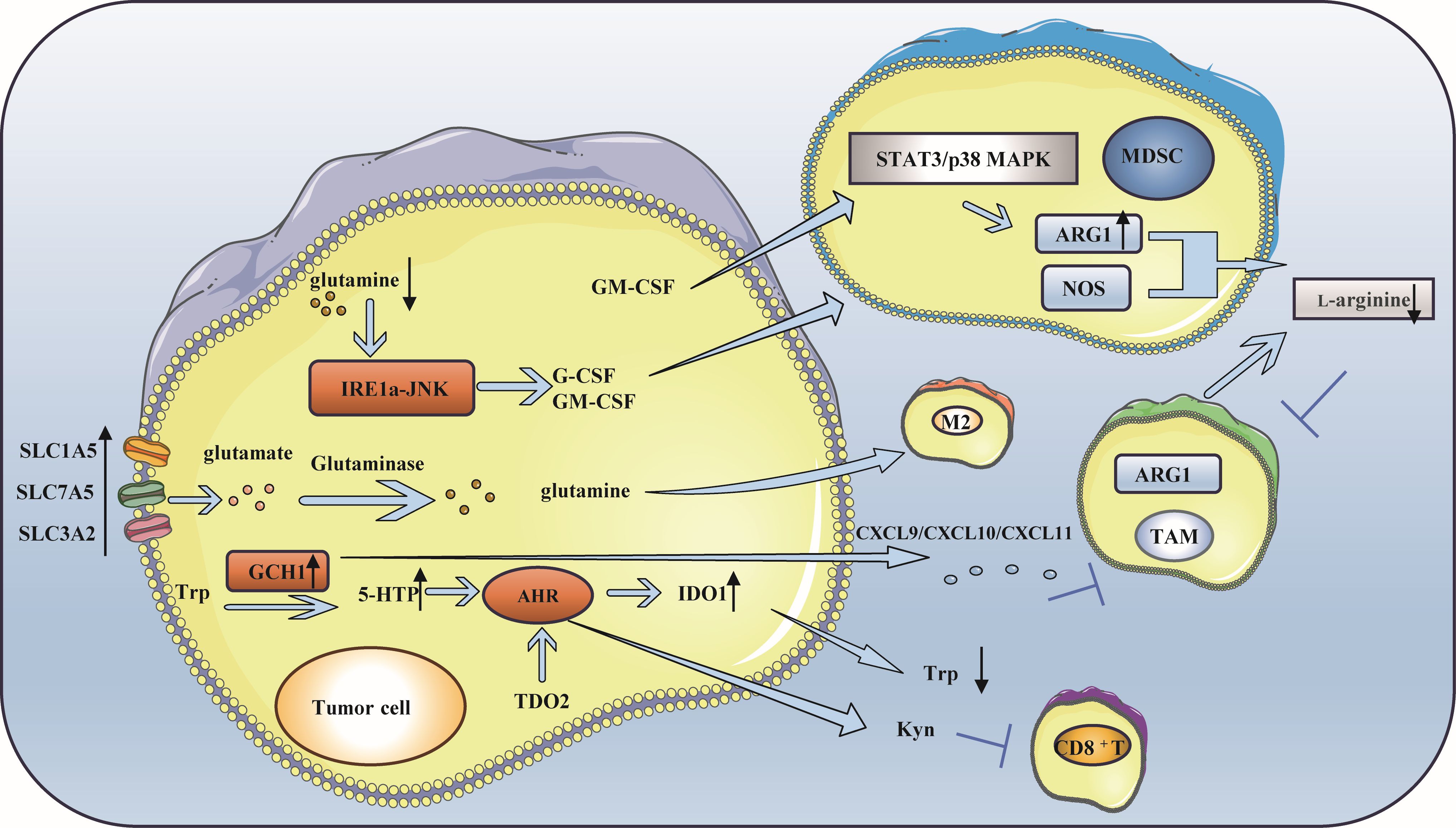
Figure 4. Amino acid metabolism is involved in immune escape of triple-negative breast cancer. (A) Glutamine metabolism participates in immune escape: A deficiency in glutamine leads to the production of MDSC, which in turn facilitates immune escape. Simultaneously, the breakdown of glutamine to produce a-KG is advantageous for sustaining the M2 phenotype of TAM. (B) Arginine metabolism participates in immune escape: L-arginine can hydrolyze and lead to the lack of available arginine in the microenvironment, which inhibits the function of immune cells. ARG 1 and NOS activated in tumor microenvironment inhibit NO production and promote tumor development. L-Arg is consumed by MDSCs, which inhibits T cell activation. Increased expression of ARG1 in breast cancer causes TAM to differentiate into M2 phenotype. (C) Tryptophan metabolism is involved in immune escape: GCH1 is highly expressed in TNBC, resulting in a low tryptophan microenvironment and immune escape. In addition, the activity of TDO2 in TNBC cells increased, which produced kynurenine and inhibited the viability of CD8+T cells.
5.1 Glutamine metabolism participates in immune escape
Although glutamine is a nonessential amino acid (NEAA), many tumor cells show a “glutamine dependence” in the TME (119, 120). The enhancement of glutamine metabolism in breast cancer alters the function and state of immune cells within the TME, which plays a crucial role in maintaining the balance between Teffs and Tregs and in immune escape (121–125). Research has shown that when glutamine deficiency occurs, G-CSF and GM-CSF are expressed in breast cancer cells through the IRE1 α-JNK pathway to induce the production of MDSCs, resulting in immune escape (126). Moreover, the overexpression of glutamine transporter proteins (such as recombinant solute carrier family 1, member 5 (SLC1A5), SLC7A5, and SLC3A2) in cancer cells directly influences glutamine metabolism, produces specific subtypes of inflammatory infiltration, and impacts the differentiation of TAMs toward a pro-carcinogenic phenotype (121). The α-ketoglutarate (α-KG) generated during glutamine catabolism supports the maintenance of a TAM M2-like phenotype (127, 128).
5.2 Arginine metabolism participates in immune escape
One of the key enzymes involved in arginine metabolism is ARG1, which is highly expressed in TNBC (129). Arginase plays a crucial role in promoting immune escape through several mechanisms: The content of L-arginine is closely related to the survival and proliferation of T cells (130–132). L-arginine is hydrolyzed to L-ornithine and urea under the catalysis of ARG1, which leads to a lack of available arginine in the microenvironment (104), which suppresses the function of immune cells and results in immune escape. Activated ARG1 and nitric oxide synthase (NOS) in the tumor microenvironment affect TAMs and inhibit the production of nitric oxide (NO), which can promote the differentiation of M1 macrophages, thus facilitating tumor development. The coexpression of ARG1 and inducible nitric oxide synthase (iNOS) enhances the production of ROS and reactive nitrogen species, which further inhibits the function of T cells within tumor cells (133, 134). The kinase GCN affects MDSCs during l-arginine depletion, and the L-arginine (L-Arg) is consumed by NOS and ARG-1 produced by MDSCs through the kinase GCN2, leading to amino acid starvation, thus inhibiting T-cell activation. MDSCs disrupt innate immunity and affect antitumor immunity by interacting with macrophages, NK cells, and NK T cells (135). In breast cancer, the expression of ARG1 increases when GM-CSF stimulates the STAT3 or p38MAPK signaling pathway, and immune cells in the microenvironment obtain less L-arginine, which increases the phenotypic differentiation of TAMs to M2 macrophages, reduces the infiltration of CD8+ T cells and ultimately promotes immune escape (134).
5.3 Tryptophan metabolism is involved in immune escape
Tryptophan catabolism plays an important role in immune escape in TNBC (118, 136). Its metabolism is closely associated with enzymes such as indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO) (137–141). Guanosine triphosphate cyclohydrolase 1 (GCH1), which is highly expressed in TNBC, regulates the metabolism of tryptophan (Trp), increases the level of 5-hydroxytryptophan (5-HTP), activates the aryl hydrocarbon receptor (AHR), and enhances IDO1 activity. This leads to a reduction in tryptophan levels and an increase in kynurenine levels. It is worth noting that kynurenine plays a role in promoting immunosuppression in the immune system. In addition, IDO1 can upregulate FoxO3α by activating Tregs, subsequently upregulating PD-1 and inducing sustained immune suppression via the PD-1/PTEN pathway, ultimately resulting in immune escape (142–145). Moreover, some studies have suggested that GCH1 mainly induces immunosuppression in an IDO1-dependent manner; however, the expression of chemokines such as CXCL9, CXCL10 and CXCL11 decreases because of the expression of GCH1, an increase in the tryptophan metabolite kynurenine and a decrease in tryptophan, which leads to a decrease in effector T-cell aggregation and promotes immune escape (146). Other studies have shown that IDO in breast cancer mediates MDSC-induced T cell proliferation and Th1 polarization inhibition, promotes T cell apoptosis and the secretion of immunosuppressive cytokines (IL-10 and TGF-β), causing breast cancer immune escape (147). Notably, when IDO is overexpressed, TAMs tend to polarize to the M2 phenotype, and T-cell activity is inhibited (148). In addition, the increased activity of TDO2 in TNBC cells mediates the initial production of the tryptophan catabolite kynurenine by AhR, which hinders the viability of CD8+ T cells and facilitates tumor immune escape (149, 150). Ultimately, by depleting tryptophan, the tryptophan metabolite kynurenine accumulates, inhibiting the function of effector T cells and NK cells and stimulating Tregs, MDSCs and macrophages to polarize into tolerant phenotypes, resulting in an immunosuppressive TME (151).Overall, IDO1 promotes immunosuppression through tryptophan depletion and direct effects of tryptophan catabolites. However, the interaction between TDO and other immune cells remains unclear, and the specific role of TDO in regulating immune responses is not well understood (152, 153).
6 Microbiome and TNBC
The microbiome can influence tumor development and progression (154). Current research has primarily concentrated on bacterial communities within tumors. Study finds potential link between tumor microbiome and cancer development (155, 156). The intratumoral microbiota is currently shown to play an important role in the pathogenesis of cancer (157). Studies have found that the toxic protein BFT-1 secreted by enterotoxigenic Bacteroides fragilis can promote the lysosomal degradation of NUMB by binding to NOD1, which is highly expressed on breast cancer stem cells, thereby activating the NOTCH1-HEY1 pathway, promoting breast cancer cell stemness and chemotherapy resistance to docetaxel (158). In contrast, trimethylamine N-oxide (TMAO), a metabolite related to the genera under Clostridiales, was more abundant in tumors with an activated immune microenvironment. Trimethylamine N-oxide induces tumor cell pyroptosis by activating endoplasmic reticulum stress kinase PERK, thereby enhancing CD8+ T cell-mediated anti-tumor immunity in TNBC. The results of this study suggest that microbial metabolites may serve as a new therapeutic approach to improve the efficacy of immunotherapy for TNBC (159). Tumor-associated microbiota plays an important role in the occurrence and development of tumors. Researchers have currently conducted relevant research on the carcinogenic mechanism of the microbiome, but more research is still needed to further explain it. It is worth noting that the intestinal microbiota is a research hotspot, which leads to insufficient understanding of related contents such as the microbiota of other ecological niches, so there is still a long way to go for future research (160).
7 Concluding remarks
In summary, metabolic reprogramming plays a pivotal role in the development and progression of TNBC and is one of the hallmarks of tumor progression. Under hypoxia, the expression of granzyme B, IFN-γ and degranulation marker CD107a on the surface of NK cells decreased, and the expression of activated receptors NKP30, NKP46 and NKG2D on NK cells decreased, resulting in a significant decrease in the activity of NK cells. In addition, HIF-1 α is one of the important factors. HIF-1 α can affect the function of MDSCs through microRNA210, PD-L1 and VISTA, up-regulate the transcription of FoxP3 to enhance the production of Treg, and inhibit the expression of IL-18. High rates of tumor cell glycolysis activate LAP leading to the formation of MDSCs and a decrease in the population of effector CD8+ T cells. At the same time, lactic acid produced by cancer cells up-regulated VEGF and ARG1, promoting TAM differentiation into the M2 phenotype. With the change of lipid metabolism, FASN and PGE2 in cancer cells can induce TAM to M2 phenotypic polarization, while 27-HC secreted by TAMs can promote cancer cell proliferation and promote monocytes to differentiate into M2 macrophages. In addition, Leptin has been shown to upregulate the expression of IL-8 in M2 macrophages and IL-18 in TAMs, increase the expression of PLOD2, and promote the metastasis of cancer cells. Arginine plays an indispensable role in amino acid metabolism and the lack of available arginine in the microenvironment inhibits immune cell function. L-arginine, absorbed by bone marrow mesenchymal stem cells, inhibits T cell activation. The high expression of ARG1 in breast cancer leads to TAM differentiation into the M2 phenotype. Tryptophan is also involved in immune escape, with GCH1 highly expressed in TNBC resulting in a low tryptophan microenvironment and immune escape.
It is evident that metabolic reprogramming plays a crucial role in immune escape of TNBC. Due to the characteristics of high invasiveness, easy metastasis, and recurrence of TNBC, studying metabolic reprogramming is of great significance and can provide new insights for TNBC immunotherapy. With the widespread use of immunotherapy, drug resistance will be inevitable. Therefore, conducting thorough research on the tumor microenvironment and immune escape mechanisms, as well as elucidating the impact of metabolic reorganization on immune escape in TNBC, will serve as a crucial foundation for addressing drug resistance. We can identify metabolic-related targets that contribute to immune escape more precisely. By utilizing gene editing techniques, it may be possible to modify or decrease the metabolic factors responsible for immune escape, ultimately enhancing the effectiveness of immunotherapy in treating TNBC. During the patient’s treatment, finding metabolic level-related changes or targets can increase the synergistic effect through combined immunotherapy. More research is needed in the future to explore the potential of combined therapy and how to translate these mechanisms into effective clinical treatments. Through continued efforts and collaboration, more effective treatment options are expected to be provided to improve the prognosis of patients with triple-negative breast cancer.
Author contributions
RB: Writing – original draft. HQ: Data curation, Formal analysis, Writing – original draft. BL: Data curation, Investigation, Writing – original draft. KC: Data curation, Investigation, Writing – original draft. YM: Writing – review & editing. JW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Shandong Province Medical and Health Science and Technology Development Plan Project (NO. 202209030830). Scientific and Technological Innovation Plan Project for Medical System Staff in Shandong Province (SDYWZGKCJHLH2023031).
Acknowledgments
We thank the Young Elite Sponsorship Program of Shandong Provincial Medical Association (2023_GJ_0019) for offering help during the manuscript process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
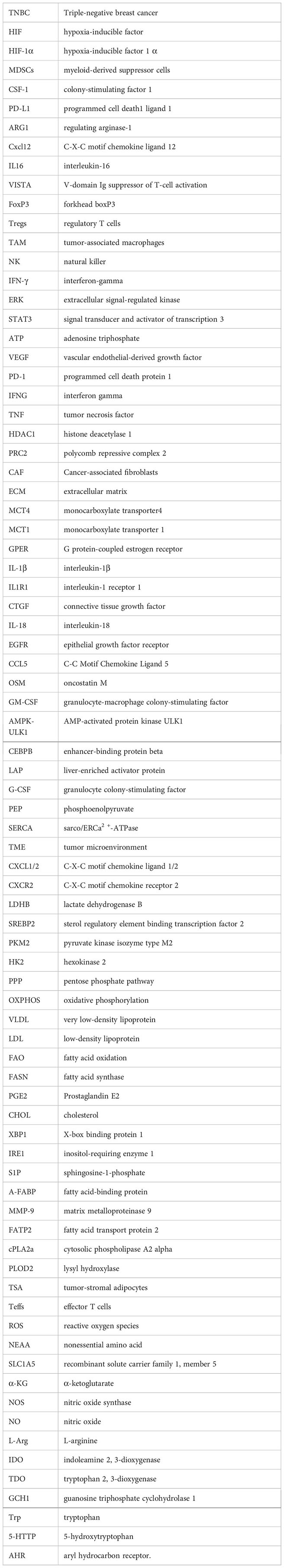
Glossary
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca: A Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Waks AG, Winer EP. Breast cancer treatment: a review. Jama. (2019) 321:288–300. doi: 10.1001/jama.2018.19323
3. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. (2010) 363:1938–48. doi: 10.1056/NEJMra1001389
4. Bou Zerdan M, Ghorayeb T, Saliba F, Allam S, Bou Zerdan M, Yaghi M, et al. Triple negative breast cancer: updates on classification and treatment in 2021. Cancers (Basel). (2022) 14:1253. doi: 10.3390/cancers14051253
5. Liu Y, Hu Y, Xue J, Li J, Yi J, Bu J, et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer. (2023) 22(1):145. doi: 10.1186/s12943-023-01850-7
6. Li Y, Zhang H, Merkher Y, Chen L, Liu N, Leonov S, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. (2022) 15:1–121. doi: 10.1186/s13045-022-01341-0
7. Miglietta F, Bottosso M, Griguolo G, Dieci MV, Guarneri V. Major advancements in metastatic breast cancer treatment: when expanding options means prolonging survival. Esmo Open. (2022) 7:100409. doi: 10.1016/j.esmoop.2022.100409
8. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. (2014) 346:256–59. doi: 10.1126/science.1256930
9. Ramon YCS, Sese M, Capdevila C, Aasen T, De Mattos-Arruda L, Diaz-Cano SJ, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med (Berl). (2020) 98:161–77. doi: 10.1007/s00109-020-01874-2
10. Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, et al. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer. (2019) 19:639–50. doi: 10.1038/s41568-019-0185-x
11. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
12. Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. (2015) 36:1219–27. doi: 10.1038/aps.2015.92
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
14. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
15. Wang Z, Jiang Q, Dong C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol Med. (2020) 17:44–59. doi: 10.20892/j.issn.2095-3941.2019.0210
16. Yan L, Tan Y, Chen G, Fan J, Zhang J. Harnessing metabolic reprogramming to improve cancer immunotherapy. Int J Mol Sci. (2021) 22:10268. doi: 10.3390/ijms221910268
17. Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to warburg hypothesis and beyond. Pharmacol Ther. (2009) 121:29–40. doi: 10.1016/j.pharmthera.2008.09.005
18. Naik A, Decock J. Lactate metabolism and immune modulation in breast cancer: a focused review on triple negative breast tumors. Front Oncol. (2020) 10:598626. doi: 10.3389/fonc.2020.598626
19. Zipinotti Dos Santos D, de Souza JC, Pimenta TM, Da Silva Martins B, Junior RSR, Butzene SMS, et al. The impact of lipid metabolism on breast cancer: a review about its role in tumorigenesis and immune escape. Cell Commun Signal. (2023) 21:161. doi: 10.1186/s12964-023-01178-1
20. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
21. Pietrobon V, Marincola FM. Hypoxia and the phenomenon of immune exclusion. J Transl Med. (2021) 19:9. doi: 10.1186/s12967-020-02667-4
22. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
23. Tutzauer J, Sjöström M, Holmberg E, Karlsson P, Killander F, Leeb-Lundberg LMF, et al. Breast cancer hypoxia in relation to prognosis and benefit from radiotherapy after breast-conserving surgery in a large, randomised trial with long-term follow-up. Br J Cancer. (2022) 126:1145–56. doi: 10.1038/s41416-021-01630-4
24. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
25. Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci. (2014) 111(20):E2120-9. doi: 10.1073/pnas.1406655111
26. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. Pd-l1 is a novel direct target of hif-1α, and its blockade under hypoxia enhanced mdsc-mediated t cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
27. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of mir-210. Cancer Res. (2015) 75:3771–87. doi: 10.1158/0008-5472.CAN-15-0405
28. Deng J, Li J, Sarde A, Lines JL, Lee Y, Qian DC, et al. Hypoxia-induced vista promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res. (2019) 7:1079–90. doi: 10.1158/2326-6066.CIR-18-0507
29. Santagata S, Napolitano M, D'Alterio C, Desicato S, Maro SD, Marinelli L, et al. Targeting cxcr4 reverts the suppressive activity of t-regulatory cells in renal cancer. Oncotarget. (2017) 8:77110–20. doi: 10.18632/oncotarget.20363
30. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via ccl28 and treg cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
31. Liu B, Wei C. Hypoxia induces overexpression of ccl28 to recruit treg cells to enhance angiogenesis in lung adenocarcinoma. J Environ Pathol Toxicol Oncol. (2021) 40:65. doi: 10.1615/JEnvironPatholToxicolOncol.2020035859
32. Hsu T, Lai M. Hypoxia-inducible factor 1α plays a predominantly negative role in regulatory t cell functions. J Leukoc Biol. (2018) 104:911–18. doi: 10.1002/JLB.MR1217-481R
33. Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, et al. Hypoxia enhances immunosuppression by inhibiting cd4+ effector t cell function and promoting treg activity. Cell Physiol Biochem. (2017) 41:1271–84. doi: 10.1159/000464429
34. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
35. Fu L, Du W, Cai M, Yao J, Zhao Y, Mou X. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
36. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, et al. Knockout of hif-1 in tumor-associated macrophages enhances m2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. (2010) 31:1863–72. doi: 10.1093/carcin/bgq088
37. Teng R, Wang Y, Lv N, Zhang D, Williamson RA, Lei L, et al. Hypoxia impairs nk cell cytotoxicity through shp-1-mediated attenuation of stat3 and erk signaling pathways. J Immunol Res. (2020) 2020:1–14. doi: 10.1155/2020/4598476
38. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Bühler L, et al. Single-cell rna sequencing of tumor-infiltrating nk cells reveals that inhibition of transcription factor hif-1α unleashes nk cell activity. Immunity. (2020) 52:1075–87. doi: 10.1016/j.immuni.2020.05.001
39. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, et al. B cells and t follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell. (2019) 179:1191–206. doi: 10.1016/j.cell.2019.10.028
40. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An hif-1α/vegf-a axis in cytotoxic t cells regulates tumor progression. Cancer Cell. (2017) 32:669–83. doi: 10.1016/j.ccell.2017.10.003
41. Chang C, Qiu J O, Sullivan D, MD B, Noguchi T, JD C, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
42. Bannoud N, Dalotto-Moreno T, Kindgard L, García PA, Blidner AG, Mariño KV, et al. Hypoxia supports differentiation of terminally exhausted cd8 t cells. Front Immunol. (2021) 12:660944. doi: 10.3389/fimmu.2021.660944
43. Ma S, Zhao Y, Lee WC, Ong L, Lee PL, Jiang Z, et al. Hypoxia induces hif1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-pd-1 immunotherapy. Nat Commun. (2022) 13(1):4118. doi: 10.1038/s41467-022-31764-9
44. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol. (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
45. Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized atm-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. Ebiomedicine. (2019) 41:370–83. doi: 10.1016/j.ebiom.2019.02.025
46. Becker LM, Connell JT O, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (Cambridge). (2020) 31:107701. doi: 10.1016/j.celrep.2020.107701
47. Lappano R, Talia M, Cirillo F, Rigiracciolo DC, Scordamaglia D, Guzzi R, et al. The il1β-il1r signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (cafs). J Exp Clin Cancer Res. (2020) 39(1):153. doi: 10.1186/s13046-020-01667-y
48. Ren J, Guo H, Wu H, Tian T, Dong D, Zhang Y, et al. Gper in cafs regulates hypoxia-driven breast cancer invasion in a ctgf-dependent manner. Oncol Rep. (2015) 33:1929–37. doi: 10.3892/or.2015.3779
49. Vuillefroy De Silly R, Ducimetière L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR. Phenotypic switch of cd8+ t cells reactivated under hypoxia toward il-10 secreting, poorly proliferative effector cells. Eur J Immunol. (2015) 45:2263–75. doi: 10.1002/eji.201445284
50. Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL, et al. Macrophage in®ltration is associated with vegf and egfr expression in breast cancer. J Pathol. (2000) 190(4):430–6. doi: 10.1002/(SICI)1096-9896(200003)190:4<430::AID-PATH538>3.0.CO;2-6
51. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic m2-polarized phenotype via hypoxic cancer cell derived cytokines oncostatin m and eotaxin. Oncotarget. (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
52. Wu J, Huang T, Hsieh Y, Wang Y, Yen C, Lee G, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. (2020) 77:213–27. doi: 10.1016/j.molcel.2019.10.023
53. Sami E, Paul BT, Koziol JA, ElShamy WM. The immunosuppressive microenvironment in brca1-iris–overexpressing tnbc tumors is induced by bidirectional interaction with tumor-associated macrophages. Cancer Res. (2020) 80:1102–17. doi: 10.1158/0008-5472.CAN-19-2374
54. Choi H, Na KJ. Different glucose metabolic features according to cancer and immune cells in the tumor microenvironment. Front Oncol. (2021) 11:769393. doi: 10.3389/fonc.2021.769393
55. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett. (2018) 413:102–09. doi: 10.1016/j.canlet.2017.10.037
56. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific cebpb isoform in triple-negative breast cancer. Cell Metab. (2018) 28:87–103. doi: 10.1016/j.cmet.2018.04.022
57. Ho P, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor t cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
58. Franklin DA, Sharick JT, Ericsson-Gonzalez PI, Sanchez V, Dean PT, Opalenik SR, et al. Mek activation modulates glycolysis and supports suppressive myeloid cells in tnbc. JCI Insight. (2020) 5(15):e134290. doi: 10.1172/jci.insight.134290
59. Thomas R, Shaath H, Naik A, Toor SM, Elkord E, Decock J. Identification of two hla-a*0201 immunogenic epitopes of lactate dehydrogenase c (ldhc): potential novel targets for cancer immunotherapy. Cancer Immunol Immunother. (2020) 69:449–63. doi: 10.1007/s00262-020-02480-4
60. Sun L, Suo C, Li S, Zhang H, Gao P. Metabolic reprogramming for cancer cells and their microenvironment: beyond the warburg effect. Biochim Et Biophys Acta Rev On Cancer. (2018) 1870:51–66. doi: 10.1016/j.bbcan.2018.06.005
61. Rizwan A, Serganova I, Khanin R, Karabeber H, Ni X, Thakur S, et al. Relationships between ldh-a, lactate, and metastases in 4t1 breast tumors. Clin Cancer Res. (2013) 19:5158–69. doi: 10.1158/1078-0432.CCR-12-3300
62. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces m2 macrophage polarization via the activation of the erk/stat3 signaling pathway in breast cancer. Cell Cycle (Georgetown Tex.). (2018) 17:428–38. doi: 10.1080/15384101.2018.1444305
63. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
64. Frank A, Raue R, Fuhrmann DC, Sirait-Fischer E, Reuse C, Weigert A, et al. Lactate dehydrogenase b regulates macrophage metabolism in the tumor microenvironment. Theranostics. (2021) 11:7570–88. doi: 10.7150/thno.58380
65. Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, et al. The lactate receptor gpr81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. (2020) 39:3292–304. doi: 10.1038/s41388-020-1216-5
66. Lim S, Li C, Xia W, Lee H, Chang S, Shen J, et al. Egfr signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. (2016) 76:1284–96. doi: 10.1158/0008-5472.CAN-15-2478
67. Elf SE, Chen J. Targeting glucose metabolism in patients with cancer. Cancer. (2014) 120:774–80. doi: 10.1002/cncr.28501
68. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. Nk cell metabolism and tumor microenvironment. Front Immunol. (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
69. Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheum. (2019) 48:1142–45. doi: 10.1016/j.semarthrit.2019.04.006
70. Yu W, Lei Q, Yang L, Qin G, Liu S, Wang D, et al. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J Hematol Oncol. (2021) 14. doi: 10.1186/s13045-021-01200-4
71. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
72. Matsushita Y, Nakagawa H, Koike K. Lipid metabolism in oncology: why it matters, how to research, and how to treat. Cancers (Basel). (2021) 13:474. doi: 10.3390/cancers13030474
73. Bleve A, Durante B, Sica A, Consonni FM. Lipid metabolism and cancer immunotherapy: immunosuppressive myeloid cells at the crossroad. Int J Mol Sci. (2020) 21:5845. doi: 10.3390/ijms21165845
74. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). (2018) 38:1–14. doi: 10.1186/s40880-018-0301-4
75. Rombaldova M, Janovska P, Kopecky J, Kuda O. Omega-3 fatty acids promote fatty acid utilization and production of pro-resolving lipid mediators in alternatively activated adipose tissue macrophages. Biochem Biophys Res Commun. (2017) 490:1080–85. doi: 10.1016/j.bbrc.2017.06.170
76. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
77. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. (2018) 78:5492–503. doi: 10.1158/0008-5472.CAN-18-1367
78. Gómez V, Eykyn TR, Mustapha R, Flores-Borja F, Male V, Barber PR, et al. Breast cancer-associated macrophages promote tumorigenesis by suppressing succinate dehydrogenase in tumor cells. Sci Signal. (2020) 13(365):eaax4585. doi: 10.1126/scisignal.aax4585
79. Yin J, Kim SS, Choi E, Oh YT, Lin W, Kim T, et al. Ars2/magl signaling in glioblastoma stem cells promotes self-renewal and m2-like polarization of tumor-associated macrophages. Nat Commun. (2020) 11(1):2978. doi: 10.1038/s41467-020-16789-2
80. Liu L, Ge D, Ma L, Mei J, Liu S, Zhang Q, et al. Interleukin-17 and prostaglandin e2 are involved in formation of an m2 macrophage-dominant microenvironment in lung cancer. J Thorac Oncol. (2012) 7:1091–100. doi: 10.1097/JTO.0b013e3182542752
81. Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJP, et al. macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. (2011) 187(3):1157–65. doi: 10.4049/jimmunol.1100889
82. Shi S, Lee E, Lin Y, Chen L, Zheng H, He X, et al. Recruitment of monocytes and epigenetic silencing of intratumoral cyp7b1 primarily contribute to the accumulation of 27-hydroxycholesterol in breast cancer. Am J Cancer Res. (2019) 9(10):2194–208. doi: 2156-6976/ajcr0099894
83. Di Conza G, Tsai C, Gallart-Ayala H, Yu Y, Franco F, Zaffalon L, et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol. (2021) 22:1403–15. doi: 10.1038/s41590-021-01047-4
84. Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G, et al. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell. (2007) 18(10):3810–9. doi: 10.1091/mbc.e06-12-1096
85. Hao J, Yan F, Zhang Y, Triplett A, Zhang Y, Schultz DA, et al. Expression of adipocyte/macrophage fatty acid–binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. (2018) 78:2343–55. doi: 10.1158/0008-5472.CAN-17-2465
86. Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang Y, et al. Fatty acid-binding protein e-fabp restricts tumor growth by promoting ifn-β responses in tumor-associated macrophages. Cancer Res. (2014) 74:2986–98. doi: 10.1158/0008-5472.CAN-13-2689
87. Gan L, Qiu Z, Huang J, Li Y, Huang H, Xiang T, et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through akt pathway. Int J Biol Sci. (2016) 12:1533–43. doi: 10.7150/ijbs.15943
88. Qin Q, Ji H, Li D, Zhang H, Zhang Z, Zhang Q. Tumor-associated macrophages increase cox-2 expression promoting endocrine resistance in breast cancer via the pi3k/akt/mtor pathway. Neoplasma. (2021) 68:938–46. doi: 10.4149/neo_2021_201226N1404
89. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. Cox2/mpges1/pge2 pathway regulates pd-l1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
90. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
91. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on nk-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
92. Terrén I, Orrantia A, Vitallé J, Astarloa-Pando G, Zenarruzabeitia O, Borrego F. Modulating nk cell metabolism for cancer immunotherapy. Semin Hematol. (2020) 57:213–24. doi: 10.1053/j.seminhematol.2020.10.003
93. Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, et al. Increased lipid metabolism impairs nk cell function and mediates adaptation to the lymphoma environment. Blood. (2020) 136:3004–17. doi: 10.1182/blood.2020005602
94. Inoue H, Miyaji M, Kosugi A, Nagafuku M, Okazaki T, Mimori T, et al. Lipid rafts as the signaling scaffold for NK cell activation: tyrosine phosphorylation and association of LAT with phosphatidylinositol 3-kinase and phospholipase C-gamma following CD2 stimulation. Eur J Immunol. (2002) 32(8):2188–98. doi: 10.1002/1521-4141(200208)32:8<2188::AID-IMMU2188>3.0.CO;2-T
95. Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer. (2019) 19(1):823. doi: 10.1186/s12885-019-6045-y
96. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. (2018) 19:1330–40. doi: 10.1038/s41590-018-0251-7
97. Carpenter KJ, Valfort A, Steinauer N, Chatterjee A, Abuirqeba S, Majidi S, et al. Lxr-inverse agonism stimulates immune-mediated tumor destruction by enhancing cd8 t-cell activity in triple negative breast cancer. Sci Rep. (2019) 9(1):19530. doi: 10.1038/s41598-019-56038-1
98. Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, et al. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Et Biophys Acta (Bba) - Mol Cell Res. (2015) 1853:3211–23. doi: 10.1016/j.bbamcr.2015.09.013
99. Lopes-Coelho F, André S, Félix A, Serpa J. Breast cancer metabolic cross-talk: fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol. (2018) 462:93–106. doi: 10.1016/j.mce.2017.01.031
100. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. Stat3 activation-induced fatty acid oxidation in cd8+ t effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. (2020) 31:148–61. doi: 10.1016/j.cmet.2019.10.013
101. Cao H, Huang Y, Wang L, Wang H, Pang X, Li K, et al. Leptin promotes migration and invasion of breast cancer cells by stimulating il-8 production in m2 macrophages. Oncotarget. (2016) 7:65441–53. doi: 10.18632/oncotarget.11761
102. Li K, Wei L, Huang Y, Wu Y, Su M, Pang X, et al. Leptin promotes breast cancer cell migration and invasion via il-18 expression and secretion. Int J Oncol. (2016) 48:2479–87. doi: 10.3892/ijo.2016.3483
103. He J, Wei X, Li S, Liu Y, Hu H, Li Z, et al. Adipocyte-derived il-6 and leptin promote breast cancer metastasis via upregulation of lysyl hydroxylase-2 expression. Cell Commun Signal. (2018) 16(1):100. doi: 10.1186/s12964-018-0309-z
104. Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, et al. The opposing effects of interferon-beta and oncostatin-m as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. (2019) 21(1):54. doi: 10.1186/s13058-019-1136-x
105. Gyamfi J, Lee Y, Eom M, Choi J. Interleukin-6/stat3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci Rep. (2018) 8:8859. doi: 10.1038/s41598-018-27184-9
106. Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, Van Den Broecke R, et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin m and jak/stat3 signaling. Cancer Res. (2014) 74:6806–19. doi: 10.1158/0008-5472.CAN-14-0160
107. García-Estevez L, González-Martínez S, Moreno-Bueno G. The leptin axis and its association with the adaptive immune system in breast cancer. Front Immunol. (2021) 12:784823. doi: 10.3389/fimmu.2021.784823
108. Martínez-Rodríguez OP, Thompson-Bonilla MDR, Jaramillo-Flores ME. Association between obesity and breast cancer: molecular bases and the effect of flavonoids in signaling pathways. Crit Rev Food Sci Nutr. (2020) 60:3770–92. doi: 10.1080/10408398.2019.1708262
109. Ruan C, Meng Y, Song H. Cd36: an emerging therapeutic target for cancer and its molecular mechanisms. J Cancer Res Clin Oncol. (2022) 148:1551–58. doi: 10.1007/s00432-022-03957-8
110. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
111. O'Sullivan SE, Kaczocha M. Fabp5 as a novel molecular target in prostate cancer. Drug Discovery Today. (2020) 2020:S1359-6446(20)30375-5. doi: 10.1016/j.drudis.2020.09.018
112. Alicea GM, Rebecca VW, Goldman AR, Fane ME, Douglass SM, Behera R, et al. Changes in aged fibroblast lipid metabolism induce age-dependent melanoma cell resistance to targeted therapy via the fatty acid transporter fatp2. Cancer Discovery. (2020) 10:1282–95. doi: 10.1158/2159-8290.CD-20-0329
113. Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, et al. Orlistat as a fasn inhibitor and multitargeted agent for cancer therapy. Expert Opin Investig Drugs. (2018) 27:475–89. doi: 10.1080/13543784.2018.1471132
114. Jin HR, Wang J, Wang ZJ, Xi MJ, Xia BH, Deng K, et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol. (2023) 16:103. doi: 10.1186/s13045-023-01498-2
115. Munir R, Lisec J, Swinnen JV, Zaidi N. Too complex to fail? Targeting fatty acid metabolism for cancer therapy. Prog Lipid Res. (2022) 85:101143. doi: 10.1016/j.plipres.2021.101143
116. Lieu EL, Nguyen T, Rhyne S, Kim J. Amino acids in cancer. Exp Mol Med. (2020) 52:15–30. doi: 10.1038/s12276-020-0375-3
117. Yan C, Yang J, Saleh N, Chen S, Ayers GD, Abramson VG, et al. Inhibition of the pi3k/mtor pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int J Mol Sci. (2021) 22:5207. doi: 10.3390/ijms22105207
118. Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discovery Today. (2017) 22:796–804. doi: 10.1016/j.drudis.2016.12.003
119. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
120. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
121. Ansari RE, Craze ML, Althobiti M, Alfarsi L, Ellis IO, Rakha EA, et al. Enhanced glutamine uptake influences composition of immune cell infiltrates in breast cancer. Br J Cancer. (2020) 122:94–101. doi: 10.1038/s41416-019-0626-z
122. Luo M, Brooks M, Wicha MS. Asparagine and glutamine: co-conspirators fueling metastasis. Cell Metab. (2018) 27:947–49. doi: 10.1016/j.cmet.2018.04.012
123. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. (2017) 36:1302–15. doi: 10.15252/embj.201696151
124. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
125. Kim S, Kim DH, Jung W, Koo JS. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr Relat Cancer. (2013) 20:339–48. doi: 10.1530/ERC-12-0398
126. Sun H, Wu W, Chen H, Xu Y, Yang Y, Chen J, et al. Glutamine deprivation promotes the generation and mobilization of mdscs by enhancing expression of g-csf and gm-csf. Front Immunol. (2021) 11:616367. doi: 10.3389/fimmu.2020.616367
127. Wang Y, Wang D, Yang L, Zhang Y. Metabolic reprogramming in the immunosuppression of tumor-associated macrophages. Chin Med J (Engl). (2022) 135:2405–16. doi: 10.1097/CM9.0000000000002426
128. Liu P, Wang H, Li X, Chao T, Teav T, Christen S, et al. A-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. doi: 10.1038/ni.3796
129. Su X, Xu Y, Fox GC, Xiang J, Kwakwa KA, Davis JL, et al. Breast cancer–derived gm-csf regulates arginase 1 in myeloid cells to promote an immunosuppressive microenvironment. J Clin Invest. (2021) 131(20):e145296. doi: 10.1172/JCI145296
130. Rodriguez PC, Quiceno DG, Antonia S, Ochoa JB, Ochoa AC, Zabaleta J, et al. Arginase i production in the tumor microenvironment by mature myeloid cells inhibits t-cell receptor expression and antigen-specific t-cell responses. Cancer Res (Chicago Ill.). (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
131. Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: a multifaceted enzyme important in health and disease. Physiol Rev. (2018) 98:641–65. doi: 10.1152/physrev.00037.2016
132. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates t cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42. doi: 10.1016/j.cell.2016.09.031
133. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
134. Xu J, Luo Y, Yuan C, Han L, Wu Q, Xu L, et al. Downregulation of nitric oxide collaborated with radiotherapy to promote anti-tumor immune response via inducing cd8+ t cell infiltration. Int J Biol Sci. (2020) 16:1563–74. doi: 10.7150/ijbs.41653
135. Cao Y, Feng Y, Zhang Y, Zhu X, Jin F. L-arginine supplementation inhibits the growth of breast cancer by enhancing innate and adaptive immune responses mediated by suppression of mdscs in vivo. BMC Cancer. (2016) 16:343. doi: 10.1186/s12885-016-2376-0
136. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discovery. (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
137. Liu Q, Zhai J, Kong X, Wang X, Wang Z, Fang Y, et al. Comprehensive analysis of the expression and prognosis for tdo2 in breast cancer. Mol Ther Oncolytics. (2020) 17:153–68. doi: 10.1016/j.omto.2020.03.013
138. Heng B, Bilgin AA, Lovejoy DB, Tan VX, Milioli HH, Gluch L, et al. Differential kynurenine pathway metabolism in highly metastatic aggressive breast cancer subtypes: beyond ido1-induced immunosuppression. Breast Cancer Res. (2020) 22(1):113. doi: 10.1186/s13058-020-01351-1
139. Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, et al. Reversal of indoleamine 2,3-dioxygenase–mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol. (2018) 36:758–64. doi: 10.1038/nbt.4180
140. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolušić E, Frédérick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci. (2012) 109:2497–502. doi: 10.1073/pnas.1113873109
141. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene bin1, potentiates cancer chemotherapy. Nat Med. (2005) 11:312–19. doi: 10.1038/nm1196
142. Wei J, Wu S, Yang Y, Xiao Y, Jin X, Xu X, et al. Gch1 induces immunosuppression through metabolic reprogramming and ido1 upregulation in triple-negative breast cancer. J Immunother Cancer. (2021) 9:e2383. doi: 10.1136/jitc-2021-002383
143. Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: tryptophan's metabolites in exercise, inflammation, and mental health. Science. (2017) 357(6349):eaaf9794. doi: 10.1126/science.aaf9794
144. Tatham AL, Crabtree MJ, Warrick N, Cai S, Alp NJ, Channon KM. Gtp cyclohydrolase i expression, protein, and activity determine intracellular tetrahydrobiopterin levels, independent of gtp cyclohydrolase feedback regulatory protein expression. J Biol Chem. (2009) 284:13660–68. doi: 10.1074/jbc.M807959200
145. Staats Pires A, Tan VX, Heng B, Guillemin GJ, Latini A. Kynurenine and tetrahydrobiopterin pathways crosstalk in pain hypersensitivity. Front Neurosci. (2020) 14:620. doi: 10.3389/fnins.2020.00620
146. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
147. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through ido expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
148. Wang X, Wang H, Wang H, Zhang F, Wang K, Guo Q, et al. The role of indoleamine 2,3-dioxygenase (ido) in immune tolerance: focus on macrophage polarization of thp-1 cells. Cell Immunol. (2014) 289:42–8. doi: 10.1016/j.cellimm.2014.02.005
149. Greene LI, Bruno TC, Christenson JL, D'Alessandro A, Culp-Hill R, Torkko K, et al. A role for tryptophan-2,3-dioxygenase in cd8 t-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res. (2019) 17:131–39. doi: 10.1158/1541-7786.MCR-18-0362
150. D'Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A tdo2-ahr signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. (2015) 75:4651–64. doi: 10.1158/0008-5472.CAN-15-2011
151. Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol. (2018) 9:353. doi: 10.3389/fimmu.2018.00353
152. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of t cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
153. Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic t cell proliferation by indoleamine 2,3-dioxygenase–expressing dendritic cells. J Exp Med. (2002) 196:447–57. doi: 10.1084/jem.20020052
154. Park EM, Chelvanambi M, Bhutiani N, Kroemer G, Zitvogel L, Wargo JA. Targeting the gut and tumor microbiota in cancer. Nat Med. (2022) 28:690–703. doi: 10.1038/s41591-022-01779-2
155. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. (2020) 368:973–80. doi: 10.1126/science.aay9189
156. Narunsky-Haziza L, Sepich-Poore GD, Livyatan I, Asraf O, Martino C, Nejman D, et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell. (2022) 185:3789–806. doi: 10.1016/j.cell.2022.09.005
157. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
158. Ma W, Zhang L, Chen W, Chang Z, Tu J, Qin Y, et al. Microbiota enterotoxigenic bacteroides fragilis-secreted bft-1 promotes breast cancer cell stemness and chemoresistance through its functional receptor nod1. Protein Cell. (2024) 15:419–40. doi: 10.1093/procel/pwae005
159. Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine n-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab. (2022) 34:581–94. doi: 10.1016/j.cmet.2022.02.010
Keywords: triple-negative breast cancer, immune escape, hypoxia, glucose metabolism, lipid metabolism, amino acid metabolism
Citation: Bao R, Qu H, Li B, Cheng K, Miao Y and Wang J (2024) The role of metabolic reprogramming in immune escape of triple-negative breast cancer. Front. Immunol. 15:1424237. doi: 10.3389/fimmu.2024.1424237
Received: 27 April 2024; Accepted: 29 July 2024;
Published: 13 August 2024.
Edited by:
Thuc Le, University of California, Los Angeles, United StatesReviewed by:
Wenzheng Guo, University of Kentucky, United StatesNilesh Kumar Sharma, Dr. D. Y. Patil Biotechnology & Bioinformatics Institute, India
Copyright © 2024 Bao, Qu, Li, Cheng, Miao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangtao Wang, d2FuZ2p0MjJAYnptYy5lZHUuY24=