 Bouchra Ghazi1,2
Bouchra Ghazi1,2 Zakia Harmak3
Zakia Harmak3 Mounir Rghioui1,2Abdou-Samad Kone3Adil El Ghanmi1,2
Mounir Rghioui1,2Abdou-Samad Kone3Adil El Ghanmi1,2 Abdallah Badou3,4,5*
Abdallah Badou3,4,5*- 1Immunopathology-Immunotherapy-Immunomonitoring Laboratory, Faculty of Medicine, Mohammed VI University of Sciences and Health, Casablanca, Morocco
- 2Mohammed VI International University Hospital, Bouskoura, Morocco
- 3Immuno-genetics and Human Pathology Laboratory, Faculty of Medicine and Pharmacy, Hassan II University, Casablanca, Morocco
- 4Mohammed VI Center for Research and Innovation, Rabat, Morocco
- 5Mohammed VI University of Sciences and Health (UM6SS), Casablanca, Morocco
Over the last decades, extracellular vesicles (EVs) have become increasingly popular for their roles in various pathologies, including cancer and neurological and immunological disorders. EVs have been considered for a long time as a means for normal cells to get rid of molecules it no longer needs. It is now well established that EVs play their biological roles also following uptake or by the interaction of EV surface proteins with cellular receptors and membranes. In this review, we summarize the current status of EV production and secretion in glioblastoma, the most aggressive type of glioma associated with high mortality. The main purpose is to shed light on the EVs as a universal mediator of interkingdom and intrakingdom communication in the context of tumor microenvironment heterogeneity. We focus on the immunomodulatory EV functions in glioblastoma-immune cross-talk to enhance immune escape and reprogram tumor-infiltrating immune cells. We critically examine the evidence that GBM-, immune cell-, and microbiome-derived EVs impact local tumor microenvironment and host immune responses, and can enter the circulatory system to disseminate and drive premetastatic niche formation in distant organs. Taking into account the current state of the art in intratumoral microbiome studies, we discuss the emerging role of bacterial EV in glioblastoma and its response to current and future therapies including immunotherapies.
1 Introduction
Brain tumors are highly aggressive and rank among the deadliest cancers (1, 2). The most common brain tumor is glioma, which is globally recognized as the most common primary brain tumor in the central nervous system (CNS) and has the greatest prevalence of all brain tumors (approximately 46%) (3). Gliomas are defined as brain tumors of glial origin (4). Depending on both histology and molecular features, gliomas have been divided into six different families in the 2021 5th edition of the WHO Classification of Tumors of the Central Nervous System (1). The first family, adult-type diffuse gliomas, constitute the majority of primary brain tumors [e.g., glioblastoma multiforme (GBM) and isocitrate dehydrogenase (IDH) wild type]. Insights gained from next-generation sequencing and DNA methylation-based profiling have prompted the characterization of the second family, pediatric-type diffuse low-grade gliomas. Under the banner of “pediatric type diffuse low-grade gliomas”, three are new tumors: diffuse astrocytoma; MYB or MYBL1-altered, polymorphous low-grade neuroepithelial tumor of the young (PLNTY); and diffuse low-grade glioma-MAPK altered. The third family, pediatric-type diffuse high-grade gliomas, is expected to behave aggressively. The fourth family, circumscribed astrocytic gliomas, encompasses a group of well-demarcated typically solid astrocytic tumors. Based on the hierarchical clustering analysis of DNA methylation profiles, the fifth family has been newly recognized, glioneuronal and neuronal tumors, which is a diverse group of tumors featuring neuronal differentiation. The sixth and last family is ependymomas, now classified according to a combination of histopathological and molecular features as well as anatomic site (1). According to the classification of the World Health Organization (WHO), glioma can be categorized into grades I–IV. The most aggressive type of glioma is GBM, classified as a grade IV brain tumor. This entity is characterized histopathologically by necrosis and endothelial growth and associated with high mortality (1, 5, 6).
Although there have been improvements in diagnostic, radiotherapy, and chemotherapy options, the prognosis of gliomas is still poor, especially for malignant and invasive gliomas (7, 8). The prognosis of glioma patients varies according to molecular subtype, with IDH-mutated gliomas generally showing a better disease course and distinct ontogeny compared with IDH wild-type gliomas. There is a clear genetic difference between IDH-mutated and wild-type IDH gliomas, and PTEN mutation is a poor prognostic factor for wild-type IDH patients (9–11). In parallel, several potential pathologic characteristics of glioma have been investigated, which include 1p/19q codeletion, IDH, epidermal growth factor receptor (EGFR), p53, PTEN/Akt pathway, Rb, Ras/MAPK pathway, extrachromosomal DNA, MGMT, TERT, and ATRX (5, 10, 12). Genetic aberrations contribute to the specific glioma subtype and a unique metabolic footprint.
On the other hand, metabolic reprogramming can, in fact, act as a driver of cancer genome modification and oncogenic pathways, through epigenetic, transcriptional, and posttranslational modifications (13). Exactly how cells acquire these hallmarks, and how they can be counteracted is the main question at stake for developing efficacious cancer treatments. Currently, the mainstay of glioma treatment is surgical resection, followed by radiotherapy and chemotherapy (5, 14). However, despite advances in diagnosis and treatment, the prognosis for gliomas remains poor, particularly for malignant and invasive gliomas. This limited efficiency may be due to the intratumoral heterogeneity of tumors. Unfortunately, glioma shows a high biological and genetic heterogeneity associated with exceptional aggressiveness. The discovery of molecular heterogeneity between tumors from different patients as well as within tumors from the same patient suggests the complexity of this cancer. Glioma cancer cells exhibit distinct biological hallmarks including extensive pseudopalisading necrosis (a configuration that is relatively unique to malignant gliomas), microvascular proliferation and angiogenesis, cellular heterogeneity, bilateral invasion, altered metabolism, immunosuppressive microenvironment and heterogeneity, and cancer stem-like cells (10–12, 15, 16). In addition, several potential pathologic hallmarks of glioma have been investigated, which include 1p/19q codeletion, IDH, EGFR, p53, PTEN/Akt pathway, Rb, Ras/MAPK pathway, extrachromosomal DNA, MGMT, TERT, and ATRX (5, 10, 12, 16). Genetic aberrations contribute to the specific glioma subtype and a unique metabolic footprint. On the other hand, metabolic reprogramming can, in fact, act as a driver of cancer genome modification and oncogenic pathways, through epigenetic, transcriptional, and posttranslational modifications (13). Exactly how cells acquire these hallmarks, and how they can be counteracted is the main question at stake for developing efficacious cancer treatments. Moreover, the emerging role of the human microbiome in modulating immune responses and tumor progression highlights the importance of addressing the complex interactions between tumor cells and microbiota. Until now, relatively little attention has been paid to the role of the human microbiome in glioma and particularly GBM. Previous studies have not been conclusive regarding the association between the human microbiome and gliomas and continuous research is required to reshape our understanding of the pathogenesis of glioma (17, 18). High secretion of EV is another characteristic of GBM. While most cells secrete EVs, human GBMs secrete EVs at significantly higher levels in vivo, approximately 10,000 EVs over a 48-h period per single GBM cell (19). The RNA-encapsulating EVs were first isolated from patient-derived glioma cells, and thereafter, glioma served as a useful model allowing EV release monitoring, cargo profiling, and intercellular communication investigation (19, 20). In this review, we summarize the current state of EVs in GBM and discuss the interkingdom cross-talk, including the communication established between intratumoral microbiome and immune host cells in the tumor microenvironment (TME). EVs provides a new insight into the pathogenesis of GBM. We also highlight the significant role of EVs in tumor progression, escape, and therapeutic response.
2 EV biogenesis, release, cargo, and uptake
EVs are phospholipid bilayer enclosed extracellular spherical structures secreted by cells into the extracellular space (21, 22). Recent advances in isolation and analytical methods have allowed the identification of an ever-increasing number of EV types: microvesicles (MVs), exosomes, apoptotic bodies, small ectosomes, migrasomes, marge oncosomes, and exophers (23–26) (Figure 1). EVs have been classified based on their biogenesis, release pathways, size, content, and function (21, 22, 27).
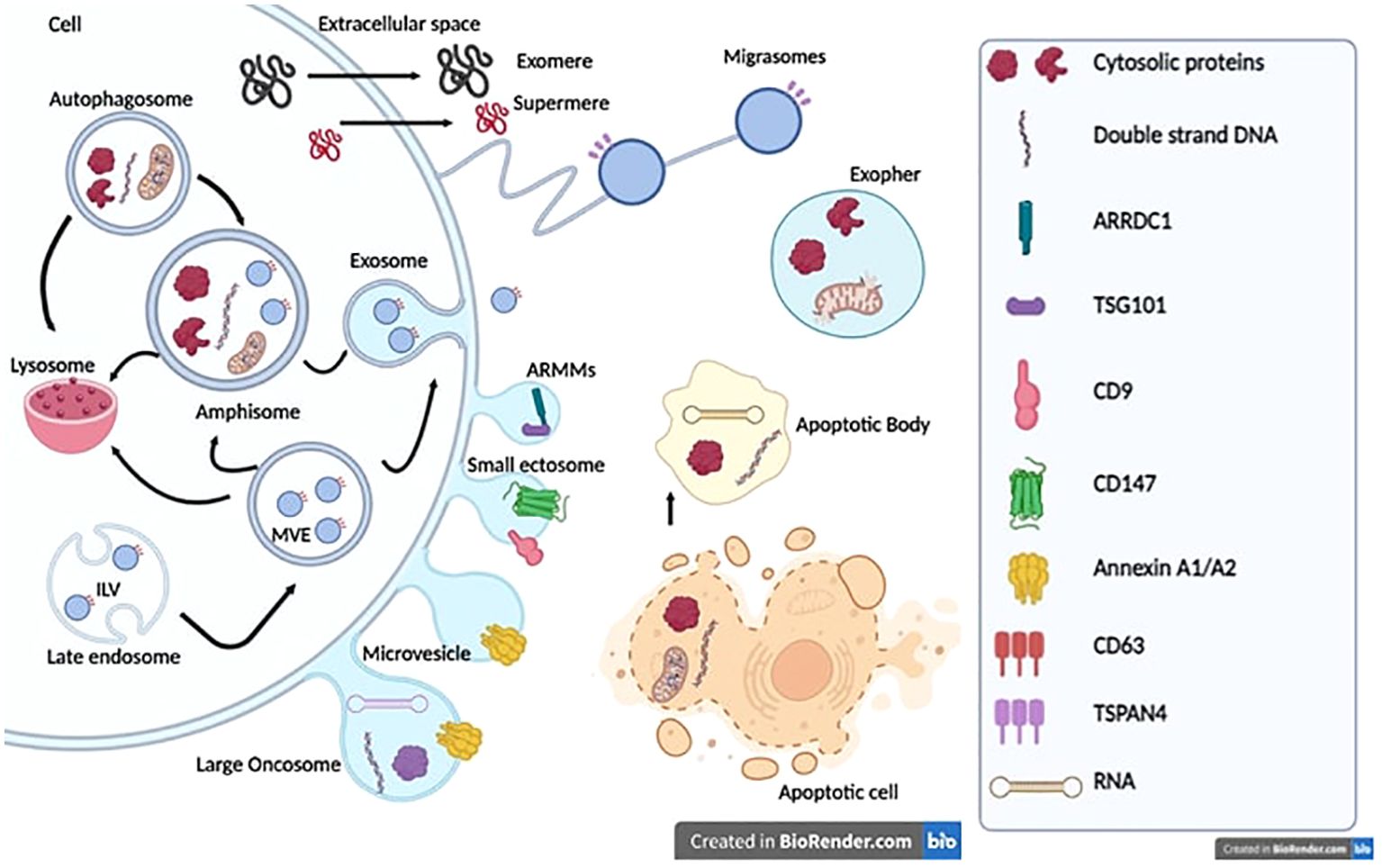
Figure 1. Extracellular vesicle biogenesis and release. Exosomes or small vesicles (30–150 nm) arise from multivesicular endosomes (MVEs) and amphisome. The maturation of the early endosome into the late endosome via inward budding of the endosomal membrane and encapsulation of intraluminal vesicles (ILVs) result in the formation of MVEs. Autophagosomes and MVEs can fuse with amphisomes or lysosomes, thus containing more proteins, nucleic acids, lipids and cytosolic components involved in degradation pathways. Microvesicles or medium/large vesicles (100–1,000 nm) emerge through direct budding of the plasma membrane straight to the extracellular space. Large oncosomes (1–5 μm) arise from tumor cells and contain oncogenic proteins and nucleic acids. ARMMs and small ectosomes (around 150 nm) originate from normal or cancer cells. Exomeres and supermeres (<50 nm) are mostly characterized by specific gene markers, especially TGFBI, ENO1, and GPC1. However, the underlying biogenesis mechanism remains unknown. Migrasomes originate from cell migration and involve structural and adhesion molecules such as Integrins. Exospheres (around 4 μm) result from the release of autophagosomes fused to lysosomes into the extracellular space. Apoptotic bodies originate from cells undergoing apoptosis and contain the remaining components of dead cells, proteins from the nucleus, mitochondria, lipids, and nucleic acids.
Cells typically communicate with each other by secreting signaling molecules, including proteins, lipids, and nucleic acids. In an effort to maintain homeostasis, influence metabolism, and regulate the immune response, cells can package different signaling molecules in EVs resulting in local and long-distance intercellular communication (21, 28). EVs contain various bioactive molecules both in the lumen and in the surface detected in all tissues and bodily fluids (23–25, 29–32).
• EVs have been considered for a long time as a means for normal cells to get rid of molecules it no longer wants, to maintain normal tissue homeostasis, or for cancer cells to promote their malignant tendencies (33, 34). It is now well established that EVs play their biological roles following their uptake by the recipient cell or by the interaction of EV surface proteins with cellular receptors and membranes (19, 26, 32, 35, 36). Indeed, secretion of specific types of EVs has been linked to numerous disease states, including cancer, neurological, and immunological disorders through aberrant signaling (36–39) (Figure 2).
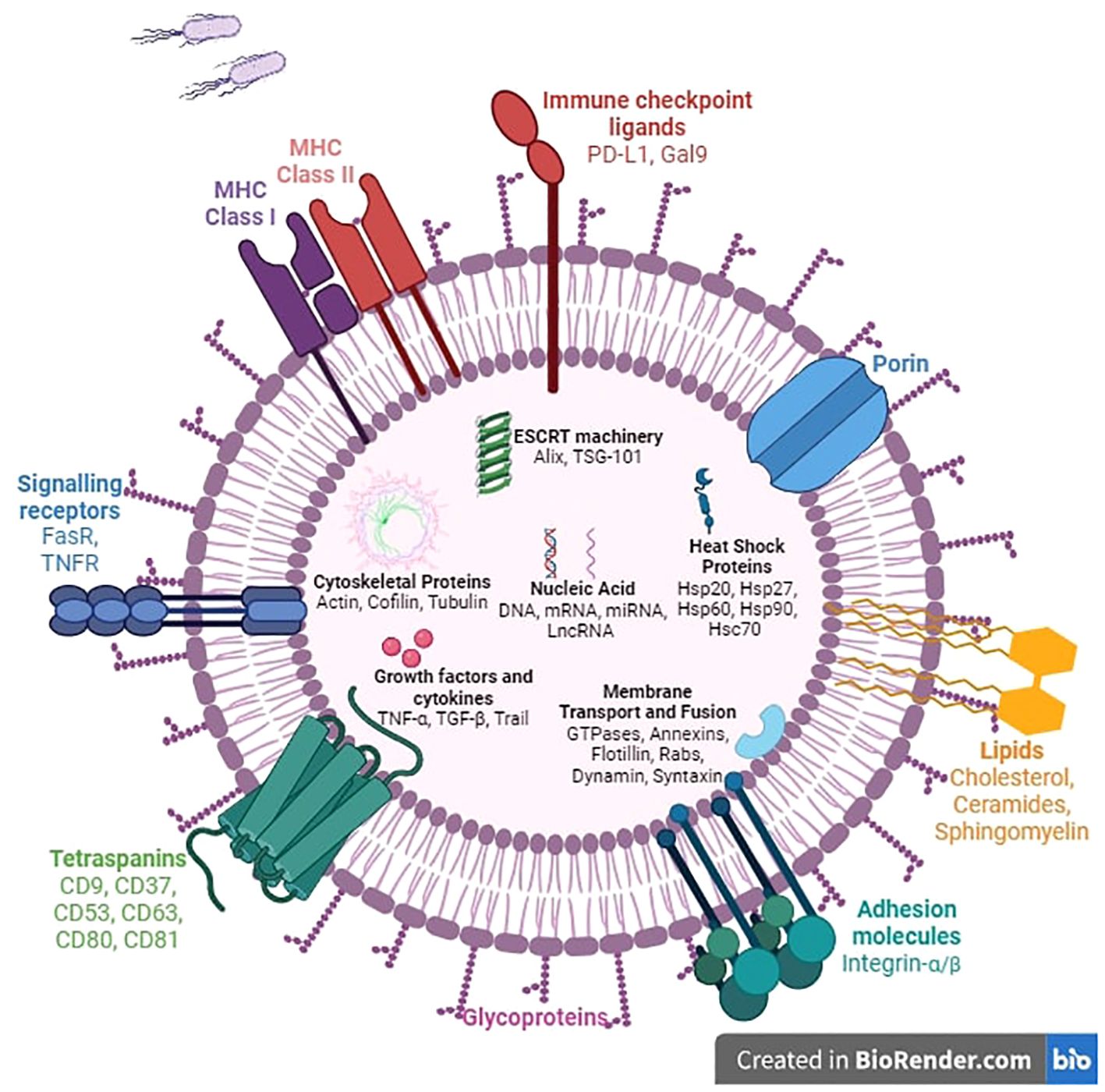
Figure 2. Composition of extracellular vesicles. The structure of extracellular vesicles includes intracellular, transmembrane, and surface components. Most intracellular molecules such as nucleic acids, growth factors, cytokines, and heat shock proteins will be released into the extracellular space and regulate numerous biological mechanisms. The ESCRT machinery, fusion proteins, and membrane transport proteins are involved in membrane remodeling to facilitate exchange between the two sides of the phospholipid bilayer. Surface proteins such as adhesion molecules, immune checkpoint ligands, MHC molecules, and death signaling receptors trigger various mechanisms including cell anchoring, immune cell regulation, and apoptosis.
Based on their origin, biogenesis, and, thus, their cargo composition, different types of EVs have been classified (21, 22, 27). At least two major modes of biogenesis are known: exosomes or small vesicles (30–150 nm), which starts with the formation of early endosomes that later fuse with the plasma membrane, and MVs or medium/large vesicles (100–1,000 nm) through direct budding of the plasma membrane straight to the extracellular space (Figure 1). The process of exosome generation starts with the formation of early endosomes that accumulate intraluminal vesicles (ILVs), by the inward budding of endosomal membranes, during their maturation towards late endosomes or multivesicular bodies (MVBs) (40). Late endosomes and MVBs are a subset of specialized endosomal compartments rich in ILVs, which encapsulate specific sorted proteins and nucleic acids, lipids, and cytosolic components. The fate of MVBs varies according to the proteins that are expressed on their surface. Some function as “delivery trucks” and get transported to plasma membrane via cytoskeletal and microtubule network and undergo exocytosis whereby the ILVs get released as exosomes into the extracellular space (41). Other MVBs function as “garbage trucks” and follow a degradation pathway either by direct fusion with lysosomes or by fusion with autophagosomes followed by lysosomes and thus promote ILV destruction and removal (42).
MVs are a heterogeneous group of membrane‐enclosed vesicles that shed by outward blebbing of the plasma membrane of various cells. These vesicles are loaded with multiple selectively sorted proteins including cytokines, chemokines, proteins involved in cellular signaling and/or migration, lipids, carbohydrates, and genetic material including messenger RNA (mRNA) and microRNAs (miRNAs) (43). Generation of MVs requires membrane lipid and actin cytoskeleton rearrangement to promote plasma membrane budding and subsequent vesicle shedding. The mechanism for classical MV biogenesis, cargo sorting, and shedding is tightly regulated by the small GTP-binding protein ADP ribosylation factor 6 (ARF6). A number of pathways including the small GTPase ARF6/phospholipase D/ERK/myosin light chain kinase pathway mediate phosphorylation of the myosin light chain resulting in actin cytoskeleton contraction at the MV necks in order to enhance myosin contractility and favor the fission and the release of the MVs (44).
Classical MVs are distinguished from the other EVs by size (150–1,000 nm) and lower flotation densities compared with small EVs, and are characterized by the expression of Annexin A1 as a specific protein marker of classical shedding MVs, distinct from both exosomes and arrestin-domain-containing protein 1 (ARRDC1)-mediated MVs (ARMMs) (23, 24, 29). ARMMs are small (inf 150 nm) arrestin domain-containing protein 1 (ARRDC1)-mediated MVs that bud directly from the plasma membrane. The budding of ARMMs requires ARRDC1, which is localized to the cytosolic side of the plasma membrane, and recruits the ESCRT-I complex protein TSG101 to the cell surface to initiate the outward membrane budding (45, 46).
Ectosome cargoes are enriched in cytoskeletal proteins, glycolytic enzymes, and integrins. Initially, they are assembled at the cytosolic face, then differentiated membrane microdomains appear at the cell surface followed by vesicle fission and rapid release to the extracellular space (47, 48). It has been shown that T cells release synaptic ectosomes (~70 nm) at the immunological synapse when they make contact with antigen-presenting cells. Thus, accumulated TCRs on the surface of extracellular MVs bud at the immunological synapse center. This process requires tumor susceptibility gene 101 (TSG101) for sorting of TCRs and inclusion in MVs, and vacuolar protein sorting 4 (VPS4) mediates scission of MVs from the T-cell plasma membrane (49, 50). Furthermore, ectosomes released by platelets induce differentiation of CD4+ T cells into Treg cells and may represent a mechanism of peripheral tolerance (51). Exposure of activated CD4+ T cells to platelet-derived ectosomes decreased their release of IFN-γ, TNFα, and interleukin-6 (IL-6), and increased the production of transforming growth factor-β1 (TGF-β1) (51). Finally, depending again on VPS4, perivascular dendritic cells (DCs) release antigen-bearing ectosomes to share antigen with mast cells and elicit anaphylaxis (52). Thereby, once the IgE-bound mast cells contacted an allergen on the surface of DC-derived ectosomes, they degranulated, releasing their inflammatory mediators (52). This ability of DCs to distribute antigen-bearing ectosomes to immune cells in the perivascular space potentiates inflammatory and rapid immune responses to blood-borne antigens.
Large oncosomes are a class of atypically large 1- to 5-μm MVs carrying abnormal and transforming macromolecules such as oncogenic proteins and nucleic acids (53). They have first been identified in highly migratory and invasive prostate cancer cells and have not been detected in benign tissues (53–56). Their release from tumor cells can be induced by overexpression or constitutive activity of oncoproteins (53, 56–58). Large oncosomes can participate in tumor progression through extracellular matrix degradation and exporting oncogenic content to other tumor or stroma cells, thus reprogramming their phenotype (transcriptomic, metabolism, etc.) and creating a tumor growth-supporting microenvironment (55, 56, 59).
Indeed, other vesicles like apoptotic bodies are formed in a similar way to MVs and considered as an important mediator of extracellular interactions. Apoptotic cell-derived EVs are released from cells entering apoptosis and formed through a process termed apoptotic cell disassembly. They contain the remaining components of dead cells, which include proteins from the nucleus, mitochondria, and plasma membrane; lipids; and nucleic acids like mRNA, long non-coding RNA (lncRNA), ribosomal RNA (rRNA), and miRNA (60–62). Apoptotic EVs have been divided into larger apoptotic bodies (1,000–5,000 nm) and smaller apoptotic vesicles (50–1,000 nm) (63–65). It has been suggested that the molecular cargo of apoptotic EVs differs based on size (66–69). By transporting bioactive molecules, proteins, lipids, and nucleic acids, apoptotic EVs are thought to promote regeneration in skin, bone, and muscle. They also function in inflammation and immune regulation within the TME (61, 70–77).
• Upon secretion into the cellular space, EVs can affect the target cells nearby and further away. Currently, the mechanisms and determinants of EV targeting are not fully elucidated yet (78, 79). There are four major pathways by which EVs can enter a recipient cell: macropinocytosis, lipid raft-mediated uptake, phagocytosis (phagocytosis, micropinocytosis, and lipid raft-, clathrin-, or caveolin-mediated endocytosis), and membrane fusion (26, 78, 80–82). Once the vesicle is internalized, its cargo can be degraded or released into the cytoplasm and transported to the nucleus or the cell membrane. However, it should be noted that the EV functionality does not require internalization, as surface proteins can interact with receptors on the plasma membrane of the recipient cell and initiate intracellular signaling cascades (83). The molecular mechanisms of exosome surface molecules, like tetraspanins, immunoglobulins, proteoglycans, and lectin receptors, binding to target cells are largely unknown (84–86). Exosomal ligands programmed death ligand-1 (PD-L1), TNF, FasL, and TRAIL are interesting potential targets for cancer therapies since their receptors are present on the cancer cell surface (87).
EV uptake and interactions trigger various intracellular signaling pathways that can induce epigenetic modifications in the recipient cells, through transfer of bioactive molecules, and affect cellular behavior and function. Even if EVs are a common communication channel of many cell types in many different contexts and pathologies, the processes involved and the messages they convey are highly personalized. It has been suggested that various aspects of tumor–host interactions are mediated through EVs. From an immunosurveillance point of view, it is becoming increasingly evident that EVs play key roles in cancer progression and drug resistance via promoting cancer-intrinsic pathways as well as immune microenvironment editing toward a pro-tumoral activities (88).
3 Pro-tumor roles of GBM-derived EVs in oncogenesis
The RNA-encapsulating EVs were first isolated from patient-derived glioma cells, and thereafter glioma served as a useful model allowing EV release monitoring, cargo profiling, and intercellular communication investigation (19, 20). It has been recognized that functional extracellular RNAs carried by EVs [such as miRNA, mRNA, rRNA, transfer RNA (tRNA), small RNA (sRNA), and lncRNA (89)] play important roles in intercellular communication.
While most cells secrete EVs, human GBMs secrete EVs at significantly higher levels in vivo, approximately 10,000 EVs over a 48-h period per single GBM cell (19). Furthermore, the presence of MVBs and exosomes inside GBM tissues has been demonstrated by electron microscopy (90). GBM-derived EVs are enriched in a wide variety of signaling molecules, functional RNAs, and lipids that modulate cell–microenvironment communication, support GBM progression, recurrence, and drug resistance through the establishment of a pro-tumoral microenvironment, thereby stimulating GBM cell growth, survival, and invasiveness (91–93). In addition, GBM-derived EVs modulate diverse aspects of the microenvironment like brain endothelial cells reprogramming toward an enhanced and disturbed angiogenesis, altering neighboring normal cells by propagating their oncogenic content, promoting the immunosuppressive properties of microglia, skewing the differentiation of peripheral blood-derived monocytes to activated M2-type macrophages with tumor supportive behavior, and suppressing T cell-mediated immune responses by acting on monocyte maturation and differentiation (94–101) (Figure 3).
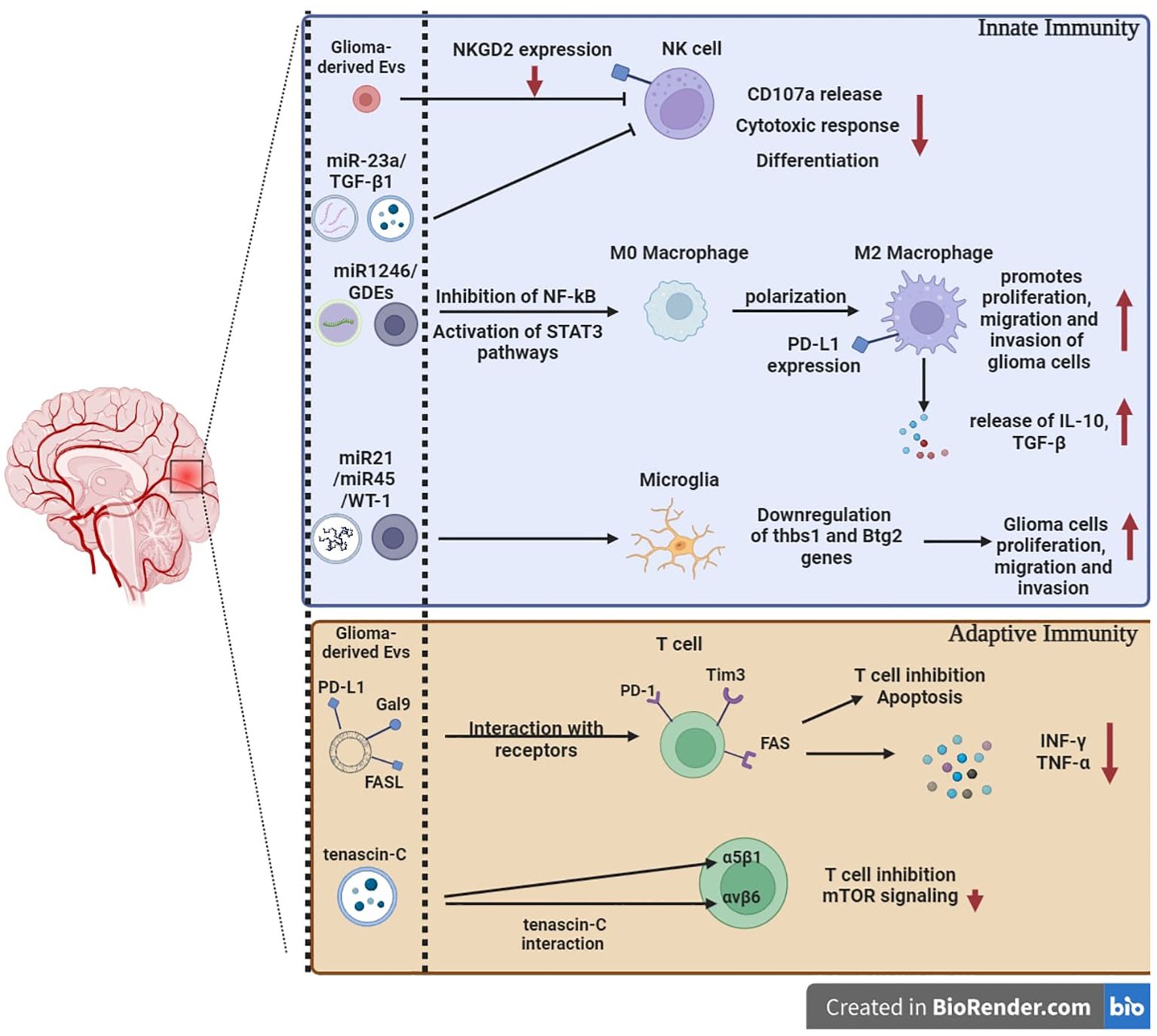
Figure 3. The immunomodulation of glioblastoma-derived extracellular vesicles. GBM-derived EVs include nucleic acids and intracellular and surface proteins. Specific miRNAs and GDEs can either inhibit effector immune cells or promote tumor progression by downregulating co-stimulatory receptors, upregulating co-inhibitory ligands, and activating M2 macrophages that release pro-tumor molecules. GBM-derived EVs expressing co-inhibitory ligands and death signaling ligands can act directly on effector adaptive immune cells and promote apoptosis and T cells’ inhibition. The regulation and differentiation of T cells can be impaired by direct interaction of tenascin-C with integrins involved in T cells’ effector function. miRNAs: MicroRNAs. GDEs: glioma stem-like cell-derived exosomes. Thbs1: thrombospondin 1. Btg2: BTG anti-proliferation factor 2.
It has been shown that EVs can serve as a means for short- and long-distance altered receptor transfer in GBM. Chief among specific genetic alterations in GBM is EGFR (102). The estimated rate of EGFR amplification in GBM ranges from 25% to 40%, and many contain the EGFRvIII variant, which is not expressed in normal brain (103–105). GBM-derived EVs containing EGFRvIII can merge with the plasma membranes of cancer cells lacking the receptor and share it with the recipient cells. This event leads to the transfer of oncogenic activity, including activation of transforming signaling pathways (MAPK and Akt), changes in expression of EGFRvIII-regulated genes [VEGF, Bcl-x(L), and p27], morphological transformation, and increase in anchorage-independent growth capacity, resulting in a subsequent transformation of the recipient cells that originally lacked the receptor (91).
It has been demonstrated that hypoxic GBM cells release small EVs (50–200 nm) with pro-angiogenic capacity, due to their enrichment with hypoxia-induced proteins including matrix metalloproteinase-9 (MMP-9), IL-8, platelet-derived growth factors (PDGFs), insulin-like growth factor binding protein (IGFBP)-1 and -3, and caveolin-1, which are associated with poor prognosis in glioma (94). To expand, GBM EVs may also have an immunomodulatory effect, modulating the TME to promote tumor growth via immune escape. In this regard, the cargo of GBM EVs comprised several immunomodulating molecules (i.e., TGF-β, IL-10, and heat shock proteins) as well as PD-L1, which binds to the PD-1 receptor on the surface of tumor-infiltrating lymphocytes (TILs) and leads to cancer immune evasion through the inhibition of T-cell responses and, in turn, decreased survival outcomes in cancer patients (106–108).
It has been suggested that the communication between GBM and surrounding cells in the microenvironment enhances the process of transformation and thereby feed continuously the tumor with newly transformed neoplastic cells. Glioma-derived EVs can be taken up by virtually every cell type in the brain microenvironment, including astrocytes, microglia, and microvascular cells, and therefore skewing their phenotypes toward tumor-promoting cells and thereby supporting the tumor progression or recurrence (19, 96, 109–114). For example, the immunosuppressive properties of microglia can be promoted after taking up EVs from GBM cells, underpinned partially by RNA-mediated mechanisms (98, 99). GBM-derived EVs also enhance the neovascularization capacity of human brain endothelial cells, by reprogramming brain endothelial cells toward highly distinct gene regulatory responses that converge on malignant vasculature, a hallmark of the GBM (95). A variety of mutated or amplified oncoproteins in glioma, such as P53, TERT, and RAS, can transform astrocytes to neoplastic cells in vitro and in vivo. It has been demonstrated that glioma-derived EVs are efficiently transferred to astrocytes, which provide a niche for glioma-initiating cell production in the brain microenvironment. EVs supports the self-renewal, proliferation, and anchorage-independent growth of human astrocytes, by enhancing aberrant signaling pathways commonly observed in GBM: activated Ras, telomerase, or simultaneously inactivated p53 and pRb pathways (102, 115). Furthermore, GBM EV-treated normal human astrocytes exhibit increased migratory capacity and enhanced cytokine production, which lead to increased tumor cell growth. GBM EV-treated normal human astrocytes also acquire tumor-like signaling pathways and exhibited colony-forming behaviors, suggesting that GBM EVs drive astrocytes to a tumorigenic phenotype that could impact the local environment to benefit the tumor itself (110). The transcriptomic analysis of the recipient astrocytes suggested dynamic changes of metabolic genes upon EV uptake, particularly factors of glycolysis, associated with activation of mitochondrial respiration and glycolysis in these cells (116–118).
Recently, it has been proposed that mRNAs encoding glycolytic enzymes and mitochondrial oxidative phosphorylation (OXPHOS) system factors secreted by glioma cells in EVs reprogram the metabolism of the GBM microenvironment (97). The direct transfer of mRNAs encoding metabolic factors may explain part of the observed metabolic alterations induced in astrocytes (97). Several classes of mRNAs, with the complete open reading frames and protein-coding potential, have been identified to be highly enriched in GBM EVs and have been suggested to exert functional effects in the recipient cells. Notably, transcripts for ribosomal proteins (RPs), mitochondrial OXPHOS system, and glycolytic factors represent the dominant fraction of the GBM EV-mRNA species. Ribosome activity is a critical regulator of growth and metabolism as ribosomal availability affects glycolysis and mitochondrial function (119). For instance, enolase-1 mRNA is encapsulated in both GBM stem cells’ MVs and exosomes. This mRNA encodes alpha-enolase, a key glycolytic enzyme, frequently overexpressed in glioma and multiple other cancers (120). Intriguingly, with its direct role in glycolysis, enolase-1 promotes cell proliferation by regulating the PI3K/AKT signaling pathway and is associated with glioma progression (120).
The EV-mediated transfer of oncogenes may contribute to the dysregulated proliferative and metabolic phenotypes observed in the astrocytes. Zeng and colleagues have shown that c-Myc and CCND3 mRNAs were encapsulated in glioma MVs (97). It is evident that Myc deregulation in cancer is a dramatic event in the cell. The MYC oncogene encodes a transcription factor, c-Myc, which tightly controls metabolic pathways to maintain cellular homeostasis in nontransformed cells. c-Myc is often genetically deregulated in cancer and correlates with the grade of glioma malignancy (121). Deregulated cancer metabolism impacts Myc expression and function. Consistently, it is no longer surprising that Myc operates at the intersection between metabolic pathway activation and gene expression. Furthermore, the uncontrolled growth of gliomas can be driven by frequent mutation and transcriptional dysregulation of cell cycle factors, such as cyclin D3 encoded by CCND3, and involved in the control of G1/S phase transition (122, 123).
To conclude, GBM-derived EVs mirror the molecular features of the tumor and its microenvironment (124–127). The expression level of several GBM-derived EV miRNAs and proteins has been linked to GBM pathogenesis and progression. Direct transfer of these mRNAs and proteins from tumor cells to normal cells within the brain microenvironment may aid/enhance their metabolic reprogramming and drive neoplastic transformation. Transformed cells adapt malignant mechanisms, through protein synthesis and metabolism, to support tumor growth and recurrence via EV-mediated horizontal mRNA transfer.
4 Role of EVs in the cross-talk between cancer cells and immune cells
The main infiltrating immune cell populations within the GBM microenvironment are tumor-associated macrophages (TAMs), immunosuppressive myeloid-derived suppressor cells (MDSCs), and CD4+CD25+Foxp3+T-regulatory cells (Tregs) that function as tumor growth promoters and induce T-cell dysfunction (128). However, despite the reduced proportion of GBM-infiltrating T cells, these are among the most critical cells in the antitumor response (129).
4.1 Effects on innate immune cells
Innate immune cells present in the GBM microenvironment are represented by cytotoxic NK cells and myeloid cells. It has been shown in vitro that GBM-derived EVs can impair the antitumor function of NK cells by suppression of NKG2D activating receptor expression and, thus, NK cell activation (129). It is well known that tumor-derived MVs secreted under hypoxic conditions compromise NK cell cytotoxic responses (130–132). Using multiple tumor models, it has been shown that hypoxic tumor-derived MVs contain two immunosuppressive factors, TGF-β1 and miR-23a, involved in the impairment of NK cell cytotoxicity. Following hypoxic tumor-derived MV uptake by NK cells, the transferred TGF-β1 decreases the NK cell surface expression of the activating receptor NKG2D, thus resulting in NK cell function inhibition. Similarly, miR-23a in hypoxic MVs reinforces the immunosuppression by targeting the expression of CD107a in NK cells (132). On the other hand, activated NK-derived EVs contain the cytotoxic proteins, perforin, granulysin, and granzymes A and B, and are known to induce dose-dependent apoptosis in neuroblastoma by caspase-dependent apoptotic pathways, which is possibly the same mechanism in other tumors like GBM (133).
Growing evidence reveals a central role for myeloid cells in the GBM microenvironment, including DCs, monocytes, macrophages, and microglia, and comprising around one-third of cells of the GBM tumor mass. The proportion of these tumor-associated cells is correlated with the clinical outcome in GBM and other solid cancers (134). Macrophages are of particular interest, as they can acquire different phenotypes according to microenvironment conditions. In solid tumors, including GBM, it is believed that the M1/M2 macrophage paradigm plays a key role in tumor progression. Historically, polarized M1 macrophages are deemed as antitumor cells, because of their enhanced capacity of phagocytosis, cytotoxicity, antigen presentation, and secretion of inflammatory cytokines. On the other hand, the M2-polarized macrophages are commonly regarded as tumor-associated macrophages (TAMs), and associated to pro-tumorigenic outcomes through angiogenic and lymphangiogenic regulation, immune suppression, EV production, hypoxia induction, tumor cell proliferation, and metastasis (135).
In GBM, recent observations suggest that non-polarized M0 macrophages, part of the so-called glioma-associated macrophages (GAMs), are present in the microenvironment (136).
From research within the last few years, released GBM-derived EVs were shown to promote a tumor-supportive macrophage phenotype. In vitro, GBM cell line-derived EVs were able to polarize blood-derived monocytes to M2‐like macrophages in vitro (99, 100, 137). Moreover, EVs from the hypoxic zones of GBM tumors induce M2 macrophage polarization, in vitro, which promote glioma proliferation, migration and invasion. Interestingly, it was shown that a polarization switch towards M2 phenotype exists through EV-mediated delivery of miR1246, which inhibit NF-kB and activate STAT3 pathways in macrophages (138, 139). Furthermore, glioma stem-like cell-derived exosomes (GDEs) are predilected toward monocytes and skew them toward the immunosuppressive M2 phenotype, including PD-L1 expression. GDEs contain members of the signal transducer and activator of transcription 3 (STAT3) pathway that functionally mediate this immune suppressive switch. Mass spectrometry analysis demonstrated that the GDEs are enriched in ephrin and axonal guidance signaling proteins, which are directly transferred to the cytoplasm of the monocytes (140). Glioma-derived exosomes suppress CD3+ and CD4+ T-cell activation and responses by acting on monocyte maturation and formation of monocytic MDSCs rather than on direct interaction with T cells (101). The use of EVs with immune checkpoints is one of the most important mechanisms leading to tumor immune escape and growth in many solid tumors, potentially including GBM. One such mechanism is the receptor Tim-3 that could be engaged by its natural ligand Galectin-9 and lead to immunosuppressive pathways (141). Indeed, it has been shown that cerebrospinal fluid-derived GBM EVs are enriched in Galectin-9 and decrease the antigen-presenting properties of DCs in vitro, in a Tim-3-dependent pathway (142).
Glioma-derived EVs exert pro-tumorigenic functions in monocytes and promote their conversion into suppressor cells involved in inhibition of activated CD4+ T cells through upregulation of suppressive cytokines, PD-L1, and lymphocyte antigen six complex (Ly6C), and downregulation of proinflammatory cytokines, MHC II, and costimulatory molecule expression (143).
4.2 Effects of EV on T-cell function
In one of the first studies to investigate GBM EVs’ effects on cytotoxic activity of immune cells, it has been reported that mouse GBM EVs promoted tumor growth and inhibited CD8+ T-cell cytolytic activity (144). Interestingly, high and low concentrations of GBM-derived EVs were shown to induce differential modulatory effects on peripheral blood mononuclear cells. Data provided by Hellwinkel et al. revealed that EVs at high concentrations induce selective tolerance associated with decreased IFN-γ secretion and migration capacities in peripheral blood mononuclear cells from healthy donors (145). Accordingly, EV-derived signals can act to suppress different aspects of T-cell responses. Indeed, in vitro secreted GSC-derived EVs were shown to be enriched in Tenascin-C that inhibits T-cell proliferation through interaction with α5β1 and αvβ6 integrins on T lymphocytes, associated with reduced mTOR signaling (146). The mTOR signaling pathway plays an essential regulatory role in the differentiation and function of both innate and adaptive immune cells (147). GBM-derived EVs exert an important role in immune evasion through the PD-1/PD-L1 axis (106). The EVs express PD-L1, which binds to PD-1 on activated T cells, resulting in the suppression of T-cell activation and proliferation. This leads to immune escape in glioblastoma patients (148), which confirm the critical function of EVs in facilitating intercellular communication during cancer development (149). Likewise, the expression of PD-L1 on EVs is associated with the mesenchymal GBM subtype and is identified in distinct niches of GBM samples, suggesting a possible involvement in tumor growth (106). Furthermore, treatment of IFN-γ in glioblastoma cells increases expression of PD-L1 and indoleamine 2,3-dioxygenase 1 (IDO1) in EVs, without affecting their size or frequency. IFN-γ-exposed GBM-derived EVs lead to higher differentiation of immunosuppressive MDSCs and NCMs in healthy donor monocytes when compared to naive GBM EVs. Monocytes treated with IFN-γ-exposed GBM EVs exhibit greater suppression of T-cell growth versus those treated with naive GBM EVs. Knocking down PD-L1 and/or IDO1 in GBM cells removes the immunosuppressive effect of IFN-γ-exposed GBM EVs on monocytes, suggesting that these molecules could be considered as possible therapeutic targets to combat GBM EV-mediated immunosuppression (143). It was shown that human GSC-derived EVs inhibited TCR-mediated T-cell activation and proliferation and that these effects arise through direct PD-L1/PD-1 interactions (106). By binding to PD-1 expressed on the surface of activated T cells, PD-L1, expressed by GBM cells and myeloid cells, induce T cell-mediated immune tolerance in tumor local microenvironment, leading to tumor immune escape and tumor growth stimulation (150).
Furthermore, PD-L1 on GBM-derived EVs in combination with other immunosuppressive molecules, FasL, CTLA-4, and CD39, suppresses CD4+ T-cell activation and induces apoptosis in CD8 T cells associated with reduced IFN-γ and TNF-α production, as well as an inhibition of NK cell and CD4+ T-cell response (129). This significant role in immunosuppression can be at least partially mediated by FasL, suggesting that both FasL expressed on GBM cells (by cell–cell contact) and FasL expressed on GBM-derived EVs inhibit T-cell functions (151). In GBM, it seems that the two immunosuppressive mechanisms are involved in T-cell inhibition (1): direct interaction of cancer cell-derived EVs with T cells in the TME, and (2) myeloid cell-dependent T-cell inhibition (101, 152–155). Owing to the natural origin, small size, and short half-life of EVs, monitoring whether in vitro results are representative of direct EV-mediated GBM/T-cell interactions in vivo remains extremely challenging.
5 Emerging role of bacterial extracellular vesicles in cancer
Microbe–host interactions are complex processes that directly and indirectly influence host health by modulating, among other mechanisms, immune responses, metabolism, and integrity of the intestinal barrier (156–162).
Bacteria communicate and interact with nearby bacteria, their environment, and the cells of their host through direct contact and secretion of soluble factors, such as metabolites, lipoglycans, nucleic acids, and proteins (163, 164). Bacteria also communicate via bacterial extracellular vesicles (BEVs), which are likely to be a highly efficient, robust, and economic manner of exchanging information between cells. BEVs are spherical membrane-enveloped particles ranging in size from 20 to 400 nm secreted by both pathogenic and non-pathogenic bacteria. Several lines of evidence show that BEVs can enter the systemic circulation and be detected in human body fluids that disseminate part of the molecular content of the parent bacterium into the extracellular milieu (165, 166). A combination of proteomic and biochemical analyses has demonstrated that BEVs carry a dynamic range of membrane-bound and periplasmic proteins, metabolites, nucleic acids (DNA and RNA), enzymes and toxins, polysaccharides, and peptidoglycan, and their cargo is controlled by specific molecular sorting machineries (165, 167, 168). Accumulating data now indicate that BEVs are heterogeneous in their structure, size, density, molecular cargo composition, and function, with different subtypes that vary based on their different biogenesis routes, the membrane envelope structure, the genetic background of the parental bacterium, and the environmental growth conditions (166). Chromosomal DNA in released BEVs from various Gram-negative pathogenic bacteria like Pseudomonas aeruginosa, Porphyromonas gingivalis, and Salmonella typhimurium is mainly extraluminal with a small fraction in the intraluminal space (169). It has been suggested that external DNA acts in biofilm formation while internal BEV DNA is involved in intercellular cross-talk and horizontal gene transfer of virulence, stress response, antibiotic resistance, and metabolism (169). In addition to innate immune response modulation, pathogenic BEV-derived DNA can be found inserted in the host genome in the nucleus of non-phagocytic cells (e.g., epithelial cells) (169). Like DNA viruses, the possibility that bacterial genetic material could be transferred to human somatic cells and integrated into the host genome is intriguing. The mechanisms underlying these integration events are still poorly characterized. The first proof-of-concept evidence that bacterial DNA sequences integrate into the human genome of cancer cells was reported by Riley and colleagues in gastrointestinal tumors with close proximity to the gut microbiome, suggesting a bacterial DNA role in carcinogenesis (170).
The mechanisms by which bacteria and BEVs impact carcinogenesis as well as tumor progression and therapy response are largely unknown. Several mechanisms have been advanced, including direct tumor-promoting mechanisms such as induction of genomic instability or indirect ones such as generation of proinflammatory and immunosuppressive TME (171, 172). For BEVs, almost all we know about their pathological potential is based on inflammatory disease studies (165, 173). The interkingdom cross-talk, either mutually tumor promoting or tumor inhibiting, between the intestinal, intratumoral microbiome, and host cells in the TME can be mediated through secreted microbial metabolites such as short-chain fatty acids or BEVs. Moreover, it is admitted that gut microbiome-derived BEVs can enter the circulatory system to disseminate to distant organs and tissues and interact with various resident immune cell populations like DCs, neutrophils, and macrophages. The potential role of circulating BEVs have been largely discussed as immunomodulators or even a key driver of premetastatic niche formation in distant organs, a conducive microenvironment to the survival and outgrowth of tumor cells before their arrival at these sites (174). In the same line, a retrospective pan-cancer examination of whole-genome sequencing datasets in the TCGA for microbial reads found unique microbial signatures in tissue and blood that could discriminate between and within most major types of cancer (175). This was confirmed with a study demonstrating that pancreatic adenocarcinoma microbiome composition, which cross-talks to the gut microbiome, influences the host immune response and predicts long- versus short-term survival (176).
The role of BEVs on oncogenesis and tumor progression is likely to be context-dependent. Based on studies of BEVs in infectious diseases, it has been suggested that microbial dysbiosis in cancer could enhance the systemic release of microbiome-derived BEVs, which could promote tumor progression by immunosuppressive reprogramming of the TME. BEVs can drive suppressive cellular monocytic differentiation and indirectly induce T-cell anergy, in a TLR-dependent manner (171). On the other hand, BEVs are able to interact with host cells in distant organs through engaging their microbe-associated molecular pattern (MAMPs) to initiate proinflammatory signaling and drive alterations in the immune landscape, particularly myeloid cells, to foster pre-metastatic niches.
Gut bacteria-derived BEVs have been shown to prime the host innate immune system with subsequent activation of T-cell responses, in a strain-specific manner. Specific immunomodulatory effects were due, in part, to the differential regulation of miRNAs (177). It has been shown that exosomes released by BEV-activated DCs were enriched in surface proteins involved in antigen presentation and T-cell activation, but differed in the content of immune-related miRNA, depending on the origin of the BEVs (177). From a therapeutic standpoint, to identify new candidates for inclusion in the acellular vaccine formulations, spontaneously released outer membrane vesicles (OMVs) were used as a potential source of key adhesins (178). Adhesins are virulence factors that are surface-bound protein or polysaccharide molecules that confer tissue-specific binding during microbial pathogenesis (179). The clinically approved OMV-based 4CMenB vaccine (4CMenB; Bexsero, GSK) is a four-component protein-based meningococcal B vaccine that was licensed in the European Union in 2013. This vaccine is composed of three highly immunogenic recombinant antigens (factor H–binding protein, Neisseria heparin–binding antigen, and Neisseria adhesin A), as well as OMV containing Porin A subtype P1.4 from the strain NZ98/254 (180). Interestingly, the OMV-based 4CMenB was shown to confer a broad protective antibody response against different Neisseria meningitidis and provide a level of cross-protection against Neisseria gonorrhoeae because of the molecular similarities shared between the two pathogens (181–183) (Figure 4).
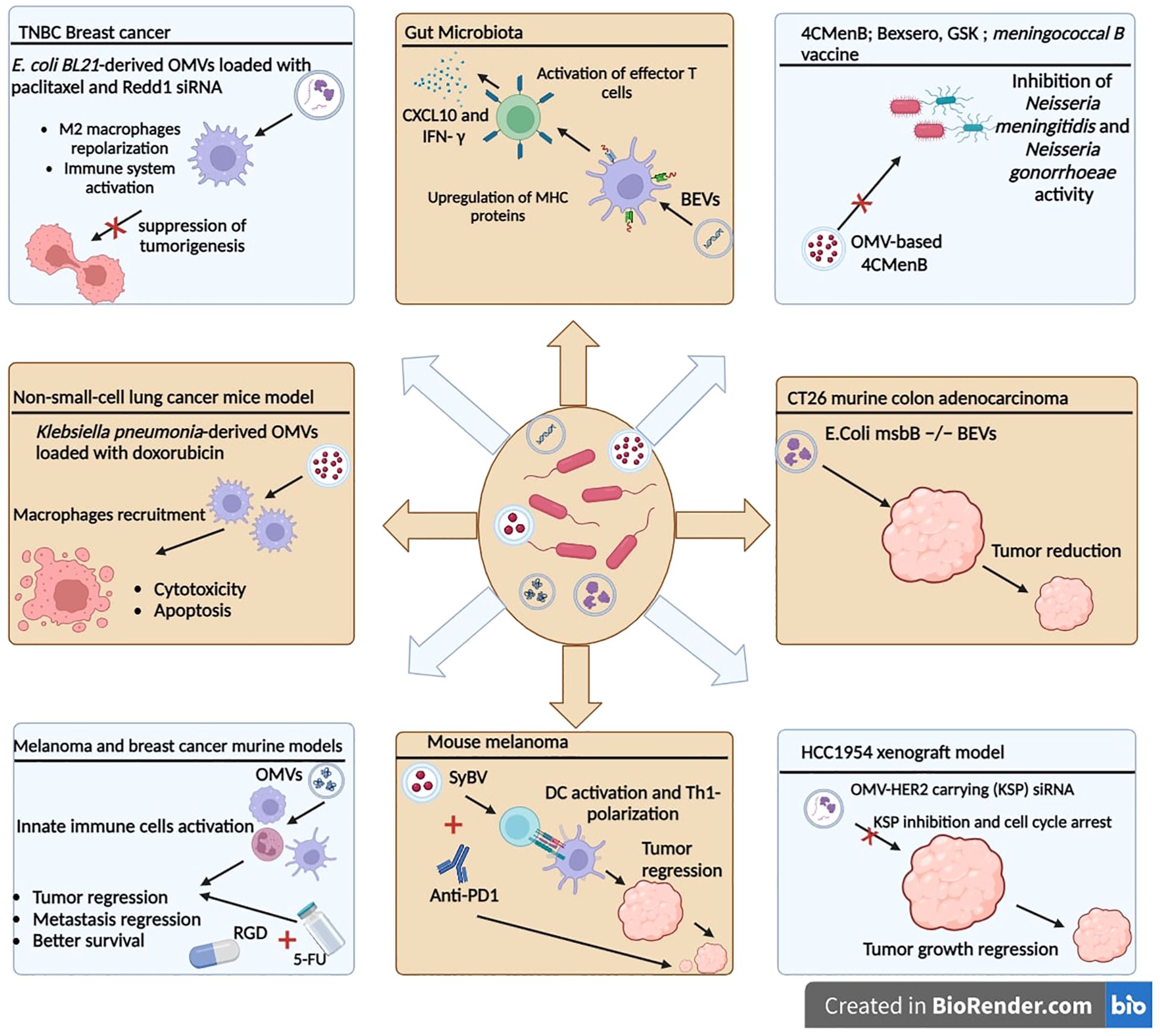
Figure 4. Therapeutic applications of bacterial extracellular vesicles. OMVs can be used in therapeutic strategies and combined with chemotherapy agents or siRNAs to improve the antitumor response. OMVs: outer membrane vesicles. SyBV: synthetic bacterial vesicles. BEVs: bacterial extracellular vesicles.
As biologically derived entities, the properties of BEVs, like endotoxicity, can be easily determined through molecular biology and genetic engineering approaches. It is expected that, in the future, BEVs will be used as cancer immunotherapeutic agents or cancer vaccines in conjunction with other therapeutic forms, to elicit durable antitumor immune responses. Kim et al. showed the greater immunogenic potential of OMVs over bacteria and the evidence to explore this further in animal models. In a recent report, they showed that systemic intravenous administration of BEVs, from the genetically modified Escherichia coli msbB −/− strain (endotoxin-free), in CT26 murine colon adenocarcinoma transplanted mice significantly reduced the tumor volume in a concentration-dependent manner. Biodistribution studies indicated a selective tropism for tumor tissue, which was attributed to the enhanced permeability and retention (EPR) effect given the nano-size range of these OMVs (38.7 ± 4.2 nm) (184). The EPR effect is a property in which the appropriate sizes of nanoparticles leak preferentially into tumor tissue through permeable tumor vessels and are then retained in the tumor bed due to reduced lymphatic drainage (185). The remarkable capability of inducing long-term antitumor immune responses was associated with CXCL10 and interferon-γ cytokine production that can fully eradicate established tumors without notable adverse effects (184). A mutant E. coli strain that exhibits less immunogenicity and consequently less toxicity toward human cells was engineered to generate OMVs displaying a cellular selectivity, incorporated genetically with a human epidermal growth factor receptor 2 (HER2)-specific affibody in the membrane as a targeting ligand. The authors used the approach of endogenous loading of antigens to the OMV lumen to generate OMV-HER2 carrying kinesin spindle protein (KSP) siRNA, which is an overexpressed protein in rapidly dividing cancer cells. KSP inhibition cause cell cycle arrest during mitosis by inhibiting KSP function, ultimately leading to cell death. In vivo studies in the HCC1954 xenograft model showed that siKSP-packaged OMVs caused targeted gene silencing and induced highly significant tumor growth regression. Interestingly, passive siKSP-loaded OMVs, without HER2 targeting, showed partial tumor regression, offered by the EPR effect, which increases extravasation and retention within the tumor bed (186). Another approach to reduce toxicity and avoid excessive activation of the immune system is the synthetic bacterial vesicles (SyBVs), which are spherical synthetic bacterial vesicles with similar morphology and size to natural bacterial OMVs, but carry less proteins and nucleic acids. The better toxicity profile of SyBVs, compared to OMVs, is due to the limited cytosolic molecular content. Furthermore, SyBVs are capable of engaging cells of the immune system such as DCs and eliciting an adaptive immune response. Co-immunization with SyBV and mouse melanoma derived EVs enhances tumor regression in melanoma-bearing mice in a Th1-dependant manner. Moreover, the immunotherapeutic effect of SyBV was synergistically enhanced by anti-PD-1 inhibitor (187).
Recently, combination therapy using OMVs, as a nanoparticle coating, in tandem with conventional cancer therapies was evaluated. The combination of attenuated Salmonella-derived OMVs with chemotherapeutics has been evaluated (188). First, the OMV coating approach has been used to elicit an innate immune response as it travels to the TME, followed by targeting the tumor cells via the arginyl-glycyl-aspartic acid (RGD) peptide on the surface, and subsequently delivering the chemotherapeutic prodrug 5-fluorouracil (5-FU) tegafur (188). The study’s promising results show that successive pretreatment in the mouse model protects against tumor challenge and seems to act like a vaccine. The therapeutic efficacy was also confirmed in melanoma and breast cancer murine models, eliciting repressed tumor growth, reduced metastatic nodules, and better survival than control and tegafur-treated mice groups. In the non-small-cell lung cancer mouse model, apoptotic and cytotoxic effects have been observed with passively loaded doxorubicin in attenuated Klebsiella pneumonia-derived OMVs. This antitumor effect of doxorubicin-loaded OMVs was synergized by macrophage recruitment in the TME and enhanced immunogenicity (189). In another study using a triple-negative breast cancer model, genetically engineered E. coli BL21-derived OMVs were loaded with paclitaxel and Redd1 siRNA, to enhance immune system activation and chemotherapeutic drug delivery (190). Redd1 is a negative regulator of mTOR signaling and is defined as a key metabolic regulator suppressing tumorigenesis (191). The results show that, after reaching the TME, the paclitaxel is released, followed by OMV-associated siRNA uptake by M2 macrophages leading to tumor-associated macrophage repolarization and tumor immune response activation (190).
Li et al. generated modified OMVs expressing the ectodomain of PD-1 on the surface (OMV-PD-1) capable of inducing a proinflammatory immune response in DCs, and interacting with PD-L1 on the tumor surface (192). Importantly, the engineered OMV-PD-1 can bind to PD-L1 on the tumor cell surface and facilitate its internalization, thereby protecting T cells from the PD-1/PD-L1 immune inhibitory axis. Moreover, in the colon carcinoma cell line CT26 model, 40% of mice exhibited complete tumor regression associated with increased levels of pro-inflammatory cytokines IFN-γ, IL-6, and TNF-α in tumor and serum, and enhanced CD8+ T-cell infiltration. More broadly, the study illustrates the potential of OMVs as a promising agent for cancer immunotherapy capable of regulating the TME and subsequently increasing antitumor therapy efficacy.
Finally, the interaction of OMVs with the host immune system makes them an exciting option for therapeutic cancer vaccines (193–195). Engineered OMVs have elicited an efficient cytotoxic CD8+ T-cell activation by DCs (196). A recent study has demonstrated that OMVs conjugated to antigenic epitope tyrosinase-related protein 2 (TRP2) drive antitumor immunity by eliminating tumor metastasis and inducing a strong cytotoxic T-cell response. These OMVs accumulate in the lymph nodes and carry the potential to efficiently present antigens to the DCs, bringing us one step closer to personalized cancer vaccines (193).
While fecal microbiota transplantation seems to hold promise for many diseases, including cancers, recent events have triggered a greater need to monitor the transfer of antibiotic resistance, which is the main significant risk directly related to fecal microbiota transplantation. Moreover, other causes of deaths following fecal microbiota transplantation have been attributed to heart attack and associated with increased amount of trimethylamine oxide, a metabolite produced by gut bacteria that was shown to be involved in cardiovascular diseases (197). A safer and more controlled way of utilizing the immune-modulatory effect of microbial parts would be to use BEVs. Systemic administration of BEVs directly to tumor-bearing hosts may constitute one of the promising directions of BEVs-based cancer therapy, and could represent a superior alternative to fecal microbiota transplantation (198). The intrinsic properties of BEVs including immunogenicity, a cell-free system, the non-replicative nature, and, thus, safety and nanoscale structure made them become a potential candidate for cancer treatment. Based on their inbuilt adjuvanticity, thermostability, and resistance to low pH and enzymatic degradation, and immunomodulatory properties, several studies tried to evaluate BEVs use for vaccination against infectious pathogens (165, 199).
6 EVs in interkingdom communication in the immune tumor microenvironment
The horizontal EV transfer is a new form of intercellular communication that operates at both short and long distances to regulate gene expression, angiogenesis, immune responses, and cell metabolism (112, 200). In cancer, the transfer of EV-associated biomolecules delivers complex biological messages from one cell to another and thereby spread malignant traits across the microenvironment (113). It has been shown in multiple studies that GBM-released EVs are incorporated by neighboring cells in the brain microenvironment, including endothelial cells and microglia, leading to altered phenotypes and functionality, and creating a more supportive TME (95, 98, 109). Bidirectional EV communication shares functional molecules between cancer and stromal cells to facilitate intercellular communication and regulation within the TME.
Recent research has revealed that EVs have a role in the progression of GBM and in the reconstruction of the TME (201) through the interaction with stromal cells, monocytes, macrophages, mast cells, microglia, T cells, astrocytes, and oligodendrocytes (202). GBM-derived EVs also regulate many cellular and extracellular components of the TME, leading to GBM growth and progression (203). GBM-derived EV-mediated interactions may allow TME cells to become activated, notably fibroblasts, microglia, and macrophages. The latter may also adopt either M1 or M2 phenotypes. On the other hand, this cross-talk could potentially result in lineage conversion towards more aggressive phenotypes, such as anaplastic astrocytoma arising from astrocytoma or oligodendroglioma (204). Furthermore, EVs influence other types of cells in the CNS to support the TME. For example, GBM EV-treated astrocytes demonstrate enhanced migration and cytokine production, leading to a tumor-supporting phenotype with a senescence-associated secretory profile (205). Moreover, during treatment with GBM-derived EVs, normal astrocytes demonstrate enhanced migration rates and heightened release of cytokines and growth factors, which could then cooperate with EGF in recruiting precursor cells of mesenchymal origin (110).
Several studies have shown that microglial cells or astrocytes play a critical role in GBM progression (206). Based on recent findings, the complex network of interaction between microglial/astrocytes cells and GBM constitute a potential therapeutic target (207). One reason for this is that because of the glioma-derived EV uptake by astrocytes, the cells possessing high transformation capacity to glioma provide a pool of glioma-initiating cells in the brain microenvironment. It has been shown that EVs enhance self-renewal, proliferation, and anchorage-independent growth properties of human astrocytes, by triggering Ras and telomerase activation or simultaneously p53 and pRb inactivation pathways, the most common signaling aberrations observed in GBM (102, 115). The astrocyte transformation is related to the malignant characteristics of GBM-derived EVs that can elicit additional effects on astrocytes, such as promoting their migration (110).
It has been shown that GBM-derived EVs regulate immune cell activity in the TME (208). Notably, GBM-derived EVs generated upon tumor apoptotic cells may bind to adjacent cells and change their phenotype to become more aggressive. Additionally, these EVs help create an immunosuppressive environment that prevents GBM from antigen-specific detection and death by T cells (208). For example, it has been demonstrated that GBM-derived EVs can facilitate recruitment of Tregs along with additional suppressor cells (209). MDSCs, like other kinds of immune cells, are impacted by GBM and GBM-derived EVs. In vitro treatment of PBMCs with GBM-derived EVs raises the MDSC population that exhibit more pronounced immunosuppressive phenotypes and aberrant miRNA profiles approximately 1.5-fold (210, 211). In spite of the increased number and activated state of DCs in GBM patients’ cerebrospinal fluid, the majority of these cells are unable to adequately present tumor antigens. In several situations, GBM-derived EVs severely reduced the antigen-presenting ability of DC as mentioned earlier (142). GBM-derived EV specifically impacts cells of the monocytic lineage, such as monocytes, macrophages, and microglia (100). In fact, GBM-derived EVs can cause peripheral blood monocytes to differentiate into alternatively activated M2-type macrophages. This impact is seen in EVs derived from established cell lines as well as initial cultures of GBM stem-like cells (GSC) (100). Furthermore, GSC-derived EVs influenced primary human microglia, resulting in elevated production of membrane type 1-matrix metalloproteinase, a hallmark for tumor-supportive microglia (100). Moreover, modulatory effects on PBMCs were determined through differential low and high EV concentration effects. These findings suggest that high concentration of EVs can cause specific immune tolerance within the TME (145). Overall, GBM-derived EVs negatively impact the TME immune cells, resulting in an immunosuppression that promotes tumor growth.
Additionally, in glioma, accumulated data suggest that high expression of glycolytic signature genes predicts unfavorable prognosis and immunological heterogeneity (212–217). Interestingly, the GBM-derived EVs can reprogram the metabolism of the recipient pre-transformed astrocytes by activating both glycolysis and OXPHOS, providing a dynamic cross-talk between cancer cells and neighboring cells of the TME. Notably, according to proteomic studies, exosomes contain several key glycolytic enzymes, such as GAPDH, enolase, pyruvate kinase, and phosphoglycerate kinase (108, 218). Several classes of mRNAs are highly enriched in GBM and, most remarkably, transcripts for RP, OXPHOS, and glycolytic factors account for more than 50% of the EV-abundant mRNA (97). In addition to these described means, EV-mediated transfer of oncogenes is another mechanism driving the proliferative and metabolic phenotypes observed in the astrocytes. The full complete coding sequences of c-Myc and CCND3 mRNAs were shown to be encapsulated in glioma-derived EVs (97). The transcription factor c-Myc, which correlates with the grade of glioma malignancy, is known to modulate metabolic reprogramming in the pathogenesis of glioma. c-Myc is also important for the proliferation, growth, and survival of glioma cancer stem cells (121).
It is now well established that the gut microbiome affects the behavior of tumors through blood circulation, bacterial metabolites, and enterohepatic circulation (219–221). Mucosal barriers of the gastrointestinal tract are the hub for interspecies and even interkingdom communication. It is now well established that the gut microbiota is one of the key elements implicated in cancer and shown to modulate anticancer drug efficacy. EVs released by host eukaryotic cells and from prokaryotic symbiotic and/or pathogenic cells, fungi, and parasite-derived EVs meet in intraluminal space and interact constantly with intestinal host cells (222, 223). Interspecies communication between nematodes and host intestinal cells has been recently reported in a mouse model showing that Heligmosomoides polygyrus secreted miRNA-loaded EVs suppress host immune response after being internalized by host mice cells (224).
Production of EVs from human parasites, such as trematodes and nematodes, or parasitized cells has been described for a number of parasitic infections (225–227). Recognizing the presence of invading pathogens by germline-encoded pattern recognition receptors is key to mounting an effective innate immune response (228). For example, circulating exosomal miRNAs act as ligands of Toll-like receptors (TLRs) after internalization by target host cells (229). In the same line, mice TLR13 recognize the 23S ribosomal RNA molecule of bacterial pathogen Staphylococcus aureus (230). Recently, bacterial DNA integration into the human genome has become a hot topic as it has been found around cancers, such as pancreatic cancer, breast cancer, and colorectal cancer. New lines of evidence support the hypothesis that bacterial integrations and related mutagenesis through lateral gene transfer occur in the human somatic genome and play a role in carcinogenesis (170, 231, 232).
In esophageal squamous cell carcinoma (ESCC), it has been hypothesized that intratumoral microbiota constitute a bridge between digestive tract microbiota and the tumor immune microenvironment, which inevitably influence esophageal carcinogenesis (233, 234). In a study published by Zhang et al., the characterization of the ESCC TME unveils a high abundance of intratumoral Lactobacillus and bacterial alpha-diversity, associated with the formation of the immunosuppressive TME depicted by the upregulated PD-L1 expression on epithelial cells and TAMs, and reduced infiltration of NK cells and activated cytotoxic T lymphocytes (235). The authors speculate that intratumoral microbiota might influence patients’ outcomes through the immunosuppressive TME (235, 236).
Moreover, tumor molecular mimicry by gut and extra-gut microbial species producing epitopes that resemble tumor neoantigen epitopes is likely to influence the quality and strength of the immune anticancer response (237). Molecular mimicry occurs when similarities between foreign and self-peptides favor an activation of T or B cells (238). Molecular mimicry can lead to the formation of cross-reactive antigens and/or T-lymphocyte activation and proliferation. Furthermore, epitope spreading, defined as the diversification of epitope specificity from the initial dominant epitope-specific immune response directed against a self or foreign protein, damages healthy tissue and induces apoptosis and concomitant presentation of self- and microbial antigens (239, 240). In an elegant work of Fluckiger et al., the authors reported that MHC-I epitopes derived from a prophage in the gut microbiomes are cross-reactive tumor antigens that enhance immunotherapeutic efficacy in both the preclinical murine model and cancer patients (241). These data highlight the important role of microbiome in modulating antitumor responses and that one of the mechanisms is molecular mimicry.
In CRC, it has been hypothesized that EV-derived proteomes from gastrointestinal tract cancer cells match gut microbiome protein sequences. To investigate this, the CRC EV proteome has been compared with protein sequences from different commensal bacteria and viruses and a number of matching microbial sequences were identified (242–245). Strikingly, the pseudokinase domain sequence in the B. fragilis genome matches the PDGFR-α sequence. The oncogenic mutations of PDGFRs and overexpression of PDGF/PDGFRs members are implicated in cancers and are associated with the stage, grade, and poor outcomes of various cancers (246–249). In CRC, it has been suggested that the presence of pseudokinase with activation loop and homology to PDGFR-α in Bacteroides spp. may be related to PDGFRα’s role in CRC pathogenesis (250). Hence, matching protein sequences from the host cell-derived EV proteome with the protein sequences of microbiome will help to identify new similarities between bacteria and host cells, including cancer and immune cells and proteins, and understand their functional role in cancer pathogenesis.
Exosomes offer numerous options to study physiological processes and pathologies. Aside from their innate cargo, exosomes from several taxonomic kingdoms have been shown to be loadable with therapeutic agents, acting as nanocarrier for drug delivery (251). Some of the exosomes’ advantages regarding therapeutic purposes are their biocompatibility, stability, low toxicity, penetration into deep tissues, a characteristic zeta potential allowing prolonged circulation, and their intrinsic cell-targeting properties (251–253). Medical potential applications of exosomes include therapeutic approaches such as anticancer therapies, regenerative medicine, microbial vaccines with low immunogenicity that help avoid autoimmunity, cancer vaccines, drug delivery systems, and biomarkers in early diagnosis and therapy monitoring (251).
GBM-derived EVs play vital roles in the induction of the TME, which in the GBM context involves the relationship between GBM tumors and adjacent cells, inducing immunosuppression and stimulating cancer cell proliferation within the brain. Furthermore, in the TME, EVs can serve as a vehicle for both paracrine and endocrine signaling, to adjust metabolic pathways of cells to fit into the objective of the TME.
7 Conclusions and future perspectives
To conclude, it is now well established that EV release by cancer cells and other cells within the GBM microenvironment, as well as their presence in biological fluids, is an incontrovertible feature of GBM biology. However, further research efforts are needed to understand and address the functional properties of EVs to potentially gain from the GBM TME-derived EVs.
GBM-derived EVs show strong therapeutic translation potential. Such EVs carry biomolecules that are similar to those that might be secreted by tumor cells, which makes these EVs useful for both diagnostic and therapeutic purposes in GBM. Investigations have shown their ability to cross the blood–brain barrier, allowing imaging agents and treatments to be delivered directly to GBM lesions. Furthermore, EV cargo acts as a pharmacodynamic reporter, providing information about drug distribution and target interaction, which may improve early-phase clinical trials of new therapies. Large amount of data highlights the critical significance of GBM-EVs in improving diagnosis and therapy relating to this challenging brain cancer.
In order to understand the very complex interactions between cancer cells, immune cells, and microbes in the TME, the biologically active concentrations of EVs that actually reach the different intratumoral structures of GBM (e.g., enhancing tumor, necrotic, edema, and non-enhancing tumor) remain to be determined.
Characterizing EV-producing cells provide opportunities to modulate EV biogenesis, release, and cargo content, such as a bioactive proteins or miRNAs. Different strategies can be considered to modulate EV content and biological activities, including biochemical stimuli and genetic modification of the producing cells to overexpress specific proteins or miRNAs.
However, the cargo molecule that will be therapeutically targeted would need to be chosen based on using the relevant non-clinical models for rigorous functional testing of recipient cell responses, ultimately in vivo. By gaining a better understanding of the biological functions of EVs and manipulating their biogenesis, research methods in GBM need to be ahead of the game to control their pathophysiological effects in tumor development and produce populations of EVs with antitumor effects.
Finally, isolation and characterization of distinct subsets of EVs from plasma, CSF, or urine have established the proof of principle of using EVs as liquid biopsy biomarkers for the early detection, prognosis, and monitoring of various cancers. Moreover, the non-invasive use of EVs from blood of GBM patients should be envisaged for the monitoring of cancer therapy response efficacy and overcoming cancer drug resistance, notably in immunotherapy clinical trials, with appropriate use of precise and robust EV and cargo characterization.
Finally, further research needs to be performed in this area to comprehensively characterize EV biogenesis and release, and cope with complexity that exists in the interactions between organisms on interspecies and interkingdom levels.
Author contributions
BG: Conceptualization, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. ZH: Software, Writing – original draft. MR: Visualization, Writing – review & editing. A-SK: Software, Writing – original draft. AE: Validation, Visualization, Writing – original draft. AB: Conceptualization, Investigation, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Moroccan Ministry of Higher Education, Research and innovation through a “PPR1” project and by the Moroccan Ministry of Higher Education, Research and innovation and the Digital Development Agency "ADD” through an “Al-khawarizmi” project and Intra-Africa Academic Mobility Grant provided by the European Commission, ref number 624289-PANAF-1- 2020-1-KEPANAFMOBAF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ARF6: ADP ribosylation factor 6
ARMMs: Arrestin-domain-containing protein 1-mediated microvesicles
ARRDC1: Arrestin-domain-containing protein 1
BEVs: Bacterial extracellular vesicles
CNS: Central nervous system
CRC: Colorectal cancer
DCs: Dendritic cells
EGFR: Epidermal growth factor receptor
ESCC: Esophageal squamous cell carcinoma
EVs: Extracellular vesicles
GAMs: Glioma-associated macrophages
GBM: Glioblastoma multiforme
GDEs: Glioma stem-like cell-derived exosomes
HER2: Human epidermal growth factor receptor 2
IDH: Isocitrate dehydrogenase
IGFBP: Insulin-like growth factor binding protein
IL: Interleukin
ILVs: Intraluminal vesicles
KSP: Kinesin spindle protein
lncRNA: Long non-coding RNA
Ly6C: Lymphocyte antigen 6 complex
MAMPs: Microbe-associated molecular pattern
miRNAs: MicroRNAs
MMP-9: Metalloproteinase-9
mRNA: Messenger RNA
MSDC: Myeloid-derived suppressor cells
MVBs: Multivesicular bodies
MVs: Microvesicles
OMV: Outer membrane vesicles
OXPHOS: Oxidative phosphorylation system
PDGFs: Platelet-derived growth factors
PD-L1: Programmed death ligand-1
PLNTY: Polymorphous low-grade neuroepithelial tumor of the young
RGD: Arginyl-glycyl-aspartic acid
RP: Ribosomal protein
rRNA: Ribosomal RNA
sRNA: Small RNA
SyBV: Synthetic bacterial vesicles
TAMs: Tumor-associated macrophages
TGF-b1: Transforming growth factor-b1
TILs: Tumor-infiltrating lymphocytes
TLRs: Toll-like receptors
TME: Tumor microenvironment
Tregs: Regulatory T cells
tRNA: Transfer RNA
TRP2: Tyrosinase-related protein 2
TSG101: Tumor susceptibility gene 101
VPS4: Vacuolar protein sorting 4
WHO: World Health Organization
5-FU: 5-Fluorouracil
References
1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
2. Kone A-S, Ghouzlani A, Qandouci A, Issam Salah NEI, Bakoukou Y, Lakhdar A, et al. High expression of BTN3A1 is associated with clinical and immunological characteristics and predicts a poor prognosis in advanced human gliomas. Front Immunol. (2024) 15:1397486. doi: 10.3389/fimmu.2024.1397486
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA A Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
4. Li Z-H, Guan Y-L, Liu Q, Wang Y, Cui R, Wang Y-J. Astrocytoma progression scoring system based on the WHO 2016 criteria. Sci Rep. (2019) 9:96. doi: 10.1038/s41598-018-36471-4
5. Liang J, Li T, Zhao J, Wang C, Sun H. Current understanding of the human microbiome in glioma. Front Oncol. (2022) 12:781741. doi: 10.3389/fonc.2022.781741
6. Rafii S, Ghouzlani A, Naji O, Ait Ssi S, Kandoussi S, Lakhdar A, et al. A2AR as a prognostic marker and a potential immunotherapy target in human glioma. Int J Mol Sci. (2023) 24:6688. doi: 10.3390/ijms24076688
7. Boulhen C, AIT SSIS, Benthami H, Razzouki I, Lakhdar A, Karkouri M, et al. TMIGD2 as a potential therapeutic target in glioma patients. Front Immunol. (2023) 14:1173518. doi: 10.3389/fimmu.2023.1173518
8. Ghouzlani A, Lakhdar A, Rafii S, Karkouri M, Badou A. The immune checkpoint VISTA exhibits high expression levels in human gliomas and associates with a poor prognosis. Sci Rep. (2021) 11:21504. doi: 10.1038/s41598-021-00835-0
9. Li S, Lai M, Zhou J, Zhen J, Cai L. Path-22. Genetic variation between idh mutant and idh wild-type glioma. Neuro Oncol. (2021) 23:vi119. doi: 10.1093/neuonc/noab196.474
10. Rong Y, Durden DL, Van Meir EG, Brat DJ. Pseudopalisading” necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. (2006) 65:529–39. doi: 10.1097/00005072-200606000-00001
11. Mills BN, Albert GP, Halterman MW. Expression profiling of the MAP kinase phosphatase family reveals a role for DUSP1 in the glioblastoma stem cell niche. Cancer Microenviron. (2017) 10:57–68. doi: 10.1007/s12307-017-0197-6
12. Vitovcova B, Skarkova V, Rudolf K, Rudolf E. Biology of glioblastoma multiforme—Exploration of mitotic catastrophe as a potential treatment modality. Int J Mol Sci. (2020) 21:5324. doi: 10.3390/ijms21155324
13. Björkblom B, Wibom C, Eriksson M, Bergenheim AT, Sjöberg RL, Jonsson P, et al. Distinct metabolic hallmarks of WHO classified adult glioma subtypes. Neuro Oncol. (2022) 24:1454–68. doi: 10.1093/neuonc/noac042
14. Wang G, Zhou H, Tian L, Yan T, Han X, Chen P, et al. A prognostic DNA damage repair genes signature and its impact on immune cell infiltration in glioma. Front Oncol. (2021) 11:682932. doi: 10.3389/fonc.2021.682932
15. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
16. Torrisi F, Alberghina C, D’Aprile S, Pavone AM, Longhitano L, Giallongo S, et al. The hallmarks of glioblastoma: heterogeneity, intercellular crosstalk and molecular signature of invasiveness and progression. Biomedicines. (2022) 10:806. doi: 10.3390/biomedicines10040806
17. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science. (2020) 368:973–80. doi: 10.1126/science.aay9189
18. Zhao J, He D, Lai HM, Xu Y, Luo Y, Li T, et al. Comprehensive histological imaging of native microbiota in human glioma. J Biophotonics. (2022) 15:e202100351. doi: 10.1002/jbio.202100351
19. Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. (2008) 10:1470–6. doi: 10.1038/ncb1800
20. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
21. Yáñez-Mó M, Siljander PR -M, Andreu Z, Bedina Zavec A, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracellular Vesicle. (2015) 4:27066. doi: 10.3402/jev.v4.27066
22. Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. BioScience. (2015) 65:783–97. doi: 10.1093/biosci/biv084
23. Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, et al. Reassessment of exosome composition. Cell. (2019) 177:428–445.e18. doi: 10.1016/j.cell.2019.02.029
24. Jeppesen DK, Zhang Q, Franklin JL, Coffey RJ. Extracellular vesicles and nanoparticles: emerging complexities. Trends Cell Biol. (2023) 33:667–81. doi: 10.1016/j.tcb.2023.01.002
25. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
26. Mathieu M, Martin-Jaular L, Lavieu G, Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. (2019) 21:9–17. doi: 10.1038/s41556-018-0250-9
27. Borges FT, Reis LA, Schor N. Extracellular vesicles: structure, function, and potential clinical uses in renal diseases. Braz J Med Biol Res. (2013) 46:824–30. doi: 10.1590/1414-431X20132964
28. Wang J, Yao Y, Chen X, Wu J, Gu T, Tang X. Host derived exosomes-pathogens interactions: Potential functions of exosomes in pathogen infection. BioMed Pharmacother. (2018) 108:1451–9. doi: 10.1016/j.biopha.2018.09.174
29. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. (2016) 113:E968-77. doi: 10.1073/pnas.1521230113
30. Merchant ML, Rood IM, Deegens JKJ, Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. (2017) 13:731–49. doi: 10.1038/nrneph.2017.148
31. Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, et al. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. (2012) 10:5. doi: 10.1186/1479-5876-10-5
32. van Niel G, Carter DRF, Clayton A, Lambert DW, Raposo G, Vader P. Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat Rev Mol Cell Biol. (2022) 23:369–82. doi: 10.1038/s41580-022-00460-3
33. Ostenfeld MS, Jeppesen DK, Laurberg JR, Boysen AT, Bramsen JB, Primdal-Bengtson B, et al. Cellular disposal of miR23b by RAB27-dependent exosome release is linked to acquisition of metastatic properties. Cancer Res. (2014) 74:5758–71. doi: 10.1158/0008-5472.CAN-13-3512
34. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. (1985) 101:942–8. doi: 10.1083/jcb.101.3.942
35. Yates AG, Pink RC, Erdbrügger U, Siljander PR, Dellar ER, Pantazi P, et al. In sickness and in health: The functional role of extracellular vesicles in physiology and pathology in vivo: Part I: Health and Normal Physiology. J Extracellular Vesicle. (2022) 11:e12151. doi: 10.1002/jev2.12151
36. Yates AG, Pink RC, Erdbrügger U, Siljander PR, Dellar ER, Pantazi P, et al. In sickness and in health: The functional role of extracellular vesicles in physiology and pathology in vivo: Part II: Pathology. J Extracellular Vesicle. (2022) 11:e12190. doi: 10.1002/jev2.12190
37. Buzas EI, György B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. (2014) 10:356–64. doi: 10.1038/nrrheum.2014.19
38. Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527:329–35. doi: 10.1038/nature15756
39. Sahoo S, Adamiak M, Mathiyalagan P, Kenneweg F, Kafert-Kasting S, Thum T. Therapeutic and diagnostic translation of extracellular vesicles in cardiovascular diseases: roadmap to the clinic. Circulation. (2021) 143:1426–49. doi: 10.1161/CIRCULATIONAHA.120.049254
40. Pan B-T, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell. (1983) 33:967–78. doi: 10.1016/0092-8674(83)90040-5
41. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
42. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367:eaau6977. doi: 10.1126/science.aau6977
43. Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. (2010) 78:838–48. doi: 10.1038/ki.2010.278
44. Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. (2009) 19:1875–85. doi: 10.1016/j.cub.2009.09.059
45. Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci USA. (2012) 109:4146–51. doi: 10.1073/pnas.1200448109
46. Wang Q, Lu Q. Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat Commun. (2017) 8:709. doi: 10.1038/s41467-017-00767-2
47. Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. (2015) 25:364–72. doi: 10.1016/j.tcb.2015.01.004
48. Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
49. Choudhuri K, Llodrá J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, et al. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature. (2014) 507:118–23. doi: 10.1038/nature12951
50. Saliba DG, Céspedes-Donoso PF, Bálint Š, Compeer EB, Korobchevskaya K, Valvo S, et al. Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. eLife. (2019) 8:e47528. doi: 10.7554/eLife.47528
51. Sadallah S, Amicarella F, Eken C, Iezzi G, Schifferli J. Ectosomes released by platelets induce differentiation of CD4+ T cells into T regulatory cells. Thromb Haemost. (2014) 112:1219–29. doi: 10.1160/th14-03-0281
52. Choi HW, Suwanpradid J, Kim IH, Staats HF, Haniffa M, MacLeod AS, et al. Perivascular dendritic cells elicit anaphylaxis by relaying allergens to mast cells via microvesicles. Science. (2018) 362:eaao0666. doi: 10.1126/science.aao0666
53. Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. (2009) 69:5601–9. doi: 10.1158/0008-5472.CAN-08-3860
54. Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, et al. Focus on extracellular vesicles: new frontiers of cell-to-cell communication in cancer. IJMS. (2016) 17:175. doi: 10.3390/ijms17020175
55. Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. (2012) 181:1573–84. doi: 10.1016/j.ajpath.2012.07.030
56. Minciacchi VR, Spinelli C, Reis-Sobreiro M, Cavallini L, You S, Zandian M, et al. MYC mediates large oncosome-induced fibroblast reprogramming in prostate cancer. Cancer Res. (2017) 77:2306–17. doi: 10.1158/0008-5472.CAN-16-2942
57. Bertolini I, Terrasi A, Martelli C, Gaudioso G, Di Cristofori A, Storaci AM, et al. A GBM-like V-ATPase signature directs cell-cell tumor signaling and reprogramming via large oncosomes. EBioMedicine. (2019) 41:225–35. doi: 10.1016/j.ebiom.2019.01.051
58. Morello M, Minciacchi V, de Candia P, Yang J, Posadas E, Kim H, et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle. (2013) 12:3526–36. doi: 10.4161/cc.26539
59. Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria P-J, et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget. (2015) 6:11327–41. doi: 10.18632/oncotarget.3598
60. Holmgren L, Szeles A, Rajnavölgyi E, Folkman J, Klein G, Ernberg I, et al. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood. (1999) 93:3956–63. doi: 10.1182/blood.V93.11.3956
61. Pavlyukov MS, Yu H, Bastola S, Minata M, Shender VO, Lee Y, et al. Apoptotic cell-derived extracellular vesicles promote Malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell. (2018) 34:119–135.e10. doi: 10.1016/j.ccell.2018.05.012
62. Li M, Liao L, Tian W. Extracellular vesicles derived from apoptotic cells: an essential link between death and regeneration. Front Cell Dev Biol. (2020) 8:573511. doi: 10.3389/fcell.2020.573511
63. Park SJ, Kim JM, Kim J, Hur J, Park S, Kim K, et al. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc Natl Acad Sci USA. (2018) 115:E11721-30. doi: 10.1073/pnas.1811432115
64. Schiller M, Parcina M, Heyder P, Foermer S, Ostrop J, Leo A, et al. Induction of type I IFN is a physiological immune reaction to apoptotic cell-derived membrane microparticles. J Immunol. (2012) 189:1747–56. doi: 10.4049/jimmunol.1100631
65. Ainola M, Porola P, Takakubo Y, Przybyla B, Kouri VP, Tolvanen TA, et al. Activation of plasmacytoid dendritic cells by apoptotic particles – mechanism for the loss of immunological tolerance in Sjögren’s syndrome. Clin Exp Immunol. (2018) 191:301–10. doi: 10.1111/cei.13077
66. Dieudé M, Bell C, Turgeon J, Beillevaire D, Pomerleau L, Yang B, et al. The 20 S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. (2015) 7(318):318ra200. doi: 10.1126/scitranslmed.aac9816
67. Hristov M, Erl W, Linder S, Weber PC. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood. (2004) 104:2761–6. doi: 10.1182/blood-2003-10-3614
68. Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz H-M. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. (2008) 15:183–91. doi: 10.1038/sj.cdd.4402239
69. Tucher C, Bode K, Schiller P, Claßen L, Birr C, Souto-Carneiro MM, et al. Extracellular vesicle subtypes released from activated or apoptotic T-lymphocytes carry a specific and stimulus-dependent protein cargo. Front Immunol. (2018) 9:534. doi: 10.3389/fimmu.2018.00534
70. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just debris. Front Immunol. (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
71. Gregory CD, Dransfield I. Apoptotic tumor cell-derived extracellular vesicles as important regulators of the onco-regenerative niche. Front Immunol. (2018) 9:1111. doi: 10.3389/fimmu.2018.01111
72. Gregory CD, Paterson M. An apoptosis-driven ‘onco-regenerative niche’: roles of tumour-associated macrophages and extracellular vesicles. Phil Trans R Soc B. (2018) 373:20170003. doi: 10.1098/rstb.2017.0003
73. Laurenzana I, Lamorte D, Trino S, De Luca L, Ambrosino C, Zoppoli P, et al. Extracellular vesicles: A new prospective in crosstalk between microenvironment and stem cells in hematological Malignancies. Stem Cells Int. (2018) 2018:1–11. doi: 10.1155/2018/9863194
74. Muhsin-Sharafaldine M-R, McLellan AD. Tumor-derived apoptotic vesicles: with death they do part. Front Immunol. (2018) 9:957. doi: 10.3389/fimmu.2018.00957
75. Muhsin-Sharafaldine M, McLellan AD. Apoptotic vesicles: deathly players in cancer-associated coagulation. Immunol Cell Biol. (2018) 96:723–32. doi: 10.1111/imcb.12162
76. Grant LR, Milic I, Devitt A. Apoptotic cell-derived extracellular vesicles: structure–function relationships. Biochem Soc Trans. (2019) 47:509–16. doi: 10.1042/BST20180080
77. Battistelli M, Falcieri E. Apoptotic bodies: particular extracellular vesicles involved in intercellular communication. Biology. (2020) 9:21. doi: 10.3390/biology9010021
78. Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracellular Vesicle. (2014) 3:24641. doi: 10.3402/jev.v3.24641
79. O’Brien K, Ughetto S, Mahjoum S, Nair AV, Breakefield XO. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. (2022) 39:110651. doi: 10.1016/j.celrep.2022.110651
80. Montecalvo A, Larregina AT, Shufesky WJ, Beer Stolz D, Sullivan MLG, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. (2012) 119:756–66. doi: 10.1182/blood-2011-02-338004
81. Horibe S, Tanahashi T, Kawauchi S, Murakami Y, Rikitake Y. Mechanism of recipient cell-dependent differences in exosome uptake. BMC Cancer. (2018) 18:47. doi: 10.1186/s12885-017-3958-1
82. Tian T, Zhu Y-L, Zhou Y-Y, Liang G-F, Wang Y-Y, Hu F-H, et al. Exosome Uptake through Clathrin-mediated Endocytosis and Macropinocytosis and Mediating miR-21 Delivery. J Biol Chem. (2014) 289:22258–67. doi: 10.1074/jbc.M114.588046
83. Tankov S, Walker PR. Glioma-derived extracellular vesicles – far more than local mediators. Front Immunol. (2021) 12:679954. doi: 10.3389/fimmu.2021.679954
84. Gurung S, Perocheau D, Touramanidou L, Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal. (2021) 19:47. doi: 10.1186/s12964-021-00730-1
85. Sung P-S, Huang T-F, Hsieh S-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
86. Tkach M, Kowal J, Zucchetti AE, Enserink L, Jouve M, Lankar D, et al. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. (2017) 36:3012–28. doi: 10.15252/embj.201696003
87. Krylova SV, Feng D. The machinery of exosomes: biogenesis, release, and uptake. IJMS. (2023) 24:1337. doi: 10.3390/ijms24021337
88. Moloudizargari M, Asghari MH, Jørgensen MM, Reiter RJ, Kabelitz D. Editorial: Extracellular vesicles in cancer immunosurveillance. Front Immunol. (2022) 13:993967. doi: 10.3389/fimmu.2022.993967
89. Turchinovich A, Drapkina O, Tonevitsky A. Transcriptome of extracellular vesicles: state-of-the-art. Front Immunol. (2019) 10:202. doi: 10.3389/fimmu.2019.00202
90. Del Bene M, Osti D, Faletti S, Beznoussenko GV, DiMeco F, Pelicci G. Extracellular vesicles: The key for precision medicine in glioblastoma. Neuro-Oncology. (2022) 24:184–96. doi: 10.1093/neuonc/noab229
91. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. (2008) 10:619–24. doi: 10.1038/ncb1725
92. Whitehead CA, Kaye AH, Drummond KJ, Widodo SS, Mantamadiotis T, Vella LJ, et al. Extracellular vesicles and their role in glioblastoma. Crit Rev Clin Lab Sci. (2020) 57:227–52. doi: 10.1080/10408363.2019.1700208
93. Setti M, Osti D, Richichi C, Ortensi B, Del Bene M, Fornasari L, et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget. (2015) 6:31413–27. doi: 10.18632/oncotarget.5105
94. Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringnér M, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci USA. (2013) 110:7312–7. doi: 10.1073/pnas.1220998110
95. Lucero R, Zappulli V, Sammarco A, Murillo OD, Cheah PS, Srinivasan S, et al. Glioma-derived miRNA-containing extracellular vesicles induce angiogenesis by reprogramming brain endothelial cells. Cell Rep. (2020) 30:2065–2074.e4. doi: 10.1016/j.celrep.2020.01.073
96. Hallal S, Mallawaaratchy DM, Wei H, Ebrahimkhani S, Stringer BW, Day BW, et al. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and MYC signaling pathways. Mol Neurobiol. (2019) 56:4566–81. doi: 10.1007/s12035-018-1385-1
97. Zeng A, Wei Z, Rabinovsky R, Jun HJ, El Fatimy R, Deforzh E, et al. Glioblastoma-derived extracellular vesicles facilitate transformation of astrocytes via reprogramming oncogenic metabolism. iScience. (2020) 23:101420. doi: 10.1016/j.isci.2020.101420
98. Abels ER, Maas SLN, Nieland L, Wei Z, Cheah PS, Tai E, et al. Glioblastoma-associated microglia reprogramming is mediated by functional transfer of extracellular miR-21. Cell Rep. (2019) 28:3105–3119.e7. doi: 10.1016/j.celrep.2019.08.036
99. van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, Oakley D, et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. (2016) 18:58–69. doi: 10.1093/neuonc/nov244
100. de Vrij J, Maas SLN, Kwappenberg KMC, Schnoor R, Kleijn A, Dekker L, et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Intl J Cancer. (2015) 137:1630–42. doi: 10.1002/ijc.29521
101. Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, Manini I, et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS One. (2017) 12:e0169932. doi: 10.1371/journal.pone.0169932
102. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
103. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. (2009) 16:748–54. doi: 10.1016/j.jocn.2008.12.005
104. Wang J, Bettegowda C. Genomic discoveries in adult astrocytoma. Curr Opin Genet Dev. (2015) 30:17–24. doi: 10.1016/j.gde.2014.12.002
105. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. (2013) 45:1141–9. doi: 10.1038/ng.2734
106. Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv. (2018) 4:eaar2766. doi: 10.1126/sciadv.aar2766
107. Graner MW, Cumming RI, Bigner DD. The heat shock response and chaperones/heat shock proteins in brain tumors: surface expression, release, and possible immune consequences. J Neurosci. (2007) 27:11214–27. doi: 10.1523/JNEUROSCI.3588-07.2007
108. Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. (2009) 23:1541–57. doi: 10.1096/fj.08-122184
109. Gao X, Zhang Z, Mashimo T, Shen B, Nyagilo J, Wang H, et al. Gliomas interact with non-glioma brain cells via extracellular vesicles. Cell Rep. (2020) 30:2489–2500.e5. doi: 10.1016/j.celrep.2020.01.089
110. Oushy S, Hellwinkel JE, Wang M, Nguyen GJ, Gunaydin D, Harland TA, et al. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Phil Trans R Soc B. (2018) 373:20160477. doi: 10.1098/rstb.2016.0477
111. Bian E, Chen E, Xu Y, Yang Z, Tang F, Ma C, et al. Exosomal lncRNA−ATB activates astrocytes that promote glioma cell invasion. Int J Oncol. (2018) 54(2):713–21. doi: 10.3892/ijo.2018.4644
112. Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol. (2018) 14:482–95. doi: 10.1038/s41582-018-0025-8
113. Nakano I, Garnier D, Minata M, Rak J. Extracellular vesicles in the biology of brain tumour stem cells – Implications for inter-cellular communication, therapy and biomarker development. Semin Cell Dev Biol. (2015) 40:17–26. doi: 10.1016/j.semcdb.2015.02.011
114. Taheri B, Soleimani M, Aval SF, Memari F, Zarghami N. C6 glioma-derived microvesicles stimulate the proliferative and metastatic gene expression of normal astrocytes. Neurosci Lett. (2018) 685:173–8. doi: 10.1016/j.neulet.2018.08.034
115. The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. (2008) 455:1061–8. doi: 10.1038/nature07385
116. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
117. Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci USA. (2019) 116:3909–18. doi: 10.1073/pnas.1816391116
118. Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. (2017) 77:1564–74. doi: 10.1158/0008-5472.CAN-16-2074
119. Sousa FL, Nelson-Sathi S, Martin WF. One step beyond a ribosome: The ancient anaerobic core. Biochim Biophys Acta (BBA) - Bioenergetics. (2016) 1857:1027–38. doi: 10.1016/j.bbabio.2016.04.284
120. Song Y, Luo Q, Long H, Hu Z, Que T, Zhang X, et al. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol Cancer. (2014) 13:65. doi: 10.1186/1476-4598-13-65
121. Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-myc is required for maintenance of glioma cancer stem cells. PLoS One. (2008) 3:e3769. doi: 10.1371/journal.pone.0003769
122. Büschges R, Weber RG, Actor B, Lichter P, Collins VP, Reifenberger G. Amplification and expression of cyclin D genes ( CCND1 CCND2 and CCND3 ) in human Malignant gliomas. Brain Pathol. (1999) 9:435–42. doi: 10.1111/j.1750-3639.1999.tb00532.x
123. Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann Oncol. (2017) 28:1457–72. doi: 10.1093/annonc/mdx106
124. Giusti I, Francesco M, Dolo V. Extracellular vesicles in glioblastoma: role in biological processes and in therapeutic applications. CCDT. (2017) 17:221–35. doi: 10.2174/1568009616666160813182959
125. Figueroa JM, Skog J, Akers J, Li H, Komotar R, Jensen R, et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro-Oncology. (2017) 19:1494–502. doi: 10.1093/neuonc/nox085
126. Hallal S, Ebrahimkhani S, Shivalingam B, Graeber MB, Kaufman KL, Buckland ME. The emerging clinical potential of circulating extracellular vesicles for non-invasive glioma diagnosis and disease monitoring. Brain Tumor Pathol. (2019) 36:29–39. doi: 10.1007/s10014-019-00335-0
127. Santiago-Dieppa DR, Steinberg J, Gonda D, Cheung VJ, Carter BS, Chen CC. Extracellular vesicles as a platform for ‘liquid biopsy’ in glioblastoma patients. Expert Rev Mol Diagnostics. (2014) 14:819–25. doi: 10.1586/14737159.2014.943193
128. Qi Z, Long X, Liu J, Cheng P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front Cell Neurosci. (2022) 16:819363. doi: 10.3389/fncel.2022.819363
129. Azambuja JH, Ludwig N, Yerneni S, Rao A, Braganhol E, Whiteside TL. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neuro-Oncology Adv. (2020) 2:vdaa056. doi: 10.1093/noajnl/vdaa056
130. Viel S, Marçais A, Guimaraes FS-F, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9(415):ra19. doi: 10.1126/scisignal.aad1884
131. Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. (2004) 294:15–22. doi: 10.1016/j.jim.2004.08.008
132. Berchem G, Noman MZ, Bosseler M, Paggetti J, Baconnais S, Le cam E, et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. OncoImmunology. (2016) 5:e1062968. doi: 10.1080/2162402X.2015.1062968
133. Jong AY, Wu C, Li J, Sun J, Fabbri M, Wayne AS, et al. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracellular Vesicle. (2017) 6:1294368. doi: 10.1080/20013078.2017.1294368
134. Zhang Q, Liu L, Gong C, Shi H, Zeng Y, Wang X, et al. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS One. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
135. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. (2008) 216:15–24. doi: 10.1002/path.2370
136. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. (2016) 1(2):e85841. doi: 10.1172/jci.insight.85841
137. Pello OM, Chèvre R, Laoui D, De Juan A, Lolo F, Andrés-Manzano MJ, et al. In vivo inhibition of c-MYC in myeloid cells impairs tumor-associated macrophage maturation and pro-tumoral activities. PLoS One. (2012) 7:e45399. doi: 10.1371/journal.pone.0045399
138. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. (2018) 17:428–38. doi: 10.1080/15384101.2018.1444305
139. Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene. (2020) 39:428–42. doi: 10.1038/s41388-019-0996-y
140. Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. OncoImmunology. (2018) 7:e1412909. doi: 10.1080/2162402X.2017.1412909
141. Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel β-galactoside-binding mammalian lectin. J Biol Chem. (1997) 272:6078–86. doi: 10.1074/jbc.272.9.6078
142. Wang M, Cai Y, Peng Y, Xu B, Hui W, Jiang Y. Exosomal LGALS9 in the cerebrospinal fluid of glioblastoma patients suppressed dendritic cell antigen presentation and cytotoxic T-cell immunity. Cell Death Dis. (2020) 11:896. doi: 10.1038/s41419-020-03042-3
143. Luong N, Lenz JA, Modiano JF, Olson JK. Extracellular vesicles secreted by tumor cells promote the generation of suppressive monocytes. ImmunoHorizons. (2021) 5:647–58. doi: 10.4049/immunohorizons.2000017
144. Liu Z-M, Wang Y-B, Yuan X-H. Exosomes from murine-derived GL26 cells promote glioblastoma tumor growth by reducing number and function of CD8+T cells. Asian Pacific J Cancer Prev. (2013) 14:309–14. doi: 10.7314/APJCP.2013.14.1.309
145. Hellwinkel JE, Redzic JS, Harland TA, Gunaydin D, Anchordoquy TJ, Graner MW. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. (2016) 18:497–506. doi: 10.1093/neuonc/nov170
146. Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, et al. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. OncoImmunology. (2018) 7:e1478647. doi: 10.1080/2162402X.2018.1478647
147. Mafi S, Mansoori B, Taeb S, Sadeghi H, Abbasi R, Cho WC, et al. mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front Immunol. (2022) 12:774103. doi: 10.3389/fimmu.2021.774103
148. Ghouzlani A, Kandoussi S, Tall M, Reddy KP, Rafii S, Badou A. Immune checkpoint inhibitors in human glioma microenvironment. Front Immunol. (2021) 12:679425. doi: 10.3389/fimmu.2021.679425
149. Lawler SE, Nowicki MO, Ricklefs FL, Chiocca EA. Immune escape mediated by exosomal PD-L1 in cancer. Adv Biosyst. (2020) 4:e2000017. doi: 10.1002/adbi.202000017
150. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
151. Saas P, Walker PR, Hahne M, Quiquerez AL, Schnuriger V, Perrin G, et al. Fas ligand expression by astrocytoma in vivo: maintaining immune privilege in the brain? J Clin Invest. (1997) 99:1173–8. doi: 10.1172/JCI119273
152. Iorgulescu JB, Ivan ME, Safaee M, Parsa AT. The limited capacity of Malignant glioma-derived exosomes to suppress peripheral immune effectors. J Neuroimmunology. (2016) 290:103–8. doi: 10.1016/j.jneuroim.2015.11.025
153. Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. (2019) 10:3000. doi: 10.1038/s41467-019-10979-3
154. Maybruck BT, Pfannenstiel LW, Diaz-Montero M, Gastman BR. Tumor-derived exosomes induce CD8+ T cell suppressors. J immunotherapy Cancer. (2017) 5:65. doi: 10.1186/s40425-017-0269-7
155. Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol. (2009) 183:3720–30. doi: 10.4049/jimmunol.0900970
156. Macia L, Nanan R, Hosseini-Beheshti E, Grau GE. Host- and microbiota-derived extracellular vesicles, immune function, and disease development. IJMS. (2019) 21:107. doi: 10.3390/ijms21010107
157. Yang J, Kim EK, McDowell A, Kim Y-K. Microbe-derived extracellular vesicles as a smart drug delivery system. Transl Clin Pharmacol. (2018) 26:103. doi: 10.12793/tcp.2018.26.3.103
158. Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. (2017) 356:j831. doi: 10.1136/bmj.j831
159. Takiishi T, Fenero CIM, Câmara NOS. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers. (2017) 5:e1373208. doi: 10.1080/21688370.2017.1373208
160. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. (2018) 33:570–80. doi: 10.1016/j.ccell.2018.03.015
161. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
162. Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science. (2018) 359:1366–70. doi: 10.1126/science.aar6918
163. Hughes DT, Sperandio V. Inter-kingdom signalling: communication between bacteria and their hosts. Nat Rev Microbiol. (2008) 6:111–20. doi: 10.1038/nrmicro1836
164. Tulkens J, De Wever O, Hendrix A. Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat Protoc. (2020) 15:40–67. doi: 10.1038/s41596-019-0236-5
165. Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol. (2015) 15:375–87. doi: 10.1038/nri3837
166. Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. (2019) 17:13–24. doi: 10.1038/s41579-018-0112-2
167. Orench-Rivera N, Kuehn MJ. Environmentally controlled bacterial vesicle-mediated export: Environmentally controlled bacterial vesicle-mediated export. Cell Microbiol. (2016) 18:1525–36. doi: 10.1111/cmi.12676
168. Lee E-Y, Bang JY, Park GW, Choi D-S, Kang JS, Kim H-J, et al. Global proteomic profiling of native outer membrane vesicles derived from. Escherichia coli. Proteomics. (2007) 7:3143–53. doi: 10.1002/pmic.200700196
169. Bitto NJ, Chapman R, Pidot S, Costin A, Lo C, Choi J, et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci Rep. (2017) 7:7072. doi: 10.1038/s41598-017-07288-4
170. Riley DR, Sieber KB, Robinson KM, White JR, Ganesan A, Nourbakhsh S, et al. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. (2013) 9:e1003107. doi: 10.1371/journal.pcbi.1003107
171. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discovery. (2018) 8:403–16. doi: 10.1158/2159-8290.CD-17-1134
172. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nat. (2020) 580:269–73. doi: 10.1038/s41586-020-2080-8
173. Yu Y, Wang X, Fan G-C. Versatile effects of bacterium-released membrane vesicles on mammalian cells and infectious/inflammatory diseases. Acta Pharmacol Sin. (2018) 39:514–33. doi: 10.1038/aps.2017.82
174. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
175. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. (2020) 579:567–74. doi: 10.1038/s41586-020-2095-1
176. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. (2019) 178:795–806.e12. doi: 10.1016/j.cell.2019.07.008
177. Diaz-Garrido N, Badia J, Baldomà L. Modulation of dendritic cells by microbiota extracellular vesicles influences the cytokine profile and exosome cargo. Nutrients. (2022) 14:344. doi: 10.3390/nu14020344
178. Gasperini G, Biagini M, Arato V, Gianfaldoni C, Vadi A, Norais N, et al. Outer membrane vesicles (OMV)-based and proteomics-driven antigen selection identifies novel factors contributing to bordetella pertussis adhesion to epithelial cells. Mol Cell Proteomics. (2018) 17:205–15. doi: 10.1074/mcp.RA117.000045
179. Raynes JM, Young PG, Proft T, Williamson DA, Baker EN, Moreland NJ. Protein adhesins as vaccine antigens for Group A Streptococcus. Pathog Dis. (2018) 76(2):fty016. doi: 10.1093/femspd/fty016
180. Castilla J, García Cenoz M, Abad R, Sánchez-Cambronero L, Lorusso N, Izquierdo C, et al. Effectiveness of a meningococcal group B vaccine (4CMenB) in children. N Engl J Med. (2023) 388:427–38. doi: 10.1056/NEJMoa2206433
181. Vesikari T, Esposito S, Prymula R, Ypma E, Kohl I, Toneatto D, et al. Immunogenicity and safety of an investigational multicomponent, recombinant, meningococcal serogroup B vaccine (4CMenB) administered concomitantly with routine infant and child vaccinations: results of two randomised trials. Lancet. (2013) 381:825–35. doi: 10.1016/S0140-6736(12)61961-8
182. Petousis-Harris H, Paynter J, Morgan J, Saxton P, McArdle B, Goodyear-Smith F, et al. Effectiveness of a group B outer membrane vesicle meningococcal vaccine against gonorrhoea in New Zealand: a retrospective case-control study. Lancet. (2017) 390:1603–10. doi: 10.1016/S0140-6736(17)31449-6
183. Petousis-Harris H, Radcliff FJ. Exploitation of Neisseria meningitidis Group B OMV Vaccines Against N. gonorrhoeae to Inform the Development and Deployment of Effective Gonorrhea Vaccines. Front Immunol. (2019) 10:683. doi: 10.3389/fimmu.2019.00683
184. Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, et al. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat Commun. (2017) 8:626. doi: 10.1038/s41467-017-00729-8
185. Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug delivery: is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjugate Chem. (2016) 27:2225–38. doi: 10.1021/acs.bioconjchem.6b00437
186. Gujrati V, Kim S, Kim S-H, Min JJ, Choy HE, Kim SC, et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano. (2014) 8:1525–37. doi: 10.1021/nn405724x
187. Park K, Svennerholm K, Crescitelli R, Lässer C, Gribonika I, Lötvall J. Synthetic bacterial vesicles combined with tumour extracellular vesicles as cancer immunotherapy. J Extracellular Vesicle. (2021) 10:e12120. doi: 10.1002/jev2.12120
188. Chen Q, Bai H, Wu W, Huang G, Li Y, Wu M, et al. Bioengineering bacterial vesicle-coated polymeric nanomedicine for enhanced cancer immunotherapy and metastasis prevention. Nano Lett. (2020) 20:11–21. doi: 10.1021/acs.nanolett.9b02182
189. Kuerban K, Gao X, Zhang H, Liu J, Dong M, Wu L, et al. Doxorubicin-loaded bacterial outer-membrane vesicles exert enhanced anti-tumor efficacy in non-small-cell lung cancer. Acta Pharm Sin B. (2020) 10:1534–48. doi: 10.1016/j.apsb.2020.02.002
190. Guo Q, Li X, Zhou W, Chu Y, Chen Q, Zhang Y, et al. Sequentially triggered bacterial outer membrane vesicles for macrophage metabolism modulation and tumor metastasis suppression. ACS Nano. (2021) 15:13826–38. doi: 10.1021/acsnano.1c05613
191. Zhidkova EM, Lylova ES, Grigoreva DD, Kirsanov KI, Osipova AV, Kulikov EP, et al. Nutritional sensor REDD1 in cancer and inflammation: friend or foe? IJMS. (2022) 23:9686. doi: 10.3390/ijms23179686
192. Li Y, Zhao R, Cheng K, Zhang K, Wang Y, Zhang Y, et al. Bacterial outer membrane vesicles presenting programmed death 1 for improved cancer immunotherapy via immune activation and checkpoint inhibition. ACS Nano. (2020) 14:16698–711. doi: 10.1021/acsnano.0c03776
193. Cheng K, Zhao R, Li Y, Qi Y, Wang Y, Zhang Y, et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug-and-Display technology. Nat Commun. (2021) 12:2041. doi: 10.1038/s41467-021-22308-8
194. Wang S, Gao J, Wang Z. Outer membrane vesicles for vaccination and targeted drug delivery. WIREs Nanomed Nanobiotechnol. (2019) 11:e1523. doi: 10.1002/wnan.1523
195. Zhang Y, Fang Z, Li R, Huang X, Liu Q. Design of outer membrane vesicles as cancer vaccines: A new toolkit for cancer therapy. Cancers. (2019) 11:1314. doi: 10.3390/cancers11091314
196. Schetters STT, Jong WSP, Horrevorts SK, Kruijssen LJW, Engels S, Stolk D, et al. Outer membrane vesicles engineered to express membrane-bound antigen program dendritic cells for cross-presentation to CD8+ T cells. Acta Biomaterialia. (2019) 91:248–57. doi: 10.1016/j.actbio.2019.04.033
197. Al-Ali D, Ahmed A, Shafiq A, McVeigh C, Chaari A, Zakaria D, et al. Fecal microbiota transplants: A review of emerging clinical data on applications, efficacy, and risks (2015–2020). Qatar Med J. (2021) 2021(1):5. doi: 10.5339/qmj.2021.5
198. McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. (2019) 20:e77–91. doi: 10.1016/S1470-2045(18)30952-5
199. Fonseca S, Carvalho AL, Miquel-Clopés A, Jones EJ, Juodeikis R, Stentz R, et al. Extracellular vesicles produced by the human gut commensal bacterium Bacteroides thetaiotaomicron elicit anti-inflammatory responses from innate immune cells. Front Microbiol. (2022) 13:1050271. doi: 10.3389/fmicb.2022.1050271
200. Thakur A, Wei Z, Chen HJ. Editorial: Extracellular vesicles and cell-cell communication in normal cellular processes and cancer. Front Mol Biosci. (2023) 10:1172797. doi: 10.3389/fmolb.2023.1172797
201. Chen Y, Jin Y, Wu N. Role of tumor-derived extracellular vesicles in glioblastoma. Cells. (2021) 10:512. doi: 10.3390/cells10030512
202. Han L, Lam EW-F, Sun Y. Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol Cancer. (2019) 18:59. doi: 10.1186/s12943-019-0980-8
203. Yekula A, Yekula A, Muralidharan K, Kang K, Carter BS, Balaj L. Extracellular vesicles in glioblastoma tumor microenvironment. Front Immunol. (2019) 10:3137. doi: 10.3389/fimmu.2019.03137
204. Indira Chandran V, Gopala S, Venkat EH, Kjolby M, Nejsum P. Extracellular vesicles in glioblastoma: a challenge and an opportunity. NPJ Precis Onc. (2024) 8:1–8. doi: 10.1038/s41698-024-00600-2
205. Low JJW, Sulaiman SA, Johdi NA, Abu N. Immunomodulatory effects of extracellular vesicles in glioblastoma. Front Cell Dev Biol. (2022) 10:996805. doi: 10.3389/fcell.2022.996805
206. Virtuoso A, Giovannoni R, De Luca C, Gargano F, Cerasuolo M, Maggio N, et al. The glioblastoma microenvironment: morphology, metabolism, and molecular signature of glial dynamics to discover metabolic rewiring sequence. IJMS. (2021) 22:3301. doi: 10.3390/ijms22073301
207. Matias D, Balça-Silva J, da Graça GC, Wanjiru CM, Macharia LW, Nascimento CP, et al. Microglia/astrocytes–glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front Cell Neurosci. (2018) 12:235. doi: 10.3389/fncel.2018.00235
208. Musatova OE, Rubtsov YP. Effects of glioblastoma-derived extracellular vesicles on the functions of immune cells. Front Cell Dev Biol. (2023) 11:1060000. doi: 10.3389/fcell.2023.1060000
209. Epple LM, Griffiths SG, Dechkovskaia AM, Dusto NL, White J, Ouellette RJ, et al. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS One. (2012) 7:e42064. doi: 10.1371/journal.pone.0042064
210. Jung M-Y, Aibaidula A, Brown DA, Himes BT, Cumba Garcia LM, Parney IF. Superinduction of immunosuppressive glioblastoma extracellular vesicles by IFN-γ through PD-L1 and IDO1. Neuro-Oncology Adv. (2022) 4:vdac017. doi: 10.1093/noajnl/vdac017
211. Ridder K, Sevko A, Heide J, Dams M, Rupp A-K, Macas J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. (2015) 4:e1008371. doi: 10.1080/2162402X.2015.1008371
212. Liang X, Wang Z, Dai Z, Zhang H, Zhang J, Luo P, et al. Glioblastoma glycolytic signature predicts unfavorable prognosis, immunological heterogeneity, and ENO1 promotes microglia M2 polarization and cancer cell Malignancy. Cancer Gene Ther. (2022) 30:481–96. doi: 10.1038/s41417-022-00569-9
213. Wolf A, Agnihotri S, Guha A. Targeting metabolic remodeling in glioblastoma multiforme. Oncotarget. (2010) 1:552–62. doi: 10.18632/oncotarget.190
214. Chen C, Shi Y, Li Y, He Z-C, Zhou K, Zhang X-N, et al. A glycolysis-based ten-gene signature correlates with the clinical outcome, molecular subtype and IDH1 mutation in glioblastoma. J Genet Genomics. (2017) 44:519–30. doi: 10.1016/j.jgg.2017.05.007
215. Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front Cell Dev Biol. (2017) 5:43. doi: 10.3389/fcell.2017.00043
216. Stadlbauer A, Zimmermann M, Doerfler A, Oberndorfer S, Buchfelder M, Coras R, et al. Intratumoral heterogeneity of oxygen metabolism and neovascularization uncovers 2 survival-relevant subgroups of IDH1 wild-type glioblastoma. Neuro-Oncology. (2018) 20:1536–46. doi: 10.1093/neuonc/noy066
217. Ippolito JE, Yim AK-Y, Luo J, Chinnaiyan P, Rubin JB. Sexual dimorphism in glioma glycolysis underlies sex differences in survival. JCI Insight. (2017) 2:e92142. doi: 10.1172/jci.insight.92142
218. Göran Ronquist K. Extracellular vesicles and energy metabolism. Clinica Chimica Acta. (2019) 488:116–21. doi: 10.1016/j.cca.2018.10.044
219. He Y, Fu L, Li Y, Wang W, Gong M, Zhang J, et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab. (2021) 33:988–1000.e7. doi: 10.1016/j.cmet.2021.03.002
220. Cullin N, Azevedo Antunes C, Straussman R, Stein-Thoeringer CK, Elinav E. Microbiome and cancer. Cancer Cell. (2021) 39:1317–41. doi: 10.1016/j.ccell.2021.08.006
221. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science. (2018) 360:eaan5931. doi: 10.1126/science.aan5931
222. Simbari F, McCaskill J, Coakley G, Millar M, Maizels RM, Fabriás G, et al. Plasmalogen enrichment in exosomes secreted by a nematode parasite versus those derived from its mouse host: implications for exosome stability and biology. J Extracellular Vesicle. (2016) 5:30741. doi: 10.3402/jev.v5.30741
223. Mu J, Zhuang X, Wang Q, Jiang H, Deng Z, Wang B, et al. Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Mol Nutr Food Res. (2014) 58:1561–73. doi: 10.1002/mnfr.201300729
224. Buck AH, Coakley G, Simbari F, McSorley HJ, Quintana JF, Le Bihan T, et al. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat Commun. (2014) 5:5488. doi: 10.1038/ncomms6488
225. Barteneva NS, Maltsev N, Vorobjev IA. Microvesicles and intercellular communication in the context of parasitism. Front Cell Infect Microbiol. (2013) 3:49. doi: 10.3389/fcimb.2013.00049
226. Wang L, Li Z, Shen J, Liu Z, Liang J, Wu X, et al. Exosome-like vesicles derived by Schistosoma japonicum adult worms mediates M1 type immune- activity of macrophage. Parasitol Res. (2015) 114:1865–73. doi: 10.1007/s00436-015-4373-7
227. Nowacki FC, Swain MT, Klychnikov OI, Niazi U, Ivens A, Quintana JF, et al. Protein and small non-coding RNA-enriched extracellular vesicles are released by the pathogenic blood fluke Schistosoma mansoni. J Extracellular Vesicle. (2015) 4:28665. doi: 10.3402/jev.v4.28665
228. Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. (2013) 13:551–65. doi: 10.1038/nri3479
229. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA. (2012) 109(31):E2110-6. doi: 10.1073/pnas.1209414109
230. Oldenburg M, Krüger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23 S rRNA devoid of erythromycin resistance–forming modification. Science. (2012) 337:1111–5. doi: 10.1126/science.1220363
231. Sieber KB, Bromley RE, Dunning Hotopp JC. Lateral gene transfer between prokaryotes and eukaryotes. Exp Cell Res. (2017) 358:421–6. doi: 10.1016/j.yexcr.2017.02.009
232. Robinson KM, Dunning Hotopp JC. Mobile elements and viral integrations prompt considerations for bacterial DNA integration as a novel carcinogen. Cancer Lett. (2014) 352:137–44. doi: 10.1016/j.canlet.2014.05.021
233. Xie Y, Xie F, Zhou X, Zhang L, Yang B, Huang J, et al. Microbiota in tumors: from understanding to application. Advanced Sci. (2022) 9:2200470. doi: 10.1002/advs.202200470
234. Liu A-Q, Vogtmann E, Shao D-T, Abnet CC, Dou H-Y, Qin Y, et al. A comparison of biopsy and mucosal swab specimens for examining the microbiota of upper gastrointestinal carcinoma. Cancer Epidemiology Biomarkers Prev. (2019) 28:2030–7. doi: 10.1158/1055-9965.EPI-18-1210
235. Zhang S, Zhang S, Ma X, Zhan J, Pan C, Zhang H, et al. Intratumoral microbiome impacts immune infiltrates in tumor microenvironment and predicts prognosis in esophageal squamous cell carcinoma patients. Front Cell Infect Microbiol. (2023) 13:1165790. doi: 10.3389/fcimb.2023.1165790
236. Pan C, Wang Y, Liu Q, Hu Y, Fu J, Xie X, et al. Phenotypic profiling and prognostic significance of immune infiltrates in esophageal squamous cell carcinoma. OncoImmunology. (2021) 10:1883890. doi: 10.1080/2162402X.2021.1883890
237. Boesch M, Baty F, Rothschild SI, Tamm M, Joerger M, Früh M, et al. Tumour neoantigen mimicry by microbial species in cancer immunotherapy. Br J Cancer. (2021) 125:313–23. doi: 10.1038/s41416-021-01365-2
238. Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular mimicry and autoimmunity. J Autoimmun. (2018) 95:100–23. doi: 10.1016/j.jaut.2018.10.012
239. Ruff WE, Kriegel MA. Autoimmune host–microbiota interactions at barrier sites and beyond. Trends Mol Med. (2015) 21:233–44. doi: 10.1016/j.molmed.2015.02.006
240. Hintermann E, Holdener M, Bayer M, Loges S, Pfeilschifter JM, Granier C, et al. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J Autoimmun. (2011) 37:242–53. doi: 10.1016/j.jaut.2011.06.005
241. Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, et al. Cross-reactivity between tumor MHC class I–restricted antigens and an enterococcal bacteriophage. Science. (2020) 369:936–42. doi: 10.1126/science.aax0701
242. Barteneva NS, Baiken Y, Fasler-Kan E, Alibek K, Wang S, Maltsev N, et al. Extracellular vesicles in gastrointestinal cancer in conjunction with microbiota: On the border of Kingdoms. Biochim Biophys Acta (BBA) - Rev Cancer. (2017) 1868:372–93. doi: 10.1016/j.bbcan.2017.06.005
243. Choi D-S, Yang J-S, Choi E-J, Jang SC, Park S, Kim OY, et al. The protein interaction network of extracellular vesicles derived from human colorectal cancer cells. J Proteome Res. (2012) 11:1144–51. doi: 10.1021/pr200842h
244. Choi D, Park JO, Jang SC, Yoon YJ, Jung JW, Choi D, et al. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics. (2011) 11:2745–51. doi: 10.1002/pmic.201100022
245. Choi D, Choi D, Hong B, Jang S, Kim D, Lee J, et al. Quantitative proteomics of extracellular vesicles derived from human primary and metastatic colorectal cancer cells. J Extracellular Vesicle. (2012) 1:18704. doi: 10.3402/jev.v1i0.18704
246. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. (2013) 19:460–73. doi: 10.1016/j.molmed.2013.05.002
247. Manzat Saplacan RM, Balacescu L, Gherman C, Chira RI, Craiu A, Mircea PA, et al. The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflammation. (2017) 2017:1–9. doi: 10.1155/2017/4708076
248. Mantur M, Koper O, Snarska J, Sidorska A, Kruszewska-Wnorowska K. Evaluation of PDGF-AB and sP-selectin concentrations in relation to platelet count in patients with colorectal cancer before and after surgical treatment. Pol Arch Med Wewn. (2008) 118:345–50. doi: 10.20452/pamw.410
249. Yu J-H, Ustach C, ChoiKim H-R. Platelet-derived growth factor signaling and human cancer. BMB Rep. (2003) 36:49–59. doi: 10.5483/BMBRep.2003.36.1.049
250. Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. (2006) 16:443–52. doi: 10.1016/j.tcb.2006.07.003
251. Schuh CMAP, Cuenca J, Alcayaga-Miranda F, Khoury M. Exosomes on the border of species and kingdom intercommunication. Trans Res. (2019) 210:80–98. doi: 10.1016/j.trsl.2019.03.008
252. Gangadaran P, Hong CM, Ahn B-C. An update on in vivo imaging of extracellular vesicles as drug delivery vehicles. Front Pharmacol. (2018) 9:169. doi: 10.3389/fphar.2018.00169
Keywords: extracellular vesicles, glioblastoma, immune responses, immune tumor microenvironment, intratumoral microbiome, tumor progression, immunotherapies
Citation: Ghazi B, Harmak Z, Rghioui M, Kone A-S, El Ghanmi A and Badou A (2024) Decoding the secret of extracellular vesicles in the immune tumor microenvironment of the glioblastoma: on the border of kingdoms. Front. Immunol. 15:1423232. doi: 10.3389/fimmu.2024.1423232
Received: 25 April 2024; Accepted: 06 August 2024;
Published: 29 August 2024.
Edited by:
Bo Li, Southern Medical University, ChinaReviewed by:
Daniele Vergara, University of Salento, ItalyShi Hu, Second Military Medical University, China
Copyright © 2024 Ghazi, Harmak, Rghioui, Kone, El Ghanmi and Badou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdallah Badou, YWJkYWxsYWguYmFkb3VAdW5pdmgyYy5tYQ==