 Qiaoyan Si
Qiaoyan Si Mingjian Bai
Mingjian Bai Xiaolong Wang
Xiaolong Wang Tianyu Wang
Tianyu Wang Yan Qin
Yan Qin- 1School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing, China
- 2School of Biomedical Engineering, Affiliated Cancer Hospital & Institute of Guangzhou Medical University, State Key Laboratory of Respiratory Disease, Guangzhou Medical University, Guangzhou, China
- 3Institute of Biophysics, Chinese Academy of Sciences, Beijing, China
With the outbreak of the coronavirus disease 2019 (COVID-19), reductions in T-cell function and exhaustion have been observed in patients post-infection of COVID-19. T cells are key mediators of anti-infection and antitumor, and their exhaustion increases the risk of compromised immune function and elevated susceptibility to cancer. Non-small cell lung cancer (NSCLC) is the most common subtype of lung cancer with high incidence and mortality. Although the survival rate after standard treatment such as surgical treatment and chemotherapy has improved, the therapeutic effect is still limited due to drug resistance, side effects, and recurrence. Recent advances in molecular biology and immunology enable the development of highly targeted therapy and immunotherapy for cancer, which has driven cancer therapies into individualized treatments and gradually entered clinicians’ views for treating NSCLC. Currently, with the development of photosensitizer materials, phototherapy has been gradually applied to the treatment of NSCLC. This review provides an overview of recent advancements and limitations in different treatment strategies for NSCLC under the background of COVID-19. We discuss the latest advances in phototherapy as a promising treatment method for NSCLC. After critically examining the successes, challenges, and prospects associated with these treatment modalities, their profound prospects were portrayed.
1 Introduction
Lung cancer is one of the most common and deadly cancer types in the world (1). According to the cell source, lung cancer can be divided into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), the latter accounting for approximately 85% of all lung cancer cases (2, 3). However, NSCLC can be further subdivided into lung adenocarcinoma, lung squamous cell carcinoma, and large cell carcinoma according to histological features (4). There are many risk factors for lung cancer, among which smoking (second-hand smoke) is the main risk factor for lung cancer (5). Approximately 80%–90% of lung cancer patients are related to active or passive smoking (6). Epidemiological studies indicate a significant age-related increase in lung cancer incidence (7). However, due to the pandemic of SARS-CoV-2 in 2020 (8), the elderly have become one of the most susceptible groups (9). Moreover, patients with lung cancer are at a higher risk of SARS-CoV-2 infection, and the mortality of lung cancer patients increased after COVID-19 infection (10–12). At the same time, those patients infected with COVID-19 exhibited a reduced total number of T cells and increased concentrations of IL-6 and IL-10, which are risk factors that aggravate the severity of COVID-19 in patients with lung cancer (13). Additionally, COVID-19 may alter the tumor microenvironment, promoting cancer cell proliferation and dormant cancer cell (DCC) reawakening. DCCs reawakened upon infection with SARS-CoV-2 can populate the premetastatic niche in the lungs and other organs, leading to tumor dissemination (14). Therefore, the treatment of cancer has become the focus of multidisciplinary research.
There are more and more treatment options for lung cancer. In addition to traditional surgery (15), radiotherapy (16), and chemotherapy (17), targeted therapy (18), immunotherapy (19), phototherapy (20) (Figure 1), and other treatment methods have significant effects on lung cancer. In this paper, targeted therapy, immunotherapy, and phototherapy for NSCLC are summarized in detail, and treatments for NSCLC after the COVID-19 epidemic are prospected.
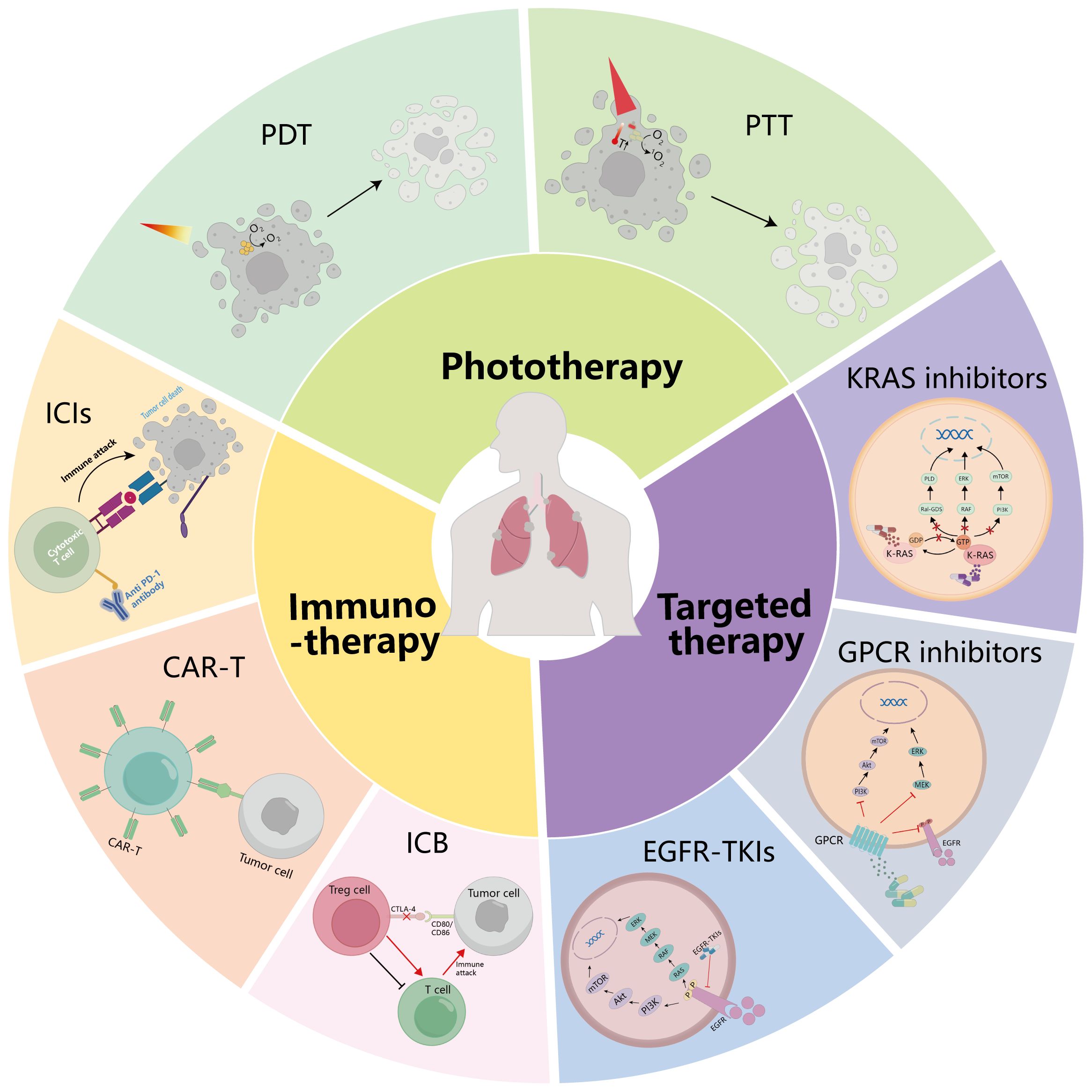
Figure 1. Summary of treatment modalities for NSCLC. The treatment methods of NSCLC are summarized: targeted therapy, immunotherapy, and phototherapy. GPCR, G protein-coupled receptor; EGFR, epidermal growth factor receptor; ICIs, immune checkpoint inhibitors; CAR-T, chimeric antigen receptor (CAR)-modified T cells; ICB, immune checkpoint blockade; PDT, photodynamic therapy; PTT, photothermal therapy.
2 Targeted therapy for non-small cell lung cancer
Targeted therapy is a means of precise treatment at the molecular level, which is a common way to treat NSCLC. However, the impact of COVID-19 on the efficacy of targeted therapy remains unclear. Nevertheless, new targeted therapy is still the focus of attention in the treatment of NSCLC. In this section, we review three common modalities of targeted therapy for NSCLC, namely, EGFR-TKI, KRAS inhibitor therapy, and GPCR inhibitor therapy, and look forward to new research directions for these three treatment modalities.
2.1 EGFR-TKI
Epidermal growth factor receptor (EGFR) is a receptor for promoting mammalian epidermal growth factor, which mainly exists on the surface of human keratinocytes, epithelial cells, glial cells, fibroblasts, and other cell membranes (21). EGFR can be activated through pathways such as phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) pathway, rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPK) pathway, janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, and phospholipase C-protein kinase C (PLC-PKC) pathway (22–25). These pathways transmit signals for cell proliferation and differentiation (26), which makes EGFR play a key role in tumor proliferation and survival. EGFR mutation is a key genetic alteration in non-small cell lung cancer (NSCLC), with mutation rate as high as 30% among NSCLC patients and approximately 16% among those with advanced lung adenocarcinoma (27). Most EGFR mutations in NSCLC patients occur in exons 18–21 of the receptor tyrosine kinase domain (28). The L858R mutation in exon 21 and deletion in exon 19 account for approximately 85% of EGFR mutations (29, 30). These mutations are recognized as oncogenic drivers of NSCLC and enhance the sensitivity to EGFR tyrosine kinase inhibitors (EGFR-TKIs).
The drugs for the treatment of NSCLC caused by EGFR mutations, epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), have gone through three generations of targeted drugs and are expected to usher in a fourth generation of targeted drugs (31) (Table 1). Among them, first-generation EGFR-TKI targeted drugs, such as gefitinib (32), erlotinib (33), and icotinib (34), targeting the typical mutations of EGFR, namely, the deletion mutation in exon 19 (delE746-A750) (35) and the point mutation in exon 21 (L858R) (36), have a good therapeutic effect. The clinical application of second-generation EGFR-TKIs, including afatinib (37) and dacomitinib (38), has been limited due to modest efficacy improvements and more serious side effects compared to first-generation drugs. These two generations of EGFR-TKIs inhibit the activation of EGFR by mimicking the structure of ATP and competitively interact with the binding site of the EGFR kinase domain, thereby inhibiting the occurrence of cancer. Concomitant drug use inevitably leads to the emergence of acquired drug resistance, and the T790M mutation is the most common, accounting for approximately 50%–60% of NSCLC cases (39, 40). The T790M mutation caused drug resistance due to the substitution of threonine (T) by methionine (M) at position 790 of EGFR, which hindered the binding of EGFR-TKI to EGFR. Meanwhile, the T790M mutation enhances the affinity between EGFR and ATP, which is also the reason for the decreased efficacy of EGFR-TKI (39, 41).
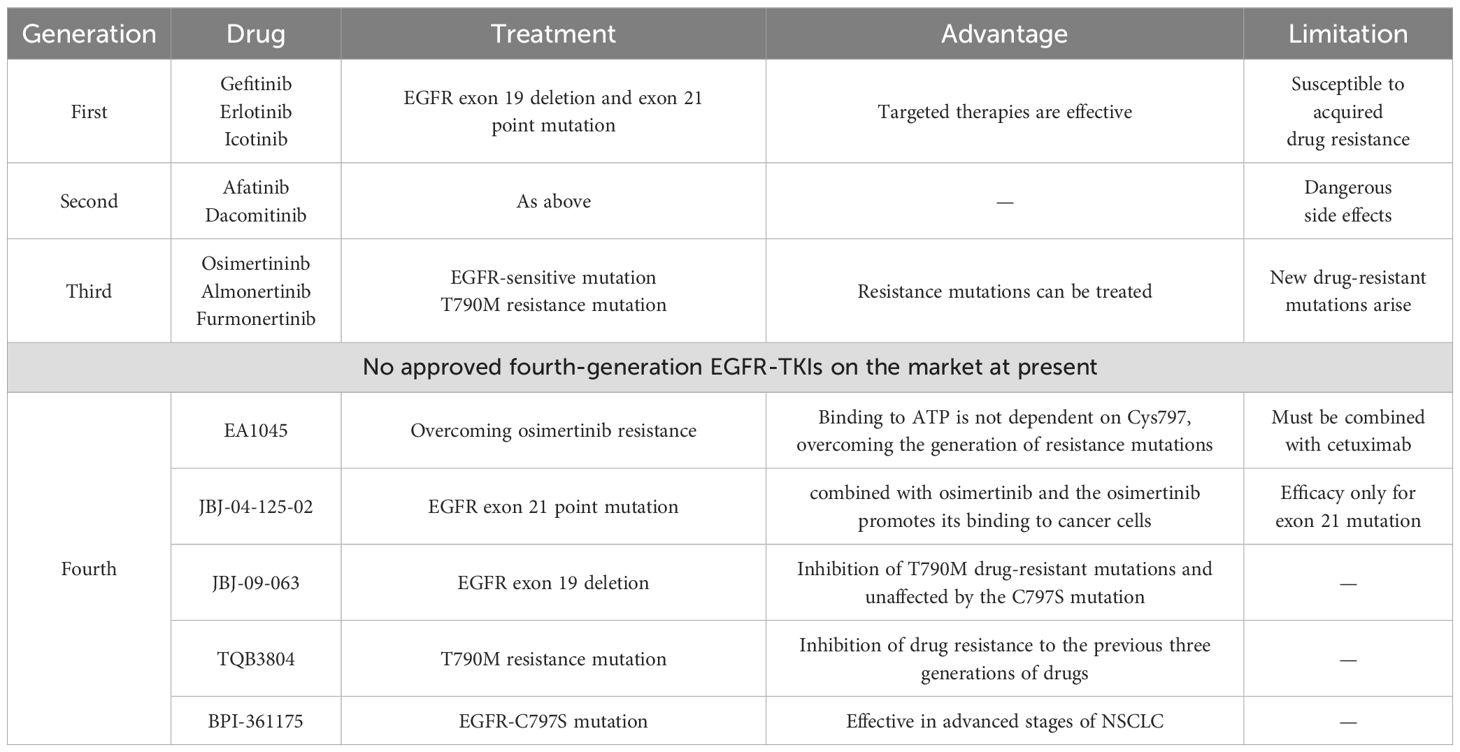
Table 1. Advantages and disadvantages of EGFR-TKIs targeted agents in the treatment of NSCLC induced by EGFR mutations.
To solve the T790M acquired resistance mutation, the third-generation of EGFR-TKIs has been developed, including the famous osimertinib (42), almonertinib (43), and furmonertinib (44), which have been approved for marketing in recent years. At present, the third-generation EGFR-TKIs have been recommended by the authoritative lung cancer diagnosis and treatment guidelines of the United States and China as the first choice for the treatment of NSCLC with EGFR-sensitive mutation and T790M resistance mutation (45). Due to the widespread use of the drug, the development of resistance to the third-generation EGFR-TKI is also expected, and EGFR C797S mutation is the most common resistance mutation to osimertinib.
The fourth-generation EGFR-TKIs have been designed to treat patients who were resistant to first-generation EGFR-TKIs harboring the T790M mutation or to treat patients who were resistant to third-generation EGFR-TKIs harboring the C797S mutation. However, no fourth-generation EGFR-TKI has been approved for clinical application worldwide. Therefore, the treatment of EGFR-mutated non-small cell lung cancer should focus on solving EGFR-TKI resistance mutations.
2.2 KRAS inhibitors
The protein encoded by the KRAS gene is a small GTPase membrane-binding protein that plays a major role in cell growth regulation through signal transduction, and it belongs to the RAS superfamily of superproteins (46, 47). KRAS gene mutation accounts for 75% of the total number of RAS gene mutations (48), which is also a very common type of oncogene mutation. Specifically, KRAS gene mutation accounts for 20%–30% of the total number of NSCLCs, which is higher in lung adenocarcinoma and rare in lung squamous cell carcinoma (49, 50). In contrast, H-Ras mutations account for less than 1% (51, 52), and most H-Ras mutations occur on codon 61 (53). Due to the very low proportion of H-Ras mutations in NSCLC patients, there are relatively few research works on the clinical treatment of H-Ras mutations. Known targeted therapeutics for H-Ras mutation mainly focus on tipifarnib, a farnesyl transferase (FT) inhibitor, which is one of the most promising candidates (54, 55). However, no significant benefit was seen in people treated with tipifarnib, so tipifarnib is classified as a pan-Ras targeted drug (56). Therefore, the KRAS subtype is more concerned. KRAS G12C is the most common type of KRAS mutation (57). KRAS is a small GTPase, which cycles between inactivation (binding to GDP) and activation (binding to GTP) (58, 59). When KRAS is activated by binding to GTP, activated KRAS promotes RAF recruitment (60) and PI3K activation (61). Thus, it promotes cell proliferation, differentiation, and survival (62) (Figure 2). In addition, Ral-GDS has also been found to be an effector of KRAS, which is involved in vesicle trafficking and cytoskeleton composition (63). When KRAS mutation occurs, the ability of KRAS to hydrolyze GTP or interact with GDP will decrease, resulting in the continuous activation of KRAS and its downstream signaling factors and promoting tumorigenesis (64). At the same time, KRAS also influences ribosome production (65) through PI3K/Akt/mTOR, and the expression of ribosomal translated p53 protein is increased in cancer cells, contributing to both cancer generation and progression (66) (Figure 3).
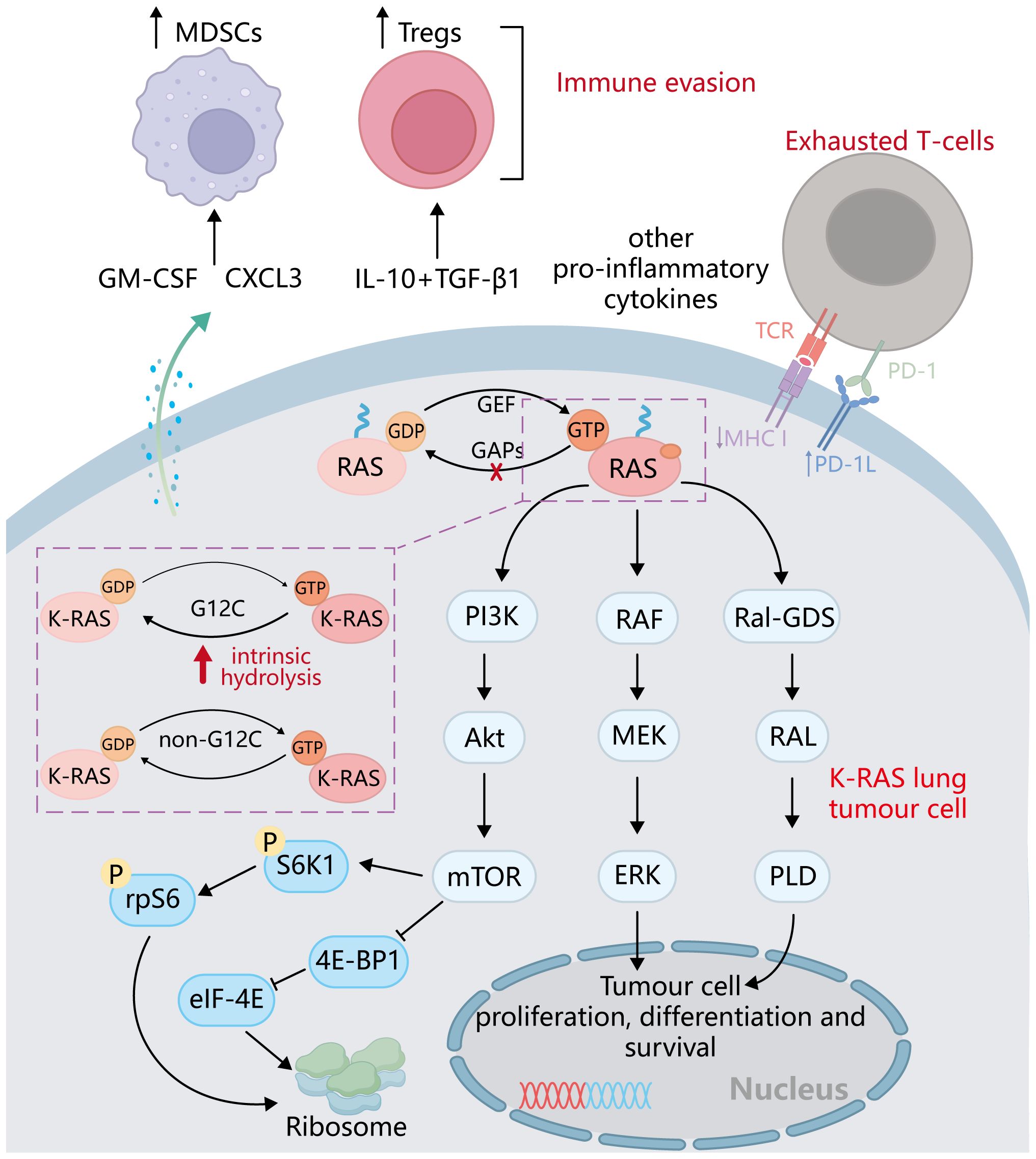
Figure 2. Major downstream pathways after RAS family activation: PI3K/Akt/mTOR, RAF/MEK/ERK, and RAL-GDS /RAL/PDL signaling pathways. After KRAS mutation is activated, it regulates the proliferation, differentiation, and survival of tumor cells through the above-mentioned major pathways and also affects the synthesis of ribosomes.
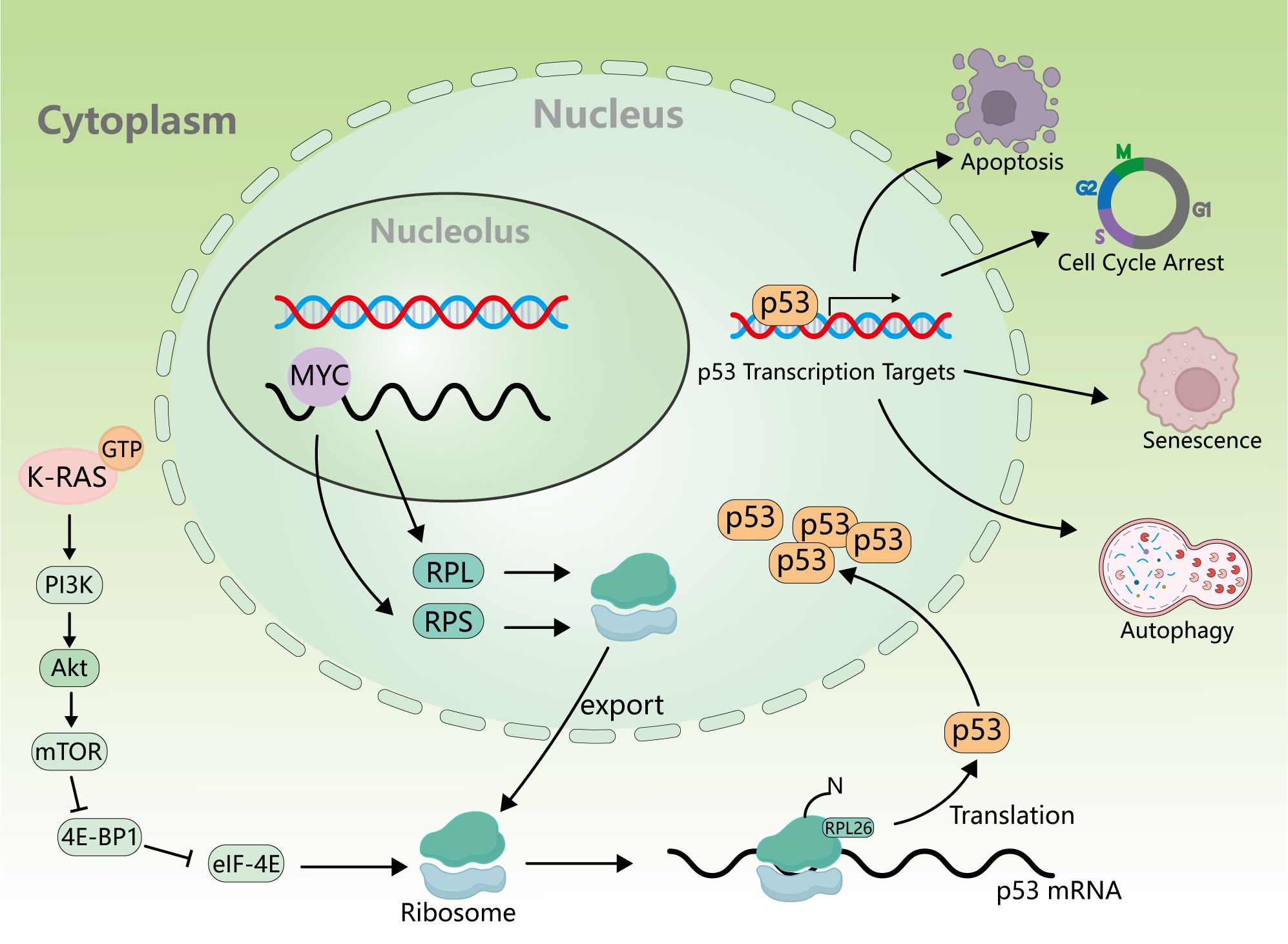
Figure 3. Relationship between KRAS activation and ribosomes and p53. KRAS activation affects ribosome synthesis through the PI3K/Akt/mTOR signaling pathway, and p53mRNA is translated on the ribosome to produce p53 protein, which further promotes apoptosis, cell cycle arrest, senescence, and autophagy.
For a long time, there was no good strategy to treat tumors caused by KRAS mutations. Until the introduction of inhibitors of the KRAS G12C mutation, a solution to tumors arising from KRAS mutation can be available. The FDA approved sotorasib (AMG510) for the first time for the treatment of tumors carrying KRAS G12C mutation (67). According to the results of clinical trials, AMG510 has a high disease control rate and good safety in NSCLC (68). Moreover, adagrasib (MRTX849) is a specifically optimized oral inhibitor of KRAS G12C mutant (69) by irreversibly and selectively binding to KRAS G12C in the inactive state, preventing it from sending cell growth signals and causing cancer cell death (70).
In addition to KRAS inhibitors targeting the G12C mutation, inhibitors targeting other mutations, such as G12D and G12V mutations, also have certain therapeutic effects on NSCLC caused by KRAS mutation (71, 72). However, these inhibitors are still in the stage of clinical trials, and there is still a long way to go before they can be approved for marketing. At the same time, the discovery of pan-KRAS inhibitors also provides another treatment for NSCLC (73) (Table 2). Because of the role of KRAS in ribosome formation, the combination of tumor-associated protein drugs and KRAS inhibitors could represent a novel approach to treating KRAS mutation-induced NSCLC.
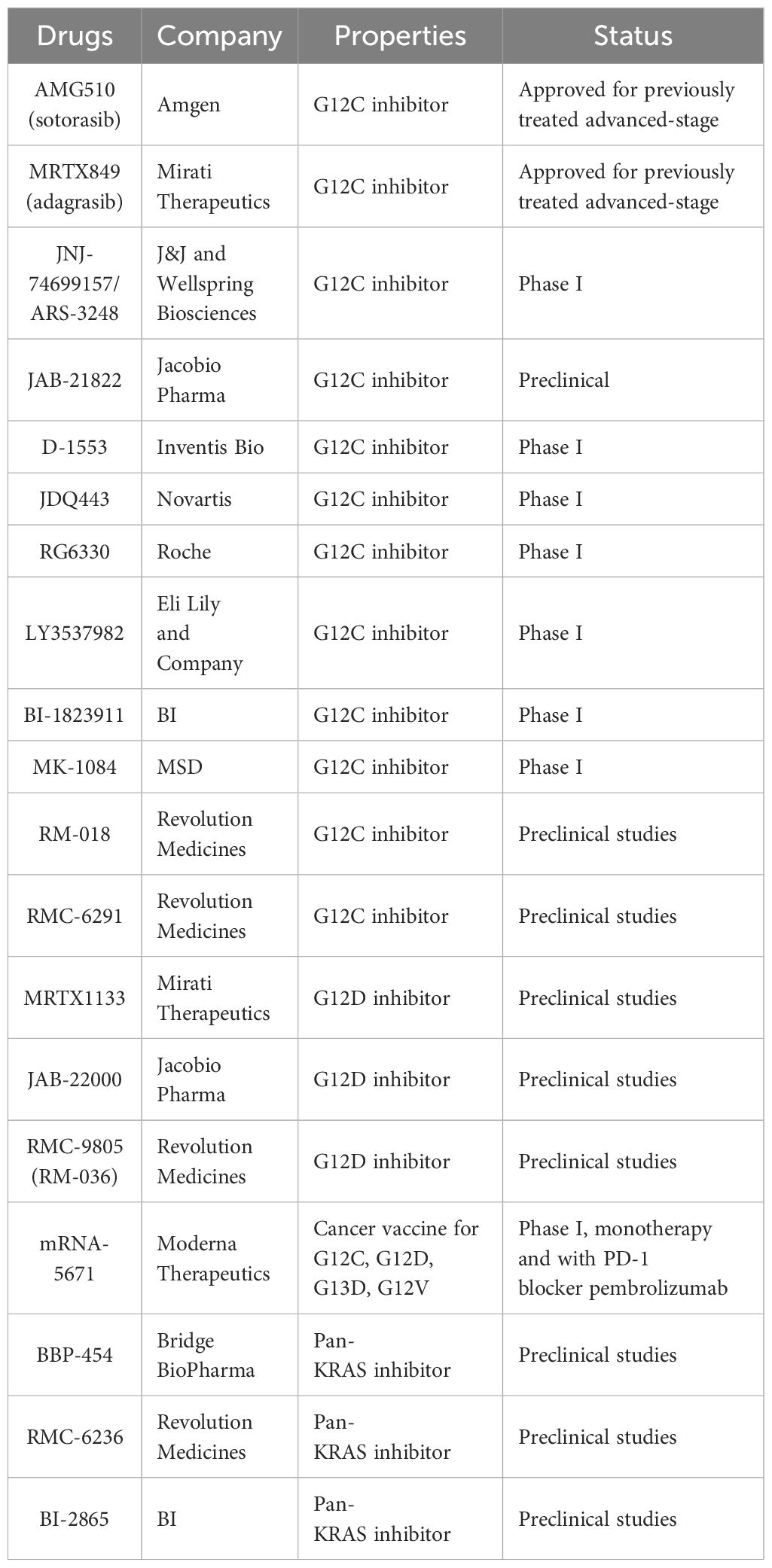
Table 2. Different types of drugs targeting KRAS mutations and their research stages.
2.3 GPCR inhibitors
Seven transmembrane GPCRS constitute one of the largest families of extracellular signaling proteins in the human genome and regulate a large number of physiological and pathological processes. Therefore, they have also become one of the most attractive therapeutic targets for cancer treatment (74). GPCR can couple with G proteins to transmit a large number of extracellular signals produced by hormones, neurotransmitters, ions, photons, trend factors, and so on. Furthermore, GPCR can interact with heterotrimeric G proteins composed of α, β, and γ subunits, inducing the exchange of guanosine diphosphate (GDP) and guanosine triphosphate (GTP) at the Gα subunit. Then, Gα dissociates from Gβγ transduction signals to different downstream effectors, regulating the downstream signal transduction pathway (75). Thus, GPCRs serve as signaling hubs in cancer development by participating in cell proliferation, invasion, and migration and influencing immune cell-mediated functions (76, 77). Considerable evidence has shown that GPCR activation can promote the phosphorylation of its huge downstream corresponding factors, such as PI3k/Akt/mTOR (78) and PCK/RAF/MEK/ERK (79), thereby promoting the development of NSCLC—for example, Opsin 4/opiomelanocortin (OPN4) (79), a member of the Opsin family of GPCRs, is overexpressed in NSCLC patients and correlated with survival. The mechanism of OPN4 in the development of NSCLC has also been clearly elucidated, that is, OPN4 first binds to Gα11 to activate PCK, and then PCK phosphorylation activates its downstream signaling pathway BRAF/MEK/ERKs, which mediates NSCLC cell proliferation and migration. Moreover, Opsin4 inhibitor can effectively inhibit the proliferation and invasion of NSCLC cells.
In addition, much evidence suggests that GPCR ligand activation can also induce EGFR reactivation in various types of cancer cells, which is an important mechanism for regulating GPCR- and EGFR-mediated tumorigenesis (80, 81). Activated GPCR can upregulate EGFR ligand levels, thereby activating EGFR and its downstream signaling pathways (76, 82). In addition to their dependence on binding ligand activation, several adaptor proteins such as c-Src and β-arrestin were found to potentially participate in EGFR reactivation via GPCR (83, 84). Indeed GPCR interactions with EGFR contribute to the initiation and progression of colon, breast, ovary, prostate, and head and neck tumors (85). In NSCLC cells, GPCR transactivation of EGFR plays an important role in proliferation, migration, and drug resistance (86)—for example, protease-activated receptors (PARs), a subfamily of G protein-coupled receptors (GPCR), play important roles in hemostasis, thrombosis, embryonic development, wound healing, and inflammation (87). Among them, PAR2 activation is not only associated with arthritic pain, skin heat allergy, and paclitaxel-induced neuropathic pain (88, 89) but regulates a variety of downstream signaling pathways and cellular functions in a variety of cancer cells, including liver and renal cancer cells (90, 91). Meanwhile, many studies suggest that PAR2–EGFR interaction leads to COX2 expression, which promotes the proliferation of colon cancer cells (92, 93). However, PAR2 expression was significantly enhanced during the development of NSCLC and related to NSCLC patients’ survival (94). Meanwhile, in NSCLC, the activation of PAR2 leads to the reactivation of EGFR, which promotes the epithelial–mesenchymal transition (EMT) process of lung cancer cells through the ERK signaling pathway and then promotes the proliferation and migration of NSCLC cells (95). The β-blocker is an important regulator of PAR2-mediated ERK signaling activation following EGFR trans-activation. Therefore, PAR2 can promote the proliferation and migration of NSCLC through the β-arrestin-EGFR-ERK signaling pathway. At the same time, activation of ERK signaling and dysregulation of EMT are both considered to be the molecular mechanisms of resistance to osimertinib (94). Moreover, PAR2 plays an important role in the regulation of drug resistance in NSCLC by mediating the ERK signaling pathway. The use of the PAR2 inhibitor P2pal-18S can significantly attenuate the EMT process of NSCLC cells, reduce PD-L1 expression, and regulate cell viability, cell migration, and apoptosis. The simultaneous use of PAR2 inhibitors and NSCLC drugs, such as osimertinib and gefitinib, has demonstrated promising therapeutic effects on NSCLC even in cases of drug resistance (96). Therefore, the development of highly selective GPCR inhibitors has become a new frontier in targeted therapy NSCLC.
Certainly, targeted therapy also faces many challenges, especially drug resistance. In addition to the major challenge of drug resistance, other issues are summarized in Table 3. Firstly, because targeted therapy works by inhibiting a specific biomarker that is necessary for cancer progression, its effectiveness is limited to patients whose tumors express that biomarker (97). Secondly, even though targeted therapies can be effective for cancer therapy, unpredictable side effects are always inevitable. Common adverse events associated with targeted therapies include rash, diarrhea, hypertension, hypothyroidism, proteinuria, depigmentation, and hepatotoxicity (98). These side effects have both physical and mental impact on the treated patients. These problems continue to inspire researchers to innovate research to explore new ways of targeted therapy aimed at overcoming these existing limitations.
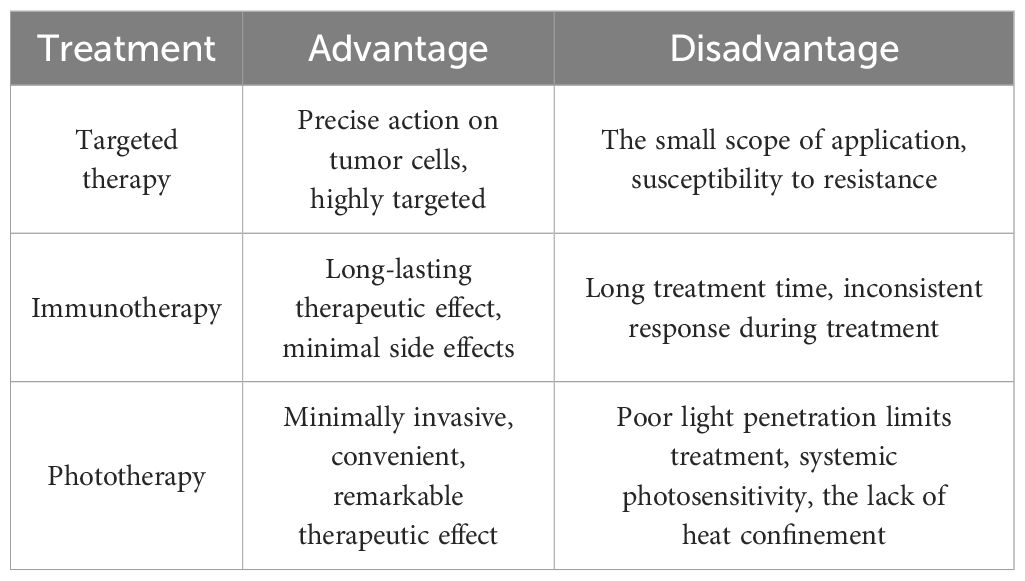
Table 3. Advantages and disadvantages of the three described treatment modalities for NSCLC (targeted therapy, immunotherapy, and phototherapy).
3 Immunotherapy for non-small cell lung cancer
Immunotherapy has revolutionized the treatment landscape for NSCLC, particularly in advanced or metastatic stages. It has significantly improved the survival rates and quality of life for many patients. Currently, immunotherapy options for NSCLC mainly include the following approaches: immune checkpoint inhibitors (ICIs), immune checkpoint blockade (ICB), and adoptive cell therapy (99).
3.1 ICI/ICB
With the rapid development of tumor immunotherapy, there are a variety of immunotherapy options for the first-line treatment of NSCLC, among which ICIs and ICB are the most promising (100). They were able to restore the ability of T cells to recognize and attack tumor cells by blocking immune checkpoint molecular interactions between tumor cells and T cells. Immune checkpoint molecules, such as programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), play a crucial role in regulating the immune response (101). They act as “brakes” to prevent excessive immune activation against healthy cells and tissues. However, cancer cells can exploit these immune checkpoints to evade the immune system and continue growing (102) (Figure 4). Therefore, ICI/ICB drugs block these immune checkpoint proteins, releasing the brakes on the immune system and allowing it to recognize and attack cancer cells more effectively (103).
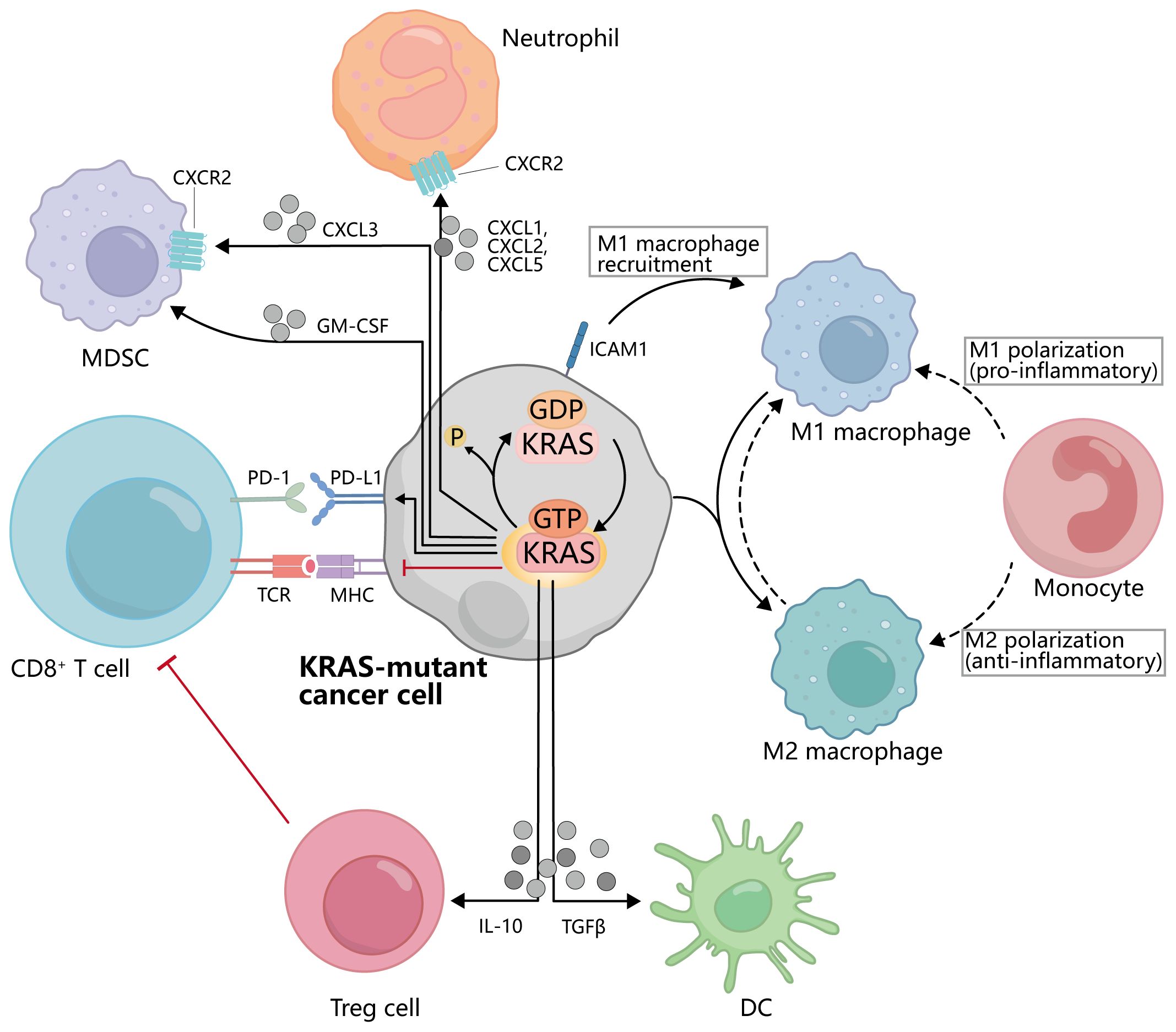
Figure 4. Interactions between KRAS-mutated cancer cells and immune cells. KRAS activation inhibits the binding of MHC on the cancer cell to TCR on CD8+T cells and promotes the binding of PD-1 to PD-L1. KRAS mutant cancer cells can secrete IL-10, TGFβ, GM-CSF, CXCL3, CXCL1, etc., to affect the activation and recruitment of Treg cells, DC, MDSC, and other cells. Meanwhile, the activation of KRAS also promoted the polarization of macrophages from M1 to M2.
The development of immunotherapies, particularly antibodies that inhibit the PD-1/PD-L1 pathways, has revolutionized the treatment of patients worldwide with advanced or metastatic NSCLC and improved their survival outcomes (104). Among ICIs, those targeting the PD-1/PD-L1 axis are the most promising, exerting potent anti-tumor effects by reversing T cell exhaustion and releasing anti-tumor immune responses (105, 106). PD-1 is a cell surface receptor expressed on activated T cells, B cells, and natural killer cells. Its ligands, PD-L1 and PD-L2, are primarily expressed in antigen-presenting cells (APCs) and tumor cells. The binding of PD-1 to PD-L1 or PD-L2 transmits an inhibitory signal to T cells, dampening their activation and effector functions (107). This signaling pathway plays a crucial role in maintaining immune homeostasis and preventing excessive immune responses. In the context of cancer, tumor cells often exploit the PD-1/PD-L1 pathway as an immune evasion mechanism. By expressing PD-L1, tumor cells can suppress and downregulate the immune response directed against them and escape destruction by cytotoxic T cells (108). The development of ICI/ICB targeting the PD-1/PD-L1 pathway has revolutionized cancer treatment, including NSCLC. For FDA-approved antibodies, two antibodies against PD-1 (nivolumab and pembrolizumab) and two antibodies against PD-L1 (atezolizumab and durvalumab) (109–112) have shown significant clinical benefits in NSCLC patients, with improved overall survival and progression-free survival compared to traditional chemotherapy in certain patient subgroups (113). These inhibitors have become standard treatment options, either monotherapy or in combination with other therapies, for advanced or metastatic NSCLC. However, it is worth noting that the response to PD-1/PD-L1 inhibitors varies among patients and tumor types (114). Factors such as tumor PD-L1 expression, tumor mutational burden, and the presence of other immune cells in the tumor microenvironment can influence the efficacy of PD-1/PD-L1 inhibitors (115).
Targeting the PD-1/PD-L1 pathway with immune checkpoint inhibitors has emerged as a promising therapeutic strategy for NSCLC and other cancers. These inhibitors have demonstrated significant clinical benefits by enhancing the immune response against tumor cells. Ongoing research aims to optimize the selection of patients to maximize the efficacy of PD-1/PD-L1 inhibitors and to develop strategies to overcome therapies’ resistance.
CTLA-4 is another immune checkpoint receptor that plays a critical role in regulating immune responses. It is expressed on the surface of T cells, where it acts as a negative regulator of T-cell activation. CTLA-4 binds to the B7 ligands (CD80 and CD86) expressed on APCs. This interaction leads to the inhibition of T-cell activation and the suppression of immune responses (116). By engaging CTLA-4, T cells become less responsive to antigen stimulation, leading to a dampening of the immune response and preventing excessive immune activation (117). Similar to the PD-1/PD-L1 pathway, cancer cells can exploit the CTLA-4 pathway to evade immune destruction by upregulating the expression of B7 ligands or expressing soluble CTLA-4, thus suppressing T cell activity and promoting tumor immune evasion (118). In NSCLC, although PD-1/PD-L1 inhibitors have shown remarkable efficacy in treatment, CTLA-4 is also considered a potential treatment target. Several clinical trials have investigated the efficacy of CTLA-4 inhibitors alone (such as ipilimumab) or in combination with PD-1/PD-L1 inhibitors in NSCLC patients. The results have shown significant clinical benefits in certain patient subgroups (NCT03527251). However, compared to other cancer types such as melanoma, the response to CTLA-4 inhibitors alone in NSCLC patients is not as prominent. This may be related to the differences in immune characteristics and antigen expression of NSCLC. Some studies have found that NSCLC patients with a high tumor mutational burden (TMB) may respond better to CTLA-4 inhibitors (NCT03527251 and NCT03913923). Tumors with high TMB have more nonsynonymous mutations, which may generate more neoantigens and trigger a stronger immune response (119). Therefore, high TMB may be an important indicator to predict the response to CTLA-4 inhibitors in NSCLC patients. Additionally, the therapeutic potential of CTLA-4 inhibitors is still under further investigation compared to PD-1/PD-L1 inhibitors. Currently, ongoing clinical trials are exploring the combination of CTLA-4 and PD-1/PD-L1 inhibitors to determine if it can produce better treatment outcomes in NSCLC patients (NCT03527251). Although the efficacy of CTLA-4 inhibitors alone in NSCLC patients is limited, combination therapy with PD-1/PD-L1 inhibitors may lead to improved treatment outcomes. By targeting both PD-1/PD-L1 and CTLA-4, it is possible to address different activation and inhibition mechanisms in T cells, further enhancing the anti-tumor immune response. Further research is still needed to identify which NSCLC patients are most suitable for receiving CTLA-4 inhibitor treatment and to make progress in defining treatment regimens and predicting patient responses.
Compared with conventional chemotherapy, ICI/ICB treatment significantly improved the survival outcomes in patients with advanced NSCLC. However, overall, there remains a subset of patients who do not respond to ICI/ICB therapy. Therefore, future clinical studies are needed to develop strategies to overcome immunotherapy resistance by identifying patients who are likely to respond to ICI/ICB therapy, explore new immune checkpoints and develop new immune checkpoint inhibitors or blockers for the treatment of NSCLC patients, and achieve a better understanding of the safety profile of ICI/ICB therapy to determine the proportion of patients who would benefit from expanding the ICI/ICB therapy (120, 121).
3.2 Adoptive cell therapy for non-small cell lung cancer
Adoptive cell therapy (ACT) involves utilizing immune cells (especially different types of T cells) that react to tumors. These cells are cultivated, grown, and genetically engineered outside the patient’s body before being re-administered as a therapy to identify and target cancer cells. The two most frequently employed forms of ACT are chimeric antigen receptor (CAR)-modified T cells (CAR T) therapy and tumor-infiltrating lymphocytes (TILs) therapy (122, 123) (Figure 5).
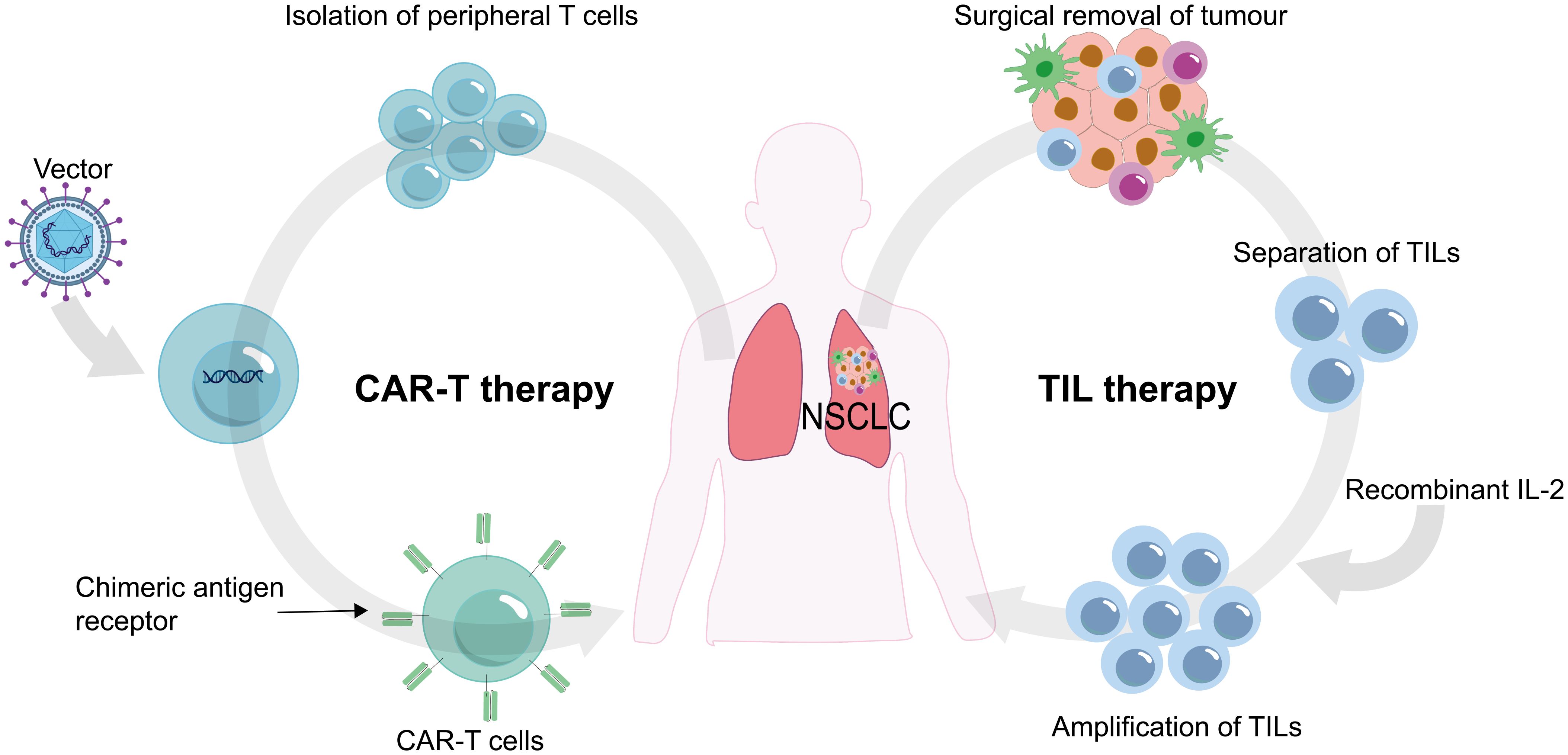
Figure 5. Chimeric antigen receptor (CAR) T cells and tumor-infiltrating lymphocytes (TILs) are procured from the patient's own body, subjected to laboratory processing, and subsequently reinfused back into the patient.
3.2.1 CAR-T
CAR-T (chimeric antigen receptor T-cell immunotherapy) is an emerging tumor immunotherapy method (124–126). CAR-T involves genetically engineering T cells, which are immune cells in the human body, to specifically recognize and attack tumor cells (127). Researchers install a CAR (chimeric antigen receptor) onto the T cells, acting as a navigation system to identify and bind to specific antigens on the surface of tumor cells (128). Once modified, these CAR-T cells are expanded in large quantities and then infused back into the patient’s body to exert their anti-tumor effects. CAR-T therapy has shown significant efficacy in treating various types of cancers, particularly in hematologic malignancies, offering new hope and treatment options for patients. In addition, it has shown promising results in the treatment of various types of cancer, including NSCLC. In CAR-T cell therapy, T cells are extracted from a patient’s blood and modified in the laboratory to express CAR on their surface. The CAR is designed to specifically recognize a protein antigen that is expressed on the surface of cancer cells. Once the CAR-T cells are infused back into the patient, they can recognize and bind to the cancer cells, leading to their destruction (129, 130). CAR is an artificial fusion protein consisting of an extracellular antigen binding domain, an extracellular spacer sequence motif, a transmembrane (TM) domain, and an intracellular signaling domain. The TM structure and the hinge region fix the CAR to the cell membrane. The signal region is an intracellular T-cell activation complex composed of the signal module CD3ζ and many co-stimulatory molecules (131–133). They trigger antigen binding by regulating downstream signaling cascades activated by T cells. Co-stimulatory molecules include CD27, CD28, OX40 (CD134), and 4-1BB (CD137), inducing co-stimulatory molecules (CD278), and glucocorticoid-induced tumor necrosis factor (TNF) receptor-associated protein (GITR) (134). T cells modified with synthetic CAR vectors can recognize antigens specific to many human malignancies, including solid tumors (135–137), thus playing a therapeutic role in tumors. At present, CAR vectors have been upgraded to the fifth generation, differing mainly in their specific co-stimulatory molecular modifications.
The current fifth-generation CAR contains IL-2Rβ fragment modification. The IL-2Rβ fragment can induce the production of Janus kinase and transcriptional signal transducer and activator of transcription (STAT)-3/5 (138, 139) to enhance the efficacy of CAR-T therapy. The safety and efficacy of the fifth-generation CAR is being tested (140).
In solid tumors, CAR-T cell research mostly focuses on NSCLC (141). The most common antigens targeted by NSCLC include EGFR (142), mesothelin (MSLN) (143), carcinoembryonic antigen (CEA) (144), PD-L1 (145), human epidermal growth factor receptor 2 (HER2) (146), etc. CAR T cells bind to the antigens of NSCLC to treat NSCLC. In a study, the authors constructed CAR T cells targeting PD-L1 and evaluated their efficacy in the treatment of NSCLC. The results showed that PD-L1-CAR T cells effectively cleared PD-L1 NSCLC cells and xenograft tumors. Furthermore, the combination of PD-L1-CAR T cell injection with local radiotherapy enhanced the efficacy of PD-L1-CAR T cells against PD-L1 NSCLC cells and tumors (147).
CAR-T cell therapy offers several advantages over other forms of treatment. Firstly, CAR-T cells can specifically target cancer cells, sparing healthy cells from damage. Secondly, CAR-T cells can persist in the body and continue to attack cancer cells over a prolonged period. Lastly, CAR-T cell therapy can potentially induce long-term remission or even cure in some patients (122). However, there are still many obstacles in the application of CAR-T cell therapy in NSCLC, especially the specificity of antigens expressed by tumor cells. Therefore, improving the safety and efficacy of CAR-T cell therapy by optimizing CAR vectors is the direction of future development.
3.2.2 TILs
TILs (tumor-infiltrating lymphocytes) are a subset of immune cells, primarily T cells, that migrate from the bloodstream and infiltrate the microenvironment of the tumor. These TILs can be found within the tumor mass itself as well as in the surrounding adjacent tissues (148). Once inside the tumor, TILs engage in intricate interactions with cancer cells and the surrounding tumor microenvironment. These interactions enable TILs to recognize specific antigens expressed by the cancer cells, subsequently triggering an immune response for combating the tumor (149). Compared to peripheral blood T cells, TILs often exhibit a higher concentration of T cell clones that target tumor antigens. This heightened specificity makes TILs a valuable resource for TILs therapy (150). This immunotherapy approach involves isolating TILs from the patient’s tumor, expanding them ex vivo with the assistance of recombinant IL-2 (rIL-2), and re-introducing amplified TILs into the patient to proliferate, recognize, and effectively eliminate the tumor cells (123).
A previous study examined the potential therapeutic effects of utilizing TILs as a post-operative intervention in patients diagnosed with stage II–III NSCLC. The study involved the surgical extraction of tissue samples from the primary lung lesions of these patients (151). After the isolation of lymphocytes, they were cultivated in a medium supplemented with rIL-2. The resulting TILs were then administered to patients who were stratified based on their disease stage. In addition, patients received daily subcutaneous injections of IL-2 until the maximum tolerable dose was reached. Notably, the TILs groups exhibited a more positive median survival outcome in comparison to the standard care treatment groups. These promising findings were further supported by a recent phase I trial characterized by a reduced tumor sample size. The trial demonstrated that TILs therapy, administered in doses ranging from 20 × 109 to 20 × 1010 CD3+ cells and combined with IL-2, represented a viable treatment option with a manageable safety profile (152). It is important to note that the isolation and expansion of T cells from tumor tissue require advanced laboratory skills and a controlled environment. The preparation process is highly intricate and time-consuming, demanding a significant workforce. These reasons contribute to the high cost of treatment required for TILs therapy.
Immunotherapy can use the ability of the immune system to attack lung cancer cells and achieve therapeutic effects (153). Its advantages include a lower incidence of serious side effects than traditional therapy, longer efficacy than traditional therapy for specific patients, and the ability to be combined with traditional therapy. The disadvantages of immunotherapy include the need to test specific biomarkers to determine a patient’s suitability and the risk of triggering an autoimmune response. Immunotherapy is still in the early stage of development, and its efficacy needs to be confirmed by more studies (101, 154, 155) (Table 3).
4 Phototherapy for non-small cell lung cancer
Chemotherapy has been regarded as the first choice of standard lung cancer treatment, but resistance to chemotherapy is the biggest challenge in the treatment of lung cancer. The molecular mechanisms contributing to chemotherapy resistance include reduced drug activity and impaired drug uptake and delivery (156). These factors prevent the drug from reaching the desired concentration at the treatment site, thereby limiting its therapeutic efficacy (97). Chemotherapy drugs that do not reach the treatment site will throughout circulate the body and even appear in healthy organs and tissues, causing adverse reactions in patients with lung cancer and even endangering their lives (157, 158). To overcome the limitations of chemotherapy, researchers have discovered phototherapy. Phototherapy is a minimally invasive and effective treatment, a convenient method of ablating tumors with light irradiation in clinical practice (159). Phototherapy, including photodynamic therapy (PDT) and photothermal therapy (PTT), has attracted many researchers to study because of its being non-invasive, operability, and minimal dark toxicity in cancer treatment (160). Notably, PDT/PTT can potentially overcome chemotherapy resistance. Firstly, differently from chemotherapy, the effect of PDT/PTT can be dependent on the photophysical properties or photothermal characteristics of photosensitive drugs. The activity of PDT/PTT drugs cannot be decreased after multiple treatments (157, 161), and the cancer cells targeting of general photosensitizers (such as porphyrin derivatives) has been widely proven. Therefore, there is no decreased drug uptake for PDT (162). Most importantly, photodynamic therapy could significantly reduce the expression of ATP-binding cassette subfamily G member 2 (ABCG2), which is responsible for drug resistance in chemotherapy, resulting in enhanced chemotherapy effects and achieving reduced drug resistance (163). For cancer patients after COVID-19, due to the decrease in the number of T cells, the immune function is relatively weakened. However, it is unclear whether the efficacy of phototherapy for NSCLC will be diminished due to COVID-19. Therefore, this section mainly reviews photodynamic therapy and photothermal therapy for NSCLC in the hope that the impact of COVID-19 on human immunity can be considered when designing drugs for phototherapy in the future so as to maximize the efficacy of drugs.
4.1 PDT
In PDT, photosensitizers undergo photoinduced electron transfer, usually in a physiological environment, to generate various ROS, including superoxide anion radical (·O2-), hydrogen peroxide (H2O2), and hydroxyl radical (·HO) (164, 165). There are three main cell death pathways in PDT, which are apoptosis, necrosis, and autophagy-related cell death pathways, of which apoptosis is the most commonly recognized (166, 167). At present, some small-molecule photosensitizers have been approved for clinical treatment, such as indocyanine green (ICG) (168), chloroe6 (Ce6) (169), porphyrin (170, 171), and other phototherapy drugs (172–174). However, its poor systemic circulation and limited targeting ability limit the output of its therapeutic effect. Therefore, more and more studies have been conducted to modify small-molecule photosensitizers with nano-agents to enhance their therapeutic effects and biological safety (175). A multifunctional nanoprobe GNPs@PEG/Ce6-PD-L1 peptide containing gold nanoprisms with chlorin e6 (GNPs@PEG/Ce6-P) (Figure 6) (176) was developed to exploit the targeting of PD-L1 for imaging-guided photothermal/photodynamic therapy. Moreover, the experiments in vitro and in vivo demonstrated that the nanoprobe GNPs@PEG/Ce6-P not only enabled real-time visualization via fluorescence and photoacoustic imaging but also served as a phototherapeutic agent for synergistic photodynamic therapy (PDT). In addition, the treatment of tumors derived from human lung cancer cells showed that the nanoprobe GNPs@PEG/Ce6-P could significantly inhibit tumor growth by PDT of GNPs and Ce6. It can be seen that the prepared novel nanoprobes have significant dual-mode imaging targeting ability and enhanced PDT effect on lung cancer, showing good prospects for nanomedicines (176).

Figure 6. Schematic diagram of the synthesis process of GNPs@PEG/Ce6-P and its application in mice.
Among various photodynamic therapy materials, nanomedicine based on cell membrane biomimetic strategy has received much attention due to its excellent ability to reduce the clearance of the reticuloendothelial system and enhance tumor targeting (177, 178)–for example, porphyrin cholesterol conjugate (TPPC) for synergistic photodynamic therapy (PDT) immunotherapy in lung cancer (Figure 7A) (179). Porphyrin derivatives have high generation efficiency of reactive oxygen species (ROS) and have been widely used as photosensitizers in clinical practice (169), while cholesterol is one of the main components of cell membranes. Porphyrin–cholesterol conjugates can be assembled into nanoparticles (NPS) in the absence of surfactants or amphiphilic polymers. Moreover, TPPC NP-mediated PDT can accumulate at the tumor site and induce immunogenic cell death, thereby stimulating and recruiting antigen-presenting cells to mature and activating T cells, making cancer cells more sensitive to ICI (Figure 7B) (179).
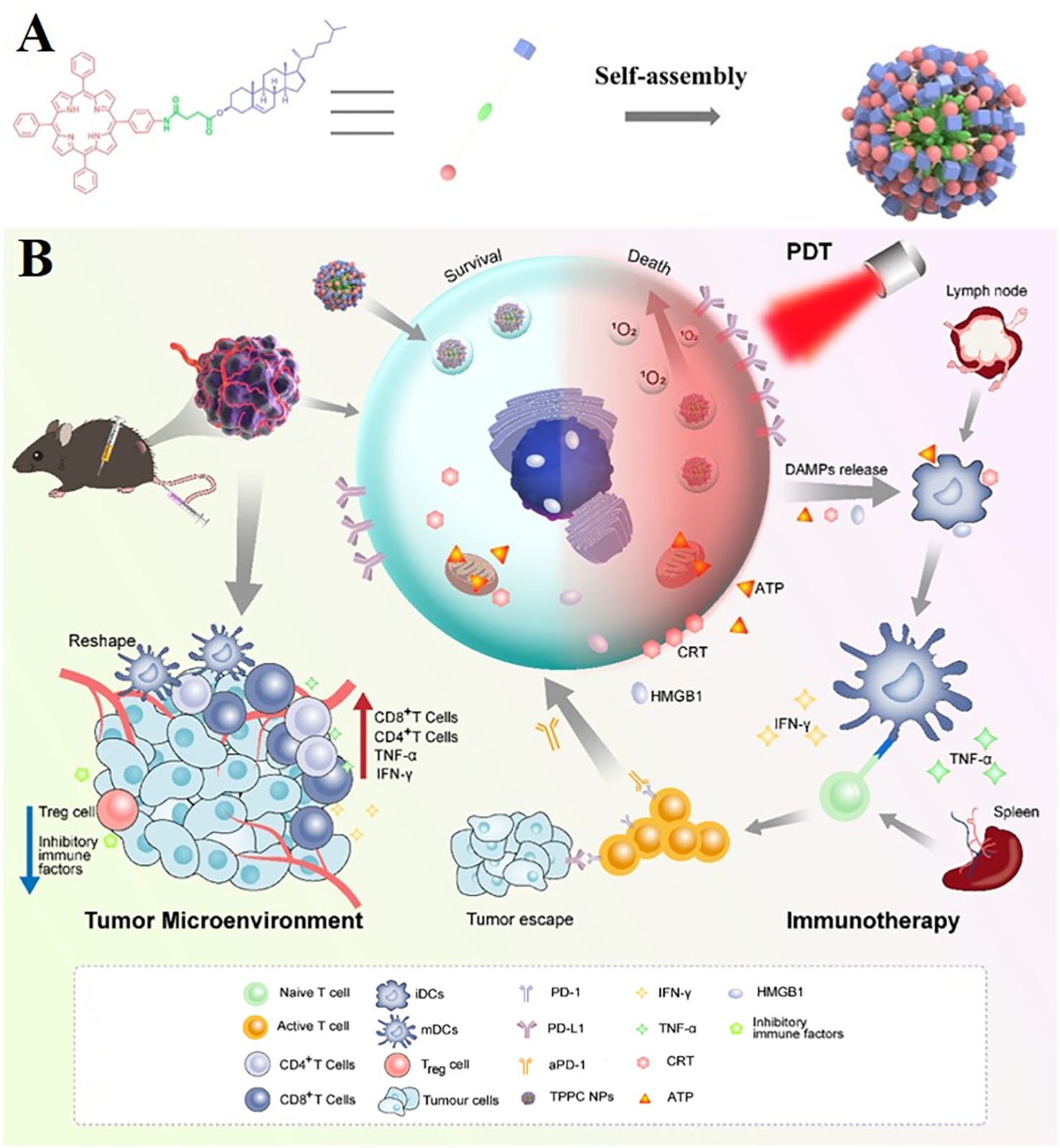
Figure 7. Porphyrin cholesterol conjugates (TPPC) for enhanced photodynamic immunotherapy toward non-small cell lung cancer. (A) Schematic illustration of preparation processes of TPPC NPs. (B) Schematic illustration of preparation and comparison of photodynamic immunotherapy of TPPC NPs.
In addition to the modification of cell membrane components, hybrid membranes can also be used to modify nanomedicines. Except for the function of drug delivery to the body, hybrid membranes prepared by mixing and remodeling of different cell membranes can also be used for the homologous targeting of tumors through the homing effect of cancer cell membranes or immune escape of nanomedicines through macrophage membranes (180). PDT has many advantages, such as that mutations resistant to chemotherapy or radiotherapy do not limit PDT efficacy, and PDT treatments do not affect future therapy options. However, the limitations of PDT include the patient’s photosensitivity after treatment, bronchial tumor invasion of the pulmonary artery is not suitable for PDT treatment, and the treatment of thoracic malignant tumors depends on the location and size of the tumor (181).
4.2 PTT
PTT can reduce tumor burden by thermal ablation and has become an attractive option in cancer treatment (182). The high temperatures (>42°C) generated by photothermal agents by the local application of NIR light irradiation can lead to apoptosis of cancer cells by damaging cell membranes, disrupting the cytoskeleton, and inhibiting DNA synthesis (183–185). At the same time, PTT can trigger host immunity, revealing a broad prospect for combination with immunotherapy (186, 187). In addition, tumors are more sensitive to heat than normal tissues due to hypoxia and a weakly acidic microenvironment. Thus, this enhances the tumor selectivity of PTT (188).
The key to achieving tumor photoablation with PTT lies in the design of photothermal agents, which must meet the requirements of strong near-infrared light absorption, high photothermal conversion efficiency, good biocompatibility, and sufficient accumulation in the tumor. Currently, various types of inorganic and organic photothermal agents have been developed for their ability to perform PTT on tumors. Inorganic photothermal agents mainly include gold nanomaterials, such as Fe-based nanosphere (189), gold nanostars (AuNSs) (190, 191), and so on. Sulfide metal nanoparticles include CuS, MnS2, black phosphorus, etc. (192). In general, they are characterized by high photothermal conversion efficiency and good photothermal stability. Their photothermal properties can be tuned by changing their size, shape, or doping with other elements. Organic photothermal agents typically include small-molecule dyes such as indocyanine green (ICG) and IR780 as well as polydopamine (pD), polyaniline (PANI), and polypyrrole nanoparticles (193). These organic photothermal agents are generally degradable and biocompatible. In addition, photothermal agents are typically designed as nanoplatforms. These photothermal agents can be passively or actively targeted to tumors to enhance tumor accumulation owing to nanoscale size or surface modifications of targeting ligands such as antibodies, folate, peptides, and hyaluronic acid (194–196). In addition, they can simultaneously act as nanocarriers to load drugs, antigens, or adjuvants, showing the potential for combination therapy with other modalities (197, 198).
For example, core-shell Au@Pt−Se nanoprobes (Au@Pt−Se NPs) linked to peptide chains via Pt–Se bonds were designed and synthesized, which can be used to target NSCLC biomarker protein calcium-activated neutral protease 2 (CAPN2) and achieve photothermal therapy (PTT) enhancement (199). The rapidly grown tumors can exceed their vascular system, resulting in hypoxia at the tumor site (200). Although a variety of transcription factors are involved in the cellular response to hypoxia, hypoxia-inducible factor-1α (HIF-1α) is considered to be the most important transcription factor, which promotes tumor angiogenesis by upregulating pro-angiogenic genes such as vascular endothelial growth factor (VEGF) (201–203). Several studies have shown that blocking the activation of HIF-1α can significantly inhibit tumor angiogenesis and progression (204, 205). The platinum (Pt) shell has a catalase-like property and catalyzes the generation of oxygen from endogenous hydrogen peroxide within the tumor, thereby reducing the level of hypoxia-inducible factor-1α (HIF-1α) and alleviating the hypoxic environment at the tumor site (206–209). The Au@Pt−Se nanoparticles exhibit a strong absorption band, enabling PTT in the near-infrared II region (NIR II) (Figure 8). Moreover, according to the cell activity detection, the cell death rate of the NPs + H2O2 group and the laser group increased significantly, and the cell death rate was significantly higher than that of the other groups. This was consistent with the results of apoptosis rates measured by using flow cytometry (199).
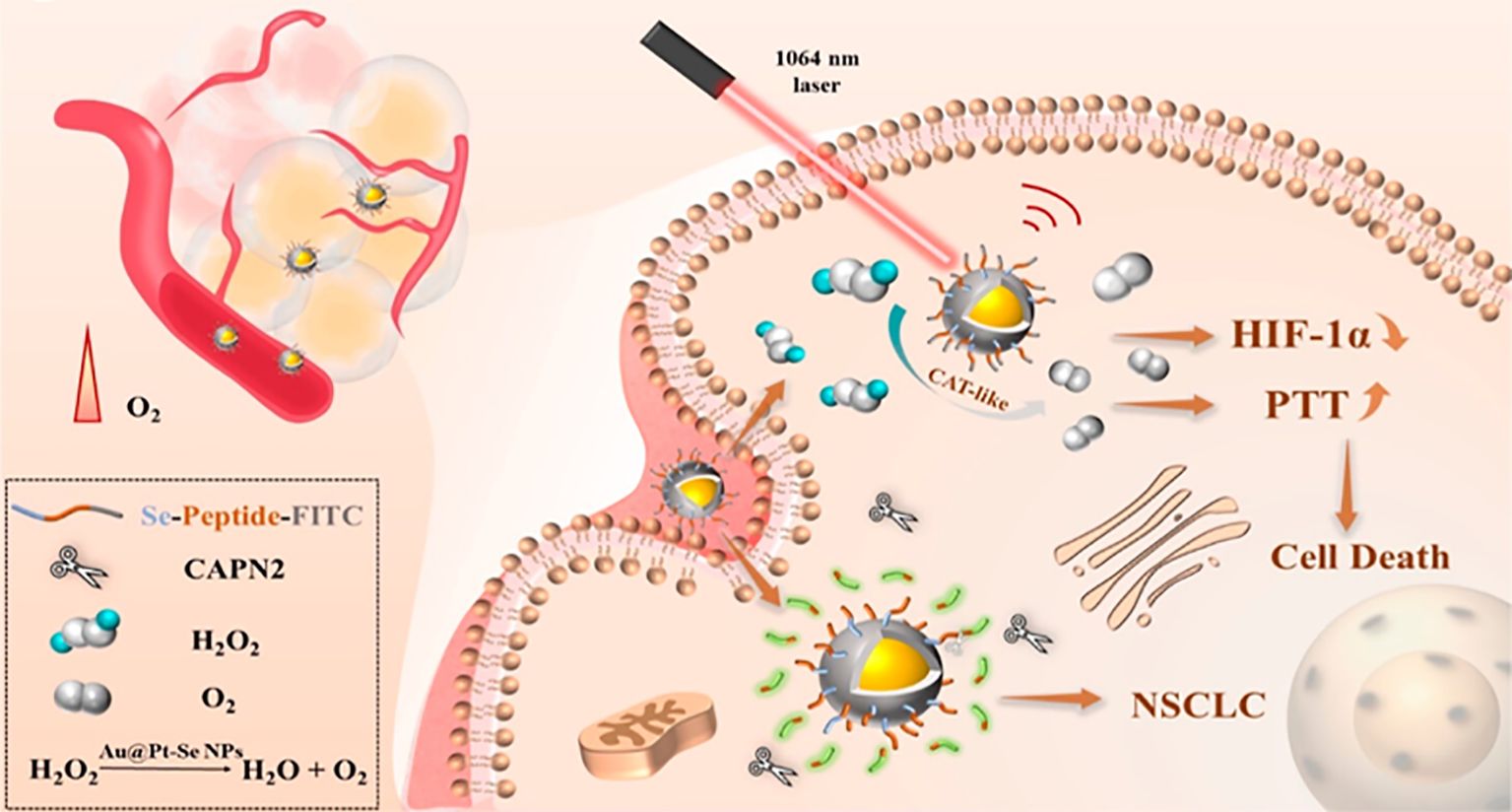
Figure 8. Schematic diagram of the effects of Au@Pt−Se NPs in NSCLS cells. Au@Pt-Se nanoparticles catalyze the generation of O2 from endogenous H2O2, alleviate the hypoxic environment, reduce the level of hypoxia-inducible factor-1α (HIF-1α), and improve the efficacy of PTT.
Although PTT can rapidly reduce tumor volume, it is usually difficult to completely eradicate tumors with PTT alone, so PTT can be used in combination with other therapies to enhance the therapeutic effect on tumors.
For example, the integration of PTT and gene therapy (GT) on a single nanoplatform shows great potential in cancer therapy. Porous iron oxide nanoparticles (PIONs) are widely used as magnetic nanoparticles for drug delivery and can also be used as photothermal nanoparticles for photothermal therapy (210–213). However, the therapeutic effects of PION-mediated GT have not been investigated. Long non-coding RNA (lncRNA) CRYBG3 (LNC CRYBG3) is a lncRNA produced by heavy ion irradiation in lung cancer cells. It has been reported that it can directly bind to globular actin (G-actin), leading to cytoskeleton degradation and blocking cytodivision, indicating its strong gene therapy effect (214, 215). The potential of PION-mediated PTT and LNC crybg3-mediated GT to damage non-small cell lung cancer (NSCLC) cells in vitro and in vivo has been jointly applied (216). The combination therapy showed a high tumor-cell-killing effect and a better cure rate than PTT or GT alone. As a magnetic nanoagent, PIONs can be used for magnetic resonance imaging (MRI) and photoacoustic imaging (PAI) in vitro and in vivo (Figure 9). In addition, the tumor growth was almost completely inhibited in the PIONs@pDNA + NIR group in the animal experiment. These findings suggest that the combination of photothermal therapy (PTT) and gene therapy (GT) has a high potential for cancer treatment (216).
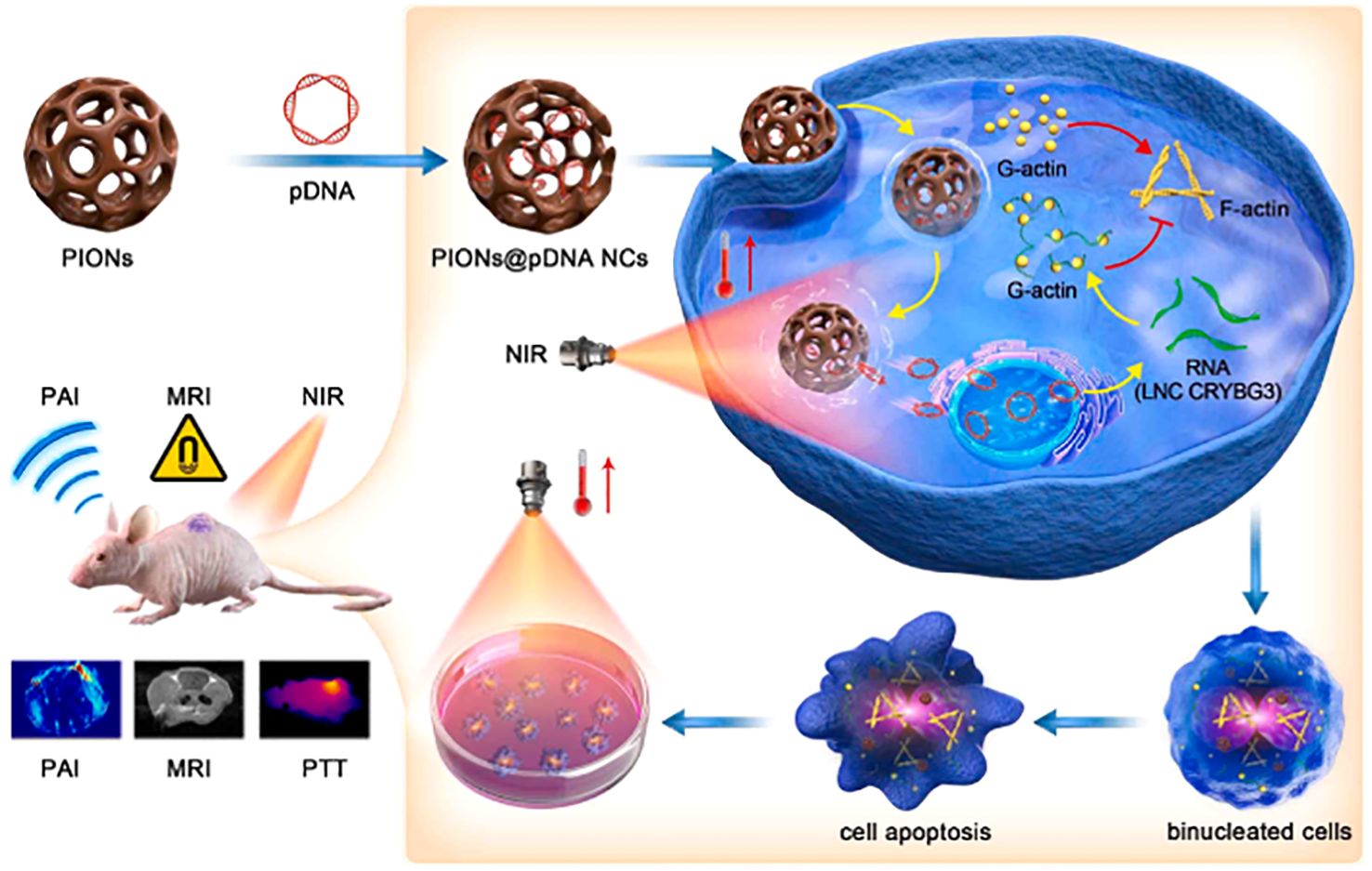
Figure 9. Schematic diagram of photothermal therapy using PIONs loaded pcDNA3.1-LNC CRYBG3 to synthesize PIONs nanocomplexes (PIONs@pDNA NCs) as photosensitizers.
In addition to the above-mentioned specific examples, many various materials have been applied in PDT/PTT (Table 4).
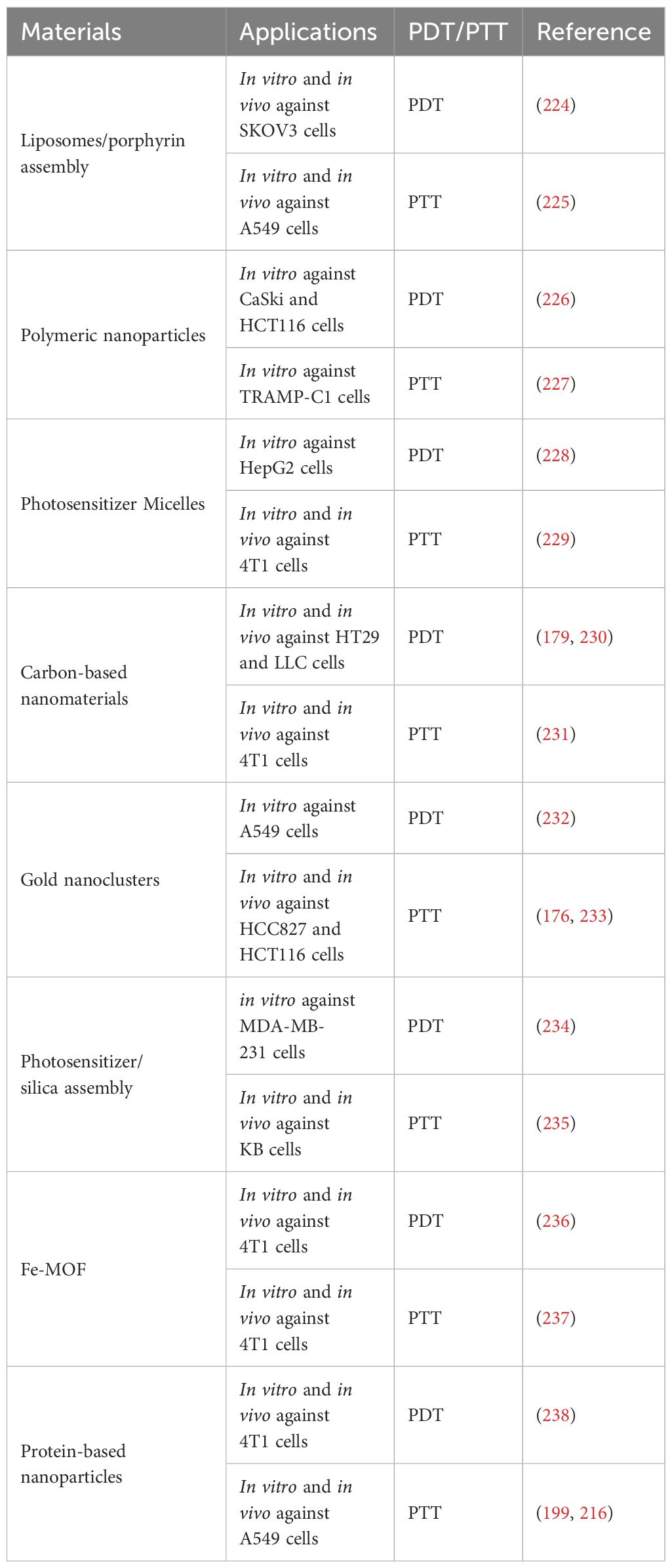
Table 4. Various types of nanocarriers for PDT/PTT.
PDT/PTT therapy does not simply kill tumor cells by generating singlet oxygen or generating high temperatures. PDT/PPT can induce immunogenic cell death (ICD) by releasing calreticulin and exposing dead tumor cell debris, which triggers immunogenicity, leading to enhanced antigen presentation and T-cell activation to kill residual tumors (217–219). Moreover, the ROS (reactive oxygen species) generated during PDT treatments contributes to ICD and immune response (220).
Nevertheless, PDT/PTT alone, of course, is not the perfect way for the treatment of NSCLC. Firstly, the main limitation of any light therapy is the penetration limitation of the laser, which limits the role of PDT/PTT in treatment. PDT/PTT can only achieve a good therapeutic effect in the treatment of shallow epidermal tumors with rich blood vessels but cannot play a role in deep tumor therapy (221). Secondly, during PDT/PTT treatment, there are some difficulties in assessing the intratumoral drug concentration. Therefore, it may be complicated to evaluate the pharmacokinetics of some PDT/PTT drugs and subsequently interfere with the accurate clinical application of PDT/PTT (222). Finally, some safety problems exist in PDT/PTT treatment, such as general thermal constraints and lack of sensitivity of patients (223) (Table 3).
5 Discussion
After COVID-19, the treatments for NSCLC patients are encountering new changes and challenges. We have summarized and reviewed the main treatment methods for NSCLC and found that the treatment methods and therapeutic drugs for NSCLC are constantly updated, but drug resistance is still the biggest challenge in the treatment process. Therefore, the hot spot of NSCLC drug development in the future is still to break the bondage of drug resistance. Moreover, the therapeutic effect of single therapy for NSCLC patients is still limited, and combination therapy is the most favorable treatment for NSCLC patients. In addition, phototherapy, as a new treatment modality, has become the focus of research for the treatment of NSCLC. The biggest challenge for phototherapy to be used in the clinic in the future is whether the laser required for phototherapy can penetrate the skin to stimulate the photosensitizer to work. Therefore, exploring new phototherapy materials that can be applied in the clinic is the goal of researchers.
Author contributions
QS: Writing – original draft. MB: Writing – original draft. XW: Conceptualization, Data curation, Methodology, Software, Supervision, Writing – review & editing. TW: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – review & editing. YQ: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Guangzhou Medical University High-level Talent Funding Project - Scientific Research Work Funding - School of Biomedical Engineering - Yan Qin (06-445-1121), The National Key R&D Program of China (2018YFA0106900, 2018YFA0106901), National Natural Science Foundation of China (31830043, 22072004).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang S, Sun K, Zheng R, Zeng H, Wang S, Chen R, et al. Cancer incidence and mortality in China, 2015. J Natl Cancer Center. (2021) 1:2–11. doi: 10.1016/j.jncc.2020.12.001
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in globocan 2012. Int J Cancer. (2015) 136:E359–E86. doi: 10.1002/ijc.29210
3. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA-Cancer J Clin. (2022) 72:7–33. doi: 10.3322/caac.21708
4. Halliday PR, Blakely CM, Bivona TG. Emerging targeted therapies for the treatment of non-small cell lung cancer. Curr Oncol Rep. (2019) 21:12. doi: 10.1007/s11912-019-0770-x
5. Gao SG, Li N, Wang SH, Zhang F, Wei WQ, Li N, et al. Lung cancer in people’s republic of China. J Thorac Oncol. (2020) 15:1567–76. doi: 10.1016/j.jtho.2020.04.028
6. Fillon M. Pairing smoking cessation with lung cancer screening may save lives. CA-Cancer J Clin. (2021) 71:283–4. doi: 10.3322/caac.21675
7. Bates JHT, Hamlington KL, Garrison G, Kinsey CM. Prediction of lung cancer risk based on age and smoking history. Comput Meth Programs BioMed. (2022) 216:6. doi: 10.1016/j.cmpb.2022.106660
8. Huang CL, Wang YM, Li XW, Ren LL, Zhao JP, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/s0140-6736(20)30183-5
9. Elkrief A, Hennessy C, Kuderer NM, Rubinstein SM, Wulff-Burchfield E, Rosovsky RP, et al. Geriatric risk factors for serious covid-19 outcomes among older adults with cancer: A cohort study from the covid-19 and cancer consortium. Lancet Healthy Longev. (2022) 3:e143–e52. doi: 10.1016/s2666-7568(22)00009-5
10. Calabrò L, Rossi G, Covre A, Morra A, Maio M. Covid and lung cancer. Curr Oncol Rep. (2021) 23(11). doi: 10.1007/s11912-021-01125-8
11. Rogado J, Pangua C, Serrano-Montero G, Obispo B, Marino AM, Pérez-Pérez M, et al. Covid-19 and lung cancer: A greater fatality rate? Lung Cancer. (2020) 146:19–22. doi: 10.1016/j.lungcan.2020.05.034
12. Rolfo C, Meshulami N, Russo A, Krammer F, García-Sastre A, Mack PC, et al. Lung cancer and severe acute respiratory syndrome coronavirus 2 infection: identifying important knowledge gaps for investigation. J Thorac Oncol. (2022) 17:214–27. doi: 10.1016/j.jtho.2021.11.001
13. Wu L, Zhang C, Zhao X. The impact of covid-19 pandemic on lung cancer community. World J Oncol. (2021) 12:1–6. doi: 10.14740/wjon1367
14. Aramini B, Masciale V, Samarelli AV, Tonelli R, Cerri S, Clini E, et al. Biological effects of covid-19 on lung cancer: can we drive our decisions. Front Oncol. (2022) 12:1029830. doi: 10.3389/fonc.2022.1029830
15. Yaqub F. Thoracoscopic lobectomy versus thoracotomy for nsclc. Lancet Oncol. (2014) 15:E535–E. doi: 10.1016/s1470-2045(14)71024-1
16. Moreno AC, Fellman B, Hobbs BP, Liao ZX, Gomez DR, Chen A, et al. Biologically effective dose in stereotactic body radiotherapy and survival for patients with early-stage nsclc. J Thorac Oncol. (2020) 15:101–9. doi: 10.1016/j.jtho.2019.08.2505
17. Herbst RS, Sznol M. Diminished but not dead: chemotherapy for the treatment of nsclc. Lancet Oncol. (2016) 17:1464–5. doi: 10.1016/s1470-2045(16)30524-1
18. Hutchinson L. Targeted therapies: defining the best-in-class in nsclc. Nat Rev Clin Oncol. (2017) 14:457. doi: 10.1038/nrclinonc.2017.99
19. Gridelli C, Casaluce F. Lung cancer frontline immunotherapy for nsclc: alone or not alone? Nat Rev Clin Oncol. (2018) 15:593–4. doi: 10.1038/s41571-018-0070-7
20. Kato H, Harada M, Ichinose S, Usuda J, Tsuchida T, Okunaka T. Photodynamic therapy (Pdt) of lung cancer: experience of the Tokyo medical university. Photodiagnosis Photodyn Ther. (2004) 1:49–55. doi: 10.1016/s1572-1000(04)00008-0
21. Baselga J. Targeting the epidermal growth factor receptor with tyrosine kinase inhibitors: small molecules, big hopes. J Clin Oncol. (2002) 20:2217–9. doi: 10.1200/jco.2002.20.9.2217
22. Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao DW, Habib AA. Ligand-independent egfr signaling. Cancer Res. (2015) 75:3436–41. doi: 10.1158/0008-5472.Can-15-0989
23. Huang LH, Fu LW. Mechanisms of resistance to egfr tyrosine kinase inhibitors. Acta Pharm Sin B. (2015) 5:390–401. doi: 10.1016/j.apsb.2015.07.001
24. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. (2007) 7:169–81. doi: 10.1038/nrc2088
25. Yarden Y, Shilo BZ. Snapshot: egfr signaling pathway. Cell. (2007) 131:2. doi: 10.1016/j.cell.2007.11.013
26. Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. (2001) 7(10):2958–70.
27. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. (2009) 361:958–U38. doi: 10.1056/NEJMoa0904554
28. Hsu WH, Yang JCH, Mok TS, Loong HH. Overview of current systemic management of egfr-mutant nsclc. Ann Oncol. (2018) 29:I3–9. doi: 10.1093/annonc/mdx702
29. Ahsan A. Mechanisms of resistance to egfr tyrosine kinase inhibitors and therapeutic approaches: an update. In: Ahmad A, Gadgeel S, editors. Lung Cancer and Personalized Medicine: Current Knowledge and Therapies. Advances in Experimental Medicine and Biology, vol. 893. Springer International Publishing Ag, Cham (2016). p. 137–53.
30. Gazdar AF. Activating and resistance mutations of egfr in non-small-cell lung cancer: role in clinical response to egfr tyrosine kinase inhibitors. Oncogene. (2009) 28:S24–31. doi: 10.1038/onc.2009.198
31. Remon J, Steuer CE, Ramalingam SS, Felip E. Osimertinib and other third-generation egfr tki in egfr-mutant nsclc patients. Ann Oncol. (2018) 29:I20–I7. doi: 10.1093/annonc/mdx704
32. Campbell L, Blackhall F, Thatcher N. Gefitinib for the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. (2010) 11:1343–57. doi: 10.1517/14656566.2010.481283
33. Yang JCH, Gadgeel SM, Sequist LV, Wu CL, Papadimitrakopoulou VA, Su WC, et al. Pembrolizumab in combination with erlotinib or gefitinib as first-line therapy for advanced nsclc with sensitizing egfr mutation. J Thorac Oncol. (2019) 14:553–9. doi: 10.1016/j.jtho.2018.11.028
34. Lyu X, Zeng LZ, Shi J, Ming ZJ, Li W, Liu BX, et al. Essential role for stat3/foxm1/atg7 signaling-dependent autophagy in resistance to icotinib. J Exp Clin Cancer Res. (2022) 41:14. doi: 10.1186/s13046-022-02390-6
35. Kaneda T, Hata A, Tomioka H, Tanaka K, Kaji R, Fujita S, et al. Possible differential egfr-tki efficacy among exon 19 deletional locations in egfr-mutant non-small cell lung cancer. Lung Cancer. (2014) 86:213–8. doi: 10.1016/j.lungcan.2014.09.014
36. Kwon CS, Lin HM, Crossland V, Churchill EN, Curran E, Forsythe A, et al. Non-small cell lung cancer with egfr exon 20 insertion mutation: A systematic literature review and meta-analysis of patient outcomes. Curr Med Res Opin. (2022) 38:1341–50. doi: 10.1080/03007995.2022.2083326
37. Nelson V, Ziehr J, Agulnik M, Johnson M. Afatinib: emerging next-generation tyrosine kinase inhibitor for nsclc. OncoTargets Ther. (2013) 6:135–43. doi: 10.2147/ott.S23165
38. Sidaway P. Dacomitinib delays disease progression. Nat Rev Clin Oncol. (2017) 14:712–. doi: 10.1038/nrclinonc.2017.162
39. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. Egfr mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. (2005) 352:786–92. doi: 10.1056/NEJMoa044238
40. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to egfr-tki therapy in 155 patients with egfr-mutant lung cancers. Clin Cancer Res. (2013) 19:2240–7. doi: 10.1158/1078-0432.Ccr-12-2246
41. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790m mutation in egfr kinase causes drug resistance by increasing the affinity for atp. Proc Natl Acad Sci USA. (2008) 105:2070–5. doi: 10.1073/pnas.0709662105
42. Cerbone L, Benitez JC, Planchard D, Genova C. An overview of osimertinib as a treatment of non-small cell lung cancer (Nsclc): an update. Expert Opin Pharmacother. (2021) 22:12. doi: 10.1080/14656566.2021.1888928
43. Yang JCH, Camidge DR, Yang CT, Zhou JY, Guo RH, Chiu CH, et al. Safety, efficacy, and pharmacokinetics of almonertinib (Hs-10296) in pretreated patients with egfr-mutated advanced nsclc: A multicenter, open-label, phase 1 trial. J Thorac Oncol. (2020) 15:1907–18. doi: 10.1016/j.jtho.2020.09.001
44. Deeks ED. Furmonertinib: first approval. Drugs. (2021) 81:1775–80. doi: 10.1007/s40265-021-01588-w
45. Jukna A, Montanari G, Mengoli MC, Cavazza A, Covi M, Barbieri F, et al. Squamous cell carcinoma “Transformation” Concurrent with secondary T790m mutation in resistant egfr-mutated adenocarcinomas. J Thorac Oncol. (2016) 11:E49–51. doi: 10.1016/j.jtho.2015.12.096
46. Fan GJ, Lou LL, Song ZD, Zhang XL, Xiong XF. Targeting mutated gtpase kras in tumor therapies. Eur J Med Chem. (2021) 226:18. doi: 10.1016/j.ejmech.2021.113816
47. Parikh K, Banna G, Liu SV, Friedlaender A, Desai A, Subbiah V, et al. Drugging kras: current perspectives and state-of-art review. J Hematol Oncol. (2022) 15:22. doi: 10.1186/s13045-022-01375-4
48. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res. (2020) 80:2969–74. doi: 10.1158/0008-5472.Can-19-3682
49. Dogan S, Shen RL, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular epidemiology of egfr and kras mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related kras-mutant cancers. Clin Cancer Res. (2012) 18:6169–77. doi: 10.1158/1078-0432.Ccr-11-3265
50. Finn SP, Addeo A, Dafni U, Thunnissen E, Bubendorf L, Madsen LB, et al. Prognostic impact of kras G12c mutation in patients with nsclc: results from the European thoracic oncology platform lungscape project. J Thorac Oncol. (2021) 16:990–1002. doi: 10.1016/j.jtho.2021.02.016
51. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
52. Mendiratta G, Ke E, Aziz M, Liarakos D, Tong M, Stites EC. Cancer gene mutation frequencies for the U.S. Population. Nat Commun. (2021) 12(1). doi: 10.1038/s41467-021-26213-y
53. Mathiot L, Herbreteau G, Robin S, Fenat C, Bennouna J, Blanquart C, et al. Hras Q61l mutation as a possible target for non-small cell lung cancer: case series and review of literature. Curr Oncol. (2022) 29:3748–58. doi: 10.3390/curroncol29050300
54. End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. (2001) 61(1):131–7.
55. Roskoski R. Blockade of mutant ras oncogenic signaling with a special emphasis on kras. Pharmacol Res. (2021) 172. doi: 10.1016/j.phrs.2021.105806
56. Moore AR, Rosenberg SC, McCormick F, Malek S. Ras-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. (2020) 19:533–52. doi: 10.1038/s41573-020-0068-6
57. Wood K, Hensing T, Malik R, Salgia R. Prognostic and predictive value in kras in non-small-cell lung cancer a review. JAMA Oncol. (2016) 2:805–12. doi: 10.1001/jamaoncol.2016.0405
58. Cherfils J, Zeghouf M. Regulation of small gtpases by gefs, gaps, and gdis. Physiol Rev. (2013) 93:269–309. doi: 10.1152/physrev.00003.2012
59. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily gefs and gaps: validated and tractable targets for cancer therapy? Nat Rev Cancer. (2010) 10:842–57. doi: 10.1038/nrc2960
60. Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, et al. Normal and oncogenic P21ras proteins bind to the amino-terminal regulatory domain of C-raf-1. Nature. (1993) 364:308–13. doi: 10.1038/364308a0
61. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-oh kinase as a direct target of ras. Nature. (1994) 370:527–32. doi: 10.1038/370527a0
62. Sjolander A, Yamamoto K, Huber BE, Lapetina EG. Association of P21ras with phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. (1991) 88:7908–12. doi: 10.1073/pnas.88.18.7908
63. Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT. Ralgds family members interact with the effector loop of ras P21. Mol Cell Biol. (1994) 14:7483–91. doi: 10.1128/mcb.14.11.7483
64. Menyhárd DK, Pálfy G, Orgován Z, Vida I, Keseru GM, Perczel A. Structural impact of gtp binding on downstream kras signaling. Chem Sci. (2020) 11:9272–89. doi: 10.1039/d0sc03441j
65. Wang Q, Wang Y, Li S, Zhou A, Qin Y. Organelle biogenesis: ribosomes as organizer and performer. Sci Bull. (2022) 67:1614–7. doi: 10.1016/j.scib.2022.07.023
66. Kang J, Brajanovski N, Chan KT, Xuan JC, Pearson RB, Sanij E. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct Target Ther. (2021) 6:22. doi: 10.1038/s41392-021-00728-8
67. American Association for Cancer Research. Fda approves first kras inhibitor: sotorasib. Cancer Discov. (2021) 11:OF4. doi: 10.1158/2159-8290.Cd-nb2021-0362
68. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with kras P. G12c Mutation. N Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
70. Jaenne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SHI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a krasG12c mutation. N Engl J Med. (2022) 387:120–31. doi: 10.1056/NEJMoa2204619
71. Christensen JG, Hallin J. The krasG12d inhibitor mrtx1133 elucidates kras-mediated oncogenesis. Nat Med. (2022) 28:2017–8. doi: 10.1038/s41591-022-02008-6
72. Tammaccaro SL, Prigent P, Le Bail JC, Dos-Santos O, Dassencourt L, Eskandar M, et al. Tead inhibitors sensitize krasG12c inhibitors via dual cell cycle arrest in krasG12c-mutant nsclc. Pharmaceuticals. (2023) 16:20. doi: 10.3390/ph16040553
73. Kim D, Herdeis L, Rudolph D, Zhao YL, Böttcher J, Vides A, et al. Pan-kras inhibitor disables oncogenic signalling and tumour growth. Nature. (2023) 619:160. doi: 10.1038/s41586-023-06123-3
74. Bar-Shavit R, Maoz M, Kancharla A, Nag JK, Agranovich D, Grisaru-Granovsky S, et al. G protein-coupled receptors in cancer. Int J Mol Sci. (2016) 17:16. doi: 10.3390/ijms17081320
75. Vilardaga JP, Bünemann M, Feinstein TN, Lambert N, Nikolaev VO, Engelhardt S, et al. Minireview: gpcr and G proteins: drug efficacy and activation in live cells. Mol Endocrinol. (2009) 23:590–9. doi: 10.1210/me.2008-0204
76. Nagareddy PR, Chow FL, Hao L, Wang X, Nishimura T, MacLeod KM, et al. Maintenance of adrenergic vascular tone by mmp transactivation of the egfr requires pi3k and mitochondrial atp synthesis. Cardiovasc Res. (2009) 84:368–77. doi: 10.1093/cvr/cvp230
77. Wang WJ, Qiao YH, Li ZJ. New insights into modes of gpcr activation. Trends Pharmacol Sci. (2018) 39:367–86. doi: 10.1016/j.tips.2018.01.001
78. Zhang XW, Li L, Hu WQ, Hu MN, Tao Y, Hu H, et al. Neurokinin-1 receptor promotes non-small cell lung cancer progression through transactivation of egfr. Cell Death Dis. (2022) 13:15. doi: 10.1038/s41419-021-04485-y
79. Wang QS, Zhang TS, Chang XY, Wang KK, Lee MH, Ma WY, et al. Targeting opsin4/melanopsin with a novel small molecule suppresses pkc/raf/mek/erk signaling and inhibits lung adenocarcinoma progression. Mol Cancer Res. (2020) 18:1028–38. doi: 10.1158/1541-7786.Mcr-19-1120
80. Bhola NE, Freilino ML, Joyce SC, Sen M, Thomas SM, Sahu A, et al. Antitumor mechanisms of targeting the pdk1 pathway in head and neck cancer. Mol Cancer Ther. (2012) 11:1236–46. doi: 10.1158/1535-7163.Mct-11-0936
81. Jiang YH, Lim J, Wu KC, Xu WJ, Suen JY, Fairlie DP. Par2 induces ovarian cancer cell motility by merging three signalling pathways to transactivate egfr. Br J Pharmacol. (2021) 178:913–32. doi: 10.1111/bph.15332
82. Kodali R, Hajjou M, Berman AB, Bansal MB, Zhang SH, Pan JJ, et al. Chemokines induce matrix metalloproteinase-2 through activation of epidermal growth factor receptor in arterial smooth muscle cells. Cardiovasc Res. (2006) 69:706–15. doi: 10.1016/j.cardiores.2005.09.012
83. Kallifatidis G, Munoz D, Singh RK, Salazar N, Hoy JJ, Lokeshwar BL. B-arrestin-2 counters cxcr7-mediated egfr transactivation and proliferation. Mol Cancer Res. (2016) 14:493–503. doi: 10.1158/1541-7786.Mcr-15-0498
84. Kraus S, Benard O, Naor Z, Seger R. Retraction: C-src is activated by the epidermal growth factor receptor in a pathway that mediates jnk and erk activation by gonadotropin-releasing hormone in cos7 cells (Retraction of vol 278, pg 32618, 2003). J Biol Chem. (2017) 292:8851–. doi: 10.1074/jbc.A117.303886
85. Wetzker R, Böhmer FD. Transactivation joins multiple tracks to the erk/mapk cascade. Nat Rev Mol Cell Biol. (2003) 4:651–7. doi: 10.1038/nrm1173
86. Zia F, Fagarasan M, Bitar K, Coy DH, Pisegna JR, Wank SA, et al. Pituitary adenylate cyclase activating peptide receptors regulate the growth of non-small cell lung cancer cells. Cancer Res. (1995) 55(21):4886–91.
87. Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (Grac), 3rd edition. Br J Pharmacol. (2008) 153:S1–S209. doi: 10.1038/sj.bjp.0707746
88. Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. (2011) 193:440–51. doi: 10.1016/j.neuroscience.2011.06.085
89. Russell FA, Schuelert N, Veldhoen VE, Hollenberg MD, McDougall JJ. Activation of par2 receptors sensitizes primary afferents and causes leukocyte rolling and adherence in the rat knee joint. Br J Pharmacol. (2012) 167:1665–78. doi: 10.1111/j.1476-5381.2012.02120.x
90. Pawar NR, Buzza MS, Antalis TM. Membrane-anchored serine proteases and protease-activated receptor-2-mediated signaling: co-conspirators in cancer progression. Cancer Res. (2019) 79:301–10. doi: 10.1158/0008-5472.Can-18-1745
91. Sun L, Li PB, Yao YF, Xiu AY, Peng Z, Bai YH, et al. Proteinase-activated receptor 2 promotes tumor cell proliferation and metastasis by inducing epithelial-mesenchymal transition and predicts poor prognosis in hepatocellular carcinoma. World J Gastroenterol. (2018) 24:1120–33. doi: 10.3748/wjg.v24.i10.1120
92. Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer - trypsin-induced mapk phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. (2004) 279:20927–34. doi: 10.1074/jbc.M401430200
93. Darmoul D, Gratio V, Devaud H, Peiretti F, Laburthe M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol Cancer Res. (2004) 2:514–22. doi: 10.1158/1541-7786.514.2.9
94. Jiang YH, Zhuo X, Fu XJ, Wu Y, Mao CQ. Targeting par2 overcomes gefitinib resistance in non-small-cell lung cancer cells through inhibition of egfr transactivation. Front Pharmacol. (2021) 12:625289. doi: 10.3389/fphar.2021.625289
95. Köse M. Gpcrs and egfr - cross-talk of membrane receptors in cancer. Bioorg Med Chem Lett. (2017) 27:3611–20. doi: 10.1016/j.bmcl.2017.07.002
96. Jiang Y, Zhuo X, Wu Y, Fu X, Mao C. Par2 blockade reverses osimertinib resistance in non-small-cell lung cancer cells via attenuating erk-mediated emt and pd-L1 expression. Biochim Biophys Acta (BBA) - Mol Cell Res. (2022) 1869(1). doi: 10.1016/j.bbamcr.2021.119144
97. Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. (2018) 834:188–96. doi: 10.1016/j.ejphar.2018.07.034
98. Liu S, Kurzrock R. Toxicity of targeted therapy: implications for response and impact of genetic polymorphisms. Cancer Treat Rev. (2014) 40:883–91. doi: 10.1016/j.ctrv.2014.05.003
99. Osmani L, Askin F, Gabrielson E, Li QK. Current who guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (Nsclc): moving from targeted therapy to immunotherapy. Semin Cancer Biol. (2018) 52:103–9. doi: 10.1016/j.semcancer.2017.11.019
100. Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, Bisht B, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol Cancer. (2023) 22:37. doi: 10.1186/s12943-023-01740-y
101. Rolfo C, Caglevic C, Santarpia M, Araujo A, Giovannetti E, Gallardo CD, et al. Immunotherapy in nsclc: A promising and revolutionary weapon. In: Naing A, Hajjar J, editors. Immunotherapy. Advances in Experimental Medicine and Biology, vol. 995 . Springer International Publishing Ag, Cham (2017). p. 97–125.
102. Zhang N, Tu JY, Wang X, Chu Q. Programmed cell death-1/programmed cell death ligand-1 checkpoint inhibitors: differences in mechanism of action. Immunotherapy. (2019) 11:429–41. doi: 10.2217/imt-2018-0110
103. Azoury SC, Straughan DM, Shukla V. Immune checkpoint inhibitors for cancer therapy: clinical efficacy and safety. Curr Cancer Drug Targets. (2015) 15:452–62. doi: 10.2174/156800961506150805145120
104. Wu YL, Wang CL, Liao ML, Guan ZZ, Gao CY, Lu S, et al. A consensus on immunotherapy from the 2017 Chinese lung cancer summit expert panel. Transl Lung Cancer Res. (2018) 7:428–36. doi: 10.21037/tlcr.2018.04.15
105. Cui PF, Li RX, Huang ZW, Wu ZZ, Tao HT, Zhang SJ, et al. Comparative effectiveness of pembrolizumab vs. Nivolumab in patients with recurrent or advanced nsclc. Sci Rep. (2020) 10:7. doi: 10.1038/s41598-020-70207-7
106. Xia LL, Liu YY, Wang Y. Pd-1/pd-L1 blockade therapy in advanced non-small-cell lung cancer: current status and future directions. Oncologist. (2019) 24:S31–41. doi: 10.1634/theoncologist.2019-IO-S1-s05
107. Pu YZ, Ji Q. Tumor-associated macrophages regulate pd-1/pd-L1 immunosuppression. Front Immunol. (2022) 13:874589. doi: 10.3389/fimmu.2022.874589
108. Ghosh C, Luong G, Sun Y. A snapshot of the pd-1/pd-L1 pathway. J Cancer. (2021) 12:2735–46. doi: 10.7150/jca.57334
109. Dang TO, Ogunniyi A, Barbee MS, Drilon A. Pembrolizumab for the treatment of pd-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev Anticancer Ther. (2016) 16:13–20. doi: 10.1586/14737140.2016.1123626
110. Garon EB, Rizvi NA, Hui RN, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
111. Lim SH, Sun JM, Lee SH, Ahn JS, Park K, Ahn MJ. Pembrolizumab for the treatment of non-small cell lung cancer. Expert Opin Biol Ther. (2016) 16:397–406. doi: 10.1517/14712598.2016.1145652
112. Sul J, Blumenthal GM, Jiang XP, He K, Keegan P, Pazdur R. Fda approval summary: pembrolizumab for the treatment of patients with metastatic non-small cell lung cancer whose tumors express programmed death-ligand 1. Oncologist. (2016) 21:643–50. doi: 10.1634/theoncologist.2015-0498
113. Eguren-Santamaria I, Sanmamed MF, Goldberg SB, Kluger HM, Idoate MA, Lu BY, et al. Pd-1/pd-L1 blockers in nsclc brain metastases: challenging paradigms and clinical practice. Clin Cancer Res. (2020) 26:4186–97. doi: 10.1158/1078-0432.Ccr-20-0798
114. Masuda K, Horinouchi H, Tanaka M, Higashiyama R, Shinno Y, Sato J, et al. Efficacy of anti-pd-1 antibodies in nsclc patients with an egfr mutation and high pd-L1 expression. J Cancer Res Clin Oncol. (2021) 147:245–51. doi: 10.1007/s00432-020-03329-0
115. Jardim DL, Goodman A, Gagliato DD, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. (2021) 39:154–73. doi: 10.1016/j.ccell.2020.10.001
116. Liu Y, Zheng P. Preserving the ctla-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. (2020) 41:4–12. doi: 10.1016/j.tips.2019.11.003
117. Van Coillie S, Wiernicki B, Xu J. Molecular and cellular functions of ctla-4. In: Xu J, editor. Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy. Advances in Experimental Medicine and Biology, vol. 1248 . Springer International Publishing Ag, Cham (2020). p. 7–32.
118. Zhang H, Dai ZY, Wu WT, Wang ZY, Zhang N, Zhang LY, et al. Regulatory mechanisms of immune checkpoints pd-L1 and ctla-4 in cancer. J Exp Clin Cancer Res. (2021) 40:22. doi: 10.1186/s13046-021-01987-7
119. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. (2019) 30:44–56. doi: 10.1093/annonc/mdy495
120. Guo HF, Li LY, Cui JW. Advances and challenges in immunotherapy of small cell lung cancer. Chin J Cancer Res. (2020) 32:115–28. doi: 10.21147/j.issn.1000-9604.2020.01.13
121. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. (2020) 52:17–35. doi: 10.1016/j.immuni.2019.12.011
122. Sterner RC, Sterner RM. Car-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:11. doi: 10.1038/s41408-021-00459-7
123. Wang SH, Sun JW, Chen K, Ma PW, Lei Q, Xing SJ, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. (2021) 19:7. doi: 10.1186/s12916-021-02006-4
124. Levin A, Shah NN. Chimeric antigen receptor modified T cell therapy in B cell non-hodgkin lymphomas. Am J Hematol. (2019) 94:S18–23. doi: 10.1002/ajh.25403
125. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.Cd-12-0548
126. Song EZ, Milone MC. Pharmacology of chimeric antigen receptor-modified T cells. Annu Rev Pharmacol Toxicol. (2021) 61:805–29. doi: 10.1146/annurev-pharmtox-031720-102211
127. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
128. Yan T, Zhu LF, Chen J. Current advances and challenges in car T-cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. (2023) 12(1). doi: 10.1186/s40164-023-00373-7
129. Srivastava S, Riddell SR. Engineering car-T cells: design concepts. Trends Immunol. (2015) 36:494–502. doi: 10.1016/j.it.2015.06.004
130. Ying ZT, Huang XF, Xiang XY, Liu YL, Kang X, Song YQ, et al. A safe and potent anti-cd19 car T cell therapy. Nat Med. (2019) 25:947. doi: 10.1038/s41591-019-0421-7
131. Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen LP, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res. (2019) 25:4592–602. doi: 10.1158/1078-0432.Ccr-18-1538
132. Firor AE, Jares A, Ma YP. From humble beginnings to success in the clinic: chimeric antigen receptor-modified T-cells and implications for immunotherapy. Exp Biol Med. (2015) 240:1087–98. doi: 10.1177/1535370215584936
133. Hu ZM, Zheng XH, Jiao DF, Zhou YG, Sun R, Wang BL, et al. Lunx-car T cells as a targeted therapy for non-small cell lung cancer. Mol Ther-Oncolytics. (2020) 17:361–70. doi: 10.1016/j.omto.2020.04.008
134. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. Car-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:12. doi: 10.1038/s41419-018-0278-6
135. Li J, Li WW, Huang KJ, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (Car-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. (2018) 11:18. doi: 10.1186/s13045-018-0568-6
136. Titov A, Valiullina A, Zmievskaya E, Zaikova E, Petukhov A, Miftakhova R, et al. Advancing car T-cell therapy for solid tumors: lessons learned from lymphoma treatment. Cancers. (2020) 12:22. doi: 10.3390/cancers12010125
137. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. Car T-cell therapy of solid tumors. Immunol Cell Biol. (2017) 95:356–63. doi: 10.1038/icb.2016.128
138. Kim DW, Cho JY. Recent advances in allogeneic car-T cells. Biomolecules. (2020) 10:15. doi: 10.3390/biom10020263
139. Zhao LJ, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
140. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation car T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
141. Kiesgen S, Chicaybam L, Chintala NK, Adusumilli PS. Chimeric antigen receptor (Car) T-cell therapy for thoracic Malignancies. J Thorac Oncol. (2018) 13:16–26. doi: 10.1016/j.jtho.2017.10.001
142. Ke EE, Wu YL. Egfr as a pharmacological target in egfr-mutant non-small-cell lung cancer: where do we stand now? Trends Pharmacol Sci. (2016) 37:887–903. doi: 10.1016/j.tips.2016.09.003
143. Kachala SS, Bograd AJ, Villena-Vargas J, Suzuki K, Servais EL, Kadota K, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res. (2014) 20:1020–8. doi: 10.1158/1078-0432.Ccr-13-1862
144. Grunnet M, Sorensen JB. Carcinoembryonic antigen (Cea) as tumor marker in lung cancer. Lung Cancer. (2012) 76:138–43. doi: 10.1016/j.lungcan.2011.11.012
145. Liu M, Wang X, Li W, Yu XF, Flores-Villanueva P, Xu-Monette ZJY, et al. Targeting pd-L1 in non-small cell lung cancer using car T cells. Oncogenesis. (2020) 9:11. doi: 10.1038/s41389-020-00257-z
146. Hirsh V. Next-generation covalent irreversible kinase inhibitors in nsclc: focus on afatinib. Biodrugs. (2015) 29:167–83. doi: 10.1007/s40259-015-0130-9
147. Hou AJ, Chang ZNL, Lorenzini MH, Zah E, Chen YY. Tgf-B-responsive car-T cells promote anti-tumor immune function. Bioeng Transl Med. (2018) 3:75–86. doi: 10.1002/btm2.10097
148. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. (2021) 18:842–59. doi: 10.1038/s41423-020-00565-9
149. Lin BS, Du LK, Li HM, Zhu X, Cui L, Li XS. Tumor-infiltrating lymphocytes: warriors fight against tumors powerfully. BioMed Pharmacother. (2020) 132:9. doi: 10.1016/j.biopha.2020.110873
150. Veatch JR, Simon S, Riddell SR. Tumor-infiltrating lymphocytes make inroads in non-small-cell lung cancer. Nat Med. (2021) 27:1339–41. doi: 10.1038/s41591-021-01445-z
151. Ratto GB, Zino P, Mirabelli S, Minuti P, Aquilina R, Fantino G, et al. A randomized trial of adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 versus standard therapy in the postoperative treatment of resected nonsmall cell lung carcinoma. Cancer. (1996) 78:244–51. doi: 10.1002/(sici)1097-0142(19960715)78:2<244::Aid-cncr9>3.0.Co;2-l
152. Creelan B, Wang C, Teer J, Toloza E, Mullinax J, Yao JQ, et al. Durable complete responses to adoptive cell transfer using tumor infiltrating lymphocytes (Til) in non-small cell lung cancer (Nsclc): A phase I trial. Cancer Res. (2020) 80:2. doi: 10.1158/1538-7445.Am2020-ct056
153. Naylor EC, Desani JK, Chung PK. Targeted therapy and immunotherapy for lung cancer. Surg Oncol Clinics North America. (2016) 25:601. doi: 10.1016/j.soc.2016.02.011
154. Martinez P, Peters S, Stammers T, Soria JC. Immunotherapy for the first-line treatment of patients with metastatic non-small cell lung cancer. Clin Cancer Res. (2019) 25:2691–8. doi: 10.1158/1078-0432.Ccr-18-3904
155. Patel SA, Weiss J. Advances in the treatment of non-small cell lung cancer: immunotherapy. Clinics Chest Med. (2020) 41:237. doi: 10.1016/j.ccm.2020.02.010
156. Gonzalez-Molina J, Moyano-Galceran L, Single A, Gultekin O, Alsalhi S, Lehti K. Chemotherapy as a regulator of extracellular matrix-cell communication: implications in therapy resistance. Semin Cancer Biol. (2022) 86:224–36. doi: 10.1016/j.semcancer.2022.03.012
157. El-Hussein A, Manoto SL, Ombinda-Lemboumba S, Alrowaili ZA, Mthunzi-Kufa P. A review of chemotherapy and photodynamic therapy for lung cancer treatment. Anti-Cancer Agents Med Chem. (2020) 21:149–61. doi: 10.2174/1871520620666200403144945
158. Estanqueiro M, Amaral MH, Conceição J, Sousa Lobo JM. Nanotechnological carriers for cancer chemotherapy: the state of the art. Colloids Surfaces B: Biointerfaces. (2015) 126:631–48. doi: 10.1016/j.colsurfb.2014.12.041
159. Xiao Q, Mai B, Nie Y, Yuan C, Xiang M, Shi Z, et al. In vitro and in vivo demonstration of ultraefficient and broad-spectrum antibacterial agents for photodynamic antibacterial chemotherapy. ACS Appl Mater Interfaces. (2021) 13:11588–96. doi: 10.1021/acsami.0c20837
160. Xie ZJ, Fan TJ, An J, Choi W, Duo YH, Ge YQ, et al. Emerging combination strategies with phototherapy in cancer nanomedicine. Chem Soc Rev. (2020) 49:8065–87. doi: 10.1039/d0cs00215a
161. Lin K, Ma Z, Li J, Tang M, Lindstrom A, Ramachandran M, et al. Single small molecule-assembled mitochondria targeting nanofibers for enhanced photodynamic cancer therapy in vivo. Adv Funct Mater. (2020) 31(10). doi: 10.1002/adfm.202008460
162. Janas K, Boniewska-Bernacka E, Dyrda G, Słota R. Porphyrin and phthalocyanine photosensitizers designed for targeted photodynamic therapy of colorectal cancer. Bioorganic Med Chem. (2021) 30. doi: 10.1016/j.bmc.2020.115926
163. Chen M, Liang X, Gao C, Zhao R, Zhang N, Wang S, et al. Ultrasound triggered conversion of porphyrin/camptothecin-fluoroxyuridine triad microbubbles into nanoparticles overcomes multidrug resistance in colorectal cancer. ACS Nano. (2018) 12:7312–26. doi: 10.1021/acsnano.8b03674
164. Allison RR, Downie GH, Cuenca R, Hu XH, Childs CJH, Sibata CH. Photosensitizers in clinical pdt. Photodiagnosis Photodyn Ther. (2004) 1:27–42. doi: 10.1016/s1572-1000(04)00007-9
165. Triesscheijn M, Baas P, Schellens JHM, Stewart FA. Photodynamic therapy in oncology. Oncologist. (2006) 11:1034–44. doi: 10.1634/theoncologist.11-9-1034
166. Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. (2002) 2:277–88. doi: 10.1038/nrc776
167. Oleinick NL, Morris RL, Belichenko T. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem Photobiol Sci. (2002) 1:1–21. doi: 10.1039/b108586g
168. Ji B, Wei MJ, Yang B. Recent advances in nanomedicines for photodynamic therapy (Pdt)-driven cancer immunotherapy. Theranostics. (2022) 12:434–58. doi: 10.7150/thno.67300
169. Sun XK, Sun J, Lv JK, Dong B, Liu M, Liu JS, et al. Ce6-C6-tpz co-loaded albumin nanoparticles for synergistic combined pdt-chemotherapy of cancer. J Mat Chem B. (2019) 7:5797–807. doi: 10.1039/c9tb01346f
170. Lupu M, Maillard P, Mispelter J, Poyer F, Thomas CD. A glycoporphyrin story: from chemistry to pdt treatment of cancer mouse models. Photochem Photobiol Sci. (2018) 17:1599–611. doi: 10.1039/c8pp00123e
171. Wang J, Yang B, Lv C, Chen T, Sun L, Sun L, et al. Amino porphyrin-peptide assemblies induce ribosome damage and cancer stem cell inhibition for an enhanced photodynamic therapy. Biomaterials. (2022) 289. doi: 10.1016/j.biomaterials.2022.121812
172. Lou L, Zhou S, Tan S, Xiang M, Wang W, Yuan C, et al. Amplifying the efficacy of ala-based prodrugs for photodynamic therapy using nanotechnology. Front Pharmacol. (2023) 14:1137707. doi: 10.3389/fphar.2023.1137707
173. Xiang M, Zhou Q, Shi Z, Wang X, Li M, Jia Y, et al. A review of light sources and enhanced targeting for photodynamic therapy. Curr Med Chem. (2021) 28:6437–57. doi: 10.2174/0929867328666210121122106
174. Xiao Q, Wu J, Pang X, Jiang Y, Wang P, Leung AW, et al. Discovery and development of natural products and their derivatives as photosensitizers for photodynamic therapy. Curr Med Chem. (2018) 25:839–60. doi: 10.2174/0929867324666170823143137
175. Shao W, Yang C, Li FY, Wu JH, Wang N, Ding Q, et al. Molecular design of conjugated small molecule nanoparticles for synergistically enhanced ptt/pdt. Nano-Micro Lett. (2020) 12:14. doi: 10.1007/s40820-020-00474-6
176. Liu B, Qiao G, Han Y, Shen E, Alfranca G, Tan H, et al. Targeted theranostics of lung cancer: pd-L1-guided delivery of gold nanoprisms with chlorin E6 for enhanced imaging and photothermal/photodynamic therapy. Acta Biomaterialia. (2020) 117:361–73. doi: 10.1016/j.actbio.2020.09.040
177. Li Q, Zhou YX, He WD, Ren XM, Zhang MZ, Jiang Y, et al. Platelet-armored nanoplatform to harmonize janus-faced ifn-Γ against tumor recurrence and metastasis. J Control Release. (2021) 338:33–45. doi: 10.1016/j.jconrel.2021.08.020
178. Zhu L, Zhong Y, Wu S, Yan M, Cao Y, Mou NL, et al. Cell membrane camouflaged biomimetic nanoparticles: focusing on tumor theranostics. Mater Today Bio. (2022) 14:16. doi: 10.1016/j.mtbio.2022.100228
179. Zhao M, Hao D, Wu Q, Li Y, Pei Q, Sun T, et al. Porphyrin cholesterol conjugates for enhanced photodynamic immunotherapy toward lung cancer. ACS Appl Mater Interfaces. (2023) 15:35927–38. doi: 10.1021/acsami.3c05825
180. Tang HL, Xue YA, Li BW, Xu XJ, Zhang F, Guo JJ, et al. Membrane-camouflaged supramolecular nanoparticles for co-delivery of chemotherapeutic and molecular-targeted drugs with sirna against patient-derived pancreatic carcinoma. Acta Pharm Sin B. (2022) 12:3410–26. doi: 10.1016/j.apsb.2022.02.007
181. Simone CB, Friedberg JS, Glatstein E, Stevenson JP, Sterman DH, Hahn SM, et al. Photodynamic therapy for the treatment of non-small cell lung cancer. J Thorac Dis. (2012) 4:63–75. doi: 10.3978/j.issn.2072-1439.2011.11.05
182. Ahmad R, Fu J, He NY, Li S. Advanced gold nanomaterials for photothermal therapy of cancer. J Nanosci Nanotechnol. (2016) 16:67–80. doi: 10.1166/jnn.2016.10770
183. Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun. (2018) 9:13. doi: 10.1038/s41467-018-03473-9
184. Oei AL, Vriend LEM, Crezee J, Franken NAP, Krawczyk PM. Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol. (2015) 10:13. doi: 10.1186/s13014-015-0462-0
185. Yata T, Takahashi Y, Tan MM, Nakatsuji H, Ohtsuki S, Murakami T, et al. DNA nanotechnology-based composite-type gold nanoparticle-immunostimulatory DNA hydrogel for tumor photothermal immunotherapy. Biomaterials. (2017) 146:136–45. doi: 10.1016/j.biomaterials.2017.09.014
186. Zhang F, Lu GH, Wen XL, Li F, Ji XY, Li QQ, et al. Magnetic nanoparticles coated with polyphenols for spatio-temporally controlled cancer photothermal/immunotherapy. J Control Release. (2020) 326:131–9. doi: 10.1016/j.jconrel.2020.06.015
187. Zhang XG, Tang JJ, Li C, Lu Y, Cheng LL, Liu J. A targeting black phosphorus nanoparticle based immune cells nano-regulator for photodynamic/photothermal and photo-immunotherapy. Bioact Mater. (2021) 6:472–89. doi: 10.1016/j.bioactmat.2020.08.024
188. Wang S, Zhang Q, Luo XF, Li J, He H, Yang F, et al. Magnetic graphene-based nanotheranostic agent for dual-modality mapping guided photothermal therapy in regional lymph nodal metastasis of pancreatic cancer. Biomaterials. (2014) 35:9473–83. doi: 10.1016/j.biomaterials.2014.07.064
189. Yang B, Zhang Y, Sun L, Wang J, Zhao Z, Huang Z, et al. Modulated ultrasmall Γ-fe2o3 nanocrystal assemblies for switchable magnetic resonance imaging and photothermal-ferroptotic-chemical synergistic cancer therapy. Adv Funct Mater. (2022) 33(5). doi: 10.1002/adfm.202211251
190. Li J, Zhang CT, Gong SM, Li XC, Yu MY, Qian CG, et al. A nanoscale photothermal agent based on a metal-organic coordination polymer as a drug-loading framework for effective combination therapy. Acta Biomaterialia. (2019) 94:435–46. doi: 10.1016/j.actbio.2019.06.014
191. Wang YF, Barhoumi A, Tong R, Wang WP, Ji TJ, Deng XR, et al. Batio3-core au-shell nanoparticles for photothermal therapy and bimodal imaging. Acta Biomaterialia. (2018) 72:287–94. doi: 10.1016/j.actbio.2018.03.029
192. Ye XY, Liang X, Chen Q, Miao QW, Chen XL, Zhang XD, et al. Surgical tumor-derived personalized photothermal vaccine formulation for cancer immunotherapy. ACS Nano. (2019) 13:2956–68. doi: 10.1021/acsnano.8b07371
193. Fernandes N, Rodrigues CF, Moreira AF, Correia IJ. Overview of the application of inorganic nanomaterials in cancer photothermal therapy. Biomater Sci. (2020) 8:2990–3020. doi: 10.1039/d0bm00222d
194. Agrawal S, Dwivedi M, Ahmad H, Chadchan SB, Arya A, Sikandar R, et al. Cd44 targeting hyaluronic acid coated lapatinib nanocrystals foster the efficacy against triple-negative breast cancer. Nanomed-Nanotechnol Biol Med. (2018) 14:327–37. doi: 10.1016/j.nano.2017.10.010
195. Huang ZG, Lv FM, Wang J, Ca SJ, Liu ZP, Liu Y, et al. Rgd-modified pegylated paclitaxel nanocrystals with enhanced stability and tumor-targeting capability. Int J Pharm. (2019) 556:217–25. doi: 10.1016/j.ijpharm.2018.12.023
196. Meng FF, Wang JP, Ping QN, Yeo Y. Camouflaging nanoparticles for ratiometric delivery of therapeutic combinations. Nano Lett. (2019) 19:1479–87. doi: 10.1021/acs.nanolett.8b04017
197. Kumar S, Mongia A, Gulati S, Singh P, Diwan A, Shukla S. Emerging theranostic gold nanostructures to combat cancer: novel probes for combinatorial immunotherapy and photothermal therapy. Cancer Treat Res Commun. (2020) 25:100258. doi: 10.1016/j.ctarc.2020.100258
198. Wu HS, Gu DH, Xia SJ, Chen FH, You CQ, Sun BW. One-for-all intelligent core-shell nanoparticles for tumor-specific photothermal-chemodynamic synergistic therapy. Biomater Sci. (2021) 9:1020–33. doi: 10.1039/d0bm01734e
199. Chang Z, Niu T, Shao Q, Yue J, Zhang H, Tong L, et al. Pt–se-bonded nanoprobe for high-fidelity detection of non-small cell lung cancer and enhancement of nir ii photothermal therapy. Anal Chem. (2023) 95:18426–35. doi: 10.1021/acs.analchem.3c03511
200. Höckel M, Vaupel P. Biological consequences of tumor hypoxia. Semin Oncol. (2001) 28:36–41. doi: 10.1016/s0093-7754(01)90211-8
201. Park SY, Jang WJ, Yi EY, Jang JY, Jung Y, Jeong JW, et al. Melatonin suppresses tumor angiogenesis by inhibiting hif-1α Stabilization under hypoxia. J Pineal Res. (2010) 48:178–84. doi: 10.1111/j.1600-079X.2009.00742.x
202. Semenza GL. Hif-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. (2000) 88:1474–80. doi: 10.1152/jappl.2000.88.4.1474
203. Tang N, Wang LC, Esko J, Giordano FJ, Huang Y, Gerber HP, et al. Loss of hif-1α in endothelial cells disrupts a hypoxia-driven vegf autocrine loop necessary for tumorigenesis. Cancer Cell. (2004) 6:485–95. doi: 10.1016/j.ccr.2004.09.026
204. Nordgren IK, Tavassoli A. Targeting tumour angiogenesis with small molecule inhibitors of hypoxia inducible factor. Chem Soc Rev. (2011) 40:4307–17. doi: 10.1039/c1cs15032d
205. Yang QC, Zeng BF, Shi ZM, Dong Y, Jiang ZM, Huang J, et al. Inhibition of hypoxia-induced angiogenesis by trichostatin a via suppression of hif-1a activity in human osteosarcoma. J Exp Clin Cancer Res. (2006) 25(4):593–9.
206. Chen Y-Z, Wang ZU, Wang H, Lu J, Yu S-H, Jiang H-L. Singlet oxygen-engaged selective photo-oxidation over pt nanocrystals/porphyrinic mof: the roles of photothermal effect and pt electronic state. J Am Chem Soc. (2017) 139:2035–44. doi: 10.1021/jacs.6b12074
207. Jung N, Sohn Y, Park JH, Nahm KS, Kim P, Yoo SJ. High-performance ptcux@Pt core-shell nanoparticles decorated with nanoporous pt surfaces for oxygen reduction reaction. Appl Catalysis B: Environ. (2016) 196:199–206. doi: 10.1016/j.apcatb.2016.05.028
208. Xiang Z-P, Tan A-D, Fu Z-Y, Piao J-H, Liang Z-X. Oxygen reduction reaction on single pt nanoparticle. J Energy Chem. (2020) 49:323–6. doi: 10.1016/j.jechem.2020.02.051
209. Yu J, Liu S, Wang Y, He X, Zhang Q, Qi Y, et al. Synergistic enhancement of immunological responses triggered by hyperthermia sensitive pt nps via nir laser to inhibit cancer relapse and metastasis. Bioact Mater. (2022) 7:389–400. doi: 10.1016/j.bioactmat.2021.05.030
210. Chang MY, Hou ZY, Wang M, Li CX, Lin J. Recent advances in hyperthermia therapy-based synergistic immunotherapy. Adv Mater. (2021) 33:29. doi: 10.1002/adma.202004788
211. Chang MY, Hou ZY, Wang M, Wang MF, Dang PP, Liu JH, et al. Cu2Mos4/au heterostructures with enhanced catalase-like activity and photoconversion efficiency for primary/metastatic tumors eradication by phototherapy-induced immunotherapy. Small. (2020) 16:14. doi: 10.1002/smll.201907146
212. Kepp O, Marabelle A, Zitvogel L, Kroemer G. Oncolysis without viruses - inducing systemic anticancer immune responses with local therapies. Nat Rev Clin Oncol. (2020) 17:49–64. doi: 10.1038/s41571-019-0272-7
213. Rong L, Zhang Y, Li WS, Su ZG, Fadhil JI, Zhang C. Iron chelated melanin-like nanoparticles for tumor-associated macrophage repolarization and cancer therapy. Biomaterials. (2019) 225:9. doi: 10.1016/j.biomaterials.2019.119515
214. Mao WD, Guo ZY, Dai YC, Nie J, Li BY, Pei HL, et al. Lnc crybg3 inhibits tumor growth by inducing M phase arrest. J Cancer. (2019) 10:2764–70. doi: 10.7150/jca.31703
215. Pei HL, Hu WT, Guo ZY, Chen HY, Ma J, Mao WD, et al. Long noncoding rna crybg3 blocks cytokinesis by directly binding G-actin. Cancer Res. (2018) 78:4563–72. doi: 10.1158/0008-5472.Can-18-0988
216. Huang H, Yuan G, Xu Y, Gao Y, Mao Q, Zhang Y, et al. Photoacoustic and magnetic resonance imaging-based gene and photothermal therapy using mesoporous nanoagents. Bioact Mater. (2022) 9:157–67. doi: 10.1016/j.bioactmat.2021.07.025
217. Deng H, Zhou Z, Yang W, L-s L, Wang S, Niu G, et al. Endoplasmic reticulum targeting to amplify immunogenic cell death for cancer immunotherapy. Nano Lett. (2020) 20:1928–33. doi: 10.1021/acs.nanolett.9b05210
218. Yang W, Zhang F, Deng H, Lin L, Wang S, Kang F, et al. Smart nanovesicle-mediated immunogenic cell death through tumor microenvironment modulation for effective photodynamic immunotherapy. ACS Nano. (2019) 14:620–31. doi: 10.1021/acsnano.9b07212
219. Yang W, Zhu G, Wang S, Yu G, Yang Z, Lin L, et al. In situ dendritic cell vaccine for effective cancer immunotherapy. ACS Nano. (2019) 13:3083–94. doi: 10.1021/acsnano.8b08346
220. Zou J, Li L, Yang Z, Chen X. Phototherapy meets immunotherapy: A win–win strategy to fight against cancer. Nanophotonics. (2021) 10:3229–45. doi: 10.1515/nanoph-2021-0209
221. Kim MM, Darafsheh A. Light sources and dosimetry techniques for photodynamic therapy. Photochem Photobiol. (2020) 96:280–94. doi: 10.1111/php.13219
222. Damasco JA, Ravi S, Perez JD, Hagaman DE, Melancon MP. Understanding nanoparticle toxicity to direct a safe-by-design approach in cancer nanomedicine. Nanomaterials. (2020) 10(11). doi: 10.3390/nano10112186
223. Overchuk M, Weersink RA, Wilson BC, Zheng G. Photodynamic and photothermal therapies: synergy opportunities for nanomedicine. ACS Nano. (2023) 17:7979–8003. doi: 10.1021/acsnano.3c00891
224. Jiang YF, Liu Y, Fang SY, Ji M. Gold nanoshells coated 5-aminolevulinic liposomes for photothermal-photodynamic antitumor therapy. J Nanosci Nanotechnol. (2020) 20:1–14. doi: 10.1166/jnn.2020.168860
225. Gao G, Jiang YW, Guo Y, Jia HR, Cheng X, Deng Y, et al. Enzyme-mediated tumor starvation and phototherapy enhance mild-temperature photothermal therapy. Adv Funct Mater. (2020) 30(16). doi: 10.1002/adfm.201909391
226. Lu IL, Liu T-I, Lin H-C, Chang S-H, Lo C-L, Chiang W-H, et al. Ir780-loaded zwitterionic polymeric nanoparticles with acidity-induced agglomeration for enhanced tumor retention. Eur Polymer J. (2020) 122. doi: 10.1016/j.eurpolymj.2019.109400
227. Ha J-H, Kim Y-J. Photodynamic and cold atmospheric plasma combination therapy using polymeric nanoparticles for the synergistic treatment of cervical cancer. Int J Mol Sci. (2021) 22(3). doi: 10.3390/ijms22031172
228. Tong H, Wang Y, Li H, Jin Q, Ji J. Dual ph-responsive 5-aminolevulinic acid pseudopolyrotaxane prodrug micelles for enhanced photodynamic therapy. Chem Commun. (2016) 52:3966–9. doi: 10.1039/c6cc00450d
229. Peng J, Xiao Y, Li W, Yang Q, Tan L, Jia Y, et al. Photosensitizer micelles together with ido inhibitor enhance cancer photothermal therapy and immunotherapy. Adv Sci. (2018) 5(5). doi: 10.1002/advs.201700891
230. Xiao Q, Lin H, Wu J, Pang X, Zhou Q, Jiang Y, et al. Pyridine-embedded phenothiazinium dyes as lysosome-targeted photosensitizers for highly efficient photodynamic antitumor therapy. J Med Chem. (2020) 63:4896–907. doi: 10.1021/acs.jmedchem.0c00280
231. Chen C, Zhang W, Ouyang Y, Tan Y, Zhao S, Chen Y, et al. Hemoglobin-decorated boron-carbon nanosheets with catalytic ability and near-infrared ii light response for tumor photothermal-chemodynamic therapy. ACS Appl Nano Mater. (2023) 6:7572–81. doi: 10.1021/acsanm.3c00726
232. Srinivasulu YG, Mozhi A, Goswami N, Yao Q, Xie J. Traceable nanocluster–prodrug conjugate for chemo-photodynamic combinatorial therapy of non-small cell lung cancer. ACS Appl Bio Mater. (2021) 4:3232–45. doi: 10.1021/acsabm.0c01611
233. Park S, Kim H, Lim SC, Lim K, Lee ES, Oh KT, et al. Gold nanocluster-loaded hybrid albumin nanoparticles with fluorescence-based optical visualization and photothermal conversion for tumor detection/ablation. J Control Release. (2019) 304:7–18. doi: 10.1016/j.jconrel.2019.04.036
234. Bharathiraja S, Moorthy MS, Manivasagan P, Seo H, Lee KD, Oh J. Chlorin E6 conjugated silica nanoparticles for targeted and effective photodynamic therapy. Photodiagnosis Photodyn Ther. (2017) 19:212–20. doi: 10.1016/j.pdpdt.2017.06.001
235. Peng J, Zhao L, Zhu X, Sun Y, Feng W, Gao Y, et al. Hollow silica nanoparticles loaded with hydrophobic phthalocyanine for near-infrared photodynamic and photothermal combination therapy. Biomaterials. (2013) 34:7905–12. doi: 10.1016/j.biomaterials.2013.07.027
236. Wan X, Zhong H, Pan W, Li Y, Chen Y, Li N, et al. Programmed release of dihydroartemisinin for synergistic cancer therapy using a caco3 mineralized metal–organic framework. Angewandte Chemie Int Edition. (2019) 58:14134–9. doi: 10.1002/anie.201907388
237. Chen Z, Sun Y, Wang J, Zhou X, Kong X, Meng J, et al. Dual-responsive triple-synergistic fe-mof for tumor theranostics. ACS Nano. (2023) 17:9003–13. doi: 10.1021/acsnano.2c10310
Keywords: non-small cell lung cancer (NSCLC), photodynamic therapy (PDT), Kras mutants and targeted drugs, mammalian target of rapamycin (mTOR) signaling, ribosome, immunotherapy
Citation: Si Q, Bai M, Wang X, Wang T and Qin Y (2024) Photonanozyme–Kras–ribosome combination treatment of non-small cell lung cancer after COVID-19. Front. Immunol. 15:1420463. doi: 10.3389/fimmu.2024.1420463
Received: 20 April 2024; Accepted: 14 August 2024;
Published: 06 September 2024.
Edited by:
Romina Vuono, University of Kent, United KingdomReviewed by:
Dalin Zhang, Stanford University, United StatesQicai Xiao, Sun Yat-sen University, China
Vadim V. Sumbayev, University of Kent, United Kingdom
Copyright © 2024 Si, Bai, Wang, Wang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Qin, cWlueUBpYnAuYWMuY24=; Tianyu Wang, dHdhbmdAdXN0Yi5lZHUuY24=; Xiaolong Wang, czF4aWFvY2hvbmdAMTI2LmNvbQ==